

Abstract
T cell suppression contributes to immune dysfunction in sepsis. However, the underlying mechanisms are not well-defined. Here, we show that exposure of human peripheral blood mononuclear cells to bacterial lipopolysaccharide (LPS) can rapidly and dose-dependently suppress interleukin-2 (IL-2) production and T cell proliferation. We also report that these effects depend on monocytes. LPS did not prevent the interaction of monocytes with T cells, nor did it induce programmed cell death protein 1 (PD-1) signaling that causes T cell suppression. Instead, we found that LPS stimulation of monocytes led to the accumulation of extracellular ATP that impaired mitochondrial function, cell migration, IL-2 production, and T cell proliferation. Mechanistically, LPS-induced ATP accumulation exerted these suppressive effects on T cells by activating the purinergic receptor P2Y11 on the cell surface of T cells. T cell functions could be partially restored by enzymatic removal of extracellular ATP or pharmacological blocking of P2Y11 receptors. Plasma samples obtained from sepsis patients had similar suppressive effects on T cells from healthy subjects. Our findings suggest that LPS and ATP accumulation in the circulation of sepsis patients suppresses T cells by promoting inappropriate P2Y11 receptor stimulation that impairs T cell metabolism and functions. We conclude that inhibition of LPS-induced ATP release, removal of excessive extracellular ATP, or P2Y11 receptor antagonists may be potential therapeutic strategies to prevent T cell suppression and restore host immune function in sepsis.
Keywords: T cell, lipopolysaccharide (LPS), monocyte, purinergic receptor, sepsis, adaptive immunity, ATP release, immune dysregulation, purinergic signaling, T cell suppression
Introduction
Sepsis is defined as dysregulated host immune response to infection that results in multiple organ dysfunction syndrome (1). Sepsis, septic shock, and multiple-organ dysfunction syndrome are leading causes of death among intensive care patients (2). Despite substantial progress in the clinical management of sepsis patients, overall clinical outcome has remained unsatisfactory due to the lack of effective pharmacological interventions that prevent the dysregulated host immune response (3). The immune response in sepsis involves both pro- and anti-inflammatory processes that culminate in profound immunosuppression, which increases the risk of nosocomial infections and further promotes the progression of sepsis (4). Immunosuppression and immune paralysis are accompanied by T cell apoptosis, by a reduction in T cell numbers, and by T cell dysfunction (4, 5). T cell dysfunction is caused by increased expression of inhibitory co-receptors such as programmed cell death protein 1 (PD-1)4 and cytotoxic T lymphocyte–associated protein 4 (CTLA4), by immunosuppressive cytokines such as transforming growth factor-β and IL-10, and by the shift of T cell populations to immunosuppressive and regulatory T cell subsets (6, 7). However, the mechanisms that lead to T cell dysfunction and the roles that antigen-presenting cells (APCs) play as instigators of T cell suppression in sepsis are still not fully characterized.
Lipopolysaccharides (LPS) are endotoxins found in the outer cell wall of Gram-negative bacteria. LPS activates immune cells by binding to Toll-like receptor 4 (TLR4). TLR4 signaling contributes to the deleterious host immune response in sepsis. TLR4 is highly expressed in monocytes and other APCs. Stimulation of TLR4 by LPS alters cell metabolism, causes NLRP3 inflammasome activation, and leads to ATP release from activated APCs (8, 9). LPS can affect T cells by promoting regulatory T cell phenotypes with suppressive properties or by impairing the antigen-presenting capacity of APCs (10–12). These roles of APCs as instigators of T cell suppression are supported by the findings that monocytes in sepsis patients express reduced levels of the human leukocyte antigen-DR (HLA-DR) that is required for T cell stimulation and that this decrease goes hand-in-hand with the loss of antimicrobial resistance of patients in the end stages of sepsis (13). However, T cells themselves can also express TLR4 and may thus respond directly to LPS (14). Understanding the mechanisms by which LPS impairs T cell functions is necessary for the development of effective therapeutic strategies to treat patients with sepsis.
We have previously shown that LPS stimulation of monocytes and macrophages involves rapid ATP release that is required for IL-1β production and other APC functions (15). Purinergic signaling is a recently discovered ubiquitous cell activation mechanism that regulates the functions of various leukocyte populations, including T cells (16–19). T cell activation requires several steps, including cell migration, immune synapse (IS) formation, and the interactions of T cells with APCs that lead to T cell receptor (TCR) and CD28 co-receptor stimulation. All of these processes involve ATP release and autocrine stimulation of purinergic P2 receptors that regulate cellular Ca2+ influx and mitochondrial activation (20–23). Recently, we reported that localized stimulation of P2X4 receptors promotes Ca2+ influx and selective activation of mitochondria at the leading edge of T cells during cell migration and IS signaling (23). These synchronized actions of P2X4 receptors and mitochondria orchestrate the complex processes that lead from T cell migration to effector functions, such as IL-2 production and T cell proliferation. Exogenous ATP, however, can inhibit T cells (24–26). We hypothesized that LPS-induced ATP release from APCs may cause T cell suppression by interfering with the localized purinergic signaling mechanisms that regulate T cell functions.
Here, we show that external ATP that is released from LPS-stimulated monocytes does indeed suppress T cells. Specifically, we demonstrate that external ATP causes excessive activation of P2Y11 receptors that impair mitochondrial function and deplete the endogenous purinergic signaling system that is needed for cell polarization, cell migration, IS formation, and T cell activation by APCs.
Results
LPS rapidly and dose-dependently suppresses T cell activation
Sepsis-induced T cell suppression is characterized by impaired T cell proliferation and cytokine production (27). Previous studies have shown that overnight incubation of peripheral blood mononuclear cell (PBMC) cultures with LPS suppresses TCR-induced T cell proliferation (10, 12). In agreement with those studies, we found that LPS dose-dependently inhibited the proliferation of CD4 T cells (Fig. 1A and Fig. S1) and the production of IL-2 (Fig. 1B) in human PBMC cultures. LPS significantly suppressed T cell functions at concentrations as low as 100 pg/ml, which is well within the range of the LPS levels found in the peripheral blood of sepsis patients (28). We stimulated T cells with soluble anti-CD3 antibodies, which forces T cells to search for and interact with APCs so as to receive co-stimulatory CD28 signals that are required for full-fledged T cell responses. As expected, there were no measurable T cell responses in monocyte-depleted PBMC cultures (Fig. 1C). In contrast to previous studies (10, 12), we found that prolonged pretreatment of PBMCs with LPS was not required to achieve T cell suppression. Instead, LPS blocked IL-2 production with equal efficiency regardless of whether it was added 10 min before or at the same time of T cell stimulation (Fig. 1D). In fact, the addition of LPS shortly before or during TCR stimulation had a stronger suppressive effect than pretreatment with LPS for 24 h prior to TCR stimulation. Moreover, LPS was still able to suppress IL-2 production even when it was added as late as 2 h after TCR stimulation (Fig. 1D). Taken together, these results demonstrate that LPS can rapidly suppress T cells, apparently without the need for de novo gene expression.
Figure 1.
