

Abstract
The view on antimicrobials has dramatically changed due to the increased knowledge on the importance of microbiota composition in different body parts. Antimicrobials can no longer be considered only beneficial, but also potentially deleterious for favourable bacterial populations. Still, the use of metaphylactic antimicrobial treatment at early stages of life is a practice in use in porcine production. Many reports have shown that antibiotics can critically affect the gut microbiota, however the effect of perinatal antimicrobial treatment on the nasal microbiota has not been explored yet. To gain insights on the potential changes in nasal microbial composition due to antimicrobial treatments, piglets from two different farms were sampled at weaning. The nasal microbiota was analysed when antimicrobial treatment was used early in life, and later, when no antimicrobial treatment was used during the lactation period. Removal of perinatal antimicrobials resulted in an increased bacterial diversity in nasal microbiota at weaning. Concurrently, elimination of antimicrobials produced an increase in the relative abundance of Prevotella and Lactobacillus, and a decrease in Moraxella and Bergeyella. These changes in microbiota composition were accompanied by an improvement of the piglets’ health and a higher productivity in the nursery phase.
Subject terms: Microbial communities, Classification and taxonomy
Introduction
The use of antimicrobials to control bacterial diseases is a common practice in porcine production. Despite the undeniable fact that usage of antimicrobials plays a vital role in the production of food-animals and in protection of public health1, they have proved to negatively affect the beneficial microbiota2. The microbiota is a key factor for the well function and homeostasis of different body systems in animals. Key functions of the microbiota include the correct development of the immune system and resistance against pathogens colonization or pathogenicity reduction3–5. The interest on animal microbiota has increased in recent years due to its impact in health. Knowledge on microbiota composition can result in the identification of potential bacterial groups associated with health5–7, although many factors can interfere in the establishment of a so-called adequate microbiota, such as environment, pig production system, pig genetics and antimicrobial treatments8. Microbial dysbiosis, which may appear secondarily to antimicrobial use, can facilitate pathogen infections and enhance the tissue damage inflicted by pathogenic bacteria9. The use of metaphylactic antimicrobials, especially early in life, can have a deleterious impact in animal health through the alteration of the gastrointestinal tract (GIT) microbiota composition10. Many efforts have been made to elucidate the GIT microbiota composition and the relationship of dysbiosis with many diseases, however, there is scarce knowledge on the nasal microbiota composition in animals. In pigs, it has been demonstrated that the microbial communities inhabiting the nasal cavities at weaning may influence the development of Glässer’s disease later in life6 and perinatal antimicrobials are sometimes used to reduce the risk of this disease. However, whether the antibiotic treatment early in life has an effect on the bacterial communities from the piglet’s nose, has not been explored yet. Here, we analysed the nasal microbiota composition of 3–4 week old piglets at weaning regarding antimicrobial administration in the lactation phase, with the aim of detecting the effect on the microbiota compositions and the association with health status later in life.
Results
Diversity and species richness from the nasal microbiota
With the aim of analysing the effect of perinatal antibiotic treatment on the nasal microbiota composition, 3–4 week old piglets from two farms (MT and MC farms) from Spain were sampled. Nasal swabs were taken when both farms were using perinatal antimicrobial treatments (MC1 and MT1), and total DNA was extracted and subjected to individual massive sequencing. The same sampling was repeated one productive cycle after elimination of perinatal antibiotics in both farms (MC2 and MT2). We obtained a total of 8,883,783 joint reads after filtering for both farms (MC1: 876,010; MC2: 3,178,022; MT1: 2,533,426; MT2: 2,296,325). The OTUs were identified in the samples that passed the quality-based filters (MT1, n = 7; MT2, n = 8; MC1, n = 8; MC2, n = 11) through clustering sequences at 97% sequence homology. At phylum level, 98.2% of the sequences were assigned to 7 phyla, while 93% of the sequences were assigned at family level, to 41 families. At genus level, 75.5% of the reads were assigned to 68 genera. The relative abundance of the OTUs found at genus level in the nasal microbiota is represented in Fig. 1 (for the full list of OTUs assigned at genera level, please refer to Additional File 1; see also Additional File 2 for other taxonomical levels).
Figure 1.

Nasal microbiota of piglets from farms MT and MC before (1) and after (2) antimicrobial treatment removal. Microbiota rarefaction curve generated using Shannon diversity estimator with samples from farms MT (A) and MC (B). Samples have been rarefied at an even depth of 40,000 sequences per sample. Error bars indicate the 95% confidence intervals. The mean relative abundance (%) of OTUs found in nasal swabs of piglets from farms MT (C) and MC (D) is presented at genera level.
