

Abstract
The centrosome, consisting of two centrioles surrounded by a dense network of proteins, is the microtubule-organizing center of animal cells. Polo-like kinase 4 (PLK4) is a Ser/Thr protein kinase and the master regulator of centriole duplication, but it may play additional roles in centrosome function. To identify additional proteins regulated by PLK4, we generated an RPE-1 human cell line with a genetically engineered “analog-sensitive” PLK4AS, which genetically encodes chemical sensitivity to competitive inhibition via a bulky ATP analog. We used this transgenic line in an unbiased multiplex phosphoproteomic screen. Several hits were identified and validated as direct PLK4 substrates by in vitro kinase assays. Among them, we confirmed Ser-78 in centrosomal protein 131 (CEP131, also known as AZI1) as a direct substrate of PLK4. Using immunofluorescence microscopy, we observed that although PLK4-mediated phosphorylation of Ser-78 is dispensable for CEP131 localization, ciliogenesis, and centriole duplication, it is essential for maintaining the integrity of centriolar satellites. We also found that PLK4 inhibition or use of a nonphosphorylatable CEP131 variant results in dispersed centriolar satellites. Moreover, replacement of endogenous WT CEP131 with an S78D phosphomimetic variant promoted aggregation of centriolar satellites. We conclude that PLK4 phosphorylates CEP131 at Ser-78 to maintain centriolar satellite integrity.
Keywords: centrosome, centriole, microtubule, phosphoproteomics, mass spectrometry (MS), chemical biology, cell division, centriole duplication, chemical genetics, chromosome segregation, PLK4, CEP131, centriolar satellite
Introduction
The centrosome is the major microtubule-organizing center of animal cells and consists of two orthogonal centrioles, which are cylindrical, 9-fold symmetric arrays of microtubules that help to nucleate microtubule arrays. Centrioles are crucial for accurate chromosome segregation (1), primary cilia formation, and neural development (2–4). Centriolar satellites are small, dynamic structures surrounding the centrosome. These satellites are important for the recruitment of proteins involved in microtubule organization (5), ciliogenesis (6), and centriole duplication (7).
Polo-like kinase 4 (PLK4)3 is a key regulator of centriole duplication (8, 9). Depletion of PLK4 impairs duplication, resulting in loss of centrioles, whereas overexpression causes overduplication and increased centriole number (9–11). On a molecular level, the formation of a new (daughter) centriole in proximity to the preexisting (maternal) centriole is initiated by CEP152- and CEP192-mediated recruitment of PLK4 at the G1/S transition (12, 13) and involves the interactions of several additional centrosomal proteins, including SASS6, STIL, CPAP, CEP135, and CP110 (14). After the initiation of procentriole formation by PLK4 recruitment, cartwheel formation is initiated. The 9-fold symmetric structure of the cartwheel is derived from the intrinsic 9-fold symmetry of SAS6 oligomers that form the cartwheel (15). The next step is elongation. Last, the plus end of the centriole is capped by CP110 and associated proteins (16). In this manner, PLK4 regulates key steps of centriole duplication.
Independent of its role in duplicating centrioles, PLK4 is implicated in other critical roles in centrosome biology, such as ciliogenesis (17, 18) and centriolar satellite maintenance (19). Regarding the latter, PLK4 phosphorylates PCM1, which partly regulates the integrity of centriolar satellite integrity (19). These data support a role for PLK4 in other centrosome functions, but a comprehensive list of substrates or functions is not available.
A number of centrosomal PLK4 substrates relevant to highly regulated centriole duplication are known (Table 1). Notably, PLK4 autophosphorylation results in ubiquitination and subsequent proteasomal degradation (20–23), which is critical for limiting PLK4-mediated centriole duplication to once per cell cycle. Substrates mediating centriole duplication include FBXW5 (24), GCP6 (25), and CP110 (26). Moreover, the substrates SAS6 (27) and STIL/Ana2 (28–31) mediate recruitment and stabilization of SAS6 at the centrosome during centriole duplication. Additionally, phosphorylation of CPAP allows for procentriole assembly and centriole elongation, and phosphorylation of CEP135 allows for recruitment of asterless/CEP152 to centrioles (32). Other known substrates of PLK4 mediate centriolar satellite integrity (PCM1 (19)), trophoblast differentiation (HAND1 (33)), and cell motility (APR2 (34)). Additionally, CEP152 (28, 35), CDC25C (36), CHK2 (37), and ECT2 (38) have been identified as PLK4 substrates, although the functions of these phosphorylation events remain unclear. Three previous studies have reported MS-based screens to identify PLK4 interactors. One study purified Plx4 from Xenopus egg extracts by affinity chromatography and analyzed its associated binding partners by MS (35), the second study performed PLK4 BioID to identify proximity interactions (39), and the third study immunoprecipitated tagged PLK4 from HeLa cells and examined binding partners by MS (28). To our knowledge, analysis of PLK4-regulated phosphorylation sites in human cells has not been reported. Here, we employed a chemical genetic system to perform an unbiased MS-based analysis of PLK4 phosphorylation targets in nontransformed human cells to discover novel substrates of PLK4. We identify CEP131 as a novel substrate of PLK4 and find that PLK4 phosphorylation of CEP131 is an important contributor to maintaining centriolar satellite integrity.
Table 1.
Previously identified substrates of PLK4
| Gene | Uniprot ID | Other Names and Orthologs | Residue(s) | Protein function | Effect of phosphorylation | Ref |
|---|---|---|---|---|---|---|
| PLK4 | O00444 | Sak, STK18, ZYG-1, Plx4 | Thr-170, Ser-305; Ser-293 and Thr-297 in Drosophila | Coordinates centriole duplication | Results in ubiquitination and degradation, limiting centriole duplication to once per cell cycle | 20–23, 42, 80 |
| FBXW5 | Q969U6 | F-box/WD repeat-containing protein 5, FBW5, PP3971 | Ser-151 | Substrate recognition component of both SCF (SKP1-CUL1-F-box protein) and DCX (DDB1-CUL4-X-box) E3 ubiquitin–protein ligase complexes | Prevents its ability to phosphorylate SAS6, keeping SAS6 at the centrosome and enabling centriole duplication | 24 |
| TUBGCP6 | Q96RT7 | GCP6, KIAA1669 | Undetermined | Microtubule nucleation | Required for centriole duplication | 25 |
| SAS6 | Q6UVJ0 | SASS6, HsSAS-6, MCPH14 | Ser-123 (in C. elegans) | Centriole cartwheel formation | Allows maintenance of SAS6 and emerging centriole | 27 |
| HAND1 | O96004 | BHLHA27, EHAND | Thr-107, Ser-109 | Transcription factor that plays an essential role in both trophoblast–giant cell differentiation and in cardiac morphogenesis | Frees HAND1 from the nucleolus, allowing for cell differentiation | 33 |
| ECT2 | Q9H8V3 | Epithelial cell-transforming 2, ARHGEF31 | Undetermined | Guanine nucleotide exchange factor (GEF), facilitates myosin contractile ring formation during cytokinesis | Unclear, only in vitro kinase assay performed | 38 |
| STIL | Q15468 | SCL/TAL1-interrupting locus, SIL, MCPH7, Ana2, SAS5 | Ser-1108 and Ser-1116 in human STIL; Ser-318, Ser-365, Ser-370, Ser-373 in Drosophila Ana2 | Recruits SAS6, allowing for proper centriole duplication | Allows for SAS6 recruitment | 28, 30, 31 |
| CEP110 | O43303 | CCP110, CP110, KIAA0419 | Ser-98 | Centriole duplication, caps mother centriole | Promotes centriole duplication, may stabilize SAS6 | 26 |
| CEP152 | O94986 | KIAA0912, MCPH9, SCKL5, MCPH4, asterless, Asl | Undetermined | Scaffold for recruitment of PLK4 and CENPJ | Unclear | 35 |
| CDC25C | P30307 | M-phase inducer phosphatase 3 | Undetermined | Tyrosine protein phosphatase required for progression of the cell cycle | Unclear, only in vitro kinase | 36 |
| CHK2 | O96017 | Checkpoint kinase 2, CHEK2, CDS1, RAD53 | Undetermined | DNA damage response and cell cycle arrest | Unclear, only in vitro kinase assay performed | 37 |
| PCM1 | Q15154 | Pericentriolar material 1, PTC4 | Ser-372 | Centrosome assembly, ciliogenesis, forms centriolar satellites | Required for centriolar satellite formation and ciliogenesis | 19 |
| CPAP | Q9HC77 | Centromere protein J, CENPJ, LAP, LIP1 | Ser-595 | Recruits centrosome components, inhibits microtubule nucleation from centrosome | Allows for procentriole assembly and centriole elongation | 81 |
| CEP135 | Q66GS9 | CEP4, KIAA0635 | Undetermined | Centriole duplication, recruitment of centriolar satellite proteins | Allows for recruitment of asterless to centrioles | 32 |
| ARP2 | P61160 | ACTR2 | Thr-237, Thr-238 | Actin organization | Enhances cell motility | 34 |
Results
Generation of PLK4 analog-sensitive cell line
PLK4 is the master regulator of centriole duplication (8, 9) and plays additional roles, but few substrates have been identified. To identify new substrates, we engineered a PLK4 conditional knockout cell line and a PLK4 analog-sensitive (AS) cell line. First, recombinant adeno-associated virus was used to insert loxP sites around exons 3 and 4 of one endogenous PLK4 locus. The other genomic locus of PLK4 was deleted, so the genotype of this cell line is PLK4WT/Δ. Two clones were identified and validated by PCR (Fig. S1, A and B) and by examining the disappearance of centrosome components by immunofluorescence after treatment with adenoviral Cre (AdCre; Fig. S1, C and D). The efficiency of centrosome loss is similar to a previous report of 39% of cells losing centrosomes 2 days after treatment with PLK4 siRNA (9). As expected in p53 WT cells (40, 41), loss of PLK4 significantly reduced colony formation (Fig. S1E).
Next, we used recombinant adeno-associated virus to engineer an AS cell line in immortalized, nontransformed RPE-1 cells. We targeted exons 3 and 4 to generate a point mutation at the conserved gatekeeper residue (L89G), enlarging the ATP-binding pocket for selective inhibition by the bulky ATP analog 3-MB-PP1 (42). To ensure that only PLK4AS was expressed, the floxed exons 3 and 4 were deleted from the PLK4WT locus by AdCre treatment, and clones were screened by PCR (Fig. S1, F and G). The genotype of this cell line is PLK4AS/Δ, which is henceforth referred to as “AS” throughout.
PLK4 inhibition is expected to prevent new centriole assembly without disassembling preexisting centrioles. Consistent with this, inhibition of AS with 3-MB-PP1 for 5 days yields about 65% of cells with fewer than two centrioles (Fig. 1, A and B), consistent with previous reports (40, 41).
