

Abstract
Site-specific suppressors of superoxide production (named S1QELs) in the quinone-reaction site in mitochondrial respiratory complex I during reverse electron transfer have been previously reported; however, their mechanism of action remains elusive. Using bovine heart submitochondrial particles, we herein investigated the effects of S1QELs on complex I functions. We found that the inhibitory effects of S1QELs on complex I are distinctly different from those of other known quinone-site inhibitors. For example, the inhibitory potencies of S1QELs significantly varied depending on the direction of electron transfer (forward or reverse). S1QELs marginally suppressed the specific chemical modification of Asp160 in the 49-kDa subunit, located deep in the quinone-binding pocket, by the tosyl chemistry reagent AL1. S1QELs also failed to suppress the binding of a photoreactive quinazoline-type inhibitor ([125I]AzQ) to the 49-kDa subunit. Moreover, a photoaffinity labeling experiment with photoreactive S1QEL derivatives indicated that they bind to a segment in the ND1 subunit that is not considered to make up the binding pocket for quinone or inhibitors. These results indicate that unlike known quinone-site inhibitors, S1QELs do not occupy the quinone- or inhibitor-binding pocket; rather, they may indirectly modulate the quinone-redox reactions by inducing structural changes of the pocket through binding to ND1. We conclude that this indirect effect may be a prerequisite for S1QELs' direction-dependent modulation of electron transfer. This, in turn, may be responsible for the suppression of superoxide production during reverse electron transfer without significantly interfering with forward electron transfer.
Keywords: bioenergetics, mitochondria, Complex I, ubiquinone, enzyme inhibitor, chemical biology, photoaffinity labeling, oxidative stress, electron transfer chain
Introduction
Excessive production of reactive oxygen species (ROS)2 from mitochondria is associated with a wide range of age-associated diseases, such as diabetes, cardiovascular disease, neurodegeneration, and cancer (1–5). However, a growing body of evidence indicates that ROS produced in mitochondria serve as a critical cellular signal in physiological pathways (6–9). Progress in defining the sites of ROS production revealed that many sites exist in mitochondria, such as respiratory complexes (complexes I-III), the electron-transferring flavoprotein-ubiquinone oxidoreductase, and sn-glycerol-3-phosphate dehydrogenase (10). The contribution of particular mitochondrial sites varies with different respiratory substrates due to different entry points of reducing equivalents during metabolism and the resulting different redox states of the enzymes and a quinone/quinol pool. It is likely that mitochondria in resting skeletal muscle produce ROS primarily from four sites: site IQ (the ubiquinone-binding site in complex I active during reverse electron transport), site IF (the flavin (FMN) site in complex I), site IIF (the flavin (FAD) site in complex II), and site IIIQo (the quinol oxidation site in complex III) (11). Although sites IF and IIIQo are generally recognized as major sites of ROS production, sites IQ and IIF are also considered to have the potential to significantly produce ROS under specific conditions (10).
In respiratory complex I, site IQ generates ROS at high rates during reverse electron transfer from ubiquinol in a ubiquinone/ubiquinol pool, which is supported by a high proton-motive force formed across the inner mitochondrial membrane (12). Inhibition of site IQ by the so-called “quinone-site inhibitors” (such as rotenone and piericidin A) completely suppresses ROS production from this site by preventing ubiquinol oxidation. In contrast, site IF is a prominent ROS production site during forward electron transfer from the NADH pool in the mitochondrial matrix and has a much lower rate of ROS production than site IQ in intact mitochondria (12). The presence of quinone-site inhibitors increases ROS production from site IF by enhancing the reduction state of FMN (12). It should be noted that some research groups argue that ROS are predominantly produced at site IF by a single, unified mechanism that is applied during both forward and reverse electron transfer (13, 14). The inhibition of complex I by rotenone or neurotoxic N-methyl-4-phenylpyridinium has been considered to be associated with parkinsonism in both rodents and humans, suggesting a link between ROS production due to dysfunctional complex I and neurodegeneration (15, 16). Comparative analyses suggested an inverse relationship between maximal ROS production from site IQ, but not site IF, and the maximum lifespan across vertebrate species (17, 18). Taking these into consideration, selective modulators of ROS production from a definite site in the respiratory complexes would become powerful chemical tools to investigate the roles of mitochondrial ROS in biochemical and physiological studies.
In this context, antioxidants are also useful chemical tools for this aim. Many years of research on antioxidants have provided many natural and synthetic chemicals, which modulate ROS with different selectivity and efficiency within different cellular compartments depending on their diverse chemical properties (19–22). However, because antioxidants scavenge ROS downstream from production sites and, hence, are unable to fully suppress the production of ROS, they cannot substitute for the selective modulators of ROS production mentioned above.
Brand and colleagues (23, 24) developed a panel of high-throughput screening assays to identify low-molecular-weight chemicals, which suppress ROS production during reverse electron transfer through complex I (from site IQ) in mitochondria isolated from rat skeletal muscle without significant concomitant interferences with bioenergetic functions of mitochondria (including forward electron transfer (i.e. NADH-quinone oxidoreductase activity)). They named the chemicals “S1QEL,” suppressor of site IQ electron leak (23, 24). Through screening of 635,000 compounds, they discovered two structural classes of S1QELs, named S1QEL1 (thiazole-type) and S1QEL2 (piperazine-type) families (24). They showed that S1QEL1 and S1QEL2 analogues protect against stress-induced stem cell hyperplasia in Drosophila intestine in vivo and against ischemia-reperfusion injury in the perfused mouse heart (24). Although the detailed mechanism of action of S1QELs remains elusive, their unique action may be explained by considering that each S1QEL only modulates ubiquinol oxidation (reverse electron transfer) and not quinone reduction (forward electron transfer) in a definite concentration range. However, referring to the architecture of the quinone/inhibitor-access channel in mammalian complex I modeled by single-particle cryo-electron microscopy (25–27), this leads to a critical question of how S1QELs selectively modulate one of the two opposite quinone-redox reactions that take place inside a common narrow channel (note that we recently questioned whether the quinone/inhibitor-access channel models fully reflect physiologically relevant states present throughout the catalytic cycle (28)). Brand et al. (24) did not investigate the binding position of S1QELs in complex I; however, this is absolutely necessary to fully define the mechanism of action of these unique chemicals.
Here, we synthesized some S1QELs as reported in Ref. 24 (Fig. 1) in our laboratory and investigated their effects on the functions of complex I in bovine heart SMPs. To identify the binding position of S1QELs, we carried out photoaffinity labeling experiments with photoreactive derivatives that were synthesized using original S1QEL as a template (Fig. 1). We found that all S1QELs examined have the potential to inhibit both forward and reverse electron transfers. However, their inhibitory effects were unique and distinctly different from those observed for known quinone-site inhibitors such as quinazoline and bullatacin; therefore, we concluded that S1QELs are a new type of inhibitor of complex I. Based on the results obtained in the present study, we discuss the causal connection between the unique inhibitory actions of S1QELs and their behavior as suppressors of superoxide production during reverse electron transfer.
Figure 1.

Structures of S1QELs and their derivatives studied in the present study. S1QEL1.1, S1QEL1.5, S1QEL2.1, and S1QEL2.3 were reported in Ref. 24. S1QEL1.1_D1, S1QEL1.1_D2, S1QEL1.1_D3, and S1QEL1.5_D1 were derived from corresponding parent S1QELs. Photolabile [125I]S1QEL1.1_PD1 and [125I]S1QEL1.1_PD2 were used for photoaffinity labeling experiments.
Results
Syntheses of S1QEL analogues
Among S1QELs discovered by Brand et al. (24), we picked up S1QEL1.1/S1QEL1.5 and S1QEL2.1/S1QEL2.3 from S1QEL1 (thiazole-type) and S1QEL2 (piperazine-type) families, respectively. We synthesized these four compounds in our laboratory by the methods described under Schemes S1 and S2. We also synthesized three derivatives of S1QEL1.1 (S1QEL1.1_D1, S1QEL1.1_D2, and S1QEL1.1_D3, Scheme S3) and one derivative of S1QEL1.5 (S1QEL1.5_D1, Scheme S1) to examine the structure-activity relationship (Fig. 1), although these derivatives were not reported in the earlier work (24). To conduct photoaffinity labeling experiments, we synthesized [125I]S1QEL1.1_PD1 (Scheme S4) and [125I]S1QEL1.1_PD2 (Scheme S5), which possess an azido group and 125I as a photolabile group and a detecting tag, respectively (Fig. 1).
Inhibition of forward electron transfer by S1QELs
Brand et al. (24) reported that S1QEL1.1, S1QEL1.5, S1QEL2.1, and S1QEL2.3 elicit no inhibitory effect on respiration driven by succinate plus rotenone (covering complexes II, III, and IV) and by glutamate plus malate (covering complexes I, III, and IV) in mitochondria isolated from rat skeletal muscle at 10 μm or 20 × IC50 (i.e. 20-fold of the IC50 value that is the molar concentration required to suppress superoxide production from site IQ by 50%). S1QEL1.1 had the lowest IC50 value (0.07 μm) among S1QELs that they discovered (24). We examined the effects of S1QELs and their derivatives on NADH oxidase activity in bovine heart SMPs (covering complexes I, III, and IV). SMPs were incubated with a test compound for 4 min before initiating the reaction by adding NADH (final concentration: 50 μm).
The inhibitory potencies of S1QELs, in terms of the EC50 value, which is the molar concentration needed to reduce the control NADH oxidase activity by 50%, are listed in Table 1. Their inhibitory potencies widely varied depending on their structural features. S1QEL1.1 was the most potent inhibitor of forward electron transfer (EC50 = 0.059 μm). The extent of inhibition by S1QEL1.5 and S1QEL1.5_D1 was just ∼10–30% even at 50 μm, as shown in Fig. 2 (trace b) taking S1QEL1.5 as an example. The residual NADH oxidase activity in the presence of these compounds was completely inhibited (>95%) by the addition of other quinone-site inhibitors, such as bullatacin and quinazoline. Considering marked structural similarities between S1QEL1.1 and S1QEL1.5/S1QEL1.5_D1, these results seem to be peculiar at first glance.