One cycle after elimination of perinatal antimicrobials, nasal microbiota of all the weaning piglets analysed showed a significant increase in bacterial diversity, as indicated by the analysis of alpha diversity through Shannon diversity index (P = 0.002). When farms MC and MT were analysed individually (Fig. 1), this increase in alpha diversity was also observed, showing to be statistically significant through non-parametric two-sample t-test (999 permutations, P ≤ 0.05) only in farm MT (Fig. 1A,B). The mean of observed OTUs richness was also measured in rarefied samples at maximum depth (40,000) at the two sampling times in both farms, with means values of 1,465 (MC1) and 2,237 (MC2) OTUs for MC farm, and 1,221 (MT1) and 4,619 (MT2) OTUs for MT farm. The most predominant bacterial genera across all the samples at first sampling time was Moraxella for both farms (34.22 ± 6.30% for MT1 and 34.02 ± 5.79% for MC1, Fig. 1C,D) followed by Bergeyella (23.27 ± 3.6% for MT1 and 8.21 ± 0.7% for MC1, Fig. 1C,D), while the most abundant genera after antimicrobial elimination in both farms changed to Prevotella (14.06 ± 4.46% for MT2 and 16.81 ± 3.26% for MC2).
Analysis of the beta diversity both before and after elimination of antimicrobials, showed remarkable differences in the bacterial population of the nasal microbiota (Fig. 2). Antimicrobial treatment explained 22.63% of the differences observed (P = 0.001). The five most abundant genera explaining the divergence between the nasal microbiota before and after elimination of perinatal antimicrobials were: Prevotella, Moraxella, Bergeyella, Haemophilus and Mycoplasma (Fig. 2). When the beta diversity was analysed in each farm individually, significant differences were observed after elimination of the antimicrobial treatment in both farms (adonis, R2 = 0.2186, P = 0.001 for MC and R2 = 0.1935, P = 0.005 for MT). Elimination of antimicrobials caused significant changes in the relative abundance of several genera. Among others, Lactobacillus, Phascolarctobacterium, Megasphaera, Roseburia and Campylobacter were increased in both farms. The mean of the relative abundance of the Prevotella genus increased also after elimination of the antimicrobials, but this increase was only statistically significant for MC farm (Table 1). On the contrary, the relative abundance of Moraxella and Bergeyella significantly decreased, while Haemophilus and Mycoplasma showed a decreased tendency when antimicrobials were removed in farm MT, but increased in farm MC (Table 1). Finally, the relative abundance of Neisseria decreased after removal of antimicrobials, showing to be significant in MC farm (Table 1).
Figure 2.
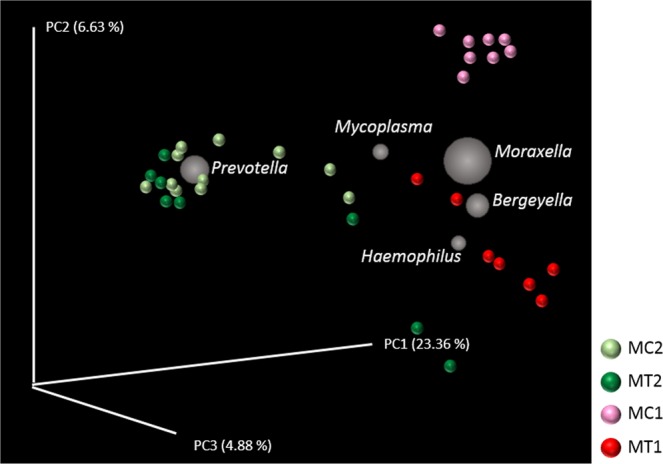
Principal Component Plots (jackknifed) representing beta diversity on rarefied samples. Beta diversity of nasal samples of piglets was computed through unweighted UniFrac analysis for both farms, MT and MC, in two different sampling times, before (1) and after (2) elimination of perinatal antibiotic treatment. The biplot shows grey spheres corresponding to the five most abundant genera visualized in the PCA space. The size of the spheres is proportional to their relative abundance across all the samples.
Table 1.
