

Abstract
Lysyl oxidase-generated intermolecular cross-links are essential for the tensile strength of collagen fibrils. Two cross-linking pathways can be defined, one based on telopeptide lysine aldehydes and another on telopeptide hydroxylysine aldehydes. Since the 1970s it has been accepted that the mature cross-linking structures on the lysine aldehyde pathway, which dominates in skin and cornea, incorporate histidine residues. Here, using a range of MS-based methods, we re-examined this conclusion and found that telopeptide aldol dimerization is the primary mechanism for stable cross-link formation. The C-telopeptide aldol dimers formed labile addition products with glucosylgalactosyl hydroxylysine at α1(I)K87 in adjacent collagen molecules that resisted borohydride reduction and after acid hydrolysis produced histidinohydroxylysinonorleucine (HHL), but only from species with a histidine in their α1(I) C-telopeptide sequence. Peptide MS analyses and the lack of HHL formation in rat and mouse skin, species that lack an α1(I) C-telopeptide histidine, revealed that HHL is a laboratory artifact rather than a natural cross-linking structure. Our experimental results also establish that histidinohydroxymerodesmosine is produced by borohydride reduction of N-telopeptide allysine aldol dimers in aldimine intermolecular linkage to nonglycosylated α1(I) K930. Borohydride reduction of the aldimine promotes an accompanying base-catalyzed Michael addition of α1(I) H932 imidazole to the α,β-unsaturated aldol. These aldehydes are intramolecular at the N terminus but at the C terminus they can be both intramolecular and intermolecular according to present and earlier findings.
Keywords: collagen, cornea, protein cross-linking, lysyl oxidase, mass spectrometry (MS), skin, tendon, connective tissue, allysine aldol
Introduction
Collagen is the most abundant vertebrate protein. The highly evolved collagen family includes over 40 different gene products and many additional splice variants with diverse tissue-dependent roles (1, 2). Fibril-forming collagens make up the bulk of an organism's collagen mass and are responsible for the underlying strength of most tissues throughout metazoan evolution. The strength of individual collagen fibrils rests heavily on covalent cross-links being formed between individual molecules by the action of lysyl oxidase during fibril assembly and growth (3, 4). The content, placement, and chemistry of the resulting cross-linking amino acids is highly regulated and variable, dependent on the tissue type and in particular on the quality of the post-translational modifications of the newly synthesized collagen molecules (5, 6). Covalent cross-links can vary among tissues even across a single genetic type of collagen such as collagen type I, the most abundant molecular type in vertebrates (7, 8).
In essence, two pathways of lysyl oxidase-mediated cross-linking operate in fibril-forming collagens, one based on telopeptide lysine aldehydes and the other on telopeptide hydroxylysine aldehydes (3). The latter features in bone, cartilages, and many other skeletal and tough connective tissues, whereas the former is characteristic of skin, cornea, and certain tendons, notably rodent tail tendons. The aldehyde side chains created by lysyl oxidase initially interact with specific helical domain hydroxylysines on neighboring collagen molecules to form divalent cross-links. These undergo further reactions to produce mature, chemically more stable cross-linking structures. On a purely hydroxylysine-aldehyde pathway these are the trivalent pyridinolines, hydroxylysyl pyridinoline (HP),2 and lysyl pyridinoline. In tissues featuring purely a lysine-aldehyde pathway, the only known stable natural maturation product is histidinohydroxylysinonorleucine (HHL), which is prominent on analysis of mature human and bovine skin (9–12), but not rat or mouse skin or tail tendon type I collagen (11–14). Originally this cross-linking structure had been misidentified as hydroxyaldol-histidine (15), which later studies corrected (11–14).
Bone type I collagen in contrast has a unique cross-linking pattern that depends on the reactions between a mix of both telopeptide lysine and telopeptide hydroxylysine aldehydes and their helical domain partners. The mature structures in bone include a mix of trivalent pyridinolines and pyrroles and divalent ketoimine cross-links (16, 17). This chemistry is largely controlled during collagen synthesis in the endoplasmic reticulum of osteoblasts by chaperone protein complexes that regulate the activity of lysyl hydroxylase isozymes against telopeptide and triple-helical domain substrate sites (5, 8, 18).
The well-recognized initial reaction products on the lysine aldehyde pathway after lysyl oxidase has converted telopeptide lysines to aldehyde side chains are intramolecular aldol dimers formed between α1 and α2 and α1 and α1 N-telopeptides within the individual type I collagen molecules (19). These aldols and remaining telopeptide lysine aldehyde monomers form aldimine cross-links intermolecularly with adjacent helical domain hydroxylysine side chains, which on borohydride reduction of tissue produce tetravalent histidinohydroxymerodesmosine (HHMD) and the reduced divalent aldimine cross-link hydroxylysinonorleucine (HLNL) (20, 21). The reactions progress spontaneously in the fibril in vivo, catalyzed by their local protein environment within the highly ordered collagen polymer. The original methods by which the lysyl oxidase catalyzed cross-linking products were identified require borohydride reduction to stabilize labile aldimines as secondary amines. After acid hydrolysis the novel cross-linking amino acids were resolved by column chromatography and their structures were determined by MS, NMR, and other analytical methods (22–29). The essential elements of lysyl oxidase-mediated cross-linking in collagen and elastin were thus established in the late 1960s through the 1970s (30). There remained some controversy as to the source and origin of HHMD and the structurally related aldol histidine (31) on borohydride reduction, whether the histidine was added as an artifact of base-catalyzed Michael addition (21–28) or not (29). The controversy was never clearly resolved by methods available at the time.
Peptide MS is a powerful tool for revealing site-specific collagen post-translational modifications including collagen cross-link analysis (32). It helped us discover effects on cross-linking in bone collagen in various human genetic variants and mouse models of osteogenesis imperfecta and in skin and tendon collagens of mice with null genes that affect collagen cross-linking (8, 17, 32). From these studies we discovered that the allysine aldol dimer, the first cross-link to be identified in collagen (19), which is responsible for the prominent β-chain dimers seen on SDS-PAGE of type I collagen extracts from skin and other soft tissues, was also formed between α1(I) C-telopeptide lysine aldehydes, both intramolecularly and intermolecularly (34).
None of our cross-linking findings on skin, however, were consistent with the proposed origin of the stable maturation product, HHL, predicted to be an additional product between the α1(I) C-telopeptide to K87 helix aldimine cross-link, dehydro-HLNL, and a third chain histidine residue at α2(I) H92 (10). To understand better the nature of the mature cross-links on the lysine aldehyde pathway we applied MS and different proteolytic cleavage and detection methods to examine the structure of cross-linked peptides containing C-telopeptide and N-telopeptide cross-linking sites from various skin, cornea, and tendon collagens that are known to be particularly rich in or to lack HHL cross-links (35).
The findings show that allysine aldol dimers form the major stable cross-linking bonds at both ends of the type I collagen molecule in tissues that use the lysine aldehyde pathway. Together with literature data they provide evidence for a mechanism by which HHL is produced on acid hydrolysis artifactually from a C-telopeptide allysine aldol in labile linkage to glcgal Hyl at α1(I) K87, but only in those species that have a conserved histidine in their α1(I)-C-telopeptide sequence.
Results
Comparison of cross-linked α-chain SDS-PAGE patterns across tissue collagen extracts
Some quantitative differences in extractability of type I collagen are evident between tissues that depend on lysine aldehyde cross-linking (Fig. 1), but the patterns of α, β, γ, and higher oligomers are very similar. In contrast, an extract of demineralized bone was run as a contrasting type I collagen-based matrix that is cross-linked by a mix of lysine aldehydes and hydroxylysine aldehydes (17). The results show a consistent pattern of β-chain dimers being more prominent than α-chains in extracts of skin, cornea, and tail tendon collagens compared with bone collagen. The skin, cornea and tendon extracts also reveal higher order oligomers of which γ112 is the most prominent in nondenaturing, acetic acid extracts. The latter γ112 trimer was previously shown to be derived from extracted native type I collagen molecules that contain an allysine aldol between the α1(I) N- and the α2(I) N-telopeptide and between the two α1(I) C-telopeptides. Under the denaturing conditions of SDS-PAGE this runs as the γ112 band.
