

Abstract
Upon phagocytosis into macrophages, the intracellular bacterial pathogen Legionella pneumophila secretes effector proteins that manipulate host cell components, enabling it to evade lysosomal degradation. However, the bacterial proteins involved in this evasion are incompletely characterized. Here we show that the L. pneumophila effector protein RavD targets host membrane compartments and contributes to the molecular mechanism the pathogen uses to prevent encounters with lysosomes. Protein–lipid binding assays revealed that RavD selectively binds phosphatidylinositol-3-phosphate (PI(3)P) in vitro. We further determined that a C-terminal RavD region mediates the interaction with PI(3)P and that this interaction requires Arg-292. In transiently transfected mammalian cells, mCherry-RavD colocalized with the early endosome marker EGFP-Rab5 as well as the PI(3)P biosensor EGFP-2×FYVE. However, treatment with the phosphoinositide 3-kinase inhibitor wortmannin did not disrupt localization of mCherry-RavD to endosomal compartments, suggesting that RavD's interaction with PI(3)P is not necessary to anchor RavD to endosomal membranes. Using superresolution and immunogold transmission EM, we observed that, upon translocation into macrophages, RavD was retained onto the Legionella-containing vacuole and was also present on small vesicles adjacent to the vacuole. We also report that despite no detectable effects on intracellular growth of L. pneumophila within macrophages or amebae, the lack of RavD significantly increased the number of vacuoles that accumulate the late endosome/lysosome marker LAMP-1 during macrophage infection. Together, our findings suggest that, although not required for intracellular replication of L. pneumophila, RavD is a part of the molecular mechanism that steers the Legionella-containing vacuole away from endolysosomal maturation pathways.
Keywords: host–pathogen interaction, phosphoinositide, infection, virulence factor, bacterial pathogenesis, bacterial effectors, cellular localization, immune evasion, Legionella pneumophila, Legionnaires' disease
Introduction
Legionella pneumophila is a facultative intracellular bacterial pathogen that commonly infects protozoa, and in humans it causes a life-threatening pneumonia known as Legionnaires' disease (1). Following inhalation of contaminated aerosols, the bacterium reaches the lungs, where it infects resident alveolar macrophages as well as neutrophils (2, 3). These professional phagocytes ingest microbes by phagocytosis, sealing them into a plasma membrane–derived compartment, the nascent phagosome, which gradually acquires microbicidal properties through progressive fusions with endocytic compartments and, finally, with the lysosome (4). This process, referred to as phagosome maturation, is critical for microbial degradation; however, L. pneumophila and related species of bacterial pathogens have the capacity to arrest this process (5). Upon phagocytosis, L. pneumophila gradually converts the phagosome into a replication-permissive niche resembling the endoplasmic reticulum, termed the Legionella-containing vacuole (LCV)2 (6–8). This entails remodeling of the vacuolar membrane and depends on the function of a specialized Dot/Icm type IVb secretion system that delivers L. pneumophila effector proteins (over 300) directly into the host cell (9–12). Repurposing the degradation-destined phagosome into a bacterial replication shelter is crucial for intracellular survival of L. pneumophila, but the effector proteins responsible for disrupting phagosome maturation remain largely unknown.
Phagosome maturation upon engulfment of microbes entails a tightly coordinated sequence of events (13). Phosphoinositides, a class of lipids derived from phosphatidylinositol, play a key role in regulating these events (14). Seven phosphoinositide species, each specifically enriched on distinct cellular membrane compartments, arise from reactions catalyzed by enzymes that phosphorylate or dephosphorylate the D-myo-inositol headgroup of phosphatidylinositol (at positions 3′, 4′, and/or 5′) (15). Phosphoinositide distribution, therefore, is largely determined by the localization of enzymes that regulate their metabolism. Because of their preferential distribution, phosphoinositides help impart organelle identity, serving as signposts for the recruitment and activation of protein complexes (16). The sequential appearance of phosphoinositides on the phagosome is centrally important for the regulation of phagosome maturation (14, 17). Phosphatidylinositol-3-phosphate (PI(3)P) accumulates on early phagosomes, signaling the onset of maturation (18), and depletion of PI(3)P impairs progression through phagosome maturation. L. pneumophila alters this dynamic during infection, as shown by live-cell tracking of phosphoinositides on the LCV, which revealed that PI(3)P levels rise on the vacuolar membrane soon after sealing and following an initial brief PI(4)P wave, but then PI(3)P levels gradually diminish, concurrent with resurgence of PI(4)P (19). This dynamic depended on the bacterium's ability to translocate effector proteins, as the vacuoles harboring translocation-deficient mutants did not lose PI(3)P and did not appear to accumulate PI(4)P. Previous studies showed that L. pneumophila effector proteins actively deplete PI(3)P on the vacuolar membrane either directly or by recruiting host cell enzymes that metabolize phosphoinositides (20–22). Beyond altering the phosphoinositide composition of the LCV, L. pneumophila effectors harbor regions that selectively bind phosphoinositides to target particular membrane compartments or to latch on to the cytosolic façade of the vacuolar membrane, essentially acting as molecular carabiners (23, 24). Further understanding the roles phosphoinositide-binding effectors play during infection will provide a clearer perspective on how effectors capitalize on host phosphoinositides to support intracellular survival.
In this study, we showed that the effector protein RavD selectively interacts with PI(3)P via a C-terminal region and that it is bound to the vacuolar surface and adjacent vesicles beginning early during infection. Despite no distinguishable intracellular growth defects, the absence of RavD led to a significant increase in the number of LCVs that colocalized with LAMP-1, consistent with the hypothesis that RavD is contributes to phagolysosomal avoidance.
Results
RavD associates with host membranes and preferentially binds PI(3)P
RavD (Lpg0160) was first identified as a translocated effector through a SidC fusion translocation assay (25). Because RavD has not yet been experimentally characterized, we set out to determine its subcellular localization in mammalian cells. To this end, we transiently transfected HEK293T cells with a plasmid producing either mCherry or mCherry-RavD. By cellular fractionation, we found mCherry-RavD both in the cytosol and on membranes, whereas mCherry was only present in the cytosolic fractions, as expected (Fig. 1A). A growing number of L. pneumophila effectors bind host membrane compartments by recognizing specific phosphoinositides, and therefore, we next asked whether RavD also targets host membranes in this way. To address this question, we examined the cellular distribution of RavD in relation to the tandem FYVE domains of the early endosomal antigen 1 (EEA1), commonly used as a PI(3)P marker (26). HeLa cells were transiently co-transfected with plasmids encoding mCherry-RavD and EGFP-2×FYVE or EGFP alone. By confocal microscopy, we determined that mCherry-RavD was present in both the cytosol as well as on cellular structures enriched in PI(3)P (Fig. 1B). To determine the extent of colocalization between mCherry-RavD and either EGFP or EGFP-2×FYVE, we quantified the overlap between the red and green fluorescence signals by calculating the Mander's overlap coefficient (MOC, where a value of 1 represents complete overlap and 0 represents random localization). A total of 15 cells were analyzed for each condition. A Kruskal–Wallis test was run to compare Mander's overlap coefficients obtained under each condition (Kruskal–Wallis, H = 18, p < 1–13). We then used Dunn–Sidak post hoc tests to statistically determine which groups differed while correcting for multiple comparisons. Given the nonparametric distribution, MOCs were expressed as median and interquartile range. Based on these statistical tests, we determined that the overlap between mCherry-RavD and EGFP-2×FYVE signals (0.60, IQR 0.50–0.68) was significantly higher than that between mCherry-RavD and EGFP (0.22, IQR 0.21–0.28).
