

Abstract
The dominant model for Alzheimer’s disease (AD) is the amyloid cascade hypothesis, in which the accumulation of excess amyloid-β (Aβ) leads to inflammation, excess glutamate and intracellular calcium, oxidative stress, tau hyperphosphorylation and tangle formation, neuronal loss, and ultimately dementia. In a cascade, AD proceeds in a unidirectional fashion, with events only affecting downstream processes. Compelling evidence now exists for the presence of positive feedback loops in AD, however, involving oxidative stress, inflammation, glutamate, calcium, and tau. The pathological state of AD is thus a system of positive feedback loops, leading to amplification of the initial perturbation, rather than a linear cascade. Drugs may therefore be effective by targeting numerous points within the loops, rather than concentrating on upstream processes. Anti-inflammatories and anti-oxidants may be especially valuable, since these processes are involved in many loops and hence would affect numerous processes in AD.
Keywords: Aggregation, amyloid, amyloid-β protein precursor, directed acyclic graph, drug discovery, peptide, systems biology
THE AMYLOID CASCADE HYPOTHESIS
Since 1992 [1], the amyloid cascade hypothesis has played the prominent role in seeking to explain the onset, progression, and pathogenesis of Alzheimer’s disease (AD). It proposes that the over-production and/or decreased degradation and subsequent deposition of amyloid-β (Aβ) is the initial pathological event in AD. A range of subsequent events then follow: Aβ oligomer and plaque formation, activation of microglia and astrocytes, chronic inflammation, increased levels of the neurotransmitter glutamate, elevated intracellular calcium, oxidative stress, synaptic dysfunction, tau hyperphosphorylation and tangle formation, neuronal loss, and ultimately dementia. A simple, though typical, representation of the amyloid cascade is shown in Fig. 1. A wealth of evidence supports the involvement of all these processes, and more, in AD. While there is thus little doubt that each of the steps shown in Fig. 1 are key events in AD, it is now apparent that numerous positive feedback loops also occur, requiring modification of the amyloid cascade hypothesis to a system of loops, rather than a linear process.
Fig.1.
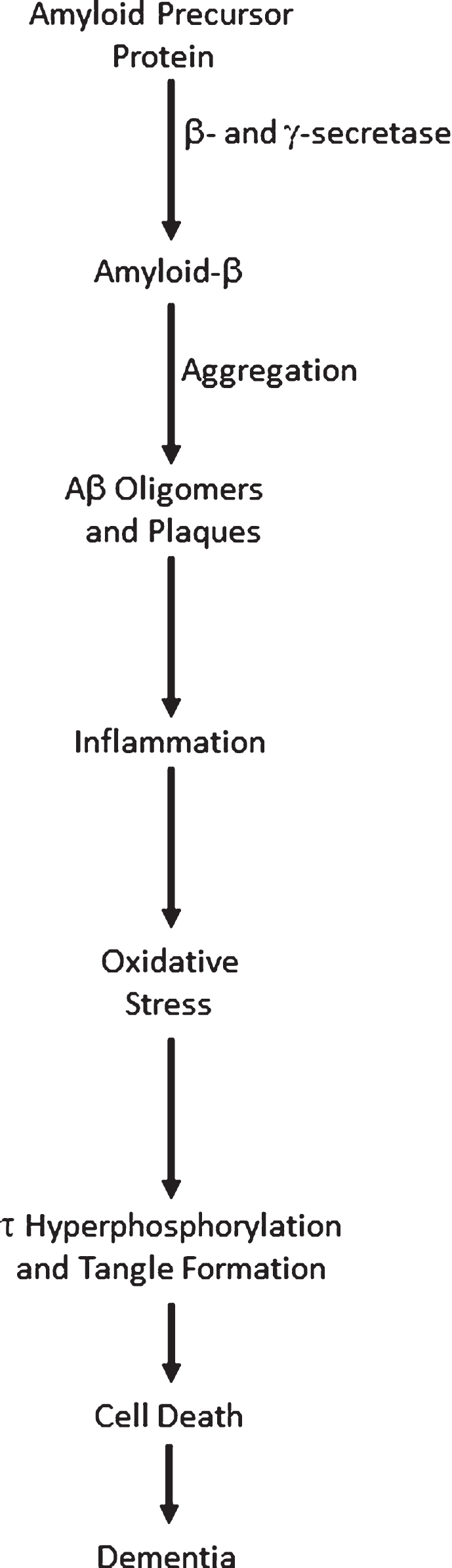
The Amyloid cascade.
A cascade is defined as: “a small waterfall, typically one of several that fall in stages down a steep rocky slope”, or, more pertinently here, “A succession of devices or stages in a process, each of which triggers or initiates the next.” (https://en.oxforddictionaries.com/definition/cascade). Both these definitions imply that information flow can only travel in one direction, as water can only flow downhill, otherwise known as a directed acyclic graph. The amyloid cascade is thus a unidirectional process, where initial stress causes an increase in total Aβ (Aβ42 in particular), leading to a succession of processes and eventually dementia. In this model, perturbations to the healthy system can only lead to changes downstream of the location of the perturbation. For example, increased oxidative stress could cause tau hyperphosphorylation and tangle formation, but not remove Aβ oligomers. This concept of unidirectional flow of information is inaccurate, however, since it is now apparent that there are a variety of positive feedback loops in AD, where processes downstream in the cascade can stimulate processes upstream, leading to amplification of the initial perturbation. It is thus no longer tenable to frame AD as a cascade. Here I give examples of positive feedback loops in AD to illustrate this. The aim of this review is not to give a comprehensive overview of AD pathways— rather it is to show that diverse positive feedback loops are present in AD and to discuss their implications for the disease and its possible therapies.
