

Abstract
Oxidative modifications of cysteine residues are an important component in signaling pathways, enzymatic regulation, and redox homeostasis. Current direct and indirect methods detect specific modifications and a general binary population of “free” or “oxidized” cysteines, respectively. In an effort to combine both direct and indirect detection strategies, here we developed a method that we designate isotopic tagging of oxidized and reduced cysteines (iTORC). This method uses synthetic molecules for rapid isotopic coding of sulfenic acids, reduced cysteines, and disulfides in cells. Our approach utilizes isotopically distinct benzothiazine and halogenated benzothiazine probes to sequentially alkylate sulfenic acids and then free thiols and, finally, after a reduction step, cysteines oxidized to disulfides or other phosphine-reducible states. We ascertained that the iodinated benzothiazine probe has reduced cross-reactivity toward primary amines and is highly reactive with the cysteine of GSH, with a calculated rate constant of 2 × 105 m−1 s−1 (pH 8.0, 23 °C) (i.e. 10–20 times faster than N-ethylmaleimide). We applied iTORC to a mouse hepatocyte lysate to identify known sulfenylated and disulfide-bonded proteins, including elongation factor 1-α1 and mouse serum albumin, and found that iTORC reliably detected their expected oxidation status. This method can be easily employed to study the effects of oxidants on recombinant proteins and cell and tissue extracts, and the efficiencies of the alkylating agents enable completion of all three labeling steps within 2 h. In summary, we demonstrate here that halogenated benzothiazine-based alkylating agents can be utilized to rapidly measure the cellular thiol status in cells.
Keywords: thiol, disulfide, mass spectrometry (MS), proteomics, oxidative stress, oxidation-reduction (redox), cysteine, isotopic tagging of oxidized and reduced cysteines (iTORC), oxidative damage, sulfenic acid
Introduction
Cysteine residues are known targets of oxidation due to the nucleophilic and reactive nature of sulfur atoms. Reaction of the cysteine thiol with a variety of oxidants (e.g. H2O2) has proven to be an important cellular signaling event that can elicit a diverse number of responses, including transcriptional changes and enzymatic regulation (1). Previously studied redox-sensitive proteins include peroxiredoxins, epidermal growth factor receptor, and protein-tyrosine phosphatase 1B (2–5). Detecting and quantitating oxidatively modified cysteines is crucial to understanding their biological importance. One such oxidative modification is cysteine sulfenylation (Cys-SOH formation). Sulfenic acids can be reduced back to free thiols by reductants, and these protein modifications have short cellular half-lives (6). Additionally, sulfenic acids are involved in regulatory pathways and can directly function as enzymatic redox switches (7). This oxidative modification readily participates in a variety of reactions including disulfide bond formation with free thiols and further oxidation to sulfinic (Cys-SO2−) and sulfonic (Cys-SO3−) acids (8). Detection of sulfenic acids with 1,3-dicarbonyl moieties, particularly dimedone, has been known for over 40 years (9).
Synthetic efforts have recently been made to improve the detection of sulfenic acids, with several successes (10–12). Specifically, benzothiazines are fast, selective alkylators of sulfenic acids and promising reagents for discovery of sites of sulfenylation (1, 13). We used a straightforward chemical procedure to synthesize three stable, distinct benzothiazine isotopomers, and two were further iodinated to react with reduced thiol moieties (Fig. 1). These iodinated reagents proved to be very efficient alkylators of cysteine thiols. Additionally, three structurally identical isotopically unique reagents provide a combination of two heavily used techniques to study thiol redox status (14, 15). Our approach, isotopic tagging of oxidized and reduced cysteines (iTORC),2 links the techniques of direct detection of sulfenic acids and the indirect detection of free and reducible thiols in a quantitative fashion (Fig. 1). iTORC uses sequential additions of readily synthesized molecules, 1-([2H7]-propyl)-1H-[13C6]-benzo[c][1,2]thiazin-4(3H)-one-4a,5,6,7,8,8a-2,2-dioxide (13C6-d7-pBTD), 3-iodo-1-(propyl-d7)-1H-benzo[c][1,2]thiazin-4(3H)-one 2,2-dioxide (d7-ipBTD), and then d0-ipBTD to alkylate sulfenic acids, free thiols, and reducible thiols, respectively (Fig. 1). Because of the reactivity and efficiency of these alkylating agents, all three labeling steps can be completed within 2 h.
Figure 1.
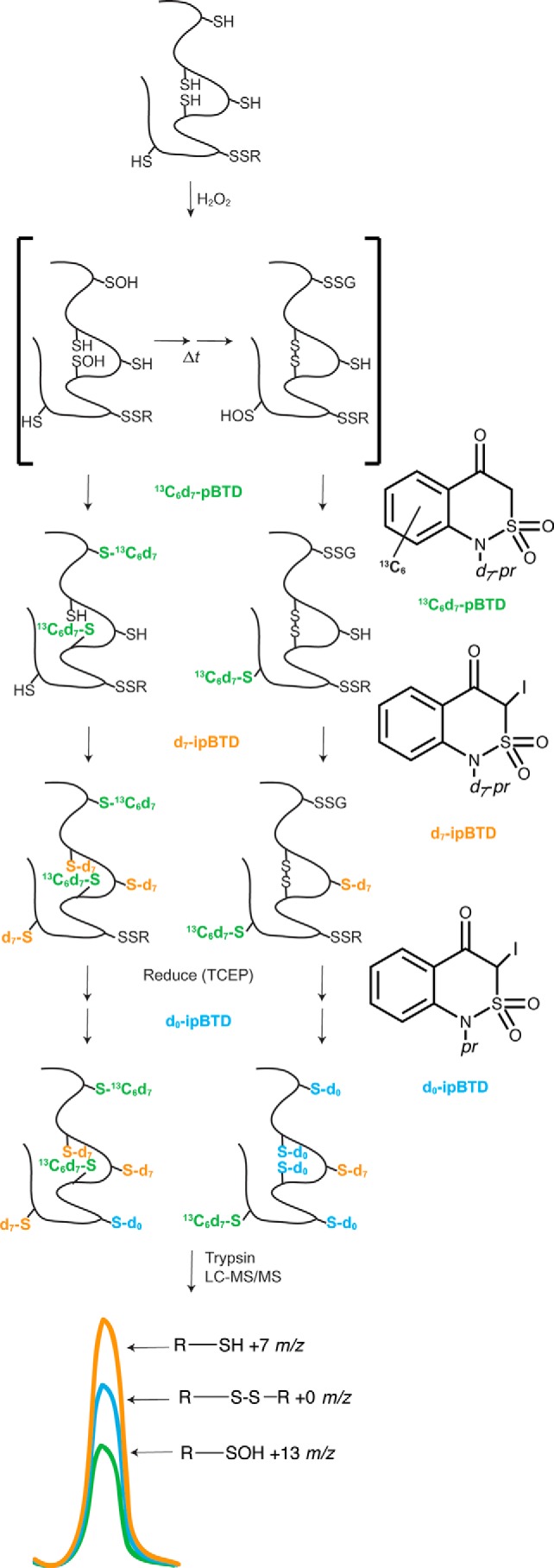
iTORC methodology. When proteins are exposed to an oxidant (e.g. H2O2), sulfenic acids can be formed, and these sulfenylated cysteines react with thiols to form disulfides or other oxidized moieties. 13C6-d7-pBTD reacts with these proteins to alkylate sulfenic acids. d7-ipBTD is then added to the protein to alkylate free thiols. The sample is then reduced, and d0-ipBTD is added to alkylate the oxidized cysteines (-SSR and other TCEP-reducible thiol species) after their reduction. Digestion with trypsin and subsequent LC-MS/MS analysis identifies cysteine-containing peptides labeled with 13C6-d7-, d7-, and d0-pBTD moieties, and relative quantitation can be performed. pr, propyl. The mass of pBTD is 239, so the addition of this entity adds 237 atomic mass units to a thiol group and thus 237, 244, or 250 atomic mass units, depending on the isotopic substitution.