Figure 1.

RavD localizes to membrane compartments positive for PI(3)P and binds PI(3)P in vitro. A, RavD is cytosolic and membrane-bound. HEK293T cells producing mCherry-RavD or mCherry alone were homogenized, and the post-nuclear supernatant (PNS) was subjected to cellular fractionation. An anti-mCherry antibody was used to detect the presence of RavD. Antibodies against tubulin and calnexin were used to mark the cytosolic (Cyto) and membrane (Mem) fractions, respectively. B, confocal images of HeLa cells transiently co-transfected with plasmids encoding either EGFP-2×FYVE or EGFP and mCherry-RavD or mCherry. Scale bars = 10 μm (insets, 2 μm). C, protein–lipid overlay assay showing that GST-RavD specifically recognizes PI(3)P. A nitrocellulose membrane prespotted with 100 pmol of each indicated lipid was incubated with purified GST-RavD. RavD retained on the membrane was detected by incubation with anti-GST and an HRP-conjugated secondary anti-rabbit antibody. LPA, lysophosphatidic acid; LPC, lysophosphocholine; PtdIns, phosphatidylinositol; PE, phosphatidylethanolamine; PC, phosphatidylcholine; SIP, sphingosine-1-phosphate; PI, phosphatidylinositol; P, phosphate; P2, biphosphate; P3, triphosphate; PA, phosphatidic acid; PS, phosphatidylserine. D, liposomes containing the indicated lipids were incubated with GST-RavD, GST-SidM, or GST alone and subjected to ultracentrifugation to separate bound from unbound protein. Input and float samples were separated on 4–15% TGX stain–free SDS-PAGE gels. Results are representatives of at least two independent experiments with similar outcomes.
To determine whether RavD binds phosphoinositides directly, we performed an in vitro protein–lipid overlay assay using purified GST-RavD (Fig. S2) and a nitrocellulose membrane prespotted with all seven phosphoinositides and several other lipids (Fig. 1C). We found that GST-RavD preferentially recognized PI(3)P. We further confirmed this finding by performing a liposome flotation assay using liposomes containing 10% phosphatidylcholine, 80% phosphatidylserine (PS), and 10% PI(3)P or PI(4)P (Fig. 1D). Purified proteins (Fig. S2) were incubated with liposomes for 30 min, after which density gradient centrifugation was performed to separate liposomes from unbound protein. In agreement with the protein–lipid overlay data, GST-RavD was recovered only in liposome samples containing PI(3)P and not PI(4)P. As expected based on previous findings, GST-SidM co-floated predominantly with liposomes containing PI(4)P. Together, these results demonstrate that RavD selectively interacts with PI(3)P.
A C-terminal region of RavD facilitates interaction with PI(3)P
L. pneumophila effectors that associate with membrane compartments are typically multidomain proteins, and when present, the phosphoinositide-binding region is often harbored closer to the C terminus (23). To determine which region within RavD was responsible for PI(3)P binding, we generated two truncated variants of RavD containing amino acids 1–189 (RavD1–189) and 190–325 (RavD190–325). The boundary between these fragments was selected based on secondary structure predictions so that secondary structures were not disrupted (Fig. 2A). These fragments were fused to an N-terminal GST or mCherry tag to perform protein-lipid overlay or subcellular localization assays, respectively. The protein-lipid overlay assay revealed that the GST-RavD190–325 fragment retained the ability to bind PI(3)P whereas GST-RavD1–189 did not (Fig. 2C). We next asked whether this C-terminal fragment also colocalizes with PI(3)P-rich endosomes. To this end, HeLa cells were transiently transfected with plasmids encoding EGFP-2×FYVE and either of the two truncations, mCherry-RavD1–189 or mCherry-RavD190–325 (Fig. 2D). Although mCherry-RavD190–325 displayed a punctate pattern, the mCherry-RavD1–189 fluorescence signal was mostly cytosolic (Fig. 2D). Quantification of the signal overlap indicated that mCherry-RavD190–325 colocalized with EGFP-2×FYVE (0.63, IQR 0.50–0.67) whereas mCherry-RavD1–189 did not (0.09, IQR 0.01–0.17), supporting the notion that RavD binds PI(3)P and PI(3)P-positive membrane compartments via a 135-amino acid C-terminal region.
Figure 2.

The PI(3)P binding region of RavD is positioned within a C-terminal region. A, schematic of the predicted secondary structure of RavD (obtained using the JPred Protein Secondary Structure Prediction Server) (blue, α helix; yellow, β sheet); the position of relevant amino acid (aa) residues is marked. B, alignment of RavD's conserved arginine residues in the indicated Legionella species. C, protein–lipid overlay assays show that the GST-RavD190–325 and GST-RavDR237A variants recognize PI(3)P whereas GST-RavD1–189 and GST-RavDR292A do not. LPA, lysophosphatidic acid; LPC, lysophosphocholine; PtdIns, phosphatidylinositol; PE, phosphatidylethanolamine; PC, phosphatidylcholine; SIP, sphingosine-1-phosphate; PI, phosphatidylinositol; P, phosphate; P2, biphosphate; P3, triphosphate; PA, phosphatidic acid; PS, phosphatidylserine. D, confocal images of HeLa cells transiently co-transfected with plasmids encoding either EGFP-2×FYVE and variants mCherry-RavD190–325, mCherry-RavD1–189, GST-RavDR237A, or RavDR292A. Scale bars = 10 μm (insets, 2 μm). Confocal images and assays are representative of at least two independent experiments with similar outcomes. E, Mander's overlap coefficient for fluorescence signals of EGFP-tagged and mCherry-tagged proteins, as specified. The plot shows the median (black vertical line) and interquartile range (25–75) (gray box) from 15 different cells for each condition. Individual dots represent the Mander's overlap coefficient obtained from a single cell. ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant.