Relative abundance of OTUs before and after elimination of antibiotic treatment in two different farms (>1% relative abundance).
| OTU | Relative abundance (%) | |||||
|---|---|---|---|---|---|---|
| MT1 | MT2 | P value | MC1 | MC2 | P value | |
| Acinetobacter | 1.090 | 0.835 | 0.132 | <1 | <1 | NA |
| Actinobacillus | 0.837 | 1.123 | 0.728 | <1 | <1 | NA |
| Afipia | <1 | <1 | NA | 0.000 | 1.089 | 0.001* |
| Alistipes | <1 | <1 | NA | 1.242 | 0.006 | <0.001* |
| Alloprevotella | 0.048 | 1.902 | 0.064 | 0.444 | 1.635 | 0.021* |
| Anaerovibrio | 0.011 | 1.009 | 0.037* | 0.048 | 1.094 | 0.010* |
| Bacteroides | <1 | <1 | NA | 1.694 | 0.366 | <0.001* |
| Barnesiella | 0.104 | 2.001 | 0.049* | 1.172 | 2.562 | 0.160 |
| Bergeyella | 23.275 | 4.370 | 0.003* | 8.215 | 0.009 | <0.001* |
| Bosea | <1 | <1 | NA | 0 | 1.153 | <0.001* |
| Campylobacter | 0.007 | 1.883 | 0.021* | 0.030 | 2.882 | 0.001* |
| Clostridium sensu stricto | <1 | <1 | NA | 1.207 | 0.736 | 0.039* |
| Clostridium IV | <1 | <1 | NA | 2.473 | 1.283 | 0.017* |
| Clostridium XI | <1 | <1 | NA | 1.341 | 0.723 | 0.017* |
| Clostridium XlVa | <1 | <1 | NA | 2.416 | 1.195 | 0.017* |
| Escherichia/Shigella | 0.100 | 1.174 | 0.418 | 0.528 | 2.212 | 0.058* |
| Faecalibacterium | 0.026 | 1.411 | 0.011* | 0.098 | 1.633 | 0.001* |
| Gemmiger | 0.019 | 1.186 | 0.015* | <1 | <1 | NA |
| Haemophilus | 13.896 | 2.927 | 0.015* | 0.905 | 2.195 | 0.026* |
| Klebsiella | <1 | <1 | NA | 8.810 | 0.011 | <0.001* |
| Lachnospiracea_incerta_sedis | 0.068 | 1.233 | 0.132 | 0.328 | 1.089 | 0.117 |
| Lactobacillus | 0.328 | 3.453 | 0.004* | 1.400 | 4.406 | 0.032* |
| Megasphaera | 0.010 | 2.164 | 0.008* | 0.033 | 4.971 | 0.001* |
| Moraxella | 34.220 | 9.185 | 0.015* | 34.024 | 3.189 | 0.001* |
| Mycoplasma | 3.051 | 0.577 | 0.011* | 6.718 | 10.324 | 0.509 |
| Neisseria | 5.09 | 4.26 | 0.780 | 1.00 | 0.02 | 0.003* |
| Oscillibacter | 0.119 | 1.426 | 0.165 | 1.586 | 1.483 | 0.680 |
| Paraprevotella | 0.010 | 1.067 | 0.028* | 0.039 | 1.131 | <0.001* |
| Phascolarctobacterium | 0.043 | 3.013 | 0.004* | 0.106 | 4.072 | <0.001* |
| Prevotella | 0.364 | 14.066 | 0.083 | 1.319 | 16.805 | 0.002* |
| Psychrobacter | 0.276 | 5.518 | 0.563 | <1 | <1 | NA |
| Roseburia | 0.020 | 2.466 | 0.005* | 0.080 | 1.972 | 0.001* |
| Rothia | 0.558 | 1.406 | 0.817 | <1 | <1 | NA |
| Selenomonas | 0.007 | 1.113 | 0.028* | <1 | <1 | NA |
| Staphylococcus | 1.493 | 0.029 | 0.002* | <1 | <1 | NA |
| Streptococcus | 1.584 | 1.028 | 0.418 | 1.002 | 0.982 | 0.457 |
| Succinivibrio | 0.012 | 1.058 | 0.037* | 0.083 | 1.706 | 0.137 |
| Treponema | 0.047 | 2.666 | 0.105 | 0.083 | 3.554 | <0.001* |
*Significantly different.
NA, not available. When the relative abundance was <1% in both sampling times, the P value was not estimated. The OTU was included for comparison with the other farm that showed higher abundance (>1%).
Stability of microbiota composition after antimicrobial elimination
In order to evaluate the stability of these changes in the nasal microbiota composition, a third sampling was performed in farm MT (MT3). In this run, we obtained a total of 4,003,077 joint reads after filtering (MT3, n = 12). Analysis of alpha diversity showed no further increase in diversity after additional time without perinatal antimicrobials in the farm (Fig. 3A, for a full list of OTUs assigned at genera level, please see Additional File 3). However, the samples after 5 cycles without antimicrobials showed a more similar composition demonstrated with the lower P value obtained through the comparison (Fig. 3A; Shannon index, P = 0.003). Besides, the beta diversity analysis, showed a distinct clustering at the three sampling times tested (Fig. 3B, adonis R2 = 0.15643, P < 0.001). Sixteen genera were involved in the long-term changes observed after elimination of the antimicrobials (Table 2 and Fig. 3C). Nine of these genera were increased at the third sampling point after 5 productive cycles without antimicrobials, such as Faecaliabacterium and Bacteroides.