Figure 1.

Validation of PLK4 chemical genetic system. A, representative images of RPE PLK4 AS cells treated with 10 μm 3-MB-PP1 for 5 days. Individual cells are outlined with dotted lines in the centrin image, and the arrow points to the one cell in the image with centrioles. Scale bar, 5 μm. B, centrosomes were quantified after treatment with 3-MB-PP1 for the indicated times. Bars represent the average of 3 biological replicates of 100 cells each ± S.E. (error bars). C, PLK4AS/Δ and PLK4WT/Δ were subcloned by limiting dilution, and cloning efficiency was assessed using a Poisson correction. D, proliferation was assessed by culturing cells in the indicated concentration of 3-MB-PP1 for 7 days and then staining with crystal violet. E, 1000 cells were plated per well in a 6-well plate (day −2), and 3-MB-PP1 was added on day 0. Cells were counted every 2 days. Each point represents the average of three technical replicates ± S.E. F, cells were cultured for 7 days with DMSO or 3-MB-PP1, and apoptosis was assessed by both trypan blue exclusion assay and staining with propidium iodide and anti-annexin V followed by flow cytometric detection. The percentage of cells staining negative for both propidium iodide and annexin V is plotted. G, senescence was assessed by β-gal staining after 14 days of culture with 3-MB-PP1. The micrographs are representative images of the staining, and the bar graphs quantify the percentage of senescent cells. All experiments in this figure utilized RPE cells. *, p < 0.05.
Coincident with centriole loss, there was a proliferation defect in the PLK4 AS cells after 5 days of incubation in 10 μm 3-MB-PP1 (Fig. 1, C–E). Importantly, the effect was specific to PLK4 AS cells, as PLK4 WT cells had normal cloning efficiency and proliferated well in the presence of 3-MB-PP1 (Fig. 1, C–E). Further characterization of the proliferation defect revealed cell cycle arrest, but not cell death, as evidenced by negative trypan blue and annexin V staining (Fig. 1F) and positive β-gal staining (Fig. 1G). These findings are consistent with previous reports indicating that cells lacking centrioles undergo cell cycle arrest but not apoptosis (40, 41). Thus, this PLK4 AS cell line allows for specific inhibition by 3-MB-PP1, resulting in loss of centrioles and cell cycle arrest.
CEP131 is a substrate of PLK4
To identify novel substrates of PLK4 using this system, we employed quantitative high-resolution MS for phosphoproteome analysis of PLK4 AS cells arrested at the G1/S boundary (Fig. 2A) with or without 3-MB-PP1 challenge. Isobaric labeling with 6-plex tandem mass tags (TMTs) allowed us to run three replicates each of AS cells with and without 3-MB-PP1 in a single MS experiment (43–46). The G1/S boundary was selected for this analysis (Fig. 2A) because centriole duplication occurs at this phase of the cell cycle (14, 47). Using this approach, we identified 11,501 phosphopeptides (Table S1), corresponding to 3711 proteins, of which 15 were significantly down-regulated (p value <0.05 and -fold change >1.5) by inhibition of PLK4. The most strongly regulated phosphopeptides were found in RUNX1, PTPN12, IL6ST, TRIM3, and SCRIB. Given that none of these proteins are known to localize to the centrosome or play any role in centrosome or microtubule biology, these phosphorylation events are likely to be indirectly regulated by PLK4. To increase the likelihood of identifying direct substrates, we therefore focused on proteins known to localize to the centrosome (Fig. 2B) and reduced our -fold change and statistical significance thresholds, as PLK4 is a low-abundance kinase with rare phosphorylation events that may not reach robust -fold change or statistical significance thresholds. We considered proteins to be centrosomal if their Uniprot entry mentioned localization to the centrosome, centriole, or minus ends of microtubules or if the protein was identified in a phosphoproteomic screen of purified centrosomes (48) (Fig. 2C). Many such phosphopeptides had only modest changes in abundance, possibly due to the moderating effect of feedback regulation of PLK4 catalytic activity (42). Two centrosome proteins identified in our screen, CEP152 and PCM1, were previously identified as PLK4 substrates (Table 1), providing credence that this screening approach identified PLK4 substrates. From the remaining list, we selected the proteins known to be associated with centriole duplication: CEP131 (7, 49), CEP215/CDK5RAP2 (50), and CEP350 (51). None of these proteins have been previously identified as PLK4 substrates. Among the potential phosphorylation sites, we identified residues that are evolutionarily conserved: Ser-78 and Ser-89 on CEP131, Ser-1238 on CEP215, and Ser-1648 on CEP350 (Fig. S2). To evaluate these as potential phosphorylation sites, direct phosphorylation was tested with GST-tagged 13-mer peptide fragments with the serine of interest located centrally (sequences listed in Table S2). STIL (Ser-1108 ± 6 amino acids) was used as a positive control (29). Of the potential fragments identified, only CEP131 Ser-78 produced a strong signal when incubated with WT PLK4 (Fig. 2D). We also incubated PLK4 with a larger N-terminal fragment of CEP131 (amino acids 1–250) and observe that PLK4 is capable of phosphorylating this fragment (Fig. S3). Importantly, phosphorylation is abolished when the PLK4 inhibitor, centrinone B (40), is added to the reaction (Fig. 2D). Moreover, a CEP131 fragment containing the S78A mutation is not phosphorylated (Fig. 2E). We conclude that CEP131 Ser-78 is directly phosphorylated by PLK4 in vitro. To assess which domain of CEP131 interacts with PLK4, we performed co-immunoprecipitation experiments. Full-length PLK4 was co-expressed with different domains of CEP131. We find that PLK4 interacts with the N terminus of CEP131 (Fig. S5).
Figure 2.

PLK4 phosphorylates CEP131 Ser-78. A, cell cycle profile of cells arrested with aphidicolin to enrich RPE PLK4 AS cells in the G1/S transition, which is when centriole duplication occurs and therefore when PLK4 is active. These cells were utilized for MS. B, volcano plot of −log10(p value) versus log2(-fold change) for the centrosome components (red dots) that were identified in the MS screen. C, diagram of the centrosome showing where each of these proteins is thought to localize; derived from Ref. 82. D, GST-tagged peptides and PLK4 kinase domain were purified from Escherichia coli and utilized for in vitro kinase assays. GST serves as a negative control, and STIL serves as a positive control. Centrinone B is a PLK4 inhibitor. E, an S78A mutation was created in the CEP131 peptide and purified for in vitro kinase assay to confirm that PLK4 phosphorylates CEP131 Ser-78.
CEP131 Ser-78 phosphorylation is required for centriolar satellite integrity but is dispensable for centriole duplication and ciliogenesis
CEP131 is a component of the centrosome and centriolar satellites and was previously identified as a PLK4 interactor in BioID experiments (39). Furthermore, CEP131 and PLK4 co-localize (Fig. S4). CEP131 is known to have three major functions in the cell: centriole duplication, ciliogenesis, and centriolar satellite formation. Moreover, PLK4 activity is implicated in each of these activities (8, 9, 19). Therefore, we tested whether PLK4 phosphorylation of CEP131 is required for these functions. To do this, we inhibited PLK4 in RPE-1 AS cells with 3-MB-PP1 and in HeLa cells using centrinone B. As expected, inhibition of PLK4 blocked centriole duplication (Fig. S6, A and B), ciliogenesis (Fig. S6, C and D), and centriolar satellite organization (Fig. S3, E and F).
To test whether CEP131 Ser-78 phosphorylation is important for any of these three functions, we expressed siRNA-resistant, enhanced GFP–tagged, nonphosphorylatable (S78A) or phosphomimetic (S78D) CEP131 mutants in HeLa cells. These mutants properly localize to centrioles and centriolar satellites (Fig. 3A), suggesting that this phosphorylation event is not required for CEP131 localization. Next, we depleted endogenous CEP131 with siRNA (Fig. 3B). Knockdown-addback efficiency was also assessed by quantitative RT-PCR (qRT-PCR) (Fig. 3C).
Figure 3.

PLK4 phosphorylation of CEP131 maintains centriolar satellite integrity. A, localization of WT CEP131 and S78A and S78D mutants in WT HeLa cells. The dotted white box is enlarged for the adjacent gray scale images showing GFP, PCM1, and centrin at the centrosome. Scale bar sizes are indicated on the images. B, knockdown addback experiment timeline. C, quantitative RT-PCR experiment demonstrating the efficiency of knockdown and addback of CEP131. Asterisks demonstrate significant enrichment of GFP-tagged CEP131 versus endogenous GFP (black bars) or significant depletion of endogenous CEP131 versus untreated cells (gray bars). D and E, images and quantification of centriole duplication. Each dot represents one cell, and bars represent means ± S.D. (error bars) of at least 100 cells/condition. Centrioles were identified by counting centrin foci. For example, the image in D shows two centrosomes with four total centrioles. F and G, images and quantification of ciliogenesis. Cilia were marked by staining for acetylated tubulin. At least 300 total cells were analyzed per condition. H and I, images and quantification of centriolar satellites. At least 300 total cells were analyzed per condition. J, schema of a more quantitative approach to assess centriolar satellites. The yellow circle measures the PCM1 intensity within a 2.5-μm radius of the center of the centrosome, whereas the blue circle measures the PCM1 intensity in the entire cell. K, quantification of centriolar satellites using the schema described in J. Nocodazole is used as a positive control for centriolar satellite dispersion. All experiments in this figure utilize HeLa cells. Bars, means ± S.E. (error bars) from three independent experiments. Scale bars, 10 μm. *, p < 0.05. NS, not significant.
We next analyzed the ability of these cells to duplicate centrioles and nucleate primary cilia. No defects in centriole numbers were observed among the conditions (Fig. 3, D and E). With regard to primary cilia formation, CEP131-depleted cells exhibited a reduction in primary cilia formation, consistent with previous reports (52, 53). However, defects in primary cilia formation did not occur in either the nonphosphorylatable (S78A) or phosphomimetic (S78D) mutant cell lines (Fig. 3, F and G). Combined, these data suggest that PLK4 phosphorylation of CEP131 Ser-78 is dispensable for centriole duplication and primary cilia formation.
PCM1 comprises a structural platform for centriolar satellites (54), and PLK4 phosphorylation of PCM1 is required for proper organization of centriolar satellites (19). To test whether CEP131 Ser-78 phosphorylation is also important for centriolar satellite integrity, we quantified PCM1 as either normal, dispersed, or aggregated, as has been reported previously (19, 55). Nocodazole served as positive control for dispersion (56). We find that depletion of CEP131 increases the dispersion of centriolar satellites (Fig. 3, H and I). Additionally, addback of WT and S78D CEP131 rescues this phenotype, but addback of the S78A nonphosphorylatable mutant does not rescue this phenotype. Furthermore, we devised a more objective method of quantifying centriolar satellite organization by computing the percentage of PCM1 intensity found within 2.5 μm of the center of the centrosome (Fig. 3J). Similarly, we find that addback of the S78A nonphosphorylatable mutant does not rescue centriolar satellite dispersion (Fig. 3K). Additionally, addback of the S78D phosphomimetic mutant causes increased aggregation of centriolar satellites. These data support a model where phosphorylation of Ser-78 by PLK4 helps restrain dispersion of centriolar satellites.