Aβ PEPTIDES AND AβPP FRAGMENTS
Aβ40 and Aβ42 are generally regarded as the most important amyloid-β protein precursor (AβPP) derivatives: Aβ40 because it is the most abundant in senile plaques and Aβ42 because it is also abundant and more toxic than Aβ40. Hence the Aβ42:Aβ40 ratio is often seen as a critical measure of AD stress, in addition to absolute concentrations of these peptides. Very many other AβPP fragments and derivatives are also likely to be involved in AD progression, however, such as soluble AβPP α and β (sAβPPα and sAβPPβ), C83, C99, P3, amyloid precursor protein intracellular domain (AICD), N3-pyroglutamate [2], isoaspartyl, phosphoserine and other post-translational modifications, and Aβ extended at both N- and C-termini [3, 4]. Even pure Aβ42 can aggregate into a host of oligomers (dimers, trimers, tetramers, hexamer, dodecamers etc.) with most likely multiple structures for each oligomer. We thus have a vast host of Aβ derivatives, with varying N- and C-termini, aggregated species of different sizes, and oligomers formed from more than one type of peptide, of differing populations, lifetimes, potential interactions with other molecules and toxicities. Hunter and Brayne have summarized how some of these can interact with each other in many ways, providing numerous feedback loops even when just considering interactions between Aβ species [5]. Aggregation is highly dependent on concentration, solution conditions, interactions with other molecules (e.g., seeds) and often shows variable lag times waiting for the formation of an aggregation nucleus. We thus have a highly complex situation, even considering only AβPP products in vitro. Hunter and Brayne discuss many of these issues in more detail [6]. How closely we can model the brain of a human with dementia, using a mouse model overexpressing an AβPP derivative, or cells with Aβ added extracellularly, remains to be seen.
OXIDATIVE STRESS
Oxidative stress arises when there is an imbalance in the generation of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), hydroxyl radical (HO·), and superoxide (O2·–), over their removal. Life in an environment exposed to O2 causes a constant production of ROS. There is evidence for the complex involvement of oxidative stress at numerous points in AD pathogenesis [7–9], making it unclear to what extent oxidative stress is a cause or consequence of AD [9]. Metal ions such as zinc, iron, and copper have been found in amyloid plaques [10]. In particular, iron and copper can generate ROS, since they commonly exist in oxidation states differing by a single electron, thus readily generating free radicals. Elevated levels of Aβ have been reported to be associated with increased levels of oxidation products in proteins, lipids, and nucleic acids in AD hippocampus and cortex [11]. Such oxidation products may include Aβ itself, in the form of the sulphoxide at Met35 [12] or dityrosine linked dimers [13]. Mitochondria are major sources of ROS [14] and mitochondrial dysfunction may play a major, early role in AD [15–18]. Increased levels of Aβ may disrupt the electron transfer chain, increasing ROS production [19–21].
Some antioxidants, such as epigallocatechin-3-gallate (EGCG) and curcumin, have been reported to show beneficial effects in AD models and to decrease Aβ deposition [22–26]. These could work by multiple mechanisms, including: binding to Aβ directly, chelating metals [27], and acting as reducing agents or free radical scavengers. As such, disentangling the effects of anti-oxidants on AD pathways is difficult.
Here, I concentrate on evidence for feedback via oxidative stress in AD. There are several plausible mechanisms for this:
LRP1
Low density lipoprotein receptor-related protein 1 (LRP1) is involved in receptor-mediated endocytosis. In particular, it helps clear Aβ from the brain to the blood, across the blood-brain barrier [28–30]. LRP activity is compromised by increased levels of oxidized LRP, which does not bind Aβ [31]. Levels of protein oxidation can be monitored by protein carbonyls and protein resident 3-nitrotyrosine (3NT); lipid peroxidation can be monitored by protein-bound 4-hydroxy-2-nonenal (HNE). LRP1 is oxidized in AD, shown by a 60%increase in the levels of the HNE-bound LRP1 β-subunit in AD hippocampus compared to age-matched controls, though there is no change in 3NT levels [32]. Aβ induced oxidative stress thus oxidizes LRP1, decreasing LRP1 activity, disrupting Aβ clearance [32] and causing Aβ accumulation in the brain.
Oxysterols
End-products of cholesterol metabolism, named oxysterols, have been implicated in a wide range of diseases, including: atherosclerosis, inflammatory bowel diseases, age-related macular degeneration, and other pathological conditions characterized by altered cholesterol uptake and/or metabolism [33]. Non-enzymatic oxidative stress can form the oxysterols 7-ketocholesterol, 7α-hydroxycholesterol, 4β-hydroxycholesterol, 5α,6α-epoxycholesterol, and 5β,6β-epoxycholesterol. These have been detected in postmortem human AD brain and changes in their levels are associated with AD progression [34, 35]. Plasma levels of 24-hydroxycholesterol may be a marker of brain atrophy in AD patients [36].
Specific oxysterols have been suggested to be involved in AD in several ways: 3β-hydroxy-5-oxo-5,6-secocholestan-6-al can be converted into its aldol form and then react with an amine in Aβ to form a Schiff base (C = N double bond) [37]. This covalent modification of Aβ increases its amyloidogenicity [38], leading to the formation of neurotoxic spherical aggregates [39].
The oxysterol 24-hydroxycholesterol is produced in the brain by cholesterol 24-hydroxylase [40]. In contrast, 27-hydroxycholesterol is mostly synthesized elsewhere and enters the brain by crossing the blood-brain barrier [41]. An increased ratio of 27-hydroxycholesterol to 24-hydroxycholesterol has been observed in AD brains [42] where increased brain levels of 27-hydroxycholesterol may affect the production of Aβ [34]. In a human neuroblastoma cell line (SK-N-BE) differentiated into neuron-like cells, 1 μM of both oxysterols induced AβPP overexpression and increased β-secretase activity, leading to amyloidogenesis [43].
Advanced glycation endproducts (AGEs)
AGEs are proteins or lipids that are non-enzymatically glycated by sugars (typically glucose), a process enhanced by oxidative stress. AGEs are commonly found in ageing brains, particularly those with AD. Increased AGE levels may explain many features of AD, such as crosslinking in Aβ and tau, glial activation and neuronal cell death. AGEs produce superoxide radicals directly by the transition metal-catalyzed oxidation of glycated proteins. AGE specific receptors (RAGE) bind Aβ and are upregulated in AD [44, 45]. The interaction of AGEs with cells increases oxidative stress by binding of AGEs to RAGEs on the cell surface and subsequent diffusion of ROS across the membrane or by interactions between a radical producing enzyme (e.g., NADPH-oxidase) and RAGE [45]. The combination of AGEs and RAGE can cause oxidative stress, secretion of cytokines, TNFα, heme oxygenase-1, and the activation of nuclear transcription factor κB (NFκB) [46–48], thus linking ROS to inflammation [49] and forming another positive feedback loop. Stimulation of inducible nitric oxide synthase by cytokines not only results in the synthesis of toxic nitric oxide, but also modifies Aβ via nitration, where nitrated Aβ suppresses long-term potentiation of synapses more effectively than Aβ alone [50, 51].
