

Abstract
Missing in metastasis (MIM), an inverse Bin–Amphiphysin–Rvs (I-BAR) domain protein, promotes endocytosis of C-X-C chemokine receptor 4 (CXCR4) in mammalian cells. In response to the CXCR4 ligand stromal cell–derived factor 1 (SDF-1 or CXCL12), MIM associates with RAS-related GTP-binding protein 7 (RAB7) 30 min after stimulation. However, RAB7's role in MIM function remains undefined. Here we show that RNAi-mediated suppression of RAB7 expression in human HeLa cells has little effect on the binding of MIM to RAB5 and on the recruitment of CXCR4 to early endosomes but effectively abolishes MIM-mediated CXCR4 degradation, chemotactic response, and sorting into late endosomes and lysosomes. To determine whether I-BAR domain proteins interact with RAB7, we examined cells expressing insulin receptor tyrosine kinase substrate (IRTKS), an I-BAR domain protein bearing an Src homology 3 (SH3) domain. We observed that both MIM and IRTKS interact with RAB5 at an early response to SDF-1 and that IRTKS binds poorly to RAB7 but strongly to RAB11 at a later time point. Moreover, IRTKS overexpression reduced CXCR4 internalization and enhanced the chemotactic response to SDF-1. Interestingly, deletion of the SH3 domain in IRTKS abolished the IRTKS–RAB11 interaction and promoted CXCR4 degradation. Furthermore, the SH3 domain was required for selective targeting of MIM–IRTKS fusion proteins by both RAB7 and RAB11. Hence, to the best of our knowledge, our results provide first evidence that the SH3 domain is critical in the regulation of specific endocytic pathways by I-BAR domain proteins.
Keywords: endocytosis, Rab, chemotaxis, chemokine, C-X-C chemokine receptor type 4 (CXCR-4), cell signaling, endosome, insulin receptor tyrosine kinase substrate (IRTKS), missing in metastasis (MIM), RAB11, RAB7, SH3
Introduction
The human MIM3 gene is a putative tumor suppressor because it is often down-regulated in a subset of advanced malignant cells. However, the molecular role of MIM in cancer progression remains unknown. MIM is abundantly expressed in the cerebellum, the kidneys, the spleen, and the heart. Disruption of the genomic MIM in mice has generated several modest phenotypes, including kidney dysfunction (1, 2), impairment in the formation of neuronal dendritic spines (3), a tendency to develop B cell malignancies only in aged mice (4), and multiple spinocerebellar ataxias (5). Recent large-scale, genome-wide, and whole-exome sequencing analyses have revealed that genomic variations of the MIM gene are associated with left ventricular hypertrophy (6, 7). Therefore, the current data indicate that MIM plays a diverse role in different pathophysiological contexts.
The mammalian MIM gene encodes a protein with an N-terminal motif homologous to the Bin–Amphiphysin–Rvs (BAR) domain, which is present in many intracellular membrane-binding proteins through a curved interface (8). Unlike most BAR domains, which have a positively charged concave surface, the MIM BAR domain displays an inverse or convex exterior (9, 10). This feature is also shared with four other mammalian proteins, including insulin receptor substrate p53 (IRSP53), IRTKS, actin-bunding protein with BAIAP2 (ABBA), and planar intestinal and kidney-specific BAR protein (PINKBAR), which, together with MIM, make up the inverse BAR (I-BAR) protein family. These proteins can be further divided into two subgroups based on whether they contain an internal SH3 domain, which is present in IRSP53, IRTKS, and PINKBAR but absent in MIM and ABBA (11). Despite the difference, all I-BAR domain proteins bind to the inner leaflet of phospholipid membranes and sense or generate membrane protrusions when they are overexpressed. In addition to binding to membranes, MIM and most other I-BAR domain proteins interact with small GTPases such as RAC via its I-BAR domain (12–15). Furthermore, I-BAR domains have a high affinity for phosphatidylinositol (4,5)-biphosphate, a phospholipid that is abundant in the plasma membrane (16). Double knockout of Irtks and Irsp53 caused embryonic lethality in mice, at least partially because of abnormal development of trophoblasts (17). Trophoblasts invade the endometrium, probably through the podosome, a protrusive membrane structure that is formed by actin polymerization driven by small GTPases (18, 19), highlighting the physiological role of I-BAR domain proteins in membrane deformation in concert with small GTPases.
Recent advances have also evidenced implication of I-BAR domain proteins in the modulation of intracellular membranes. The I-BAR domain is evolutionally conserved in the UNIKONT supergroup (Amoebozoa, Fungi, and Metazoa). In the genome of yeast, there is only one single I-BAR-like gene, which encodes a protein with a strong affinity for Ypt7 (yeast homologue of RAB7) and Vps33 and is thus called Ivy1 (20). Interestingly, Ivy1 locates in vacuoles (yeast lysosomes) in a Ypt7-dependent manner (21), and overexpression of Ivy1 causes accumulation of multivesicular bodies and abnormal sorting of vacuolar proteins (20). On the other hand, lack of Ivy1 in combination with deletion of vacuolar-type H+-ATPase causes hypersensitivity to rapamycin along with expansion of the vacuolar membrane, presumably because of a defect in a microautophagy program (21). Conversely, overexpression of Ivy1 suppresses sensitivity to rapamycin in yeast lacking Ypt6 (the yeast homolog of RAB6) (21, 22). The genome of Dictyostelium also encodes a single I-BAR protein, called IBARa, which contains an SH3 domain (23). Instead of localizing at the cell leading edge or in filopodia, IBARa accumulates in clathrin-coated pits just before they are dissolved into endosomes. Similarly, the MIM homolog of Drosophila interferes with the interaction between cortactin and endophilin/CD2AP, components of the endocytic machinery, and inhibits endocytosis (24). We reported recently that cells derived from the bone marrow of a MIM-deficient mouse strain were impaired in ligand-mediated internalization of CXCR4, a chemokine that directs the interaction of hematopoietic stem cells with their niches in the bone marrow and the metastasis of certain malignant cells (25). Although the detailed mechanism of CXCR4 internalization remains elusive, the major events following exposure to its ligand SDF-1 include phosphorylation at the C terminus of the receptor and activation of the ubiquitin E3 ligase AIP4, which further leads to endocytic sorting of the receptor from early to late endosomes (26). Significantly, MIM interacts with the complex of AIP4 and CXCR4 upon SDF-1 stimulation and promotes CXCR4 ubiquitination and its sorting into late endosomes (27). Also, MIM associates sequentially with RAB5 and RAB7 during the response to SDF-1. However, the precise role of RABs in MIM-mediated sorting of CXCR4 into endocytic vesicles has not yet been established. In this study, we investigated the role of RAB7 in MIM-mediated CXCR4 internalization and observed that RAB7 is required for MIM and CXCR4 to be sorted into late endosomes and for the chemotactic response to SDF-1. Unexpectedly, we found that IRTKS has a low affinity for RAB7 and a high affinity for RAB11 in SDF-1–stimulated cells. In addition, we provided evidence of a critical role of the SH3 domain in the interaction of IRTKS with RAB11. Hence, our data establish, for the first time, a functional link between RABs and I-BAR domain proteins in different intracellular trafficking pathways.