Conserved lipid-binding domains such as FYVE, pleckstrin homology, and Bin, Amphiphysin, Rvs contain pockets of positively charged amino acids that form electrostatic interactions with phosphoinositides through their negatively charged phosphate groups (26). We hypothesized that RavD relied on this type of interaction to bind PI(3)P. RavD did not display similarity to any known phosphoinositide-binding domains, and thus we sought to determine whether RavD possesses any conserved lysines, arginines, or histidines that could be involved in binding PI(3)P. A multisequence alignment focusing on the C-terminal region of RavD revealed two conserved arginine residues at positions 237 and 292 (Fig. S1 and Fig. 2B). Mutation of arginine 292 to alanine abolished PI(3)P binding in the protein–lipid overlay assay (Fig. 2C). Further, we observed that mCherry-RavDR292A displayed a cytosolic distribution in HeLa cells and decreased localization with EGFP-2×FYVE (0.21, IQR 0.14–0.29) (Fig. 2D). The R237A point mutant retained the ability to bind PI(3)P in the protein-lipid overlay assay, and in HeLa cells, it colocalized with EGFP-2×FYVE to an extent similar to that of WT RavD (0.62, IQR 0.51–0.67). These results indicate that Arg-292 is a key residue responsible for RavD's ability to bind PI(3)P in vitro and to localize to PI(3)P-positive membrane compartments.
RavD localizes to the LCV and surrounding vesicles during infection
Given that RavD was capable of binding host membranes, we next addressed whether RavD localized to the LCV during infection. We infected RAW264.7 murine macrophages with a ΔravD strain carrying the pMMB207c-4×HA-ravD plasmid (ΔravD+pHA-RavD) at an m.o.i. of 20 (Fig. 3A and Fig. S3). At 5 h post-infection, we performed immunostaining using an HA antibody, and by structured illumination microscopy, we observed high fluorescence intensity surrounding the bacterium. We obtained similar results upon infection of either U937 human monocytes or the ameba Acanthamoeba castellanii (Fig. S4), confirming that RavD localizes to the LCV regardless of the type of host cell infected. Furthermore, by immunogold transmission EM, we found that, during RAW264.7 infection, HA-RavD also bound vesicular structures surrounding the LCV (Fig. 3B).
Figure 3.
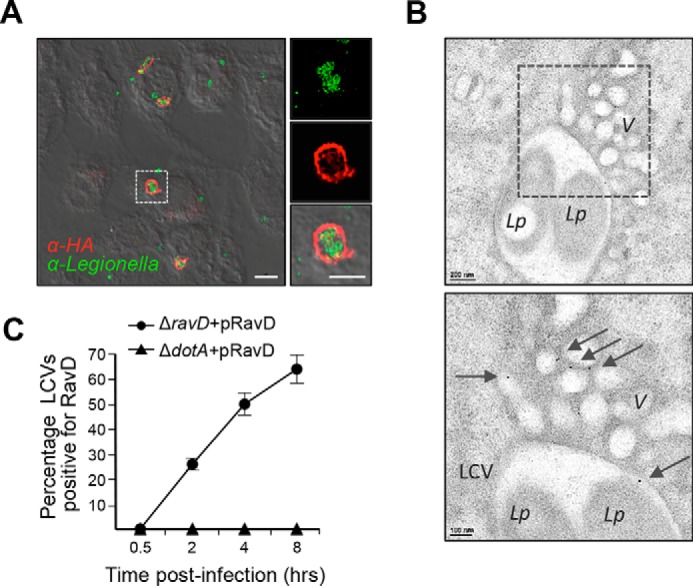
RavD localizes to the LCV and surrounding vesicles. A, superresolution structured illumination microscopy image of RAW264.7 macrophages infected at an m.o.i. of 20, fixed 5 h post-infection, and stained with anti-L. pneumophila (green) and anti-HA (red) antibodies. Scale bars = 5 μm (insets, 2 μm). B, representative transmission electron microscopy images showing immunogold localization of HA-RavD in RAW264.7 macrophages. Areas highlighted by rectangles (dashed line) in the top panels are magnified in the bottom panels. HA-RavD localized on the LCV membrane and on vesicles (V) surrounding the LCV. L.p., L. pneumophila. Scale bars = 200 nm in the top panels and 100 nm in the bottom panels. C, the percentage of RavD-positive LCVs gradually increased post-infection. RAW264.7 macrophages were infected with the ΔravD+pHA-RavD or ΔdotA+pHA-RavD for 0.5, 2, 4, and 8 h, and the percentage of HA-RavD-positive vacuoles was determined for each time point. Data are the average and standard deviation from two independent experiments where at least 50 different vacuoles in different cells were assessed.
To narrow in on the timing of RavD's localization to the LCV, we infected RAW264.7 macrophages with the ΔravD+pHA-RavD strain for 0.5, 2, 4, and 8 h, and, after immunostaining with an HA antibody, we determined the percentage of LCVs that colocalized with HA-RavD (Fig. 3C). Although no signal was detected 30 min post-infection, at 2 h, on average, 26% of the LCVs were positive, and the percentage continued to rise to 50% 4 h and 64% 8 h post-infection (Fig. 3C). Thus, RavD associates with the vacuolar membrane relatively early and during the midstage of infection.
Association with the LCV suggested that RavD may play a role in vacuole maintenance and could potentially impact intracellular survival of L. pneumophila. We therefore compared intracellular growth of WT, ΔravD, and the translocation-deficient ΔdotA strains upon infection of RAW264.7 macrophages and A. castellanii (Fig. 4). The ΔravD mutant strain grew similarly to WT L. pneumophila, whereas ΔdotA showed impaired growth in both host cell types, as expected. It is possible that because L. pneumophila effectors are functionally redundant, RavD's function could be masked by other effectors with similar functional properties; thus, intracellular growth may not be the most sensitive approach to reveal RavD's function (27).
Figure 4.
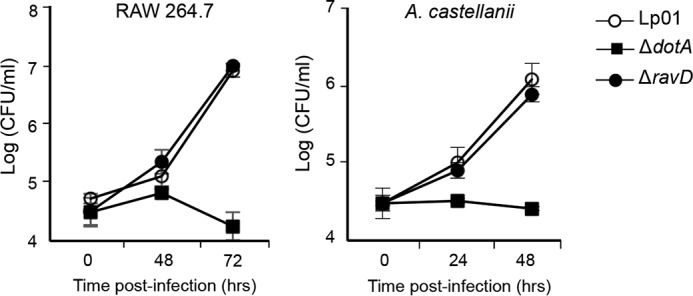
RavD is dispensable for intracellular growth. Monolayers of RAW264.7 macrophages or A. castellanii were infected with WT Lp01, ΔdotA, or ΔravD strains at an m.o.i. of 1 or 0.3, and cells were maintained at 37 °C or 25 °C. The number of intracellular bacteria was determined by recording the number of colony-forming units per milliliter. The mean and standard deviation from three independent experiments is displayed for each time point.