Figure 1.
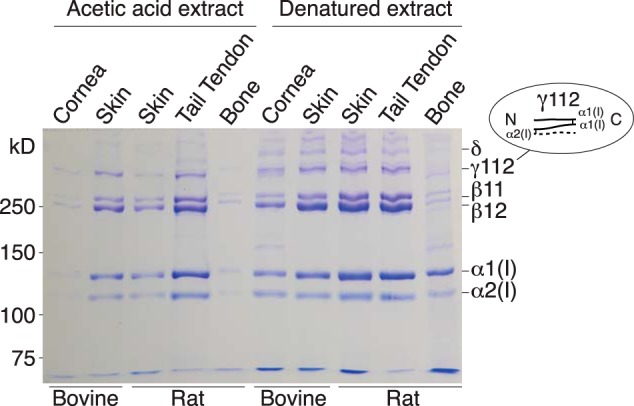
SDS-PAGE showing similar α, β, γ, and oligomeric cross-linked chain profiles for collagen extracts from skin, cornea, and tendon. Native collagen molecules extracted by 3% acetic acid were run in the left five lanes and heat denatured collagen from a separate tissue sample in the right five lanes. Loads based on the following original tissue weights extracted were: bovine cornea, 60 μg; bovine skin, 30 μg; rat skin, 30 μg; tail tendon, 15 μg; demineralized bone, 300 μg.
Tissue-dependent hydroxylation and glycosylation differences between helical domain cross-linking lysines
In-gel trypsin and Lys-C digestion were also used to measure the post-translational status of the known triple-helical domain cross-linking sites in α1(I) and α2(I) chains from the various tissues. The results are summarized in Fig. 2. Type I collagen of skin and cornea differs from that of tail tendon in having K87 fully hydroxylated and glycosylated as glcgal-Hyl, whereas in tail tendon K87 is fully hydroxylated but is not glycosylated. Bone collagen has primarily a mix of glcgal-Hyl and gal-Hyl at K87, the ratio of which varies between species (17).
Figure 2.

Summary of tissue-dependent differences in hydroxylation and glycosylation of helical domain cross-linking in type I collagen. The four known lysine sites of helical domain cross-linking were interrogated for post-translational status by in-gel trypsin or endo Lys-C digestion followed by LC-MS. A, summary of the results for bovine cornea and skin, rat skin, tail tendon, and bone. B, example of the mass spectra from cornea for the trypsin-digested α1(I) K87 peptide and (D) its fragmentation pattern. C, example of the mass spectrum from cornea of the Endo Lys-C–digested α1(I) K933 peptide and (E) its fragmentation pattern.
Western blot analysis of CNBr-digested tissue collagens for C-telopeptide cross-linked structures
The β-chain dimers, β11 and β12 (Fig. 1), were the first established products of lysyl oxidase cross-linking in collagen, the result of allysine aldol intramolecular dimerization of N-telopeptides (19). Only recently were C-telopeptide aldol dimers discovered, initially from studying the tail tendon collagen of fibromodulin-null mice (34) but subsequently in the skin and tendons of other mutant and WT mice (8, 33). These studies showed evidence for both intramolecular and intermolecular C-telopeptide aldol cross-link formation. We therefore designed an analytical screen to profile all forms of C-telopeptide-containing structures in CNBr digests of tissue collagen. To do this a mAb, mAb 1G7, that recognizes an epitope specific to the α1(I) C-telopeptide was applied to SDS-PAGE resolved tissue digests. The results are shown in Fig. 3.
Figure 3.

Western blotting detection of α1(I) C-telopeptide cross-linked structures from CNBr-digested skin, cornea, and tendon gives a similar, characteristic pattern distinct to that from bone. A, illustration of the cross-linking interaction sites between telopeptides and helical domains of 4D-staggered neighboring collagen molecules packed in fibrils. Underneath, a line drawing shows the position of CNBr peptides defined by methionine residues (marked by vertical lines) in rat and bovine α1(I) and α2(I) chains. The mAb 1G7 recognizes an epitope at the C terminus of the α1(I) C-telopeptide whether or not its cross-linking lysine is hydroxylated, oxidized to aldehyde by lysyl oxidase, or cross-linked in oligomeric peptides. B, aliquots of CNBr-digested samples of bovine cornea and skin, rat skin, tail tendon, and demineralized bone were run on 12.5% SDS-PAGE, stained with Coomassie Blue (left panel) or transblotted to polyvinylidene difluoride membrane and α1(I) C-telopeptide-containing bands detected by probing with mAb 1G7 and chemiluminescence development (right panel). Lane loads as follows were adjusted to give approximately equal stained band intensities across the left gel (bovine cornea, 10 μg; bovine skin, 9 μg; rat skin 8 μg; tail tendon, 5 μg; bone, 7 μg) and the right immunoblot (bovine cornea, 10 μg; bovine skin, 2.5 μg; rat skin 8 μg; tail tendon, 6 μg; bone, 2 μg).
Although immunoreactive yields vary somewhat between individual tissue sources as seen from the lane intensities (which may depend on efficiency of C-telopeptide epitope generation by trypsin), all tissues show similar patterns except bone. The fastest positive band in each lane from skin, cornea, and tendon is the linear α1(I)CB6 sequence that ends with the C-telopeptide. Looking at the rat skin lane as an example, the next slower band is an aldol dimer of CB6 and above that is the linear CNBr partial cleavage product, α1CB7,6. The same bands repeat in the lanes for rat tail tendon, bovine cornea, and skin. In the latter two lanes, with stronger overall band intensities, the higher band is an α1CB6 × α1CB7,6 dimeric partial cleavage product. No other significant cross-linked forms of CB6 are revealed. For example, no band that would be consistent with the proposed molecular origin of HHL as a mature cross-link linking the three CB peptides, α1CB6 × α1CB5 × α2CB4 (10), was recovered from skin or cornea.
The lane from rat bone collagen gives a different band pattern that is nevertheless consistent with the known cross-linking mechanism of C-telopeptides in bone type I collagen. Stable ketoimine divalent cross-links are responsible for the α1CB6 × CB5 band and pyridinoline plus pyrrole trivalent cross-links for the α1(CB6 × CB6 × CB5) band running above the α1CB6 dimer and α1CB7,6 bands from skin, cornea, and tendon.
Mass spectral identification of CB-peptides
To confirm the CB-peptide band identities, histidine-containing peptides were enriched from tissue CNBr digests by an immobilized metal ion affinity column, then fractionated on a C8 reverse-phase column (Fig. 4). As shown, collected fractions were run serially across lanes of an SDS-PAGE gel. Individual bands were excised and subjected to in-gel trypsin digestion and tandem LC-MS analysis, which established their indicated CB-peptide identities. These results also helped confirm the identities of the bands revealed by Western blot analysis in Fig. 3.
Figure 4.

Mass spectral identification of α1(I)CB6 (C-telopeptide containing) linear, cross-linked and partial cleavage forms in a CNBr digest of bovine cornea fractionated by C8-HPLC/serial fraction SDS-PAGE. An IMAC column was used to enrich histidine-containing peptides from a CNBr digest of bovine cornea. This pool was run on C8-HPLC as shown in the lower profile, and serial collected fractions were run on 12.5% SDS-PAGE and stained with Coomassie Blue (inset panel). All bands were excised, digested with trypsin in-gel, and the digests run on LC-MS to identify them. Identities of all individual and partial cleavage linear peptides are shown, as well as a peptide identified as the cross-linked dimer of α1(I)CB6. No α1(I)CB6-containing peptides other than the monomer, dimer, and their 7,6 partial cleavage forms were found.