Results
MIM promotes CXCR4 internalization in human malignant cells
We recently demonstrated that MIM promotes CXCR4 internalization in either primary mouse bone marrow cells or HeLa cells (25, 27). As aged MIM-deficient mice were prone to lymphatic malignancies (4), we were interested in whether MIM plays a similar role in human lymphatic cells and analyzed the internalization of CXCR4 in several lymphocytic malignant cells, including Reh (acute lymphocytic leukemia), Raji (Burkitt's lymphoma), and Daudi (Burkitt's lymphoma) cells, and patient-derived human B lymphatic (PBL) cells. Immunoblot analysis revealed that MIM expression was relatively low in Daudi and Raji cells compared with Reh and PBL cells (Fig. 1A). Consistently, CXCR4 was readily internalized in response to SDF-1 in Reh and PBL cells compared with Daudi and Raji cells (Fig. 1B). To verify the function of MIM in lymphatic cells, we transfected Raji cells with a plasmid encoding MIM protein tagged by GFP (MIM-GFP) or a control plasmid encoding GFP only and then analyzed CXCR4 internalization in transfected cells by measuring surface CXCR4 30 min after exposure to SDF-1 at different concentrations. At 500 ng/ml, only 20% of cells expressing MIM-GFP showed surface CXCR4, which was significantly lower than in control cells, 50% of which remained to express surface CXCR4 under the same condition (Fig. 1C). Thus, overexpression of MIM-GFP promoted CXCR4 internalization. As the surface CXCR4 impacts chemotactic responses to its ligand, we also examined the chemotactic capacity of Raji cells by using Transwell plates. Control Raji cells expressing GFP showed a nearly 17-fold increase in chemotaxis at 300 or 400 ng/ml SDF-1, whereas cells expressing MIM-GFP increased chemotaxis by only 6-fold under the same condition (Fig. 1D). In addition, we also examined MDA-MB-231 breast cancer cells expressing MIM-GFP and obtained a similar result (Fig. S1A). Taken together, these data are consistent with our previous finding based on HeLa cells and mouse cells that MIM down-regulates CXCR4-mediated chemotaxis (27).
Figure 1.

RAB7 is required for MIM-mediated CXCR4 internalization. A, immunoblot analysis of endogenous MIM protein in Raji, Reh, Daudi, and PBL cells. The expression levels were quantified based on the ratio of MIM to β-actin. B, malignant B cells were stimulated with SDF-1 at the indicated concentrations for 30 min. Surface expression of CXCR4 was analyzed by flow cytometry and normalized to that of control cells without SDF-1 treatment. C, Raji cells were transiently transfected with GFP or MIM-GFP and stimulated for 30 min with SDF-1 at different concentrations. The percentage of cells expressing surface CXCR4 was determined by flow cytometry. D, Raji cells expressing MIM-GFP or GFP were plated on Transwell plates in which the lower chamber was filled with medium containing SDF-1 at the indicated concentrations. After 24 h, cells were fixed and stained with 0.1% crystal violet. The number of cells that migrated to the lower chamber was compared with that of cells without SDF-1 treatment. E, HeLa cells expressing GFP or MIM-GFP were treated with siRAB7 or Ct-siRNA for 48 h. The treated cells were then subjected to a Transwell assay for their chemotactic response to 500 ng/ml SDF-1. F, HeLa cells expressing either MIM-GFP or GFP were treated with siRAB7 or Ct-SiRNA. The treated cells were further incubated with 500 μg/ml cycloheximide for 30 min prior to exposure to 150 ng/ml SDF-1 for the indicated times. The total amounts of CXCR4 protein in treated cells at different times were estimated by immunoblot and used to calculate t½ using Prism software. All data represent mean ± S.E.M. (n = 3). ***, p < 0.001.
RAB7 is required for MIM to down-regulate CXCR4
MIM binds to RAB7 in response to SDF-1 (27) (Fig. S3A). However, the role of RAB7 in the MIM-mediated regulation of CXCR4 has not yet been determined. To evaluate the role of RAB7 in detail, we utilized established HeLa cell populations stably expressing MIM-GFP or GFP at an efficiency of nearly 100% (data not shown). Although intrinsic fluorescent signals of these GFP proteins were microscopically weak after a conventional fixation procedure (Fig. S2), they were readily detectable by indirect immunofluorescence using anti-GFP antibody (Fig. S2, A and D) or by Western blotting (Fig. S3A). Expression of RAB7 in those cells was suppressed by 80% by a set of siRNAs against RAB7a (siRAB7) compared with cells treated with a siRNA with a scrambled sequence (Ct-siRNA) (Fig. S3B). When siRAB7 was used to treat HeLa cells for 48 h prior to SDF-1, the attenuated chemotaxis of MIM-GFP cells was apparently restored (Fig. 1E). siRAB7 treatment also caused a slight increase in the motility of cells expressing GFP only, presumably because of its inhibition of the uptake of basal CXCR4 on the cell surface. Thus, MIM-mediated inhibition of the chemotactic response to SDF-1 requires RAB7.
To further analyze the role of RAB7 in the function of MIM, we measured CXCR4 degradation, the event that occurs within lysosomes following internalization. In cells treated with Ct-siRNA, overexpression of MIM-GFP reduced the half-life of CXCR4 from 4.1 h to nearly 2.1 h (Fig. 1F). However, siRAB7 treatment restored the half-life of CXCR4 in MIM-GFP cells up to 4.3 h.
RAB7 is required for recruitment of CXCR4 and MIM to late endosomes
RAB7 is known to regulate late endosomes (28). Thus, we examined the impact of RAB7 deficiency on the recruitment of CXCR4 into late endosomes based on colocalization of CXCR4 and CD63, a marker of late endosomes. Based on indirect immunofluorescence staining, we observed that, prior to SDF-1 treatment, about 12% of CD63 puncta (red) were colocalized with CXCR4 (green) in GFP cells that had been pretreated with Ct-siRNA (Fig. 2, A and E) and 18% in MIM-GFP cells (Fig. 2, C and E). Upon SDF-1 stimulation, about 23% of CD63 was colocalized with CXCR4 in GFP cells and 72% in MIM-GFP cells (Fig. 2E). Remarkably, the degree of colocalization of CD63 and CXCR4 in MIM-GFP cells was dramatically reduced to 17% when treated with siRAB7 prior to SDF-1 (Fig. 2, C versus D, also E). This dramatic reduction was not observed with control GFP cells. Also, in unstimulated cells either expressing MIM-GFP or GFP, colocalization of CD63 and CXCR4 was low and not significantly impacted by siRAB7. MIM promotes the formation of late endosomes (27), as indicated by increased CD63 puncta (Fig. 2F). To examine the impact of siRAB7 on MIM-mediated late endosome assembly, we examined CD63 puncta in siRAB7-treated cells. As shown in Fig. 2F, the increased number of CD63 puncta in MIM-GFP cells was diminished by siRAB7. However, siRAB7 had little impact on CD63 puncta in control cells (Fig. 2F), suggesting that depletion of RAB7 did not impair the basal level of late endosomes.
