

Abstract
Hsp70 and Hsp90 chaperones are critical for protein quality control in the cytosol, whereas organelle-specific Hsp70/Hsp90 paralogs provide similar protection for mitochondria and the endoplasmic reticulum (ER). Cytosolic Hsp70/Hsp90 can operate sequentially with Hsp90 selectively associating with Hsp70 after Hsp70 is bound to a client protein. This observation has long suggested that Hsp90 could have a preference for interacting with clients at their later stages of folding. However, recent work has shown that cytosolic Hsp70/Hsp90 can directly interact even in the absence of a client, which opens up an alternative possibility that the ordered interactions of Hsp70/Hsp90 with clients could be a consequence of regulated changes in the direct interactions between Hsp70 and Hsp90. However, it is unknown how such regulation could occur mechanistically. Here, we find that the ER Hsp70/Hsp90 (BiP/Grp94) can form a direct complex in the absence of a client. Importantly, the direct interaction between BiP and Grp94 is nucleotide-specific, with BiP and Grp94 having higher affinity under ADP conditions and lower affinity under ATP conditions. We show that this nucleotide-specific association between BiP and Grp94 is largely due to the conformation of BiP. When BiP is in the ATP conformation its substrate-binding domain blocks Grp94; in contrast, Grp94 can readily associate with the ADP conformation of BiP, which represents the client-bound state of BiP. Our observations provide a mechanism for the sequential involvement of BiP and Grp94 in client folding where the conformation of BiP provides the signal for the subsequent recruitment of Grp94.
Keywords: heat shock protein 90 (Hsp90), chaperone, nucleotide, fluorescence resonance energy transfer (FRET), fluorescence anisotropy, nuclear magnetic resonance (NMR), allostery, BiP, Grp94, structural dynamics
Introduction
The ATP-dependent chaperones Hsp703 and Hsp90 play a central role in cellular health via their protective influence on client protein folding and stability. Metazoans express four Hsp90 paralogs: Hsp90α and Hsp90β in the cytosol, Trap-1 in mitochondria, and Grp94 in the ER. Numerous Hsp70 paralogs reside in the cytosol, whereas mitochondria and the ER each house one canonical Hsp70 (Mortalin and BiP, respectively). It has long been known that the eukaryotic Hsp70/Hsp90 system in the cytosol is coordinated by a cytosol-specific cochaperone (Hsp70/Hsp90-organizing protein (HOP)) (1) that facilitates a sequential interaction where Hsp70 binds to the client first and Hsp90 binds subsequently (2). The sequential activity of Hsp70 and Hsp90 in client folding has been observed for both model clients like luciferase and natural clients like the glucocorticoid receptor (3, 4). A reasonable interpretation is that Hsp70 has a preference for interacting with early folding states and Hsp90 has a preference for interacting with late folding states. However, studies with Hsp90 and client proteins have not yielded consistent evidence that Hsp90 has a strong preference for a specific folding state (5). Indeed, the mechanism that enables Hsp70 and Hsp90 to operate sequentially is not known. A complicating factor in addressing this issue for the eukaryotic system is that Hsp70/HOP/Hsp90 is both conformationally and compositionally heterogeneous (6–9).
Recent work has shown that cytosolic Hsp70/Hsp90 homologs can directly interact in the absence of a client. For example, although prokaryotes do not encode a HOP-like cochaperone, the bacterial Hsp70/Hsp90 pair (DnaK/HtpG) interacts directly (10, 11), can function more effectively together than in isolation (12), and shows evidence of DnaK interacting with clients prior to HtpG (13). An additional sign of the coordination between DnaK and HtpG is that DnaK stimulates the ATPase activity of HtpG (12). A collection of disruptive mutations on the HtpG middle domain (MD) and the DnaK nucleotide-binding domain (NBD) have suggested an interaction surface (10, 14). Similar mutations disrupt coordination between the yeast cytosolic Hsp70/Hsp90 pair (15), suggesting that the location of this direct interaction between Hsp70 and Hsp90 may be widely conserved. These observations open up a possibility that the sequential involvement of Hsp70/Hsp90 in client folding could be a consequence of regulated changes in the direct interactions between Hsp70 and Hsp90 and not necessarily a consequence of Hsp90 preferentially binding clients when they are in their later folding states.
Similar to the bacterial system, the ER chaperones BiP and Grp94 lack a bridging cochaperone. Previous findings in the field are consistent with a direct interaction between BiP and Grp94. For example, recombinant BiP can pull down Grp94 in rat ER lysate (16), and BiP and Grp94 both associate with unassembled immunoglobulin chains (17–19). Similar to the cytosolic system, BiP has been suggested to bind to the immunoglobulin heavy chain prior to Grp94 (18). These observations motivated our investigation of whether BiP and Grp94 directly interact and how their interaction is regulated.
Like other Hsp90 family members, Grp94 cycles between open and closed states through successive rounds of ATP binding and hydrolysis (20, 21). Mechanistic studies of BiP are complicated by oligomerization (22–24); however, BiP is predominantly monomeric in the client-bound form (22). Single-molecule analysis of BiP shows nucleotide-dependent conformational changes characteristic of other Hsp70 homologs (25). Specifically, the NBD and substrate-binding domain (SBD) of BiP are docked together under ATP conditions (26) and dissociated under ADP conditions (25). Like other Hsp70 homologs, BiP can trap peptide substrates in the ADP conformation (25–27). Fig. 1 shows structures of BiP and Grp94 with the locations of previously identified mutations that disrupt interactions between bacterial Hsp70 and Hsp90 (10, 14). Importantly, the mutation sites on BiP are exposed in the ADP conformation and largely buried in the ATP conformation, suggesting that Grp94 might bind poorly to the ATP conformation of BiP. This possible conformation-specific interaction mechanism has also been suggested from docking analysis of DnaK and HtpG (14).
Figure 1.

Overview of Grp94 and BiP conformations. Mutation sites that disrupt binding between HtpG and DnaK are shown in red spheres. A, these sites are exposed on the Grp94 open conformation (ADP; Protein Data Bank (PDB) code 2O1V) and the closed conformation (ATP; PDB code 5ULS). B, in contrast, these sites are mostly exposed in the BiP ADP conformation (modeled from PDB code 2KHO) and blocked in the ATP conformation (PDB code 5E84).