Figure 3.

Stability of the nasal microbiota of piglets from farm MT through three different sampling time-points. (A) Alpha diversity using Shannon diversity index over rarefied samples is compared before perinatal antibiotic elimination (MT1) and after 1 (MT2) or 5 productive cycles (MT3). (B) Beta diversity analysis (jackknified principal component analysis) of the nasal microbiota composition among samples collected before (MT1) and after 1 (MT2) or 5 productive cycles (MT3) antimicrobial treatment removal. The two principal axis are shown with the percentage of variation explained between brackets. (C) The mean relative abundance (%) at genera level for farm MT after 5 productive cycles without using perinatal antibiotic treatment. Color-coding for each representative OTU are the same that in Fig. 1.
Table 2.
Relative abundance of OTUs at third sampling time in MT farm compared with the previous samplings.
| OTU | Relative abundance MT3 (%) | MT1 vs MT3 P value | MT2 vs MT3 P value |
|---|---|---|---|
| Acinetobacter | 0.013# | 0.002* | NA |
| Actinobacillus | 0.521# | NA | 0.482 |
| Alistipes | 2.043 | 0.002* | <0.001* |
| Alloprevotella | 0.222# | NA | 0.354 |
| Anaerovibrio | 0.044# | NA | 0.165 |
| Bacteroides | 15.104 | 0.002* | <0.001* |
| Barnesiella | 0.895# | NA | 0.354 |
| Bergeyella | 15.109 | 0.09 | 0.011* |
| Campylobacter | 0.023# | NA | 0.105 |
| Clostridium sensu stricto | 1.452 | 0.527 | 0.217 |
| Clostridium XlVa | 2.064 | 0.002* | 0.007* |
| Escherichia/Shigella | 0.431# | NA | 0.938 |
| Faecalibacterium | 2.903 | 0.002* | 0.017* |
| Gemmiger | 0.479# | NA | 0.354 |
| Haemophilus | 6.954 | 0.343 | 0.031* |
| Lachnospiracea_incertae_sedis | 1.665 | 0.002* | 0.217 |
| Lactobacillus | 1.751 | 0.008* | 0.076 |
| Megasphaera | 0.130# | NA | 0.076 |
| Moraxella | 12.308 | 0.015* | 0.189 |
| Mycoplasma | 0.086# | 0.002* | NA |
| Oscillibacter | 1.067 | 0.002* | 0.487 |
| Parabacteroides | 1.386 | 0.002* | <0.001* |
| Paraprevotella | 0.301# | NA | 0.354 |
| Phascolarctobacterium | 0.733# | NA | 0.354 |
| Prevotella | 4.347 | 0.002* | 0.354 |
| Psychrobacter | 0.027# | NA | 0.537 |
| Roseburia | 1.181 | 0.002* | 0.938 |
| Rothia | 1.838 | 0.002* | 0.105 |
| Selenomonas | 0.009# | NA | 0.013* |
| Staphylococcus | 0.037# | 0.002* | NA |
| Streptococcus | 1.023 | 0.035* | 0.354 |
| Succinivibrio | 0.029# | NA | 0.105 |
| Treponema | 0.074# | NA | 0.142 |
*Significantly different.
#The OTU is included in the list although having relative abundance less than 1% for comparison with previous samplings.
NA, not available. When OTUs appeared at less than 1% of relative abundance in both sampling times, the P value was not estimated.
Health and production parameters in the nursery phase
Concomitantly to the increase in microbiota diversity, the MT farm experienced an improvement of health and productivity. Analysis of production parameters in the nursery phase from 2015 to 2017, indicated that the elimination of antimicrobial treatments resulted in a significant reduction in medication cost by pig and mortality rate, while the reduction observed in feed conversion ratio (FCR) was not statistically significant (P = 0.110) (Table 3). In farm MC, the FCR (P = 0.261) did not show a clear improvement, but the mortality rate (P = 0.130) and especially the cost in medication per pig (P = 0.05) decreased during the study period.
Table 3.
Production data collected from the nursery phases of MT and MC farms in three consecutive years.
| MT | P value* | MC | P value* | |||||
|---|---|---|---|---|---|---|---|---|
| 2015 | 2016 | 2017 | 2015 | 2016 | 2017 | |||
| Medication cost (€/pig) | 0.870 | 0.543 | 0.515 | 0.037 | 0.843 | 0.478 | 0.390 | 0.050 |
| Mortality rate (%) | 5.46 | 3.24 | 3.19 | 0.050 | 2.83 | 3.05 | 2.58 | 0.130 |
| Feed conversion ratio | 1.659 | 1.487 | 1.477 | 0.110 | 1.675 | 1.471 | 1.559 | 0.261 |
*P values were estimated through Krukal-Wallis non-parametric test.
