

Abstract
Growth differentiation factor 8 (GDF8; also known as myostatin) and GDF11 are closely related members of the transforming growth factor β (TGF-β) family. GDF8 strongly and negatively regulates skeletal muscle growth, and GDF11 has been implicated in various age-related pathologies such as cardiac hypertrophy. GDF8 and GDF11 signaling activities are controlled by the extracellular protein antagonists follistatin; follistatin-like 3 (FSTL3); and WAP, follistatin/kazal, immunoglobulin, Kunitz, and netrin domain–containing (WFIKKN). All of these proteins contain a follistatin domain (FSD) important for ligand binding and antagonism. Here, we investigated the structure and function of the FSD from murine WFIKKN2 and compared it with the FSDs of follistatin and FSTL3. Using native gel shift and surface plasmon resonance analyses, we determined that the WFIKKN2 FSD can interact with both GDF8 and GDF11 and block their interactions with the type II receptor activin A receptor type 2B (ActRIIB). Further, we solved the crystal structure of the WFIKKN2 FSD to 1.39 Å resolution and identified surface-exposed residues that, when substituted with alanine, reduce antagonism of GDF8 in full-length WFIKKN2. Comparison of the WFIKKN2 FSD with those of follistatin and FSTL3 revealed differences in both the FSD structure and position of residues within the domain that are important for ligand antagonism. Taken together, our results indicate that both WFIKKN and follistatin utilize their FSDs to block the type II receptor but do so via different binding interactions.
Keywords: transforming growth factor beta (TGF-β), protein structure, X-ray crystallography, inhibitor, myostatin, structural biology, muscle development, tissue homeostasis, GDF8, GDF11, WFIKKN, follistatin, antagonism
Introduction
Growth differentiation factor 8 (GDF8),4 also known as myostatin, is a member of the TGF-β superfamily of ligands (1). GDF8 is a potent negative regulator of skeletal muscle growth where genetic deletion results in a hypermuscular phenotype (2, 3). In the adult animal, several studies have established that down-regulation, or inhibition, of GDF8 induces muscle hypertrophy (2–7). In contrast, transgenic overexpression of GDF8 causes muscle wasting, consistent with the idea that certain tumors increase GDF8 levels and contribute to cancer cachexia (8–11). GDF11, which shares 90% identity at the amino acid level, has distinct functions and is important for proper anterior-posterior patterning (12, 13). Recent evidence supports a beneficial role of GDF11 in neurogenesis; however, excessive treatment of mice with GDF11 results in muscle wasting, similar to GDF8 (14–16). Given that GDF8 and GDF11 have prominent roles in tissue homeostasis, there has been a strong interest in developing therapeutics that modulate their signaling, especially ones that aim to boost muscle by targeting GDF8 in muscle-related pathologies (17–22).
To signal, TGF-β ligands bind to, and coordinate the assembly of, two type II and two type I serine-threonine kinase receptors (1). GDF8 and GDF11 signal by using a select combination of receptors, ActRIIA or ActRIIB (type II) coupled with Alk4 or Alk5 (type I) (23–25). Ligand activity is selectively controlled by extracellular protein antagonists that bind to ligands and interfere with receptor binding. Although TGF-β ligands are structurally similar, antagonists have variable sizes and domain architectures (23, 26–30). This structural diversity allows different antagonists to selectively inhibit subsets of TGF-β family members. Over the years, structural studies have provided insight into how different antagonists can adopt different mechanisms to bind and neutralize ligands. However, it appears that in most cases, antagonists block both the type I and type II receptor-binding interfaces (23, 29–32).
A number of antagonists have been shown to bind and block GDF8 signaling, including follistatin splice variants follistatin 288 (Fs288) and follistatin 315 (Fs315), FSTL3 (follistatin-like 3), decorin, and WFIKKN (23, 26–28, 33, 34). Whereas follistatin and FSTL3 antagonize multiple ligands, including activin A and activin B, WFIKKN is exceptionally specific for GDF8 and GDF11 (26, 34–36).
WFIKKN is named from the conserved multidomain architecture: whey acidic protein (WAP), follistatin domain (FSD), Immunoglobulin domain (Ig), two tandem Kunitz (K1, K2) domains, and a netrin (N) domain (Fig. 1a). Most animals have two related versions of WFIKKN, WFIKKN1 and WFIKKN2, which share 56% identity (37, 38). Differences in potency and binding stoichiometry have previously been reported, where WFIKKN2 has an IC50 of 0.26 nm for GDF8 antagonism and is nearly 100-fold more potent than WFIKKN1 (38). In addition, WFIKKN2 forms a 1:1 complex with GDF8/11 (1 WFIKKN2 and 1 ligand dimer), whereas WFIKKN1 forms a 2:1 complex. Interestingly, removing the Kunitz 2 and netrin domains both reduces the potency of WFIKKN2 (IC50 = 7.2 nm) and shifts the binding stoichiometry to that of full-length WFIKKN1 (38). Whereas multiple domains are thought to interact with GDF8 and GDF11, previous studies have determined that the FSD plays a significant role in antagonism (34, 38).
Figure 1.
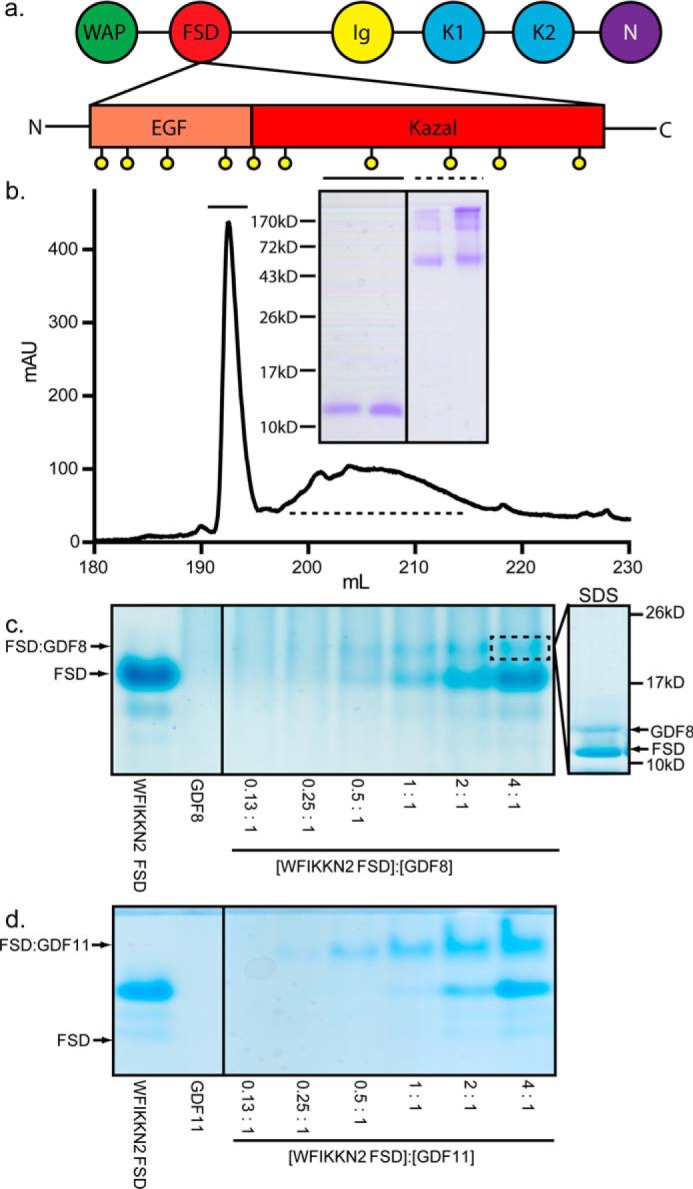
WFIKKN2 domain architecture and FSD purification. a, domain architecture of the WFIKKN FSD. Conserved cysteines are represented as yellow spheres. b, chromatograph depicting the C18 column purification of WFIKKN2 FSD. Inset, respective peak fractions run on an SDS-polyacrylamide gel under nonreducing conditions. Solid line, WFIKKN2 FSD; dotted line, misfolded FSD and contaminants. c and d, native PAGE of WFIKKN2 FSD and GDF8/11 complex formation. Molar ratio of WFIKKN2 to ligand is indicated. Inset, band (dotted box) that was excised and analyzed using SDS-PAGE under reducing conditions.