Figure 2.

RAB7 is essential for MIM to promote CXCR4 sorting into late endosomes. A–D, HeLa cells expressing GFP (A and B) or MIM-GFP (C and D) were plated in 6-well plates, treated with Ct-siRNA (A and C) or siRAB7 (B and D) for 48 h, and stimulated with 500 ng/ml SDF-1 for 30 min. The treated cells were costained with monoclonal CXCR4 antibody (green) and polyclonal CD63 antibody (red) and inspected by confocal microscopy. The boxed areas of images were amplified and are presented below. E, co-localization of CXCR4 and CD63 was quantitively analyzed based on Manders' coefficient, which represents the proportion of CD63 colocalized with CXCR4. F, the amount of CD63 puncta of the acquired images was also quantified. Scale bars = 10 μm. ***, p < 0.001 (n = 10).
We also analyzed the effect of siRAB7 on the association of CXCR4 with early endosomes, as detected by EEA1 (Fig. S4), and with lysosomes detected by LAMP2 (Fig. S5). As reported previously (27), overexpression of MIM-GFP caused a substantial increase in the association of CXCR4 with early endosomes in response to SDF-1. However, such an increase was not significantly affected by siRAB7 (Fig. S4). In contrast, siRAB7 abolished the increase of CXCR4 deposition in lysosomes of MIM-GFP cells (Fig. S5). Therefore, RAB7 is required for MIM-mediated sorting of CXCR4 into both late endosomes and lysosomes but not required for sorting into early endosomes.
Finally, we analyzed the effect of RAB7 depletion on the association of MIM with late endosomes. In control cells, GFP showed as diffused staining within the cytoplasm of SDF-1–treated or untreated cells (Fig. 3, A and B). In contrast, MIM-GFP displayed as puncta in the cytoplasm. In particular, about 70% of late endosomes were colocalized with MIM-GFP in SDF-1–treated cells (Fig. 3, C and E). However, this colocalization was nearly diminished in cells that had been pretreated with siRAB7 (Fig. 3, D and E). Hence, RAB7 is required for sorting of both CXCR4 and MIM to late endosomes.
Figure 3.
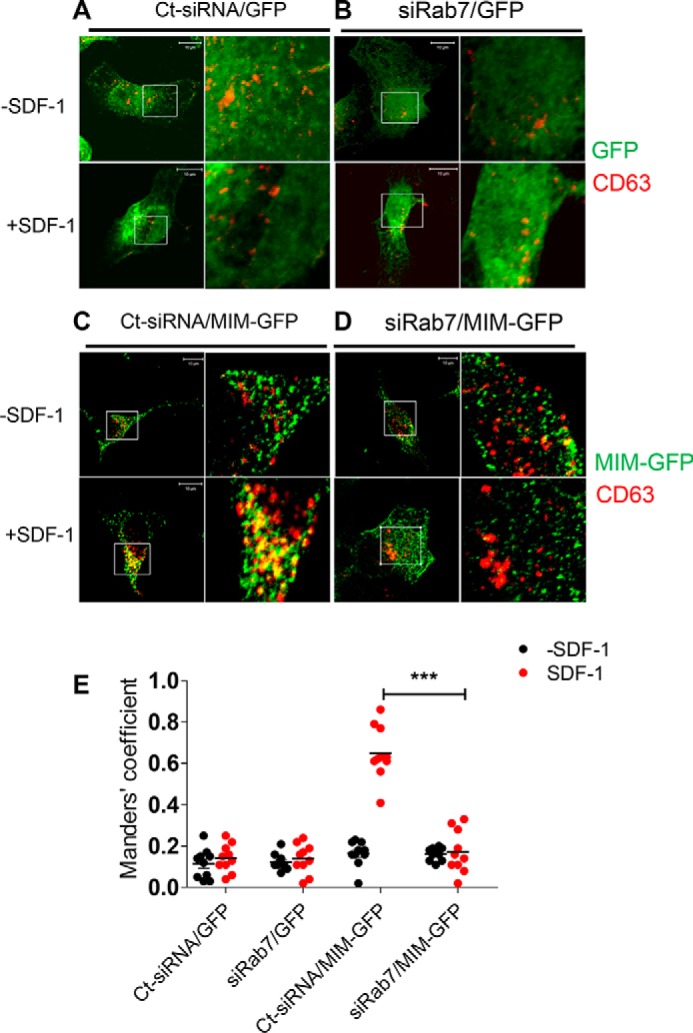
RAB7 is indispensable for recruitment of MIM into late endosomes. A–D, HeLa cells expressing GFP (A and B) or MIM-GFP (C and D) were treated with control siRNA (A and C) or siRAB7 (B and D) for 2 days prior to treatment with 500 ng/ml SDF-1 for 30 min. The treated cells were co-stained with GFP antibody (green) and CD63 antibody (red) and inspected by confocal microscopy. The boxed areas were amplified and are presented to the left of the corresponding images. E, co-localization of CD63 and GFP or MIM-GFP was quantified as described in the legend of Fig. 4. Data represent mean. ***, p < 0.001 (n = 10). Scale bars = 10 μm.
MIM and IRTKS bind to different RABs and regulate CXCR4 internalization differently
We were interested in whether interaction with RABs is a common property of I-BAR domain proteins and thus analyzed HeLa cells expressing GFP-conjugated IRTKS (IRTKS-GFP). We chose IRTKS because we had found previously that it regulates cellular motility differently than MIM does (29) and that it is structurally distinct from MIM by having an internal SH3 domain (Fig. 4A). Like MIM-GFP, IRTKS-GFP promptly associated with RAB5 during an early response (5 min) to SDF-1 (Fig. 4B). However, unlike MIM-GFP, IRTKS-GFP bound poorly to RAB7 when cells were exposed to SDF-1 for 30 min (Fig. 4C). We also analyzed the interaction with RAB11, which plays an important role in the pathway leading to receptor recycling (30). Surprisingly, IRTKS-GFP bound strongly to RAB11 at 30 min, whereas MIM-GFP failed to do so under the same condition (Fig. 4D).
Figure 4.