Table 1.
Summary of the inhibitory potencies (EC50) of S1QELs
The EC50 value is the molar concentration (μm) needed to reduce the control NADH oxidase activity (forward electron transfer) or ubiquinol-NAD+ oxidoreductase activity (reverse electron transfer) in SMPs by 50%, except for S1QEL1.5 and S1QEL1.5_D1. SMPs concentrations are 30 and 100 μg of proteins/ml for forward and reverse electron transfer measurement, respectively. The EC50 values that were normalized by protein concentrations (nmol of inhibitor/mg of proteins) are also listed in the parentheses. Values are mean ± S.E. (n = 3–4).
| S1QELs | Forward electron transfer EC50 (μm) (nmol inhibitor/mg of proteins) | Reverse electron transfer EC50 (μm) (nmol inhibitor/mg of proteins) | Ratioa (forward/reverse) |
|---|---|---|---|
| S1QEL1.1 | 0.059 ± 0.006 (2.0 ± 0.20) | 0.038 ± 0.005 (0.38 ± 0.05) | 5.2 |
| S1QEL1.5 | 1.9 ± 0.38b (63 ± 13) | 0.31 ± 0.07c (3.1 ± 0.7) | 20 |
| S1QEL2.1 | 0.29 ± 0.044 (9.7 ± 1.5) | 0.060 ± 0.003 (0.60 ± 0.03) | 16 |
| S1QEL2.3 | 0.54 ± 0.08 (18 ± 2.8) | 0.13 ± 0.02 (1.3 ± 0.20) | 14 |
| S1QEL1.5_D1 | 1.4 ± 0.16b (47 ± 5.2) | 0.24 ± 0.047c (2.4 ± 0.47) | 20 |
| S1QEL1.1_D1 | 0.89 ± 0.04 (30 ± 1.0) | 1.25 ± 0.06 (12.5 ± 0.6) | 2.4 |
| S1QEL1.1_D2 | 0.88 ± 0.07 (29 ± 2.0) | 0.50 ± 0.02 (5.0 ± 0.2) | 5.8 |
| S1QEL1.1_D3 | 0.32 ± 0.02 (11 ± 0.6) | 0.43 ± 0.04 (4.3 ± 0.4) | 2.5 |
| Bullatacin | 0.0020 ± 0.0002 (0.070 ± 0.007) | 0.0046 ± 0.0006 (0.046 ± 0.006) | 1.4 |
a The ratio of EC50 (forward)/EC50 (reverse) values in terms of nanomole of inhibitor/mg of proteins.
b The EC50 value was expediently evaluated from the inhibition observed at ∼1 min after the addition of a test compound during catalytic turnover; namely, we read a slope of the dotted line indicated for trace c in Fig. 2.
c Because the maximum inhibition by this compound was ∼40% (Fig. 4B), the molar concentration that attained 20% inhibition was determined.
Figure 2.
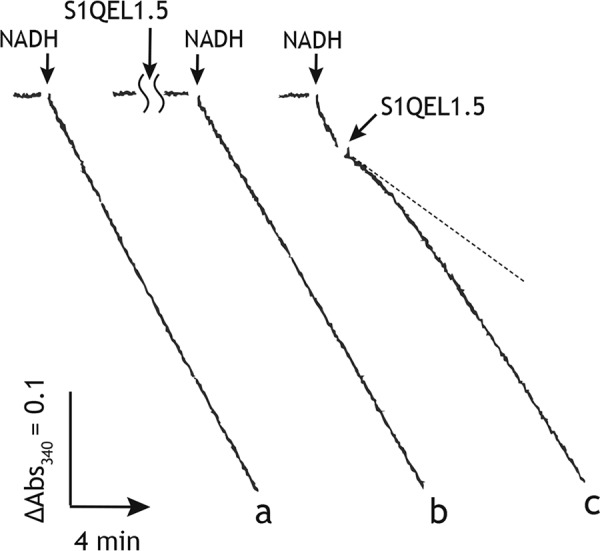
The inhibition of the NADH oxidase activity in SMPs by S1QEL1.5. Trace a, the NADH oxidase activity without inhibitor (control); trace b, the NADH oxidase activity initiated by adding NADH (200 μm) after 4 min incubation with S1QEL1.5 (50 μm); trace c, S1QEL1.5 (50 μm) was added to SMPs at ∼1 min after initiation of the reaction by adding NADH (200 μm). The final mitochondrial protein concentration was 10 μg/ml.
To characterize the inhibitory actions of S1QEL1.5 and S1QEL1.5_D1, they were added to SMPs at ∼2 min after initiation of the reaction by adding NADH (i.e. during catalytic turnover). Unexpectedly, both compounds exhibited definite inhibition of the NADH oxidase activity, as shown in Fig. 2 (trace c) taking S1QEL1.5 (50 μm) as an example. Furthermore, their inhibitory effects gradually diminished with time and significant inhibition was no longer observed ∼6 min after the addition (Fig. 2). These results strongly suggest that S1QEL1.5 and S1QEL1.5_D1 gradually decomposed during incubation with SMPs. This may be a cause of the weak inhibitory effects when they were previously incubated with SMPs before initiating the reaction (trace b). In support of this, the longer the incubation period, the weaker their inhibitory effects (Fig. 3A). Note that the inhibitory effects of other S1QELs were not affected by changing the order of addition of inhibitor and NADH.
Figure 3.
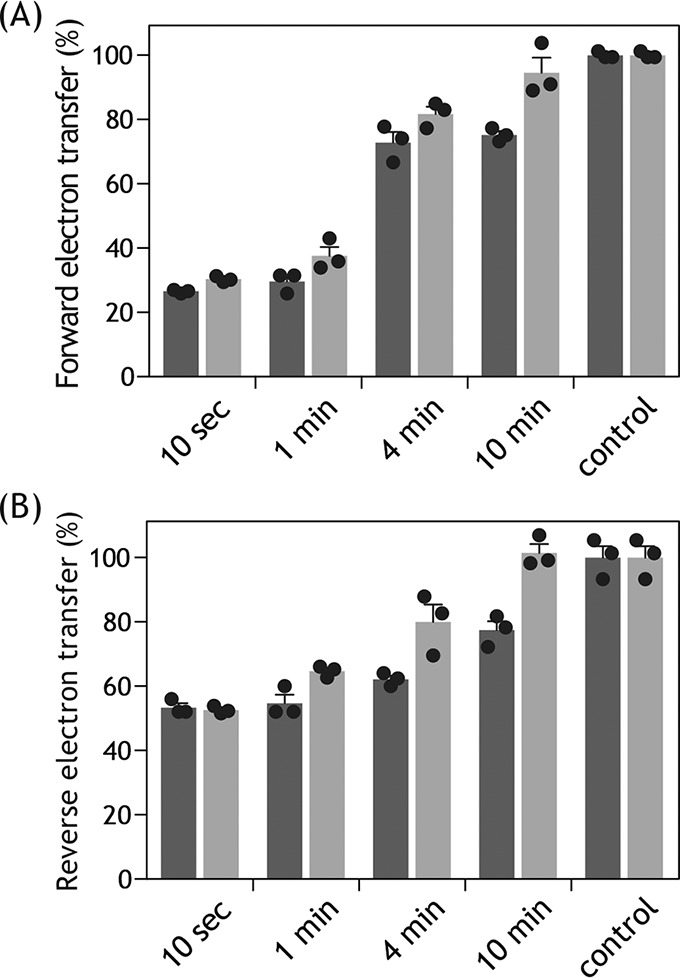
Time-dependent disappearance of the inhibitory effects of S1QEL1.5 and S1QEL1.5_D1. A, the inhibition of NADH oxidase activity (forward electron transfer) by S1QEL1.5 (5.0 μm, black bars) or S1QEL1.5_D1 (5.0 μm, gray bars) was determined after different incubation periods with SMPs at 30 °C. The averaged control forward electron transfer activity was 580 ± 55 nmol of NADH/min/mg of proteins. The final mitochondrial protein concentration was 30 μg/ml. B, the inhibition of ubiquinol-NAD+ oxidoreductase activity (reverse electron transfer) by S1QEL1.5 (6.3 μm, black bars) or S1QEL1.5_D1 (6.3 μm, gray bars) was determined after different incubation periods with SMPs at 30 °C. The averaged control reverse electron transfer activity was 50 ± 6.0 nmol of NADH/min/mg of proteins. The final mitochondrial protein concentration was 100 μg/ml. Values in graphs are mean ± S.E. (n = 3).
For comparison of the inhibitory potencies of S1QEL1.5 and S1QEL1.5_D1 with those of other S1QELs, their EC50 values, which were expediently evaluated from the inhibition observed at 1 min after the addition of these compounds during the catalytic turnover (i.e. a dotted line on trace c in Fig. 2), are listed in Table 1. Under the experimental conditions, the maximum inhibition of the NADH oxidase activity by S1QEL1.5 and S1QEL1.5_D1 was ∼80%. It should be noted that although S1QEL2.1 and S1QEL2.3 elicited inhibition at lower than single digit micromolar level, they also did not achieve complete inhibition (>95%) of the NADH oxidase activity: ∼20–30% enzyme activity remained even in the presence of excess compounds (Fig. 4A, open and closed circles). These residual enzyme activities are greater than those observed with known quinone-site inhibitors such as piericidin A and bullatacin, which generally attain >95% inhibition, as shown in Fig. 4A taking bullatacin (open triangles) as an example.
Figure 4.