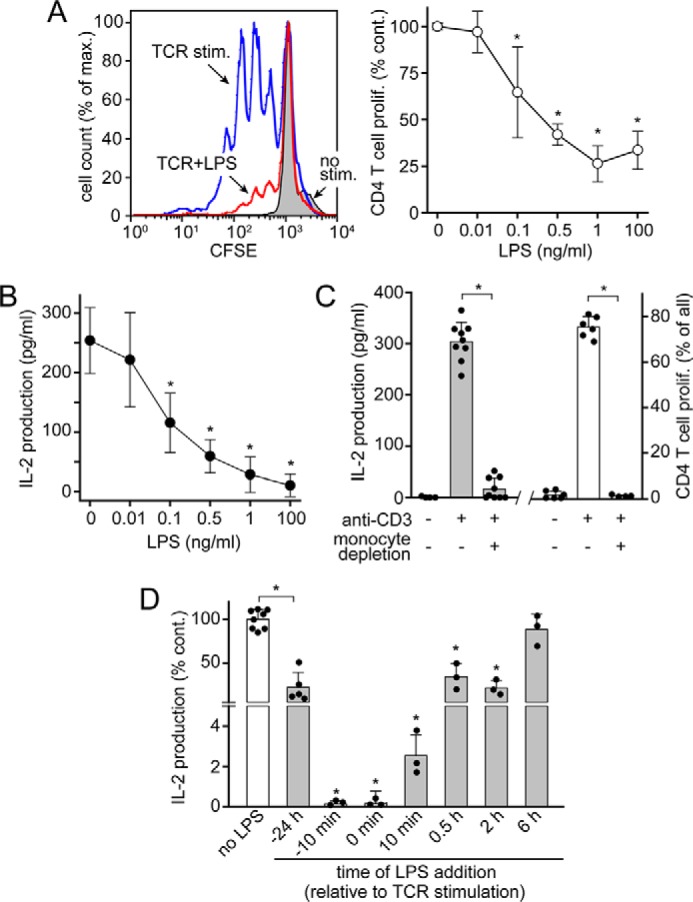
LPS rapidly and dose-dependently suppresses T cell activation. A, LPS dose-dependently suppresses proliferation of CD4 T cells in PBMC cultures stimulated with soluble anti-CD3 antibodies. PBMCs were labeled with CFSE, treated with LPS for 10 min, and stimulated with anti-CD3 antibodies for 3 days. CD4 T cell proliferation was measured by assessing CFSE dye dilution with flow cytometry as shown in Fig. S1. Representative histograms are displayed in the left panel (LPS, 1 ng/ml), and mean values ± S.D. (error bars) of n ≥ 4 independent experiments with cells from different healthy subjects are shown in the right panel. *, p < 0.05 versus no LPS, Kruskal–Wallis test. B, LPS dose-dependently inhibits IL-2 production. PBMCs were treated with LPS for 10 min prior to stimulation with anti-CD3 antibodies. IL-2 concentrations in the supernatants were determined with ELISA after 18 h. Data are means ± S.D. of n ≥ 3 experiments. *, p < 0.05 versus no LPS, one-way ANOVA. C, PBMCs or monocyte-depleted PBMCs were stimulated with soluble anti-CD3 antibodies in flat-bottom well plates, which necessitates migration of T cells to interact with APCs. IL-2 production (left) and CD4 T cell proliferation (right) were measured after 18 or 72 h, respectively. Data are means ± S.D. of n ≥ 4 experiments. *, p < 0.05, t test. D, LPS suppresses T cells in minutes, not hours. LPS (10 ng/ml) was added to PBMC cultures at the indicated times before, simultaneously with, or after stimulation of T cells with soluble anti-CD3 antibodies. IL-2 released into the culture supernatants was measured after 18 h. Data are shown as mean ± S.D. of n ≥ 3 experiments. *, p < 0.05 versus no LPS, one-way ANOVA.
Monocytes need access to the immune synapse to suppress T cells
LPS can influence T cells directly or indirectly via modulation of APC functions (10–12, 14). We found that LPS-induced T cell suppression depends on the presence of monocytes (Fig. 2A). In the absence of monocytes, LPS failed to suppress IL-2 production of T cells that were stimulated by TCR/CD28-cross-linking antibodies. LPS also failed to suppress T cells when PBMCs were stimulated with anti-CD3/CD28 antibody–coated beads (Fig. 2, A and B). This mode of T cell stimulation provides direct co-stimulation whereby T cells bind tightly to beads and form pseudo-immune synapses that are inaccessible to monocytes. However, monocytes were able to suppress T cells when PBMCs were stimulated by TCR/CD28 cross-linking. This mode of stimulation allows monocytes to access the IS, where they may deliver inhibitory signals that impair T cell function. LPS was most effective in suppressing T cells when PBMCs were stimulated with soluble anti-CD3 antibodies, which requires that T cells migrate to interact with monocytes and obtain costimulatory CD28 receptor signals (Fig. 2, A and B). Based on these findings, we concluded that monocytes are responsible for LPS-induced T cell suppression, that they require physical access to the IS, and that this access enables APCs to impair the migration and activation of T cells.
Figure 2.

LPS suppresses T cells via monocytes that must have direct access to the immune synapse. A, PBMCs or monocyte-depleted PBMCs were stimulated with anti-CD3 antibodies (TCR), with soluble anti-CD3 and anti-CD28 antibodies that were cross-linked with anti-IgG antibodies, or with anti-CD3/CD28 antibody–coated microbeads (beads) in the presence or absence of LPS (10 ng/ml). IL-2 production was measured after 18 h using ELISA. Data represent mean values ± S.D. (error bars) of n ≥ 4 experiments with cells from different donors. *, p < 0.05 versus no LPS, t test. B, PBMCs were labeled with CFSE and stimulated with anti-CD3 antibodies, with crosslinked anti-CD3 and anti-CD28 antibodies, or with beads coated with anti-CD3 and anti-CD28 antibodies in the presence or absence of LPS (10 ng/ml). CD4 T cell proliferation was measured after 3 days. Data represent mean values ± S.D. of n ≥ 4 experiments. #, p < 0.05; *, p < 0.05 versus no LPS, t test. C–E, LPS-induced T cell suppression does not involve PD-1/PD-L1 signaling. C, PBMCs were treated with LPS (10 ng/ml) for the indicated times, and PD-L1 expression on monocytes was measured by flow cytometry. Data represent mean values ± S.E. (n = 5–7). *, p < 0.5 versus no LPS (Kruskal–Wallis test). D and E, PBMCs were treated or not with LPS (10 ng/ml) and/or anti-PD1 antibodies (1 μg/ml) and stimulated with anti-CD3 antibodies for 18 h (D) or for 5 days (E), and IL-2 production or proliferation of CD4 T cells were analyzed; mean ± S.D., n = 2 (D) or 6 (E). *, p < 0.05 versus untreated control, one-way ANOVA. F and G, LPS does not prevent interactions of T cells and monocytes. PBMCs were labeled with anti-CD4-allophycocyanin and anti-CD11b-Alexa488 antibodies, stimulated with anti-CD3 antibodies in the presence or absence of LPS (10 ng/ml), and briefly pelleted in Eppendorf tubes to facilitate cell-to-cell interactions, and the percentage of monocytes that formed conjugates with CD4 T cells was determined by flow cytometry after 1 h. Representative dot plots (F) and averaged results ± S.D. (G) of n = 8 experiments are shown. *, p < 0.05 versus no stimulation, Kruskal–Wallis test.
LPS-stimulated monocytes do not require PD-1 signaling to suppress T cells
Monocytes can suppress T cells by stimulating the inhibitory PD-1 coreceptors of T cells via programmed-death ligand 1 (PD-L1) that is expressed on the cell surface of monocytes (29, 30). LPS and sepsis induce PD-L1 expression on monocytes, and blockade of PD-1/PD-L1 signaling was shown to improve outcome in sepsis (30, 31). Interestingly, we found that PD-L1 expression on the cell surface of monocytes increased within minutes of LPS stimulation, indicating a transcription-independent release of prestored receptor molecules in the early activation phase (Fig. 2C). PD-1 colocalizes with CD28 receptors at the IS of T cells (32). Therefore, we hypothesized that LPS-stimulated monocytes may inhibit T cell activation by stimulation of PD-1 receptors at the IS. To test this hypothesis, we used blocking antibodies that prevent the stimulation of PD-1 on T cells (33). PD-1 antibodies had no effect on T cell functions and failed to prevent LPS-induced T cell suppression (Fig. 2, D and E). Thus, PD-1 signaling does not appear to be involved in the suppression of T cells by LPS-stimulated monocytes.
LPS does not prevent physical interactions of monocytes with T cells
The findings shown above suggest that monocytes must access the IS to suppress T cells but that PD-1 signaling is not involved in LPS-induced T cell suppression. Another possible mechanism by which LPS could block T cell activation is by preventing IS formation between T cells and monocytes. Therefore, we examined whether LPS impairs cell-to-cell interactions between monocytes and T cells. PBMCs were incubated with anti-CD3 antibodies in microcentrifuge tubes and gently centrifuged to pellet cells. This step minimizes the requirement for cell migration to facilitate the interaction of T cells with monocytes. Pretreatment with LPS did not alter the percentages of monocytes and T cells that interacted in response to anti-CD3 antibody stimulation (Fig. 2, F and G). These findings suggest that LPS does not impair the ability of monocytes to physically interact with T cells and form monocyte/T cell clusters that are required for IS formation.
