

Abstract
Background
Hand eczema is an inflammation of the skin of the hands that tends to run a chronic, relapsing course. This common condition is often associated with itch, social stigma, and impairment in employment. Many different interventions of unknown effectiveness are used to treat hand eczema.
Objectives
To assess the effects of topical and systemic interventions for hand eczema in adults and children.
Search methods
We searched the following up to April 2018: Cochrane Skin Group Specialised Register, CENTRAL, MEDLINE, Embase, AMED, LILACS, GREAT, and four trials registries. We checked the reference lists of included studies for further references to relevant trials.
Selection criteria
We included randomised controlled trials (RCTs) that compared interventions for hand eczema, regardless of hand eczema type and other affected sites, versus no treatment, placebo, vehicle, or active treatments.
Data collection and analysis
We used standard methodological procedures expected by Cochrane. Primary outcomes were participant‐ and investigator‐rated good/excellent control of symptoms, and adverse events.
Main results
We included 60 RCTs, conducted in secondary care (5469 participants with mild to severe chronic hand eczema). Most participants were over 18 years old. The duration of treatment was short, generally up to four months. Only 24 studies included a follow‐up period. Clinical heterogeneity in treatments and outcome measures was evident. Few studies performed head‐to‐head comparisons of different interventions. Risk of bias varied considerably, with only five studies at low risk in all domains. Twenty‐two studies were industry‐funded.
Eighteen trials studied topical corticosteroids or calcineurin inhibitors; 10 studies, phototherapy; three studies, systemic immunosuppressives; and five studies, oral retinoids. Most studies compared an active intervention against no treatment, variants of the same medication, or placebo (or vehicle). Below, we present results from the main comparisons.
Corticosteroid creams/ointments: when assessed 15 days after the start of treatment, clobetasol propionate 0.05% foam probably improves participant‐rated control of symptoms compared to vehicle (risk ratio (RR) 2.32, 95% confidence interval (CI) 1.38 to 3.91; number needed to treat for an additional beneficial outcome (NNTB) 3, 95% CI 2 to 8; 1 study, 125 participants); the effect of clobetasol compared to vehicle for investigator‐rated improvement is less clear (RR 1.43, 95% CI 0.86 to 2.40). More participants had at least one adverse event with clobetasol (11/62 versus 5/63; RR 2.24, 95% CI 0.82 to 6.06), including application site burning/pruritus. This evidence was rated as moderate certainty.
When assessed 36 weeks after the start of treatment, mometasone furoate cream used thrice weekly may slightly improve investigator‐rated symptom control compared to twice weekly (RR 1.23, 95% CI 0.94 to 1.61; 1 study, 72 participants) after remission is reached. Participant‐rated symptoms were not measured. Some mild atrophy was reported in both groups (RR 1.76, 95% CI 0.45 to 6.83; 5/35 versus 3/37). This evidence was rated as low certainty.
Irradiation with ultraviolet (UV) light: local combination ultraviolet light therapy (PUVA) may lead to improvement in investigator‐rated symptom control when compared to local narrow‐band UVB after 12 weeks of treatment (RR 0.50, 95% CI 0.22 to 1.16; 1 study, 60 participants). However, the 95% CI indicates that PUVA might make little or no difference. Participant‐rated symptoms were not measured. Adverse events (mainly erythema) were reported by 9/30 participants in the narrow‐band UVB group versus none in the PUVA group. This evidence was rated as moderate certainty.
Topical calcineurin inhibitors: tacrolimus 0.1% over two weeks probably improves investigator‐rated symptom control measured after three weeks compared to vehicle (14/14 tacrolimus versus 0/14 vehicle; 1 study). Participant‐rated symptoms were not measured. Four of 14 people in the tacrolimus group versus zero in the vehicle group had well‐tolerated application site burning/itching.
A within‐participant study in 16 participants compared 0.1% tacrolimus to 0.1% mometasone furoate but did not measure investigator‐ or participant‐rated symptoms. Both treatments were well tolerated when assessed at two weeks during four weeks of treatment.
Evidence from these studies was rated as moderate certainty.
Oral interventions: oral cyclosporin 3 mg/kg/d probably slightly improves investigator‐rated (RR 1.88, 95% CI 0.88 to 3.99; 1 study, 34 participants) or participant‐rated (RR 1.25, 95% CI 0.69 to 2.27) control of symptoms compared to topical betamethasone dipropionate 0.05% after six weeks of treatment. The risk of adverse events such as dizziness was similar between groups (up to 36 weeks; RR 1.22, 95% CI 0.80 to 1.86, n = 55; 15/27 betamethasone versus 19/28 cyclosporin). The evidence was rated as moderate certainty.
Alitretinoin 10 mg improves investigator‐rated symptom control compared with placebo (RR 1.58, 95% CI 1.20 to 2.07; NNTB 11, 95% CI 6.3 to 26.5; 2 studies, n = 781) and alitretinoin 30 mg also improves this outcome compared with placebo (RR 2.75, 95% CI 2.20 to 3.43; NNTB 4, 95% CI 3 to 5; 2 studies, n = 1210). Similar results were found for participant‐rated symptom control: alitretinoin 10 mg RR 1.73 (95% CI 1.25 to 2.40) and 30 mg RR 2.75 (95% CI 2.18 to 3.48). Evidence was rated as high certainty. The number of adverse events (including headache) probably did not differ between alitretinoin 10 mg and placebo (RR 1.01, 95% CI 0.66 to 1.55; 1 study, n = 158; moderate‐certainty evidence), but the risk of headache increased with alitretinoin 30 mg (RR 3.43, 95% CI 2.45 to 4.81; 2 studies, n = 1210; high‐certainty evidence). Outcomes were assessed between 48 and 72 weeks.
Authors' conclusions
Most findings were from single studies with low precision, so they should be interpreted with caution. Topical corticosteroids and UV phototherapy were two of the major standard treatments, but evidence is insufficient to support one specific treatment over another. The effect of topical calcineurin inhibitors is not certain. Alitretinoin is more effective than placebo in controlling symptoms, but advantages over other treatments need evaluating.
Well‐designed and well‐reported, long‐term (more than three months), head‐to‐head studies comparing different treatments are needed. Consensus is required regarding the definition of hand eczema and its subtypes, and a standard severity scale should be established.
The main limitation was heterogeneity between studies. Small sample size impacted our ability to detect differences between treatments.
Plain language summary
Treatments for hand eczema
Review question
We reviewed evidence on the effects of topical and systemic (oral or injected medicines that work throughout the entire body) treatments for hand eczema when compared against placebo (an identical but inactive treatment), no treatment, vehicle (inactive ingredients that help deliver an active treatment), or another treatment. We included 60 randomised trials (5469 participants) published up to April 2018.
Background
Hand eczema is an inflammation of the skin of the hands that can be caused by contact allergens (i.e. substances that cause an allergic reaction) such as rubber chemicals, but other external factors (e.g. irritants such as water or detergents) and atopic predisposition are often important triggers. Hand eczema can cause a reduction in quality of life leading to many work‐related problems. Various types of hand eczema exist, and different topical (creams, ointments, or lotions) and systemic treatments with unknown effectiveness can be used.
Study characteristics
Most participants were hospital outpatients over 18 years of age with mild to severe chronic hand eczema. Treatment was usually given for up to four months, and outcomes were mainly assessed after treatment. A large variety of treatments were studied and compared to no treatment, variants of the same medication, placebo, or vehicle. Twenty‐two studies were funded by pharmaceutical companies.
Key results
Limited data are available to support the best way of managing hand eczema due to varying study quality and inability to pool data from studies with similar interventions. Corticosteroid creams/ointments and phototherapy (irradiation with UV light) are the major treatment options, although comparisons between these options are lacking. Below, we present results for the main comparisons of interest.
Corticosteroid creams/ointments: clobetasol propionate foam probably increases participant‐rated good/excellent control of hand eczema when compared to vehicle (516 versus 222 per 1000), but the difference between groups was less clear for investigator‐rated control, and more adverse events were reported with clobetasol propionate (178 versus 79 per 1000) (all based on moderate‐certainty evidence).
Mometasone furoate cream used thrice weekly may slightly improve investigator‐rated good/excellent control compared to twice weekly treatment, and participant‐rated control was not measured. Mild skin thinning occurred in both groups, but cases were few (all based on low‐certainty evidence).
Irradiation with UV light: various types of irradiation (i.e. exposure to radiation) were compared. Local PUVA may improve investigator‐rated good/excellent control compared to narrow‐band UVB (400 versus 200 per 1000); however, we are uncertain of this finding because results also show that local PUVA may make little or no difference. Participant‐rated symptoms were not measured. Nine out of 30 participants in the narrow‐band UVB group reported adverse events (mainly redness) compared to none in the PUVA group (all based on moderate‐certainty evidence).
Topical calcineurin inhibitors: people receiving tacrolimus are probably more likely to achieve improved investigator‐rated good/excellent symptom control compared to those given vehicle (14/14 participants with tacrolimus compared to none with vehicle), but participant‐rated control of symptoms was not measured. Four of 14 people in the tacrolimus group versus zero in the vehicle group had well‐tolerated application site burning/itching. One small study compared tacrolimus to mometasone furoate, which were both well tolerated, but did not measure investigator‐ or participant‐rated control (all based on moderate‐certainty evidence).
Oral interventions: oral immunosuppressant (a drug that hinders the immune response) cyclosporin probably slightly improves investigator‐ or participant‐rated control of good/excellent symptoms compared to topical betamethasone cream (a corticosteroid). The risk of adverse events such as dizziness was similar between groups (all based on moderate‐certainty evidence).
The oral vitamin A derivative (retinoid) alitretinoin (10 mg) achieved investigator‐rated good/excellent symptom control in 307 compared to 194 participants per 1000 with placebo, and alitretinoin 30 mg achieved investigator‐rated control in 432 compared to 157 participants per 1000 with placebo. Similar results were shown for participant‐rated control (high‐certainty evidence). When the dosage of alitretinoin was increased to 30 mg, risk of headache was higher compared to placebo (74 versus 251 per 1000; high‐certainty evidence), but this probably does not differ between alitretinoin 10 mg and placebo (based on moderate‐certainty evidence).
Quality of the evidence
The quality of evidence was mainly moderate, with most analyses based on single studies that had small sample sizes; therefore, some results should be interpreted with care.
Summary of findings
Background
Please note that unfamiliar terms may be listed in Appendix 1 ('Glossary of medical terms').
Future research would involve comparing different treatment groups. Focus on subgroups would provide reliable evidence for informed decisions about which treatment is effective in managing hand eczema.
The overall quality of evidence was very low. Many trials included in this review, particularly older ones, were of low quality with methodological weaknesses in design (small studies, short duration) or were biased (not blinded, sponsored by pharmaceuticals). Most included participants with chronic hand eczema in secondary care settings; some included only specific subtypes of hand eczema, thereby limiting direct application of study findings. Most analyses were based on single studies of small sample size and imprecise results.
Description of the condition
Definition and epidemiology
Hand eczema is an inflammation of the skin (dermatitis) that is confined to the hands. Hand eczema is a common condition with a point prevalence varying between 1% and 5% in the general population. When mild cases are included, one‐year prevalence can reach 10% (Meding 2004; Thyssen 2010; Yngveson 2000). Thyssen et al conducted a review of seven epidemiological studies on hand eczema and concluded the median incidence rate of hand eczema was 5.5 cases/1000 person‐years. For women, the incidence rate of hand eczema was 9.6 cases/1000 person‐years, and for men, 4.0 cases/1000 person‐years (Thyssen 2010). A possible explanation for this sex difference is greater exposure of women to wet work, such as cleaning, nursing, and hair dressing, for example (Mollerup 2014; Nilsson 1985). The incidence of notified (i.e. usually more severe) occupation‐related cases is estimated to be above 0.7 per 1000 people per year, with much higher incidences (up to 1 in 100) in high‐risk populations such as hairdressers (Diepgen 2003). Decreased prevalence has been observed in Swedish adults and was attributed to a decline in occupational exposure to irritants (Meding 2002).
Over the years, several authors have proposed a workable definition of hand eczema, whereby different subtypes have been recognised (Menné 2000). Hand eczema can be classified according to aetiological (causative) factors, clinical‐morphological typology, or a combination of both (Coenraads 2012; Diepgen 2009a). However, due to multi‐causality, it is difficult to assess the influence of each causative factor; therefore only one aetiological diagnosis might be insufficient. The Danish Contact Dermatitis Group developed a classification system based on morphology with clear definitions for each classification and one or more aetiological diagnoses (Menné 2011). This might facilitate the classification of hand eczema and was demonstrated to be a useful tool in general practice (Johansen 2011). However, there is an obvious need for international consensus regarding the classification of subgroups of hand eczema.
In the current literature, different names can be used for the same subgroups, or the same name can be used for different subgroups. An example of this is vesicular hand eczema (Veien 2009): this might be called pompholyx, dyshidrotic eczema, dyshidrosis, or vesicular eczema; no consensus has been reached regarding the definition. The original definition of 'pompholyx' states "an eruption of vesicles and bullae on the palms, which is accompanied by pain and severe itching". Fox 1873 hypothesised that pompholyx was caused by sweating of the palms and introduced the term 'dyshidrosis' (hydrosis from sweating); both terms were used for the same clinical vesicular type. Later, Kutzner 1986 demonstrated that sweat glands are not altered in vesicular hand eczema and discussed the histological features of eczema. However, despite this evidence, the term 'dyshidrosis' is still used in current literature.
Hand eczema may be accompanied by similar skin changes on the feet.
Causes
In many people, hand eczema has more than one cause and both predisposing and external factors play a part. Being atopic (a tendency to develop asthma, hay fever, or eczema) is a major predisposing factor responsible for hand eczema; one‐third to one‐half of people with hand eczema can be considered atopic (Coenraads 1998; Meding 1990; Svensson 1988). The role of genetic factors, especially the association between filaggrin (FLG) mutations and hand eczema, is still under investigation (Heede 2016; Kaae 2012; Molin 2015).
The most common external causes of hand eczema include contact with mild toxic agents or irritants (for instance, water and soaps). The resulting irritant contact dermatitis can be distinguished from allergic contact dermatitis, which is caused by skin contact with allergens. Allergic contact dermatitis is less common than irritant contact dermatitis, and it occurs only in persons who have developed a contact allergy to a specific substance such as rubber, nickel, or perfumes. Ingested allergens (e.g. nickel) may occasionally provoke hand eczema (Jensen 2006). Little evidence suggests that inhalation of house dust mites may increase the severity of vesicular hand eczema (Schuttelaar 2013). The relevance of psychosomatic factors remains speculative (Menné 2000). In many people with chronic hand eczema, a combination of the above‐mentioned factors plays a role. In addition, for several types of hand eczema, the cause is still unknown.
Impact
Itch is common among those with hand eczema. The itch caused by hand eczema can be intense, leading to sleep loss in the sufferer and in other family members. A vicious cycle of symptoms causing skin damage can develop, the so‐called itch/scratch/itch cycle. Cracks and blisters can be painful. Cracking, hyperkeratosis (callus‐like thickening), and inflexibility of the hands are also problematic and may limit mobility of the hands.
A visible skin disease can be a great burden and can lead to a social stigma. The hands are important organs of communication and expression; therefore any visible skin disease on the hands may result in major psychosocial problems (e.g. anxiety, low self‐esteem, social phobia).
Painful cracks and blisters, besides their negative effects on daily life outside work, can impede an individual's ability to carry out manual work, leading to significant disability and huge economic losses for both individuals and society. A systematic review estimated the mean annual total cost per hand eczema patient at between €1712 and €9792 (Politiek 2016). Hand eczema accounts for an estimated 90% of occupational skin disease. Patients have substantial use of sick leave due to their hand eczema. Studies in patients with chronic severe hand eczema have reported job loss up to 20% (Cvetkovski 2005). Quality of life assessments have shown an impact on daily life and on employment (Agner 2008; Moberg 2009). A comparison between the generic quality of life instrument Short Form Health Survey (SF‐36) and the skin‐related Dermatology Life Quality Index (DLQI) revealed slightly higher impact of hand eczema on women compared to men for specific sub‐items (Wallenhammar 2004). A comparison of physician‐rated versus participant‐rated assessments of severity showed a poor correlation, indicating that patients may evaluate several aspects of their hand eczema (including degree of erythema, vesicles, and fissures) differently from physicians (van Coevorden 2006).
Prognosis
Previous studies have suggested that hand eczema tends to run a chronic relapsing course, with the vast majority of people experiencing negative psychosocial consequences (Hald 2009; Meding 2005; Petersen 2014; Veien 2008).
Description of the intervention
Many diverse therapies are used to control the disease, such as:
skin protection measures, including gloves;
topical treatments (bland emollients, corticosteroid creams/ointments, calcineurin inhibitors, coal tar and derivatives, irradiation with ultraviolet (UV) light or X‐rays); and
systemic treatments (oral corticosteroids, oral retinoids, or other immunosuppressants such as cyclosporin).
The main groups of interventions covered by this review are topical corticosteroids, topical calcineurin inhibitors (immunomodulators), irradiation with UV light, and oral retinoids or systemic immunosuppressants.
Overall, after proper education and counselling, including the recommendation of emollients, application of topical corticosteroids remains the mainstream treatment for hand eczema (nationaleczema.org).
How the intervention might work
Theoretically, identifying and eliminating an allergic contact factor (e.g. nickel or rubber allergy) could result in cure of hand eczema, provided this is the sole cause. In clinical practice, however, such cases are rare, as hand eczema is often due to a combination of irritant and allergic contact exposure, as well as to endogenous factors.
This review deals with a great variety of interventions. Major types of interventions are topical corticosteroids, topical immunomodulators, irradiation with UV light, and oral retinoids.
Topical corticosteroids are the most frequently prescribed treatments for hand eczema (Soost 2012). They have overlapping mechanisms of action: like oral immunosuppressants (e.g. corticosteroids), they inhibit inflammation (anti‐inflammatory) and production of inflammatory substances (immunosuppressive) (Ahluwalia 1998; Sakuma 2001; Schleimer 1993).
Topical immunomodulators, such as tacrolimus and pimecrolimus, are non‐steroidal immunosuppressants that are more selective in their mode of action than corticosteroids. They inhibit the production of inflammatory substances in the body (such as synthesis and release of inflammatory cytokines from T‐lymphocytes, and release of inflammatory mediators from mast cells). Calcineurin is present during activation of T‐lymphocytes, and since tacrolimus and pimecrolimus block this step, they are called 'calcineurin inhibitors' (de Paulis 1992; Sakuma 2001).
Topical moisturisers or emollients can relieve dryness of the skin, can improve the skin barrier function, and can influence transepidermal water loss (depending on the composition of the emollient) (Lodén 2012b; Rawlings 2004). Moisturisers are available in various compositions such as oil‐in‐water, water‐in‐oil, lotions, gels, and emulsions, among others, and various adjuvants such as urea or salicylic acid can be added to reduce thickness and scaling of the skin.
Coal tar has been used to treat eczema since ancient times. It is claimed to increase epidermal differentiation and to up‐regulate various key barrier proteins such as filaggrin, thus improving the skin barrier function (McLean 2013; van den Bogaard 2013). Moreover coal tar suppresses the Th2 cytokine response (McLean 2013; van den Bogaard 2013).
Irradiation with UV light can be performed with different types of UVA and UVB, depending on the wavelength. UVA treatment overall is combined with a topical or oral agent (psoralen) to make the skin more sensitive to UVA. Examples of different types of phototherapy include broad‐spectrum UVB (280 to 315 nm), small‐spectrum UVB (311 to 313 nm, also known as TL‐01 or narrow‐band UVB), UVA‐1 (340 to 400 nm), and topical and oral psoralen combined with UVA (PUVA; 315 to 400 nm). UVA‐1 phototherapy can be used at high (HD; 130 J/cm²), medium (MD; 50 J/cm²), and low doses (LD; 10 J/cm²) (Hönigsmann 2003). The mechanism of photo(chemo)therapy is multi‐factorial. In general, UV light locally decreases the activity of the immune system and inhibits the quantity of inflammatory cells. It suppresses the antigen‐presenting function of the Langerhans cells and induction of apoptosis of T‐cells (Majoie 2009). In addition, photo(chemo)therapy results in an increase in the amount of stratum corneum; in other words, the skin gets thicker (Jekler 1990). Finally, UVB reduces the number of microbes on the skin, including Staphylococcus aureus (Faergemann 1987).
Oral retinoids are vitamin A derivatives. Retinoids are thought to interfere at different steps in the inflammatory process. They have immunomodulatory properties and interfere with the epidermal differentiation process in various ways (Blair 2016; Kislat 2014; Schmitt‐Hoffmann 2012). Both alitretinoin and acitretin are retinoids, although their mechanism of action is slightly different. Alitretinoin is thought to have anti‐inflammatory and immunomodulatory effects on the skin. Alitretinoin binds with high affinity to both retinoic acid receptor (RAR) and retinoid X receptor (RXR) and presents anti‐inflammatory and immunomodulatory activity, and acitretin binds only selectively to RAR, although both retinoids are thought to reduce inflammation (Blair 2016; Kislat 2014; Schmitt‐Hoffmann 2012).
Hand eczema is a chronic condition that might be accompanied by flares and might improve as a result of the natural course; therefore, we believe a minimum treatment duration of three months is required to document important data such as duration and frequency of disease relapse.
Why it is important to do this review
The high prevalence of hand eczema, along with its poor prognosis and associated disability with economic losses and impairment of quality of life, makes hand eczema an important disease to study from an individual and a societal perspective. This, coupled with the long list of diverse treatments of unknown effectiveness and several conflicting studies (Diepgen 2007; van Coevorden 2004b), suggests that a systematic review is needed. Even if methodological constraints do not permit sufficient clarification of existing conflicts to provide clear guidance in clinical practice, this review will be an important step in identifying research gaps and consequently providing directions for future research.
The plans for this review were published as a protocol "Interventions for hand eczema" (van Coevorden 2009). Differences between the review and the protocol are stated in the section Differences between protocol and review.
Objectives
To assess the effects of topical and systemic interventions for hand eczema in adults and children.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) of interventions for hand eczema regardless of hand eczema type and other affected localisations.
Types of participants
People (adults and children, occupational and non‐occupational) with the diagnosis of hand eczema, regardless of the underlying assumed cause, were eligible. We also included participants with other parts of the body affected in addition to the hand. The terms 'eczema' and 'dermatitis' were acceptable whenever they referred to the hands. Other terms such as 'pompholyx', 'dyshidrosis', and 'pulpitis' were also deemed acceptable. We included participants with different types of hand eczema, for example, chronic hand eczema, hyperkeratotic palmar (also know as tylotic) hand eczema, and vesicular eczema (also known as dyshidrotic hand eczema or pompholyx).
We included in this review studies that included participants with other diagnoses besides hand eczema only when we were able to obtain separate data for hand eczema participants.
Types of interventions
We included only studies comparing the intervention versus no treatment, placebo, vehicle, or other active treatments. We considered all types of interventions, except interventions to prevent hand eczema (primary prevention). We excluded studies that focused on prevention of hand eczema and studies that investigated integrated care programmes or educational programmes (non‐pharmacological interventions).
We considered studies comparing different interventions, for example, topical corticosteroids versus topical calcineurin inhibitors or oral cyclosporin versus topical corticosteroids, as most clinically relevant. For 'Summary of findings' tables, we included the following comparisons.
Mometasone furoate cream on different treatment schedules.
Local narrow‐band UVB versus local PUVA.
Tacrolimus 0.1% ointment versus vehicle.
Tacrolimus 0.1% ointment versus mometasone furoate ointment.
Oral cyclosporin versus topical betamethasone dipropionate.
Oral alitretinoin at 10 mg and 30 mg a day versus placebo.
When a study reported on treatment during a remission‐ or clearance‐induction phase for participants before they were randomised to a follow‐up or maintenance phase, we considered only the latter (randomised) phase for this review.
Types of outcome measures
We extracted the following primary and secondary outcomes from the included studies.
Primary outcomes
Percentage of participants with self‐rated good/excellent control of symptoms.
Percentage of participants with investigator‐rated good/excellent control of symptoms.
Adverse events: adverse effects (long‐ and short‐term) of the intervention. Long‐term adverse events are defined as adverse events occurring after completion of the treatment phase; short‐term adverse events occur during the treatment phase.
Secondary outcomes
Reduction in severity (participant‐rated).
Reduction in severity (investigator‐rated).
Time until relapse, defined as the number of days/weeks until the participant reported worsening of symptoms after initial response.
Dose reduction: reduction in treatment dose per time unit or cumulative prescribed treatment dose. For example, a decrease in daily topical medication, or a decrease in weekly photo irradiation.
We did not exclude studies from the review that did not include these outcomes.
We believe that three months is the minimum study duration required to document important data such as duration and frequency of disease relapse.
Search methods for identification of studies
We aimed to identify all relevant RCTs regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
The Cochrane Skin Information Specialist searched the following databases up to 19 April 2018, using strategies based on the draft strategy for MEDLINE presented in our published protocol (van Coevorden 2009).
Cochrane Skin Group Specialised Register (search strategy in Appendix 2).
Cochrane Central Register of Controlled Trials (CENTRAL; 2018, Issue 3), in the Cochrane Library (search strategy in Appendix 3).
MEDLINE via Ovid (from 1946) (search strategy in Appendix 4).
Embase via Ovid (from 1974) (search strategy in Appendix 5).
Allied and Complementary Medicine (AMED) via Ovid (from 1985) (search strategy in Appendix 6).
Latin American and Caribbean Health Science Information database (LILACS) (from 1982) (search strategy in Appendix 7).
Global Resource of Eczema Trials. Centre of Evidence Based Dermatology (accessed at http://www.greatdatabase.org.uk on 19 April 2018), using the following terms in the title of the records: hand* or finger* or palm or palms.
Trials registries
We (WAC and PJC) searched the following trials registries up to 21 April 2018, using the following search terms: hand and (eczema or dermatitis).
International Standard Randomized Controlled Trials Number (ISRCTN) registry (www.isrctn.com).
ClinicalTrials.gov (www.clinicaltrials.gov).
Australian New Zealand Clinical Trials Registry (www.anzctr.org.au).
World Health Organization International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/).
Searching other resources
Correspondence with authors
If we needed clarification regarding studies, we contacted study authors using the correspondence options stated in their papers (for studies published since 1999). If email addresses did not work, we tried to find recent publications by the same corresponding author with more recent contact data, or we searched Facebook, LinkedIn, and the Internet to connect with these authors. In addition, we tried to contact all authors of studies that included other dermatoses among hand eczema, to obtain separate data for hand eczema participants. We listed in the 'notes' section of the Characteristics of included studies tables whether we contacted study authors, and if they responded. We have not included in the review complete correspondence with all studies, but we have shown the relevant citations in the Characteristics of included studies tables. The full correspondence with study authors is available upon request.
References from published studies
We checked the bibliographies of included studies for further references to relevant trials.
Adverse events
We did not perform a separate search for adverse events. However, we did examine data on adverse events from the included studies.
Unpublished literature
We contacted authors and pharmaceutical companies in relation to ongoing trials that were recently completed according to the trial registries mentioned under Electronic searches. When results were published on the trial register websites, we included these in the results, and we tried to contact study authors for additional information if necessary.
Conference proceedings
We searched the conference proceedings of annual conferences of the European Academy of Dermatology and Venereology (EADV) from 2000 to 2011 for further relevant RCTs. Some were available from the JEADV; however, some others had to be obtained from the organisation itself, from which we requested the material on CD‐ROM.
Handsearching
We handsearched using the terms 'eczema', 'dermatitis', 'hand(s)', 'palmoplantar', and 'inflammatory' in 16 English, two German, one Italian, one French, and one Dutch dermatology journal (all journals 1977 through 2003). We searched the journals listed in Appendix 8.
Data collection and analysis
Selection of studies
Three review authors (PJC, JLB, and WAC) independently checked titles and abstracts identified from the searches. Three review authors (PJC, TD, and ÅS) conducted an additional handsearch. If it was clear that the study did not refer to a randomised controlled trial on hand eczema, we excluded it. We retrieved all potential trials as full‐text articles for further independent examination by two review authors (TD and ÅS). These two review authors decided which trials conformed to the inclusion criteria and resolved discrepancies by discussion in consensus meetings. We obtained missing data from the trial authors when possible. Whenever we found duplicate publications of the same trial, we used the paper with the most relevant data (usually we had a conference abstract and a full article) as the primary reference and listed the other publication in the additional references following the reference section.
Data extraction and management
Three review authors (PJC, TD, and ÅS) extracted data independently, using a standardised data extraction form. These review authors and future reviewers piloted the data extraction form during a meeting of the European Dermato‐Epidemiology Network, in July 2000. This form was based on a preceding systematic review of psoriasis interventions and was later updated according to Cochrane recommendations. We resolved discrepancies and uncertainties in a series of consensus meetings, which were led by one review author (PJC).
Two other review authors (JLB and WAC) entered into Review Manager 5.3 and checked the outcome data extracted from the included studies (RevMan).
Assessment of risk of bias in included studies
Two review authors (ÅS and TD) independently assessed the risk of bias in included studies following the domain‐based evaluation described in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions and, using the Cochrane risk of bias tool, assessed all included studies from the following aspects for potential risk of bias (Higgins 2011b).
Random sequence generation, which refers to selection bias due to inadequate generation of a randomised sequence.
Allocation concealment, which also refers to selection bias but due to inadequate concealment of the allocation sequence before assignment.
Blinding of participants and personnel, which refers to performance bias due to knowledge of intervention allocation by participants or personnel.
Blinding of outcome assessment, which refers to detection bias due to knowledge of intervention allocation by the outcome assessor.
Incomplete outcome data, which refers to the quantity, nature, or manner in which incomplete outcome data were handled.
Selective reporting, which refers to reporting bias due to selective reporting.
Other source of bias, which refers to any other types of bias not covered above, including inclusion of baseline comparisons, certainty of the diagnosis, and premature ending.
Whenever we encountered disagreement regarding assessment of risk of bias, we resolved this in a consensus meeting with a third review author (PJC or HW). Two review authors (JLB and WAC) assessed completed 'Risk of bias' forms and entered the data into RevMan.
Measures of treatment effect
We employed risk ratios (RR) with 95% confidence intervals (CIs) to measure the effect of a treatment for dichotomous outcomes. We expressed results as number needed to treat for an additional beneficial outcome (NNTB) when appropriate, along with different rates of baseline risk. We expressed results from analyses of continuous data as mean differences (MDs), along with CIs and respective P values. Whenever a small study (fewer than 30 participants) included zero events in one arm, we used Fisher's exact test to calculate the P value, and we provided numerical data for the numerator/denominator for each treatment (Grainge 2013). We calculated Fisher's exact test using GraphPad software (GraphPad).
We interpreted numerical data in charts and tables when possible. We tried to extract numerical data from graphical presentations by using a ruler, or we contacted study authors for recent trials if the data were unclear. For data that had been extracted from a graph, we added remarks.
For studies that exclusively presented median values for a particular outcome, we substituted the median for the mean, provided that data were not too skewed. When standard deviations were not available from a paper, we tried to calculate these from other available data. When confidence intervals were provided, we used the formula given in Chapter 7.7.3.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
For multi‐arm studies, we analysed each arm in comparison with placebo when possible.
Unit of analysis issues
Cluster randomised trials
We checked cluster randomised trials (groups of individuals instead of individuals randomised to intervention or control) for unit of analysis errors based on advice provided in Section 16.3.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011d).
Cross‐over studies
In cross‐over studies (with each participant allocated to a sequence of interventions, instead of to only one intervention), unit of analysis issues can arise when participants have been randomised to multiple treatments over multiple periods, or when there has been an inadequate washout period. We dealt with cross‐over studies by analysing only the first treatment period as a simple parallel‐group study.
Within‐participant studies (self‐controlled, left‐right designs)
Given that analysis of paired data was not possible with RevMan, we summarised the data from within‐participant studies in the text. The unit of analysis in within‐participant studies was one hand per participant, whereas in parallel‐group studies, the unit of analysis was per participant. Relevant data were presented in the analysis as "other data", in table format.
Studies with multiple arms
For studies including multiple arms (more than two) in the analyses, we plotted the different comparisons in different forest plots when possible.
Dealing with missing data
For trials published from 1999 onwards and with uncertainty, we tried to contact trial authors if we felt that this may yield essential additional information. In these cases, we contacted the first author or, when stated, the corresponding author of the article. For the current review, we did not make any assumption or imputation to missing data. We extracted all outcome data as they were reported in the original studies. We stated when authors were contacted and whether additional information was provided under Characteristics of included studies.
Assessment of heterogeneity
We had planned to explore reasons for heterogeneity amongst studies and, if necessary, to carry out sensitivity analyses to examine the effects of excluding study subgroups (e.g. children versus adults, atopic versus allergic contact hand eczema) or studies with high risk of bias.
Clinical heterogeneity (or clinical diversity) is considered as variability among participants, interventions, and outcomes. In future updates of this review, we plan to assess clinical heterogeneity by examining characteristics of the studies and similarity between types of participants, interventions, and outcomes. If studies were sufficiently similar, we achieved statistical pooling by using a weighted treatment effect.
We used random‐effects model meta‐analysis because of anticipated differences across studies in, amongst other things, the participant base included. Statistical heterogeneity was investigated with the I² test. If the I² statistic had been greater than 50%, reasons for heterogeneity in studies would have been explored.
Assessment of reporting biases
We planned on including statistical methods for detecting publication bias (e.g. Begg's funnel plots). However, funnel plots are recommended by the Cochrane Handbook for Systematic Reviews of Interventions, Section 10.4 (Higgins 2011c), when at least a substantial number of studies (10 or more) are included in the meta‐analysis. This was not feasible due to the heterogeneity of included studies. For reporting bias, we studied the study authors and institutions involved (pharmaceutical companies or not), funding, sponsorship of commercially available supplements, and, finally, conflicts of interest.
Data synthesis
When data permitted, we had planned to conduct statistical pooling, using a random‐effects model whenever studies appeared sufficiently similar.
Subgroup analysis and investigation of heterogeneity
We conducted no pre‐planned subgroup analyses in the current review, but in future updates, we will carry out analyses, if data permit, to examine the effects of including specific study subgroups (e.g. children versus adults, recurrent vesicular versus hyperkeratotic hand eczema).
Sensitivity analysis
We conducted no pre‐planned sensitivity analyses in this review, but for future updates, we will consider performing sensitivity analyses for pooled analysis involving only studies at low risk of bias.
'Summary of findings' tables
We included in the 'Summary of findings' tables all primary outcomes and the secondary outcome 'investigator‐rated reduction in severity' for the clinically most relevant studies (Ryan 2016). We assessed clinical relevance based on the clinical experiences of study authors. We tried to include studies from every group of interventions (topical corticosteroids, topical calcineurin inhibitors, UV therapy, and systemic treatments), and to keep the total number of included studies to a minimum. We therefore aimed to include studies that compared different groups of comparisons or studies that answered the questions that authors ask themselves on a regular basis in everyday practice. The 'Summary of findings' tables are based on the GRADE principles (GRADEPro, version 3.6.1). The GRADE approach is a sequential process that evaluates the quality of a body of evidence by considering the following domains.
Study limitations, which refers to risk of bias in either study design or conduct that could lead to biased estimation of treatment effect.
Inconsistency of results, which refers to unexplained heterogeneity of results.
Indirectness of evidence, which refers to directness of comparisons of target populations, interventions, comparators, and outcomes of the included studies compared to those of the planned PICO of the systematic review.
Imprecision, because results are generally imprecise when the study includes few participants, few events, or a wide confidence interval of the effect estimate.
Publication bias.
Randomised controlled trials (RCTs) began as high‐quality/certainty evidence, but If concerns were identified in the above domains, certainty was rated down by one or two levels depending on the severity of the concern. The GRADE approach completes assessments of the body of evidence by grading it in the high, moderate, low, or very low certainty category.
A duration of longer than three months was preferred for evaluating a clinically relevant effect. We considered interventions comparing different groups of interventions and studies providing different treatment regimens with regards to dosages or frequency as clinically most relevant. Therefore we included the following comparisons in the 'Summary of findings' tables.
Clobetasol foam compared to vehicle (Table 1).
Mometasone furoate cream in different treatment schedules (Table 2).
Local narrow‐band UVB compared to local PUVA (Table 3).
Tacrolimus 0.1% ointment compared to mometasone furoate ointment (Table 4).
Tacrolimus 0.1% ointment compared to vehicle (Table 5).
Oral cyclosporin compared to topical betamethasone dipropionate (Table 6).
Oral alitretinoin compared to placebo at 10 mg and 30 mg a day (Table 7; Table 8).
Summary of findings for the main comparison. Corticosteroid creams/ointments: clobetasol propionate foam compared to vehicle foam for hand eczema.
| Corticosteroid creams/ointments: clobetasol propionate foam compared to vehicle foam for hand eczema | ||||||
| Patient or population: participants with moderate to severe hand eczema Setting: secondary care with outpatients in Northern America Intervention: clobetasol propionate 0.05% foam twice a day for 14 days Comparison: vehicle/placebo foam twice a day for 14 days | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed riska | Corresponding risk | |||||
| Risk with vehicle foam | Risk with clobetasol propionate foam | |||||
|
Primary: investigator‐rated good/excellent control of symptoms Follow‐up: day 15 |
Study population | RR 1.43 (0.86 to 2.40) | 125 (1 RCT) | ⊕⊕⊕⊝ Moderateb | ‐ | |
| 270 per 1000 | 386 per 1000 (232 to 648) | |||||
|
Primary: participants with self‐rated good/excellent control of symptoms Follow‐up: day 15 |
Study population | RR 2.32 (1.38 to 3.91) | 125 (1 RCT) | ⊕⊕⊕⊝ Moderatec | NNTB 3 (95% CI 2 to 8) | |
| 222 per 1000 | 516 per 1000 (307 to 869) | |||||
|
Primary: adverse events ‐ at least 1 adverse event Follow‐up: day 15 |
Study population | RR 2.24 (0.82 to 6.06) | 125 (1 RCT) | ⊕⊕⊕⊝ Moderatec | ‐ | |
| 79 per 1000 | 178 per 1000 (65 to 481) | |||||
|
Primary: adverse events ‐ any adverse event treatment‐related (application site pruritus) Follow‐up: day 15 |
Study population | RR 1.02 (0.06 to 15.89) | 125 (1 RCT) | ⊕⊕⊕⊝ Moderated | ‐ | |
| 16 per 1000 | 16 per 1000 (1 to 252) | |||||
|
Secondary: reduction in severity, participant‐rated scoring Follow‐up: day 15 |
Study population | RR 1.57 (1.21 to 2.04) | 125 (1 RCT) | ⊕⊕⊕⊝ Moderateb | NNTB 3 (95% CI 2 to 7) | |
| 524 per 1000 | 822 per 1000 (634 to 1000) | |||||
|
Secondary: reduction in severity, investigator‐rated scoring ‐ improvement at least 2 grades Follow‐up: day 15 |
Study population | RR 1.47 (0.90 to 2.39) | 125 (1 RCT) | ⊕⊕⊕⊝ Moderateb | ‐ | |
| 286 per 1000 | 420 per 1000 (257 to 683) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; NNTB: number needed to treat for an additional beneficial outcome; RCT: randomised controlled trial; RR: risk ratio. Kircik 2013 | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe assumed risk is the mean control group risk.
bDowngraded by one level to moderate‐certainty evidence for imprecision. Small sample size and small number of events.
cDowngraded by one level to moderate‐certainty evidence for imprecision. Wide confidence interval with small sample size and small number of events.
dDowngraded by one level to moderate‐certainty evidence for imprecision. Summary effect contains both appreciable benefit and harm; wide confidence interval with small sample size and small number of events.
Summary of findings 2. Corticosteroid creams/ointments: mometasone furoate cream 3 times/week versus 2 times/week for hand eczema.
| Corticosteroid creams/ointments: mometasone furoate cream thrice a week versus twice a week | ||||||
|
Patient or population: people (all patch‐tested) with hand eczema > 6 months that had cleared upon daily treatment for a maximum of 9 weeks with mometasone furoate cream
Settings: secondary care with outpatients from hospitals in Denmark
Intervention: mometasone furoate cream 3 times/week up to 36 weeks Comparision: mometasone furoate cream 2 times/week up to 36 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed riska | Corresponding risk | |||||
| Risk with mometasone furoatetwice a week | Risk with mometasone furoate thrice a week | |||||
| Primary: investigator‐rated good/excellent control of symptoms Follow‐up: 36 weeks | Study population | RR 1.23 (0.94 to 1.61) | 72 (1 RCT) | ⊕⊕⊝⊝ Lowb | ‐ | |
| 676 per 1000 | 831 per 1000 (635 to 1000) | |||||
| Primary: participant‐rated good/excellent control of symptoms Not measured | See comment | See comment | Not estimable | ‐ | See comment | No data available |
| Primary: adverse events Follow‐up: 36 weeks | Study population | RR 1.76 (0.45 to 6.83) | 72 (1 RCT) | ⊕⊕⊝⊝ Lowc | ‐ | |
| 81 per 1000 | 143 per 1000 (36 to 554) | |||||
|
Secondary: investigator‐rated reduction in severity Not measured |
See comment | See comment | Not estimable | ‐ | See comment | No data available |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. Veien 1999 | ||||||
| GRADE Working Group grades of evidence. High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe assumed risk is the mean control group risk.
bDowngraded by two levels to low‐certainty evidence. Imprecision downgraded by one level: the event number was small as was the sample size. Downgraded one level for risk of bias, given the high risk of detection and performance bias.
cDowngraded by two levels to low‐certainty evidence. Imprecision downgraded by one level: the summary effect contains both appreciable benefit and harm; sample size was small as was the event rate. Downgraded one level for risk of bias, given the high risk of detection and performance bias.
Summary of findings 3. Irradiation with UV light: local narrow‐band UVB compared to local PUVA for hand eczema.
| Irradiation with UV light: local narrow‐band UVB compared to local PUVA for hand eczema | ||||||
| Patient or population: people with hand eczema unresponsive to clobetasol propionate Setting: secondary care with outpatients in the United Kingdom. Intervention: local narrow‐band UVB twice weekly for 12 weeks Comparison: immersion PUVA twice weekly for 12 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed riska | Corresponding risk | |||||
| Risk with local PUVA | Risk with local narrow‐band UVB | |||||
|
Primary: investigator‐rated good/excellent control of symptoms Follow‐up: 12 weeks |
Study population | RR 0.50 (0.22 to 1.16) | 60 (1 RCT) | ⊕⊕⊕⊝ Moderateb | ‐ | |
| 400 per 1000 | 200 per 1000 (88 to 464) | |||||
|
Primary: participant‐rated good/excellent control of symptoms Not measured |
See comment | See comment | Unable to estimate treatment effect | ‐ | See comment | No data reported |
|
Primary: adverse events ‐ reported adverse events, mainly erythema Follow‐up: 12 weeks |
See comment | See comment | RR 19.00 (1.16 to 312.42) | 60 (1 RCT) | ⊕⊕⊕⊝ Moderatec |
PUVA: No adverse events reported (0/30) Narrow‐band UVB: 9 out of 30 participants reported an adverse event, mainly erythema Fisher's exact test P = 0.0019 |
|
Secondary: investigator‐rated reduction in severity in mTLSSd Follow‐up: 12 weeks |
‐ | ‐ | Unable to estimate treatment effect | 43 (1 RCT) | ⊕⊕⊕⊝ Moderatee |
Reduction in mTLSS PUVA: Median mTLSS of 8.5 (range 0 to 16) and 8 (range 3 to 15) for the left and right hand, to a median mTLSS 3 (range 0 to 13) and 3 (range 0 to 14) (n = 23) Reduction mTLSS local narrow‐band UVB group: Median mTLSS of 7 (range 0 to 16) and 8.5 (range 1 to 15) to a median mTLSS5 (range 0 to 11) and 4.5 (range 0 to 11) (n = 20) |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; mTLSS: modified total lesion symptom score; PUVA: oral psoralen combined with UVA; RCT: randomised controlled trial; RR: risk ratio; UV: ultraviolet. 2015 | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe assumed risk is the mean control group risk.
bDowngraded by one level to moderate‐certainty evidence for imprecision. Small sample size, small number of events, and high dropout rate.
cDowngraded by one level to moderate‐certainty evidence for imprecision. Wide confidence interval with small sample size, small number of events, and high dropout rate.
dThe Modified Total Lesion Symptom Score (mTLSS) is the sum of seven items (erythema, oedema, vesiculation, scaling, lichenification/hyperkeratosis, fissures, and pruritus/pain) scored on a 4‐point scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe). A high mTLSS represents severe hand eczema.
eDowngraded by one level to moderate‐certainty evidence for imprecision. Small sample size based on single study.
Summary of findings 4. Topical calcineurin inhibitors: tacrolimus 0.1% ointment compared to mometasone furoate ointment for vesicular hand eczema.
| Topical calcineurin inhibitors: tacrolimus 0.1% ointment compared to mometasone furoate ointment for vesicular hand eczema | ||||||
| Patient or population: people with moderate to severe chronic relapsing dyshidrotic eczema on hands Setting: secondary care setting at a single dermatology department in Germany Intervention: topical calcineurin inhibitors tacrolimus 0.1% ointment twice daily during 4 weeks Comparison: topical corticosteroid mometasone furoate ointment twice daily during 4 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Risk with mometasone furoate ointment | Risk with topical calcineurin inhibitor tacrolimus 0.1% ointment | |||||
|
Primary: investigator‐rated good/excellent control of symptoms ‐ Not measured |
See comment | See comment | Not estimable | ‐ | See comment | Not measured |
|
Primary: participant‐rated good/excellent control of symptoms ‐ Not measured |
See comment | See comment | Not estimable | ‐ | See comment | Not measured |
| Primary: adverse events Follow‐up: 2 weeks | See comment | See comment | Not estimable | 16 pairs of hands (1 RCT) | ⊕⊕⊕⊝ Moderatea | Within‐participant design None of the participants dropped out because of adverse events |
|
Secondary: investigator‐rated reduction in severity ‐ DASIb Follow‐up: 2 weeks |
See comment | See comment | Not estimable | 16 pairs of hands (1 RCT) | ⊕⊕⊕⊝ Moderatea | Within‐participant design Tacrolimus group: Mean DASI from 18 (SD 12.68) to 6.6 (SD 6.18) Mometasone furoate group: Mean DASI from 18.5 (SD 14.09) to 6.9 (SD 7.7) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; DASI: Dyshydrotic Eczema Area and Severity Index; RCT: randomised controlled trial; RR: risk ratio; SD: standard deviation. Schnopp 2002 | ||||||
| GRADE Working Group grades of evidence. High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded by one level to moderate‐certainty evidence for imprecision: small sample size in a single study and small number of events.
bDASI: Dyshydrotic Eczema Area and Severity Index is an assessment of severity combining objective (vesicles, erythema, and desquamation) and subjective (itch) evaluations on a scale from 0 (no eczema) to 60 (severe hand eczema).
Summary of findings 5. Topical calcineurin inhibitors: tacrolimus 0.1% ointment versus vehicle for hand eczema.
| Topical calcineurin inhibitor tacrolimus 0.1% ointment compared to vehicle for hand eczema | ||||||
|
Patient or population: people with moderate to severe nickel sulphate‐induced allergic contact dermatitis based on clinical history (hand eczema) and proven by patch testing, resistant to topical corticosteroids
Settings: secondary care setting in a single‐centre study in Italy
Intervention: topical calcineurin inhibitor tacrolimus 0.1% ointment twice daily for 2 weeks Comparison: vehicle twice daily for 2 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Risk with vehicle | Risk with tacrolimus 0.1% ointment | |||||
|
Primary: investigator‐rated good/excellent control of symptoms Follow‐up: 3 weeks |
See comment | See comment | RR 29.00 (1.90 to 443.25) | 28 (1 RCT) | ⊕⊕⊕⊝ Moderatea | Unable to calculate assumed risk as no events in the control group ‐ 14/14 participants in the tacrolimus group had good/excellent control of symptoms. Fisher's exact test P = 0.0001, NNTB 1, 95% CI 1 to 1 |
| Primary: participant‐rated good/excellent control of symptoms Not measured | See comment | See comment | Not estimable | ‐ | See comment | No data reported |
|
Primary: adverse events ‐ burning/itching at application site Follow‐up: 3 weeks |
See comment | See comment | RR 9.00 (0.53 to 152.93) | 28 (1 RCT) | ⊕⊕⊕⊝ Moderatea | Unable to calculate assumed risk as no events in the control group ‐ 4/14 participants in the tacrolimus group had burning/itching at the application site. Fisher's exact test P = 0.1129, RR 9.00, 95% CI 0.53 to 152.93 No data on "all adverse events" |
|
Secondary: investigator‐rated reduction in severity ‐ Not measured |
See comment | See comment | Not estimable | ‐ | See comment | No data reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; NNTB: number needed to treat for an additional beneficial outcome; RCT: randomised controlled trial; RR: risk ratio. Pacor 2006 | ||||||
| GRADE Working Group grades of evidence. High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded by one level to moderate‐certainty evidence for imprecision: very small sample size, low event rate, and very large confidence intervals.
Summary of findings 6. Oral immunosuppressants: oral cyclosporin versus topical betamethasone dipropionate.
| Oral cyclosporin compared to topical betamethasone for patient with hand eczema | ||||||
| Patient or population: people with hand eczema, continuously for 6 months, significant disability, inadequate response to conventional treatment, confirmation by histopathology Setting: secondary care setting at a single centre in Finland Intervention: oral cyclosporin 3 mg/kg/d and placebo cream for 6 weeks Comparison: topical betamethasone dipropionate 0.05% cream and placebo capsules for 6 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed riska | Corresponding risk | |||||
| Risk with topical betamethasone | Risk with oral cyclosporin | |||||
|
Primary: investigator‐ rated good/excellent control of symptomsb Follow‐up: 6 weeks |
Study population | RR 1.88 (0.88 to 3.99) | 34 (1 RCT) | ⊕⊕⊕⊝ Moderatec | ‐ | |
| 333 per 1000 | 627 per 1000 (293 to 1000) | |||||
|
Primary: participant‐rated good/excellent control of symptomsb Follow‐up: 6 weeks |
Study population | RR 1.25 (0.69 to 2.27) | 34 (1 RCT) | ⊕⊕⊕⊝ Moderatec | ‐ | |
| 500 per 1000 | 625 per 1000 (345 to 1000) | |||||
|
Primary: adverse events ‐ at least 1 adverse event Follow‐up: 36 weeks |
Study population | RR 1.22 (0.80 to 1.86) | 55d (1 RCT) | ⊕⊕⊕⊝ Moderatec | Because of partial cross‐over design, a different number of participants is given for this outcome | |
| 556 per 1000 | 678 per 1000 (444 to 1000) | |||||
|
Secondary: investigator‐rated reduction in severityb Follow‐up: 6 weeks |
Mean investigator‐rated reduction in severity in total disease activity score after 6 weeks of treatment was 5.7 | MD 0.30 higher (2.50 lower to 3.10 higher) | ‐ | 34 (1 RCT) | ⊕⊕⊕⊝ Moderatec | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. Granlund 1996 | ||||||
| GRADE Working Group grades of evidence. High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe assumed risk is the mean control group risk.
bObserver‐rated disease activity score: grading 0 to 3 (0 = none, 1 = mild, 2 = moderate, 3 = severe) on erythema, scaling, infiltration, excoriation, crusting, and vesicles for both hands. A high score represents severe hand eczema.
cDowngraded by one level to moderate‐certainty evidence. Imprecision downgraded by one level: small sample size.
dThe number of participants varies between different outcomes because this is a cross‐over study, and adverse events were included from all different phases of the trial.
Summary of findings 7. Oral retinoids: alitretinoin 30 mg versus placebo for hand eczema.
| Oral retinoids: alitretinoin 30 mg versus placebo for hand eczema | ||||||
|
Patient or population: people with moderate to severe chronic hand eczema
Settings: secondary care with outpatients in an international multi‐centre setting
Intervention: oral retinoid alitretinoin 30 mg for 12 to 24 weeks Comparison: oral placebo for 12 to 24 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed riska | Corresponding risk | |||||
| Risk with placebo | Risk with alitretinoin 30 mg | |||||
|
Primary: investigator‐rated good/excellent control of symptoms Follow‐up: 48 weeks to 72 weeks |
Study population | RR 2.75 (2.20 to 3.43) | 1210 (2 RCTs) | ⊕⊕⊕⊕ Highb | NNTB 4, 95% CI 3 to 5 | |
| 157 per 1000 | 432 per 1000 (346 to 539) | |||||
|
Primary: participant‐rated good/excellent control of symptoms Folluw‐up: 48 weeks to 72 weeks |
Study population | RR 2.75 (2.18 to 3.48) | 1210 (2 RCTs) | ⊕⊕⊕⊕ Highb | NNTB 4, 95% CI 3 to 5 | |
| 143 per 1000 | 394 per 1000 (312 to 498) | |||||
|
Primary: adverse events ‐ headache Folluw‐up: 48 weeks to 72 weeks |
Study population | RR 3.43 (2.45 to 4.81) | 1210 (2 RCTs) | ⊕⊕⊕⊕ Highb | All adverse events not stated in Ruzicka 2008 NNTH 6, 95% CI 4 to 11 |
|
| 74 per 1000 | 251 per 1000 (179 to 352) | |||||
| Secondary: investigator‐rated reduction in severity in TLSSc and mTLSSd | See comment | See comment | Not estimable | ‐ | See comment | Only incomplete data reported; therefore we were unable to extract these data |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; RCT: randomised controlled trial; RR: risk ratio. Ruzicka 2008; Fowler 2014 | ||||||
| GRADE Working Group grades of evidence. High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe assumed risk is the mean control group risk.
bRelatively high number of dropouts, although analysed via intention‐to‐treat analysis. Risk of bias was low, the two included studies were consistent, and the evidence is applicable to patients with (moderate to) severe hand eczema. Risk of publication bias was considered low, although the studies were sponsored by a pharmaceutical company.
cThe total lesion symptom score (TLSS) is the sum of seven items (erythema, oedema, vesicles, desquamation, hyperkeratosis, fissures, and pruritus/pain) scored on a 4‐point scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe). A high TLSS represents severe hand eczema.
dThe modified total lesion symptom score (mTLSS) is the sum of seven items (erythema, oedema, vesiculation, scaling, lichenification/hyperkeratosis, fissures, and pruritus/pain) scored on a 4‐point scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe). A high mTLSS represents severe hand eczema.
Summary of findings 8. Oral retinoids: alitretinoin 10 mg versus placebo for hand eczema.
| Oral retinoids: alitretinoin 10 mg versus placebo for hand eczema | ||||||
|
Patient or population: people with moderate to severe chronic hand eczema
Settings: secondary care with outpatients in an international multi‐centre setting
Intervention: oral retinoid alitretinoin 10 mg for 12 to 24 weeks Comparison: oral placebo for 12 to 24 weeks | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed riska | Corresponding risk | |||||
| Risk with placebo | Risk with alitretinoin | |||||
|
Primary: investigator‐rated good/excellent control of symptoms Follow‐up: up to 48 weeks |
Study population | RR 1.58 (1.20 to 2.07) | 781 (2 RCTs) | ⊕⊕⊕⊕ Highb | NNTB 11, 95% CI 6.3 to 26.5 | |
| 194 per 1000 | 307 per 1000 (233 to 402) | |||||
|
Primary: participant‐rated good/excellent control of symptoms Follow‐up: up to 48 weeks |
Study population | RR 1.73 (1.25 to 2.40) | 765 (2 RCTs) | ⊕⊕⊕⊕ Highb | NNTB 9, 95% CI 6 to 20 | |
| 144 per 1000 | 249 per 1000 (180 to 345) | |||||
|
Primary: all adverse events Follow‐up: up to 48 weeks |
Study population |
RR 1.01 (0.66 to 1.55) |
158 (1 RCT) | ⊕⊕⊕⊝ Moderatec | NNTH 260, 95% CI ‐14.47 to 15.24 | |
| 346 per 1000 |
350 per 1000 (228 to 537) |
|||||
|
Secondary: investigator‐rated reduction in severity of TLSSd Follow‐up: up to 48 weeks |
See comment | See comment | ‐ | 158 (1 RCT) | ⊕⊕⊕⊝ Moderatec | Median % change in score from baseline (95% CI) Placebo group: ‐25% (95% CI ‐42 to ‐14) Aitretinoin 10 mg: –59 (95% CI –73 to –33) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
CI: confidence interval; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; RCT: randomised controlled trial; RR: risk ratio; TLSS: total lesion symptom score. Ruzicka 2004; Ruzicka 2008 | ||||||
| GRADE Working Group grades of evidence. High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aThe assumed risk is the mean control group risk.
b Both studies were at low risk of bias and results were precise.
cDowngraded by one level to moderate‐certainty evidence: imprecision downgraded by one level: small sample size.
dThe total lesion symptom score (TLSS) is the sum of seven items (erythema, oedema, vesicles, desquamation, hyperkeratosis, fissures, and pruritus/pain) scored on a 4‐point scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe). A high TLSS represents severe hand eczema.
Results
Description of studies
We included 60 RCTs on different interventions for hand eczema.
Results of the search
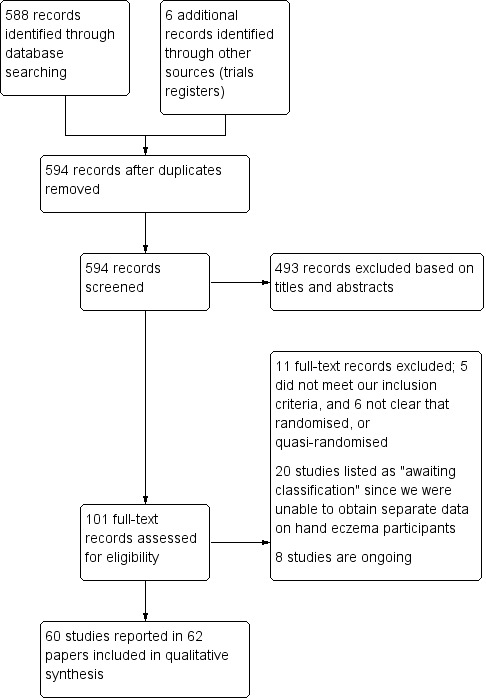
Our searches of the databases yielded 588 records (Electronic searches). Our searches of the trials registries identified six further studies. We therefore had a total of 594 records.
No duplicate records were identified, hence we screened 594 references. We excluded 493 records based on titles and abstracts. We obtained the full text of the remaining 101 records. We excluded 11 studies (Characteristics of excluded studies). We added 20 records to Characteristics of studies awaiting classification because we were unable to extract separate data on hand eczema patients. We identified eight ongoing studies (Characteristics of ongoing studies).
We included 60 studies reported in 62 references. For a further description of our screening process, see the study flow diagram (Figure 1).
1.
Study flow diagram.
Included studies
Details of the 60 included studies with a total of 5469 participants are summarised in the Characteristics of included studies table. We included studies published from May 1967 to April 2018, as well as unpublished data from studies registered in trial registries up to April 2018.
Design
Of the 60 RCTs, 18 were within‐participant studies (i.e. having a left‐right design, comparing one hand with the other) (Adams 2007; Baskan 2005; Cartwright 1987; Chu 2009; Faghihi 2008; Fairris 1984; Fairris 1985; Fredriksson 1975; Grattan 1991; Kemper 1998; King 1984; Lindelöf 1987; Möller 1983; Odia 1996; Schnopp 2002; Sezer 2007; Sheehan‐Dare 1989; Uggeldahl 1986). In total, 41 studies used a parallel‐group design. Fowler 2005 used a parallel‐group design but within each group chose a within‐participant design as well. Two of these parallel‐group studies used a cross‐over design (Burrows 1986; Granlund 1996), but they were parallel before cross‐over.
Participants
The original protocol stipulated diagnosis by a physician. Although only one of the identified studies stated this explicitly, all studies were based on participants being outpatients at hospitals. Therefore, we assumed that the diagnosis was established by a physician for all participants. Some studies included a specific subgroup of hand eczema, while others excluded these subgroups, for example, vesicular (or dyshidrotic) hand eczema was included by 11 studies (Adams 2007; Grattan 1991; Odia 1996; Pigatto 1990; Polderman 2003; Said 2010; Schnopp 2002; Sezer 2007; Sharma 2006; Sheehan‐Dare 1989; Tzaneva 2009), and vesicular hand eczema was excluded by three studies (Bleeker 1989; Chu 2009; Hordinsky 2010). The same was true for atopic dermatitis and atopic dermatitis on the hands: six studies targeted atopic eczema specifically (Bauer 2012; Fowler 2005; Lauriola 2011; NCT01231854; Veien 1995; Yousefi 2012), while six other studies excluded participants with characteristics of atopic eczema (Bleeker 1989; Burrows 1986; Chu 2009; Hordinsky 2010; Katsarou 2012; Lodén 2012a).
All studies were performed in a secondary setting and included participants who had hand eczema for at least several weeks to months; therefore the review did not include acute hand eczema.
The studies included participants with different grades of severity, and not all studies included a severity grade as an inclusion criterion. Moderate to severe hand eczema was included in the following studies: Baskan 2005; Kircik 2013; Krejci‐Manwaring 2008; Pacor 2006; Ruzicka 2004; Schnopp 2002; Tzaneva 2009; van Coevorden 2004a. Chu 2009 included only mild hand eczema. Mild to moderate hand eczema was included in Belsito 2004,Hordinsky 2010,Kucharekova 2003,Lauriola 2011, and Odia 1996. Cherill 2000,Fowler 2005,Hanifin 2004, and Uggeldahl 1986 included moderate hand eczema. Fowler 2014,NCT01231854, and Ruzicka 2008 included only severe hand eczema, and Bauer 2012 included moderate to very severe hand eczema. In two studies, the included severity was not completely clear (Veien 1995; Veien 1999).
Another inclusion criterion was poor response or resistance to conventional therapies such as topical corticosteroids (Adams 2007;Brass 2015; Cartwright 1987; Fairris 1984; Fairris 1985;Fowler 2014; Granlund 1996; King 1984; Lindelöf 1987; NCT01231854; Odia 1996; Pacor 2006; Ruzicka 2004; Ruzicka 2008; Sezer 2007; Sheehan‐Dare 1989; Sjövall 1987; Tzaneva 2009). Only one study included a minimally affected area of hand eczema (Bayerl 1999). Disabling hand eczema was an inclusion criterion in two studies (Granlund 1996; Grattan 1991).
Overall, children were not included as a study population. One study included participants between 1.5 and 70 years of age (Uggeldahl 1986), another study included participants at least 10 years of age (Boroujeni 2017), four studies included participants 12 years of age and older (Faghihi 2008; Jowkar 2011; Jowkar 2014; Kircik 2013), and one study had a minimum inclusion age of 16 years (Grattan 1991). The remaining studies included only adults. A few studies excluded older participants. Two studies used an upper age limit of 60 years (Jowkar 2011; Yousefi 2012), three studies 65 years (Agarwal 2013; Bleeker 1989; Fowler 2005), three studies 70 years (Granlund 1996; Ruzicka 2004; Uggeldahl 1986), and three studies 75 years (Fowler 2014; NCT01231854; Ruzicka 2008).
One study included female participants exclusively (Kaaber 1983). The remaining studies included both female and male participants. Pregnant or lactating women, or both, were excluded from about half of the studies (32 studies).
Overall, participants were in general good health, and studies often excluded systemic diseases such as diabetes and renal or hepatic disease.
Sample size calculation
A total of 5469 participants were enrolled. Most studies were relatively small (12 to 158 participants), and sample size calculations often were not stated. A large proportion of the 5469 participants were included in five trials (Belsito 2004; Fowler 2014; Hordinsky 2010; Ruzicka 2004; Ruzicka 2008). Twelve studies included fewer than 25 participants (Burrows 1986; Fairris 1984; Grattan 1991; Kemper 1998; King 1984; Lindelöf 1987; Odia 1996; Pigatto 1990; Schnopp 2002; Sezer 2007; Sharma 2006; Sjövall 1987). In 27 studies, between 25 and 50 participants were included (Baskan 2005; Bauer 2012; Bayerl 1999; Cartwright 1987; Cherill 2000; Faghihi 2008; Fairris 1985; Fredriksson 1975; Granlund 1996; Gupta 1993; Hanifin 2004; Jowkar 2011; Kaaber 1983; Katsarou 2012; Krejci‐Manwaring 2008; Kucharekova 2003; Lauriola 2011; Lodén 2012a; Odia 1996; Pacor 2006; Polderman 2003; Said 2010; Sheehan‐Dare 1989; Thestrup‐Pedersen 2001; Tzaneva 2009; Veien 1995; Whitaker 1996). Between 50 and 100 participants were included in eight studies (Bleeker 1989; Boroujeni 2017; Brass 2015; Fowler 2005; Jowkar 2014; Möller 1983; Uggeldahl 1986; Yousefi 2012). Between 100 and 500 participants were included in eight studies (Agarwal 2013; Belsito 2004; Bissonnette 2010; Hill 1998; Kircik 2013; Ruzicka 2004; van Coevorden 2004a; Veien 1999). Three studies included more than 500 participants (Fowler 2014; Hordinsky 2010; Ruzicka 2008).
NCT01231854 aimed to include 78 participants based on a sample size calculation; however the study was ended prematurely and included only 15 participants.
Setting
None of the studies were conducted in a primary care setting. As far as we know, all studies were conducted in a secondary care setting and included outpatients from hospitals. About half of the studies were conducted as multi‐centre studies, usually within the same country. Six studies were international multi‐centre studies (Belsito 2004; Bissonnette 2010; Cherill 2000; Hordinsky 2010; Ruzicka 2004; Ruzicka 2008).
Although most studies did not declare the country in which the study was conducted, we assumed that they were conducted in the hospitals of the investigators. Based on this assumption, most studies were conducted in North America and Europe. A substantial number of studies were conducted in the United Kingdom (Brass 2015; Burrows 1986; Cartwright 1987; Fairris 1984; Fairris 1985; Grattan 1991; Hill 1998; King 1984; Sheehan‐Dare 1989), Sweden (Bleeker 1989; Fredriksson 1975; Lindelöf 1987; Möller 1983; Sjövall 1987), Germany (Adams 2007; Bauer 2012; Bayerl 1999; Bissonnette 2010; NCT01231854; Odia 1996; Schnopp 2002), Denmark (Kaaber 1983; Thestrup‐Pedersen 2001; Veien 1995; Veien 1999), and the Netherlands (Kemper 1998; Kucharekova 2003; Polderman 2003; van Coevorden 2004a).
A few studies were conducted in other parts of the world, including Iran (Boroujeni 2017; Faghihi 2008; Jowkar 2011; Jowkar 2014; Yousefi 2012), India (Agarwal 2013; Sharma 2006), Turkey (Baskan 2005; Sezer 2007), Singapore (Said 2010), South Africa (Whitaker 1996), and Taiwan (Chu 2009).