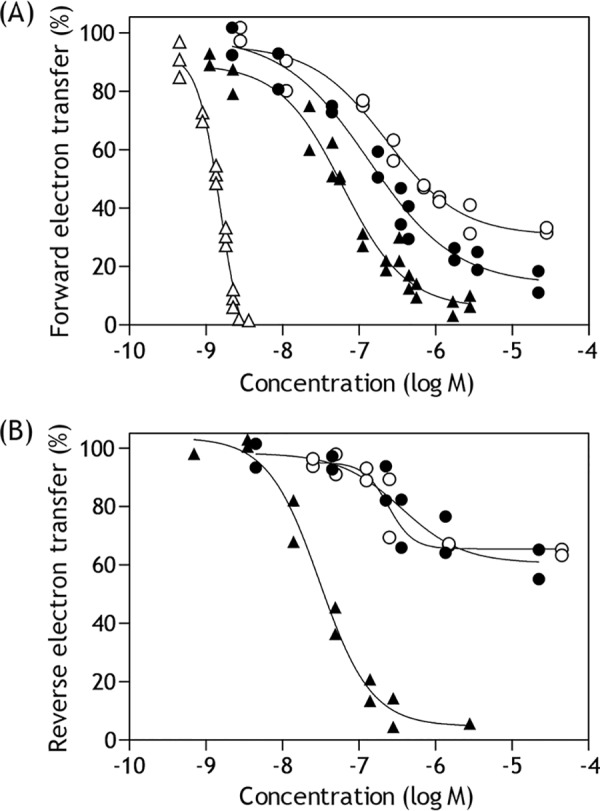
Dose-response curves for the inhibition of electron transfer in complex I. A, the inhibition of NADH oxidase activity by S1QELs. The reaction was initiated by adding NADH (final concentration: 50 μm) after incubation of SMPs with each test compound for 4 min at 30 °C. S1QEL2.1, closed circles; S1QEL2.3, open circles; S1QEL1.1, closed triangles; bullatacin, open triangles. The final mitochondrial protein concentration was 30 μg/ml. B, the inhibition of reverse electron transfer by S1QELs. The reaction was initiated by adding ATP (final concentration: 1.0 mm) after incubation of SMPs with each compound for 4 min at 30 °C. S1QEL1.5, closed circles; S1QEL1.5_D1, open circles; S1QEL1.1, closed triangles. The final mitochondrial protein concentration was 100 μg/ml.
S1QEL1.1 elicited the most potent inhibition of the forward electron transfer (Table 1) and achieved almost complete inhibition (Fig. 4A, closed triangles). To know which structural factors are important for the inhibitory activity of S1QEL1.1, we synthesized its derivatives S1QEL1.1_D1, S1QEL1.1_D2, and S1QEL1.1_D3. As shown in Table 1, modifications of the acetylamino group in S1QEL1.1 resulted in a significant decrease in the inhibitory potencies, indicating that this functional group is important for tight interaction with complex I.
Inhibition of reverse electron transfer by S1QELs
We next examined the effects of S1QELs on the reverse electron transfer (the quinol-NAD+ oxidoreductase activity) driven by succinate oxidation with support of proton-motive force due to ATP hydrolysis in bovine SMPs (Table 1). Because reverse electron transfer is considerably slower than forward electron transfer, a higher concentration of SMPs was used in the reverse electron transfer assay (100 versus 30 μg of proteins/ml). Accordingly, to compare the inhibitory potencies between the forward and reverse events, the EC50 values that were normalized by protein concentrations (nanomole inhibitor/mg of proteins) are also listed in parentheses in Table 1. As for forward electron transfer, each test compound was incubated with SMPs for 4 min before the reverse reaction started by the addition of ATP (final concentration: 1.0 mm). We preliminary confirmed that reverse electron transfer is completely blocked in the presence of SF6847 (a protonophoric uncoupler) or oligomycin (FoF1-ATPase inhibitor).
S1QEL1.1, S1QEL2.1, S1QEL2.3, and S1QEL1.1 analogues (S1QEL1.1_D1−S1QEL1.1_D3) achieved almost complete inhibition (>90%) of reverse electron transfer, as shown in Fig. 4B taking S1QEL1.1 (closed triangles) as an example. Their inhibitory potencies determined for reverse electron transfer, in terms of EC50 (mole inhibitor/mg of proteins), were stronger than those for forward electron transfer (>5-fold), indicating a direction-dependent inhibition. It is noteworthy that the extent of inhibition by S1QEL1.5 (closed circles) or S1QEL1.5_D1 (open circles) was saturated at ∼40% (Fig. 4B). The substantial residual activity was completely blocked by the addition of known inhibitors such as quinazoline and bullatacin. For these compounds, the molar concentrations that achieved 20% inhibition (in place of 50% inhibition) are tentatively listed in Table 1.
The inhibitory effects of S1QEL1.5 and S1QEL1.5_D1 on the reverse electron transfer also gradually disappeared during incubation with SMPs (Fig. 3B), as observed with forward electron transfer. If the disappearance of inhibitory activities is attributable to their decomposition, a possible mechanism would be hydrolysis at -NH-CO-CO-NH-, because there is no other partial structure that is anticipated to be unstable under the experimental conditions, although we have not experimentally verified the mechanism. The substantial residual activity observed for S1QEL1.5 and S1QEL1.5_D1 may not be explained solely by their decomposition because the extent of inhibition was ∼40% even after a short incubation (10 s, Fig. 3B) and saturated at this level irrespective of further increases in concentrations of these compounds added (Fig. 4B).
The order of the suppressive efficiencies of the four S1QELs against ROS production from site IQ, in terms of the IC50 values, was S1QEL1.1 > S1QEL2.1 > S1QEL2.3 > S1QEL1.5 (24). This order is identical to that of the inhibitory potencies (EC50) against the reverse electron transfer determined in the present study (Table 1), although complex I samples differ between the two studies (rat skeletal muscle mitochondria versus bovine heart SMPs).
Effects of S1QELs on the alkynylation of 49 kDa-Asp160 by AL1
Ligand-directed tosyl (LDT) chemistry (29, 30), which is based on a principle of affinity labeling, provides a powerful means to covalently attach a synthetic tag of choice to target protein with a high location-specificity. Using this technique, we previously demonstrated that Asp160 in the 49-kDa subunit (49 kDa-Asp160), which is located deep inside the predicted quinone-access channel (25–27), can be specifically alkynylated (Asp160(COO)-(CH2)2-C CH) by incubating SMPs with acetogenin-type ligand AL1 (Figs. S1 and S2) (31, 32). This alkynylation was completely suppressed by various quinone-site inhibitors, such as quinazoline, bullatacin, and fenpyroximate (31, 32). To elucidate the action manner of S1QELs, we examined the effects of S1QELs on the alkynylation of 49 kDa-Asp160 by AL1.
CH) by incubating SMPs with acetogenin-type ligand AL1 (Figs. S1 and S2) (31, 32). This alkynylation was completely suppressed by various quinone-site inhibitors, such as quinazoline, bullatacin, and fenpyroximate (31, 32). To elucidate the action manner of S1QELs, we examined the effects of S1QELs on the alkynylation of 49 kDa-Asp160 by AL1.
Alkynylation by AL1 (0.1 μm) can be visualized by conjugating a fluorescent tag TAMRA-N3 to [Asp160(COO)-(CH2)2-CCH] via click chemistry after solubilizing SMPs (Fig. S2) (31, 32), as shown in Fig. 5A (control lane). We confirmed that bullatacin (10 μm) completely suppressed the alkynylation (Fig. 5A). The extents of suppression by S1QELs (10 μm each, 100-hold of AL1) varied depending on their structures (Fig. 5A), but none exhibited a remarkable suppressive effect like bullatacin. Even S1QEL1.1, with the most potent inhibitory activity among the S1QELs tested, suppressed the alkynylation by ∼70% at far over the concentration giving full inhibition of forward electron transfer. This incomplete suppression is not due to an insufficient amount of S1QEL1.1 added because the extent of suppression by S1QEL1.1 had already reached a plateau (∼70%) by ∼5 μm and did not further increase with increasing concentrations up to 50 μm (Fig. 5B). S1QEL1.5 and S1QEL1.5_D1 hardly effected the alkynylation, consistent with the fact that these inhibitors markedly lost their inhibitory effects during incubation with SMPs (Figs. 2 and 3). Taken together, it is obvious that the binding positions of S1QELs and known quinone-site inhibitors are considerably different.
Figure 5.