TRPM2 and zinc
Transient receptor potential melastatin-related 2 (TRPM2) is a Ca2+-permeable channel primarily located on the cell surface, activated by ROS. In hippocampal neurons, exposure to Aβ42 generates ROS via protein kinase C and NADPH-dependent oxidases. ROS induces activation of mitogen-activated protein kinases and poly(ADP-ribose) polymerase-1, generation of ADP-ribose and activation of the TRPM2 channel. This causes lysosomal dysfunction and Zn2+ release to the cytosol. Zn2+ is highly neurotoxic [52], enhancing oxidative stress by impairing mitochondrial function and inducing mitochondrial generation of ROS, thus forming a positive feedback loop, driving loss of lysosomal and mitochondrial function and ultimately neuronal death [53].
INFLAMMATION
There is strong evidence for the presence of feedback loops in AD via inflammation. Chronic (neuro)inflammation is prevalent in AD [51, 54–56]. AD brains show inflammation at and around the sites of plaques, tangles and afflicted neurons. The aggregation and deposition of Aβ plaques cause activation of microglia and astrocytes, accompanied by overexpression of complement system. Long-term use of non-steroidal anti-inflammatory drugs (NSAIDs) shows a reduction in the incidence of AD [57–60], perhaps via feedback to decrease Aβ deposition. Links from inflammation to Aβ are now apparent via several systems:
Aβ in the periphery
Platelets are anucleate cells responsible for hemostasis and thrombosis. Circulating blood platelets are the second largest source of the AβPP protein after the brain, accounting for around 90%of circulating AβPP and are responsible for the majority of Aβ released into the plasma [61, 62]. Proteolytic cleavage of AβPP in platelets can occur at the cell surface or in intracellular secretory pathways. sAβPPα and Aβ peptides are stored in α-granules until platelets are activated, which leads to exocytosis via calcium-dependent degranulation [63]. Increased Aβ levels in the periphery may lead to increased Aβ in the brain via transport across the blood-brain barrier (the controversial peripheral sink hypothesis [31, 64–66]).
Aβ interacting with endothelial cells leads to a plethora of inflammatory factors being released. Resultant effects include: reduced anti-thrombotic activity, apoptosis, angiogenesis and accelerated tau aggregation. RAGE are also upregulated leading to over expression at the blood-brain barrier. This causes an increase in Aβ peptide deposition in the cerebral intracellular space. Contact between Aβ peptides and platelets causes further Aβ42 release by platelets, giving positive feedback.
COX-2 is one of two cyclooxygenases found in humans (the other being COX-1), responsible for the production of prostaglandins. In AD, Aβ binding to endothelial cells increases the production and release of COX-2, with a subsequent increase in COX-2 concentration. An accumulation of COX-2 above normal levels is also seen in the regions of the brain associated with AD (cerebral cortex and hippocampus). This additional COX-2 is stimulated by inflammatory cytokines, mitogenic stimuli and glutamate, in addition to Aβ [67]. COX2-expressing cells show elevated levels of AβPP mRNA leading to increased Aβ synthesis [68]. The product of COX2 activity, the unstable PGH2, rearranges into PGE2 and PGD2, as well as into the highly reactive γ-ketoaldehydes levuglandin E2 and LGD2 [69]. These levuglandins prevent Aβ proteolysis and accelerate Aβ oligomerization [70].
In human platelets, thrombin and PGE2 regulate the processing and secretion of Aβ peptides and sAβPP. The thrombin receptor is activated by Aβ, which leads to a cascade of events resulting in the stimulation of cPLA2. cPLA2 catalyzes the release of arachidonic acid, allowing synthesis of TxA2, activation of platelets and release of Aβ42 [71, 72].
Aβ contact can also lead to membrane disorganization, shrinking of platelets, and induction of apoptosis via ROS and inflammatory responses. Continued platelet activation and Aβ release results in chronic inflammation and further endothelial cell stress [71]. Hence, platelets are likely to accelerate and increase the severity of AD [73], via several positive feedback loops.
Inflammasome
Inflammasomes are innate immune system receptors and sensors that regulate the activation of caspase-1 and release of pro-inflammatory proteins IL-1β and IL-18, thus inducing inflammation in response to infection or disease modified molecules derived from host proteins [74]. In particular, inflammasomes can respond to Aβ [75, 76]. Human brains with AD or mild cognitive impairment show strongly enhanced active caspase-1 expression via NLRP3 inflammasome activity [77]. Nlrp3–/– or Casp1–/– mice carrying mutations associated with familial AD showed less memory loss, reduced brain caspase-1 and IL-1β activation, and enhanced Aβ clearance. NLRP3 inflammasome deficiency resulted in the decreased deposition of Aβ in the AβPP/PS1 model of AD [77], thus showing positive feedback between Aβ deposition and inflammation, via the inflammasome.
Intracellular inflammasome formation is followed by the release from microglia of complexes of the ASC protein called ASC specks. Recent work provides clear evidence for feedback from the inflammasome to Aβ deposition via ASC. ASC interacts directly with Aβ, as it readily seeds formation of amyloid-like aggregates in vitro, a process inhibited by ASC antibodies. AD mice genetically engineered to lack ASC had less deposition of Aβ in the brain and improved memory, compared to when ASC was present. Brain extracts from aged AβPP/PS1 mice induced the formation of Aβ deposits in the brains of young AβPP/PS1 mice, but only if ASC was present [78, 79].
