

Abstract
The kinesin-3 family member KIF1A plays a critical role in site-specific neuronal cargo delivery during axonal transport. KIF1A cargo is mislocalized in many neurodegenerative diseases, indicating that KIF1A's highly efficient, superprocessive motility along axonal microtubules needs to be tightly regulated. One potential regulatory mechanism may be through posttranslational modifications (PTMs) of axonal microtubules. These PTMs often occur on the C-terminal tails of the microtubule tracks, act as molecular “traffic signals” helping to direct kinesin motor cargo delivery, and include C-terminal tail polyglutamylation important for KIF1A cargo transport. KIF1A initially interacts with microtubule C-terminal tails through its K-loop, a positively charged surface loop of the KIF1A motor domain. However, the role of the K-loop in KIF1A motility and response to perturbations in C-terminal tail polyglutamylation is underexplored. Using single-molecule imaging, we present evidence that KIF1A pauses on different microtubule lattice structures, linking multiple processive segments together and contributing to KIF1A's characteristic superprocessive run length. Furthermore, modifications of the KIF1A K-loop or tubulin C-terminal tail polyglutamylation reduced KIF1A pausing and overall run length. These results suggest a new mechanism to regulate KIF1A motility via pauses mediated by K-loop/polyglutamylated C-terminal tail interactions, providing further insight into KIF1A's role in axonal transport.
Keywords: molecular motor, single-molecule biophysics, kinesin, post-translational modification (PTM), microtubule, cytoskeleton, K-loop, KIF1A, neuron, polyglutamylation
Introduction
Axonal transport is a critical process for neuronal viability and function involving the highly choreographed, long-distance trafficking of cargo. This process is facilitated in part by members of the kinesin superfamily of motors that utilize the mechanochemical coupling of ATP hydrolysis (1) to carry motor-specific cargo in the anterograde direction along axonal microtubules.
The neuron-specific kinesin-3 family member KIF1A (2) is a key mediator of axonal transport, delivering neuronal cargo, such as dense core vesicles (3) and synaptic vesicle proteins (2), in a spatiotemporally regulated manner along axonal microtubules (4, 5). Upon cargo-mediated dimerization and release of steric autoinhibition, KIF1A motors are reported to exhibit “superprocessive” motility behavior, traveling comparatively long distances on microtubules when compared with conventional kinesin (6, 7). The discovery of this superprocessive behavior supports KIF1A as an important player in long-distance axonal transport, providing further insight into KIF1A's role in many neuronal processes such as synaptogenesis (8) and neurogenesis (9). The importance of tightly regulated KIF1A cargo delivery is highlighted in neurodegenerative diseases where irregular KIF1A cargo accumulation is observed (10–12). However, our understanding of this disease-state presentation is limited by the gap in knowledge of the mechanisms that regulate KIF1A motility.
Beyond serving as tracks for kinesin motors, microtubules can direct subcellular trafficking by providing directional cues for motor specificity on select microtubule populations. These “traffic signs” are frequently introduced via microtubule posttranslational modifications (PTMs),2 often on the C-terminal tails of microtubules (13, 14). Although there are many recent discoveries of how microtubule PTMs regulate different kinesin families (15–17), how this concept can be extended to kinesin-3 motors such as KIF1A is relatively unexplored. One potential PTM regulating KIF1A trafficking is the heavy polyglutamylation of both α- and β-tubulin subunits in neurons (18) via the tubulin tyrosine ligase-like family of enzymes (19, 20). KIF1A's dependence on optimal levels of polyglutamylation has been observed in the ROSA22 mouse model, characterized by a loss of neuronal α-tubulin polyglutamylation leading to altered KIF1A cargo trafficking and reduced KIF1A affinity for microtubules (21). These physiological repercussions of altered KIF1A function highlight the importance of further studying interactions between KIF1A and the polyglutamylation of C-terminal tails of tubulin at the molecular level.
Polyglutamylation may impact KIF1A motility via a lysine-rich surface loop (loop-12) that is characteristic of the kinesin-3 motor family (22, 23). Previous work has demonstrated that this “K-loop” is a critical component for optimal KIF1A function. From a catalytic perspective, it is thought that the K-loop is necessary for KIF1A's structural interaction with the microtubule during the ATP-hydrolysis cycle (24). Furthermore, it is known that the K-loop interacts electrostatically with the glutamic acid–rich C-terminal tail of tubulin (25, 26), tethering the motor to the microtubule track (22). This K-loop/C-terminal tail relationship, combined with the physiological detriment of reduced microtubule polyglutamylation, illustrates that specific interactions between the KIF1A K-loop and C-terminal tails of tubulin are essential for optimal KIF1A function. Therefore, we hypothesized that C-terminal tail polyglutamylation regulates KIF1A motility and behavior on microtubules mediated by interactions with the KIF1A K-loop structure. Using single-molecule total internal reflection fluorescence microscopy, we first tested our hypothesis by quantifying the motility and behavioral response of KIF1A on various microtubule lattices and then specifically perturbed the K-loop/C-terminal tail interaction. Our findings demonstrate that KIF1A pauses on the microtubule lattice and suggest that this behavior is caused by a polyglutamylation-sensitive K-loop/C-terminal tail interaction.
Results
KIF1A exhibits pausing behavior and superprocessive motility on Taxol-stabilized and GMPCPP microtubules assembled from brain tubulin
Initial single-molecule total internal reflection fluorescence (TIRF) microscopy observations revealed that dimeric KIF1A motors possess the novel ability to pause during a run on Taxol-stabilized microtubules (Fig. 1A and Movie S1). While pausing, the dimeric motor appears to be spatially constrained as it does not exhibit the highly diffusive behavior of the KIF1A monomer (26). Although other kinesin motors have been shown to exhibit transient pauses (27, 28), the high pause frequency (Table 1) and substantial number of pauses per distance (Fig. 2F) of KIF1A have not been previously characterized, leading us to investigate this behavior further.
Figure 1.

KIF1A pauses during runs along the microtubule. A, representative kymograph with corresponding nomenclature used to describe novel KIF1A motility behavior. B and C, representative kymographs demonstrating that at high motor concentrations, pausing becomes obscured by surrounding motor motility (B), whereas at low motor concentrations, extensive pausing behavior (white arrows) is revealed at stochastic positions on the microtubule (C).
Table 1.
Summary of KIF1A motility and behavior on microtubules across all conditions
Pause durations were fit to a single-exponential decay, and the time constant of the fit is reported in seconds. All other metrics are reported as mean ± S.D. *, p < 0.001 relative to KIF1A on Taxol-stabilized microtubules.
| Overall run length | Overall speed | Continuous run length | Continuous speed | Pause frequency | Pauses per distance | Pause duration | Landing rate | |
|---|---|---|---|---|---|---|---|---|
| μm | μm/s | μm | μm/s | no. pauses/overall run | no. pauses/μm | s | events/μm/min | |
| Taxol | 6.24 ± 2.09 | 1.35 ± 0.41 | 2.95 ± 1.07 | 2.01 ± 0.53 | 0.95 | 0.14 | 1.23 | 7.93 ± 1.25 |
| GMPCPP | 6.01 ± 2.17 | 1.27 ± 0.28 | 2.81 ± 0.91 | 2.06 ± 0.54 | 1.02 | 0.16 | 0.66 | 4.86 ± 0.88* |
| Subtilisin | 3.31 ± 1.34* | 1.58 ± 0.38 | 2.91 ± 1.16 | 1.94 ± 0.50 | 0.18* | 0.02* | 0.68 | 2.80 ± 0.42* |
| 80 mm PIPES | 5.98 ± 2.14 | 1.34 ± 0.30 | 3.14 ± 0.98 | 1.99 ± 0.47 | 0.90 | 0.14 | 0.78 | 3.28 ± 0.61* |
| Tri-Ala | 2.41 ± 1.42* | 1.66 ± 0.45 | 2.00 ± 1.11* | 1.72 ± 0.45 | 0.23* | 0.05* | 0.47 | 0.64 ± 0.14* |
| HeLa tubulin | 4.12 ± 1.48* | 1.70 ± 0.41 | 2.98 ± 0.96 | 1.87 ± 0.40 | 0.40* | 0.07* | 0.59 | 0.49 ± 0.13* |
Figure 2.