CEP131 Ser-78 was previously reported to be phosphorylated in response to UV irradiation by mitogen-activated protein kinase-activated protein kinase 2 (known as MK2 or MAPKAP2) (57). In the presence of UV and other cellular stresses, centriolar satellites disperse in a manner dependent on p38-mediated MK2 phosphorylation of CEP131 at both Ser-47 and Ser-78, thereby allowing 14-3-3 proteins to bind and sequester CEP131 to the cytoplasm. Given our findings, we tested whether PLK4 is important for this stress-induced remodeling. Indeed, p38 is required for this remodeling in response to UV, but PLK4 activity is dispensable (Fig. 4). This suggests that PLK4 may also regulate centriolar satellite integrity via CEP131 phosphorylation in a manner similar to MK2, albeit under different conditions.
Figure 4.
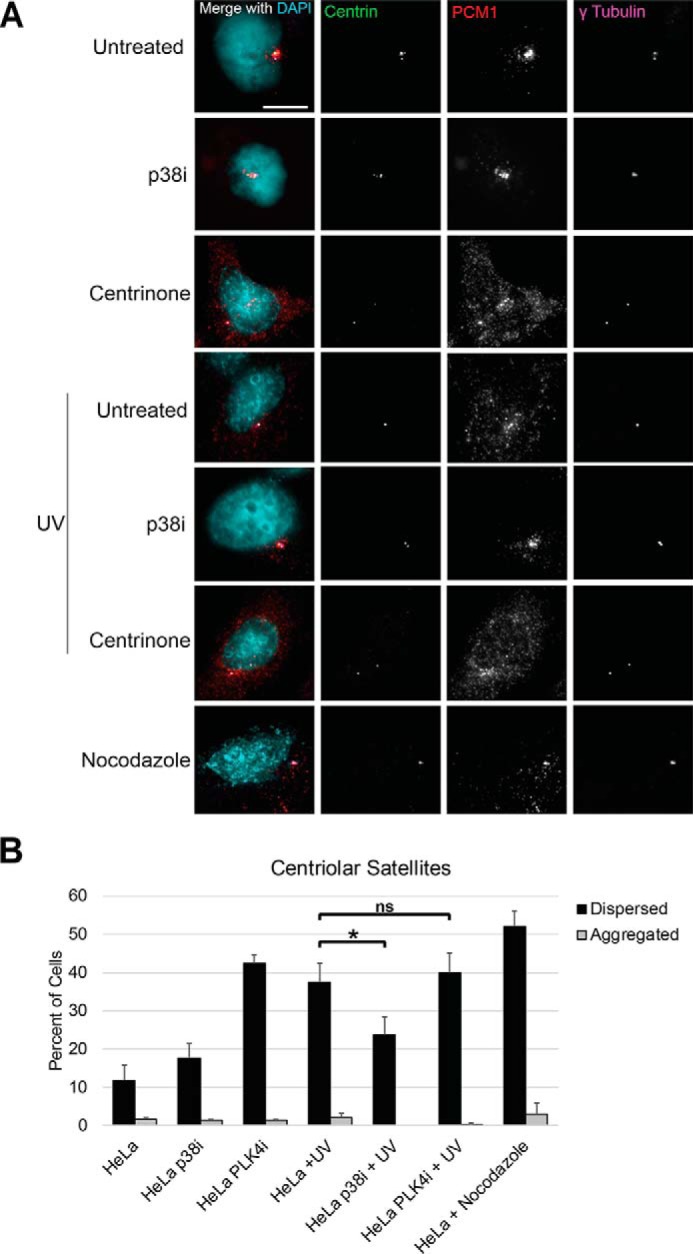
PLK4 activity is not required for centriolar satellite remodeling in response to UV. A, representative images of centriolar satellites (PCM1 staining) in HeLa cells under the indicated conditions. The p38 inhibitor (p38i) utilized was SB203580. Scale bar, 10 μm. B, quantification of centriolar satellites as aggregated or dispersed. DAPI, 4′,6-diamidino-2-phenylindole. Bars, averages ± S.D. (error bars) for one independent experiment. *, p < 0.05; ns, not significant. Scale bar, 5 μm.
CEP131 is known to enhance CEP152 recruitment to the centrosome. CEP152 then recruits WDR62 and CEP63 and promotes the centrosomal localization of CDK2 (7). We therefore tested whether CEP131 Ser-78 phosphorylation alters the recruitment of CEP152 to the centrosome by quantitative immunofluorescence in our knockdown-depletion experiment. However, we did not observe any differences in CEP152 localization to the centrosome (Fig. 5).
Figure 5.

Effects of S78A and S78D mutations on CEP131 interaction with CEP152. A, HeLa cells were simultaneously transfected with CEP131 siRNA and the indicated GFP-tagged CEP131 construct. Cells were then fixed and stained for CEP152. The amount of CEP152 at the centrosome was quantified and is shown in the adjacent dot plot. Insets in the CEP152 images show magnified CEP152 at the centrosome. B, WT HeLa cells and CEP131 knockout cell lines (KO4E and KO9) were stained for CEP152, and the amount of CEP152 at the centrosome was quantified. Insets, magnifications of the centrosome. Each dot represents a single cell. Bars, means ± S.D. (error bars). Scale bars, 10 μm. ***, p < 0.0001; ns, not significant.
CEP131 knockout cell lines exhibit reduced centriolar satellite organization and increased sensitivity to nutrient stresses
To assess the effects of chronic loss of endogenous CEP131, we engineered CEP131 knockout cell lines using CRISPR/Cas9 editing; this is possible as it is not essential (58). As expected, we recovered monoclonal CEP131 knockout cell lines, and confirmed these by CEP131 immunoblotting (Fig. 6A) and immunofluorescence (Fig. 6B). We utilized two different knockout clones for our experiments, denoted KO4E and KO9. As expected, knockout cells had dispersed centriolar satellites, as visualized by PCM1 (Fig. 6, C and D), a decrease in CEP152 expression at the centrosome (Fig. 5B), and an increase in mitotic errors and multinuclei (Fig. S7), consistent with previous reports (7, 49). However, no differences were seen in centriole number (Fig. 6E) or formation of primary cilia (Fig. 6, F and G).
Figure 6.

Chronic CEP131 depletion causes dispersion of centriolar satellites and is rescued by CEP131 phosphorylation. A, CEP131 Western blotting in WT HeLa cells compared with two CEP131 knockout cell lines, KO4E and KO9. B, immunofluorescent images showing loss of CEP131 in the knockout cell lines. C, immunofluorescent imaging of centrioles and centriolar satellites in the knockout cell lines. D, quantification of centriolar satellites in WT HeLa cells and the two CEP131 knockout lines. E, quantification of centrioles (centrin foci) in WT and CEP131 knockout HeLa lines. F, representative images of primary cilia in WT and CEP131 knockout HeLa lines. G, quantification of primary cilia in WT HeLa cells and the two CEP131 knockout lines. Scale bars, 10 μm. H, cell counts over 12 days in WT HeLa cells compared with two CEP131 knockout cell lines. I and J, proliferation in cells treated with serum-free medium (I) or with 1% serum (J). Proliferation was normalized to untreated cells within each cell line. K, representative images of centriolar satellites (marked by PCM1) in CEP131 knockout cells transfected with either WT, S78A, or S78D CEP131 transgenes. L, quantification of the percentage of cells with dispersed and aggregated PCM1. M, quantification of the percentage of PCM1 intensity within a 2.5-μm radius of the center of the centrosome. Nocodazole is used as a positive control for centriolar satellite dispersion. All experiments in this figure utilize HeLa cells. Bars, means ± S.E. (error bars). Scale bars, 10 μm. *, p < 0.05; ns, not significant.
Next, we assessed proliferation in CEP131 knockout cell lines and found slightly reduced proliferation (Fig. 6H), as shown previously by CEP131 knockdown (49). Centriolar satellites are important for response to cellular stresses (54, 57, 59). Therefore, we tested whether our knockout cell lines were more sensitive to nutrient stresses. Indeed, both knockout cell lines are more sensitive to both serum-free medium (Fig. 6I) and 1% serum medium (Fig. 6J), as the CEP131 knockout cell lines demonstrated a greater decrease in cell proliferation in response to these treatments.
To identify the role of phosphorylation at Ser-78, we expressed GFP-tagged WT, S78A, and S78D CEP131 constructs in the CEP131 knockout cell lines and evaluated the integrity of centriolar satellites. Only cells with visible GFP signal were analyzed. Consistent with previous experiments, addback of WT and S78D rescues the dispersed satellites, whereas addback of the S78A mutant does not (Fig. 6, K–M). Additionally, there is an increase in centriolar satellite aggregation in the S78D addback. To confirm that this effect observed using PCM1 as a centriolar satellite marker is indeed a disruption in centriolar satellite integrity versus other causes (e.g. disruption in CEP131 interaction with PCM1), we assessed centriolar satellites in these same conditions using a different marker of centriolar satellites: CEP72. Similarly, we find that CEP72 is dispersed in CEP131 knockout cells and is rescued by addback of WT or S78D transgenes but not S78A (Fig. 7).
Figure 7.

CEP72 staining recapitulates regulation of centriolar satellite integrity by CEP131 Ser-78 phosphorylation status. A, to ensure that our results regarding PLK4 phosphorylation of CEP131 regulating centriolar satellite integrity were not specific to PCM1, we also analyzed another marker of centriolar satellites, CEP72. WT or CEP131 KO HeLa cell lines were transfected with GFP-tagged CEP131 constructs (WT, S78A, and S78D) versus GFP-only control. Nocodazole treatment served as a positive control for satellite dispersion. Representative images are shown. B, CEP72 was assessed as either dispersed or aggregated, just as done for PCM1 staining of centriolar satellites in other figures. All experiments in this figure utilize HeLa cells. DAPI, 4′,6-diamidino-2-phenylindole. Bars, means ± S.D. (error bars) from three replicates. Scale bars, 10 μm. At least 300 cells were analyzed for each condition. *, p < 0.05.