RIPK1
A positive feedback loop between inflammation and Aβ may also act via RIPK1 kinase. Aβ accumulation in mice activates RIPK1 in disease-associated microglia, causing lysosomal defects and impaired phagocytic clearance of extracellular Aβ by microglia, thus giving a positive-feedback loop between neurons and microglia. Activated RIPK1 also causes excessive release of TNFα and IL-6 cytokines [80, 81].
GLUTAMATE
The anaphase promoting complex/cyclosome (APC/C) is a large protein complex forming an E3 ubiquitin ligase, targeting numerous proteins for degradation, particularly those involved in cell cycle regulation in proliferating cells [82, 83]. Cdh1 is an activator subunit of APC/C, expressed in post-mitotic neurons [84]. Aberrant accumulation of some of the targets of APC/C-Cdh1 has been associated with neurodegeneration [85]. For example, an increase in the APC/C-Cdh1 target cyclin B1 [86] affects control of the cell cycle in neurons [87]. Aβ oligomers cause proteasome-dependent degradation of Cdh1 in primary neuronal culture. This leads to a subsequent accumulation of glutaminase, an enzyme that converts glutamine to glutamate [88] and a degradation target of APC/C-Cdh1. Increased levels of glutaminase have been found in the prefrontal cortex of AD patients [89]. Elevated glutamate levels cause increases in intraneuronal Ca2+ levels, resulting in neuronal apoptosis. Excitotoxic glutamate stimulus causes APC/C-Cdh1 inactivation by decreasing Cdh1 [86]. This causes further accumulation of glutaminase, creating a positive feedback loop [88] (Fig. 2).
Fig.2.

Feedback Loops in the Amyloid Cascade. Blue arrows show pathways in healthy cells; black arrows show AD pathways; pathways activated in AD are in green; pathways inhibited in AD are in red. The dashed line shows a tentative connection from hyperphosphorylated τ to increased AβPP processing to Aβ.
CALCIUM
Ca2+ signaling plays many roles in cellular function as a second messenger, involving entry and release channels, clearance mechanisms, and intracellular stores. Ca2+ dysregulation plays a central role in AD [90–94].
Several positive feedback loops are present in AD that go through Ca2+, where increased Ca2+ promotes a pathogenic state, which in turn further increases Ca2+ levels [93]. Application of Aβ to cultured cells triggers flux of Ca2+ through the plasma membrane, either by acting on ion channels, disrupting membranes or forming pores [93, 95, 96]. Increased Ca2+ levels can lead to mitochondrial Ca2+ overload, oxidative stress by generation of superoxide radicals, and production of pro-apoptotic mitochondrial proteins such as caspases and cytochrome c [90]. As above, oxidative stress can promote the generation of more Aβ. In addition, mutations in proteins that control cytosolic Ca2+ concentrations are known to affect Aβ levels and susceptibility to AD [97].
TAU HYPERPHOSPHORYLATION
The hyperphosphorylation of tau, leading to its dissociation from microtubules and formation of tangles, is invariably shown downstream of Aβ deposition. Much evidence from in vitro and in vivo models, and from biomarkers in patients, show Aβ-induced acceleration of tau pathology as a critical trigger for AD, though the exact mechanisms linking Aβ to tau hyperphosphorylation and accumulation are unclear [98–100]. AD is thus a disease of both Aβ and tau aggregation, whereas tauopathies, caused by mutations in the MAPT gene, show only tau pathology. Nevertheless, intriguing work on the effects of knocking out the MAPT gene in transgenic mice expressing mutant AβPP and PS1 suggest that tau may also be able to exert upstream effects on Aβ [101]. As would be expected if tau plays a key role in AD, AβPP/PS1/tau++ mice had reduced survival, neuronal and synaptic loss, and developed spatial memory impairments. These deficits were rescued in AβPP/PS1/tau–/– mice, showing that tau contributes to AD pathology. Remarkably, however, levels of soluble and insoluble Aβ40 and Aβ42, the Aβ42/Aβ40 ratio and the amyloid plaque burden were decreased in the cortex and the spinal cord of the AβPP/PS1/tau–/–mice. Levels of phosphorylated AβPP, of β-C-terminal fragments (CTFs), and of β-secretase 1 (BACE1) were also lowered, suggesting that β-secretase cleavage of AβPP was reduced in AβPP/PS1/tau–/– mice. The tau deletion therefore had a protective effect against amyloid induced toxicity, not just by reducing tau pathology directly, but also by reducing the production of Aβ, thus showing feedback from tau to Aβ production. Positive feedback may, therefore, reach all the way from tau to AβPP processing, provided that these results generalize from this mouse model.
CONCLUSIONS: IMPLICATIONS FOR AD AND AD THERAPIES
The purpose of this review is not to give a comprehensive overview of all the pathways involved in AD. Rather, it is to show that there is compelling evidence for positive feedback loops in AD pathogenesis and hence that the simple concept of a cascade, with information flow only in one direction, is unsustainable.
Figure 2 shows where such loops can be added to the amyloid cascade. In particular, inflammation and oxidative stress can affect Aβ production, deposition, and clearance in multiple ways. Such loops may help explain the onset of the disease. Most people never get AD; the physiological processes relevant to AD are maintained in a state of homeostasis. For AD pathogenesis to start, it is likely that multiple stresses are needed to move the system far enough away from the healthy state so that positive feedback loops operate [102], committing the body to the AD pathway and subsequent dementia. The feedback loops mean that the body is in an either/or state of health or AD, since the cusp between the two states is unstable— either homeostasis or AD feedback loops will pull the system to one state or the other.
Multiple stresses, such as an ApoE4 genotype [103], age, inefficient protein degradation [104], autophagic/lysosomal dysfunction [105], iron dysregulation [27], deficiencies in blood coagulation via bacterial lipopolysaccharides [106, 107], vascular conditions [108], mitochondrial dysfunction [17], viral infection [109–111], microbial infection [112], and head injury [113], are almost certainly required to initiate progress into AD. Even dominant mutations that cause early onset AD are insufficient in themselves, as AD is not present from birth and decades of ageing are also necessary. Regarding AβPP as the single entry point to AD is thus an oversimplification, since factors such as prolonged mitochondrial dysfunction, impaired protein degradation or chronic inflammation can also be involved.