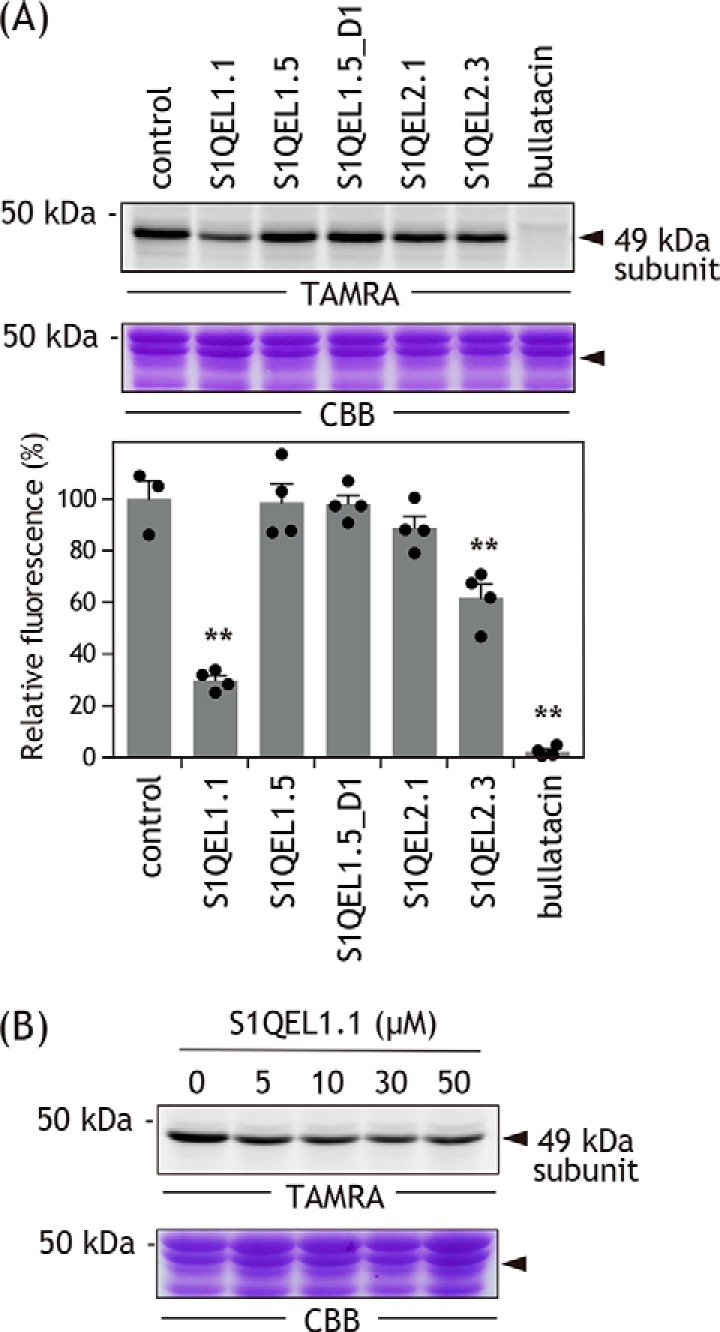
Effects of S1QELs on alkynylation of 49 kDa-Asp160 by AL1.
A, alkynylation of 49 kDa-Asp160 in intact complex I was achieved via LDT chemistry using AL1 (0.1 μm). The alkynylation can be visualized by conjugating a fluorescent tag TAMRA-N3 to [Asp160(COO)-(CH2)2-CCH] via click chemistry after solubilizing SMPs (see “Experimental procedures” and Fig. S2). Approximately 30 μg of proteins were loaded into each well. Upper panel, gel image of SDS-PAGE analysis used for LDT chemistry; lower panel, the extent of suppression by S1QELs (10 μm each, 100-fold of AL1). Values in graphs are mean ± S.E. (n = 3–4). **, p < 0.001 compared with control (one-way ANOVA followed by Dunnet's test). B, suppression of the alkynylation by different concentrations of S1QEL1.1 (5.0–50 μm). Data are representative of three independent experiments.
Effects of S1QELs on the binding of [125I]AzQ to the 49-kDa subunit
We previously demonstrated that quinazoline-type inhibitor [125I]AzQ (Fig. S1) binds at the interface of the 49-kDa and ND1 subunits in bovine complex I, although radioactivity was predominantly distributed into the former (33, 34). The labeled regions in the 49-kDa and ND1 subunits were the N-terminal (Asp41–Arg63 in mature sequence numbering) and the third matrix loop connecting transmembrane helices (TMHs) 5 and 6 (Asp199–Lys262), respectively. The binding of [125I]AzQ to the enzyme was completely suppressed by an excess of other quinone-site inhibitors such as rotenone, piericidin A, and bullatacin (33, 34).
We examined whether S1QELs (5.0 μm each) suppress the binding of [125I]AzQ (5.0 nm) to the predominant target, the 49-kDa subunit. We confirmed, as a control, that bullatacin (5.0 μm) completely suppressed the binding of [125I]AzQ (Fig. 6). It is noteworthy that none of the S1QELs (1000-fold of [125I]AzQ) efficiently suppressed the binding of [125I]AzQ (Fig. 6). Although S1QEL1.1 moderately suppressed the alkynylation of 49 kDa-Asp160 by AL1 (Fig. 5A), this inhibitor hardly suppressed the binding of [125I]AzQ. These results also strongly suggest that the binding position of S1QELs significantly differ from that of ordinary quinone-site inhibitors.
Figure 6.
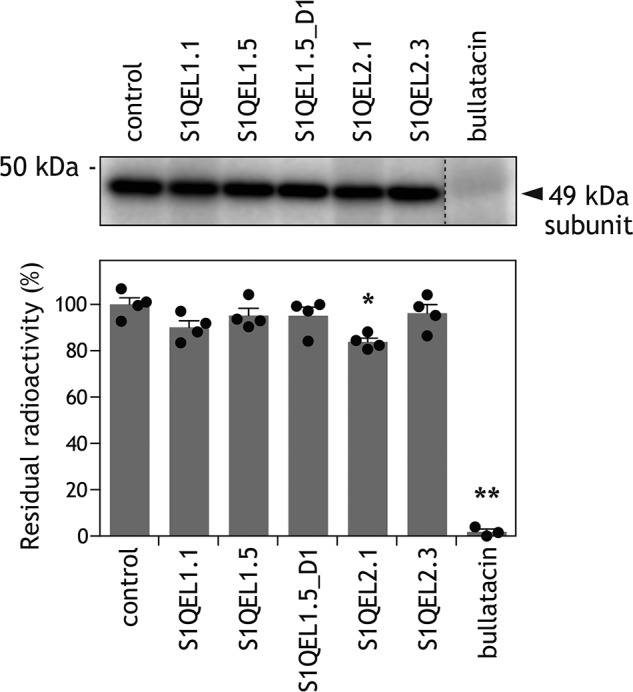
Effects of S1QELs on specific binding of [125I]AzQ to the 49-kDa subunit. The photoaffinity labeling of the 49-kDa subunit in intact complex I using [125I]AzQ (5.0 nm) was carried out in the presence of different S1QELs (5.0 μm each, 1000-fold). Approximately 30 μg of proteins were loaded into each well. Upper panel, gel image of SDS-PAGE analysis used for the photoaffinity labeling; lower panel, the extent of suppression by S1QELs. Values in graphs are mean ± S.E. (n = 3–4). *, p < 0.01; **, p < 0.001 compared with control (one-way ANOVA followed by Dunnet's test).
The binding position of S1QELs in complex I
To identify the binding position of S1QELs in complex I, we performed photoaffinity labeling experiments using two photoreactive derivatives ([125I]S1QEL1.1_PD1 and [125I]S1QEL1.1_PD2, Fig. 1), which were derived using S1QEL1.1 as a synthetic template. We selected S1QEL1.1 (the most efficient S1QEL reported in Ref. 24) to produce as potent derivatives as possible to minimize nonspecific labeling, which is a primary cause of false-positive results. The EC50 values of [125I]S1QEL1.1_PD1 and [125I]S1QEL1.1_PD2, determined with the NADH oxidase assay using their nonradioactive derivatives, were 23 (±3.0) and 290 (±42) nm, respectively. Introduction of an azido (−N=N+=N−) group into the left benzene ring was significantly unfavorable for the inhibition. This is because the interaction between this moiety and the enzyme is critical for the inhibitory action, as demonstrated by the structure-activity relationship study above.
SMPs (4.0 mg of proteins/ml) were incubated with [125I]S1QEL1.1_PD1 (10 nm), irradiated with a UV lamp on ice, and the radiolabeled complex I was isolated by BN-PAGE, followed by the resolution on doubled SDS gel (35) using 10 and 16% Schägger-type SDS gels (Fig. 7A, left). The labeling of complex I by [125I]S1QEL1.1_PD1 gave a major ∼30 kDa radioactive spot above a diagonal axis (Fig. 7A, center), which corresponds to the position of the hydrophobic ND1 subunit (identified by MS, Table S1).
Figure 7.

Photoaffinity labeling of bovine complex I by photoreactive [125I]S1QEL1.1 derivatives. A, SMPs (4.0 mg of proteins/ml) were cross-linked by [125I]S1QEL1.1_PD1 or [125I]S1QEL1.1_PD2 (5.0 nm each), followed by the purification of complex I by BN-PAGE and electroelution. Isolated complex I was resolved by doubled SDS-PAGE, and the SDS gel was subjected to silver staining or autoradiography. The black arrow indicates an ADP/ATP carrier, which was labeled by an azidophenyl group (28, 36). Proteins equivalent to ∼200 μg of SMPs were loaded into each gel. All data are representative of three independent experiments. B, localization of the region labeled by [125I]S1QEL1.1_PD1 or [125I]S1QEL1.1_PD2. The ND1 subunit labeled by [125I]S1QEL1.1_PD1 or [125I]S1QEL1.1_PD2 was exhaustively digested with Lys-C or Asp-N. The digests were resolved on a 16% Schägger-type SDS gel (16% T, 6% C, containing 6.0 m urea), followed by autoradiography. Proteins equivalent to ∼50 μg of SMPs were loaded into each well. C, schematic representation of the exhaustive digestion of the ND1 subunit with Lys-C or Asp-N. The TMHs were assigned according to the structures of bovine (25) or ovine (26) complex I. Predicted cleavage sites are denoted by arrowheads and marked with their residue numbers in the matured sequences of the bovine ND1 subunit (SwissProt entry P03887).
We previously showed that complex I inhibitors having a photoreactive azidophenyl group concomitantly bind to the ADP/ATP carrier and 3-hydroxybutyrate dehydrogenase for an unknown reason (28, 33, 36), although the labeling yields varies depending on chemical frameworks of inhibitors. The weak radioactive spot at ∼35 kDa on a diagonal axis (Fig. 7A, center) was identified as a ADP/ATP carrier by MS (Table S1).
[125I]S1QEL1.1_PD2 also cross-linked to the ND1 subunit, although incorporated radioactivity into the protein was 10–20% of that by [125I]S1QEL1.1_PD1 (Fig. 7A, right). This is the reason why [125I]S1QEL1.1_PD2 appears to predominantly cross-link to AAC and to a lesser extent to ND1. In light of the EC50 values (290 versus 23 nm), the lower radioactivity observed for [125I]S1QEL1.1_PD2 must be primarily due to its lower binding affinity to the enzyme. Altogether, S1QEL1s turned out to exclusively bind to the ND1 subunit.