Discussion
The effect of perinatal antimicrobial treatments in the gut microbiota of piglets has been already documented11,12, but less information is available on the effect of these treatments on the respiratory tract microbiota. In our study, elimination of perinatal antimicrobials resulted in an increase in nasal microbiota diversity, which was accompanied by a general health improvement in the piglets. This observation is in agreement with previous reports showing an association between health and microbiota diversity10,13. Although not addressed here, another additional benefit expected after elimination of metaphylactic antimicrobial treatments, would be the reduction in the abundance of resistance genes, as reported in the pig GIT microbiome10. Worth to mention is that a more similar microbiota composition among the individuals was found in the piglets after 5 cycles without using antimicrobials, indicating a developed stability in the microbiota composition. On the contrary, dissimilarity in microbiota composition within groups has been considered a measurement of gut dysbiosis associated with poor health status14. Importantly, a lower relative abundance of several potentially pathogenic bacteria in the nasal microbiota was also demonstrated, while beneficial bacteria were more abundant in both farms when no perinatal antimicrobial treatment was applied. Our results agree with those reported by Janczyk et al. (2007) for the impact of amoxicillin on the intestinal microbiota of piglets12. Higher relative abundance of potential pathogens and a reduction of beneficial bacteria, such as Lactobacillus, was observed when long-lasting amoxicillin was administered to piglets, although no direct connection with health status was reported12. Recently, the effect of antimicrobial administration was evaluated in older pigs (8 week old) under experimental conditions, where no differences were found on bacterial diversity15. Apparent incongruences with that study could be explained considering the age of the animals, as it was reported that the nasal microbiota reaches stability 2–3 weeks after weaning16. Thus, the effect of antimicrobial treatment is not expected to be the same in adult animals with an established nasal microbiota, than in new-born piglets where the microbiota composition is still developing and can be more easily influenced.
Under farm conditions, the use of perinatal antimicrobials has been reported to be detrimental to the health of piglets, which may be linked to the higher abundance of potential pathogens17. Although we cannot rule out that a reduced diversity caused a poorer development of the immune system11, the healthier condition observed in the present study can be associated to the decreased colonization of the upper respiratory tract by potential pathogens, which may be farm specific in some cases (e.g., Mycoplasma or Haemophilus). Nasal colonization of piglets by Mycoplasma hyopneumoniae at weaning has been associated to a higher risk of disease development in growing pigs18. Additionally, M. hyopneumoniae plays a role in the porcine respiratory disease complex and has been associated to increase disease severity19. Some potential pathogens, such as Haemophilus parasuis, are highly heterogeneous, and while the presence of virulent strains in the nasal cavity of piglets can be considered a risk factor for disease development, the presence of non-virulent strains could confer protection against disease20. In other cases, the role of a genus in disease is less clear. The genus Moraxella is highly abundant in the nasal microbiota of healthy pigs6, but some Moraxella species have been isolated from systemic lesions, indicating that they have pathogenic potential21–23. Although there is no available confirmation that Bergeyella can be pathogenic to pigs, some species has been associated to human septicaemia and endocarditis24. Experimental support to the putative pathogenicity of Bergeyella in pigs has been obtained in in vitro studies with nasal isolates from this bacterial genus25. The reduction in the abundance of pathogens reported may be the result of direct competition of the microbiota against pathogen colonization or indirectly by an improved maturation of the immune system11. Either way, the resulting reduction in pathogen load would most probably result in an improved-health status. On the other hand, the relative abundance of some potential pathogens was increased, as observed for Campylobacter and Treponema. However, the role of these genera in disease needs deeper analysis at species and strain level to proper assess their virulence potential. For instance, Campylobacter comprises a high diversity of strains, which are becoming apparent by studying Campylobacter populations in different animal species26.
The balance in the composition of the microbiota community needs to be also considered. Thus, while the abundance of some pathogenic bacterial genera decreased, the opposite was observed for some of the genera linked to beneficial effects on animal health. Besides the well-known probiotic genus Lactobacillus, other potential beneficial genera were detected in this study, such as Prevotella and Bacteroides. Prevotella has been reported previously as a beneficial and/or protective bacteria or at least an important component of healthy microbiota of animals and humans27,28. Although some Bacteroides have been considered opportunist pathogens29–31, at least one strain within the species Bacteroides fragilis has been identified as a potential probiotic for human health32. In general, an in-depth analysis of the specific species and strains is needed to define the specific role of each taxon within each genus and its potential role in animal health.