Follistatin domains are also functionally important for the antagonists follistatin and FSTL3. FSTL3 and follistatin contain two and three FSDs, respectively, which have been shown through multiple X-ray crystal structures to directly contact the ligand (23, 32, 39). Interestingly, each of the FSDs (FSD1–3) within follistatin/FSTL3 adopt different molecular conformations and functions differently at the ligand interface, indicating that the FSDs are not functionally redundant. In fact, biochemical experiments showed that changing the order of the FSDs can severely alter ligand binding and specificity (27, 40).
Whereas WFIKKN selectively inhibits GDF8 and GDF11, the molecular basis for ligand selectivity has not been established. Given that WFIKKN and follistatin/FSTL3 each have FSDs important for binding, whether they exhibit unique or common ligand mechanisms has not been determined. To investigate this, we characterized the WFIKKN FSD and contrasted the binding features of the follistatin FSDs with the WFIKKN FSD. We found that the FSD of WFIKKN blocks the type II receptor-binding interface similar to the FSD2 of follistatin and FSTL3. However, WFIKKN2 FSD displays different structural features and residues that interact with the ligand that map to a different location of the FSD, indicating a different binding mode when compared with the FSDs of follistatin and FSTL3.
Results
Production of WFIKKN2 follistatin domain
Recombinant murine WFIKKN2 FSD, containing residues 104–172, was produced in bacteria. WFIKKN2 FSD protein formed inclusion bodies, which were solubilized and subjected to oxidative refolding to induce disulfide bond formation. Properly folded material was purified using reverse-phase chromatography, as depicted in Fig. 1b. First, native PAGE was used to analyze the refolded WFIKKN2 FSD and to evaluate binding interactions with GDF8. FSD alone migrated as a single band, indicating the isolation of a single refolded species. Similar to previously published results, GDF8 alone does not enter the native PAGE and is not visible (25). Upon mixing WFIKKN2 FSD with GDF8, an additional band appeared, indicating complex formation (Fig. 1c). Titration of GDF8 with increasing concentrations of WFIKKN2 FSD showed an increase in the complex band intensity. Analysis of the newly formed band by SDS-PAGE shows that both WFIKKN2 FSD and GDF8 are present, supporting complex formation (Fig. 1c). Similar results were observed with GDF11 where the complex band was significantly sharper (Fig. 1d). These results indicate that WFIKKN2 FSD is properly folded and can bind both GDF8 and GDF11, as previously implicated (34).
Comparison of WFIKKN2 FSD and WFIKKN2 full-length protein
We next wanted to compare the WFIKKN2 FSD with full-length (FL) WFIKKN2 for their ability to bind and antagonize GDF8 and GDF11. Surface plasmon resonance (SPR) was used to measure binding affinity, where GDF8 and GDF11 were coupled to a CM5 SPR chip. Binding analysis was performed by injecting increasing concentrations of WFIKKN2 FL or WFIKKN2 FSD (Fig. 2, a–d). Similar to previous reports, WFIKKN2 FL bound GDF8 and GDF11 with a high affinity, as demonstrated by the slow dissociation rate (34, 35). Consistent with the native PAGE analysis, WFIKKN2 FSD exhibited a significant interaction with GDF8 and GDF11, albeit with a much faster dissociation rate as compared with WFIKKN2 FL. The association and dissociation rate constants and equilibrium constant, KD, are reported in Table 1. Overall, WFIKKN2 FL exhibited a high-affinity interaction with GDF8 (KD = 0.74 nm) and GDF11 (0.24 nm), whereas the WFIKKN2 FSD was ∼1000-fold weaker (KD = 0.66 μm for GDF8 and 0.12 μm for GDF11). WFIKKN2 FL and WFIKKN2 FSD were able to bind TGFβ1, albeit weakly, but have no affinity for activin A, consistent with previous studies (35). We next sought to determine whether the affinity of WFIKKN2 FSD for GDF8 and GDF11 was sufficient to inhibit signaling. Using a luciferase reporter assay responsive to GDF8 and GDF11, we titrated increasing concentrations of WFIKKN2 FL and WFIKKN2 FSD. Whereas WFIKKN2 FL inhibited GDF8 and GDF11 signaling with an IC50 value of 0.34 and 0.13 nm, respectively, WFIKKN2 FSD exhibited a significantly reduced inhibition of GDF8 and GDF11 with an IC50 value of 0.85 and 0.28 μm, respectively (Fig. 2d), similar to the 1000-fold difference in binding affinity determined by SPR. This demonstrates that whereas the WFIKKN2 FSD binding affinity is severely reduced compared with full-length WFIKKN2, it is still able to antagonize GDF8 and GDF11, albeit with weaker potency.
Figure 2.

Binding and antagonism of WFIKKN2 FSD to GDF8. a–d, SPR of WFIKKN2 FL and WFIKKN2 FSD over immobilized GDF8 and GDF11. Red, experimental binding trace; black, data fit using a 1:1 binding model. a, WFIKKN2 FL applied to immobilized GDF8; b, WFIKKN2 FSD applied to immobilized GDF8; c, WFIKKN2 FL applied to immobilized GDF11; d, WFIKKN2 FSD applied to immobilized GDF11; e, WFIKKN2 FL (red) or WFIKKN2 FSD (black) applied to immobilized TGF-β1. Shown is WFIKKN2 FL (gray) or WFIKKN2 FSD (orange) applied to immobilized activin A. f, luciferase assay using (CAGA)12 HEK293 cells treated with GDF8/GDF11 alone or titrated with increasing concentrations of WFIKKN2 FL or WFIKKN2 FSD. Data were fit using nonlinear regression to determine an IC50 and plotted as mean ± S.D. (error bars) conducted at least twice with each point measured in triplicate.
Table 1.
Surface plasmon resonance of WFIKKN2 interactions with GDF8
Values shown represent the association constant (ka), dissociation constant (kd), and equilibrium dissociation constant (KD) of WFIKKN2 FL or the FSD injected over primary amine-coupled GDF8 or GDF11.
| WFIKKN2 | GDF8 ka | GDF8 kd | GDF8 KD | GDF11 ka | GDF11 kd | GDF11 KD |
|---|---|---|---|---|---|---|
| m−1 s−1 | s−1 | m−1 s−1 | s−1 | |||
| WFIKKN2 FL | 3.37 × 105 | 2.48 × 10−4 | 0.74 nm | 6.00 × 105 | 1.34 × 10−4 | 0.24 nm |
| WFIKKN2 FSD | 6.9 × 103 | 4.6 × 10−3 | 0.66 μm | 3.52 × 104 | 4.25 × 10−3 | 0.12 μm |
Does the WFIKKN2 FSD bind GDF8 at the type II receptor epitope?
To signal, GDF8 binds the extracellular domain of the type II receptor, ActRIIB, with high affinity (KD ∼1 nm) (41, 42). Two ActRIIB receptors are expected to bind GDF8, or GDF11, at each knuckle region of the dimeric ligand, similar to the observed binary crystal structures of ActRIIB in complex with activin A and BMP7 (43, 44). Previous studies have demonstrated that full-length WFIKKN2 interferes with type II receptor binding (26). To determine whether WFIKKN2 FSD could interfere with type II receptor binding, we performed a competition analysis using native PAGE. GDF11 mixed with WFIKKN2 FSD was titrated with increasing concentrations of the extracellular domain of ActRIIB (ActRIIB-ECD) and analyzed by native PAGE (Fig. 3a). GDF11 was used for analysis because the complex bands bound to WFIKKN2 FSD and ActRIIB-ECD are more distinct than those with GDF8. Our results show that ActRIIB-ECD easily displaces WFIKKN2 FSD, leading to the formation of the ActRIIB-ECD:GDF11 complex (Fig. 3a). A higher-order complex that contained all three components was not visible, indicating that the WFIKKN2 FSD and ActRIIB-ECD were mutually exclusive and likely bind to the same position on the ligand. Bands corresponding to WFIKKN2 FSD:GDF11 and ActRIIB-ECD:GDF11 were excised and verified by SDS-polyacrylamide gel under reducing conditions (Fig. 3b). These results indicate that the WFIKKN2 FSD and ActRIIB compete for the same binding interface of GDF11.
Figure 3.