Results
Synthesis of benzothiazine probes
pBTD and ipBTD, with the appropriate isotopic substitutions, were readily synthesized in 32 and 20% overall yield, respectively, using the methods described. The synthesis of pBTD is a simple three-step sequence (Scheme S1), and the one-step iodination of pBTD yielded ipBTD in 70% yield.
Second-order rate constants for modifications
pBTD was reacted with the sulfenic acid-derivative of a dipeptide (N-carbobenzoxy-Cys-Val-OCH3), which is oxidized to a cyclic sulfenamide and ring-opens to form a sulfenic acid under aqueous conditions (Fig. S1) (11). The rate constant for the reaction of pBTD with the model sulfenic acid–containing dipeptide (Fig. S2B) was measured to be 1.7 × 103 m−1 s−1, which can be compared with the value estimated for dimedone (12 m−1 s−1) and agrees with previously calculated rates reported for benzothiazine probes (11) (Fig. S2B, pH 8.0, 23 °C). The estimated rate constant for the reaction of ipBTD with the model peptide glutathione (GSH) was 2.0 × 105 m−1 s−1. Compared with rate constants previously reported for N-ethylmaleimide (NEM) and 2-mercaptoethanol, ipBTD was found to alkylate cysteines 10–20-fold faster (Fig. S2C, pH 8.0, 23 °C) (16). To directly compare this rate with that of other alkylating agents in a pH-independent manner, ipBTD was reacted with 2-nitro-5-thiobenzoate, which has a pKa of 4.4 (Fig. S2D, pH 7.4, 23 °C) (17). The rate constant was measured to be 2.8 × 105 m−1 s−1, compared with 2.2 m−1 s−1 reported for iodoacetamide (IAM) and 46 m−1 s−1 for NEM (17). Comparing ipBTD with IAM and NEM provides ∼120,000- and 6,000-fold rate differences, respectively. A rate constant of only 17 m−1 s−1 was estimated for amine modification using the model N-α-acetyl-l-lysine, demonstrating a 104-fold selectivity for thiol over amine moieties for ipBTD (Fig. S2E, pH 8.0, 23 °C). Reaction of ipBTD with the sulfenic acid dipeptide mentioned earlier provided a rate constant of 4.3 × 103 m−1 s−1, indicating 100-fold selectivity toward free thiols (Fig. S2F, pH 8.0, 23 °C). Such cross-reactivity would produce a sulfoxide product (+16 Da), but using pBTD with an initial incubation time of 1 h to alkylate sulfenic acids limits the cross-reactivity. Sulfoxide formation can also occur after the alkylation step, during sample handling, but proteins with all three labels (d0-, d7-, and 13C6-d7-pBTD–modified cysteines) should be oxidized at a similar rate under these conditions.
The high reactivity of ipBTD allows for fast and efficient labeling of reduced cysteines but comes with the drawback of instability. As with most α-haloketone alkylating agents, ipBTD is light-sensitive. The reagent should be prepared fresh in acetonitrile in amber glass before each experiment. Adding 2 mm ipBTD in a reaction provides an ∼100-fold excess of ipBTD over cysteine. Under our reaction conditions (100 mm HEPPS, pH 8.0), a degradation rate of 0.0009 s−1 was calculated, corresponding to a half-life of 13 min (Fig. S2G). In the course of a 15-min alkylation procedure, 37% of the reagent would be remaining, still providing a high molar excess. From our time course experiment (Fig. 3), ∼90% of cysteines were labeled within 2.5 min, suggesting that under these conditions, enough of the probe remains active to complete alkylation in 15 min.
Figure 3.
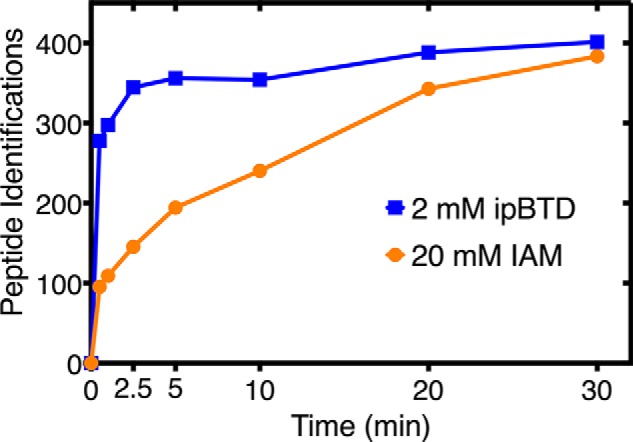
Comparison of rates of reactions of alkylating agents and fluorescence visualization of pBTD-labeled proteins. E. coli lysate was reacted with either IAM (20 mm) or ipBTD (2 mm) for the indicated times, and the number of labeled cysteine-containing peptide spectrum matches was reported. The average total number of peptide spectrum matches was 4,223 and 4,513 for IAM and ipBTD, respectively.