Treatment duration
Overall the studies were of relatively short duration. One study had a duration of only one week (Gupta 1993). The treatment episode was less than one month in 22 studies (Belsito 2004; Bleeker 1989; Boroujeni 2017; Chu 2009; Faghihi 2008; Fowler 2005; Fredriksson 1975; Hill 1998; Jowkar 2011; Jowkar 2014; Kemper 1998; King 1984; Kircik 2013; Lauriola 2011; Lodén 2012a; Odia 1996; Pacor 2006; Polderman 2003; Schnopp 2002; Sharma 2006; Uggeldahl 1986; Yousefi 2012), and it was less than two months (eight weeks) in 12 studies (Adams 2007; Baskan 2005; Bauer 2012; Bayerl 1999; Cherill 2000; Grattan 1991; Kucharekova 2003; Lindelöf 1987; Said 2010; Sheehan‐Dare 1989; Sjövall 1987; Thestrup‐Pedersen 2001); nine studies had a treatment duration between two and four months (Brass 2015; Cartwright 1987; Fairris 1984; Fairris 1985; Katsarou 2012; Krejci‐Manwaring 2008; Pigatto 1990; Sezer 2007; van Coevorden 2004a).
Only 11 studies had a duration of active treatment longer than four months (Agarwal 2013; Bissonnette 2010; Fowler 2014; Hanifin 2004; NCT01231854; Ruzicka 2004; Ruzicka 2008; Tzaneva 2009; Veien 1995; Veien 1999; Whitaker 1996).
Studies with a cross‐over design had an active treatment phase of six weeks for both drugs (Burrows 1986; Granlund 1996), and Hordinsky 2010 had an active treatment phase of six weeks, followed by an open‐label phase.
The total duration of active treatment was unclear in two studies (Kaaber 1983; Möller 1983).
Follow‐up
Most studies did not include a follow‐up period. Only 24 studies included a follow‐up period (Baskan 2005; Cartwright 1987; Fairris 1984; Fairris 1985; Fowler 2014; Granlund 1996; Grattan 1991; Jowkar 2011; Krejci‐Manwaring 2008; Lindelöf 1987; NCT01231854; Pacor 2006; Polderman 2003; Ruzicka 2004; Ruzicka 2008; Said 2010; Schnopp 2002; Sezer 2007; Sharma 2006; Sheehan‐Dare 1989; Sjövall 1987; Tzaneva 2009; van Coevorden 2004a; Whitaker 1996). This period varied from a week to several months and involved scheduled visits or just a single follow‐up questionnaire. Veien 1999 clearly states that the treatment episode was 30 weeks, although data in the survival analyses suggest follow‐up to 250 days.
Two studies were ended prematurely (Burrows 1986; NCT01231854).
Interventions and comparisons
In most studies, an active intervention was compared to no treatment, variants of the same medication, or placebo (or vehicle). Very few studies compared two different classes of interventions: one study compared coal tar paste with a corticosteroid (Kemper 1998), one study phototherapy (PUVA) with X‐rays (Sheehan‐Dare 1989), one study phototherapy (UVA‐1) with a topical corticosteroid (Said 2010), two studies a calcineurin inhibitor with a corticosteroid (Katsarou 2012; Schnopp 2002), one study cyclosporin with a topical corticosteroid (Granlund 1996), and one study cromoglycate with a diet (Pigatto 1990). One study compared oral cyclosporin to oral alitretinoin (NCT01231854). We organised the remaining trials into the categories described below and provide details of the various dose regimens. Full details of interventions and comparisons for each included study are given in the Characteristics of included studies.
I. Skin protection measures, including gloves
These were not included in this review.
II. Topical treatments
A. Bland emollients
One study (Table 9) compared effects of two different emollients ‐ an emollient with ceramides (Locobase Repair) in 17 participants versus a regular petrolatum‐based emollient (Vaseline‐lanette) in 15 participants ‐ as adjuvants in the treatment of hand eczema over two months (Kucharekova 2003).
1. Overview of studies on bland emollients: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comment |
| Kucharekova 2003 | Ceramide‐containing emollient versus regular petrolatum‐based emollient | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Chu 2009 | E‐DO lotion versus vehicle lotion | E‐DO: 37 responders, 12 of whom responded to E‐DO only (19%), and 25 to both (39.7%) Vehicle: 36 responders, 11 of whom responded to vehicle only (17.5%), and 25 to both (39.7%) Investigator‐rated RR 1.06 (95% CI 0.54 to 2.10) |
Within‐patient study |
E‐DO is a trade/product name.
One within‐participant study compared an emollient with E‐DO lotion once daily to vehicle lotion. E‐DO claims to be a potential agent for revitalising skin cells to regain their moisture retention capacity and might improve wound healing and inhibition of Staphylococcus aureus and Propionibacterium acnes, according to the study authors (Chu 2009).
B. Corticosteroid creams or ointments
Nine studies evaluated topical corticosteroids as the main intervention (Bleeker 1989; Faghihi 2008; Fowler 2005; Gupta 1993; Kircik 2013; Lodén 2012a; Möller 1983; Uggeldahl 1986; Veien 1999).
Bleeker 1989 compared two topical corticosteroids to determine whether the less potent fluprednidene (Cortoderm) cream was as effective as the more potent betamethasone‐17‐valerate (Betnovate) cream. Each product was applied once daily, in the evenings, for a study period of three weeks. In both study groups, a specific emollient was used if required.
In a within‐participant study (Fowler 2005), the effectiveness of hydrocortisone butyrate (HB) 0.1% cream was compared with three other medium‐potency corticosteroid creams (fluticasone propionate 0.05% cream (FP), prednicarbate emollient 0.1% cream (PC), and mometasone furoate 0.1% cream (MF)) for treatment of chronic atopic and hand dermatitis. Participants were randomised to one of three treatment groups: HB versus FP, HB versus PC, or HB versus MF. Subsequently, participants applied twice‐daily HB to one hand, and FP, PC, or MF to the other hand, for a duration of two weeks.
A double‐blind within‐participant study investigated whether the addition of zinc sulphate to clobetasol cream is effective in the treatment of chronic hand eczema (Faghihi 2008). Forty‐seven participants were randomised and subsequently were treated twice daily with clobetasol + zinc sulphate cream on one hand and clobetasol 'only' cream on the other hand for two weeks.
In Gupta 1993, one group received betamethasone dipropionate polyacrylic film‐forming lotion (Occlucort) twice a day for seven days. The other group received a traditional betamethasone dipropionate (Diprosone) lotion, slightly thickened to resemble the consistency of the other product.
In Kircik 2013, participants received clobetasol propionate 0.05% foam or vehicle foam twice daily for a period of 15 days.
In a double‐blind randomised clinical trial (Lodén 2012a), twice‐daily application of betamethasone‐valerate 0.1% cream (BV group) was compared to once‐daily application of betamethasone‐valerate 0.1% cream in combination with once‐daily application of a moisturiser cream containing 5% urea (BV + M group). The study duration was two weeks. Both groups were allowed to use urea 5% cream for additional hand treatment.
A multi‐centre study was designed to investigate whether twice weekly application of a steroid was effective in keeping hand eczema, which had been brought into remission, under control (Möller 1983). To induce remission, 61 participants with symmetrical hand eczema of at least six months duration were treated with clobetasol propionate (Dermovate) cream twice weekly. Then, the 55 (out of 61) participants who were healed were included in a maintenance study and were followed for a mean period of 138 days (range 55 to 193 days); this occurred in the form of an RCT that compared one hand (receiving clobetasol (Dermovate) cream) with the other hand (receiving fluprednidene (Cortoderm) cream). When relapse occurred during the maintenance phase, the cream allocated to that hand could be applied more frequently; if this failed, the cream for the other (best) hand could be used temporarily. Participants were allowed to use an emollient (Essex cream) as needed.
Two strengths of the same topical corticosteroid were compared in a within‐participant design (Uggeldahl 1986). Forty‐six participants were treated twice daily with desonide (Tridesilon) cream 0.1% on one hand and desonide (Apolar) cream 0.05% on the other for two weeks. Participants had not been treated for eczema for at least one week before the study began.
The aim of one study was to compare mometasone (Elocon) ointment ("fatty cream") applied three times per week versus two times per week (Veien 1999). Initially, all participants were treated for three weeks with daily application of mometasone furoate to bring their dermatitis under control. This RCT investigated 106 participants whose dermatitis was brought under control. They were randomised to three parallel study groups for up to 36 weeks: treatment with mometasone furoate ointment once daily three times a week, treatment with mometasone furoate ointment once daily two times a week, and treatment with only emollients. In this study, in case of obvious bacterial infection, a course of oral antibiotics or potassium permanganate soaks, or both, was permitted. All participants were given an emollient to be used freely. Clinical evaluations were carried out after 3, 6, 12, 18, 24, and 30 weeks of maintenance treatment.
For an overview of the outcome 'Investigator‐rated good/excellent control' in these studies, see the additional tables section (Table 10).
2. Overview of studies on topical corticosteroids: investigator‐rated good/excellent control.
| Study | Comparison of topical corticosteroids | Investigator‐rated good/excellent control in RR (95% CI) | Comment |
| Möller 1983 | Intermittent clobetasol versus fluprednidene | Clobetasol better since 32/46 versus 14/46 hands remained in remission; investigator‐rated | Within‐patient study |
| Uggeldahl 1986 | Desonide 0.1% versus desonide 0.05% | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Bleeker 1989 | Fluprednidene versus betamethasone | RR 0.59 (95% CI 0.28 to 1.23); investigator‐rated | ‐ |
| Gupta 1993 | Betamethasone film versus betamethasone lotion | RR 10.24 (95% CI 0.59 to 176.56); investigator‐rated | ‐ |
| Veien 1999 | Mometasone 3 times/week versus mometasone 2 times/week | RR 1.23 (95% CI 0.94 to 1.61); investigator‐rated | ‐ |
| Fowler 2005 | Hydrocortisone butyrate versus fluticasone propionate twice daily
Hydrocortisone butyrate versus prednicarbate emollient twice daily Hydrocortisone butyrate 0.1% cream versus mometasone furoate twice daily |
No data regarding the primary outcome investigator‐rated good/excellent control | Three parallel treatment groups Each group separately within‐patient |
| Faghihi 2008 | 0.05% clobetasol cream versus 0.05% clobetasol + zinc sulphate cream | Clobetasol + zinc sulphate better in terms of respectively scaling (25/47 versus 3/47), redness (41/47 versus 1/41), and lichenification (24/47 versus 7/47); investigator‐rated | Within‐patient study |
| Lodén 2012a | Betamethasone‐valerate 0.1% cream twice daily versus betamethasone‐valerate 0.1% cream once daily + urea 5% cream once daily | RR 0.75 (0.55 to 1.03); investigator‐rated | ‐ |
| Kircik 2013 | Clobetasol propionate 0.05% foam twice daily versus vehicle foam twice daily | RR 1.43 (0.86 to 2.40); investigator‐rated | ‐ |
CI: confidence interval.
RR: risk ratio.
C. Coal tar and derivatives
Kemper 1998 investigated the efficacy of coal tar paste (pix lithanthracis) compared to zinc oxide paste and betamethasone‐valerate. Nineteen participants with symmetrical hand eczema were included and were treated with coal tar paste on one hand and betamethasone‐valerate ointment 0.1% or zinc oxide paste on the other hand. Participants were instructed to wear gloves on both hands for protection and bandage. Clinical evaluation of the hands was carried out once a week, and at that same visit, the corresponding treatment was applied, again to the hands. Treatment duration was four weeks.
For an overview of the outcome 'Investigator‐rated good/excellent control' in this study, see the additional tables section (Table 11).
3. Overview of studies on coal tar and derivatives: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comment |
| Kemper 1998 | Coal tar paste versus betamethasone‐valerate or zinc oxide once a week | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
D. Irradiation with UV light
Variations in UV phototherapy (UVA, UVB, PUVA) were investigated in 10 studies (Adams 2007; Bayerl 1999; Brass 2015; Grattan 1991; Polderman 2003; Said 2010; Sezer 2007; Sjövall 1987; Tzaneva 2009; van Coevorden 2004a).
Said 2010 compared the efficacy of topical betamethasone‐valerate 0,1% cream twice daily to UVA‐1 phototherapy thrice weekly for six weeks. Twenty‐four participants with chronic vesicular hand eczema were treated with phototherapy, and 23 participants were treated with topical corticosteroids.
Treatment with a portable UVB phototherapy unit, to be used at home, was compared with treatment by non‐specific topical treatment in a study among 48 participants with occupational hand dermatitis (Bayerl 1999). It seems that the UVB‐treated group also applied this non‐specific topical treatment. The UVB‐treated group irradiated their hands at home five days per week for eight weeks according to a predetermined dosage scheme.
Two studies compared oral PUVA with topical bath PUVA: van Coevorden 2004a and Tzaneva 2009.
Tzaneva 2009 compared oral PUVA versus bath PUVA. Immediately after immersion for 15 minutes, the hands and feet were exposed to UVA irradiation. The irradiation doses in both groups were increased depending on the degree of erythematous response. Treatment was given three to four times a week until complete clearance, or over a maximum period of 20 weeks. After clearing, participants were maintained on PUVA twice weekly for two weeks and then once weekly for another four weeks.
van Coevorden 2004a compared a randomised controlled parallel study of oral PUVA phototherapy whereby the hands were irradiated by participants themselves at home with bath PUVA; the hands were soaked in a psoralen (trioxsalen) solution followed by UVA in the clinic. The aim was to demonstrate equal clinical efficacy, assuming that costs for home treatment would be substantially lower. Treatment was given for 10 weeks, and there was follow‐up after the end of treatment for another eight weeks. Emollients were allowed in both groups.
Sezer 2007 compared UVA with UVB: 12 participants received local narrow‐band UVB three times a week on one hand and local PUVA on the other hand for nine weeks. The initial dose was 150 mJ/cm² for each participant. A 20% increasing dose schedule was used until a final dose of 2000 mJ/cm² was reached versus local PUVA three times a week during nine weeks on 12/15 contralateral hands. The initial dose of psoralen plus UVA irradiation was 1.0 J/cm² with an increase of 0.5 J/cm² in every second session until a final dose of 7.5 J/cm² was achieved.
Brass 2015 compared the efficacy of narrow‐band UVB with localised PUVA. Sixty participants received immersion PUVA or narrow‐band UVB twice a week for 12 weeks.
Studies that employed UVA treatment were Adams 2007 and Grattan 1991.
A within‐participant study compared the effectiveness of middle‐dose UVA‐1 irradiation to topical cream PUVA therapy (Adams 2007). UVA‐1 is a newer form of UV therapy that contains only long‐wavelength UVA‐1 radiation (340 to 400 nm) and thus reduces the risk of burning. Participants with chronic relapsing dyshidrotic hand eczema received one treatment modality on one hand and one treatment modality on the other hand. Treatment was given three times a week during a period of five weeks (middle‐dose UVA‐1 irradiation three times a week during five weeks (cumulative dose of 600 J/cm²) versus local 8‐MOP‐cream‐PUVA irradiation three times a week during five weeks (cumulative dose of 17.4 J/cm²)). 8‐MOP‐crème was applied 30 minutes before the start of irradiation. Grattan 1991 used topical PUVA three times weekly for eight weeks versus UVA (with placebo psoralen paint). The PUVA treatment was performed by applying a liquid ("paint") containing methoxypsoralen to one hand. On the contralateral hand, an inactive paint was applied, whereupon both hands were irradiated with UVA. Moisturisers were allowed on both hands, and both hands received a small fraction of UVB from UVA lamps.
Polderman 2003 used UVA‐1 (long‐wavelength UV radiation) irradiation 40 J/cm² on the hands five times weekly for three weeks versus placebo (simulated blue light). Emollients seem to have been allowed in both groups.
Sjövall 1987 used UVB irradiation only on the hands four times a week for eight weeks in six participants versus a placebo for UVB (filtered light) on the hands four times a week for eight weeks in six participants versus hand UVB followed by whole‐body UVB + UVA four times a week during eight weeks in six participants. Their 'ordinary topical treatment' was permitted in all groups. Emollients were allowed in both groups.
For an overview of studies with UV therapy, see Table 12.
4. Overview of studies on irradiation with UV light: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comment |
| Bayerl 1999 | UVB versus no UVB | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Sjövall 1987 | Local UVB versus placebo | RR 2.0 (95% CI 0.26 to 15.6) | ‐ |
| Sjövall 1987 | Local UVB hand versus whole body + hand UVB | RR 2.2 (95% CI 0.83 to 5.8) | ‐ |
| Sjövall 1987 | Local UVB hand versus whole body versus placebo | RR 3.67 (95% CI 0.90 to 14.97) | ‐ |
| van Coevorden 2004a | Oral PUVA versus topical bath PUVA | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Grattan 1991 | Topical PUVA versus UVA | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Polderman 2003 | UVA‐1 versus placebo | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Brass 2015 | Local NB‐UVB versus topical PUVA | RR 0.50 (95% CI 0.22 to 1.16) | ‐ |
| Sezer 2007 | Local NB‐UVB versus topical PUVA | NB‐UVB was effective in 2/12 hands and topical PUVA was effective in 1/12 hands | Within‐patient study |
| Tzaneva 2009 | Oral PUVA versus bath PUVA | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Adams 2007 | UVA‐1 versus topical cream PUVA | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Said 2010 | Local UVA‐1 versus topical betamethasone‐valerate cream | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
CI: confidence interval.
RR: risk ratio.
NB‐UVB: narrow‐band ultraviolet B.
PUVA: psoralen + ultraviolet A.
UVB: ultraviolet B.
UVA‐1: a subtype of ultraviolet A.
E. Irradiation with X‐rays (ionising radiation)
X‐rays/radiotherapy/Grenz rays were studied in five publications (Cartwright 1987; Fairris 1984; Fairris 1985; King 1984; Lindelöf 1987). One study compared conventional superficial radiotherapy to UV phototherapy (Sheehan‐Dare 1989). All these studies used within‐participant designs (i.e. comparing one hand with the contralateral hand).
Two of these studies used superficial X‐rays 300 Rad as active treatment (Fairris 1984; King 1984).
King 1984 included 20 participants and treated one hand with three fractionated doses of 100 Rad (i.e. a total of 300 Rad) at 45 kV given at one‐week intervals; Fairris 1984 treated participants with a combination of topical therapy and superficial X‐ray therapy, and assessed them at 6, 9, and 18 weeks after the start of X‐ray therapy. One hand was treated with 100 Rad at 50 kV on three occasions at intervals of 21 days (i.e. total 300 Rad), and the other hand with placebo. Participants continued treatment with tar paste or steroid ointments on both hands throughout the trial.
Lindelöf 1987 gave six fractionated doses of 3 Gy at one‐week intervals for six weeks. Placebo therapy was achieved by allowing the apparatus to hum without emitting radiation.
In Cartwright 1987, one hand was irradiated three times with 3 Gy of Grenz rays (total 900 Rad), and the contralateral hand was treated in an exactly similar manner with sham radiation. Treatments were repeated at 21‐day intervals for a total of three visits. Evaluations were performed by the doctor and the participant at 3, 6, 9, 12, 15, and 18 weeks after initial treatment.
One study compared superficial X‐ray and Grenz ray irradiation (Fairris 1985). Both radiation therapies were given in three divided doses at 21‐day intervals. One hand received 1 Gy of conventional superficial X‐ray 50 kV, the other 3 Gy of Grenz ray 10 kV.
One study compared X‐ray irradiation to UV phototherapy (Sheehan‐Dare 1989). Superficial X‐ray irradiation (0.9 Gy at 50 kV administered on three occasions at 21‐day intervals) on one hand was compared with topical PUVA therapy (three times a week for six weeks) on the contralateral hand in 25 participants. Assessments were performed before and at 6, 9, and 18 weeks after the start of treatment.
For an overview of studies including irradiation with X‐rays, see Table 13.
5. Overview of studies on X‐rays (ionising radiation): investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comments |
| King 1984 | X‐rays 300 rad versus placebo | No difference after 6 months. Grenz ray effective in 11/15 hands versus 8/15 hands with placebo. | Within‐patient study |
| Fairris 1984 | X‐rays 300 rad versus placebo | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Lindelöf 1987 | Grenz rays 1800 rad versus placebo | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Cartwright 1987 | Grenz rays 300 rad versus placebo | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Fairris 1985 | X‐rays 1 Gy versus Grenz rays 3 Gy | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Sheehan‐Dare 1989 | X‐rays versus PUVA | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
CI: confidence interval.
Grenz rays: a type of X‐rays (ionising radiation).
Gy: Gray, a unit of radiation dose.
RR: risk ratio.
F. Topical calcineurin inhibitors
Tacrolimus was studied in four papers (Katsarou 2012; Krejci‐Manwaring 2008; Pacor 2006; Schnopp 2002). Pimecrolimus was evaluated in three papers (Bauer 2012; Belsito 2004; Hordinsky 2010), as well as in two conference abstracts (Baskan 2005; Cherill 2000).
Topical tacrolimus 0.1% ointment (FK506) twice daily was compared with the topical corticosteroid mometasone furoate 0.1% ointment in a within‐participant design (Schnopp 2002). Participants were encouraged to use emollients in addition. Treatment duration was four weeks, and treatment was followed by a washout period of two weeks. Tacrolimus ointment 0.1% twice daily during four weeks versus mometasone furoate 0.1% ointment twice daily was also used in Katsarou 2012 (with tapering mometasone furoate dose in the mometasone arm of the trial).
Katsarou 2012 compared topical tacrolimus 0.1% twice daily for 30 days and once daily for 31 to 90 days in 15 participants to mometasone furoate ointment twice daily for one week, once daily during week two and week three, once daily three times a week for weeks four and five, and once daily two times a week during the rest of the study (for 90 days) in 15 participants.
Two studies addressed tacrolimus ointment versus vehicle (Krejci‐Manwaring 2008; Pacor 2006).
Twice‐daily application of tacrolimus ointment was compared to its vehicle to study its effectiveness in keeping hand eczema in remission (Krejci‐Manwaring 2008). Remission was induced after a three‐week taper of prednisone. Simultaneous to the prednisone taper, participants started with tacrolimus or its vehicle for a total treatment duration of 12 weeks.
The aim of another trial was to evaluate the efficacy of 0.1% tacrolimus ointment for nickel sulphate‐induced allergic contact dermatitis of the hands (Pacor 2006). Participants were randomised to twice‐daily treatment with either 0.1% tacrolimus ointment or its vehicle during 14 days.
Five of our included studies addressed the use of pimecrolimus cream: Baskan 2005; Bauer 2012; Belsito 2004; Cherill 2000; Hordinsky 2010.
A large multi‐centre study with 294 participants compared twice‐daily application of pimecrolimus 1% cream to twice‐daily application of vehicle in a three‐week study (Belsito 2004). In both groups, the evening application was followed by six‐hour occlusion. Time to relapse was compared between pimecrolimus 1% cream and vehicle in a randomised controlled parallel study (Bauer 2012). Before commencement, participants with atopic hand eczema used mometasone furoate for one to three weeks until symptoms had cleared. This was followed by an eight‐week maintenance period with pimecrolimus versus vehicle cream. Another published abstract reporting a placebo‐controlled randomised trial comparing pimecrolimus 1% cream with vehicle over eight weeks found pimecrolimus to be effective in suppressing all clinical signs of hand eczema apart from vesiculation (Baskan 2005). Only limited data could be extracted from one study comparing pimecrolimus 1% cream (with or without occlusion) to vehicle because this study was published as a conference abstract (Cherill 2000). In a large multi‐centre study (Hordinsky 2010), 652 adults were randomised to pimecrolimus 1% or vehicle cream twice daily with overnight occlusion for six weeks.
For an overview of studies including topical calcineurin inhibitors, see Table 14.
6. Overview of studies on topical calcineurin inhibitors: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comments |
| Schnopp 2002 | Tacrolimus ointment versus mometasone furoate | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Katsarou 2012 | Tacrolimus ointment versus mometasone furoate | No data regarding the primary outcome investigator‐rated good/excellent control | Improvement was reported separately for subcategories of clinical signs |
| Krejci‐Manwaring 2008 | Tacrolimus ointment versus vehicle | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Pacor 2006 | Tacrolimus ointment versus vehicle | RR 29.0 (95% CI 1.9 to 443.25) | ‐ |
| Belsito 2004 | Pimecrolimus cream versus vehicle | RR 1.53 (95% CI 0.99 to 2.36) | ‐ |
| Hordinsky 2010 | Pimecrolimus cream versus vehicle | RR 1.28 (95% CI 0.99 to 1.66) | ‐ |
| Bauer 2012 | Pimecrolimus cream versus vehicle | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Baskan 2005 | Pimecrolimus cream versus vehicle | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Cherill 2000 | Pimecrolimus cream versus vehicle | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
CI: confidence interval.
RR: risk ratio.
G. Other topical interventions
Two antibacterial agents ‐ clioquinol cream and fusidic acid cream ‐ each combined with a corticosteroid (betamethasone‐valerate) were compared in a multi‐centre study on 120 hand eczema participants with confirmed or suspected secondary infection of their eczema (Hill 1998). The unblinded study had a duration of four weeks.
One study investigated urea cream (Fredriksson 1975), that is, Aquacare HP cream, a moisturising emulsion containing multi‐sterols, phospholipids, and fatty diols (pH 6), twice a day (morning and evening) for four weeks, versus control of Calmurid cream containing betaine and lactic acid (pH 3), twice a day for four weeks.
Bexarotene, a novel type of retinoid, was evaluated in 55 participants by a three‐arm unblinded (phase I to II open label) study lasting 22 weeks (Hanifin 2004). The intervention was bexarotene 1% gel applied in a stepwise accumulation every two weeks from once every other day to three times daily (bexarotene only group). Comparators were bexarotene application in combination with mometasone furoate (B + MF group) and in combination with hydrocortisone (B + HC group). All three groups used emollients.
One study compared topical furpalmate‐containing cream (0.3%) with a topical corticosteroid (hydrocortisone acetate 0.5%) twice a day (Lauriola 2011).
Jowkar 2014 studied the efficacy of topical fumaric acid 5% cream twice daily compared to triamcinolone 0.1% cream twice daily in 92 participants.
Three studies investigated herbal topical treatments: one study compared a 2% oil extract of Nigella sativa L. to betamethasone ointment 0.1% and Eucerin (Yousefi 2012). Nigella sativa L. (family Ranunculaceae) is an annual flowering plant that grows in south and southwest Asia, of which the seeds can be used as spice. Another study compared a cream with 4% Fumaria parviflora Lam. twice daily to vehicle cream (Jowkar 2011). Fumaria parviflora Lam. extract (family Papaveraceae) is a Persian herbal medicine that is called 'Shahtareh' in Iran. The plants were dried, and from them an abstract was made for the cream. Finally, twice‐daily application with an oil‐in‐water emulsion‐based herbal cream containing fenugreek seeds 5%, marshmallow 5%, chamomile 5%, and walnut leaves 5% was compared with twice‐daily application of the topical steroid fluocinolone acetonide cream 2%, in the study of Boroujeni 2017.
In a within‐participant study, pulsed direct iontophoresis on one hand was compared with no iontophoresis on the contralateral hand (Odia 1996), in which one of the participants' hands received pulsed direct current iontophoresis, 20 times 15 minutes each during three weeks in 20 hands, or as a control, no iontophoresis on contralateral hands for three weeks. Both hands received steroid‐free tar solution and zinc paste.
For an overview of other topical interventions, see Table 15.
7. Overview of studies on other topical interventions: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comments |
| Hill 1998 | Betamethasone‐valerate + clioquinol versus betamethasone‐valerate + fusidic acid | RR 1.03 (95% CI 0.74 to 1.43) | ‐ |
| Fredriksson 1975 | Aquacare HP cream versus calmurid cream containing betaine and lactic acid | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Odia 1996 | Iontophoresis versus no iontophoresis | No data regarding the primary outcome investigator‐rated good/excellent control | Within‐patient study |
| Boroujeni 2017 | Herbal cream containing fenugreek seeds 5%, marshmallow 5%, chamomile 5%, and walnut leaves 5% versus fluocinolone acetonide cream 2% twice daily | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Hanifin 2004 | Bexarotene 1% gel versus bexarotene with either mometasone furoate or hydrocortisone | RR 0.85 (95% CI 0.40 to 1.80) for bexarotene only versus B + MF; 1.83 (95% CI 0.61 to 5.53) for bexarotene only versus B + HC; and 2.15 (95% CI 0.67 to 6.89) for B + MF versus B + HC | ‐ |
| Jowkar 2014 | Fumaric acid 5% cream versus triamcinolone 0.1% cream | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Lauriola 2011 | Furpalmate versus hydrocortisone acetate cream | RR 0.90 (95% CI 0.76 to 1.07) | ‐ |
| Jowkar 2011 | 4% Fumaria Parviflora Lam cream versus vehicle cream twice daily | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Yousefi 2012 | Nigella sativa L. versus betamethasone ointment versus Eucerin | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
B: bexarotene 1% gel.
CI: confidence interval.
Fumaria Parviflora Lam: Fumaria Parviflora Lamarck.
HC: hydrocortisone.
MF: mometasone furoate.
Nigella sativa L: Nigella sativa Linne.
RR: risk ratio.
III. Systemic treatments
A. Oral corticosteroids
We identified no RCTs addressing oral corticosteroids.
B. Immunosuppressants
We found two publications on cyclosporin (Granlund 1996), but these studies were based on the same trial, which had three phases. Oral cyclosporin 3 mg/kg/d and placebo cream for six weeks was compared with topical betamethasone dipropionate 0.05% cream and placebo capsules identical to cyclosporin. This was a cross‐over trial, in which participants who failed to respond to their intervention in phase I were crossed over to the alternative intervention. The use of own emollients was allowed in both groups.
Agarwal 2013 investigated a low dose of azathioprine combined with topical clobetasol 0.05% cream compared to topical clobetasol 0.05% cream alone during 24 weeks.
NCT01231854 compared the effects of oral cyclosporin 2.7 to 4.0 mg/kg to those of alitretinoin 30 mg/d during 24 weeks. Please see Table 16.
8. Overview of studies on immunosuppressants: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comments |
| Granlund 1996 | Oral cyclosporin versus topical betamethasone | RR 1.88 (95% CI 0.88 to 3.99) | ‐ |
| Agarwal 2013 | Oral azathioprine and clobetasol propionate 0.05% cream twice daily versus topical clobetasol propionate 0.05% cream twice daily | RR 2.33 (95% CI 1.61 to 3.38) | ‐ |
| NCT01231854 | Oral cyclosporin versus alitretinoin | RR 1.50 (95% CI 0.35 to 6.40) | Study terminated prematurely and included 15 participants only |
CI: confidence interval.
RR: risk ratio.
C. Oral retinoids
We identified six studies evaluating oral retinoids (Bissonnette 2010; Fowler 2014; NCT01231854; Ruzicka 2004; Ruzicka 2008; Thestrup‐Pedersen 2001). Ruzicka 2004 had previously been presented in part as a conference abstract (Larsen 2003, listed under Ruzicka 2004).
Three studies investigated the effect of 10 mg oral alitretinoin (Bissonnette 2010; Ruzicka 2004; Ruzicka 2008)
Two large multi‐centre studies compared a total of four different oral doses of a novel retinoid (alitretinoin) with placebo capsules (Ruzicka 2004; Ruzicka 2008). In Ruzicka 2004, three groups, each receiving respectively, 10, 20, or 40 mg per day, were compared with a placebo group. The trial lasted 12 weeks. The other study (Ruzicka 2008; also known as Benefit of Alitretinoin in Chronic Hand Eczema or BACH study) compared two groups receiving, respectively, oral alitretinoin 10 or 30 mg once daily versus placebo up to 24 weeks. In both studies, participants were allowed to use a standard emollient.
The large multicenter study of Fowler 2014 compared alitretinoin 30 mg/d to placebo in 596 participants with severe chronic hand eczema. The treatment duration was 24 weeks, and afterwards participants were followed up for a substantial period of time.
NCT01231854 aimed to compare the effectiveness and safety of 30 mg alitretinoin to cyclosporin during 24 weeks in 78 participants.
In Bissonnette 2010, 117 participants suffering from chronic hand eczema were included who had been successfully treated with alitretinoin in an earlier study (Ruzicka 2008), and who had relapsed within the 24‐week observation period after treatment. These 117 relapsed participants were randomised to receive their previous treatment or placebo in a 2:1 ratio. A total of 73 participants were included who had been treated with 30 mg alitretinoin in the previous BACH study (Ruzicka 2008). No other topical or systemic medication for hand eczema was allowed during the treatment period. Dose reductions of study medication were not allowed.
Thestrup‐Pedersen 2001 compared acitretin given orally at 30 mg daily for eight weeks to placebo capsules given for eight weeks. Both groups were allowed to use topical emollients.
For an overview of studies on oral retinoids, see Table 17.
9. Overview of studies on oral retinoids: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐ or participant‐rated good/excellent control in RR (95% CI) | Comments |
| Thestrup‐Pedersen 2001 | Oral acitretin versus placebo | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Ruzicka 2004 | Oral alitretinoin (20 mg and 40 mg) versus placebo |
40 mg
Participant‐rated
RR 3.51 (95% CI 1.80 to 6.82)
Investigator‐rated
RR 1.97 (95% CI 1.3 to 3.0) 20 mg Participant rated RR 2.74 (95% CI 1.37 to 5.46) Investigator‐rated RR 1.49 (95% CI 0.94 to 2.34) |
‐ |
| Ruzicka 2008; Fowler 2014 | Oral alitretinoin 30 mg versus placebo | 30 mg Participant‐rated RR 2.75 (95% CI 2.18 to 3.48) Investigator‐rated RR 2.75 (95% CI 2.20 to 3.43) | ‐ |
| Ruzicka 2004; Ruzicka 2008 | Oral alitretinoin 10 mg versus placebo |
10 mg Participant‐rated RR 1.73 (95% CI 1.25 to 2.40) Investigator‐rated RR 1.58 (95% CI 1.20 to 2.07) |
‐ |
| Bissonnette 2010 | Re‐treatment with oral alitretinoin (30 mg and 10 mg) versus placebo |
30 mg
Investigator‐rated
RR 9.55 (95% CI 2.51 to 36.27) 10 mg Investigator‐rated RR 4.76 (95% CI 0.70 to 32.25) |
‐ |
CI: confidence interval.
RR: risk ratio.
D. Other oral interventions
This group included six studies (Table 18) ‐ one on triethylenetetramine (Burrows 1986), two on disulphiram (Kaaber 1983; Sharma 2006), one comparison of a low‐nickel diet versus oral treatment with disodium cromoglycate (Pigatto 1990), one on oral ranitidine (Veien 1995), and one on evening primrose oil (Whitaker 1996).
10. Overview of other oral interventions: investigator‐rated good/excellent control.
| Study | Comparison | Investigator‐rated good/excellent control in RR (95% CI) | Comments |
| Burrows 1986 | Oral triethylenetetramine versus placebo | Trientine was effective in 6/20 participants versus 10/20 in the placebo group | Unclear whether participant‐ or investigator‐rated |
| Kaaber 1983 | Oral tetraethylthiuram disulphide versus placebo |
Investigator‐rated RR 2.95 (95% CI 0.71 to 12.34) |
‐ |
| Pigatto 1990 | Oral disodium cromoglycate without dietary restriction versus a low‐nickel diet | No data regarding the primary outcome investigator‐rated good/excellent control | ‐ |
| Sharma 2006 | Low‐nickel diet and disulphiram versus a normal diet and placebo |
Investigator‐rated RR 9.09 (95% CI 1.40 to 58.91) |
‐ |
| Veien 1995 | Ranitidine versus placebo | RR 2.22 (95% CI 1.20 to 4.10) | Unclear whether participant‐ or investigator‐rated |
| Whitaker 1996 | Oral gamma‐linoleic acid (GLA; evening primrose oil) versus placebo | No data regarding the primary outcome investigator‐rated good/excellent control |
CI: confidence interval.
RR: risk ratio.
Three studies aimed specifically to intervened on the imputed role of nickel allergy in hand eczema, and included exclusively nickel‐sensitive participants (Burrows 1986; Kaaber 1983; Sharma 2006).
Burrows 1986 compared oral triethylenetetramine (Trientine) 300 mg daily for six weeks to placebo and was designed as a cross‐over study, but this trial was terminated prematurely (23 participants had been included) because of literature reports on teratogenicity in rats.
Kaaber 1983 compared oral tetraethylthiuram disulphide 50 mg/d first week, increasing to 200 mg/d for at least six weeks, to placebo, and was performed in 30 nickel‐sensitive (patch test‐positive) women with pompholyx‐type hand eczema. Half of the participants (n = 15) received tetraethylthiuram disulphide (Antabuse) with gradually increasing dosage (up to 20 mg/d) for "at least six weeks"; probably this maximum dose was given for six weeks. The other 15 women received placebo tablets. Both groups were allowed to use a topical corticosteroid (desoximethasone) and emollients.
Twenty‐one nickel‐sensitive participants (proven by means of patch testing) with vesicular hand eczema were included in a single‐blinded trial and were randomised into two treatment groups (Sharma 2006); a low‐nickel diet in combination with disulphiram was compared with a normal diet in combination with placebo.
Pigatto 1990 compared a low‐nickel diet in eight participants to oral disodium cromoglycate (DSCG) 1500 to 2000 mg three times a day in nine participants to no treatment in seven participants for a period of three months; however this last group was not randomised and therefore was not included in the analyses.
Veien 1995 compared oral ranitidine 300 mg twice daily to placebo tablets in a trial of probably 16 weeks. Both groups were allowed to use betamethasone cream/ointment and emollients.
Evening primrose oil (GLA ‐ gamma linolenic acid) 50 mg in 20 participants was compared to placebo capsules in 19 participants for 16 weeks (Whitaker 1996). Both groups were allowed to use unlimited qualities of emollients and a limited amount of group III corticosteroids. Participants were followed up for eight weeks after the end of treatment, resulting in a total study duration of 24 weeks.
For an overview, see Table 18.
Outcomes
The 60 included RCTs reported diverse outcomes. About half of these studies (n = 33) included our primary outcome good/excellent control either participant‐ or investigator‐rated, although not all included reproducible data. Almost all studies reported our primary outcome 'adverse events' (n = 55). There was substantial heterogeneity between the studies in terms of outcome measures, duration, and timing of outcome assessments.
Most studies used a scale to score the (change in) severity of hand eczema or the rate of clearance. However, many different scoring systems were composed to score different items on different scales, and some did, while others did not, include the affected area. Most scoring systems were unnamed, non‐validated, and self‐created, and combined objective and subjective scores; in these cases, we provided a narrative account of study results and did not attempt quantitative analyses.
The Hand Eczema Severity Index (HECSI) is an assessment of the clinical severity of hand eczema that includes the extent and severity of hand eczema. The hands are divided into five areas (fingertips, fingers, palms, backs of hands and wrists). For each of these areas, the intensity of the six following clinical signs is scored on a grade from 0 (no skin changes) to 3 (severe changes): erythema, induration/papulation, vesicles, fissures, scaling, and oedema were graded on this scale. Moreover, the affected area for each is scored from 0 to 4 (0 = 0%; 1 = 1% to 25%; 2 = 26% to 50%; 3 = 51% to 75%; 4 = 76% to 100%) for the extent of clinical symptoms. Finally, the scores given for each extent location are multiplied by the total sum of the intensity of each clinical feature, resulting in a score of 0 (no hand eczema symptoms) to a maximum severity score of 360 points (very severe hand eczema). Scores above 28 represent severe hand eczema. This is a validated scoring system with excellent agreement for both interobserver and intraobserver reliability (Held 2005). Four studies used the HECSI (Agarwal 2013; Bauer 2012; NCT01231854; Yousefi 2012); three studies awaiting classification (EUCTR2005‐005793‐75‐DE; IRCT201112018263N1; NCT01950494), as well as six ongoing studies, included this outcome parameter (IRCT2014012916412N1; ISRCTN80206075; NCT02664805; NCT03026907; NCT03026946; PACTR201704002194318).
The Dyshydrotic Eczema Area and Severity Index (DASI) is an assessment of severity combining objective (vesicles, erythema, and desquamation) and subjective (itch) evaluations. Each item has to be assessed on a scale ranging from 0 to 3 (0 = absent, 1 = mild, 2 = moderate, 3 = severe), and the grading must be representative for all affected areas. The severity grading is multiplied by a number representative of the total affected area. DASI score = (Vesicles + Erythema + Desquamation + Itch) × Area score points. This results in a DASI ranging from 0 to 60. A DASI score between 0 and 15 represents mild dyshidrotic eczema, 16 to 30 is moderate, and between 31 and 60 represents severe dyshidrotic eczema. The DASI was first described by Odia (Odia 1996). It was used as primary outcome in five studies (Adams 2007; Odia 1996; Polderman 2003; Said 2010; Schnopp 2002); however, this regularly used instrument is not validated.
The Hand Eczema Area and Severity Score (HEAS) is used to assess clinical severity, corrected for the percentage of affected skin area (Simons 1997). The score ranges from 0 (no hand eczema) to 96 (very severe hand eczema) points. Two studies used the HEAS score, although this score is not validated for hand eczema (Chu 2009; Kucharekova 2003).
The Hand Eczema Extent Score (HEES) is a simple clinical score that is not validated (Meding 1989). The HEES scores only the extent of the presence of eczema signs on different locations of the hands, without including intensity of the lesions, with a range of 0 (no hand eczema) to 74 (very severe hand eczema) points. One study included the HEES (Lodén 2012a).
The Total Lesion Symptom Score (TLSS) is the sum of seven items (erythema, oedema, vesicles, desquamation, hyperkeratosis, fissures, and pruritus/pain) scored on a 4‐point scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe). The TLSS was used in Ruzicka 2004, and afterwards a slightly modified version (modified Total Lesion Symptom Score ‐ mTLSS) was used in Ruzicka 2008, Bissonnette 2010, Fowler 2014, and Brass 2015; the seven items were erythema, oedema, vesiculation, scaling, lichenification/hyperkeratosis, fissures, and pruritus/pain. A high mTLSS represents severe hand eczema. The mTLSS relates to the Physician Global Assessment, and a photographic guide has been developed to train observers.
Some studies used scoring systems derived and validated for atopic dermatitis. For example, the validated Eczema Area and Severity Index (EASI) was used (Jowkar 2011;Jowkar 2014), and this scoring system was adjusted to a Hand Eczema Area and Severity Index (HEASI) score (Hanifin 2004). NCT01231854 included the validated SCORing Atopic Dermatitis (SCORAD) next to the HECSI for participants with atopic hand eczema.
The Hand Eczema Area and Severity score is adapted from the well known EASI. The HEASI is calculated by (sum of severity scores for signs) × (involved hand area integer), whereby for the area, 1 = < 10% involvement, 2 = 10% to 29%, 3 = 30% to 49%, 4 = 50% to 69%, 5 = 70% to 89%, and 6 = 90% to 100%. Severity score is 0 = none, 1 = mild, 2 = moderate, 3 = moderately severe, and 4 = severe for the following signs: erythema, scaling, oedema, lichenification, vesiculation, and fissuring.
Investigator and Physician Global assessments (PGA and IGA) or variants of this scoring system (such as the Investigator's Static Global Assessment (ISGA)) were used in different studies, on a 4‐ or 5‐point scale for both hands overall (Bauer 2012; Belsito 2004; Bissonnette 2010; Bleeker 1989; Brass 2015; Cartwright 1987; Chu 2009; Fairris 1984; Fairris 1985; Fowler 2014; Grattan 1991; Gupta 1993; Hill 1998; Hordinsky 2010; King 1984; Kircik 2013; Krejci‐Manwaring 2008; Kucharekova 2003; Lauriola 2011; NCT01231854; Pacor 2006; Ruzicka 2004; Ruzicka 2008; Sheehan‐Dare 1989; Sjövall 1987). In general, a low IGA or PGA score represents well‐controlled hand eczema, whereas a high score represents severe hand eczema. PGA scores have been extensively studied and compared to, for example, HECSI and HEAS (Coenraads 2005; Van Der Valk 2013).
Itch was scored as subjective parameter in most studies (Agarwal 2013; Bayerl 1999; Bleeker 1989; Boroujeni 2017; Brass 2015; Cherill 2000; Chu 2009; Faghihi 2008; Fowler 2005; Fowler 2014; Granlund 1996; Gupta 1993; Hanifin 2004; Hill 1998; Hordinsky 2010; Jowkar 2014; Katsarou 2012; Kemper 1998; Kircik 2013; Krejci‐Manwaring 2008; Kucharekova 2003; Lauriola 2011; Lindelöf 1987; Odia 1996; Pacor 2006; Pigatto 1990; Polderman 2003; Ruzicka 2004; Ruzicka 2008; Schnopp 2002; Sezer 2007; Sharma 2006; Thestrup‐Pedersen 2001; Uggeldahl 1986; van Coevorden 2004a; Veien 1995).
Ten studies included quality of life as an outcome parameter; especially more recent studies and studies in trial registries included quality of life. The extensively studied and validated Dermatology Life Quality Index (DLQI from Finlay 1994) was used in seven studies (Bauer 2012; Brass 2015; Chu 2009; Kircik 2013; Lodén 2012a; Ruzicka 2004; Yousefi 2012). The DLQI contains 10 questions regarding the impact of skin conditions and their treatment on participants' lives, which are answered on a scale ranging from 0 (not at all) to 3 (very much). A total DLQI score between 0 and 1 represents no or minimal effect at all on a participant's life. A DLQI score of 10 or greater represents significant impact on life quality, whereas a score over 21 represents an extremely large effect on quality of life. A change in DLQI score of at least 4 points is considered clinically relevant in inflammatory skin conditions (Basra 2008; Basra 2015).
Another quality of life questionnaire that was used is the Eczema Disability Index (EDI) (Granlund 1996). The EDI includes 15 questions representing different dimensions of quality of life on a scale from 0 (not at all) to 6 (very much). The Skindex‐29 was used by one study (Fowler 2014). Finally the Skindex‐17 was used in another study (NCT01231854). The Skindex‐17 is a dermatological health‐related quality of life instrument that is derived from the Skindex‐29 and includes only 17 items instead of 29, and a 3‐point scale for answers instead of a 5‐point scale. A high score on the Skindex represents the huge impact of a skin condition on quality of life.
Economic losses such as sick days or out‐of‐pocket expenses were rarely registered as outcome parameters and were not included as outcome parameters before 2004. NCT01231854 and Brass 2015 contained an extensive cost‐effectiveness analysis based on the EQ‐5D. In addition, out‐of‐pocket expenses were registered in NCT01231854. van Coevorden 2004a registered travel expenses and time off work for participants. Two studies included the influence of hand eczema on work impairment with the Work Productivity and Activity Impairment Questionnaire (WPAI) (Kircik 2013; NCT01231854).
Cosmetic acceptability was considered as another outcome parameter in four studies (Fowler 2005; Fredriksson 1975; Kucharekova 2003; Lauriola 2011).
One of our secondary outcomes was 'dose reduction' ‐ reduction in treatment dose per time unit or cumulative prescribed treatment dose. None of the included studies provided reproducible data regarding this outcome.
Funding
For many older studies, it is unclear who funded the study. More recent studies often declared funding for the study or clearly stated relationships with pharmaceutical companies. In total, 22 studies were funded by pharmaceutical industries or were (co‐)authored by employees of pharmaceutical companies (Bauer 2012; Belsito 2004; Bissonnette 2010; Bleeker 1989; Cherill 2000; Chu 2009; Fowler 2005; Fowler 2014; Granlund 1996; Gupta 1993; Hill 1998; Hordinsky 2010; Kircik 2013; Krejci‐Manwaring 2008; Lodén 2012a; Möller 1983; Ruzicka 2004; Ruzicka 2008; Uggeldahl 1986; Veien 1995; Veien 1999; Whitaker 1996). Thirteen studies were sponsored by governmental organisations, universities, or hospitals (Baskan 2005; Brass 2015; Faghihi 2008; Jowkar 2011; Jowkar 2014; Katsarou 2012; NCT01231854; Pacor 2006; Schnopp 2002; Sharma 2006; Tzaneva 2009; van Coevorden 2004a; Yousefi 2012).
Excluded studies
The excluded studies are summarised under Characteristics of excluded studies. The 11 excluded studies comprised studies that were excluded for different reasons such as:
study on 'slightly irritated hands' in employees, which we did not accept as being hand eczema (Berndt 2001);
quasi‐randomised study, or unclear whether the study was randomised (Aertgeerts 1985; Güler Özden 2004; HogenEsch 1998; Petering 2004; Rosén 1987; Zimmerman 1967);
study that did not examine hand eczema but rather colonisation with a bacterium ‐ Staphylococcus aureus (Grivcheva‐Panovska 2013);
study without a comparator (Zeichner 2018);
study on prevention of hand eczema after initial treatment of hand eczema (Gergovska 2017); and
study from which we were unable to extract separate data on hand eczema because the study combined data on hand and foot eczema (Chen 2015).
Ongoing studies
The search yielded eight ongoing studies whose content we have summarised under Characteristics of ongoing studies:
Three studies are focused on topical treatments.
NCT02664805: comparing the efficacy of twice daily applications of LEO 124249 ointment with LEO 124249 ointment vehicle for up to eight weeks for treatment of chronic hand eczema.
IRCT2014012916412N1: comparing the efficacy of pumpkin ointment twice daily with betamethasone ointment twice daily, and almond ointment twice daily and Eucerin ointment twice daily.
IRCT2017070922965N10: evaluating the effect of topical atorvastatin as adjuvant therapy for treatment of hand eczema.
One study is examining palmar botulinum toxin injections.
PACTR201704002194318: evaluating the efficacy and tolerability of botulinum toxin type A for treatment of hand eczema.
Four are exploring systemic treatments.
JPRN‐UMIN000003326: determining the effect of olopatadine on itching in hand eczema.
ISRCTN80206075: comparing alitretinoin 30 mg with PUVA twice weekly as first‐line treatment for severe chronic hand eczema.
NCT03026946: comparing the efficacy of alitretinoin 30 mg and cyclosporine for treatment for severe recurrent vesicular hand eczema.
NCT03026907: comparing alitretinoin 30 mg with azathioprine in severe non‐hyperkeratotic hand eczema.
Data from ongoing trials that have been completed at the time of the next update will be included in the review, if those results are available.
Studies awaiting classification
We added 20 records to Characteristics of studies awaiting classification. These include a lot of studies on different topical treatments such as hand creams and sanitisers for the treatment of hand eczema that were listed in different trial registries and, although they seem completed, results have never been posted and we were unable to obtain these results.
This section also contains studies that included different dermatoses among hand eczema, but for which we were unable to obtain separate data for hand eczema despite contacting the study investigator.
Risk of bias in included studies
Many studies were at high or unclear risk of bias in one or more components of trial design. We assessed only six studies as having low risk of bias in all components of trial design (Baskan 2005; Bauer 2012; Fowler 2005; Lindelöf 1987; Pacor 2006; Ruzicka 2004). Eight studies had only one unclear risk of bias with remaining domains rated as low risk (Bissonnette 2010; Fairris 1984; Fairris 1985; Fowler 2014; Kircik 2013; Ruzicka 2008; Sheehan‐Dare 1989; Yousefi 2012). We rated 29 studies as having high risk of bias in at least one domain (Adams 2007; Agarwal 2013; Bayerl 1999; Brass 2015; Burrows 1986; Cartwright 1987; Fredriksson 1975; Hanifin 2004; Hill 1998; Jowkar 2014; Katsarou 2012; Kemper 1998; King 1984; Krejci‐Manwaring 2008; Kucharekova 2003; Lauriola 2011; Möller 1983; NCT01231854; Odia 1996; Pigatto 1990; Said 2010; Schnopp 2002; Sharma 2006; Sjövall 1987; Thestrup‐Pedersen 2001; Tzaneva 2009; van Coevorden 2004a; Veien 1995; Veien 1999).
Further information can be found in the risk of bias tables for each included study and in Figure 2 and Figure 3.
2.
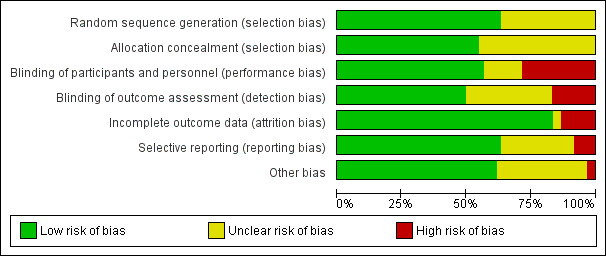
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
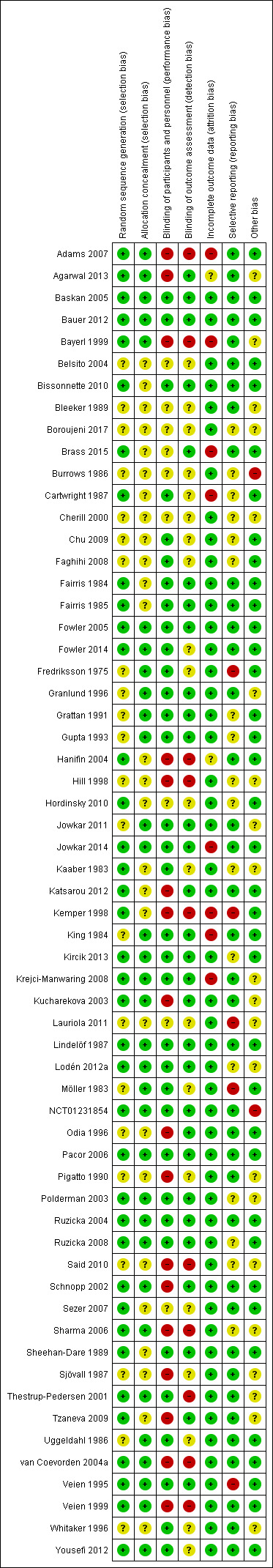
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Randomisation procedure
We judged the procedure as adequate (low risk of bias) when the allocation sequence was able to protect against biased allocation of comparison groups. If no details were given about the methods of sequence generation (i.e. if there was doubt about the adequacy of sequence generation), we judged studies as having unclear risk. We considered systematic methods that allow biased allocations, such as alternation or assignment based on day of admission, as inadequate (high risk of bias). References to a lottery system, throwing dice, or using a computer programme were considered as adequate (low risk of bias).
The randomisation procedure was unclear in 22 studies. For 25 studies, we judged the randomisation procedure as adequate based on the article (Adams 2007; Bauer 2012; Bayerl 1999; Cartwright 1987; Fairris 1984; Fairris 1985; Fowler 2014; Hanifin 2004; Hordinsky 2010; Kaaber 1983; Katsarou 2012; Kemper 1998; Kircik 2013; Krejci‐Manwaring 2008; Lindelöf 1987; Lodén 2012a; NCT01231854; Polderman 2003; Ruzicka 2004; Ruzicka 2008; Sezer 2007; Sheehan‐Dare 1989; Tzaneva 2009; van Coevorden 2004a; Yousefi 2012). For 13 studies, we were unable to base judgement on the article, but personal communication with study authors clarified that the randomisation procedure was adequate (Agarwal 2013; Baskan 2005; Bissonnette 2010; Brass 2015; Fowler 2005; Jowkar 2014; Kucharekova 2003; Pacor 2006; Schnopp 2002; Sharma 2006; Thestrup‐Pedersen 2001; Veien 1995; Veien 1999). In total, we judged the randomisation procedure as adequate in 38 studies.
Concealment of allocation
We judged this as adequate (low risk of bias) when clinicians and participants were unaware of future allocations before participants gave consent to the study. Examples of these include randomisation by a third party or use of sequentially numbered, opaque, sealed envelopes. We judged this as unclear risk of bias if insufficient details are given about methods of allocation concealment. We judged the allocation inadequate (high risk of bias) when there was a possibility of knowledge of the next assignment, so when investigators could have successfully guessed the allocation before the participant gave consent.
Of the above‐mentioned 38 studies with an appropriate randomisation procedure, concealment of allocation was adequate in 25 (Adams 2007; Agarwal 2013; Baskan 2005; Bauer 2012; Bayerl 1999; Fowler 2005; Fowler 2014; Jowkar 2014; Kircik 2013; Krejci‐Manwaring 2008; Kucharekova 2003; Lindelöf 1987; Lodén 2012a; NCT01231854; Pacor 2006; Polderman 2003; Ruzicka 2004; Ruzicka 2008; Schnopp 2002; Sharma 2006; Thestrup‐Pedersen 2001; van Coevorden 2004a; Veien 1995; Veien 1999; Yousefi 2012).
For eight studies, concealment of allocation was clear, but the randomisation procedure was unclear (Fredriksson 1975; Granlund 1996; Grattan 1991; Gupta 1993; Jowkar 2011; King 1984; Möller 1983; Uggeldahl 1986).
In total, in 33 studies the method used to conceal allocation was judged as adequate. In the remaining 27 studies, it is unclear if allocation was concealed.
Blinding
Performance bias
Performance bias refers to systematic differences between groups in the care provided, or in exposure to factors other than the interventions of interest (Higgins 2011a). After enrolment into the study, blinding of participants and site staff can reduce the risk that knowledge of which intervention was received, rather than the intervention itself, affects outcomes. Effective blinding can also ensure that the compared groups receive similar amounts of attention, ancillary treatment, and diagnostic investigation. With regards to performance bias, blinding of participants and of study personnel was judged separately. Use of identical looking study and control drugs (vehicle or placebo) was considered an adequate method of blinding, if the study was double‐blind.
If study authors made every attempt to blind the study to the best of their abilities, we judged the study as low risk. When, for example, the radiographer was the only person aware of treatment allocation but the study could not have been done in another way, we judged this as low risk of bias because we considered this the best possible way to minimise the risk of performance bias.
For 27 studies, both participants and staff were blinded in an adequate manner (Baskan 2005; Bauer 2012; Bissonnette 2010; Cartwright 1987; Chu 2009; Faghihi 2008; Fowler 2014; Fredriksson 1975; Granlund 1996; Grattan 1991; Gupta 1993; Jowkar 2011; Jowkar 2014; King 1984; Kircik 2013; Krejci‐Manwaring 2008; Lindelöf 1987; Lodén 2012a; NCT01231854; Pacor 2006; Ruzicka 2004; Ruzicka 2008; Sheehan‐Dare 1989; Thestrup‐Pedersen 2001; Veien 1995; Whitaker 1996; Yousefi 2012). Seven studies were only participant‐blinded (Fairris 1984; Fairris 1985; Fowler 2005; Kaaber 1983; Möller 1983; Polderman 2003; Uggeldahl 1986). Nine studies had unclear risk of blinding (Belsito 2004; Bleeker 1989; Boroujeni 2017; Brass 2015; Burrows 1986; Cherill 2000; Hordinsky 2010; Lauriola 2011; Sezer 2007).
For 17 studies, no blinding of participants was attempted, so the risk of performance bias was considered high (Adams 2007; Agarwal 2013; Bayerl 1999; Hanifin 2004; Hill 1998; Katsarou 2012; Kemper 1998; Kucharekova 2003; Odia 1996; Pigatto 1990; Said 2010; Schnopp 2002; Sharma 2006; Sjövall 1987; Tzaneva 2009; van Coevorden 2004a; Veien 1999). Some studies claimed a double‐blind design but this was not feasible because participants had to follow lifestyle interventions such as a low‐nickel diet (e.g. Pigatto 1990; Sharma 2006), or because treatment groups received completely different treatment such as whole‐body irradiation versus local radiation (e.g. Sjövall 1987).
Detection bias
'Detection bias' refers to systematic differences between groups in how outcomes are determined. Blinding of outcome assessors reduces the risk that knowledge of which intervention was received, rather than the intervention itself, affects outcome measurement (Higgins 2011a). We judged the procedure as having low risk of bias for detection when the outcome assessor was unaware of the allocation. When an article states only that the study was investigator‐blinded or double‐blinded, we considered this as too little information by which to judge the risk of bias and concluded that risk was unclear. We judged low risk of detection bias for studies that used independent observers, that received study drugs packed and dispensed by a third party, or that described another adequate method used to blind the observer.
The observer was blinded in an adequate manner in 30 studies (Agarwal 2013; Baskan 2005; Bauer 2012; Bissonnette 2010; Brass 2015; Fairris 1984; Fairris 1985; Fowler 2005; Granlund 1996; Grattan 1991; Gupta 1993; Jowkar 2011; Jowkar 2014; Katsarou 2012; King 1984; Kircik 2013; Krejci‐Manwaring 2008; Kucharekova 2003; Lindelöf 1987; Lodén 2012a; NCT01231854; Odia 1996; Pacor 2006; Polderman 2003; Ruzicka 2004; Ruzicka 2008; Schnopp 2002; Sheehan‐Dare 1989; Tzaneva 2009; Veien 1995).
Ten studies made no attempt to blind the observer (Adams 2007; Bayerl 1999; Hanifin 2004; Hill 1998; Kemper 1998; Said 2010; Sharma 2006; Thestrup‐Pedersen 2001; van Coevorden 2004a; Veien 1999). For the remaining 20 studies, it is unclear whether the observer was truly blinded.
Incomplete outcome data
We tried to minimise the quantity of missing data by contacting all study authors from 1999 forward. We contacted them through personal communication by email, letters, or social media features such as LinkedIn. We asked questions with regard to uncertainty in the assessment of risks of bias or trial design. We also tried to search other sources such as trial registries, which may provide additional information with regards to study design, or we compared the study to similar studies done by the same authors.
We judged attrition bias as low risk in 50 studies (Baskan 2005; Bauer 2012; Belsito 2004; Bissonnette 2010; Bleeker 1989; Boroujeni 2017; Burrows 1986; Cherill 2000; Chu 2009; Faghihi 2008; Fairris 1984; Fairris 1985; Fowler 2005; Fowler 2014; Fredriksson 1975; Granlund 1996; Grattan 1991; Gupta 1993; Hill 1998; Hordinsky 2010; Jowkar 2011; Kaaber 1983; Katsarou 2012; Kircik 2013; Kucharekova 2003; Lauriola 2011; Lindelöf 1987; Lodén 2012a; Möller 1983; NCT01231854; Odia 1996; Pacor 2006; Pigatto 1990; Polderman 2003; Ruzicka 2004; Ruzicka 2008; Said 2010; Schnopp 2002; Sezer 2007; Sharma 2006; Sheehan‐Dare 1989; Sjövall 1987; Thestrup‐Pedersen 2001; Tzaneva 2009; Uggeldahl 1986; van Coevorden 2004a; Veien 1995; Veien 1999; Whitaker 1996; Yousefi 2012). We judged attrition bias as unclear in two studies (Agarwal 2013; Hanifin 2004), and as high in eight studies (Adams 2007; Bayerl 1999; Brass 2015; Cartwright 1987; Jowkar 2014, Kemper 1998; King 1984; Krejci‐Manwaring 2008).
Loss to follow‐up and intention‐to‐treat (ITT) analysis
We judged the risk of attrition bias (incomplete outcome bias) as adequate (low risk of bias) when more than 80% of participants were followed up and analysed in the groups to which they were originally randomised. In addition, we considered an intention‐to‐treat (ITT) analysis as having low risk of bias for the attrition bias. When more than 20% of participants dropped out and no ITT analysis was carried out, we considered the study element to have high risk of bias.
A total of 13 studies reported no dropouts (Cherill 2000; Faghihi 2008; Fredriksson 1975; Katsarou 2012; Lauriola 2011; Lodén 2012a; Odia 1996; Pacor 2006; Pigatto 1990; Schnopp 2002; Sharma 2006; Thestrup‐Pedersen 2001; Veien 1999).
Despite loss of participants during follow‐up, data were analysed according to the ITT analysis principle in 14 studies (Bauer 2012; Belsito 2004; Bissonnette 2010; Chu 2009; Fowler 2014; Granlund 1996; Hill 1998; Hordinsky 2010; Kircik 2013; Polderman 2003; Ruzicka 2004; Ruzicka 2008; van Coevorden 2004a; Veien 1995). NCT01231854 included an ITT analysis but included only 14 of the 78 planned participants due to early termination.
For 23 studies, at least 80% of participants were followed up and were included in the analyses (Agarwal 2013; Baskan 2005; Bleeker 1989; Boroujeni 2017; Burrows 1986; Fairris 1984; Fairris 1985; Fowler 2005; Grattan 1991; Gupta 1993; Jowkar 2011; Kaaber 1983; Kucharekova 2003; Lindelöf 1987; Möller 1983; Said 2010; Sezer 2007; Sheehan‐Dare 1989; Sjövall 1987; Tzaneva 2009; Uggeldahl 1986; Whitaker 1996; Yousefi 2012).
The highest dropout rates were, respectively, 40%, 39%, 37%, and 37% (Cartwright 1987 ; Krejci‐Manwaring 2008; Kemper 1998; Jowkar 2014). Eight studies analysed less than 80% (Adams 2007; Bayerl 1999; Brass 2015; Cartwright 1987; Jowkar 2014, Kemper 1998; King 1984; Krejci‐Manwaring 2008).
For one study, it is unclear how many participants were analysed because the text states that less than 80% finished the protocol; however all participants seem to have been analysed without mention of ITT analyses (Hanifin 2004).
Selective reporting
We found a total 38 studies that we judged as having low risk of bias (Adams 2007; Agarwal 2013; Baskan 2005; Bauer 2012; Bayerl 1999; Belsito 2004; Bissonnette 2010; Bleeker 1989; Brass 2015; Fairris 1984; Fairris 1985; Fowler 2005; Fowler 2014; Granlund 1996; Hanifin 2004; Jowkar 2011; Jowkar 2014; Katsarou 2012; King 1984; Krejci‐Manwaring 2008; Kucharekova 2003; Lindelöf 1987; NCT01231854; Odia 1996; Pacor 2006; Pigatto 1990; Ruzicka 2004; Schnopp 2002; Sezer 2007; Sheehan‐Dare 1989; Sjövall 1987; Thestrup‐Pedersen 2001; Tzaneva 2009; Uggeldahl 1986; van Coevorden 2004a; Veien 1999; Whitaker 1996; Yousefi 2012). We judged 17 studies as having unclear risk of reporting bias (Boroujeni 2017; Burrows 1986; Cartwright 1987; Cherill 2000; Chu 2009; Faghihi 2008; Grattan 1991; Gupta 1993; Hill 1998; Hordinsky 2010; Kaaber 1983; Kircik 2013; Lodén 2012a; Polderman 2003; Ruzicka 2008; Said 2010; Sharma 2006), and five as having high risk of reporting bias (Fredriksson 1975; Kemper 1998; Lauriola 2011; Möller 1983; Veien 1995). High risk of bias was assigned whenever we found severe discrepancies between the Materials and Methods section and the study protocol and Results section, when the stated primary outcome was neglected (Fredriksson 1975), and when significance levels were reached on subscores or on other scores, or were not stated at all (Kemper 1998; Lauriola 2011; Möller 1983). For example, Fredriksson 1975 used an unclear severity scale ranging from 0 to 5 and did not state the results of this outcome at all.
Many, especially older studies, did not register before commencement of the trial, so that the correspondence between actually reported outcomes and outcomes intended to be reported could not be assessed for most included studies. We found trial registration for 13 studies (Adams 2007; Bauer 2012; Bissonnette 2010; Brass 2015; Chu 2009; Fowler 2014; Hordinsky 2010; Jowkar 2011; Kircik 2013; Lodén 2012a; Ruzicka 2008; NCT01231854; Yousefi 2012), and we found discrepancies in Chu 2009,Hordinsky 2010, Kircik 2013,Lodén 2012a, and Ruzicka 2008 with regards to additional or missing outcome parameters. We found no major discrepancies between protocol and report in eight studies (Adams 2007; Bauer 2012; Bissonnette 2010; Brass 2015; Fowler 2014; Jowkar 2011; NCT01231854; Yousefi 2012), although one study was registered two years after the recruitment start date (Yousefi 2012).