Prominence of allysine aldol cross-linked N- and C-telopeptide dimers in bacterial collagenase-digested cornea collagen
To dissect the cross-linking sites more finely down to smaller peptides using mass spectrometric analysis, the same tissues were digested with bacterial collagenase under conditions that generated reproducible and informative cross-linked peptide structures (8, 16). The digests were fractionated by reverse-phase HPLC and individual fractions analyzed by LC-MS and their structures were determined manually from the MS/MS fragmentation patterns (8, 16). In Fig. 5, the elution positions and the structures of the major N-telopeptide and C-telopeptide cross-linked dimers are identified.
Figure 5.
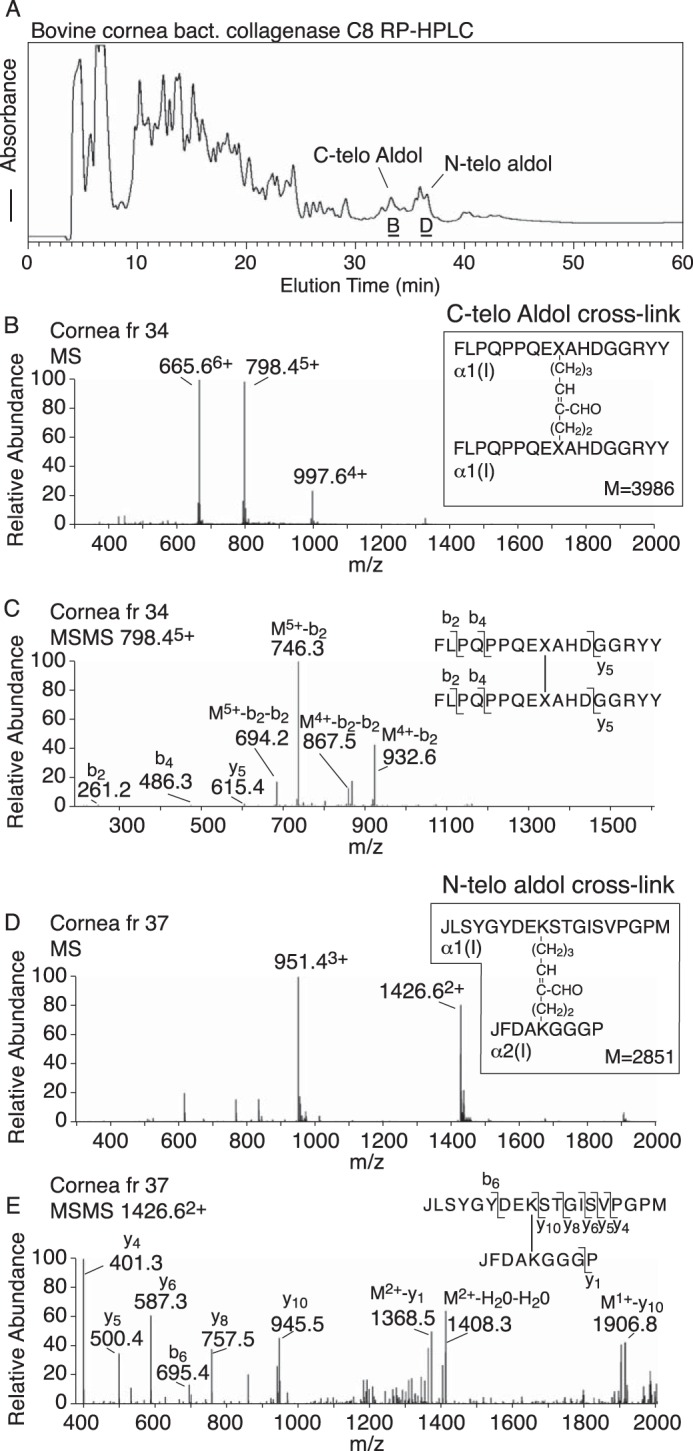
Allysine aldol cross-linked dimers of N-telopeptides and C-telopeptides are major products in a bacterial collagenase digest of corneal collagen if unreduced by sodium borohydride. Bovine corneal tissue was completely digested with bacterial collagenase under conditions that preserved reproducible telopeptide and helical domain cross-linking sequences. A, elution profile of the resulting peptides on C8 reverse phase HPLC. The late-eluting peaks labeled C-telo aldol and N-telo aldol were shown by MS and MS/MS (B–E) to have the structures shown.
Effect of borohydride reduction on telopeptide aldol dimeric peptide yields
Reduction of tissue with sodium borohydride failed to change the yield of C-telopeptde aldol dimeric peptides but N-telopeptide dimers were no longer found. Instead the same N-telopeptide dimeric peptides were recovered in covalent linkage to hydroxylysine at K930 in a peptide fragment from the α1(I) chain.
The masses and fragmentation profile of these three chained cross-linked structures (Fig. 6) are exactly consistent with reduction of the aldimine bond formed between the α,β-unsaturated allysine aldol aldehyde and the ϵ-amino group of K930 hydroxylysine. Apparently on tissue reduction, the N-telopeptide aldol adducted to the helical domain hydroxylysine is susceptible to borohydride reduction but the C-telopeptide aldol adduct is not.
Figure 6.

Mass spectral identification of N-telopeptide aldol dimers linked to a peptide containing α1(I) K930 from bacterial collagenase-digested, sodium borohydride-reduced mouse skin and tail tendon collagens. C8 RP-HPLC chromatography of tissue digests prepared as described in the legend to Fig. 5 gave the same prominent α1(I)N × α2(I)N × α1(I) helix (A and B) and α1(I)N × α1(I)N × α1(I) helix (C and D) peptides from mouse skin and tail tendon, which were identified by their mass spectra. E, MS/MS fragmentation pattern of the 1200.58+ parent ion of α1(I)N × α1(I)N × α1(I) helix from mouse skin, shown as an example.
Identification of an α1(I) C-telopeptide dimer in labile linkage to glycosylated K87 in bacterial collagenase-digested cornea collagen
In further analyses of borohydride-reduced and nonreduced cornea after bacterial collagenase digestion, a large peptide was recovered spread across several fractions of the C8 HPLC chromatogram. The peptide was recovered with the same characteristic properties whether tissue had been reduced or not (Fig. 7). The structure was determined manually based on fragmentation properties of the parent ions and of MS3 of the helical arm throw off (Fig. 7, C and D). The parent ion was unusual in our experience with collagen cross-linked peptides in that the intact helical peptide arm (ions 1082.61+ and 541.82+) containing K87 hydroxylysine with sugars (glucosylgalactosyl) attached was released on MS/MS as a major, intact fragment ion. This implied a labile covalent bond between the allysine aldol cross-linking residue of the C-telopeptide dimer and the glycosylated K87 hydroxylysine. The 10494+ prominent ion (Fig. 7B) matches the mass of the C-telopeptide dimer being released as the α,β-unsaturated allysine aldol peptide dimer.
Figure 7.

Mass spectral identification of a C-telopeptide aldol dimer in labile linkage to a peptide containing glycosylated α1(I) K87 from bacterial collagenase-digested corneal collagen. Further mass spectral analysis of fractions from the C8 HPLC chromatogram shown in Fig. 5 revealed a larger peptide spread through fractions 35–40 underlying the aldol dimeric peptides. A, mass spectrum and determined structure of the trimeric peptide. B, MS/MS (ms2) fragmentation of the parent 880.26+ ion followed by ms3 fragmentation patterns from the helical arm fragment GMKGHR with (C) and without (D) the originally attached glucosylgalactosyl modification. In the helical domain sequence shown, K is hydroxylysine, as confirmed by parent and fragment ion masses.