Validation of pBTD sulfenic acid and ipBTD thiol alkylation by LC-MS/MS
13C6-d7-pBTD was reacted with the model sulfenic acid dipeptide (N-carbobenzoxy-Cys (SOH)-Val-OCH3), and d7- and d0-ipBTD were reacted with the same dipeptide under reducing conditions (1 mm TCEP, to form the free thiol). The reaction mixtures were combined in equal parts and subjected to LC-MS/MS analysis (Fig. 2). Extracted ion chromatograms corresponding to the d0-, d7-, and 13C6-d7-pBTD–modified dipeptide co-chromatographed, with the deuterated compounds being expectedly retarded (Fig. 2, A, B, and C, respectively). Smaller peaks with exactly the same mass (and UV spectra, not shown) eluted later and are likely diastereomers (due to the chiral center of carbon-3 of the reagent). Additionally, MS2 spectra revealed mass shifts corresponding to M+0, M+7, and M+13 for the d0-, d7-, and 13C6-d7-pBTD–modified dipeptide, respectively (Fig. 2, D, E, and F, respectively). These results validate the ability of these reagents to selectively alkylate thiols or sulfenic acids and co-elute in HPLC. The use of the ratios of these three alkylating agents provides relative quantitation of reduced and oxidized thiols.
Figure 2.

Alkylation of dipeptide with 13C6-d7-pBTD and d7- and d0-ipBTD. A synthetic sulfenic acid dipeptide (11) was reacted with 13C6-d7-pBTD and with d7- or d0-ipBTD in the presence of TCEP. The three separate reactions were combined, and LC-MS/MS was performed. Extracted ion chromatograms are shown as follows: d0- (A), d7- (B), and 13C6-d7-pBTD (C). The corresponding fragmentation patterns are shown: MS/MS of 604.2 m/z d0- (D), MS/MS of 611.2 m/z d7- (E), and MS/MS of 617.2 m/z 13C6-d7-pBTD (F).
Time course of alkylation by ipBTD and IAM
Either 20 mm IAM or 2 mm ipBTD was reacted with reduced Escherichia coli lysate for the indicated times; the proteins were digested with trypsin, and LC-MS/MS analysis was performed on the labeled peptides (Fig. 3). Even with the lower concentration of reagent (2 mm), ipBTD alkylation was ∼90% complete within 2.5 min, whereas iodoacetamide alkylation was not complete until 20 min, further demonstrating the high reactivity of ipBTD with free thiols.
Fluorescence visualization of ipBTD alkylation
BSA was reduced and alkylated with ipBTD. The labeled BSA was separated via SDS-PAGE alone or with an unlabeled E. coli lysate background and excited using a UV transilluminator (commonly used for DNA agarose gels) (Fig. 4A). The emission of the labeled BSA was readily visible. The gel was then stained with Coomassie Brilliant Blue dye for comparison (Fig. 4B). ipBTD-labeled E. coli lysate was also separated via SDS-PAGE and excited via UV fluorescence (Fig. 4C) and then stained with Coomassie dye (Fig. 4D).
Figure 4.

Fluorescence visualization of pBTD-labeled proteins. A, BSA was alkylated with ipBTD and separated via SDS-PAGE alone (left lane) or in the presence of unlabeled E. coli lysate (right lane) and visualized with a UV transilluminator. B, the gel was then stained with Coomassie Brilliant Blue for comparison. C, ipBTD-alkylated E. coli lysate was separated via SDS-PAGE and visualized with a UV transilluminator. D, the same gel was then stained with Coomassie Brilliant Blue for comparison. E, BSA was reacted with ipBTD under nonreducing conditions: no ipBTD added (lane 1) and BSA reacted with ipBTD (lane 2). Under reducing conditions (1 mm TCEP), BSA was reacted with IAM (50 mm) for 30 min, the IAM was then quenched with DTT (25 mm), and, after 15 min, ipBTD was added for 15 min: BSA reduced but no ipBTD added (lane 3), BSA reduced and incubated with IAM before adding ipBTD (lane 4), and BSA reduced and treated with ipBTD (lane 5). F, the same gel (as in E) was then stained with Coomassie Brilliant Blue for comparison.
To address the possibility of amine reactivity of ipBTD, we performed another fluorescence experiment (Fig. 4, E and F). BSA was reacted with ipBTD under nonreducing conditions, and no alkylation (fluorescence) was observed (Fig. 4E). Under reducing conditions (1 mm TCEP), BSA was reacted with IAM (50 mm), the residual IAM was then quenched with DTT (25 mm), and ipBTD was added (Fig. 4E). No fluorescence was observed, suggesting that ipBTD is highly selective for reduced thiols under these conditions. Utilizing the fluorescent nature of these alkylating agents provides a facile method to ensure proper labeling and to identify proteins of interest for further analysis.
iTORC labeling of mouse hepatocytes
Freshly isolated hepatocytes from four mice were lysed, subjected to iTORC labeling, and submitted for LC-MS/MS peptide analysis. A total of 639 sulfenylated peptides (13C6-d7-pBTD–labeled) corresponding to 1,073 spectra were identified (Table S1). A total of 2,785 peptides were identified as d7-ipBTD–labeled, corresponding to 12,612 spectra, and 1,020 peptides were identified as d0-ipBTD–labeled, corresponding to 3,764 spectra (Table S1). Following analysis of several of the resulting peptides with Skyline, two proteins were selected to highlight the utility of iTORC (Fig. 4). Elongation factor 1-α1 (Eef1a1) is known to oligomerize in a thiol-dependent fashion, with Cys-411 contributing in disulfide bond formation (presumably through sulfenic acid formation) and Cys-234 playing less of a role in this oligomerization (18). iTORC analysis showed a larger population of Cys-411 being oxidized, with some sulfenylation, and Cys-234 existing mostly as a free thiol (Fig. 4A), in agreement with the findings of Liu et al. (18). Eef1a1 plays a role in oxidative stress–induced apoptosis, and measuring the redox status of its cysteines in cells using iTORC could help to further elucidate the mechanism. Additionally, three peptides from mouse serum albumin (a protein synthesized for secretion in hepatocytes) were found to be >95% oxidized, consistent with their known oxidation state (i.e. in disulfide bonds) (Fig. 4B) (19).
Discussion
In summary, halogenated benzothiazine-based alkylating agents were synthesized and utilized to measure cellular thiol status. ipBTD has high reactivity toward thiol moieties and has a considerably faster reaction rate than the most commonly used alkylating agents, IAM and NEM. pBTD-alkylated proteins can be excited with UV (360-nm) fluorescence, adding to the utility of these reagents.