The binding position of S1QELs in the ND1 subunit
To localize the position labeled by photoreactive S1QEL1s in the ND1 subunit, the ND1 labeled by [125I]S1QEL1.1_PD1 or [125I]S1QEL1.1_PD2 was isolated from the SDS gel and digested with Lys-C or Asp-N, whose theoretical cleavage sites (Lys and Asp) are relatively few (Fig. 7C). The digests of the ND1 labeled by [125I]S1QEL1.1_PD1 was resolved on a 16.5% Schägger-type SDS gel to provide single radioactive bands at ∼7 and ∼18 kDa, respectively (Fig. 7B). On the basis of the theoretical cleavage sites, these digests must be assigned to the peptides Glu59–Lys126 (7.4 kDa) and Asp51–Phe198 (16.3 kDa), respectively. These results indicate that the position labeled by [125I]S1QEL1.1_PD1 is within the Glu59–Lys126 region (Fig. 7C), which includes part of the matrix-side first loop and TMHs 2–3.
The digestion of the ND1 labeled by [125I]S1QEL1.1_PD2 gave major digests at ∼16 and ∼14 kDa, respectively (Fig. 7B). The Lys-C and Asp-N digests appeared to be peptides Tyr127–Lys262 (15.2 kDa) and Asp199–Thr318 (13.8 kDa, containing a uncleavable site at Asp283), respectively. The results indicate that the position labeled by [125I]S1QEL1.1_PD2 is assigned to the Asp199–Lys262 region (Fig. 7C). This region includes the matrix-side third loop connecting TMHs 5–6, which is adjacent to the N-terminal domain of the 49-kDa subunit. Taken together, it was revealed that irrespective of substituted positions of a photolabile azido group, S1QEL1s bind to the ND1 subunit that is situated in the inner mitochondrial membrane. This finding strongly suggests that the whole S1QEL1 molecule is buried in this hydrophobic subunit.
Effects of different inhibitors on the binding of [125I]S1QEL1.1_PD1 to the ND1 subunit
We next examined the suppressive effects of different inhibitors on the binding of [125I]S1QEL1.1_PD1 to the ND1 subunit. Because radioactivity incorporated into ND1 by [125I]S1QEL1.1_PD2 was ∼10–20% of that by [125I]S1QEL1.1_PD1 (Fig. 7A), we used [125I]S1QEL1.1_PD1 to obtain definite results in the competition tests (Fig. 8). The concentration of all test compounds was set at 5.0 μm, 1000-fold of [125I]S1QEL1.1_PD1 (5.0 nm).
Figure 8.
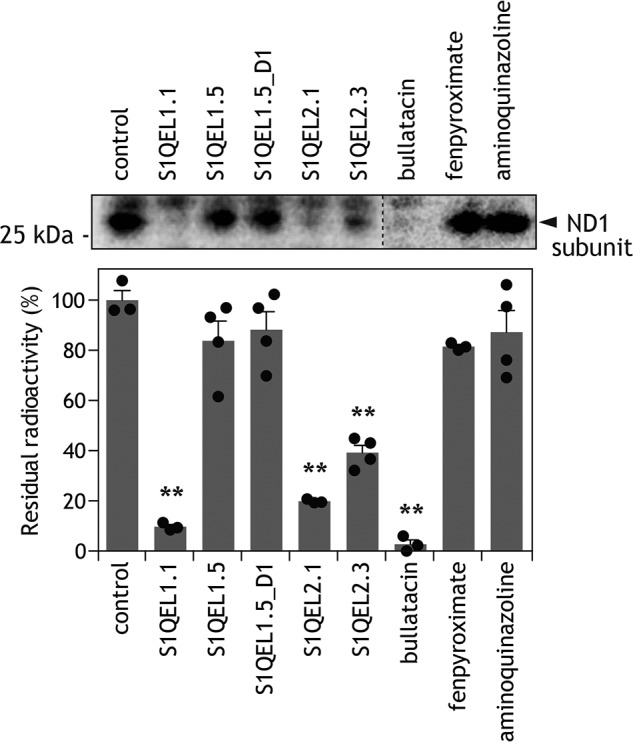
Effects of different inhibitors on the labeling of the ND1 subunit by [125I]S1QEL1.1_PD1. The photoaffinity labeling of the ND1 subunit by [125I]S1QEL1.1_PD1 (5.0 nm) was carried out in the presence of different inhibitors (5.0 μm each, 1000-fold of [125I]S1QEL1.1_PD1), as described under “Experimental procedures.” Proteins equivalent to ∼50 μg of SMPs were loaded into each well. Upper panel, gel image of SDS-PAGE analysis used for photoaffinity labeling; lower panel, the extent of suppression (values in graphs are mean ± S.E. (n = 3–4)). **, p < 0.001 compared with control (one-way ANOVA followed by Dunnet's test).
An important finding from the competition tests is that the suppressive effects of known quinone-site inhibitors were significantly different. Namely, although bullatacin completely suppressed the labeling by [125I]S1QEL1.1_PD1, fenpyroximate and amino-quinazoline failed to suppress the labeling. These differences are not explained by their different binding affinities to the enzyme because the three compounds are equally very potent inhibitors. The marked suppression by bullatacin is consistent with the fact that this inhibitor also binds to the ND1 subunit (37). Such significant differences in the suppressive efficiencies among quinone-site inhibitors are the first for photoaffinity labeling experiments conducted so far in our laboratory (28, 33, 34, 36, 37) and will be discussed in connection with the binding position of S1QELs in the ND1 subunit later.
Although we did not perform the photoaffinity labeling experiments using an analogue derived from the S1QEL2 family (piperazine-type), S1QEL2.1 and S1QEL2.3 moderately suppressed the labeling by [125I]S1QEL1.1_PD1 (Fig. 8). These results strongly suggest that the binding positions of S1QEL1 and S1QEL2 families, the structural frames of which are considerably different, are not identical but close to each other. S1QEL1.5 and S1QEL1.5_D1 hardly affected the labeling by [125I]S1QEL1.1_PD1 because of decomposition during incubation with SMPs in the labeling experiments.
Effects of BSA on the inhibitory action of S1QEL1.5
The results obtained in this study raise another question: although S1QEL1.5 was decomposed during incubation with SMPs for a few minutes (Figs. 2 and 3), why did it pass a panel of high-throughput screening with mitochondria isolated from rat skeletal muscle (24)? In these screening assays, incubation periods varied depending on the assays (15–30 min) but were longer than that adopted in the present study. Because the mechanism of decomposition of S1QEL1.5 (also S1QEL1.5_D1) in SMPs remains elusive, we have no definite answer to this question at present. Nevertheless, it should be pointed out that Brand et al. (24) used assay medium containing 0.3% (w/v) fatty acid-free bovine serum albumin (BSA) for assays of mitochondrial functions to prevent mitochondrial uncoupling (38). Because BSA tends to absorb hydrophobic chemicals, we cannot rule out the possibility that BSA protected S1QEL1.5 from decomposition by moderately or transiently absorbing it.
To examine this possibility, we determined the inhibition of forward electron transfer by S1QELs in reaction buffer containing 0.3% fatty acid-free BSA. The concentrations of S1QELs were set to around their EC50 values. As shown in Fig. 9A, the extents of inhibition by S1QELs were generally reduced in the presence of BSA, indicating that BSA absorbs S1QELs. In this experiment, the inhibition by S1QEL1.1, S1QEL2.1, and S1QEL2.3 was determined after incubation with SMPs for 4 min, and the inhibition by S1QEL1.5 and S1QEL1.5_D1 was evaluated at ∼1 min after addition of the compound to respiring SMPs (cf. trace c in Fig. 2). Furthermore, to examine whether BSA protects S1QEL1.5 and S1QEL1.5_D1 from decomposition, the inhibition by these compounds was determined after incubation with SMPs for 4 min in the presence of 0.3% BSA. As a result, BSA did not significantly enhance their inhibitory activities (Fig. 9B). These results indicate that BSA absorbs S1QELs, but does not necessarily protect them from decomposition. Thus, it remains unclear how S1QEL1.5 maintained its inhibitory effects in the previous high-throughput screening with isolated mitochondria (24).
Figure 9.