Next, we assessed whether this aggregation defect observed in S78D mutants depends on dynein. Previous reports have demonstrated that PCM1 (5, 56, 60) and CEP131 (49) localization to centriolar satellites is dynein-dependent. We expressed WT CEP131 and CEP131 S78D phosphomimetic mutant in CEP131 knockout cells and treated with the dynein inhibitor ciliobrevin D. Ciliobrevin D caused dispersion of PCM1 and/or accumulation of PCM1 aggregates outside of the centrosome (Fig. 8, A and B). Further, treatment with ciliobrevin D in CEP131 knockout cells expressing the S78D mutant significantly reduced the aggregation of PCM1 at the centrosome (Fig. 8, C and D). These findings suggest that aggregation of satellites by PLK4 phosphorylation of CEP131 is dynein-dependent.
Figure 8.

Dynein is required for satellite aggregation with CEP131 S78D phosphomimetic. A, representative images of HeLa cells treated with nocodazole and the dynein inhibitor ciliobrevin D. B, quantification of centriolar satellites (PCM1 staining) in the conditions shown in A. C, representative images of WT HeLa cells transfected with CEP131 S78D and treated with ciliobrevin D. D, quantification of centriolar satellites (PCM1 staining) in the conditions shown in C. Bars, means ± S.D. (error bars) from three replicates. Scale bars, 10 μm. At least 300 cells were analyzed for each condition. *, p < 0.05.
To verify that these observed effects of CEP131 Ser-78 mutants depend on PLK4 activity, we overexpressed WT, S78A, or S78D constructs and GFP control in WT HeLa cells (Fig. 9A), treated with the PLK4 inhibitor centrinone B, and observed the effects on centriolar satellite organization by PCM1 immunofluorescence (Fig. 9B). Consistent with previous findings, PLK4 inhibition increased the dispersion of centriolar satellites, and the S78D phosphomimetic mutant restored clustered centriolar satellites in the presence of PLK4 inhibition (Fig. 9, C and D).
Figure 9.

PLK4 phosphorylation of CEP131 at Ser-78 is critical for proper centriolar satellite integrity. A, WT HeLa cells were transfected with GFP or GFP-tagged WT CEP131 or S78A or S78D mutant. Western blotting demonstrating transfection efficiency. B, images of centriolar satellites in these transfected cells after treatment with the PLK4 inhibitor centrinone B. Scale bar, 10 μm. C, qualitative assessment of centriolar satellites as either dispersed or aggregated. D, quantitative assessment of centriolar satellite dispersion/aggregation. All experiments in this figure utilize WT HeLa cells. Bars, means ± S.E. (error bars). Scale bars, 10 μm. *, p < 0.05; **, p < 0.01; ***, p < 0.0001; ns, not significant.
Additive effects of PLK4 phosphorylation of PCM1 and CEP131 on centriolar satellite organization
The effect of PLK4 phosphorylation of CEP131 on centriolar satellite organization is reminiscent of the previously reported effect of PLK4 phosphorylation of PCM1 (19). Therefore, we tested whether these two phosphorylation events cooperate to control centriolar satellite integrity. We overexpressed both phosphomimetic versions of FLAG-CEP131 (S78D) and GFP-PCM1 (S372D) in WT HeLa cells (Fig. 10A) and treated with centrinone B. We observed an increase in centriolar satellite aggregation in cells co-expressing both CEP131 S78D and PCM1 S372D phosphomimetic constructs compared with cells expressing just one of these phosphomimetic constructs (Fig. 10, B and C), suggesting that phosphorylations of both CEP131 and PCM1 by PLK4 are important for centriolar satellite integrity. In summary, PLK4 phosphorylates CEP131 and PCM1 to maintain the integrity of centriolar satellites in interphase (Fig. 10D).
Figure 10.

PLK4 phosphorylation of CEP131 cooperates with its phosphorylation of PCM1. A, Western blotting demonstrating transfection efficiency of the GFP-PCM1 and FLAG-CEP131 constructs. B, images of centriolar satellites in the transfected cell before and after PLK4 inhibition with centrinone B. Scale bar, 10 μm. C, quantification of dispersed and aggregated centriolar satellites. *, p < 0.05. Error bars, S.D. All experiments in this figure utilize HeLa cells. D, diagram summarizing the regulation of centriolar satellite integrity by PLK4 phosphorylation of CEP131 and PCM1. Red, cytoplasm; light blue, DNA; green, centrosome; yellow, centrioles; dark blue, centriolar satellites.
Discussion
Herein, we have coupled a chemical genetic system of selective PLK4 inhibition and an unbiased, multiplex phosphoproteomic screen to identify novel substrates of PLK4. We identify CEP131 Ser-78 as a substrate of PLK4 and find that PLK4 phosphorylation of this site is important for maintaining the integrity of centriolar satellites in interphase. Centriolar satellites are typically dispersed during mitosis (61), so we speculate that a cellular phosphatase regulates this process by reversing phosphorylation events prior to mitotic onset. However, other regulatory mechanisms are also possible. Additionally, our phosphoproteomic screen has uncovered many other potential substrates that could be explored.
This is the first study to our knowledge to characterize cellular knockout of human PLK4 protein using a Cre/lox conditional system versus prior work that focused solely on chemical inhibition or knockdown. However, the phenotype is highly similar to knockdown PLK4 with RNAi (8), auxin-inducible degradation (41), or inhibited PLK4 catalytic activity (29, 40). This is most consistent with PLK4 playing a primarily catalytic role without additional structural roles that would impart additional differences in phenotype between chemical inhibition and depletion or knockout.
PLK4 regulates other centrosomal activities independent of centriole duplication, including phosphorylation of PCM1 to maintain centriolar satellite integrity and primary cilia formation (19) and phosphorylation of CEP135 to regulate its interaction with CEP152 and influence the radial positioning of CEP152 on centrioles (32). Herein we have contributed to our understanding of one such noncanonical role of PLK4: the organization of centriolar satellites. Centriolar satellites are structures located around the centrosome and are important for proper microtubule nucleation, centrosome assembly, and primary cilia formation (5, 54, 56). Our data suggest that PLK4 phosphorylation of both PCM1 and CEP131 is required to maintain centriolar satellite organization and/or integrity. However, PLK4 phosphorylation of CEP131 is dispensable for centriole duplication and primary cilia formation. Given the previously reported findings that intact, functional centriolar satellites and/or their components are important for centriole duplication (7, 49) and ciliogenesis (19, 62), it is likely that different methods of centriolar satellite perturbations affect these pathways differently. It will be important to determine whether it is centriolar satellites or the individual components themselves that are important for these processes.
A number of centriolar satellite components are important for accurate mitosis. For example, we and others (49) have demonstrated that depletion of CEP131 increases multipolar spindles and chromosome missegregation. Given this increase in chromosomal instability with loss of CEP131, it might be suspected that CEP131 is a tumor suppressor. However, data from the Cancer Genome Atlas indicate that genomic deletion of CEP131 or decreased CEP131 gene expression are extremely rare events in cancer and are likely not drivers of chromosomal instability in cancer. Furthermore, increased CEP131 expression has been reported in hepatocellular carcinoma and correlates with worse outcomes (63). The specific mechanism by which centriolar satellites regulate proper chromosome segregation remains to be elucidated.
As the cell enters mitosis, centriolar satellites disperse, and upon completion of mitosis and entry into G1, the satellites reorganize around the centrosome (61). PLK4 phosphorylation of PCM1 and CEP131 may help regulate the reorganization of centriolar satellites in G1 in preparation for centriole duplication. Previous evidence has suggested that centriolar satellites are required for recruiting many key regulators of centriole duplication to the centrosome. Other possible mechanisms explaining centriolar satellite dispersion at the onset of mitosis include NEK2A kinase-dependent loss of C-NAP1 from the centrosome leading to reduced centriolar satellite density (64). There may be additional mechanisms of centriolar satellite dispersion in mitosis and reorganization in G1 that remain to be elucidated, and this is an important area of future research. One additional question is why and how some centriolar satellite proteins remain at the centrosome when centriolar satellites are dispersed in mitosis and with microtubule depolymerization (e.g. OFD1 and CEP290), whereas others (e.g. BBS4 and PCM1) do not remain at the centrosome under these conditions (6). Further work is required to elucidate this mechanism of centriolar satellite dispersion and aggregation during the cell cycle and its functional significance.
Centriolar satellites are also important for response to stresses (54), such as UV, heat stock, proteotoxic reagents, and nutrient deprivation. In response to serum starvation, MIB1 E3 ubiquitin ligase activity is reduced, which decreases PCM1 and CEP131 ubiquitination (59). Furthermore, loss of centriolar satellites by depletion of PCM1 sensitizes glioblastoma cells to temozolomide (65), a DNA-alkylating agent commonly used to treat glioblastoma. Further work will be needed to further elucidate these molecular mechanisms regarding centriolar satellites and response to cellular stresses. For example, because PLK4 inhibition causes centriolar satellite dispersion, does PLK4 inhibition also sensitize glioblastoma and other cancer types to temozolomide and other stress-inducing agents? As PLK4 inhibition is a potential strategy to treat cancer (40, 66–68), this is an important area of future research.
Another stressor, UV light, induces restructuring of centriolar satellites, including the displacement of PCM1, CEP131, and CEP290 from satellites (57, 59). Phosphorylation of CEP131 at the Ser-47 and Ser-78 residues by the p38 effector kinase MK2 promotes centriolar satellite remodeling and dissociation of CEP131 from PCM1 in response to UV light (57). Surprisingly, this previous report found that expression of both the nonphosphorylatable and phosphomimetic CEP131 prevented dispersion in response to UV light, whereas we have found that the nonphosphorylatable mutant causes centriolar satellite dispersion, whereas the phosphomimetic mutant promotes centriolar satellite aggregation. Although the reason for the discrepancy for the function of the phosphomimetic is unclear, we speculate that the regulatory mechanisms during DNA damage stresses, such as UV, may differ from non-DNA damage cellular stresses.
CEP131 is thought to be involved in centriole duplication or, at least, controlling centriole numbers. However, one report demonstrated that CEP131 depletion increases centriole numbers (49), whereas another report demonstrated that CEP131 depletion decreases centriole numbers (7). Additional evidence from Cep131 mutant MEFs found no differences in centrin foci or centrosome amplification (percentage of cells with more than two γ-tubulin foci) (52). Furthermore, USP9X is a deubiquitinase that was shown to stabilize CEP131. This prior report also found that USP9X is required for centriole duplication, providing some indirect evidence that CEP131 may be required for centriole duplication (69). However, herein we find that depletion of CEP131 does not significantly alter centriole number, regardless of the degree or duration of knockdown.