LPS-stimulated monocytes release ATP that can affect T cells
Based on the results shown above, we wondered whether LPS-stimulated monocytes employ soluble mediators to impair T cell functions. LPS is known to trigger ATP release from monocytes and macrophages (34–36). We have recently shown that monocytes and macrophages release ATP within seconds of LPS stimulation (15). Therefore, we hypothesized that LPS-stimulated monocytes could affect T cells by rapid ATP release that impairs T cell functions. To test this hypothesis, we labeled PBMCs with fluorescent anti-CD4 antibodies and a membrane-anchoring ATP probe that binds to the surface of cells, where it acts as a fluorescence indicator of ATP release (37). Then we stimulated these labeled PBMCs with anti-CD3 antibodies to induce the formation of immune synapses. We found that LPS triggered ATP release that was most pronounced at cell contact sites between monocytes and T cells (Fig. 3A and Video S1). In agreement with previous reports (15), we found that stimulation of purified monocyte cultures with LPS triggered rapid accumulation of extracellular ATP (Fig. 3B). Interestingly, stimulation of PBMC cultures with LPS resulted in significantly higher ATP concentrations when compared with purified monocytes. Moreover, the resulting ATP levels in PBMC cultures remained elevated for at least 30 min, whereas ATP levels in monocyte culture supernatants returned to baseline levels within 1 min after LPS stimulation (Fig. 3B). Extracellular ATP itself has been shown to induce cellular ATP release from human peripheral blood leukocytes (38). Therefore, it seems possible that ATP release in PBMC cultures is triggered by LPS-stimulated monocytes and amplified by ATP-induced ATP release from other cells in the PBMC cultures.
Figure 3.
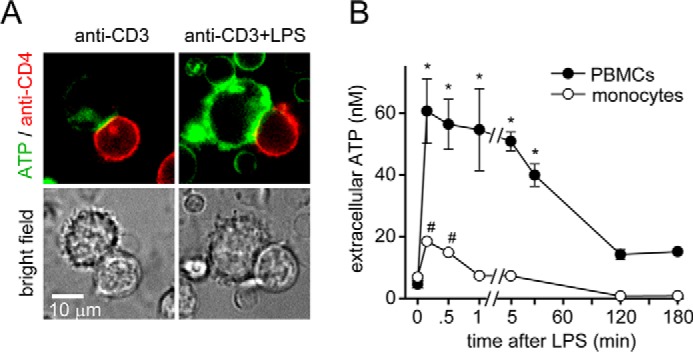
LPS-stimulated monocytes release ATP that can affect T cells. A, LPS-stimulated monocytes release excessive ATP. PBMCs were stained with anti-CD4-allophycocyanin antibodies. Cells were allowed to attach to fibronectin-coated glass-bottom coverslip dishes. Then they were labeled with the fluorescent membrane–anchoring ATP probe 2-2Zn. Immune synapse formation between monocytes and CD4 T cells was initiated by stimulation with anti-CD3 antibodies, and ATP release in response to LPS (10 ng/ml) was observed with live-cell fluorescence microscopy. Representative images of n = 7–10 T cell/monocyte conjugates derived from three different experiments are shown; ×100 objective (NA 1.4). B, PBMCs release more ATP than monocytes in response to LPS. Purified human monocytes or PBMC cultures were stimulated with LPS (10 ng/ml), and ATP concentrations in the cell supernatant were measured at the indicated times. Data are means ± S.D. (error bars) of n = 4 (monocytes) or 6 (PBMCs) experiments. * and #, p < 0.05 versus no LPS controls, one-way ANOVA.
Exogenous ATP impairs migration of T cells and their activation by monocytes
We have previously shown that ATP release and autocrine stimulation of P2X4 receptors are essential for T cell migration and TCR/CD28 signaling at the IS (21, 23). However, external ATP can cause T cell suppression (24, 25). Therefore, we tested whether treatment of PBMCs with exogenous ATP or with the nonhydrolysable ATP analog ATPγS affects T cell functions. ATP and ATPγS dose-dependently blocked T cell migration, IL-2 production, and T cell proliferation in response to TCR stimulation (Fig. 4, A–D). These findings support the concept that LPS suppresses T cells by inducing ATP accumulation in PBMC cultures that interferes with T cell functions.
Figure 4.

Exogenous ATP impairs T cell migration and activation by monocytes. A and B, exogenous ATP impairs T cell migration. CD4 effector T cells were seeded onto fibronectin-coated glass-bottom dishes, and spontaneous cell migration was recorded for 30 min in the presence or absence of ATP or the nonhydrolysable ATP analog ATPγS using live-cell time-lapse microscopy. Migration paths of single cells were analyzed to determine migration speed (A) and the proportion of cells that maintained cell migration without interruption (B). Data are means ± S.D. (error bars) of n ≥ 3 (ATP) or n = 2 (ATPγS) experiments, each comprising at least 20 analyzed cells. * and #, p < 0.05 versus untreated control, one-way ANOVA. C and D, exogenous ATP impairs IL-2 production and T cell proliferation. PBMCs were treated with the indicated concentrations of ATP or ATPγS and stimulated with anti-CD3 antibodies, and IL-2 production (C) and CD4 T cell proliferation (D) were analyzed after 18 h and 3 days, respectively. Data represent mean values ± S.D., n = 3–6. * and #, p < 0.05 versus control, one-way ANOVA.
LPS-induced ATP accumulation impairs T cells by stimulation of P2Y11 receptors
Human CD4 T cells express primarily P2X4 receptors, but P2Y11 receptors are also highly expressed (21, 39). Endogenous stimulation of P2X4 is essential for T cell migration and for TCR/CD28 signaling at the IS (23). P2X4 receptors are ATP-gated Ca2+ channels that accumulate with mitochondria at the leading edge and IS of T cells, suggesting that P2X4 receptors regulate mitochondrial metabolism and T cell functions in a spatially and temporally defined manner (21, 23). P2Y11 receptors are ATP-selective G protein–coupled receptors that can couple to both Gq and Gs proteins that activate PLC and intracellular cAMP/PKA signaling, respectively (40). Various T cell functions are inhibited by cAMP/PKA signaling (41). Therefore, we examined whether and how stimulation of P2Y11 receptors contributes to LPS-induced T cell suppression. We added the ATP scavenger apyrase at low concentrations that were sufficient to remove exogenous ATP without depleting endogenous ATP signaling that occurs at the T cell surface. This treatment was able to fully restore the ability of T cells to undergo cell migration and partially alleviated the suppressive effect of LPS on T cell proliferation (Fig. 5A). Inhibition of P2Y11 receptors with the antagonist NF340, but not of P2X4 receptors with 5-BDBD, restored T cell polarization and migration in the presence of exogenous ATP (Fig. 5B and Fig. S2, A and B). Blocking of P2Y11 receptors also reduced the suppressive effects of exogenous ATP or LPS on IL-2 production (Fig. 5, C and D). Similar effects could be achieved with the PKA inhibitor H89 (Fig. S2C), suggesting that P2Y11 receptors suppress T cell functions via cAMP/PKA signaling. Taken together, these findings suggest that LPS-induced accumulation of extracellular ATP suppresses T cells by inappropriate stimulation of P2Y11 receptors, thereby interfering with the endogenous purinergic signaling that is required for cell polarization, T cell migration, and the interaction of T cells with APCs.
Figure 5.