KIF1A pausing behavior occurs on Taxol-stabilized and GMPCPP microtubules. A and B, representative kymographs of KIF1A motility on Taxol-stabilized (A) or GMPCPP (B) microtubules (MTs). C, quantification demonstrates no significant differences in KIF1A overall run length (RL) (6.24 ± 2.09 (n = 165) and 6.01 ± 2.17 μm (n = 171), respectively) and continuous run length (2.95 ± 1.07 (n = 282), and 2.81 ± 0.91 μm (n = 305), respectively). Run length values are reported as mean ± S.D. and were calculated as reported previously (71). KIF1A pause duration is 1.23 s (CI, 1.00–1.42 s) on Taxol-stabilized microtubules (n = 147) (D) and 0.66 s (CI, 0.59–0.73 s) on GMPCPP microtubules (n = 152) (E). F, KIF1A exhibits a similar pause per distance on Taxol-stabilized (0.14 pause/μm (n = 165)) and GMPCPP (0.16 pause/μm (n = 171)) microtubules. Pause durations were fit to a single-exponential decay and are represented as cumulative frequency distributions; the time constant of fit is reported in seconds with a 95% confidence interval. All other metrics are reported as mean ± S.D. Kinesin-1 was used as an experimental control across all conditions (Figs. S2–S5). Each condition is representative of at least four independent experiments.
The discovery of this novel pausing behavior led us to redefine our nomenclature of KIF1A motility by dissecting KIF1A motility into three segments (Fig. 1A). First, we define the continuous run length as segments where, within a single event, KIF1A is moving at a constant velocity. Next, continuous run lengths are connected by pauses that occur at the beginning of continuous runs, in between continuous runs, or after continuous runs. To characterize KIF1A pausing, we have quantified both the pause frequency (number of pauses/overall run) and pauses per distance (number of pauses/μm of microtubule). Although representative KIF1A kymographs include long pauses (Figs. 1C and 2, A and B), it is important to note that the majority of pausing events are less than 1.5 s and occur at stochastic locations on the microtubule (Fig. 1C and Table 1). Lastly, we define the overall run length as the sum of continuous run lengths during a single motility event on the microtubule. KIF1A pausing behavior (Fig. 1, B and C) is best revealed at motor concentrations lower than those utilized in previous work (22).
To confirm that this behavior was not dependent on the facilitated dimerization of our leucine zipper construct (KIF1A-LZ-3xmCitrine), we repeated our single-molecule experiments with a non-leucine zipper construct (KIF1A-GFP). We confirmed that in the absence of the leucine zipper, KIF1A exhibits extensive pausing behavior and superprocessive motility (Fig. S1). Additionally, we observed an increase in purely diffusive events with this construct (Fig. S1A), likely resulting from KIF1A monomers (6, 7). Our results demonstrate that the KIF1A-LZ-3xmCitrine construct is an appropriate model for dimeric KIF1A behavior. As our current investigations are focused on the processive, dimeric KIF1A motor, we performed all other experiments with the LZ construct.
We next investigated KIF1A's pausing on GMPCPP microtubules, which contain tubulin subunits in the GTP state rather than the GDP state and are known to have more protofilaments than Taxol-stabilized microtubules (29–31). KIF1A pausing occurred on both microtubule lattices (Fig. 2, A and B) with similar frequencies of 0.95 pause/overall run (Taxol (n = 165)) versus 1.02 pauses/overall run (GMPCPP (n = 171)) and pauses per distance of 0.14 (Taxol) versus 0.16 pause/μm (GMPCPP) (Fig. 2F and Table 1). However, there was a 46% decrease in pause duration from 1.23 (Taxol) to 0.66 s (GMPCPP) (Fig. 2, D and E, and Table 1). With respect to other motility characteristics, we observed no significant change in overall run length (6.24 ± 2.09 (n = 165) and 6.01 ± 2.17 μm (n = 171)) or continuous run length (2.95 ± 1.07 (n = 282) and 2.81 ± 0.91 μm (n = 305)) along Taxol-stabilized versus GMPCPP microtubules, respectively (Fig. 2C and Table 1). We also observed no change in the overall speed (1.35 ± 0.41 and 1.27 ± 0.28 μm/s, respectively; Table 1 and Fig. S2) or continuous speed (2.01 ± 0.53 and 2.06 ± 0.54 μm/s, respectively; Table 1 and Fig. S3) along Taxol-stabilized versus GMPCPP microtubules, respectively. However, there was a significant decrease in KIF1A's ability to land on GMPCPP microtubules in motility buffer with the presence of ADP with 4.86 ± 0.88 (n = 958) compared with 7.93 ± 1.25 events/μm/min (n = 1152) on Taxol microtubules (Fig. 7 and Table 1). Thus, although KIF1A displays a decrease in pause duration and a decreased landing rate on GMPCPP microtubules (Table 1), these results confirm that KIF1A's unique pausing behavior occurs on microtubules of varying protofilament and nucleotide composition.
Figure 7.
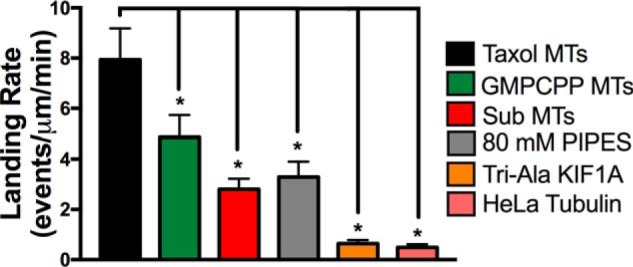
KIF1A landing is influenced by microtubule state and the K-loop/C-terminal tail interaction. Quantification of KIF1A landing rate in the ADP state on the indicated microtubule (MT) subsets (mean ± S.D.) is shown. On Taxol-stabilized microtubules, KIF1A had a landing rate of 7.93 ± 1.25 events/μm/min (n = 1552). On GMPCPP microtubules, the KIF1A landing rate was significantly reduced to 4.86 ± 0.88 events/μm/min (n = 958). Removal of the tubulin C-terminal tails (Sub MTs) significantly reduced the KIF1A landing rate to 2.80 ± 0.42 events/μm/min (n = 717). Increasing the salt concentration (80 mm PIPES) of the motility buffer reduced the landing rate to 3.28 ± 0.61 events/μm/min (n = 592). Mutation of the KIF1A K-loop (Tri-Ala KIF1A) significantly reduced the landing rate to 0.64 ± 0.14 event/μm/min (n = 527). On HeLa microtubules with reduced polyglutamylation, the KIF1A landing rate was significantly reduced to 0.49 ± 0.13 event/μm/min (n = 429). Each condition is representative of at least four independent experiments. Mean ± S.D. is reported. *, p < 0.001.
Interaction with tubulin C-terminal tails mediates KIF1A pausing behavior, landing rate, and superprocessive motility
To investigate a potential mechanism facilitating KIF1A pausing, we considered the known interaction of KIF1A's lysine-rich K-loop with tubulin's glutamate-rich C-terminal tail projections (22, 23). Previous work has shown that the positively charged surface loop-12 (K-loop) of the KIF1A motor domain interacts with the microtubule C-terminal tails, helping to anchor KIF1A to the microtubule (25) and mediate KIF1A landing on the microtubule (22). We thus asked whether subtilisin-mediated proteolytic removal (32) (Fig. 3A) of the tubulin C-terminal tails could influence KIF1A motility and pausing behavior. On subtilisin-treated microtubules, we observed a 47% reduction of KIF1A overall run length (3.31 ± 1.34 μm (n = 171)) when compared with untreated microtubules (Fig. 3, B and C, and Table 1). Subtilisin treatment did not significantly change the continuous run length (2.91 ± 1.16 μm (n = 205)) of KIF1A (Fig. 3C and Table 1); however, we did observe a 45% decrease in pause duration (from 1.23 to 0.68 s), a 79% reduction in pause frequency (from 0.95 to 0.18 pause/overall run), and an 85% reduction in pauses per distance (from 0.14 to 0.02 pause/μm) (Fig. 3, D and E, and Table 1). Due to the decreased pausing events, KIF1A's overall speed increased on subtilisin-treated microtubules from 1.35 ± 0.41 (untreated) to 1.58 ± 0.38 μm/s (Table 1 and Fig. S2). However, continuous speed remained relatively unchanged between untreated and subtilisin-treated microtubules (2.01 ± 0.53 and 1.94 ± 0.50 μm/s, respectively) (Table 1 and Fig. S3).