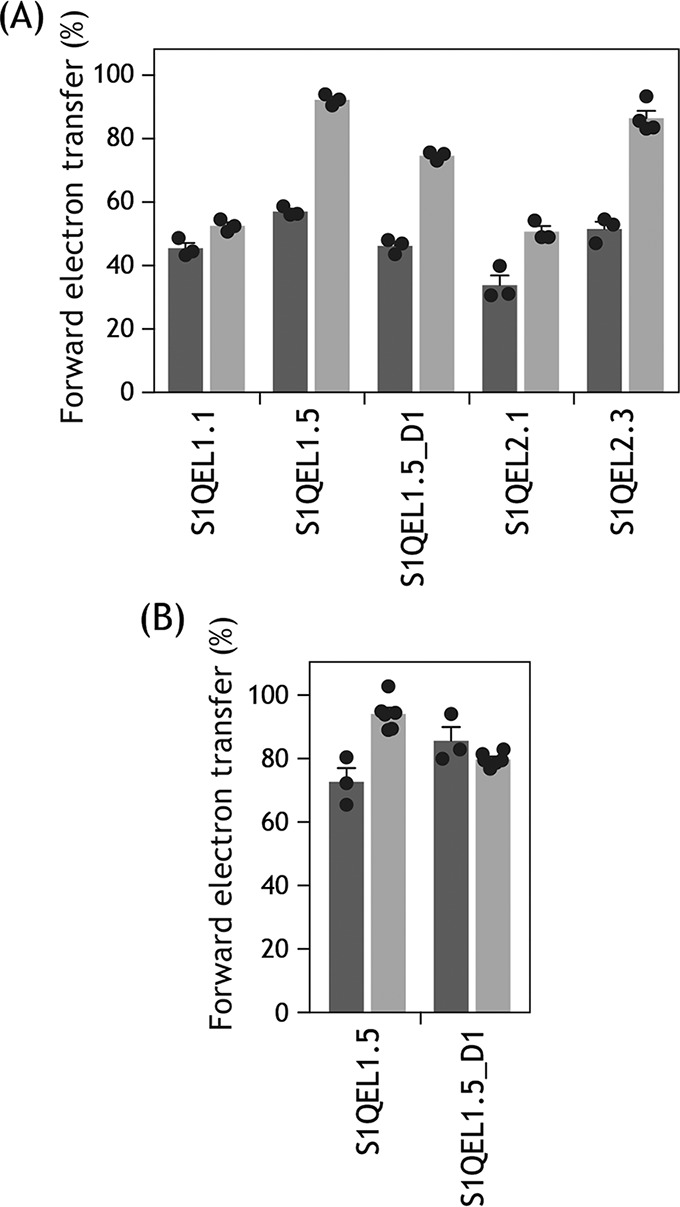
Effects of BSA on the inhibitory action of S1QELs. A, the inhibition of forward electron transfer by S1QELs was examined after incubation with SMPs for 4 min in the absence (black bars) or presence (gray bars) of 0.3% fatty acid-free BSA in assay buffer, except for S1QEL1.5 and S1QEL1.5_D1. The inhibition by S1QEL1.5 and S1QEL1.5_D1 was evaluated at ∼1 min after addition of the compound to respiring SMPs (cf. trace c in Fig. 2). The concentrations of S1QELs were: S1QEL1.1 (0.062 μm), S1QEL1.5 (1.2 μm), S1QEL1.5_D1 (2.0 μm), S1QEL2.1 (0.28 μm), and S1QEL2.3 (0.37 μm). B, the inhibition by S1QEL1.5 (1.2 μm) and S1QEL1.5_D1 (2.0 μm) was examined after incubation with SMPs for 4 min in the absence (black bars) or presence (gray bars) of 0.3% fatty acid-free BSA in assay buffer. Values in graphs are mean ± S.E. (n = 3–6).
Discussion
Brand et al. (24) discovered S1QELs as specific modulators of superoxide production at site IQ during reverse electron transfer in mitochondria isolated from rat skeletal muscle. S1QELs exhibited no inhibition against mitochondrial enzymes and transporters presiding over ATP production via oxidative phosphorylation at 10 μm or 20 × IC50. The present study was conducted to address the question of how each S1QEL selectively modulates reverse electron transfer (quinol oxidation) but not forward electron transfer (quinone reduction) in complex I at a definite concentration range, even though both take place inside a common reaction pocket. To this end, we examined their inhibitory action using bovine heart SMPs in detail, and found that regardless of family types (S1QEL1 or S1QEL2), S1QELs inherently have the potential to inhibit forward and reverse electron transfers in complex I. However, the features of their inhibitory actions were distinctly different from those of various known quinone-site inhibitors. We will discuss the potential relationship between the unique inhibitory action of S1QELs and their function as the site IQ-specific suppressors of superoxide production from two aspects: one is the direction-dependent inhibition of electron transfer, and the other is the indirect effect on the quinone-redox reactions (although the two factors are inseparably related to each other).
First, the inhibitory potencies of all S1QELs examined varied depending on the direction of electron transfer (forward or reverse). Their potencies observed with the reverse electron transfer, in terms of EC50 (mole inhibitor/mg of proteins), were considerably stronger than those observed with forward electron transfer (>5-fold, Table 1). Such distinct differences have not been reported for known quinone-site inhibitors. It is likely that a structural difference in the ND1 subunit between forward and reverse electron transfer is a primary cause of different binding affinities of each S1QEL between the two events. Accordingly, there may be a concentration range for each S1QEL, where they only modulate reverse electron transfer without distinct interference with forward electron transfer. Moreover, it is important to note that to evaluate the inhibitory effects of test compounds on forward electron transfer, Brand et al. (24) measured the NADH oxidase activity using glutamate/malate as substrates in the presence of a test compound solely at a one-point concentration (10 μm or 20 × IC50), and not a range of concentrations, with a high throughput Seahorse XF24 analyzer. Overall, it is conceivable that test compounds solely modulating reverse electron transfer at 10 μm or 20 × IC50 without distinct interference with forward electron transfer were passed as S1QELs in the screening. In contrast, test compounds that did not elicit such definite direction-dependent inhibition at 10 μm or 20 × IC50 might have been discarded in the screening.
Next, we consider a second point. Some S1QELs did not achieve complete (>95%) inhibition of the forward and/or reverse electron transfer, even though they elicited the inhibition at or lower than single digit micromolar levels. In particular, as much as ∼60% of the reverse electron transfer activity remained in the presence of excess S1QEL1.5 or S1QEL1.5_D1. Moreover, S1QELs were unable to efficiently suppress the alkynylation of 49 kDa-Asp160 by AL1 and the binding of [125I]AzQ to the 49-kDa subunit. These results strongly suggest that S1QELs do not occupy the binding pocket for quinone and ordinary quinone-site inhibitors; rather, they may indirectly modulate the quinone-redox reactions through inducing structural changes of the pocket by binding to the enzyme.
In support of this, the photoaffinity labeling experiments revealed that [125I]S1QEL1.1_PD1 and [125I]S1QEL1.1_PD2 label the Glu59–Lys126 (including TMHs 2–3) and Asp199–Lys262 regions (including TMHs 6–7), respectively, in the ND1 subunit (Figs. 7 and 10) and that some quinone-site inhibitors (fenpyroximate and quinazoline) failed to suppress the binding of [125I]S1QEL1.1_PD1 to ND1 (Fig. 8). Although we were unable to identify their binding positions at a single amino acid level, comprehensive interpretation of these results as well as the above findings strongly suggest that the binding position of thiazole-type S1QELs is located “under” the matrix-side third loop connecting TMHs 5–6 of the ND1 subunit. This loop is adjacent to the N-terminal domain of the 49-kDa subunit, to which fenpyroximate and quinazoline bind (33, 34, 36). Referring to the structural models (25–27), this predicted position is not considered to make up the quinone-access channel cavity (Fig. 10). Bullatacin also binds to the ND1 subunit (37) and, in fact, significantly suppressed the labeling of this subunit by [125I]S1QEL1.1_PD1 (Fig. 8). However, because bullatacin completely suppresses both the alkynylation of 49 kDa-Asp160 by AL1 (31, 32) and the labeling of the 49-kDa subunit by [125I]AzQ (33, 34) (also see Figs. 5 and 6), it is obvious that the binding positions of S1QELs and bullatacin in ND1 somewhat differ from each other. Taken together, it is highly likely that S1QELs indirectly modulate the quinone-redox reactions through inducing structural changes of the pocket by binding to the bottom of ND1. This indirect effect may be a prerequisite for the direction-dependent inhibition mentioned above; for example, susceptibility of the quinone-redox reactions against the structural changes may considerably differ between quinol oxidation and quinone reduction.
Figure 10.

The regions labeled by [125I]S1QEL1.1_PD1 and [125I]S1QEL1.1_PD2. [125I]S1QEL1.1_PD1 and [125I]S1QEL1.1_PD2 labeled the Glu59–Lys126 (yellow spheres, including TMHs 2–3) and Asp199–Lys262 regions (orange spheres, including TMHs 6–7), respectively, in the ND1 subunit (orange). The labeled regions are shown in a structural model of ovine complex I (Protein Data Bank entry 5LNK). Here, we used the ovine enzyme (26) because the matrix-side third loop (Thr201–Ala217) connecting the TMHs 5–6 of the ND1 subunit is disordered in the deactive state of the bovine enzyme (25). For reference, the third loop (Thr201–Ala217) of ND1 is presented in red spheres (right figure). The 49-kDa and PSST subunits are in pink and blue, respectively. The quinone/inhibitor-access channel was generated using MOLE (https://mole.upol.cz)3 (46) and is shown in black. The hydrophilic domain above the dotted line shown in a was deleted in b for clarity.
We previously proposed that in contrast to the predicted quinone-access channel models in complex I (25–27, 39, 40), the binding pocket for quinone and quinone-site inhibitors is considerably “open” to allow a wide range of ligands access to deep inside (28). The possible entrance of this open space may be the area where the regions labeled by different types of photoreactive inhibitors are in contact or close to one other: the labeled regions are the N terminus Asp41–Glu67 of 49 kDa, Lys33–Tyr67 of PSST, the loop connecting TMHs 5–6 of ND1, and the C terminus Thr227–Lys283 of 39 kDa. Based on the structural models (25–27), the binding position of thiazole-type S1QELs proposed above is located under this possible entrance (Fig. 10). This binding position is consistent with the finding that S1QELs hardly affected the reactions that take place above the loop connecting TMH5–6 of the ND1 subunit (i.e. alkynylation of 49 kDa-Asp160 by AL1 and labeling of the 49-kDa subunit by [125I]AzQ). It is also reasonable that fenpyroximate and quinazoline, both of which bind to the region above this loop of ND1 (28, 33, 34, 36), failed to suppress the labeling of ND1 by [125I]S1QEL1.1_PD1 (Fig. 8). Thus, the present study is the first to demonstrate the existence of inhibitors that modulate terminal electron transfer in complex I without occupying the binding pocket for quinone and the so-called quinone-site inhibitors.
In conclusion, the present study was aimed at defining how S1QELs selectively modulate one of the two opposite quinone-redox reactions that take place inside a common binding pocket in complex I. Our results revealed that in contrast to various known quinone-site inhibitors, S1QELs do not occupy the binding pocket for the quinone/inhibitor; rather, they may indirectly modulate the quinone-redox reactions through inducing structural changes of the pocket by binding to the bottom of the ND1 subunit. This indirect effect may be a prerequisite for the direction-dependent modulation of electron transfer, which, in turn, is responsible for the suppression of superoxide production during reverse electron transfer without distinct interference with forward electron transfer.