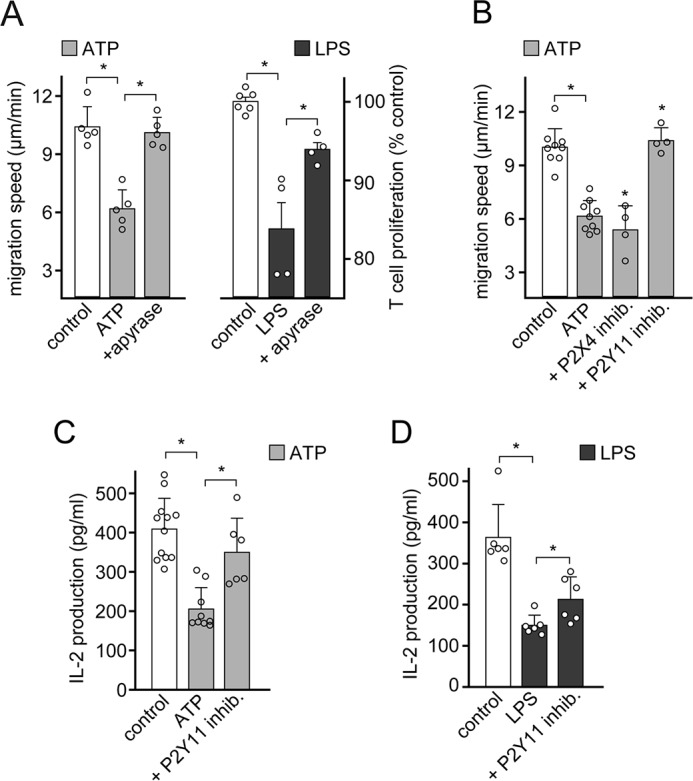
LPS-induced ATP release impairs T cells via P2Y11 receptors. A, scavenging of extracellular ATP restores T cell migration and proliferation. CD4 effector T cells were exposed to exogenous ATP (10 μm; left), and PBMCs were stimulated with LPS (100 pg/ml) and anti-CD3 antibodies (right) in the presence or absence of apyrase (1 microunit/ml). T cell migration was assessed by time-lapse microscopy, and CD4 T cell proliferation was measured with flow cytometry after 3 days. Data are means ± S.D. (error bars) of n = 5 experiments (left; each experiment comprising the analysis of at least 20 individual cells) or n = 4 experiments (right). *, p < 0.05, one-way ANOVA. B, stimulation of P2Y11 receptors by exogenous ATP blocks T cell migration. CD4 effector T cells exposed to exogenous ATP (10 μm) were treated with selective antagonists of P2X4 (5-BDBD; 10 μm) or P2Y11 (NF340; 10 μm) receptors, and T cell migration was assessed by time-lapse microscopy. Data are means ± S.D. of n ≥ 4 separate experiments, each comprising the analysis of at least 20 individual cells. *, p < 0.05, one-way ANOVA. C and D, LPS and exogenous ATP impair T cell activation via P2Y11 receptors. PBMCs were pretreated with the P2Y11 receptor antagonist NF340 (100 nm), exposed to exogenous ATP (100 μm) or LPS (10 ng/ml), and stimulated with anti-CD3 antibodies, and IL-2 production was determined after 18 h. Data represent mean ± S.D. of n ≥ 6 experiments. Values are corrected for the effect of inhibitors alone. *, p < 0.05 versus control (one-way ANOVA or Kruskal–Wallis test).
Exogenous ATP impairs mitochondrial activity in T cells by intercepting Ca2+ signaling
Mitochondria in T cells generate the ATP that fuels the endogenous purinergic signaling mechanisms that regulate cell functions (22, 42). We found that localized ATP release and autocrine feedback via P2X4 receptors regulates Ca2+ signaling and mitochondrial function (23). Therefore, we studied whether inappropriate stimulation of T cells by exogenous ATP could interfere with these endogenous signaling mechanisms. Indeed, we found that exposure of cells to exogenous ATP or ATPγS time-dependently reduced the mitochondrial membrane potential and mitochondrial activity as assessed by reactive oxygen species (ROS) production in CD4 T cells (Fig. 6A). These findings indicate that exogenous ATP reduces oxidative phosphorylation, which results in the shutdown of mitochondrial activity in T cells. Oxidative phosphorylation depends on the uptake of Ca2+ into the mitochondrial matrix (43). We expressed a mitochondrial Ca2+ biosensor in Jurkat T cells to study whether exogenous ATP impairs mitochondrial Ca2+ uptake. Treatment of these cells with exogenous ATP almost completely abolished mitochondrial Ca2+ uptake in response to TCR/CD28 stimulation (Fig. 6B). Similarly, exposure of unstimulated and TCR/CD28-stimulated CD4 T cells or Jurkat cells to exogenous ATP dose-dependently impaired cytosolic Ca2+ signaling (Fig. 6, C and D). Taken together, these findings indicate that LPS-induced accumulation of exogenous ATP suppresses T cells by interfering with their purinergic receptors and the coordinated mitochondrial and Ca2+ signaling responses that regulate T cell functions.
Figure 6.
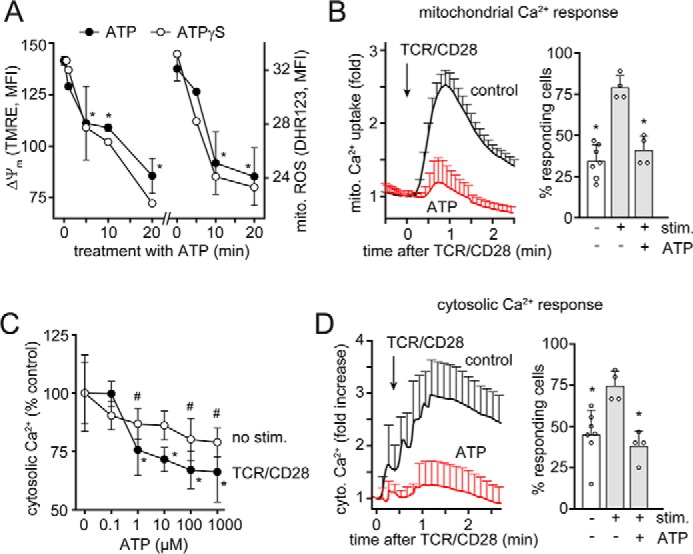
Exogenous ATP impairs mitochondrial activity and Ca2+ signaling in T cells. A, blood leukocytes were treated with ATP (100 μm) or ATPγS (100 μm) for the indicated periods of time, and mitochondrial membrane potential (ΔΨm) and ROS production of CD4 T cells were analyzed by flow cytometry. Data are shown as mean ± S.D. (error bars) (n = 3). *, p < 0.05 versus untreated control. B, Jurkat T cells expressing the mitochondrial Ca2+ biosensor mito-CAR-GECO1 were treated or not (control) with ATP (100 μm) for 20 min, and mitochondrial Ca2+ uptake following stimulation via TCR/CD28 cross-linking was monitored by time-lapse fluorescence microscopy. Mean fluorescent values ± S.E. of n = 150–200 cells analyzed in 4–7 separate experiments are shown in the left panel, and the percentage of cells responding with an increase in mitochondrial Ca2+ uptake is shown on the right (mean ± S.D.; *, p < 0.05 versus stimulated control, one-way ANOVA). C, anti-CD4–labeled leukocytes were loaded with Fluo-4 and treated for 20 min with ATP, and intracellular Ca2+ levels were assessed before and 3 min after TCR/CD28 cross-linking; mean ± S.D. (n = 3). * and #, p < 0.01 versus untreated control, one-way ANOVA. D, Jurkat cells expressing the cytosolic Ca2+ indicator G-GECO1.1 were treated as described in B, and changes in cytosolic Ca2+ were recorded by video fluorescence microscopy. Representative results (mean ± S.D.) of one of n = 4–6 experiments (each comprising 15–25 single cells) are shown in the left panel, and the mean ± S.D. percentage of responding cells is shown in the right panel. *, p < 0.05 versus untreated control, one-way ANOVA.
Stimulation of P2Y11 receptors by exogenous ATP impairs T cells in sepsis
Conditions that are associated with sepsis, such as inflammation, ischemia, and hypoxia, increase ATP levels in the extracellular space (44). Mice with polymicrobial sepsis have significantly elevated circulating plasma ATP concentrations (45). LPS levels in the plasma of sepsis patients are significantly increased (28). Therefore, it is likely that LPS in the plasma of sepsis patients triggers monocyte-induced ATP release that generates sufficient exogenous ATP to suppress T cells. We tested this concept by incubating PBMCs of healthy human subjects with plasma samples obtained from sepsis patients or from healthy control subjects (Fig. 7, A and B). Exposure of CD4 T cells from healthy subjects to plasma samples from sepsis patients but not from healthy controls significantly reduced mitochondrial membrane potential, basal and TCR/CD28-stimulated cytosolic Ca2+ signaling, and T cell migration (Fig. 7, A and B). Removal of extracellular ATP with apyrase or inhibition of P2Y11 receptors with the antagonist NF340 prevented the suppressive effect of sepsis plasma on T cell migration (Fig. 7B). Taken together, these findings suggest that soluble mediators like LPS in the plasma of sepsis patients suppress T cells by generating the accumulation of exogenous ATP that interferes through P2Y11 receptors with the coordinated action by which mitochondria and purinergic signaling regulate T cell functions.
Figure 7.