Concerns can be raised about the multistep labeling procedure due to possible thiol oxidation during the initial labeling of sulfenic acids and the potential for thiol–disulfide exchange during the reaction with pBTD. We have considered these issues and do not believe that they are factors, for five reasons.
First, our lysis buffer, in which the sulfenic acid labeling is performed, contains 100 units/ml catalase, which should remove the vast majority of oxidizing (H2O2) species. This enzyme limits the possibility of artifactual sulfenic acid formation after lysis.
Second, other labeling strategies for sulfenic acids also employ multistage labeling (e.g. the procedure reported by Seo and Carroll (12) with dimedone and iododimedone, which we have employed ourselves in the past (20, 21)). Thus, the sulfenic acid–specific reagents (either dimedone or pBTD) should be added first. Reisz et al. (22) report a series of rates and rate constants for reactions with Cys-46 of the peroxiredoxin protein AhpC, utilizing the classic reagents IAM and dimedone. Both the thiol and sulfenic acid (forms of Cys-46) reacted with IAM with very similar rate constants (0.15 and 0.10 m−1 s−1, respectively). Very similar rate constants were observed for the reaction of the protein (or a mutant) with dimedone. Thus, the proposal to mix reagents is not realistic. We are of the opinion that using faster-reacting reagents is very advantageous in competing with the rates of thiol–sulfenic acid reaction (i.e. disulfide formation) that occur during the labeling procedure.
Adding selective reagents for sulfenic acids and free thiols at the same time, in principle, would be most ideal for prevention of post-lysis oxidation and thiol–disulfide interchange. To this end, a thiol-selective reagent was recently reported and is a very exciting advancement in specific reagents (23). However, our goal in designing our method was to compare differences in thiol oxidation using a global proteomics approach. MS quantitation is much more reliable for labeled peptides or whole proteins with isotopomeric ions because these species co-chromatograph, fragment identically, and have the same ionization efficiency and therefore can be measured easily and confidently by mass difference using high resolution MS (HRMS). Additionally, a time course experiment recommended by one of the reviewers was conducted, and we saw very little change in oxidation between 20 and 60 min, suggesting that 20 min is a sufficient preincubation time in the future (Fig. S4).
Third, rates of reaction of sulfenic acids with thiols have been measured and reported. The most complete and relevant set of rate constants was done with human serum albumin containing a single cysteine sulfenic acid, which was reacted with a variety of thiols (at pH 7.4) (24, 25). 2-Nitro-5-thiobenzoate had a rate constant of 105 m−1 s−1, and the alkyl thiols (cysteine derivatives) yielded rate constants of 2.9–55 m−1 s−1. Dimedone reacted with a sulfenic acid in albumin with a rate constant of 0.027 m−1 s−1. Thus, labeling with dimedone has a deficiency of poor competition with thiols (which will generate disulfides). The main point we emphasize here is that the rapid reaction of pBTD removes the sulfenic acid rapidly (k = 1,700 m−1 s−1) before it can react with a thiol. Moreover, the high concentration of pBTD used (2–5 mm) makes this reaction even more competitive with any thiols in the sample (e.g. with the 10-fold dilution, the GSH concentration would be ≤0.5 mm). Thus, we are of the opinion that the iTORC method is more quantitative in trapping sulfenic acids than dimedone. Admittedly, the rate of reaction of a sulfenic acid with a thiol within a peroxiredoxin or similar antioxidant enzyme is much faster due to approximation (2), and such labile intermediates would not be trapped with our method. However, these faster disulfide-forming reactions will be detected by quantifying differences between the d7- and d0-ipBTD–modified cysteines.
Additionally, comparisons were made between cysteines detected with iTORC and recent studies on sulfenylated proteins and reversibly oxidized cysteine sites utilizing other probes. Sulfenylated proteins (Table S1, cited by gene name) detected with 13C6-d7-modifications showed 5 and 3% overlap with the results of use of the 1H-benzo[c][1,2]thiazin-4(3H)-one 2,2-dioxide probes from Gupta et al. (26) and 4-(pent-4-yn-1-yl)cyclohexane-1,3-dione (DYn-2) from Yang et al. (27), respectively. Considering that these two data sets both utilized the human RKO adenocarcinoma cell line and had a 26% overlap in sulfenylated protein identifications between each other, the 3–5% overlap observed with mouse hepatocytes in this study is indicative of similarity between basally oxidized proteins across cells of diverse origins and species. A comparison of proteins with basal reversible redox modifications from mouse RAW 264.7 macrophages as reported in Guo et al. (28) showed a 12% overlap with proteins detected with d0-modifications (i.e. reversibly oxidized). Of those shared proteins, 38% of cysteine sites were detected to be reversibly oxidized by both methods. Overall, these results demonstrate that the modifications with iTORC are comparable with previously published data sets.
Fourth, if the reaction of the sulfenic acid with ipBTD occurs, we would expect to observe a product with an added oxygen (M+16) in our labeling procedure (Scheme S2). However, we have reanalyzed our proteomic data with thiol modifications corresponding to the thioether sulfoxides of each of the three isotopic labels and detected only very low amounts of peptides with such modification (Table S2). If the d7-ipBTD reagent had reacted with any sulfenic acids, we would expect a significant amount of oxidized thiols identified (Cys plus additional mass of 260.1 Da) compared with the reduced alkylation product (Cys plus additional mass of 244.1 Da). We did see some identification with this modification (4.2%), but the level was similar (in percentage) to the oxidized modifications identified in the d0 channel, suggesting that this oxidation likely happened during the subsequent SDS-PAGE and digestion procedure. Additionally, there was very little overlap of cysteines (1.3%, 31 sites) detected having d7- (2,367 total sites) and d0+[O]- (89 total sites) modifications, further supporting the conclusion that oxidation occurs at cysteine sites reduced after TCEP incubation.
The fifth and final point, regarding concern about interchange of the thiols and disulfides, is addressed in our albumin results (Fig. 5B). Albumin (including mouse albumin) is well-known to have all of its thiols in disulfide form except for Cys-34 (numbering for human). In general, rates of nonspecific thiol interchanges are known to be low (e.g. 0.01–2 m−1 s−1, except for reported high rates due to their production in enzymatic reactions (29). If thiol–disulfide exchange (including low-molecular weight thiols such as those formed from GSH) was prevalent during the 1-h incubation with pBTD, we would observe a much greater population of “free” thiols at sites that are known to be contributing in disulfide bonds. However, relative quantitation for three separate peptides showed >87% reversibly oxidized thiol, suggesting that these thiols are contributing in disulfide bonds.
Figure 5.