Figure 3.

Removal of the tubulin C-terminal tail reduces KIF1A pausing. A, cartoon depiction of tubulin C-terminal tail (CTT) cleavage upon addition of subtilisin. B, representative kymographs of KIF1A motility on untreated microtubules (top) versus subtilisin-treated microtubules (bottom). C, the overall run length (RL) of KIF1A was reduced from 6.24 ± 2.09 to 3.31 ± 1.34 μm (n = 171) upon subtilisin (Sub) treatment of microtubules, whereas continuous run length was not significantly changed (2.95 ± 1.07 and 2.91 ± 1.16 μm (n = 205), respectively). Control values for KIF1A overall and continuous run lengths are the same as reported for Fig. 2C. D, KIF1A pause duration is 0.68 s (CI, 0.57–0.83 s) on subtilisin-treated microtubules (n = 34) compared with untreated microtubules (1.23 s; Fig. 2D). E, KIF1A pause per distance was significantly decreased from 0.14 to 0.02 pause/μm upon subtilisin treatment of Taxol-stabilized microtubules (MTs). Run length values and standard deviations were calculated as reported previously (71). Kinesin-1 was used as an experimental control across all conditions (Figs. S2–S5). Each condition is representative of at least four independent experiments. Pause durations were fit to a single-exponential decay and are represented as cumulative frequency distributions; the time constant of fit is reported in seconds with a 95% confidence interval. All other metrics are reported as mean ± S.D. *, p < 0.001.
It has been previously reported that the perturbation of KIF1A's interaction with the C-terminal tail reduces the motor's landing rate (22). Measuring KIF1A landing rate in ADP-containing motility buffer confirms the necessity of this interaction, showing that subtilisin treatment of microtubules reduces the landing rate of KIF1A by 65% (from 7.93 ± 1.25 to 2.80 ± 0.42 events/μm/min; Fig. 7 and Table 1). Taken together, these results expose KIF1A's reliance on the microtubule C-terminal tail to engage with the microtubule, initiate pausing behavior, and generate superprocessive overall run lengths.
Conserved lysine residues of the K-loop are key mediators in KIF1A pausing and motility
Given the influence of the tubulin C-terminal tails on KIF1A pausing behavior, we further investigated the influence of the electrostatic interaction between the KIF1A K-loop and the microtubule C-terminal tails. First, we assessed the dependence of KIF1A pausing on the local electrostatic environment by increasing the salt concentration of our motility buffer from 12 to 80 mm PIPES. We observed a decrease in pause duration (from 1.23 to 0.78 s; Fig. 4C) with no significant changes in pause frequency (0.90 pause/overall run), pauses per distance (0.14 pause/μm), or any motility parameters (Fig. 4, A and B, and Figs. S2 and S3). Furthermore, in 80 mm PIPES we observed a decrease in KIF1A landing rate to 3.28 ± 0.61 events/μm/min (n = 592) (Fig. 7 and Table 1).
Figure 4.

KIF1A behavior and motility in 80 mm PIPES motility buffer on Taxol-stabilized microtubules. A, KIF1A exhibits a similar overall run length (RL) (5.98 ± 2.14 μm (n = 164)) and a continuous run length (3.14 ± 0.98 μm (n = 280)) in 80 mm PIPES as compared with standard motility buffer. B, KIF1A exhibits a similar number of pauses per distance in motility buffer (0.14 pause/μm) and 80 mm PIPES (0.14 pause/μm). C, in 80 mm PIPES, KIF1A pause duration is 0.78 s (CI, 0.66–0.91 s) (n = 132) compared with motility buffer (1.23 s; Fig. 2D). Pause durations were fit to a single-exponential decay and are represented as cumulative frequency distributions; the time constant of fit is reported in seconds with a 95% confidence interval. All other metrics are reported as mean ± S.D.
Second, we investigated how positively charged residues within the K-loop contribute to KIF1A pausing and motility. It has been previously reported that mutating all six lysines of the K-loop to alanine residues abolishes KIF1A motility and interaction with the microtubule (22), highlighting the importance of the K-loop/microtubule interaction to the intrinsic motility properties of KIF1A. Therefore, we mutated the three lysines most conserved across the kinesin-3 family to maintain the ability of KIF1A motors to engage with the microtubule surface (Fig. 5A). The KIF1A K-loop mutants (Tri-Ala KIF1A) displayed a marked reduction in both motility and pausing behavior (Fig. 5, B–E). Specifically, both the overall run length (2.41 ± 1.42 μm (n = 173)) and continuous run length (2.00 ± 1.11 μm (n = 207)) were significantly reduced by 61 and 32%, respectively, when compared with WT KIF1A on Taxol-stabilized microtubules (Fig. 5C and Table 1). Due to fewer pausing events, there was a 23% increase in overall speed (1.66 ± 0.45 μm/s) and a continuous speed of 1.72 ± 0.45 μm/s (Table 1 and Figs. S2 and S3). Tri-Ala KIF1A mutants also exhibited impaired pausing behavior quantified by a 62% reduction in pause duration (from 1.23 to 0.47 s), a 75% decrease in pause frequency (from 0.95 to 0.23 pause/overall run), and a 64% decrease in pauses per distance (from 0.14 to 0.05 pause/μm) (Fig. 5, D and E, and Table 1). As expected from previous work (22), the Tri-Ala KIF1A mutant also had a dramatic, 92% reduction in landing rate when compared with WT KIF1A on Taxol-stabilized microtubules (0.64 ± 0.14 event/μm/min (n = 527) for Tri-Ala and 7.93 ± 1.25 events/μm/min (n = 1552) for WT) in motility buffer supplemented with ADP (Fig. 7, Fig. S7, and Table 1) but no change in microtubule interaction in motility buffer supplemented with AMPPNP (1.47 ± 0.34 events/μm/min (n = 574) for Tri-Ala and 2.01 ± 0.49 events/μm/min (n = 917) for WT) (Fig. S7). These results confirm that the KIF1A K-loop/microtubule C-terminal tail interaction plays a critical role not only in the landing rate as described previously (22) but also for the pausing behavior of KIF1A.
Figure 5.

The KIF1A K-loop regulates pausing. A, amino acid sequence alignment of the KIF1A loop-12 (K-loop) of WT KIF1A and the Tri-Ala KIF1A mutant. For the Tri-Ala mutant, the three most conserved lysines among the kinesin-3 family have been mutated to alanine residues (yellow text). B, representative kymographs of WT KIF1A motility (top) versus Tri-Ala KIF1A motility (bottom). C, the Tri-Ala mutation of the KIF1A K-loop significantly reduced both overall run length (RL) (from 6.24 ± 2.09 to 2.41 ± 1.42 μm (n = 173)) and continuous (from 2.95 ± 1.07 to 2.00 ± 1.11 μm (n = 207)) run length. D, KIF1A pause duration is 0.47 s (CI, 0.41–0.53 s) upon mutation of the K-loop (n = 36) compared with WT KIF1A (1.23 s; Fig. 2D). E, mutation of the K-loop significantly decreased the number of pauses per distance from 0.14 (WT KIF1A) to 0.05 pause/μm (Tri-Ala KIF1A). Run length values and standard deviations were calculated as reported previously (71). Kinesin-1 was used as an experimental control across all conditions (Figs. S2–S5). Each condition is representative of at least four independent experiments. Pause durations were fit to a single-exponential decay and are represented as cumulative frequency distributions; the time constant of fit is reported in seconds with a 95% confidence interval. All other metrics are reported as mean ± S.D. *, p < 0.001.