LC-MS analysis of HHL, HHMD, and total cross-linking amino acids after acid hydrolysis
The LC total ion-current profiles of the cross-linking amino acid products of acid hydrolysis of borohydride-reduced bovine tendon, skin, and cornea, and mouse skin are shown in Fig. 8A, together with mass spectra from scrolled regions of these profiles. The results confirm that bovine cornea and skin are rich sources of HHL and HHMD, whereas mouse skin yields only HHMD but no HHL. Bovine flexor tendon reveals products of cross-linking from both the lysine aldehyde and hydroxylysine aldehyde pathways as expected, with hydroxylysyl pyridinoline (HP), HHMD, HHL, and the reduced divalent cross-links HLNL and LNL all present. From skin, desmosine (Des, from elastin) is also detected. Samples of the same tissues hydrolyzed without prior borohydride reduction gave HHL but no HHMD, HLNL, or LNL as expected.
Figure 8.
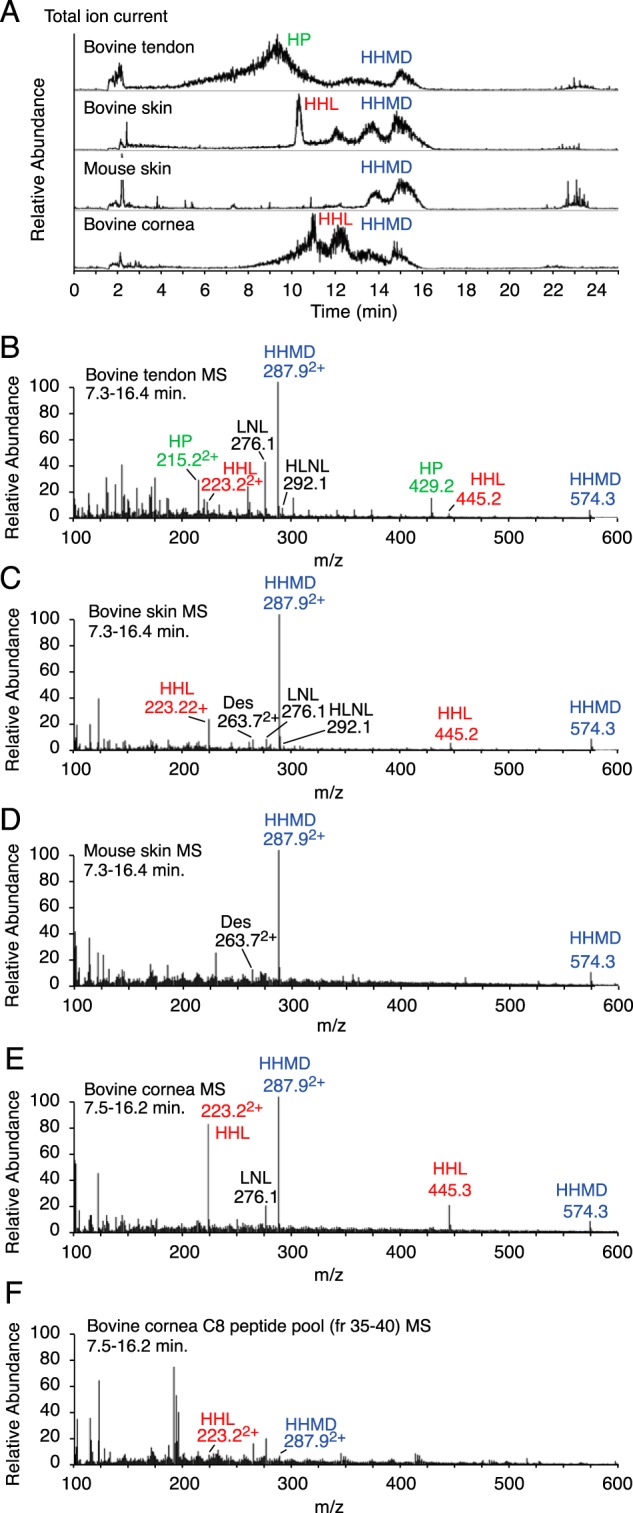
LC-MS analysis of collagen and elastin cross-link derivatives in acid-hydrolyzed cornea, skin, and tendon. Cross-linking amino acid products in acid hydrolysates of whole tissues were separated from bulk amino acids by organic solvent partition on a hydrated cellulose column, then resolved and analyzed by electrospray LC-MS on the LTQ XL using a Cogent 4 diamond hydride column essentially according to the method of Naffa et al. (37). A, total ion current elution profile from the LC column for hydrolysates of sodium borohydride-reduced samples of bovine tendon, bovine skin, mouse skin, and bovine cornea. The elution positions of the various major collagen cross-linking amino acids, HP, HHMD, HHL, HLNL, LNL, lysinonorleucine, and Des, desmosine (from elastin), are shown. B–E, mass spectra scrolled across the regions of the LC eluent for each tissue sample showing the masses of the various cross-linking entities. Both HHL and HHMD are prominent in bovine skin and cornea but absent from mouse skin. F, acid-hydrolyzed pooled fractions from the C8 RP-HPLC chromatogram of bacterial collagenase-digested cornea containing the C-telopeptide aldol dimer to α1(I) K87 adduct (Fig. 6) reveal a low but significant yield of HHL, as did acid hydrolysates of whole bacterial collagenase-digested cornea samples.
Discussion
The SDS-PAGE patterns of extracted collagen chains from skin, cornea, and tendon are essentially similar, with more prominent β-chain dimers than α-chains (Fig. 1). The cross-linked trimer, γ112, is also consistently present. We have shown that this is the product of C-telopeptide aldol dimer formation in mouse tail tendon and skin collagen, and is especially prominent in fibromodulin-null mice due to increased telopeptide lysine oxidation by lysyl oxidase (34). Similar, abnormally high levels of γ112 and C-telopeptide aldol dimeric cross-links in skin collagen were also observed in mice with null genes encoding Sc65 or P3h3, two members of the prolyl 3-hydroxylase gene family (8, 33). The latter two mouse models phenocopy a biochemical abnormality in the post-translational modification of type I collagen that results in underhydroxylation of triple-helical domain lysines at cross-linking sites α1(I) and α2(I) K87 and α1(I) K930 and α2(I) K933 in all tissues but especially skin (8). This results in mostly lysine and little glcgal-Hyl at α1(I) K87 in skin collagen. The increase in γ112 and intramolecular C-telopeptide aldol dimers in skin collagen was attributed to the lack of sugar residues at K87 allowing intramolecular aldols to form when both α1(I) C-telopeptides in the same molecule had their lysines oxidized to aldehydes by lysyl oxidase. In WT skin, intermolecular aldols at the C terminus appeared to be the norm based on the γ and higher cross-linked chain patterns (8). For the present study, this implies that the state of glycosylation at K87 may sterically determine the reaction properties of C-telopeptide allysines in the growing fibril during spontaneous cross-link formation.