Results
Because BiP oligomerization imposes constraints on experimental design, we first performed analytical gel filtration to determine conditions in which BiP is primarily monomeric. As shown in Fig. 2A BiP forms a mixture of oligomeric states that become increasingly populated at higher concentration as the population of the monomeric form decreases. Similar to previous observations, we find that ATP suppresses oligomerization (22, 24). Under ADP conditions an intermediate level of oligomerization is observed. The percentage of monomer as calculated from peak areas (Fig. 2B and Fig. S1) shows that BiP can be maintained in a predominantly monomeric state under all nucleotide conditions at BiP concentrations below 100 nm.
Figure 2.

A, example of concentration-dependent BiP oligomerization. The BiP monomer elutes at 6.8 min. B, BiP can be maintained in a predominantly monomeric state at concentrations below 100 nm under all nucleotide conditions. Error bars are the S.E. of the mean for at least three measurements. C, design of a previously established BiP FRET pair (25). D, the BiP fluorescence spectrum shows anticorrelated changes at donor and acceptor emission under ADP and ATP conditions. mAU, milliabsorbance units; cps, counts per second.
To confirm nucleotide-dependent conformational changes of BiP, we tested a previously established FRET pair used in single-molecule analysis (25). The donor and acceptor pair positions are shown in Fig. 2C. In the ADP conformation the fluorophores are expected to be closer than the Förster distance (R0 = 51 Å), whereas in the ATP conformation the pairs are further apart. Fig. 2D indeed shows lower FRET under ATP conditions and higher FRET under ADP conditions, as indicated by anticorrelated changes in the donor and acceptor emissions at 567 and 668 nm, respectively.
The interaction between BiP and Grp94 is nucleotide-specific
Analytical gel filtration shows that BiP and Grp94 elute independently when incubated with ATP, thereby showing no indication of tight binding. In contrast, under ADP conditions a new elution is observed at 5.7 min (Fig. S1C). Although these results show nucleotide-specific binding between BiP and Grp94, BiP oligomerization makes gel filtration poorly suited for a more detailed analysis.
We developed a fluorescence depolarization binding assay to quantify BiP/Grp94 affinity (Fig. 3). Here, FITC-labeled BiP can be maintained at a low concentration (50 nm) to suppress oligomerization. We find that BiP/Grp94 bind ∼20-fold more tightly with ADP (Kd,ADP of 0.69 ± 0.13 μm) than with ATP (Kd,ATP of 12.2 ± 0.5 μm when fit with a plateau value that is fixed at the saturating value under ADP conditions). Results with ATP are similar with an added ATP-regenerating system, indicating a negligible contribution of accumulated ADP from ATP hydrolysis. The interaction affinities measured under ADP conditions are salt-dependent (Kd of 0.69 ± 0.13 μm at 50 mm KCl and 4.0 ± 0.4 μm at 150 mm KCl), suggesting an electrostatic contribution. In all binding experiments the Grp94 concentration is expressed in monomer units. These results are consistent with a mechanism in which the nucleotide-specific conformations of BiP and/or Grp94 control their association.
Figure 3.
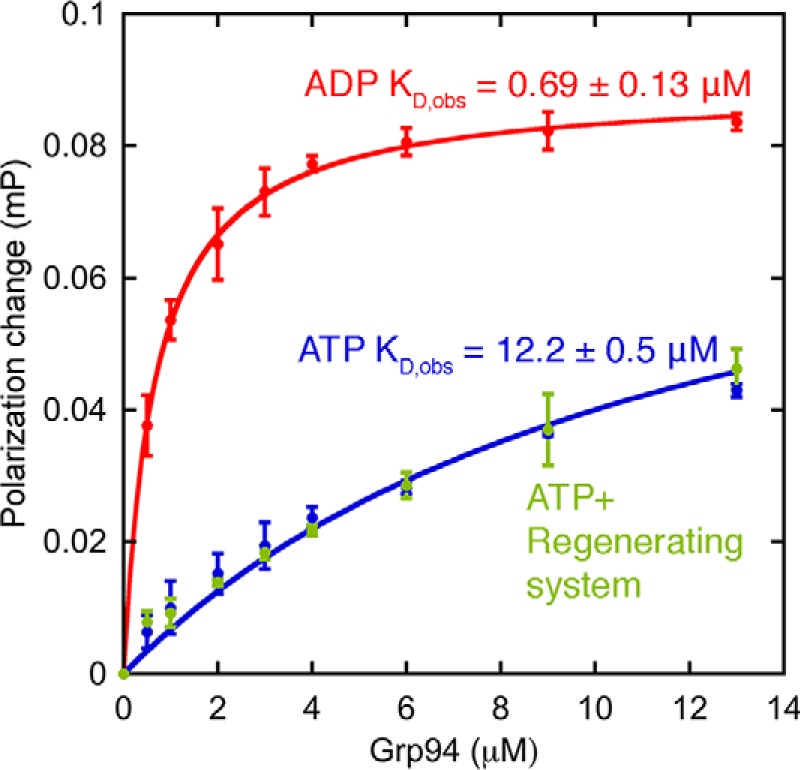
Fluorescence depolarization binding measurements show strong binding between BiP and Grp94 under ADP conditions and weak binding under ATP conditions. Error bars are the S.E. of the mean for at least three measurements. Buffer conditions were 25 mm Tris, pH 7.5, 50 mm KCl, 1 mm MgCl2, 1 mm ADP or ATP, and 1 mg/ml BSA at 37 °C. mP, millipolarization units.
The direct interaction between BiP and Grp94 shows evolutionary conservation to the cytosolic Hsp70/Hsp90 system
Work with the cytosolic Hsp70/Hsp90 systems from Escherichia coli and yeast (10, 15) has shown that 1) the Hsp70/Hsp90 interaction is mediated by the Hsp70 NBD and Hsp90 MD, 2) specific mutations on these domains disrupt the Hsp70/Hsp90 association, and 3) Hsp70/Hsp90 synergistically increase their ATPase activities. We next tested whether BiP/Grp94 share these characteristics.