MIM and IRTKS were targeted by different Rabs. A, schematic of the MIM and IRTKS proteins. WH2, WASP homology 2; PRD, proline-rich domain; PPLP, proline-proline-leucine-proline residue sequence. B–D, HeLa cells expressing MIM-GFP or IRTKS-GFP were treated with 150 ng/ml SDF-1 for 5 min (B) or 30 min (C and D). Interactions between I-BAR domain proteins with RAB5 (B), RAB7 (C), and RAB11 (D) were analyzed by co-immunoprecipitation (IP), followed by immunoblot (IB) with the indicated antibodies. E, HeLa cells expressing GFP or IRTKS-GFP were plated in Transwell plates, treated with SDF-1 at different concentrations, and analyzed for chemotaxis as described in the legend of Fig. 1D. F, HeLa cells expressing GFP, MIM-GFP, and IRTKS-GFP were incubated with 500 μg/ml cycloheximide for 30 min prior to treatment with 150 ng/ml SDF-1 for the indicated times. The levels of CXCR4 on the surface of treated cells were estimated by flow cytometry. All data represent mean ± S.E.M. (n = 3). ***, p < 0.001
To analyze whether IRTKS has any influence on the function of CXCR4, we examined the chemotactic response of IRTKS-GFP cells to SDF-1. Unlike MIM-GFP cells, IRTKS-GFP cells showed increases in the chemotactic response to SDF-1 at concentrations ranging from 50 to 300 ng/ml (Fig. 4E). As RAB11 is associated with nondegradation endocytic pathways, we measured surface CXCR4 in the presence of cycloheximide, which inhibits protein synthesis. Thus, changes in surface CXCR4 after initial uptake would reflect a dynamic process of the receptor. Under this condition, by 30 min, only 20% of CXCR4 remained on the surface of MIM-GFP cells, 53% on control cells, and 62% on IRTKS-GFP cells. However, after 30 min, the level of surface CXCR4 in IRTKS-GFP cells was significantly recovered up to 80% (Fig. 4F). The recovery of surface CXCR4 was also correlated with increased recruitment of CXCR4 to RAB11-associated vesicles at 30 min (Fig. S6). Although the reason for the recovery is currently unknown, the data suggest that IRTKS has distinct role in CXCR4 degradation.
The SH3 domain determines the interaction with RAB11
Because the major difference between MIM and IRTKS is that the latter contains an SH3 domain, we were interested in whether the SH3 domain could play a role in the interaction of IRTKS with RABs and thus examined cells expressing a mutant with deletion of the SH3 domain (IRTKSΔSH3) (Fig. 5A). Although the mutant maintained the ability to interact with RAB5 5 min after exposure to SDF-1, it failed to bind to RAB11 at 30 min (Fig. 5B). Instead, it promptly interacted with RAB7 at this time. The acquired interaction with RAB7 was apparently correlated with the mutant's ability to induce CXCR4 internalization at an efficiency compatible with that of MIM-GFP (Fig. 5C). The sequence of MIM responsible for binding to RAB7 is located within the I-BAR domain (27). To examine whether the I-BAR domain determines binding to RAB7, we analyzed a chimeric mutant in which the MIM I-BAR domain was fused with the C-terminal part of IRTKS (MIM–I-BAR–IRTKS) (Fig. 5A). Interestingly, the fusion protein showed a poor ability to bind to RAB7 but a strong affinity for RAB11 in response to SDF-1 (Fig. 5D). We also analyzed a chimeric mutant in which the IRTKS I-BAR domain was fused with the C-terminal part of MIM. This mutant (IRTKS–I-BAR–MIM) interacted poorly with RAB11 but strongly with RAB7 (Fig. 5D). Overall, these data indicate that, although the I-BAR domain provides a binding site for RABs, the SH3 domain specifies the interaction with RAB11 during intracellular trafficking of CXCR4.
Figure 5.

The SH3 domain determines recruitment of IRTKS to RAB11. A, schematic of MIM, IRTKS, and their fusion mutants. WH2, WASP homology 2. B, HeLa cells expressing GFP-tagged IRTKS or IRTKSΔSH3 proteins were stimulated with 150 ng/ml SDF-1 for 30 min. The cell lysates were subjected to immunoprecipitation (IP) with GFP antibody, followed by immunoblot (IB) using anti-RAB11, RAB5, or RAB7, as indicated. C, cells expressing GFP-tagged MIM, IRTKS, and IRTKSΔSH3 were treated with 500 ng/ml SDF-1 for up to 45 min. The levels of surface CXCR4 were estimated by flow cytometry. The data represent mean ± S.E.M. (n = 3). *, p < 0.05; ***, p < 0.001; referring to the differences between cells expressing IRTKS-GFP and those expressing IRTKSΔSH3-GFP. D, cells expressing MIM-GFP or its fusions were treated with 150 ng/ml SDF-1 for 30 min. The interactions of MIM and its fusions with RAB7 and RAB11 were analyzed as described in B.
Discussion
The data presented here demonstrate that RAB7 is necessary for MIM-mediated CXCR4 internalization and degradation. Consistent with this result, cells with depletion of RAB7 were impaired in sorting of both MIM and CXCR4 into late endosomes. The impaired recruitment of MIM or CXCR4 to late endosomes could be due to reduced formation of endocytic vesicles in the absence of RAB7. However, we observed that depletion of RAB7 had little effect on the association of CXCR4 with early endosomes or the formation of late endosomes in resting cells, suggesting that RAB7 is neither necessary for the initial sorting of intracellular CXCR4 nor essential for the formation of late endosomes. As MIM forms a stable complex with AIP4 and ubiquitinated CXCR4 after exposure to SDF-1 (27), the finding described here is consistent with the view that MIM acts as an effector of RAB7 and a carrier of ubiquitinated CXCR4. In this experiment, endogenous CXCR4 proteins were detected by indirect immunofluorescence using a secondary antibody conjugated with a green dye. We argue that this procedure is valid because endogenous GFP or MIM-GFP yielded few green fluorescent signals detected by microscopy after fixation (Fig. S2). However, we could not rule out the possibility that the detected signals could be due to cross-reactivity of the antibodies used.
In addition to RAB7, MIM also bound RAB5 prior to RAB7 during an early response to SDF-1 (27). Conversion from RAB5 to RAB7 on endosomes is driven by Mon1-Ccz1, a RAB guanine exchange factor (GEF) that is ubiquitously expressed (31). However, binding of I-BAR domain proteins to RAB5 does not seem to be sufficient for their subsequent interactions with other RABs, as MIM and IRTKS show distinct affinities for RAB7 and RAB11 after binding to RAB5. Specifically, MIM bound to RAB7, whereas IRTKS bound to RAB11, which often participates in a pathway for endocytic recycling (32, 33). Consistently, MIM promoted CXCR4 degradation along with inhibition of chemotaxis, whereas IRTKS enhanced the cellular response to SDF-1 along with promotion of CXCR4 surface expression. Hence, I-BAR domain proteins may assign endocytic cargoes to different vesicles by selectively targeting different RABs, including RAB7 and RAB11. Such selectivity could be due to the function of either the I-BAR domain itself or the SH3 domain, which is not present in MIM. It has been reported that the interaction between the I-BAR domain of IRSP53 and IRTKS with membranes is through electrostatic interactions, whereas that of MIM's I-BAR domain involves insertion of an amphipathic helix into the membrane layer (10). Such a difference in membrane binding is not likely to be responsible for choosing a specific RAB during receptor trafficking, as we found that MIM and IRTKS have a similar affinity for RAB5 at an early response. Also, deletion of the SH3 domain abolished the ability of IRTKS to bind to RAB11 but rendered it able to associate with RAB7. Likewise, fusion proteins of MIM and IRTKS bound to RAB7 and RAB11 differently depending on the presence of the SH3 domain. Therefore, our data strongly suggest that the specific binding of IRTKS to RAB11 is mainly determined by the SH3 domain during a late response to SDF-1, whereas binding to RAB7 appears to be a default property of the I-BAR domain itself.