A limitation of our experiments is that PLK4 has low abundance with rare phosphorylation events, making it particularly challenging to detect direct substrates. As a result, we reduced our -fold change and statistical significance thresholds and focused on centrosomal proteins. Because of this, any future use of our phosphoproteomic data set to determine potential substrates of PLK4 will also require robust in vitro and in vivo validation. Furthermore, our analysis did not identify some of the previously identified PLK4 substrates, such as STIL. Despite this limitation, we detected other known substrates, including CEP152 and PCM1, as well as CEP131, which is validated as a direct substrate in biochemical assays. Another limitation of our work is that we narrowly focused our potential substrates (e.g. evolutionary conservation, role in centriole duplication), and these criteria may have been too strict to uncover novel PLK4 functions and substrates. Last, the identified phosphosite on CEP131, Ser-78, does not conform to the canonical PLK4 consensus motif (Ser/Thr followed by two hydrophobic residues) (70). However, this consensus motif does not hold in all cases. Despite this, we did validate that direct phosphorylation occurs with purified proteins.
In summary, we developed a chemical genetic method to interrogate PLK4 signaling and utilized multiplex phosphoproteomics to discover novel substrates of PLK4. We identified CEP131 as a novel substrate of PLK4 and found that PLK4 phosphorylation of CEP131 Ser-78 is important for proper organization and integrity of centriolar satellites and may help explain the changes seen in centriolar satellite organization during the cell cycle.
Experimental procedures
Generation of PLK4 conditional knockout and analog-sensitive cell lines
Gene editing was performed using adeno-associated virus in RPE-1 (ATCC, Manassas, VA) an immortalized, nontransformed human epithelial cell line. Procedures for preparation of infectious adeno-associated virus particles, transduction of RPE-1 cells, and isolation of properly targeted clones were performed as described previously (71, 72). Briefly, a targeting vector was made by inserting a neo cassette surrounded by FRT sites in the intron upstream of exons 3 and 4 of PLK4, all flanked by loxP sites (Fig. S1). To generate adenovirus producing this targeting vector, HEK293 cells were transfected with pRC and pHelper. RPE-1 cells were infected with adenovirus collected from the supernatant and subcloned in the presence of G418 (0.4 μg/ml) and screened/verified. This cell line was targeted in two ways: 1) to make a conditional knockout cell line and and 2) to make an analog sensitive cell line. To make the conditional knockout cell line, one copy of PLK4 was deleted by treating with Cre, and then cells were retargeted with the same initial vector (neo cassette, exons 3 and 4, all flanked by loxP sites). To make the analog-sensitive cell line, cells were transfected with pFLIPe to remove the neo cassette, subcloned, and retargeted with the initial targeting vector in which the gatekeeper residue was mutated to a glycine (L89G) to enlarge the ATP-binding pocket and genetically encode chemical sensitivity to the bulky ATP analog 3-MB-PP1 (42) (Fig. S1). The resulting ASflox/WTflox cell lines were treated with Cre and subcloned in the presence G418. Clones were screened by PCR with primers both inside and outside the neo cassette (Fig. S1). In addition, RNA was isolated from the AS cells and converted to cDNA, and exon 3 was PCR-amplified and sequenced to confirm the presence of only the mutant (L89G).
Cell culture and assays
All cell lines were propagated at 37 °C in 5% CO2. RPE-1–derived cell lines were grown in a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium supplemented with 2.5 mm l-glutamine, with 10% fetal bovine serum and 100 units/ml penicillin-streptomycin. HeLa cells (ATCC) were grown in high-glucose Dulbecco's modified Eagle's medium supplemented with 2.5 mm l-glutamine, with 10% fetal bovine serum and 100 units/ml penicillin-streptomycin. For stable retroviral transduction, constructs were co-transfected with a VSV-G envelope plasmid into Phoenix cells. Fresh medium was applied at 24 h post-transfection, harvested 24 h later, clarified by centrifugation and filtration through a 0.45-μm membrane to remove cell debris, and diluted 1:1 with complete medium containing 10 μg/ml Polybrene. Target cells were infected at 40–60% confluence for 24 h. Polyclonal transductants were further purified by limiting dilution to obtain individual clones.
To test for a proliferative defect, subcloning was performed in the presence of 10 μm 3-MB-PP1. Poisson correction was used to determine cloning efficiency. Cellular senescence was assayed using a pH-dependent β-gal staining kit (Cell Signaling Technology) according to the manufacturer's instructions.
Chemicals used in this study include aphidicolin (Sigma), 3-MB-PP1 (Toronto Research Chemicals), doxycycline (Fisher), puromycin (Thermo), SB203580 (p38 inhibitor, MedChem Express), etoposide (obtained from UW Oncology Pharmacy), doxorubicin (Fisher), paclitaxel (Fisher), vincristine (Fisher), thapsigargin (Fisher), tunicamycin (Fisher), chloroquine (Fisher), and bafilomycin A1 (Fisher).
siRNA directed against CEP131 (5′-AGGCCCTCAAGGCCAACAA-3′) was purchased from GE Healthcare, and control siRNA (Universal Negative Control 1) was purchased from Thermo Scientific.
For CEP131 knockdown-addback experiments, HeLa cells were grown to 80–90% confluence in 6-well plates. 2 μg of CEP131 cDNA plasmid and 4 μl of 20 μm siRNA were co-transfected simultaneously using Lipofectamine 2000 according to the manufacturer's instructions. Cells were given fresh medium 4 h after the transfection. 24 h after the transfection, cells were either transferred to coverslips for fixation and immunofluorescence or harvested for Western blotting and qRT-PCR.
For ciliogenesis, 5 × 104 cells were plated on coverslips, allowed to adhere overnight, washed three times with Hank's balanced salt solution, given serum-free medium for 48 h, and then fixed with methanol and stained for acetylated tubulin to mark primary cilia.
CRISPR/Cas9
The CRISPR/Cas9 system was used to create CEP131 knockout cell lines in HeLa cells. Four guide RNAs targeting CEP131 were cloned into lentiCRISPRv2 (73, 74) from Feng Zhang (Addgene plasmid 52961). The four guides used were GCGCTCCGGGACGCTGCCGA, CCGGGACGCTGCCGATGGCC, TTGACTCACCAGCGGGCCCG, and GGCACTGGAACTCCGGGCAT. Cutting efficiency of each guide was assessed by a surveyor assay (IDT, 706025) following PCR of 600-bp fragments from genomic DNA encompassing the guide RNA locus. lentiCRISPRv2 vectors were transfected into HeLa cells using Fugene (Promega). Prior to transfection, medium was replaced with serum- and antibiotic-free medium. 24 h after transfection, cells were selected with puromycin (1 μg/ml) for 72 h and then allowed to recover for 24 h in fresh medium. The guide RNA yielding the greatest cutting by surveyor assay (TTGACTCACCAGCGGGCCCG) was transfected again into HeLa cells, which were then subcloned by limiting dilution. Colonies were selected and screened by PCR of the region flanking the cut site and sequencing. Verification of knockout lines was performed with CEP131 Western blotting and immunofluorescence.
Immunofluorescence and microscopy
Immunofluorescence and imaging were carried out as described previously (75, 76). Cells were seeded on glass coverslips in 24-well plates and fixed with 100% ice-cold methanol for 30 min. Fixed cells were then blocked for 30 min in 3% BSA and 0.1% Triton X-100 in PBS (PBSTx + BSA). Primary antibodies were incubated in PBSTx + BSA for 1 h at room temperature and washed three times in PBSTx, followed by secondary antibody incubation in PBSTx + BSA for 30 min at room temperature and two washes with PBSTx. Cells were counterstained with 4′,6-diamidino-2-phenylindole and mounted on glass slides with Prolong Gold antifade medium (Invitrogen). Image acquisition was performed on a Nikon Eclipse Ti inverted microscope and Hamamatsu ORCA Flash 4.0 camera. Where indicated in the figure legends, optical sections were taken at 0.2-μm intervals and deconvolved using Nikon Elements. Where appropriate, the observer was blinded to treatment conditions during image acquisition and analysis. Images were processed and analyzed using Nikon Elements.
Qualitative analysis of centriolar satellites was performed by categorizing cells as either normal, dispersed, or aggregated, as described previously (19). Further, quantitative analysis of centriolar satellites was performed by taking z stack images, deconvolving, and quantifying the percentage of PCM1 or CEP72 intensity within a 2.5-μm radius of the center of the centrosome (identified by γ-tubulin).
Primary antibodies used were CEP131 (Thermo, PA5-38978, 1:500), CEP72 (Proteintech, 19928–1-AP, 1:500), CEP152 (Bethyl, A302-480A, 1:500), α-tubulin (Abcam, ab4074, 1:5000), acetylated tubulin (Santa Cruz Biotechnology, sc-23950, 1:1000), pericentrin (Abcam, ab4448, 1:1000), polyglutamylated tubulin (Adipogen, AG-20B-0020, 1:500), PCM1 (Cell Signaling, 5259, 1:500), centrin (Millipore, 04-1624, 1:500), γ tubulin (Abcam, ab27074, 1:1000), β-actin (Abcam, ab6276, 1:5000), FLAG (Sigma, F1804, 1:1000), and GFP (Invitrogen, A-11120, 1:1000). Alexa Fluor–conjugated secondary antibodies were used at 1:350 (Invitrogen).
Western blotting and immunoprecipitation
Cells were lysed in buffer (50 mm HEPES, pH 7.5, 100 mm NaCl, 0.5% Nonidet P-40, 10% glycerol) containing phosphatase inhibitors (10 mm sodium pyrophosphate, 5 mm β-glycerol phosphate, 50 mm NaF, 0.3 mm Na3VO4), 1 mm phenylmethylsulfonyl fluoride, protease inhibitor mixture (Thermo Scientific), and 1 mm DTT. Samples were sonicated and heated in SDS buffer. Proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride membrane (Millipore), and blocked for at least 30 min in 5% milk and 0.1% Tween 20 in TBS, pH 7.4 (TBST + milk). Membranes were incubated 1 h at room temperature or overnight at 4 °C with primary antibodies diluted in TBST + 5% milk, washed three times with TBST, and incubated for 1 h at room temperature in secondary antibodies conjugated to horseradish peroxidase in TBST + 5% milk. Membranes were washed and developed with luminol/peroxide (Millipore) and visualized with film.
Antibodies utilized for immunoblotting include actin (DSHB, JLA20, 1:1000), CEP131 (Thermo, PA5–38978, 1:1000), FLAG (Sigma, F1804, 1:5000), GFP (Invitrogen, A-11120, 1:1000), and α-tubulin (DSHB, 12G10, 1:1000). Horseradish peroxidase–conjugated secondary antibodies were used (Jackson, 1:5000). Relative intensities of bands were calculated from scanned images using ImageJ and normalized to their respective loading control.
For immunoprecipitation experiments, T75 flasks of HeLa cells were transfected with FLAG-PLK4 and GFP-tagged CEP131 fragments. Two days after transfection, cells were lysed in the aforementioned lysis buffer used for Western blotting. Extracts were incubated with anti-FLAG M2 agarose beads (Sigma). Beads were washed three times with lysis buffer, and immunoprecipitated protein complexes were analyzed by SDS-PAGE and immunoblotting with anti-FLAG and anti-GFP antibodies.