Microtubule C-terminal tail polyglutamylation regulates KIF1A pausing behavior and motility
We next aimed to identify a specific property of the tubulin C-terminal tail that could influence KIF1A pausing and motility. It has been previously established that reduced levels of α-tubulin polyglutamylation result in improper KIF1A localization and insufficient cargo delivery in hippocampal and superior cervical ganglia neurons of ROSA22 mice (21). As neuronal microtubules are subjected to extensive C-terminal tail polyglutamylation (18), we hypothesized that this posttranslational modification is important for regulation of KIF1A motility behavior, including pausing. We thus assessed KIF1A's pausing and superprocessivity on tubulin purified from HeLa cells, which are known to contain markedly less polyglutamylation than neuronal tubulin (Fig. 6A) due to low expression of the tubulin tyrosine-like ligase enzymes (33–35). It is important to note that this change in tubulin polyglutamylation alters the local charge of the microtubule surface as compared with neuronal tubulin. KIF1A overall run length was reduced by 34% on Taxol-stabilized HeLa microtubules (4.12 ± 1.48 μm (n = 164)) when compared with Taxol-stabilized neuronal microtubules with no significant reduction in continuous run length (Fig. 6C and Table 1). In regard to pausing behavior, we observed a 58% reduction in pause frequency (from 0.95 to 0.40 pause/overall run), a 50% reduction in pauses per distance (from 0.14 to 0.07 pause/μm), and a 51% decrease in pause duration (from 1.23 to 0.59 s) (Fig. 6, B, D, and E, and Table 1). Due to the reduction in pausing events, the overall speed of KIF1A on HeLa tubulin (1.70 ± 0.41 μm/s) increased by 26% when compared with KIF1A on neuronal microtubules, whereas continuous speed decreased by only 7% (Table 1 and Figs. S2 and S3). We also observed a substantial, 94% reduction in KIF1A landing rate on HeLa microtubules when compared with neuronal microtubules (7.93 ± 1.25 (n = 1552) versus 0.49 ± 0.13 event/μm/min (n = 429)) (Fig. 7 and Table 1). Taken together, these results confirm that microtubule C-terminal tail polyglutamylation dictates KIF1A's ability to pause and thus mediates superprocessive behavior.
Figure 6.

KIF1A pausing is regulated by the polyglutamylation state of the microtubule. A, Western blot revealing higher levels of polyglutamylation (PolyE; top) on purified neuronal (Brain) tubulin compared with purified HeLa tubulin, known to have little tubulin polyglutamylation (33–35). The total α-tubulin loading control is shown in the bottom blot. B, representative kymographs of KIF1A motility on neuronal microtubules (top) versus HeLa microtubules (middle and bottom). C, the overall run length (RL) of KIF1A was reduced from 6.24 ± 2.09 to 4.12 ± 1.48 μm (n = 164) on HeLa microtubules, whereas continuous run length was not significantly changed (2.95 ± 1.07 μm on HeLa microtubules versus 2.98 ± 0.96 μm (n = 222) on brain microtubules). Control values for KIF1A overall and continuous run lengths are the same as reported for Fig. 2C. D, KIF1A pause duration is 0.59 s (CI, 0.54–0.64 s) on HeLa microtubules (n = 58) compared with neuronal microtubules (1.23 s; Fig. 2D). E, KIF1A pause per distance was significantly decreased from 0.14 pause/μm on brain microtubules to 0.07 pause/μm on HeLa microtubules. Run length values and standard deviations were calculated as reported previously (71). Kinesin-1 was used as an experimental control across all conditions (Figs. S2–S5). Each condition is representative of at least four independent experiments. Pause durations were fit to a single-exponential decay and are represented as cumulative frequency distributions; the time constant of fit is reported in seconds with a 95% confidence interval. All other metrics are reported as mean ± S.D. *, p < 0.001.
Discussion
The superprocessivity of the kinesin-3 family member KIF1A has been established previously (7), supporting its known role in long-distance axonal transport (3, 10). Here, we present a novel molecular mechanism that influences KIF1A's superprocessive motion. First, we demonstrate that KIF1A engages in previously unreported pauses on the microtubule lattice. KIF1A walks a superprocessive distance (∼3 μm on average) before initiating a pause, supporting the notion that although pausing is rare, it is related to the superprocessive nature of the motor itself. Pausing has several effects on the overall motility of dimeric KIF1A motors. Pauses enable KIF1A to string together multiple continuous runs to generate a longer overall run. Of note, even in the absence of pauses, KIF1A remains a superprocessive motor, with continuous run lengths of ∼3 μm. KIF1A's overall speed is also decreased in the absence of pauses, with no effect on continuous speed. This response is expected, considering that a reduction in pause frequency will reduce a slower population of motors undergoing pauses.
Furthermore, we show that pauses are mediated by interactions between the KIF1A K-loop and tubulin C-terminal tails. When the K-loop/C-terminal tail interaction is interrupted (e.g. by subtilisin treatment or K-loop mutation), KIF1A shows a reduced pause frequency and duration on the microtubule lattice. Disruption of the K-loop/C-terminal tail interaction also results in a dramatic reduction in the landing rate of dimeric KIF1A motors, consistent with previous work (22). The K-loop is also critical for monomeric KIF1A motors to diffuse along the surface of microtubules (23). In addition, cellular work has shown that loop-12 is important for regulation of KIF1A dendritic cargo sorting (36). As the K-loop is conserved across members of the kinesin-3 family, our newly identified pausing mechanism is likely to be a conserved feature of this class of motor.
Finally, we demonstrate that polyglutamylation of the tubulin C-terminal tail is a posttranslational modification that regulates KIF1A pausing behavior and subsequent motility. Along HeLa microtubules, which lack polyglutamylation, KIF1A displayed significantly reduced landing rate, pause frequency, and pause duration. Electrostatic interactions between the K-loop and C-terminal tail play an important role as increasing the salt concentration in our motility buffer resulted in a reduced landing rate and pause duration. That the increased salt concentration did not influence pause frequency suggests that the factors that influence pausing are not completely based on electrostatics. That tubulin C-terminal tail polyglutamylation influences KIF1A behavior at the single-molecule level provides a mechanistic understanding of how polyglutamylation can regulate KIF1A cargo trafficking (21). The levels of tubulin C-terminal tail polyglutamylation are highly tuned during neural development (18, 37) and could also influence KIF1A during synaptogenesis (38) and interkinetic nuclear migration (39). Compartmentalizing neuronal tubulin polyglutamylation into localized hot spots (40) may contribute an advantageous level of regulation to fine-tune KIF1A function during these developmental processes.
With a molecular regulatory mechanism identified in regard to KIF1A pausing and motility, a question still remains: how is KIF1A able to pause in between processive segments (continuous runs)? Our proposed mechanism states that pausing is mediated in part by a KIF1A/C-terminal tail interaction. A structural mechanism responsible for KIF1A pausing can be proposed based on past work examining the flexibility and structural conformation of the K-loop during ATP hydrolysis (24). This study demonstrated that when KIF1A is bound to AMPPNP, loop-12 is positioned up and away from the microtubule surface, whereas when KIF1A is bound to ADP, loop-12 adopts a “downward” position to interact with the C-terminal tails on the microtubule surface. From this, we propose that pauses are initiated by loop-12 interaction with the microtubule C-terminal tail when KIF1A is in the ADP state. Importantly, a pause would happen when both motor domains are in the same nucleotide state at the same time, making this a catalytically rare event and explaining why pausing occurs on the magnitude of every ∼3 μm. We speculate that with both loops-12 in a downward position, KIF1A exhibits a spatially constrained form of diffusion close to the resolution limit of our TIRF microscope as compared with the previously observed highly diffusive behavior of the KIF1A monomer (26).
The mechanistic importance of loop-12 positioning is further supported by our data comparing the landing rate of WT and Tri-Ala motors in the presence of ADP or AMPPNP in the motility buffer. Under ADP conditions, the landing rate of the K-loop mutant (Tri-Ala) is significantly reduced when compared with WT KIF1A (Fig. S7), consistent with previous work (22). However, under AMPPNP conditions, there is no significant difference in landing rates between WT and Tri-Ala KIF1A motors (Fig. S7). These findings support our proposed structural mechanism of pausing in which the downward positioning of KIF1A loop-12 when bound to ADP enables loop-12 to interact with the microtubule C-terminal tails and facilitate pausing. Furthermore, the significant reduction of Tri-Ala KIF1A pausing and continuous run length in motility assays (Fig. 5, C and E) highlights the importance of the K-loop for KIF1A motility. Further testing of how KIF1A catalytic activity and structural states influence pausing is an enticing future direction beyond the scope of this current study.