How does the SH3 domain recruit IRTKS to RAB11 is currently unclear, as the intrinsic partners of the SH3 domain are poorly characterized. Nevertheless, IRTKS is most closely related to IRSP53, which shares a redundant function with IRTKS in embryonic development (34) and links, via its SH3 domain, to Wiskott–Aldrich syndrome protein (WASP) and WASP family verprolin homologous (WAVE) proteins (12). In fact, direct interaction of IRTKS with WAVE has also been reported (35). Both WASP and WAVE proteins can activate the ARP2/3 complex, the primary actin nucleator responsible for assembly of branched actin filaments, which are enriched in the lamellipodia and endocytic vesicles and associated with RAB5 and RAB11 (36). As RAB11 binds to Myosin Vb, a nonconventional myosin that assists with movement of endocytic cargos to the cell periphery (37), IRTKS-mediated actin assembly may provide functional coordination between morphogenesis of endosomes and their movements toward the plasma membrane rather than fusion with lysosomes. Another possibility is that the SH3 domain facilitates interaction with RAB11 by linking to an unknown intracellular protein that regulates RAB11. In this regard, it is worth noting that SH3BP5 (also REI-I), a newly identified GEF of RAB11, is an SH3 binding protein and contains an F-BAR domain (38). Whether the link to actin assembly or an RAB11 GEF is responsible for the selective interaction of IRTKS with RAB11 requires further investigation. Based on the data presented here and previous reports from us and others, we propose a model for the function of MIM and IRTKS in cellular trafficking of CXCR4 (Fig. 6). Briefly, in the early response to SDF-1, MIM binds AIP4 and forms a complex with CXCR4, resulting in CXCR4 ubiquitination. The resulting cargo complex is initially recruited to early endosomes because of the interaction with RAB5 and then to late endosomes because of binding to RAB7. Such interactions may provide a mechanism for specific membrane remodeling that has yet to be identified. As for IRTKS, although it takes a similar pathway at an early response to SDF-1, it is likely recruited to other vesicles in a late response because of its interaction with RAB11, which may be mediated by either actin assembly or an unidentified GEF of RAB11. Whether these vesicles represent recycling endosomes, which are associated with RAB11, needs to be further verified.
Figure 6.

A model depicting the function of I-BAR domain proteins in the regulation of CXCR4. CXCR4 is a G protein–coupled receptor with seven transmembrane domains. Upon SDF-1 stimulation, CXCR4 is phosphorylated at its C terminus by a G protein–coupled receptor kinase (GRK) (1). The phosphorylated CXCR4 allows its association with the E3 ubiquitin ligase AIP4, which is promoted by the I-BAR domain protein MIM or IRTKS. This results in formation of a complex of the CXCR4, AIP4, and I-BAR proteins (2) and a subsequent increase in ubiquitination of the receptor (3). Because of the ability to bind to RAB5 and RAB7, MIM facilitates sorting of the ubiquitinated receptor complex from early endosomes (EE) to late endosomes (LE) (5). On the other hand, IRTKS facilitates sorting of the receptor complex into a type of RAB11-coated endocytic vesicles, such as recycling vesicles (RE) (6). This step may be facilitated by actin assembly or an unidentified GEF of RAB11.
Discrete interaction with different RABs implies that MIM and IRTKS may antagonize each other within cells. However, MIM and IRTKS have distinct expression profiles in most types of tissues, according to the portal for the Genotype-Tissue Expression Project. For example, MIM is highly expressed in the cerebellum, spleen, and tibia, tissues where IRTKS is nearly silent. In contrast, IRTKS is highly expressed in the stomach and prostate, where MIM expression is extremely low. Also, a search of a public microarray database indicates that MIM is highly expressed in naïve B cells and memory B cells but nearly silent in centroblasts and centrocytes, whereas IRTKS is low in naïve B cells but highly elevated in centroblasts and centrocytes (Fig. S7). Centroblasts and centrocytes are B cells associated with the germinal center, the area in lymphoid organs responsible for differentiation of naïve B cells into antibody-producing cells. Importantly, recruitment of naïve B cells to the dark zone of the germinal center is mainly driven by CXCR4 (39). Hence, the expression pattern of MIM and IRTKS and their reciprocal role in CXCR4 internalization imply that they are coordinated in the regulation of B cell differentiation. Consistent with expression profiles in B cells, MIM expression is significantly low in a cohort (n = 104) of diffuse large B cell lymphoma cells, which are mainly derived from B cells in the germinal center. In contrast, IRTKS expression is elevated in diffuse large B cell lymphoma cells (Fig. S7). It should also be pointed out that, unlike CXCR4, which is present in most types of hematopoietic cells, IRTKS is poorly expressed in cells other than germinal center-associated cells. Thus, it is unlikely that IRTKS plays a universal role in CXCR4 hemostasis. We speculate that receptor surface expression in other types of cells may involve different SH3-containing BAR domain proteins.
Experimental procedures
Cell lines and transfection
MDA-MB-231 cells and HeLa cells were cultured in DMEM (Corning). Daudi, Raji, and Reh cells and primary B lymphatic cells derived from a patient were a gift from Dr. Curt Civin. All B cells were cultured at 37 °C under 5% CO2 in RPMI 1640 medium supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 100 unit/ml penicillin, and 100 μg/ml streptomycin. DNA-mediated transfection was performed with FuGENE transfection reagent (Active Motif Co., Carlsbad, CA). Stably transfected cells were selected and maintained in medium containing G418, as described previously (27). Raji cells were transfected with pMIM-GFP using Raji Cell Avalanch transfection reagent (EZ Biosystems, catalog no. EZT-RAJI-1). The expression of transfected genes was verified by fluorescence microscopy and immunoblot analysis. siRNA-mediated transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's recommendations.
Antibodies, chemicals, and plasmids
All chemicals, unless otherwise indicated, were purchased from Sigma-Aldrich. The polyclonal MIM antibody (PA517047), polyclonal GFP antibody (A11122), monoclonal GFP antibody (33-2600), polyclonal IRTKS antibody (PA5-22026), Alexa Fluor 488 goat anti-rabbit IgG antibody (A-11008), Alexa Fluor 568 goat anti-rabbit IgG (A11004 or A21134), Alexa Flour 488 goat anti-mouse IgG (A32723), Alexa Flour 568 goat anti-mouse IgG (A11011), and protein A/G–agarose beads (20423) were purchased from Thermo Scientific (Rockford, IL). The polyclonal CD63 antibody (sc-15363), monoclonal CXCR4 antibody (sc-9046), and normal mouse IgG (sc-2025) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Normal rabbit IgG (2729S) was from Cell Signaling Technology (Danvers, MA). The monoclonal EEA1 antibody (610456) and monoclonal RAB11 antibody (610656) were from BD Biosciences. The SDF-1 (581206) and PE-conjugated anti-human CXCR4 antibodies (306506) were purchased from BioLegend (San Diego, CA). The monoclonal RAB7 antibody (R8779), a set of siRNAs targeting human RAB7a at 16 different sites, and control siRNA with a scrambled sequence were from Sigma-Aldrich (St. Louis, MO). PBS was from Corning. The plasmids encoding MIM-GFP, IRTKS-GFP, IRTKSΔSH3-GFP, IRTKS-IBAR-MIM-GFP, and MIM-IBAR-IRTKS-GFP have been described previously (29).