Analysis of the state of hydroxylation of lysine and glycosylation of hydroxylysine at the triple-helical domain cross-linking sites established that in skin and cornea collagens K87 was fully glycosylated, but in tail tendon type I collagen K87 was exclusively Hyl with no attached sugars (Fig. 2). This lack of glycosylation of tendon type I collagen has been noted before (35). To determine whether this had any tissue-dependent, selective effect on C-telopeptide cross-linking interactions, not evident by SDS-PAGE analysis of extracted collagen chains, we next looked at CNBr digests of whole tissue, matrix collagen. The mAb 1G7, which recognizes the C terminus of the C-telopeptide sequence, EXAHDGGR, where K can be Lys, Hyl, or a cross-link to one or more additional peptides, provides a useful screen on Western blotting for all linear and cross-linked structures that have this terminal peptide sequence. Whole tissue was treated with trypsin under nondenaturing conditions to enhance the yield of the C-terminal neoepitope that 1G7 recognizes. All tissues, except a bone control, were selected for comparison based on their collagen being cross-linked exclusively by the lysine aldehyde pathway. Bovine skin and particularly cornea were known to be rich in HHL (36), whereas this cross-link was reported absent from rat and mouse skin and tail tendon collagens (10, 12–14). The results in Fig. 3 showing essentially the same patterns of linear and cross-linked forms of CB6 from skin, cornea, and tendons is not consistent with the proposed intermolecular sites linked by HHL residues. The origin of HHL as a trivalent cross-link of three chains, an α1(I) C-telopeptide in aldimine linkage α1(I) Hyl87 adducted to α2(I) H92, was proposed based on N-terminal Edman sequencing analyses of a partially purified tryptic peptide prepared from bovine dermis (10). No band fitting the size of such a combination of CB-peptide fragments is evident in the 1G7 Western blots from bovine skin or cornea, the latter tissue collagen in our hands being the richest source of HHL at about 1 mol/mol of collagen (36). The only cross-linked form of CB6 on SDS-PAGE from all tissues except bone was a CB6 dimer and a larger CB7,6 partial cleavage form of it (Fig. 3). From a bone CB-digest run as a control, the main cross-linked forms of CB6 are the divalent ketoimine cross-linked peptide, CB6 × α1(I)CB5, and trivalent pyridinoline/pyrrole cross-linked CB6 × CB6 × α1(I)CB5. The latter serves as a useful size marker to compare with the smaller, faster running CB6 aldol dimer from skin, cornea, and tendon CNBr digests.
To further refine the identity of the native cross-linked structures, smaller peptides were prepared by bacterial collagenase digestion. On MS these gave prominent yields of the same C-telopeptide and N-telopeptide aldol dimeric peptides from all skin, cornea, and tendon digests (Fig. 5 shows the results for bovine cornea), whether or not the tissue had been reduced with sodium borohydride. In addition, the bovine cornea digest revealed a larger peptide consisting of the CB6 dimer in labile linkage to α1(I) Hyl87 contained in a short segment of helix (Fig. 7). This adduct was recovered consistently from repeat digests of cornea and also from skin but not rat or mouse tail tendon (data not shown), but in relatively low and variable yields compared with the free C-telopeptide aldol dimeric peptide (Fig. 5). The mass spectral fragmentation behavior of this larger peptide with a strong throw off of the intact GMK(galglc)GHRG K87 arm is consistent with an interpretation that this was an adduct in labile linkage to the allysine aldol aldehyde group of the C-telopeptide dimer. Variable yields of this peptide together with the more abundant free C-telopeptide dimer were recovered from digests whether or not the tissue was previously treated with sodium borohydride.
This was in sharp contrast to the behavior of the N-telopeptide dimers that were absent from digests of borohydride-reduced tissue and quantitatively converted to the reduced aldimine trimeric peptides identified in Fig. 6. Whether the tissue was reduced or not, the labile C-telopeptide trimer behaved the same on MS, with the characteristic throw off of the intact helical arm shown in Fig. 7. Again in sharp contrast, the MS/MS profile of the borohydride-reduced N-telopeptide trimeric peptides did not yield any intact helical arm fragmentation products (Fig. 6). The low yields of the C-telopeptide aldol/K87 adduct on bacterial collagenase digestion, and of HHL on acid hydrolysis of bacterial collagenase-digested cornea (Fig. 8), are consistent with the C-telopeptide aldol forming a labile adduct with glcgal-Hyl at K87 in vivo, which can break on proteolysis but produces HHL on acid hydrolysis. The mAb 1G7 Western blotting results are consistent with this conclusion (Fig. 3).
The present findings can explain why skin collagen from certain species yield HHL on acid hydrolysis but rat and mouse skin do not. Specifically, bovine, human, and most vertebrates contain the α1(I) C-telopeptide sequence, EKAHDGGR (NCBI accession P02453.3), with histidine (H18c) two residues C-terminal to the cross-linking lysine, whereas the rat and mouse sequence, EKSQDGGR (NCBI accession numbers P02454.5 and P11087.4), does not (Fig. 9). Skin from other species (sheep, goat, deer, as well as cow) all yield HHL on hydrolysis (37) and have the EKAHDGGR sequence conserved. From the present structural results, it is clear that it is this telopeptide histidine imidazole that is incorporated into HHL on acid hydrolysis of native fibrils. If so, this raises the question, what happens to the C-telopeptide aldol dimers in rat and mouse skin on acid hydrolysis? Without the neighboring telopeptide histidine, these native aldol complexes are probably broken down on acid hydrolysis as early studies of allysine aldol properties in collagen concluded (38).
Figure 9.

Protein sequence alignment of the α1(I) C-telopeptide. The cross-linking lysine (K16C) is shown in red. The histidine (H18C), which is incorporated on acid hydrolysis into HHL, is shown in blue, and the arginine (R22C) that helps catalyze the cross-link formation is shown in green. It is notable that both mouse and rat lack the histidine residue and do not form HHL. Sequences are from Ensembl databases.
The intramolecular aldol dimers formed between N-telopeptides on the other hand clearly behave differently from the C-telopeptide aldol dimers in their chemical reaction properties in native fibrils, being quantitatively and stably reduced as aldimine adducts to K930 of an α1(I) chain by sodium borohydride (Fig. 6). Although we did not recover equivalent reduced aldol dimer peptide adducts to α2(I) K933 they are possible and we cannot rule them out.
Proposed origin and mechanism of formation of the histidine-containing adducts HHL and HHMD
The above properties support the proposed mechanisms outlined in Fig. 10 whereby the C-telopeptide aldol dimer resists borohydride reduction as an α1(I) K87-linked enamine tautomer, whereas N-telopeptide aldol aldimine adducts are sodium borohydride-reduced producing HHMD, which is released on acid hydrolysis. The speculated retro-aldol mechanism for generating HHL from the enamine tautomer on acid hydrolysis is proposed based on current knowledge of the base-catalyzed mechanisms that stabilize Schiff's base to enamine intermediates at the active sites of a range of aldolase enzymes (39–42), together with the following unique sequence features of the C-telopeptide to helix cross-linking site.
Figure 10.

Proposed mechanism of formation of HHL from C-telopeptide aldols compared with HHMD from N-telopeptide aldols. A, HHL formation on acid hydrolysis is restricted to skin and cornea from species that have a conserved α1(I) C-telopeptide histidine residue. Both this histidine and glucosylgalactosyl hydroxylysine at α1(I) K87 appear to be necessary for HHL formation from tissue on acid hydrolysis. The proposed reaction mechanism involves a retro-aldol cleavage and covalent addition of the telopeptide histidine imidazole to give the accepted structure for HHL (10, 47). B, HHMD is a product of borohydride reduction of the aldimine addition product between N-telopeptide α,β-unsaturated aldol cross-links and K930 hydroxylysine together with the Michael adduction of the histidine at H932 in the same helical sequence through its imidazole group. The peptide structures obtained from borohydride-reduced, bacterial collagenase-digested skin and tail tendon collagen show that the histidine has to be from this same chain, not another α-chain. If the latter occurred, more complex peptides should have been found. The accepted structure of HHMD with the ring nitrogen of histidine imidazole added to the β-carbon of the aldol is shown (10, 37).