RavD localizes on early endosomes and is involved in avoidance of phagosome maturation
Next we hypothesized that, given its ability to bind PI(3)P and associate with membranes, RavD targets compartments of the endocytic pathway. To test this hypothesis, we co-transfected HeLa cells with a plasmid encoding mCherry-RavD and a plasmid encoding EGFP-Rab5, a marker for early endosomes. Image analysis indicated that there was extensive colocalization between the two proteins (0.71, IQR 0.70–0.78). In turn, mCherry-RavD did not colocalize with EGFP-Rab1, an endoplasmic reticulum-to-Golgi marker (0.15, IQR 0.11–0.19) (Fig. S5). We also analyzed colocalization of EGFP-Rab5 with truncated forms of RavD and found extensive colocalization with mCherry-RavD190–325 (0.74, IQR 0.65–0.78) but not with mCherry-RavD1–189 (0.12, IQR 0.10–0.13). These observations provide further support for the notion that the C-terminal region is responsible for governing the localization of the protein to endosomes.
The cytosolic leaflet of early endosomes is PI(3)P-rich. To determine whether RavD's localization to early endosomes depends on PI(3)P binding, we used the PI3K inhibitor wortmannin (28) to deplete PI(3)P pools within transfected HeLa cells. When control cells producing both mCherry-RavD and EGFP-2×FYVE were treated with wortmannin, the EGFP-2×FYVE signal diffused throughout the cytosol, but full-length and mCherry-RavD190–325 persisted in localizing to membrane compartments (Fig. 5A). In cells co-transfected with plasmids encoding EGFP-Rab5 and mCherry-RavD, although treated cells seemed to show a modest reduction in colocalization, the MOC median values of untreated (0.71, IQR 0.70–0.78) and treated (0.70, IQR 0.63–0.72) cells were not significantly different (Fig. 5B). We obtained a similar result for cells producing EGFP-Rab5 and mCherry-RavD190–325 (untreated, 0.74, IQR 0.65–0.78; treated, 0.62, IQR 0.57–0.71). These results suggest that PI(3)P binding is not required for RavD localization to early endosomes. It is possible that RavD binds early endosomes by recognizing both PI(3)P and another membrane component.
Figure 5.

Wortmannin does not disrupt colocalization of RavD with endocytic vesicles. A and B, confocal images of HeLa cells transiently co-transfected with plasmids encoding mCherry-RavD or mCherry-RavD190–325 and EGFP-2×FYVE (A) or EGFP-Rab5 (B). 18 h post-transfection, cells were incubated with or without wortmannin. Scale bars = 5 μm.
RavD's extensive colocalization with PI(3)P-positive endosomes prompted us to ask whether it is involved in the molecular mechanisms that mediate evasion of phagosome maturation. To test this, we infected RAW264.7 macrophages with the following L. pneumophila strains: WT, ΔdotA, ΔravD, and ΔravD+pHA-RavD. 1, 3, 5, and 10 h post-infection, macrophages were fixed, immunostained with an antibody against LAMP-1 (Fig. 6A), and quantified with regard to the number of LAMP-1–positive LCVs (Fig. 6B). The majority of vacuoles containing WT did not acquire LAMP-1 1, 3, 5, and 10 h post-infection, whereas vacuoles containing ΔdotA acquired LAMP-1 within the first hour of infection. Most vacuoles containing ΔravD were not associated with LAMP-1 at 1 h. Macrophages infected with the WT strain for 3 h displayed 29% ± 4% of LAMP-1–positive vacuoles, whereas macrophages infected with the avirulent ΔdotA strain displayed 76% ± 3%. Host cells infected with the ΔravD mutant or the complemented ΔravD+pHA-RavD strain displayed 61% ± 9% and 37% ± 6% LAMP-1–positive vacuoles, respectively (Fig. 6). The percentage of ΔravD-containing vacuoles associated with LAMP-1 continued to increase at subsequent time points (3 h, 61% ± 9%; 5 h, 68% ± 9%, and 10 h, 70% ± 8%). These results suggest that RavD is involved in preventing fusion of the LCV with late endosome and/or lysosomes. How the ΔravD mutant survives in a vacuole that has seemingly embarked on the path of phagosome maturation is an important question and remains to be determined in future studies.
Figure 6.

RavD prevents accumulation of LAMP-1 on Legionella-containing vacuoles. A, RAW264.7 macrophages were infected with WT, ΔdotA, ΔravD, or ΔravD+pHA-RavD for 1, 3, 5, and 10 h, and the percentage of LAMP-1–positive vacuoles for each condition was determined. The bar graph displays the average and standard deviation for the percentage of LCVs that were LAMP-1–positive at each time point. Data were obtained from three independent experiments where at least 100 different vacuoles were assessed. Asterisks denote data that showed a significant difference from the control (WT) in an unpaired Student's t test (***, p ≤ 0.001; **, p ≤ 0.01). B, representative confocal microscopy images of LAMP1-stained cells infected with the indicated strains. Scale bars = 2 μm (insets, 0.5 μm).
Discussion
Phosphoinositides are key lipids that direct vesicle traffic in eukaryotic cells by facilitating recruitment of proteins required for membrane fusion, fission, and transport (15). During phagocytosis, phosphoinositide conversion, closely regulated by kinases and phosphatases, serves as a gatekeeping mechanism that guides the newly formed phagosome through sequential fusions with endocytic compartments as it matures (14). Given this key role, phosphoinositides can be lucrative targets for bacterial pathogens seeking to survive within human cells. Here we show that the L. pneumophila effector RavD selectively binds PI(3)P. We provide evidence that it localizes to the LCV and is involved in preventing maturation of the LCV.
Previous work has demonstrated that L. pneumophila effectors exploit host phosphoinositides, directly affecting their metabolism to alter the lipid composition of host membranes or using them as molecular landmarks to selectively target host membrane compartments. In our in vitro assays, RavD selectively recognized PI(3)P, and in transiently transfected mammalian cells, it colocalized with the FYVE domain, widely used as a PI(3)P biosensor (26, 29). We determined that recognition of PI(3)P was mediated by a C-terminal region and found that mutation of the conserved arginine 292 to alanine abolished PI(3)P binding in vitro and dramatically diminished RavD's colocalization with the FYVE domain, pointing to this residue's key role in facilitating RavD's binding of PI(3)P. Positioning of the phosphoinositide-binding region closer to the C terminus end of the protein aligns with observations made for previously identified effectors that latch on to the LCV through interactions with PI(3)P and/or PI(4)P (23, 30). Recent work revealed that several effector proteins harbor one of two conserved PI(4)P-binding domains in their C-terminal region (31). Intriguingly, despite identification of multiple PI(3)P binding proteins (i.e. LidA, SetA, RidL, LpnE, LotA) a conserved PI(3)P-binding domain has yet to emerge. We could speculate that perhaps this is because phosphoinositide-binding is mediated by small, variable motifs or that bacterial lipid binding domains may be quite diverse, as is the case for eukaryotic proteins (26).
Given RavD's clear preference for binding PI(3)P, we asked whether this lipid was required for RavD's localization to early endosomes. We used treatment with wortmannin to deplete PI(3)P in transiently transfected cells co-producing the early endosome marker EGFP-Rab5 and either full-length RavD or the C-terminal truncated fragment RavD190–325. Both mCherry-RavD and mCherry-RavD190–325 remained predominantly localized to early endosomes, whereas, under similar conditions, in control cells producing EGFP-2×FYVE, fluorescence shifted to a cytosolic distribution in the presence of wortmannin. Thus, RavD localization to these PI(3)P-rich compartments occurred through a mechanism independent of PI(3)P.