Cell migration assay
Chemotaxis was evaluated by Transwell assay as described previously (27). Briefly, serum-free DMEM was added to the lower chamber of a Transwell plate with an insert of 8-μm pore size (Corning, catalog no. 3422). The plate was incubated overnight at 37 °C and under 5% CO2. Cells were plated on the upper chamber at 2 × 105 cells/well in serum-free DMEM, and SDF-1 at various concentrations was applied to the lower chamber. After incubation for 16 h, the plate was washed with PBS, and the cells were fixed with 4% paraformaldehyde for 5 min, followed by washing twice with PBS. To counter cells, the plate was added with 200 μl methanol, followed by incubation at room temperature for 20 min. The cells were then stained with 0.1% crystal violet for 15 min. After washing twice with PBS, the cells on the upper surface of the insert were scraped with cotton swabs. The migrated cells were inspected microscopically at ×100 magnification, and cells within nine microscopic fields were counted for each Transwell plate membrane.
Flow cytometry analysis
CXCR4 on the cell surface was evaluated by flow cytometry assay as described previously (27).
Immunofluorescence microscopy
Cells were transfected with RAB7 siRNAs or control siRNA and stimulated with 500 ng/ml SDF-1 for 30 min. The treated cells were then stained with antibodies against proteins of interest, inspected by confocal microscopy, and quantified as described previously (27).
Protein degradation assay
Cells were plated in a 6-well plate at a density of 8 × 105 cells/well and transfected with siRNAs against RAB7 or control siRNA using FuGENE transfection reagent (Active Motif) according to manufacturer's instructions. The transfected cells were treated with 500 μg/ml cycloheximide for 30 min at 37 °C prior to incubation with 150 ng/ml SDF-1 for different times. The treated cells were then placed on ice and lysed with RIPA buffer supplemented with protease inhibitor mixture (Sigma). The protein content of the cell lysates was estimated using a BCA assay kit (Thermo Fisher Scientific), and lysates containing equal amounts of proteins were loaded onto 12% SDS-PAGE. CXCR4 proteins were detected by immunoblot. The same blots were stripped and reblotted with β-actin antibody as loading controls. After three independent experiments, quantification was performed with ImageJ software through the densitometry assay.
Western blotting
Cells were plated in a 6-well plate at a density of 8 × 105 cells/well and treated with 150 ng/ml SDF-1 for different times at 37 °C and 5% CO2. The treated cells were then placed on ice and lysed with RIPA buffer (25 mm Tris (pH 8.0), 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Triton X-100) supplemented with a protease inhibitor mixture (Roche) and an anti-phosphatase mixture tablet (Sigma-Aldrich). The protein content of the cell lysates was estimated using a BCA assay kit (Thermo Fisher Scientific), and lysates containing equal amounts of proteins were loaded on to 12% (v/v) SDS-PAGE. Proteins separated in the gel were transferred to a PVDF membrane and blocked in 5% milk–containing TBST buffer (10 mm Tris (pH 8.0), 150 mm NaCl, and 0.1% Tween 20) at room temperature for 1 h, incubated with 1 μg/ml primary antibody for 12 h at 4 °C with agitation, rinsed with TBST, and then incubated with 5% milk–containing TBST supplemented with horseradish peroxidase–conjugated secondary antibody at room temperature for 2 h. After washing three times with TBST, enhanced chemiluminescence detection solution (Pierce) was applied to the membrane. The proteins probed were visualized by X-autoradiography. Densitometry analysis was performed with gel images from three independent experiments using ImageJ software. To determine the loading control, the same membrane was stripped and reblotted with β-actin antibody at 5000× dilution.
Immunoprecipitation assay
The immunoprecipitation assay was performed as described previously (27). Briefly, after treatment with SDF-1, cells stably expressing GFP, MIM-GFP, or IRTKS-GFP were lysed in RIPA buffer supplemented with an anti-protease tablet (Roche) and anti-phosphatase cocktails (Sigma-Aldrich). The cell lysates were subsequently incubated with 20 μl of a 50% protein A/G–agarose slurry for 90 min at 4 °C, followed by centrifugation at 2000 rpm for 30 s. The supernatants were then incubated with 5 μg/ml polyclonal anti-GFP overnight at 4 °C before addition of 100 μl of a protein A/G slurry. After incubation for 2 h at 4 °C, the mixtures were centrifuged at 2000 rpm for 30 s. The pellets were washed three times with 0.5 ml of lysis buffer and then dissolved and boiled in 60 μl of SDS sample buffer for 10 min. The precipitated proteins were analyzed by Western blotting using 1 μg/ml monoclonal primary antibodies against RAB5, RAB7, RAB11, and GFP, respectively. As an input control, aliquots of cell lysates prior to immunoprecipitation were also analyzed by Western blotting using antibodies against the above antigens and β-actin in a parallel experiment.
Cell staining and confocal immunofluorescence microscopy
To analyze the effect of RAB7 on the colocalization of CXCR4 and CD63, sterilized glass coverslips were placed in a 6-well plate and incubated with 5 μg/ml fibronectin (Life Technologies) for 30 min at room temperature. 2 × 105 cells that stably expressed GFP or GFP-MIM were plated into each well and cultured overnight in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were then transfected with siRNAs against RAB7 or control siRNA using FuGENE transfection reagent (Active Motif) according to the manufacturer's introduction. After 16 h, cells were cultured in DMEM supplemented with 10% FBS for 24 h, and the medium was replaced with DMEM and incubated for 2 h. The starved cells were incubated with 100 nm SDF-1 (or vehicle as control) for 30 min. After washing twice with PBS, cells were fixed with 4% paraformaldehyde at room temperature for 10 min and permeabilized with 0.05% saponin for 10 min. The fixed cells were blocked with 200 μl of PBS supplemented with 5% goat serum and incubated with 100 μl of 5 μg/ml monoclonal anti-CXCR4 and polyclonal anti-CD63 antibodies in blocking buffer for 1 h at room temperature. After rinsing three times with PBS, the cells were incubated with PBS containing 1 μg/ml secondary antibodies (Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 568 goat anti-rabbit IgG) for 1 h. In some experiments, 4′,6-diamidino-2-phenylindole was also included along with secondary antibodies. After three washes with PBS, the treated coverslip was mounted on a glass slide with 20 μl of mounting medium (Kirkegaard & Perry Laboratories, Inc.). The slide was sealed with nail polish, and the stained cells were inspected under a Zeiss LSM 510 laser-scanning confocal imaging system using a Plan-Apo ×63/1.4 numerical aperture oil lens. The digital images were captured at an acquisition setting that was applied to all samples analyzed in parallel. In each group, all images were taken and presented at the same settings for brightness and contrast. Protein colocalization was quantified based on Mander's overlapping coefficient (40), which was calculated using an ImageJ plugin. The value of Mander's overlapping coefficient ranges from 0 to 1 and represents the proportion of red fluorescence intensity in the green channel. Quantification of stained puncta was conducted using ImageJ software.