Preparation of samples for mass spectrometry
PLK4 AS cells were cultured in T175 plates. At 75% confluence, cells were synchronized at the G1-S phase with aphidicolin (5 μm) for 24 h. To assess the cell cycle, a fraction of cells from MS sample preparations were taken for flow cytometry. Cell pellets were fixed in cold 70% EtOH for at least 24 h, washed once in PBS, and resuspended in PBS with 0.5 mg/ml RNase A and 50 mg/ml propidium iodide. Samples were incubated at 4 °C overnight and analyzed on a flow cytometer (FACSCalibur, BD Biosciences). 3-MB-PP1 (10 μm) was added for the last 6 h to half of the plates. Three independent replicates were utilized for each of the two conditions. Cells were collected, pelleted, snap-frozen in liquid nitrogen, and stored at −80 °C until they were ready for lysis. Cell pellets were then lysed in buffer (8 m urea, 50 mm Tris, pH 8.0, 100 mm CaCl2) containing dissolved protease and phosphatase inhibitor tablets (Roche Applied Science) and then sonicated for 20 min. Protein concentrations were determined using a BCA kit (Thermo Pierce). Cell lysates were then reduced by the addition of DTT (final concentration of 5 mm), incubated at 37 °C for 45 min, and alkylated by adding iodoacetamide (15 mm final concentration). Next, lysates were incubated for 45 min in the dark at room temperature, and remaining iodoacetamide was quenched by bringing each lysate back to a final 5 mm DTT concentration. Lysates were diluted to a 1.5 m urea concentration using a 50 mm Tris and 100 mm CaCl2 solution. Trypsin was added to each lysate in a 50:1 (protein/enzyme) ratio and digested overnight at ambient temperature. The six samples were desalted using 100-mg C18 Sep-Pak cartridges (Waters) and dried down using a vacuum centrifuge to obtain tryptically digested peptides. 1 mg of peptides for each of the six samples was incubated with 6-plex TMT reagents (Thermo Scientific) for 3 h at room temperature. An aliquot of each sample was mixed in a 1:1 ratio and run on an Orbitrap Elite mass spectrometer (Thermo Scientific) to ensure complete TMT peptide labeling. The six samples were mixed in a final 1:1 ratio across all six TMT channels and desalted using a 500-mg C18 Sep-Pak cartridge (Waters) to produce a single pooled sample containing chemically labeled peptides from all six samples. The pooled sample was fractionated using strong cation exchange chromatography to produce 12 total peptide fractions, which were subsequently lyophilized and desalted. The resultant 12 peptide fractions were each enriched using immobilized metal affinity chromatography nickel-nitrilotriacetic acid magnetic agarose beads (Qiagen), leading to 12 final strong cation exchange–fractionated enriched phosphopeptide and unenriched peptide fractions. Each fraction was dried down using a vacuum centrifuge and resuspended in 0.2% formic acid for MS analyses.
LC-MS/MS
Each sample was introduced to an Orbitrap fusion mass spectrometer (Thermo Scientific) during a 90-min nano-LC separation using a nanoAcquity UPLC (Waters). A “Top N” fusion method was used to analyze eluting peptides, using a 15,000 resolving power survey scan followed by MS/MS scans collected at 15,000 resolving power. Peptides were fragmented using higher-energy collisional dissociation (HCD) at a normalized collision energy of 35%. Phosphopeptide fractions were analyzed with 200-ms maximum injection times for MS scans and 120-ms maximum injection times for MS/MS scans, whereas unenriched fractions were analyzed with 100-ms maximum injection times for MS scans and 75-ms maximum injection times for MS/MS scans. Only peptides with charge states from +2 to +8 were selected for MS/MS with an exclusion duration of 30 s. The 12 phosphopeptide samples were run in duplicate.
MS data analysis
Data were searched using Proteome Discoverer version 1.4.1.14 (Thermo Fisher Scientific) with the Sequest search algorithm. Thermo RAW files were searched against a Homo sapiens target-decoy database (UniProt, downloaded November 6, 2014). Peptide and phosphopeptide data sets were searched using a 50-ppm precursor mass tolerance and 0.02-Da fragment tolerance for b- and y-type ions produced by HCD fragmentation. All fractions were searched with static carbamidomethyl of cysteine residues, static TMT 6-plex modifications of peptide N termini and lysines, dynamic methionine oxidation, and dynamic TMT 6-plex modification of tyrosine residues. Phosphopeptide fractions were searched with additional dynamic phosphorylation modifications of serine, threonine, and tyrosine residues. Resulting peptide identifications were filtered to 1% false discovery rate and exported to tab-delimited text files compatible with the COMPASS software suite (77). COMPASS calculated the 6-plex TMT protein and phosphopeptide quantitation for all of the 12 peptide fractions and 12 phosphopeptide fractions. Peptides were mapped back to their parent proteins using COMPASS, and Phospho RS (78) was used to localize phosphorylation to amino acid residues with a fragment tolerance of 0.02 Da, automatically considering neutral loss peaks for HCD and considering a maximum of 200 maximum position isoforms per phosphopeptide. The raw 6-plex reporter ion intensities of localized phosphopeptide isoforms were log2-transformed and normalized against inhibited PLK4 (+3-MB-PP1) to obtain the relative phosphopeptide and protein quantitation for each cell line and condition.
Kinase assays
The kinase domain (amino acids 1–390) of human PLK4 was cloned into pGEX-6P-1 vector (GE Healthcare). The catalytically inactive mutant, K41M, was generated using Phusion site-directed QuikChange mutagenesis. Fifteen-amino acid fragments of the identified potential PLK4 substrates (putative phosphosite ± 7 amino acids) were also cloned into pGEX-6P-1. Sequences of these peptides can be found in Table S2. Protein was expressed in Rosetta DE3 bacteria, extracted with GSH Sepharose 4B (GE Healthcare, 17-0756-01), and eluted by the addition of GSH for the production of the intact GST fusion proteins.
All kinase assay reactions were incubated at 30 °C for 30 min in buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 0.1 mm NaF, 10 μm Na3VO4) with 1 mm DTT, 1 μm cold ATP, 2 μCi of [γ-32P]ATP, 5 μg of substrate, and 100 ng of PLK4. Reactions were resolved by SDS-PAGE. Dried gels were exposed to a storage phosphor screen (GE Healthcare), and γ-32P incorporation was visualized by a Typhoon TRIO imager (GE Healthcare).
To confirm that CEP131 Ser-78 is a substrate of PLK4, the S78A mutant (15-amino acid fragment, as above) was generated using Phusion site-directed mutagenesis, purified, and utilized in kinase assays, as described above.
Molecular biology
CEP131 cDNA was obtained as a kind gift from Drs. Spencer Collis and Katie Myers (corresponds to Uniprot isoform 2, Q9UPN4-2). Phusion site-directed mutagenesis was used to generate S78A and S78D mutants and to make CEP131 siRNA-resistant (mutated AGGCCCTCAAGGCCAACAA to AAGCATTGAAAGCAAATA).
qRT-PCR
RNA was isolated from cells using TRI Reagent (Molecular Research Center) per the manufacturer's protocol. DNase treatment was used to remove DNA contamination (Promega). RNA was then converted to cDNA using the Applied Biosystems High Capacity cDNA Reverse Transcription Kit and the manufacturer's protocol. Primer sequences are provided in Table S3. Primers were used at a final concentration of 0.67 μm each, and 25 ng of cDNA was used per reaction. A StepOne Plus (Applied Biosystems) real-time PCR thermal cycler was used for amplification with iQ SYBR Green Supermix (Bioard) per the manufacturer's protocol. Quantification of CEP131 mRNA was normalized to three housekeeping genes (RRN18S, GAPDH, and ACTB). The ΔΔCT method was employed to calculate the -fold change in expression (79).
Drug screening
To assess the sensitivity of CEP131 knockout cell lines to chemicals, 1000 cells/well were plated in 96-well plates and allowed to adhere for 24 h. Chemicals were added the next day. Four days later, medium was removed, and cell viability was determined using Vita-Orange cell viability reagent (Biotool) according to the manufacturer's instructions. The Vita-Orange reagent detects viable, metabolically active cells. Relative proliferation was determined by subtracting absorbance values of wells containing only Vita-Orange reagent from the experimental wells and then normalizing them to untreated control wells for each cell line.
Statistics
Statistical evaluations were performed using Prism software (GraphPad). Two-tailed t tests and one-way analysis of variance were used for comparisons. p values < 0.05 were considered significant for all tests, and statistical significance is indicated in the figures.
Author contributions
R. A. D., J. J. C., and M. E. B. conceptualization; R. A. D., G. K. P., J. J. C., and M. E. B. data curation; R. A. D., G. K. P., and M. E. B. formal analysis; R. A. D., J. J. C., and M. E. B. funding acquisition; R. A. D., M. M. S., J. M. J., G. K. P., and A. C. investigation; R. A. D. and J. M. J. visualization; R. A. D., M. M. S., J. M. J., G. K. P., A. C., J. J. C., and M. E. B. methodology; R. A. D. writing-original draft; R. A. D., M. M. S., J. M. J., A. C., J. J. C., and M. E. B. writing-review and editing; G. K. P. and J. J. C. validation; J. J. C. and M. E. B. resources; J. J. C. and M. E. B. supervision; J. J. C. and M. E. B. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Spencer Collis (University of Sheffield), Katie Myers (University of Sheffield), Tim Miller (University of Wisconsin), Takashi Toda (Hiroshima University), Jun Wan (University of Wisconsin), and Beth Weaver (University of Wisconsin) for reagents and formative discussions; Roshan Norman (University of Wisconsin) for assistance with determination of homology/conservation; and Rob Lera for critical review of the manuscript.
This work is supported by National Institutes of Health (NIH) Grants R01 GM097245 (to M. E. B.), F30CA203271 (to R. A. D.), the National Center for Quantitative Biology of Complex Systems (NIH Grant P41 GM108538 to J. J. C.), and University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520. M. E. B. is on the medical advisory board of Strata Oncology and receives research funding from Abbvie, Genentech, Puma, and Loxo Oncology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S3 and Figs. S1–S7.
- PLK4
- Polo-like kinase 4
- AS
- analog-sensitive
- KO
- knockout
- TMT
- tandem mass tags
- qRT-PCR
- quantitative RT-PCR
- TMT
- tandem mass tag
- HCD
- higher-energy collisional dissociation.