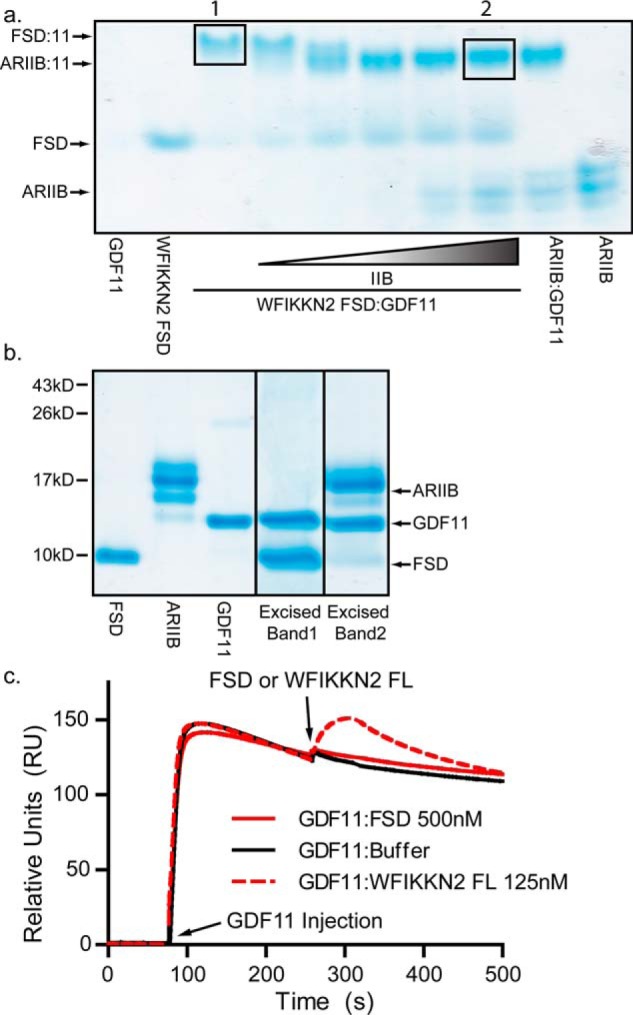
Competitive binding between WFIKKN2 FSD and ActRIIB to GDF11. a, native PAGE analysis using preformed WFIKKN2 FSD:GDF11 complex mixed with increasing amounts of ActRIIB (ARIIB). b, reduced SDS gel of WFIKKN2 FSD, ARIIB, and GDF11 and excised bands represented in a by a black box. c, co-injection SPR binding experiment. First, ActRIIB-Fc was captured onto a protein A sensor chip, and baseline was normalized. Next, GDF11 was injected to form the binary ActRIIB:GDF11 complex. Subsequently, WFIKKN2 FL or FSD was injected, and binding to the ActRIIB:GDF11 complex was monitored.
To further test the idea that WFIKKN2 FSD and ActRIIB bind GDF8 and GDF11 at similar locations, competition experiments were performed using SPR. Experiments were conducted using the extracellular domain of ActRIIB fused to the Fc portion of an antibody (ActRIIB-Fc). ActRIIB-Fc was captured onto a protein A chip, followed by an injection of GDF11 to form the binary ActRIIB-Fc:GDF11 complex. We next injected WFIKKN2 FSD at 500 nm and observed no increase in binding, indicating that the WFIKKN2 FSD could not bind GDF11 in the presence of ActRIIB (Fig. 3c). Interestingly, when similar experiments were performed with WFIKKN2 FL, a noticeable mass increase was observed, indicating that WFIKKN2 FL was able to bind GDF11 that was already in complex with ActRIIB (Fig. 3c).
Crystallization and structural determination of WFIKKN2 FSD
Because the WFIKKN2 FSD can bind and antagonize GDF8 and GDF11, we next wanted to determine the molecular structure and draw comparisons with the FSD of follistatin and FSTL3. The X-ray crystal structure of WFIKKN2 FSD was solved by single isomorphous replacement with anomalous scattering to 1.39 Å resolution. Data collection and refinement statistics are presented in Table 2. Fig. 4a depicts the overall structure of the WFIKKN2 FSD. Similar to other FSDs, the structure contains two subdomains: an N-terminal EGF-like portion followed by a Kazal-like protease inhibitor subdomain. The EGF-like portion contains anti-parallel β-strands (β1 and β2), whereas the Kazal-like protease inhibitor subdomain contains a central helix (α1) and another set of anti-parallel β-strands (β3 and β4) that caps the Kazal subdomain. Similar to the FSDs from follistatin and FSTL3, the WFIKKN2 FSD contains five conserved disulfide bonds. Two are positioned in the EGF subdomain that connect the β1 and β2 strands (Fig. 4a). The other three are located in the Kazal subdomain; two link the α1 to the segment connecting the EGF and Kazal subdomains, and one links β3 to the C terminus.
Table 2.
X-ray crystallographic statistic for structural determination
Overall statistics of native and heavy atom data sets for WFIKKN2 FSD structural determination. Values for the highest-resolution shell are shown in parenthesis.
| Native | K2PtCl4 | |
|---|---|---|
| Data collection | ||
| Resolution (Å) | 41.80–1.39 (1.42–1.39) | 41.85–1.54 (1.56–1.54) |
| Wavelength | 0.98 | 1.07 |
| No. of observations | 277,020 | 259,934 |
| No. of unique reflections | 21,677 | 29,887 |
| No. of heavy atom derivatives (Pt) | 9 | |
| Space group | P43 21 2 | P43 21 2 |
| Unit cell | ||
| a, b, c (Å) | 46.5, 46.5, 95.7 | 46.6, 46.6, 95.1 |
| α, β, γ (degrees) | 90, 90, 90 | 90, 90, 90 |
| Completeness (%) | 99.7 (99.8) | 99.9 |
| Redundancy | 12.8 | 24.1 |
| Anomalous completeness (%) | 99.9 | |
| Anomalous redundancy | 13.4 | |
| Rmeasa | 0.063 (0.184) | 0.239 (3.508) |
| Rpimb | 0.024 (0.080) | 0.05 (0.943) |
| Mean ((I)/σ(I)) | 25.5 (8.7) | 13.2 (1.1) |
| BAYES-CC | 55.5 ± 15.0 | |
| FOMc initial (after DMd) | 0.357 (0.61) | |
| Refinement | ||
| Resolution (Å) | 41.80–1.39 (1.42–1.39) | |
| Reflections (total/free) | 21,708 | |
| Cutoff for refinement | F > 0σ | |
| Rwork/Rfree (%) | 16.45/18.01 | |
| Atoms total/protein | 770/598 | |
| Root mean square deviations | ||
| Bonds (Å) | 0.004 | |
| Angles (degrees) | 0.75 | |
| Average B factors (Å2) | 17 | |
| Amino acids | 13.97 | |
| Ligands | 25.6 | |
| Water | 32.26 | |
| Wilson B factors (Å2) | 11.5 | |
| Ramachandran plot | ||
| Favored (%) | 98.68 | |
| Allowed (%) | 1.32 | |
| Outliers (%) | 0.00 | |
| Clashscore | 2.57 | |
a Overall measure of error between multiple measurements of a reflection within I+/I−, independent of redundancy. Rmeas = (Σhkl√(n/(n − 1))Σj = 1n|Ihklj − 〈Ihkl〉|)/(ΣhklΣjIhklj).
b S.E. for intensity measurements within I+/I−. Rpim = (Σhkl√(1/(n − 1))Σj = 1n|Ihklj − 〈Ihkl〉|)/(ΣhklΣjIhklj).
c Figure of merit.
d Density modification.
Figure 4.