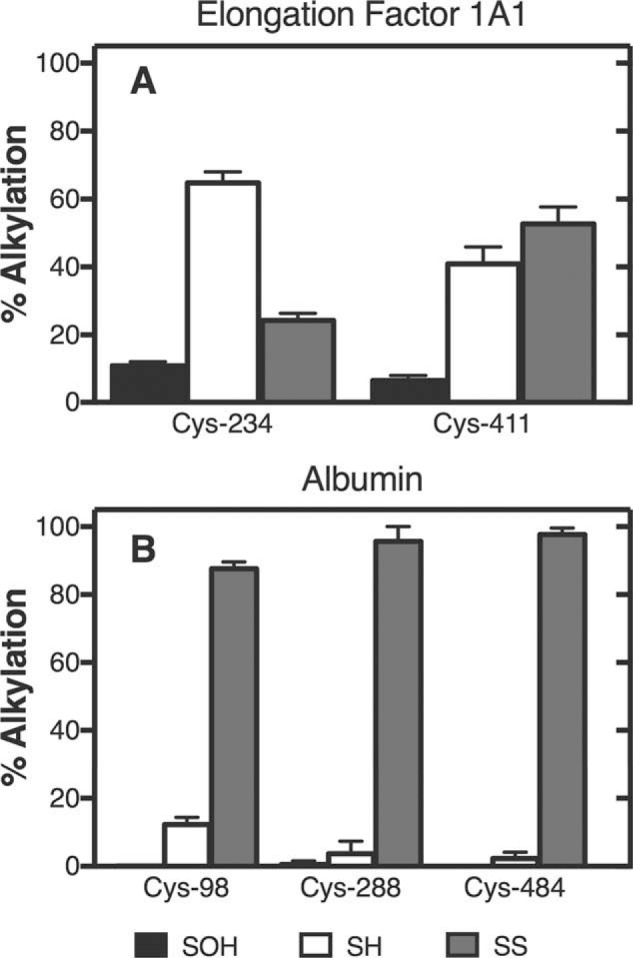
iTORC labeling of isolated mouse hepatocytes. A, elongation factor 1A1; B, albumin. Results are presented as mean ± S.D. (range) of duplicate assays.
To further address concerns about potential thiol–disulfide interchange, iTORC was employed in two time-course experiments in which lysis of mouse hepatocytes was performed with or without 13C6-d7-pBTD and then d7-ipBTD was added at varying time points after lysis. The change in the proportion of spectra modified with each probe was ≤5% relative to the reported workflow timeline with d7-ipBTD added 60 min after lysis (Fig. S4). Furthermore, the variation was lower in the time course with all three reagents added, suggesting that labeling by 13C6-d7-pBTD first may limit disulfide exchange by alkylating the reactive sulfenic acid species (Fig. S4A).
Sequentially alkylating hepatocyte lysates with 13C6-d7-pBTD, d7-, and d0-ipBTD is a novel method, termed iTORC, which combines approaches for detecting the redox status of proteins and produces a structurally identical modification with isotopic differences easily separated by MS. This methodology is a powerful tool for determining the effect of redox perturbations on protein thiols in vitro and in cell or tissue extracts.
Experimental procedures
Mouse models
The Vanderbilt University Animal Care and Use Committee approved all procedures. Mice were maintained in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Control male C57BL/6J mice were provided free access to rodent chow (PicoLabTM Laboratory Rodent Diet 5L0D, Purina, Richmond, IN) and water. Mice were housed in a temperature- and humidity-controlled room on a 12-h/12-h light/dark cycle.
Chemical synthesis
Ethyl-2-amino-[13C6]benzoate
13C6-Anthranilic acid (1.0 g, 7.0 mmol, Cambridge Isotope Laboratories, Cambridge, MA) was dissolved in 20 ml of C2H5OH and sparged with HCl gas until saturated. The reaction was heated under reflux (60 °C, 1 h) and then evaporated to dryness in vacuo. The solid was then neutralized with 100 mm aqueous NaOH (75 ml), and the product was extracted three times with 50 ml of ethyl acetate. The liquid was then dried over anhydrous MgSO4, filtered, and evaporated to dryness, yielding a brown oil (80% yield). 1H NMR (CDCl3, 600 MHz): δ 7.90 (qq, 1H, J = 26.5 Hz), 7.28 (quiqui, 1H, J = 21.1 Hz), 6.67 (m, 1H, J = 20.6 Hz), 5.74 (bs, 2H), 4.36 (q, 2H, J = 7.3 Hz), 1.41 (t, 3H, J = 7.2 Hz); 13C NMR (CDCl3, 150 MHz): δ 168.2, 150.4, 134.0, 131.2, 116.4, 111.1, 60.3, 14.4.
pBTD
pBTD was synthesized in a straightforward three-step sequence (Scheme S1) as described previously, with minor alterations (26, 30). 1-Iodopropane, or 1-d7-iodopropane for d7-pBTD, was used (as the alkylating agent) to produce the n-propyl sulfonamide prior to the cyclization step. The product of the cyclization step was neutralized with 100 mm NH4HCO3 to prevent sultam ring opening, which improved the yield to 70%. Ethyl-2-amino[13C6]benzoate (see above) was used in place of ethyl-2-aminobenzoate for the synthesis of 13C6-d7-pBTD.
ipBTD
ipBTD synthesis was based on a procedure described previously (31). pBTD (2.5 mmol in 4 ml of CH3CN) was reacted with N-iodosuccinimide (3 mmol in 4 ml of CH3CN) for 30 min at 25 °C. The reaction was extracted with 50 ml of ethyl acetate, and the product (in the upper organic layer) was washed three times with 50 ml of H2O, followed by one 50-ml wash with brine. The ethyl acetate layer was dried over anhydrous MgSO4, filtered, and evaporated to dryness in vacuo. The crude reaction was purified by silica gel column chromatography using 15% ethyl acetate/hexanes (v/v), in 70% yield. 1H NMR (CDCl3, 600 MHz): δ 8.31 (dd, 1H), 7.71 (dt, 1H), 7.29 (dt, 1H), 7.25 (d, 1H), 5.92 (bs, 1H), 4.06 (t, 2H), 2.07 (qt, 2H), 1.14 (t, 3H); 13C-NMR (CDCl3, 150 MHz): δ 181.1, 142.4, 136.5, 132.0, 124.5, 117.8, 117.1, 53.4, 34.9, 23.9, 11.1 (Fig. S3). HRMS m/z 363.9540 (MH+, Δ 8 ppm).