The present study confirms the benefits of the elimination of perinatal antimicrobial usage in the health of piglets, through modulation of the nasal microbiota composition. Based on the long-term results presented herein, antimicrobial treatments should be carefully applied especially at early stages in life, when the cross-talk with commensal microorganisms will determine the microbiota establishment throughout life.
Material and Methods
Farms and sampling
Nasal swabs from 3–4 week old piglets were collected at different time points, from two different farms (MT and MC) located in the area of Catalonia, Spain. First sampling was done on May-June 2015 when both farms were using perinatal antimicrobial treatments, labelled as MT1 and MC1, for MT and MC farms respectively. MT farm used penicillin and streptomycin at 3 days after birth and tulatromycin one week later; while MC farm used ceftiofur at 3 days and tulatromycin one week later. A second sampling was done after one farrowing interval without usage of any perinatal antimicrobial treatment, in October-November 2015, labelled as MT2 and MC2. In MT farm, a third sampling was performed in March 2017 (labelled as MT3). The MT farm was a 3300-sows multi-site pig production system and MC farm was a 480-sows farrow-to-finish production system. MC farm was negative to porcine reproductive and respiratory syndrome virus (PRRSV) while MT was PRRS-positive, although no circulation of the virus was diagnosed throughout the study. All the animals were vaccinated against porcine circovirus type 2 (PCV2) and Mycoplasma hyopneumoniae. During the study period no other management practices were modified in any farm, except for the antibiotic treatment elimination. Production data were collected during the period of the study. Collection of nasal swabs (sterile with aluminium handle) was done one day prior weaning from both nostrils (5–6 cm depth) using the same swab. The swabs were kept refrigerated until arrival within 24 hours to the laboratory, where they were stored in PBS (0.5 mL) at −20 °C.
Sampling of piglets was done under institutional authorization and followed good veterinary practices. According to European (Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes) and Spanish (Real Decreto 53/2013) normative, this procedure did not require specific approval by an Ethical Committee. Nasal sampling was performed only once to each piglet and is not likely to cause pain, suffering, distress or lasting harm equivalent to, or higher than, that caused by the introduction of a needle in accordance with good veterinary practice (Chapter I, Article 1, 5 (f) of 2010/63/EU).
DNA extraction and sequencing
Total genomic DNA was extracted using Machinery Nigel Kit (GmbH & Co, Düren; Germany), following manufacter’s instructions and resuspended in 50 µl of elution buffer. Quality and quantity was evaluated on a BioDrop DUO (BioDrop Ltd, Cambridge. UK). The library preparation for sequencing was performed within 24 h after the DNA extraction at Servei de Genòmica, Universitat Autònoma de Barcelona. Sequencing of the V3-V4 region of 16S rDNA gene was done with Illumina MiSeq pair-end 2 × 250 bp technology following the manufacturer instructions (MS-102–2003 MiSeq® Reagent Kit v2, 500 cycle). The region targeted to perform the 16S amplification was the one spanning the V3 and V4 region of 16S rRNA gene selected from Klindworth et al.33. Interest-specific primers targeting this region were the ones recommended by Illumina with overhang adapters attached:
16S Forward Primer 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG
16S Reverse Primer 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC.
Cycling conditions were the same reported previously6. The PCR products were purified and checked to verify its size on a Bioanalyzer DNA 1000 chip (Agilent).
Read filtering and data analysis
For the taxonomic analysis, reads were quality-filtered (Q > 25) before paired-end joining by QIIME v1.9 software34, using fastq join35,36 under default values. Sequences were clustered into OTUs at 97% similarity using UCLUST algorithm37 and the Greengenes database38. Chimeric detection and removal was done with USEARCH 6.137,39 against the ChimeraSlayer reference database40. To minimize the inflation of rare OTUs in the community analysis, we also exclude singletons for further processing. Taxonomic assignment was done Naïve Bayes classification against RDP database41. For each taxon, the Kruskal Wallis test was perform to compare OTU frequencies in sample groups and to ascertain whether or not there are statistically significant differences between the OTU abundance in the different sample groups, P values were FDR-corrected for multiple hypotheses testing. Single rarefaction, based on the sample with the lowest number of reads, was used for alpha-diversity analysis. Diversity indexes were calculated on rarefied 16S rRNA gene sequence data for all samples at 97% similarity. Alpha diversity between groups was compared through two-sample non-parametric t-tests (Monte Carlo method) at maximum depth in rarefied samples (with 999 permutations). In addition, equal number of samples was subsampled to assess the significant differences between sample types using UniFrac weighted and unweighted distances42,43. The percentage of variation between grouped samples was measured by R2, using adonis function of the vegan package44 in R software. Estimation of P values was done through Monte Carlo test with 999 random permutations of the data set. Preliminarily, combined data from both farms were compared before and after antimicrobial treatment elimination to explore the data. However, each farm was analysed separately at the different time points throughout all the study. Samples were considered to be significantly different when the accompanying P value was ≤0.05. Results were confirmed with SAS software through PROC GLM analysis.