Our results demonstrate that KIF1A responds not only to posttranslational modification of the microtubule surface but also to the nucleotide state of the microtubule. Specifically, we demonstrate that KIF1A has a reduced landing rate (Fig. 7) and pause duration (Fig. 2D and Table 1) on GMPCPP microtubules as compared with Taxol-stabilized microtubules. This is potentially due to a nucleotide-specific difference in positioning of the C-terminal tails on the microtubule surface. Whether the motor is influenced by protofilament number and/or nucleotide state of the GMPCPP lattice will require further study. Recent compelling work has demonstrated that microtubule-associated proteins (MAPs) and septins can also influence KIF1A motility and function (36, 41–43). Pausing may allow KIF1A to navigate around MAPs and other obstacles on the microtubule. The necessity to regulate kinesin cargo transport through pausing is a pre-established concept on a cellular level as many neuronal cargo have been observed to halt processive movement to navigate around obstacles in the crowded cellular environment (44–47). Conversely, microtubule C-terminal tail polyglutamylation can regulate the microtubule affinity of MAPs such as MAP1A, MAP1B, MAP2, and Tau (48, 49). For example, Tau interacts with tubulin C-terminal tails in a unique diffusive behavior that is important for Tau function on a cellular and systemic level (50). Tau could thus compete with KIF1A for microtubule C-terminal tails and thereby regulate KIF1A pausing and motility. This concept is a compelling topic for further study.
Understanding the mechanisms responsible for KIF1A motility is critical when one considers the extensive range of axonal cargo that KIF1A transports along neuronal microtubules (3, 9, 11, 51–57). Furthermore, until we advance our understanding of KIF1A regulation, we are limited in our ability to address the disease state manifestations of altered KIF1A function. The regulatory capabilities of Tau's binding behavior on KIF1A cargo transport are critical to understanding KIF1A's role in diseases known for impaired axonal transport such as frontotemporal dementia where changes in Tau isoform expression have been correlated to disease progression (58). For example, pathological overexpression (59–61) of the more diffusive Tau isoforms (62) and reduced transport of cargo known to be transported by KIF1A are observed in frontotemporal dementia (11, 12, 63). As we speculate that the diffusive binding state of Tau may compete with KIF1A for the microtubule C-terminal tail, this is an enticing concept for further investigation on the molecular and cellular levels. In summary, our discovery of a microtubule C-terminal tail polyglutamylation-mediated mechanism for regulation of KIF1A motility provides crucial insight as to how KIF1A cargo delivery is regulated during axonal transport and how this process may become altered in the disease state.
Experimental procedures
Tubulin isolation, microtubule preparation, and labeling
Neuronal tubulin was isolated from bovine brains donated from Vermont Livestock Slaughter and Processing (Ferrisburgh, VT) using a high molarity PIPES buffer (1 m PIPES, pH 6.9 at room temperature, 10 mm MgCl2, and 20 mm EGTA) as described previously (64). Purified tubulin was clarified using ultracentrifugation for 20 min at 95,000 rpm at 4 °C in an Optima TLX ultracentrifuge (Beckman). After clarification, tubulin concentration was calculated using the tubulin extinction coefficient of 115,000 cm−1 m−1 and read at 280 nm in a spectrophotometer.
Tubulin was purified from HeLa Kyoto cells with TOG affinity column chromatography using a gravity-flow setup (65, 66). Cells were resuspended in BRB80 (80 mm PIPES, 1 mm EGTA, and 1 mm MgCl2, pH 6.8) supplemented with 1 mm DTT and protease inhibitors (cOmpleteTM, Mini, EDTA-free (Sigma-Aldrich) and 0.2 mm PMSF) and sonicated. Cleared lysate was loaded onto a TOG column in which ∼15 mg of bacterially purified GST-TOG1/2 protein was conjugated with 1 ml of NHS-activated Sepharose 4 Fast Flow resin (GE Healthcare). Washing, elution, desalting, and concentration were carried out as described in Hotta et al. (66). Glycerol was not added to the purified tubulin. Aliquoted tubulin was snap frozen in liquid nitrogen and stored at −80 °C.
Clarified tubulin was supplemented with 1 mm GTP (Sigma-Aldrich) or GMPCPP (Jena Bioscience, Jena, Germany). To label microtubules, unlabeled tubulin was mixed with rhodamine-labeled tubulin (Cytoskeleton, Denver, CO) at a ratio of 100:1. Microtubules were stabilized with GMPCPP following methods reported previously (67) or polymerized at 37 °C for 20 min and stabilized with 20 μm paclitaxel (Taxol; Sigma-Aldrich) in DMSO. Microtubules were diluted to a working concentration of 1 μm in P12 Buffer (12 mm PIPES, 1 mm MgCl2, and 1 mm EGTA, pH 6.8, supplemented with 20 μm paclitaxel) (22).
Plasmids, mutagenesis, and cell lysate motor expression
KIF1A(1–393)-LZ-3xmCitrine plasmid was a generous gift from Kristen Verhey (University of Michigan, Ann Arbor, MI), and the KIF1A(1–396)-GFP (Addgene plasmid number 45058) mammalian plasmid was a gift from Gary Banker (Oregon Health and Science University). TriAla-KIF1A-LZ-3xmCitrine was generated from the KIF1A(1–393)-LZ-3xmCitrine plasmid using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). COS-7 monkey kidney fibroblasts (American Type Culture Collection, Manassas, VA) were cultured in DMEM-GlutaMAXTM with 10% fetal bovine serum at 37 °C with 5% CO2. Cells were transfected with 1 μg of either WT KIF1A-LZ-3xmCitrine, Tri-Ala KIF1A-LZ-3xmCitrine, or KIF1A-GFP mammalian plasmid using the Lipofectamine 2000 delivery system (Thermo Fisher Scientific, Waltham, MA) and incubated in Opti-MEMTM (Thermo Fisher Scientific) medium with 4% fetal bovine serum. The next day, cells were harvested, pelleted, and washed with DMEM-GlutaMAX. The pellet was vigorously resuspended in lysis buffer (25 mm HEPES, 11 mm K+ acetate, 5 mm Na+ acetate, 5 mm MgCl2, 0.5 mm EGTA, 1% Triton X-100, 1 mm PMSF, 1 mg/ml pepstatin, 10 μg/ml leupeptin, and 5 μg/ml aprotinin) and centrifuged at room temperature for 15 min at 14,000 rpm. Relative amounts of protein between preparations and motor constructs were determined using densitometry, and supernatant containing expressed motor protein was aliquoted and stored at −80 °C until further use.
Subtilisin treatment
To remove C-terminal tails, 5 μm paclitaxel-stabilized microtubules was treated with 0.05 μm subtilisin A (Sigma-Aldrich), resuspended in P12 Buffer, for 45 min at 25 °C. The reaction was stopped by the addition of 5 mm phenylmethanesulfonyl fluoride. Subtilisin treatment was confirmed by Coomassie staining of an SDS-polyacrylamide denaturing gel. Tubulin concentration was determined as described previously.
In vitro single-molecule TIRF
Flow chambers used in in vitro TIRF experiments were constructed as described previously (68). Flow chambers were incubated with monoclonal anti-β III (neuronal) antibodies at 33 μg/ml for 5 min, then washed twice with 0.5 mg/ml bovine serum albumin (BSA; Sigma-Aldrich), and incubated for 2 min. 1 μm microtubules (any experimental condition) was administered and incubated for 8 min. Nonadherent microtubules were removed with a P12 Buffer wash supplemented with 20 μm paclitaxel. Kinesin motors in motility buffer consistent with past literature (12 mm PIPES, 1 mm MgCl2, 1 mm EGTA, 10 mm DTT, 10 mg/ml BSA, 2 mm ATP, and an oxygen scavenger system (5.8 mg/ml glucose, 0.045 mg/ml catalase, and 0.067 mg/ml glucose oxidase; Sigma-Aldrich) supplemented with 20 μm paclitaxel) (22) or a high-salt motility buffer (same as motility buffer with the only change being that the PIPES concentration was raised to 80 mm) supplemented with 2 mm ATP were added to the flow cell just before image acquisition. For landing rate assays, in vitro KIF1A motility assays were prepared as described above with the only change being that motility buffer was supplemented with 2 mm ADP. Control Drosophila melanogaster biotin-tagged kinesin-1 motors were labeled with streptavidin-conjugated Qdot 655 (Life Technologies) at a 1:4 motor:Qdot ratio as described previously (68, 69).