pBTD and ipBTD reaction with cyclic sulfenamide dipeptide
13C6-d7-pBTD (100 μm, from a 1 mm stock in DMSO) was reacted with a 100 μm solution of a previously described cyclic sulfenamide derivative of the dipeptide (N-carbobenzoxy-Cys-Val-OCH3 (Fig. S1), prepared as described (11)) for 15 min. The cyclic sulfenamide dipeptide (100 μm, reduced with 100 μm TCEP) was reacted with either 100 μm d7-ipBTD or d0-ipBTD (1 mm stock in CH3CN) for 15 min. The three samples were then mixed in equal parts and subjected to LC-MS/MS analysis. The mixed sample (10 μl) was loaded onto a Agilent Poroshell 120 SB-C18 column (2.1 mm × 100 mm, 2.7 μm) and separated using an Acquity UPLC (Waters) at a flow rate of 0.4 ml min−1 with the following linear gradient: 5% solvent B (CH3CN, 0.1% HCO2H (v/v)), 95% solvent A (H2O, 0.1% HCO2H (v/v)) to 100% B over 8 min. The UPLC was interfaced with a Thermo Orbitrap mass spectrometer via an electrospray ionization source, in the negative ion mode. MS1 was collected at a resolution of 30,000. Three MS2 fragmentation scans were acquired in the linear ion trap throughout the gradient corresponding to the m/z for the 13C6-d7-, d7-pBTD, or d0-pBTD alkylated dipeptide (617.2, 611.2, and 604.2, respectively).
ipBTD and IAM reactions with E. coli lysate
E. coli (DH5α) was grown overnight in 100 ml Terrific Broth medium (Difco). Cells were pelleted by centrifugation (8,000 × g, 10 min), resuspended in 5 ml of lysis buffer (100 mm HEPES, 100 mm NaCl, pH 7.4), and probe-sonicated (50% amplitude) for three 10-s bursts. Protein concentration was determined by a bicinchoninic acid assay (Bio-Rad). The lysate (100 μg in 100 μl) was reacted with either 20 mm IAM (100 mm stock, H2O) or 2 mm ipBTD (10 mm stock, CH3CN) for the indicated times. Samples were quenched by the addition of 30 mm DTT (1 m stock, H2O) and then counteralkylated with 125 μl of 200 mm NEM for 30 min. Samples were then loaded onto an SDS-polyacrylamide gel (10% BisTris, NuPAGE, Invitrogen), separated at 200 V, and stained with SimplyBlue SafeStain (Invitrogen). These samples were excised and subjected to in-gel trypsin digestion and LC-MS/MS analysis (32).
UV fluorescence visualization of SDS-polyacrylamide gels
BSA (2 mg/ml, Pierce) was reduced with 1 mm TCEP for 30 min at 55 °C, and thiols were subsequently alkylated with 2 mm ipBTD (10 mm stock, CH3CN) for 10 min. The reaction was quenched with 100 mm DTT (1 m stock, H2O). BSA (5 μg) alone (or added to 15 μg of unlabeled E. coli lysate) was separated via SDS-PAGE and visualized with a UV transilluminator (360 nm) (Bio-Rad). The fluorophore pBTD was initially determined to have an excitation wavelength of 380 nm and an emission wavelength of 420 nm.
In a separate experiment, BSA was reacted with ipBTD (2 mm) under nonreducing conditions, as above. Under reducing conditions (1 mm TCEP), BSA was reacted with IAM (50 mm) for 30 min. The IAM was then quenched with DTT (25 mm) for 15 min, and ipBTD (2 mm) was added for 15 min. The different BSA samples (5 μg) were separated via SDS-PAGE and visualized with a UV transilluminator as above.
iTORC labeling of mouse hepatocytes
Hepatocytes from male C57BL/6J mice were isolated as described previously (33). Cells from each mouse (3 × 106, n = 4) were pelleted via centrifugation (50 × g) and resuspended in 1.2 ml of lysis buffer (50 mm sodium HEPES (pH 7.6) containing 150 mm NaCl, 1% (v/v) IGEPAL, CA-630 (formerly Nonidet NP-40), and 100 units/ml catalase) with 5 mm 13C6-d7-pBTD (50 mm stock, DMSO) at 37 °C for 1 h to alkylate sulfenic acids. d7-ipBTD (2 mm, from 10 mm stock, CH3CN) was then added and reacted at 25 °C in the dark for 15 min to alkylate free thiols. The reaction was quenched by the addition of 50 mm DTT (1 m stock, H2O) and precipitated with the addition of 2 volumes of CH3CN. Proteins were precipitated by centrifugation (5,000 × g, 5 min). The pellet was washed with CH3OH/H2O (1:1, v/v) and briefly resuspended with one 10-s burst of probe sonication. After centrifugation (5,000 × g, 5 min), the pellet was resuspended in 500 μl of 100 mm HEPPS buffer (pH 8.0) containing 5% SDS (w/v) and 1 mm TCEP, probe-sonicated for one 10-s burst, and allowed to react for 30 min at 55 °C. d0-ipBTD (2 mm, from 10 mm stock, in CH3CN) was then added to alkylate the initially oxidized thiols. This reaction was again quenched with 50 mm DTT (1 m stock, in H2O). SDS-PAGE sample-loading buffer was added, and 100 μg of each sample was loaded onto an SDS-polyacrylamide gel (10% BisTris, NuPAGE, Invitrogen). Proteins were separated at 200 V and stained with SimplyBlue SafeStain (Invitrogen). These samples were excised and subjected to in-gel trypsin digestion prior to LC-MS/MS analysis (11).
LC-MS/MS analysis
Extracted E. coli lysate peptides were analyzed on a nanoLC Ultra system (Eksigent Technologies, Dublin, CA) interfaced with an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, San Jose, CA). Approximately 5 μg of digested E. coli lysate was reconstituted in 0.1% HCO2H (v/v) and pressure-loaded (1.5 μl min−1) onto a 360-μm outer diameter × 100-μm inner diameter microcapillary analytical column packed with Jupiter octadecylsilane (C18) (3 μm, 300 Å; Phenomenex) and equipped with an integrated electrospray emitter tip. Peptides were then separated with a linear gradient formed with 0.1% HCO2H in H2O (solvent A) and 0.1% HCO2H in CH3CN (solvent B) (all v/v) by increasing from 2 to 45% B (v/v) over a period of 0–80 min at a flow rate of 500 nl min−1. The spray voltage was set to 2.0 kV, and the heated capillary temperature was at 200 °C. Collision-induced dissociation (CID) MS/MS spectra were recorded in the data-dependent mode using a Top 7 method. MS1 spectra were measured with a resolution of 70,000, an AGC target of 1e6, and a mass range from m/z 300 to 1,500. CID MS/MS spectra were acquired with a normalized collision energy of 30. Peptide m/z values that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 20 s, with a repeat count of 1.