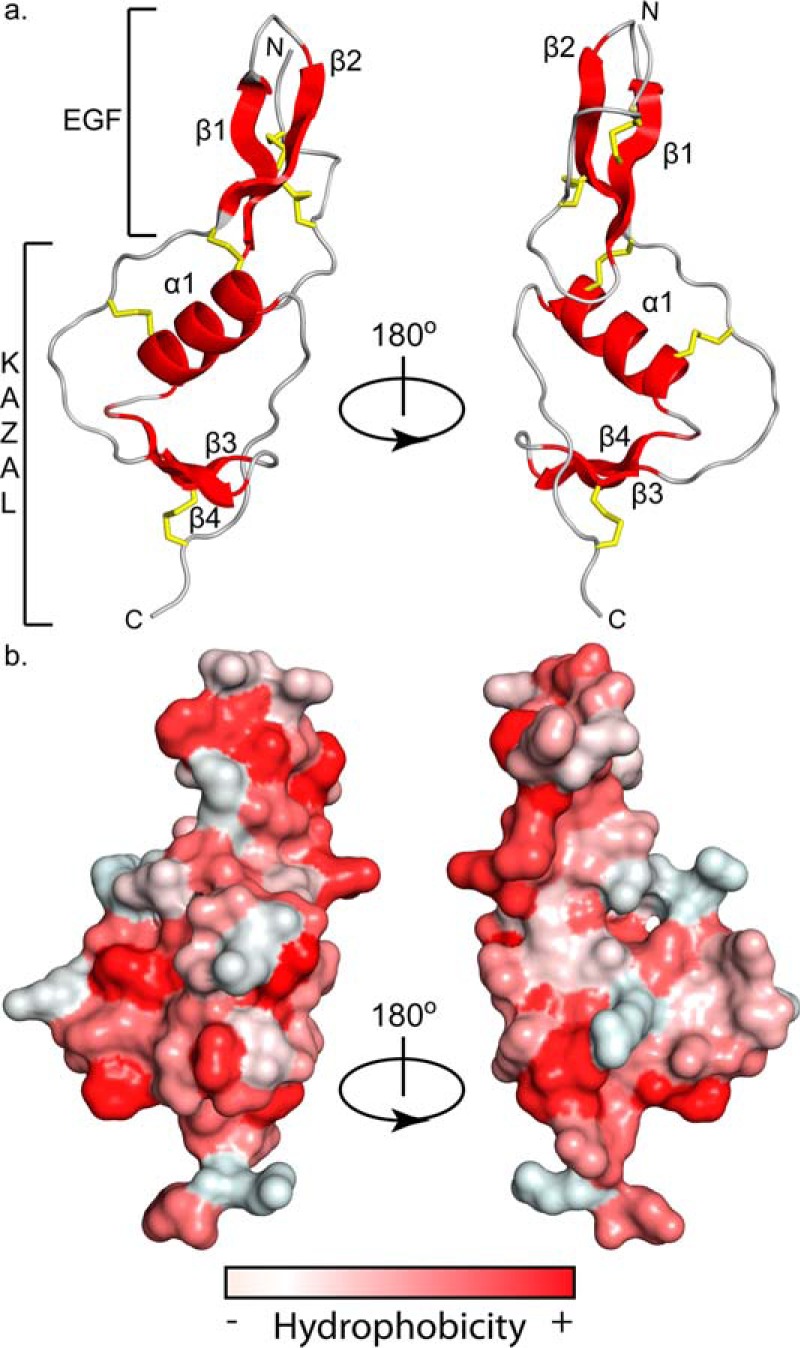
WFIKKN2 FSD structure and surface hydrophobicity. a, ribbon diagram depicting WFIKKN2 FSD with structural components (α-helix and β-sheets) in red, flexible loops in gray, and disulfide bonds shown as yellow sticks rotated about the y axis 180º. b, hydrophobicity of the GASP1 Fs domain surface, red being the most hydrophobic and white being the least hydrophobic using the Color_H PyMOL script (46). Structures are in the same orientation as shown in a.
WFIKKN2 FSD mutagenesis and inhibition
Utilizing the structure, we wanted to identify residues that are important for the interaction with GDF8 and GDF11. TGF-β ligands, including BMP2, BMP4, BMP7, activin A, activin B, GDF8, and GDF11, predominantly interact with receptors and extracellular antagonists using hydrophobic interactions (23, 45). Therefore, we hypothesized that surface-exposed hydrophobic residues of WFIKKN2 FSD might be important for ligand binding and antagonism. Several surface-exposed hydrophobic residues in both the EGF and Kazal subdomains were readily apparent (Fig. 4b) (46). Residues were selected for alanine mutagenesis, including two that are positioned in the EGF domain (Phe-109 and Trp-121) and three positioned within the Kazal subdomain (Phe-139, Phe-153, and Ile-163) (Fig. 5a). To determine whether these residues were important for WFIKKN2 antagonism, we generated single point mutations of the selected residues in the full-length WFIKKN2. WT and mutant versions of WFIKKN2 FL were expressed in human embryonic kidney cells (HEK293F), purified, and tested for GDF8 inhibition in a cell-based luciferase reporter assay. HEK293T-CAGA cells were treated with 0.62 nm GDF8 and titrated with increasing concentrations of either purified WT or mutant WFIKKN2 FL (Fig. 5b). Data were fit to a dose-response curve to determine an IC50 for WFIKKN2 FL and mutations. Mutations within the EGF domain (F109A and W121A) resulted in little to no change in the antagonism of GDF8. However, WFIKKN2 mutants tested within the Kazal subdomain (F139A, F153A, and I163A) all displayed weaker GDF8 inhibition. The most striking effects were seen with the mutations F139A and F153A, which were 10- and 18-fold weaker than WT WFIKKN2 (Fig. 5). Thus, the three residues that had a negative impact on GDF8 antagonism are located on the highly hydrophobic face of the WFIKKN2 FSD Kazal subdomain, as shown in Fig. 4.
Figure 5.

Full-length WFIKKN2 mutant selection and inhibition. a, ribbon diagram of WFIKKN2 FSD; labeled sticks represent residues mutated to alanine and colored based on the inhibition curve. b, IC50 curves generated via luciferase assay with increasing amounts of WFIKKN2 mutants titrated against a constant concentration of GDF8. c, IC50 values of mutants tested in b and depicted on the WFIKKN2 FSD structure in a. Error bars, S.D.
Comparison of the WFIKKN2 FSD with the FSDs of follistatin
Using the structure of WFIKKN2 FSD, we can draw structural comparisons to the different FSDs in FS288 and FSTL3. Because the FSD1 and FSD2 in both FS288 and FSTL3 share similar structures, the comparison focuses on the FSDs within FS288. The WFIKKN2 FSD and the FSDs from FS288 and FSTL3 contain a similar domain architecture with EGF and Kazal protease inhibitor subdomains, including the spacing and alignment of the five disulfide bonds. In addition, all of the FSDs contain two highly conserved residues, Thr-146 and Tyr-147, highlighted in Fig. 6a, which are conserved in the broader Kazal domain of protease inhibitors (e.g. ovomucoids and serine protease inhibitor Kazal-type 1) (47–49). Despite these conservations, there are differences in the relative position of the subdomains, making it challenging to perform an overall alignment of the FSDs. However, the Kazal subdomains exhibit similar structures, where alignment results in a root mean square of 3.1, 2.1, and 2.9 Å2 for FSD1, FSD2 and FSD3 of follistatin, respectively (40 α positions), when aligned to WFIKKN2 FSD. The moderate differences of the Cα positions between the Kazal subdomains localize in the loops connecting the α1-helix and the position of β-strands 3 and 4. Despite their similar structure, the Kazal subdomain of WFIKKN2 FSD only contains two of the three β-strands found in other FSDs (Fig. 6b).
Figure 6.

Comparison of WFIKKN2 FSD and Fs288 FSD1–3. a, sequence alignment of WFIKKN2 FSD to Fs288 FSDs with the EGF and Kazal subdomains indicated. Rectangles, β strands; cylinders, α helices; black bars, conserved disulfide bonds; asterisks, conserved residues. Residues determined to be important for GDF8 binding are highlighted. Underlined residues are involved in the EGF–Kazal interaction. b, WFIKKN2 FSD (red) to FSD1 (blue), FSD2 (green), and FSD3 (yellow) of Fs288 were aligned using the Kazal subdomain. Top left, schematic representation depicting the relative orientation of the Kazal and EGF subdomains. The inset shows the 2Fo − Fc (2α) map of the EGF–Kazal interaction of WFIKKN2 FSD. Dotted lines, missing segments in the crystal structures. Residues involved in the EGF–Kazal interaction are indicated in ball-and-stick representations. Far right, alignment of WFIKKN2 and Fs288 FSD1–3 only depicting the α-helix and β1–2 of the EGF. The relative difference in orientation between the tip of the EGF domain and α-helix is shown by a black line.
Whereas the individual subdomains are structurally similar, there are significant conformational differences in the relative position of the subdomains. In retrospect, this explains why using the available FSD structures (FSD1–3) as search models failed to provide a molecular replacement solution for the WFIKKN2 FSD X-ray diffraction data. Thus, we aligned the structures using only the Kazal subdomain (Fig. 6b). Comparison of the structures shows that different FSDs can adopt either an “open” or “closed” conformation. An open conformation is observed in FSD1 and WFIKKN2 FSD, where the EGF β-strands are extended away from the Kazal domain, which creates a more linear appearance of the FSD domain. In contrast, a closed, more compact conformation is observed in FSD2 and FSD3, where the EGF domain, specifically β-strand 1, interacts with the Kazal domain. In FSD2, Thr-178, Val-180, and Val-181 within β-strand 1 of the EGF domain form hydrophobic interactions with the Kazal subdomain (Fig. 6b). Similarly, in FSD3, Lys-256, Leu-258, and Phe-261 of β-strand 1 in the EGF domain interact with the Kazal domain. Whereas the FSD of WFIKKN2 is in the open conformation, it should be noted that the N terminus contains tandem glutamine residues (Gln-113 and Gln-114) that wrap back into the Kazal subdomain to interact with the α1-helix. Residues at the interfaces of all of the FSDs are highly conserved across species, including Gln-113 and Gln-114, suggesting that they might serve a role to stabilize the different conformations of the FSDs.