TIRF microscopy was performed at room temperature using an inverted Eclipse Ti-E microscope (Nikon, Melville, NY) with a 100× Apo TIRF objective lens (1.49 numerical aperture) and dual iXon Ultra electron-multiplying charge-coupled device cameras running NIS Elements version 4.51.01. Rhodamine-labeled microtubules were excited with a 561 nm laser and a 590/50 filter. KIF1A-LZ-3xmCitrine (WT or Tri-Ala) motors were excited with a 488 nm laser and a 525/50 filter. Qdot 655–conjugated kinesin-1 motors were excited with a 640 nm laser and 655 nm filter. All movies were recorded with an acquisition time of 200 ms for 500 frames (100-s observation).
Photobleaching assay
As an additional control, a photobleaching assay was performed to confirm that KIF1A run lengths were not underestimated due to photobleaching of the C-terminal 3xmCitrine tag (Fig. S6). Motors were adhered to microtubules in the presence of AMPPNP (Sigma-Aldrich) and washed once to remove unattached motors. Using ImageJ, a region of interest (ROI) was drawn around each fluorescent spot, and the average intensity of each pixel within the ROI was measured over time. Intensity was background-corrected, and a motor was considered photobleached when the ROI average intensity was 0.
Western blot analysis
Purified tubulin protein was separated by electrophoresis on Mini-PROTEAN® TGXTM gels (Bio-Rad) and transferred to a polyvinylidene fluoride membrane (Bio-Rad). Membranes were blocked in in a 1:1 solution of phosphate-buffered saline (PBS; 155 mm NaCl, 3 mm Na2HPO4, and 1 mm KH2PO4, pH 7.4) and Odyssey® blocking reagent (Li-COR Biosciences, Lincoln, NE). A mouse anti-polyglutamylated tubulin antibody (1:8,000; GT335, AdipoGen, San Diego, CA) or a mouse anti-α-tubulin antibody (1:10,000; DM1A, Sigma-Aldrich) was administered to membrane followed by a secondary DyLight 800 anti-mouse IgG antibody (1:10,000; Thermo Fisher Scientific). Secondary antibody fluorescence was detected using an Odyssey CLx (Li-COR Biosciences).
Data analysis
Motility events were analyzed as reported previously (69, 70). In brief, overall run length motility data were measured using the ImageJ MTrackJ plug-in for a frame-by-frame quantification of KIF1A motility. Pauses are defined as segments where the average velocity is less than 0.2 μm/s over three frames or more. Continuous events were identified at the boundaries of pausing events. Average overall/continuous speeds were plotted as a histogram, with mean and S.D. reported in Table 1. KIF1A pause duration events were fit to a single-exponential decay, represented as cumulative frequency plots with confidence intervals (CIs) of 95%. Pause duration is reported as the inverse of the decay rate, representing the lifetime of pause duration events.
Kymographs of motor motility were created using the MultipleKymograph ImageJ plug-in, with a set line thickness of 3. To correct for microtubule track length effects on motor motility, overall/continuous run length data were resampled to generate cumulative frequency plots (99% CI) using methods reported previously (71). After data resampling, statistical significance between motor run length and speed data sets was determined using a paired t test.
To determine the landing rate of KIF1A on various microtubule conditions, the total number of motor landing events on the microtubule was divided by the length of the microtubule and further divided by the duration of the movie (events/μm/min). To be considered a landing rate event, a motor must remain on the microtubule for three consecutive frames under ADP- or AMPPNP-state conditions.
Author contributions
D. V. L., K. J. V., R. O., and C. L. B. conceptualization; D. V. L. and O. J. Z. data curation; D. V. L. and O. J. Z. formal analysis; D. V. L., T. H., and C. L. B. supervision; D. V. L., O. J. Z., and C. L. B. investigation; D. V. L., O. J. Z., T. H., K. J. V., R. O., and C. L. B. methodology; D. V. L. and C. L. B. writing-original draft; D. V. L., O. J. Z., T. H., K. J. V., R. O., and C. L. B. writing-review and editing; T. H., K. J. V., and R. O. resources; T. H. and C. L. B. validation; K. J. V., R. O., and C. L. B. funding acquisition; C. L. B. project administration.
Supplementary Material
Acknowledgments
We thank David Warshaw and Guy Kennedy for training and use of the TIRF microscope at the University of Vermont. We especially thank Vermont Livestock Slaughter and Processing (Ferrisburgh, VT) for supporting our work. We thank Gary Banker and Marvin Bentley for the gifted pBa.Kif1a 1–396.GFP (Addgene plasmid number 45058), William Hancock for the use of the kinesin-1 construct, and Adam Hendricks for support.
This work was supported by NIGMS, National Institutes of Health Grants GM101066 (to C. L. B.), GM070862 (to K. J. V), and GM086610 (to R. O.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7 and Movie S1.
- PTM
- posttranslational modification
- TIRF
- total internal reflection fluorescence
- LZ
- leucine zipper
- GMPCPP
- guanosine 5′-[(α,β)-methyleno]triphosphate sodium salt
- AMPPNP
- adenylyl imidodiphosphate
- MAP
- microtubule-associated protein
- PMSF
- phenylmethylsulfonyl fluoride
- TOG
- tumor overexpressed gene
- DMEM
- Dulbecco's modified Eagle's medium
- ROI
- region of interest
- CI
- confidence interval.