For the other studies, we examined discrepancies between the Materials and Methods section and the Results section and noted no major discrepancies in most (Agarwal 2013; Baskan 2005; Bayerl 1999; Belsito 2004; Bleeker 1989; Fairris 1984; Fairris 1985; Fowler 2005; Granlund 1996; Hanifin 2004; Jowkar 2014; Katsarou 2012; King 1984; Krejci‐Manwaring 2008; Kucharekova 2003; Lindelöf 1987; Odia 1996; Pacor 2006; Pigatto 1990; Schnopp 2002; Sezer 2007; Sheehan‐Dare 1989; Sjövall 1987; Thestrup‐Pedersen 2001; Tzaneva 2009; Uggeldahl 1986; van Coevorden 2004a; Veien 1999; Whitaker 1996), although in some studies we did find severe discrepancies, mainly involving missing information (Fredriksson 1975; Kemper 1998; Lauriola 2011; Möller 1983; Veien 1995).
Other potential sources of bias
Baseline comparison for severity of disease and diagnostic certainty
When assessing other potential sources of bias, we considered several aspects, namely, baseline balance for severity of disease and/or participants, diagnostic certainty, and whether the study was completed or ended prematurely.
Diagnostic certainty, meaning that the diagnosis was confirmed by a physician, was applicable to almost all studies. We judged this as low risk in all but one study (Said 2010).
For within‐participant studies, we considered a baseline comparison dispensable. This was true for 18 within‐participant studies (i.e. having a left‐right design, comparing one hand with the other) (Adams 2007; Baskan 2005; Cartwright 1987; Chu 2009; Faghihi 2008; Fairris 1984; Fairris 1985; Fredriksson 1975; Grattan 1991; Kemper 1998; King 1984; Lindelöf 1987; Möller 1983; Odia 1996; Schnopp 2002; Sezer 2007; Sheehan‐Dare 1989; Uggeldahl 1986). For three studies, we found significant differences at baseline and therefore declared them as having unclear risk of bias (Granlund 1996; Hill 1998; Krejci‐Manwaring 2008). Sixteen studies did not state baseline comparisons (Agarwal 2013; Bayerl 1999; Boroujeni 2017; Burrows 1986; Cherill 2000; Kaaber 1983; Kucharekova 2003; Lauriola 2011; Lodén 2012a; Pigatto 1990; Polderman 2003; Said 2010; Sharma 2006; Sjövall 1987; Thestrup‐Pedersen 2001; Tzaneva 2009), and these were unclear in three studies (Bleeker 1989; Jowkar 2011; Whitaker 1996). For the remaining studies, baseline comparisons were clearly stated (Adams 2007; Bauer 2012; Belsito 2004; Bissonnette 2010; Brass 2015; Fowler 2005; Fowler 2014; Gupta 1993; Hanifin 2004; Hordinsky 2010; Jowkar 2014; Katsarou 2012; Kircik 2013; NCT01231854; Pacor 2006; Ruzicka 2004; Ruzicka 2008; van Coevorden 2004a; Veien 1995; Veien 1999; Yousefi 2012).
Studies ending prematurely
Two studies were ended prematurely (Burrows 1986; NCT01231854). Burrows 1986 ended because teratogenicity in rats was reported during the study, and NCT01231854 ended because the investigator‐initiated study was unable to include the planned number of participants. For this, we judged high risk of bias.
In total, we judged the risk of other potential sources of bias as high in two studies, unclear in 21 studies, and low in the remaining 37 studies.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8
In the text below, where it has been possible to calculate an effect size, we have reported these with 95% confidence intervals. If no data were available for these analyses, we removed the result from this section and mentioned this in Table 9, Table 10, Table 11, Table 12, Table 13, Table 14, Table 15, Table 16, Table 17, and Table 18 for the primary outcome investigator‐rated good/excellent control.
We considered statistical pooling, but the studies were too heterogeneous in terms of design, types of particular treatment compared, assessment of outcomes, duration of the trial, and presentation of data. The exceptions were two studies that compared 10 mg alitretinoin as active treatment versus placebo (Ruzicka 2004; Ruzicka 2008), along with studies regarding topical calcineurin inhibitors with regard to adverse events (Bauer 2012; Belsito 2004; Hordinsky 2010). Beside results of the pooled analysis, these studies also reported heterogeneity statistics. We considered, for example, pooling Brass 2015 and Sezer 2007, although treatment intensity (twice weekly versus thrice weekly) and study duration were too different. Moreover, only limited information was available on Brass 2015. Finally, Sezer 2007 was a within‐participant study, and Brass 2015 was a parallel‐group study.
In the additional tables section, we have tabulated the primary outcome investigator‐rated good/excellent control for the different categories of interventions, that is, corticosteroids, irradiation with UV light, and irradiation with X‐rays (respectively, Table 10Table 12 and Table 13).
I. Skin protection measures, including gloves
We identified no randomised controlled trials.
II. Topical treatments
Comparison 1. Bland emollients: ceramide‐containing emollients
A comparison was made between an emollient with ceramides (Locobase Repair) in 17 participants versus a regular petrolatum‐based emollient (Vaseline‐lanette) in 15 participants (Kucharekova 2003). Results were mainly depicted in graphic presentation (bar diagrams), and exact numbers cannot be extracted.
Primary outcome: adverse events
One participant in the ceramide group experienced an exacerbation of hand dermatitis, as did two participants in the comparison group (risk ratio (RR) 0.44, 95% confidence interval (CI) 0.04 to 4.39; P = 0.49; Analysis 1.1). This showed no clear difference between groups.
1.1. Analysis.
Comparison 1 Bland emollients: ceramide‐containing emollient versus regular petrolatum‐based emollient, Outcome 1 Primary: adverse events: exacerbation resulting in dropout.
Comparison 2. Bland emollients: emollient E‐DO versus vehicle
Primary outcome: percentage of participants with self‐rated good/excellent control at week 4
Chu 2009 conducted a within‐participant study. After four weeks, the percentage of hands with a self‐rated reduction of at least 50% on the participants' global assessment (PaGA) was 34.92% (22 hands) in the E‐DO group, and 36.51% (23 hands) in the vehicle group (reported as 'other data'; see Analysis 2.1).
2.1. Analysis.
Comparison 2 Bland emollients: emollient E‐DO versus vehicle, Outcome 1 Primary: percentage of participants with self‐rated good/excellent control at week 4.
| Primary: percentage of participants with self‐rated good/excellent control at week 4 | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Total number of events | Total number of pairs of hands analysed |
| Chu 2009 | Emollient E‐DO | 22 | 67 |
| Chu 2009 | Vehicle | 23 | 67 |
Primary outcome: percentage of participants with investigator‐rated good/excellent control at week 4
We identified one within‐participant study on E‐DO (Chu 2009). After four weeks, 12 (19.0%) hands responded to E‐DO only, 11 (17.5%) responded to vehicle only, and 25 (39.7%) responded to both. The overall response rate was 37 (58.73%) E‐DO hands and 36 (57.14%) vehicle hands (reported as 'other data'; see Analysis 2.2). Also see Table 9.
2.2. Analysis.
Comparison 2 Bland emollients: emollient E‐DO versus vehicle, Outcome 2 Primary: percentage of participants with investigator‐rated good/excellent control at week 4.
| Primary: percentage of participants with investigator‐rated good/excellent control at week 4 | |||
|---|---|---|---|
| Study | Group‐ within‐participant study | Event number | Total number of pairs of hands randomised |
| Chu 2009 | Emollient E‐DO | 37 | 67 |
| Chu 2009 | Vehicle | 36 | 67 |
Primary outcome: adverse events
At least one adverse event was reported by 19.4% of participants; 12 occurred on the E‐DO hand (17.9%), and eight (11.9%) on the vehicle hand. Prurirtus was recorded for six E‐DO hands and for two vehicle hands (reported as 'other data'; see Analysis 2.3). No serious adverse events were reported.
2.3. Analysis.
Comparison 2 Bland emollients: emollient E‐DO versus vehicle, Outcome 3 Adverse events.
| Adverse events | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of hands pairs of analysed |
| At least 1 adverse event | |||
| Chu 2009 | Emollient E‐DO | 12 | 67 |
| Chu 2009 | Vehicle | 8 | 67 |
| Pruritus | |||
| Chu 2009 | Emollient E‐DO | 6 | 67 |
| Chu 2009 | Vehicle | 2 | 67 |
Comparison 3. Corticosteroid creams or ointments: fluprednidene acetate cream versus betamethasone‐valerate
For an overview of studies on topical corticosteroids for the outcome investigator‐rated good/excellent control, see the additional tables section (Table 10). That overview presents only the primary outcomes as defined according to our protocol (i.e. participant‐ or investigator‐rated good or excellent control); consequently, this does not necessarily reflect the primary outcome that may be stated in the study report.
The point estimates (RRs) and confidence intervals (CIs), if available, are based on the per‐protocol evaluation of participants, and are not based on an ITT analysis, unless stated otherwise. We considered statistical pooling, but the studies were too heterogeneous in terms of design, types of corticosteroid, assessment of outcomes, and presentation of data.
Primary outcome: percentage of participants with investigator‐rated good/excellent control (healed) after three weeks of treatment
In a parallel study (Bleeker 1989), 14 out of 38 participants in the betamethasone group healed, as did 8 of 37 in the fluprednidene group. There was no clear difference between groups (RR 0.59, 95% CI 0.28 to 1.23; Analysis 3.1;Table 10).
3.1. Analysis.
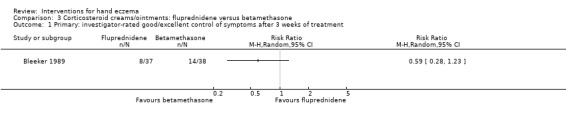
Comparison 3 Corticosteroid creams/ointments: fluprednidene versus betamethasone, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms after 3 weeks of treatment.
Primary outcome: adverse events
Eight participants in the betamethasone group and seven in the fluprednidene group reported adverse events such as redness, smarting, swelling, irritation, or dryness (RR 0.90, 95% CI 0.36 to 2.23; Analysis 3.2), showing no clear differences between groups.
3.2. Analysis.
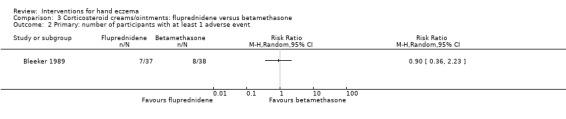
Comparison 3 Corticosteroid creams/ointments: fluprednidene versus betamethasone, Outcome 2 Primary: number of participants with at least 1 adverse event.
Secondary outcome: reduction in severity, investigator‐rated scoring improvement > 50% after three weeks
After three weeks of treatment, 23 of 38 participants in the betamethasone group and 27 of 37 participants in the fluprednidene group showed improvement greater than 50% (Bleeker 1989). There were no clear differences between groups (RR 1.21, 95% CI 0.87 to 1.67; Analysis 3.3).
3.3. Analysis.
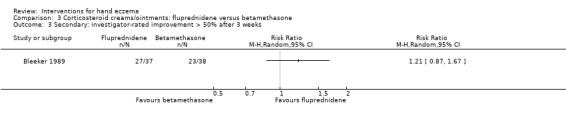
Comparison 3 Corticosteroid creams/ointments: fluprednidene versus betamethasone, Outcome 3 Secondary: investigator‐rated improvement > 50% after 3 weeks.
Comparison 4. Corticosteroid creams/ointments: betamethasone‐dipropionate film‐forming lotion versus betamethasone‐dipropionate thick lotion
For this comparison, we found only one relevant trial (Gupta 1993).
Primary outcome: percentage of participants with investigator‐rated good/excellent control of symptoms at day 7
Five of the 28 people in the betamethasone‐dipropionate film‐forming lotion group achieved good/excellent symptom control compared to zero of 26 in the control group. Fisher's exact test results in a P value of 0.051 (Analysis 4.1; Table 10).
4.1. Analysis.
Comparison 4 Corticosteroid creams/ointments: betamethasone‐dipropionate film‐forming lotion versus betamethasone‐dipropionate thick lotion, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms at day 7.
| Primary: investigator‐rated good/excellent control of symptoms at day 7 | |||
|---|---|---|---|
| Study | Group | Number of participants with good/excellent control | Total number of participants |
| Gupta 1993 | B‐film forming lotion | 5 | 28 |
| Gupta 1993 | B‐thick lotion | 0 | 26 |
Primary outcome: adverse events
No clear difference was found in relation to the occurrence of at least one adverse event (RR 1.33, 95% CI 0.33 to 5.44; Analysis 4.2).
4.2. Analysis.
Comparison 4 Corticosteroid creams/ointments: betamethasone‐dipropionate film‐forming lotion versus betamethasone‐dipropionate thick lotion, Outcome 2 Primary: adverse events.
In the betamethasone‐dipropionate film‐forming lotion group, two participants had stinging at the application site, one stinging in the eyes when opening the bottle close to the face, and one a "melting" feeling during the sauna visit compared to no application site reactions in the control group (Fisher's exact test P = 0.11). In the thickened lotion group, one participant experienced headache (probably not related to the study drug), and two had an exacerbation of hand eczema, compared to none in the control group (Fisher's exact test P = 1.00 and 0.49; also see Analysis 4.2). Zero events were reported in one of the arms for all of these three subgroups, and the confidence interval around the point of estimate was wide.
Secondary outcome: reduction in severity, investigator‐rated scoring (not specified), and in overall severity at day 7
At day 7, 23 out of 28 participants in the film‐forming group improved compared to 10 out of 26 participants in the thickened lotion group. There may be a difference between corticosteroid creams/ointments in favour of the betamethasone‐dipropionate film‐forming lotion at day 7 (RR 2.30, 95% CI 1.35 to 3.93; number needed to treat for an additional beneficial outcome (NNTB) 2, 95% CI 1 to 5; Analysis 4.3).
4.3. Analysis.
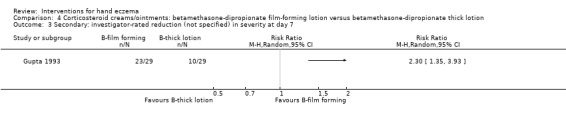
Comparison 4 Corticosteroid creams/ointments: betamethasone‐dipropionate film‐forming lotion versus betamethasone‐dipropionate thick lotion, Outcome 3 Secondary: investigator‐rated reduction (not specified) in severity at day 7.
Secondary outcome: reduction in severity, investigator‐rated global improvement, of eczema
The global comparison between treatments at day 7 showed improved eczema in 23 of 28 participants treated with polyacrylic film‐forming lotion versus 18 of 26 participants treated with thickened lotion (RR 1.19, 95% CI 0.87 to 1.62; Analysis 4.4), indicating no clear difference in improvement between the two treatments.
4.4. Analysis.
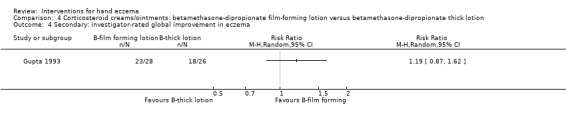
Comparison 4 Corticosteroid creams/ointments: betamethasone‐dipropionate film‐forming lotion versus betamethasone‐dipropionate thick lotion, Outcome 4 Secondary: investigator‐rated global improvement in eczema.
Comparison 5. Corticosteroid creams/ointments: clobetasol propionate cream versus intermittent fluprednidene acetate cream
Primary outcome: percentage of participants with investigator‐rated good/excellent control
No relapses were observed in 32 of 46 (70%) hands treated with clobetasol propionate cream and in 14 of 46 (30%) hands treated with fluprednidene acetate cream (Möller 1983). This is reported as 'other data'; see Analysis 5.1 and Table 10.
5.1. Analysis.
Comparison 5 Corticosteroids creams/ointments: clobetasol propionate versus flupredniden acetate, Outcome 1 Primary: percentage of participants with investigator‐rated good/excellent control.
| Primary: percentage of participants with investigator‐rated good/excellent control | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Möller 1983 | Clobetasol | 32 | 46 |
| Möller 1983 | Flupredniden | 14 | 46 |
Primary outcome: adverse events
Adverse events occurred in four participants treated with clobetasol and in three participants treated with fluprednidene (reported as 'other data' in Analysis 5.2). One participant reported an adverse event from both glucocorticoids.
5.2. Analysis.
Comparison 5 Corticosteroids creams/ointments: clobetasol propionate versus flupredniden acetate, Outcome 2 Adverse events.
| Adverse events | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| At least 1 adverse event | |||
| Möller 1983 | Clobetasol | 4 | 46 |
| Möller 1983 | Flupredniden | 3 | 46 |
| Burning sensation | |||
| Möller 1983 | Clobetasol | 2 | 46 |
| Möller 1983 | Flupredniden | 2 | 46 |
| Reversible atrophy | |||
| Möller 1983 | Clobetasol | 1 | 46 |
| Möller 1983 | Flupredniden | 0 | 46 |
| Brittle skin | |||
| Möller 1983 | Clobetasol | 1 | 46 |
| Möller 1983 | Flupredniden | 1 | 46 |
Comparison 6. Corticosteroid creams/ointments: clobetasol propionate foam 0.05% versus vehicle foam
This comparison included one study of 125 participants (Kircik 2013).
Primary outcome: percentage of participants with investigator‐rated good/excellent control at day 15
In the clobetasol group, 38.7% (24/62 participants) had an Investigator Static Global Assessment (ISGA) score of 0 or 1 versus 27% (17/63 participants) in the vehicle group. There was no clear difference between groups (RR 1.43, 95% CI 0.86 to 2.40; Analysis 6.1;Table 1; Table 10).
6.1. Analysis.
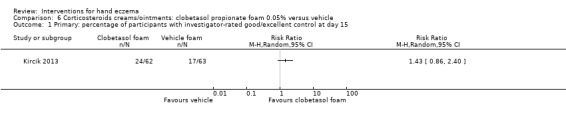
Comparison 6 Corticosteroids creams/ointments: clobetasol propionate foam 0.05% versus vehicle, Outcome 1 Primary: percentage of participants with investigator‐rated good/excellent control at day 15.
Primary outcome: percentage of participants with self‐rated good/excellent control at day 15
At the end of the study on day 15, 51.6% (32/62 participants) in the clobetasol group graded their hand eczema as clear or almost clear versus 22.2% (14/63 participants) in the vehicle group using the subject's global assessment (SGA). The relative risk of 2.32 favours clobetasol propionate foam over vehicle (RR 2.32, 95% CI 1.38 to 3.91; NNTB 3, 95% CI 2 to 8; Analysis 6.2; Table 1).
6.2. Analysis.
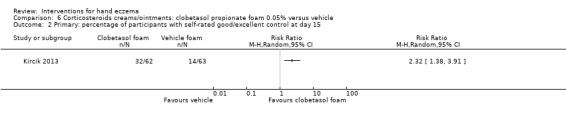
Comparison 6 Corticosteroids creams/ointments: clobetasol propionate foam 0.05% versus vehicle, Outcome 2 Primary: percentage of participants with self‐rated good/excellent control at day 15.
Primary outcome: adverse events
Adverse events were reported in 18% (11/62) of participants in the clobetasol propionate foam group and in 8% (5/63) of those in the vehicle foam group (RR 2.24, 95% CI 0.82 to 6.06; Analysis 6.3; Table 1). One participant reported application site burning/pruritus after clobetasol foam application. Three participants in the clobetasol group reported nasopharyngitis compared to one participant in the control group (RR 3.05, 95% CI 0.33 to 28.52; Analysis 6.3). No serious adverse events were reported in the clobetasol propionate foam group, and one participant in the vehicle group discontinued due to severe fissures (RR 0.34, 95% CI 0.01 to 8.16; Analysis 6.3). The wide confidence interval in this case could in part be the result of zero events in the clobetasol propionate foam group.
6.3. Analysis.
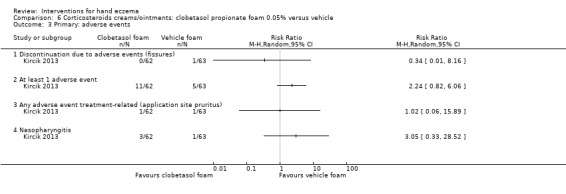
Comparison 6 Corticosteroids creams/ointments: clobetasol propionate foam 0.05% versus vehicle, Outcome 3 Primary: adverse events.
Secondary outcome: reduction in severity, participant‐rated scoring, at day 15
At the end of treatment, 51 out of 62 participants (82.3%) had at least one grade improvement in SGA score, compared to 33 out of 63 participants (52.4%) in the vehicle group (RR 1.57, 95% CI 1.21 to 2.04; NNTB 3, 95% CI 2.2 to 7; Analysis 6.4; Table 1). This difference is statistically significant, but we have reduced confidence in it is clinical significance due to small sample size and limitation in study design.
6.4. Analysis.
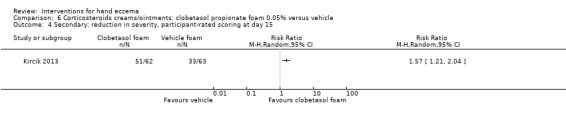
Comparison 6 Corticosteroids creams/ointments: clobetasol propionate foam 0.05% versus vehicle, Outcome 4 Secondary: reduction in severity, participant‐rated scoring at day 15.
Secondary outcome: reduction in severity, investigator‐rated scoring, at day 15
In Kircik 2013, 26 out of 62 participants (41.9%) in the clobetasol group versus 18 out of 63 (28.6%) in the control group improved by two grades or more in ISGA score. There was no clear difference between groups (RR 1.47, 95% CI 0.90 to 2.39; Analysis 6.5).
6.5. Analysis.
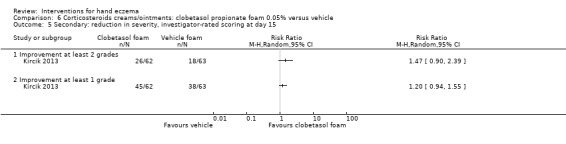
Comparison 6 Corticosteroids creams/ointments: clobetasol propionate foam 0.05% versus vehicle, Outcome 5 Secondary: reduction in severity, investigator‐rated scoring at day 15.
In total, 45 participants (72.6%) in the clobetasol group versus 38 (60.3%) in the control group improved by at least one grade in ISGA score after 15 days of treatment. Again, there was no clear difference between groups (RR 1.20, 95% CI 0.94 to 1.55; Analysis 6.5).
Comparison 7. Corticosteroid creams/ointments: desonide cream 0.1% versus desonide cream 0.05%
Two strengths of the same topical corticosteroid were compared in a study using a within‐participant (left/right) design (Uggeldahl 1986). Forty‐six participants were treated twice daily with desonide (Tridesilone) cream 0.1% on one hand and desonide (Apolar) cream 0.05% on the other hand. These participants had not been treated for eczema for at least one week before the study. The duration of the study was only 14 days.
Primary outcome: adverse events
Two participants reported stinging upon application of desonide 0.05% cream (reported as 'other data'; Analysis 7.1).
7.1. Analysis.
Comparison 7 Corticosteriods creams/ointments: desonide cream 0.1% versus desonide cream 0.05%, Outcome 1 Adverse events.
| Adverse events | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Uggeldahl 1986 | Desonide cream 0.1% | 0 | 50 |
| Uggeldahl 1986 | Desonide cream 0.05% | 2 | 50 |
Comparison 8. Corticosteroid creams/ointments: intermittent treatment with topical mometasone furoate at different frequencies (long term)
Veien 1999 included participants with chronic hand eczema that had cleared upon daily treatment for a maximum of 9 weeks with mometasone furoate cream.
Primary outcome: percentage of participants with investigator‐rated good/excellent control
Mometasone furoate 3 times/week versus mometasone furoate 2 times/week
For this subgroup, we found only one relevant trial (n = 72) (Veien 1999). Among participants treated with mometasone three times a week, 29 out of 35 (83%) had no recurrences, compared to 25 out of 37 (68%) of those treated with mometasone two times a week. Mometasone furoate cream used thrice weekly may slightly improve investigator‐rated good/excellent control of symptoms when compared to twice weekly application; however, the 95% confidence interval does include 1 (RR 1.23, 95% CI 0.94 to 1.61; Analysis 8.1; Table 2Table 10).
8.1. Analysis.
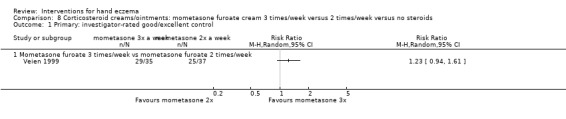
Comparison 8 Corticosteroid creams/ointments: mometasone furoate cream 3 times/week versus 2 times/week versus no steroids, Outcome 1 Primary: investigator‐rated good/excellent control.
Mometasone furoate 3 times/week versus emollients only
For this subgroup, we found only one relevant trial (n = 69) (Veien 1999). We noted a clear difference between corticosteroid creams/ointments: mometasone furoate cream 3 times weekly versus no steroids (RR 3.13, 95% CI 1.75 to 5.59; NNTB 2, 95% CI 1 to 3; Analysis 8.2;Table 10), but the difference may not be clinically significant due to imprecision of results caused by small sample size and limitations in study design/conduct.
8.2. Analysis.
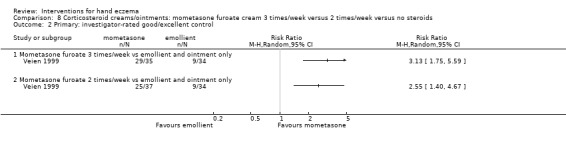
Comparison 8 Corticosteroid creams/ointments: mometasone furoate cream 3 times/week versus 2 times/week versus no steroids, Outcome 2 Primary: investigator‐rated good/excellent control.
Mometasone furoate 2 times/week versus emollients only
For this subgroup, we found only one relevant trial (n = 71) (Veien 1999). There was a statistically significant difference between corticosteroid creams/ointments: mometasone furoate cream 2 times weekly versus no steroids (RR 2.55, 95% CI 1.40 to 4.67; NNTB 2, 95% CI 2 to 5; Analysis 8.2), i.e. mometasone furoate twice a week was better than emollient only, but again, the difference may not be clinically significant due to imprecision of results caused by small sample size and limitations in study design/conduct.
Primary outcome: adverse events
Mometasone furoate 3 times/week versus mometasone furoate 2 times/week
In 10 participants, mild skin atrophy was noted at some point during the study. In five participants, atrophy disappeared during the study, and five participants had mild atrophy at the end of the study. The difference between groups was not clear (RR 1.76, 95% CI 0.45 to 6.83; Analysis 8.3; Table 2).
8.3. Analysis.
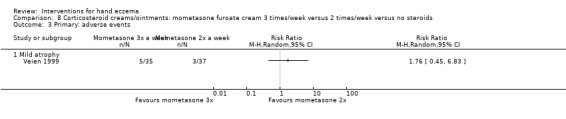
Comparison 8 Corticosteroid creams/ointments: mometasone furoate cream 3 times/week versus 2 times/week versus no steroids, Outcome 3 Primary: adverse events.
Comparison 9. Corticosteroid creams/ointments: 0.05% clobetasol and 2.5% zinc sulphate cream versus 0.05% clobetasol cream
Faghihi 2008 investigated whether zinc sulphate added to clobetasol cream is effective in the treatment of chronic hand eczema (n = 47 hands).
Primary outcome: percentage of participants with investigator‐rated good/excellent control
After eight weeks, 25 out of 47 hands (53%) treated with clobetasol + zinc sulphate cream were clear from scaling compared to three hands (6%) treated with clobetasol cream alone (presented as 'other data' in Analysis 9.1;Table 10). Regarding erythema, 41 hands (87%) treated with clobetasol + zinc sulphate cream were clear after eight weeks compared to one hand (2%) treated with clobetasol cream alone (reported as 'other data' in Analysis 9.1). Last, 24 hands (51%) treated with clobetasol + zinc sulphate cream and seven hands (15%) treated with clobetasol cream alone were clear of lichenification (reported as 'other data' in Analysis 9.1). Absence of scaling, redness, and/or lichenification was seen as investigator‐rated good/excellent control. An overall assessment of good/excellent control was not possible.
9.1. Analysis.
Comparison 9 Corticosteroid creams/ointments: clobetasol and zinc sulphate cream versus clobetasol cream, Outcome 1 Primary outcome: percentage of participants with investigator‐rated good/excellent control.
| Primary outcome: percentage of participants with investigator‐rated good/excellent control | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Scaling | |||
| Faghihi 2008 | Clobetasol cream | 3 | 47 |
| Faghihi 2008 | Clobetasol cream Clobetasol & Zinc sulphate cream | 25 | 47 |
| Redness | |||
| Faghihi 2008 | Clobetasol cream | 1 | 47 |
| Faghihi 2008 | Clobetasol cream Clobetasol & Zinc sulphate cream | 41 | 47 |
| Lichenification | |||
| Faghihi 2008 | Clobetasol cream | 7 | 47 |
| Faghihi 2008 | Clobetasol cream Clobetasol & Zinc sulphate cream | 24 | 47 |
Primary outcome: adverse events
Treatments were well tolerated and no significant adverse events were reported or observed by participants in both groups. Trial authors concluded that treatments were generally well tolerated (no exact data given).
Comparison 10. Corticosteroid creams/ointments: betamethasone‐valerate 0.1% cream twice daily versus betamethasone‐valerate 0.1% cream and urea 5% cream
Lodén 2012a compared the application of betamethasone‐valerate 0.1% cream twice daily versus the application of betamethasone‐valerate 0.1% cream in the morning and a moisturiser containing urea 5% cream in the evening.
Primary outcome: percentage of participants with investigator‐rated good/excellent control (cleared)
For this outcome, we found only one relevant trial (Lodén 2012a; n = 44). Clearance was defined as a score ≤ 3 on the HEES. There was no clear difference between betamethasone‐valerate 0.1% cream (15 out of 22) and urea 5% cream (20 out of 22) (RR 0.75, 95% CI 0.55 to 1.03; Analysis 10.1;Table 10).
10.1. Analysis.
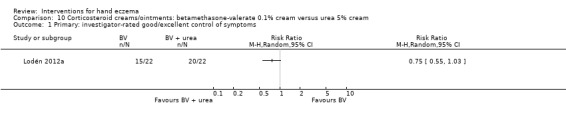
Comparison 10 Corticosteroid creams/ointments: betamethasone‐valerate 0.1% cream versus urea 5% cream, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms.
Secondary outcome: reduction in severity, participant‐rated
For this outcome, we found only one relevant trial (Lodén 2012a; n = 44). The average reduction in VAS (mm) was 36.3 in the betamethasone‐valerate 0.1% cream (BV) twice daily treatment group compared with 54.0 in the betamethasone‐valerate and urea (BV + M) group. The mean difference with regards to the VAS score was ‐17.70, although the relatively wide confidence interval did borderline include zero; therefore the results should be interpreted with caution (mean difference (MD) ‐17.70, 95% CI ‐35.42 to 0.02; Analysis 10.2).
10.2. Analysis.
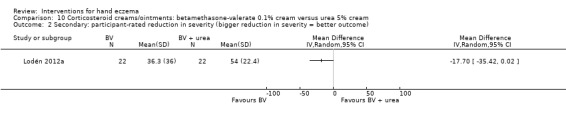
Comparison 10 Corticosteroid creams/ointments: betamethasone‐valerate 0.1% cream versus urea 5% cream, Outcome 2 Secondary: participant‐rated reduction in severity (bigger reduction in severity = better outcome).
Secondary outcome: reduction in severity, investigator‐rated
For this outcome, we found only one relevant trial (Lodén 2012a; n = 44). After two weeks, the average reduction in HEES was 12.5 (standard deviation (SD) 13.9) in the BV group compared to 10.5 (SD 9.0) in the BV + M group. There was no clear difference between groups (MD 2.00, 95% CI ‐4.92 to 8.92; Analysis 10.3).
10.3. Analysis.
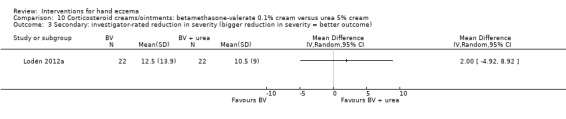
Comparison 10 Corticosteroid creams/ointments: betamethasone‐valerate 0.1% cream versus urea 5% cream, Outcome 3 Secondary: investigator‐rated reduction in severity (bigger reduction in severity = better outcome).
Comparison 11. Topical others: coal tar paste versus betamethasone‐valerate ointment 0.1% versus zinc oxide paste
In an unblinded randomised within‐participant study (Kemper 1998), the efficacy of pix lithanthracis (coal tar paste) compared to zinc oxide paste and betamethasone‐valerate was investigated (n = 19). Also see Table 11.
Primary outcome: adverse events
Six participants dropped out because they experienced problems with wearing gloves (the specific type of problem is not identified). One participant dropped out due to pompholyx as a result of allergy to 5% pix lithanthracis (reported as 'other data' in Analysis 11.1).
11.1. Analysis.
Comparison 11 Topical others: coal tar versus betamethasone‐valerate, Outcome 1 Primary outcome: adverse events.
| Primary outcome: adverse events | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Kemper 1998 | Betamethasone valerate 0.1% cream | 0 | 19 |
| Kemper 1998 | Coal tar paste | 1 | 19 |
Comparison 12. Irradiation with UV light: UVB versus no UVB
For the phototherapy studies (UVA, UVB, PUVA), pooling was considered for two studies with data comparing UVB with no UVB or placebo (Bayerl 1999; Sjövall 1987); however, we found these studies too heterogeneous in terms of design, outcome assessment, and presentation of data to do so.
Treatment with a portable UVB phototherapy unit, to be used at home, was compared to no UVB treatment in a study among 48 participants with occupational hand dermatitis (Bayerl 1999).
Primary outcome: adverse events
In both groups, two participants showed an exacerbation. Other adverse events were stinging and burning sensations in some participants, which limited the increase in UVB therapy (RR 1.00, 95% CI 0.15 to 6.53; Analysis 12.1).
12.1. Analysis.
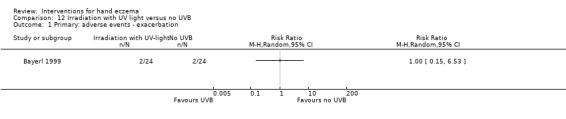
Comparison 12 Irradiation with UV light versus no UVB, Outcome 1 Primary: adverse events ‐ exacerbation.
Comparison 13. Irradiation with UV light: whole body UVB versus placebo or local UVB hands
Primary outcome: percentage of participants with investigator‐rated good/excellent control (cleared)
Local UVB hands alone versus placebo
For this subgroup, we found only one relevant trial. Three groups were compared in a trial of 18 participants with chronic hand eczema, and data for 10 participants were available (Sjövall 1987). Among participants receiving local UVB, two cleared, while in the group receiving filtered light (placebo UVB), one participant cleared (RR 2.00, 95% CI 0.26 to 15.62; Analysis 13.1;Table 12), but the confidence interval for the RR did not indicate clear differences between groups.
13.1. Analysis.
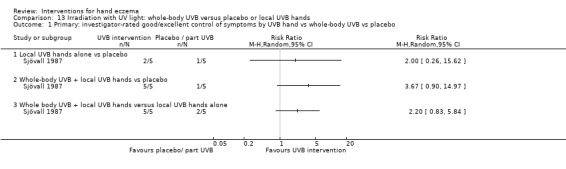
Comparison 13 Irradiation with UV light: whole‐body UVB versus placebo or local UVB hands, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms by UVB hand vs whole‐body UVB vs placebo.
Whole‐body UVB + local UVB hands versus placebo
For this subgroup, we found only one relevant trial (Sjövall 1987; n = 10). Five out of five participants in the whole‐body UVB group showed good symptom control compared to one in the control group (RR 3.67, 95% CI, 0.90 to 14.97; Analysis 13.1;Table 12). However, the confidence interval around the effect estimate was wide and imprecise.
Whole‐body UVB + local UVB hands versus local UVB hands alone
Five participants in the whole‐body UVB irradiation group had good symptom control compared to two in the control group, but due to the small sample size, the intervention group did not demonstrate clear advantage over the group given local UVB of the hands alone (RR 2.20, 95% CI 0.83 to 5.84; Analysis 13.1;Table 12).
Primary outcome: adverse events
Adverse events were not seen in either group.
Secondary outcome: time until relapse (low score = better outcome)
A postal follow‐up questionnaire three months after completion of treatment asked participants about the course of their hand dermatitis: the number of weeks in remission was presented in a descriptive way. In the local UVB group, two participants were still in remission after 15 weeks. The other three participants relapsed after 1 to 12 weeks (median 5 weeks). In the UVB local + whole‐body group, all participants relapsed within 3 to 10 weeks (median 6 weeks). The participant in the placebo group who had reached remission relapsed after three weeks. This difference was not statistically significant for local UVB hands alone versus placebo (MD 4.10, 95% CI ‐3.25 to 11.45); for whole‐body UVB + local UVB hands versus placebo (MD 0.50, 95% CI ‐4.98 to 5.98); nor for whole body UVB + local UVB hands versus local UVB hands alone (MD ‐3.60, 95% CI ‐9.68 to 2.48) (Analysis 13.2).
13.2. Analysis.
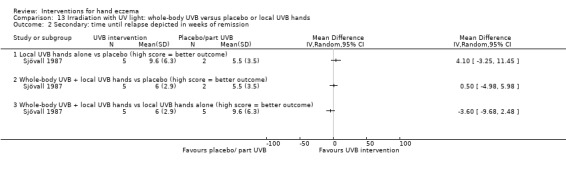
Comparison 13 Irradiation with UV light: whole‐body UVB versus placebo or local UVB hands, Outcome 2 Secondary: time until relapse depicted in weeks of remission.
Comparisons 14 and 15. Irradiation with UV light: local narrow‐band UVB versus local PUVA
For this outcome, we found two relevant trials (Sezer 2007, n = 24; and Brass 2015, n = 60). Brass 2015 was a parallel‐group study that investigated local narrow‐band UVB twice weekly compared to local PUVA twice weekly over a period of 12 weeks. Sezer 2007 studied local narrow‐band UVB thrice weekly for nine weeks compared to local PUVA thrice weekly in a left‐right study.
Primary outcome: percentage of participants with investigator‐rated good/excellent control (clearance) in UVB versus PUVA
In Brass 2015, six out of 30 participants treated with narrow‐band UVB improved compared to 12 out of 30 participants on local PUVA after 12 weeks (RR 0.50, 95% CI 0.22 to 1.16; Analysis 14.1;Table 3; Table 12).
14.1. Analysis.
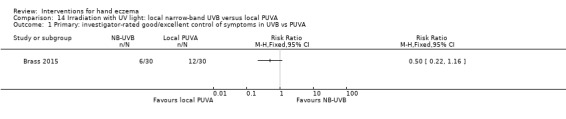
Comparison 14 Irradiation with UV light: local narrow‐band UVB versus local PUVA, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms in UVB vs PUVA.
In Sezer 2007, two out of 12 hands treated with UVB cleared (17%). On the PUVA‐treated side, one hand cleared (8%), as presented in Analysis 15.1 and in Table 12.
15.1. Analysis.
Comparison 15 Irradiation with UV light: local narrow‐band UVB versus local PUVA, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms in UVB vs PUVA.
| Primary: investigator‐rated good/excellent control of symptoms in UVB vs PUVA | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Sezer 2007 | NB‐UVB | 2 | 12 |
| Sezer 2007 | Local PUVA | 1 | 12 |
Primary outcome: adverse events
Brass 2015 reported no serious treatment‐related adverse events. An adverse event (mainly erythema) was reported in nine participants treated with local narrow‐band UVB and in none of the participants treated with local PUVA (Fisher's exact test P = 0.0019; RR 19.00, 95% CI 01.16 to 312.42; Analysis 14.2; Table 3).
14.2. Analysis.
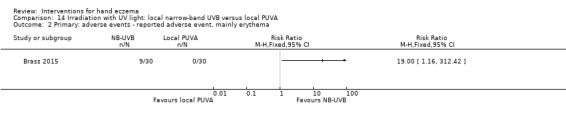
Comparison 14 Irradiation with UV light: local narrow‐band UVB versus local PUVA, Outcome 2 Primary: adverse events ‐ reported adverse event, mainly erythema.
In Sezer 2007, one participant dropped out because of an exacerbation of eczema in both hands (unclear from which group). Palmar hyperpigmentation due to PUVA was observed in three participants (see Analysis 15.2).
15.2. Analysis.
Comparison 15 Irradiation with UV light: local narrow‐band UVB versus local PUVA, Outcome 2 Primary: adverse events.
| Primary: adverse events | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Palmar hyperpigmentation | |||
| Sezer 2007 | NB‐UVB | 0 | 12 |
| Sezer 2007 | Local PUVA | 3 | 12 |
Secondary outcome: reduction in severity, investigator‐rated, by local narrow‐band UVB versus local PUVA
The data for Brass 2015 were not reproducible; however in the PUVA group, the mTLSS was reduced from a median of 8.5 (range 0 to 16) and 8 (range 3 to 15) for the left and right hand, to a median of 3 (range 0 to 13) and 3 (range 0 to 14) (n = 23). In the local narrow‐band UVB group, the median mTLSS was reduced from 7 (range 0 to 16) and 8.5 (range 1 to 15) to 5 (range 0 to 11) and 4.5 (range 0 to 11) after 12 weeks of treatment (n = 20) (Table 3).
We identified Sezer 2007 as the only relevant trial for this comparison (Sezer 2007, n = 24). For both treatments, researchers observed a marked clinical improvement in nine out of 12 hands (75%). The difference in total clinical scores between irradiation with UV light, local narrow‐band UVB, and local PUVA was not clear (see Analysis 15.3).
15.3. Analysis.
Comparison 15 Irradiation with UV light: local narrow‐band UVB versus local PUVA, Outcome 3 Secondary: investigator‐rated improvement by local narrow‐band UVB vs local PUVA.
| Secondary: investigator‐rated improvement by local narrow‐band UVB vs local PUVA | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Sezer 2007 | NB‐UVB | 9 | 12 |
| Sezer 2007 | Local PUVA | 9 | 12 |
Comparison 16. Irradiation with UV light: oral PUVA versus topical bath PUVA
Two studies investigated oral PUVA and bath PUVA (Tzaneva 2009; van Coevorden 2004a); however because the designs of these studies were substantially different, and because van Coevorden 2004a mainly focused on the at‐home versus hospital‐based version, we did not pool these studies. Also see Table 12.
Primary outcome: adverse events
van Coevorden 2004a included only adverse events that constituted a reason to discontinue. From the oral/home PUVA group, three participants dropped out because of adverse events (nausea). From the hospital/bath PUVA group, one dropped out because of adverse events (burn). There were no clear differences between groups (nausea Fisher's exact test P = 0.1180; RR 7.18, 95% CI 0.38 to 136.71; burn Fisher's exact test P = 1.00; RR 0.34, 95% CI 0.01 to 8.26; Analysis 16.1).
16.1. Analysis.
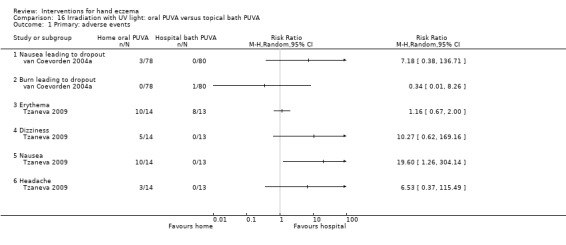
Comparison 16 Irradiation with UV light: oral PUVA versus topical bath PUVA, Outcome 1 Primary: adverse events.
In Tzaneva 2009, investigators gave oral PUVA (8‐methoxypsoralen capsules) to 14 participants and topical bath PUVA therapy with 8‐methoxypsoralen to 13 participants. Erythema occurred in 10 participants (71%) in the oral PUVA group, and in eight participants (62%) in the bath PUVA group (Fisher's exact test P = 0.49; RR 1.16, 95% CI 0.67 to 2.00; Analysis 16.1). In the oral PUVA group, 10 participants reported nausea (Fisher's exact test P = 0.0002; RR 19.60, 95% CI 1.26 to 304.14; number needed to treat for an additional harmful outcome (NNTH) 1, 95% CI 1 to 2), five reported dizziness (Fisher's exact test P = 0.04; RR 10.27, 95% CI 0.62 to 169.16), and three reported headache (Fisher's exact test P = 0.22; RR 6.53, 95% CI 0.37 to 115.49). Adverse events were observed most often at the beginning of therapy and improved during subsequent treatments. None of these adverse events led to dropouts.
With the exception of 'erythema', all other subgroups in this outcome had zero events in one of the arms, which could have been responsible in part for the wide confidence interval around the point of estimate, and for which FIsher's exact test was used.
Secondary outcome: reduction in severity, investigator‐rated improvement, in mean eczema scores at week 10
For this outcome, we found only one relevant trial (van Coevorden 2004a, n = 158). At the end of the treatment phase (10 weeks) in the home PUVA group, 56/78 participants (72%) showed improvement (mean 3.3, SD 3.8) versus 49/80 participants (61%) in the hospital/bath PUVA group (mean 2.5, SD 3.4) (MD 0.80, 95% CI ‐0.33 to 1.93; Analysis 16.2).
16.2. Analysis.
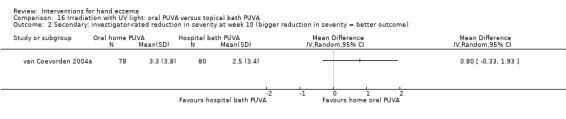
Comparison 16 Irradiation with UV light: oral PUVA versus topical bath PUVA, Outcome 2 Secondary: investigator‐rated reduction in severity at week 10 (bigger reduction in severity = better outcome).
Secondary outcome: reduction in severity, investigator‐rated improvement, in mean scores at week 18 (low score = better outcome)
For this outcome, we found only one relevant trial (van Coevorden 2004a, n = 158). At eight weeks after the treatment phase, the reduction in mean score from baseline was 3.1 (SD 4.05) versus 2.7 (SD 3.4), respectively; there was no clear difference between irradiation with UV light: oral PUVA and topical bath PUVA (MD 0.40, 95% CI ‐0.77 to 1.57; Analysis 16.3).
16.3. Analysis.
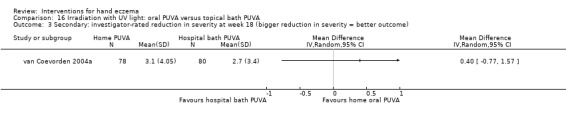
Comparison 16 Irradiation with UV light: oral PUVA versus topical bath PUVA, Outcome 3 Secondary: investigator‐rated reduction in severity at week 18 (bigger reduction in severity = better outcome).
Comparison 17. Irradiation with UV light: topical PUVA versus UVA
In a 16‐week within‐participant (left‐right) study, topical PUVA was compared with UVA (Grattan 1991) in 15 participants (n = 30 hands).
Primary outcome: adverse events
Only one participant who completed the study experienced a burning sensation on the back of his PUVA‐treated hand (see Analysis 17.1). Probably two participants had to be withdrawn due to exacerbation of eczema ‐ one from each group (see Analysis 17.1).
17.1. Analysis.
Comparison 17 Irradiation with UV light: topical PUVA versus UVA, Outcome 1 Primary: adverse events.
| Primary: adverse events | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Discontinuation due to adverse events | |||
| Grattan 1991 | Topical PUVA | 1 | 15 |
| Grattan 1991 | UVA | 1 | 15 |
| Burning | |||
| Grattan 1991 | Topical PUVA | 1 | 15 |
| Grattan 1991 | UVA | 0 | 15 |
| Exacerbation of eczema | |||
| Grattan 1991 | Topical PUVA | 1 | 15 |
| Grattan 1991 | UVA | 0 | 15 |
Comparison 18. Irradiation with UV light: UVA‐1 versus betamethasone‐valerate 0.1% cream
Irradation with UVA‐1 three times a week was compared to topical betamethasone‐valerate 0.1% twice a day over a six‐week period in 47 participants (Said 2010).
Primary outcome: adverse events
Tolerance of both treatments was good. The only adverse event noted was post‐phototherapy pigmentation, which occurred in 18 of the 24 participants treated with UVA‐1 compared to none of the participants in the control group (Fisher's exact test P = 0.0001; RR 35.52, 95% CI 2.26 to 557.08; NNTH 1, 95% CI 1 to 2; Analysis 18.1). Zero events in the control group is likely to explain the wide confidence interval.
18.1. Analysis.
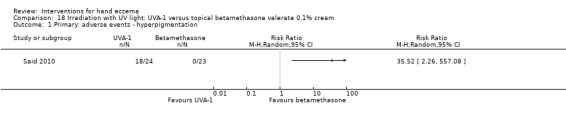
Comparison 18 Irradiation with UV light: UVA‐1 versus topical betamethasone valerate 0.1% cream, Outcome 1 Primary: adverse events ‐ hyperpigmentation.
Comparison 19. Irradiation with UV light: UVA‐1 versus placebo
UVA‐1 irradiation for three weeks in 15 participants with dyshidrotic hand eczema was compared with placebo (simulated blue light) in 13 participants (Polderman 2003).
Primary outcome: adverse events
Apart from some minor erythemal reactions, no adverse events occurred. Three of 13 participants in the placebo group dropped out after two weeks because of exacerbation, but no clear differences was identified between groups (Fisher's exact test P = 0.2258; RR 0.13, 95% CI 0.01 to 2.22; Analysis 19.1). Zero events in the intervention group is likely to explain the wide confidence interval.
19.1. Analysis.
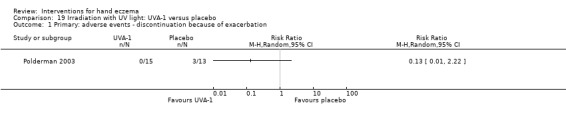
Comparison 19 Irradiation with UV light: UVA‐1 versus placebo, Outcome 1 Primary: adverse events ‐ discontinuation because of exacerbation.
Secondary outcome: reduction in severity of itch, participant‐rated decrease in VAS, at week 3 (higher score = greater reduction)
For this outcome, we found only one relevant trial (Polderman 2003, n = 28). Although there was a notable difference between irradiation with UV light: UVA‐1 (mean 2.31, SD 2.01) and placebo (mean ‐1.37, SD 4.05) with regards to VAS scores for itch (MD 3.68, 95% CI 1.25 to 6.11; Analysis 19.2), we have low confidence about the strength of the finding due to limited sample size (i.e. imprecision).
19.2. Analysis.
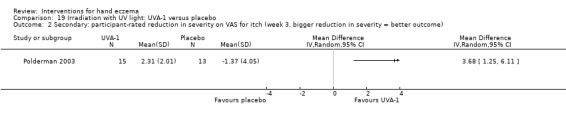
Comparison 19 Irradiation with UV light: UVA‐1 versus placebo, Outcome 2 Secondary: participant‐rated reduction in severity on VAS for itch (week 3, bigger reduction in severity = better outcome).
Secondary outcome: reduction in severity, investigator‐rated improvement in DASI, at week 3 (higher score = greater reduction)
In the same group of participants (Polderman 2003), the severity score on the Dyshydrotic eczema Area and Severity Index (DASI) decreased in the UVA‐1 group (mean 8.67, SD 6.72) compared to the placebo group (mean ‐0.38, SD 8.87) in week 3 (MD 9.05, 95% CI 3.15 to 14.95; Analysis 19.3).
19.3. Analysis.
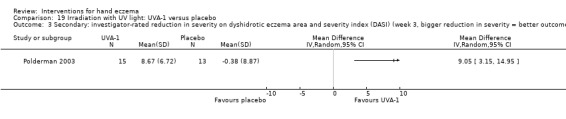
Comparison 19 Irradiation with UV light: UVA‐1 versus placebo, Outcome 3 Secondary: investigator‐rated reduction in severity on dyshidrotic eczema area and severity index (DASI) (week 3, bigger reduction in severity = better outcome).
Comparison 20. Irradiation with UV light: PUVA versus UVA‐1
In a within‐participant study, the effectiveness of middle‐dose UVA‐1 irradiation was compared with topical cream PUVA therapy in 15 participants with chronic relapsing dyshidrotic hand eczema (Adams 2007).
Primary outcome: adverse events
Burning occurred in three participants in the topical cream PUVA group and in one participant in the UVA‐1 group, and increased pruritus occurred in five participants in the topical PUVA group versus three in the UVA‐1 group (Analysis 20.1).
20.1. Analysis.
Comparison 20 Irradiation with UV light: PUVA versus UVA‐1, Outcome 1 Primary: adverse events.
| Primary: adverse events | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Burning | |||
| Adams 2007 | PUVA | 3 | 11 |
| Adams 2007 | UVA‐1 | 1 | 11 |
| Itching | |||
| Adams 2007 | PUVA | 5 | 11 |
| Adams 2007 | UVA‐1 | 3 | 11 |
Comparison 21. Irradiation with X‐rays (ionising radiation)
Among trials evaluating the effects of ionising radiation (X‐rays), we considered pooling the results of four studies comparing X‐rays with placebo irradiation (Cartwright 1987; Fairris 1984; King 1984; Lindelöf 1987), but dosages, presentation of results, and follow‐up times were considered too heterogeneous in most cases. Moreover, all these studies used a within‐participant design (i.e. comparing one hand versus the contralateral hand). Superficial X‐ray irradiation on one hand was compared with topical PUVA on the contralateral hand in 25 participants (Sheehan‐Dare 1989). Also see Table 13.
Comparison 21A. Irradiation with X‐rays: X‐rays versus placebo
Primary outcomes: percentage of participants with investigator‐rated good/excellent control
After one month in seven out of 15 participants, hands treated with X‐rays were categorised as showing good response (defined as 'clear' or 'nearly clear'), whereas all 15 placebo‐treated hands were categorised as showing poor response (defined as 'partly clear', 'no change', or 'relapse') (King 1984; see Analysis 21.1 and Table 13). After three months, ten irradiated hands and six placebo‐treated hands were categorised as showing good response (see Analysis 21.1), and after six months there was a good response in 11 irradiated and eight placebo‐treated hands (see Analysis 21.1). There were no clear differences between groups.
21.1. Analysis.
Comparison 21 Irradiation with Grenz ray, Outcome 1 Primary: investigator‐rated improvement good/excellent control.
| Primary: investigator‐rated improvement good/excellent control | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| after 1 month | |||
| King 1984 | Grenz Ray | 7 | 15 |
| King 1984 | Placebo | 0 | 15 |
| after 3 months | |||
| King 1984 | Grenz Ray | 10 | 15 |
| King 1984 | Placebo | 6 | 15 |
| after 6 months | |||
| King 1984 | Grenz Ray | 11 | 15 |
| King 1984 | Placebo | 8 | 15 |
| Hyperkeratotic eczema after 6 months | |||
| King 1984 | Grenz Ray | 4 | 8 |
| King 1984 | Placebo | 2 | 6 |
| Pompholyx after 6 months | |||
| King 1984 | Grenz Ray | 7 | 7 |
| King 1984 | Placebo | 6 | 7 |
| Chronic palmar eczema after 6 months | |||
| King 1984 | Grenz Ray | 11 | 15 |
| King 1984 | Placebo | 0 | 15 |
Primary outcome: adverse events
No systemic or local adverse events were noted.
Comparison 21B. Irradiation with X‐rays: Grenz ray
The effect of 3 Gy Grenz ray therapy six times in weekly intervals was investigated in within‐participant studies (Lindelöf 1987; 24 participants, 48 hands in a within‐participant design; Cartwright 1987; 30 participants, 60 hands in a within‐participant design).
Primary outcome: adverse events
Six participants had hyperpigmentation in treated hands, and no participants in the placebo group experienced adverse events (see Analysis 21.2); however, there is no clear difference between groups.
21.2. Analysis.
Comparison 21 Irradiation with Grenz ray, Outcome 2 Primary: adverse events ‐ hyperpigmentation.
| Primary: adverse events ‐ hyperpigmentation | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Cartwright 1987 | Grenz | 1 | 30 |
| Cartwright 1987 | Placebo | 0 | 30 |
| Lindelöf 1987 | Grenz | 5 | 24 |
| Lindelöf 1987 | Placebo | 0 | 24 |
Comparison 21C. Irradiation with X‐rays: X‐rays versus Grenz rays
A within‐participant study on 25 participants compared superficial X‐ray and Grenz ray irradiation (Fairris 1985).
Primary outcome: adverse events
Fairris 1985 reported no adverse events from either therapy.
Comparison 22. Topical calcineurin inhibitors: tacrolimus ointment versus mometasone furoate
An overview of all of the studies on topical calcineurin inhibitors can be found in Table 14.
The current comparison included two studies (Schnopp 2002, n = 16; Katsarou 2012, n = 30).
Primary outcome: adverse events
Both treatments were well tolerated. None of the participants in Schnopp 2002 dropped out because of adverse events.
Katsarou 2012 investigated adverse events but did not report any.
Secondary outcome: reduction in severity, investigator‐rated
Although the reduction in mean DASI equalled improvement in scores for both treatments after two weeks, no clear difference was found between groups. The mean DASI score was reduced from 18 (SD 12.68) to 6.6 (SD 6.18) in the tacrolimus group, and from 18.5 (SD 14.09) to 6.9 (SD 7.7) in the mometasone furoate group, respectively (Schnopp 2002; see Analysis 22.1 and Table 4).
22.1. Analysis.
Comparison 22 Topical calcineurin inhibitors: tacrolimus 0.1% ointment versus mometasone furoate, Outcome 1 Secondary: reduction in investigator‐rated severity ‐ DASI.
| Secondary: reduction in investigator‐rated severity ‐ DASI | ||||
|---|---|---|---|---|
| Study | Group ‐ within‐participant study | Mean | SD | Total number of pairs of hands analysed |
| Schnopp 2002 | Tacrolimus | 6.6 | 6.18 | 8 |
| Schnopp 2002 | Mometasone | 6.9 | 7.7 | 8 |
Comparison 23. Topical calcineurin inhibitors: tacrolimus 0.1% ointment versus vehicle cream
Two studies addressed this comparison (Krejci‐Manwaring 2008, n = 32; Pacor 2006, n = 28).
Primary outcome: percentage of participants with investigator‐rated good/excellent control (remarkable improvement/complete remission)
Pacor 2006: In the tacrolimus group, complete remission at the end of treatment was observed in six participants (6/14), and remarkable improvement in eight participants (8/14). Treatment with vehicle cream did not lead to remarkable improvement (0/14) and led to only mild improvement in 4 of 14 participants (Fisher's exact test P = 0.0001; RR 29.00, 95% CI 1.90 to 443.25; NNTB 1, 95% CI 1 to 1; Analysis 23.1; Table 5Table 14). Zero events in the control group is likely to explain the wide confidence interval.
23.1. Analysis.
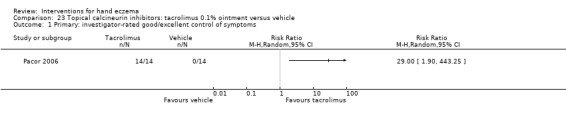
Comparison 23 Topical calcineurin inhibitors: tacrolimus 0.1% ointment versus vehicle, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms.
Primary outcome: adverse events
In Krejci‐Manwaring 2008, researchers observed one case of each of the following adverse events: acute contact dermatitis at the site of the necklace, flare of atopic dermatitis on the foot, acne‐like rash on the face, leg cramps, and worsening of hand dermatitis. Stinging was not reported.
In Pacor 2006, four participants (4/14) in the tacrolimus group experienced transient burning and itching at the application site, which was well tolerated (Fisher's exact test P = 0.1129; RR 9.00, 95% CI 0.53 to 152.93; Analysis 23.2; Table 5).
23.2. Analysis.
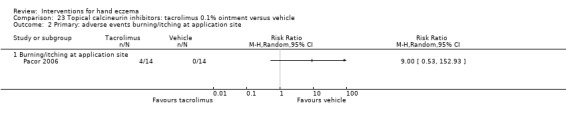
Comparison 23 Topical calcineurin inhibitors: tacrolimus 0.1% ointment versus vehicle, Outcome 2 Primary: adverse events burning/itching at application site.
RR given above is based on zero events in one arm, which is likely to explain the wide confidence interval.
Comparison 24. Topical calcineurin inhibitors: pimecrolimus 1% cream versus vehicle
Five of the included studies addressed this comparison: Belsito 2004 (n = 294), Hordinsky 2010 (n = 652), Bauer 2012 (n = 36), Cherill 2000 (n = 48), and Baskan 2005 (n = 25).
Primary outcome: percentage of participants with investigator‐rated good/excellent control (clear or almost clear) with pimecrolimus cream versus vehicle at three weeks
For this subgroup, we found only one relevant trial (Belsito 2004, n = 294). In all, 42 of 151 versus 26 of 143 participants had good investigator‐rated symptom control in intervention and control groups, respectively. The favourable outcome for pimecrolimus was borderline because the confidence interval includes 1 and therefore should be interpreted with care (RR 1.53, 95% CI 0.99 to 2.36; NNTB 10, 95% CI 5 to 1111; Analysis 24.1). When the subgroups were analysed based on aetiology, we did not find significant differences for irritant, allergic, or endogenous hand eczema (Analysis 24.1;Table 14).
24.1. Analysis.
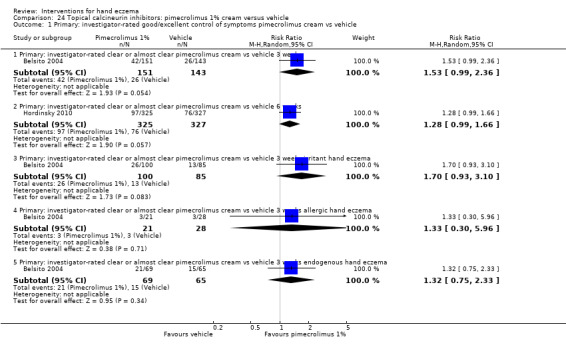
Comparison 24 Topical calcineurin inhibitors: pimecrolimus 1% cream versus vehicle, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms pimecrolimus cream vs vehicle.
Primary outcome: percentage of participants with investigator‐rated good/excellent control (clear or almost clear) with pimecrolimus cream versus vehicle at six weeks
For this subgroup, we found only one relevant trial (Hordinsky 2010, n = 652). Treatment success (IGA score 0 = clear and 1 = almost clear) was achieved in 97 of 325 participants (29.8%) in the pimecrolimus cream 1% group and in 76 of 327 participants in the vehicle group. Favourable outcomes for pimecrolimus were borderline significant because the confidence interval included 1 and should be interpreted with care (RR 1.28, 95% CI 0.99 to 1.66; Analysis 24.1).
Primary outcome: adverse events
Bauer 2012, Belsito 2004, and Hordinsky 2010 reported adverse events (Analysis 24.2). Hordinsky 2010 found no clear differences between groups in terms of treatment‐related erythema or irritation (RR 0.56, 95% CI 0.30 to 1.06; n = 652); itching (RR 0.89, 95% CI 0.52 to 1.53; n = 652); warmth, stinging, and burning (RR 0.82, 95% CI 0.52 to 1.29; n = 652); or herpes simplex infection (RR 0.60, 95% CI 0.15 to 2.51; n = 652). No adverse events were stated in Cherill 2000 and Baskan 2005.
24.2. Analysis.
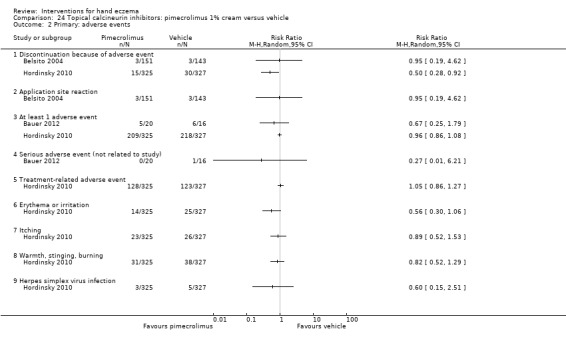
Comparison 24 Topical calcineurin inhibitors: pimecrolimus 1% cream versus vehicle, Outcome 2 Primary: adverse events.
Secondary outcome: reduction in severity, participant‐rated
For pruritus relief between pimecrolimus 1% and vehicle, we found only one relevant trial (Hordinsky 2010, n = 652). There appears to be greater pruritus relief in the intervention group (pimecrolimus 1% cream) than in the vehicle group (RR 1.15, 95% CI 1.06 to 1.25; NNTB 9, 95% CI 6 to 22; Analysis 24.3); however, benefit relative to the control group appears to be marginal.
24.3. Analysis.
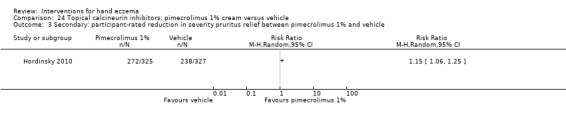
Comparison 24 Topical calcineurin inhibitors: pimecrolimus 1% cream versus vehicle, Outcome 3 Secondary: participant‐rated reduction in severity pruritus relief between pimecrolimus 1% and vehicle.
Secondary outcome: time until relapse
Time to relapse was compared between pimecrolimus 1% cream and vehicle in Bauer 2012 (n = 36). Time to relapse did not differ significantly between groups according to the trial authors (pimecrolimus: 39.35 days; vehicle: 33.19 days); this was represented in a survival graph. We were unable to reproduce these analyses.
Comparison 25. Topical antibacterial agents: betamethasone‐valerate/clioquinol cream versus betamethasone‐valerate/fusidic acid
Primary outcomes: percentage of participants with investigator‐rated good/excellent control (intention‐to‐treat) after four weeks
For this outcome, we found one relevant trial (Hill 1998, n = 120). In the ITT analysis, 34 of 62 participants (54.8%) in the betamethasone‐valerate/clioquinol group and 31 of 58 (53,4%) in the betamethasone‐valerate/fusidic acid group had a good response (RR 1.03, 95% CI 0.74 to 1.43; Analysis 25.1;Table 15).
25.1. Analysis.
Comparison 25 Topical antibacterial agents: betamethasone‐valerate/clioquinol cream versus betamethasone‐valerate/fusidic acid, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms (intention‐to‐treat).
Primary outcome: adverse events
In the clioquinol group, 11 of 62 participants experienced adverse events versus nine of 58 participants in the fusidic acid group (RR 1.14, 95% CI 0.51 to 2.56; Analysis 25.2). Several other adverse events were observed, including chest infection (1/62 versus 0/58), application‐related irritation (5/62 versus 5/58), deterioration of eczema (4/62 versus 4/58), eye watering (1/62 versus 0/58), hands coloured yellow (1/62 versus 0/58), hands feeling thick (0/62 versus 1/58), and vesicle on the hands (0/62 versus 1/58), but none of these showed between‐group differences (Analysis 25.2).
25.2. Analysis.
Comparison 25 Topical antibacterial agents: betamethasone‐valerate/clioquinol cream versus betamethasone‐valerate/fusidic acid, Outcome 2 Primary: adverse events.
As shown above, quite a few subgroups under this outcome had zero events in one of the arms; this is likely to explain the wide 95% confidence interval.
Comparison 26. Topical retinoids: bexarotene 1% gel versus bexarotene with corticosteroids
Primary outcomes: percentage of participants with investigator‐rated good/excellent control (> 90% clearance on physician response rates)
Treatment success (> 90% clearance) was achieved by 39% in the bexarotene only group, by 46% in the B + MF group, and by 21% in the B + HC group.
Bexarotene only versus bexarotene + mometasone
For this subgroup, we found only one relevant trial (Hanifin 2004, n = 41). There was no clear difference between topical retinoids: bexarotene 1% gel and bexarotene with mometasone (RR 0.85, 95% CI 0.40 to 1.8; Analysis 26.1;Table 15).
26.1. Analysis.
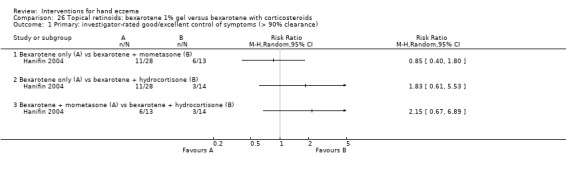
Comparison 26 Topical retinoids: bexarotene 1% gel versus bexarotene with corticosteroids, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms (> 90% clearance).