The presence of feedback loops shows that the term cascade is misleading, since it implies a unidirectional progression from upstream processes, such as overproduction and deposition of Aβ, to dementia. While Aβ undoubtedly plays a key role in AD, it is an oversimplification to think that Aβ causes all other processes in AD. Instead, a complex network of positive feedback loops reinforces AD processes once the body is committed to the disease. Given this complexity, a systems biology approach that quantifies fluxes within these pathways may well be essential to understand effects of perturbing the system [114–116]. Poorly liganded iron can generate hydroxyl radicals via positive feedback loops in many pathological conditions, including AD [117].
Multiple phenotypes associated with AD are deleterious. Hence, Fig. 2 shows that the totality of AD processes contributes to cell death, rather than there being a direct link to cell death only from tau, as in Fig. 1. Ideally, one would want to reverse all these phenotypes, restoring then to normal levels. Combinatorial therapies may be essential [117–119], since hitting a single target in a pathway may well have a negligible effect on its total flux [120].
If AD is a cascade, however, then interventions cannot affect upstream processes, if the steps are irreversible. Anti-inflammatories or inhibitors of the kinases that hyperphosphorylate tau would then not affect Aβ production and deposition, for example. A naïve interpretation of Fig. 1 is that drugs should seek to intervene as early as possible in the cascade to beneficially affect as many adverse phenotypes as possible. Intense work on β-and γ-secretase inhibitors, and antibodies targeting Aβ has implicitly followed this strategy, with little success to date.
Alternatively, if AD is driven by feedback loops, then it may not be the case that drugs should preferentially hit upstream targets. Many processes thought to be downstream of Aβ actually appear to affect AβPP processing, and Aβ deposition or clearance. Intervention at any point, or better still multiple points, in a loop may thus cause favorable phenotypic changes throughout the cascade. Blocking feedback loops may allow the body to escape AD processes and restore healthy homeostasis, by clearing excess Aβ once its overproduction is no longer stimulated. We should therefore perhaps take seriously a much wider range of drug targets in AD, particularly combinations of anti-inflammatories and anti-oxidants. Even targeting tau may be able to restore excess Aβ. This prospect of a much wider range of AD targets gives hope for developing new AD therapies.
ACKNOWLEDGMENTS
I thank Douglas Kell, Nigel Hooper, David Brough, Sally Hunter, Laura Castro Aldrete, Monica Seif, and Nishtha Chandra for suggestions on the manuscript and helpful discussions. Previous work that has led to this review has been funded by Alzheimer’s Research UK and the BBSRC (grant code BB/C008219/1).
The author’s disclosure is available online (https://www.j-alz.com/manuscript-disclosures/18-0583r2).
REFERENCES
- [1]. Hardy JA, Higgins GA (1992) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- [2]. Jawhar S, Wirths O, Bayer TA (2011) Pyroglutamate amyloid-beta (A beta): A hatchet man in Alzheimer disease. J Biol Chem 286, 38825–38832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, Hornburg D, Evans LDB, Moore S, Daria A, Hampel H, Muller V, Giudici C, Nuscher B, Wenninger-Weinzierl A, Kremmer E, Heneka MT, Thal DR, Giedraitis V, Lannfelt L, Muller U, Livesey FJ, Meissner F, Herms J, Konnerth A, Marie H, Haass C (2015) Eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Roher AE, Kokjohn TA, Clarke SG, Sierks MR, Maarouf CL, Serrano GE, Sabbagh MS, Beach TG (2017) APP/A beta structural diversity and Alzheimer’s disease pathogenesis. Neurochem Int 110, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Hunter S, Brayne C (2012) Relationships between the amyloid precursor protein and its various proteolytic fragments and neuronal systems. Alzheimers Res Therapy 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Hunter S, Brayne C (2014) Integrating the molecular and the population approaches to dementia research to help guide the future development of appropriate therapeutics. Biochem Pharmacol 88, 652–660. [DOI] [PubMed] [Google Scholar]
- [7]. Huang WJ, Zhang X, Chen WW (2016) Role of oxidative stress in Alzheimer’s disease. Biomed Rep 4, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. Feng Y, Wang XC (2012) Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev 2012, 472932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]. Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018) Oxidative stress and the amyloid β peptide in Alzheimer’s disease. Redox Biol 14, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158, 47–52. [DOI] [PubMed] [Google Scholar]
- [11]. Butterfield DA, Lauderback CM (2002) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress Free Radic Biol Med 32, 1050–1060. [DOI] [PubMed] [Google Scholar]
- [12]. Butterfield DA, Galvan V, Lange MB, Tang HD, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang JL, Sultana R, Bredesen DE (2010) In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med 48, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Al-Hilaly YK, Williams TL, Stewart-Parker M, Ford L, Skaria E, Cole M, Bucher WG, Morris KL, Sada AA, Thorpe JR, Serpell LC (2013) A central role for dityrosine crosslinking of Amyloid-beta in Alzheimer’s disease. Acta Neuropathol Commun 1, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Swerdlow RH, Khan SM (2004) A “mitochondrial cascade hypthesis” for sporadic Alzheimer’s disease. Med Hypotheses 63, 8–20. [DOI] [PubMed] [Google Scholar]
- [16]. Moreira PI, Carvalho C, Zhu XW, Smith MA, Perry G (2010) Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta 1802, 2–10. [DOI] [PubMed] [Google Scholar]
- [17]. Swerdlow RH, Burns JM, Khan SM (2010) The Alzheimer’s disease mitochondrial cascade hypothesis. , S265-S. J Alzheimers Dis 20, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Swerdlow RH (2018) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis 62, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP (1997) Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid β-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem 69, 273–284. [DOI] [PubMed] [Google Scholar]
- [20]. Abramov AY, Canevari L, Duchen MR (2004) β-Amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15, 1437–1449. [DOI] [PubMed] [Google Scholar]