Instead, a possible explanation for these results is that RavD binds host membranes by simultaneously interacting with PI(3)P and another host membrane component. This mechanism, termed coincident detection, has been described for a number of eukaryotic proteins. Existing evidence clearly shows that L. pneumophila effectors can simultaneously bind host lipids and proteins; however, binding of either of these components is typically sufficient to localize to the target membrane (32, 33).
Many of the previously identified PI(3)P- and PI(4)P-binding effectors localize to the cytosolic surface of the vacuolar membrane (23). Similarly, we found that RavD localized on the LCV in both RAW 264.7 macrophages and A. castellanii. In macrophages, RavD was present on the LCVs as early as 1 h post-infection, and the percentage of RavD-positive vacuoles increased throughout the course of infection. A closer look at the LCV within infected macrophages, facilitated by immunogold transmission electron microscopy, revealed that, in addition to localizing onto the vacuolar membrane, RavD can be found on small vesicles adjacent to the LCV 5 h post-infection. The nature of these vesicles remains to be determined in future studies.
Given RavD's ability to bind endosomal compartments in transfected cells, we asked whether, during host infection, RavD may participate in the molecular mechanism to prevent trafficking of the bacteria to lysosomes. We found that, in the absence of RavD, the late endosome/lysosome marker LAMP-1 accumulated on the LCV 3 h post-infection; 10 h post-infection, the percentage of ΔravD-containing vacuoles positive for LAMP-1 was significantly higher than that of WT-containing vacuoles. Because this observation is consistent with the notion that the ΔravD mutant is unable to avoid endocytic maturation, it would be expected that this mutant strain would be deficient for intracellular growth in host cells. Contrary to these expectations, the ΔravD mutant replicated as efficiently as the WT strain in both RAW264.7 macrophages and in A. castellanii. It is well known that effectors share a high degree of functional redundancy. Thus, intracellular growth assays may not be the most appropriate assay to determine whether an effector protein contributes to LCV biogenesis because effects caused by the absence of one effector could be masked by other effectors. Instead, other phenotypes, such as accumulation of late endosomes/lysosomes, could provide clues regarding which effector proteins are contributing to evasion of phagosome maturation. Using this type of observation, in future studies, we may be able to identify effector proteins that function in the molecular mechanisms that steer the Legionella-containing vacuole away from endolysosomal maturation pathways.
Experimental procedures
Strains, media, and reagents
L. pneumophila strain Lp01 (hsdR rpsL) and Lp01ΔdotA (type IVb secretion system−) are strains derived from L. pneumophila strain Philadelphia-1 (9), and they were cultured as described previously (34). All strains used in this study are listed in Table S1. HEK293T cells (ATCC, CRL-1573) and RAW264.7 murine macrophages (ATCC, TIB-71) were cultured in DMEM supplemented with 2 mm l-glutamine and 10% FBS and incubated in 5% CO2 at 37 °C. HeLa and U937 cells (ATCC, CRL-1593.2) were cultured in RPMI 1640 medium supplemented with 2 mm l-glutamine and 10% FBS and incubated as above. U937 monocytes were differentiated into macrophage-like cells by supplementing the medium with 10 ng/ml 12-O-tetradecanoylphorbol-13-acetate. A. castellanii (ATCC 50739) was cultured in PYG 712 medium (2% proteose peptone, 0.1% yeast extract, 0.1 m glucose, 4 mm MgSO4, 0.4 m CaCl2, 0.1% sodium citrate dihydrate, 0.05 mm Fe(NH4)2(SO4)2·6H2O, 2.5 mm NaH2PO3, and 2.5 mm K2HPO3) at 25 °C. Wortmannin was purchased from Acros Organic (AC32859). Antibodies were purchased from Thermo Fisher Scientific (goat anti-rabbit Alexa 568, A-11036; goat anti-mouse Alexa 488, A-11001; goat anti-rat Texas Red-X, T-6392; mouse monoclonal anti-GST, MA4-004; rabbit polyclonal anti-mCherry, Pa5-34974, HRP-conjugated anti-mouse antibody, NA931; and HRP-conjugated anti-rabbit antibody, 31460), Enzo (rabbit polyclonal Calnexin, ADI-SPA-860), ProteinTech (mouse monoclonal α-tubulin, 66031-1-Ig and rabbit polyclonal isocitrate dehydrogenase), and Sigma (rat anti-HA 11867423001A). LAMP-1 rat mAb (1D4B) was obtained from the Hybridoma Bank at the University of Iowa. Polyclonal rabbit antibodies directed against L. pneumophila were a kind gift from Matthias Machner (National Institutes of Health).
Construction of expression clones
The plasmids and oligonucleotides used in this study are listed in Tables S1 and S2. Full-length and fragments of ravD were cloned into the destination vectors, pDEST15 and 362 pCS Cherry DEST, by GatewayTM cloning technology to generate translational fusion with either GST or mCherry, respectively. The plasmid pMMB207c-4×HA, carrying a hemagglutinin tag under an isopropyl 1-thio-β-d-galactopyranoside–inducible promoter was a kind gift from Dr. Gunnar Schroeder (Queen's University Belfast). The pMMB207c-4×HA-ravD plasmid was generated by cloning the ravD gene into pMMB207c-4×HA at the BamHI and HindIII restriction sites. The 362 pCS Cherry DEST plasmid was a gift from Nathan Lawson (Addgene plasmid 13075) (35).
Construction of the L. pneumophila ΔravD deletion mutant
An in-frame deletion of ravD was obtained by allelic exchange using the pNTPS138 plasmid, which carries a chloramphenicol resistance cassette and the sacB gene (sucrose sensitivity). The pNTPS138 plasmid was a gift from Howard Steinman (Addgene plasmid 41891). A total of ∼1057 nt adjacent to the start and stop codons of ravD were PCR-amplified and cloned into the HindIII and EcoRI restriction sites of pNTPS138. This plasmid was then transformed into the Lp01 strain by electroporation as described previously (36), and single recombinants were selected on CYE agar plates containing chloramphenicol. Selected clones were then plated on CYE agar with 8% sucrose, and sucrose selection was used to identify clones in which the plasmid was lost. Deletion of the WT allele was identified by PCR analysis and confirmed by sequencing.
Multiple sequence alignments
We collected sequences related to lpg0160 using the sequence Q5ZZ51 as a query for PSI-BLAST (37) against the Nonredundant database filtered for low complexity (e value cutoff, 0.005; five iterations). Collected proteins (63 sequences from various Legionella species as well as Fluoribacter gormanii) corresponding to the C-terminal region of lpg0160 were aligned using the PROMALS3D web server (38) (Fig. S1).