All vertebrate α1(I) C-telopeptides including rat and mouse have a conserved arginine 6 residues C-terminal (R22C) to the cross-linking lysine (see Fig. 9). Also in skin and cornea, α1(I) K87 is almost all glucosylgalactosyl hydroxylysine, whereas α1(I) K930 and α2(I) K933 are not glycosylated. In contrast to skin and cornea, neither K87 nor K930/933 are glycosylated in rodent tail tendon type I collagen. We believe these chemical features are responsible for the distinctive tissue-dependent reactivity properties of C-telopeptide aldehydes, and their ensuing aldol dimers, compared with N-telopeptides. Specifically, we propose that R22C, which will be in close proximity to the aldehyde in the hairpin loop conformation of each C-telopeptide (43), base-catalyzes both aldol formation and the subsequent enamine tautomer structure as the dominant tautomeric form of the C-telopeptide aldol reaction product with K87. When in addition, K87 is glcgal Hyl, as it is in skin and cornea, the product is HHL on acid hydrolysis. The hexoses may provide the latent, local reducing agent required by the proposed reaction mechanism. In support of this mechanism, it has been established that short synthetic peptides containing the α,β-unsaturated aldehyde cinnamaldehyde inhibit certain classes of phosphatases by forming a reversible covalent enamine adduct with the guanido group of an arginine residue in their Src homology 2 domain active site (44, 45). As enamines, these adducts resisted borohydride reduction. We propose that R22C in α1(I) C-telopeptides can similarly interact with the aldol formed between two K16C allysines and, in native fibrils, influence the equilibrium of the addition reactions between K16C allysines and K87 helix hydroxylysines, favoring the enamine tautomer shown in Fig. 10.
Significance
Our findings show that the long accepted collagen cross-linking pathway that involves histidine addition to produce the mature cross-links in skin, cornea, and other tissues is incorrect. In considering how tissues that use this pathway grow and remodel their fibrillar extracellular matrix the new concept has important implications.
We conclude that aldol dimers are the main, stable cross-linking bonds formed by C-telopeptides and N-telopeptides in skin, cornea, and certain tendon type I collagens. Their further reaction products form relatively labile intermolecular cross-links. If the aldol-forming bonds are all intramolecular at the N terminus and mostly intramolecular at the C terminus, as is the case with rodent tail tendon, this can explain why rodent tail tendon collagen is highly soluble as native monomers in dilute acetic acid (8, 34), because the intermolecular cross-links will be mostly acid-labile aldimines and enamines (Fig. 10). In normal skin collagen, most of the C-terminal aldol bonds appear to be intermolecular (8), which can explain the lower acid solubility of skin collagen versus tail tendon (8).
The findings are also significant when considering how collagenous tissues that depend on the lysine-aldehyde cross-linking pathway for strength can build and remodel their extracellular fabric (46). Growing tendons, for example, may benefit from having labile intermolecular cross-links that allow collagen fibril remodeling to occur through bond breakage and reformation. In this way, new collagen molecules could be added to increase the diameter of existing fibrils, and fibrils could fuse laterally, whereas the tissue fabric as a whole could continue its mechanical function as it grows with the skeleton and remodels as a mature tissue.
Cornea is a unique tissue that depends on the lysine aldehyde pathway for the natural strength of its highly organized type I collagen fibrillar array (47). The new insight that aldol dimers are the principal source of stable cross-linking bonds in cornea is potentially significant with respect to the recognized clinical benefits from artificially cross-linking keratoconus corneas with riboflavin and then UV-A light (48). The underlying molecular mechanism by which this treatment strengthens corneal tissue and prevents bulging is unclear, but there is experimental evidence that endogenous carbonyl groups are the reactive components that form new cross-links (49). Because cornea type I collagen contains roughly 2 mol of allysine aldol per mole, this is a strong candidates for being the proposed reactive species when corneas are treated with riboflavin and UV-A light.
Experimental procedures
Tissue sources and preparation
Adult bovine tissues (skin, eyes, and superficial digital flexor tendon) were purchased from Sierra for Medical Science (Whittier, CA). Adult normal Sprague-Dawley laboratory rats and adult normal (C57B6/j) mice were obtained as byproducts from approved and completed animal studies. Tissues were scraped clean and washed in saline (0.15 m NaCl, 0.05 m Tris), pH 7.4, with protease inhibitors (2 mm EDTA, 5 mm benzamidine, 10 mm 1,10-phenanthroline, and 2 mm phenylmethylsulfonyl fluoride) for 24 h at 4 °C. Tendon and skin were defatted with chloroform/methanol (1:1, v/v) for 24 h at room temperature. Cornea was extracted in 4 m quanidine HCl, 0.05 m Tris, pH 7.4, with protease inhibitors overnight at 4 °C to remove proteoglycans before collagen extraction. Bone was demineralized in 0.1 m HCl, 24 h at 4 °C.
Collagen extraction and proteolysis
Type I collagen was solubilized from 2 to 3-mg samples of prepared bone, tendon, skin, and cornea by heat denaturation for 3 min at 90 °C in Laemmli buffer (denatured extract), or 3% acetic acid at 4 °C for 24 h, or cyanogen bromide digestion in 70% formic acid at room temperature for 24 h. Prepared tissues were also digested with bacterial collagenase as described (16) with the addition of 10% (v/v) acetonitrile to the digestion buffer, and collagenase-generated peptides were separated by reversed-phase HPLC (C8, Brownlee Aquapore RP-300, 4.6 mm × 25 cm) with a linear gradient of acetonitrile/n-propyl alcohol (3:1, v/v) in aqueous 0.1% (v/v) TFA and monitored by absorbance at 220 nm.
Cornea CNBr peptide chromatography
An IMAC (immobilized metal ion affinity chromatography) column was prepared by charging HiTrap chelating HP beads (Amersham Biosciences) with CuCl2. CNBr-digested cornea was loaded onto the IMAC column in 0.05 m Tris-HCl, 0.15 m NaCl, 2 m guanidine HCl, pH 8.0, and the bound peptides were eluted with 0.1 m sodium acetate, pH 4.6, containing 2 m guanidine HCl and 0.2 m imidazole. Eluted CNBr peptides were run on reversed-phase HPLC and individual fractions run on 12.5% SDS-PAGE.
SDS-PAGE
The method of Laemmli (50) was used with 5 or 12.5% gels for extracts of tissue collagen. Type I α-chains and cyanogen bromide peptides were cut from gels and digested with trypsin or Endo Lys-C (Mass Spec Grade, Promega, Madison, WI) in-gel, for mass spectral analyses.
Immunoblotting with mAb 1G7
A mouse mAb (1G7 isotype IgG1,k) was generated against the synthetic peptide EKAHDGGR that recognizes a C-terminal neoepitope of the α1(I) collagen C-telopeptide. This can be generated by trypsin digestion of native collagen. Peptide conjugation, immunization, and hybridoma selection were done as described in Ref. 51 using RBF/DnJ mice. The synthetic peptide EKAHDGGR was used for screening and characterization. Using variable length C-telopeptide synthetic peptides, we determined that the C-terminal arginine is necessary for 1G7 antibody recognition. Tissue samples were activated with 0.1 mg/ml of trypsin (trypsin, proteomics grade, Roche Diagnostics) in 0.1 m NH4HCO3 for 24 h then digested with CNBr in 70% formic acid both at room temperature for 24 h. CNBr peptides where separated by 12.5% SDS-PAGE and transblotted to polyvinylidene difluoride membrane. Membranes were blocked in 5% nonfat milk, probed with IG7 mAb (1:5,000 dilution), and detected with an horseradish peroxidase-conjugated secondary antibody (1:10,000, anti-mouse IgG). Blots were developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific).
LC-MS peptide analysis
Electrospray LC-MS/MS was performed on tryptic and Lys-C peptides generated from gel bands and individual collagenase HPLC fractions using an LTQ XL ion-trap mass spectrometer (Thermo Scientific) equipped with in-line LC using a C4 5-μm capillary column (300 μm × 150 mm; Higgins Analytical RS-15M3-W045) eluted at 4.5 μl min. The LC mobile phase consisted of buffer A (0.1% formic acid in MilliQ water) and buffer B (0.1% formic acid in 3:1 acetonitrile/n-propyl alcohol, v/v). The LC sample stream was introduced into the mass spectrometer by electrospray ionization (ESI) with a spray voltage of 4 kV. Proteome Discoverer search software (Thermo Scientific) was used for peptide identification using the NCBI protein database. Proline and lysine modifications were examined manually by scrolling or averaging the full scan over several minutes so that all of the post-translational variations of a given peptide appeared together in the full scan. Cross-linked peptides were identified manually by calculating theoretical parent ion masses and possible MS/MS ions and matching these to the actual parent mass and MS/MS spectrum.