Bexarotene only versus bexarotene + hydrocortisone
For this subgroup, we found only one relevant trial (Hanifin 2004, n = 42). There was no clear difference between topical retinoids: bexarotene 1% gel and bexarotene with hydrocortisone (RR 1.83, 95% CI 0.61 to 5.53; Analysis 26.1;Table 15).
Bexarotene + mometasone versus bexarotene + hydrocortisone
For this subgroup, we found only one relevant trial (Hanifin 2004, n = 27). There was no clear difference between topical retinoids: bexarotene 1% gel and mometasone versus bexarotene with hydrocortisone (RR 2.15, 95% CI 0.67 to 6.89; Analysis 26.1;Table 15).
Primary outcome: adverse events
Forty‐one participants (75%) had one or more adverse events during the study, of whom 27 (49%) had one or more events possibly related to the study drugs. The bexarotene group had irritation/rash in eight participants; stinging/burning in two; and dermatitis flare in five. The B + MF group had irritation/rash in four participants and stinging/burning in four participants. The B + HC group had irritation/rash in four participants; stinging/burning in four participants, and dermatitis flare in zero participants (which is likely to explain the wide 95% confidence interval). None of the adverse events occurred significantly more often in a study or control group (Analysis 26.2; Analysis 26.3).
26.2. Analysis.
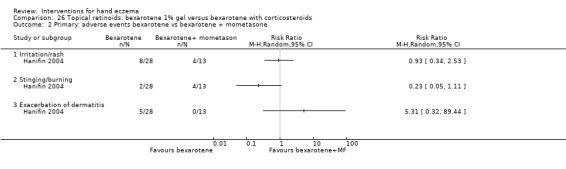
Comparison 26 Topical retinoids: bexarotene 1% gel versus bexarotene with corticosteroids, Outcome 2 Primary: adverse events bexarotene vs bexarotene + mometasone.
26.3. Analysis.
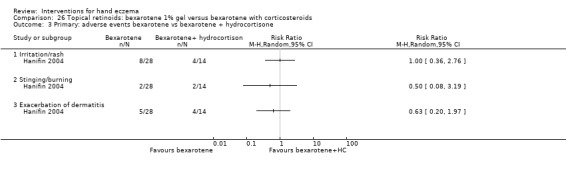
Comparison 26 Topical retinoids: bexarotene 1% gel versus bexarotene with corticosteroids, Outcome 3 Primary: adverse events bexarotene vs bexarotene + hydrocortisone.
Secondary outcome: reduction in severity, investigator‐rated: > 90% and > 50% reduction in hand eczema area and severity index (HEASI)
The percentage with > 90% reduction in Hand Eczema Area and Severity Index (HEASI) score in the bexarotene only group was 36%, in the B + MF group 38%, and in the B + HC group 14%. But there was no clear difference between groups according to the study authors. For > 50% reduction in HEASI, the percentages were, respectively, 79%, 85%, and 64%.
Bexarotene only versus bexarotene + mometasone
For this subgroup, we found only one relevant trial (Hanifin 2004, n = 41). There was no clear difference between topical retinoids: bexarotene 1% gel and bexarotene with mometasone (RR 0.93, 95% CI 0.69 to 1.26; Analysis 26.4).
26.4. Analysis.
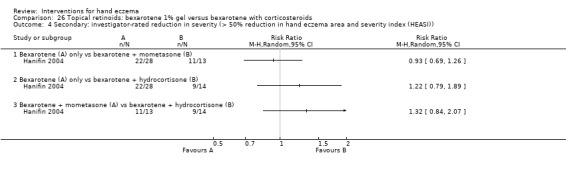
Comparison 26 Topical retinoids: bexarotene 1% gel versus bexarotene with corticosteroids, Outcome 4 Secondary: investigator‐rated reduction in severity (> 50% reduction in hand eczema area and severity index (HEASI)).
Bexarotene only versus bexarotene + hydrocortisone
For this subgroup, we found only one relevant trial (Hanifin 2004, n = 42). There was no clear difference between topical retinoids: bexarotene 1% gel and bexarotene with hydrocortisone (RR 1.22, 95% CI 0.79 to 1.89; Analysis 26.4).
Bexarotene + mometasone versus bexarotene + hydrocortisone
For this subgroup, we found only one relevant trial (Hanifin 2004, n = 27). There was no clear difference between topical retinoids: bexarotene 1% gel and mometasone versus bexarotene with hydrocortisone (RR 1.32, 95% CI 0.84 to 2.07; Analysis 26.4).
Comparison 27. Other topical agents: calmurid versus Aquacare
A within‐participant study compared topical Aquacare HP cream to a calmurid cream (Fredriksson 1975).
Primary outcome: adverse events
In the calmurid group, 13 participants experienced a burning sensation upon application compared to no adverse events in the Aqua HP group (see Analysis 27.1).
27.1. Analysis.
Comparison 27 Other topical agents: calmurid versus aquacare, Outcome 1 Primary: adverse events: burning.
| Primary: adverse events: burning | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event number | Total number of pairs of hands analysed |
| Fredriksson 1975 | Aquacare | 0 | 30 |
| Fredriksson 1975 | Calmurid | 13 | 30 |
Comparison 28. Fumaric acid 5% cream versus triamcinolone 0.1% cream
This study compared topical fumaric acid twice daily to triamcinolone cream twice daily for four weeks in 58 participants (Jowkar 2014).
Primary outcome: adverse events
Erythema and pruritus were noted in two participants in each treatment group (RR 0.93, 95% CI 0.14 to 6.18; Analysis 28.1).
28.1. Analysis.
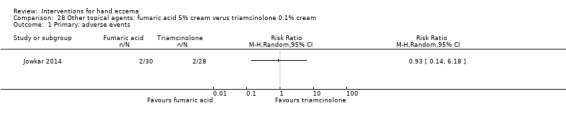
Comparison 28 Other topical agents: fumaric acid 5% cream verus triamcinolone 0.1% cream, Outcome 1 Primary: adverse events.
Comparison 29. Furpalmate 0.3% cream versus hydrocortisone acetate 0.5% cream
Primary outcome: percentage of participants with investigator‐rated and/or self‐rated good/excellent control (complete remission)
For this outcome, we found only one relevant trial (Lauriola 2011, n = 40). In the study report, treatments were shown to be equally effective in "curing" or "improving" hand dermatitis after 14 days. In the furpalmate group, 18 of 20 participants (90%) were cured or improved after 14 days, and this occurred in 20 of 20 participants in the hydrocortisone group (100%) (RR 0.90, 95% CI 0.76 to 1.07; Analysis 29.1;Table 15).
29.1. Analysis.
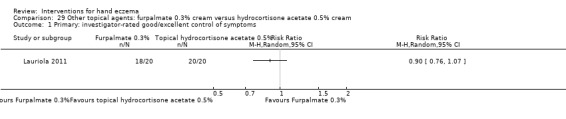
Comparison 29 Other topical agents: furpalmate 0.3% cream versus hydrocortisone acetate 0.5% cream, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms.
Comparison 30. Fumaria parviflora versus vehicle cream
Studies using a parallel‐group design compared use of 2% Nigella sativa L. (family Ranunculaceae) ointment (a traditional medicine) twice daily with 0.1% betamethasone ointment twice daily and with Eucerin cream twice daily in 60 participants over four weeks (Yousefi 2012, n = 60).
A parallel‐group study (Jowkar 2011, n = 44) compared the effect of an extract of 4% Fumaria parviflora Lam. cream twice a day versus vehicle cream (placebo) twice daily in 44 participants for four weeks.
Primary outcome: adverse events
Yousefi 2012 reported no adverse events for treatment with Nigella sativa L. and Eucerin cream.
In Jowkar 2011, one participant dropped out due to development of redness and papules in the Fumaria parviflora Lam. cream group (RR 3.00, 95% CI 0.13 to 69.87; Analysis 30.1). Zero events in the vehicle group is likely to explain the wide confidence interval and Fisher's exact test results with a P value of 1.00.
30.1. Analysis.
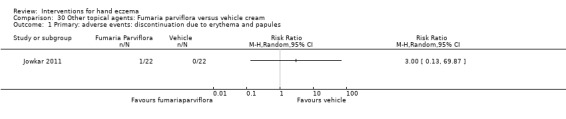
Comparison 30 Other topical agents: Fumaria parviflora versus vehicle cream, Outcome 1 Primary: adverse events: discontinuation due to erythema and papules.
III. Systemic treatments
We identified no randomised controlled trials on oral corticosteroids.
Comparison 31. Oral immunosuppressants: oral azathioprine and topical clobetasol propionate versus topical clobetasol propionate only
Agarwal 2013 compared oral azathioprine with topical clobetasol propionate 0.05% cream to topical clobetasol propionate 0.05% cream alone in 108 participants; 91 participants completed the trial.
Primary outcome: percentage of participants with investigator‐rated good/excellent control
After eight weeks, 36.95% in the clobetasol only group had a good response (defined as 75% improvement in signs and symptoms) versus 73.3% in the clobetasol with additional azathioprine 50 mg group (RR 1.98, 95% CI 1.31 to 3.01; NNTB 3, 95% CI 2 to 6; Analysis 31.1). After 24 weeks, 39.13% in the clobetasol only group improved, as did 91.1% in the clobetasol and azathioprine group (RR 2.33, 95% CI 1.61 to 3.38; NNTB 2, 95% CI 1 to 3; Analysis 31.1; Table 16).
31.1. Analysis.
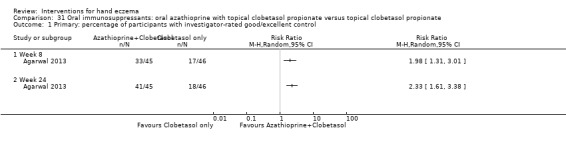
Comparison 31 Oral immunosuppressants: oral azathioprine with topical clobetasol propionate versus topical clobetasol propionate, Outcome 1 Primary: percentage of participants with investigator‐rated good/excellent control.
Primary outcome: adverse events
No adverse events that would require reduction of dosage or discontinuation of treatment were reported.
Secondary outcomes: reduction in severity, investigator‐rated (higher score = greater reduction)
This was measured by the hand eczema severity index (HECSI): after 24 weeks, 64.66% in the control group showed improvement, as did 91.29% in the intervention group (MD 10.79, 95% CI 4.77 to 16.81; Analysis 31.2).
31.2. Analysis.
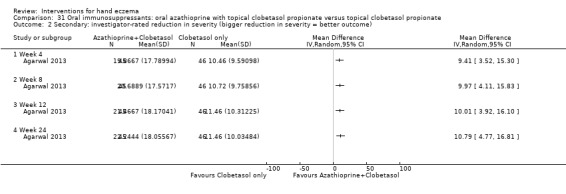
Comparison 31 Oral immunosuppressants: oral azathioprine with topical clobetasol propionate versus topical clobetasol propionate, Outcome 2 Secondary: investigator‐rated reduction in severity (bigger reduction in severity = better outcome).
Secondary outcome: reduction in severity of itch, participant‐rated (higher score = greater reduction)
After 24 weeks, the itch score difference was 6.04 (SD 2.35) in the intervention group, and 4.56 (SD 2.26) in the control group (MD 1.48, 95% CI 0.53 to 2.43; Analysis 31.3). This is a participant‐rated outcome, measured on a numerical scale from 0 to 10.
31.3. Analysis.
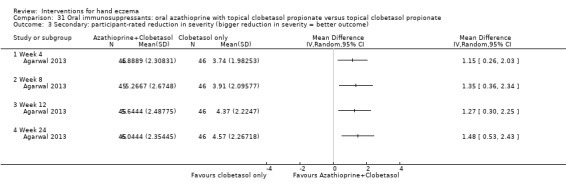
Comparison 31 Oral immunosuppressants: oral azathioprine with topical clobetasol propionate versus topical clobetasol propionate, Outcome 3 Secondary: participant‐rated reduction in severity (bigger reduction in severity = better outcome).
Comparison 32. Oral immunosuppressants: oral cyclosporin versus topical betamethasone dipropionate
Primary outcome: percentage of participants with investigator‐rated good/excellent control
Overall assessment of good/very good efficacy was 60% in the cyclosporin group and 31% in the betamethasone group (Granlund 1996). There was no apparent difference between groups (RR 1.88, 95% CI 0.88 to 3.99; n = 34; Analysis 32.1; Table 6; Table 16).
32.1. Analysis.
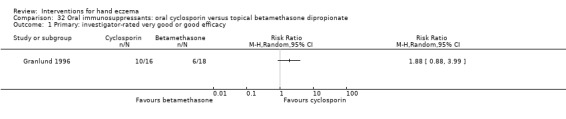
Comparison 32 Oral immunosuppressants: oral cyclosporin versus topical betamethasone dipropionate, Outcome 1 Primary: investigator‐rated very good or good efficacy.
Primary outcome: percentage of participants with self‐rated good/excellent control
One study addressed this (Granlund 1996, n = 34; the original randomised number was n = 41, but seven people left the study early; hence data were available for only 34 people). Overall assessment of good/very good efficacy was 60% in the cyclosporin group and 48% in the betamethasone group; the difference between groups was unclear (RR 1.25, 95% CI 0.69 to 2.27; Analysis 32.2; Table 6).
32.2. Analysis.
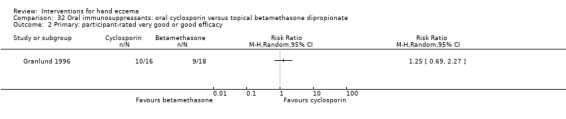
Comparison 32 Oral immunosuppressants: oral cyclosporin versus topical betamethasone dipropionate, Outcome 2 Primary: participant‐rated very good or good efficacy.
Primary outcome: adverse events
"Some kind of adverse event" occurred in 19 of 28 participants on cyclosporin and in 15 of 27 participants in the betamethasone group (RR 1.22, 95% CI 0.80 to 1.86; Analysis 32.3; Table 6). In the cyclosporin group, one participant experienced dizziness, vomiting, and facial oedema versus zero events in the control group (Fisher's exact test P = 1.00; RR 2.90, 95% CI 0.12 to 68.15; Analysis 32.3). In the betamethasone group, one participant had insomnia versus zero events in the cyclosporin group (Fisher's exact test P = 0.49; RR 0.32, 95% CI 0.01 to 7.57; Analysis 32.3) (Granlund 1996). Two people in the cyclosporin group had an increase in serum creatinine of greater than 30% versus zero events in the betamethasone group (Fisher's exact test P = 0.49; RR 4.83, 95% CI 0.24 to 96.16; Analysis 32.3). Zero events in some of the above analyses is likely to explain the wide 95% confidence interval.
32.3. Analysis.
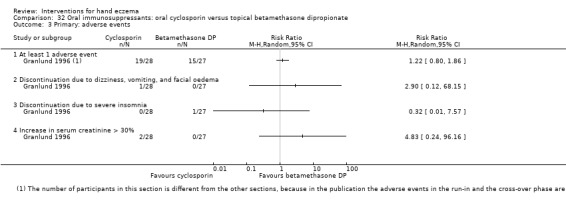
Comparison 32 Oral immunosuppressants: oral cyclosporin versus topical betamethasone dipropionate, Outcome 3 Primary: adverse events.
The number of participants in this section is different from that in the other sections because in the publication, adverse events in the run‐in and cross‐over phases are combined.
Secondary outcome: reduction in severity, investigator‐rated total disease activity score (six weeks; higher score = greater reduction)
For this outcome, we found only one relevant trial (n = 34) (Granlund 1996).
The mean total decrease in total disease activity score was 6.0 (SD 4.3) in the cyclosporin group and 5.7 (SD 4.0) in the betamethasone group (MD 0.30, 95% CI ‐2.50 to 3.10; Analysis 32.4; Table 6).
32.4. Analysis.
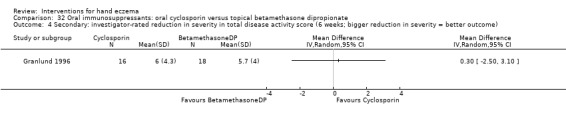
Comparison 32 Oral immunosuppressants: oral cyclosporin versus topical betamethasone dipropionate, Outcome 4 Secondary: investigator‐rated reduction in severity in total disease activity score (6 weeks; bigger reduction in severity = better outcome).
Comparison 33. Oral immunosuppressants: oral cyclosporin versus alitretinoin
NCT01231854 compared cyclosporin to alitretinoin but was ended prematurely due to inability to include the total number of participants. According to the sample size calculation, 78 participants should have been included; however, only 15 participants were included and 14 were analysed.
Primary outcome: percentage of participants with investigator‐rated good/excellent control (IGA) after 24 weeks
In the cyclosporin group, three out of seven participants (42.9%) reached complete or nearly complete clearance of hand eczema according to the Investigator Global Assessment (IGA), as did two out of seven participants (28.6%) in the alitretinoin group, after 24 weeks. There was no apparent difference between groups (Fisher's exact test P = 1.00; RR 1.50, 95% CI 0.35 to 6.40; Analysis 33.1; Table 16).
33.1. Analysis.
Comparison 33 Oral immunosuppressants: oral cyclosporin versus alitretinoin, Outcome 1 Primary: investigator‐rated good/excellent control (IGA) after 24 weeks.
Primary outcomes: percentage of participants with participant‐rated good/excellent control (PGA) after 24 weeks
In the cyclosporin group, four out of seven participants (57.1%) reached complete or nearly complete clearance of hand eczema according to the Patient Global Assessment (PGA), as did two out of seven participants (28.6%) in the alitretinoin group. There was no apparent difference between groups (Fisher's exact test P = 0.59; RR 2.00, 95% CI 0.53 to 7.60; Analysis 33.2).
33.2. Analysis.
Comparison 33 Oral immunosuppressants: oral cyclosporin versus alitretinoin, Outcome 2 Primary: participant‐rated good/excellent control (PGA) after 24 weeks.
Primary outcome: adverse events
Six adverse events were documented, of which two were possibly related to the use of cyclosporin (fatigue, bone ache, dry lips in one participant, and exacerbation of atopic eczema in another participant). No serious adverse events were recorded throughout the trial. At least one adverse event occurred in 3 of 7 cyclosporin participants and in 2 of 7 alitretinoin participants (Fisher's exact test P = 1.00; RR 1.50, 95% CI 0.35 to 6.40; Analysis 33.3).
33.3. Analysis.
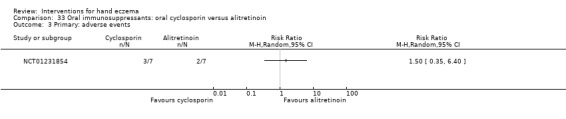
Comparison 33 Oral immunosuppressants: oral cyclosporin versus alitretinoin, Outcome 3 Primary: adverse events.
Secondary outcome: time until relapse
None of the participants relapsed during the 24 weeks of follow‐up (0 of 7 versus 0 of 7).
Comparison 34. Oral retinoids: acitretin versus placebo
Oral acitretin was compared with placebo capsules in a study that enrolled 29 participants with hyperkeratotic dermatitis of the palms (Thestrup‐Pedersen 2001). Fourteen participants were allocated to 30 mg acitretin once daily for eight weeks, and 15 participants received identical looking placebo capsules. This study did not provide useable data for analysis, as only subscale mean score was available, without SD.
Primary outcome: adverse events
No adverse events were reported and all biochemical parameters were within normal limits in both groups.
Secondary outcome: reduction in severity, investigator rated, after four and eight weeks
Trial authors used a score system composed of subscales with hyperkeratosis, fissures, scaling, itch, and redness. After four weeks of treatment, a 51% reduction in all symptoms was seen in the acitretin group compared to a 9% reduction in the placebo group. No further improvement was seen after eight weeks of treatment (Thestrup‐Pedersen 2001). No reproducible data were given.
Secondary outcome: reduction in severity, participant‐rated number of participants with improvement in itch
After eight weeks of treatment, itch was reduced by 41% in the acitretin group compared to 19% in the placebo group (Thestrup‐Pedersen 2001). No reproducible data were given.
Comparison 34A. Oral retinoids: alitretinoin versus placebo
Four studies investigated the effect of oral alitretinoin: Bissonnette 2010,Fowler 2014,Ruzicka 2004, and Ruzicka 2008.
Primary outcome: percentage of participants with investigator‐rated good/excellent control (clear or almost clear) at week 12, at week 24, or at end of treatment
In Ruzicka 2004 and Ruzicka 2008, clearance or almost clearance of eczema occurred more often in all groups treated with alitretinoin compared to placebo after 12 weeks. Fowler 2014 studied this after 24 weeks.
Alitretinoin 40 mg versus placebo
For this subgroup, we found only one relevant trial (Ruzicka 2004, n = 159). There might be a difference between groups, as 43 out of 81 participants in the 40 mg group had clear or almost clear status for PaGA compared to 21 of 78 in the placebo group (RR 1.97, 95% CI 1.30 to 3.00; NNTB 4, 95% CI 2 to 9; Analysis 34.1; Table 17).
34.1. Analysis.
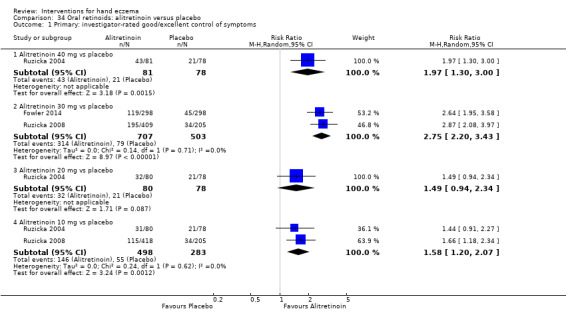
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms.
Alitretinoin 30 mg versus placebo
For this subgroup, we found two relevant trials (Ruzicka 2008, n = 614; Fowler 2014, n = 596). There was a clear difference between alitretinoin 30 mg and placebo, and the alitretinoin group was 2.75 times more likely to achieve symptom‐clear status compared to the placebo group (RR 2.75, 95% CI 2.20 to 3.43; NNTB 4, 95% CI 3 to 5; Analysis 34.1; Table 7; Table 17).
Alitretinoin 20 mg versus placebo
For this subgroup, we found only one relevant trial (Ruzicka 2004, n = 158). There was no clear difference between groups, as 32 out of 80 participants in the 20 mg group had clear or almost clear status for PaGA, compared to 21 of 78 in the placebo group (RR 1.49, 95% CI 0.94 to 2.34; Analysis 34.1; Table 17).
Alitretinoin 10 mg versus placebo
For this subgroup, we found two relevant trials (n = 781). According to both studies (Ruzicka 2004; Ruzicka 2008), alitretinoin 10 mg was more effective for this outcome (respectively, 39% and 28%) compared to placebo (RR 1.58, 95% CI 1.20 to 2.07; NNTB 11, 95% CI 6.3 to 26.5; Analysis 34.1; Table 8; Table 17). There might be a difference between groups, but we are uncertain of the strength of the evidence due to imprecision of the estimates.
Primary outcome: percentage of participants with self‐rated good/excellent control (clear or almost clear) with PaGA at week 12, at week 24, or at end of treatment
Ruzicka 2004 shows that for all doses of alitretinoin, statistically significantly more participants rated their eczema as clear or almost clear compared to those given placebo.
Alitretinoin 40 mg versus placebo
For this subgroup, we found only one relevant trial (Ruzicka 2004, n = 147). Of 74 participants in the 40 mg group, we judged that 32 had clear or almost clear status for PaGA compared to nine of 73 in the placebo group (RR 3.51, 95% CI 1.80 to 6.82; NNTB 3, 95% CI 2 to 6; Analysis 34.5). There might be a difference between groups, but we are uncertain of the strength of the evidence due to imprecision of the estimates.
34.5. Analysis.
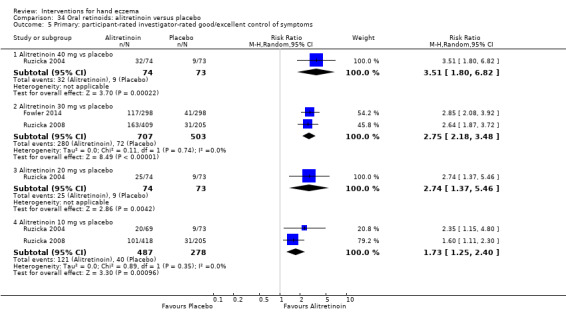
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 5 Primary: participant‐rated investigator‐rated good/excellent control of symptoms.
Alitretinoin 30 mg versus placebo
For this subgroup, we found two relevant trials (Ruzicka 2008, n = 614; Fowler 2014, n = 596). There might be a difference in the study of Ruzicka 2008: 163 out of 409 participants in the 30 mg group were judged as having clear or almost clear status for PaGA after 200 days or at the end of treatment, compared to 31 of 205 in the placebo group (RR 2.64, 95% CI 1.87 to 3.72; Analysis 34.5), but we are uncertain of the strength of the evidence due to imprecision of the estimates.
Pooling the data for alitretinoin 30 mg (heterogeneity statistics: Chi² test = 0.11, P = 0.74; I² = 0) gives an effect estimate that clearly favours the intervention group and demonstrates that the alitretinoin group was 2.75 times more likely to achieve improvement relative to the placebo group (RR 2.75, 95% CI 2.18 to 3.48; NNTB 4, 95% CI 3 to 5; Analysis 34.5; Table 7).
Alitretinoin 20 mg versus placebo
For this subgroup, we found only one relevant trial (Ruzicka 2004, n = 147). Of 74 participants in the 20 mg group, 25 were judged to have clear or almost clear status for PaGA compared to 9 of 73 in the placebo group (RR 2.74, 95% CI 1.37 to 5.46; NNTB 5, 95% CI 3 to 13; Analysis 34.5). There might be a difference between groups, but we are uncertain of the strength of the evidence due to imprecision of the estimates.
Alitretinoin 10 mg versus placebo
For this subgroup, we found two relevant trials (n = 765). Both studies found that 10 mg alitretinoin was more effective (respectively, 29% and 24% clear or almost clear) than placebo (Ruzicka 2004; Ruzicka 2008). Pooling these data for 10 mg alitretinoin (heterogeneity statistics: Chi² test = 0.89, P = 0.35; I² = 0) shows there might be a difference between groups, but we are uncertain of the strength of the evidence due to imprecision of the estimate (RR 1.73, 95% CI 1.25 to 2.40; NNTB 9, 95% CI 6 to 20; Analysis 34.5; Table 8).
Primary outcome: adverse events
Studies listed in detail the adverse events observed; headache was one of the most frequent events (22 in 40 mg group, eight in 20 mg group, four in 10 mg group, and seven in the placebo group in Ruzicka 2004; and 87 of 296 and 81 of 409 participants using alitretinoin 30 mg in Fowler 2014 and Ruzicka 2008, respectively). There was no clear difference between groups for 10 mg (Analysis 34.6), 20 mg (Analysis 34.7), or 40 mg (Analysis 34.9) versus placebo. However, the 30 mg versus placebo subgroup comparison produced a few notable between‐group differences (Analysis 34.8), specifically for the following adverse events: headache (RR 3.43, 95% CI 2.45 to 4.81; NNTH 6, 95% CI 4 to 11), flushing (RR 7.28, 95% CI 2.05 to 25.86; NNTH 25, 95% CI 17 to 50), erythema (RR 5.79, 95% CI 2.09 to 16.06; NNTH 25, 95% CI 14 to 100), nausea (RR 3.82, 95% CI 1.67 to 8.76; NNTH 27, 95% CI 18 to 56), elevated blood triglycerides (RR 7.05, 95% CI 1.89 to 26.28; NNTH 33, 95% CI 20 to 50), vomiting (RR 8.00, 95% CI 1.01 to 63.57; NNTH 50, 95% CI 23 to 250), and tinnitus (RR 4.33, 95% CI 1.25 to 15.05; NNTH 33, 95% CI 17 to 100). With the exception of headache, we have limited confidence in the clinical significance of the differences mentioned above because in most of these analyses, the number of events was too small; hence, this reduced the precision of the effect estimates. Limitations in the quality of the trial further compromised our confidence in this finding.
34.6. Analysis.
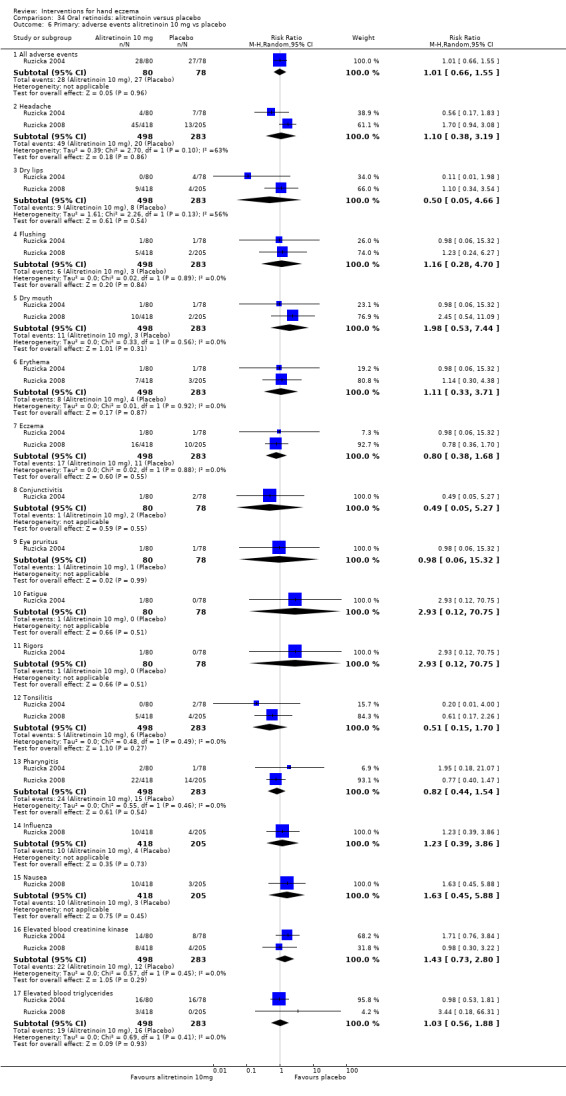
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 6 Primary: adverse events alitretinoin 10 mg vs placebo.
34.7. Analysis.
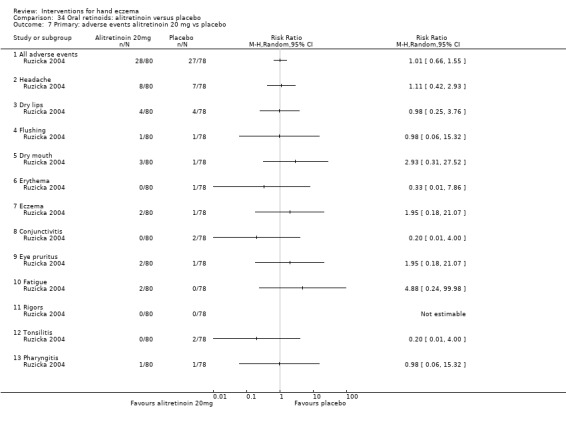
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 7 Primary: adverse events alitretinoin 20 mg vs placebo.
34.9. Analysis.
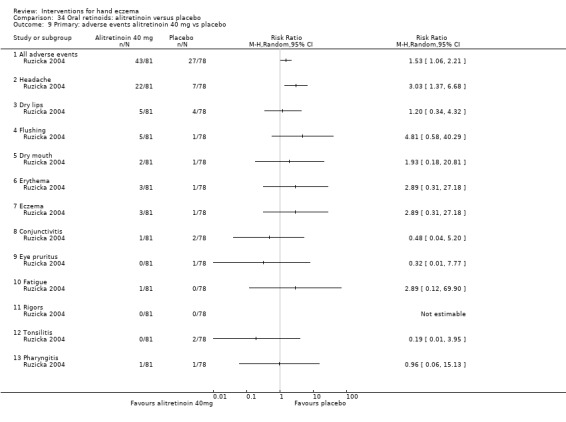
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 9 Primary: adverse events alitretinoin 40 mg vs placebo.
34.8. Analysis.
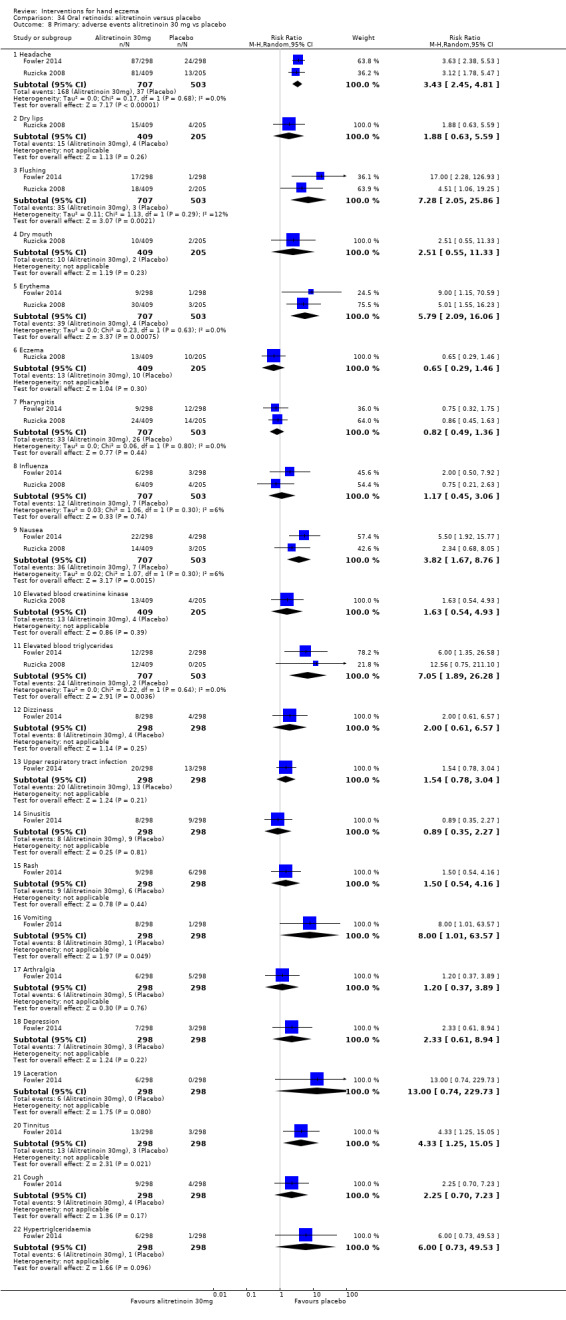
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 8 Primary: adverse events alitretinoin 30 mg vs placebo.
Some of the outcomes above had zero events in one arm, which is likely to explain the wide 95% confidence interval. These outcomes are dry lips, fatigue, rigours, tonsillitis, and elevated blood triglycerides.
Secondary outcome: reduction in severity, investigator‐rated, in total lesion symptom score
Ruzicka 2004 observed a higher median % reduction in total lesion symptom score for all doses of alitretinoin compared to placebo: 25% in the placebo group (stated 95% CI ‐42 to ‐14) versus 52% in the 20 mg group (stated 95% CI ‐73 to ‐42) and 71% in the 40 mg group (stated 95% CI ‐80 to ‐44; Analysis 34.10). The difference between alitretinoin and placebo was apparent for both doses according to study authors (Analysis 34.10). There was also reporting of a decrease in extent of disease in all groups, but no details were given. We have plotted these data (Analysis 34.11) based on the medians. Because the original data were not available to us, we were unable to assess whether the data were skewed; therefore, it is uncertain whether the medians are close to the means.
34.10. Analysis.
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 10 Secondary: investigator‐rated reduction in severity in total lesion symptom score.
| Secondary: investigator‐rated reduction in severity in total lesion symptom score | |
|---|---|
| Study | |
| Alitretinoin 40 mg | |
| Ruzicka 2004 | Median of % change from baseline: ‐70.5% (95% CI ‐44 to ‐80). |
| Ruzicka 2004 | Significant more reduction than placebo (P < 0.001; Kruskal‐Wallis test). |
| Alitretinoin 30 mg | |
| Ruzicka 2008 | Median of % change from baseline: ‐75% |
| Ruzicka 2008 | Significant more reduction than placebo (P < 0.001; Kruskal‐Wallis test). |
| Alitretinoin 20 mg | |
| Ruzicka 2004 | Median of % change from baseline: ‐52 (95% CI ‐42 to ‐73) |
| Ruzicka 2004 | Significant more reduction than placebo (P < 0.01; Kruskal‐Wallis test). |
| Alitretinoin 10 mg | |
| Ruzicka 2004 | Median of % change from baseline: ‐25% (95% CI ‐14 to ‐42) |
| Ruzicka 2004 | Significant more reduction than placebo (P < 0.01; Kruskal‐Wallis test). |
| Ruzicka 2008 | Median of % change from baseline: ‐56% |
| Ruzicka 2008 | Significant more reduction than placebo (P < 0.01; Kruskal‐Wallis test). |
34.11. Analysis.
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 11 Secondary: investigator‐rated reduction in severity in total lesion symptom score.
| Secondary: investigator‐rated reduction in severity in total lesion symptom score | ||||
|---|---|---|---|---|
| Study | Group | Median | SD | N |
| Alitretinoin 40 mg vs placebo | ||||
| Ruzicka 2004 | Alitretinoin 40 mg | 70.5 | 81.407 | 81 |
| Ruzicka 2004 | Placebo | 25.0 | 62.13 | 78 |
| Alitretinoin 20 mg vs placebo | ||||
| Ruzicka 2004 | Alitretinoin 20 mg | 52.0 | 80.9 | 80 |
| Ruzicka 2004 | Placebo | 25.0 | 62.13 | 78 |
| Alitretinoin 10 mg vs placebo | ||||
| Ruzicka 2004 | Alitretinoin 10 mg | 59.0 | 89.892 | 80 |
| Ruzicka 2004 | Placebo | 25.0 | 62.13 | 78 |
Alitretinoin 40 mg versus placebo
For this subgroup, we found only one relevant trial (Ruzicka 2004, n = 159). Only median data were available; hence, we reported these as 'other data' in a table (Analysis 34.11). The median of the alitretinoin group is evidently higher than that of the placebo group; however, we are unsure of the clinical importance of the observed difference.
Alitretinoin 20 mg versus placebo
For this subgroup, we found only one relevant trial (Ruzicka 2004, n = 158). Only median data were available; hence, we reported these as 'other data' in a table (Analysis 34.11). Similar to the previous analysis, the median of the alitretinoin group is evidently higher than that of the placebo group; however, we are unsure of the clinical importance of the observed difference.
Alitretinoin 10 mg versus placebo
For this subgroup, we found only one relevant trial (Ruzicka 2004, n = 158). Only median data were available; hence, we reported these as 'other data' in a table (Analysis 34.11). The median of the alitretinoin group is evidently higher than that of the placebo group, as in the previous analysis, and we are unsure of the clinical importance of the observed difference.
Secondary outcome: reduction in severity, investigator‐rated, in modified total lesion symptom score
Fowler 2014: the modified total lesion symptom score showed a change of ‐53.99% in the alitretinoin 30 mg group after 24 weeks and a change of ‐29.86% in the placebo group. For the mean difference in reduction, these numbers were inverted, so the mean difference in reduction of severity was 24.13 (MD 24.13, 95% CI 17.87 to 30.39; Analysis 34.12).
34.12. Analysis.
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 12 Secondary: reduction in severity, investigator‐rated in modified total lesion symptom score (bigger reduction in severity scored negative = better outcome).
Ruzicka 2008: the median reduction in the modified total lesion symptom score was 75% in the 30 mg group and 56% in the 10 mg group, compared to 39% in the placebo group (Analysis 34.10).
Secondary outcome: time to relapse
Fowler 2014 included a follow‐up phase up to 48 weeks after end of treatment. The median time to relapse after end of treatment was 83.0 weeks, with a 95% CI of 48.3 to 83.0, according to trial authors.
For Ruzicka 2008, the median time to relapse was 5.5 months for alitretinoin 30 mg, 6.2 months for alitretinoin 10 mg, and 5.4 months for placebo.
Comparison 35. Oral retinoids: re‐treatment with alitretinoin versus placebo
In Bissonnette 2010, 117 participants with chronic hand eczema were successfully treated with alitretinoin in an earlier study (Ruzicka 2008); 24 withdrew.
Primary outcome: percentage of participants with investigator‐rated good/excellent control (clear or almost clear)
Alitretinoin 30 mg versus placebo
For this subgroup, we found only one relevant trial (Bissonnette 2010, n = 73). A total of 39 out of 49 participants (80%) who were re‐treated with 30 mg alitretinoin were rated as 'clear' or 'almost clear' according to the PGA, compared to 2 of 24 participants (8%) who were re‐treated with placebo. There appears to be a large effect favouring the intervention group (RR 9.55, 95% CI 2.51 to 36.27; NNTB 1, 95% CI 1 to 2; Analysis 35.1; Table 17); however, we have limited confidence in this finding due to the small sample size and risk of bias in the study itself.
35.1. Analysis.
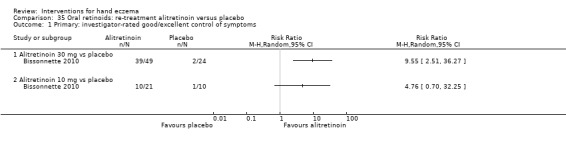
Comparison 35 Oral retinoids: re‐treatment alitretinoin versus placebo, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms.
Alitretinoin 10 mg versus placebo
For this subgroup, we found only one relevant trial (Bissonnette 2010, n = 31). Ten out of 21 participants were cleared or almost cleared again under re‐treatment with 10 mg alitretinoin in comparison to 1 out of 10 participants who were re‐treated with placebo (10%) (RR 4.76, 95% CI 0.70 to 32.25; Analysis 35.1; Table 17). In the group that was re‐treated with placebo, 9 out of 13 participants (69%) responded again.
Primary outcome: adverse events
Headache was the most frequently reported adverse event in the 30 mg group; 7 of 50 participants reported headache in the intervention group compared with zero events in the placebo group (Fisher's exact test P = 0.0129; RR 13.82, 95% CI 0.81 to 235.45; Analysis 35.3). None of the participants in the alitretinoin 10 mg group or in the placebo group reported headache. Adverse events occurred similarly in both groups (Analysis 35.2; Analysis 35.3). Three serious adverse events were reported: one case of acute cardiac failure with fatal outcome in the 10 mg group, which was not related to the study drug; one case of aortic aneurysm and one case of coronary artery disease (both in the 30 mg group) were assessed as having a remote relationship to the study drug.
35.3. Analysis.

Comparison 35 Oral retinoids: re‐treatment alitretinoin versus placebo, Outcome 3 Primary: adverse events 30 mg vs placebo.
35.2. Analysis.
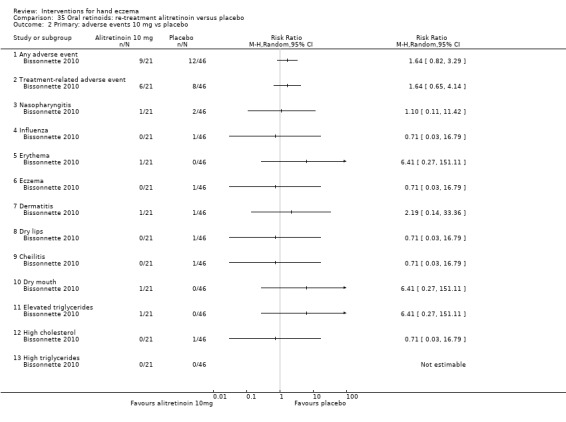
Comparison 35 Oral retinoids: re‐treatment alitretinoin versus placebo, Outcome 2 Primary: adverse events 10 mg vs placebo.
Comparison 36. Other oral interventions: oral triethylenetetramine versus placebo
Burrows 1986 (n = 23) studied oral triethylenetetramine versus placebo and included exclusively nickel‐sensitive participants.
Primary outcome: percentage of participants with investigator‐ and participant‐rated good/excellent control
We found one study for this outcome (Burrows 1986), including 23 participants in a cross‐over design, of which 20 were analysed. Because the data before cross‐over are not available, rather than analysing the post‐cross‐over data, we have presented them in a table for the readers' review (Analysis 36.1; Table 18). This outcome was based on a global assessment (improved/no change/deterioration) by participant and doctor, probably by consensus, and included both phases of the cross‐over study.
36.1. Analysis.
Comparison 36 Other oral interventions: oral triethylenetetramine versus placebo, Outcome 1 Primary: investigator‐ and/or participant‐rated improvement.
| Primary: investigator‐ and/or participant‐rated improvement | |||
|---|---|---|---|
| Study | Group ‐ within‐participant study | Event | Total number of participants analysed |
| Burrows 1986 | Trientine | 6 | 20 |
| Burrows 1986 | Placebo | 10 | 20 |
Primary outcome: adverse events
None of the participants in Burrows 1986 reported adverse events; however, this trial was ended prematurely due to increased teratogenicity among rats who received trientine.
Comparison 37. Other oral interventions: tetraethylthiuram disulfide (TETDS) versus placebo
Kaaber 1983 studied oral tetraethylthiuram disulfide (TETDS) versus placebo in 24 nickel‐sensitive participants.
Primary outcome: percentage of participants with investigator‐rated good/excellent control during treatment period
For this outcome, we found only one relevant trial (Kaaber 1983, n = 24). Among participants receiving the active compound, 5 out of 11 'healed' versus 2 out of 13 in the placebo group. Analysis 37.1 shows no clear difference between the two groups in that the 95% CI includes 1 and is wide (Fisher's exact test P = 0.1819; RR 2.95, 95% CI 0.71 to 12.34). Also see Table 18.
37.1. Analysis.
Comparison 37 Other oral interventions: oral tetraethylthiuram disulfide (TETDS) versus placebo, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms during treatment period.
Primary outcome: adverse events
In the group receiving tetraethylthiuram disulphide, hepatic toxicity was experienced in two participants (Fisher's exact test P = 0.48; RR 5.00, 95% CI 0.26 to 96.13; Analysis 37.2) and headache in one (Fisher's exact test P = 1.00; RR 3.00, 95% CI 0.13 to 68.26; Analysis 37.2). Two participants had mild acne, but it is not clear to which group they were assigned. In the intervention group, there was one case of discontinuation due to depression and one case of discontinuation due to dyspepsia, whereas neither event occurred in the control group (Fisher's exact test P = 1.00; Analysis 37.2). There were zero events in placebo group for all of the above subgroups; this is likely to explain the wide 95% confidence intervals.
37.2. Analysis.
Comparison 37 Other oral interventions: oral tetraethylthiuram disulfide (TETDS) versus placebo, Outcome 2 Primary: adverse events.
Comparison 38. Other oral interventions: low‐nickel diet (LND) and disulphiram versus normal diet and placebo
Sharma 2006 (n = 21) compared a low‐nickel diet combined with disulphiram versus a normal diet and placebo and included exclusively nickel‐sensitive participants.
Primary outcomes: percentage of participants with self‐rated good/excellent control (clearance of eczema) after four weeks
For this outcome, we found only one relevant trial (Sharma 2006, n = 21). Ten of the 11 participants in the LND group reached good/excellent control compared to 1 of 10 in the control group (Fisher's exact test P = 0.0003; RR 9.09, 95% CI 1.40 to 58.91; NNTB 1, 95% CI 1 to 2; Analysis 38.1).
38.1. Analysis.
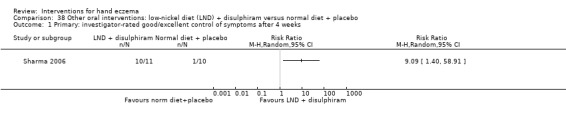
Comparison 38 Other oral interventions: low‐nickel diet (LND) + disulphiram versus normal diet + placebo, Outcome 1 Primary: investigator‐rated good/excellent control of symptoms after 4 weeks.
Primary outcome: adverse events
Three out of 11 participants treated with disulphiram experienced a metallic taste (Fisher's exact test P = 0.2143; RR 6.42, 95% CI 0.37 to 110.71; Analysis 38.2), and two had mild drowsiness (Fisher's exact test P = 0.4762; RR 4.58, 95% CI 0.25 to 85.33; Analysis 38.2 ). Three participants treated with disulphiram showed mild elevation of liver enzymes (Fisher's exact test P = 0.2143; RR 6.42, 95% CI 0.37 to 110.71; Analysis 38.2).
38.2. Analysis.
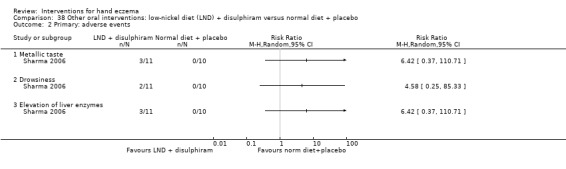
Comparison 38 Other oral interventions: low‐nickel diet (LND) + disulphiram versus normal diet + placebo, Outcome 2 Primary: adverse events.
Comparison 39. Other oral interventions: oral evening primrose oil versus placebo
Secondary outcome: reduction in severity, investigator‐rated score at week 24
For this outcome we only found one relevant trial (Whitaker 1996, n = 34) on oral gamma‐linoleic acid (GLA, evening primrose oil, Epogam). Mean and SD of the evening primrose oil group is 18 ± 12.37, and for the placebo group is 30.4 ± 23.36. There was no clear difference between the oral interventions (MD ‐12.40, 95% CI ‐25.46 to 0.66, Analysis 39.1).
39.1. Analysis.
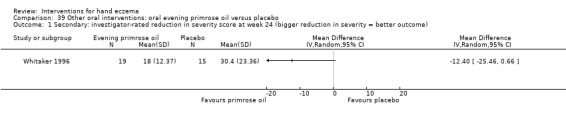
Comparison 39 Other oral interventions: oral evening primrose oil versus placebo, Outcome 1 Secondary: investigator‐rated reduction in severity score at week 24 (bigger reduction in severity = better outcome).
Comparison 40. Other oral interventions: ranitidine versus placebo
Primary outcome: percentages of participants with self‐ and/or investigator‐rated good/excellent control (clearance / marked alleviation)
For this outcome, we found only one relevant trial (Veien 1995, n = 47). Although it is not clear whether this was participant‐ or investigator‐rated, 17 out of 23 with ranitidine cleared or were markedly improved versus 8 out of 24 receiving placebo (RR 2.22, 95% CI 1.20 to 4.10; NNTB 2, 95% CI 2 to 7; Analysis 40.1; Table 18).
40.1. Analysis.
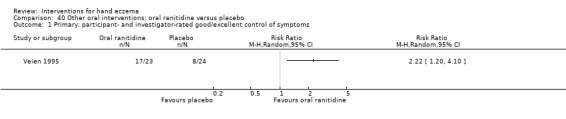
Comparison 40 Other oral interventions: oral ranitidine versus placebo, Outcome 1 Primary: participant‐ and investigator‐rated good/excellent control of symptoms.
Primary outcome: adverse events
No adverse events were reported in the ranitidine or placebo group.
Comparison 41. Other oral interventions: disodium cromoglycate diet (DSCG) versus low‐nickel diet
For this comparison, we found one study (Pigatto 1990), which included 16 participants in three different treatment groups (disodium cromoglycate diet (DSCG) versus low‐nickel diet versus a non‐randomised control for eight participants who did not give consent for the study and were only followed up). Because participants were not randomised, this subgroup is deleted from the review.
Primary outcomes: number of participants with self‐rated good/excellent control of itch after three months
For this outcome, we found only one relevant trial (n = 16) (Pigatto 1990). The numbers of events in the disodium cromoglycate and low‐nickel groups were 5 of 8 and 1 of 8, respectively (Fisher's exact test P = 0.1189; RR 5.00, 95% CI 0.74 to 33.78; Analysis 41.1).
41.1. Analysis.
Comparison 41 Other oral interventions: disodium cromoglycate diet (DSCG) versus low‐nickel diet, Outcome 1 Primary: participant‐rated good/excellent control of symptoms (itch) after 3 months of itch in DSCG versus diet.
Discussion
Summary of main results
Hand eczema is a common condition. In light of the high prevalence of hand eczema, it is striking that the results of all 60 identified randomised controlled trials (RCTs) are based on approximately 5469 participants, whereby about half of them (n = 2893) were enrolled in five RCTs: three on the oral retinoid alitretinoin (Fowler 2014; Ruzicka 2004; Ruzicka 2008), and two on the topical calcineurin inhibitor pimecrolimus (Belsito 2004; Hordinsky 2010).
Although many systematic reviews focus on a single treatment modality or its closely related variants, we have tried to include all interventions in this review in an attempt to determine which therapy would reflect current standard treatment and the extent to which there is evidence for its effectiveness. The wide range of available treatments underlines the fact that there does not seem to be a single candidate for standard therapy. Topical corticosteroids and ultraviolet (UV) phototherapy are the major treatment options for chronic hand eczema, although in this review, we found little strong or consistent evidence that one intervention for hand eczema should be recommended over the other.
About half of the studies (n = 33) included our primary outcome of good/excellent control of symptoms rated by participants or by investigators. The definition of good/excellent control varied across studies because a wide variety of outcome measures were used. Most studies included the primary outcome adverse events (n = 55). None of the adverse events were life‐threatening, and most were mild (local irritation with stinging, erythema, and burning).
Of the nine trials on topical corticosteroids, each dealt with a different type of steroid. The duration of six studies was rather short, namely, one week (Gupta 1993), two weeks (Faghihi 2008; Fowler 2005; Kircik 2013; Lodén 2012a; Uggeldahl 1986), and three weeks (Bleeker 1989). Treatment in Veien 1999 lasted up to 36 weeks, and treatment duration in Möller 1983 is unknown. Three trials compared two different corticosteroids. The comparators used in the remaining trials were the same corticosteroids as those used for the intervention, the same treatment but in a different vehicle, or a different dosage, or they were applied at a different frequency or were combined with zinc sulphate or urea, or consisted of vehicle alone.
Based on one study (125 participants), which compared clobetasol propionate foam with vehicle, clobetasol probably improves participant‐rated good/excellent symptom control more than vehicle; however, the difference between groups on observer‐rated scales is less clear (moderate‐certainty evidence) (Kircik 2013). Another study (72 participants) compared mometasone furoate cream used thrice weekly compared to twice weekly, and mometasone furoate cream used thrice weekly may slightly improve investigator‐rated good/excellent control of symptoms (low‐certainty evidence); participant‐rated symptoms were not measured (Veien 1999). See Table 1 and Table 2.
The 10 trials on UV phototherapy were too heterogeneous for pooling. Three studies provided UVB as the main intervention, three gave UVA‐1, and six used oral psoralen combined with UVA (PUVA) as the main intervention or comparator. Other comparators included no treatment, placebo, UVB, the same treatment as the intervention but at different sites, UVA, and topical betamethasone‐valerate cream. One study had a treatment duration of less than one month (Polderman 2003), five had a treatment duration of less than two months (Adams 2007; Bayerl 1999; Grattan 1991; Said 2010; Sjövall 1987), three had a treatment duration of two to four months (Brass 2015; Sezer 2007; van Coevorden 2004a), and one had a treatment duration greater than four months (Tzaneva 2009).
In one of the studies comparing local narrow‐band UVB to local PUVA, results showed that PUVA may lead to an improvement in investigator‐rated good/excellent symptom control (60 participants), but the 95% confidence interval indicates that local PUVA might make little or no difference (moderate‐certainty evidence). Participant‐rated symptoms were not measured (Brass 2015). See Table 3.
The topical calcineurin inhibitors were studied in nine RCTs, and almost all studies compared tacrolimus or pimecrolimus to vehicle. Based on one small study comparing tacrolimus over two weeks to vehicle, investigator‐rated good/excellent symptom control is probably more likely to be achieved in those treated with tacrolimus (14/14 participants in the tacrolimus group versus zero people in the vehicle group), but participant‐rated good/excellent control of symptoms was not measured (28 participants) (moderate‐certainty evidence) (Pacor 2006). Tacrolimus was compared to mometasone in a within‐participant trial but did not measure investigator‐ or participant‐rated good/excellent symptoms (Schnopp 2002). See Table 4 and Table 5.
Three studies assessed immunosuppressants, which were compared against a steroid or a retinoid. In one cross‐over RCT comparing oral cyclosporin to topical betamethasone dipropionate, cyclosporin probably slightly improves participant‐ or investigator‐rated good/excellent control of symptoms (34 participants) (moderate‐certainty evidence) (Granlund 1996). See Table 6.
A relatively new treatment option is oral alitretinoin, which has been compared with placebo in three large trials, with a total enrolment of 1947 participants (Fowler 2014; Ruzicka 2004; Ruzicka 2008). These trials investigated, in addition to other dosages, a daily dosage of 10 mg. Ruzicka 2004 and Ruzicka 2008 were considered sufficiently equivalent to pool the data for 10 mg daily, which showed that alitretinoin was more effective than placebo in both investigator‐ and participant‐rated good/excellent control of symptoms (high‐certainty evidence). Even larger risk ratios were observed when a higher dosage of alitretinoin (30 mg) was compared to placebo for both outcomes (Fowler 2014; Ruzicka 2008) (high‐certainty evidence). See Table 7 and Table 8.
Oral alitretinoin has not yet been compared to other treatment modalities such as corticosteroids or UV phototherapy. Unfortunately, the study that was expected to further clarify the position of systemic treatments with retinoids or systemic immunosuppressants (alitretinoin versus cyclosporin) in the treatment of hand eczema was ended prematurely (NCT01231854). Although this study shows low risk of bias in all other domains, it included only 15 of the required 78 participants.
Adverse events were reported by 55 of the 60 studies; they were generally mild and similar between groups. Mild atrophy was reported with mometasone furoate thrice weekly or twice weekly (low‐certainty evidence), but more adverse events (e.g. application site burning/pruritus after intervention application, nasopharyngitis, one incident of severe fissures) were noted when clobetasol propionate foam was compared to vehicle placebo (moderate‐certainty evidence). In the study comparing local narrow‐band UVB to local PUVA, only the narrow‐band UVB group reported adverse events (mainly erythema) (moderate‐certainty evidence). When tacrolimus was compared to vehicle, well‐tolerated burning/itching was reported only in the tacrolimus group (moderate‐certainty evidence). With systemic treatment, the risk of adverse events with oral cyclosporin compared to topical betamethasone was similar, and dizziness was reported (moderate‐certainty evidence). The occurrence of headaches and flushing was similar when alitretinoin 10 mg was compared to placebo (moderate‐certainty evidence), but risk of headache was greater with alitretinoin 30 mg than with placebo (high‐certainty evidence).
Overall completeness and applicability of evidence
We included 60 RCTs with a total of 5469 participants. Overall, studies included adults of both genders in general good health, which in our opinion is applicable to an important part of the hand eczema population, since hand eczema can be related to occupation.
The applicability of evidence is limited by several methodological weaknesses of the included studies; one of the most prominent of these is the varied definition of hand eczema. Furthermore, the definition of hand eczema was different in almost all trials. Studies defined 'chronic hand eczema' as duration longer than six months or longer than three months, or did not include a minimal duration of disease at all. We intended to conduct subgroup analyses, but a minority of the trials defined subgroups (e.g. Ruzicka 2004 and Ruzicka 2008 did include subgroups). In general, it is not clear which participants had hyperkeratotic hand eczema or vesicular hand eczema, and clinical experience suggests that the clinical subtype might influence treatment success. Without logical and comprehensive definitions of hand eczema with clear diagnostic criteria for hand eczema and its subgroups, RCTs are seriously flawed, which is one of the main pitfalls of this review.
Furthermore, in this review, we found a wide range of severity scoring systems for hand eczema, which prevented meaningful data pooling.
Finally, some studies, especially older studies, did collect useable data with regard to the effectiveness of treatment but did not report these data (Fredriksson 1975). Since these were all single‐study results, which could not have been pooled anyway, we do not believe that this influenced the overall completeness of the evidence.
Of all treatment categories, the largest number of studies focus on topical steroids (nine RCTs) and UV therapy (10 RCTs). Nevertheless, most trials do not include one of these treatments as a comparator. In fact, most trials provide placebo, vehicle, or a variant of the intervention as a comparator, making it difficult to draw conclusions on the comparative advantages of different treatments. We did identify some ongoing studies, for example, ISRCTN80206075,NCT03026907, and NCT03026946; results of these trials might eventually help to fill some of the gaps. With regard to phototherapy, it is difficult to compare different studies because different treatment regimens were used. Although in daily practice the treatment regimens are highly dependent on patient skin type and on the occurrence of adverse events, it might be challenging to align treatment protocols. Topical steroids were assessed in nine studies, although different treatment regimens were not investigated intensively, nor was the strength of different corticosteroids. Topical calcineurin inhibitors were investigated in nine studies. Topical calcineurin inhibitors were compared with placebo and with active treatment (topical corticosteroids), although this last comparator might have been used more often. Alitretinoin was examined in well‐designed studies with a substantial number of participants. Other oral treatments such as cyclosporin, methotrexate, or acitretin were barely/not investigated, which is a severe shortcoming in the overall completeness of evidence.
With regard to outcomes, our primary outcomes percentage of participants with investigator‐rated and/or self‐rated good/excellent control of symptoms and/or adverse events were reported in most of the included studies. However, the secondary outcome dose reduction was not stated in any of the included studies. Moreover, various studies did not report on time until relapse.
The enrolled participants had typical long‐standing eczema. Studies included overall chronic hand eczema with long‐lasting disease and included patients in secondary care settings; therefore acute eczema is not included in these studies. Consequently, the results are less applicable for the primary care setting. This review included participants of all ages; however, most of the included studies did not include children. Only four studies included participants under the age of 16 years and did not provide separate results for this subgroup; therefore, the results of this review may not be applicable to children. With regard to external validity, these studies were conducted all over the world, supporting the generalisability of the results.
The objective of this Cochrane Review was to assess the effects of topical and systemic interventions for hand eczema in adults and children. Because of the above‐mentioned implications, it is difficult to answer our review question with a single answer. As stated before, we cannot comment on children based on the included studies, which mainly include adults. However, this review does provide a clear overview of different studies on potential topical and systemic interventions for adult patients with chronic hand eczema. A pitfall is the lack of head‐to‐head studies, which makes it impossible to know whether one treatment is favoured over another.
Quality of the evidence
We found some serious limitations in the quality of reporting and aimed to discuss these according to the GRADE considerations(study limitations, consistency of effect, imprecision, indirectness, and publication bias).
Limitations in study design and implementation
We included only RCTs in this review. Overall, the older studies had more shortcomings with regard to risk of bias, and we judged them as having 'high' or 'unclear' risk of bias with regard to allocation concealment, blinding, and/or loss to follow‐up. Frequent shortcomings included missing information on randomisation and blinding, no justification of the number of participants, and no analysis of dropouts. Studies that were conducted more recently had an overall low or unclear risk of bias for most of the risks (allocation concealment, blinding, intention‐to‐treat analysis, and selective reporting of outcomes), although they sometimes were sponsored by pharmaceutical industries. Over a third of the studies used a within‐participant design (left‐right studies). Although these studies show strengths in terms of power to obtain statistically significant results with small numbers of participants, this is done at the expense of problems in interpreting studies finding no difference in effect. This might be a consequence of cross‐contamination of topical interventions, possible systemic effects of topical preparations, or both. In general, we consider the body of evidence in this review as having 'unclear risk of bias'.
As a consequence, we downgraded evidence only for one of our main comparisons (mometasone furoate cream three times per week versus two times per week; Table 2), as we considered the included study to be at high risk of detection and performance bias (Veien 1999).
Indirectness of evidence
Overall the included studies were of relatively small sample size and short duration. Although hand eczema usually has a chronically relapsing course, less than half of the studies had a duration longer than three months, which in our opinion is the minimum duration required to document important data such as duration and frequency of disease relapse. Therefore, this is considered as a form of indirectness. This review analysed the efficacy of many different interventions, of which various included a placebo. Moreover, because the number of studies comparing different groups of interventions (e.g. corticosteroids, oral retinoids, phototherapy) is limited and the number of participants for each intervention is limited (with the exception of alitretinoin), the evidence is mostly indirect. Overall, participants in a secondary care setting with chronic eczema were included. As mentioned above, the definition of 'chronic hand eczema' was not always clear; this could be defined as having a minimal duration of six weeks to six months. Some studies included participants with specific subtypes of hand eczema such as recurrent vesicular hand eczema, whereas others excluded this subgroup. We were unable to pool the data for different subgroups of hand eczema, for example, to focus on hyperkeratotic palmar hand eczema, since the data for specific subgroups often were not stated. A wide range of outcome parameters was presented, most of which were not validated. Some studies used a validated outcome measure such as the Hand Eczema Severity Index (HECSI), whereas most created their own non‐validated, un‐named scoring system. Another limitation arose from the comparators used: most interventions were compared to an inactive placebo, which is less effective than standard treatment in most settings. We decided not to downgrade the evidence in our main comparisons for indirectness, as we judged this to be a less serious concern than imprecision (see below).
Consistency of results
It is difficult to judge the consistency of the results because we were unable to pool the study results for most of the outcomes assessed because only a single study was available, or because of clinical heterogeneity in interventions (and co‐interventions), treatment duration, comparison groups, and outcomes measured or reported. As a direct consequence of the overwhelming diversity in study characteristics (i.e. clinical heterogeneity), most of the comparisons are based on single studies, hence making it difficult for the review authors to draw any firm conclusions with confidence. We can interpret this review only as a scoping review. Hence, we could not downgrade any evidence for inconsistency.
Imprecision of results
Most of the analyses are based on a single study of small sample size, and often with low event rates (in some cases, zero events), and the 95% confidence interval of effect estimates was often very wide, resulting in a low‐precision assessment. Hence, we downgraded most of the outcomes included in the summary of findings tables for imprecision because we believe the small sample size means there was not enough power to detect any differences between groups. The effectiveness outcomes in two of our main comparisons comparing alitretinoin 10 mg or 30 mg versus placebo were not downgraded for imprecision because the analyses included two studies equalling a larger sample size, and the results had fairly narrow 95% confidence intervals, which did not include one showing high‐certainty evidence supporting the effectiveness of alitretinoin (see Table 7 and Table 8).
Probability of publication bias
We did not produce funnel plots due to insufficient numbers of included trials for all given outcomes. Publication bias may especially be present in the wide range of studies on different moisturisers to treat hand eczema: we did find various registered trials in trial registries that were not (yet) published. Contact with study authors in some cases revealed that the results were minimal and would not be published, or study authors did not respond to our writings at all. In other cases, study authors were not at liberty to disclose results but referred us to pharmaceutical sponsors, who often remained unresponsive.
Potential biases in the review process
We acknowledge that there was potential for bias at all stages of the review process, but we made various attempts to restrict the level of bias.
We comprehensively searched for randomised controlled trials from a wide range of databases to avoid the risk of publication bias, and we used clinically relevant outcome measures. We tried to compare respective trial registrations with published trials to ascertain whether there was lack of correspondence between what was intended to be an outcome and actually reported outcomes. We attempted to be as inclusive as possible in our search strategy and included studies reported in languages other than English. The different language backgrounds of review authors enabled us to include Dutch ‐ Kemper 1998 ‐ and German articles ‐ Adams 2007; Bayerl 1999. We translated a Turkish article to minimise language bias (Baskan 2005). Nevertheless, the studies included in this review were predominantly conducted in European or North American countries and were published in European or American journals.