The production data parameters were statistically analysed through Krukal-Wallis non-parametric test using R software45.
Supplementary information
Acknowledgements
Nuria Galofré-Milà is acknowledged for her technical work. The authors are also grateful to the Centres de Recerca de Catalunya (CERCA) Programme. This study was supported by the Ministry of Economy and Competitiveness (MINECO) of the Spanish Government (Grant Number AGL2016-77361-R).
Author Contributions
F.C.F. analysed and interpreted the data, drafted the manuscript. J.M.G. made statistical analysis and drafted the manuscript. F.I.: participated in the sampling and collected production data. V.A.: conceived the study, study design and coordination, and drafted the manuscript. All the authors reviewed and approved the final manuscript.
Data Availability
The entire sequence dataset is available at the NCBI database, SRA accession PRJNA495126.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-43022-y.
References
- 1.Hao, H. et al. Benefits and risks of antimicrobial use in food-producing animals. Front. Microbiol. 5 (2014). [DOI] [PMC free article] [PubMed]
- 2.Ghaisas S, Maher J, Kanthasamy A. Gut microbiome in health and disease: Linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2016;158:52–62. doi: 10.1016/j.pharmthera.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013;13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ubeda C, et al. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect. Immun. 2013;81:965–973. doi: 10.1128/IAI.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsey, M. M., Freire, M. O., Gabrilska, R. A., Rumbaugh, K. P. & Lemon, K. P. Staphylococcus aureus shifts toward commensalism in response to Corynebacterium species. Front. Microbiol. 7 (2016). [DOI] [PMC free article] [PubMed]
- 6.Correa-Fiz, F., Fraile, L. & Aragon, V. Piglet nasal microbiota at weaning may influence the development of Glässer’s disease during the rearing period. BMC Genomics17 (2016). [DOI] [PMC free article] [PubMed]
- 7.Espinosa-Gongora, C., Larsen, N., Schønning, K., Fredholm, M. & Guardabassi, L. Differential Analysis of the Nasal Microbiome of Pig Carriers or Non-Carriers of Staphylococcus aureus. PLoS One11 (2016). [DOI] [PMC free article] [PubMed]
- 8.Niederwerder MC. Role of the microbiome in swine respiratory disease. Vet. Microbiol. 2017;209:97–106. doi: 10.1016/j.vetmic.2017.02.017. [DOI] [PubMed] [Google Scholar]
- 9.Thomason CA, Mullen N, Belden LK, May M, Hawley DM. Resident Microbiome Disruption with Antibiotics Enhances Virulence of a Colonizing Pathogen. Sci. Rep. 2017;7:16177. doi: 10.1038/s41598-017-16393-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Looft T, et al. Bacteria, phages and pigs: the effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014;8:1566–1576. doi: 10.1038/ismej.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schokker D, et al. Early-life environmental variation affects intestinal microbiota and immune development in new-born piglets. PloS One. 2014;9:e100040. doi: 10.1371/journal.pone.0100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janczyk P, et al. Parenteral long-acting amoxicillin reduces intestinal bacterial community diversity in piglets even 5 weeks after the administration. ISME J. 2007;1:180–183. doi: 10.1038/ismej.2007.29. [DOI] [PubMed] [Google Scholar]
- 13.Ober RA, et al. Increased microbiome diversity at the time of infection is associated with improved growth rates of pigs after co-infection with porcine reproductive and respiratory syndrome virus (PRRSV) and porcine circovirus type 2 (PCV2) Vet. Microbiol. 2017;208:203–211. doi: 10.1016/j.vetmic.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 14.Gresse R, et al. Gut microbiota dysbiosis in postweaning piglets: Understanding the Keys to Health. Trends Microbiol. 2017;25:851–873. doi: 10.1016/j.tim.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Zeineldin M, Aldridge B, Blair B, Kancer K, Lowe J. Microbial shifts in the swine nasal microbiota in response to parenteral antimicrobial administration. Microb. Pathog. 2018;121:210–217. doi: 10.1016/j.micpath.2018.05.028. [DOI] [PubMed] [Google Scholar]