The interacting domains of BiP and Grp94 were identified by NMR. The BiP SBD and NBD can be purified at high levels and do not oligomerize (26, 28). Because the Grp94 NTD and MD have high-quality TROSY-HSQC spectra, the following chemical shift perturbation experiments were performed: [15N]Grp94 NTD with unlabeled BiP NBD or SBD and [15N]Grp94 MD with unlabeled BiP NBD or SBD. Of these four combinations, a direct interaction is only observed between the BiP NBD and Grp94 MD (Fig. 4A and Fig. S2).
Figure 4.

A, direct binding between the BiP NBD and Grp94 MD shown by chemical shift perturbations ([15N]Grp94 MD alone, black, 150 μm, 32 scans; [15N]Grp94 MD with BiP NBD, red, 200 μm, 32 scans). B, the BiP NBD and full-length BiP have comparable affinities for Grp94 under ADP conditions. C, the Grp94 MD is sufficient for binding the BiP NBD, but full affinity requires both the MD and NTD. D, mutants that disrupt binding between the bacterial Hsp70 and Hsp90 system also disrupt binding between BiP and Grp94. Error bars are the S.E. of the mean for at least three measurements. Buffer conditions for binding experiments are the same as in Fig. 3. mP, millipolarization units.
To quantify the BiP NBD affinity contribution in binding Grp94, we generated an NBD construct that is FITC-labeled at the same site used for full-length BiP. Fig. 4B shows that the NBD and full-length BiP bind Grp94 with almost identical affinities under ADP conditions. The negligible influence of the BiP SBD under ADP conditions is in stark contrast to the role of the SBD under ATP conditions (discussed later).
To further dissect the individual contributions to BiP binding, we examined domains of Grp94 (Fig. 4C). The MD of Grp94 is sufficient for binding to the BiP NBD but with reduced affinity relative to the NTD + MD (NM) fragment, which suggests that a second BiP interaction surface is present on the Grp94 NTD. However, the NTD by itself does not bind, which is consistent with the NMR experiments in Fig. S3B showing no chemical shifts on [15N]Grp94 NTD from addition of unlabeled BiP NBD. The NM fragment of Grp94 has a similar affinity as the full-length construct, indicating a negligible influence of the C-terminal dimerization domain. Grp94 constructs with and without the charged linker have comparable affinity for BiP (0.92 ± 0.07 and 0.69 ± 0.13 μm, respectively).
The direct interaction between the BiP NBD and the Grp94 MD is consistent with findings from the cytosolic Hsp70/Hsp90 systems. Wickner and co-workers (10, 14) have identified mutations on these domains that cause both in vivo defects and in vitro interaction defects. We selected DnaK/HtpG mutations with severe interaction defects to test with BiP/Grp94. The Grp94 mutations R466A and K467A correspond to the HtpG mutations K354A and R355A. The BiP mutation E243A corresponds to the DnaK mutation E217A. Fig. 4D shows the critical influence of Glu-243 on BiP and Lys-467 on Grp94 and a smaller influence of Arg-466 on Grp94.
Although the above results indicate that BiP Glu-243 plays a direct role in binding Grp94, an alternative explanation is that the E243A substitution exerts an indirect influence by shifting BiP to the ATP conformation with a weak affinity for Grp94. Because Glu-243 is close to the BiP SBD, we considered this a possible confounding factor. Therefore, we constructed the E243A variant of the BiP NBD as a control. The NBD E243A shows minimal binding to Grp94 (Fig. 4D), which rules out the possibility that E243A disrupts Grp94 binding through a shifted conformational equilibrium of BiP. Collectively, the above results show that the binding surface of BiP/Grp94 is similar to the binding surface of cytosolic Hsp70/Hsp90.
BiP activates the ATPase of Grp94
Bacterial Hsp70/Hsp90 show enhanced ATPase activity when together versus their additive contributions in isolation (12). BiP and Grp94 also exhibit a higher ATPase activity together compared with their additive individual contributions (Fig. 5). The BiP NBD elicits a greater increase in ATPase versus full-length BiP. This is likely due to Grp94 having a higher affinity to BiP NBD compared with full-length BiP under ATP conditions as explained later. To determine whether the activity increase is due to BiP or Grp94 we constructed a BiP mutant that can bind but not hydrolyze ATP (T229G). This variant shows that the BiP NBD directly stimulates Grp94's ATPase, increasing the activity 8-fold. The Grp94 inhibitor radicicol decreases the total ATPase to the BiP-only level, further verifying that the activation comes from Grp94. As controls we tested binding-impaired mutants of BiP and Grp94 (BiP NBD E243A and Grp94 K467A), and in both cases no synergistic increase in activity is observed. Collectively, our results show that the direct interaction between BiP and Grp94 has many characteristics that are evolutionarily conserved with the cytosolic Hsp70/Hsp90 system (10, 14, 15).
Figure 5.
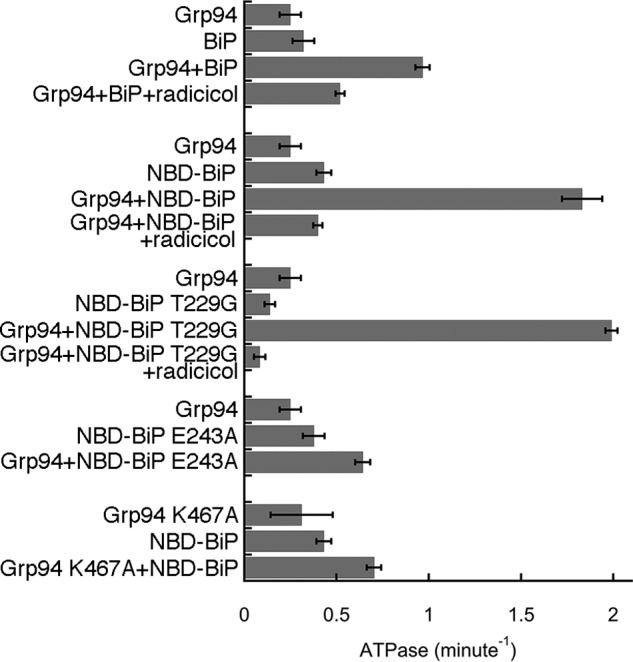
BiP activates the ATPase of Grp94 ATPase. The ATPase-deficient construct of BiP (T229G) shows that the increase in activity is due to Grp94. The binding-deficient mutants (BiP E243A and Grp94 K467A) show no activation, yielding roughly additive ATPase results. Error bars are the S.E. of the mean for at least three measurements. Buffer conditions were 25 mm Tris, pH 7.5, 50 mm KCl, 1 mm MgCl2, 1 mm ATP, and 1 mg/ml BSA at 37 °C.