The authors of this review independently assessed the eligibility of studies for inclusion in this review; two other review authors extracted data and assessed risk of bias to minimise the potential for additional bias beyond that detailed in the 'Risk of bias in included studies’ tables. Discrepancies between review authors were resolved by discussion to reach consensus. However, we acknowledge that our assessments may occasionally have been subjective, for example, in the case of the not‐blinded radiographers (Cartwright 1987; Fairris 1984; Fairris 1985; King 1984). Therefore, readers may not agree with all of our decisions.
Review authors who were involved as trialists for certain studies were not involved in selection, assessment, and data extraction for those studies. Pieter‐Jan Coenraads was involved in the studies of Ruzicka 2004, Ruzicka 2008, and van Coevorden 2004a. Thomas L Diepgen was involved in the studies of Bauer 2012 and Ruzicka 2008 (Declarations of interest).
The authors of this review are aware that some differences between protocol and review (see Differences between protocol and review) may have been a source of bias. The protocol was published in 2009, and Cochrane guidance has since developed. Such differences include changing adverse events to a primary outcome, adding a time point of a minimum of three months for measuring outcomes such as relapse, changing the way measures of treatment effect are expressed, and making changes to the literature search. We tried to not make these decisions based on the data we had extracted, but rather on the new Cochrane guidance.
We judged a lot of studies to have unclear risk of bias, especially with regard to selection bias, since a substantial number of studies did not describe the way allocation concealment and sequence generation were performed. To obtain more clarity on this matter, we contacted all authors from studies published after 1999 by email or through other forms of social media such as LinkedIn. Study authors with a personal or professional relation to one of the authors of this review may have been reached more easily and might have been more prone to respond to our requests. Therefore, these studies may have been judged more often as having low risk of bias. For studies pre‐1999 and for reports for which study authors were unresponsive, we had less information and had to deal with more ambiguity, which we were unable to resolve; this may have contributed to some bias in assessments of these studies, and these studies were more often judged as having unclear risk of bias.
The time frame for the studies included in this review inevitably shows that there is a time trend in treatments that are evaluated: earlier studies tend to focus on corticosteroids, UV phototherapy, or X‐rays, and more recent trials evaluate the effects of novel medicaments such as oral retinoids and topical calcineurin inhibitors.
The fact that 20 studies have not yet been incorporated into this review may be a source of potential bias.
Agreements and disagreements with other studies or reviews
This Cochrane Review studies a wide range of treatments that have been evaluated by RCTs since 1967. Within the same time frame, many uncontrolled and non‐randomised controlled studies have been published. van Coevorden 2004b conducted a review to describe study design and the quality of studies on hand eczema, covering the time period from 1977 up to 2003. These review authors included 90 studies, of which 44 were case series, 15 non‐randomised controlled trials, and 31 RCTs. In total, 11 different categories of treatment were found, and most trials studied ultraviolet irradiation (n = 32) or corticosteroids (n = 13). This review concluded that the overall quality of reporting on hand eczema was poor, and most hand eczema trials were not considered adequate to guide clinical practice. Since the current Cochrane Review was conducted in part by the same review authors and incorporated the same RCTs, it is not surprising that the results and conclusions of both reviews overlap. However, since we tried to obtain more information by contacting authors in this Cochrane Review, and since we sought additional published and unpublished data, we had fewer uncertainties with regard to the quality of evidence. We also included RCTs that were conducted after 2003, and in general these studies are of better quality than the older studies. We maintain that the overall quality of reporting in hand eczema is low, and that there is a need for well‐designed head‐to‐head studies of adequate duration, reported according to the CONSORT guidelines.
Over the years, various groups have composed guidelines for the management of (chronic) hand eczema (Veien 2003; Diepgen 2007; Diepgen 2009b; English 2009; Lynde 2010; Menné 2011). These guidelines have in common that they all acknowledge the lack of RCTs. All guidelines recommend topical corticosteroids as one of the first steps in pharmacological treatment for all types of hand eczema. Thereafter, the guidelines recommend different steps, in which the subtype of hand eczema can be a leading factor, usually starting with topical treatments (e.g. more potent or prolonged use of corticosteroids or calcineurin inhibitors).
For severe hand eczema that is unresponsive to topical treatment, basically all guidelines recommend a treatment regimen with tar, phototherapy, and systemic (oral) treatment (acitretin, alitretinoin, cyclosporin, corticosteroids, or others). Since alitretinoin was recently licensed for the treatment of hand eczema in Europe and Canada (not yet in the United States), the more recent guidelines include this treatment option for severe chronic hand eczema (Diepgen 2009b; English 2009; Lynde 2010; Menné 2011).
English 2009 published a consensus statement on the management of chronic hand eczema in the view of general practitioners and dermatologists. The authors did not conduct a systematic review but based their statement on a mix of clinical experience and a variety of RCTs and non‐RCTs on hand eczema and atopic dermatitis. In general, they advise a skin protection programme and topical treatment with corticosteroids or calcineurin inhibitors in a primary care setting whenever possible. For referrals to secondary care (dermatologist), PUVA, cyclosporin, azathioprine, and alitretinoin are preferred treatment options for hyperkeratotic and vesicular hand eczema, with emphasis on the importance of patient preference and local availability. Furthermore, PUVA (also in our review a well‐studied intervention) is recommended for hyperkeratotic hand eczema. Methotrexate and mycophenolate are recommended after failure of other systemic interventions; however, this recommendation is not supported because of lack of RCTs examining these interventions.
The German Dermatologic Society stresses the importance of education and prevention (Diepgen 2009b). Topical corticosteroids, topical calcineurin inhibitors, and iontophoresis are the first treatment steps. For moderate to severe hand eczema, highly potent corticosteroids, UV therapy, and alitretinoin are recommended, while other systemic treatment options such as cyclosporin are the final resort. This recommendation is based largely on the fact that alitretinoin is registered for the treatment of hand eczema, while cyclosporin is an off‐label therapy.
The Canadian guideline states that treatment of hand eczema can be difficult and unsatisfactory (Lynde 2010). Researchers distinguish three important clinical types: irritant contact dermatitis, allergic contact dermatitis, and atopic hand eczema. This guideline provides a clear flow diagram for acute hand eczema and chronic hand eczema, which is divided into mild, moderate, and severe. Topical corticosteroids are the mainstream of treatment, and phototherapy is recommended for moderate chronic hand eczema unresponsive to topical corticosteroids. For severe cases that are unresponsive to potent topical corticosteroids, phototherapy and alitretinoin are recommended. When this is insufficient as well, cyclosporin can be considered. Because the comparative study NCT01231854 was ended prematurely, we do not know whether alitretinoin should be preferred over cyclosporin.
Danish guidelines state that treatment for hand eczema should be tailored to the individual and that skin care education is very important (Menné 2011). They classify hand eczema into six different clinical types: chronic fissured hand eczema, recurrent vesicular hand eczema, hyperkeratotic palmar hand eczema, pulpitis, interdigital eczema, and nummular hand eczema. Furthermore, they distinguish between mild/moderate hand eczema and severe hand eczema. Mild/moderate hand eczema should be treated with topical corticosteroids, potentially in rotation with calcineurin inhibitors. For severe hand eczema, a step‐up with topical corticosteroids and "possibly potassium permanganate baths" for vesicular hand eczema and "silver nitrate solutions" for hyperkeratotic eczema is recommended. However, our review did not find evidence for these treatment options. If topical treatment is insufficient, a further step‐up regimen is recommended with tar, phototherapy, and systemic treatment (acitretin, alitretinoin, cyclosporin, corticosteroids, or others), although the guideline does not given an order of priority and does not make further recommendations regarding the different subtypes of hand eczema.
The American Academy of Dermatology has published guidelines on the use of topical glucocorticoids ‐ the mainstay of treatment for hand eczema (Drake 1996). The British Photodermatology Group developed a guideline on phototherapy and included a comment on the use of phototherapy in hand eczema (Halpern 2000). Although the evidence for topical PUVA over oral PUVA is scarce, this group suggests a commonsense approach, which is not contradictory to the findings of this review.
The studies included in this review regarding alitretinoin did find an increase in the number of participants reporting headache while taking alitretinoin compared to placebo with a high level of evidence (Bissonnette 2010; Ruzicka 2004; Ruzicka 2008), which is in line with multiple daily life studies that have reported headache as a well known side effect of alitretinoin (Diepgen 2012; Augustin 2016).
Overall, we can conclude that most guidelines do not give a single recommendation based on the current literature, which is consistent with the main finding of this review.
Authors' conclusions
Implications for practice.
The results of this review cannot be used to inform clinical practice with regard to the best way of managing hand eczema, especially in the long term. Until such data are forthcoming, physicians will be tempted to use an array of treatments.
For the comparison of clobetasol propionate versus vehicle foam, the percentage of participants with self‐rated good/excellent control of symptoms probably improves with clobetasol propionate, but the effect is less clear for investigator‐rated symptoms (moderate‐quality evidence). Mometasone furoate cream thrice weekly may slightly improve investigator‐rated symptoms compared to twice weekly application (low‐quality evidence); participant‐rated control was not measured.
Tacrolimus ointment probably improves investigator‐rated good/excellent control of symptoms compared to vehicle foam (moderate‐quality evidence); participant‐rated control was not measured.
A relatively new systemic treatment (an oral retinoid called alitretinoin) for patients with severe chronic hand eczema showed clearance or almost clearance of about half the participants in three large RCTs (Fowler 2014; Ruzicka 2004, Ruzicka 2008). We found high‐quality evidence that relative to placebo, people who are given alitretinoin were more likely to achieve good symptom control (investigator or participant rated). The benefit became more apparent with increased dosage (10 mg versus 30 mg).
Local PUVA may lead to improvement compared to local narrow‐band UVB; however, the 95% confidence interval indicates that local PUVA might make little or no difference (moderate‐quality evidence). Participant‐rated control was not measured.
Oral cyclosporin probably slightly improves investigator‐/participant‐rated control of symptoms compared with topical betamethasone dipropionate (moderate‐quality evidence).
For the comparison tacrolimus 0.1% ointment versus mometasone furoate ointment, investigator‐rated symptoms and participant‐rated control was not measured.
Adverse events: adverse (long‐ and short‐term) effects of the interventions
Clobetasol propionate led to more adverse events (including application site burning/pruritus after intervention application, nasopharyngitis, and one incident of severe fissures) compared to vehicle foam (moderate‐quality evidence)
With regard to mometasone furoate cream used thrice weekly compared to twice weekly, mild atrophy was reported in both groups (low‐quality evidence)
When compared to local PUVA, adverse events (mainly erythema) were reported in the local narrow‐band UVB group only (moderate‐quality evidence)
With regard to tacrolimus ointment compared to mometasone furoate ointment, both treatments were well tolerated; none of the participants dropped out due to adverse events (moderate‐quality evidence)
When compared to vehicle foam, adverse events (well‐tolerated burning/itching at the application site) were reported in the group taking tacrolimus ointment only (moderate‐quality evidence)
The risk of adverse events such as dizziness was fairly similar between those taking oral cyclosporin and those taking topical betamethasone dipropionate (moderate‐quality evidence)
The 20 studies listed under Studies awaiting classification may alter the conclusions of the review once assessed.
Implications for research.
The most important implication of this review is the need to conduct high‐quality RCTs of people with hand eczema to compare commonly used interventions by using simple outcome measures that can be understood by participants and clinicians.
E (Evidence): current evidence for managing hand eczema is mainly of low to moderate certainty and especially head‐to‐head trials are missing. Recently, head‐to‐head trials for different (systemic) treatments have been registered in trial registries (ISRCTN80206075; NCT03026907; NCT03026946), which might alter the outcomes of this review in the near future.
P (Population): people with chronic (longer than six months) moderate to severe hand eczema should be included in future trials. Subgroup analyses on participants with different variants of hand eczema are recommended, although lack of consensus regarding the classification of hand eczema is a major limitation. We need international consensus regarding the definition of (chronic) hand eczema and subgroups of hand eczema, based on morphology or aetiology. Subgroups of especial interest include participants with hyperkeratotic hand eczema and participants with recurrent vesicular hand eczema. Studies on acute hand eczema were not included in this review but may be of interest, especially in primary care settings. Not many children were included in these studies, and this is a potential subgroup of interest for future studies.
I (Intervention): all sources of treatment can be included, although we would recommend including the main interventions (topical corticosteroids, UV therapy, topical calcineurin inhibitors, acitretin, alitretinoin, and cyclosporin).
C (Comparison): head‐to‐head trials, in which different groups of commonly used interventions are compared, are highly desirable, for example, cyclosporin versus alitretinoin or UVA therapy versus topical corticosteroids. If an RCT includes placebo (or vehicle or inactive treatment) as the only comparator instead of an established treatment modality, this should be clearly and convincingly justified.
O (Outcome): at the moment, international consensus on a standard severity scale for hand eczema is lacking. Many of the scales used were not validated, and validation of commonly used scoring systems is needed. Alternatively, a simple global rating measure with, for example, photographic anchors is highly recommended (Charman 2005; Weistenhöfer 2010). We would like to recommend the same procedure as is currently ongoing in atopic dermatitis: Harmonising Outcome Measures for Eczema (HOME) (Schmitt 2010). The HOME group is a worldwide initiative with the aim of developing a consensus‐based set of core outcome domains for trials and clinical record keeping in atopic dermatitis. This is important, to allow comparison of data across trials ‐ one of the difficulties that we encountered in this review on interventions for hand eczema. Duration of remission, the way the disease is brought under control, adverse events, focus on patient‐reported outcomes, and simple outcome measures applicable to all participants are preferable. Hand eczema is known to influence quality of life; therefore quality of life should be an important outcome. In addition, trials should focus on economic consequences, since hand eczema is a common occupational disease. A major limitation of almost all reviewed trials is that no measure of effect size including precision is given. This is necessary to enable judgement of whether advantages of treatments are not only statistically significant but also meaningful.
T (Time stamp): our latest search was conducted in April 2018. Older studies focused mainly on topical corticosteroids, UV therapy, and irradiation, and more recent (namely, industry‐funded) studies focus on topical calcineurin inhibitors (pimecrolimus and tacrolimus) and alitretinoin. The included studies were predominantly of short duration. Future studies should have adequate treatment duration, preferably longer than three months, which in our opinion is the minimum duration required to document important data such as duration and frequency of disease relapse. Furthermore, studies on chronic hand eczema should include a follow‐up period of at least equal duration. Acute hand eczema, especially the allergic type, tends to respond quickly to treatment and needs only a short follow‐up, in which case a few weeks of treatment and follow‐up should be sufficient.
It is obvious in many of the reviewed trials that the approach to statistical analyses was limited. Several parametric and non‐parametric statistical procedures that are able to model both within (person and/or time) and between subject (treatment) factors simultaneously have been offered by most statistical packages for many years. A major limitation of many of the treatment comparisons is that they did not control for baseline variation. In addition, omnibus factorial designs (allowing contrasts to be specified a priori) reduce the type 1 error rate because they test several hypotheses at the same time. Post‐hoc comparisons would be necessary only should the data reveal surprising results. These analyses, of course, would have to be viewed in an explorative fashion. Future studies need to overcome said limitations.
Many deficiencies in trial reporting thus far can be avoided if all specialist dermatology journals adopt the CONSORT guidelines (Moher 2001), especially since many of the 'unclear' risks, turned out to be based on missing information in the report instead of flaws in the study design. All future studies should adhere to these guidelines. Future studies should ensure they are adequately powered to detect any differences between treatment groups and to reduce imprecision.
Practical recommendations for upcoming studies include the above‐mentioned recommendations on chronic hand eczema. Studies that are highly recommended include the comparison of phototherapy (e.g. bath‐PUVA) versus alitretinoin 30 mg in a large cohort of participants with chronic hand eczema with a duration of at least three months and follow‐up of at least equal length. This study is already registered (ISRCTN80206075), and results of this trial might influence the outcomes of this review in future updates.
Another recommendation would be to compare alitretinoin 30 mg to cyclosporin in participants with vesicular hand eczema and with hyperkeratotic hand eczema, since participants with vesicular hand eczema seemed to respond less to alitretinoin in the included trials on alitretinoin. This study design is already registered for vesicular hand eczema as well, and we are awaiting the results (NCT03026946). Other potential research options include comparison of a potent topical corticosteroid, since this is the mainstream of treatment, to alitretinoin 30 mg or to phototherapy. The comparative advantage over other treatments needs further evaluation, since the only study that did compare alitretinoin to another immunosuppressant (cyclosporin) was ended prematurely (NCT01231854).
Acknowledgements
The steering committee of EDEN (European Dermato‐Epidemiology Network) has made valuable suggestions, and has participated in a feasibility study for this review.
The review authors would like to thank the Cochrane Review Group for its support.
Cathy Bennett Systematic Research Ltd. provided comments on draft versions of the review and provided great support throughout the process.
We would like to acknowledge all authors and trialists who provided additional information for this review: Dr. Uma Shankar Agarwal; Prof. Dr. Emel Bülbül Baskan; Christiane Bayerl; Chia‐Yu Chu; John English; Dr. Joubert G. Gama; Dr. Phil Hampton, PhD, FRC; P.F. Jowkar; Prof. Alan J. Fleischer; Prof. C.L. Goh; Dr. M. van Geel‐Kucháreková; Dr. J.M. Hanifin; Prof. A. Laumann; Prof. Gabriele Di Lorenzo; Claire Macdonald; Philippe Marchessault; T. Ruzicka; Prof. J. Schmitt; Dr. Nasrin Saki; Dr. Christina Schnopp; A.D.Sharma, MD; and A. Tanew, on behalf of S. Tzaneva, K. Thestrup‐Pedersen, Prof. N. Veien, and Dr. Vijaya Jaiswal.
We would like to acknowledge Marco van Coevorden for his contributions in drafting the protocol of this review, and Uwe Matterne for his contributions in searching and identifying studies, as well as performing data entry.
The Cochrane Skin editorial base wishes to thank Robert Boyle, who was the Cochrane Dermatology Editor for this review; Matthew Grainge, who was the Statistical Editor; Ching‐Chi Chi, who was Methods Editor; external content expert Miriam Wittmann; and the consumer referee, Amanda Roberts. We would also like to thank Dolores Matthews for copy‐editing the review.
Appendices
Appendix 1. Glossary of medical terms
| Medical term | Explanation |
| Acrovesicular eczema | Form of vesicular hand eczema. (Large) vesicle eruptions on the palms that usually tend to recur. Also called dyshidrotic eczema or pompholyx |
| Betamethasone | Topical corticosteroid, high potency |
| Clobetasol propionate | Topical corticosteroid, very high potency |
| Desonide | Topical corticosteroid, low potency |
| Dyshidrotic hand eczema | Form of vesicular hand eczema. (Large) vesicle eruptions on the palms that usually tend to recur. Also called dyshidrotic eczema, pompholyx or acro vesicular eczema |
| Fluprednidene acetate | Topical corticosteroid, medium potency |
| Heterogeneity | Differences in which studies have been undertaken with regard to methods and/or materials |
| Hydrocortisone butyrate | Topical corticosteroid, low potency |
| Hyperkeratotic hand eczema | Form of hand eczema with areas of thick scaling on the palms, also called tylotic hand eczema |
| IGA | Investigator global assessment: global assessment of disease severity usually on a 5‐point scale |
| Immunomodulator | Drug which changes the immune response such as tacrolimus |
| Immunosuppressor | Drug which suppresses the immune response such ad topical corticosteroids |
| Iontophoresis | Treatment by which the skin is soaked in (tap) water through which a weak electric current is passed |
| Mometasone furoate cream | Potent steroid cream |
| NB‐UVB | Narrow‐band ultraviolet B |
| Nummular hand eczema | Round ("coin sized") eczematous patches on the back of the hands |
| Palmar | Hand palms, the inside surface of the hands |
| Palmoplantar | Hand palms and foot soles |
| Phase I clinical trial | A clinical trial of a new drug or therapy. Phase I trials are conducted in small groups of participants |
| Phase II clinical trial | A clinical trial of a new drug or therapy. Phase II trials are conducted in larger groups of participants than phase I trials |
| Pimecrolimus | Topical calcineurin inhibitor, also known as "elidel" |
| Placebo | Simulated or otherwise medically ineffective treatment |
| Pompholyx | Form of vesicular hand eczema. (Large) vesicle eruptions on the palms that usually tend to recur. Also called dyshidrotic eczema, pompholyx or acro vesicular eczema |
| Potency | Strength |
| Prevalence | The proportion of a population having a particular condition or characteristic: e.g. the percentage of people in a city with hand eczema, or the proportion of people who smoke |
| Primary care | Health care provided at the principal point of consultation for patients within a healthcare system, e.g. GP |
| Pruritus | Itch |
| Psychosomatic disorder | A disorder in which physical symptoms originate from mental or emotional causes |
| Pulpitis | A dry, fissured, scaling dermatitis of the fingertips with occasional episodes of vesicles. Also known as fingertip dermatitis |
| PUVA | (Topical and oral) psoralen combined with UVA |
| Randomised control trials | A study in which a number of similar people are randomly assigned to two (or more) groups to test a specific drug, treatment or other intervention. (National Institute for Health and Care Excellence) |
| RR | Relative risk |
| Secondary care | Health care provided by medical specialists and other health professionals, including dermatologists, who generally do not have first contact with patients. This contains hospital and out‐patient care |
| Systemic treatment | Treatment which does not pertain to a certain surface area but might affect the entire body, usually taken by mouth or injection. |
| Tacrolimus | Topical calcineurin inhibitor, also known as "protopic" |
| Teratogenicity | Developmental abnormalities in the foetus |
| Therapy | A treatment that helps someone feel better, grow stronger, etc., especially after an illness (Cambridge dictionary) |
| Topical treatment | Treatment pertaining to a certain surface area (usually the skin) and only affecting the area to which it is applied |
| Transepidermal water loss (TEWL) | The amount of water that moves from inside the body to the surrounding atmosphere through the epidermal layer of the skin by means of diffusion and evaporation. |
| Tylotic hand eczema | Form of hand eczema with areas of thick scaling on the palms, also called hyperkeratotic hand eczema |
| UVA‐1 | Form of UV‐phototherapy which only uses the longer UV wavelengths (340 to 400 nm) and reduces the risk of burning, which is associated with the shorter‐wavelength UVA2 (320 to 340 nm) and UVB (290 to 320 nm). |
| UVB | Ultraviolet B |
| VAS (Visual Analogue Scale) | Continious scale to measure a (subjective) response |
| Vehicle | Something used to transport people or goods (Cambridge dictionary), in this case something to help the treatment get transport in/on the skin, but a vehicle alone (without the active substance) can be used as placebo. |
Appendix 2. Cochrane Skin Specialised Register (CRSW)
pompholyx or cheiropompholyx or acrodermatitis or "hand eczema" or ((eczema or dermatitis) and (dyshidro* or dyshydro* or dishidro* or dishydro* or tylotic or hyperkeratotic or microbial or discoid or nummular or pulpitis or pulpite) and (hand* or finger* or palm*))
Appendix 3. CENTRAL (Cochrane Library) search strategy
#1 tylotic or hyperkeratotic or nummular or microbial or discoid #2 MeSH descriptor: [Eczema] explode all trees #3 MeSH descriptor: [Dermatitis] explode all trees #4 eczema or dermatitis #5 #2 or #3 or #4 #6 MeSH descriptor: [Hand Dermatoses] explode all trees #7 MeSH descriptor: [Eczema, Dyshidrotic] explode all trees #8 hand eczema #9 MeSH descriptor: [Acrodermatitis] explode all trees #10 pompholyx #11 cheiropompholyx #12 {or #6‐#11} #13 MeSH descriptor: [Hand] explode all trees #14 (hand* or finger* palm*) #15 #13 or #14 #16 #1 and #5 and #15 #17 pulpitis or pulpite #18 #15 and #17 #19 dyshidro* or dyshydro* or dishidro* or dishydro* #20 #5 and #19 #21 #12 or #16 or #18 or #20
Appendix 4. MEDLINE (Ovid) search strategy
1. exp *Hand Dermatoses/ 2. exp Eczema, Dyshidrotic/ 3. hand eczema.mp. 4. exp *Acrodermatitis/ 5. pompholyx.mp. 6. cheiropompholyx.mp. 7. 1 or 2 or 3 or 4 or 5 or 6 8. exp Eczema/ or eczema.mp. 9. exp Dermatitis/ or dermatitis.mp. 10. 8 or 9 11. (tylotic or hyperkeratotic).mp. 12. (nummular or microbial or discoid).mp. 13. (pulpitis or pulpite).mp. 14. (dyshidro$ or dyshydro$ or dishidro$ or dishydro$).mp. 15. 11 or 12 16. exp Hand/ 17. (hand$ or finger$ or palm$).mp. 18. 16 or 17 19. 13 and 18 20. 10 and 15 and 18 21. 10 and 14 22. 7 or 19 or 20 or 21 23. randomised controlled trial.pt. 24. controlled clinical trial.pt. 25. randomized.ab. 26. placebo.ab. 27. clinical trials as topic.sh. 28. randomly.ab. 29. trial.ti. 30. 23 or 24 or 25 or 26 or 27 or 28 or 29 31. exp animals/ not humans.sh. 32. 30 not 31 33. 22 and 32
[Lines 23‐32: Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision)]
Appendix 5. Embase (Ovid) search strategy
1. exp *pompholyx/ 2. hand eczema.mp. 3. exp *acrodermatitis/ 4. cheiropompholyx.mp. 5. pompholyx.mp. 6. exp *hand eczema/ 7. or/1‐6 8. eczema.mp. or exp *eczema/ 9. exp *dermatitis/ or dermatitis.mp. 10. 8 or 9 11. (tylotic or hyperkeratotic).mp. 12. (nummular or microbial or discoid).mp. 13. (pulpitis or pulpite).mp. 14. (dyshidro$ or dyshydro$ or dishidro$ or dishydro$).mp. 15. 11 or 12 16. exp hand/ 17. (hand$ or finger$ or palm$).mp. 18. 16 or 17 19. 13 and 18 20. 10 and 15 and 18 21. 10 and 14 22. 7 or 19 or 20 or 21 23. crossover procedure.sh. 24. double‐blind procedure.sh. 25. single‐blind procedure.sh. 26. (crossover$ or cross over$).tw. 27. placebo$.tw. 28. (doubl$ adj blind$).tw. 29. allocat$.tw. 30. trial.ti. 31. randomized controlled trial.sh. 32. random$.tw. 33. or/23‐32 34. exp animal/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 35. human/ or normal human/ 36. 34 and 35 37. 34 not 36 38. 33 not 37 39. 22 and 38
Appendix 6. AMED (Ovid) search strategy
1. random allocation/ 2. double blind method/ 3. single blind method.mp. 4. exp Clinical trials/ 5. (clin$ adj25 trial$).mp. 6. ((singl$ or doubl$ or trebl$ or tripl$) adj25 (blind$ or mask$ or dummy)).mp. 7. (placebo$ or random$).mp. 8. research design/ or clinical trials/ or comparative study/ or double blind method/ or random allocation/ 9. prospective studies.mp. 10. cross over studies.mp. 11. Follow up studies/ 12. control$.mp. 13. (multicent$ or multi‐cent$).mp. 14. ((stud or design$) adj25 (factorial or prospective or intervention or crossover or cross‐over or quasi‐experiment$)).mp. 15. Randomized controlled trials/ 16. or/1‐15 17. hand eczema.mp. 18. hand dermatoses.mp. 19. acrodermatitis.mp. 20. pompholyx.mp. 21. or/17‐20 22. exp Eczema/ or eczema.mp. 23. dermatitis.mp. or exp Dermatitis/ 24. 22 or 23 25. exp Hand/ or hand.mp. 26. (hand$ or finger$ or palm$).mp. 27. 25 or 26 28. 24 and 27 29. (dyshidro$ or dyshydro$ or dishidro$ or dishydro$).mp. 30. 24 and 29 31. (tylotic or hyperkeratotic).mp. 32. (nummular or microbial or discoid).mp. 33. (pulpitis or pulpite).mp. 34. 27 and 33 35. 31 or 32 36. 24 and 27 and 35 37. 21 or 28 or 30 or 34 or 36 38. 16 and 37
Appendix 7. LILACS search strategy
(pompholyx or ponfolix or cheiropompholyx or acrodermatitis or ((eczema or eccema or dermatitis) and (hand$ or finger$ or palm$ or mano$)))
In LILACS we searched using the above terms and the Controlled clinical trials topic‐specific query filter.
Appendix 8. Journals handsearched
1. Acta Dermato‐Venereologica 2. Archives of Dermatological Research 3. Archives of Dermatology 4. British Journal of Dermatology 5. British Medical Journal 6. Clinical and Experimental Dermatology 7. Contact Dermatitis 8. Cutis 9. Dermatology (formerly Dermatologica) 10. Environmental Dermatology 11. Journal of Investigative Dermatology 12. Journal of the American Academy of Dermatology 13. Journal of the American Medical Association 14. Lancet 15. New England Journal of Medicine 16. Der Hautarzt 17. Giornale Italiano di Dermatologia e Venereologia 18. Nederlands Tijdschrift voor Dermatologie en Venereologie 19. H+G Zeitschrift für Hautkrankheiten 20. Annales de Dermatologie et Venerelogie 21 Journal of Dermatologic Treatment
Data and analyses
Comparison 1. Bland emollients: ceramide‐containing emollient versus regular petrolatum‐based emollient.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events: exacerbation resulting in dropout | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 2. Bland emollients: emollient E‐DO versus vehicle.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: percentage of participants with self‐rated good/excellent control at week 4 | Other data | No numeric data | ||
| 2 Primary: percentage of participants with investigator‐rated good/excellent control at week 4 | Other data | No numeric data | ||
| 3 Adverse events | Other data | No numeric data | ||
| 3.1 At least 1 adverse event | Other data | No numeric data | ||
| 3.2 Pruritus | Other data | No numeric data |
Comparison 3. Corticosteroid creams/ointments: fluprednidene versus betamethasone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms after 3 weeks of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: number of participants with at least 1 adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Secondary: investigator‐rated improvement > 50% after 3 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 4. Corticosteroid creams/ointments: betamethasone‐dipropionate film‐forming lotion versus betamethasone‐dipropionate thick lotion.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms at day 7 | Other data | No numeric data | ||
| 2 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 At least 1 adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Application site reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Exacerbation eczema leading to withdrawal | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Secondary: investigator‐rated reduction (not specified) in severity at day 7 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Secondary: investigator‐rated global improvement in eczema | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 5. Corticosteroids creams/ointments: clobetasol propionate versus flupredniden acetate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: percentage of participants with investigator‐rated good/excellent control | Other data | No numeric data | ||
| 2 Adverse events | Other data | No numeric data | ||
| 2.1 At least 1 adverse event | Other data | No numeric data | ||
| 2.2 Burning sensation | Other data | No numeric data | ||
| 2.3 Reversible atrophy | Other data | No numeric data | ||
| 2.4 Brittle skin | Other data | No numeric data |
Comparison 6. Corticosteroids creams/ointments: clobetasol propionate foam 0.05% versus vehicle.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: percentage of participants with investigator‐rated good/excellent control at day 15 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: percentage of participants with self‐rated good/excellent control at day 15 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Discontinuation due to adverse events (fissures) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 At least 1 adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Any adverse event treatment‐related (application site pruritus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Nasopharyngitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Secondary: reduction in severity, participant‐rated scoring at day 15 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Secondary: reduction in severity, investigator‐rated scoring at day 15 | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Improvement at least 2 grades | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Improvement at least 1 grade | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 7. Corticosteriods creams/ointments: desonide cream 0.1% versus desonide cream 0.05%.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Adverse events | Other data | No numeric data |
Comparison 8. Corticosteroid creams/ointments: mometasone furoate cream 3 times/week versus 2 times/week versus no steroids.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Mometasone furoate 3 times/week vs mometasone furoate 2 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Primary: investigator‐rated good/excellent control | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Mometasone furoate 3 times/week vs emollient and ointment only | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Mometasone furoate 2 times/week vs emollient and ointment only | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Mild atrophy | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 9. Corticosteroid creams/ointments: clobetasol and zinc sulphate cream versus clobetasol cream.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome: percentage of participants with investigator‐rated good/excellent control | Other data | No numeric data | ||
| 1.1 Scaling | Other data | No numeric data | ||
| 1.2 Redness | Other data | No numeric data | ||
| 1.3 Lichenification | Other data | No numeric data |
Comparison 10. Corticosteroid creams/ointments: betamethasone‐valerate 0.1% cream versus urea 5% cream.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Secondary: participant‐rated reduction in severity (bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Secondary: investigator‐rated reduction in severity (bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 11. Topical others: coal tar versus betamethasone‐valerate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary outcome: adverse events | Other data | No numeric data |
Comparison 12. Irradiation with UV light versus no UVB.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events ‐ exacerbation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 13. Irradiation with UV light: whole‐body UVB versus placebo or local UVB hands.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms by UVB hand vs whole‐body UVB vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Local UVB hands alone vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Whole‐body UVB + local UVB hands vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Whole body UVB + local UVB hands versus local UVB hands alone | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Secondary: time until relapse depicted in weeks of remission | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 Local UVB hands alone vs placebo (high score = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Whole‐body UVB + local UVB hands vs placebo (high score = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Whole‐body UVB + local UVB hands vs local UVB hands alone (high score = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 14. Irradiation with UV light: local narrow‐band UVB versus local PUVA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms in UVB vs PUVA | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Primary: adverse events ‐ reported adverse event, mainly erythema | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 15. Irradiation with UV light: local narrow‐band UVB versus local PUVA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms in UVB vs PUVA | Other data | No numeric data | ||
| 2 Primary: adverse events | Other data | No numeric data | ||
| 2.1 Palmar hyperpigmentation | Other data | No numeric data | ||
| 3 Secondary: investigator‐rated improvement by local narrow‐band UVB vs local PUVA | Other data | No numeric data |
Comparison 16. Irradiation with UV light: oral PUVA versus topical bath PUVA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Nausea leading to dropout | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Burn leading to dropout | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Dizziness | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Secondary: investigator‐rated reduction in severity at week 10 (bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Secondary: investigator‐rated reduction in severity at week 18 (bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 17. Irradiation with UV light: topical PUVA versus UVA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events | Other data | No numeric data | ||
| 1.1 Discontinuation due to adverse events | Other data | No numeric data | ||
| 1.2 Burning | Other data | No numeric data | ||
| 1.3 Exacerbation of eczema | Other data | No numeric data |
Comparison 18. Irradiation with UV light: UVA‐1 versus topical betamethasone valerate 0.1% cream.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events ‐ hyperpigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 19. Irradiation with UV light: UVA‐1 versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events ‐ discontinuation because of exacerbation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Secondary: participant‐rated reduction in severity on VAS for itch (week 3, bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Secondary: investigator‐rated reduction in severity on dyshidrotic eczema area and severity index (DASI) (week 3, bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 20. Irradiation with UV light: PUVA versus UVA‐1.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events | Other data | No numeric data | ||
| 1.1 Burning | Other data | No numeric data | ||
| 1.2 Itching | Other data | No numeric data |
Comparison 21. Irradiation with Grenz ray.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated improvement good/excellent control | Other data | No numeric data | ||
| 1.1 after 1 month | Other data | No numeric data | ||
| 1.2 after 3 months | Other data | No numeric data | ||
| 1.3 after 6 months | Other data | No numeric data | ||
| 1.4 Hyperkeratotic eczema after 6 months | Other data | No numeric data | ||
| 1.5 Pompholyx after 6 months | Other data | No numeric data | ||
| 1.6 Chronic palmar eczema after 6 months | Other data | No numeric data | ||
| 2 Primary: adverse events ‐ hyperpigmentation | Other data | No numeric data |
Comparison 22. Topical calcineurin inhibitors: tacrolimus 0.1% ointment versus mometasone furoate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Secondary: reduction in investigator‐rated severity ‐ DASI | Other data | No numeric data |
Comparison 23. Topical calcineurin inhibitors: tacrolimus 0.1% ointment versus vehicle.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: adverse events burning/itching at application site | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Burning/itching at application site | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 24. Topical calcineurin inhibitors: pimecrolimus 1% cream versus vehicle.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms pimecrolimus cream vs vehicle | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Primary: investigator‐rated clear or almost clear pimecrolimus cream vs vehicle 3 weeks | 1 | 294 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.99, 2.36] |
| 1.2 Primary: investigator‐rated clear or almost clear pimecrolimus cream vs vehicle 6 weeks | 1 | 652 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.99, 1.66] |
| 1.3 Primary: investigator‐rated clear or almost clear pimecrolimus cream vs vehicle 3 weeks irritant hand eczema | 1 | 185 | Risk Ratio (M‐H, Random, 95% CI) | 1.7 [0.93, 3.10] |
| 1.4 Primary: investigator‐rated clear or almost clear pimecrolimus cream vs vehicle 3 weeks allergic hand eczema | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.30, 5.96] |
| 1.5 Primary: investigator‐rated clear or almost clear pimecrolimus cream vs vehicle 3 weeks endogenous hand eczema | 1 | 134 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.75, 2.33] |
| 2 Primary: adverse events | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Discontinuation because of adverse event | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Application site reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 At least 1 adverse event | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Serious adverse event (not related to study) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 Treatment‐related adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 Erythema or irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.7 Itching | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.8 Warmth, stinging, burning | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.9 Herpes simplex virus infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Secondary: participant‐rated reduction in severity pruritus relief between pimecrolimus 1% and vehicle | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 25. Topical antibacterial agents: betamethasone‐valerate/clioquinol cream versus betamethasone‐valerate/fusidic acid.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms (intention‐to‐treat) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 At least 1 adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Chest infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Irritation associated with application | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Deterioration of eczema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 Eyes watering | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 Hands coloured yellow | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.7 Hands felt thick | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.8 Vesicle on hand | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 26. Topical retinoids: bexarotene 1% gel versus bexarotene with corticosteroids.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms (> 90% clearance) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Bexarotene only (A) vs bexarotene + mometasone (B) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Bexarotene only (A) vs bexarotene + hydrocortisone (B) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Bexarotene + mometasone (A) vs bexarotene + hydrocortisone (B) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Primary: adverse events bexarotene vs bexarotene + mometasone | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Irritation/rash | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Stinging/burning | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Exacerbation of dermatitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Primary: adverse events bexarotene vs bexarotene + hydrocortisone | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Irritation/rash | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Stinging/burning | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Exacerbation of dermatitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Secondary: investigator‐rated reduction in severity (> 50% reduction in hand eczema area and severity index (HEASI)) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Bexarotene (A) only vs bexarotene + mometasone (B) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Bexarotene (A) only vs bexarotene + hydrocortisone (B) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Bexarotene + mometasone (A) vs bexarotene + hydrocortisone (B) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 27. Other topical agents: calmurid versus aquacare.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events: burning | Other data | No numeric data |
Comparison 28. Other topical agents: fumaric acid 5% cream verus triamcinolone 0.1% cream.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 29. Other topical agents: furpalmate 0.3% cream versus hydrocortisone acetate 0.5% cream.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 30. Other topical agents: Fumaria parviflora versus vehicle cream.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: adverse events: discontinuation due to erythema and papules | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 31. Oral immunosuppressants: oral azathioprine with topical clobetasol propionate versus topical clobetasol propionate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: percentage of participants with investigator‐rated good/excellent control | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Week 8 | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Week 24 | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Secondary: investigator‐rated reduction in severity (bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 Week 4 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Week 8 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Week 12 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Week 24 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Secondary: participant‐rated reduction in severity (bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Week 4 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Week 8 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Week 12 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Week 24 | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 32. Oral immunosuppressants: oral cyclosporin versus topical betamethasone dipropionate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated very good or good efficacy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: participant‐rated very good or good efficacy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 At least 1 adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Discontinuation due to dizziness, vomiting, and facial oedema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Discontinuation due to severe insomnia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Increase in serum creatinine > 30% | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Secondary: investigator‐rated reduction in severity in total disease activity score (6 weeks; bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 33. Oral immunosuppressants: oral cyclosporin versus alitretinoin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control (IGA) after 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: participant‐rated good/excellent control (PGA) after 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 34. Oral retinoids: alitretinoin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Alitretinoin 40 mg vs placebo | 1 | 159 | Risk Ratio (M‐H, Random, 95% CI) | 1.97 [1.30, 3.00] |
| 1.2 Alitretinoin 30 mg vs placebo | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 2.75 [2.20, 3.43] |
| 1.3 Alitretinoin 20 mg vs placebo | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [0.94, 2.34] |
| 1.4 Alitretinoin 10 mg vs placebo | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 1.58 [1.20, 2.07] |
| 2 Primary: investigator‐rated good/excellent control of symptoms hyperkeratotic eczema | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Alitretinoin 40 mg vs placebo hyperkeratotic eczema | 1 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 2.61 [1.61, 4.23] |
| 2.2 Alitretinoin 30 mg vs placebo hyperkeratotic eczema | 1 | 519 | Risk Ratio (M‐H, Random, 95% CI) | 3.94 [2.60, 5.97] |
| 2.3 Alitretinoin 20 mg vs placebo hyperkeratotic eczema | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 1.72 [1.02, 2.90] |
| 2.4 Alitretinoin 10 mg vs placebo hyperkeratotic eczema | 2 | 662 | Risk Ratio (M‐H, Random, 95% CI) | 2.05 [1.47, 2.86] |
| 3 Primary: investigator‐rated good/excellent control of symptoms pompholyx | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Alitretinoin 40 mg vs placebo pompholyx | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.66 [0.58, 4.72] |
| 3.2 Alitretinoin 30 mg vs placebo pompholyx | 1 | 166 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [1.06, 3.91] |
| 3.3 Alitretinoin 20 mg vs placebo pompholyx | 1 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 0.9 [0.26, 3.08] |
| 3.4 Alitretinoin 10 mg vs placebo pompholyx | 2 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.70, 2.39] |
| 4 Primary: investigator‐rated good/excellent control of symptoms fingertip | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Alitretinoin 40 mg vs placebo fingertip | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [0.81, 3.86] |
| 4.2 Alitretinoin 30 mg vs placebo fingertip | 1 | 297 | Risk Ratio (M‐H, Random, 95% CI) | 2.49 [1.59, 3.89] |
| 4.3 Alitretinoin 20 mg vs placebo fingertip | 1 | 53 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.50, 2.77] |
| 4.4 Alitretinoin 10 mg vs placebo fingertip | 2 | 330 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.99, 2.29] |
| 5 Primary: participant‐rated investigator‐rated good/excellent control of symptoms | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Alitretinoin 40 mg vs placebo | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 3.51 [1.80, 6.82] |
| 5.2 Alitretinoin 30 mg vs placebo | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 2.75 [2.18, 3.48] |
| 5.3 Alitretinoin 20 mg vs placebo | 1 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 2.74 [1.37, 5.46] |
| 5.4 Alitretinoin 10 mg vs placebo | 2 | 765 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [1.25, 2.40] |
| 6 Primary: adverse events alitretinoin 10 mg vs placebo | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 All adverse events | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 6.2 Headache | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.38, 3.19] |
| 6.3 Dry lips | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.05, 4.66] |
| 6.4 Flushing | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.28, 4.70] |
| 6.5 Dry mouth | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [0.53, 7.44] |
| 6.6 Erythema | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.33, 3.71] |
| 6.7 Eczema | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.38, 1.68] |
| 6.8 Conjunctivitis | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.05, 5.27] |
| 6.9 Eye pruritus | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.06, 15.32] |
| 6.10 Fatigue | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 2.93 [0.12, 70.75] |
| 6.11 Rigors | 1 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 2.93 [0.12, 70.75] |
| 6.12 Tonsilitis | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.15, 1.70] |
| 6.13 Pharyngitis | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.44, 1.54] |
| 6.14 Influenza | 1 | 623 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.39, 3.86] |
| 6.15 Nausea | 1 | 623 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [0.45, 5.88] |
| 6.16 Elevated blood creatinine kinase | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.73, 2.80] |
| 6.17 Elevated blood triglycerides | 2 | 781 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.56, 1.88] |
| 7 Primary: adverse events alitretinoin 20 mg vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 All adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Dry lips | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Flushing | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Dry mouth | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.6 Erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.7 Eczema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.8 Conjunctivitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.9 Eye pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.10 Fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.11 Rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.12 Tonsilitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.13 Pharyngitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Primary: adverse events alitretinoin 30 mg vs placebo | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Headache | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 3.43 [2.45, 4.81] |
| 8.2 Dry lips | 1 | 614 | Risk Ratio (M‐H, Random, 95% CI) | 1.88 [0.63, 5.59] |
| 8.3 Flushing | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 7.28 [2.05, 25.86] |
| 8.4 Dry mouth | 1 | 614 | Risk Ratio (M‐H, Random, 95% CI) | 2.51 [0.55, 11.33] |
| 8.5 Erythema | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 5.79 [2.09, 16.06] |
| 8.6 Eczema | 1 | 614 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.29, 1.46] |
| 8.7 Pharyngitis | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.49, 1.36] |
| 8.8 Influenza | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.45, 3.06] |
| 8.9 Nausea | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 3.82 [1.67, 8.76] |
| 8.10 Elevated blood creatinine kinase | 1 | 614 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [0.54, 4.93] |
| 8.11 Elevated blood triglycerides | 2 | 1210 | Risk Ratio (M‐H, Random, 95% CI) | 7.05 [1.89, 26.28] |
| 8.12 Dizziness | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.61, 6.57] |
| 8.13 Upper respiratory tract infection | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.78, 3.04] |
| 8.14 Sinusitis | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.35, 2.27] |
| 8.15 Rash | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.54, 4.16] |
| 8.16 Vomiting | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 8.00 [1.01, 63.57] |
| 8.17 Arthralgia | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 1.2 [0.37, 3.89] |
| 8.18 Depression | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 2.33 [0.61, 8.94] |
| 8.19 Laceration | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 13.0 [0.74, 229.73] |
| 8.20 Tinnitus | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 4.33 [1.25, 15.05] |
| 8.21 Cough | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 2.25 [0.70, 7.23] |
| 8.22 Hypertriglceridaemia | 1 | 596 | Risk Ratio (M‐H, Random, 95% CI) | 6.0 [0.73, 49.53] |
| 9 Primary: adverse events alitretinoin 40 mg vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 All adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Dry lips | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 Flushing | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.5 Dry mouth | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.6 Erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.7 Eczema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.8 Conjunctivitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.9 Eye pruritus | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.10 Fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.11 Rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.12 Tonsilitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.13 Pharyngitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Secondary: investigator‐rated reduction in severity in total lesion symptom score | Other data | No numeric data | ||
| 10.1 Alitretinoin 40 mg | Other data | No numeric data | ||
| 10.2 Alitretinoin 30 mg | Other data | No numeric data | ||
| 10.3 Alitretinoin 20 mg | Other data | No numeric data | ||
| 10.4 Alitretinoin 10 mg | Other data | No numeric data | ||
| 11 Secondary: investigator‐rated reduction in severity in total lesion symptom score | Other data | No numeric data | ||
| 11.1 Alitretinoin 40 mg vs placebo | Other data | No numeric data | ||
| 11.2 Alitretinoin 20 mg vs placebo | Other data | No numeric data | ||
| 11.3 Alitretinoin 10 mg vs placebo | Other data | No numeric data | ||
| 12 Secondary: reduction in severity, investigator‐rated in modified total lesion symptom score (bigger reduction in severity scored negative = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
34.2. Analysis.
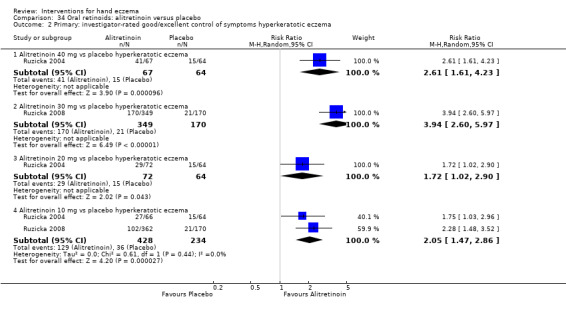
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 2 Primary: investigator‐rated good/excellent control of symptoms hyperkeratotic eczema.
34.3. Analysis.
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 3 Primary: investigator‐rated good/excellent control of symptoms pompholyx.
34.4. Analysis.
Comparison 34 Oral retinoids: alitretinoin versus placebo, Outcome 4 Primary: investigator‐rated good/excellent control of symptoms fingertip.
Comparison 35. Oral retinoids: re‐treatment alitretinoin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Alitretinoin 30 mg vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Alitretinoin 10 mg vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Primary: adverse events 10 mg vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Any adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Treatment‐related adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Nasopharyngitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Influenza | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 Erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 Eczema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.7 Dermatitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.8 Dry lips | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.9 Cheilitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.10 Dry mouth | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.11 Elevated triglycerides | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.12 High cholesterol | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.13 High triglycerides | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Primary: adverse events 30 mg vs placebo | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Any adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Treatment‐related adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Treatment‐related serious adverse event | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Nasopharyngitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Rhinitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Bronchitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Upper respiratory tract infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 Influenza | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.9 Erythema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.10 Eczema | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.11 Dermatitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.12 Dry skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.13 Headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.14 Dry lips | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.15 Nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.16 Cheilitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.17 Dry mouth | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.18 Elevated creatinine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.19 TSH high | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.20 TSH low | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.21 High cholesterol | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.22 High triglycerides | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 36. Other oral interventions: oral triethylenetetramine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐ and/or participant‐rated improvement | Other data | No numeric data |
Comparison 37. Other oral interventions: oral tetraethylthiuram disulfide (TETDS) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms during treatment period | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Discontinuation due to depression | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Discontinuation due to dyspepsia | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Hepatic toxicity | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Headache requiring dose reduction | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 38. Other oral interventions: low‐nickel diet (LND) + disulphiram versus normal diet + placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: investigator‐rated good/excellent control of symptoms after 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Primary: adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Metallic taste | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Drowsiness | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Elevation of liver enzymes | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 39. Other oral interventions: oral evening primrose oil versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Secondary: investigator‐rated reduction in severity score at week 24 (bigger reduction in severity = better outcome) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 40. Other oral interventions: oral ranitidine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: participant‐ and investigator‐rated good/excellent control of symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 41. Other oral interventions: disodium cromoglycate diet (DSCG) versus low‐nickel diet.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary: participant‐rated good/excellent control of symptoms (itch) after 3 months of itch in DSCG versus diet | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adams 2007.
| Methods | Within‐participant, randomised controlled study (left‐right design). This study was carried out in the secondary care setting; it was a single‐centre study conducted in Germany |
|
| Participants | 15 participants at least 18 years old suffering from chronic relapsing dyshidrotic hand eczema with a minimal duration of 1 month that was resistant to conventional therapies Dropouts: 4 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Middle‐dose UVA‐1 irradiation 3 times a week (cumulative dose of 600 J/cm²) in 11/15 hands during 5 weeks • Local 8‐MOP‐cream‐PUVA irradiation 3 times a week during 5 weeks (cumulative dose of 17.4 J/cm²) in 11/15 contralateral hands. 8‐MOP‐crème was applied 30 minutes before the start of irradiation Duration 5 weeks |
|
| Outcomes |
Primary outcomes of the trial • Observer‐rated assessment of improvement (DASI score) Other outcomes • Adverse events |
|
| Notes | Therapeutic efficacy was shown with relatively low cumulative doses of UVA and UVA‐1 The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included but did not provide reproducible data Study authors were contacted by mail on 6 March 2014 and responded 10 March 2014 Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The article states that randomisation was done by an independent third person. No information regarding random sequence generation appeared in the article; however personal communication clarified that this was done appropriately: "cards with the characterisation "A" and "B" were enclosed in envelopes by a third person, mixed like a card play by a third person, then numbered consecutively by a third person and opened by the study doctor consecutively after informed consent to the study" |
| Allocation concealment (selection bias) | Low risk | Quote: "randombriefe wurden nach Wűrfeln von einer von der Studie unabhängigen Person erstellt" (free translation: the randomisation letter was created by an independent person after throwing dices) Comment: adequate allocation concealment as randomisation was accomplished by a third party |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "eine Verblindung der Studie erfolgte nicht" (free translation: the study was not blinded) Comment: no attempts were made to blind participants or personnel |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "eine Verblindung der Studie erfolgte nicht" (free translation: the study was not blinded) Comment: observers were not blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (11 of 15 = less than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however all outcomes listed in Materials and Methods are given in the Results section |
| Other bias | Low risk | Baseline comparisons revealed no significant differences between groups, as within‐participant study was not applicable Diagnostic certainty: yes The study was completed |
Agarwal 2013.
| Methods | Parallel‐group, randomised controlled trial This study was probably carried out in a secondary care setting as a single‐centre study at a Department of Dermatology in India |
|
| Participants | 108 participants with clinically diagnosed hand eczema included; 91 completed the study Dropouts: 17 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Topical clobetasol propionate 0.05% cream twice daily with oral azathioprine 50 mg daily in 46 participants for 24 weeks Control intervention • Topical clobetasol propionate 0.05% cream twice daily alone in 45 participants for 24 weeks Participants were instructed to use the topical clobetasol intermittently and to stop application whenever the signs and symptoms disappeared and restart when the complaints returned Duration 24 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes • Reduction in severity, investigator‐rated scoring measured by the Hand Eczema Scoring Index (HECSI) at 2, 4, 8, 12, and 24 weeks • Reduction in severity of itch, participant‐rated, measured on a numerical scale from 0 to 10 • Number of exacerbations • Adverse events |
|
| Notes | The total quantity of corticosteroids used was not registered Study authors were contacted on 27 February 2014 by email and responded 1 March 2014 Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomised into 2 groups using block randomization" Comment: unclear from the article how these blocks were generated; however contact with study authors clarified that a valid computer‐generated table was used |
| Allocation concealment (selection bias) | Low risk | No information is provided in the article on how allocation was concealed from participants and investigators, but personal communication revealed that randomisation was done by a third person |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "patients paid for the medicine themselves" Comment: participants were not blinded because they had to buy their own study drugs, and no placebo was used |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "an observer blinded randomized comparative trial" Comment: study authors stated that the study was observer blinded but gave no details as to how this was achieved. Contact with study authors clarified that observers were independent and were not involved in treatment nor in dispensation of study drugs |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 91 of the 108 (84.3%) included participants were analysed. No intention‐to‐treat analysis. Dropouts were not evenly distributed between the 2 groups |
| Selective reporting (reporting bias) | Low risk | No trial registration found. However, all mentioned outcomes are described in the Results section |
| Other bias | Unclear risk | No baseline comparisons were conducted or reported Diagnostic certainty: yes The study was completed |
Baskan 2005.
| Methods | Within‐participant, placebo‐controlled, randomised controlled trial The study was conducted in the secondary setting at a single centre in Turkey |
|
| Participants | 25 participants with moderate to severe bilateral hand dermatitis with a minimal duration of 6 months Dropouts: 1 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Pimecrolimus 1% cream twice daily in 24/25 hands presumably for 8 weeks Control intervention • Placebo cream twice daily presumably for 8 weeks in 24/25 hands • Participants were followed up for the same period Duration 16 weeks (8 weeks active treatment, 8 weeks follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes • Clinical response to therapy; erythema, desquamation, lichenification, oedema, vesiculation, and fissuring were scored between 0 and 4 and were controlled at 2nd, 4th, 6th, and 8th weeks of therapy • Clinical response to therapy for pruritus • At the end of therapy, participants were followed up for the same period to observe recurrences • Adverse events |
|
| Notes | Conference abstract from which only limited information can be extracted Study authors were contacted for additional information, which led to review of an additional full‐text article in Turkish. The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included but did not provide reproducible data Declarations of interest: not stated Funding: not stated in the paper, but personal communication clarified that study authors did not receive any funding Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Study authors stated randomised but gave no clear description of how this was attained. Personal communication with study authors clarified that they used a card drawing system Quote: "a simple randomisation method by card drawing was performed" |
| Allocation concealment (selection bias) | Low risk | No information on how allocation was concealed from participants and investigators is provided in the abstract Personal communication clarified that one investigator drew the cards after participants gave their consent. The topical drugs were packed in yellow and red boxes, and staff gave participants the corresponding boxes for treatment, without knowledge about the content of those boxes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Not clear from the abstract; however personal communication clarified that dispensation of study drug was done by a third person in boxes labelled for each hand. The vehicle and pimecrolimus were packed in similar boxes and were indistinguishable for participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not clear from the abstract; however personal communication clarified that observation was done by another physician who was not involved in randomisation or drug dispensation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Unclear whether an intention‐to‐treat analysis was conducted; however 24 of the 25 included participants completed the study, which is more than 80% |
| Selective reporting (reporting bias) | Low risk | No trial registration found. All relevant clinical signs were scored in the symptom score, and all described outcomes were depicted in the Results section |
| Other bias | Low risk | No baseline comparisons were conducted or reported, as within‐participant study was not applicable Diagnostic certainty: yes The study was completed |
Bauer 2012.
| Methods | Parallel‐group, randomised controlled trial. Multi‐centre study at the outpatient clinics of the University Hospitals Heidelberg and Dresden, Germany |
|
| Participants | 40 adult outpatients Dropouts: 4. Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions | Participants were randomly allocated in a ratio of 1:1 to receive the following Intervention • Pimecrolimus 1% cream twice a day for 8 weeks after clinical response (IGA 2) to 1 to 3 weeks of treatment with mometasone furoate 0.1% in 20/20 participants Control intervention • Vehicle twice a day for 8 weeks after clinical response (IGA 2) to 1 to 3 weeks of treatment with mometasone furoate 0.1% in 16/20 participants To control for compliance, study medication was weighed at every visit Duration 8 weeks after 1 to 3 weeks of start‐up treatment |
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
Other outcomes
|
|
| Notes | Secondary outcomes ‐ reduction in severity, participant‐ and observer‐rated ‐ were included but did not provide reproducible data Study authors were contacted on 27 February 2014 and provided additional information on 27 February 2014 Funding: the study was funded by a grant from Novartis Pharma GmbH, Nurnberg, Germany, the manufacturer of the study drug Study authors gave lectures for Novartis, although they claim this is not directly related to the study. One study author was an employee of Novartis Pharma, the manufacturer of the pimecrolimus cream Sample size rationale: adequate |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was performed by allocation of the consecutive patients to the lowest available number from the randomisation list. The allocation sequence was generated by use of a permutated block randomisation list in blocks of 4 with equal allocation to pimecrolimus and vehicle" Comment: this is considered as adequate random sequence generation |
| Allocation concealment (selection bias) | Low risk | This was unclear from the article. Personal communication with study authors clarified that the investigators had no access to the randomisation code and used sealed envelopes for the allocation procedure |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "verum and vehicle creams were prepared from the commercial product and blinded labelled for this study by Novartis Pharma GmbH. Patients and investigators were blinded to assignment of patients during the entire study period until the closing of the data bank" Comment: participants and personnel of the study at the study site (except personnel for Novartis, which was located somewhere else) were unaware of the allocation during the study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "verum and vehicle creams were prepared from the commercial product and blinded labelled for this study by Novartis Pharma GmbH. Patients and investigators were blinded to assignment of the patients during the entire study period until the closing of the data bank" Comment: investigators were blinded during the entire study, and only the investigators from Novartis were aware of allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "ITT analysis was performed" Comment: intention‐to‐treat analysis for primary outcome as well as per protocol (35/36 ≥ 80%) |
| Selective reporting (reporting bias) | Low risk | The trial was registered under Eudra CT Nr 2005‐0003644‐59 before the start of the study. We found no discrepancy between outcomes stated in the protocol and reported in the publication |
| Other bias | Low risk | Baseline comparison: no statistically significant differences were found between the pimecrolimus (n = 20) and vehicle (n = 16) groups with regards to age, sex, body weight, height, and ethnicity, and in IGA at baseline Diagnostic certainty: yes The study was completed |
Bayerl 1999.
| Methods | Parallel‐group, randomised controlled trial The study was conducted in the secondary setting at 2 different dermatology departments in Germany |
|
| Participants | 48 participants with chronic hand eczema (21 irritant, 18 allergic, 9 atopic) > 3 months' duration, more than 30% of the hands involved. All had occupation‐related hand eczema: 41% were in a wet occupation
Dropouts: 12 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • UV‐B phototherapy 5 days/week for 8 weeks in 19/24 participants Control intervention • No UVB for 8 weeks in 17/24 participants Both groups were allowed to use non‐specific creams/emollients Duration 8 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Study authors rightly state that this is a pilot study. Only graphic presentation of a few components of some outcome parameters. Not clear, but assumed, that 24 participants were randomised to each group. The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included but did not provide reproducible data Study authors were contacted on 7 March 2014 and responded 10 March 2014 Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The article states only that the study is randomised, without details about the method of randomisation. Personal communication clarified that the same method was used as in Adams 2007: "cards with the characterisation "A" and "B" were enclosed in envelopes by a third person, mixed like a card play by a third person, then numbered consecutively by a third person and opened by the study doctor consecutively after informed consent to the study" This is an adequate method |
| Allocation concealment (selection bias) | Low risk | No details about whether the allocation was concealed from participants and investigators in the article, but personal communication clarified that this was done appropriately by a third person, and that the study doctor and participants opened the consecutively numbered envelopes after informed consent was retrieved |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were not blinded during the study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Observers were not blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (36 of 48 = less than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however outcomes described in Materials and Methods are depicted in the Results section and are adequate |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons regarding group differences (randomisation check) Diagnostic certainty: yes The study was completed |
Belsito 2004.
| Methods | Parallel‐group, randomised controlled trial The study was carried out in a secondary care setting This was a multi‐centre study that was conducted in Brazil, Canada, and the USA |
|
| Participants | 294 participants with mild to moderate chronic hand eczema (117 irritant, 94 endogenous, 32 irritant + endogenous, 32 irritant + allergic, 9 allergic, 4 allergic + endogenous, 4 irritant + allergic + endogenous, 2 unknown aetiology)
Dropouts: 22 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Pimecrolimus 1% cream twice daily with 6 hours of love occlusion in the evenings for 3 weeks in 140/151 participants Control intervention • Vehicle cream twice daily with 6 hours of glove occlusion in the evenings for 3 weeks in 132/143 participants Barrier creams or emollients were allowed in both groups if applied more than 1 hour before study cream Duration 3 weeks |
|
| Outcomes |
Primary outcomes of the trial
Other outcomes
|
|
| Notes | Overall efficacy (proportion of treatment successes) for both groups at end of study presented as graph (bar chart); exact figures not given. In separate table, exact figures for treatment successes, with strata of selected groups overlapping ("to identify groups highly responsive") Study authors contacted by email and LinkedIn 27 February 2014, but we were unable to obtain additional information Declarations of interest: some study authors were employees of pharmaceutical companies Funding: the study was supported by a grant from Novartis Pharmaceuticals Corporation, East Hanover, New Jersey, which is the manufacturer of pimecrolimus cream. Four study authors had an ongoing financial relationship with Novartis, and four were employees of Novartis Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "a multicenter, randomized, vehicle‐controlled 3 week study..." Comment: the article states only that subjects were randomised, without further information. Insufficient information provided to judge the risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "study design ‐ the study was a double‐blind, multicenter, vehicle‐controlled trial of ...." Comment: unclear; participants and personnel probably blinded as this is stated in the paper, but no further details are given. It is unclear whether vehicle and placebo were identical in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "study design ‐ the study was a double‐blind, multicenter, vehicle‐controlled trial of ...." Comment: no details regarding blinding of observers |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the intention‐to‐treat (ITT) and safety populations consisted of all randomized patients who received the study medication, and the per‐protocol population included all patients from the ITT population who did not violate the protocol in ways that would affect efficacy evaluations" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however all outcomes listed in the Methods section are described in the Results section |
| Other bias | Low risk | Baseline comparisons: no significant differences in demographic or disease characteristics between groups Diagnostic certainty: yes The study was completed |
Bissonnette 2010.
| Methods | Parallel‐group, placebo‐controlled, randomised study with participants who initially responded in a previous BACH study (Ruzicka 2008), but who relapsed in the observational period This study was carried out in a secondary care setting This was a multi‐centre study conducted in Canada, France, and Germany |
|
| Participants | 117 participants with chronic hand eczema who relapsed after they were successfully treated with alitretinoin 30 mg, 10 mg, or placebo Dropouts: 24 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Alitretinoin 10 mg once daily for 12 to 24 weeks in 21 participants • Alitretinoin 30 mg once daily for 12 to 24 weeks in 49 participants Control intervention • Placebo for 12 to 24 weeks in 47 participants Participants were re‐treated with the same dose that they had received in the previous study, or they were treated with placebo Participants who were initially treated with placebo in the first trial were treated with placebo again No other topical or systemic therapy for hand eczema was allowed Duration 12 to 24 weeks |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | Study included participants who relapsed after successful treatment in a previous study (Ruzicka 2008). No other active treatment as comparator. Analysis of efficacy based on intention‐to‐treat principle. Study included a safety assessment by careful medical and laboratory monitoring. The primary outcome percentages of participants with self‐rated good/excellent and secondary outcome reduction in severity, investigator‐rated scoring were included in the study, but we were unable to reproduce the data Study authors were contacted and referred us for further information to GSK, which provided additional information Declarations of interest: various study authors were investigators or consultants for Basilea Pharmaceutica International Ltd. Funding: the study was supported and funded by Basilea Pharmaceutica International Ltd, Basel, Switzerland, the manufacturer of alitretinoin. Study authors were investigators in Basilea clinical trials, or were consultants or employees of Basilea Pharmaceutica International Ltd. Sample size rationale: not stated Quote: "the sample size was not prespecified, and all relapsing patients from the BACH trial were eligible for trial screening" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomised to the same dose they received in the BACH study or to placebo" Comment: no further details given. In personal communication, the study authors clarified: "the study was a follow on from the BACH study BAP00089, which was a randomised double blind placebo controlled study of subjects in a 2:2:1 randomisation of treatment to alitretinoin 30 mg, alitretinoin 10 mg, or placebo, respectively. Patients who responded in study BAP00089 and relapsed during the posttreatment observation period were assigned to the same dose they had received or to placebo in a 2:1 ratio. Responding patients who had received placebo in study BAP00089 were assigned to continue receiving placebo. Each was assigned a coded allocation of study drug containing either placebo or a dosage of active drug. The randomisation was computer generated" |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators. In personal communication, the study authors stated: "it is unclear how this knowledge was imparted, but it is clear from the protocols that those subjects who had received placebo in the original trial BAP00089, and who had been successfully treated but had subsequently relapsed, would upon entering this study be given placebo again, as it was considered unethical to expose them unnecessarily to drug" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the investigator, sponsor and all participants remained blinded throughout the course of the BACH and re‐treatment studies" Comment: Study authors stated that they conducted a double‐blind study but made no statements about how this was done Quote: "the placebo and active drug were indistinguishable and packaged in the same way" Comment: use of identical packages is a sufficient form of blinding; however it is unclear whether site staff were also blinded because the study was a follow‐up study in which most participants received the same treatment as in the previous study Personal communication clarified: "a list of treatment assignments was sealed and kept in a central repository by the Biometrics Department and by the Drug Safety Department. No open key to the code was available at the Study Center, or to monitors or members of the project team" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were blinded throughout the study; however no details regarding blinding were given. The observers might have been the same as in the study of Ruzicka 2008. Personal communication clarified: "the investigator had access to coded, sealed envelopes for each participant to be used in an emergency that would have required knowledge of the study medication to manage the emergency. If the investigator wished to know the identity of the treatment given to study subjects for any other purpose, this request was first to be discussed with Basilea Pharmaceutica" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "efficacy evaluations are based on the intention‐to‐treat population, which included all randomized patients" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | The trial was registered at clinicialtrials.gov (NCT00124436 and BAP00091 and EUCTR2004‐000432‐85‐HU). No major changes in primary or secondary outcomes. The only difference is that the trial registration states enrolment of 300 participants, and the actual trial included only 117 participants; this was done so all participants from the BACH study who relapsed could be included |
| Other bias | Low risk | Baseline comparison of disease severity in table, but no significance of differences in tests provided. Personal communication clarified that there were no statistically significant differences Diagnostic certainty: yes The study was completed |
Bleeker 1989.