Discussion
WFIKKN proteins are evolutionarily conserved in all vertebrates and can be found in other organisms, such as sea urchins and worms (37). When compared with other extracellular protein antagonists of the TGF-β family, WFIKKN proteins are remarkably specific for the ligands GDF8 and GDF11 (26, 35). Although the basis for this specificity remains unknown, it is clear that the FSD plays an important role in binding and antagonism (34, 38). For example, a construct containing only the WF domains of WFIKKN2 is sufficient for antagonism of GDF8, and the FSD of WFIKKN1 has a higher affinity for GDF8 than the Kunitz 2 and netrin domain (34, 38). Interestingly, comparing the different domains of WFIKKN2, the FSD is the most conserved domain across mammalian species, with the lowest rate of substitution (37).
In other ligand antagonists, such as follistatin and FSTL3, FSDs play an important role in ligand binding and antagonism (32, 40, 50). However, whether the FSD from WFIKKN serves a similar role remains to be determined. To address this, we produced recombinant WFIKKN2 FSD and characterized its interaction with GDF8 and GDF11.
A complex between WFIKKN2 FSD and GDF8 or GDF11 was observed using native PAGE analysis, which SPR experiments revealed to involve submicromolar-affinity interactions. Using native PAGE, we also demonstrated that the addition of the type II receptor, ActRIIB, was able to dissociate a WFIKKN2 FSD:GDF11 complex, suggesting a direct competition between WFIKKN2 FSD and ActRIIB. Consistent with these results, SPR experiments showed that GDF11 in complex with ActRIIB-Fc was unable to bind WFIKKN2 FSD. Therefore, similar to their role in follistatin and FSTL3, the FSD of WFIKKN2 appears to block or at least compete for type II receptor binding on the ligand. However, full-length WFIKKN2 retained the ability to bind the GDF11:ActRIIB-Fc complex. This interaction is likely due to other domains of WFIKKN2 associating with regions of GDF8 and GDF11 apart from the type II–binding site. Thus, whereas the other domains of WFIKKN2 are important for potent antagonism, the WFIKKN2 FSD is sufficient to antagonize GDF8 and GDF11 by blocking type II receptor binding. Whether WFIKKN2 FSD or full-length WFIKKN2 interferes with type I receptor binding has not been resolved and is complicated by the low-affinity interaction of the type I receptor with GDF8 and GDF11 (25).
To help draw comparisons with the FSD of follistatin, we solved the crystal structure of WFIKKN2 FSD. The structure reveals a similar domain architecture with an N-terminal EGF subdomain and a C-terminal Kazal subdomain, as observed previously for other FSDs (23, 32, 40, 51). Using the structure, we were able to identify hydrophobic residues at the surface that contribute to GDF8 antagonism. Single point mutations within the FSD of WFIKKN2 FL reduced the capacity to inhibit GDF8, presumably by reducing the ability of WFIKKN2 FL to interact with the type II receptor binding interface. These results are consistent with antagonists across the family that use hydrophobic interactions to engage the ligand surfaces important for binding type II receptors. However, it is possible that the residues identified could be important for interacting with other domains within WFIKKN2 FL, functioning to stabilize the WFIKKN2:ligand complex.
Comparison of WFIKKN2 FSDs across 54 species reveals a high sequence identity (68%) (Fig. S1). Interestingly, the residues identified (Phe-139, Phe-153, and Ile-163) within the FSDs as important for antagonism of GDF8 are highly conserved. Further, these residues are not conserved in other FSDs (Fig. 6), indicating that they are not necessary to the structural integrity of the Kazal subdomain. Thus, the decrease in antagonism is not likely due to a misfolded FSD within WFIKKN2. Taken together, these results support a functional rather than structural role of Phe-139, Phe-153, and Ile-163 in the binding and antagonism of GDF8 and GDF11.
Interestingly, when compared with follistatin and FSTL3, WFIKKN2 interacts with the ligands through a different surface of the FSD. The WFIKKN2 FSD residues, Phe-139, Phe-153, and Ile-163, are located within the Kazal subdomain, centered on the hydrophobic face of the α1 opposite of the N-terminal loop (Figs. 4 and Fig. 7). In FSD1 and FSD2, residues that interact with the ligands are located in a completely different location of the domain, especially for FSD2, where the interaction surface is on the opposite side as compared with WFIKKN2 FSD (Fig. 7).
Figure 7.
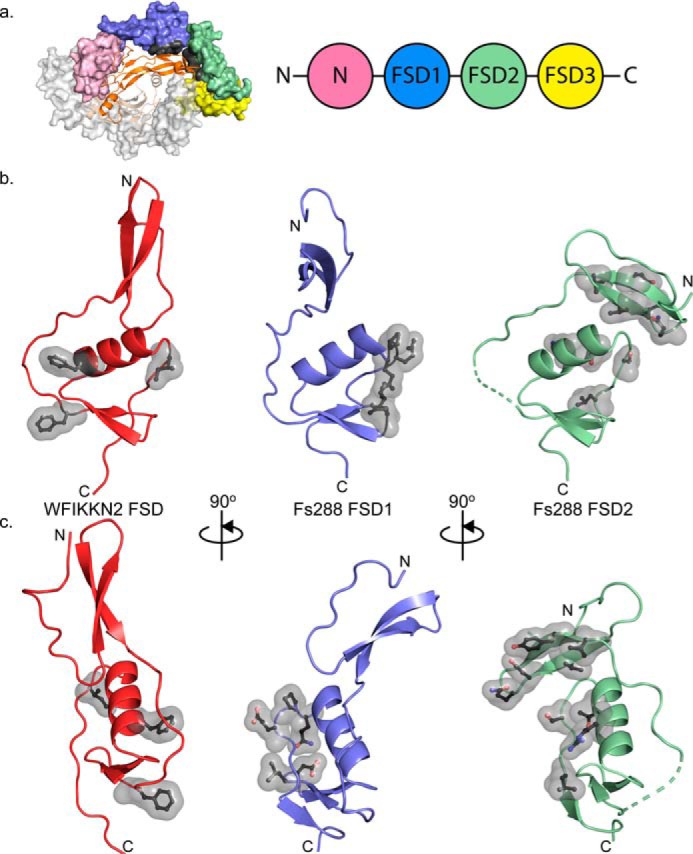
Binding interface of WFIKKN2 FSD is distinct from the FSDs of FS288. a, structure of Fs288 bound to GDF8 (Protein Data Bank entry 3HH2 (23)). The central GDF8 dimer is depicted as ribbon and colored orange and wheat. One Fs288 molecule (white) is shown as a surface representation. The second FS288 molecule is shown as a surface and colored based on the schematic. The FS288 FSD1-FSD2 surface that interacts with GDF8 is colored gray. b, WFIKKN2 FSD (red), FS288 FSD1 (blue), and FS288 FSD2 (green) are shown in similar orientation based on alignment of the Kazal subdomain. Residues within each FSD important for GDF8 antagonism are displayed. c, rotation of b along the vertical axis by 90º.
Although it is possible that the conformational differences observed between the EGF and Kazal subdomains of FSDs are related to crystallization, the linker region is well-ordered in the crystal lattice and exhibits similar temperature factors throughout the structure, suggesting that the conformation of the domain is stable. Consistent with the FSD having a stable structure, the open conformation of the follistatin FSD1 was observed in both the bound and unbound state, indicating that the domain structure is not affected by ligand binding and/or alternative crystallization conditions.
In addition to WFIKKN, follistatin, and FSTL3, FSDs are found in several proteins with various roles in TGF-β signaling. For instance, FSDs are also found in agrin, tomoregulin, and FSTL1, all of which are implicated in regulating TGF-β ligand activity (52–54). This raises a question as to the general function of the FSD and whether the domain serves a common role in ligand interactions. Whereas the FSD in both WFIKKN and follistatin/FSTL3 both interact at the type II receptor interface, differences are readily apparent in both their structures and the placement of residues utilized for this interaction. Thus, it does not appear that the FSD represents a common binding domain, but rather a stable scaffold that allows the presentation of surface-exposed hydrophobic residues that can be important for protein–protein interactions.
Given the differences in domain architecture and the differences in the position of surface residues important for ligand binding, we anticipate that the FSD of WFIKKN2 does not resemble the ligand binding scheme of any of the three FSDs of follistatin. Certainly, resolution of WFIKKN2 FSD or FL in complex with a ligand will ultimately resolve these differences and help us to understand how WFIKKN specifically engages GDF8 and GDF11 over other TGF-β ligands.