LC-MS of cross-linking amino acids
Samples of freeze-dried tissues both untreated and reduced with sodium borohydride (36), bacterial digested tissue, and subsequent chromatographic peptide pools were hydrolyzed in 6 n HCl at 108 °C for 24 h. Cross-linking amino acids were enriched from hydrolysates by partition on a hydrated cellulose column (37). Briefly, a column was prepared by pouring a 5% cellulose (CF1, Whatman) slurry in butanol/water/acetic acid (4:1:1, v/v/v) into a 10-ml syringe plugged at the bottom with glass wool. The column was washed three times with butanol/water/acetic acid (4:1:1, v/v/v) before the sample dissolved in a 5% cellulose/butanol/water/acetic acid slurry was applied. Free amino acids were removed by washing the column with butanol/water/acetic acid solution. The multicharged bound cross-linking amino acids were eluted with water and freeze-dried for mass spectral analysis. Electrospray LC-MS/MS was performed on the LTQ XL using a Cogent 4 diamond hydride column (15 cm × 1 mm; Microsolv Technology, 70000-15P-1) eluted at 50 μl min. The LC mobile phase consisted of buffer A (0.1% formic acid in MilliQ water) and buffer B (0.1% formic acid in 80% acetonitrile).
Author contributions
D. R. E. conceptualization; D. R. E. resources; D. R. E. and M. W. supervision; D. R. E. funding acquisition; D. R. E., M. W., and J. R. methodology; D. R. E. and M. W. writing-original draft; D. R. E., M. W., and J. R. writing-review and editing; M. W. data curation; M. W. formal analysis; M. W. and J. R. investigation.
This work was supported in whole or in part by National Institutes of Health NIAMS Grants AR037318 and AR036794 (to D. E.) and NICHD Grant HD070394 (to D. E.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HP
- hydroxylysyl pyridinoline
- HHL
- histidinohydroxylysinonorleucine
- HHMD
- histidinohydroxymerodesmosine
- HLNL
- hydroxylysinonorleucine
- LNL
- lysinonorleucine
- IMAC
- immobilized metal ion affinity chromatography
- Hyl
- hydroxylysine
- glcgal
- glucosylgalactosyl
- Des
- desmosine.
References
- 1. Ricard-Blum S. (2011) The collagen family. Cold Spring Harb. Perspect. Biol. 3, a004978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bella J., and Hulmes D. J. (2017) Fibrillar collagens. Subcell. Biochem. 82, 457–490 10.1007/978-3-319-49674-0_14 [DOI] [PubMed] [Google Scholar]
- 3. Eyre D. R., Paz M. A., and Gallop P. M. (1984) Cross-linking in collagen and elastin. Annu. Rev. Biochem. 53, 717–748 10.1146/annurev.bi.53.070184.003441 [DOI] [PubMed] [Google Scholar]
- 4. Rodriguez-Pascual F., and Slatter D. A. (2016) Collagen cross-linking: insights on the evolution of metazoan extracellular matrix. Sci. Rep. 6, 37374 10.1038/srep37374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gjaltema R. A., and Bank R. A. (2017) Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 52, 74–95 10.1080/10409238.2016.1269716 [DOI] [PubMed] [Google Scholar]
- 6. Yamauchi M., and Sricholpech M. (2012) Lysine post-translational modifications of collagen. Essays Biochem. 52, 113–133 10.1042/bse0520113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hudson D. M., and Eyre D. R. (2013) Collagen prolyl 3-hydroxylation: a major role for a minor post-translational modification? Connect. Tissue Res. 54, 245–251 10.3109/03008207.2013.800867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hudson D. M., Weis M., Rai J., Joeng K. S., Dimori M., Lee B. H., Morello R., and Eyre D. R. (2017) P3h3-null and Sc65-null mice phenocopy the collagen lysine under-hydroxylation and cross-linking abnormality of Ehlers-Danlos syndrome type VIA. J. Biol. Chem. 292, 3877–3887 10.1074/jbc.M116.762245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamauchi M., London R. E., Guenat C., Hashimoto F., and Mechanic G. L. (1987) Structure and formation of a stable histidine-based trifunctional cross-link in skin collagen. J. Biol. Chem. 262, 11428–11434 [PubMed] [Google Scholar]
- 10. Mechanic G. L., Katz E. P., Henmi M., Noyes C., and Yamauchi M. (1987) Locus of a histidine-based, stable trifunctional, helix to helix collagen cross-link: stereospecific collagen structure of type I skin fibrils. Biochemistry 26, 3500–3509 10.1021/bi00386a038 [DOI] [PubMed] [Google Scholar]
- 11. Yamauchi M., Woodley D. T., and Mechanic G. L. (1988) Aging and cross-linking of skin collagen. Biochem. Biophys. Res. Commun. 152, 898–903 10.1016/S0006-291X(88)80124-4 [DOI] [PubMed] [Google Scholar]
- 12. Woodley D. T., Yamauchi M., Wynn K. C., Mechanic G., and Briggaman R. A. (1991) Collagen telopeptides (cross-linking sites) play a role in collagen gel lattice contraction. J. Invest. Dermatol. 97, 580–585 10.1111/1523-1747.ep12481920 [DOI] [PubMed] [Google Scholar]
- 13. Reiser K. M. (1994) Influence of age and long-term dietary restriction on enzymatically mediated crosslinks and nonenzymatic glycation of collagen in mice. J. Gerontol. 149, B71–B79 [DOI] [PubMed] [Google Scholar]
- 14. Paul R. G., Tarlton J. F., Purslow P. P., Sims T. J., Watkins P., Marshall F., Ferguson M. J., and Bailey A. J. (1997) Biomechanical and biochemical study of a standardized wound healing model. Int. J. Biochem. Cell Biol. 29, 211–220 10.1016/S1357-2725(96)00134-3 [DOI] [PubMed] [Google Scholar]
- 15. Housley T., Tanzer M. L., Henson E., and Gallop P. M. (1975) Collagen crosslinking: isolation of hydroxyaldol-histidine, a naturally-occurring crosslink. Biochem. Biophys. Res. Commun. 67, 824–830 10.1016/0006-291X(75)90887-6 [DOI] [PubMed] [Google Scholar]
- 16. Hanson D. A., and Eyre D. R. (1996) Molecular site specificity of pyridinoline and pyrrole cross-links in type I collagen of human bone. J. Biol. Chem. 271, 26508–26516 10.1074/jbc.271.43.26508 [DOI] [PubMed] [Google Scholar]
- 17. Eyre D. R., and Weis M. A. (2013) Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta. Calcif. Tissue Int. 93, 338–347 10.1007/s00223-013-9723-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamauchi M., Taga Y., Hattori S., Shiiba M., and Terajima M. (2018) Analysis of collagen and elastin cross-links. Methods Cell Biol. 143, 115–132 10.1016/bs.mcb.2017.08.006 [DOI] [PubMed] [Google Scholar]
- 19. Bornstein P., Kang A. H., and Piez K. A. (1966) The nature and location of intramolecular cross-links in collagen. Proc. Natl. Acad. Sci. U.S.A. 55, 417–424 10.1073/pnas.55.2.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanzer M. L., Housley T., Berube L., Fairweather R., Franzblau C., and Gallop P. M. (1973) Structure of two histidine-containing crosslinks from collagen. J. Biol. Chem. 248, 393–402 [PubMed] [Google Scholar]
- 21. Robins S. P., and Bailey A. J. (1973) The chemistry of the collagen cross-links: the characterization of fraction C, a possible artifact produced during the reduction of collagen fibres with borohydride. Biochem. J. 135, 657–665 10.1042/bj1350657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rojkind M., Blumenfeld O. O., and Gallop P. M. (1966) Localization and partial characterization of an aldehydic component in tropocollagen. J. Biol. Chem. 241, 1530–1536 [PubMed] [Google Scholar]