| Methods | Parallel‐group, randomised controlled trial This study was conducted as a multi‐centre study at dermatology departments and private clinics in Sweden |
|
| Participants | 76 participants (22 male, 54 female; ages 18 to 65) with different subtypes of hand eczema. Vesicular and infected dermatitis excluded
Dropouts: 1 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Fluprednidene cream once daily in the evening for 3 weeks in 37/38 participants • Betamethasone cream once daily in the evening for 3 weeks in 38/38 participants Emollient (Unguentum Merck) was allowed in both groups if required Duration 3 weeks |
|
| Outcomes |
Primary outcomes of the trial Not stated Other outcomes
|
|
| Notes | Unclear whether severity score as stated in Methods was used in analysis. Aim was to study equivalency of treatment effect The secondary outcome ‐ reduction in severity, participant‐rated ‐ was included but did not provide reproducible data Declarations of interest: not stated Funding: E. Merck A.B. Sweden, which was the manufacturer of unguentum Merck, supported the study Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "each patient was allocated a patient number and assigned at random to one of the two treatment groups" Comment: the article states only that each participant was allocated a participant number and was assigned at random, without further details |
| Allocation concealment (selection bias) | Unclear risk | Quote: "each patient was allocated a patient number and assigned at random to one of the two treatment groups" Comment: no details about how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a double blind study design was used" Comment: participants and personnel probably blinded, as this is stated in the paper, but no details are given as to how this was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "a double blind study design was used" Comment: the article states a double‐blind design; however no details are given regarding blinding of observers |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (75 of 76 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however all important clinical outcomes described in Materials and Methods are depicted in the Results. The only drawback is that with regards to the participant‐rated general assessment, the Results section states only that this was "significantly improved (P < 0.001) after 1 week in both treatment groups"; however no details are given |
| Other bias | Unclear risk | Baseline comparison: a baseline comparison of disease severity is depicted in a table, but no significance of differences in tests is provided Diagnostic certainty: yes The study was completed |
Boroujeni 2017.
| Methods | Parallel‐group, randomised controlled study The study was conducted at a single centre in Iran |
|
| Participants | 64 participants with hand eczema
Dropouts: 4 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Twice‐daily application with an oil‐in‐water emulsion‐based herbal cream. Concentrations of plant material were as follows: fenugreek seeds 5%, marshmallow 5%, chamomile 5%, and walnut leaves 5% • Twice‐daily application of fluocinolone acetonide cream 2% Duration Two weeks |
|
| Outcomes |
Primary outcomes of the trial Reduction in severity of symptoms such as burning, itching, erythema level, papules and vesicles bumps, and fissures of the skin (method and scale not stated) Other outcomes
|
|
| Notes | Study authors were contacted for additional information in April 2018 but remained unresponsive Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "64 patients with hand eczema were randomly divided into two groups of 32 patients each" Comment: no further details on randomisation procedure |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "this study is a double blind clinical trial" Comment: no details regarding blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "this study is a double blind clinical trial" Comment: no details regarding observer blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (60 of 64 = more than 80%) |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found We did not find major differences between what was stated in the Methods and Result sections; however it is unclear how the outcomes were scored |
| Other bias | Unclear risk | Baseline comparison: not stated Diagnostic certainty: yes The study was completed |
Brass 2015.
| Methods | Parallel‐group, randomised controlled pilot study The study was conducted at a single centre in the United Kingdom |
|
| Participants | 60 participants with hand eczema unresponsive to clobetasol propionate Dropouts: 17 Inclusion criterion
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Immersion PUVA twice weekly for 12 weeks with 4‐weekly assessments in 30 participants • NB‐UVB twice weekly for 12 weeks with 4‐weekly assessments in 30 particiBpants Duration 12 weeks |
|
| Outcomes |
Primary outcomes of the trial Number of participants achieving a 'clear' or 'almost clear' treatment response at 12 weeks Other outcomes
|
|
| Notes | Data are based on a conference abstract with limited data. Study authors were contacted and provided some additional information Additional information was extracted from the trial register (ISRCTN18213910) Declarations of interest: not stated Funding: the study was sponsored by Newcastle upon Tyne Hospitals NHS Foundation Trust (UK) Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Personal communication with study authors clarified that a computerised randomisation programme was used |
| Allocation concealment (selection bias) | Unclear risk | Personal communication with study authors stated that before recruitment no one knew what the treatment allocation would be, as the trial was randomised |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "we performed a randomized, observer‐blinded pilot study of PUVA vs. NB‐UVB for the treatment of hand eczema" Comment: no data available |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "we performed a randomized, observer‐blinded pilot study of PUVA vs. NB‐UVB for the treatment of hand eczema" Comment: the abstract states that observers were blinded Personal communication clarified that only the research co‐ordinator was aware of the treatment allocation. All observers carrying out assessments were blind |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (43 of 60 = less than 80%) |
| Selective reporting (reporting bias) | Low risk | The trial was registered at http://www.isrctn.com/ISRCTN18213910 before the start. No significant differences between the trial register and the abstract were found |
| Other bias | Low risk | Baseline comparison: a baseline comparison of disease severity was provided Diagnostic certainty: yes The study was completed |
Burrows 1986.
| Methods | Parallel‐group, randomised controlled study; cross‐over design This multi‐centre study was conducted in Ireland and in the UK in a secondary setting at 3 different centres |
|
| Participants | 23 participants with chronic eczema on palms or dorsa, with positive patch test to nickel
Dropouts: 3 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Triethylenetetramine (Trientene) 300 mg daily for 6 weeks in an unknown number of participants Control intervention • Placebo for 6 weeks Cross‐over after 4‐week washout. The total expected duration of the study was thus 16 weeks; however the trial was terminated prematurely Duration 6 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | The trial was terminated due to a literature report on potential adverse events (teratogenicity). Study results were based on participants entered before termination. Results table is difficult to interpret in view of the cross‐over; probably based on 20 participants Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were allocated randomly" Comment: no further details on randomisation procedure |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a multicentre, double‐blind, crossover trial was initiated to test the ability of...." Comment: participants and staff were probably blinded, as this is stated in the paper, and a placebo was used, but no details are given as to how this was achieved. It is not clear whether placebo was identical in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "a multicentre, double‐blind, crossover trial was initiated to test the ability of...." Comment: no details regarding observer blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (20 of 23 = more than 80%) |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. The outcome parameters used are very concise. The Materials and Methods section describes that observer‐ and participant‐rated scores would be used with regard to improvement/no change/deterioration; however, only one table shows improvement versus no improvement and worse, and it is unclear whether this was participant‐ or observer‐rated |
| Other bias | High risk | No baseline comparisons Diagnostic certainty: yes The study was ended prematurely |
Cartwright 1987.
| Methods | Within‐participant, randomised controlled trial This was a single‐centre study, conducted in the UK |
|
| Participants | 30 participants with bilateral symmetrical constitutional hand eczema, resistant to previous treatment
Dropouts: 12 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Superficial X‐ray 300 Rad 10 kV 3 times with a 21‐day interval in 18/30 hands Control intervention • Placebo‐radiation in 18/30 contralateral hands 3 times with a 21‐day interval Participants were followed up for 18 weeks after initial treatment Participants continued application of tar paste or steroid ointments throughout the trial Duration 21 weeks (3 weeks active treatment, 18 weeks follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined. Other outcomes
|
|
| Notes | Secondary outcomes ‐ reduction in severity, investigator‐ and participant‐rated scoring ‐ were included but provided no reproducible data. Only graphic representation of outcome scores. Graphs in Figures 1 and 2 seem to have been exchanged. High dropout: 12 out of 30. Reasons given for the 12 dropouts: unwilling to attend, mostly because eczema improved Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "according to a predetermined random code known by the radiographer..." Reference to a predetermined random code known only by the radiographer |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators, although the article states that only the radiographer knew the randomisation code |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "according to a predetermined random code known by the radiographer and unknown to patient and observer, one hand was irradiated with 300 rad (3 Gy) of Grenz rays (...) and the other hand treated in an exactly similar manner, except that sham therapy was given" Comment: participants were truly blinded and received placebo‐radiation. The radiographer was the only one aware of the randomisation code in that he had to programme the radiation; however in our opinion, this study could not have been done in another fashion; therefore we judged this trial to have low risk, although not all staff were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "according to a predetermined random code known by the radiographer and unknown to patient and observer..." Comment: the observer was unaware of the allocation; however no further details are provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (18 of 30 = less than 80%) |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. Outcomes mentioned in the Methods section are depicted in graphs and tables in the Results section; however participant‐ and observer‐rated graphs probably are switched because results in the participant section range from 0 to 3, and results for the observer range from 0 to 7 |
| Other bias | Low risk | Baseline comparisons: within‐participant study was not applicable Diagnostic certainty: yes The study was completed |
Cherill 2000.
| Methods | Parallel‐group (4 groups), randomised controlled trial; proof of concept Single‐centre study, probably conducted in the USA, although not clear from abstract |
|
| Participants | 48 adult participants with chronic irritant hand dermatitis of moderate severity
No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Pimecrolimus 1% cream twice daily for 6 weeks in 12 participants • Pimecrolimus 1% cream under occlusion twice daily for 6 weeks in 12 participants Control intervention • Vehicle twice daily for 6 weeks in 12 participants • Vehicle under occlusion for 6 weeks in 12 participants Duration 6 weeks |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | Study was published as a conference abstract; therefore information on quality issues is limited. Study authors were contacted by email and LinkedIn but were unresponsive. Similar abstract published in JEADV 2000;14(S1):128 The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included but did not provide reproducible data Declarations of interest: some study authors were employee of Novartis Pharma AG Funding: study authors were employees of Novartis Pharmaceuticals Corp., East Hanover, New Jersey, USA, and Novartis Pharma AG, Basel, Switzerland, the manufacturer of the study drug Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "within each treatment group, patients were randomly assigned (1:1) to occlusion or no occlusion" Comment: not stated whether participants were randomly assigned to pimecrolimus or vehicle ‐ only that participants in both groups were randomly assigned to occlusion or no occlusion. No further details are given |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a 6‐week, randomized, double‐blind, vehicle controlled, single center study" Comment: title suggests double‐blinded; however, no details about how this was accomplished. Participants used pimecrolimus or vehicle: it is unclear whether these were similar in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "a 6‐week, randomized, double‐blind, vehicle controlled, single center study" Comment: no details regarding blinding of observers |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All included participants were analysed |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found, and only limited information could be extracted from the abstract. In the abstract, only the overall scores were given ‐ not the key scores for each item |
| Other bias | Unclear risk | No baseline comparisons conducted or reported Diagnostic certainty: yes The study was completed |
Chu 2009.
| Methods | Within‐participant, randomised controlled trial This study was conducted at a single centre in Taiwan |
|
| Participants | 67 participants with chronic hand eczema Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • E‐DO (HK‐03) topical lotion, once daily (evening), for 4 weeks, applied to 1 hand in 67 participants Control intervention • Placebo applied once daily on the contralateral hand for 4 weeks, on 67 contralateral hands Duration 4 weeks |
|
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
|
| Notes | This study is (not yet) published but was registered on clinicaltrials.gov, and Dr. Chu released the results in personal communication after obtaining consent from HenKan Pharmaceutical The secondary outcomes ‐ reduction in severity, investigator‐rated and participant‐rated ‐ were included but did not provide reproducible data Declarations of interest: not stated, although the study was sponsored by HenKan Pharmaceutical Co. Funding: the study was sponsored by HenKan Pharmaceutical Co., Ltd., and results of this negative study (E‐DO was not statistically significant better than vehicle) were not published, although HenKan Pharmaceuticals did give Dr. Chai‐Yu consent to release the results Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The report and the trial register claim a randomised design; however it is unclear how randomisation was done Personal communication did not reveal further information |
| Allocation concealment (selection bias) | Unclear risk | Not described in the protocol; personal communication did not reveal further details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "randomized, double‐blind, vehicle controlled..." Comment: double‐blind study in which a placebo vehicle was used |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "randomized, double‐blind, vehicle controlled..." Comment: double‐blind study; unclear how this was done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "sixty‐three subjects received at least one dose of each investigational product (..) and [having] at least one post‐baseline assessment on both hands were included in the ITT population" Comment: intention‐to‐treat analysis was carried out on all participants who received the study drug. Only 63 of the 67 randomised participants received the study drug (94%) |
| Selective reporting (reporting bias) | Unclear risk | The trial was registered at clinicaltrials.gov (NCT00556855). We found no major discrepancies between the trial registration and the final study report; however for most of the secondary outcomes (quality of life, TEWL, HEAS, pain score), the report states only that no statistically significant differences were found and does not provide actual numbers |
| Other bias | Low risk | Baseline comparisons: a baseline comparison with regards to disease severity is provided; however because this trial used a within‐participant design, this is not further applicable Diagnostic certainty: yes The study was completed |
Faghihi 2008.
| Methods | Within‐participant, randomised controlled trial This study was conducted in a secondary setting at 2 different centres in Iran |
|
| Participants | 47 participants with nearly symmetrical chronic hand eczema with a duration > 4 weeks No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • 0.05% clobetasol + 2.5% zinc sulphate cream on 1 hand in 47 participants for 2 weeks • 0.05% clobetasol cream alone on the other hand in 47 participants for 2 weeks Duration 2 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Overall severity of hand eczema was not an outcome. Use of the Mann‐Whitney U‐test for statistical analysis appears incorrect, as the data were related (within‐subject design) The secondary outcomes ‐ reduction in severity, participant‐rated, and time until relapse ‐ were included but did not provide reproducible data Study authors were contacted by mail on 28 February 2014 but remained unresponsive Declarations of interest: not stated Funding: the study was funded and supported by Isfahan University of Medical Sciences Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "... right or left hand of them were randomised to be treated..." Comment: no further details given |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the patients and investigators were blinded to type of treatment" Comment: the drugs were made in "similar shape" by a third party; this is considered an adequate way to blind participants Quote: "drugs were made by the Isfahan Pharmacy School in the similar shape, and the patients and investigators were blinded to the type of treatment" Comment: the code of drugs was revealed only at the end of the study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "drugs were made by the Isfahan Pharmacy School in the similar shape, and the patients and investigators were blinded to the type of treatment. The code of drugs was revealed only at the end of the study" Comment: no details regarding blinding of outcome assessors, although study authors mention a double‐blind design; this is insufficient information to judge the risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "overall, 47 patients (94 samples) were evaluated and all of them completed the study" Comment: all participants completed the study and were included in the analyses |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. All outcomes described in Subjects and Methods are described in the Results section, although for itch, only the statistical significance level is stated, but the other outcomes are stated in tables with exact numbers |
| Other bias | Low risk | Baseline comparisons revealed no significant differences between groups in terms of erythema, scaling, lichenification, and pruritus; further within‐participant design Diagnostic certainty: yes The study was completed |
Fairris 1984.
| Methods | Within‐participant, randomised controlled trial The study was conducted at a single centre in the UK and was carried out in a secondary care setting |
|
| Participants | 24 participants with chronic constitutional therapy‐resistant hand eczema
Dropouts: 1 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Ionising radiation 100 rad 50 kV 3 times with 21‐day interval in 23/24 hands Control intervention • Placebo radiation 3 times with a 21‐day interval in 23/24 contralateral hands Participants were followed up until 18 weeks after initial treatment Duration 21 weeks (3 weeks active treatment, 18 weeks follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | The secondary outcomes ‐ reduction in severity, participant‐rated and investigator‐rated ‐ were included but did not provide reproducible data. Only graphic presentation of scores with statistical significance Declarations of interest: not stated Funding: not stated. Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the radiographer (D.P.M.) treating the patient gave the active radiation or placebo treatment according to a predetermined code" Comment: reference to a predetermined code |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators; only that the code was broken after the end of the trial |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind study: participants were unaware of which hand received treatment and which one placebo due to the placebo‐irradiation. The radiographer did know the code of randomisation and gave placebo‐X‐ray therapy to participants that was indistinguishable from actual X‐ray therapy. Although the radiographer (staff) was aware of the treatment, this study could not have been done in another fashion; therefore we judged low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the observer (G.M.F.) did not know which hand was receiving X‐ray therapy until the code was broken at the end of the trial" Comment: the observer was unaware of the treatment group, and we judged this as low risk |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (23 of 24 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. However all outcomes described in the Methods section are clearly described in the Results section |
| Other bias | Low risk | Baseline comparisons: within‐participant study was not applicable Diagnostic certainty: yes The study was completed |
Fairris 1985.
| Methods | Within‐participant, randomised controlled trial The study was conducted at a single centre in the UK and was carried out in secondary care setting |
|
| Participants | 25 participants with chronic constitutional therapy‐resistant hand eczema Dropouts: 5 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Superficial X‐ray 300 Rad 10 kV 3 times with 21‐day interval in 20/25 hands • 100 Rad 50 kV 3 times with 21‐day interval in 20/25 contralateral hands Participants were followed for 18 weeks after initial treatment Duration 21 weeks (3 weeks active treatment, 18 weeks follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | The secondary outcomes ‐ reduction in severity, participant‐rated and investigator‐rated ‐ were included but did not provide reproducible data. Only graphic presentation of scores with statistical significance Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "one hand received 100 rad (1 Gy) of conventional superficial X‐ray (...), the other 300 rad (3 Gy) of Grenz ray (...) according to a predetermined random code operated by the radiographer..." Comment: reference to a predetermined random code operated by the radiographer |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...operated by the radiographer and unknown to the observer" Comment: no information about participant blinding; however the difference between grenz ray and X‐ray therapy is indistinguishable for a participant. Although the radiographer (staff) was aware of the treatment, this study could not have been done in another fashion; therefore we judged low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...operated by the radiographer and unknown to the observer" Comment: the study claims to be double‐blinded and thus observer‐blinded, which was probably adequate as in Fairris 1984, because the study designs are similar |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (20 of 25 = 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however, all outcomes described in the Methods section are clearly described in the Results section |
| Other bias | Low risk | Baseline comparisons: within‐participant study was not applicable Diagnostic certainty: yes The study was completed |
Fowler 2005.
| Methods | Within‐participant, randomised controlled trial of 3 parallel groups This was a multi‐centre study conducted in the USA and carried out in a secondary care setting |
|
| Participants | 86 participants with chronic hand eczema
Dropouts: 4 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention
• Hydrocortisone butyrate 0.1% cream on the one hand vs fluticasone propionate 0.05% cream twice daily on the other hand for 2 weeks in 26 participants
• Hydrocortisone butyrate 0.1% cream on the one hand vs prednicarbate emollient 0.1% cream twice daily on the other hand for 2 weeks in 28 participants
• Hydrocortisone butyrate 0.1% cream on the one hand vs mometasone furoate 0.1% cream twice daily on the other hand for 2 weeks in 31 participants Duration 2 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Three participants with atopic dermatitis participated in the study. Percentage of hand involvement was the only outcome whereby exclusively participants with hand eczema were analysed. Each intervention group had a within‐participant design. in addition, the difference in efficacy between the 3 groups was evaluated The study did include a participant‐ and investigator‐rated severity score, but we were unable to use the data. The study also included adverse events, but we were unable to use this information because only numbers for both treatment groups combined were stated Study authors were contacted on 4 March 2014 and replied 6 March 2014 Declarations of interest: 2 study authors acted as consultants Funding: the study was funded by Ferndale Laboratories, Inc., manufacturer of the study drugs. Two study authors were investigator and consultant for Ferndale Laboratories, Inc. Sample size rationale: not stated; personal communication clarified this was not conducted |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomised in balanced cohorts to 3 parallel treatment groups" Comment: no further details given in the article; personal communication with the study author clarified that a computer programme was used to create the randomisation code |
| Allocation concealment (selection bias) | Low risk | No details in the article about how allocation was concealed from participants and investigators. Personal communication revealed that allocation was conducted by the sponsor, who was at a remote site. Participants were enrolled without knowledge of the expected treatment group |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the medication[s] were dispensed to the subjects in blind‐labelled tubes that were clearly marked with the subject's identification number and the word left or right" Comment: study authors state double‐blind design. The sponsor and the study co‐ordinator had access to the randomisation code list; treating physicians and participants were unaware of this |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind study, which includes observer blinding. Observers had no access to the randomisation code and were truly blinded (personal communication) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (82 of 86 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however all outcomes described in the Methods section are described in the Results |
| Other bias | Low risk | Baseline comparisons: no significant differences among the 3 groups in terms of age, gender, race, and eczema severity Diagnostic certainty: yes The study was completed |
Fowler 2014.
| Methods | Randomised, parallel‐group, placebo‐controlled trial This study was carried out in a secondary care setting This was a multi‐centre study conducted at 70 centres in the United States The study consisted of 4 phases: screening, run‐in, treatment, and follow‐up |
|
| Participants | 596 participants with severe chronic hand eczema refractory to potent topical corticosteroids
Dropouts during treatment phase: 307 participants 162 participants entered the follow‐up period after achieving a PGA rating of clear or almost clear Dropouts during follow‐up: 35 A total of 467 entered the safety follow‐up phase Dropouts during safety follow‐up: 116 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral alitretinoin 30 mg once daily for 24 weeks in 298 participants Control intervention • Placebo capsules once daily for 24 weeks in 298 participants Duration 72 weeks (24 weeks of active treatment and follow‐up up to 48 weeks after the end of treatment) |
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
Other outcomes
|
|
| Notes | The treatment phase was included in this review Study authors were contacted for additional information Declarations of interest: the first study author was a sponsored investigator and served as consultant to GSK The other study authors were employed by Stiefel, a GSK Company Funding: the study was supported and funded by Stiefel, a GlaxoSmithKline Company (GSK), manufacturer of the study drug. No information was provided about external monitoring or quality control Sample size rationale: adequate |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "participants were randomized 1:1 to receive once‐daily alitretinoin 30 mg or placebo through a central randomization system that used an interactive voice response system" Comment: randomisation method adequate |
| Allocation concealment (selection bias) | Low risk | Quote: "investigators, study personnel, patients and statisticians were unaware of assigned study treatment" Comment: the central randomisation point was used at a distance location |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote from the clinical trial register: "patients receive matching placebo for up to 24 weeks" Comment: placebo was used to achieve blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not clear who assessed the outcome ‐ probably the 70 providers of the treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the intent‐to‐treat (ITT) population (randomised patients who were dispensed medication) was used for efficacy analyses" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | The study was registered at clinicaltrials.gov NCT00817063. Except for some small differences in the exclusion criteria, no substantial differences between protocol and study report were found |
| Other bias | Low risk | Baseline comparison: no significant differences were reported Diagnostic certainty: yes The study was completed |
Fredriksson 1975.
| Methods | Within‐participant, randomised controlled trial The study was probably conducted at a single centre in Sweden |
|
| Participants | 30 participants with bilateral eczematous dermatitis of the hands were selected from a clinical pool No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Aquacare HP cream, a moisturising emulsion containing multi‐sterols, phospholipids, and fatty diols (pH 6) twice a day (morning and evening) for 4 weeks in 30 hands Control intervention • Calmurid cream containing betaine and lactic acid (pH 3) twice a day for 4 weeks in 30 contralateral hands Duration 4 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | The last outcome is unclear, and results are not depicted in the article. Study authors state only that Aquacare was statistically significantly more effective over Calmurid; no exact results or data are given Declarations of interest: not stated. Funding: not stated. Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "tubes containing 90 gpm of Aqaucare HP cream and Calmurid cream were packed into identical cartons after being randomly marked left and right" Comment: no clarification on how random marking of left and right was done; therefore risk was judged as unclear |
| Allocation concealment (selection bias) | Low risk | Quote: "tubes containing 90 gpm of Aqaucare HP cream and Calmurid cream were packed into identical cartons after being randomly marked left and right. These were dispensed in a double‐blind fashion" Comment: the drugs were dispensed in identical looking cartons that at random were marked with left or right, without any organisation; therefore physicians and participants were unaware of treatment allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double‐blind fashion..." Comment: study authors stated double‐blinded design. Randomly marked tubes were dispensed in a "double‐blind fashion", which is considered an adequate way to blind participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...double‐blind fashion..." Comment: double‐blind study |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All included participants were analysed |
| Selective reporting (reporting bias) | High risk | No trial registration found. The article describes an unclear severity scale ranging from 0 to 5; however the results for this outcome are not registered in the article |
| Other bias | Low risk | No baseline comparisons; however within‐participant study was not applicable Diagnostic certainty: yes The study was completed |
Granlund 1996.
| Methods | Randomised, parallel‐group design, with a partial cross‐over design in the second phase Randomisation procedure unclear The study was carried out in a secondary care setting This was a single‐centre study conducted in Finland |
|
| Participants | 41 participants with hand eczema, continuously for 6 months, significant disability, inadequate response to conventional treatment, confirmation by histopathology
Dropouts: 6 in the first phase, 1 in the second phase Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral cyclosporin 3 mg/kg/d and placebo cream for 6 weeks in 17/20 participants • Topical betamethasone dipropionate 0.05% cream and placebo capsules identical to cyclosporin in 19/21 participants At week 6, cross‐over of those who had treatment failure in the first 6 week phase: 8 participants switched to betamethasone, and 6 to cyclosporin In the third phase, a 24‐week follow‐up period without intervention Use of own emollients was allowed in both groups Duration Maximum 36 weeks with 6 to 12 weeks of active treatment |
|
| Outcomes |
Grandlund 1996: Primary outcomes of the trial Not defined Other outcomes
Grandlund 1997: Primary outcome of the trial
Other outcomes
|
|
| Notes |
Grandlund 1996: study had 3 phases, which were partially overlapping. The second phase dealt with participants who had treatment failure in phase 1. In this second phase, participants were switched over to the alternative intervention. The third phase includes only participants who had treatment success in phase 1. Outcome assessment based on intention‐to‐treat analysis. This review deals with only phase 1 and phase 3 Granlund 1997: paper is based on the same trial (same participants) as Granlund 1996, but deals only with phases 1 and 2. The study had 3 phases, which were partially overlapping. The second phase dealt with participants who had treatment failure in phase 1. In this second phase, participants were switched over to the alternative intervention. The third phase includes only participants who had treatment success in phase 1. In this review, only results of the first phase will be discussed. Outcome assessment was based on intention‐to‐treat analysis The secondary outcomes ‐ reduction in severity, participant‐rated scoring, time until relapse, and dose reduction ‐ were included in the study but did not provide reproducible data Declarations of interest: not stated Funding: the study was supported by Sandoz Pharmaceuticals, Switzerland (manufacturer of the study drug) and Finland, and by a grant from Finska Läkaresällskapet, Finland Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were given numbers 1‐41 in consecutive order, which had been reassigned to treatment with..." Comment: however, neither article clarifies how this reassignment was done |
| Allocation concealment (selection bias) | Low risk | Quote: "the codes were not opened until all participants had finished all parts of the study" Comment: the study used identical placebos for topical and oral treatment, and participants were given consecutive numbers |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "soft gelatine capsules containing 25, 50 or 100 mg and identical placebo capsules were supplied by ..." "Identical 100 tubes were used for the creams" Comment: study authors stated double‐blinded design. Sufficient information provided about how participant blinding was achieved and identical tubes and placebos were used |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the codes were not opened until all patients had finished all parts of the study" Comment: double‐blind design; because identical placebos were used and the randomisation code was not broken before the end of the study, it is unlikely that assessors were aware of the treatment group |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "results were analyzed on an intention to treat basis" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however all relevant outcomes are addressed in the Materials and Methods sections of the 2 articles and are depicted in graphs in the Results section |
| Other bias | Unclear risk | Baseline comparisons revealed a significant difference between groups in terms of pre‐study antibiotics treatment Diagnostic certainty: yes The study was completed |
Grattan 1991.
| Methods | Within‐participant, randomised controlled trial The study was probably conducted in a secondary care setting at a single centre in the UK |
|
| Participants | 15 participants with vesicular hand eczema for at least 6 months
Dropouts: 3 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Topical PUVA 3 times a week for 8 weeks on 12/15 hands Control intervention • UVA (with placebo psoralen paint) on 12/15 contralateral hands Moisturisers were allowed on both hands, and both hands received a small fraction of UVB from UVA lamps During an unclear follow‐up period, participants received a questionnaire Duration 8 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Small number of participants. The secondary outcomes ‐ reduction in severity, investigator‐rated and participant‐rated, and time until relapse ‐ were included but did not provide reproducible data. Exact figures for main outcomes are not given; instead there are graphic presentations. Questionnaire assessment was performed after completion of the study, but duration of follow‐up in this questionnaire assessment remains unclear Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "psoralen and placebo were randomised and coded by one independent investigator (GJS) and supplied in bottles labelled left and right" Comment: no further details |
| Allocation concealment (selection bias) | Low risk | Quote: "psoralen and placebo were randomised and coded by one independent investigator (GJS) and supplied in bottles labelled left and right" Comment: randomisation and coding were accomplished by an independent investigator, and bottles were supplied labelled 'left' and 'right'; therefore the physician was unaware of allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "psoralen and placebo were randomised and coded by one independent investigator (GJS) and supplied in bottles labelled left and right" Quote: "the placebo was...." Comment: double‐blind study with a similar looking placebo; it was not possible for the participant to distinguish these |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the code was not broken until completion of the study" Comment: double‐blind design in which an independent investigator supplied the treatments. It was not possible for observers to know the treatment groups |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (12 of 15 = 80%) |
| Selective reporting (reporting bias) | Unclear risk | No trial register found. No major differences between outcomes described in the Methods section and outcomes described in the Results section; however although the article text claims that separate scores for the T‐120 are described, we were unable to find a score for separate items |
| Other bias | Low risk | Baseline comparisons: as within‐participant study not applicable Diagnostic certainty: yes The study was completed |
Gupta 1993.
| Methods | Parallel‐group, randomised controlled trial The study was conducted at a single dermatology centre in Canada |
|
| Participants | 58 participants with steroid‐responsive dermatitis limited to the hands Evaluable: 54 Dropouts: 6, of whom 4 permitted a protocol violation and 2 ended prematurely because of an exacerbation of hand eczema Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Betamethasone dipropionate film‐forming lotion in 28/29 participants daily for 7 days • Betamethasone dipropionate thickened lotion in 26/29 participants for 7 days Duration 1 week |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Very short study of only 7 days. Unclear about withdrawals in lotion group. Exact number allocated to each treatment not specified. Among the different outcomes, unclear how change in overall severity was calculated Declarations of interest: not stated. Funding: the study was supported in part by a grant from GenDerm Corporation, Montreal, Canada, manufacturer of the study drugs Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "participants were randomly assigned to 2 groups..." Comment: no further details |
| Allocation concealment (selection bias) | Low risk | Quote: "bottles were dispensed in their marketed containers with identical overlabels and the contents were not known to the patients or the investigator who assessed the results. Only the study coordinator was aware of the contents of the bottles" Comment: sequentially numbered drug containers of identical appearance are considered as adequate allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "bottles were dispensed in their marketed containers with identically appearing overlabels and the contents were not known to the patients or the investigator who assessed the results. Only the study coordinator was aware of the contents of the bottles" Comment: double‐blind study; identical looking containers were used |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "bottles were dispensed in their marketed containers with identically appearing overlabels and the contents were not known to the patients or the investigator who assessed the results. Only the study coordinator was aware of the contents of the bottles" Comment: observers were unaware of the study drug, which was identical in appearance |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (54 or 52 of 58 = more than 80%) |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. No major differences between the Methods and Results sections found; however for erythema and pruritus, study authors state only that no significant differences were found |
| Other bias | Low risk | Baseline comparisons: at baseline, significant differences in scaling between groups, but controlled for by statistical procedure. No significant differences at baseline with regard to age, gender, race, erythema, induration, pruritus, or area of eczematous involvement Diagnostic certainty: yes The study was completed |
Hanifin 2004.
| Methods | A phase I‐II, open‐label, randomised controlled, parallel‐group (3 groups) study The study was conducted at various dermatology departments in the USA |
|
| Participants | 55 participants with chronic severe hand dermatitis (32 atopic, 18 irritant, 5 dyshidrotic or other): duration at least 6 months and severity score 3 or 4
Dropouts: 13 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Bexarotene 1% gel escalated stepwise from 1× every other day to 3× daily in 28 participants for 22 weeks • Bexarotene gel stepwise plus mometasone furoate 0.1% ointment 2× daily in 13 participants for 22 weeks • Bexarotene gel stepwise plus hydrocortisone 1% ointment 2× daily in 14 participants for 22 weeks In all 3 groups, daily use of emollients was allowed Duration 22 weeks |
|
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
Other outcomes
|
|
| Notes | Phase I to II open‐label study. Intention‐to‐treat principle not stated, but the proportion of participants with treatment success is based on the number of all participants enrolled in each treatment group. Of the 12 dropouts/withdrawals, it is unknown to which treatment group they belong The secondary outcome ‐ reduction in severity, participant‐rated scoring ‐ was included but no reproducible data were provided Study authors were contacted by email and LinkedIn; however they were unable to answer all of our questions Declarations of interest: one of the study authors was an employee of Ligand Pharmaceuticals, San Diego, USA Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized at 2:1:1 into three treatment arms..." Comment: no further details given in the article; personal communication clarified that a computer‐generated scheme was used |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "a phase I‐II open label randomized clinical study...." Comment: no blinding of participants or observers as open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "a phase I‐II open label randomized clinical study...." Comment: observers were not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No intention‐to‐treat analysis according to the text; however in the tables, 55 participants seems to be included, where the text clearly states that 13 participants withdrew before completing the 22 weeks of the study 42 of 55 = less than 80% |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however, relevant outcomes were described, such as HEASI score, and we found no major discrepancies between participants in the Methods section and the Results section |
| Other bias | Low risk | Baseline comparisons: no significant difference among groups in demographic or disease characteristics Diagnostic certainty: yes The study was completed |
Hill 1998.
| Methods | Randomised, parallel‐group design. The study was conducted in a secondary care setting at different dermatology departments in the UK |
|
| Participants | 120 participants with diagnosis of eczema on one or both hands, and with suspected or confirmed infection
Dropouts: 10 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Betamethasone‐valerate 0.1% + clioquinol 3% cream twice daily for 4 weeks in 57/61 participants • Betamethasone‐v 0.1% + fusidic acid 2% cream twice daily in 53/55 participants for 4 weeks Duration 4 weeks |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | Primary outcome assessed at last on‐treatment visit: probably for most participants at week 4, but unclear how much earlier for dropouts (graph suggests after week 4). Not clear if data for secondary outcome number 2 (participant‐rated response) are presented The primary outcome percentage of participants with self‐rated good/excellent control and the secondary outcomes ‐ reduction in severity, investigator‐ and participant‐rated scoring ‐ were included but provided no reproducible data Declarations of interest: one study author was an employee of Leo Pharmaceuticals, Princes Risborourg, UK Funding: the study was designed and sponsored by Leo Pharmaceuticals, Princes Risborough, UK, manufacturer of the study drug Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "treatment was allocated at random" Comment: the article states only that treatment was allocated at random, without further details |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "this was a multicentre, prospective, randomized, open‐parallel‐group comparison" Comment: not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "this was a multicentre, prospective, randomized, open‐parallel‐group comparison" Comment: not blinded, which might have affected observer‐rated outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "... and were included in an intention‐to‐treat analysis in respect of the primary efficacy criterion only" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. We did not find major differences between what was stated in the Methods section and in the Results section; however the subscores for clinical signs and symptoms were not given, and it was stated only whether they were statistically significantly different |
| Other bias | Unclear risk | Baseline comparisons: more 'severe' classification of signs in the betamethasone/fusidic acid group; unclear whether this constitutes a significant difference and was controlled for Diagnostic certainty: yes The study was completed |
Hordinsky 2010.
| Methods | Randomised controlled, parallel‐group design This was a multi‐centre study conducted at 57 centres in 7 countries (Austria, Canada, Denmark, Hungary, Italy, Norway, USA) |
|
| Participants | 652 (246 male, 406 female) with diagnosis of mild to moderate hand dermatitis as defined by IGA 555 participants completed the double‐blind phase, 544 (269 in the pimecrolimus group and 275 in vehicle group) entered the open extension phase, and 512 (248 and 264, respectively) completed the study Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Pimecrolimus 1% ointment twice daily with daily occlusion by use of vinyl gloves of at least 6 hours after second (evening) application for up to 43 weeks in 325 participants Control intervention • Vehicle ointment twice daily with daily occlusion by use of vinyl gloves of at least 6 hours after second (evening) application for up to 43 weeks in 327 participants Duration Up to 43 weeks |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | Participants could enter open‐label phase before 42nd day if hand dermatitis had remained cleared on 2 consecutive weekly assessments. However, efficacy comparisons were made at day 42 in intention‐to‐treat analysis. Not clear how many participants were blind to treatment during assessments at days 29, 36, and 43, as open‐label phase could already have started We were unable to obtain additional information from study authors Declarations of interest: one study author was an employee of Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA Funding: the study was supported by Novartis Pharma AG, manufacturer of the study drug Sample size rationale: provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "eligible patients were randomized in a 1:1 ratio to receive..." Quote: "randomization was performed using a validated automated system and was stratified by baseline IGA score at each centre" Comment: randomisation method was considered adequate |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "...double‐blind and vehicle‐controlled..." Comment: study authors state double‐blinded design, although unclear whether pimecrolimus and vehicle were identical in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...double‐blind and vehicle‐controlled..." Comment: double‐blind study; however, insufficient details are given about investigator blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all the efficacy assessments were done in the intent‐to‐treat population using a last observation carried forward approach" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Trial registration was found on clinicaltrials.gov (NCT00226707) Work productivity and activity impairment questionnaires are included in the trial registration but are not mentioned in the article The trial register stated that the primary outcome was IGA on day 43, although this is not clearly stated in the article |
| Other bias | Low risk | Baseline comparisons conducted: "there were no clinically relevant differences in baseline demographic characteristics or disease history between the pimecrolimus cream 1% and vehicle groups" Diagnostic certainty: yes The study was completed |
Jowkar 2011.
| Methods | Randomised controlled, parallel‐group design This study was conducted at a single dermatology centre in Iran |
|
| Participants | 44 "healthy patients with hand eczema that did not use topical medication in 2 weeks ago or systemic medication in 1 month ago were enrolled" (participants aged 12 to 60 years with hand eczema) No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • 4% topical cream of Fumaria parviflora L. alcoholic extract for 4 weeks twice daily, 10 grams on hand surface skin in probably 22 participants, although this is not clearly described in the article Control intervention • Placebo twice daily in probably 22 participants for 4 weeks Participants were followed up until 2 weeks after the end of treatment Duration 6 weeks (4 weeks active treatment, 2 weeks follow‐up) |
|
| Outcomes |
Primary outcome of the trial
Other outcomes of the trial
|
|
| Notes | The number of participants in each group is not described, and the ratio intervention vs placebo is unclear Because the data are presented in a graphical manner, they are difficult to reproduce. The secondary outcome ‐ reduction in severity investigator‐rated ‐ was included but did not provide reproducible data Study authors were contacted on 28 February 2014 and replied the same day Declarations of interest: none declared Funding: the study was supported by Shiraz University of Medical Science Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomization was conducted based on block randomization design" Comment: randomisation block design suggests the use of a randomisation code list; however study authors denied the existence of a randomisation list in personal communication |
| Allocation concealment (selection bias) | Low risk | No details about allocation concealment in the article; however personal communication clarified that treatment allocation was done by a third person |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind (patient‐physician) placebo‐controlled study" Comment: no additional information is provided in the article. Personal communication clarified that placebo and actual treatment were the same in appearance, and the secretary (third party, not involved in actual treatment) dispensed the study drugs |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "both dermatologist and patients were blind to study groups. Data were recorded by an assessor" Comment: physicians were blinded and unaware of treatment allocation, which was done by a third person |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "a total of 44 patients completed the study" Comment: one patient was excluded from the study due to side effects; however more than 80% completed the study |
| Selective reporting (reporting bias) | Low risk | No discrepancy between the registered trial (IRCT 1388103030741N1) and the original article with regard to outcomes |
| Other bias | Unclear risk | Baseline characteristics depicted in graphs; not stated whether there was a significant difference between groups Diagnostic certainty: yes The study was completed |
Jowkar 2014.
| Methods | Randomised controlled, parallel‐group study This study was conducted at teaching dermatology clinics of the Shiraz University of Medical Sciences | |
| Participants | 92 consecutive patients with a clinical diagnosis of hand eczema Dropouts: 58 participants were analysed, of whom 4 were excluded due to adverse events; unclear what happened to the remaining 34 participants Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Interventions • Topical fumaric acid 5% cream twice a day for 1 month in 30 participants • Triamcinolone 0.1% cream twice a day for 1 month in 28 participants Duration 4 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes of the trial
|
|
| Notes | 92 consecutive patients were recruited; a substantial portion of these patients were lost to follow‐up Study authors were contacted for additional information by email and provided additional information regarding design and risk of bias Declarations of interest: none declared Funding: the study was sponsored by Shiraz University of Medical Sciences. Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a list of randomized coded groups was given to the study investigator, and as patients consecutively were enrolled in the study, they were assigned to the next available randomized group on the list" Comment: consecutive patients were enrolled, and the investigator was given a list of randomised coded groups Study authors clarified they used a random number table generated by a statistical computer programme |
| Allocation concealment (selection bias) | Low risk | No details about how allocation was concealed from participants and clinicians besides the above stated. Study authors clarified that assignment of study medication was done by a third party ‐ the hospital pharmacy |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind study" Comment: no additional information is provided in the article, but study authors stated that study drugs were prescribed in similar looking bottles of 40 grams |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double‐blind study" Comment: no additional information is provided in the article, but study authors explained that observers were unaware of the allocation and were not involved in application or distrubution of study drugs |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (58 of 92 = less than 80%). A substantial number of the 92 participants were lost to follow‐up, and 4 left the study due to adverse events (explanation of the study author) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however all outcomes listed in the Materials and Methods section are also listed in the Results section |
| Other bias | Low risk | Baseline comparisons were conducted, and no differences were found regarding age, gender, and duration of disease Diagnostic certainty: yes The study was completed |
Kaaber 1983.
| Methods | Randomised controlled, parallel‐group design This study was probably conducted in a secondary care setting at 2 Danish departments of dermatology |
|
| Participants | 30 female participants with pompholyx more than 6 months, and positive patch test to nickel
Dropouts: 6 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral tetraethylthiuram disulphide (TETDS) 50 mg/d first week, increasing to 200 mg/d for at least 6 weeks in 11/15 participants for at least 6 weeks Control intervention • Placebo tablets in 13/15 The total duration of the study was probably 8 weeks (?); however run‐in time and total duration of treatment are not completely clear Both groups were allowed to use desoximetasone ointment and emollients Duration Probably 8 weeksIS |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Study duration unclear. Timing of outcome assessments not clear. Comparison based on slopes of linear regression of scores. The secondary outcomes ‐ reduction in severity, investigator‐rated, and dose reduction ‐ were included but did not provide reproducible data Declarations of interest: not stated Funding: Hoechst Danmark and Dumex Ltd. Danmark supplied the study drugs Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "referring to a system of random numbers, the patients received..." Comment: reference to a system of random numbers |
| Allocation concealment (selection bias) | Unclear risk | Unclear if this concerned an open list and unclear how allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the tablets were identical in appearance" Comment: study authors stated double‐blinded design; this is considered an adequate way to blind participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind design. No information is given about how observer blinding was achieved |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (24 of 30 = 80%) |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. For observer‐rated severity score, the Results section states only that this was not statistically significant, only with regards to scaling and the frequency of flares. The Results section is very concise |
| Other bias | Unclear risk | Baseline comparisons: not stated Diagnostic certainty: yes The study was completed |
Katsarou 2012.
| Methods | Randomised controlled, parallel‐group study This study was conducted in a specialised unit for contact dermatitis at a single centre in Greece |
|
| Participants | 30 participants with chronic hand eczema and a relevant allergic contact dermatitis No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Tacrolimus 0.1% twice daily for 30 days and once daily for 31 to 90 days in 15 participants • Mometasone furoate ointment twice daily for 1 week, once daily during week 2 and week 3, once daily 3 times a week for weeks 4 and 5, and once daily 2 times a week during the rest of the study until 90 days in 15 participants Duration 90 days |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | The secondary outcome ‐ reduction in severity, participant‐rated ‐ was included but not provide reproducible data. The conducted analyses are inappropriate and insufficient, and in consequence, the conclusions are invalid Study authors were contacted on 28 February 2014 but were not responsive Declarations of interest: none declared Funding: no financial support received Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "according to the study design, patients were randomized according to random numbers in a computerized way" Comment: the study used randomisation according to random numbers generated in a computerised way |
| Allocation concealment (selection bias) | Unclear risk | Quote: "an investigator’s assistant enrolled and assigned the treatment of the participants while the clinical evaluation was performed by a group of three investigators..." Comment: an investigator's assistant enrolled and assigned treatment; unclear whether this person was aware of treatment allocations |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants were not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "an investigator’s assistant enrolled and assigned the treatment of the participants while the clinical evaluation was performed by a group of three investigators, in order to make the assessments more objective as the investigators were unaware of the patient’s group" Comment: assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All included participants completed the study |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however all outcomes listed in the Materials and Methods section are also listed in the Results section |
| Other bias | Low risk | Baseline comparison: there were no statistical differences in baseline variables according to study authors Diagnostic certainty: yes The study was completed |
Kemper 1998.
| Methods | Within‐participant, randomised controlled study This study was conducted in a secondary care setting at 2 Dutch departments of dermatology |
|
| Participants | 19 participants with hand eczema (all types)
Dropouts: 7 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Coal tar 5% paste (pix lithanthracis) once weekly on the one hand in 12 participants for 4 weeks • Betamethasone valerate 0.1% ointment once weekly in 6 contralateral hands for 4 weeks • Zinc oxide paste once weekly during 4 weeks in the remaining 6 contralateral hands All participants had to wear gloves for an entire week after application of the ointment Use of oral antihistamines was allowed in all groups Duration 4 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes of the trial
|
|
| Notes | The only trial included studying the effect of coal tar paste. Small number of participants with relatively high dropout rate. The secondary outcomes ‐ reduction in severity, investigator‐rated and participant‐rated ‐ were included but did not provide reproducible data. The results are listed as overall mean scores. No exact data are given Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "door middel van loting werd bepaald welke hand..." (free translation: "by means of a draw was decided which hand...") Comment: the paper refers to a lottery system |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded, which is also difficult given the colour and smell of coal tar treatment |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not blinded, which might have affected the investigator‐rated outcome |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (12 of 19 = less than 80%) |
| Selective reporting (reporting bias) | High risk | No trial registration found. In the Methods section, study authors list a scoring system based on erythema, vesicles, papules, scaling/hyperkeratosis, and lichenification multiplied by the affected area, and refer to 2 different references with scoring systems; however in the Results section, a total sum score is given, along with scores for inflammation, hyperproliferation, and lichenification; these are not explained in the article, nor in the references |
| Other bias | Low risk | Baseline comparisons: as within‐participant study not applicable Diagnostic certainty: yes The study was completed |
King 1984.
| Methods | Within‐participant, randomised controlled study This study was conducted at a single centre in a secondary care setting in the UK |
|
| Participants | 20 participants with chronic palmar eczema Evaluable: 15 (8 hyperkeratotic, 7 pompholyx). Dropouts: 5 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Superficial ionising radiation fractionated 100 rad at 45 kV once weekly for 3 weeks; total dose 300 rad in 15/20 hands Control intervention • Placebo radiation once weekly for 3 weeks in 15/20 contralateral hands In both groups, the topical medication was continued unchanged Duration 3 weeks' active treatment with follow‐up until 6 months |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Outcome 2 (photographs) was not used in the presentation of results Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the radiographer randomly selected one palm..." Comment: no further details are given |
| Allocation concealment (selection bias) | Low risk | Quote: "neither patient nor doctor was aware which hand had received treatment, but a record was kept by a radiographer in a sealed envelope" Comment: the study used sealed envelopes. This is a clear description of an adequate allocation concealment procedure |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "neither patient nor doctor was aware which hand had received treatment, but a record was kept by a radiographer in a sealed envelope" Comment: double‐blinded. One hand received actual radiotherapy; the other received "simulated radiotherapy" in the same regimen. This was considered as an adequate way to blind participants. The radiographer (staff) was aware of the treatment arm, but we consider this the best possible way to blind participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "neither patient nor doctor was aware which hand had received treatment, but a record was kept by a radiographer in a sealed envelope" Comment: double‐blind design. At the end of the study, the records of the radiographer were studied. Observers had no direct access to the randomisation code due to the sealed envelope and were not involved in administration of treatment. This is considered an adequate method to blind outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (15 of 20 = less than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. We did not find discrepancies between Methods and Results sections |
| Other bias | Low risk | Baseline comparisons: as within‐participant study not applicable Diagnostic certainty: yes The study was completed |
Kircik 2013.
| Methods | Parallel‐group, randomised controlled study This study was probably conducted in North America |
|
| Participants | 125 participants with moderate to severe hand eczema Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Clobetasol propionate 0.05% foam twice a day for 14 days in 62 participants Control intervention • Vehicle/Placebo foam twice a day for 14 days in 63 participants Duration 2 weeks |
|
| Outcomes |
Primary outcome of the trial
Other outcomes of the trial
|
|
| Notes | Sponsor: Stiefel, a GSK company We contacted study authors for additional information; however they remained unresponsive Declarations of interest: the primary study author has worked as consultant, researcher, or speaker for GSK Stiefel; 2 other study authors are employees of Stiefel, a GSK company Funding: the study was supported by Stiefel, a GSK company Sample size rationale: adequate |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "subjects were randomized 1:1 at baseline using a computerized‐generated schedule..." Comment: subjects were randomised 1:1 at baseline according to a computer‐generated schedule, which is considered as low risk |
| Allocation concealment (selection bias) | Low risk | Quote: "study product assignments were unavailable to study personnel..." Comment: allocation was likely concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "both products were identical in packaging, labelling and ingredients except for the presence or absence of clobetasol propionate" Comment: the study claims to be double‐blind. Identical looking placebos were used, which we consider an adequate method of blinding of participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "study product assignments were unavailable to study personnel until after all data had been collected and validated following applicable standard operating procedures" Comment: observers were unaware of treatment allocation and had no direct access to the randomisation code |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the intent‐to‐treat (ITT) population included all randomized subjects who were dispensed the study product. The per‐protocol (PP) analysis set was used for the primary and key secondary analyses (which were also performed for the ITT analysis set)" Comment: an intention‐to‐treat analysis was carried out next to a per‐protocol analysis |
| Selective reporting (reporting bias) | Unclear risk | The study design and results were registered on clinicaltrials.gov (NCT01323673). In the article, substantially more outcomes are listed that were not registered in the trial register, for example, quality of life and work impairment |
| Other bias | Low risk | Baseline comparisons: an extensive baseline comparison was given Diagnostic certainty: yes The study was completed |
Krejci‐Manwaring 2008.
| Methods | Parallel‐group, randomised controlled study This study was conducted in a secondary care setting at a single centre in the USA |
|
| Participants | 32 participants with moderate to severe hand dermatitis who did not use topical tacrolimus during the previous 28 days and did not use topical corticosteroids, non‐steroidal immunosuppressants, or light treatment during the last 7 days
Dropouts: 13 (including 1 participant who dropped out before the intervention was started) Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Topical tacrolimus twice daily for 12 weeks in addition to a daily dose of prednisone during 3 weeks; 30 mg in week 1, 20 mg in week 2, 10 mg in week 3 in 14/21 participants Control intervention • Vehicle ointment applied twice daily for 12 weeks; in addition, a daily dose of prednisone during 3 weeks: 30 mg in week 1, 20 mg in week 2, 10 mg in week 3 in 6/11 participants Participants were followed up at 5‐week intervals until week 14 after initial treatment Duration 14 weeks (3 weeks active treatment, 11 weeks follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Pilot study on maintenance therapy. Exact numbers of results for main outcomes not given ‐ only whether there was a statistically significant difference between the 2 interventions The study did include the secondary outcomes reduction in severity, investigator and participant‐rated and time until relapse, but we were unable to reproduce these data Declarations of interest: study authors received research, speaking, and/or consulting support from various pharmaceutical companies Funding: the study was supported by a grant from Astellas Pharma Inc. Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the randomization code list, correlating the kit number with the content of each kit, was kept on file at Fuijsawa Healthcare Medical Information Department until the time of analysis" Comment: reference to a randomisation code list |
| Allocation concealment (selection bias) | Low risk | Quote: "the vehicle and tacrolimus ointments were packaged in identical containers labelled with the subject number, so neither the subject, coordinator, nor the investigator knew which treatment the patient received" Comment: randomisation was remote from the participant‐recruitment centre. Vehicle and tacrolimus were packaged in pre‐labelled identical containers corresponding to a participant number |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the vehicle and tacrolimus ointments were packaged in identical containers labelled with the subject number, so neither the subject, coordinator, nor the investigator knew which treatment the patient received" Comment: double‐blinded; this is considered an adequate blinding method |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the vehicle and tacrolimus ointments were packaged in identical containers labelled with the subject number, so neither the subject, coordinator, nor the investigator knew which treatment the patient received" Comment: by the use of pre‐labelled and identical containers, observers were blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No intention‐to‐treat analysis but per protocol (20 of 33 = less than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. All relevant outcomes described in the Materials section are described in the Results section |
| Other bias | Unclear risk | Baseline comparisons: at baseline, significant difference between groups for demographic characteristics was given. All participants had a combined symptom severity score of 5 to 16 Diagnostic certainty: yes The study was completed |
Kucharekova 2003.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary care setting; it was a single‐centre study. This study was conducted in the Netherlands |
|
| Participants | 32 participants with bilateral chronic hand dermatitis for more than 6 months, with mild to moderate severity and good response to topical steroids
Dropouts: 6 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Emollient with ceramides twice daily for 2 months in 14/17 participants • Traditional pet‐based emollient in 12/15 participants Both groups were allowed to use triamcinolone ointment in case of active dermatitis Duration 2 months |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Unclear about 2 dropouts. Study authors state that this is a pilot study. Analysis may have been intention‐to‐treat, but procedure unclear. Results presented graphically, without exact numbers. Accuracy of the statistics is unclear because all between‐group comparisons were conducted at each time individually rather than comparing difference scores between groups. The primary outcomes percentage of participants with self‐rated and observer‐rated improvement and the secondary outcomes reduction in severity, investigator‐rated and participant‐rated, were included in the study, although no useable data were provided. Data were given in a graphic presentation; no exact figures were given Study authors were contacted for additional information on 4 March 2014 and responded 10 March 2014 Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomised into 2 groups" Comment: no further details given in the article. Personal communication with the study author revealed that sealed, numbered envelopes were used |
| Allocation concealment (selection bias) | Low risk | No details in the article about how allocation was concealed from participants and clinicians; personal communication clarified that the study author used sealed envelopes that were distributed after informed consent was obtained. Participants did not know the randomisation before signing informed consent but became aware of the allocation afterwards |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Observer‐blinded, but not participant‐blinded. Participants were aware of their treatment, and the study nurse who distributed study drugs was aware of the treatment arms |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the same study investigator blindly assessed the dermatitis at various time‐points" Comment: observer‐blinded. Personal communication clarified that the study nurse was responsible for distribution of study drugs; outcomes were observed by a third person |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (26 of 32 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; no major differences between Methods and Results sections |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons regarding group differences (randomisation check) Diagnostic certainty: yes The study was completed |
Lauriola 2011.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary care setting at a single centre in Italy |
|
| Participants | 40 participants with mild to moderate atopic dermatitis of hands No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Furpalmate‐containing creams (0.3%) twice a day for 14 days in 20 participants • Corticosteroid (hydrocortisone acetate 0.5%) twice a day for 14 days in 20 participants Duration 2 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Conference abstract, from which only limited information can be extracted The secondary outcomes ‐ reduction in severity, participant‐ and investigator‐rated ‐ were included but did not provide reproducible data Study authors were contacted on 28 February 2014 but were not responsive Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "a single‐center, randomized, prospective, investigator blinded, controlled trial..." Quote: "...patients were randomly allocated..." Comment: study authors stated randomised but gave no clear description of how this was attained |
| Allocation concealment (selection bias) | Unclear risk | No details of how allocation was concealed from participants and investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a single‐center, randomized, perspective, investigator blinded, controlled trial..." Comment: study authors stated investigator‐blinded; participants were not blinded (open‐label?), although this is not clear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "a single‐center, randomized, prospective, investigator blinded, controlled trial..." Comment: the article states only that the study was done in an investigator‐blinded way. We considered this as insufficient information to judge this risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all patients completed the study period" Comment: all participants completed the study |
| Selective reporting (reporting bias) | High risk | No trial registration found. The abstract is very concise, and not all outcomes listed in the Methods section are described in the Results section, for example, cosmetic aspects or adverse events |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons regarding group differences (randomisation check) Diagnostic certainty: yes The study was completed |
Lindelöf 1987.
| Methods | Within‐participant, randomised controlled study This study was carried out in a secondary care setting at a single centre. The study was conducted in Sweden |
|
| Participants | 24 participants with chronic hand eczema (13 allergic, 5 atopic, 3 irritant, 2 tylotic, 1 pompholyx)
Dropouts: 1 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Ionising radiation (Grenz rays, 300 rad) 1× weekly for 6 weeks in 23/24 hands Control intervention • Placebo radiation once a week for 6 weeks in 23/24 contralateral hands Participants were followed up to 10 weeks after initial treatment Duration 10 weeks (6 weeks active treatment, 4 weeks follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included but provided no reproducible data. Total scores are only graphically presented, without statistical analysis Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the treatments were administered by a nurse according to a predetermined randomized code unknown to both patients and doctors" Comment: reference to a predetermined randomisation code |
| Allocation concealment (selection bias) | Low risk | Quote: "the treatments were administered by a nurse according to a predetermined randomized code unknown to both patients and doctors" Comment: by including a third person, neither the physician/observer, nor the participants can be aware of the treatment allocation. Therefore we considered this as low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the treatments were administered by a nurse according to a predetermined randomised code unknown to both patients and doctors" Comment: double‐blinded. Placebo therapy was achieved by "allowing the apparatus to hum without emitting radiation", which could be considered as adequate; however the treatments were administered by a nurse who was aware of the predetermined randomised code. Although one might argue that part of the staff was aware of the treatment allocation, we decided this is the best way to blind participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the treatments were administered by a nurse according to a predetermined randomized code unknown to both patients and doctors" Comment: the observer was blinded, and treatment was administered by someone else |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (23 of 24 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major differences between the Methods and Results sections |
| Other bias | Low risk | Baseline comparisons: as within‐participant study not applicable Diagnostic certainty: yes The study was completed |
Lodén 2012a.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary setting at 4 outpatient clinics in Norway This study consists of 2 parts (Lodén 2010); only the second part is included in this review |
|
| Participants | 44 participants with a clinically proven history of hand eczema and a recent relapse participated in part 1 of this trial No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Betamethasone 0.1% cream twice daily in 22 participants during 2 weeks • Betamethasone 0.1% cream once daily + urea 5% cream once daily in 22 participants during 2 weeks Duration 2 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Short duration of 2 weeks Declarations of interest: study authors were paid consultants or employees of ACO Hud Nordic AB Funding: the study was funded by ACO Hud Nordic AB (manufacturer of the study drug) and by Knowledge Foundation, Stockholm, Sweden Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the coded tubes were sequentially numbered according to a randomization list which was prepared and retained by the contract research organization" Comment: reference to a prepared randomisation list |
| Allocation concealment (selection bias) | Low risk | Quote: "the coded tubes were sequentially numbered according to a randomization list which was prepared and retained by the contract research organization" Comment: the randomisation list was prepared and retained by the contract research organisation. The tubes were coded and sequentially numbered, and the clinicians who dispensed the tubes to participants were blinded. This is considered as low risk of selection bias because randomisation was done at a remote site |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the patients, the clinicians, those assessing the outcomes and those making the data analyses were blinded" Quote: "the treatment was double‐blinded and combined with a moisturizer cream (M) (5% urea, Canoderm, ACO Hud AB, Sweden). All patients received two coded tubes; one for evening applications, labelled ‘evening’ and containing BV and one for morning applications, labelled ‘morning’ and containing either BV or M. The creams had a similar texture, were white and did not contain perfume" Comment: double‐blind design. The different creams were identical in appearance and were labelled by a contract research organisation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the patients, the clinicians, those assessing the outcomes and those making the data analyses were blinded" Comment: the observations and the data analysis were conducted by blinded assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all included participants received treatment and were analysed" Comment: no dropouts |
| Selective reporting (reporting bias) | Unclear risk | The trial was registered on clinicaltrials.gov (NCT00576550) as part 2. The primary outcomes registered in the trial register are for part 1 of the study (Lodén 2010), not for part 2. Therefore it is difficult to judge the risk of reporting bias In the article, there are no major discrepancies between the Methods and Results sections |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons regarding group differences (randomisation check) Diagnostic certainty: yes The study was completed |
Möller 1983.
| Methods | Within‐participant, randomised controlled trial This study was carried out in a secondary care setting; it was a multi‐centre study involving 14 dermatological centres in Sweden |
|
| Participants | 55 participants with chronic symmetrical hand eczema > 6 months, who had been treated with clobetasol twice daily in a preceding 3‐week healing phase; 46 participants completed the trial Dropouts: unclear on status of 9 withdrawals Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Clobetasol propionate cream twice weekly for unclear duration (55 to 193 days) in 46/55 hands • Fluprednidene acetate cream twice weekly in 46/55 contralateral hands Emollients were allowed on both hands When relapse occurred during the maintenance phase, the cream allocated to that hand could be applied more frequently; if this failed, the cream of the other (best) hand could be used temporarily Duration Unclear |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Study on maintenance therapy. Handling of dropouts unclear: 9 participants were withdrawn because of unsatisfactory results (this could be an outcome). Study duration unclear. Difficult to interpret results for participants with relapses. Unclear which of the 2 treatments was the intervention or the comparator The secondary outcomes ‐ reduction in severity investigator‐rated and time until relapse ‐ were included in the study but did not provide reproducible data Declarations of interest: 2 authors were employees of Glaxo Läkemedel AB, Mölndal, Sweden Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the side distribution was randomly allocated..." Comment: no further details given |
| Allocation concealment (selection bias) | Low risk | Quote: "the side distribution was randomly allocated and the creams were provided in coded samples of identical appearance except for the pairing in ‘left’ and ‘right’" Comment: allocation was likely concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...identical appearance...." Comment: double‐blind study in which an identical looking placebo was used |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...double‐blind study..." Comment: although the study claims to be double‐blind, no information was given about how observers were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (46 of 55 = more than 80%) |
| Selective reporting (reporting bias) | High risk | No trial registration found. It is not clear from the Methods section which are outcomes from the study, and the Results section contains various parameters that are not described in the Methods section |
| Other bias | Low risk | Baseline comparisons: within‐participant study not applicable Diagnostic certainty: yes The study was completed |
NCT01231854.
| Methods | Parallel‐group, randomised controlled trial This study was conducted at a single centre in Germany |
|
| Participants | The study planned on 78 participants with severe atopic hand dermatitis but ended prematurely Included: 15 Dropouts: 6 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral cyclosporin depending on body weight: 50 to 74.9 kg: daily dosage 200 mg; 75 to 100 kg: daily dosage 300 mg (7 participants) • Oral alitretinoin 30 mg once daily in 8 participants Duration 24 weeks |
|
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
|
| Notes | The study was ended prematurely. According to the sample size calculation, 78 participants should have been included; however only 15 participants were included, and 14 were analysed. Results are not yet published. The study author released the preliminary study results in personal communication and is aware of the fact that the data are used in this review. The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included, but we were unable to reproduce the data Declarations of interest: not stated Funding: TU Dresden, Germany Sample size rationale: adequate, although the needed number of participants was not included |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "during the baseline visit patients underwent central randomization with the use of a randomization table with constant length of blocks and stratification according to the patients’ body weights (50‐74.9 kg vs. 75‐100 kg) with equal allocation to ciclosporin and alitretinoin. The allocation sequence was generated by the KKS Dresden utilizing the trial software MACRO 3.0, and stored by the clinical trials pharmacist at the Technical University Dresden" Comment: random sequence generation method was considered adequate |
| Allocation concealment (selection bias) | Low risk | Quote: "treatment packs were prepared and labelled at the pharmacy. The research assistants used consecutively numbered packs to allocate new participants to treatment groups" Comment: study authors declared that they were blinded during allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "participants and assessors were blinded to group assignment during collection of the data" Comment: drug dispensation was done by a third party (the pharmacist). Unclear whether drugs were identical in appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "participants and assessors were blinded to group assignment during collection of the data. Database lock was on January 28, 2013. Unblinding occurred on March 07, 2013 before data analyses" Comment: the article claims that the observer had no access to the randomisation list, and a third party was used for drug dispensation. Unblinding occurred before data analyses |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all analyses were performed on the Intention‐to‐Treat (ITT) population. This population includes all patients that completed the baseline visit and used the trial medication at least once during the study" The study aimed to include 78 participants but was terminated early due to inability to include this number of participants. Finally, 15 participants were randomised and 1 withdrew before the study drug was used. The intention‐to‐treat analysis included all participants who received the study drug (14 of 15 participants) Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | The trial was registered under NCT01231854 before it was begun. We found no major discrepancies between the trial register and the final study report |
| Other bias | High risk | Baseline comparison: groups were comparable at baseline with regards to disease severity Diagnostic certainty: yes The study was ended prematurely |
Odia 1996.