Plasma of sepsis patients impairs mitochondrial activity and migration of T cells by stimulation of P2Y11 receptors. A, enriched white blood cells of healthy subjects were treated with plasma from healthy controls or from sepsis patients (20%, v/v) for 60 min, and the mitochondrial membrane potential (ΔΨm) and cytosolic Ca2+ levels were analyzed in CD4 T cells by flow cytometry. Ca2+ levels were measured with Fluo-4 before and 3 min after cell stimulation by TCR/CD28 cross-linking (mean ± S.E. (error bars), n = 10–15; * and #, p < 0.05, t test). B, plasma of sepsis patients blocks T cell migration. CD4 effector T cells of healthy human donors were incubated with plasma of healthy controls or sepsis patients in the presence or absence of apyrase (1 microunit/ml) or the P2Y11 receptor antagonist NF340 (10 μm), and migration was observed by time-lapse microscopy. Data are means ± S.D. of n ≥ 3 separate experiments, each comprising the analysis of at least 20 individual cells; * and #, p < 0.05, one-way ANOVA. C, proposed model of LPS-induced T cell suppression. T cells must assume polarized cell shapes that facilitate efficient cell migration in response to chemokines such as the CXCR4 ligand SDF-1α. Cell migration allows T cells to interact with and become stimulated by APCs. Cell migration is regulated by purinergic signaling clusters at the leading edge, which consist of P2X4 receptors, pannexin-1 channels (panx1), and mitochondria. Autocrine stimulation of P2X4 receptors fuels a feed-forward signaling loop involving Ca2+ influx, mitochondrial activation and ATP synthesis, and ATP release that stimulates P2X4 receptors at the front and P2Y11 receptors at the back of cells. Endogenous stimulation of P2Y11 receptors at the back promotes uropod retraction, stabilizes cell polarity, and prevents mitochondrial activation and aberrant pseudopod formation at the back. LPS stimulates TLR4 receptors on monocytes, resulting in the accumulation of large amounts of ATP in the extracellular space where this systemic ATP interferes with the endogenous ATP signaling mechanisms that regulate T cell functions. Excessive P2Y11 receptor stimulation leads to high intracellular cAMP levels that cause the global shutdown of mitochondria in T cells and prevent the polarization, migration, and activation of T cells.
Discussion
Restoring the function of T cells can ameliorate clinical outcome in critically ill patients (3, 4, 7). Our current data suggest that LPS-induced ATP release from monocytes contributes to T cell suppression by generating elevated systemic ATP levels that interfere with T cell metabolism and functions. It is well known that inflammatory conditions increase extracellular ATP concentrations and that systemic ATP can act as a danger signal that contributes to poor clinical outcome in inflammatory and infectious diseases (44). However, it is less well-understood how LPS-induced ATP release contributes to the dysregulated host immune response and clinical course in sepsis patients.
LPS can induce ATP release from monocytes and macrophages (15, 35). Most previous studies focused on the late phase of ATP release that occurs hours after TLR stimulation and coincides with NLRP3 inflammasome activation (34, 36). Our previous work demonstrated that pannexin-1–dependent ATP release occurs within seconds after the stimulation of monocytes and macrophages with LPS and that this early phase of ATP release initiates a cascade of events that ultimately culminates in inflammasome activation (15).
ATP release and autocrine purinergic signaling regulate the functions of many other immune cell populations besides monocytes and macrophages (17, 18). In CD4 T cells, stimulation of the chemokine receptor CXCR4 triggers rapid ATP release that regulates T cell migration through autocrine feedback via P2X4 receptors (23). T cell migration is an essential feature that allows T cells to interact with APCs and recognize matching antigens (46). This antigen recognition process also involves autocrine stimulation of P2X4 receptors that accumulate at the IS along with pannexin-1 ATP release channels and mitochondria, resulting in a stimulatory complex that promotes Ca2+ influx, T cell activation, cytokine production, and T cell proliferation (20–22). Here, we found that LPS-induced ATP release from monocytes blocks these T cell responses. Our findings are in agreement with previous reports from several other groups that demonstrated suppressive effects of extracellular ATP on T cell functions (25, 47, 48). In the current study, we show that this suppression is caused at least in part by P2Y11 receptors. P2Y11 receptors can signal through Gq and Gs proteins that increase intracellular Ca2+ and cAMP levels, respectively (40). Our data suggest that excessive stimulation of P2Y11 receptors by LPS-induced ATP accumulation blocks T cells via cAMP/PKA-dependent mechanisms. This conclusion is in agreement with other reports that have shown that extracellular ATP can increase intracellular cAMP levels in CD4 T cells (25).
The exact mechanisms by which P2Y11 receptors regulate T cell functions are not well-defined. However, our findings demonstrate that overstimulation of P2Y11 receptors with exogenous ATP results in the shutdown of mitochondrial activity and Ca2+ signaling, which impairs cell migration and T cell activation. Cell migration depends on the polarization of cells, which is accomplished by excitatory signals that promote pseudopod protrusion at the front of cells and complementary inhibitory signals that facilitate uropod retraction at the back. We hypothesize that endogenous P2Y11 receptor signaling provides this inhibitory signal needed for T cell migration and that systemic ATP interferes with these regulatory functions of P2Y11 receptors. Roles for P2Y11 receptors in T cell regulation are also suggested by recent findings that P2Y11 receptors regulate T cell functions and T cell viability in autoimmune diseases (39, 49). In addition to P2Y11 receptors, T cells also express A2a and A2b adenosine receptors that also couple to Gs and have been implicated in immunosuppression. Further studies will be needed to determine whether A2 receptors can substitute for P2Y11 receptors and how these receptors contribute to T cell functions and the inhibition of these functions by external ATP. This is particularly important for animal models using mice that do not express P2Y11 receptors (50).
Based on our current findings and previous work, we propose the following mechanism by which LPS suppresses T cells (Fig. 7C). Under normal physiological conditions, spatiotemporally defined and localized signaling events downstream of P2X4 and P2Y11 receptors regulate T cell migration and T cell activation by APCs. In the presence of LPS or other inflammatory mediators, excessive accumulation of systemic ATP results in inappropriate P2Y11 receptor stimulation that disrupts T cell polarity, blocks cell migration, and interferes with subsequent T cell responses that are required for host defense. We found that removal of systemic ATP with apyrase can prevent these disruptive effects of exogenous ATP. Interestingly, apyrase has been shown to significantly improve bacterial clearance and survival in animal models of inflammation and sepsis (51, 52). The beneficial effect of apyrase can be partially explained by the restoration of neutrophil chemotaxis that is impaired by exogenous ATP (53, 54). However, apyrase may have similarly beneficial effects on T cells by restoring their ability to undergo cell migration, which raises the question of whether apyrase treatment could help reduce defects in both the innate and adaptive immune responses that contribute to morbidity and mortality in sepsis.
The host immune response in sepsis is complex and involves an inflammatory phase that causes tissue damage and an anti-inflammatory phase that impairs antimicrobial host defenses (55). Since the discovery of TLR signaling, most effort has been focused on targeting the inflammatory component, but with little clinical success (56). In recent years, attention has turned to the anti-inflammatory phase that suppresses T cells (4, 7, 57). However, the inflammatory and anti-inflammatory phases are both critical components of sepsis. Our work suggests that both phases are linked by ATP release and that targeting the excessive accumulation of ATP may be a possible therapeutic approach to restore both innate and adaptive immune responses in sepsis.
Experimental procedures
Reagents
Carboxyfluorescein succinimidyl ester (CFSE), dihydrorhodamine 123 (DHR123), tetramethylrhodamine ethyl ester (TMRE), and Fluo-4 AM were purchased from Molecular Probes (Thermo Fisher Scientific). Anti-human CD4-allophycocyanin (clone OKT4), anti-human PD-L1-allophycocyanin (clone 29E.2A3), anti-human CD11b-Alexa Fluor 488 (clone ICRF44), and LEAF-purified anti-human PD-1 antibodies (clone EH12.2H7) were purchased from Biolegend (San Diego, CA). Mouse anti-human CD3 (clone HIT3a) and anti-human CD28 (clone CD28.2) antibodies were from BD Pharmingen (Franklin Lakes, NJ), and goat anti-mouse IgG Fc antibodies were from Pierce (Thermo Fisher Scientific). NF340, 5-BDBD, and H89 were purchased from Tocris (R&D Systems). LPS from Escherichia coli (O111:B4, purified by gel-filtration chromatography) and all other reagents were from Sigma-Aldrich if not otherwise stated.
Preparation and culture of primary cells
All studies involving human subjects were approved by the institutional review board of the Beth Israel Deaconess Medical Center and conducted in compliance with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all participants or a legal representative. PBMCs and CD4 T cells were isolated from heparinized blood of healthy volunteers and maintained as described previously (22). CD4 effector T cells were generated by culturing CD4 T cells for 3 days with anti-CD3/CD28 antibody-coated Dynabeads (Thermo Fisher Scientific) at a cell/bead ratio of 1:1. For some experiments, monocytes were depleted from PBMC cultures by plastic adherence in serum-free RPMI medium for 60 min. Enriched white blood cell preparations were obtained by allowing heparinized blood to spontaneously sediment over a Ficoll-Paque (GE Healthcare, Chicago, IL) gradient for 30 min.