Experimental procedures
WFIKKN2 FSD production and purification
Mouse WFIKKN2 FSD (residues 104–172, 97% identical to human) with a cleavable N-terminal His6 tag was cloned into pET28a(+) and expressed in BL21 Rosetta cells. Cells were spun down and resuspended in PBS before sonication for cell lysis. Pellets were washed two times with PBS, 0.1% Triton X-100, followed by a final wash with PBS. Inclusion bodies containing the WFIKKN2 FSD were solubilized using 10 mm sodium tetrathionate, 100 mm sodium sulfite, 100 mm Tris, pH 8.5, 8 m urea, and 100 mm DTT. Solubilized inclusion bodies were dialyzed into 4 m urea, 50 mm sodium acetate, pH 4.5, 100 mm NaCl, 50 mm Tris, 15 mm imidazole and rapidly diluted into refolded buffer containing 100 mm Tris, pH 8.5, 150 mm NaCl, 1 mm EDTA, 0.1 mm oxidized GSH, 0.5 mm reduced GSH, and 0.5 m arginine to a final concentration of 0.1 mg/ml. After 3 days, protein was diluted 1:5 in nickel-nitrilotriacetic acid running buffer (50 mm Tris, pH 8, 500 mm NaCl, and 15 mm imidazole) and applied to a HisTrapTM Excel column (GE Healthcare). WFIKKN2 FSD was eluted with 500 mm imidazole and dialyzed into 10 mm HCl prior to separation on a C18 reverse-phase column (Phenomenex).
ActRIIB-ECD production and purification
ActRIIB-ECD was produced within SF+ insect cells using pFastBac expression plasmid. Purification was conducted as described previously (44). In short, ActRIIB-ECD containing a His6 tag was purified from SF+ conditioned medium using a nickel-nitrilotriacetic acid affinity column. Bound protein was eluted with 20 mm Tris, pH 7.4, 500 mm NaCl containing 500 mm imidazole and subsequently applied to an S75 size exclusion column. Fractions containing the ActRIIB-ECD were pooled and used for native PAGE analysis.
Native PAGE and Western blotting
3 μg of GDF11/8 was mixed with WFIKKN2 FSD at different molar ratios, starting with 4:1 (WFIKKN2 FSD:GDF11/8), and WFIKKN2 FSD was serially diluted 1:2 five times for a final ratio of 0.13:1. Native polyacrylamide gels (12%) were run at 20 °C for 140 min at 110 V and stained using colloidal Coomassie Blue. For staining, gels were fixed using 40% EtOH and 10% acetic acid for at least 1 h before washing three times using distilled H2O. A working dye solution was used containing 80% colloidal Coomassie Blue and 20% methanol and stained overnight.
Native PAGE to SDS-PAGE transfer
Native polyacrylamide gels were run as described above. However, rather than a titration, one condition was repeated in seven lanes. One lane was cut, stained, and realigned to the unstained gel. The desired stained band was used as a guide to excise the other six unstained bands. Excised gel was then placed in a dialysis bag with 1 ml of SDS running buffer and electro-eluted with 180 V for 1 h. The buffer containing the eluted band was then concentrated and subjected to SDS-PAGE under reducing conditions followed by colloidal staining as described above.
Luciferase assay
Luciferase assays were conducted in HEK293 (CAGA)12 cells as described previously by our laboratory (25, 38, 55). For the assay, 20,000 cells were seeded in growth medium in a 96-well format on poly-d-lysine–coated plates (catalog no. 655940, Greiner Bio-One GmbH). Cells were grown at 37 °C with 5% CO2 until reaching 75–85% confluence. Medium was then removed and treated with 100 μl of serum-free medium containing GDF8 in the presence or absence of antagonists. GDF8 was kept at a constant concentration (0.62 nm) while antagonists (WFIKKN2 FSD and FL) were titrated in using 1:2 dilutions. After 1 day, cells were lysed using 1× passive lysis buffer (E1941, Promega) on a plate shaker (800 rpm, 20 min, 20 °C). Lysates were transferred to black and white 96-well plates, and 40 μl of LAR (E1501 and E1960, Promega) was added. Firefly luminescence was measured using the Synergy H1 Hybrid Plate Reader (BioTek). All experiments were conducted independently at least twice with all data points being done in triplicate. The concentration of antagonists at which 50% of GDF8 activity is lost, or IC50, was calculated using nonlinear regression with variable slope using GraphPad Prism version 5 software.
Surface plasmon resonance
SPR experiments were conducted on the Biacore T200 microfluidic system. Ligands were primary amine–coupled to the GE series S CM5 sensor chip (catalog no. BR-1005-30) using the manufacturer's protocols. GDF8, GDF11, activin A, and TGFβ1 at 1 μg/ml was sufficient to achieve ∼500 response units bound to the chip. SPR experiments were conducted in HBS-EP buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.005% P-20 surfactant (Biacore AB)). WFIKKN2 full-length or WFIKKN2 FSD was diluted in HBS-EP buffer to a concentration of 500 nm and then serially diluted 2-fold 10 times and applied to the chip at 15 μl/min. Association was measured for 180 s followed by 120 s of buffer to measure dissociation. The dissociation equilibrium constant (KD) was calculated using Biacore T200 software using a 1:1 binding model. For the receptor-binding experiments, ActRIIB-Fc at a concentration of 312.5 ng/ml (∼4.2 nm) was immobilized on a protein A chip. GDF11 was then applied at a concentration of 250 nm for 180 s. Subsequently, binding of 125 and 500 nm WFIKKN2 full-length or WFIKKN2 FSD to the receptor:ligand complex was measured.
Structural determination and experimental phasing
Crystals of WFIKKN2 FSD were grown in 2.5 m AmNO3 and 0.1 m sodium citrate, pH 4.6, via hanging drop vapor diffusion at 4 mg/ml. Rectangular crystals grew to a size of 100 × 200 μm. Diffraction data were collected at Advanced Photon Source (APS) beamline GM/CA 23-ID-D at Argonne National Laboratory. Molecular replacement techniques for phasing were unsuccessful, requiring experimental phasing via single isomorphous replacement with anomalous scattering (SIRAS). In short, crystals were soaked in 5 mm KCl4Pt for 96 h before being transferred to a cryogenic solution containing mother liquor and 35% PEG 550 and flash-frozen. Heavy atom positions were identified using phenix AutoSol (56, 57). Phasing and model refinement were carried out using AutoSol and phenix.refine (58). Model validation was performed using the program MolProbity (59, 60). Coordinates are available at the Protein Data Bank, accession code 6MAA. Hydrophobicity depicted in Fig. 4 was determined using the Color_H PyMOL script (46).
WFIKKN2 FSD and Fs288 alignment and structural representation
Sequence alignments between WFIKKN2 FSD and other FSDs were conducted using T-Coffee (61). Structural alignments were conducted using ce-alignment within PyMOL (62). Alignments were conducted between the Kazal subdomain of each FSD starting from the conserved cysteine (residue 132 in WFIKKN2) to the end of the FSD (residue 172 in WFIKKN2). Residues used for each Fs288 are as follows: FSD1, 119–166; FSD2, 194–242; FSD3, 271–318. Supplemental sequence alignments were conducted using CLUSTAL O (version 1.2.4) (63).
Mutagenesis and purification of full-length WFIKKN2
Full-length WFIKKN2 was purified as described previously (38). In short, conditioned medium from CHO cells stably expressing WFIKKN2 was collected and subjected to butyl-Sepharose and heparin columns. Elutions containing WFIKKN2 were dialyzed into 50 mm Tris, pH 7.4, 20 mm NaCl, 1 mm EDTA and applied to a MonoQ 10/100 GL column and eluted with a linear gradient of NaCl. Following Mono Q, elutions were subjected to S2000 size-exclusion chromatography to obtain pure WFIKKN2. Full-length WFIKKN2 was cloned into pcDNA4 mammalian expression vector and subjected to site-directed mutagenesis. WFIKKN2 mutants were transfected and produced via HEK293F cells. Following expression, WFIKKN2 mutants were purified similarly to full-length WFIKKN2 produced in CHO cells.
Author contributions
J. C. M., R. G. W., and T. B. T. designed experiments. J. C. M. solved the FSD structure and conducted SPR, luciferase, and native PAGE analysis. R. G. W. conducted WFIKKN mutagenesis and luciferase assays with N. H. M. J. C. M. wrote the paper with T. B. T.