| Methods | Within‐participant, randomised controlled trial This study was most likely conducted at a single centre in Germany, although this is not clear from the article |
|
| Participants | 20 participants with bilateral dyshidrotic hand eczema (13 male, 7 female). Atopic 7 and 9 with nickel allergy (4/9 also atopic)
No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • One hand pulsed direct current iontophoresis, 20 times of 15 minutes each during 3 weeks in 20 hands Control intervention • No iontophoresis on 20 contralateral hands for 3 weeks Both hands received steroid‐free tar solution and zinc paste Duration 3 weeks |
|
| Outcomes |
Primary outcomes of the trial
|
|
| Notes | Unclear at which point in time outcome was assessed. Same scoring system (DASI) was used in Adams 2007, Polderman 2003, and Schnopp 2002 The report did not provide any useful data for analyses, although the secondary outcomes ‐ reduction in severity, participant‐rated and investigator‐rated ‐ were included Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...were subjected to tap water iontophoresis in a randomised one‐side comparison..." Comment: no further details given |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants received unilateral iontophoresis, so one side was left untreated. Participants were allowed to use additional tar solutions and zinc paste. This might have influenced the amount of additional topical therapy used and thus the performance of the study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "scoring was performed by a second investigator, who did not know which side had been treated with iontophoresis" Comment: observer‐blinded: scorings were performed by a third person who was not involved in the treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All included participants were analysed. (20 of 20 = 100%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major discrepancies between Methods and Results sections |
| Other bias | Low risk | Baseline comparisons: within‐participant study not applicable Diagnostic certainty: yes The study was completed |
Pacor 2006.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary care setting. It was a single‐centre study conducted in Italy |
|
| Participants | 28 participants with moderate to severe nickel sulphate‐induced allergic contact dermatitis based on clinical history (hand eczema) and proven by patch testing, resistant to topical corticosteroids
No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • 0.1% tacrolimus ointment twice daily for 2 weeks in 14 participants Control intervention • Vehicle twice daily for 2 weeks in 14 participants 2 weeks of treatment was followed by 1 week of follow‐up Duration 3 weeks (2 weeks active treatment, 1 week follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Unclear from the abstract whether all participants had hand eczema, but contact with study authors confirmed that all participants had active hand eczema at the beginning of the trial. Treatment started after a run‐in period of 7 days The primary outcome percentage of participants with participant‐rated good/excellent control and the secondary outcome ‐ reduction in severity, investigator‐rated ‐ were included in the study, but no reproducible data were provided Study authors were contacted on 28 February 2014 and replied 2 March 2014 Declarations of interest: none declared Funding: the study was supported by grants from the Ministero Italiano Universita e Ricerca (MIUR) Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a randomized, double‐blind, placebo‐controlled, parallel‐group study design..." Comment: study authors state this is a randomised study but gave no details in the article Personal communication clarified that StatsDirect Statistical software was used, which is considered to provide adequate random sequence generation |
| Allocation concealment (selection bias) | Low risk | No details in the article about how allocation was concealed from participants and clinicians. Personal communication revealed that the personnel recruiting participants were unaware of treatment allocation because this was done by a third party (the hospital pharmacist) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the test compounds were contained in opaque syringes and the treatment was not distinguishable from placebo and was blinded for both investigator and patients" Comment: the authors state that the study uses a double‐blind design. The placebo ointment of tacrolimus was made of the same components as the study drug, only without the active component. All personnel involved in direct contact with participants were unaware of treatment allocation. Only the pharmacist was aware of the allocation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the test compounds were contained in opaque syringes and the treatment was not distinguishable from placebo and was blinded for both investigator and patients" Comment: the article claims to be double‐blind. Personal communication with the study author clarified that the outcomes were observed by a blind observer and were analysed in blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study (28 of 28 = 100%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major discrepancies between Methods and Results sections |
| Other bias | Low risk | Baseline comparisons: no significant difference between groups in terms of age, gender, and severity of hand eczema Diagnostic certainty: yes The study was completed |
Pigatto 1990.
| Methods | Parallel‐group, randomised controlled trial (3 groups) This study was carried out in a secondary care setting at 1 centre in Italy |
|
| Participants | 16+8 participants with dyshidrotic eczema (pompholyx) and positive patch test to nickel, confirmed by reaction on oral challenge with nickel No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Low‐nickel diet for 3 months in 8 participants • Oral disodium cromoglycate (DSCG) 1500 to 2000 mg 3× daily for 3 months in 8 participants Control intervention • No treatment for 3 months in 8 participants ‐ not included in the review Duration 3 months |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Unclear which of the 2 is intervention and which is comparator. The third group consisted of participants who did not give consent for the interventions and was observed without undergoing any treatment. This group was not randomised and therefore was not included in the analysis Unclear how the outcome 'Degree of itching' was assessed. In addition, an intestinal permeability study was performed in 5 DSCG and 5 diet participants The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included but no reproducible data were provided The article states different numbers of participants included in intervention and control groups (8;8;8 vs 8;9;7) Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the patients were then randomly divided into 3 groups" Comment: groups 1 and 2 were probably randomised; however, the third group consisted of patients who did not give informed consent for the study and therefore were not randomised. Group 3 is not included in the analysis |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "the patients avoided eating the foods indicated on a list, reduced the quantity of vegetables and of dairy products, and avoided using stainless steel utensils and ornaments" Comment: because the intervention group in group A had to follow a low‐nickel diet with strict dietary restrictions for 3 months, blinding was not possible |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "the patients were evaluated blind by an observer...." Comment: observer blinded but no further details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None of the participants dropped out, and all included participants were analysed (16 of 16 = 100%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major differences between Methods and Results sections |
| Other bias | Unclear risk | Baseline comparisons: no baseline analyses regarding group differences (randomisation check) Diagnostic certainty: yes The study was completed |
Polderman 2003.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary care setting at a single dermatology clinic in the Netherlands |
|
| Participants | 28 participants with dyshidrotic hand eczema, with duration of 4 months to 34 years
Dropouts: 3 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • UVA‐1 irradiation 40 J/cm² on the hands in 15/15 participants 5 times weekly for 3 weeks Control intervention • Placebo (simulated blue light) in 10/13 participants Emollients probably were allowed in both groups Participants were followed until 6 weeks after the end of treatment Duration 9 weeks (3 weeks active treatment, 6 weeks follow‐up) |
|
| Outcomes |
Primary outcomes of the trial
Other outcomes
|
|
| Notes | Primary outcome probably at week 3 (i.e. at end of treatment). Analysis based on intention‐to‐treat principle. There was a follow‐up 6 weeks after treatment, but only summary data were given for the treatment group Study authors were contacted on 28 February 2014 but remained not responsive Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "they were randomly assigned to either UVA‐1 (n=15) or placebo treatment (n=13) by an independent investigator using a lottery system" Comment: reference to a lottery system, which is an adequate way to prevent selection bias |
| Allocation concealment (selection bias) | Low risk | Quote: "they were randomly assigned to either UVA‐1 (n=15) or placebo treatment (n=13) by an independent investigator using a lottery system" Comment: participants were assigned to different study arms by an independent investigator using a lottery system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "placebo treatment comprised of TL tubes, emitting visible light, covered with a blue plastic plate to mimic the blue UVA‐1 light" Comment: participants: placebo design to mimic the intervention; participants wore protective eyewear and protection on their forearms during both interventions. For participants, the placebo is probably indistinguishable from the actual treatment; however personnel who delivered the treatment were not blinded. Given that the staff had to administer the treatment, and we could not think of a better way to blind participants and staff, this was considered as low risk |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "a blinded investigator was responsible for the evaluation of the parameters" Comment: observer‐blinded; this was another person, then the one who assigned participants to treatment arms |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "analysis was performed according to the intention‐to‐treat principle" Comment: intention‐to‐treat analyses |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. No major differences between Methods and Results sections for results during the treatment phase, except that results 6 weeks after treatment are very scarce. Study authors remarked that for ethical reasons, a lot of participants were prescribed topical corticosteroids; therefore these results are less reliable and probably are poorly reported |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons regarding group differences (randomisation check) Diagnostic certainty: yes The study was completed |
Ruzicka 2004.
| Methods | Randomised controlled, parallel‐group design study of 1 placebo group and 3 treatment groups given different doses of the same (oral) medicament This study was carried out in a secondary care setting This was a multi‐centre study involving 43 clinics in Belgium, Denmark, Finland, France, Germany, Holland, Hungary, Poland, Switzerland, and the United Kingdom |
|
| Participants | 319 participants (235 male, 84 female) with moderate or severe chronic hand dermatitis of at least 3 months' duration and refractory to standard therapy. All types of hand dermatitis
Dropouts: 75 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral alitretinoin 10 mg daily for 12 weeks in 62/80 participants • Oral alitretinoin 20 mg/d in 67/80 participants for 12 weeks • Oral alitretinoin 40 mg/d in 63/81 participants for 12 weeks Control intervention • Placebo capsules in 62/78 participants for 12 weeks Standard emollients were allowed in all treatment groups Responders were followed up for 3 months Duration 6 months (12 weeks active treatment, 3 months follow‐up) |
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
|
| Notes | No other active treatment as comparator. More males enrolled because of exclusion of women of child‐bearing potential. Study included a safety assessment by careful medical and laboratory monitoring. Analysis of efficacy based on intention‐to‐treat principle
Of the 127 responders, 117 were followed up for another 12 weeks after end of treatment; only summary data about this extra follow‐up are presented Declarations of interest: some study authors were employees, received grants, or had received consultancy fees from Basilea Pharmaceutica Funding: the study was supported and funded by Basilea Pharmaceutica Ltd., Basel, Switzerland, manufacturer of the study drug Sample size rationale: provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "eligible patients were randomized to treatment by center, in blocks of 4 without stratification, by use of computer‐generated randomization codes...." Comment: computer‐generated randomisation codes |
| Allocation concealment (selection bias) | Low risk | Quote: "eligible patients were randomized to treatment by center, in blocks of 4 without stratification, by use of computer‐generated randomization codes provided by the study sponsor (Basilea Pharmaceutica Ltd, Basel, Switzerland) and incorporated into double‐blind coded drug packaging" Comment: the site staff had no direct access to randomisation codes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "placebo and active drug (as soft gelatine capsules) and packaging were indistinguishable" Comment: participant blinding. The double‐blind coded packages were provided by the sponsor, which blinded site personnel and participants sufficiently |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "design: multicenter, randomized, double‐blind, placebo‐control, prospective trial" Comment: observers were blinded during the study because the identical looking package of study drugs was provided by a third party. One might argue that the observer could have guessed the treatment group due to headache and dry mucosa; however this was also seen in the control group and therefore was not conclusive. The trial was designed in such a way to minimise risk of bias; we agree that this could not have been done in a better way |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all statistical tests were 2 sided and based on a level of .05 and were carried out using SAS (version 8.1; SAS Institute Inc, Cary, NC) with the intention‐to‐treat population" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No trial register found. No major discrepancies between Methods and Results sections |
| Other bias | Low risk | Baseline comparisons: no significant differences between groups in demographic or disease characteristics Diagnostic certainty: yes The study was completed |
Ruzicka 2008.
| Methods | Parallel‐group, randomised controlled trial including 1 placebo group and 2 treatment groups given different doses of the same (oral) retinoid This study was carried out in a secondary care setting This was a multi‐centre study at 111 clinics in Europe and Canada |
|
| Participants | 1032 participants (582 male, 450 female) with severe chronic hand dermatitis of at least 6 months' duration and refractory to standard therapy. All types of hand dermatitis
Dropouts: 273 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral alitretinoin 10 mg once daily for 12 or 24 weeks (depending on moment of response according to the PGA) in 319/418 participants • Oral alitretinoin 30 mg/d in 303/409 participants for 12 or 24 weeks Control intervention • Placebo capsules in 137/205 participants for 12 or 24 weeks Standard emollient in all treatment groups All participants were followed up for 4 weeks, and responders were observed for relapses for 24 weeks after end of treatment Duration Up to 48 weeks (12 to 24 weeks of active treatment, up to 24 weeks of follow‐up) |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | No other active treatment as comparator. Analysis of efficacy based on intention‐to‐treat principle. Study included a safety assessment by careful medical and laboratory monitoring. More males were enrolled because of exclusion of women of child‐bearing potential Declarations of interest: some study authors were employees, received grants, or had received consultancy fees from Basilea Pharmaceutica Funding: see above item Sample size rationale: provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "eligible patients were randomized to treatment by centre, in blocks of 5 without stratification, by the use of computer‐generated randomization codes provided by the study sponsor (Basilea Pharmaceutica)..." Comment: computer‐generated randomisation codes |
| Allocation concealment (selection bias) | Low risk | Quote: "eligible patients were randomized to treatment by centre, in blocks of 5 without stratification, by the use of computer‐generated randomization codes provided by the study sponsor (Basilea Pharmaceutica) and incorporated into double‐blind coded drug packaging. Placebo, active drug and packaging were indistinguishable. Investigators allocated consecutively numbered packages of medication to patients in their order of enrolment" Comment: codes provided by study sponsor. Sequentially numbered packages of different treatment modalities of identical appearance were used |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "... incorporated into double‐blind coded drug packaging. Placebo, active drug and packaging were indistinguishable" Comment: double‐blinded; the identical looking packages were provided by the sponsor and by site staff, and participants were unaware of the treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Observers were blinded during the study because the identical looking package of study drugs was provided by a third party. One might argue that the observer could have guessed the treatment group due to headache and dry mucosa; however these were also seen in the control group and therefore were not conclusive. The trial was designed in such a way as to minimise risk of bias; we agree that this could not have been done in a better way |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all efficacy evaluations were based on the intent‐to‐treat population. All randomized patients were included in this population, and were analysed according to their randomization with last observation carried forward (LOCF) in cases of missing data" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Unclear risk | Trial registration on clinicaltrials.gov (NCT00124475). No differences in primary outcomes; however small discrepancies in other outcomes between trial registration and article |
| Other bias | Low risk | Baseline comparisons: no significant differences between groups in demographic or disease characteristics Diagnostic certainty: yes The study was completed |
Said 2010.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary care setting in a single dermatology department in Singapore |
|
| Participants | 47 participants with chronic vesicular hand eczema Dropouts: 7 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Interventions • UVA‐1 phototherapy 3 times a week for 6 weeks in 24 participants • Betamethasone‐valerate 0.1% cream twice a day for 6 weeks in 23 participants Duration 6 weeks active treatment, 6 weeks follow‐up. Total duration: 12 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Conference abstract, from which only limited information can be extracted We were unable to contact the study authors Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "twenty‐four patients were randomly assigned to..." Comment: insufficient data |
| Allocation concealment (selection bias) | Unclear risk | Insufficient data |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "open‐label study" Comment: no blinding |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "open‐label study" Comment: no blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | More than 80% of participants were analysed (40/47) |
| Selective reporting (reporting bias) | Unclear risk | No trial register was found |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons regarding group differences (randomisation check) Diagnostic certainty: no The study was completed |
Schnopp 2002.
| Methods | Within‐participant, randomised controlled study This study was carried out in a secondary care setting at a single dermatology department in Germany |
|
| Participants | 16 participants with moderate to severe chronic relapsing dyshidrotic eczema on hands No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Tacrolimus 0.1% ointment twice daily on 12/12 hands for 4 weeks • Mometasone furoate 0.1% ointment twice daily on 12/12 contralateral hands for 4 weeks Follow‐up period was up to 8 weeks after the end of treatment Duration 12 weeks (4 weeks of active treatment, up to 8 weeks of follow‐up) |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | Originally, 20 participants with hand and/or foot involvement, 4 of whom were excluded due to poor disease control during the trial‐preceding washout phase. Study in 16 participants, of whom 12 had their hands involved. The limited data on the 4‐week post‐treatment follow‐up period are difficult to interpret. Outcome scores at week 4 presented graphically, without exact numbers Scoring of outcome (DASI) same as the study by Odia The secondary outcomes ‐ reduction in severity, participant‐rated, and time until relapse ‐ were included but did not provide reproducible data Study authors were contacted 3 March 2014 and replied 4 March 2014 Declarations of interest: none declared Funding: no funding Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned....." Comment: no further details were given in the article; however personal communication with study authors clarified that they threw dice to create a randomisation list |
| Allocation concealment (selection bias) | Low risk | No details in the article about how allocation was concealed from participants and clinicians. Contact with study authors clarified that the randomisation list was composed by a third person. This person was involved in the distribution of study drugs, but not in the recruiting. The third person held office in a different building of the hospital that was not accessible for physicians |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "this study was a randomized, observer‐blinded, intraindividual comparison study..." Comment: participants were not blinded during the study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "all assessments were performed by an independent observer on separate sheets on different premises. Patients were instructed not to talk about treatment modalities" Comment: study authors clearly described how they tried to prevent detection bias. Observers were blinded adequately |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None of the participants dropped out during the study; all participants were included in the analyses (16 of 16 = 100%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found; however the DASI is a valid score for hand eczema and was described in the Methods and Results sections without major discrepancies |
| Other bias | Low risk | Baseline comparisons: within‐participant study not applicable Diagnostic certainty: yes The study was completed |
Sezer 2007.
| Methods | Within‐participant, randomised controlled study This study was conducted in a secondary care setting at a single centre in Turkey |
|
| Participants | 15 participants with chronic hand eczema of the dry or dyshidrotic type were randomised
Dropouts: 3 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Local narrow‐band UVB 3 times a week for 9 weeks in 12/15 hands. The initial dose was 150 mJ/cm² for each participant. A 20% increasing dose schedule was used until a final dose of 2000 mJ/cm² was reached • Local PUVA 3 times a week during 9 weeks in 12/15 contralateral hands. The initial dose of psoralen plus UVA irradiation was 1.0 J/cm² with an increase of 0.5 J/cm² at every second session until a final dose of 7.5 J/cm² was achieved Participants who completed the treatment sessions were followed up for 10 weeks after the last therapy Duration 19 weeks (9 weeks of active treatment, 10 weeks of follow‐up) |
|
| Outcomes |
Primary outcomes of the trial
|
|
| Notes | Unblinded study with a small number of participants Study author was contacted on 4 March 2014 by email, but we were unable to obtain additional information Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the NB‐UVB and PUVA treatments were randomly assigned to the left or right hand. The hand treated was selected using a computer‐based program..." Comment: treatment was randomly assigned to the left or right hand using a computer‐based programme |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on whether participant‐ and/or observer‐blinded study |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "clinical assessments were performed by the same investigator (E.S.) every 3 weeks during the 9‐week treatment period" Comment: unclear. All observations were made by the same investigator, although it is unknown whether this observer was aware of the treatment modalities |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (12 of 15 = 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major differences between Methods and Results sections |
| Other bias | Low risk | Baseline comparisons: at baseline, no significant differences between groups in total clinical scores; however within‐participant study not applicable Diagnostic certainty: yes The study was completed |
Sharma 2006.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary care setting at a single centre in India |
|
| Participants | 21 participants with proven nickel allergy by patch testing No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Low‐nickel diet and disulphiram 125 mg daily in the first 2 weeks and 250 mg daily in weeks 3 and 4 in 11 participants Control intervention • Normal diet and placebo tablet (lactose) for 4 weeks in 10 participants Participants were followed up for 2 to 12 weeks after end of treatment Duration Up to 16 weeks (4 weeks of active treatment, 2 to 12 weeks of follow‐up) |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | Two weeks after the start of a low‐nickel diet in the experimental group, disulphiram was started for a duration of 4 weeks. Participants in the control group were treated only with a placebo tablet during those 4 weeks. In addition, the low‐nickel diet was continued during follow‐up (i.e. 12 weeks after disulphiram was stopped) The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included, but the article did not provide reproducible data Study authors were contacted for additional information with regards to the risk of bias table Declarations of interest: none declared Funding: no funding Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "all the 21 patients were randomly divided in 2 groups" Comment: no further details in the article. Personal communication with study authors clarified that they had used a lottery system |
| Allocation concealment (selection bias) | Low risk | No details about how allocation was concealed from participants and clinicians. Personal communication clarified that this was done by a third person |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "it was a comparative study and participants were not aware if they belonged to study group or control group (single blind trial)" Comment: participant blinding attempted with the use of placebo tablets (lactose tablets), although participants in the control group were allowed to continue with their normal diet, while the intervention group remained on the low‐nickel diet. Site personnel probably were not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "it was a comparative study and participants were not aware if they belonged to study group or control group (single blind trial)" Comment: observers were not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None of the participants dropped out, and all participants were included in the analyses (21 of 21 = 100%) |
| Selective reporting (reporting bias) | Unclear risk | No trial registration found. In the Methods section, no clear description is given of the outcome parameters; in the Results section, outcomes are listed that are not described in the Materials section |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons Diagnostic certainty: yes The study was completed |
Sheehan‐Dare 1989.
| Methods | Within‐participant, randomised controlled trial. Hands were unit of randomisation and analysis This study was conducted at a single department of dermatology in the UK |
|
| Participants | 25 participants with chronic constitutional hand eczema; participants with irritant or allergic contact dermatitis were excluded
Dropouts: 4 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Topical PUVA thrice weekly for 6 weeks in 21/24 hands • Radiotherapy 90 Rad 50 KV 3 times with 21‐day interval in 21/24 contralateral hands for 6 weeks Participants were followed up until 18 weeks after initial treatment Duration 18 weeks (6 weeks of active treatment, 12 weeks of follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | The primary outcome adverse events and the secondary outcomes reduction in severity, participant‐rated and investigator‐rated, were included but did not provide reproducible data. Means of outcome scores were not given as exact figures but in a graphical presentation Declarations of interest: not stated Funding: not stated Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly allocated topical PUVA to one hand and superficial radiotherapy to the other using a pre‐determined code" Comment: participants were randomly allocated using a predetermined code |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the procedure was carried out in such a way that patients were unable to tell which hand had received active treatment" Quote: "the topical PUVA treated hand received sham radiotherapy during which the X‐ray machine appeared to function normally but the power supply to the tube was interrupted such that no X‐rays were received by the patient" Quote: "the superficial radiotherapy treated hand was treated with a sham PUVA procedure. This consisted of an application of the organic solvent base without psoralen 5 min prior to exposure to the light source" Comment: participant blinding. We consider this an adequate way to blind participants, although personnel probably were not blinded to perform the procedures |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "patients were assessed by 2 observers (R.S‐D and M.G) who were unaware of the treatment status of each hand until the codes were broken at the end of the study" Comment: observer blinding; independent observers are considered an adequate method for detection bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (21 of 25 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No discrepancies between the Materials and Results sections |
| Other bias | Low risk | Baseline comparisons: within‐participant design not applicable Diagnostic certainty: yes The study was completed |
Sjövall 1987.
| Methods | Parallel‐group, randomised controlled trial (3 groups) This study was carried out in a secondary care setting and was probably a single‐centre study in Sweden |
|
| Participants | 18 participants (3 male, 15 female) with chronic hand eczema of different types resistant to conventional therapy (11 patch test‐proven relevant allergy, 4 atopic, 3 endogenous)
Dropouts: 3 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • UVB irradiation only on hands 4 times a week for 8 weeks in 6 participants • Filtered light (placebo UVB, no UVB) on the hands 4 times a week for 8 weeks in 6 participants • Hand UVB followed by whole‐body UVB + UVA 4 times a week during 8 weeks in 6 participants Their 'ordinary topical treatment' was permitted in all groups Three months after end of treatment, participants were mailed a questionnaire regarding the course of their hand eczema and their opinions on treatment Duration 8 weeks with an email follow‐up after 3 months |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Small number of participants. Main table unclear: results at 8 weeks or at 20 weeks? Follow‐up at 3 months presented in a descriptive way, without exact details Declarations of interest: not stated Funding: the study was supported by grants from Alfred Österlund and Finsen Foundations Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the patients were randomly divided..." Comment: no further details |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "the whole device was covered by green clothes, thus making it possible to perform a double blind trial between the patients in group 1 and 2" Comment: the investigators covered the machine with green clothes; by this method, 2 groups (A and B) were blinded; however the third group of participants was not blinded because they received whole‐body irradiation. Staff probably was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The investigators state a partly double‐blind design; however it is unclear how observers were blinded, and if they were independent observers |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (15 of 18 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major differences between Materials and Methods sections |
| Other bias | Unclear risk | Baseline comparisons: no baseline comparisons Diagnostic certainty: yes The study was completed |
Thestrup‐Pedersen 2001.
| Methods | Parallel‐group, randomised controlled trial This study was conducted in a secondary care setting at 4 dermatology departments or clinics in Denmark |
|
| Participants | 29 participants (21 male, 8 female) with hyperkeratotic eczema on palms, patch‐test negative or irrelevant
No dropouts Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Acitretin orally 30 mg daily for 8 weeks in 14/14 participants Control intervention • Placebo capsules for 8 weeks in 15/15 participants Both groups were allowed to use topical emollients Duration 8 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | We contacted the study author for additional information by letter; however he was unable to respond to all of our questions No overall scores were presented as outcomes. Details of biochemical parameters were not given A proper between‐group comparison was not conducted; only within‐group comparisons with Wilcoxon‐rank sum test were conducted The study did include the secondary outcomes ‐ reduction in severity, participant‐ and investigator‐rated ‐ although we were unable to include these data because of missing data Declarations of interest: none declared Funding: Roche A/S, Copenhagen, supplied the study drug free of charge, but the investigators did not receive financial support nor consultant fees from Roche Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "they were asked to take three 10‐mg capsules of acitretin once daily for 8 weeks or identically looking placebo capsules" Comment: from the article, it is unclear whether the study was randomised at all. Personal communication with study authors clarified that randomisation was done by a third party according to a pre‐defined randomisation list |
| Allocation concealment (selection bias) | Low risk | No details about how allocation was concealed from participants and clinicians. Personal communication clarified that the sponsor shipped 4 identical boxes to all participating centres, which could at random be dispensed to participants. The investigators were unaware of the content of the boxes; therefore we judged this as low risk |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "they were asked to take three 10‐mg capsules of acitretin once daily for 8 weeks or identically looking placebo capsules" Comment: the study contained an identical looking placebo in an attempt to blind participants, and randomisation and dispensation of drugs were done at a remote site by a third party. Therefore we judged this as adequate blinding of participants |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "as several patients in the active treatment group experienced dryness of the lips, we have called our study single‐blind" Comment: study authors declared this a single‐blind study because the observers could have guessed the acitretin group due to adverse events of acitretin |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None of the participants dropped out, and all participants were included in the analyses |
| Selective reporting (reporting bias) | Low risk | No trial registration found. However we found no major discrepancies between Methods and Results sections |
| Other bias | Unclear risk | Baseline comparisons: not stated whether there was a significant difference in disease severity between groups Diagnostic certainty: yes The study was completed |
Tzaneva 2009.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary care setting at a dermatology department in Austria |
|
| Participants | 29 participants with moderate to severe dyshidrotic or hyperkeratotic palmar and/or plantar eczema, with symmetrical distribution
Dropouts: 2 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral PUVA: 8‐MOP at a dose of 0.6 mg/kg 1 hour before irradiation with UVA in 14 participants. Irradiation dose: 1.5 J/cm² for the palms and soles and 1.0 J/cm² for the backs of the hands and feet in participants with skin type III/IV. Respective doses for skin type I/II were 1.2 and 0.8 J/cm² • Bath PUVA: 2 mL of a 0.5% 8‐MOP concentration of 5 mg/L in 13/15 participants. Immediately after immersion for 15 minutes, the hands and feet were exposed to UVA irradiation. Skin type III/IV: initial dose of 0.7 J/cm² for the backs of the hands and feet. Skin type I/II: 0.8 and 0.55 J/cm², respectively Irradiation doses in both groups were increased depending on the degree of an erythematous response. Treatment was given 3 to 4 times a week until complete clearance or over a maximum period of 20 weeks. After clearing, participants were maintained on PUVA twice weekly for 2 weeks, then once weekly for another 4 weeks At the end of treatment, participants were followed up until relapse, or for a maximum of 40 months Duration Up to 40 months (up to 20 weeks of active treatment, up to 40 months of follow‐up) |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | From the article, it is unclear whether all participants had hand eczema. After writing to study authors, it became clear that all participants had hand eczema, and some also had plantar eczema Study authors were contacted with an additional request for allocation on 4 March 2014, but we were unable to obtain further data The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was used, and study authors did not find a statistically significant difference in dyshidrotic hand eczema (stated P = 0.67; multi‐factorial ANOVA) and a significant difference in the hyperkeratotic group (stated P = 0.03; multi‐variate ANOVA), although we were unable to reproduce these data because the standard deviation was not available The secondary outcome ‐ time until relapse ‐ was included, but we were unable to reproduce these data Declarations of interest: none declared Funding: no clear indication of funding that might lead to conflict of interest Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly allocated to receive either oral or bath PUVA therapy by means of a computer‐generated sequentially numbered randomization list" Comment: randomisation method was adequate |
| Allocation concealment (selection bias) | Unclear risk | Insufficient details provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "...observer‐blinded study..." Comment: participants and personnel were not blinded, and no placebo was used |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "eczema severity was determined by a blinded investigator (A. T.) at baseline and every 4 weeks..." Comment: a single, blinded observer assessed all outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (27 of 29 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major discrepancies between Methods and Results sections. The Methods section gives a clear description of the definitions of relapse and clearance |
| Other bias | Unclear risk | Baseline comparisons: not stated Diagnostic certainty: yes The study was completed |
Uggeldahl 1986.
| Methods | Within‐participant, randomised controlled trial This study was conducted in a secondary care setting, probably at 2 centres in Finland |
|
| Participants | 50 (46) participants (1.5 to 70 years) with bilateral moderate hand/wrist/lower arm eczema, with left‐right comparable severity, were included; 4 were excluded because of asymmetrical hand eczema
Dropouts: 2 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Desonide cream 0.1% twice daily for 2 weeks in 44/46 hands • Desonide cream 0.05% twice daily in 44/46 contralateral hands for 2 weeks Duration 2 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | In fact, 50 participants were randomised, but 4 participants were excluded at the start. Not clear whether inclusion criteria of the trial (hand/wrist/lower arm) stipulated that the hands had to be involved in all participants. The youngest participant was 1.5 years old Aim was to study equivalency, but this was not reflected in the analysis The secondary outcomes ‐ reduction in severity, investigator‐rated and participant‐rated ‐ were included but did not provide reproducible data Declarations of interest: 2 study authors were employees of the research department at Apothekernes Laboratorium A.S., Oslo, Norway, the manufacturer of the study drugs Funding: see above item Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the study consisted of a double‐blind, randomized, left‐right comparative study..." Comment: stated only that this was a randomised study. No further details provided |
| Allocation concealment (selection bias) | Low risk | Quote: "each patient was given 2 tubes identical in appearance...." Quote: "according to the double‐blind nature of the study, the creams were randomly allocated to the left and right side..." Comment: no details about how allocation was concealed from participants and clinicians. The identical tubes were randomly allocated to left and right sides |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "each patient was given 2 tubes identical in appearance...." Comment: the only difference between the 2 tubes was the concentration, wherefore this is considered as low risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "...double‐blind nature..." Comment: unclear whether observers were aware of the treatment modalities, although study authors claim double‐blind design |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (44 or 46 of 50 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. No major discrepancies between Methods and Results sections |
| Other bias | Low risk | Baseline comparisons: within‐participant study not applicable Diagnostic certainty: yes The study was completed |
van Coevorden 2004a.
| Methods | Parallel group, open‐label randomised controlled trial This study was conducted at 2 university hospital outpatient clinics in the Netherlands |
|
| Participants | 158 participants (88 male, 70 female) with chronic hand eczema of at least 1 year's duration, with at least 2 relapses of at least 3 weeks' duration, moderate to severe, grade 6 on a hand eczema score at start of treatment
Dropouts: 33 during treatment, 8 during follow‐up Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral PUVA (methoxypsoralen) phototherapy at home on both hands thrice weekly for 10 weeks in 63/78 participants • Topical bath PUVA (trioxsalen) twice weekly in hospital for 10 weeks in 62/80 participants Emollients were allowed in both groups Participants were followed up for an additional 8 weeks after the end of treatment Duration 18 weeks (10 weeks of active treatment, 8 weeks of follow‐up) |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | Blinding of participants impossible. Observers of outcomes not blinded. Analysis based on intention‐to‐treat principle. Scoring of eczema was similar to the scoring used by Rosén 1987a
Secondary outcome number 3 (number of participants improved at week 10) not specified in the Methods. Study authors mention adherence to CONSORT statement. Missing standard deviations were calculated according to formula provided in Chapter 7.7.3.2 of theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) Declarations of interest: none declared Funding: the study was supported in part by the Netherlands Healthcare Insurance Board, Amstelveen, the Netherlands Sample size rationale: provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated randomization lists with blocks of 4 were created by a secretary" Comment: randomisation method was considered adequate |
| Allocation concealment (selection bias) | Low risk | Quote: "consecutive patients were given consecutive numbers on the list and randomized accordingly by the trial’s dermatologists. The randomization sequence was kept concealed by the secretary until the end of the trial" Comment: allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "blinding of the patient and the outcome assessor was not practically feasible: patients are aware of their treatment modality and assessors can easily identify a hand treated with bath PUVA because of its rim of pigmentation" Comment: no blinding was attempted. Study authors state that blinding was not practically feasible. In addition, outcomes of this study were time off work and travel time. When participants would have received placebo in hospital or at home radiation, this outcome would not be applicable |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "hand eczema severity was assessed by one of the unblinded trial’s dermatologists (trained in assessment of hand eczema)" Quote: "blinding of the patient and the outcome assessor was not practically feasible: patients are aware of their treatment modality and assessors can easily identify a hand treated with bath PUVA because of its rim of pigmentation" Comment: no blinding; study authors state in a comment that blinding of observers was practically not feasible because observers would be able to guess the difference based on the rim of pigmentation in the bath PUVA‐treated group |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the statistical analysis was based on the intention‐to‐treat principle, using the “last value carried forward” method" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No trial registration found. The article provides a clear participant flow chart based on CONSORT guidelines. With regards to one of the important outcomes ‐ travel costs and time of work ‐ study authors stated only that "patients treating themselves at home had substantially lower travel costs and substantially less time of work"; however additional information is provided in a separate report |
| Other bias | Low risk | Baseline comparisons did not reveal significant differences in hand eczema between groups Diagnostic certainty: yes The study was completed |
Veien 1995.
| Methods | Parallel‐group, randomised controlled trial This study was conducted at 4 dermatological departments and clinics in Denmark |
|
| Participants | 47 participants (11 male and 36 female) with hand eczema of at least 6 months' duration. All had or previously had atopic dermatitis. All without positive reaction to standard patch test series
Dropouts: 9 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Oral ranitidine 300 mg twice daily (21/23 participants) for 16 weeks Control intervention • Placebo tablets (17/24 participants) for 16 weeks Both groups received betamethasone cream/ointment and emollient Duration 16 weeks |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Published as brief communication. Analysis according to intention‐to‐treat principle, but no details given The secondary outcome ‐ reduction in severity, investigator‐rated ‐ was included, although because standard deviations were missing, we were unable to reproduce the data Study authors were contacted by email but were unable to answer all of our questions because the study was conducted such a long time ago Declarations of interest: not stated Funding: the study drugs were provided by Glaxo Denmark A/S, and Glaxo provided an employee to assist with the study. The emollients were provided by Rhône‐Poulene, and statistical analyses were performed by Biomedica, Copenhagen, Denmark Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the patients were randomly selected" Comment: no further details given in the article; personal communication with study authors clarified that a computer‐generated code was used |
| Allocation concealment (selection bias) | Low risk | No details in the article about how allocation was concealed from participants and clinicians. Personal communication with study authors clarified the following: "The allocation was concealed from patients and investigators by numbers on identical boxes of tablets containing either ranitidine or placebo". Because randomisation was done by a third party, boxes were identical, and investigators received the randomisation code only in a sealed envelope, this was considered an adequate method to prevent selection bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the patients were randomly selected to receive oral ranitidine, 300 mg twice daily, or placebo tablets of identical appearance" Comment: double‐blind design. Because randomisation was done by a third party and staff received identical looking boxes for ranitidine and placebo, it was not possible for participants and staff to know the treatment arm |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "the code was broken when all the patients had completed the study and all results were recorded" Comment: randomisation was carried out by a third party; therefore observers could not have known the treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "statistical evaluations were based on the intention‐to‐treat principle" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | High risk | No trial registration found. The Results section is very concise. In the Results section, it is unclear whether the outcome was based on participants' or investigators' scores or on a combination of these, and only total scores or significance levels are given |
| Other bias | Low risk | Baseline comparisons: the 2 groups were comparable with regard to age, duration of dermatitis, eczema at other sites, and presence of other atopic symptoms Diagnostic certainty: yes The study was completed |
Veien 1999.
| Methods | Parallel‐group (3 groups), randomised controlled trial This study was conducted in a secondary care setting at 3 centres in Denmark (study was carried out at a university department as well as at 2 private dermatology clinics) |
|
| Participants | 106 participants were randomised (all patch tested) with hand eczema > 6 months that had cleared upon daily treatment for a maximum of 9 weeks with mometasone furoate cream 120 participants were recruited, and 14 dropped out during the initial phase No dropouts after randomisation (see notes) Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Mometasone furoate cream thrice weekly (Sunday/Tuesday/Thursday) for up to 36 weeks or 30 (?) in 35/35 participants • Mometasone cream twice weekly (Saturday/Sunday) for up to 36 weeks in 37/37 participants Control intervention • No corticosteroids in 34/34 participants Emollients (Essex cream and ointment) used in all groups In case of recurrence, all groups were permitted to use mometasone daily for a maximum of 3 weeks at separate period Additional treatment was permitted in all groups in case of a bacterial infection Duration Up to 36 weeks |
|
| Outcomes |
Primary outcome of the trial
Other outcomes
|
|
| Notes | All randomised participants were supposed to be free of eczema due to preceding treatment (induction of remission) with mometasone, yet recurrence was defined as a score equal to or higher than before this remission induction phase. In each group, a few participants received additional treatment. Dropout was defined as participant who had more than 2 recurrences The secondary outcome ‐ time until relapse ‐ was included in the study, but data were not reproducible Study authors were contacted on 7 March 2014 but responded on 13 March 2014 that they were unable to provide additional information Declarations of interest: not stated Funding: Schering‐Plough A/S Farum, Denmark, supplied the study drugs and covered the expenses of processing the data Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients.... were randomised into 1 of 3 groups" Comment: no further details in the article. Personal contact with the study author clarified that the randomisation table was computer generated with blocks of 5, and this was carried out by Schering‐Plough A/S, Farum, Denmark |
| Allocation concealment (selection bias) | Low risk | No details in the article about how allocation was concealed from participants and clinicians; however personal contact with study authors clarified that randomisation was created by a third party. The investigators received sealed and numbered envelopes for allocation to a treatment arm |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "ideally, the maintenance phase should have been double‐blind. This would, however, have required a very complicated distribution of the medicaments, with many different tubes for various days of the week. We felt the risks of mistakes by the patients and of poor compliance to be too great" Comment: no blinding; blinding was difficult because participants had to follow different treatment schedules |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "the investigation was carried out as an open, prospective, randomized trial" Comment: no blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the intention‐to‐treat principle was used to calculate the effect of the treatments" Comment: intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | No trial registration found. Severity of pruritus, erythema and vesicles, scaling, and fissures is described in the Materials and Methods section, but separate results for these are not given in the Results section; however, because these are not listed as outcome, we judged this as low risk. All (clearly described) outcomes listed in the Materials and Methods section are included in the Results section |
| Other bias | Low risk | Baseline comparisons: "there were no statistically significant differences in the demographic features represented in the 3 centres or in the 3 randomisation groups" Diagnostic certainty: yes The study was completed |
Whitaker 1996.
| Methods | Parallel‐group, randomised controlled trial This study was carried out in a secondary setting for outpatients at a single centre in South Africa |
|
| Participants | 39 participants with chronic stable hand eczema of > 12 months' duration
Dropouts: 5 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • GLA (gamma linolenic acid) 50 mg (in 500 mg evening primrose oil capsules) daily for 16 weeks in 19/20 participants Control intervention • Gelatine capsules with 500 mg sunflower oil daily for 16 weeks in 15/19 participants Both groups were allowed to use unlimited quantities of standard emollient and a limited quantity of group III corticosteroids Participants were followed up for 8 weeks after the end of treatment Duration 24 weeks (16 weeks of active treatment, 8 weeks of follow‐up) |
|
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
|
| Notes | Part of the study was a laboratory investigation in 10 matched healthy controls. At the beginning of the study, all participants had blood taken for laboratory parameters, as well as biopsies for histology and electron microscopy. No participant‐rated outcomes Study authors were contacted for additional information but remained unresponsive Declarations of interest: not stated Funding: the study was planned and funded by Scotia Pharmaceuticals Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "a blind random method was used..." Comment: no further details |
| Allocation concealment (selection bias) | Unclear risk | No details about how allocation was concealed from participants and clinicians, although study authors stated that they used a "blind random method" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "placebo was given to 19 patients as identical‐appearing gelatine capsules..." Comment: study authors stated double‐blinded design and included a placebo of identical appearance, which is an adequate way to blind participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "a parallel, double‐blind, placebo‐controlled trial..." Comment: study authors stated a double‐blind design but provided no information regarding observer blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (34 of 39 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | No trial registration found. All results described in the Materials and Methods section are depicted in the Results section |
| Other bias | Unclear risk | Baseline comparisons: redness was significantly more severe in the placebo group than in the intervention group Diagnostic certainty: yes The study was completed |
Yousefi 2012.
| Methods | Parallel‐group, randomised controlled trial This study was conducted in a secondary care setting at a single centre in Iran |
|
| Participants | 60 participants with chronic hand eczema Dropouts: 8 Inclusion criteria of the trial
Exclusion criteria of the trial
Study population
|
|
| Interventions |
Intervention • Nigella sativa oil extract 2% with Eucerin base applied twice a day for a period of 4 weeks in 19 participants • 0.1% betamethasone ointment applied twice a day for a period of 4 weeks in 15 participants Control intervention • Only Eucerin ointment applied twice a day for a period of 4 weeks in 18 participants Duration 4 weeks |
|
| Outcomes |
Primary outcomes of the trial
Other outcomes
|
|
| Notes | The secondary outcomes ‐ reduction in severity, participant‐rated and investigator‐rated ‐ were included but did not provide reproducible data Declarations of interest: none declared Funding: the study was planned and funded by Shahid Beheshti University of Medical Science, Tehran, Iran Sample size rationale: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "assigning patients to the treatment groups was performed based on randomly permuted blocks of size 6. The project biostatistician prepared the randomization list" Comment: this was considered an adequate method to generate a randomisation sequence |
| Allocation concealment (selection bias) | Low risk | Quote: "they were pre‐packed in tubes and consecutively numbered for each participant according to the randomization list. This was done by the project pharmacologist. Each participant was assigned an order number and the dermatology resident used the corresponding numbered packs to allocate participants to treatment groups. Both patients and dermatologists were blind to the assigned drugs due to randomization procedure" Comment: this is considered an adequate and well‐described procedure |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes (Iranian Clinical Trial Register IRCT201111266959N3): "blinding: single blind"
"For blinding the participants but not investigators, all 3 creams( Nigella, Betamethasone, Eucerin as placebo) will be prepared in the identical tubes which only labelled A, B, and C with no other information" Comment: the trial registrations stated a single‐blind study, although the original article claims that the study is double‐blind Quotes (from article): "in this randomised, controlled, double‐blind clinical trial..." "The therapeutic medications were manufactured identical in appearance, odour and other characteristics by adding ineffective ingredients. We added the essence of 0.1% mint oil to create the same smell in all ointments" "Each participant was assigned an order number and the dermatology resident used the corresponding numbered packs to allocate participants to treatment groups. Both patients and dermatologists were blind to the assigned drugs due to randomization procedure" Comment: we assume that participants were unaware of their treatment modality by this method |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (from Iranian Clinical Trial Register IRCT201111266959N3): "for blinding the participants but not investigators, all 3 creams (Nigella, Betamethasone, Eucerin as placebo) will be prepared in the identical tubes which only labelled A, B, and C with no other information" Quote (from article): "in this randomised, controlled, double‐blind clinical trial..." Comment: unclear if outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No intention‐to‐treat analysis but per protocol (52 of 60 = more than 80%) |
| Selective reporting (reporting bias) | Low risk | Trial registration found (IRCT201111266959N3), registered before publication; no major differences between trial registration and the study were found |
| Other bias | Low risk | Baseline comparison: no significant difference was observed between treatment groups with respect to disease severity at baseline as measured by HECSI and DLQI scores (P = 0.43; Welch’s ANOVA; and P = 0.99; ANOVA, respectively) Diagnostic certainty: yes The study was completed |
ALT: alanine aminotransferase.
ANOVA: analysis of variance.
AST: asparate aminotransferase.
BSI: brief symptom inventory.
CDLQI: Children's Dermatology Life Quality Index.
CI: confidence interval.
CPK: creatine phosphokinase.
DASI: Dyshidrotic eczema Area and Severity Index.
DLQI: Dermatology Life Quality Index.
DSCG: disodium cromoglycate.
EASI: Eczema Area and Severity Index.
EDI: Eczema Disability Index.
EQ‐5D: standardised index for measuring quality of life by EuroQol in 5 dimensions.
FSH: follicle‐stimulating hormone.
GLA: gamma linolenic acid.
HDL: high‐density lipoprotein.
HEAS: Hand Eczema Area and Severity Score.
HEASI: adaptation of EASI for the hands: Hand Eczema Area and Severity Index.
HECSI: hand eczema severity index.
HEES: hand eczema extent score.
IGA: investigators' global assessment.
ISGA: investigators' static global assessment.
LDH: lactate dehydrogenase.
LDL: low‐density lipoprotein.
MOP‐8: 8‐methoxypsoralene.
mTLSS: modified total lesion symptom score.
NB‐UVB: narrow‐band ultraviolet B.
PaGA: participants' global assessment.
PGA: physicians' global assessment.
PHQ: patient health questionnaire.
PSA: patient self‐assessment.
PUVA: (topical and oral) psoralen combined with UVA.
QALY: quality‐adjusted life‐year.
RR: risk ratio.
SCORAD: Scoring Atopic Dermatitis tool.
SD: standard deviation.
SGA: subjects' global assessment.
TETDS: tetraethylthiuram disulfide.
TEWL: transepidermal water loss.
TLSS: total lesion symptom score.
UVA: ultraviolet A.
UVA‐1: newer form of UV therapy that contains only long‐wavelength UVA‐1 radiation (340 to 400 nm) and thus reduces the risk of burning.
UVB: ultraviolet B.
VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aertgeerts 1985 | The study included participants with different dermatoses on arms, hands, and legs. It was not clear for which participants the hands were involved and how the outcome was in the participants with hand eczema. This within‐participant study was not randomised and therefore was excluded |
| Berndt 2001 | Participants did not have hand eczema. The study examined slightly irritated hands in nurses. The study may be included in the Skin Group Review titled "Interventions for preventing occupational irritant hand dermatitis" |
| Chen 2015 | Hand and foot eczema combined; no separate data available |
| Gergovska 2017 | Study focusses on prevention instead of treatment for active disease |
| Grivcheva‐Panovska 2013 | Study on Staphylococcus aureus colonisation instead of hand eczema |
| Güler Özden 2004 | Not clear whether participants were randomised to different treatment arms |
| HogenEsch 1998 | Pilot study, in which it is unclear how many were allocated to each intervention. Not clear if the study was randomised |
| Petering 2004 | Non‐randomised study with a within‐participant (left‐right) design. It could be argued whether randomisation is important in this left‐right study with bilateral hand eczema of similar severity. Observer of outcomes was blinded |
| Rosén 1987 | Quasi‐randomised |
| Zeichner 2018 | Single‐arm study without a comparator |
| Zimmerman 1967 | It is unclear whether this within‐participant study was randomised Study on 54 participants with "bilateral, symmetrical areas of dermatitis" and stating only the preference for betamethasone 17‐valerate or fluocinolone acetonide ointment for 8 participants with hand eczema. No other outcomes for hand eczema specified. No information on frequency, dosage, and duration of treatment |
Characteristics of studies awaiting assessment [ordered by study ID]
Beitner 1996.
| Methods | Randomised controlled trial This study was conducted at a single centre in Sweden The study probably was not blinded |
| Participants | 30 consecutive participants with different dermatological hand dermatoses including pustulosis palmoplantaris (n = 5), psoriasis (n = 1), tylotic eczema (n = 4), atopic eczema (n = 3), dyshidrotic eczema (n = 1), allergic eczema (n = 7), and non‐allergic eczema (n = 1), both allergic eczema and dyshidrotic (n = 2) Dropouts: 6 Inclusion and exclusion criteria were not stated |
| Interventions |
Intervention • Clobetsaol propionate (Dermovate Glaxo) solution under occlusion with a hydrocolloid dressing twice a week for the first 2 weeks and once a week for the next 2 weeks • Clobetasol propionate ointment under occlusion with a hydrocolloid dressing twice a week for the first 2 weeks and once a week for the next 2 weeks Follow‐up included a visit 12 weeks after the start of treatment |
| Outcomes |
Primary outcomes of the trial Not stated Other outcomes
|
| Notes | Study results for hand eczema participants were not presented separately, and we were unable to obtain additional data from the study author This study was published as a very concise letter to the editor, and a lot was uncertain, for example, visits and outcome parameters |
CTRI/2009/091/000212.
| Methods | Randomised parallel‐group open‐label study |
| Participants | 60 participants with hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Interventions • Herbavate applied on affected area 3 times a day for 4 weeks • Betamethasone + Gentamycin applied on affected area 3 times a day for 4 weeks |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Notes | Sponsor: Troikaa Pharmaceuticals Limited Study author was contacted on 20 February; however email was not‐working: medicalservices@troikaapharma.com. Contact through LinkedIn revealed that study results have not been published and provided no further details |
Draelos 2000.
| Methods | Randomised controlled trial with a within‐participant design This study was conducted at a single centre in North Carolina, USA The study was double‐blind; an intention‐to‐treat analysis was not carried out |
| Participants | 80 participants between the ages of newborn and 80 years with the following dermatological conditions: household hand dermatitis (n = 21), occupational hand dermatitis (n = 18), latex glove irritant contact dermatitis (n = 9), diaper dermatitis (n = 5), cutaneous wounds (n = 17), and allergic contact dermatitis (n = 10) Dropouts: 7 Exclusion criteria of the trial
|
| Interventions |
Intervention • The study hydrogel barrier/repair cream (Hydron) for 4 weeks in 80 participants Control • Control moisturising cream (Eucerin, Beiersdorf, Germany) for 4 weeks in 80 participants |
| Outcomes |
Primary outcomes of the trial Not stated Other outcomes
|
| Notes | RCT on different diseases, mostly on hand eczema. Number of participants with hand eczema and results for participants with this condition are impossible to reconstruct. We were unable to obtain separate data from the study investigator |
English 1989.
| Methods | Randomised controlled parallel‐group study This study was conducted at 2 dermatology departments in the United Kingdom |
| Participants | 97 outpatients with steroid‐responsive dermatoses including 63 with endogenous eczema (38 with atopic dermatitis, 19 with hand eczema, and 6 with discoid eczema) and 34 with chronic plaque psoriasis Dropouts: 12 Inclusion and exclusion criteria were not stated |
| Interventions |
Intervention • Betamethasone dipropionate cream 005% twice daily during 3 weeks • Betamethasone dipropionate cream 0.05% in the morning and base cream in the evening during 3 weeks |
| Outcomes |
Primary outcomes of the trial Not stated Other outcomes
|
| Notes | This study was part of a larger study on eczema and psoriasis. Analyses/outcomes among the 19 hand eczema participants were not given. We contacted the study author on 11 March 2014. This author responded 13 March 2014 that he was unable to provide additional data |
EUCTR2004‐002398‐22‐DE.
| Methods | Randomised placebo‐controlled double‐blind study |
| Participants | 240 participants with different dermatoses, of which dyshidrotic hand eczema was one Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • IDEA‐070 (ketoprofen in Transfersome) Control intervention • Placebo |
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
| Notes | Study conducted in Germany Sponsor: IDEA AG Current status: not recruiting |
EUCTR2005‐005793‐75‐DE.
| Methods | Double‐blind randomised parallel‐group design |
| Participants | 40 patients with chronic hand dermatitis rated mild to moderate according to Investigator’s global assessment (score, see page 20) that has persisted for longer than 6 month in spite of attempts to identify and remove the cause Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • ASF‐1075 cream Control intervention • Placebo cream |
| Outcomes |
Primary outcomes of the trial
|
| Notes | Current status: not recruiting |
EUCTR2008‐006148‐20‐DE.
| Methods | Double‐blind randomised parallel or within‐participant design |
| Participants | Individuals with mild to moderate plaque‐type psoriasis (PASI < 10) or hand and foot eczema with at least 2 symmetrical lesions |
| Interventions |
Intervention • Mometasone furoate 0.1% ointment Control intervention • Placebo ointment |
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
| Notes | Not available |
Goh 1999.
| Methods | Within‐participant randomised controlled trial This study was conducted at a single centre in Singapore in a secondary setting This was an open‐label, observer‐blinded study; an intention‐to‐treat analysis was not done |
| Participants | 60 consecutive patients with moderate to severe bilateral chronic eczema on the limbs for at least 6 months. Patients had different dermatological diseases such as hand and foot eczema, lichen simplex chronicus, discoid eczema, prurigo nodularis, and unclassifiable eczema. In 8 patients, the hands were treated Dropouts: 2 Exclusion criteria of the trial
|
| Interventions |
Intervention • Mometasone furoate cream 0.1% once daily during 3 weeks • Clobetasol propionate cream 0.05% applied twice daily during 3 weeks No other medication was allowed during the study |
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
| Notes | Study on different types of eczema in different body regions. Specific data on outcomes among the 8 hand eczema participants were not given. We contacted study authors on 11 March 2014; they responded 13 March 2014 that they were unable to obtain these data |
Grundmann 1999.
| Methods | Randomised within‐participant study This study probably was conducted at a single centre in Germany The study was not blinded, and all participants were included in the analyses |
| Participants | 12 patients with severe plaque psoriasis (n = 4), severe atopic dermatitis (n = 4), or severe hyperkeratotic eczema (n = 4) Dropouts: none Inclusion criteria
|
| Interventions |
Intervention • PUVA cream therapy with 8‐methoxypsoralen (8‐MOP) 4 times a week • PUVA bath therapy 4 times a week Participants were followed up for an additional 8 weeks after the end of treatment. Exact duration of treatment and number of treatments were not clear |
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
| Notes | RCT on different dermatoses among hand eczema. We were unable to reconstruct the results from the paper among participants with hand eczema, and we were unsuccessful in locating the study authors |
Haddican 2014.
| Methods | Randomised double‐blind vehicle‐controlled parallel‐group study |
| Participants | 60 patients with hand and/or foot eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Clobetasol propionate 0.05% foam twice‐daily application to the hands or feet for 2 weeks and retapamulin 1% ointment twice‐daily application to anterior nares and the hands or feet for 5 consecutive days • Clobetasol propionate 0.05% foam twice‐daily application to the hands or feet for 2 weeks and vehicle (placebo) ointment application twice daily to anterior nares and the hands or feet for 5 consecutive days |
| Outcomes |
Primary outcomes of the trial Proportion of participants with a PGA of clear or almost clear at day 6, day 15, and day 28 compared to baseline Other outcomes
|
| Notes | RCT on hand and foot dermatitis combined. It was not possible to extract from the paper results of hand eczema participants |
Handa 1988.
| Methods | Randomised parallel‐group study |
| Participants | 100 patients with moderate to severe eczema or other steroid‐responsive dermatoses including eczema (n = 45), lichen simplex chronicus (n = 27), atopic dermatitis (n = 14), psoriasis (n = 4), contact dermatitis (n = 2), stasis dermatitis (n = 2), seborrhoeic dermatitis (n = 1), actinic dermatoses (n = 1), unclear (n = 4) Dropouts: 4 Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Alclometasone dipropionate 0.05% ointment twice a day without occlusion for 3 weeks • Hydrocortisone 1.0% ointment twice a day without occlusion for 3 weeks |
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
| Notes | RCT on different dermatoses. It was impossible to extract from the paper the results of hand eczema participants, and we were unable to locate the study authors |
IRCT201112018263N1.
| Methods | Double‐blind randomised parallel‐group design, placebo‐controlled |
| Participants | 70 patients with moderate to severe hand eczema, aged 12 to 70 years Exclusion criteria
|
| Interventions |
Intervention • Doxepine 5% cream applied twice daily for 8 days and clobetasol 0.05% applied twice daily for 8 days Control intervention • Placebo and clobetasol 0.05% applied twice daily for 8 days |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Notes | Recruitment completed Study authors contacted for additional information on 20 February 2014, but they remained unresponsive Sponsor: Shahid Sadoughi University of Medical Sciences, Yazd |
IRCT201212303734N2.
| Methods | Randomised double‐blind parallel‐group study |
| Participants | 108 patients with hand eczema confirmed by a dermatologist Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Anti Dry cream (contains Aloe Vera essence, Geranium essence, Lavander essence, respectively, with ratio of 50:1:15 in vanishing cream), 1 fingertip unit (0.5 grams) for 10 × 10 cm², twice daily for 2 weeks • Fluocinolone acetonide cream 0.025%, twice daily, for 2 weeks |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Notes | Sponsor: Barij Essence Herbal Medicine Research Center Current status: recruiting |
Lassus 1981.
| Methods | Randomised within‐participant study Probably conducted at a single centre in Finland This study claims to be double‐blind |
| Participants | 80 patients with various symmetrical eczemas: allergic eczema (n = 27), atopic dermatitis (n = 21), toxic hand eczema (n = 10), chronic eczema (n = 7), stasis eczema (n = 5), nummular eczema (n = 4), neurodermatitis (n = 3), dyshidrotic eczema (n = 2), seborrhoeic eczema (n = 1) and 40 participants with psoriasis Dropouts: probably none Inclusion and exclusion criteria were not stated |
| Interventions | All participants were first treated with placebo for 1 week. Afterwards, they were randomised Intervention • Budesonide 0.025% ointment twice daily probably with occlusive dressing • Betamethasone‐17‐valerate 0.1% ointment twice daily probably with occlusive dressing Duration is not really clear from the article, but probably 2 weeks |
| Outcomes |
Primary outcomes of the trial Not defined Other outcomes
|
| Notes | RCT on different forms of symmetrical eczema; this study is published alongside a study on psoriasis. We were unable to reconstruct the results in participants with hand eczema, and we were unsuccessful in locating study authors |
NCT00404196.
| Methods | Double‐blind randomised parallel‐group design |
| Participants | 75 patients with clinical diagnosis of hand eczema with or without atopic aetiology/background Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • LEO19123 cream (calcipotriol 50 mcg/g and LEO80122 0.6 mg/g) for 3 weeks • LEO19123 cream (calcipotriol 15 mcg/g and LEO80122 0.2 mg/g) for 3 weeks • LEO19123 cream vehicle alone for 3 weeks |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Notes | Sponsor: LEO Pharma Study has been completed; john.english@nuh.nhs.uk was contacted on 20 February 2014. However, he was not at liberty to disclose information regarding the study. We therefore contacted LEO Pharma, which was unresponsive |
NCT00614289.
| Methods | Double‐blind randomised within‐participant design |
| Participants | 30 patients with hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions | This study is designed as a prospective, randomised, double‐blind right/left comparison of Epikeia coatings to improve hand dermatitis |
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
| Notes | Sponsor: Biomedical Development Corporation Study has been completed; however we were unable to obtain data from the study authors, who were unresponsive |
NCT00843466.
| Methods | Randomised open‐label parallel‐group study |
| Participants | 46 patients who experience mild to moderate hand dermatitis induced by frequent cleansing Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • A test product (mild, moisturising hand cleanser) for all hand cleansing needs during the duration of the study. Control intervention • No Intervention: the control group will continue to use their current cleanser for handwashing |
| Outcomes |
Primary outcome of the trial
|
| Notes | Sponsor: Wake Forest School of Medicine Study authors were contacted for additional information; they informed us that trial results were marginal and this exploratory study was the terminus of this line of investigation, but we were unable to obtain additional information |
NCT00867607.
| Methods | Double‐blind randomised cross‐over design |
| Participants | 80 patients 18 to 65 years of age with bilateral allergic contact dermatitis of the hands and forearms Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • MRX‐6 (2%) twice daily for 21 days • MRX‐6 (1%) twice daily for 21 days • MRX‐6 (0.2%) twice daily for 21 days • Steroid twice daily for 21 days |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Notes | Sponsor: Hadassah Medical Organization |
NCT00890968.
| Methods | Double‐blind randomised parallel‐group design |
| Participants | 56 adults with chronic hand dermatitis Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Triamcinolone acetonide (TAC) DuraPeel topical gel once daily (nightly) for total duration of 4 weeks Control intervention • Placebo: placebo DuraPeel topical gel once daily (nightly) for a total duration of 4 weeks |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Notes | Study has been completed; additional information was requested on 20 February 2014; however study authors were unresponsive Sponsor: ZARS Pharma Inc. |
NCT01950494.
| Methods | A randomised double‐blind placebo‐controlled parallel‐group study |
| Participants | 20 patients with moderate to severe hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • FiteBac Hand Sanitizer Control intervention • Placebor: blinded emollient therapy |
| Outcomes |
Primary outcome of the trial
Other outcomes of the trial
|
| Notes | Both contact persons were emailed for additional information on 19 February 2014 but did not respond to our request for additional information Sponsor: National Jewish Health |
8‐MOP: 8‐methoxypsoralene.
AEs: adverse events.
BCC: basal cell carcinoma.
DASI: Dyshidrotic eczema Area and Severity Index.
GCP: Good Clinical Practice.
HECSI: hand eczema severity index.
HFMD: hand foot and mouth disease.
ICH: International Conference on Harmonisation.
IGA: investigators' global assessment.
IUD: intrauterine device.
mTLSS: modified total lesion symptom score.
NSAIDs: non‐steroidal anti‐inflammatory drugs.
OTC: over‐the‐counter.
PaGA: participants' global assessment.
PASI: Psoriasis Area and Severity Index.
PGA: physicians' global assessment.
PUVA: (topical and oral) psoralen combined with UVA.
PVA: physicians visual assessment.
RCT: randomised controlled trial.
SCC: squamous cell carcinoma.
SGIC: subjects' global impression of change.
TAC: triamcinolone acetonide.
TLSS: total lesion symptom score.
UV: ultraviolet.
UVA: ultraviolet A.
UVB: ultraviolet B.
VAS: visual analogue scale.
WHO: World Health Organization.
Characteristics of ongoing studies [ordered by study ID]
IRCT2014012916412N1.
| Trial name or title | Effects of pumpkin (Moschata cucurbita) ointment on chronic hand eczema Official title: Effect of topical pumpkin, traditional medicine products, on the chronic hand eczema |
| Methods | Randomised double‐blind parallel‐group placebo‐controlled study |
| Participants | 60 patients with moderate chronic hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Betamethasone ointment twice a day for 1 month • Almond ointment twice a day for 1 month • Eucerin ointment twice a day for 1 month • Pumpkin ointment twice a day for 1 month |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | September 2014 |
| Contact information | Alemeh Khademi Imam Khomeini Hospital Islamic Republic of Iran Skin Clinic, Imam Khomeini Hospital, Keshavarz bulv Tehran 00982166595911 00989171132340 00 alemehkhadi@gmail.com |
| Notes | Sponsor: Vice Chancellor for Research at Tehran University School of Traditional Medicine |
IRCT2017070922965N10.
| Trial name or title | Effect of topical atorvastatin on hand eczema Official title: Evaluating the effect of topical atorvastatin as adjuvant therapy in treatment of hand eczema |
| Methods | Randomised double‐blind placebo‐controlled parallel‐group study |
| Participants | 70 patients with moderate to severe eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Betamethasone ointment plus atorvastatin 5% cream 2 times per day for 10 days • Control group: betamethasone ointment plus placebo atorvastatin 5% cream 2 times per day for 10 days |
| Outcomes |
Primary outcome of the trial
|
| Starting date | 2017‐08‐19 |
| Contact information | Maryam Mehrpooya Hamedan University of Medical Sciences Shahid Fahmide Avenue Hamedan Iran (Islamic Republic of) +98 81 3838 1594 m_mehrpooya2003@yahoo.com |
| Notes | Sponsor: Vice Chancellor for Research, Hamadan University of Medical Sciences |
ISRCTN80206075.
| Trial name or title | Comparison of alitretinoin with PUVA as the first line treatment in patients with severe chronic hand eczema: a randomised controlled trial |
| Methods | Randomised open‐label parallel‐group design |
| Participants | 500 to 780 participants with severe chronic hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Alitretinoin 30 mg a day for 12 to 24 weeks • Immersion PUVA (twice weekly) for 12 to 24 weeks |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | 01/01/2015 |
| Contact information | Dr. Victoria Goss (Senior Trial Coordinator) ctru‐alpha@leeds.ac.uk |
| Notes | The study protocol is published at http://www.nihr.ac.uk/__data/assets/pdf_file/0005/136994/PRO‐12‐186‐01.pdf Sponsor: Unversity of Leeds (UK) |
JPRN‐UMIN000003326.
| Trial name or title | A clinical trial to determine the effect of olopatadine on itching in hand eczema |
| Methods | Randomised single‐blind (participants are blinded) parallel‐group placebo‐controlled study |
| Participants | 50 patients with hand eczema and itch Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Olopatadine‐treated group Control intervention • Placebo olopatadine non‐treated group |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | 1 March 2010 |
| Contact information | Kaoru Takayama Tokyo Medical and Dental University Dermatology Department Dermatology Bukyou‐ku 1‐5‐45 Japan tkaoru.derm@tmd.ac.jp Telephone: +81‐358035286 |
| Notes | Sponsor: Department of Dermatology, Tokyo Medical and Dental University Current status: recruiting |
NCT02664805.
| Trial name or title | Proof of concept, twice daily applications of LEO 124249 ointment in the treatment of chronic hand eczema Official title: A phase 2a, proof of concept trial, testing twice daily applications of LEO 124249 ointment in the treatment of chronic hand eczema |
| Methods | Randomised double‐blind vehicle‐controlled parallel‐group study |
| Participants | 91 patients with chronic hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • LEO 124249 ointment twice‐daily cutaneous application for 8 weeks Control intervention • LEO 124249 ointment vehicle twice‐daily cutaneous application for 8 weeks |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | February 2016 |
| Contact information | Prof. Dr. Margitta Worm, Allergie‐Centrum‐Charité Klinik für Dermatologie, Venerologie und Allergologie Berlin, Germany 10117 |
| Notes | Sponsor: LEO Pharma |
NCT03026907.
| Trial name or title | Efficacy of oral alitretinoin versus oral azathioprine in patients with severe chronic non‐hyperkeratotic hand eczema. A randomized prospective open‐label trial with blinded outcome assessment |
| Methods | Randomised observer‐blind vehicle‐controlled parallel‐group study |
| Participants | 116 patients with severe chronic non‐hyperkeratotic hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention • Oral alitretinoin capsule of 30 mg once daily for a total of 24 weeks • Oral azathioprine tablets twice daily at a dose of 1.5 or 2.5 mg/kg/d, depending on thiopurine methyltransferase (TPMT) activity |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | May 2016 |
| Contact information | Marie‐Louise A. Schuttelaar, MD, PhD University Medical Center Groningen The Netherlands +31503612520 m.l.a.schuttelaar@umcg.nl |
| Notes | Sponsor: University Medical Center Groningen |
NCT03026946.
| Trial name or title | Efficacy of oral alitretinoin versus oral cyclosporine in patients with severe recurrent vesicular hand eczema. A randomised prospective open‐label trial with blinded outcome assessment |
| Methods | Randomised observer‐blind vehicle‐controlled parallel‐group study |
| Participants | 116 patients with severe or very severe recurrent vesicular hand eczema Inclusion criteria
Exclusion criteria Extensive list |
| Interventions |
Intervention • Oral alitretinoin capsule of 30 mg once daily for a total of 24 weeks • Oral cyclosporine A starting dose 5 mg/kg/d (split into 2 doses), with decreasing dose after 8 weeks to 3 to 3.5 mg/kg/d (split into 2 doses). The treatment period is 24 weeks |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | 1 March 2017 |
| Contact information | Marie‐Louise A Schuttelaar, MD, PhD University Medical Center Groningen The Netherlands +31503612520 m.l.a.schuttelaar@umcg.nl |
| Notes | Sponsor: University Medical Center Groningen ZonMw: The Netherlands Organisation for Health Research and Development |
PACTR201704002194318.
| Trial name or title | Assessment of botulinum toxin type A in the treatment of hand eczema |
| Methods | Randomised double‐blind placebo‐controlled parallel‐group study |
| Participants | 60 patients with hand eczema Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention: • Intradermal palmar injection of a minimum of 50 units of botulinum toxin type A combined with topical corticosteroids • Topical betamethasone twice daily |
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | 2017‐04‐15 |
| Contact information | Carmen Amin El Areesh Street, No. 2, Smouha, 3rd floor #302 21646 Alexandria Egypt 002‐01222966670 carmen271173@yahoo.com |
| Notes | Sponsor: Dermatology Department, Faculty of Medicine, University of Alexandria |
BTXA: botulinum toxin A.