For samples produced with mouse hepatocytes, an analytical column was packed with 20 cm of octadecylsilane (C18) reversed-phase material (Jupiter, 3-μm beads, 300 Å; Phenomenex) directly into a laser-pulled emitter tip. Peptides (500 ng) were loaded on the capillary reversed phase analytical column (360-μm outer diameter, 100-μm inner diameter) using a Dionex Ultimate 3000 nanoLC and autosampler. The mobile phase solvents consisted of 0.1% HCO2H, 99.9% H2O (solvent A) and 0.1% HCO2H, 99.9% CH3CN (solvent B) (v/v). Peptides were eluted at a flow rate of 400 nl min−1. The 90-min gradient consisted of the following steps: 1–3 min, 2% B (sample loading from autosampler); 3–70 min, 2–40% B; 70–78 min, 40–95% B; 78–79 min, 95% B; 79–80 min, 95–2% B; 80–90 min (column re-equilibration), 2% B (all v/v). A Q-Exactive mass spectrometer (Thermo Scientific), equipped with a nanoelectrospray ionization source, was used to analyze the eluting peptides. The instrument method consisted of MS1 using an MS AGC target value of 1e6, followed by up to 20 MS/MS scans of the most abundant ions detected in the preceding MS scan. A maximum MS/MS ion time of 100 ms was used with an MS2 AGC target of 1e5 and an intensity threshold of 5e4. Dynamic exclusion was set to 15 s, higher-energy collision dissociation collision energy was set to 27 normalized collision energy, and peptide match and isotope exclusion were enabled.
Peptide data analysis
Raw data files were analyzed using MyriMatch software (version 2.2.140) (34) against a decoy protein database consisting of a forward and reversed murine Uniprot/Swissprot database, with only reviewed proteins included (version 20150825; 16,718 entries) or an E. coli database (4,325 entries). Trypsin with fully specific digestion was used as the enzyme search parameter. The number of missed cleavages permitted was two. Precursor ion mass tolerance was set at 10 ppm, and the fragmentation tolerance was 20 ppm for the database search. Methionine oxidation (15.9949 Da, dynamic) and cysteine modifications by 13C6-d7-pBTD, d7-ipBTD, or d0-ipBTD (250.11003, 244.08990, and 237.04596 Da, respectively, dynamic) were included as variable search modifications. For the E. coli assays, an additional low-resolution search was performed with the same databases, where precursor ion mass tolerance was set at 10 ppm and fragmentation tolerance was set at 0.5 m/z. The maximum Q values of peptide spectrum matches were adjusted to achieve a peptide false discovery rate ≤ 5%, using IDPicker software (version 3.1.642.0) (35). MS1 precursor quantitation was performed using Skyline software as described previously (20).
Author contributions
M. E. A. designed and conducted the experiments and analyzed the results. S. M. G. conducted some of the experiments and analyzed the results. E. T. prepared the hepatocytes. F. P. G. and M. E. A. conceived the studies, evaluated the results, and wrote the paper.
Supplementary Material
Acknowledgments
We thank Prof. Ambra Pozzi (Vanderbilt University) for help in optimizing the method. Mass spectrometry was performed at the Vanderbilt Mass Spectrometry Resource Core (MSRC) and at the MSRC Proteomics Core Facility. We also thank K. Trisler for assistance in the preparation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM118122 (to F. P. G.), F31 HL136133 (to M. E. A.), T32 ES007028 (to F. P. G. and S. M. G.), F31 DK112553 (to E. T.), R37 DK050277 (to E. T.), and R01 DK054902 (to E. T.) and American Heart Association Predoctoral Fellowship PRE33410007 (to M. E. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Schemes S1 and S2, Table S1, and Figs. S1 and S2.
- iTORC
- isotopic tagging of oxidized and reduced cysteines
- CID
- collision-induced dissociation
- Eef1a1
- elongation factor 1-α1
- HRMS
- high-resolution MS
- IAM
- 2-iodoacetamide
- ipBTD
- 3-iodo-1-propyl-1H-benzo[c][1,2]thiazin-4(3H)-one 2,2-dioxide
- NEM
- N-ethylmaleimide
- pBTD
- 1-propyl-1H-benzo[c][1,2]thiazin-4(3H)-one 2,2-dioxide
- TCEP
- tris(2-carboxyethyl)phosphine
- HEPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- BisTris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- AGC
- automatic gain control.
References
- 1. Parvez S., Long M. J. C., Poganik J. R., and Aye Y. (2018) Redox signaling by reactive electrophiles and oxidants. Chem. Rev. 118, 8798–8888 10.1021/acs.chemrev.7b00698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Portillo-Ledesma S., Randall L. M., Parsonage D., Dalla Rizza J., Karplus P. A., Poole L. B., Denicola A., and Ferrer-Sueta G. (2018) Differential kinetics of two-cysteine peroxiredoxin disulfide formation reveal a novel model for peroxide sensing. Biochemistry 57, 3416–3424 10.1021/acs.biochem.8b00188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tavender T. J., Springate J. J., and Bulleid N. J. (2010) Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 29, 4185–4197 10.1038/emboj.2010.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Truong T. H., Ung P. M., Palde P. B., Paulsen C. E., Schlessinger A., and Carroll K. S. (2016) Molecular basis for redox activation of epidermal growth factor receptor kinase. Cell Chem. Biol. 23, 837–848 10.1016/j.chembiol.2016.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou H., Singh H., Parsons Z. D., Lewis S. M., Bhattacharya S., Seiner D. R., LaButti J. N., Reilly T. J., Tanner J. J., and Gates K. S. (2011) The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. J. Am. Chem. Soc. 133, 15803–15805 10.1021/ja2077137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta V., and Carroll K. S. (2014) Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 1840, 847–875 10.1016/j.bbagen.2013.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Devarie-Baez N. O., Silva Lopez E. I., and Furdui C. M. (2016) Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 50, 172–194 10.3109/10715762.2015.1090571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Albertolle M. E., and Guengerich F. P. (2018) The relationships between cytochromes P450 and H2O2: production, reaction, and inhibition. J. Inorg. Biochem. 186, 228–234 10.1016/j.jinorgbio.2018.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benitez L. V., and Allison W. S. (1974) The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J. Biol. Chem. 249, 6234–6243 [PubMed] [Google Scholar]