- [22]. Ono K, Hamaguchi T, Naiki H, Yamada M (2006) Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochem Biophys Acta 1762, 575–586. [DOI] [PubMed] [Google Scholar]
- [23]. Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou HY, Jeanniton D, Ehrhart J, Townsend K, Zeng J, Morgan D, Hardy J, Town T, Tan J (2005) Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci 25, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Cascella M, Bimonte S, Muzio MR, Schiavone V, Cuomo A (2017) The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect Agent Cancer 12, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy| SA, Cole GM (2005) Curcumin inhibits formation of amyloid oligomers and fibrils, binds plaques, and reduces amyloid. J Biol Chem 280, 5892–5901. [DOI] [PubMed] [Google Scholar]
- [26]. Mecocci P, Polidori MC (2012) Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochem Biophys Acta 1822, 631–638. [DOI] [PubMed] [Google Scholar]
- [27]. Kell DB (2009) Iron behaving badly: Inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Kanekiyo T, Bu G (2014) The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer’s disease. Front Aging Neurosci 6, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Storck SE, Meister S, Nahrath J, Meissner JN, Schubert N, Di Spiezio A, Baches S, Vandenbroucke RE, Bouter Y, Prikulis I, Korth C, Weggen S, Heimann A, Schwaninger M, Bayer TA, Pietrzik CU (2016) Endothelial LRP1 transports amyloid-β(1-42) across the blood-brain barrier. J Clin Invest 126, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Liu CC, Hu J, Zhao N, Wang J, Wang N, Cirrito JR, Kanekiyo T, Holtzman DM, Bu GJ (2017) Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J Neurosci 37, 4023–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu ZH, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong ZH, Zlokovic BV (2007) Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med 13, 1029–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Owen JB, Sultana R, Aluise CD, Erickson MA, Price TO, Bu G, Banks WA, Butterfield DA (2010) Oxidative modification to LDL-related receptor protein 1 (LRP1) in hippocampus from subjects with Alzheimer’s disease: Implications for Aβ accumulation in AD brain. Free Radic Biol Med 49, 1798–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Poli G, Biasi F, Leonarduzzi G (2013) Oxysterols in the pathogenesis of major chronic diseases. Redox Biol 1, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Bjorkhem I, Heverin M, Leoni V, Meaney S, Diczfalusy U (2006) Oxysterols and Alzheimer’s disease. Acta Neurol Scand 114, 43–49. [DOI] [PubMed] [Google Scholar]
- [35]. Testa G, Staurenghi E, Zerbinati C, Gargiulo S, Iuliano L, Giaccone G, Fanto F, Poli G, Leonarduzzi G, Gamba P (2016) Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol 10, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Leoni V, Caccia C (2011) Oxysterols as biomarkers in neurodegenerative diseases. Chem Phys Lipids 164, 515–524. [DOI] [PubMed] [Google Scholar]
- [37]. Usui K, Hulleman JD, Paulsson JF, Siegel SJ, Powers ET, Kelly JW (2009) Site-specific modification of Alzheimer’s peptides by cholesterol oxidation products enhances aggregation energetics and neurotoxicity. Proc Nat Acad Sci U S A 106, 18563–18568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Murray IVJ, Liu L, Komatsu H, Uryu K, Xiao G, Lawson JA, Axelsen PH (2007) Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. J Biol Chem 282, 9335–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. Zhang QH, Powers ET, Nieva J, Huff ME, Dendle MA, Bieschke J, Glabe CG, Eschenmoser A, Wentworth P, Lerner RA, Kelly JW (2004) Metabolite-initiated protein misfolding may trigger Alzheimer’s disease. Proc Nat Acad Sci U S A 101, 4752–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Bjorkhem I (2006) Crossing the barrier: Oxysterols as cholesterol transporters and metabolic modulators in the brain. J Int Med 260, 493–508. [DOI] [PubMed] [Google Scholar]
- [41]. Heverin M, Meaney S, Lutjohann D, Diczfalusy U, Wahren J, Bjorkhem I (2005) Crossing the barrier: Net flux of 27-hydroxycholesterol into the human brain. J Lipid Res 46, 1047–1052. [DOI] [PubMed] [Google Scholar]
- [42]. Heverin M, Bogdanovic N, Lutjohann D, Bayer T, Pikuleva I, Bretillon L, Diczfalusy U, Winblad B, Bjorkhem I (2004) Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res 45, 186–193. [DOI] [PubMed] [Google Scholar]
- [43]. Gamba P, Guglielmotto M, Testa G, Monteleone D, Zerbinati C, Gargiulo S, Biasi F, Iuliano L, Giaccone G, Mauro A, Poli G, Tamagno E, Leonarduzzi G (2014) Up-regulation of -amyloidogenesis in neuron-like human cells by both 24-and 27-hydroxycholesterol: Protective effect of N-acetyl-cysteine. Aging Cell 13, 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. Yan SD, Chen X, Fu J, Chen M, Zhu HJ, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691. [DOI] [PubMed] [Google Scholar]
- [45]. Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Du Yan S (2001) Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: Identification of a cellular activation mechanism. Exp Neurol 171, 29–45. [DOI] [PubMed] [Google Scholar]
- [46]. Munch G, Gerlach M, Sian J, Wong A, Riederer P (1998) Advanced glycation end products in neurodegeneration: More than early markers of oxidative stress? , S85-S. Ann Neurol 44, 88. [DOI] [PubMed] [Google Scholar]
- [47]. Thome J, Kornhuber J, Munch G, Schinzel R, Taneli Y, Zielke B, Rosler M, Riederer P (1996) A new hypothesis for the etiopathogenesis of the Alzheimer’s syndrome. Advanced glycation endproducts (AGEs). Nervenarzt 67, 924–929. [DOI] [PubMed] [Google Scholar]
- [48]. Gella A, Durany N (2009) Oxidative stress in Alzheimer disease. Cell Adh Migr 3, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49]. Schmidt AM, Du Yan S, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108, 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50]. Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R (2007) Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res 1148, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52]. Sensi SL, Paoletti P, Bush AI, Sekler I (2009) Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 10, 780-U738. [DOI] [PubMed] [Google Scholar]