DLQI: Dermatology Life Quality Index.
ECG: electrocardiogram.
EQ‐5D: standardised index for measuring quality of life by the EuroQol in 5 dimensions.
HECSI: hand eczema severity index.
mTLSS: modified total lesion symptom score.
PaGA: participants' global assessment.
PBI‐HE: patient benefit index for chronic hand eczema.
PeDeSi: Person‐Centered Dermatology Self‐Care Index.
PGA: physicians' global assessment.
PUVA: (topical and oral) psoralen combined with UVA.
QALY: quality‐adjusted life‐year.
TLSS: total lesion symptom score.
UVB: ultraviolet B.
Differences between protocol and review
The protocol for this review was published in 2009. Since that time, methodological guidance has changed including development of the MECIR standards. Hence, many sections have been edited and some new sections have been added, including Background >Description of the Intervention and Background >How the intervention might work.
Moreover, we were also required to retrospectively select the most important comparisons for our Summary of Findings (SoF) tables, and we selected them based on clinical relevance and on our own experiences.
We have changed the review question slightly and made it more precise. The original protocol stated "To assess the effects of interventions for hand eczema”, which was changed to “To assess the effects of topical and systemic interventions for hand eczema in adults and children" to make the review question a bit more concise, since this is already a very comprehensive review.
Methods >Types of outcome measures: in the original protocol, we stated primary (percentage of participants with self‐rated good/excellent control of symptoms and percentage of participants with investigator‐rated good/excellent control of symptoms), secondary (reduction in severity and time until relapse), and tertiary outcomes (adverse events and dose reduction). We adjusted the review to the most recent Cochrane guidelines and used only primary outcome measures (percentage of participants with self‐rated good/excellent control of symptoms, percentage of participants with investigator‐rated good/excellent control of symptoms, and adverse events) and secondary outcome measures (reduction in severity, time until relapse and dose reduction).The 'Side effects' were changed into the primary outcome 'Adverse events'. Adverse effects were divided into short‐term adverse events occurring during the treatment phase and long‐term adverse events occurring after completion of treatment. We also removed from the primary outcomes the conditional element 'with adequate length of follow‐up' because a substantial otherwise well‐conducted number of studies did not include long‐term follow‐up, and there is no consensus regarding 'adequate' in this context. The secondary outcome time until relapse was not defined in the protocol. In the review, time until relapse was defined as the number of days/weeks until the participant reports worsening of symptoms after initial response.
We added a recommended time point for outcome measures of a minimum of three months, which is considered the most clinically important time point for decision‐makers. Hand eczema is a chronic, relapsing condition that might improve due to the natural course. However, because of the tremendous impact of hand eczema on quality of life, we considered analyses after three months to be undesirable.
Methods > Measures of treatment effect: in the original protocol, we stated that results would be expressed as odds ratios (ORs with 95% confidence intervals (CIs)) and risk differences (RDs with 95% CIs) for dichotomous outcomes and weighted mean differences (WMDs and 95% CI) for continuous outcomes. However, during the review process, we decided that risk ratios should be used instead (RRs with 95% CIs), since these would give a more accurate estimation of relative differences between comparison groups, as in some studies the proportion of outcome events was close to one in one or both groups. In addition, risk difference is not a relative measure and is not recommended as the first choice for reporting pooled results; we decided to abandon the risk difference and include risk ratios instead. We also expressed results from analyses of continuous data as mean differences (MDs), including CIs and respective P values. If insufficient data were available for any of the two analyses, we summed up available data from the respective study including the stated P value. Subsequent to publication of the protocol, in studies where exclusively median values were presented for a particular outcome, we substituted the median for the mean, provided that data were not too skewed. Whenever standard deviations were not available from a paper, we tried to calculate them from other available data. When confidence intervals were provided, the formula given in Chapter 7.7.3.2 of the Cochrane Handbook for Systematic Reviews of Interventions was used (Higgins 2011a). The results were also expressed as number needed to treat (NNT) where appropriate with different rates of baseline risk.
Methods >Types of interventions: we encountered different studies that reported on treatment during a remission‐ or clearance‐induction phase for participants before they were randomised to a follow‐up or maintenance phase. This problem was not addressed in the protocol. In the review, we considered only the latter (randomised) phase for these studies.
Methods >Electronic searches: we tried to obtain additional data regarding unpublished and ongoing trials, or grey literature, via correspondence with study authors and pharmaceutical companies. However, the results of contacting authors and pharmaceuticals were often disappointing. We did not search Pascal and JICT‐EPLUS as planned in our protocol, as we did not have access to these databases by the time the review was written. In the review, we searched MEDLINE from 1946, rather than from 1957, as planned, and we did not use old MEDLINE, because a newer version was available and the old data were incorporated in the new database. We additionally searched LILACS and the Global Resource of EczemA Trials (GREAT) because these became available over the years. We also searched a number of trials registries, which were not part of our original plan, because over the years, the search strategy was updated, and newer ways to conduct searches became available.
Methods >Searching other resources: we contacted trial authors of articles published after 1999 for clarification of date issues. Older contact data were often obsolete, and we considered it unlikely that researchers would have saved study data longer than 15 years. We did not perform a separate search for adverse events. Although not planned in the protocol, we did examine data on adverse events from the included studies. We handsearched the following additional journals: H+G Zeitschrift für Hautkrankheiten, Annales de Dermatologie et Venerelogie, and Journal of Dermatologic Treatment, because these were expected to provide additional studies; however these journals were digitised during the time this review was compiled.
The original protocol stated that the review authors would also record methodological quality in the review. We performed a thorough assessment of risk of bias (authors PJC and AS).
The original protocol stipulated diagnosis of hand eczema by a physician. Although only one of the studies that we identified stated this explicitly, all studies were based on participants being outpatients at hospitals. Therefore, we assumed that the diagnosis was established by a physician for all participants.
The original protocol expressed the plan to conduct subgroup analyses on different classifications of hand eczema, such as recurrent vesicular hand eczema or hyperkeratotic hand eczema. However, in almost all studies, the different classifications of hand eczema were combined and we were unable to extract sufficient information to conduct subgroup analyses, or the subgroups were combined or were unclearly defined in general. This might be the topic of a future update.
Several studies reported scoring systems using un‐named, non‐validated, self‐created, and combined objective and subjective scores; in these cases we provided a narrative account of study results and summarised statistical tests reported by study investigators; we did not attempt quantitative analyses.
Methods >Types of participants: in the review, we additionally included participants who had other parts of their body affected, in addition to having hand eczema, because patients with hand eczema often have comorbidities such as atopic dermatitis. In the protocol, we stated that we would consider other terms, such as 'pompholyx', 'dyshidrosis', and 'pulpitis', as acceptable if diagnosed by a physician. However, in the review, we did not apply the need for diagnosis by a physician because all participants were included from hospitals, and although it was almost never stated in the methods, we therefore believed that the diagnosis was confirmed by a physician. Subsequent to publication of the protocol, we decided to include participants with other diagnoses besides hand eczema in the review when we were able to obtain separate data for hand eczema participants, because otherwise some studies had to be excluded although they contained potentially valuable data. We also clarified in the methods that we included participants with all types of hand eczema.
Methods >Types of participants: in the protocol, we did not impose any age limits on the participants; however, we changed this because treatment requirements and ethics for children are considerably different from adults.
Methods >Selection of studies: specified authors that we assigned in the protocol to independently check titles and abstracts were different in the review because the composition of the review author team changed over the years. The specified authors that we assigned in the protocol to independently examine the trials retrieved as full text were also different in the review because of this. In the protocol, we planned to resolve discrepancies with a third review author (PE), but instead, differences between review authors were resolved in consensus meetings because we considered these discrepancies to be substantial for the review and therefore aimed for consensus between all review authors.
Methods >Data extraction and management: specified authors that we assigned in the protocol to independently extract data were different in the review because of an organisational change in the author team. In the protocol, we planned to resolve discrepancies with a third review author (ÅS), but instead, we resolved differences between review authors in consensus meetings because consensus is preferred over partial consensus. We planned for one review author to check and enter data into Review Manager; however, in the review, three review authors did this.
Methods >Assessment of risk of bias in included studies: we updated our process for assessing methodological quality in the review, following more up‐to‐date guidance than was planned in the protocol. We used the Cochrane risk of bias tool, described in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b).
Methods > Unit of analysis issues: we did not plan for cluster randomised trials or within‐participant studies in the protocol, so the text in the Unit of analysis issues section regarding these types of trials was not envisioned at the time of development of the protocol.
Methods > Dealing with missing data: we did not state plans for dealing with missing data in the protocol, so the text in the Dealing with missing data section was not envisaged at the time of development of the protocol.
Methods > Assessment of heterogeneity: in the protocol, we did not define clinical heterogeneity as we did in the review because this was not an item at the time of writing the protocol. We did not specify in the protocol that we would investigate statistical heterogeneity using the I² test. Neither did we plan to explore reasons for heterogeneity in studies if the I² statistic was greater than 50%, because these were not issues at the time of writing the protocol.
Methods > Assessment of reporting biases: we did not state plans for assessing reporting biases in the protocol, so the text in the Assessment of reporting biases section was not envisaged at the time of development of the protocol.
Methods > Subgroup analysis and investigation of heterogeneity: had there been sufficient data, we would have examined the effects of studying or excluding study subgroups, for example, children versus adults, or recurrent vesicular versus hyperkeratotic hand eczema. This was different from the atopic versus allergic contact hand eczema analysis that we had planned in the protocol, because these subgroups often were not defined and data were not available, which were defined a priori, or those studies had high risk of bias. Future updates of this review will carry out these analyses if data permits.
Methods > Sensitivity analysis: in the protocol, we planned to undertake sensitivity analyses to examine the effects of excluding study subgroups (e.g. children versus adults, atopic versus allergic contact hand eczema) or studies with high risk of bias. But in the review, we stated, "had there been sufficient data, we had intended to perform sensitivity analyses for pooled data. Data on these subgroups were not sufficient for sensitivity analyses and often not available".
Methods > Summary of findings: we included Table 1 to Table 8 for the clinically most relevant outcomes. "Summary of findings tables" were not included in the protocol but were recommended during an update of the review.
Contributions of authors
The following contributions were made by the review authors. Linking with editorial base and co‐ordinating contributions from co‐authors: PJC. Drafting the protocol: HCW, PJC, with contributions from all. Handsearching: PJC, TD, ÅS, with help from the EDEN steering committee members J.J. Grob and L. Naldi. Performing other searches: PJC, JLB, WAC, with help from the Cochrane Information Specialist Liz Doney. Identifying relevant titles and abstracts from searches (i.e. broad screen): JLB, PJC, WAC. Obtaining copies of trials: JLB, WAC. Selecting which trials to include: TD, PJC, ÅS, WAC. Extracting data: PJC, TD, ÅS, WAC. Entering data into RevMan: JLB, WAC. Carrying out analyses: JLB, PJC, WAC, JX. Interpreting analyses: PJC, ÅS, HCW. Drafting the final review: JX, PJC, WAC, with contributions from all. Updating the review: PJC, WAC, JX.
Disclaimer
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Skin Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health.
Sources of support
Internal sources
No sources of support supplied
External sources
Foundation of Environmental and Occupational Dermatology (SMAD), Groningen, Netherlands.
-
The National Institute for Health Research (NIHR), UK.
The NIHR, UK, is the largest single funder of the Cochrane Skin Group.
Declarations of interest
Wietske Andrea Christoffers: nothing to declare.
Pieter‐Jan Coenraads was involved in the included studies Ruzicka 2004, Ruzicka 2008, and van Coevorden 2004a.
Åke Svensson has been involved in educational events for Swedish dermatologists. Novartis has sponsored this event; however, the educational group have decided the content of the education, and no payment has been paid to Åke Svensson personally. Instead, payment has been made to his hospital for his time spent working outside hospital.
Thomas L Diepgen has received lecture and consultancy fees from Leo Pharma A/S and consultancy fees from Almirall Hermal GmbH, and he has co‐authored the studies of Bauer 2012 and Ruzicka 2008.
Janine L Blok: nothing to declare.
Hywel Williams: nothing to declare.
Jun Xia: nothing to declare.
New
References
References to studies included in this review
Adams 2007 {published and unpublished data}
- Adams S, Bayerl C. Medium‐dose‐UVA‐1 irradiation ‐ and topical PUVA ‐ therapy in chronic dyshidrotic hand dermatitis ‐ a prospective randomized study [Mittel‐Dose‐UVA‐1‐ und lokale PUVA‐Therapie beim dyshidrosiformen Handekzem‐ eine prospektive randomisierte Studie]. Aktuelle Dermatologie 2007;33(4):142‐5. [CENTRAL: CN‐00642221; EMBASE: 2007235366] [Google Scholar]
Agarwal 2013 {published and unpublished data}
- Agarwal US, Besarwal RK. Topical clobetasol propionate 0.05% cream alone and in combination with azathioprine in patients with chronic hand eczema: an observer blinded randomized comparative trial. Indian Journal of Dermatology, Venerology, & Leprology 2013;79(1):101‐3. [CENTRAL: CN‐00877938; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Baskan 2005 {published and unpublished data}
- Baskan EB, Kaçar SD, Tunali S. The efficacy of topical pimecrolimus cream 1% in hand dermatitis. Journal of the European Academy of Dermatology and Venereology 2005;19(Suppl 2):267. [CENTRAL: CN‐00602438] [DOI] [PubMed] [Google Scholar]
- Baskan EB, Kaçar SD, Turan A, Tunali S. A new alternative in the treatment of hand eczema: topical pimecrolimus. Turkderm‐ Archieves of the Turkish Dermatology and Venerology 2007;41(4):125‐8. [CENTRAL: CN‐00708519] [Google Scholar]
Bauer 2012 {published and unpublished data}
- Bauer A, Lange N, Matterne U, Meurer M, Braeutigam M, Diepgen TL. Efficacy of pimecrolimus 1% cream in the long term management of atopic hand dermatitis. A double‐blind RCT. Journal der Deutschen Dermatologischen Gesellschaft [Journal of the German Society of Dermatology] 2012;10(6):426‐33. [CENTRAL: CN‐00881946; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bayerl 1999 {published and unpublished data}
- Bayerl C, Garbea A, Peiler D, Rzany B, Allgäuer T, Kleesz P, et al. Pilot study for the treatment of occupational hand dermatitis with a new portable UVB irradiation unit [Pilotstudie zur Therapie des beruflich bedingten Handekzems mit einer neuen tragbaren UVB‐Bestrahlungseinheit]. Aktuelle Dermatologie 1999;25(10):302‐5. [CENTRAL: CN‐00414680; EMBASE: 1999385287] [Google Scholar]
Belsito 2004 {published data only}
- Belsito DV, Fowler JF Jr, Marks JG Jr, Pariser DM, Hanifin J, Duarte IA, et al. Pimecrolimus cream 1%: a potential new treatment for chronic hand dermatitis. Cutis 2004;73(1):31‐8. [CENTRAL: CN‐00471734; MEDLINE: ] [PubMed] [Google Scholar]
Bissonnette 2010 {published and unpublished data}
- Bissonnette R, Worm M, Gerlach B, Guenther L, Cambazard F, Ruzicka T, et al. Successful retreatment with alitretinoin in patients with relapsed chronic hand eczema. British Journal of Dermatology 2010;162(2):420‐6. [CENTRAL: CN‐00751336; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bleeker 1989 {published data only}
- Bleeker J, Anagrius C, Iversen N, Stenberg B, Cullberg Valentin K. Double‐blind comparative study of Corticoderm® cream + unguentum Merck® and Betnovate® cream + unguentum Merck® in hand dermatitis. Journal of Dermatological Treatment 1989;1(2):87‐90. [CENTRAL: CN‐00351307; EMBASE: 1990003851] [Google Scholar]
Boroujeni 2017 {published data only}
- Boroujeni HR, Parvin N, Mirzaeian P, Marandi S. Formulation and clinical trial study of AJMT cream in treatment of eczema. IIOAB Journal 2017;8:46‐50. [CENTRAL: CN‐01372398] [Google Scholar]
Brass 2015 {published and unpublished data}
- Brass D, Farr P, Reynolds N, Stocken D, Macdonald C, Wahie S, et al. An observer‐blinded randomized controlled pilot study comparing localised psoralen‐ultraviolet A with localized narrowband ultraviolet B for the treatment of hand eczema. British Journal of Dermatology 2015;173:13. [CENTRAL: CN‐01135916; EMBASE: 71969396] [DOI] [PubMed] [Google Scholar]
- ISRCTN18213910. Study comparing localised hand psoralen immersion combined with ultraviolet A (PUVA) with localised hand narrowband ultraviolet B (UVB) for the treatment of hand eczema. www.controlled‐trials.com/ISRCTN18213910 (accessed 25 July 2017).
Burrows 1986 {published data only}
- Burrows D, Rogers S, Beck M, Kellet J, McMaster D, Merrett D, et al. Treatment of nickel dermatitis with Trientine. Contact Dermatitis 1986;15(2):55‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Cartwright 1987 {published data only}
- Cartwright PH, Rowell NR. Comparison of Grenz rays versus placebo in the treatment of chronic hand eczema. British Journal of Dermatology 1987;117(1):73‐6. [CENTRAL: CN‐00050015; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Cherill 2000 {published data only}
- Cherill R, Tofte S, MacNaul R, Maher T, Abrams B, Graeber M, et al. SDZ ASM 981 is effective in the treatment of chronic irritant hand dermatitis: a 6‐week randomized, double‐blind, vehicle‐controlled, single center study. Contact Dermatitis 2000;42 Suppl:16‐7. [CENTRAL: CN‐00400538] [Google Scholar]
Chu 2009 {published and unpublished data}
- Chu CY. A 4‐week randomized, double‐blind, placebo‐controlled, right‐left comparison study with E‐DO® in chronic hand dermatitis [Comparison study with E‐DO in chronic hand dermatitis]. Clinical Study Report 5 January 2009. [NCT00556855]
Faghihi 2008 {published data only}
- Faghihi G, Iraji F, Shahingohar A, Saidat A. The efficacy of '0.05% Clobetasol + 2.5% zinc sulphate' cream vs. '0.05% Clobetasol alone' cream in the treatment of the chronic hand eczema: a double‐blind study. Journal of the European Academy of Dermatology and Venereology 2008;22(5):531‐6. [CENTRAL: CN‐00630193; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fairris 1984 {published data only}
- Fairris GM, Mack DP, Rowell NR. Superficial X‐ray therapy in the treatment of constitutional eczema of the hands. British Journal of Dermatology 1984;111(4):445‐9. [CENTRAL: CN‐00568891; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fairris 1985 {published data only}
- Fairris GM, Jones DH, Mack DP, Rowell NR. Conventional superficial X‐ray versus Grenz ray therapy in the treatment of constitutional eczema of the hands. British Journal of Dermatology 1985;112(3):339‐41. [CENTRAL: CN‐00037293; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fowler 2005 {published and unpublished data}
- Fowler JF Jr, Fransway AF, Jackson JM, Rohowsky N. Hydrocortisone butyrate 0.1% cream in the treatment of chronic dermatitis. Cutis 2005;75(2):125‐31. [CENTRAL: CN‐00515029; MEDLINE: ] [PubMed] [Google Scholar]
Fowler 2014 {published data only}
- Fowler JF, Graff O, Hamedani AG. A phase 3, randomized, double‐blind, placebo‐controlled study evaluating the efficacy and safety of alitretinoin (BAL4079) in the treatment of severe chronic hand eczema refractory to potent topical corticosteroid therapy. Journal of Drugs in Dermatology 2014;13(10):1198‐204. [CENTRAL: CN‐01110966] [PubMed] [Google Scholar]
- NCT00817063. Efficacy and Safety of a Retinoid in the Treatment of Severe Chronic Hand Eczema (HANDEL). clinicaltrials.gov/ct2/show/NCT00817063 (accessed 26 July 2017).
Fredriksson 1975 {published data only}
- Fredriksson T, Gip L. Urea creams in the treatment of dry skin and hand dermatitis. International Journal of Dermatology 1975;14(6):442‐4. [CENTRAL: CN‐00698353; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Granlund 1996 {published data only}
- Granlund H, Erkko P, Eriksson E, Reitamo S. Comparison of cyclosporine and topical betamethasone‐17,21‐dipropionate in the treatment of severe chronic hand eczema. Acta Dermato‐Venereologica 1996;76(5):371‐6. [CENTRAL: CN‐00132709; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Granlund H, Erkko P, Reitamo S. Comparison of the influence of cyclosporine and topical betamethasone‐17,21‐dipropionate treatment on quality of life in chronic hand eczema. Acta Dermato‐Venereologica 1997;77(1):54‐8. [CENTRAL: CN‐00137396; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Grattan 1991 {published data only}
- Grattan CE, Carmichael AJ, Shuttleworth GJ, Foulds IS. Comparison of topical PUVA with UVA for chronic vesicular hand eczema. Acta Dermato‐Venereologica 1991;71(2):118‐22. [CENTRAL: CN‐00075959; MEDLINE: ] [PubMed] [Google Scholar]
Gupta 1993 {published data only}
- Gupta AK, Shear NH, Lester RS, Baxter ML, Sauder DN. Betamethasone dipropionate polyacrylic film‐forming lotion in the treatment of hand dermatitis. International Journal of Dermatology 1993;32(11):828‐9. [CENTRAL: CN‐00097910; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hanifin 2004 {published and unpublished data}
- Hanifin JM, Stevens V, Sheth P, Breneman D. Novel treatment of chronic severe hand dermatitis with bexarotene gel. British Journal of Dermatology 2004;150(3):545‐53. [CENTRAL: CN‐00470992; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hill 1998 {published data only (unpublished sought but not used)}
- Hill VA, Wong E, Corbett MF, Menday AP. Comparative efficacy of betamethasone/clioquinol (Betnovate‐C) cream and betamethasone/fusidic acid (Fucibet) cream in the treatment of infected hand eczema. Journal of Dermatological Treatment 1998;9(1):15‐9. [CENTRAL: CN‐00200945] [Google Scholar]
Hordinsky 2010 {published data only}
- Hordinsky M, Fleischer A, Rivers JK, Poulin Y, Belsito D, Hultsch T. Efficacy and safety of pimecrolimus cream 1% in mild‐to‐moderate chronic hand dermatitis: a randomized, double‐blind trial. Dermatology 2010;221(1):71‐7. [CENTRAL: CN‐00769767; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- NCT00226707. Pimecrolimus cream 1% in patients (18 years of age and over) with mild to moderate chronic hand dermatitis. www.clinicaltrials.gov/ct2/show/record/NCT00226707?term=hand+eczema&rank=11 (accessed 26 July 2017).
Jowkar 2011 {published and unpublished data}
- Jowkar F, Jamshidzadeh A, Mirzadeh Yazdi A, Pasalar M. The effects of fumaria parviflora L extract on chronic hand eczema: a randomized double‐blind placebo controlled clinical trial. Iranian Red Crescent Medical Journal 2011;13(11):824‐8. [CENTRAL: CN‐00897812; MEDLINE: ] [PMC free article] [PubMed] [Google Scholar]
Jowkar 2014 {published and unpublished data}
- Jowkar F, Saki N, Mokhtarpour A, Reza Saki M. Comparison of fumaric acid 5% cream versus triamcinolone 0.1% cream in the treatment of hand eczema. Acta Medica Iranica 2014;52(7):528‐31. [CENTRAL: CN‐00999261; PUBMED: 25135262] [PubMed] [Google Scholar]
Kaaber 1983 {published data only}
- Kaaber K, Menné T, Veien N, Hougaard P. Treatment of nickel dermatitis with Antabuse; a double blind study. Contact Dermatitis 1983;9(4):297‐9. [CENTRAL: CN‐00568885; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Katsarou 2012 {published data only}
- Katsarou A, Makris M, Papagiannaki K, Lagogianni E, Tagka A, Kalogeromitros D. Tacrolimus 0.1% vs mometasone furoate topical treatment in allergic contact hand eczema: a prospective randomized clinical study. European Journal of Dermatology 2012;22(2):192‐6. [CENTRAL: CN‐00880927; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kemper 1998 {published data only}
- Kemper MFP, Valk PGM, Arnold WP. Efficacy of coal tar paste vs betamethasone valerate ointment 0.1% in hand eczema [Effectiviteit van koolteerpasta versus betamethasonvaleraatzalf 0.1% bij handeczeem]. Nederlands Tijdschrift voor Dermatologie en Venereologie 1998;8:289‐91. [CENTRAL: CN‐00602201] [Google Scholar]
King 1984 {published data only}
- King CM, Chalmers RJ. A double‐blind study of superficial radiotherapy in chronic palmar eczema. British Journal of Dermatology 1984;111(4):451‐4. [CENTRAL: CN‐00035777; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kircik 2013 {published data only}
- Kircik LH, Eastman WJ, Gwazdauskas J. A randomized, double‐blind phase 4 study of the efficacy and safety of ethanol‐free clobetasol propionate foam, 0.05%, vs vehicle foam in the treatment of chronic hand dermatitis. Journal of Drugs in Dermatology 2013;12(3):328‐34. [CENTRAL: CN‐00876862] [PubMed] [Google Scholar]
Krejci‐Manwaring 2008 {published data only}
- Krejci‐Manwaring J, McCarty MA, Camacho F, Manuel J, Hartle J, Fleischer A Jr, et al. Topical tacrolimus 0.1% improves symptoms of hand dermatitis in patients treated with a prednisone taper. Journal of Drugs in Dermatology 2008;7(7):643‐6. [CENTRAL: CN‐00650082; MEDLINE: ] [PubMed] [Google Scholar]
Kucharekova 2003 {published and unpublished data}
- Kucharekova M, Kerkhof PC, Valk PG. A randomized comparison of an emollient containing skin‐related lipids with a petrolatum‐based emollient as adjunct in the treatment of chronic hand dermatitis. Contact Dermatitis 2003;48(6):293‐9. [CENTRAL: CN‐00470538; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lauriola 2011 {published data only}
- Lauriola MM, Pigatto PD, Pedrelli VF. A single‐center, randomized, perspective, investigator blinded, controlled trial to examine efficacy and safety of a Furpalmate‐containing cream in comparison to topical corticosteroid in atopic dermatitis of hands of 40 adult patients. 20th Congress of the European Academy of Dermatology and Venereology Lisbon (Abstracts on CD‐ROM). Geneva, 2011.
Lindelöf 1987 {published data only}
- Lindelöf B, Wrangsjö K, Lidén S. A double‐blind study of Grenz ray therapy in chronic eczema of the hands. British Journal of Dermatology 1987;117(1):77‐80. [CENTRAL: CN‐00050016; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lodén 2012a {published data only}
- Lodén M, Wirén K, Smerud KT, Meland N, Hønnås H, Mørk C, et al. The effect of a corticosteroid cream and a barrier‐strengthening moisturizer in hand eczema. A double‐blind, randomized, prospective, parallel group clinical trial. Journal of the European Academy of Dermatology & Venereology 2012;26(5):597‐601. [CENTRAL: CN‐00882835; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Möller 1983 {published data only}
- Möller H, Svartholm H, Dahl G. Intermittent maintenance therapy in chronic hand eczema with clobetasol propionate and flupredniden acetate. Current Medical Research & Opinion 1983;8(9):640‐4. [CENTRAL: CN‐00307918; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
NCT01231854 {unpublished data only}
- NCT01231854. Ciclosporin versus alitretinoin for severe atopic hand dermatitis (TocyDD). clinicaltrials.gov/show/NCT01231854 (accessed 27 February 2013).
Odia 1996 {published data only}
- Odia S, Vocks E, Rakoski J, Ring J. Successful treatment of dyshidrotic hand eczema using tap water iontophoresis with pulsed direct current. Acta Dermato‐Venereologica 1996;76(6):472‐4. [CENTRAL: CN‐00179694; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pacor 2006 {published and unpublished data}
- Pacor ML, Lorenzo G, Martinelli N, Mansueto P, Friso S, Pellitteri ME, et al. Tacrolimus ointment in nickel sulphate‐induced steroid‐resistant allergic contact dermatitis. Allergy & Asthma Proceedings 2006;27(6):527‐31. [CENTRAL: CN‐00574561; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pigatto 1990 {published data only}
- Pigatto PD, Gibelli E, Fumagalli M, Bigardi A, Morelli M, Altomare GF. Disodium cromoglycate versus diet in the treatment and prevention of nickel‐positive pompholyx. Contact Dermatitis 1990;22(1):27‐31. [CENTRAL: CN‐00568540; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Polderman 2003 {published data only}
- Polderman MC, Govaert JC, Cessie S, Pavel S. A double blind placebo‐controlled trial of UVA‐1 in the treatment of dyshidrotic eczema. Clinical & Experimental Dermatology 2003;28(6):584‐7. [CENTRAL: CN‐00458902; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ruzicka 2004 {published and unpublished data}
- Larsen F, Galewicz, Ruzicka T, Horváth A, Coenraads PJ, Zouboulis CC, et al. BAL40/9 (alitretinoin, 9‐cisretinoic acid) is effective and well tolerated in the treatment of refractory hand dermatitis. Journal of the European Academy of Dermatology and Venereology. 2003; Vol. 17:158. [Larsen 2003]
- Ruzicka T, Larsen FG, Galewicz D, Horváth A, Coenraads PJ, Thestrup‐Pedersen K, et al. Oral alitretinoin (9‐cis‐retinoic acid) therapy for chronic hand dermatitis in patients refractory to standard therapy. Results of a randomized, double‐blind placebo‐controlled, multicenter trial. Archives of Dermatology 2004;140(12):1453‐9. [CENTRAL: CN‐00503534; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ruzicka 2008 {published and unpublished data}
- Ruzicka T, Lynde CW, Jemec GB, Diepgen T, Berth‐Jones J, Coenraads PJ, et al. Efficacy and safety of oral alitretinoin (9‐cis retinoic acid) in patients with severe chronic hand eczema refractory to topical corticosteroids: results of a randomized, double‐blind, placebo‐controlled, multicentre trial. British Journal of Dermatology 2008;158(4):808‐17. [CENTRAL: CN‐00637962; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Said 2010 {published data only}
- Said NH, Lim KS, Chong WS, Theng CTS. A comparative study of UVA‐1 phototherapy versus betamethasone valerate 0.1% cream for chronic vesicular hand eczema. Annals of the Academy of Medicine Singapore 2011;40(11 Suppl 1):S28. [CENTRAL: CN‐01074005] [Google Scholar]
- Said NH, Lim KS, Chong WS, Theng CTS. A comparative study of ultraviolet light a1 phototherapy versus betamethasone valerate 0.1% cream for chronic vesicular hand eczema. Annals of the Academy of Medicine Singapore 2010;39(11 Suppl 1):S354. [CENTRAL: CN‐01074020] [Google Scholar]
Schnopp 2002 {published and unpublished data}
- Schnopp C, Remling R, Möhrenschlager M, Weigl L, Ring J, Abeck D. Topical tacrolimus (FK506) and mometasone furoate in treatment of dyshidrotic palmar eczema: a randomized, observer‐blinded trial. Journal of the American Academy of Dermatology 2002;46(1):73‐7. [CENTRAL: CN‐00376609; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sezer 2007 {published data only}
- Sezer E, Etikan I. Local narrowband UVB phototherapy vs. local PUVA in the treatment of chronic hand eczema. Photodermatology, Photoimmunology & Photomedicine 2007;23(1):10‐4. [CENTRAL: CN‐00577966; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sharma 2006 {published and unpublished data}
- Sharma AD. Disulfiram and low nickel diet in the management of hand eczema: a clinical study. Indian Journal of Dermatology, Venereology & Leprology 2006;72(2):113‐8. [CENTRAL: CN‐00565442; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sheehan‐Dare 1989 {published data only}
- Sheehan‐Dare RA, Goodfield MJ, Rowell NR. Topical psoralen photochemotherapy (PUVA) and superficial radiotherapy in the treatment of chronic hand eczema. British Journal of Dermatology 1989;121(1):65‐9. [CENTRAL: CN‐00061541; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sjövall 1987 {published data only}
- Sjövall P, Christensen OB. Local and systemic effect of UVB irradiation in patients with chronic hand eczema. Acta Dermato‐Venereologica 1987;67(6):538‐41. [CENTRAL: CN‐00053072; MEDLINE: ] [PubMed] [Google Scholar]
Thestrup‐Pedersen 2001 {published and unpublished data}
- Thestrup‐Pedersen K, Andersen KE, Menné T, Veien N. Treatment of hyperkeratotic dermatitis of the palms (eczema keratoticum) with oral acitretin. A single‐blind placebo controlled study. Acta Dermato‐Venereologica 2001;81(5):353‐5. [CENTRAL: CN‐00377187; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tzaneva 2009 {published and unpublished data}
- Tzaneva S, Kittler H, Thallinger C, Hönigsmann H, Tanew A. Oral vs. bath PUVA using 8‐methoxypsoralen for chronic palmoplantar eczema. Photodermatolology, Photoimmunology & Photomedicine 2009;25(2):101‐5. [CENTRAL: CN‐00699561; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Uggeldahl 1986 {published data only}
- Uggeldahl PE, Kero M, Ulshagen K. Comparative effect of desonide cream 0.1% and 0.05% in patients with hand eczema. Current Therapeutic Research, Clinical & Experimental 1986;40(5):969‐73. [CENTRAL: CN‐00178392; EMBASE: 1987040454] [Google Scholar]
van Coevorden 2004a {published and unpublished data}
- Coevorden AM, Kamphof WG, Sonderen E, Bruynzeel DP, Coenraads PJ. Comparison of oral psoralen‐UV‐A with a portable tanning unit at home vs hospital administered bath psoralen‐UV‐A in patients with chronic hand eczema: an open‐label randomized controlled trial of efficacy. Archives of Dermatology 2004;140(12):1463‐6. [CENTRAL: CN‐00503535; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Veien 1995 {published and unpublished data}
- Veien NK, Kaaber K, Larsen PO, Nielsen AO, Thestrup‐Pedersen K. Ranitidine treatment of hand eczema in patients with atopic dermatitis: a double blind, placebo‐controlled trial. Journal of the American Academy of Dermatology 1995;32(6):1056‐7. [CENTRAL: CN‐00114123; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Veien 1999 {published and unpublished data}
- Veien NK, Olholm Larsen P, Thestrup‐Pedersen K, Schou G. Long‐term, intermittent treatment of chronic hand eczema with mometasone furoate. British Journal of Dermatology 1999;140(5):882‐6. [CENTRAL: CN‐00278245; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Whitaker 1996 {published data only}
- Whitaker DK, Cilliers J, Beer C. Evening primrose oil (Epogam) in the treatment of chronic hand dermatitis: disappointing therapeutic results. Dermatology 1996;193(2):115‐20. [CENTRAL: CN‐00132445; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Yousefi 2012 {published data only}
- Yousefi M, Barikbin B, Kamalinejad M, Abolhasani E, Ebadi A, Younespour S, et al. Comparison of therapeutic effect of topical Nigella with Betamethasone and Eucerin in hand eczema. Journal of the European Academy of Dermatology and Venereology 2013;27(12):1498. [CENTRAL: CN‐00915004; DOI: 10.1111/jdv.12033] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aertgeerts 1985 {published data only}
- Aertgeerts P, Albring M, Klaschka F, Naseman T, Patzelt‐Wenczler R, Rauhut K, et al. Comparative testing of Kamillosan cream and steroidal (0.25% hydrocortisone, 0.75% fluocortin butyl ester) and non‐steroidal (5% bufexamac) dermatologic agents in maintenance therapy of eczematous diseases [Vergleichende Pruefung von Kamillosan Creme gegenueber steroidalen (0,25% hydrocortison, 0,75% fluorocortolon)]. Zeitschrift fur Hautkrankheiten 1985;60(3):270‐7. [MEDLINE: ] [PubMed] [Google Scholar]
Berndt 2001 {published data only}
- Berndt U, Wigger‐Alberti W, Gabard B, Elsner P. Comparison of a barrier cream and its constituents on efficacy in occupational irritant hand eczema in nurses [Vergleich einer Hautschutzcreme und ihrer Grundlage bezuglich Wirksamkeit gegen das berufsbedingte irritative handekzem bei Krankenschwestern: eine anwendungsuntersuchung]. Dermatosen in Beruf und Umwelt. Occupation and Environment 2001;49(1A):77‐80. [CENTRAL: CN‐00441979; EMBASE: 2001126306] [Google Scholar]
Chen 2015 {published data only}
- Chen Cz, Shu Xh, Yao L, Chu N. Treatment for 53 middle‐aged and old patients with chronic eczema of hand‐foot by spleen‐invigorating heat‐clearing method. Western Journal of Traditional Chinese Medicine 2015;28(8):73‐5. [CENTRAL: CN‐01434924] [Google Scholar]
Gergovska 2017 {published data only}
- Gergovska M, Briand E, Karp JM, Jacob SE, Vemula P, Gospodinov D, et al. Metal sequestering dermal cream exhibits beneficial effects in patients with dyshidrotic eczema associated with nickel induced allergic contact dermatitis ‐ a pilot study. Allergy: European Journal of Allergy and Clinical Immunology 2017;72:310. [CENTRAL: CN‐01417582] [Google Scholar]
Grivcheva‐Panovska 2013 {published data only}
- Grivcheva‐Panovska V. Staphylococcus aureus and hand eczema: to treat or not to treat. Allergy: European Journal of Allergy and Clinical Immunology 2013;68:423‐4. [EMBASE: 71369118] [Google Scholar]
Güler Özden 2004 {published data only}
- Güler Özden M, Ilter N, Gürer MA. A new treatment choice for chronic hand dermatitis: 1% pimecrolimus cream. Journal of the European Academy of Dermatology and Venereology 2004;18 Suppl:268. [Google Scholar]
HogenEsch 1998 {published data only}
- HogenEsch AJ, Coenraads PJ. Thuisbehandeling van chronisch recidiverend handeczeem met orale PUVA‐therapie. Nederlands Tijdschrift voor Dermatologie en Venereologie 1998;8:267‐8. [Google Scholar]
Petering 2004 {published data only}
- Petering H, Breuer C, Herbst R, Kapp A, Werfel T. Comparison of localized high‐dose UVA1 irradiation versus topical cream psoralen‐UVA for treatment of chronic vesicular dyshidrotic eczema. Journal of the American Academy of Dermatology 2004;50(1):68‐72. [CENTRAL: CN‐00520456; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rosén 1987 {published data only}
- Rosén K, Mobacken H, Swanbeck G. Chronic eczematous dermatitis of the hands: a comparison of PUVA and UVB treatment. Acta Dermato‐Venereologica 1987;67(1):48‐54. [CENTRAL: CN‐00047378; MEDLINE: ] [PubMed] [Google Scholar]
Zeichner 2018 {published data only}
- Zeichner JA, Berson D, Donald A. The use of an over‐the‐counter hand cream with sweet almond oil for the treatment of hand dermatitis. Journal of Drugs in Dermatology 2018;17(1):78‐82. [CENTRAL: CN‐01451024] [PubMed] [Google Scholar]
Zimmerman 1967 {published data only}
- Zimmerman EH. Betamethasone 17‐Valerate. A custom‐made topical corticosteroid. Archives of Dermatology 1967;95(5):514‐9. [MEDLINE: ] [PubMed] [Google Scholar]
References to studies awaiting assessment
Beitner 1996 {published data only}
- Beitner H. Treatment of hand dermatosis: a comparative study of a topical glucocorticoid ointment vs solution occluded with a new thin hydrocolloid dressing. Acta Dermato‐Venereologica 1996;76(5):408‐9. [CENTRAL: CN‐00332682; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
CTRI/2009/091/000212 {published data only}
- CTRI/2009/091/000212. To compare the efficacy and safety of Herbavate vs. the conventional steroid therapy in the management of hand dermatitis. apps.who.int/trialsearch/Trial.aspx?TrialID=CTRI/2009/091/000212 (accessed 20 February 2014).
Draelos 2000 {published data only}
- Draelos ZD. Hydrogel barrier/repair creams and contact dermatitis. American Journal of Contact Dermatitis 2000;11(4):222‐5. [CENTRAL: CN‐00329003; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
English 1989 {published data only (unpublished sought but not used)}
- English JS, Bunker CB, Ruthven K, Dowd PM, Greaves MV. A double‐blind comparison of the efficacy of betametasone diproprionate cream twice daily versus once daily in the treatment of steroid responsive dermatoses. Clinical & Experimental Dermatology 1989;14(1):32‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
EUCTR2004‐002398‐22‐DE {published data only}
- EUCTR2004‐002398‐22‐DE. Double‐blind, placebo‐controlled study to investigate the efficacy and safety of IDEA‐070 (ketoprofen in Transfersome®) in different dermatological diseases ‐ IDEA‐070 in different dermatological diseases. apps.who.int/trialsearch/default.aspx (accessed 20 February 2014).
EUCTR2005‐005793‐75‐DE {published data only}
- EUCTR2005‐005793‐75‐DE. Evaluation of the efficacy of a topical formulation containing ASF‐1075 in the treatment of chronic hand dermatitis ‐ hand dermatitis. apps.who.int/trialsearch/trial.aspx?trialid=EUCTR2005‐005793‐75‐DE (accessed 27 February 2013).
EUCTR2008‐006148‐20‐DE {published data only}
- EUCTR2008‐006148‐20‐DE. A vehicle‐controlled, investigator‐blinded intraindividual comparison to evaluate the safety, tolerability and efficacy of Momegalen rich cream and ointment in patients with hand and foot eczema and mild to moderate psoriasis. apps.who.int/trialsearch/trial.aspx?trialid=EUCTR2008‐006148‐20‐DE (accessed 27 February 2013).
Goh 1999 {published data only (unpublished sought but not used)}
- Goh CL, Lim JT, Leow YH, Ang CB, Kohar YM. The therapeutic efficacy of mometasone furoate cream 0.1% applied once daily vs clobetasol propionate cream 0.05% applied twice daily in chronic eczema. Singapore Medical Journal 1999;40(5):341‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Grundmann 1999 {published data only}
- Grundmann‐Kollmann M, Behrens S, Peter RU, Kerscher M. Treatment of severe recalcitrant dermatoses of the palms and soles with PUVA‐bath versus PUVA‐cream. Photodermatology, Photoimmunology & Photomedicine 1999;15(2):87‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Haddican 2014 {published data only}
- Haddican M, Linkner RV, Singer G, On SCJ, Gagliotti M, Goldenberg G. Retapamulin 1% ointment and clobetasol propionate 0.05% foam is more efficacious than vehicle ointment and clobetasol 0.05% propionate foam in the treatment of hand/foot dermatitis. Journal of Clinical and Aesthetic Dermatology 2014;7(7):32‐6. [PMC free article] [PubMed] [Google Scholar]
Handa 1988 {published data only}
- Handa F, Sharma PK, Guha A, Sharma SD. Alcometasone dipropionate 0.05% ointment versus hydrocortisone 1.0% ointment in eczema & other dermatoses. Indian Journal of Dermatology 1988;33(1):5‐8. [PUBMED: 3074045] [PubMed] [Google Scholar]
IRCT201112018263N1 {unpublished data only}
- IRCT201112018263N1. Evaluating of clobetasol 0.05% and doxepine 5% cream vs clobetasol 0.05% and placebo in the treatment of moderate to severe hand eczema. apps.who.int/trialsearch/trial.aspx?trialid=IRCT201112018263N1 (accessed 27 February 2013).
IRCT201212303734N2 {published data only}
- IRCT201212303734N2. Evaluating of anti‐dry cream vs fluocinolone acetonide 0/o25% cream in treatment of hand eczema. www.irct.ir/searchresult.php?id=3734&number=2 (accessed 20 February 2014).
Lassus 1981 {published data only}
- Lassus A, Salde L. A double‐blind comparison of two topical steroids in psoriasis and eczemas: budesonide 0.025% ointment and betamethasone‐17‐valerate 0.1% ointment. Pharmatherapeutica 1981;2(10):668‐72. [EMBASE: 1982033894] [Google Scholar]
NCT00404196 {published data only}
- NCT00404196. LEO19123 cream in the treatment of hand eczema. clinicaltrials.gov/ct2/show/NCT00404196 (accessed 27 February 2013).
NCT00614289 {published data only}
- NCT00614289. Novel topical treatment of hand dermatitis (eczema). clinicaltrials.gov/ct2/show/NCT00614289?term=NCT00614289 (accessed 27 February 2013).
NCT00843466 {published data only}
- NCT00843466. Compatibility of a mild, moisturizing hand cleanser for patients with mild to moderate hand dermatitis. clinicaltrials.gov/show/NCT00843466 and apps.who.int/trialsearch/Trial.aspx?TrialID=NCT00843466 (accessed 20 February 2014).
NCT00867607 {published data only}
- NCT00867607. Safety, tolerability, and efficacy of 21 days dermal application of MRX‐6 on mild to moderate contact dermatitis. clinicaltrials.gov/show/NCT00867607 (accessed 27 February 2013).
NCT00890968 {published data only}
- NCT00890968. Safety and efficacy study of triamcinolone acetonide DuraPeel to treat hand dermatitis (TAC‐202). clinicaltrials.gov/ct2/show/NCT00890968 (accessed 27 February 2013).
NCT01950494 {published data only}
- NCT01950494. A one month, randomized, two‐center, parallel‐group, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of FiteBac hand sanitizer TID vs emollient therapy in the management of hand dermatitis in adults. clinicaltrials.gov/ct2/show/NCT01950494 (accessed on 19 February 2014).
References to ongoing studies
IRCT2014012916412N1 {published data only}
- IRCT2014012916412N1. Effect of topical pumpkin, traditional medicine products on chronic hand eczema. www.irct.ir/searchresult.php?id=16412&number=1 (accessed 7 April 2017).
IRCT2017070922965N10 {published data only}
- IRCT2017070922965N10. Effect of topical atorvastatin on hand eczema [Evaluating the effect of topical atorvastatin as adjuvant therapy in treatment of hand eczema]. apps.who.int/trialsearch/Trial2.aspx?TrialID=IRCT2017070922965N10 (accessed 21 April 2018).
ISRCTN80206075 {published data only}
- ISRCTN80206075. Comparison of alitretinoin with PUVA as the first line treatment in patients with severe chronic hand eczema: a randomised controlled trial. www.isrctn.com/ISRCTN80206075?q=hand%20eczema&filters=&sort=&offset=3&totalResults=10&page=1&pageSize=10&searchType=basic‐search (accessed 8 April 2017).
JPRN‐UMIN000003326 {published data only}
- JPRN‐UMIN000003326. A clinical trial to determine the effect of olopatadine on itching in hand eczema. apps.who.int/trialsearch/Trial2.aspx?TrialID=JPRN‐UMIN000003326 (accessed 20 February 2014).
NCT02664805 {published data only}
- NCT02664805. A phase 2a, proof of concept trial, testing twice daily applications of LEO 124249 ointment in the treatment of chronic hand eczema. clinicaltrials.gov/ct2/show/NCT02664805 (accessed 7 April 2017).
NCT03026907 {published data only}
- NCT03026907. Efficacy of oral alitretinoin versus oral azathioprine in patients with severe chronic non‐hyperkeratotic hand eczema. A randomized prospective open‐label trial with blinded outcome assessment. www.clinicaltrials.gov/ct2/show/NCT03026907 (accessed 25 July 2017).
NCT03026946 {published data only}
- NCT03026946. Efficacy of oral alitretinoin versus oral cyclosporine in patients with severe recurrent vesicular hand eczema. A randomized prospective open‐label trial with blinded outcome assessment. clinicaltrials.gov/ct2/show/record/NCT03026946 (accessed 7 April 2017). [DOI] [PMC free article] [PubMed]
PACTR201704002194318 {published data only}
- PACTR201704002194318. Assessment of botulinum toxin type A in the treatment of hand eczema. www.pactr.org/ATMWeb/appmanager/atm/atmregistry?dar=true&tNo=PACTR201704002194318 (accessed 21 April 2018).
Additional references
Agner 2008
- Agner T, Andersen KE, Brandao FM, Bruynzeel DP, Bruze M, Frosch P, et al. Hand eczema severity and quality of life: a cross‐sectional, multicentre study of hand eczema patients. Contact Dermatitis 2008;59(1):43‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ahluwalia 1998
- Ahluwalia A. Topical glucocorticoids and the skin ‐ mechanisms of action: an update. Mediators of Inflammation 1998;7(3):183‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Augustin 2016
- Augustin M, Thaçi D, Kamps A. Impact on quality of life of alitretinoin in severe chronic hand eczema: FUGETTA real‐world study. Journal der Deutschen Dermatologischen Gesellschaft 2016;14(12):1261‐70. [DOI] [PubMed] [Google Scholar]
Basra 2008
- Basra MK, Fenech R, Gatt RM, Salek MS, Finlay AY. The Dermatology Life Quality Index 1994‐2007: a comprehensive review of validation data and clinical results. British Journal of Dermatology 2008;159(5):997‐1035. [DOI] [PubMed] [Google Scholar]
Basra 2015
- Basra MKA, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology 2015;230(1):27‐33. [DOI] [PubMed] [Google Scholar]
Blair 2016
- Blair HA, Scott LJ. Alitretinoin: a review in severe chronic hand eczema. Drugs 2016;76(13):1271‐9. [DOI] [PubMed] [Google Scholar]
Charman 2005
- Charman CR, English JS. Getting to grips with hand eczema: measuring skin disease severity objectively. British Journal of Dermatology 2005;152(2):199‐201. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Coenraads 1998
- Coenraads PJ, Diepgen TL. Risk for hand eczema in employees with past or present atopic dermatitis. International Archives of Occupational & Environmental Health 1998;71(1):7‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Coenraads 2005
- Coenraads PJ, Walle H, Thestrup‐Pedersen K, Ruzicka T, Dreno B, Loge C, et al. Construction and validation of a photographic guide for assessing severity of chronic hand dermatitis. British Journal of Dermatology 2005;152(2):296‐301. [PUBMED: 15727642] [DOI] [PubMed] [Google Scholar]
Coenraads 2012
- Coenraads PJ. Hand eczema. New England Journal of Medicine 2012;367(19):1829‐37. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Cvetkovski 2005
- Cvetkovski RS, Rothman KJ, Olsen J, Mathiesen B, Iversen L, Johansen JD, et al. Relation between diagnoses on severity, sick leave and loss of job among patients with occupational hand eczema. British Journal of Dermatology 2005;152(1):93‐8. [PUBMED: 15656807] [DOI] [PubMed] [Google Scholar]
de Paulis 1992
- Paulis A, Stellato C, Cirillo R, Ciccarelli A, Oriente A, Marone G. Anti‐inflammatory effect of FK‐506 on human skin mast cells. Journal of Investigative Dermatology 1992;99:723‐8. [DOI] [PubMed] [Google Scholar]
Diepgen 2003
- Diepgen TL. Occupational skin‐disease data in Europe. International Archives of Occupational & Environmental Health 2003;76(5):331‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Diepgen 2007
- Diepgen TL, Agner T, Aberer W, Berth‐Jones J, Cambazard F, Elsner P, et al. Management of chronic hand eczema. Contact Dermatitis 2007;57(4):203‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Diepgen 2009a
- Diepgen TL, Andersen KE, Brandao FM, Bruze M, Bruynzeel DP, Frosch P, et al. Hand eczema classification: a cross‐sectional, multicentre study of the aetiology and morphology of hand eczema. British Journal of Dermatology 2009;160(2):353‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Diepgen 2009b
- Diepgen TL, Elsner P, Schliemann S, Fartasch M, Köllner A, Skudlik C, et al. Guideline on the management of hand eczema ICD‐10 Code: L20. L23. L24. L25. L30. Journal der Deutschen Dermatologischen Gesellschaft 2009;7(Suppl 3):S1‐16. [DOI] [PubMed] [Google Scholar]
Diepgen 2012
- Diepgen TL, Pfarr E, Zimmermann T. Efficacy and tolerability of alitretinoin for chronic hand eczema under daily practice conditions: results of the TOCCATA open study comprising 680 patients. Acta Dermato‐Venereologica 2012;92(3):251‐5. [DOI] [PubMed] [Google Scholar]
Drake 1996
- Drake LA, Dinehart SM, Farmer ER, Goltz RW, Graham GF, Hordinsky MK, et al. Guidelines of care for the use of topical glucocorticosteroids. American Academy of Dermatology. Journal of the American Academy of Dermatology 1996;35(4):615‐9. [PUBMED: 8859293] [DOI] [PubMed] [Google Scholar]
English 2009
- English J, Aldridge R, Gawkrodger DJ, Kownacki S, Statham B, White JLM, et al. Consensus statement on the management of chronic hand eczema. Clinical and Experimental Dermatology 2009;34(7):761‐9. [PUBMED: 19747339] [DOI] [PubMed] [Google Scholar]
Faergemann 1987
- Faergemann J, Larkö O. The effect of UV light on human skin microorganisms. Acta Dermato‐Venereologica 1987;67:69‐72. [PubMed] [Google Scholar]
Finlay 1994
- Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI): a simple practical measure for routine clinical use. Clinical and Experimental Dermatology 1994;19(3):210‐6. [PUBMED: 8033378] [DOI] [PubMed] [Google Scholar]
Fox 1873
- Fox T. Clinical lecture on dyshidrosis: an undescribed eruption. British Medical Journal 1873;27(2):365‐6. [PUBMED: 20747210] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEPro [Computer program]
- Brozek JL, Oxman A, Schünemann HJ. GRADEpro GDT. Version 3.6.1. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Grainge 2013
- Grainge M, Leonardi‐Bee J. Reviews of conditions with low event rates. CSG Annual Meeting 2013 2013; Vol. https://skin.cochrane.org/csg‐annual‐meeting‐2013.
GraphPad [Computer program]
- Motulsky H. GraphPad Prism. San Diego: GraphPad Software, 2018.
Hald 2009
- Hald M, Agner T, Blands J, Veien NK, Laurberg G, Avnstorp C, et al. Clinical severity and prognosis of hand eczema. British Journal of Dermatology 2009;160(6):1229‐36. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Halpern 2000
- Halpern SM, Anstey AV, Dawe RS, Diffey BL, Farr PM, Ferguson J, et al. Guidelines for topical PUVA: a report of a workshop of the British Photodermatology Group. British Journal of Dermatology 2000;142(1):22‐31. [PUBMED: 10651690] [DOI] [PubMed] [Google Scholar]
Heede 2016
- Heede NG, Thyssen JP, Thuesen BH, Linneberg A, Johansen JD. Predictive factors of self‐reported hand eczema in adult Danes: a population‐based cohort study with 5‐year follow‐up. British Journal of Dermatology 2016;175(2):287‐95. [PUBMED: 26872425] [DOI] [PubMed] [Google Scholar]
Held 2005
- Held E, Skoet R, Johansen JD, Agner T. The hand eczema severity index (HECSI): a scoring system for clinical assessment of hand eczema. A study of inter and intraobserver reliability. British Journal of Dermatology 2005;152(2):302‐7. [PUBMED: 15727643] [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Deeks JJ. Chapter 7: Selecting studies and collecting data. In: Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC, on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Sterne JAC, Egger M, Moher D, on behalf of the Cochrane Bias Methods Group. Chapter 10: Addressing reporting biases. In: Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011d
- Higgins JPT, Deeks JJ, Altman DG, on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. In: Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hönigsmann 2003
- Hönigsmann H. Mechanisms of phototherapy and photochemotherapy for photodermatoses. Dermatologic Therapy 2003;16(1):23‐7. [DOI] [PubMed] [Google Scholar]
Jekler 1990
- Jekler J, Larkö O. Combined UVA‐UVB versus UVB phototherapy for atopic dermatitis: a paired‐comparison study. Journal of the American Academy of Dermatology 1990;22:49‐53. [DOI] [PubMed] [Google Scholar]
Jensen 2006
- Jensen CS, Menné T, Johansen JD. Systemic contact dermatitis after oral exposure to nickel: a review with a modified meta‐analysis. Contact Dermatitis 2006;54(2):79‐86. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Johansen 2011
- Johansen JD, Hald M, Andersen BL, Laurberg G, Danielsen A, Avnstorp C, et al. Classification of hand eczema: clinical and aetiological types. Based on the guideline of the Danish Contact Dermatitis Group. Contact Dermatitis 2011;65(1):13‐21. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kaae 2012
- Kaae J, Menné T, Carlsen BC, Zachariae C, Thyssen JP. The hands in health and disease of individuals with filaggrin loss‐of‐function mutations: clinical reflections on the hand eczema phenotype. Contact Dermatitis 2012;67(3):119‐24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kislat 2014
- Kislat A, Meller S, Mota R, Buhren BA, Gerber PA, Ruzicka T, et al. Alitretinoin, a dual RAR and RXR agonist, modulates leukocyte recruitment pathways and suppresses dendritic cell functions in vitro and in vivo. Journal of Investigative Dermatology. 2014; Vol. 134:S72.
Kutzner 1986
- Kutzner H, Wurzel RM, Wolff HH. Are acrosyringia involved in the pathogenesis of "dyshidrosis"?. American Journal of Dermatopathology 1986;8(2):109‐16. [PUBMED: 2940934] [DOI] [PubMed] [Google Scholar]
Lodén 2010
- Lodén M, Wirén K, Smerud K, Meland N, Hønnås H, Mørk G, et al. Treatment with a barrier‐strengthening moisturizer prevents relapse of hand‐eczema. An open, randomized, prospective, parallel group study. Acta Dermato‐Venereologica 2010;90(6):602‐6. [PUBMED: 21057743] [DOI] [PubMed] [Google Scholar]
Lodén 2012b
- Lodén M. Effect of moisturizers on epidermal barrier function. Clinics in Dermatology 2012;30(3):286‐96. [DOI] [PubMed] [Google Scholar]
Lynde 2010
- Lynde C, Guenther L, Diepgen TL, Sasseville D, Poulin Y, Gulliver W, et al. Canadian hand dermatitis management guidelines. Journal of Cutaneous Medicine and Surgery 2010;14(6):267‐84. [PUBMED: 21084020] [DOI] [PubMed] [Google Scholar]
Majoie 2009
- Majoie IL, Oldhoff JM, Weelden H, Laaper‐Ertmann M, Bousema MT, Sigurdsson V, et al. Narrowband ultraviolet B and medium‐dose ultraviolet A1 are equally effective in the treatment of moderate to severe atopic dermatitis. Journal of the American Academy of Dermatology 2009;60(1):77‐84. [DOI] [PubMed] [Google Scholar]
McLean 2013
- McLean WH, Irvine AD. Old King coal ‐ molecular mechanisms underlying an ancient treatment for atopic eczema. Journal of Clinical Investigation 2013;123(2):551‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Meding 1989
- Meding B, Swanbeck G. Epidemiology of different types of hand eczema in an industrial city. Acta Dermato‐Venereologica 1989;69(3):227‐33. [PUBMED: 2566226] [PubMed] [Google Scholar]
Meding 1990
- Meding B, Swanbeck G. Predictive factors for hand eczema. Contact Dermatitis 1990;23(3):154‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Meding 2002
- Meding B, Järvholm B. Hand eczema in Swedish adults ‐ Changes in prevalence between 1983 and 1996. Journal of Investigative Dermatology 2002;118(4):719‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Meding 2004
- Meding B, Järvholm B. Incidence of hand eczema ‐ a population‐based retrospective study. Journal of Investigative Dermatology 2004;122(4):873‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Meding 2005
- Meding B, Wrangsjo K, Jarvholm B. Fifteen‐year follow‐up of hand eczema: persistence and consequences. British Journal of Dermatology 2005;152(5):975‐80. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Menné 2000
- Menné T, Maibach HI. Hand Eczema. 2nd Edition. Boca Raton: CRC Press, 2000:7, 158. [084937362X] [Google Scholar]
Menné 2011
- Menné T, Johansen JD, Sommerlund M, Veien NK, Danish Contact Dermatitis Group. Hand eczema guidelines based on the Danish guidelines for the diagnosis and treatment of hand eczema. Contact Dermatitis 2011;65(1):3‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moberg 2009
- Moberg C, Alderling M, Meding B. Hand eczema and quality of life: a population‐based study. British Journal of Dermatology 2009;161(2):397‐403. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomised trials. Lancet 2001;357(9263):1191‐4. [MEDLINE: ] [PubMed] [Google Scholar]
Molin 2015
- Molin S, Merl J, Dietrich KA, Regauer M, Flaig M, Letulé V, et al. The hand eczema proteome: imbalance of epidermal barrier proteins. British Journal of Dermatology 2015;172(4):994‐1001. [PUBMED: 25244099] [DOI] [PubMed] [Google Scholar]
Mollerup 2014
- Mollerup A, Veien NK, Johansen JD. An analysis of gender differences in patients with hand eczema ‐ everyday exposures, severity, and consequences. Contact Dermatitis 2014;71(1):21‐30. [PUBMED: 24618009 ] [DOI] [PubMed] [Google Scholar]
nationaleczema.org
- National Eczema Association. Eczema Treatment. www.nationaleczema.org/eczema/treatment/ (accessed 15 March 2019).
Nilsson 1985
- Nilsson E, Mikaelsson B, Andersson S. Atopy, occupation and domestic work as risk factors for hand eczema in hospital workers. Contact Dermatitis 1985;13(4):216‐23. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Petersen 2014
- Petersen AH, Johansen JD, Hald M. Hand eczema ‐ prognosis and consequences: a 7‐year follow‐up study. British Journal of Dermatology 2014;171(6):1428‐33. [PUBMED: 25156938] [DOI] [PubMed] [Google Scholar]
Politiek 2016
- Politiek K, Oosterhaven JA, Vermeulen KM, Schuttelaar ML. Systematic review of cost‐of‐illness studies in hand eczema. Contact Dermatitis 2016;75(2):67‐76. [PUBMED: 27218305] [DOI] [PubMed] [Google Scholar]
Rawlings 2004
- Rawlings AV, Harding CR. Moisturization and skin barrier function. Dermatologic Therapy 2004;17(Suppl 1):43‐8. [DOI] [PubMed] [Google Scholar]
RevMan [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ryan 2016
- Ryan R, Santesso N, Hill S. Preparing Summary of Findings (SoF) tables. Cochrane Consumers and Communication Group. Version 1.0. cccrg.cochrane.org/author‐resources (accessed 26 July 2017).
Sakuma 2001
- Sakuma S, Higashi Y, Sato N, Sasakawa T, Sengoku T, Ohkubo Y, et al. Tacrolimus suppressed the production of cytokines involved in atopic dermatitis by direct stimulation of human PBMC system (comparison with steroids). International Immunopharmacology 2001;1:1219–26. [DOI] [PubMed] [Google Scholar]
Schleimer 1993
- Schleimer RP. An overview of glucocorticoid anti‐inflammatory actions. European Journal of Clinical Pharmacology 1993;45(Suppl 1):S3‐7; discussion S43‐4. [DOI] [PubMed] [Google Scholar]
Schmitt 2010
- Schmitt J, Williams H, HOME Development Group. HOME Development Group. Harmonising Outcome Measures for Eczema (HOME). Report from the First International Consensus Meeting (HOME 1). British Journal of Dermatology 2010;163(6):1166‐8. [PUBMED: 21137114] [DOI] [PubMed] [Google Scholar]
Schmitt‐Hoffmann 2012
- Schmitt‐Hoffmann AH, Roos B, Schoetzau A, Leese PT, Meyer I, van de Wetering J, et al. Oral alitretinoin: a review of the clinical pharmacokinetics and pharmacodynamics. Expert Reviews in Clinical Pharmacology 2012;5(4):373‐88. [DOI] [PubMed] [Google Scholar]
Schuttelaar 2013
- Schuttelaar ML, Coenraads PJ, Huizinga J, Monchy JG, Vermeulen KM. Increase in vesicular hand eczema after house dust mite inhalation provocation: a double‐blind, placebo‐controlled, cross‐over study. Contact Dermatitis 2013;68(2):76‐85. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Simons 1997
- Simons JR, Bohnen IJ, Valk PG. A left‐right comparison of UVB phototherapy and topical photochemotherapy in bilateral chronic hand dermatitis after 6 weeks' treatment. Clinical and Experimental Dermatology 1997;22(1):7‐10. [PUBMED: 9330044] [DOI] [PubMed] [Google Scholar]
Soost 2012
- Soost S, Abdollahnia M, Kostev K, Worm M. Topical therapy of hand eczema ‐ analysis of the prescription profile from dermatologists in private practice. Journal der Deutschen Dermatologischen Gesellschaft 2012;10(3):180‐4. [PUBMED: 21923729] [DOI] [PubMed] [Google Scholar]
Svensson 1988
- Svensson Å. Hand eczema: an evaluation of the frequency of atopic background and the difference in clinical pattern between patients with and without atopic dermatitis. Acta Dermato‐Venereologica 1988;68(6):509‐13. [MEDLINE: ] [PubMed] [Google Scholar]
Thyssen 2010
- Thyssen JP Johansen JD, Linneberg A, Menné T. The epidemiology of hand eczema in the general population ‐ prevalence and main findings. Contact Dermatitis 2010;62(2):75‐87. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van Coevorden 2004b
- Coevorden AM, Coenraads PJ, Svensson A, Bavinck JN, Diepgen TL, Naldi L, et al. Overview of studies of treatments for hand eczema ‐ the EDEN hand eczema survey. British Journal of Dermatology 2004;151(2):446‐51. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van Coevorden 2006
- Coevorden AM, Sonderen E, Bouma J, Coenraads PJ. Assessment of severity of hand eczema: discrepancies between patient‐ and physician‐rated scores. British Journal of Dermatology 2006;155(6):1217‐22. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van den Bogaard 2013
- Bogaard EH, Bergboer JG, Vonk‐Bergers M, van Vlijmen‐Willems IM, Hato SV, van der Valk PG, et al. Coal tar induces AHR‐dependent skin barrier repair in atopic dermatitis. Journal of Clinical Investigation 2013;123(2):917‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Der Valk 2013
- Valk PG, Gils RF, Boot CR, Evers AW, Donders R, Alkemade HA, et al. A simple tool with which to study the course of chronic hand eczema in clinical practice: a reduced‐item score. Contact Dermatitis 2013;69(2):112‐7. [PUBMED: 23869731 ] [DOI] [PubMed] [Google Scholar]
Veien 2003
- Veien NK, Menne T. Treatment of hand eczema. Skin Therapy Letters 2003;8(5):4‐7. [PUBMED: 12910323] [PubMed] [Google Scholar]
Veien 2008
- Veien NK, Hattel T, Laurberg G. Hand eczema: causes, course and prognosis II. Contact Dermatitis 2008;58(6):335‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Veien 2009
- Veien NK. Acute and recurrent vesicular hand dermatitis. Dermatologic Clinics 2009;27(3):337‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wallenhammar 2004
- Wallenhammar LM, Nyfjäll M, Lindberg M, Meding B. Health related quality of life and hand eczema ‐ a comparison of two instruments, including factor analysis. Journal of Investigative Dermatology 2004;122(6):1381‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Weistenhöfer 2010
- Weistenhöfer W, Baumeister T, Drexler H, Kütting B. An overview of skin scores used for quantifying hand eczema: a critical update according to the criteria of evidence‐based medicine. British Journal of Dermatology 2010;162(2):239‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Yngveson 2000
- Yngveson M, Svensson Å, Johannisson A, Isacsson Å. Hand dermatosis in upper secondary school pupils: 2‐year comparison and follow‐up. British Journal of Dermatology 2000;142(3):485‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
van Coevorden 2009
- Coevorden MA, Williams HC, Svensson ÅA, Diepgen TL, Elsner P, Coenraads P‐J. Interventions for hand eczema. Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD004055] [DOI] [PMC free article] [PubMed] [Google Scholar]