- 10. Carroll K. S., and Gupta V. (May 22, 2018) Targeted covalent probes and inhibitors of proteins containing redox-sensitive cysteines. United States Patent 9,977,024
- 11. Gupta V., and Carroll K. S. (2016) Profiling the reactivity of cyclic C-nucleophiles towards electrophilic sulfur in cysteine sulfenic acid. Chem. Sci. 7, 400–415 10.1039/C5SC02569A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seo Y. H., and Carroll K. S. (2011) Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew. Chem. Int. Ed. Engl. 50, 1342–1345 10.1002/anie.201007175 [DOI] [PubMed] [Google Scholar]
- 13. Fu L., Liu K., Ferreira R. B., Carroll K. S., and Yang J. (2019) Proteome-wide analysis of cysteine S-sulfenylation using a benzothiazine-based probe. Curr. Protoc. Protein. Sci. 95, e76 10.1002/cpps.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alcock L. J., Perkins M. V., and Chalker J. M. (2018) Chemical methods for mapping cysteine oxidation. Chem. Soc. Rev. 47, 231–268 10.1039/C7CS00607A [DOI] [PubMed] [Google Scholar]
- 15. Prakash A. S., Kabli A. M. F., Bulleid N. J., and Burchmore R. (2018) Mix-and-match proteomics: using advanced iodoTMT multiplexing to investigate cysteine oxidation changes with respect to protein expression. Anal. Chem. 90, 14173–14180 10.1021/acs.analchem.8b02517 [DOI] [PubMed] [Google Scholar]
- 16. Bednar R. A. (1990) Reactivity and pH dependence of thiol conjugation to N-ethylmaleimide: detection of a conformational change in chalcone isomerase. Biochemistry 29, 3684–3690 10.1021/bi00467a014 [DOI] [PubMed] [Google Scholar]
- 17. Hansen R. E., and Winther J. R. (2009) An introduction to methods for analyzing thiols and disulfides: reactions, reagents, and practical considerations. Anal. Biochem. 394, 147–158 10.1016/j.ab.2009.07.051 [DOI] [PubMed] [Google Scholar]
- 18. Liu T., Yang Y., Wang D., Xiao Y., Du G., Wu L., Ding M., Li L., and Wu C. (2015) Human eukaryotic elongation factor 1A forms oligomers through specific cysteine residues. Acta Biochim. Biophys. Sin. (Shanghai) 47, 1011–1017 10.1093/abbs/gmv113 [DOI] [PubMed] [Google Scholar]
- 19. Sigrist C. J., De Castro E., Langendijk-Genevaux P. S., Le Saux V., Bairoch A., and Hulo N. (2005) ProRule: a new database containing functional and structural information on PROSITE profiles. Bioinformatics 21, 4060–4066 10.1093/bioinformatics/bti614 [DOI] [PubMed] [Google Scholar]
- 20. Albertolle M. E., Phan T. T. N., Pozzi A., and Guengerich F. P. (2018) Sulfenylation of human liver and kidney microsomal cytochromes P450 and other drug metabolizing enzymes as a response to redox alteration. Mol. Cell. Proteomics 17, 889–900 10.1074/mcp.RA117.000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Albertolle M. E., Kim D., Nagy L. D., Yun C. H., Pozzi A., Savas Ü., Johnson E. F., and Guengerich F. P. (2017) Heme-thiolate sulfenylation of human cytochrome P450 4A11 functions as a redox switch for catalytic inhibition. J. Biol. Chem. 292, 11230–11242 10.1074/jbc.M117.792200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reisz J. A., Bechtold E., King S. B., Poole L. B., and Furdui C. M. (2013) Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 280, 6150–6161 10.1111/febs.12535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen X., Wu H., Park C. M., Poole T. H., Keceli G., Devarie-Baez N. O., Tsang A. W., Lowther W. T., Poole L. B., King S. B., Xian M., and Furdui C. M. (2017) Discovery of heteroaromatic sulfones as a new class of biologically compatible thiol-selective reagents. ACS Chem. Biol. 12, 2201–2208 10.1021/acschembio.7b00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alvarez B., Carballal S., Turell L., and Radi R. (2010) Formation and reactions of sulfenic acid in human serum albumin. Methods Enzymol. 473, 117–136 10.1016/S0076-6879(10)73005-6 [DOI] [PubMed] [Google Scholar]
- 25. Turell L., Botti H., Carballal S., Ferrer-Sueta G., Souza J. M., Durán R., Freeman B. A., Radi R., and Alvarez B. (2008) Reactivity of sulfenic acid in human serum albumin. Biochemistry 47, 358–367 10.1021/bi701520y [DOI] [PubMed] [Google Scholar]
- 26. Gupta V., Yang J., Liebler D. C., and Carroll K. S. (2017) Diverse redoxome reactivity profiles of carbon nucleophiles. J. Am. Chem. Soc. 139, 5588–5595 10.1021/jacs.7b01791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., Gupta V., Carroll K. S., and Liebler D. C. (2014) Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 5, 4776 10.1038/ncomms5776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo J., Gaffrey M. J., Su D., Liu T., Camp D. G. 2nd, Smith R. D., and Qian W. J. (2014) Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat. Protoc. 9, 64–75 10.1038/nprot.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shaked Z., Szajewski R. P., and Whitesides G. M. (1980) Rates of thiol-disulfide interchange reactions involving proteins and kinetic measurements of thiol pKa values. Biochemistry 19, 4156–4166 10.1021/bi00559a004 [DOI] [PubMed] [Google Scholar]
- 30. Volovenko Y., Volovnenko T., and Popov K. (2007) N-alkyl-4-chloro-1H-benzo [c][1,2]thiazine-3-carbaldehyde-2,2-dioxides—new functional benzothiazine derivatives. J. Heterocycl. Chem. 44, 1413–1419 10.1002/jhet.5570440627 [DOI] [Google Scholar]
- 31. Sreedhar B., Reddy P. S., and Madhavi M. (2007) Rapid and catalyst-free α-halogenation of ketones using N-halosuccinamides in DMSO. Synthetic Commun. 37, 4149–4156 10.1080/00397910701574908 [DOI] [Google Scholar]
- 32. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 33. Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., and Viollet B. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 10.1172/JCI40671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tabb D. L., Fernando C. G., and Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 10.1021/pr0604054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holman J. D., Ma Z. Q., and Tabb D. L. (2012) Identifying proteomic LC-MS/MS data sets with Bumbershoot and IDPicker. Curr. Protoc. Bioinformatics, Chapter 13, Unit 13.17 10.1002/0471250953.bi1317s37 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.