- [53]. Li X, Jiang LH (2018) Multiple molecular mechanisms form a positive feedback loop driving amyloid beta 42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis 9, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54]. Zotova E, Nicoll JAR, Kalaria R, Holmes C, Boche D (2010) Inflammation in Alzheimer’s disease: Relevance to pathogenesis and therapy. Alzheimers Res Ther 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55]. Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease-a brief review of the science and clinical literature. Cold Spring Harb Perspect Med 2, a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56]. Millington C, Sonego S, Karunaweera N, Rangel A, Aldrich-Wright JR, Campbell IL, Gyengesi E, Munch G (2014) Chronic neuroinflammation in Alzheimer’s disease: New perspectives on animal models and promising candidate drugs. Biomed Res Int 2014, 309129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57]. Imbimbo BP, Solfrizzi V, Panza F (2010) Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front Aging Neurosci 2, pii: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58]. Cole GM, Frautschy SA (2010) Mechanisms of action of non-steroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease. CNS Neurol Disord Drug Targets 9, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59]. Wang J, Tan L, Wang HF, Tan CC, Meng XF, Wang C, Tang SW, Yu JT (2015) Anti-inflammatory drugs and risk of Alzheimer’s disease: An updated systematic review and meta-analysis. J Alzheimers Dis 44, 385–396. [DOI] [PubMed] [Google Scholar]
- [60]. Deardorff WJ, Grossberg GT (2017) Targeting neuroinflammation in Alzheimer’s disease: Evidence for NSAIDs and novel therapeutics. Exp Rev Neurother 17, 17–32. [DOI] [PubMed] [Google Scholar]
- [61]. Evin G, Li Q (2012) Platelets and Alzheimer’s disease: Potential of APP as a biomarker. World J Psychiatry 2, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62]. Dawkins E, Small DH (2014) Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J Neurochem 129, 756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63]. Catricala S, Torti M, Ricevuti G (2012) Alzheimer disease and platelets: How’s that relevant. Immun Ageing 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64]. Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang LL, Casey E, Lu YF, Shiratori C, Lemere C, Duff K (2003) Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci 23, 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65]. Deane R, Bell RD, Sagare A, Zlokovic BV (2009) Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets 8, 16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66]. Georgievska B, Gustavsson S, Lundkvist J, Neelissen J, Eketjall S, Ramberg V, Bueters T, Agerman K, Jureus A, Svensson S, Berg S, Falting J, Lendahl U (2015) Revisiting the peripheral sink hypothesis: Inhibiting BACE1 activity in the periphery does not alter beta-amyloid levels in the CNS. J Neurochem 132, 477–486. [DOI] [PubMed] [Google Scholar]
- [67]. Teather LA, Packard MG, Bazan NG (2002) Post-training cyclooxygenase-2 (COX-2) inhibition impairs memory consolidation. Learn Memory 9, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68]. Xiang ZM, Ho L, Yemul S, Zhao Z, Pompl P, Kelley K, Dang A, Qing W, Teplow D, Pasinetti GM (2002) Cyclooxygenase-2 promotes amyloid plaque deposition in a mouse model of Alzheimer’s disease neuropathology. Gene Expr 10, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69]. Zagol-Ikapitte I, Masterson TS, Amarnath V, Montine TJ, Andreasson KI, Boutaud O, Oates JA (2005) Prostaglandin H-2-derived adducts of proteins correlate with Alzheimer’s disease severity. J Neurochem 94, 1140–1145. [DOI] [PubMed] [Google Scholar]
- [70]. Boutaud O, Ou JJ, Chaurand P, Caprioli RM, Montine TJ, Oates JA (2002) Prostaglandin H-2 (PGH(2)) accelerates formation of amyloid β(1-42) oligomers. J Neurochem 82, 1003–1006. [DOI] [PubMed] [Google Scholar]
- [71]. Gowert NS, Donner L, Chatterjee M, Eisele YS, Towhid ST, Munzer P, Walker B, Ogorek I, Borst O, Grandoch M, Schaller M, Fischer JW, Gawaz M, Weggen S, Lang F, Jucker M, Elvers M (2014) Blood platelets in the progression of Alzheimer’s disease. PLoS One 9, e90523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72]. Canobbio I, Abubaker AA, Visconte C, Torti M, Pula G (2015) Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front Cell Neurosci 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73]. Shen MY, Hsiao G, Fong TH, Chen HM, Chou DS, Lin CH, Sheu JR, Hsu CY (2008) Amyloid beta peptide-activated signal pathways in human platelets. Eur J Pharmacol 588, 259–266. [DOI] [PubMed] [Google Scholar]
- [74]. Guo HT, Callaway JB, Ting JPY (2015) Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med 21, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75]. Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A (2008) Amyloid-β oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J Cell Mol Med 12, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76]. Masters SL, O’Neill LAJ (2011) Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol Med 17, 276–282. [DOI] [PubMed] [Google Scholar]
- [77]. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78]. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT (2017) Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–336. [DOI] [PubMed] [Google Scholar]
- [79]. Ransohoff RM (2017) Specks of insight into Alzheimer’s disease. Nature 552, 342–343. [DOI] [PubMed] [Google Scholar]
- [80]. Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou CY, Das S, Adiconis X, Chen HB, Zhu H, Kelliher MA, Levin JZ, Yuan JY (2017) RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Nat Acad Sci U S A 114, E8788–E8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81]. Rubinsztein DC (2017) RIPK1 promotes inflammation and β-amyloid accumulation in Alzheimer’s disease. Proc Nat Acad Sci U S A 114, 10813–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82]. Schwab M, Lutum AS, Seufert W (1997) Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90, 683–693. [DOI] [PubMed] [Google Scholar]
- [83]. Hershko A (2005) The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ 12, 1191–1197. [DOI] [PubMed] [Google Scholar]
- [84]. Almeida A (2012) Regulation of APC/C-Cdh1 and its function in neuronal survival. Mol Neurobiol 46, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85]. Bolanos JP, Almeida A, Moncada S (2010) Glycolysis: A bioenergetic or a survival pathway? Trends Biochem Sci 35, 145–149. [DOI] [PubMed] [Google Scholar]