- 16.Slifierz MJ, Friendship RM, Weese JS. Longitudinal study of the early-life fecal and nasal microbiotas of the domestic pig. BMC Microbiol. 2015;15:184. doi: 10.1186/s12866-015-0512-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burrough ER, Arruda BL, Plummer PJ. Comparison of the luminal and mucosa-associated microbiota in the colon of pigs with and without swine dysentery. Front. Vet. Sci. 2017;4:139. doi: 10.3389/fvets.2017.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fano E, Pijoan C, Dee S, Deen J. Effect of Mycoplasma hyopneumoniae colonization at weaning on disease severity in growing pigs. Can. J. Vet. Res. 2007;71:195–200. [PMC free article] [PubMed] [Google Scholar]
- 19.Opriessnig T, Giménez-Lirola LG, Halbur PG. Polymicrobial respiratory disease in pigs. Anim. Health Res. Rev. 2011;12:133–148. doi: 10.1017/S1466252311000120. [DOI] [PubMed] [Google Scholar]
- 20.Brockmeier SL, et al. Virulence, transmission, and heterologous protection of four isolates of Haemophilus parasuis. Clin Vaccine Immunol. 2013;20:1466–1472. doi: 10.1128/CVI.00168-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vela AI, et al. Moraxella porci sp. nov., isolated from pigs. Int. J. Syst. Evol. Microbiol. 2010;60:2446–2450. doi: 10.1099/ijs.0.016626-0. [DOI] [PubMed] [Google Scholar]
- 22.Vela AI, et al. Moraxella pluranimalium sp. nov., isolated from animal specimens. Int J Syst Evol Microbiol. 2009;59:671–4. doi: 10.1099/ijs.0.006205-0. [DOI] [PubMed] [Google Scholar]
- 23.Larsen JL, Bille N, Nielsen NC. Occurrence and possible role of Moraxella species in pigs. Acta Pathol. Microbiol. Scand. Microbiol. Immunol. 1973;81:181–186. doi: 10.1111/j.1699-0463.1973.tb00208.x. [DOI] [PubMed] [Google Scholar]
- 24.Zamora L, Domínguez L, Fernández-Garayzábal JF, Vela AI. Bergeyella porcorum sp. nov., isolated from pigs. Syst. Appl. Microbiol. 2016;39:160–163. doi: 10.1016/j.syapm.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Lorenzo de Arriba M, Lopez-Serrano S, Galofre-Mila N, Aragon V. Characterisation of Bergeyella spp. isolated from the nasal cavities of piglets. Vet. J. 2018;234:1–6. doi: 10.1016/j.tvjl.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Sheppard SK, Maiden MCJ. The evolution of Campylobacter jejuni and Campylobacter coli. Cold Spring Harb. Perspect. Biol. 2015;7:a018119. doi: 10.1101/cshperspect.a018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiquette J, Allison MJ, Rasmussen M. Use of Prevotella bryantii 25A and a commercial probiotic during subacute acidosis challenge in midlactation dairy cows. J. Dairy Sci. 2012;95:5985–5995. doi: 10.3168/jds.2012-5511. [DOI] [PubMed] [Google Scholar]
- 28.Rajewski RA, et al. Preliminary safety evaluation of parenterally administered sulfoalkyl ether β-cyclodextrin derivatives. J. Pharm. Sci. 1995;84:927–932. doi: 10.1002/jps.2600840805. [DOI] [PubMed] [Google Scholar]
- 29.Troy EB, Kasper DL. Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front. Biosci. Landmark Ed. 2010;15:25–34. doi: 10.2741/3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazmanian SK, Kasper DL. The love-hate relationship between bacterial polysaccharides and the host immune system. Nat. Rev. Immunol. 2006;6:849–858. doi: 10.1038/nri1956. [DOI] [PubMed] [Google Scholar]
- 31.Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007;20:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng H, et al. A novel strain of Bacteroides fragilis enhances phagocytosis and polarises M1 macrophages. Sci. Rep. 2016;6:29401. doi: 10.1038/srep29401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klindworth A, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1–e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aronesty E. TOBioJ: Comparison of sequencing utility programs, 10.2174/18750362013070100001 (2013).
- 36.Aronesty E. Ea utils: Command-line tools for processing biological sequencing data (2011).
- 37.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinforma. Oxf. Engl. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 38.DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinforma. Oxf. Engl. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haas BJ, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42 (2014). [DOI] [PMC free article] [PubMed]
- 42.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okasnen J, et al. Vegan: community ecology package. R package version. 2015;2:3–0. [Google Scholar]
- 45.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952;47:583. doi: 10.1080/01621459.1952.10483441. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The entire sequence dataset is available at the NCBI database, SRA accession PRJNA495126.