Recombinant protein production and purification
Full-length and truncated forms of RavD were produced as GST fusion proteins in Escherichia coli BL21 (DE3) at 25 °C overnight after induction with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. E. coli cells producing GST variants were harvested and resuspended in PBS supplemented with 1 mm MgCl2 and 1 mm β-mercaptoethanol (PBS-MM), followed by lysis using the LV10 microfluidizer (Microfluidics). The cell lysate was centrifuged at 24,000 × g for 35 min, and the supernatant was incubated with pre-equilibrated GSH-Sepharose 4B (GE Healthcare) for 2 h at 4 °C. The resin was washed three times with PBS-MM, and proteins were eluted in 50 mm Tris-HCl (pH 8) containing 10 mm reduced GSH (Sigma). GSH was removed using a desalting Zeba column following the manufacturer's instructions (Thermo Fisher).
Protein–lipid overlay assay
Protein–lipid overlay assays were performed using commercially available PIP stripTM membranes (Echelon Biosciences Inc.). Nitrocellulose membranes prespotted with different phospholipids were blocked with 3% fat-free BSA or 3% nonfat milk in PBST (PBS and 0.1% Tween 20 (v/v) (pH 7.5)) for 1 h at room temperature. The blocked membranes were incubated with purified full-length GST-RavD, GST-RavD1–189, GST-RavD189–325, or GST alone (0.5 μg/ml in blocking buffer) overnight at 4 °C. Binding of the GST fusion protein to lipids was visualized with an anti-GST antibody (1:2000) and an HRP-conjugated anti-mouse antibody (1:10,000).
Liposome flotation assay
Phosphatidic acid, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine, and di-C16-phophatidylinositol polyphosphates were purchased from Avanti. Unilamellar liposomes were generated from a mixture of 64 μg of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (80%), 8 μg PS (10%), and 10 μg (10%) of either PI(3)P or PI(4)P, as described previously (33). Briefly, following air drying, lipid films were hydrated in 50 mm Tris (pH 7.0) and 150 mm NaCl, followed by extrusion through a 26-gauge needle. Liposome flotation assays were performed using an Optiprep (Sigma) gradient. Briefly, proteins were incubated with 20 μl of liposome suspension for 30 min at room temperature. The binding reaction was combined with 54% iodixanol forming the bottom layer. A 25% iodixanol middle layer and 5% iodixanol top layer were subsequently added. Liposomes were separated from unbound protein by ultracentrifugation at 55,000 rpm for 3 h at 20 °C in a TLS-55 ultracentrifuge rotor. 30 μl of sample from the top layer was collected, and bound proteins were analyzed by SDS-PAGE using a 4–15% TGXTM Precast stain-free gel (Bio-Rad).
Cellular fractionation assay
HEK293T cells were transfected with plasmids encoding full-length mCherry-RavD constructs for 16–20 h. Cells were scraped, washed, resuspended in PBS containing a protease inhibitor mixture (Thermo Fisher Scientific), and lysed on ice by passing the cell suspension through a 27-gauge needle. Lysates were centrifuged at 15,000 × g for 10 min at 4 °C to remove intact cells and cell debris. Cleared lysates were spun at 51,000 rpm for 45 min at 4 °C in a TLA-100 rotor in a Beckman ultracentrifuge to separate the cytosol and membrane fractions. The supernatant was collected as the cytosolic fraction, and the membrane fraction was obtained by resuspending the pellet in an equal volume of 2% Nonidet P-40 in PBS. The post-nuclear supernatant and cytosolic and membrane fractions were analyzed by Western blotting.
Confocal microscopy
Constructs based on pEGFP-C1 or 362 pCS mCherry DEST, as listed in Table S1, were transiently transfected into HeLa cells for 16–20 h using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific). Cells were fixed in PBS with 4% paraformaldehyde for 20 min at room temperature, and coverslips were mounted using ProLong diamond antifade mountant (Thermo Fisher Scientific). For wortmannin treatment experiments, HeLa cells were transfected as described previously and incubated with RPMI containing 100 nm wortmannin for 30 min before fixation. Confocal imaging was performed on a Zeiss LSM 780 laser-scanning confocal microscope using a ×63 Plan-Apochromat objective lens (numerical aperture of 1.4) and operated with ZEN software (Carl Zeiss, Inc.).
Colocalization, quantification, and statistical analysis
For each condition, we acquired confocal images for 15 cells. Quantitative colocalization analysis was performed using Volocity software (PerkinElmer Life Sciences) to calculate the Mander's overlap coefficient (39) corresponding to the fraction of green voxels overlapping with red voxels in relation to total green voxels. The normality of the distribution of variables was assessed using a Kolmogorov–Smirnov test and then confirmed visually. Data were nonparametrically distributed, and therefore, to compare coefficients across the conditions tested, we used a Kruskal–Wallis test (H = 18, p < 1–13). The median and interquartile range of coefficients were calculated for each condition. Dunn–Sidak post hoc tests were conducted to statistically determine which groups differed while correcting for multiple comparisons. Statistical significance was set at p ≤ 0.05 (Matlab, Mathworks, version 2016a, Natick, MA) was used as statistical software.
Immunogold transmission electron microscopy of macrophages infected with L. pneumophila sample preparation
Cells were grown on 1.2 mm × 200 μm high pressure freezer carriers. Just prior to freezing, the medium was removed, and the carrier was filled with 20% BSA in medium. Cells were frozen with a Leica EM Pact2 high-pressure freezer and then transferred under liquid nitrogen to a Leica AFS for freeze substitution. Samples were freeze-substituted in 0.1% uranyl acetate in 100% acetone at −90 °C for 4–5 days. Samples were warmed to −45 °C over 12 h, washed with 100% acetone, and gradually infiltrated with increasing concentrations of Lowicryl HM20 monostep resin over a period of 2 days. The samples were embedded in Lowicryl HM20 Monostep resin and polymerized under UV light for 48 h at −45 °C and for an additional 48 h at 23 °C. The cells were sectioned using a Reichert–Jung Ultracut E ultramicrotome, and ultrathin sections were collected onto 200 mesh formvar/carbon-coated nickel grids.
Immunogold labeling
Sections of resin-embedded cells were immunogold-labeled using standard procedures. Sections were blocked with a solution containing 3% BSA, 5% normal goat serum, and 0.1% cold water fish skin gelatin in Tris-buffered saline (TBS) for 30 min and then incubated with rat anti-HA primary antibody diluted 1:50 in blocking solution overnight at 4 °C in a humidified chamber. As a control, the primary antibody was substituted with an equivalent concentration of rat IgG. Grids were washed on 6 drops of TBS and then incubated with a 1:20 dilution of goat anti-rat secondary antibody conjugated to 10 nm gold (Aurion, catalog no. 25189) for 2 h at room temperature. Grids were washed on 6 drops of TBS, post-fixed with 2% glutaraldehyde in TBS for 5 min, and washed on 6 drops of water. Sections were post-stained with 2% uranyl acetate in 50% methanol. Samples were imaged with a Zeiss Libra 120 transmission electron microscope operating at 120 kV, and images were acquired with a Gatan Ultrascan 1000 CCD camera.