The BiP SBD is responsible for low Grp94 affinity under ATP conditions
We next wanted to determine the mechanism underlying the nucleotide-specific affinity between BiP and Grp94 (Fig. 3). Although the data in Fig. 4B show that the BiP SBD has a negligible influence on Grp94 binding under ADP conditions, we find that the BiP SBD plays a critical role in preventing Grp94 binding under ATP conditions (Fig. 6A). This conclusion comes from experiments performed with the NM fragment of Grp94 because this fragment can be purified at the high concentrations necessary to accurately measure the weak binding to full-length BiP when with ATP. Full-length BiP binds the NM fragment strongly under ADP conditions and weakly under ATP conditions (apparent affinities are shown in Fig. 6A; binding curves are shown in Fig. S3, A and B). In contrast, the BiP NBD has equivalent affinities for Grp94 NM under both ADP and ATP conditions. This comparison shows that the BiP SBD makes a major contribution to the low Grp94 affinity under ATP conditions.
Figure 6.

A, weak binding to Grp94 under ATP conditions is only observed in BiP constructs that have the SBD. Buffer conditions are the same as in Fig. 3. B, a docking model shows that the BiP SBD clashes with the Grp94 MD when BiP is in the ATP conformation. Grp94 is shown as a surface, and BiP is shown as a ribbon. Domains are colored separately for Grp94 (CTD, blue; MD, green; NTD, orange) and BiP (NBD, blue; SBD-β, gray; SBD-α, orange). C, docking model suggests a salt bridge between Grp94 Lys-467 and BiP Glu-243 with Arg-466 at the periphery. Docking models were built using ClusPro 2.0 (39) with no constraints.
Interestingly, a docking model shows the BiP SBD clashing with the Grp94 MD when BiP is in the ATP conformation, which provides a structural explanation for the role of BiP SBD in preventing Grp94 binding under ATP conditions (Fig. 6B). This docking model shows Glu-243 of BiP forming a salt bridge to Lys-467 of Grp94, whereas Arg-466 is at the periphery (Fig. 6C). These locations are consistent with the strong binding defect from BiP E243A and Grp94 K467A and the more modest defect from R466A (Fig. 4D). We propose that the Grp94-binding site on the BiP NBD is sterically blocked by the SBD when BiP is in the ATP conformation.
BiP and Grp94 can only associate when BiP is in the ADP conformation
The above mechanism of conformation-specific binding makes both thermodynamic and kinetic predictions. The thermodynamic prediction is that Grp94 should pull the BiP conformational equilibrium toward the ADP conformation. Single-molecule experiments show that full-length BiP exists in a mixture of conformational states under ADP conditions (25), indicating that it may be possible to observe a shift in population. Accordingly, we treat BiP as having two conformations: an “ADP” conformation (Fig. 1B, left) that has the NBD and SBD dissociated and a closed lid with high FRET and an “ATP” conformation (Fig. 1B, right) that has the NBD and SBD docked and an open lid with low FRET. Although other conformational states of BiP have been detected, we wanted to test whether this two-state description is sufficient to provide quantitative predictions for conformation-specific binding between BiP and Grp94.
Using the FRET pair described in Fig. 2, we find that BiP FRET increases with increasing Grp94 (Fig. 7A, red circles), indicating an increased population of the BiP ADP conformation. Under ATP conditions the Grp94-induced increase in FRET is much weaker (Fig. 7A, blue circles). The conformation-specific binding model demands that Grp94 mutants with weak binding will have a weak influence on the BiP conformational equilibrium, which is indeed observed for the K467A mutant of Grp94 (Fig. 7A, green circles).
Figure 7.

A, Grp94 increases BiP FRET efficiency, indicative of a higher population of the BiP ADP conformation. The binding-defective mutant Grp94 K467A loses its capability to shift the BiP conformation. Solid lines are fits using Equation 1. B, kinetic FRET measurements show BiP transitioning from the ATP conformation to the ADP conformation (green circles). A similar experiment performed with Grp94 shows similar slow changes in FRET (red circles). C, binding kinetics between BiP and Grp94 is slow when BiP starts in the ATP conformation and transitions to the ADP conformation (red circles). In contrast, binding between BiP and Grp94 is fast when BiP starts in the ADP conformation (blue circles). Solid lines in B and C are single-exponential fits. D, kinetic model of conformation-specific binding between BiP and Grp94. Buffer conditions are the same as in Fig. 3. Error bars are the S.E. of the mean for at least three measurements. mP, millipolarization units.
We tested whether the above FRET results could be quantitatively described with a linked equilibrium model (Equation 1 under “Experimental procedures”) in which BiP can only bind Grp94 when BiP is in the ADP conformation. This model has two fitting parameters: 1) the BiP conformational equilibrium, Kconf, which is influenced by the nucleotide condition, and 2) the intrinsic affinity of Grp94 for BiP when BiP is in the ADP conformation, KD,int. These parameters can be used to calculate predicted observed binding affinities between BiP and Grp94 (via Equation 2 under “Experimental procedures”), which can be compared against the experimental values from Fig. 3.
The conformation-specific binding model provides a good fit for the FRET data (Fig. 7A, solid lines), yielding predicted observed binding affinities (ADP, 0.27 ± 0.02 μm; ATP, 14.2 ± 10.3 μm) that are close to the measured values using fluorescence depolarization (Fig. 3; ADP, 0.69 ± 0.13 μm; ATP, 12.2 ± 0.5 μm). The large error for the predicted ATP affinity is from fitting uncertainty due to the lack of a saturating value at high Grp94 concentration. We conclude that the FRET data in Fig. 7A and the binding data in Fig. 3 are quantitatively consistent with Grp94 being able to bind only to the ADP conformation of BiP.