Patients
Eligible patients were adult patients (>18 years) presenting to the Emergency Department of Beth Israel Deaconess Medical Center who were clinically diagnosed with sepsis or septic shock based on published criteria (1). Heparinized blood samples were obtained within 12 h of sepsis diagnosis, and plasma was immediately separated and stored at −80 °C. Fifteen patients (60% with septic shock) were included in the study. The mean age was 70 years (range, 33–98 years), eight of the patients (53.3%) were female, and four of the patients (26.6%) died within 28 days.
Measurement of IL-2 production
PBMCs (2 × 106/ml) were cultured in 96-well flat-bottom cell culture plates. LPS, ATP, or ATPγS was added to cell cultures 10 min before stimulation with anti-CD3 antibodies (0.05 μg/ml) or at the times indicated. In some experiments, cells were stimulated with anti-CD3/CD28 antibody–coated microbeads or by TCR/CD28 cross-linking using anti-CD3, anti-CD28, and anti-mouse IgG Fc antibodies (0.05 μg/ml each). Anti-PD-1 antibodies (1 μg/ml) or the P2Y11 receptor antagonist NF340 (100 nm) were added 10 min prior to treatment with LPS or ATP. IL-2 levels in cell culture supernatants were determined 18 h after T cell stimulation using a commercially available ELISA (R&D Systems).
T cell proliferation
PBMCs were labeled with CFSE following the manufacturer's instructions, placed into flat-bottom 48-well cell culture plates (1.5 × 105/well), and stimulated with anti-CD3 antibodies in the presence or absence of LPS, ATP, or ATPγS at the indicated concentrations for 3 days. In some experiments, cells were stimulated with anti-CD3/CD28 antibody-coated microbeads or by TCR/CD28 cross-linking as described above. Apyrase (1 microunit/ml) or anti-PD-1 antibodies (1 μg/ml) were added to cells 10 min before stimulation with LPS. The percentage of proliferating (CFSElow) CD4 T cells was determined with a BD FACScalibur flow cytometer (BD Biosciences). CD4 T cells were identified based on forward and side scatter properties and CD4 labeling (Fig. S1).
PD-L1 expression
PBMCs were suspended in complete cell culture medium (2 × 106/ml), transferred to a 96-well cell culture plate, and stimulated with LPS (10 ng/ml). After different times, reactions were stopped on ice, and PD-L1 expression on monocyte cell surfaces was assessed by flow cytometry using anti-PD-L1 antibodies, forward and side scatter properties, and CD11b labeling.
Mitochondrial activity and calcium signaling
Blood leukocytes were treated with ATP, ATPγS, or plasma (20% v/v) from healthy subjects or from sepsis patients and labeled with TMRE, DHR123, or Fluo-4 AM as described previously (22). When no human plasma was present, cells were labeled in complete cell culture medium. It should be noted that TMRE labeling efficiency is affected by plasma proteins (Fig. S3). Mitochondrial membrane potential, mitochondrial ROS production, or intracellular Ca2+ levels were assessed by flow cytometry. Cytosolic Ca2+ was measured before and 3 min after stimulation by TCR/CD28 cross-linking. CD4 T cells were identified based on forward/side scatter properties and CD4 labeling.
Cell conjugates
PBMCs were stained with anti-CD4-allophycocyanin and anti-CD11b-Alexa Fluor 488 antibodies, stimulated with anti-CD3 antibodies in the presence or absence of LPS (10 ng/ml) in Eppendorf microcentrifuge tubes, and briefly pelleted at 100 × g to facilitate conjugate formation between T cells and monocytes. After 1 h at 37 °C, the percentages of monocytes that formed conjugates with CD4 T cells were determined by flow cytometry.
Imaging of ATP release
Freshly isolated PBMCs were labeled with anti-CD4-allophycocyanin antibodies and allowed to attach to fibronectin-coated (40 μg/ml) glass-bottom chamber slides (Lab-Tek, Rochester, NY). To image ATP release at the cell surface, cells were suspended in Hanks' balanced salt solution and stained for 10 min with a 500 nm concentration of a cell surface–targeting fluorescent ATP probe (2-2Zn, kind gift from Dr. Itaru Hamachi, Kyoto University, Kyoto, Japan) (37). Cells were stimulated with anti-CD3 antibodies to initiate the formation of immune synapses, and the effect of LPS (10 ng/ml) on ATP release was analyzed. Fluorescence live-cell imaging was performed with an inverted Leica DMI6000B microscope (Leica Microsystems, Wetzlar, Germany) equipped with a temperature-controlled (37 °C) stage incubator (Live Cell Instrument, Seoul, South Korea) and a Leica DFC365 FX camera. Fluorescence images were captured at 5-s intervals using a ×100 oil objective (numerical aperture (NA) 1.4), Cy5 and FITC filter sets (Leica Microsystems), and LeicaLAS microscope imaging software.
ATP measurements
Monocytes were separated from PBMCs by plastic adherence in 96-well flat-bottom culture dishes as described previously (15). Freshly isolated PBMCs were placed into Eppendorf tubes. Cell cultures were maintained in a water bath placed on a vibration isolation table to minimize mechanical cell stimulation. Cells were allowed to adjust to fresh culture medium by incubation at 37 °C for 30 min. Then cells were stimulated with LPS or vehicle control, and reactions were stopped on ice at the indicated time points. Supernatants were obtained by centrifugation (5 min, 400 × g, 0 °C), and ATP levels in the cell supernatants were measured with a bioluminescence assay kit (Invitrogen).
T cell migration
Purified human CD4 T cells were stimulated for 3 days with anti-CD3/CD28 antibody–coated beads to generate effector T cells. These cells were allowed to adhere to fibronectin-coated glass-bottom chamber slides and reconstituted with fully supplemented tissue culture medium. In some experiments, fetal bovine serum in the medium was replaced with 20% (v/v) human plasma from healthy subjects or from sepsis patients. Glass-bottom chamber slides with cells were placed into a temperature-controlled (37 °C) stage incubator and maintained in a humidified gas atmosphere at 5% CO2 and 21% O2 (Live Cell Instrument). Cell migration in the presence or absence of ATP, apyrase, or inhibitors of P2X4 (5-BDBD) or P2Y11 (NF340) receptors was recorded by time-lapse microscopy using a Leica DMIR inverted microscope and a ×20 objective (NA 0.4). Cells were monitored for 30 min, and images were captured at 45-s intervals. Migration paths of individual cells were tracked with ImageJ software (National Institutes of Health; MTrackJ plugin), and the lengths of individual cell tracks were used to calculate migration speeds. In some cases, the 30-min observation period was divided into 10 3-min intervals. When cells migrated continuously at a speed >5 μm/min in each of these observation intervals, cell migration was considered to be uninterrupted. At least 20 randomly selected cells per experiment were analyzed for each experimental condition.
Jurkat cells, transfection, and calcium imaging
The human Jurkat T cell line (clone E6-1) was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained as described previously (22). Jurkat cells transiently expressing the mitochondrial and cytosolic Ca2+ indicators mito-CAR-GECO1 and G-GECO1.1 (58) were generated by electroporation with the plasmids CMV-mito-CAR-GECO (catalog no. 46022; Addgene, Cambridge, MA) and CMV-G-GECO1.1 (catalog no. 32445; Addgene) using a Neon transfection system (Invitrogen). Electroporated cells were cultured for 4–6 h. Cells were exposed to ATP (100 μm) for 10 min and stimulated by TCR/CD28 cross-linking, and mitochondrial and cytosolic Ca2+ responses were analyzed with fluorescence time-lapse microscopy. Images were captured at 2-s intervals using a ×63 objective (NA 1.4), TRITC and FITC filter sets, and a Leica DMI6000B inverted fluorescence microscope. Image analysis was performed with ImageJ.
Statistical analysis
Unless otherwise stated, data are shown as mean values ± S.D. of n ≥ 3 experiments. Results of individual experiments are depicted by single data points. Differences between groups were tested for statistical significance using two-tailed unpaired Student's t test or one-way ANOVA followed by post hoc Holm–Sidak test if two or multiple groups were compared, respectively. Nonparametric Mann–Whitney or Kruskal–Wallis test were used accordingly for data that were not normally distributed. Differences were considered statistically significant at p < 0.05.