Assays for LAMP-1 colocalization with the LCV
For detection of LAMP-1–positive LCVs, RAW264.7 macrophages were challenged with L. pneumophila strains at an m.o.i. of 20, spun at 200 × g for 5 min, and incubated at 37 °C. Medium was supplemented with gentamicin (100 μg/ml) to eliminate extracellular bacteria. 1, 3 5, and 10 h post-infection, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 at room temperature for 20 min, and blocked in 10% normal goat serum (Sigma). Internalized bacteria were detected using anti-Legionella rabbit primary (1:6000) and Alexa 488–conjugated goat anti-rabbit secondary antibody (1:3000). LAMP-1 was detected with anti-LAMP-1 rat primary antibody (1:1000) and Texas Red–conjugated goat anti-rat IgG (1:3000). Microscopy was carried out using a confocal laser-scanning microscope (LSM780, Zeiss). The percentage of LAMP-1–positive LCVs was determined by scoring 100 cells/coverslip with three replicates for each specific condition. An unpaired two-sided Student's t test was performed with a p value cutoff of < 0.05 to determine where there was a statistically significant difference in localization to the LCV.
Assay for colocalization of the HA-RavD effector protein with the LCV
To determine localization of HA-RavD post-infection, RAW264.7, U937, or A. castellanii cells were infected with a ΔravD strain carrying a plasmid encoding HA-RavD at an m.o.i. of 20. At the specified time points, cells were fixed as mentioned above and immunostained with a rat anti-HA (1:1000) and then an Alexa Fluor 568–conjugated goat anti-rat secondary antibody (1:3000). Bacteria were immunolabeled with anti-Legionella rabbit antibodies (1:6000) and an Alexa Fluor 488-conjugated anti-rabbit secondary antibody (1:3000). Microscopy was carried out using a confocal laser-scanning microscope (LSM780, Zeiss). The percentage of HA-RavD–positive LCVs in RAW264.7 macrophages was determined by scoring 100 cells per coverslip with three replicates for each time point.
Superresolution structured illumination microscopy
Superresolution structured illumination microscopy was performed as described previously (40). The Zeiss Elyra PS.1 SIM (Carl Zeiss, Inc.) imaging system equipped with a ×63 Plan-Apochromat oil immersion objective (Numerical aperture of 1.4) was used for image acquisition. Images were generated and processed with ZEN 2011 software (Carl Zeiss, Inc.) from z-stacks containing five phase shifts and three rotations per z-slice (0.110-μm interval). Alexa Fluor 488 was excited with the 488-nm laser line, and emission was collected with a 495- to 550-nm bandpass filter. Alexa Fluor 561 was excited with the 561-nm laser line, and emission was collected with the 570- to 620-nm bandpass filter. Affine alignment of channels was carried out using images a multicolored TetraSpeck beads (100 nm, Thermo Fisher Scientific) acquired with the same settings as for the cellular sample.
Intracellular growth assay
A. castellanii cells were seeded at a density of 1 × 106 in 96-well plates and infected with WT and mutant strains at an m.o.i. of 0.03. One hour after infection, wells were rinsed three times to eliminate extracellular bacteria. 0, 24, and 48 h post-infection, cells were lysed with 0.05% saponin, diluted, and plated on CYE plates. RAW264.7 macrophages were seeded at a density of 3 × 105 in 24-well plates and infected with WT and mutant strains at an m.o.i. of 1. One hour after infection, wells were rinsed three times to eliminate extracellular bacteria. 0, 48 and 72 h post-infection, cells were lysed with 0.05% digitonin, diluted, and plated on CYE plates. The number of bacteria recovered was recorded as colony-forming units, and standard deviations were calculated based on colony-forming unit values obtained from assays carried out in triplicate.
Author contributions
C. M. P. and M. R. N. conceptualization; C. M. P., L. N. K., and M. R. N. formal analysis; C. M. P. and R. B.-A. validation; C. M. P., J. L. C., and M. R. N. visualization; C. M. P., R. B.-A., L. N. K., and M. R. N. methodology; C. M. P. and M. R. N. writing-original draft; C. M. P. and M. R. N. project administration; C. M. P., R. B.-A., L. N. K., J. L. C., and M. R. N. writing-review and editing; L. N. K. and J. L. C. software; J. L. C. and M. R. N. resources; J. L. C. and M. R. N. data curation; J. L. C. and M. R. N. funding acquisition; M. R. N. supervision.
Supplementary Material
This work was supported by NIAID, National Institutes of Health grant 1R21AI142317-01 (to M. R. N.), Delaware COBRE Program, NIGMS, National Institutes of Health Grant P20GM104316 (to M. R. N.), and Delaware INBRE Program, NIGMS, National Institutes of Health Grant P20GM103446 (to J. L. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Tables S1 and S2.
- LCV
- Legionella-containing vacuole
- PI(3)P
- phosphatidylinositol-3-phosphate
- PI(4)P
- phosphotidylinositol-4-phosphate
- EGFP
- enhanced GFP
- MOC
- Mander's overlap coefficient
- PS
- phosphatidylserine
- IQR
- interquartile range
- TBS
- Tris-buffered saline
- m.o.i.
- multiplicity of infection
- FYVE
- Fab 1, YOTB, Vac 1, EEA1
- CYE
- charcoal yeast extract.