Experimental procedures
Materials and methods
Ubiquinone-1 (UQ1) was a kind gift from Eisai (Tokyo, Japan). Bullatacin and fenpyroximate were kindly provided by J. L. McLaughlin (Purdue University, West Lafayette, IN) and Nihon Nohyaku Co., Ltd. (Tokyo, Japan). Acetogenin-type ligand (AL1) was the same sample that was used previously (31). A photoreactive [125I]AzQ was synthesized by the previously reported procedure (33). Protein standards (Precision Plus Protein Standards, Hercules, CA) for SDS-PAGE were purchased from Bio-Rad. [125I]NaI was purchased from PerkinElmer Life Sciences. Other reagents were all of analytical grade.
Preparation of bovine heart SMPs and measurement of complex I activity
Mitochondria were isolated from bovine heart. SMPs were prepared by the method of Matsuno-Yagi and Hatefi (41) and stored in buffer containing 250 mm sucrose and 10 mm Tris-HCl (pH 7.4) at −80 °C until used. The NADH oxidase activity (forward electron transfer) in SMPs was measured spectrometrically with a Shimadzu UV-3000 instrument (340 nm, ϵ = 6.2 mm−1 cm−1) at 30 °C (28). The reaction medium (2.5 ml) contained 0.25 m sucrose, 1.0 mm MgCl2, and 50 mm phosphate buffer (pH 7.4). The final mitochondrial protein concentration was 30 μg of protein/ml. The reaction was initiated by adding 50 μm NADH after the equilibration of SMPs with an inhibitor for 4 min. The EC50 values of inhibitors were calculated by Prism (version 6, GraphPad, La Jolla, CA) using sigmoid dose-response curve fitting.
Measurement of reversed electron transfer in complex I
Reverse electron transfer (the quinol-NAD+ oxidoreductase activity) was driven by the oxidation of succinate and the hydrolysis of ATP (42, 43). The reaction was measured spectrophotometrically by following the reduction of NAD+ with a Shimadzu UV-3000 (340 nm, ϵ = 6.2 mm−1 cm−1) at 30 °C. The reaction medium (2.5 ml) contained 0.25 m sucrose, 7.0 mm sodium succinate, 6.0 mm MgCl2, 1.0 mm KCN, 1.0 mm NAD+, and 50 mm Tris-HCl (pH 7.5), and the final protein concentration of SMPs was 100 μg of protein/ml. The reaction was initiated by the addition of 1.0 mm ATP after the equilibration of SMPs with inhibitor for 4 min. The activity was fully sensitive to SF6847 (protonophoric uncoupler) or oligomycin (ATP synthase inhibitor).
Alkynylation of complex I in SMPs by AL-1 (LDT chemistry)
Bovine SMPs (2.0 mg of protein/ml, 100–200 μl), suspended in a buffer containing 250 mm sucrose, 1.0 mm MgCl2, and 50 mm potassium Pi (pH 7.4), were incubated with AL1 (0.10 μm) in the absence or presence of a competitor for 24 h at 35 °C (31). SMPs were then collected by ultracentrifugation (20,000 × g, 20 min, 4 °C) and denatured in 1.0% (w/v) SDS (25–50 μl). For visualization of proteins alkynylated by AL1, they were conjugated with a fluorescent TAMRA-N3 tag via Cu+-catalyzed click chemistry using the Click-iT reaction buffer kit (Life Technologies) according to the manufacturer's protocols. Proteins were recovered by precipitation with methanol/chloroform, and subjected to Laemmli-type 12.5% SDS-PAGE, followed by the fluorescent gel imaging and Coomassie Brilliant Blue stain.
Photoaffinity labeling of complex I in SMPs by [125I]AzQ
Bovine SMPs (2.0 mg/ml, 100 μl), which were preincubated with S1QELs or bullatacin at room temperature for 10 min, were incubated with a photoreactive quinazoline-type inhibitor [125I]AzQ (6.0 nm) in buffer containing 250 mm sucrose, 50 mm potassium Pi (pH 7.4), and 5.0 mm MgCl2 at room temperature for 10 min, then 50 μm NADH was added for a further 5 min. The mixture was then irradiated with a long wavelength UV-lamp (Black-lay model B-100A, UVP, Upland, CA) on ice for 10 min at a distance of 10 cm from the light source (33). The reaction was quenched by the addition of 4× Laemmli sample buffer (34 μl). Samples were separated on a 12.5% Laemmli gel, which was stained with Coomassie Brilliant Blue, dried, exposed to an imaging plate (BAS-MS2040, Fuji Film, Tokyo, Japan), and visualized with the Bio-imaging analyzer FLA-5100 (Fuji Film) or Typhoon-FLA 9500 (GE Healthcare). The incorporated radioactivity of each band was quantified using Multi Gauge (Fuji Film) or Image Quant (GE Healthcare).
Photoaffinity labeling of complex I in SMPs by [125I]S1QEL1.1_PD1 or [125I]S1QEL1.1_PD2
Bovine SMPs (4.0 mg/ml, 100–500 μl) were labeled by [125I]S1QEL1.1_PD1 or [125I]S1QEL1.1_PD2 (6–15 nm) according to the same procedures described for [125I]AzQ. When the competition test was conducted, the mixture was incubated with other complex I inhibitors for 10 min at room temperature prior to the treatment with the [125I]S1QEL1s.
Electrophoresis
SMPs labeled by [125I]S1QELs were solubilized in sample buffer containing 50 mm BisTris-HCl (pH 7.2), 50 mm NaCl, 10% (w/v) glycerol, 1.0% (w/v) dodecyl maltoside, and 0.001% (w/v) Ponceau S on ice for 1 h, and the samples were separated by BN-PAGE (28, 36) using a 4–16% precast gel system (Life Technologies) according to the manufacturer's protocol. The isolated complex I was further solved on a Laemmli-type 12.5% SDS gel (44) or Schägger-type 10% SDS gel containing 6.0 m urea (45).
Doubled SDS-PAGE was conducted as described previously (28, 35, 36). In brief, the labeled complex I was separated on a first dimensional 10% Schägger-type gel (10% T, 3% C, containing 6.0 m urea). The gel slice was then acidified with 100 mm Tris-HCl (pH 2.0) for 30 min, followed by second dimensional separation on a 16% Schägger-type gel (16% T, 3% C). Typically, complex I equivalent to 200 μg of SMPs was separated on a mini-size gel (80 × 90 mm, 1 mm). The resolved proteins were visualized by mass spectrometry (MS)-compatible silver staining (Wako Silver stain MS kit, Wako Pure Chemicals, Osaka, Japan), followed by autoradiography.
Proteomic analysis
For exhaustive digestion of the ND1 subunit, the subunit labeled by [125I]S1QELs was recovered from the gel by electroelution or direct diffusion in 10 mm Tris-HCl buffer (pH 8.0) containing 0.025% (w/v) SDS. The partially purified ND1 subunit was digested with lysylendopeptidase (Wako Pure Chemicals, Osaka, Japan) or endoprotease Asp-N (Roche Applied Science) in 50 mm Tris-HCl buffer (pH 8.5) containing 0.1% SDS or 50 mm NaPi buffer (pH 8.0) containing 0.01% SDS, respectively. The digests were resolved on a Schägger-type SDS gel (45), followed by autoradiography. Proteins were identified by MS according to previous procedures (28) using Bruker Autoflex III Smartbeam (MALDI-TOF MS, Bruker Daltonics, Billerica, MA) or AXIMA-Performance (MALDI-TOFMS, Shimadzu).
Author contributions
A. B., A. T., H. K., M. M., and H. M. data curation; A. B., A. T., H. K., M. M., and H. M. formal analysis; A. B., A. T., H. K., M. M., and H. M. investigation; A. B., A. T., H. K., M. M., and H. M. methodology; A. B., A. T., M. M., and H. M. writing-original draft; M. M. and H. M. funding acquisition; H. M. supervision; H. M. project administration.
Supplementary Material
Acknowledgment
The experiments involving radioisotope techniques were performed at the Radioisotope Research Center, Kyoto University.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grants JP26292060 and JP18H02147 (to H. M.) and JP18K05458 (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1, Schemes S1–S5, and Figs. S1–S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- ROS
- reactive oxygen species
- 49 kDa-Asp160
- Asp160 in the 49-kDa subunit
- AL
- acetogenin ligand
- Asp-N
- endoprotease Asp-N
- AzQ
- azido-quinazoline
- BisTris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- BN-PAGE
- blue native polyacrylamide gel electrophoresis
- IC50
- the molar concentration needed to suppress superoxide production from site IQ by 50%
- LDT
- ligand-directed tosyl
- Lys-C
- lysylendopeptidase
- S1QEL
- suppressor of site IQ electron leak
- SMP
- submitochondrial particle
- TAMRA
- 6-carboxy-N,N,N′,N′-tetramethylrhodamine
- TMH
- transmembrane helix
- ANOVA
- analysis of variance.