- 23. Bailey A. J., and Peach C. M. (1969) Identification of a labile intermolecular cross-link in collagen. Biochem. J. 111, 12P–13P [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bailey A. J., Peach C. M., and Fowler L. J. (1970) Chemistry of the collagen cross-links: isolation and characterization of two intermediate intermolecular cross-links in collagen. Biochem. J. 117, 819–831 10.1042/bj1170819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kang A. H., and Gross J. (1970) Relationship between the intra and intermolecular cross-links of collagen. Proc. Natl. Acad. Sci. U.S.A. 67, 1307–1314 10.1073/pnas.67.3.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanzer M. L. (1973) Cross-linking of collagen. Science 180, 561–566 10.1126/science.180.4086.561 [DOI] [PubMed] [Google Scholar]
- 27. Bailey A. J., and Sims T. J. (1976) Nature of the cross-links in the polymorphic forms of dermal collagen during development. Biochem. J. 153, 211–215 10.1042/bj1530211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bernstein P. H., and Mechanic G. L. (1980) A natural histidine-based imminium cross-link in collagen and its location. J. Biol. Chem. 255, 10414–10422 [PubMed] [Google Scholar]
- 29. Robins S. P., and Bailey A. J. (1977) Some observations on the ageing in vitro of reprecipitated collagen fibres. Biochim. Biophys. Acta. 492, 408–414 10.1016/0005-2795(77)90092-7 [DOI] [PubMed] [Google Scholar]
- 30. Sims T. J., and Bailey A. J. (1992) Quantitative analysis of collagen and elastin cross-links using a single-column system. J. Chromatogr. 582, 49–55 10.1016/0378-4347(92)80301-6 [DOI] [PubMed] [Google Scholar]
- 31. Fairweather R. B., Tanzer M. L., and Gallop P. M. (1972) Aldol-histidine, a new trifunctional collagen crosslink. Biochem. Biophys. Res. Commun. 48, 1311–1315 10.1016/0006-291X(72)90854-6 [DOI] [PubMed] [Google Scholar]
- 32. Eyre D. R., Weis M. A., and Wu J. J. (2008) Advances in collagen cross-link analysis. Methods 45, 65–74 10.1016/j.ymeth.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heard M. E., Besio R., Weis M., Rai J., Hudson D. M., Dimori M., Zimmerman S. M., Kamykowski J. A., Hogue W. R., Swain F. L., Burdine M. S., Mackintosh S. G., Tackett A. J., Suva L. J., Eyre D. R., and Morello R. (2016) Sc65-null mice provide evidence for a novel endoplasmic reticulum complex regulating collagen Lysyl hydroxylation. PloS Genet. 12, e1006002 10.1371/journal.pgen.1006002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kalamajski S., Liu C., Tillgren V., Rubin K., Oldberg Å., Rai J., Weis M., and Eyre D. R. (2014) Increased C-telopeptide cross-linking of tendon type I collagen in fibromodulin-deficient mice. J. Biol. Chem. 289, 18873–18879 10.1074/jbc.M114.572941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Henkel W., Rauterberg J., and Stirtz T. (1976) Isolation of a cross-linked cyanogen bromide peptide from insoluble rabbit collagen. Tissue differences in glycosylation and hydroxylation of the cross-link. Eur. J. Biochem. 69, 223–231 10.1111/j.1432-1033.1976.tb10877.x [DOI] [PubMed] [Google Scholar]
- 36. Eyre D. (1987) Collagen cross-linking amino acids. Methods Enzymol. 144, 115–139 10.1016/0076-6879(87)44176-1 [DOI] [PubMed] [Google Scholar]
- 37. Naffa R., Holmes G., Ahn M., Harding D., and Norris G. (2016) Liquid chromatography-electrospray ionization mass spectrometry for the simultaneous quantitation of collagen and elastin crosslinks. J. Chromatogr. A 1478, 60–67 10.1016/j.chroma.2016.11.060 [DOI] [PubMed] [Google Scholar]
- 38. Eyre D. R., and Glimcher M. J. (1973) Evidence for intramolecular crosslinks in chicken bone collagen: the isolation of peptides containing allysine aldol. Biochim. Biophys. Acta 295, 301–307 10.1016/0005-2795(73)90097-4 [DOI] [PubMed] [Google Scholar]
- 39. Heine A., DeSantis G., Luz J. G., Mitchell M., Wong C. H., and Wilson I. A. (2001) Observation of covalent intermediates in an enzyme mechanism at atomic resolution. Science 294, 369–374; Correction (2001) Science 294, 2096 10.1126/science.1063601 [DOI] [PubMed] [Google Scholar]
- 40. Giger L., Caner S., Obexer R., Kast P., Baker D., Ban N., and Hilvert D. (2013) Evolution of a designed retro-aldolase leads to complete active site remodeling. Nat. Chem. Biol. 209, 494–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amar A., Meghezzi Boucekkine H., Kaoua A., and Kolli R B. (2010) How to drive imine-enamine tautomerism of pyronic derivatives of biological interest: a theoretical and experimental study of substituent and solvent effects. Comptes Rendus Chimie. 13, 553–560 10.1016/j.crci.2009.11.009 [DOI] [Google Scholar]
- 42. Inani H., Jha A. K., and Easwar S. (2017) An arginine-mediated protocol for the aldol addition of methyl vinyl ketone in water. Chem. Select. 2, 11666–11672 10.1002/slct.201702502 [DOI] [Google Scholar]
- 43. Malone J. P., and Veis A. (2004) Heterotrimeric type I collagen C-telopeptide conformation as docked to its helix receptor. Biochemistry 43, 15358–15366 10.1021/bi048304b [DOI] [PubMed] [Google Scholar]
- 44. Park J., Fu H., and Pei D. (2003) Peptidyl aldehydes as reversible covalent inhibitors of Src homology 2 domains. Biochemistry 42, 5159–5167 10.1021/bi034076u [DOI] [PubMed] [Google Scholar]
- 45. Park J., Fu H., and Pei D. (2004) Peptidyl aldehydes as slow-binding inhibitors of dual-specificity phosphatases. Biorg. Med. Chem. Lett. 14, 685–687 10.1016/j.bmcl.2003.11.027 [DOI] [PubMed] [Google Scholar]
- 46. Holmes D. F., Lu Y., Starborg T., and Kadler K. E. (2018) Collagen fibril assembly and function. Curr. Top. Dev. Biol. 130, 107–142 10.1016/bs.ctdb.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 47. Zhang G., Chen S., Goldoni S., Calder B. W., Simpson H. C., Owens R. T., McQuillan D. J., Young M. F., Iozzo R. V., and Birk D. E. (2009) Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J. Biol. Chem. 284, 8888–8897 10.1074/jbc.M806590200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Belin M. W., Lim L., Rajpal R. K., Hafezi F., Gomes J. A. P., and Cochener B. (2018) Corneal cross-linking: current U.S.A. status report from The Cornea Society. Cornea 37, 1218–1225 10.1097/ICO.0000000000001707 [DOI] [PubMed] [Google Scholar]
- 49. McCall A. S., Kraft S., Edelhauser H. F., Kidder G. W., Lundquist R. R., Bradshaw H. E., Dedeic Z., Dionne M. J., Clement E. M., and Conrad G. W. (2010) Mechanisms of corneal tissue cross-linking in response to treatment with topical riboflavin and long-wavelength ultraviolet radiation (UVA). Invest. Ophthalmol. Vis. Sci. 51, 129–138 10.1167/iovs.09-3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 51. Hanson D. A., Weis M. A., Bollen A. M., Maslan S. L., Singer F. R., and Eyre D. R. (1992) A specific immunoassay for monitoring human bone resorption: quantification of type I collagen cross-linking N-telopeptides in urine. J. Bone Miner. Res. 7, 1251–1258 [DOI] [PubMed] [Google Scholar]