References
- 1. Fields B. S., Benson R. F., and Besser R. E. (2002) Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15, 506–526 10.1128/CMR.15.3.506-526.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chandler F. W., Hicklin M. D., and Blackmon J. A. (1977) Demonstration of the agent of Legionnaires' disease in tissue. N. Engl. J. Med. 297, 1218–1220 10.1056/NEJM197712012972206 [DOI] [PubMed] [Google Scholar]
- 3. Copenhaver A. M., Casson C. N., Nguyen H. T., Fung T. C., Duda M. M., Roy C. R., and Shin S. (2014) Alveolar macrophages and neutrophils are the primary reservoirs for Legionella pneumophila and mediate cytosolic surveillance of type IV secretion. Infect. Immun. 82, 4325–4336 10.1128/IAI.01891-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Flannagan R. S., Jaumouillé V., and Grinstein S. (2012) The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98 10.1146/annurev-pathol-011811-132445 [DOI] [PubMed] [Google Scholar]
- 5. Clemens D. L., Lee B. Y., and Horwitz M. A. (2000) Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect. Immun. 68, 2671–2684 10.1128/IAI.68.5.2671-2684.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horwitz M. A., and Silverstein S. C. (1980) Legionnaires' disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J. Clin. Invest. 66, 441–450 10.1172/JCI109874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horwitz M. A. (1983) Formation of a novel phagosome by the Legionnaires' disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158, 1319–1331 10.1084/jem.158.4.1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nash T. W., Libby D. M., and Horwitz M. A. (1984) Interaction between the legionnaires' disease bacterium (Legionella pneumophila) and human alveolar macrophages: influence of antibody, lymphokines, and hydrocortisone. J. Clin. Invest. 74, 771–782 10.1172/JCI111493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berger K. H., and Isberg R. R. (1993) Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7, 7–19 10.1111/j.1365-2958.1993.tb01092.x [DOI] [PubMed] [Google Scholar]
- 10. Segal G., and Shuman H. A. (1999) Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect. Immun. 67, 2117–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luo Z. Q., and Isberg R. R. (2004) Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U.S.A. 101, 841–846 10.1073/pnas.0304916101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Segal G. (2013) Identification of Legionella effectors using bioinformatic approaches. Methods Mol. Biol. 954, 595–602 10.1007/978-1-62703-161-5_37 [DOI] [PubMed] [Google Scholar]
- 13. Pauwels A. M., Trost M., Beyaert R., and Hoffmann E. (2017) Patterns, receptors, and signals: regulation of phagosome maturation. Trends Immunol. 38, 407–422 10.1016/j.it.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levin R., Grinstein S., and Schlam D. (2015) Phosphoinositides in phagocytosis and macropinocytosis. Biochim. Biophys. Acta 1851, 805–823 10.1016/j.bbalip.2014.09.005 [DOI] [PubMed] [Google Scholar]
- 15. Di Paolo G., and De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- 16. Balla T. (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137 10.1152/physrev.00028.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levin R., Hammond G. R., Balla T., De Camilli P., Fairn G. D., and Grinstein S. (2017) Multiphasic dynamics of phosphatidylinositol 4-phosphate during phagocytosis. Mol. Biol. Cell 28, 128–140 10.1091/mbc.e16-06-0451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vieira O. V., Botelho R. J., Rameh L., Brachmann S. M., Matsuo T., Davidson H. W., Schreiber A., Backer J. M., Cantley L. C., and Grinstein S. (2001) Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 155, 19–25 10.1083/jcb.200107069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weber S., Wagner M., and Hilbi H. (2014) Live-cell imaging of phosphoinositide dynamics and membrane architecture during Legionella infection. mBio 5, e00839–00813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weber S. S., Ragaz C., and Hilbi H. (2009) The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell Microbiol. 11, 442–460 10.1111/j.1462-5822.2008.01266.x [DOI] [PubMed] [Google Scholar]
- 21. Toulabi L., Wu X., Cheng Y., and Mao Y. (2013) Identification and structural characterization of a Legionella phosphoinositide phosphatase. J. Biol. Chem. 288, 24518–24527 10.1074/jbc.M113.474239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong N., Niu M., Hu L., Yao Q., Zhou R., and Shao F. (2016) Modulation of membrane phosphoinositide dynamics by the phosphatidylinositide 4-kinase activity of the Legionella LepB effector. Nat. Microbiol. 2, 16236 [DOI] [PubMed] [Google Scholar]
- 23. Hilbi H., Weber S., and Finsel I. (2011) Anchors for effectors: subversion of phosphoinositide lipids by Legionella. Front. Microbiol. 2, 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weber S. S., Ragaz C., Reus K., Nyfeler Y., and Hilbi H. (2006) Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2, e46 10.1371/journal.ppat.0020046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang L., Boyd D., Amyot W. M., Hempstead A. D., Luo Z. Q., O'Connor T. J., Chen C., Machner M., Montminy T., and Isberg R. R. (2011) The E block motif is associated with Legionella pneumophila translocated substrates. Cell Microbiol. 13, 227–245 10.1111/j.1462-5822.2010.01531.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hammond G. R., and Balla T. (2015) Polyphosphoinositide binding domains: key to inositol lipid biology. Biochim. Biophys. Acta 1851, 746–758 10.1016/j.bbalip.2015.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Connor T. J., Adepoju Y., Boyd D., and Isberg R. R. (2011) Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proc. Natl. Acad. Sci. U.S.A. 108, 14733–14740 10.1073/pnas.1111678108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arcaro A., and Wymann M. P. (1993) Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 296, 297–301 10.1042/bj2960297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burd C. G., and Emr S. D. (1998) Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol. Cell 2, 157–162 10.1016/S1097-2765(00)80125-2 [DOI] [PubMed] [Google Scholar]
- 30. Kubori T., Kitao T., Ando H., and Nagai H. (2018) LotA, a Legionella deubiquitinase, has dual catalytic activity and contributes to intracellular growth. Cell Microbiol. 20, e12840 10.1111/cmi.12840 [DOI] [PubMed] [Google Scholar]
- 31. Burstein D., Amaro F., Zusman T., Lifshitz Z., Cohen O., Gilbert J. A., Pupko T., Shuman H. A., and Segal G. (2016) Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat. Genet. 48, 167–175 10.1038/ng.3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Del Campo C. M., Mishra A. K., Wang Y. H., Roy C. R., Janmey P. A., and Lambright D. G. (2014) Structural basis for PI(4)P-specific membrane recruitment of the Legionella pneumophila effector DrrA/SidM. Structure 22, 397–408 10.1016/j.str.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo X., Wasilko D. J., Liu Y., Sun J., Wu X., Luo Z. Q., and Mao Y. (2015) Structure of the Legionella virulence factor, SidC reveals a unique PI(4)P-specific binding domain essential for its targeting to the bacterial phagosome. PLoS Pathog. 11, e1004965 10.1371/journal.ppat.1004965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feeley J. C., Gibson R. J., Gorman G. W., Langford N. C., Rasheed J. K., Mackel D. C., and Baine W. B. (1979) Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 10, 437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Villefranc J. A., Amigo J., and Lawson N. D. (2007) Gateway compatible vectors for analysis of gene function in the zebrafish. Dev. Dyn. 236, 3077–3087 10.1002/dvdy.21354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen D. Q., Huang S. S., and Lu Y. J. (2006) Efficient transformation of Legionella pneumophila by high-voltage electroporation. Microbiol. Res. 161, 246–251 10.1016/j.micres.2005.09.001 [DOI] [PubMed] [Google Scholar]
- 37. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pei J., Tang M., and Grishin N. V. (2008) PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucleic Acids Res. 36, W30–W34 10.1093/nar/gkn322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manders E. M. M., Verbeek F. J., and Aten J. A. (1993) Measurement of colocalization of objects in dual-color confocal images. J. Microsc. 169, 375–382 10.1111/j.1365-2818.1993.tb03313.x [DOI] [PubMed] [Google Scholar]
- 40. Yu X., Noll R. R., Romero Dueñas B. P., Allgood S. C., Barker K., Caplan J. L., Machner M. P., LaBaer J., Qiu J., and Neunuebel M. R. (2018) Legionella effector AnkX interacts with host nuclear protein PLEKHN1. BMC Microbiol. 18, 5 10.1186/s12866-017-1147-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.