References
- 1. Schnitzer M. J., and Block S. M. (1997) Kinesin hydrolyses one ATP per 8-nm step. Nature 388, 386–390 10.1038/41111 [DOI] [PubMed] [Google Scholar]
- 2. Okada Y., Yamazaki H., Sekine-Aizawa Y., and Hirokawa N. (1995) The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81, 769–780 10.1016/0092-8674(95)90538-3 [DOI] [PubMed] [Google Scholar]
- 3. Lo K. Y., Kuzmin A., Unger S. M., Petersen J. D., and Silverman M. A. (2011) KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci. Lett. 491, 168–173 10.1016/j.neulet.2011.01.018 [DOI] [PubMed] [Google Scholar]
- 4. Hirokawa N., Niwa S., and Tanaka Y. (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638 10.1016/j.neuron.2010.09.039 [DOI] [PubMed] [Google Scholar]
- 5. Vale R. D., and Milligan R. A. (2000) The way things move: looking under the hood of molecular motor proteins. Science 288, 88–95 10.1126/science.288.5463.88 [DOI] [PubMed] [Google Scholar]
- 6. Hammond J. W., Cai D., Blasius T. L., Li Z., Jiang Y., Jih G. T., Meyhofer E., and Verhey K. J. (2009) Mammalian kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS Biol. 7, e72 10.1371/journal.pbio.1000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soppina V., Norris S. R., Dizaji A. S., Kortus M., Veatch S., Peckham M., and Verhey K. J. (2014) Dimerization of mammalian kinesin-3 motors results in superprocessive motion. Proc. Natl. Acad. Sci. U.S.A. 111, 5562–5567 10.1073/pnas.1400759111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kondo M., Takei Y., and Hirokawa N. (2012) Motor protein KIF1A is essential for hippocampal synaptogenesis and learning enhancement in an enriched environment. Neuron 73, 743–757 10.1016/j.neuron.2011.12.020 [DOI] [PubMed] [Google Scholar]
- 9. Tsai J. W., Lian W. N., Kemal S., Kriegstein A. R., and Vallee R. B. (2010) Kinesin 3 and cytoplasmic dynein mediate interkinetic nuclear migration in neural stem cells. Nat. Neurosci. 13, 1463–1471 10.1038/nn.2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hung C. O., and Coleman M. P. (2016) KIF1A mediates axonal transport of BACE1 and identification of independently moving cargoes in living SCG neurons. Traffic 17, 1155–1167 10.1111/tra.12428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siddiqui N., and Straube A. (2017) Intracellular cargo transport by kinesin-3 motors. Biochemistry 82, 803–815 10.1134/S0006297917070057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goetzl E. J., Kapogiannis D., Schwartz J. B., Lobach I. V., Goetzl L., Abner E. L., Jicha G. A., Karydas A. M., Boxer A., and Miller B. L. (2016) Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer's disease. FASEB J. 30, 4141–4148 10.1096/fj.201600816R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verhey K. J., and Gaertig J. (2007) The tubulin code. Cell Cycle 6, 2152–2160 10.4161/cc.6.17.4633 [DOI] [PubMed] [Google Scholar]
- 14. Magiera M. M., Singh P., Gadadhar S., and Janke C. (2018) Tubulin posttranslational modifications and emerging links to human disease. Cell 173, 1323–1327 10.1016/j.cell.2018.05.018 [DOI] [PubMed] [Google Scholar]
- 15. Sirajuddin M., Rice L. M., and Vale R. D. (2014) Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 16, 335–344 10.1038/ncb2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hammond J. W., Huang C. F., Kaech S., Jacobson C., Banker G., and Verhey K. (2010) Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 21, 572–583 10.1091/mbc.e09-01-0044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh-Roy A., Goncharov A., Jin Y., and Chisholm A. D. (2012) Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev Cell 23, 716–728 10.1016/j.devcel.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Audebert S., Koulakoff A., Berwald-Netter Y., Gros F., Denoulet P., and Eddé B. (1994) Developmental regulation of polyglutamylated α- and β-tubulin in mouse brain neurons. J. Cell Sci. 107, 2313–2322 [DOI] [PubMed] [Google Scholar]
- 19. Ikegami K., Mukai M., Tsuchida J., Heier R. L., Macgregor G. R., and Setou M. (2006) TTLL7 is a mammalian β-tubulin polyglutamylase required for growth of MAP2-positive neurites. J. Biol. Chem. 281, 30707–30716 10.1074/jbc.M603984200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Janke C., Rogowski K., Wloga D., Regnard C., Kajava A. V., Strub J. M., Temurak N., van Dijk J., Boucher D., van Dorsselaer A., Suryavanshi S., Gaertig J., and Eddé B. (2005) Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science 308, 1758–1762 10.1126/science.1113010 [DOI] [PubMed] [Google Scholar]
- 21. Ikegami K., Heier R. L., Taruishi M., Takagi H., Mukai M., Shimma S., Taira S., Hatanaka K., Morone N., Yao I., Campbell P. K., Yuasa S., Janke C., Macgregor G. R., and Setou M. (2007) Loss of α-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function. Proc. Natl. Acad. Sci. U.S.A. 104, 3213–3218 10.1073/pnas.0611547104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soppina V., and Verhey K. J. (2014) The family-specific K-loop influences the microtubule on-rate but not the superprocessivity of kinesin-3 motors. Mol. Biol. Cell 25, 2161–2170 10.1091/mbc.e14-01-0696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okada Y., and Hirokawa N. (2000) Mechanism of the single-headed processivity: diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc. Natl. Acad. Sci. U.S.A. 97, 640–645 10.1073/pnas.97.2.640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nitta R., Kikkawa M., Okada Y., and Hirokawa N. (2004) KIF1A alternately uses two loops to bind microtubules. Science 305, 678–683 10.1126/science.1096621 [DOI] [PubMed] [Google Scholar]
- 25. Kikkawa M., Okada Y., and Hirokawa N. (2000) 15 Å resolution model of the monomeric kinesin motor, KIF1A. Cell 100, 241–252 10.1016/S0092-8674(00)81562-7 [DOI] [PubMed] [Google Scholar]
- 26. Okada Y., and Hirokawa N. (1999) A processive single-headed motor: kinesin superfamily protein KIF1A. Science 283, 1152–1157 10.1126/science.283.5405.1152 [DOI] [PubMed] [Google Scholar]
- 27. Gramlich M. W., Conway L., Liang W. H., Labastide J. A., King S. J., Xu J., and Ross J. L. (2017) Single molecule investigation of kinesin-1 motility using engineered microtubule defects. Sci. Rep. 7, 44290 10.1038/srep44290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Padzik A., Deshpande P., Hollos P., Franker M., Rannikko E. H., Cai D., Prus P., Mågård M., Westerlund N., Verhey K. J., James P., Hoogenraad C. C., and Coffey E. T. (2016) KIF5C S176 phosphorylation regulates microtubule binding and transport efficiency in mammalian neurons. Front. Cell. Neurosci. 10, 57 10.3389/fncel.2016.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ginsburg A., Shemesh A., Millgram A., Dharan R., Levi-Kalisman Y., Ringel I., and Raviv U. (2017) Structure of dynamic, Taxol-stabilized, and GMPPCP-stabilized microtubule. J. Phys. Chem. B 121, 8427–8436 10.1021/acs.jpcb.7b01057 [DOI] [PubMed] [Google Scholar]
- 30. Hyman A.A., Chrétien D., Arnal I., and Wade R. H. (1995) Structural changes accompanying GTP hydrolysis in microtubules: information from a slowly hydrolyzable analogue guanylyl-(α,β)-methylene-diphosphonate. J. Cell Biol. 128, 117–125 10.1083/jcb.128.1.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang R., LaFrance B., and Nogales E. (2018) Separating the effects of nucleotide and EB binding on microtubule structure. Proc. Natl. Acad. Sci. U.S.A. 115, E6191–E6200 10.1073/pnas.1802637115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Knipling L., Hwang J., and Wolff J. (1999) Preparation and properties of pure tubulin S. Cell Motil. Cytoskeleton 43, 63–71 10.1002/(SICI)1097-0169(1999)43:1<63::AID-CM7>3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- 33. Regnard C., Desbruyères E., Denoulet P., and Eddé B. (1999) Tubulin polyglutamylase: isozymic variants and regulation during the cell cycle in HeLa cells. J. Cell Sci. 112, 4281–4289 [DOI] [PubMed] [Google Scholar]
- 34. Lacroix B., and Janke C. (2011) Generation of differentially polyglutamylated microtubules. Methods Mol. Biol 777, 57–69 10.1007/978-1-61779-252-6_4 [DOI] [PubMed] [Google Scholar]
- 35. Lacroix B., van Dijk J., Gold N. D., Guizetti J., Aldrian-Herrada G., Rogowski K., Gerlich D. W., and Janke C. (2010) Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell Biol. 189, 945–954 10.1083/jcb.201001024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karasmanis E. P., Phan C. T., Angelis D., Kesisova I. A., Hoogenraad C. C., McKenney R. J., and Spiliotis E. T. (2018) Polarity of neuronal membrane traffic requires sorting of kinesin motor cargo during entry into dendrites by a microtubule-associated septin. Dev. Cell 46, 204–218.e7 10.1016/j.devcel.2018.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Audebert S., Desbruyères E., Gruszczynski C., Koulakoff A., Gros F., Denoulet P., and Eddé B. (1993) Reversible polyglutamylation of α- and β-tubulin and microtubule dynamics in mouse brain neurons. Mol. Biol. Cell 4, 615–626 10.1091/mbc.4.6.615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stavoe A. K., Hill S. E., Hall D. H., and Colón-Ramos D. A. (2016) KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev Cell 38, 171–185 10.1016/j.devcel.2016.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dantas T. J., Carabalona A., Hu D. J., and Vallee R. B. (2016) Emerging roles for motor proteins in progenitor cell behavior and neuronal migration during brain development. Cytoskeleton 73, 566–576 10.1002/cm.21293 [DOI] [PubMed] [Google Scholar]