Although the BiP conformational equilibrium makes the major contribution to the observed affinities, the KD,int fit values under ADP and ATP conditions are different, suggesting that Grp94 has 5-fold lower affinity for BiP when Grp94 is in the ATP state. At first glance this result seems at odds with Fig. 6A, which shows that the NM fragment of Grp94 binds to the BiP NBD with the same apparent affinity under ADP and ATP conditions. However, we wondered whether full-length Grp94 could behave differently from the monomeric NM fragment because the Grp94 dimer can undergo ATP-driven conformational changes. Therefore, we compared binding affinities between full-length Grp94 and the BiP NBD under ADP and ATP conditions and indeed observe 5-fold weaker binding under ATP conditions (Fig. S3C), suggesting that the Grp94 conformation has a modest influence on the affinity for BiP.
We next focused on kinetic predictions associated with the proposed model of conformation-specific binding. Grp94 can pull the BiP equilibrium toward the ADP conformation by selectively binding that conformation of BiP and also potentially by accelerating the ATP-to-ADP conformational transition of BiP, potentially through accelerated ATP hydrolysis. To explore this possibility, we measured the ATP-to-ADP conformational transition rate of BiP by preincubating BiP with a small concentration of ATP and then adding an excess of ADP. The FRET change over time is slow (0.34 ± 0.07 min−1; Fig. 7B, green circles), which is within error of the steady-state BiP ATPase rate (0.41 ± 0.03 min−1; Fig. 5). These kinetics are attributed to slow ATP hydrolysis rather than nucleotide exchange (29). The ATP-to-ADP conformational transition rate of BiP is slightly slower in the presence of Grp94 (kobs,FRET = 0.21 ± 0.07 min−1; Fig. 7B, red circles), which rules out the possibility that Grp94 stabilizes the ADP conformation of BiP by accelerating the ATP-to-ADP conformational transition.
The slow kinetics of BiP transitioning from the ATP conformation to the ADP conformation enables a kinetic test of conformation-specific binding between BiP and Grp94. Specifically, if BiP starts in the ATP conformation and is transitioned to the ADP conformation, then the observed rate of Grp94 binding should become kinetically controlled by the slow rate of BiP's conformational change (kconf = 0.34 ± 0.07 min−1). In contrast, if BiP starts in the ADP conformation, then the binding to Grp94 could be much faster. The fluorescence depolarization (FP) binding assay described in Fig. 3 confirms these predictions. Specifically, when BiP starts in the ADP conformation, Grp94 binds quickly (kbind = 4.5 ± 1.3 min−1, almost within the manual mixing time). In contrast, when BiP is forced to start in the ATP conformation, the binding between BiP and Grp94 is slow (kobs,FP = 0.30 ± 0.01 min−1; Fig. 7C, red circles). Fig. 7D shows that the BiP and Grp94 binding kinetics are in quantitative agreement with predictions in which Grp94 can only bind the ADP conformation of BiP (Equation 3).
Discussion
Numerous studies have shown a functional interplay between Hsp70 and Hsp90 with Hsp70 acting upstream of Hsp90 in binding client proteins (2, 4, 10–14, 30). A natural interpretation is that Hsp90 favors associating with clients in their later stages of folding. However, studies of Hsp90 with client proteins in isolation have been inconclusive on whether Hsp90 has a strong preference for any particular client folding state. For example, three separate studies of the client p53 detected Hsp90 binding to p53 in unfolded (31), molten globule–like (32), and native (33) states. Our findings support a mechanism in which the ADP conformation of BiP specifically interacts with Grp94. As BiP can stably trap clients in its ADP conformation, our finding suggests that the client-bound conformation of BiP, not necessarily the client folding state, is a signal for the subsequent recruitment of the Grp94.
Although the eukaryotic cytosolic cochaperone HOP coordinates the assembly of Hsp70/Hsp90 complexes, studies of the bacterial chaperone system have shown that a HOP-like cochaperone is not necessary for coordination between Hsp70 and Hsp90. Subsequently, this relationship has also been demonstrated for the yeast cytosolic Hsp70/Hsp90 pair (15) and suggested for the mitochondrial Hsp70/Hsp90 pair (34). Our observations show that the mouse ER Hsp70/Hsp90 pair also can interact directly in a way that is similar to cytosolic Hsp70/Hsp90. Specifically, the BiP NBD binds at the Grp94 MD (Fig. 4, A–C), and previously established mutations that disrupt cytosolic Hsp70/Hsp90 also disrupt BiP/Grp94 (Fig. 4D). The large binding defects from K467A and E243A may have contributions from both the loss of a salt bridge and the creation of an unpaired buried charge at the binding interface. Similar to the bacterial Hsp70/Hsp90 system (12), we find that BiP activates the ATPase of Grp94 (Fig. 5). Our results indicate that the direct interaction and coordination between the Hsp70/Hsp90 pair is conserved across both the cytosolic and ER-resident family members.
Importantly, we find that BiP binds tightly to Grp94 under ADP conditions and weakly under ATP conditions (Fig. 3) and that the nucleotide-specific conformations adopted by BiP dictate the binding of Grp94. A docking model suggests a steric clash of the BiP SBD when BiP is in the ATP conformation (Fig. 6B). As demanded by this structural explanation, we show that the SBD minimally influences Grp94 binding under ADP conditions (Fig. 4B) but is required for suppressing Grp94 binding under ATP conditions (Fig. 6A). Finally, the mechanism of conformation-specific binding quantitatively rationalizes the binding and FRET data (Fig. 7). Although the conformation-specific binding model, which allows only two BiP conformations, is an oversimplification, our results lay out a quantitative foundation for building more detailed models describing how BiP and Grp94 facilitate client folding.