References
- 1. Sir J. H., Pütz M., Daly O., Morrison C. G., Dunning M., Kilmartin J. V., and Gergely F. (2013) Loss of centrioles causes chromosomal instability in vertebrate somatic cells. J. Cell Biol. 203, 747–756 10.1083/jcb.201309038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tanaka T., Serneo F. F., Higgins C., Gambello M. J., Wynshaw-Boris A., and Gleeson J. G. (2004) Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 165, 709–721 10.1083/jcb.200309025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsai J. W., Bremner K. H., and Vallee R. B. (2007) Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 10, 970–979 10.1038/nn1934 [DOI] [PubMed] [Google Scholar]
- 4. Mahjoub M. R. (2013) The importance of a single primary cilium. Organogenesis 9, 61–69 10.4161/org.25144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dammermann A., and Merdes A. (2002) Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 159, 255–266 10.1083/jcb.200204023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lopes C. A., Prosser S. L., Romio L., Hirst R. A., O'Callaghan C., Woolf A. S., and Fry A. M. (2011) Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J. Cell Sci. 124, 600–612 10.1242/jcs.077156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kodani A., Yu T. W., Johnson J. R., Jayaraman D., Johnson T. L., Al-Gazali L., Sztriha L., Partlow J. N., Kim H., Krup A. L., Dammermann A., Krogan N. J., Walsh C. A., and Reiter J. F. (2015) Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. Elife 4, 10.7554/eLife.07519 10.7554/eLife.07519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bettencourt-Dias M., Rodrigues-Martins A., Carpenter L., Riparbelli M., Lehmann L., Gatt M. K., Carmo N., Balloux F., Callaini G., and Glover D. M. (2005) SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 15, 2199–2207 10.1016/j.cub.2005.11.042 [DOI] [PubMed] [Google Scholar]
- 9. Holland A. J., Fachinetti D., Da Cruz S., Zhu Q., Vitre B., Lince-Faria M., Chen D., Parish N., Verma I. M., Bettencourt-Dias M., and Cleveland D. W. (2012) Polo-like kinase 4 controls centriole duplication but does not directly regulate cytokinesis. Mol. Biol. Cell 23, 1838–1845 10.1091/mbc.e11-12-1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Godinho S. A., Picone R., Burute M., Dagher R., Su Y., Leung C. T., Polyak K., Brugge J. S., Théry M., and Pellman D. (2014) Oncogene-like induction of cellular invasion from centrosome amplification. Nature 510, 167–171 10.1038/nature13277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Denu R. A., Zasadil L. M., Kanugh C., Laffin J., Weaver B. A., and Burkard M. E. (2016) Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 16, 47 10.1186/s12885-016-2083-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sonnen K. F., Gabryjonczyk A. M., Anselm E., Stierhof Y. D., and Nigg E. A. (2013) Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J. Cell Sci. 126, 3223–3233 10.1242/jcs.129502 [DOI] [PubMed] [Google Scholar]
- 13. Kim T. S., Park J. E., Shukla A., Choi S., Murugan R. N., Lee J. H., Ahn M., Rhee K., Bang J. K., Kim B. Y., Loncarek J., Erikson R. L., and Lee K. S. (2013) Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc. Natl. Acad. Sci. U.S.A. 110, E4849–E4857 10.1073/pnas.1319656110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fırat-Karalar E. N., and Stearns T. (2014) The centriole duplication cycle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130460 10.1098/rstb.2013.0460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kitagawa D., Vakonakis I., Olieric N., Hilbert M., Keller D., Olieric V., Bortfeld M., Erat M. C., Flückiger I., Gönczy P., and Steinmetz M. O. (2011) Structural basis of the 9-fold symmetry of centrioles. Cell 144, 364–375 10.1016/j.cell.2011.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmidt T. I., Kleylein-Sohn J., Westendorf J., Le Clech M., Lavoie S. B., Stierhof Y. D., and Nigg E. A. (2009) Control of centriole length by CPAP and CP110. Curr. Biol. 19, 1005–1011 10.1016/j.cub.2009.05.016 [DOI] [PubMed] [Google Scholar]
- 17. Martin C. A., Ahmad I., Klingseisen A., Hussain M. S., Bicknell L. S., Leitch A., Nürnberg G., Toliat M. R., Murray J. E., Hunt D., Khan F., Ali Z., Tinschert S., Ding J., Keith C., et al. (2014) Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat. Genet. 46, 1283–1292 10.1038/ng.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wheway G., Schmidts M., Mans D. A., Szymanska K., Nguyen T. T., Racher H., Phelps I. G., Toedt G., Kennedy J., Wunderlich K. A., Sorusch N., Abdelhamed Z. A., Natarajan S., Herridge W., van Reeuwijk J., et al. (2015) An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 17, 1074–1087 10.1038/ncb3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hori A., Barnouin K., Snijders A. P., and Toda T. (2016) A non-canonical function of Plk4 in centriolar satellite integrity and ciliogenesis through PCM1 phosphorylation. EMBO Rep. 17, 326–337 10.15252/embr.201541432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunha-Ferreira I., Bento I., Pimenta-Marques A., Jana S. C., Lince-Faria M., Duarte P., Borrego-Pinto J., Gilberto S., Amado T., Brito D., Rodrigues-Martins A., Debski J., Dzhindzhev N., and Bettencourt-Dias M. (2013) Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr. Biol. 23, 2245–2254 10.1016/j.cub.2013.09.037 [DOI] [PubMed] [Google Scholar]
- 21. Guderian G., Westendorf J., Uldschmid A., and Nigg E. A. (2010) Plk4 trans-autophosphorylation regulates centriole number by controlling βTrCP-mediated degradation. J. Cell Sci. 123, 2163–2169 10.1242/jcs.068502 [DOI] [PubMed] [Google Scholar]
- 22. Holland A. J., Fachinetti D., Zhu Q., Bauer M., Verma I. M., Nigg E. A., and Cleveland D. W. (2012) The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev. 26, 2684–2689 10.1101/gad.207027.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sillibourne J. E., Tack F., Vloemans N., Boeckx A., Thambirajah S., Bonnet P., Ramaekers F. C., Bornens M., and Grand-Perret T. (2010) Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 21, 547–561 10.1091/mbc.e09-06-0505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Puklowski A., Homsi Y., Keller D., May M., Chauhan S., Kossatz U., Grünwald V., Kubicka S., Pich A., Manns M. P., Hoffmann I., Gönczy P., and Malek N. P. (2011) The SCF-FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat. Cell Biol. 13, 1004–1009 10.1038/ncb2282 [DOI] [PubMed] [Google Scholar]
- 25. Bahtz R., Seidler J., Arnold M., Haselmann-Weiss U., Antony C., Lehmann W. D., and Hoffmann I. (2012) GCP6 is a substrate of Plk4 and required for centriole duplication. J. Cell Sci. 125, 486–496 10.1242/jcs.093930 [DOI] [PubMed] [Google Scholar]
- 26. Lee M., Seo M. Y., Chang J., Hwang D. S., and Rhee K. (2017) PLK4 phosphorylation of CP110 is required for efficient centriole assembly. Cell Cycle 16, 1225–1234 10.1080/15384101.2017.1325555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kitagawa D., Busso C., Flückiger I., and Gönczy P. (2009) Phosphorylation of SAS-6 by ZYG-1 is critical for centriole formation in C. elegans embryos. Dev. Cell 17, 900–907 10.1016/j.devcel.2009.11.002 [DOI] [PubMed] [Google Scholar]
- 28. Kratz A. S., Bärenz F., Richter K. T., and Hoffmann I. (2015) Plk4-dependent phosphorylation of STIL is required for centriole duplication. Biol. Open 4, 370–377 10.1242/bio.201411023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moyer T. C., Clutario K. M., Lambrus B. G., Daggubati V., and Holland A. J. (2015) Binding of STIL to Plk4 activates kinase activity to promote centriole assembly. J. Cell Biol. 209, 863–878 10.1083/jcb.201502088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dzhindzhev N. S., Tzolovsky G., Lipinszki Z., Schneider S., Lattao R., Fu J., Debski J., Dadlez M., and Glover D. M. (2014) Plk4 phosphorylates Ana2 to trigger Sas6 recruitment and procentriole formation. Curr. Biol. 24, 2526–2532 10.1016/j.cub.2014.08.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McLamarrah T. A., Buster D. W., Galletta B. J., Boese C. J., Ryniawec J. M., Hollingsworth N. A., Byrnes A. E., Brownlee C. W., Slep K. C., Rusan N. M., and Rogers G. C. (2018) An ordered pattern of Ana2 phosphorylation by Plk4 is required for centriole assembly. J. Cell Biol. 217, 1217–1231 10.1083/jcb.201605106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galletta B. J., Fagerstrom C. J., Schoborg T. A., McLamarrah T. A., Ryniawec J. M., Buster D. W., Slep K. C., Rogers G. C., and Rusan N. M. (2016) A centrosome interactome provides insight into organelle assembly and reveals a non-duplication role for Plk4. Nat. Commun. 7, 12476 10.1038/ncomms12476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martindill D. M., Risebro C. A., Smart N., Franco-Viseras M. d. M., Rosario C. O., Swallow C. J., Dennis J. W., and Riley P. R. (2007) Nucleolar release of Hand1 acts as a molecular switch to determine cell fate. Nat. Cell Biol. 9, 1131–1141 10.1038/ncb1633 [DOI] [PubMed] [Google Scholar]
- 34. Kazazian K., Go C., Wu H., Brashavitskaya O., Xu R., Dennis J. W., Gingras A. C., and Swallow C. J. (2017) Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 complex regulation of the actin cytoskeleton. Cancer Res. 77, 434–447 10.1158/0008-5472.CAN-16-2060 [DOI] [PubMed] [Google Scholar]
- 35. Hatch E. M., Kulukian A., Holland A. J., Cleveland D. W., and Stearns T. (2010) Cep152 interacts with Plk4 and is required for centriole duplication. J. Cell Biol. 191, 721–729 10.1083/jcb.201006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bonni S., Ganuelas M. L., Petrinac S., and Hudson J. W. (2008) Human Plk4 phosphorylates Cdc25C. Cell Cycle 7, 545–547 10.4161/cc.7.4.5387 [DOI] [PubMed] [Google Scholar]
- 37. Petrinac S., Ganuelas M. L., Bonni S., Nantais J., and Hudson J. W. (2009) Polo-like kinase 4 phosphorylates Chk2. Cell Cycle 8, 327–329 10.4161/cc.8.2.7355 [DOI] [PubMed] [Google Scholar]