- [86]. Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolanos JP, Almeida A (2008) Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J 27, 2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87]. Bonda DJ, Evans TA, Santocanale C, Llosa JC, Vina J, Bajic VP, Castellani RJ, Siedlak SL, Perry G, Smith MA, Lee HG (2009) Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging (Albany NY) 1, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88]. Fuchsberger T, Martinez-Bellver S, Giraldo E, Teruel-Marti V, Lloret A, Vina J (2016) A beta induces excitotoxicity mediated by APC/C-Cdh1 depletion that can be prevented by glutaminase inhibition promoting neuronal survival. Sci Rep 6, 31158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89]. Burbaeva GS, Boksha IS, Tereshkina EB, Savushkina OK, Starodubtseva LI, Turishcheva MS (2005) Glutamate metabolizing enzymes in prefrontal cortex of Alzheimer’s disease patients. Neurochem Res 30, 1443–1451. [DOI] [PubMed] [Google Scholar]
- [90]. Stutzmann GE (2007) The pathogenesis of alzheimers disease - Is it a lifelong “Calciumopathy”? . Neuroscientist 13, 546–559. [DOI] [PubMed] [Google Scholar]
- [91]. Green KN, LaFerla FM (2008) Linking calcium to A beta and Alzheimer’s disease. Neuron 59, 190–194. [DOI] [PubMed] [Google Scholar]
- [92]. Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93]. Demuro A, Parker I, Stutzmann GE (2010) Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 285, 12463–12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Alzheimer’s Association Calcium Hypothesis Workgroup (2017) Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 13, 178–182.e17. [DOI] [PubMed] [Google Scholar]
- [95]. Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280, 17294–17300. [DOI] [PubMed] [Google Scholar]
- [96]. Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26, 6011–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97]. Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao HT, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, Marambaud P (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, A beta levels, and Alzheimer’s disease risk. Cell 133, 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98]. Stancu IC, Vasconcelos B, Terwel D, Dewachter I (2014) Models of beta-amyloid induced Tau-pathology: The long and “folded” road to understand the mechanism. Mol Neurodegener 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99]. Nisbet RM, Polanco JC, Ittner LM, Gotz J (2015) Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol 129, 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100]. Bloom GS (2014) Amyloid-beta and tau the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508. [DOI] [PubMed] [Google Scholar]
- [101]. Leroy K, Ando K, Laporte V, Dedecker R, Suain V, Authelet M, Heraud C, Pierrot N, Yilmaz Z, Octave JN, Brion JP (2012) Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 Mice. Am J Pathol 181, 1928–1940. [DOI] [PubMed] [Google Scholar]
- [102]. Castrillo JI, Oliver SG (2016) Alzheimer’s as a systems-level disease involving the interplay of multiple cellular networks In Systems Biology of Alzheimer’s Disease, Castrillo JI Oliver SG, eds. Humana Press, New York, pp. 3–48. [DOI] [PubMed] [Google Scholar]
- [103]. Roses AD (1994) Apolipoprotein E affects the rate of Alzheimer’s disease expression: β-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease. J Neuropathol Exp Neurol 53, 429–437. [DOI] [PubMed] [Google Scholar]
- [104]. Wang DS, Dickson DW, Malter JS (2006) β-Amyloid degradation and Alzheimer’s disease. J Biomed Biotechnol 2006, 58406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105]. Orr ME, Oddo S (2013) Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106]. Bester J, Soma P, Kell DB, Pretorius E (2015) Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget 6, 35284–35303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107]. Pretorius E, Bester J, Kell DB (2016) A bacterial component to Alzheimer’s-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. J Alzheimers Dis 53, 1237–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108]. Attems J, Jellinger KA (2014) The overlap between vascular disease and Alzheimer’s disease - lessons from pathology. BMC Med 12, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109]. Itzhaki RF, Lin WR, Shang DH, Wilcock GK, Faragher B, Jamieson GA (1997) Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349, 241–244. [DOI] [PubMed] [Google Scholar]
- [110]. Itzhaki RF (2017) Herpes simplex virus type 1 and Alzheimer’s disease: Possible mechanisms and signposts. FASEB J 31, 3216–3226. [DOI] [PubMed] [Google Scholar]
- [111]. Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, Sano M, Liang WS, Beckmann ND, Price ND, Reiman EM, Schadt EE, Ehrlich ME, Gandy S, Dudley JT (2018) Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 11, 64–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112]. Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, Del Tredici K, Field H, Fulop T, Grassi C, Griffin WST, Haas J, Hudson AP, Kamer AR, Kell DB, Licastro F, Letenneur L, Lovheim H, Mancuso R, Miklossy J, Otth C, Palamara AT, Perry G, Preston C, Pretorius E, Strandberg T, Tabet N, Taylor-Robinson SD, Whittum-Hudson JA (2016) Microbes and Alzheimer’s disease. J Alzheimers Dis 51, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113]. Li YJ, Li YM, Li XT, Zhang S, Zhao JC, Zhu XF, Tian GZ (2017) Head injury as a risk factor for dementia and Alzheimer’s disease: A systematic review and meta-analysis of 32 observational studies. PLoS One 12, e0169650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114]. Juhasz G, Foldi I, Penke B (2011) Systems biology of Alzheimer’s disease: How diverse molecular changes result in memory impairment in AD. Neurochem Int 58, 739–750. [DOI] [PubMed] [Google Scholar]
- [115]. Castrillo JI, Oliver SG (2016), Systems Biology of Alzheimer’s Disease Totowa, Humana Press. [PubMed] [Google Scholar]
- [116]. Hunter S, Martin S, Brayne C (2015) The APP proteolytic system and its interactions with dynamic networks in Alzheimer’s disease In Systems Biology of Alzheimer’s Disease, Castrillo JI, Oliver SG, eds. Humana Press, New York, pp. 71–99. [DOI] [PubMed] [Google Scholar]
- [117]. Kell DB (2010) Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol 84, 825–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118]. Lehar J, Stockwell BR, Giaever G, Nislow C (2008) Combination chemical genetics. Nat Chem Biol 4, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119]. Lehar J, Krueger AS, Zimmermann GR, Borisy AA (2009) Therapeutic selectivity and the multi-node drug target. Discov Med 8, 185–190. [PubMed] [Google Scholar]
- [120]. Cornishbowden A, Hofmeyr JHS, Cardenas ML (1995) Strategies for manipulating metabolic fluxes in biotechnology. Bioorg Chem 23, 439–449. [Google Scholar]