Experimental procedures
Two mouse Grp94 constructs were tested for BiP binding: Grp94(72–765) and a construct with the charged linker removed (Δ287–328) (35). Grp94 domain constructs were generated based on the following domain boundaries: NTD (amino acids 72–337), MD (amino acids 338–594), and CTD (amino acids 595–765). The mouse BiP construct (amino acids 27–655) was divided into two domains: NBD (amino acids 27–411) and SBD (amino acids 418–655). Grp94, BiP, and their variants were purified via nickel-nitrilotriacetic acid, ion exchange, and gel filtration (36).
HPLC
To examine the concentration-dependent BiP oligomerization, purified BiP (12 μm BiP stored in 25 mm Tris, pH 7.5, 150 mm KCl, 5% glycerol, and 1 mm β-mercaptoethanol) was diluted into 25 mm Tris, pH 7.5, 50 mm KCl, and 1 mm MgCl2 with or without 1 mm nucleotides (ADP and ATP). After incubating for 1–3 h at 25 °C, 10 μl of each BiP sample at concentrations between 4 and 0.1 μm was injected onto an Agilent AdvanceBio SEC 300-Å, 2.7-μm, 4.6 × 300-mm analytical gel filtration column pre-equilibrated with BiP dilution buffer. Samples were run over the column at a flow rate of 0.35 ml/min. BiP's elution profile was detected by absorbance at 220 nm. The percentage of BiP monomer was calculated by integrating peak areas using Agilent OpenLab CDS software. The BiP/Grp94 nucleotide-specific interaction was measured using 1.75 μm BiP and 1.75 μm Grp94 dimer. The elution profiles were generated using the same method as the BiP oligomerization experiments.
BiP FRET
A double-cysteine BiP mutant (G518C/Y636C) was expressed and purified as above for WT BiP. After the ion-exchange step, BiP was dialyzed into 25 mm Tris, pH 7.0, 50 mm KCl, and 0.25 mm TCEP for 1 h at 25 °C. BiP G518C/Y636C was labeled simultaneously with Invitrogen Alexa Fluor 555 C2-maleimide and Alexa Fluor 647 C2-maleimide at a 2.5:1 molar ratio of each dye:BiP. The dye reaction was quenched with an excess of β-mercaptoethanol. Excess dye was separated from labeled BiP with gel filtration. The final solution for protein storage was 25 mm Tris, pH 7.0, 50 mm KCl, 5% glycerol, and 1 mm β-mercaptoethanol.
For FRET experiments, double-labeled BiP was diluted to 50 nm in buffer containing 25 mm Tris, pH 7.5, 50 mm KCl, 1 mm MgCl2, 1 mm β-mercaptoethanol, and 1 mg/ml BSA. BiP was incubated in buffer, 1 mm ADP, or 1 mm ATP for 30–55 min at 37 °C. Each spectrum was taken by exciting the donor fluorophore at 532 nm and detecting an emission spectrum from 542 to 750 nm.
For FRET kinetic experiments, double-labeled BiP was diluted to 50 nm in buffer containing 25 mm Tris, pH 7.5, 50 mm KCl, 1 mm MgCl2, 50 μm ATP, and 1 mg/ml BSA at 37 °C. After reaching equilibration, 1 mm ADP was added to initiate the conformational change of BiP. The fluorophore signal was monitored via exciting the donor fluorophore at 532 nm and detecting emission signal at 567 and 668 nm with a time interval of 10 s. Similar experiments were performed with an addition of 1 μm Grp94.
Fluorescence depolarization binding measurements
The D27C variant of BiP and the BiP NBD were labeled with a 3-fold excess of FITC under 25 mm Tris, pH 7.5, 50 mm KCl, and 0.25 mm TCEP at room temperature for 1 h. The labeling reaction was quenched by adding 5 mm β-mercaptoethanol. Labeled protein was separated from the free dye via gel filtration. The final solution for protein storage was 25 mm Tris, pH 7.5, 50 mm KCl, 5% glycerol, and 1 mm β-mercaptoethanol. Fluorescence depolarization measurements were performed with 50 nm FITC-labeled BiP on a Fluromax-4 spectrofluorometer (Jobin Yvon Technology) with an excitation wavelength of 495 nm and an emission wavelength of 517 nm. Buffer conditions were 25 mm Tris, pH 7.5, 50 mm KCl, 1 mm MgCl2, 1 mm ADP or ATP, and 1 mg/ml BSA at 37 °C. Binding experiments with an ATP-regenerating system included an additional 17 μg/ml pyruvate kinase (Sigma-Aldrich) and 0.4 mm phosphoenolpyruvate (Sigma-Aldrich).
For fluorescence depolarization kinetic measurements, FITC-labeled BiP was diluted to 50 nm in buffer containing 25 mm Tris, pH 7.5, 50 mm KCl, 1 mm MgCl2, 50 nm ATP, 1 mg/ml BSA at 37 °C, and 1 μm Grp94 dimer. After reaching equilibration, 1 mm ADP was added to initiate the conformational change of BiP. A similar experiment was performed where Grp94 was added to a preincubated BiP sample in 1 mm ADP. Kinetics were measured using the excitation and emission wavelength as described above with a time interval of 15 s.
Conformation-specific binding model
The FRET measurements in Fig. 7A were fit with a linked conformational equilibrium model (37) assuming that only the ADP conformation of BiP can bind to Grp94. The fitting has Kconf and KD,int as two floating parameters.
| (Eq. 1) |
where E0 is the FRET efficiency baseline in the BiP ATP condition, and ΔE is the FRET efficiency change from the BiP ATP baseline to the plateau of BiP ADP conformation. The error reported for Kconf and KD,int is the fitting error. The observed binding constant was calculated via Equation 2.
| (Eq. 2) |
For the BiP and Grp94 binding kinetics in Fig. 7D, the predicted observed rate constant kobs was calculated via Equation 3.
| (Eq. 3) |
NMR
15N-Labeled Grp94 NTD and 15N-labeled Grp94 MD were expressed in M9 medium containing 15NH4Cl (Cambridge Isotope Laboratories) as the sole nitrogen source. All experiments were performed on a Bruker Avance 800-Hz spectrometer. 1H-15N TROSY-HSQC spectra were collected using the standard pulse sequence (trosyetf3gpsi) in the Bruker library. All experiments were processed with TopSpin (Bruker) and analyzed with the CcpNmr package (38). Buffer conditions were 25 mm KH2PO4, pH 7.0, and 50 mm KCl. The Grp94 NTD TROSY-HSQC was collected at 20 °C, and the Grp94 MD TROSY-HSQC was collected at 40 °C.