In other experiments, cells were co-stained with monoclonal antibodies (GFP or CXCR4) and polyclonal antibodies (GFP, LAMP2, or EEA1). Proper secondary antibodies were then chosen based on the species of primary antibodies.
Analysis of the effect of IRTKS on surface CXCR4
HeLa cells stably expressing GFP, MIM-GFP, or IRTKS-GFP were plated in a 6-well plate at a density of 8 × 105 cells/well. After culture for 2 days, cells were treated with 500 μg/ml cycloheximide for 30 min at 37 °C prior to incubation with 150 ng/ml SDF-1 for different times. After treatment, cells were immediately placed on ice, followed by addition of 2 ml of prechilled PBS containing 0.75% BSA and 5 mm EDTA (FACS buffer). The treated cells were centrifuged at 1000 rpm for 3 min at 4 °C, and the cell pellet was stained with 1 μg/ml PE-conjugated anti-human CXCR4 antibody and incubated at 4 °C for 1 h in a covered box. As a control, cells were treated with PE anti-IgG antibody in parallel. After staining, 500 μl of FACS buffer was added to the cells, followed by centrifugation. The supernatant was removed by aspiration, and the pelleted cells were washed with PBS three times, fixed with 4% paraformaldehyde, and subjected to flow cytometry with a BD LSRFortessa flow cytometry system. The data were analyzed using Flowjo software version 8.8.7.
Statistical analysis
All data were analyzed using GraphPad Prism 5.0 software and subjected to two-tailed Student's t test for two-group comparisons. p < 0.01 stands for statistical significance.
Author contributions
L. L. and X. Z. conceptualization; L. L. and X. Z. data curation; L. L. formal analysis; L. L. and X. Z. investigation; L. L., S. S. B., and P. Z. methodology; L. L. and X. Z. writing-original draft; S. S. B. and X. Z. writing-review and editing; N. G. and X. Z. resources; N. G. and X. Z. funding acquisition; X. Z. supervision; X. Z. validation.
Supplementary Material
This work was supported by NCI, National Institutes of Health Grant R01 CA113809 (to X. Z.) and National Natural Science Foundation of China for Key Project of International Cooperation Grant 61420106012 (to N. G). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7.
- MIM
- missing in metastasis
- BAR
- Bin–Amphiphysin–Rvs
- IRTKS
- insulin receptor tyrosine kinase substrate
- I-BAR
- inverse BAR
- SH
- Src homology
- GEF
- guanine exchange factor
- WASP
- Wiskott–Aldrich syndrome protein
- WAVE
- WASP family verprolin homologous protein
- PE
- phosphatidylethanolamine
- RIPA
- radioimmune precipitation assay
- Ct
- control.
References
- 1. Xia S., Li X., Johnson T., Seidel C., Wallace D. P., and Li R. (2010) Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts. Development 137, 1075–1084 10.1242/dev.049437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saarikangas J., Mattila P. K., Varjosalo M., Bovellan M., Hakanen J., Calzada-Wack J., Tost M., Jennen L., Rathkolb B., Hans W., Horsch M., Hyvönen M. E., Perälä N., Fuchs H., Gailus-Durner V., et al. (2011) Missing-in-metastasis MIM/MTSS1 promotes actin assembly at intercellular junctions and is required for integrity of kidney epithelia. J. Cell Sci. 124, 1245–1255 10.1242/jcs.082610 [DOI] [PubMed] [Google Scholar]
- 3. Saarikangas J., Kourdougli N., Senju Y., Chazal G., Segerstrale M., Minkeviciene R., Kuurne J., Mattila P. K., Garrett L., Hölter S. M., Becker L., Racz I., Hans W., Klopstock T., Wurst W., et al. (2015) MIM-induced membrane bending promotes dendritic spine initiation. Dev. Cell 33, 644–659 10.1016/j.devcel.2015.04.014 [DOI] [PubMed] [Google Scholar]
- 4. Yu D., Zhan X. H., Zhao X. F., Williams M. S., Carey G. B., Smith E., Scott D., Zhu J., Guo Y., Cherukuri S., Civin C. I., and Zhan X. (2012) Mice deficient in MIM expression are predisposed to lymphomagenesis. Oncogene 31, 3561–3568 10.1038/onc.2011.509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown A. S., Meera P., Altindag B., Chopra R., Perkins E. M., Paul S., Scoles D. R., Tarapore E., Magri J., Huang H., Jackson M., Shakkottai V. G., Otis T. S., Pulst S. M., Atwood S. X., and Oro A. E. (2018) MTSS1/Src family kinase dysregulation underlies multiple inherited ataxias. Proc. Natl. Acad. Sci. U.S.A. 115, E12407–E12416 10.1073/pnas.1816177115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhi D., Irvin M. R., Gu C. C., Stoddard A. J., Lorier R., Matter A., Rao D. C., Srinivasasainagendra V., Tiwari H. K., Turner A., Broeckel U., and Arnett D. K. (2012) Whole-exome sequencing and an iPSC-derived cardiomyocyte model provides a powerful platform for gene discovery in left ventricular hypertrophy. Front. Genet. 3, 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wild P. S., Felix J. F., Schillert A., Teumer A., Chen M. H., Leening M. J. G., Völker U., Großmann V., Brody J. A., Irvin M. R., Shah S. J., Pramana S., Lieb W., Schmidt R., Stanton A. V., et al. (2017) Large-scale genome-wide analysis identifies genetic variants associated with cardiac structure and function. J. Clin. Invest. 127, 1798–1812 10.1172/JCI84840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zimmerberg J., and McLaughlin S. (2004) Membrane curvature: how BAR domains bend bilayers. Curr. Biol. 14, R250–252 10.1016/j.cub.2004.02.060 [DOI] [PubMed] [Google Scholar]
- 9. Lee S. H., Kerff F., Chereau D., Ferron F., Klug A., and Dominguez R. (2007) Structural basis for the actin-binding function of missing-in-metastasis. Structure 15, 145–155 10.1016/j.str.2006.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saarikangas J., Zhao H., Pykäläinen A., Laurinmäki P., Mattila P. K., Kinnunen P. K., Butcher S. J., and Lappalainen P. (2009) Molecular mechanisms of membrane deformation by I-BAR domain proteins. Curr. Biol. 19, 95–107 10.1016/j.cub.2008.12.029 [DOI] [PubMed] [Google Scholar]
- 11. Zhao H., Pykäläinen A., and Lappalainen P. (2011) I-BAR domain proteins: linking actin and plasma membrane dynamics. Curr. Opin. Cell Biol. 23, 14–21 10.1016/j.ceb.2010.10.005 [DOI] [PubMed] [Google Scholar]
- 12. Ahmed S., Goh W. I., and Bu W. (2010) I-BAR domains, IRSp53 and filopodium formation. Semin. Cell Dev. Biol. 21, 350–356 [DOI] [PubMed] [Google Scholar]