Author contributions
K. S., C. L., Y. S., and A. H. L. formal analysis; K. S., C. L., Y. S., and A. H. L. investigation; K. S., C. L., and W. G. J. visualization; K. S., C. L., and W. G. J. writing-original draft; C. L. and W. G. J. supervision; C. L. and W. G. J. writing-review and editing; N. I. S. and W. G. J. conceptualization; N. I. S. resources; W. G. J. funding acquisition.
Supplementary Material
This work was supported by NIAID, National Institutes of Health, Grant AI-080582 and NIGMS, National Institutes of Health, Grants GM-51477, GM-60475, GM-116162, and T32 GM-103702. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3 and Video S1.
- PD-1
- programmed cell death protein 1
- APC
- antigen-presenting cell
- 5-BDBD
- 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one
- CFSE
- carboxyfluorescein succinimidyl ester
- DHR123
- dihydrorhodamine 123
- IS
- immunological synapse
- LPS
- lipopolysaccharide(s)
- NA
- numerical aperture
- NF340
- 4,4′-(carbonylbis(imino-3,1-(4-methyl-phenylene)carbonylimino))bis(naphthalene-2,6-disulfonic acid) tetrasodium salt
- NLRP3
- NACHT, LRR, and PYD domain–containing protein 3
- PBMC
- peripheral blood mononuclear cell
- PD-L1
- programmed death-ligand 1
- ROS
- reactive oxygen species
- TCR
- T cell receptor
- TLR
- Toll-like receptor
- TMRE
- tetramethylrhodamine ethyl ester
- IL
- interleukin
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- PKA
- protein kinase A
- TRITC
- tetramethylrhodamine isothiocyanate
- ANOVA
- analysis of variance.
References
- 1. Singer M., Deutschman C. S., Seymour C. W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G. R., Chiche J. D., Coopersmith C. M., Hotchkiss R. S., Levy M. M., Marshall J. C., Martin G. S., Opal S. M., et al. (2016) The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315, 801–810 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mayr F. B., Yende S., and Angus D. C. (2014) Epidemiology of severe sepsis. Virulence 5, 4–11 10.4161/viru.27372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Delano M. J., and Ward P. A. (2016) Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J. Clin. Invest. 126, 23–31 10.1172/JCI82224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hotchkiss R. S., Monneret G., and Payen D. (2013) Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 13, 260–268 10.1016/S1473-3099(13)70001-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jensen I. J., Sjaastad F. V., Griffith T. S., and Badovinac V. P. (2018) Sepsis-induced T cell immunoparalysis: the ins and outs of impaired T cell immunity. J. Immunol. 200, 1543–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fallon E. A., Biron-Girard B. M., Chung C. S., Lomas-Neira J., Heffernan D. S., Monaghan S. F., and Ayala A. (2018) A novel role for coinhibitory receptors/checkpoint proteins in the immunopathology of sepsis. J. Leukoc. Biol. 103, 1151–1164 10.1002/JLB.2MIR0917-377R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Venet F., and Monneret G. (2018) Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol. 14, 121–137 10.1038/nrrheum.2018.8 [DOI] [PubMed] [Google Scholar]
- 8. Lachmandas E., Boutens L., Ratter J. M., Hijmans A., Hooiveld G. J., Joosten L. A., Rodenburg R. J., Fransen J. A., Houtkooper R. H., van Crevel R., Netea M. G., and Stienstra R. (2016) Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol. 2, 16246 10.1038/nmicrobiol.2016.246 [DOI] [PubMed] [Google Scholar]
- 9. Latz E., Xiao T. S., and Stutz A. (2013) Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 10.1038/nri3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bryn T., Yaqub S., Mahic M., Henjum K., Aandahl E. M., and Taskén K. (2008) LPS-activated monocytes suppress T-cell immune responses and induce FOXP3+ T cells through a COX-2-PGE2-dependent mechanism. Int. Immunol. 20, 235–245 10.1093/intimm/dxm134 [DOI] [PubMed] [Google Scholar]
- 11. Wolk K., Döcke W. D., von Baehr V., Volk H. D., and Sabat R. (2000) Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood 96, 218–223 [PubMed] [Google Scholar]
- 12. Poujol F., Monneret G., Pachot A., Textoris J., and Venet F. (2015) Altered T lymphocyte proliferation upon lipopolysaccharide challenge ex vivo. PLoS One 10, e0144375 10.1371/journal.pone.0144375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Landelle C., Lepape A., Voirin N., Tognet E., Venet F., Bohé J., Vanhems P., and Monneret G. (2010) Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. 36, 1859–1866 10.1007/s00134-010-1962-x [DOI] [PubMed] [Google Scholar]
- 14. Zanin-Zhorov A., Tal-Lapidot G., Cahalon L., Cohen-Sfady M., Pevsner-Fischer M., Lider O., and Cohen I. R. (2007) Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J. Immunol. 179, 41–44 10.4049/jimmunol.179.1.41 [DOI] [PubMed] [Google Scholar]
- 15. Lee A. H., Ledderose C., Li X., Slubowski C. J., Sueyoshi K., Staudenmaier L., Bao Y., Zhang J., and Junger W. G. (2018) Adenosine triphosphate release is required for Toll-like receptor-induced monocyte/macrophage activation, inflammasome signaling, interleukin-1β production, and the host immune response to infection. Crit. Care Med. 46, e1183–e1189 10.1097/CCM.0000000000003446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yegutkin G. G. (2014) Enzymes involved in metabolism of extracellular nucleotides and nucleosides: functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol. 49, 473–497 10.3109/10409238.2014.953627 [DOI] [PubMed] [Google Scholar]
- 17. Junger W. G. (2011) Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 11, 201–212 10.1038/nri2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burnstock G., and Boeynaems J. M. (2014) Purinergic signalling and immune cells. Purinergic Signal. 10, 529–564 10.1007/s11302-014-9427-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cekic C., and Linden J. (2016) Purinergic regulation of the immune system. Nat. Rev. Immunol. 16, 177–192 10.1038/nri.2016.4 [DOI] [PubMed] [Google Scholar]
- 20. Schenk U., Westendorf A. M., Radaelli E., Casati A., Ferro M., Fumagalli M., Verderio C., Buer J., Scanziani E., and Grassi F. (2008) Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci. Signal. 1, ra6 10.1126/scisignal.1160583 [DOI] [PubMed] [Google Scholar]
- 21. Woehrle T., Yip L., Elkhal A., Sumi Y., Chen Y., Yao Y., Insel P. A., and Junger W. G. (2010) Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 116, 3475–3484 10.1182/blood-2010-04-277707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ledderose C., Bao Y., Lidicky M., Zipperle J., Li L., Strasser K., Shapiro N. I., and Junger W. G. (2014) Mitochondria are gate-keepers of T cell function by producing the ATP that drives purinergic signaling. J. Biol. Chem. 289, 25936–25945 10.1074/jbc.M114.575308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ledderose C., Liu K., Kondo Y., Slubowski C. J., Dertnig T., Denicoló S., Arbab M., Hubner J., Konrad K., Fakhari M., Lederer J. A., Robson S. C., Visner G. A., and Junger W. G. (2018) Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J. Clin. Invest. 128, 3583–3594 10.1172/JCI120972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shinohara Y., and Tsukimoto M. (2017) Adenine nucleotides attenuate murine T cell activation induced by concanavalin A or T cell receptor stimulation. Front. Pharmacol. 8, 986 10.3389/fphar.2017.00986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duhant X., Schandené L., Bruyns C., Gonzalez N. S., Goldman M., Boeynaems J. M., and Communi D. (2002) Extracellular adenine nucleotides inhibit the activation of human CD4+ T lymphocytes. J. Immunol. 169, 15–21 10.4049/jimmunol.169.1.15 [DOI] [PubMed] [Google Scholar]
- 26. Vitiello L., Gorini S., Rosano G., and la Sala A. (2012) Immunoregulation through extracellular nucleotides. Blood 120, 511–518 10.1182/blood-2012-01-406496 [DOI] [PubMed] [Google Scholar]
- 27. Carson W. F. 4th, Cavassani K. A., Ito T., Schaller M., Ishii M., Dou Y., and Kunkel S. L. (2010) Impaired CD4+ T-cell proliferation and effector function correlates with repressive histone methylation events in a mouse model of severe sepsis. Eur. J. Immunol. 40, 998–1010 10.1002/eji.200939739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Opal S. M., Scannon P. J., Vincent J. L., White M., Carroll S. F., Palardy J. E., Parejo N. A., Pribble J. P., and Lemke J. H. (1999) Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J. Infect. Dis. 180, 1584–1589 10.1086/315093 [DOI] [PubMed] [Google Scholar]