ATPase assay
The ATPase activities of BiP and Grp94 constructs were measured using an enzyme-coupled assay as described previously with 2 μm Grp94 and BiP (36). Buffer conditions were 25 mm Tris, pH 7.5, 50 mm KCl, 1 mm MgCl2, 1 mm ATP, and 1 mg/ml BSA at 37 °C.
Author contributions
M. S., S. L., and T. O. S. conceptualization; M. S., J. L. K., S. L., and T. O. S. formal analysis; M. S., J. L. K., and T. O. S. validation; M. S., J. L. K., and S. L. investigation; M. S., J. L. K., S. L., and T. O. S. visualization; M. S., J. L. K., S. L., and T. O. S. methodology; M. S., J. L. K., and T. O. S. writing-original draft; M. S., J. L. K., and T. O. S. writing-review and editing; T. O. S. resources; T. O. S. supervision; T. O. S. funding acquisition; T. O. S. project administration.
Supplementary Material
Acknowledgments
We thank Herwig Schüler (Karolinska Institute) for the gift of BiP plasmids. We thank members of the Street laboratory for valuable discussions.
This work was supported by NIGMS, National Institutes of Health Grant R01 GM115356. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3 and Table S1.
- Hsp
- heat-shock protein
- Grp94
- glucose-regulated protein 94
- BiP
- binding immunoglobulin protein
- HOP
- Hsp70/Hsp90-organizing protein
- NBD
- nucleotide-binding domain
- SBD
- substrate-binding domain
- ER
- endoplasmic reticulum
- TROSY
- transverse relaxation-optimized spectroscopy
- HSQC
- heteronuclear single quantum coherence
- MD
- middle domain
- NTD
- N-terminal domain
- NM
- NTD + MD
- CTD
- C-terminal domain
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Chen S., Prapapanich V., Rimerman R. A., Honoré B., and Smith D. F. (1996) Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins hsp90 and hsp70. Mol. Endocrinol. 10, 682–693 10.1210/mend.10.6.8776728 [DOI] [PubMed] [Google Scholar]
- 2. Morishima Y., Murphy P. J., Li D. P., Sanchez E. R., and Pratt W. B. (2000) Stepwise assembly of a glucocorticoid receptor·hsp90 heterocomplex resolves two sequential ATP-dependent events involving first hsp70 and then hsp90 in opening of the steroid binding pocket. J. Biol. Chem. 275, 18054–18060 10.1074/jbc.M000434200 [DOI] [PubMed] [Google Scholar]
- 3. Wegele H., Wandinger S. K., Schmid A. B., Reinstein J., and Buchner J. (2006) Substrate transfer from the chaperone Hsp70 to Hsp90. J. Mol. Biol. 356, 802–811 10.1016/j.jmb.2005.12.008 [DOI] [PubMed] [Google Scholar]
- 4. Kirschke E., Goswami D., Southworth D., Griffin P. R., and Agard D. A. (2014) Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697 10.1016/j.cell.2014.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Radli M., and Rüdiger S. G. D. (2018) Dancing with the diva: Hsp90-client interactions. J. Mol. Biol. 430, 3029–3040 10.1016/j.jmb.2018.05.026 [DOI] [PubMed] [Google Scholar]
- 6. Southworth D. R., and Agard D. A. (2011) Client-loading conformation of the Hsp90 molecular chaperone revealed in the cryo-EM structure of the human Hsp90:Hop complex. Mol. Cell 42, 771–781 10.1016/j.molcel.2011.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmid A. B., Lagleder S., Gräwert M. A., Röhl A., Hagn F., Wandinger S. K., Cox M. B., Demmer O., Richter K., Groll M., Kessler H., and Buchner J. (2012) The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 31, 1506–1517 10.1038/emboj.2011.472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Röhl A., Wengler D., Madl T., Lagleder S., Tippel F., Herrmann M., Hendrix J., Richter K., Hack G., Schmid A. B., Kessler H., Lamb D. C., and Buchner J. (2015) Hsp90 regulates the dynamics of its cochaperone Sti1 and the transfer of Hsp70 between modules. Nat. Commun. 6, 6655 10.1038/ncomms7655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alvira S., Cuéllar J., Röhl A., Yamamoto S., Itoh H., Alfonso C., Rivas G., Buchner J., and Valpuesta J. M. (2014) Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nat. Commun. 5, 5484 10.1038/ncomms6484 [DOI] [PubMed] [Google Scholar]
- 10. Genest O., Hoskins J. R., Kravats A. N., Doyle S. M., and Wickner S. (2015) Hsp70 and Hsp90 of E. coli directly interact for collaboration in protein remodeling. J. Mol. Biol. 427, 3877–3889 10.1016/j.jmb.2015.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakamoto H., Fujita K., Ohtaki A., Watanabe S., Narumi S., Maruyama T., Suenaga E., Misono T. S., Kumar P. K., Goloubinoff P., and Yoshikawa H. (2014) Physical interaction between bacterial heat shock protein (Hsp) 90 and Hsp70 chaperones mediates their cooperative action to refold denatured proteins. J. Biol. Chem. 289, 6110–6119 10.1074/jbc.M113.524801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Genest O., Hoskins J. R., Camberg J. L., Doyle S. M., and Wickner S. (2011) Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proc. Natl. Acad. Sci. U.S.A. 108, 8206–8211 10.1073/pnas.1104703108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morán Luengo T., Kityk R., Mayer M. P., and Rüdiger S. G. D. (2018) Hsp90 breaks the deadlock of the Hsp70 chaperone system. Mol. Cell 70, 545–552.e9 10.1016/j.molcel.2018.03.028 [DOI] [PubMed] [Google Scholar]