- 40. Janke C., Rogowski K., and van Dijk J. (2008) Polyglutamylation: a fine-regulator of protein function? 'Protein Modifications: beyond the usual suspects' review series. EMBO Rep. 9, 636–641 10.1038/embor.2008.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monroy B. Y., Sawyer D. L., Ackermann B. E., Borden M. M., Tan T. C., and Ori-McKenney K. M. (2018) Competition between microtubule-associated proteins directs motor transport. Nat. Commun. 9, 1487 10.1038/s41467-018-03909-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gumy L.F., Katrukha E. A., Grigoriev I., Jaarsma D., Kapitein L. C., Akhmanova A, and Hoogenraad C. C. (2017) MAP2 defines a pre-axonal filtering zone to regulate KIF1- versus KIF5-dependent cargo transport in sensory neurons. Neuron 94, 347–362.e7 10.1016/j.neuron.2017.03.046 [DOI] [PubMed] [Google Scholar]
- 43. Lipka J., Kapitein L. C., Jaworski J., and Hoogenraad C. C. (2016) Microtubule-binding protein doublecortin-like kinase 1 (DCLK1) guides kinesin-3-mediated cargo transport to dendrites. EMBO J. 35, 302–318 10.15252/embj.201592929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trybus K. M. (2013) Intracellular transport: the causes for pauses. Curr. Biol. 23, R623–R625 10.1016/j.cub.2013.06.005 [DOI] [PubMed] [Google Scholar]
- 45. Maday S., Twelvetrees A. E., Moughamian A. J., and Holzbaur E. L. (2014) Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309 10.1016/j.neuron.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hendricks A. G., Perlson E., Ross J. L., Schroeder H. W. 3rd, Tokito M.., and Holzbaur E. L. (2010) Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr. Biol. 20, 697–702 10.1016/j.cub.2010.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Castle M. J., Perlson E., Holzbaur E. L., and Wolfe J. H. (2014) Long-distance axonal transport of AAV9 is driven by dynein and kinesin-2 and is trafficked in a highly motile Rab7-positive compartment. Mol Ther. 22, 554–566 10.1038/mt.2013.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bonnet C., Boucher D., Lazereg S., Pedrotti B., Islam K., Denoulet P., and Larcher J. C. (2001) Differential binding regulation of microtubule-associated proteins MAP1A, MAP1B, and MAP2 by tubulin polyglutamylation. J. Biol. Chem. 276, 12839–12848 10.1074/jbc.M011380200 [DOI] [PubMed] [Google Scholar]
- 49. Larcher J. C., Boucher D., Lazereg S., Gros F., and Denoulet P. (1996) Interaction of kinesin motor domains with α- and β-tubulin subunits at a tau-independent binding site. Regulation by polyglutamylation. J. Biol. Chem. 271, 22117–22124 10.1074/jbc.271.36.22117 [DOI] [PubMed] [Google Scholar]
- 50. Hinrichs M. H., Jalal A., Brenner B., Mandelkow E., Kumar S., and Scholz T. (2012) Tau protein diffuses along the microtubule lattice. J. Biol. Chem. 287, 38559–38568 10.1074/jbc.M112.369785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Niwa S., Lipton D. M., Morikawa M., Zhao C., Hirokawa N., Lu H., and Shen K. (2016) Autoinhibition of a neuronal kinesin UNC-104/KIF1A regulates the size and density of synapses. Cell Rep. 16, 2129–2141 10.1016/j.celrep.2016.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tanaka Y., Niwa S., Dong M., Farkhondeh A., Wang L., Zhou R., and Hirokawa N. (2016) The molecular motor KIF1A transports the TrkA neurotrophin receptor and is essential for sensory neuron survival and function. Neuron 90, 1215–1229 10.1016/j.neuron.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 53. Carabalona A., Hu D. J., and Vallee R. B. (2016) KIF1A inhibition immortalizes brain stem cells but blocks BDNF-mediated neuronal migration. Nat. Neurosci. 19, 253–262 10.1038/nn.4213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee J. R., Srour M., Kim D., Hamdan F. F., Lim S. H., Brunel-Guitton C., Décarie J. C., Rossignol E., Mitchell G. A., Schreiber A., Moran R., Van Haren K., Richardson R., Nicolai J., Oberndorff K. M., et al. (2015) De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum. Mutat. 36, 69–78 10.1002/humu.22709 [DOI] [PubMed] [Google Scholar]
- 55. Niwa S., Tanaka Y., and Hirokawa N. (2008) KIF1Bbeta- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat. Cell Biol. 10, 1269–1279 10.1038/ncb1785 [DOI] [PubMed] [Google Scholar]
- 56. Lee J. R., Shin H., Ko J., Choi J., Lee H., and Kim E. (2003) Characterization of the movement of the kinesin motor KIF1A in living cultured neurons. J. Biol. Chem. 278, 2624–2629 10.1074/jbc.M211152200 [DOI] [PubMed] [Google Scholar]
- 57. Yonekawa Y., Harada A., Okada Y., Funakoshi T., Kanai Y., Takei Y., Terada S., Noda T., and Hirokawa N. (1998) Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 141, 431–441 10.1083/jcb.141.2.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iqbal K., Liu F., Gong C. X., and Grundke-Iqbal I. (2010) Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664 10.2174/156720510793611592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kopeikina K. J., Hyman B. T., and Spires-Jones T. L. (2012) Soluble forms of tau are toxic in Alzheimer's disease. Transl. Neurosci. 3, 223–233 10.2478/s13380-012-0032-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schoch K. M., DeVos S. L., Miller R. L., Chun S. J., Norrbom M., Wozniak D. F., Dawson H. N., Bennett C. F., Rigo F., and Miller T. M. (2016) Increased 4R-Tau induces pathological changes in a human-Tau mouse model. Neuron 90, 941–947 10.1016/j.neuron.2016.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goedert M., Ghetti B., and Spillantini M. G. (2000) Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Their relevance for understanding the neurogenerative process. Ann. N.Y. Acad. Sci. 920, 74–83 [DOI] [PubMed] [Google Scholar]
- 62. McVicker D. P., Hoeprich G. J., Thompson A. R., and Berger C. L. (2014) Tau interconverts between diffusive and stable populations on the microtubule surface in an isoform and lattice specific manner. Cytoskeleton 71, 184–194 10.1002/cm.21163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rodríguez-Martín T., Pooler A. M., Lau D. H. W., Mórotz G. M., De Vos K. J., Gilley J., Coleman M. P., and Hanger D. P. (2016) Reduced number of axonal mitochondria and tau hypophosphorylation in mouse P301L tau knockin neurons. Neurobiol. Dis. 85, 1–10 10.1016/j.nbd.2015.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Castoldi M., and Popov A. V. (2003) Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expr. Purif. 32, 83–88 10.1016/S1046-5928(03)00218-3 [DOI] [PubMed] [Google Scholar]
- 65. Widlund P. O., Podolski M., Reber S., Alper J., Storch M., Hyman A. A., Howard J., and Drechsel D. N. (2012) One-step purification of assembly-competent tubulin from diverse eukaryotic sources. Mol. Biol. Cell 23, 4393–4401 10.1091/mbc.e12-06-0444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hotta T., Fujita S., Uchimura S., Noguchi M., Demura T., Muto E., and Hashimoto T. (2016) Affinity purification and characterization of functional tubulin from cell suspension cultures of Arabidopsis and tobacco. Plant Physiol. 170, 1189–1205 10.1104/pp.15.01173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McVicker D. P., Chrin L. R., and Berger C. L. (2011) The nucleotide-binding state of microtubules modulates kinesin processivity and the ability of Tau to inhibit kinesin-mediated transport. J. Biol. Chem. 286, 42873–42880 10.1074/jbc.M111.292987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stern J. L., Lessard D. V., Hoeprich G. J., Morfini G. A., and Berger C. L. (2017) Phosphoregulation of Tau modulates inhibition of kinesin-1 motility. Mol. Biol. Cell 28, 1079–1087 10.1091/mbc.e16-10-0728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hoeprich G. J., Mickolajczyk K. J., Nelson S. R., Hancock W. O., and Berger C. L. (2017) The axonal transport motor kinesin-2 navigates microtubule obstacles via protofilament switching. Traffic 18, 304–314 10.1111/tra.12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hoeprich G. J., Thompson A. R., McVicker D. P., Hancock W. O., and Berger C. (2014) Kinesin's neck-linker determines its ability to navigate obstacles on the microtubule surface. Biophys. J. 106, 1691–1700 10.1016/j.bpj.2014.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Thompson A. R., Hoeprich G. J., and Berger C. L. (2013) Single-molecule motility: statistical analysis and the effects of track length on quantification of processive motion. Biophys. J. 104, 2651–2661 10.1016/j.bpj.2013.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.