- 38. Rosario C. O., Ko M. A., Haffani Y. Z., Gladdy R. A., Paderova J., Pollett A., Squire J. A., Dennis J. W., and Swallow C. J. (2010) Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc. Natl. Acad. Sci. U.S.A. 107, 6888–6893 10.1073/pnas.0910941107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Firat-Karalar E. N., Rauniyar N., Yates J. R. 3rd, and Stearns T. (2014) Proximity interactions among centrosome components identify regulators of centriole duplication. Curr. Biol. 24, 664–670 10.1016/j.cub.2014.01.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong Y. L., Anzola J. V., Davis R. L., Yoon M., Motamedi A., Kroll A., Seo C. P., Hsia J. E., Kim S. K., Mitchell J. W., Mitchell B. J., Desai A., Gahman T. C., Shiau A. K., and Oegema K. (2015) Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 348, 1155–1160 10.1126/science.aaa5111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lambrus B. G., Uetake Y., Clutario K. M., Daggubati V., Snyder M., Sluder G., and Holland A. J. (2015) p53 protects against genome instability following centriole duplication failure. J. Cell Biol. 210, 63–77 10.1083/jcb.201502089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holland A. J., Lan W., Niessen S., Hoover H., and Cleveland D. W. (2010) Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J. Cell Biol. 188, 191–198 10.1083/jcb.200911102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Merrill A. E., and Coon J. J. (2013) Quantifying proteomes and their post-translational modifications by stable isotope label-based mass spectrometry. Curr. Opin. Chem. Biol. 17, 779–786 10.1016/j.cbpa.2013.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Richards A. L., Merrill A. E., and Coon J. J. (2015) Proteome sequencing goes deep. Curr. Opin. Chem. Biol. 24, 11–17 10.1016/j.cbpa.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Riley N. M., and Coon J. J. (2016) Phosphoproteomics in the age of rapid and deep proteome profiling. Anal. Chem. 88, 74–94 10.1021/acs.analchem.5b04123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Riley N. M., Hebert A. S., and Coon J. J. (2016) Proteomics moves into the fast lane. Cell Syst. 2, 142–143 10.1016/j.cels.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 47. Kleylein-Sohn J., Westendorf J., Le Clech M., Habedanck R., Stierhof Y. D., and Nigg E. A. (2007) Plk4-induced centriole biogenesis in human cells. Dev. Cell 13, 190–202 10.1016/j.devcel.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 48. Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., and Mann M. (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 10.1038/nature02166 [DOI] [PubMed] [Google Scholar]
- 49. Staples C. J., Myers K. N., Beveridge R. D., Patil A. A., Lee A. J., Swanton C., Howell M., Boulton S. J., and Collis S. J. (2012) The centriolar satellite protein Cep131 is important for genome stability. J. Cell Sci. 125, 4770–4779 10.1242/jcs.104059 [DOI] [PubMed] [Google Scholar]
- 50. Mori Y., Inoue Y., Tanaka S., Doda S., Yamanaka S., Fukuchi H., and Terada Y. (2015) Cep169, a novel microtubule plus-end-tracking centrosomal protein, binds to CDK5RAP2 and regulates microtubule stability. PLoS One 10, e0140968 10.1371/journal.pone.0140968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Korzeniewski N., Cuevas R., Duensing A., and Duensing S. (2010) Daughter centriole elongation is controlled by proteolysis. Mol. Biol. Cell 21, 3942–3951 10.1091/mbc.e09-12-1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hall E. A., Keighren M., Ford M. J., Davey T., Jarman A. P., Smith L. B., Jackson I. J., and Mill P. (2013) Acute versus chronic loss of mammalian Azi1/Cep131 results in distinct ciliary phenotypes. PLoS Genet. 9, e1003928 10.1371/journal.pgen.1003928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Graser S., Stierhof Y. D., Lavoie S. B., Gassner O. S., Lamla S., Le Clech M., and Nigg E. A. (2007) Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 179, 321–330 10.1083/jcb.200707181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hori A., and Toda T. (2017) Regulation of centriolar satellite integrity and its physiology. Cell. Mol. Life Sci. 74, 213–229 10.1007/s00018-016-2315-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ai R., Sun Y., Guo Z., Wei W., Zhou L., Liu F., Hendricks D. T., Xu Y., and Zhao X. (2016) NDRG1 overexpression promotes the progression of esophageal squamous cell carcinoma through modulating Wnt signaling pathway. Cancer Biol. Ther. 17, 943–954 10.1080/15384047.2016.1210734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kubo A., Sasaki H., Yuba-Kubo A., Tsukita S., and Shiina N. (1999) Centriolar satellites: molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J. Cell Biol. 147, 969–980 10.1083/jcb.147.5.969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tollenaere M. A., Villumsen B. H., Blasius M., Nielsen J. C., Wagner S. A., Bartek J., Beli P., Mailand N., and Bekker-Jensen S. (2015) p38- and MK2-dependent signalling promotes stress-induced centriolar satellite remodelling via 14-3-3-dependent sequestration of CEP131/AZI1. Nat. Commun. 6, 10075 10.1038/ncomms10075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Blomen V. A., Májek P., Jae L. T., Bigenzahn J. W., Nieuwenhuis J., Staring J., Sacco R., van Diemen F. R., Olk N., Stukalov A., Marceau C., Janssen H., Carette J. E., Bennett K. L., Colinge J., et al. (2015) Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 10.1126/science.aac7557 [DOI] [PubMed] [Google Scholar]
- 59. Villumsen B. H., Danielsen J. R., Povlsen L., Sylvestersen K. B., Merdes A., Beli P., Yang Y. G., Choudhary C., Nielsen M. L., Mailand N., and Bekker-Jensen S. (2013) A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. EMBO J. 32, 3029–3040 10.1038/emboj.2013.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Balczon R., Varden C. E., and Schroer T. A. (1999) Role for microtubules in centrosome doubling in Chinese hamster ovary cells. Cell Motil. Cytoskeleton 42, 60–72 10.1002/(SICI)1097-0169(1999)42:1<60::AID-CM6>3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 61. Balczon R., Bao L., and Zimmer W. E. (1994) PCM-1, A 228-kD centrosome autoantigen with a distinct cell cycle distribution. J. Cell Biol. 124, 783–793 10.1083/jcb.124.5.783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baron Gaillard C. L., Pallesi-Pocachard E., Massey-Harroche D., Richard F., Arsanto J. P., Chauvin J. P., Lecine P., Krämer H., Borg J. P., and Le Bivic A. (2011) Hook2 is involved in the morphogenesis of the primary cilium. Mol. Biol. Cell 22, 4549–4562 10.1091/mbc.e11-05-0405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu X. H., Yang Y. F., Fang H. Y., Wang X. H., Zhang M. F., and Wu D. C. (2017) CEP131 indicates poor prognosis and promotes cell proliferation and migration in hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 90, 1–8 10.1016/j.biocel.2017.07.001 [DOI] [PubMed] [Google Scholar]
- 64. Flanagan A. M., Stavenschi E., Basavaraju S., Gaboriau D., Hoey D. A., and Morrison C. G. (2017) Centriole splitting caused by loss of the centrosomal linker protein C-NAP1 reduces centriolar satellite density and impedes centrosome amplification. Mol. Biol. Cell 28, 736–745 10.1091/mbc.e16-05-0325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hoang-Minh L. B., Deleyrolle L. P., Nakamura N. S., Parker A. K., Martuscello R. T., Reynolds B. A., and Sarkisian M. R. (2016) PCM1 depletion inhibits glioblastoma cell ciliogenesis and increases cell death and sensitivity to temozolomide. Transl. Oncol. 9, 392–402 10.1016/j.tranon.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mason J. M., Lin D. C., Wei X., Che Y., Yao Y., Kiarash R., Cescon D. W., Fletcher G. C., Awrey D. E., Bray M. R., Pan G., and Mak T. W. (2014) Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 26, 163–176 10.1016/j.ccr.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 67. Denu R. A., Shabbir M., Nihal M., Singh C. K., Longley B. J., Burkard M. E., and Ahmad N. (2018) Centriole overduplication is the predominant mechanism leading to centrosome amplification in melanoma. Mol. Cancer Res. 16, 517–527 10.1158/1541-7786.MCR-17-0197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bedard P. L., Cescon D. W., Fletcher G., Denny T., Brokx R., Sampson P., Bray M. R., Slamon D. J., Mak T. W., and Wainberg Z. A. (2016) First-in-human phase I trial of the oral PLK4 inhibitor CFI-400945 in patients with advanced solid tumors. Cancer Res. 10.1158/1538-7445 AM2016-CT066 [DOI] [Google Scholar]
- 69. Li X., Song N., Liu L., Liu X., Ding X., Song X., Yang S., Shan L., Zhou X., Su D., Wang Y., Zhang Q., Cao C., Ma S., Yu N., et al. (2017) USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat. Commun. 8, 14866 10.1038/ncomms14866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Leung G. C., Ho C. S., Blasutig I. M., Murphy J. M., and Sicheri F. (2007) Determination of the Plk4/Sak consensus phosphorylation motif using peptide spots arrays. FEBS Lett. 581, 77–83 10.1016/j.febslet.2006.11.080 [DOI] [PubMed] [Google Scholar]
- 71. Berdougo E., Terret M. E., and Jallepalli P. V. (2009) Functional dissection of mitotic regulators through gene targeting in human somatic cells. Methods Mol. Biol. 545, 21–37 10.1007/978-1-60327-993-2_2 [DOI] [PubMed] [Google Scholar]
- 72. Burkard M. E., Randall C. L., Larochelle S., Zhang C., Shokat K. M., Fisher R. P., and Jallepalli P. V. (2007) Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc. Natl. Acad. Sci. U.S.A. 104, 4383–4388 10.1073/pnas.0701140104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelson T., Heckl D., Ebert B. L., Root D. E., Doench J. G., and Zhang F. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lasek A. L., McPherson B. M., Trueman N. G., and Burkard M. E. (2016) The functional significance of posttranslational modifications on Polo-like kinase 1 revealed by chemical genetic complementation. PLoS One 11, e0150225 10.1371/journal.pone.0150225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lera R. F., and Burkard M. E. (2012) High mitotic activity of Polo-like kinase 1 is required for chromosome segregation and genomic integrity in human epithelial cells. J. Biol. Chem. 287, 42812–42825 10.1074/jbc.M112.412544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., and Coon J. J. (2011) COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 10.1002/pmic.201000616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., and Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 10.1021/pr200611n [DOI] [PubMed] [Google Scholar]
- 79. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 80. Klebba J. E., Buster D. W., Nguyen A. L., Swatkoski S., Gucek M., Rusan N. M., and Rogers G. C. (2013) Polo-like kinase 4 autodestructs by generating its Slimb-binding phosphodegron. Curr. Biol. 23, 2255–2261 10.1016/j.cub.2013.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chang J., Cizmecioglu O., Hoffmann I., and Rhee K. (2010) PLK2 phosphorylation is critical for CPAP function in procentriole formation during the centrosome cycle. EMBO J. 29, 2395–2406 10.1038/emboj.2010.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pihan G. A. (2013) Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front. Oncol. 3, 277 10.3389/fonc.2013.00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.