References
- 1. Finkel T., and Holbrook N. J. (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247 10.1038/35041687 [DOI] [PubMed] [Google Scholar]
- 2. Lin M. T., and Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- 3. Van Gaal L. F., Mertens I. L., and De Block C. E. (2006) Mechanisms linking obesity with cardiovascular disease. Nature 444, 875–880 10.1038/nature05487 [DOI] [PubMed] [Google Scholar]
- 4. Muller F. L., Lustgarten M. S., Jang Y., Richardson A., and Van Remmen H. (2007) Trends in oxidative aging theories. Free Radic. Biol. Med. 43, 477–503 10.1016/j.freeradbiomed.2007.03.034 [DOI] [PubMed] [Google Scholar]
- 5. Nunnari J., and Suomalainen A. (2012) Mitochondria: in sickness and in health. Cell 148, 1145–1159 10.1016/j.cell.2012.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le Bras M., Clément M.-V., Pervaiz S., and Brenner C. (2005) Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 20, 205–219 [DOI] [PubMed] [Google Scholar]
- 7. Starkov A. A. (2008) The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N.Y. Acad. Sci. 1147, 37–52 10.1196/annals.1427.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hamanaka R. B., and Chandel N. S. (2010) Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35, 505–513 10.1016/j.tibs.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sena L. A., and Chandel N. S. (2012) Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167 10.1016/j.molcel.2012.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wong H.-S., Dighe P. A., Mezera V., Monternier P.-A., and Brand M. D. (2017) Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809 10.1074/jbc.R117.789271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goncalves R. L., Quinlan C. L., Perevoschchikova I. V., Hey-Mogensen M., and Brand M. D. (2015) Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 290, 209–227 10.1074/jbc.M114.619072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Treberg J. R., Quinlan C. L., and Brand M. D. (2011) Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J. Biol. Chem. 286, 27103–27110 10.1074/jbc.M111.252502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pryde K. R., and Hirst J. (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 286, 18056–18065 10.1074/jbc.M110.186841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Robb E. L., Hall A. R., Prime T. A., Eaton S., Szibor M., Viscomi C., James A. M., and Murphy M. P. (2018) Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 293, 9869–9879 10.1074/jbc.RA118.003647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Langston J. W., Ballard P., Tetrud J. W., and Irwin I. (1983) (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 10.1126/science.6823561 [DOI] [PubMed] [Google Scholar]
- 16. Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., and Greenamyre J. T. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3, 1301–1306 10.1038/81834 [DOI] [PubMed] [Google Scholar]
- 17. Lambert A. J., Boysen H. M., Buckingham J. A., Yang T., Podlutsky A., Austad S. N., Kunz T. H., Buffenstein R., and Brand M. D. (2007) Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan invertebrate homeotherms. Aging Cell 6, 607–618 10.1111/j.1474-9726.2007.00312.x [DOI] [PubMed] [Google Scholar]
- 18. Lambert A. J., Buckingham J. A., Boysen H. M., and Brand M. D. (2010) Low complex I content explains the low hydrogen peroxide production rate of heart mitochondria from the long-lived pigeon, Columba livia. Aging Cell 9, 78–91 10.1111/j.1474-9726.2009.00538.x [DOI] [PubMed] [Google Scholar]
- 19. Smith R. A., Porteous C. M., Coulter C. V., and Murphy M. P. (1999) Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 263, 709–716 10.1046/j.1432-1327.1999.00543.x [DOI] [PubMed] [Google Scholar]
- 20. Brash D. E., and Havre P. A. (2002) New careers for antioxidants. Proc. Natl. Acad. Sci. U.S.A. 99, 13969–13971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krumova K., Greene L. E., and Cosa G. (2013) Fluorogenic a-tocopherol analogue for monitoring the antioxidant status within the inner mitochondrial membrane of live cells. J. Am. Chem. Soc. 135, 17135–17143 10.1021/ja408227f [DOI] [PubMed] [Google Scholar]
- 22. Nazarewicz R. R., Dikalova A., Bikineyeva A., Ivanov S., Kirilyuk I. A., Grigor'ev I. A., and Dikalov S. I. (2013) Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid. Redox Signal. 19, 344–349 10.1089/ars.2013.5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Orr A., Ashok D., Sarantos M. R., Shi T., Hughes R. E., and Brand M. D. (2013) Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 65, 1047–1059 10.1016/j.freeradbiomed.2013.08.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brand M. D., Goncalves R. L., Orr A. L., Vargas L., Gerencser A. A., Borch Jensen M., Wang Y. T., Melov S., Turk C. N., Matzen J. T., Dardov V. J., Petrassi H. M., Meeusen S. L., Perevoshchikova I. V., Jasper H., Brookes P. S., and Ainscow E. K. (2016) Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 24, 582–592 10.1016/j.cmet.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu J., Vinothkumar K. R., and Hirst J. (2016) Structure of mammalian respiratory complex I. Nature 536, 354–358 10.1038/nature19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fiedorczuk K., Letts J. A., Degliesposti G., Kaszuba K., Skehel M., and Sazanov L. A. (2016) Atomic structure of the entire mammalian mitochondrial complex I. Nature 538, 406–410 10.1038/nature19794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agip A.-N. A., Blaza J. N., Gridges H. R., Viscomi C., Rawson S., Muench S. P., and Hirst J. (2018) Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25, 548–556 10.1038/s41594-018-0073-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Uno S., Kimura H., Murai M., and Miyoshi H. (2019) Exploring the quinone/inhibitor binding pocket in mitochondrial respiratory complex I by chemical biology approaches. J. Biol. Chem. 294, 679–696 10.1074/jbc.RA118.006056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsukiji S., Miyagawa M., Takaoka Y., Tamura T., and Hamachi I. (2009) Ligand-directed tosyl chemistry for protein labeling in vivo. Nat. Chem. Biol. 5, 341–343 10.1038/nchembio.157 [DOI] [PubMed] [Google Scholar]
- 30. Takaoka Y., Ojida A., and Hamachi I. (2013) Protein organic chemistry and applications for labeling and engineering in live-cell systems. Angew. Chem. Int. Ed. 52, 4088–4106 10.1002/anie.201207089 [DOI] [PubMed] [Google Scholar]
- 31. Masuya T., Murai M., Ifuku K., Morisaka H., and Miyoshi H. (2014) Site-specific chemical labeling of mitochondrial respiratory complex I through ligand-directed tosylate chemistry. Biochemistry 53, 2307–2317 10.1021/bi500205x [DOI] [PubMed] [Google Scholar]
- 32. Masuya T., Murai M., Morisaka H., and Miyoshi H. (2014) Pinpoint chemical modification of Asp160 in the 49 kDa subunit of bovine mitochondrial complex I via a combination of ligand-directed tosyl chemistry and click chemistry. Biochemistry 53, 7816–7823 10.1021/bi501342w [DOI] [PubMed] [Google Scholar]
- 33. Murai M., Sekiguchi K., Nishioka T., and Miyoshi H. (2009) Characterization of the inhibitor binding site in mitochondrial NADH-ubiquinone oxidoreductase by photoaffinity labeling using a quinazoline-type inhibitor. Biochemistry 48, 688–698 10.1021/bi8019977 [DOI] [PubMed] [Google Scholar]
- 34. Murai M., Mashimo Y., Hirst J., and Miyoshi H. (2011) Exploring interactions between the 49 kDa and ND1 subunits in mitochondrial NADH-ubiquinone oxidoreductase (complex I) by photoaffinity labeling. Biochemistry 50, 6901–6908 10.1021/bi200883c [DOI] [PubMed] [Google Scholar]
- 35. Rais I., Karas M., and Schägger H. (2004) Two-dimensional electrophoresis for the isolation of integral membrane proteins and mass spectrometric identification. Proteomics 4, 2567–2571 10.1002/pmic.200400829 [DOI] [PubMed] [Google Scholar]
- 36. Shiraishi Y., Murai M., Sakiyama N., Ifuku K., and Miyoshi H. (2012) Fenpyroximate binds to the interface between PSST and 49 kDa subunits in mitochondrial NADH-ubiquinone oxidoreductase. Biochemistry 51, 1953–1963 10.1021/bi300047h [DOI] [PubMed] [Google Scholar]
- 37. Nakanishi S., Abe M., Yamamoto S., Murai M., and Miyoshi H. (2011) Bis-THF motif of acetogenin binds to the third matrix-side loop of ND1 subunit in mitochondrial NADH-ubiquinone oxidoreductase. Biochim. Biophys. Acta 1807, 1170–1176 10.1016/j.bbabio.2011.05.012 [DOI] [PubMed] [Google Scholar]
- 38. Rogers G. W., Brand M. D., Petrosyan S., Ashok D., Elorza A. A., Ferrick D. A., and Murphy A. N. (2011) High throughput microplate respiratory measurements suing minimal quantities of isolated mitochondria. PLoS ONE 6, e21746 10.1371/journal.pone.0021746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baradaran R., Berrisford J. M., Minhas G. S., and Sazanov L. A. (2013) Crystal structure of the entire respiratory complex I. Nature 494, 443–448 10.1038/nature11871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zickermann V., Wirth C., Nasiri H., Siegmund K., Schwalbe H., Hunte C., and Brandt U. (2015) Mechanistic insight from the crystal structure of mitochondrial complex I. Science 347, 44–49 10.1126/science.1259859 [DOI] [PubMed] [Google Scholar]
- 41. Matsuno-Yagi A., and Hatefi Y. (1985) Studies on the mechanism of oxidative phosphorylation. J. Biol. Chem. 260, 11424–11427 [PubMed] [Google Scholar]
- 42. Ernster L., and Lee C.-P. (1967) Energy-linked reduction of NAD+ by succinate. Methods Enzymol. 10, 729–738 10.1016/0076-6879(67)10120-1 [DOI] [Google Scholar]
- 43. Murai M., Ichimaru N., Abe M., Nishioka T., and Miyoshi H. (2006) Mode of inhibitory action of Δlac-acetogenins, a new class of inhibitors of bovine heart mitochondrial complex I. Biochemistry 45, 9778–9787 10.1021/bi060713f [DOI] [PubMed] [Google Scholar]
- 44. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 45. Schägger H. (2006) Tricine-SDS-PAGE. Nat. Protoc. 1, 16–22 10.1038/nprot.2006.4 [DOI] [PubMed] [Google Scholar]
- 46. Pravda L., Sehnal D., Toušek D., Navrátilová V., Bazgier V., Berka K., Svobodová Vareková R., Koca J., and Otyepka M. (2018) MOLEonline: a web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 46, W368–W373 10.1093/nar/gky309 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.