Supplementary Material
This work was supported by NIGMS, National Institutes of Health, Grant R01 GM114640 and American Heart Association Grants 12PRE11790027 (to R. G. W.) and 18PRE33990312 (to J. C. M.). Thomas B. Thompson is a consultant for Acceleron Pharma. Ryan G. Walker is a co-founder, scientific advisor, and holder of private equity in Elevian, a company that aims to develop medicines to restore regenerative capacity. Elevian provides sponsored research support for R. G. W. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
The atomic coordinates and structure factors (code 6MAA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GDF
- growth and differentiation factor
- TGF
- transforming growth factor
- WAP
- whey acidic protein
- WFIKKN
- WAP, follistatin/kazal, immunoglobulin, Kunitz, and netrin domain–containing
- FSD
- follistatin domain
- FL
- full-length
- ActRII
- activin receptor II
- Alk
- activin-like kinase
- SPR
- surface plasmon resonance
- ECD
- extracellular domain
- EGF
- epidermal growth factor.
References
- 1. Hinck A. P., Mueller T. D., and Springer T. A. (2016) Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol. 8, a022103 10.1101/cshperspect.a022103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McPherron A. C., and Lee S. J. (1997) Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. U.S.A. 94, 12457–12461 10.1073/pnas.94.23.12457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McPherron A. C., Lawler A. M., and Lee S. J. (1997) Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 387, 83–90 10.1038/387083a0 [DOI] [PubMed] [Google Scholar]
- 4. Thomas M., Langley B., Berry C., Sharma M., Kirk S., Bass J., and Kambadur R. (2000) Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 275, 40235–40243 10.1074/jbc.M004356200 [DOI] [PubMed] [Google Scholar]
- 5. Lee S.-J. (2004) Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 20, 61–86 10.1146/annurev.cellbio.20.012103.135836 [DOI] [PubMed] [Google Scholar]
- 6. Monestier O., Brun C., Heu K., Passet B., Malhouroux M., Magnol L., Vilotte J. L., and Blanquet V. (2012) Ubiquitous Gasp1 overexpression in mice leads mainly to a hypermuscular phenotype. BMC Genomics 13, 541 10.1186/1471-2164-13-541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brun C., Périé L., Baraige F., Vernus B., Bonnieu A., and Blanquet V. (2014) Absence of hyperplasia in Gasp-1 overexpressing mice is dependent on myostatin up-regulation. Cell. Physiol. Biochem. 34, 1241–1259 10.1159/000366335 [DOI] [PubMed] [Google Scholar]
- 8. Loumaye A., de Barsy M., Nachit M., Lause P., Frateur L., van Maanen A., Trefois P., Gruson D., and Thissen J.-P. (2015) Role of activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 100, 2030–2038 10.1210/jc.2014-4318 [DOI] [PubMed] [Google Scholar]
- 9. Durieux A.-C., Amirouche A., Banzet S., Koulmann N., Bonnefoy R., Pasdeloup M., Mouret C., Bigard X., Peinnequin A., and Freyssenet D. (2007) Ectopic expression of myostatin induces atrophy of adult skeletal muscle by decreasing muscle gene expression. Endocrinology 148, 3140–3147 10.1210/en.2006-1500 [DOI] [PubMed] [Google Scholar]
- 10. Zimmers T. A., Davies M. V., Koniaris L. G., Haynes P., Esquela A. F., Tomkinson K. N., McPherron A. C., Wolfman N. M., and Lee S.-J. (2002) Induction of cachexia in mice by systemically administered myostatin. Science 296, 1486–1488 10.1126/science.1069525 [DOI] [PubMed] [Google Scholar]
- 11. Costelli P., Muscaritoli M., Bonetto A., Penna F., Reffo P., Bossola M., Bonelli G., Doglietto G. B., Baccino F. M., and Rossi Fanelli F. (2008) Muscle myostatin signalling is enhanced in experimental cancer cachexia. Eur. J. Clin. Invest. 38, 531–538 10.1111/j.1365-2362.2008.01970.x [DOI] [PubMed] [Google Scholar]
- 12. McPherron A. C., Lawler A. M., and Lee S. J. (1999) Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat. Genet. 22, 260–264 10.1038/10320 [DOI] [PubMed] [Google Scholar]
- 13. Oh S. P., Yeo C. Y., Lee Y., Schrewe H., Whitman M., and Li E. (2002) Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes Dev. 16, 2749–2754 10.1101/gad.1021802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu L., Bai X., Cao Y., Luo H., Yang X., Kang L., Shi M.-J., Fan W., and Zhao B.-Q. (2018) Growth differentiation factor 11 promotes neurovascular recovery after stroke in mice. Front. Cell Neurosci. 12, 205 10.3389/fncel.2018.00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ozek C., Krolewski R. C., Buchanan S. M., and Rubin L. L. (2018) Growth differentiation factor 11 treatment leads to neuronal and vascular improvements in the hippocampus of aged mice. Sci. Rep. 8, 17293 10.1038/s41598-018-35716-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zimmers T. A., Jiang Y., Wang M., Liang T. W., Rupert J. E., Au E. D., Marino F. E., Couch M. E., and Koniaris L. G. (2017) Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 112, 48 10.1007/s00395-017-0639-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsuchida K. (2008) Targeting myostatin for therapies against muscle-wasting disorders. Curr. Opin. Drug Discov. Devel. 11, 487–494 [PubMed] [Google Scholar]
- 18. Smith R. C., and Lin B. K. (2013) Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr. Opin. Support. Palliat. Care 7, 352–360 10.1097/SPC.0000000000000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rinaldi F., Zhang Y., Mondragon-Gonzalez R., Harvey J., Perlingeiro R. C. R. (2016) Treatment with rGDF11 does not improve the dystrophic muscle pathology of mdx mice. Skelet. Muscle 6, 21 10.1186/s13395-016-0092-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh P., Rong H., Gordi T., Bosley J., and Bhattacharya I. (2016) Translational pharmacokinetic/pharmacodynamic analysis of MYO-029 antibody for muscular dystrophy. Clin. Transl. Sci. 9, 302–310 10.1111/cts.12420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mendell J. R., Sahenk Z., Malik V., Gomez A. M., Flanigan K. M., Lowes L. P., Alfano L. N., Berry K., Meadows E., Lewis S., Braun L., Shontz K., Rouhana M., Clark K. R., Rosales X. Q., et al. (2015) A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 23, 192–201 10.1038/mt.2014.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wagner K. R., Fleckenstein J. L., Amato A. A., Barohn R. J., Bushby K., Escolar D. M., Flanigan K. M., Pestronk A., Tawil R., Wolfe G. I., Eagle M., Florence J. M., King W. M., Pandya S., Straub V., et al. (2008) A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 63, 561–571 10.1002/ana.21338 [DOI] [PubMed] [Google Scholar]
- 23. Cash J. N., Rejon C. A., McPherron A. C., Bernard D. J., and Thompson T. B. (2009) The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J. 28, 2662–2676 10.1038/emboj.2009.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rebbapragada A., Benchabane H., Wrana J. L., Celeste A. J., and Attisano L. (2003) Myostatin Signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Mol. Cell Biol. 23, 7230–7242 10.1128/MCB.23.20.7230-7242.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walker R. G., Czepnik M., Goebel E. J., McCoy J. C., Vujic A., Cho M., Oh J., Aykul S., Walton K. L., Schang G., Bernard D. J., Hinck A. P., Harrison C. A., Martinez-Hackert E., Wagers A. J., et al. (2017) Structural basis for potency differences between GDF8 and GDF11. BMC Biol. 15, 19 10.1186/s12915-017-0350-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee Y.-S., and Lee S.-J. (2013) Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc. Natl. Acad. Sci. U.S.A. 110, E3713–E3722 10.1073/pnas.1309907110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sidis Y., Mukherjee A., Keutmann H., Delbaere A., Sadatsuki M., and Schneyer A. (2006) Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 147, 3586–3597 10.1210/en.2006-0089 [DOI] [PubMed] [Google Scholar]
- 28. Miura T., Kishioka Y., Wakamatsu J., Hattori A., Hennebry A., Berry C. J., Sharma M., Kambadur R., and Nishimura T. (2006) Decorin binds myostatin and modulates its activity to muscle cells. Biochem. Biophys. Res. Commun. 340, 675–680 10.1016/j.bbrc.2005.12.060 [DOI] [PubMed] [Google Scholar]