- 29. Boussiotis V. A. (2016) Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 375, 1767–1778 10.1056/NEJMra1514296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamazaki T., Akiba H., Iwai H., Matsuda H., Aoki M., Tanno Y., Shin T., Tsuchiya H., Pardoll D. M., Okumura K., Azuma M., and Yagita H. (2002) Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 169, 5538–5545 10.4049/jimmunol.169.10.5538 [DOI] [PubMed] [Google Scholar]
- 31. Shindo Y., McDonough J. S., Chang K. C., Ramachandra M., Sasikumar P. G., and Hotchkiss R. S. (2017) Anti-PD-L1 peptide improves survival in sepsis. J. Surg. Res. 208, 33–39 10.1016/j.jss.2016.08.099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hui E., Cheung J., Zhu J., Su X., Taylor M. J., Wallweber H. A., Sasmal D. K., Huang J., Kim J. M., Mellman I., and Vale R. D. (2017) T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 10.1126/science.aaf1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Radziewicz H., Ibegbu C. C., Fernandez M. L., Workowski K. A., Obideen K., Wehbi M., Hanson H. L., Steinberg J. P., Masopust D., Wherry E. J., Altman J. D., Rouse B. T., Freeman G. J., Ahmed R., and Grakoui A. (2007) Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J. Virol. 81, 2545–2553 10.1128/JVI.02021-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferrari D., Pizzirani C., Adinolfi E., Lemoli R. M., Curti A., Idzko M., Panther E., and Di Virgilio F. (2006) The P2X7 receptor: a key player in IL-1 processing and release. J. Immunol. 176, 3877–3883 10.4049/jimmunol.176.7.3877 [DOI] [PubMed] [Google Scholar]
- 35. Piccini A., Carta S., Tassi S., Lasiglié D., Fossati G., and Rubartelli A. (2008) ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. U.S.A. 105, 8067–8072 10.1073/pnas.0709684105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Netea M. G., Nold-Petry C. A., Nold M. F., Joosten L. A., Opitz B., van der Meer J. H., van de Veerdonk F. L., Ferwerda G., Heinhuis B., Devesa I., Funk C. J., Mason R. J., Kullberg B. J., Rubartelli A., van der Meer J. W., and Dinarello C. A. (2009) Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood 113, 2324–2335 10.1182/blood-2008-03-146720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurishita Y., Kohira T., Ojida A., and Hamachi I. (2012) Organelle-localizable fluorescent chemosensors for site-specific multicolor imaging of nucleoside polyphosphate dynamics in living cells. J. Am. Chem. Soc. 134, 18779–18789 10.1021/ja308754g [DOI] [PubMed] [Google Scholar]
- 38. De Ita M., Vargas M. H., Carbajal V., Ortiz-Quintero B., López-López C., Miranda-Morales M., Barajas-López C., and Montaño L. M. (2016) ATP releases ATP or other nucleotides from human peripheral blood leukocytes through purinergic P2 receptors. Life Sci. 145, 85–92 10.1016/j.lfs.2015.12.013 [DOI] [PubMed] [Google Scholar]
- 39. Dreisig K., Sund L., Dommer M. W., Kristensen N. P., Boddum K., Viste R., Fredholm S., Odum N., Jäättelä M., Skov S., and Kornum B. R. (2018) Human P2Y(11) expression level affects human P2X7 receptor-mediated cell death. Front. Immunol. 9, 1159 10.3389/fimmu.2018.01159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Communi D., Robaye B., and Boeynaems J. M. (1999) Pharmacological characterization of the human P2Y11 receptor. Br. J. Pharmacol. 128, 1199–1206 10.1038/sj.bjp.0702909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wehbi V. L., and Taskén K. (2016) Molecular mechanisms for cAMP-mediated immunoregulation in T cells: role of anchored protein kinase A signaling units. Front. Immunol. 7, 222 10.3389/fimmu.2016.00222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ledderose C., Bao Y., Ledderose S., Woehrle T., Heinisch M., Yip L., Zhang J., Robson S. C., Shapiro N. I., and Junger W. G. (2016) Mitochondrial dysfunction, depleted purinergic signaling, and defective T cell vigilance and immune defense. J. Infect. Dis. 213, 456–464 10.1093/infdis/jiv373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Glancy B., and Balaban R. S. (2012) Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973 10.1021/bi2018909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Idzko M., Ferrari D., and Eltzschig H. K. (2014) Nucleotide signalling during inflammation. Nature 509, 310–317 10.1038/nature13085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sumi Y., Woehrle T., Chen Y., Bao Y., Li X., Yao Y., Inoue Y., Tanaka H., and Junger W. G. (2014) Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock 42, 142–147 10.1097/SHK.0000000000000180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. von Andrian U. H., and Mackay C. R. (2000) T-cell function and migration: two sides of the same coin. N. Engl. J. Med. 343, 1020–1034 10.1056/NEJM200010053431407 [DOI] [PubMed] [Google Scholar]
- 47. Wang C. M., Ploia C., Anselmi F., Sarukhan A., and Viola A. (2014) Adenosine triphosphate acts as a paracrine signaling molecule to reduce the motility of T cells. EMBO J. 33, 1354–1364 10.15252/embj.201386666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weiler M., Schmetzer H., Braeu M., and Buhmann R. (2016) Inhibitory effect of extracellular purine nucleotide and nucleoside concentrations on T cell proliferation. Exp. Cell Res. 349, 1–14 10.1016/j.yexcr.2016.05.017 [DOI] [PubMed] [Google Scholar]
- 49. Kornum B. R., Kawashima M., Faraco J., Lin L., Rico T. J., Hesselson S., Axtell R. C., Kuipers H., Weiner K., Hamacher A., Kassack M. U., Han F., Knudsen S., Li J., Dong X., et al. (2011) Common variants in P2RY11 are associated with narcolepsy. Nat. Genet. 43, 66–71 10.1038/ng.734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dreisig K., and Kornum B. R. (2016) A critical look at the function of the P2Y11 receptor. Purinergic Signal. 12, 427–437 10.1007/s11302-016-9514-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cauwels A., Rogge E., Vandendriessche B., Shiva S., and Brouckaert P. (2014) Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 5, e1102 10.1038/cddis.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Csóka B., Németh Z. H., Törő; G., Koscsó B., Kókai E., Robson S. C., Enjyoji K., Rolandelli R. H., Erdélyi K., Pacher P., and Haskó G. (2015) CD39 improves survival in microbial sepsis by attenuating systemic inflammation. FASEB J. 29, 25–36 10.1096/fj.14-253567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li X., Kondo Y., Bao Y., Staudenmaier L., Lee A., Zhang J., Ledderose C., and Junger W. G. (2017) Systemic adenosine triphosphate impairs neutrophil chemotaxis and host defense in sepsis. Crit. Care Med. 45, e97–e104 10.1097/CCM.0000000000002052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Y., Corriden R., Inoue Y., Yip L., Hashiguchi N., Zinkernagel A., Nizet V., Insel P. A., and Junger W. G. (2006) ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314, 1792–1795 10.1126/science.1132559 [DOI] [PubMed] [Google Scholar]
- 55. van der Poll T., van de Veerdonk F. L., Scicluna B. P., and Netea M. G. (2017) The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 17, 407–420 10.1038/nri.2017.36 [DOI] [PubMed] [Google Scholar]
- 56. Wenzel R. P., and Edmond M. B. (2012) Septic shock—evaluating another failed treatment. N. Engl. J. Med. 366, 2122–2124 10.1056/NEJMe1203412 [DOI] [PubMed] [Google Scholar]
- 57. Hawkins R. B., Raymond S. L., Stortz J. A., Horiguchi H., Brakenridge S. C., Gardner A., Efron P. A., Bihorac A., Segal M., Moore F. A., and Moldawer L. L. (2018) Chronic critical illness and the persistent inflammation, immunosuppression, and catabolism syndrome. Front. Immunol. 9, 1511 10.3389/fimmu.2018.01511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu J., Liu L., Matsuda T., Zhao Y., Rebane A., Drobizhev M., Chang Y. F., Araki S., Arai Y., March K., Hughes T. E., Sagou K., Miyata T., Nagai T., Li W. H., and Campbell R. E. (2013) Improved orange and red Ca2+ indicators and photophysical considerations for optogenetic applications. ACS Chem. Neurosci. 4, 963–972 10.1021/cn400012b [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.