- 14. Kravats A. N., Doyle S. M., Hoskins J. R., Genest O., Doody E., and Wickner S. (2017) Interaction of E. coli Hsp90 with DnaK involves the DnaJ binding region of DnaK. J. Mol. Biol. 429, 858–872 10.1016/j.jmb.2016.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kravats A. N., Hoskins J. R., Reidy M., Johnson J. L., Doyle S. M., Genest O., Masison D. C., and Wickner S. (2018) Functional and physical interaction between yeast Hsp90 and Hsp70. Proc. Natl. Acad. Sci. U.S.A. 115, E2210–E2219 10.1073/pnas.1719969115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jansen G., Määttänen P., Denisov A. Y., Scarffe L., Schade B., Balghi H., Dejgaard K., Chen L. Y., Muller W. J., Gehring K., and Thomas D. Y. (2012) An interaction map of endoplasmic reticulum chaperones and foldases. Mol. Cell. Proteomics 11, 710–723 10.1074/mcp.M111.016550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melnick J., Aviel S., and Argon Y. (1992) The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J. Biol. Chem. 267, 21303–21306 [PubMed] [Google Scholar]
- 18. Melnick J., Dul J. L., and Argon Y. (1994) Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 370, 373–375 10.1038/370373a0 [DOI] [PubMed] [Google Scholar]
- 19. Meunier L., Usherwood Y. K., Chung K. T., and Hendershot L. M. (2002) A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 13, 4456–4469 10.1091/mbc.e02-05-0311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dollins D. E., Warren J. J., Immormino R. M., and Gewirth D. T. (2007) Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell 28, 41–56 10.1016/j.molcel.2007.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huck J. D., Que N. L., Hong F., Li Z., and Gewirth D. T. (2017) Structural and functional analysis of GRP94 in the closed state reveals an essential role for the pre-N domain and a potential client-binding site. Cell Rep. 20, 2800–2809 10.1016/j.celrep.2017.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Preissler S., Chambers J. E., Crespillo-Casado A., Avezov E., Miranda E., Perez J., Hendershot L. M., Harding H. P., and Ron D. (2015) Physiological modulation of BiP activity by trans-protomer engagement of the interdomain linker. Elife 4, e08961 10.7554/eLife.08961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freiden P. J., Gaut J. R., and Hendershot L. M. (1992) Interconversion of three differentially modified and assembled forms of BiP. EMBO J. 11, 63–70 10.1002/j.1460-2075.1992.tb05028.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carlino A., Toledo H., Skaleris D., DeLisio R., Weissbach H., and Brot N. (1992) Interactions of liver Grp78 and Escherichia coli recombinant Grp78 with ATP: multiple species and disaggregation. Proc. Natl. Acad. Sci. U.S.A. 89, 2081–2085 10.1073/pnas.89.6.2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marcinowski M., Höller M., Feige M. J., Baerend D., Lamb D. C., and Buchner J. (2011) Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 18, 150–158 10.1038/nsmb.1970 [DOI] [PubMed] [Google Scholar]
- 26. Yang J., Nune M., Zong Y., Zhou L., and Liu Q. (2015) Close and allosteric opening of the polypeptide-binding site in a human Hsp70 chaperone BiP. Structure 23, 2191–2203 10.1016/j.str.2015.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Munro S., and Pelham H. R. (1986) An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 46, 291–300 10.1016/0092-8674(86)90746-4 [DOI] [PubMed] [Google Scholar]
- 28. Wisniewska M., Karlberg T., Lehtiö L., Johansson I., Kotenyova T., Moche M., and Schüler H. (2010) Crystal structures of the ATPase domains of four human Hsp70 isoforms: HSPA1L/Hsp70-hom, HSPA2/Hsp70-2, HSPA6/Hsp70B′, and HSPA5/BiP/GRP78. PLoS One 5, e8625 10.1371/journal.pone.0008625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mayer M., Reinstein J., and Buchner J. (2003) Modulation of the ATPase cycle of BiP by peptides and proteins. J. Mol. Biol. 330, 137–144 10.1016/S0022-2836(03)00556-4 [DOI] [PubMed] [Google Scholar]
- 30. Schumacher R. J., Hansen W. J., Freeman B. C., Alnemri E., Litwack G., and Toft D. O. (1996) Cooperative action of Hsp70, Hsp90, and DnaJ proteins in protein renaturation. Biochemistry 35, 14889–14898 10.1021/bi961825h [DOI] [PubMed] [Google Scholar]
- 31. Rudiger S., Freund S. M., Veprintsev D. B., and Fersht A. R. (2002) CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc. Natl. Acad. Sci. U.S.A. 99, 11085–11090 10.1073/pnas.132393699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park S. J., Borin B. N., Martinez-Yamout M. A., and Dyson H. J. (2011) The client protein p53 adopts a molten globule-like state in the presence of Hsp90. Nat. Struct. Mol. Biol. 18, 537–541 10.1038/nsmb.2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Müller L., Schaupp A., Walerych D., Wegele H., and Buchner J. (2004) Hsp90 regulates the activity of wild type p53 under physiological and elevated temperatures. J. Biol. Chem. 279, 48846–48854 10.1074/jbc.M407687200 [DOI] [PubMed] [Google Scholar]
- 34. Sung N., Lee J., Kim J. H., Chang C., Tsai F. T., and Lee S. (2016) 2.4 Å resolution crystal structure of human TRAP1NM, the Hsp90 paralog in the mitochondrial matrix. in Acta Crystallogr. D Struct. Biol. 72, 904–911 10.1107/S2059798316009906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu S., and Street T. O. (2016) 5′-N-Ethylcarboxamidoadenosine is not a paralog-specific Hsp90 inhibitor. Protein Sci. 25, 2209–2215 10.1002/pro.3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Halpin J. C., Huang B., Sun M., and Street T. O. (2016) Crowding activates heat shock protein 90. J. Biol. Chem. 291, 6447–6455 10.1074/jbc.M115.702928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fersht A. (1999) Structure and Mechanism in Protein Science, pp. 127–129, W. H. Freeman and Company, New York [Google Scholar]
- 38. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 39. Kozakov D., Hall D. R., Xia B., Porter K. A., Padhorny D., Yueh C., Beglov D., and Vajda S. (2017) The ClusPro web server for protein-protein docking. Nat. Protoc. 12, 255–278 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.