- 29. Nolan K., Kattamuri C., Rankin S. A., Read R. J., Zorn A. M., and Thompson T. B. (2016) Structure of Gremlin-2 in complex with GDF5 gives insight into DAN-family-mediated BMP antagonism. Cell Rep. 16, 2077–2086 10.1016/j.celrep.2016.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nolan K., and Thompson T. B. (2014) The DAN family: modulators of TGF-β signaling and beyond. Protein Sci. 23, 999–1012 10.1002/pro.2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kattamuri C., Luedeke D. M., Nolan K., Rankin S. A., Greis K. D., Zorn A. M., and Thompson T. B. (2012) Members of the DAN family are BMP antagonists that form highly stable noncovalent dimers. J. Mol. Biol. 424, 313–327 10.1016/j.jmb.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cash J. N., Angerman E. B., Kattamuri C., Nolan K., Zhao H., Sidis Y., Keutmann H. T., and Thompson T. B. (2012) Structure of myostatin·follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J. Biol. Chem. 287, 1043–1053 10.1074/jbc.M111.270801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Amthor H., Nicholas G., McKinnell I., Kemp C. F., Sharma M., Kambadur R., and Patel K. (2004) Follistatin complexes myostatin and antagonises myostatin-mediated inhibition of myogenesis. Dev. Biol. 270, 19–30 10.1016/j.ydbio.2004.01.046 [DOI] [PubMed] [Google Scholar]
- 34. Kondás K., Szláma G., Trexler M., and Patthy L. (2008) Both WFIKKN1 and WFIKKN2 have high affinity for growth and differentiation factors 8 and 11. J. Biol. Chem. 283, 23677–23684 10.1074/jbc.M803025200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szláma G., Kondás K., Trexler M., and Patthy L. (2010) WFIKKN1 and WFIKKN2 bind growth factors TGFβ1, BMP2 and BMP4 but do not inhibit their signalling activity. FEBS J. 277, 5040–5050 10.1111/j.1742-4658.2010.07909.x [DOI] [PubMed] [Google Scholar]
- 36. Hill J. J., Qiu Y., Hewick R. M., and Wolfman N. M. (2003) Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol. Endocrinol. 17, 1144–1154 10.1210/me.2002-0366 [DOI] [PubMed] [Google Scholar]
- 37. Monestier O., Brun C., Cocquempot O., Petit D., and Blanquet V. (2012) GASP/WFIKKN proteins: evolutionary aspects of their functions. PLoS One 7, e43710 10.1371/journal.pone.0043710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walker R. G., Angerman E. B., Kattamuri C., Lee Y.-S., Lee S.-J., and Thompson T. B. (2015) Alternative binding modes identified for growth and differentiation factor-associated serum protein (GASP) family antagonism of myostatin. J. Biol. Chem. 290, 7506–7516 10.1074/jbc.M114.624130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thompson T. B., Lerch T. F., Cook R. W., Woodruff T. K., and Jardetzky T. S. (2005) The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 9, 535–543 10.1016/j.devcel.2005.09.008 [DOI] [PubMed] [Google Scholar]
- 40. Cash J. N., Angerman E. B., Keutmann H. T., and Thompson T. B. (2012) Characterization of follistatin-type domains and their contribution to myostatin and activin A antagonism. Mol. Endocrinol. 26, 1167–1178 10.1210/me.2012-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sako D., Grinberg A. V., Liu J., Davies M. V., Castonguay R., Maniatis S., Andreucci A. J., Pobre E. G., Tomkinson K. N., Monnell T. E., Ucran J. A., Martinez-Hackert E., Pearsall R. S., Underwood K. W., Seehra J., and Kumar R. (2010) Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J. Biol. Chem. 285, 21037–21048 10.1074/jbc.M110.114959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heinecke K., Seher A., Schmitz W., Mueller T. D., Sebald W., and Nickel J. (2009) Receptor oligomerization and beyond: a case study in bone morphogenetic proteins. BMC Biol. 7, 59 10.1186/1741-7007-7-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greenwald J., Groppe J., Gray P., Wiater E., Kwiatkowski W., Vale W., and Choe S. (2003) The BMP7/ActRII Extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol. Cell 11, 605–617 10.1016/S1097-2765(03)00094-7 [DOI] [PubMed] [Google Scholar]
- 44. Thompson T. B., Woodruff T. K., and Jardetzky T. S. (2003) Structures of an ActRIIB:activin A complex reveal a novel binding mode for TGF-β ligand:receptor interactions. EMBO J. 22, 1555–1566 10.1093/emboj/cdg156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walker R. G., Poggioli T., Katsimpardi L., Buchanan S. M., Oh J., Wattrus S., Heidecker B., Fong Y. W., Rubin L. L., Ganz P., Thompson T. B., Wagers A. J., and Lee R. T. (2016) Biochemistry and biology of GDF11 and myostatin: similarities, differences, and questions for future investigation. Circ. Res. 118, 1125–1141 10.1161/CIRCRESAHA.116.308391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eisenberg D., Schwarz E., Komaromy M., and Wall R. (1984) Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179, 125–142 10.1016/0022-2836(84)90309-7 [DOI] [PubMed] [Google Scholar]
- 47. Ibrahim B. S., and Pattabhi V. (2004) Crystal structure of trypsin–turkey egg white inhibitor complex. Biochem. Biophys. Res. Commun. 313, 8–16 10.1016/j.bbrc.2003.11.082 [DOI] [PubMed] [Google Scholar]
- 48. Johansson M. W., Keyser P., and Söderhall K. (1994) Purification and cDNA cloning of a four-domain Kazal proteinase inhibitor from crayfish blood cells. Eur. J. Biochem. 223, 389–394 10.1111/j.1432-1033.1994.tb19005.x [DOI] [PubMed] [Google Scholar]
- 49. Pariani S., Contreras M., Rossi F. R., Sander V., Corigliano M. G., Simón F., Busi M. V., Gomez-Casati D. F., Pieckenstain F. L., Duschak V. G., and Clemente M. (2016) Characterization of a novel Kazal-type serine proteinase inhibitor of Arabidopsis thaliana. Biochimie 123, 85–94 10.1016/j.biochi.2016.02.002 [DOI] [PubMed] [Google Scholar]
- 50. Lee S.-J., Lee Y.-S., Zimmers T. A., Soleimani A., Matzuk M. M., Tsuchida K., Cohn R. D., and Barton E. R. (2010) Regulation of muscle mass by follistatin and activins. Mol. Endocrinol. 24, 1998–2008 10.1210/me.2010-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keutmann H. T., Schneyer A. L., and Sidis Y. (2004) The role of follistatin domains in follistatin biological action. Mol. Endocrinol. 18, 228–240 10.1210/me.2003-0112 [DOI] [PubMed] [Google Scholar]
- 52. Geng Y., Dong Y., Yu M., Zhang L., Yan X., Sun J., Qiao L., Geng H., Nakajima M., Furuichi T., Ikegawa S., Gao X., Chen Y.-G., Jiang D., and Ning W. (2011) Follistatin-like 1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling antagonist in controlling mouse lung development. Proc. Natl. Acad. Sci. U.S.A. 108, 7058–7063 10.1073/pnas.1007293108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patthy L., and Nikolics K. (1993) Functions of agrin and agrin-related proteins. Trends Neurosci. 16, 76–81 10.1016/0166-2236(93)90021-D [DOI] [PubMed] [Google Scholar]
- 54. Chang C., Eggen B. J., Weinstein D. C., and Brivanlou A. H. (2003) Regulation of nodal and BMP signaling by tomoregulin-1 (X7365) through novel mechanisms. Dev. Biol. 255, 1–11 10.1016/S0012-1606(02)00075-1 [DOI] [PubMed] [Google Scholar]
- 55. Walker R. G., McCoy J. C., Czepnik M., Mills M. J., Hagg A., Walton K. L., Cotton T. R., Hyvönen M., Lee R. T., Gregorevic P., Harrison C. A., and Thompson T. B. (2018) Molecular characterization of latent GDF8 reveals mechanisms of activation. Proc. Natl. Acad. Sci. U.S.A. 115, E866–E875 10.1073/pnas.1714622115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Terwilliger T. C., Adams P. D., Read R. J., McCoy A. J., Moriarty N. W., Grosse-Kunstleve R. W., Afonine P. V., Zwart P. H., and Hung L.-W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65, 582–601 10.1107/S0907444909012098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., and IUCr. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B. 3rd, Snoeyink J., Adams P. D., Lovell S. C., et al. (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J.-M., Taly J.-F., and Notredame C. (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13–W17 10.1093/nar/gkr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. DeLano W. L. (2018) The PyMOL Molecular Graphics System, version 2.2.0, Schroedinger, LLC, New York [Google Scholar]
- 63. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.