- 13. Dawson J. C., Bruche S., Spence H. J., Braga V. M., and Machesky L. M. (2012) Mtss1 promotes cell-cell junction assembly and stability through the small GTPase Rac1. PLoS ONE 7, e31141 10.1371/journal.pone.0031141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng D., Niu S., Yu D., Zhan X. H., Zeng X., Cui B., Chen Y., Yoon J., Martin S. S., Lu X., and Zhan X. (2010) Abba promotes PDGF-mediated membrane ruffling through activation of the small GTPase Rac1. Biochem. Biophys. Res. Commun. 401, 527–532 10.1016/j.bbrc.2010.09.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sudhaharan T., Sem K. P., Liew H. F., Yu Y. H., Goh W. I., Chou A. M., and Ahmed S. (2016) The Rho GTPase Rif signals through IRTKS, Eps8 and WAVE2 to generate dorsal membrane ruffles and filopodia. J. Cell Sci. 129, 2829–2840 10.1242/jcs.179655 [DOI] [PubMed] [Google Scholar]
- 16. Mattila P. K., Pykäläinen A., Saarikangas J., Paavilainen V. O., Vihinen H., Jokitalo E., and Lappalainen P. (2007) Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes by an inverse BAR domain-like mechanism. J. Cell Biol. 176, 953–964 10.1083/jcb.200609176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel A., and Dash P. R. (2012) Formation of atypical podosomes in extravillous trophoblasts regulates extracellular matrix degradation. Eur. J. Cell Biol. 91, 171–179 10.1016/j.ejcb.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oikawa T., and Matsuo K. (2012) Possible role of IRTKS in Tks5-driven osteoclast fusion. Commun. Integr. Biol. 5, 511–515 10.4161/cib.21252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oikawa T., Okamura H., Dietrich F., Senju Y., Takenawa T., and Suetsugu S. (2013) IRSp53 mediates podosome formation via VASP in NIH-Src cells. PLoS ONE 8, e60528 10.1371/journal.pone.0060528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lazar T., Scheglmann D., and Gallwitz D. (2002) A novel phospholipid-binding protein from the yeast Saccharomyces cerevisiae with dual binding specificities for the transport GTPase Ypt7p and the Sec1-related Vps33p. Eur. J. Cell Biol. 81, 635–646 10.1078/0171-9335-00290 [DOI] [PubMed] [Google Scholar]
- 21. Numrich J., Péli-Gulli M. P., Arlt H., Sardu A., Griffith J., Levine T., Engelbrecht-Vandré S., Reggiori F., De Virgilio C., and Ungermann C. (2015) The I-BAR protein Ivy1 is an effector of the Rab7 GTPase Ypt7 involved in vacuole membrane homeostasis. J. Cell Sci. 128, 2278–2292 10.1242/jcs.164905 [DOI] [PubMed] [Google Scholar]
- 22. Itoh Y., Kida K., Hanawa-Suetsugu K., and Suetsugu S. (2016) Yeast Ivy1p is a putative I-BAR-domain protein with pH-sensitive filament forming ability in vitro. Cell Struct. Funct. 41, 1–11 10.1247/csf.15014 [DOI] [PubMed] [Google Scholar]
- 23. Veltman D. M., Auciello G., Spence H. J., Machesky L. M., Rappoport J. Z., and Insall R. H. (2011) Functional analysis of Dictyostelium IBARa reveals a conserved role of the I-BAR domain in endocytosis. Biochem. J. 436, 45–52 10.1042/BJ20101684 [DOI] [PubMed] [Google Scholar]
- 24. Quinones G. A., Jin J., and Oro A. E. (2010) I-BAR protein antagonism of endocytosis mediates directional sensing during guided cell migration. J. Cell Biol. 189, 353–367 10.1083/jcb.200910136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhan T., Cao C., Li L., Gu N., Civin C. I., and Zhan X. (2016) MIM regulates the trafficking of bone marrow cells via modulating surface expression of CXCR4. Leukemia 30, 1327–1334 10.1038/leu.2016.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., and Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 10.1016/S1534-5807(03)00321-6 [DOI] [PubMed] [Google Scholar]
- 27. Li L., Baxter S. S., Gu N., Ji M., and Zhan X. (2017) Missing-in-metastasis protein downregulates CXCR4 by promoting ubiquitylation and interaction with small Rab GTPases. J. Cell Sci. 130, 1475–1485 10.1242/jcs.198937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vanlandingham P. A., and Ceresa B. P. (2009) Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 284, 12110–12124 10.1074/jbc.M809277200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L., Liu H., Baxter S. S., Gu N., Ji M., and Zhan X. (2016) The SH3 domain distinguishes the role of I-BAR proteins IRTKS and MIM in chemotactic response to serum. Biochem. Biophys. Res. Commun. 479, 787–792 10.1016/j.bbrc.2016.09.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizuno-Yamasaki E., Rivera-Molina F., and Novick P. (2012) GTPase networks in membrane traffic. Annu. Rev. Biochem. 81, 637–659 10.1146/annurev-biochem-052810-093700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nordmann M., Cabrera M., Perz A., Bröcker C., Ostrowicz C., Engelbrecht-Vandré S., and Ungermann C. (2010) The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr. Biol. 20, 1654–1659 10.1016/j.cub.2010.08.002 [DOI] [PubMed] [Google Scholar]
- 32. Sönnichsen B., De Renzis S., Nielsen E., Rietdorf J., and Zerial M. (2000) Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149, 901–914 10.1083/jcb.149.4.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ward E. S., Martinez C., Vaccaro C., Zhou J., Tang Q., and Ober R. J. (2005) From sorting endosomes to exocytosis: association of Rab4 and Rab11 GTPases with the Fc receptor, FcRn, during recycling. Mol. Biol. Cell 16, 2028–2038 10.1091/mbc.e04-08-0735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chou A. M., Sem K. P., Lam W. J., Ahmed S., and Lim C. Y. (2017) Redundant functions of I-BAR family members, IRSp53 and IRTKS, are essential for embryonic development. Sci. Rep. 7, 40485 10.1038/srep40485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goh W. I., Sudhaharan T., Lim K. B., Sem K. P., Lau C. L., and Ahmed S. (2011) Rif-mDia1 interaction is involved in filopodium formation independent of Cdc42 and Rac effectors. J. Biol. Chem. 286, 13681–13694 10.1074/jbc.M110.182683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Duleh S. N., and Welch M. D. (2010) WASH and the Arp2/3 complex regulate endosome shape and trafficking. Cytoskeleton 67, 193–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lapierre L. A., Kumar R., Hales C. M., Navarre J., Bhartur S. G., Burnette J. O., Provance D. W. Jr., Mercer J. A., Bähler M., and Goldenring J. R. (2001) Myosin vb is associated with plasma membrane recycling systems. Mol. Biol. Cell 12, 1843–1857 10.1091/mbc.12.6.1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sakaguchi A., Sato M., and Sato K. (2016) REI-1, a Novel Rab11 GEF with a SH3BP5 domain. Commun. Integr. Biol. 9, e1208325 10.1080/19420889.2016.1208325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Silva N. S., and Klein U. (2015) Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 15, 137–148 10.1038/nri3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dunn K. W., Kamocka M. M., and McDonald J. H. (2011) A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300, C723–742 10.1152/ajpcell.00462.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.