

Abstract
SecA belongs to the large class of ATPases that use the energy of ATP hydrolysis to perform mechanical work resulting in protein translocation across membranes, protein degradation, and unfolding. SecA translocates polypeptides through the SecY membrane channel during protein secretion in bacteria, but how it achieves directed peptide movement is unclear. Here, we use single‐molecule FRET to derive a model that couples ATP hydrolysis‐dependent conformational changes of SecA with protein translocation. Upon ATP binding, the two‐helix finger of SecA moves toward the SecY channel, pushing a segment of the polypeptide into the channel. The finger retracts during ATP hydrolysis, while the clamp domain of SecA tightens around the polypeptide, preserving progress of translocation. The clamp opens after phosphate release and allows passive sliding of the polypeptide chain through the SecA‐SecY complex until the next ATP binding event. This power‐stroke mechanism may be used by other ATPases that move polypeptides.
Keywords: AAA ATPase, protein translocation, SecA, SecY, single‐molecule FRET
Subject Categories: Membrane & Intracellular Transport
Introduction
Many processes in the cell involve AAA family ATPases that perform mechanical work to remodel or relocate proteins. Examples include hexameric ATPases, such as the p97 ATPase (Cdc48 in yeast), which extracts proteins from membranes or tight complexes, the Clp's and the ATPases of the 26S proteasome, which push polypeptides into a proteolytic chamber, and the NSF protein, which disassembles SNARE complexes involved in membrane fusion (for review, see Zhao et al, 2007; Bodnar & Rapoport, 2017; Ye et al, 2017; Yedidi et al, 2017). Another important member of this ATPase family is SecA, which translocates polypeptides through the plasma membrane in bacteria (for review, see Corey et al, 2016; Rapoport et al, 2017; Cranford‐Smith & Huber, 2018). SecA acts a monomer (Or et al, 2005) and uses the energy of ATP hydrolysis to move its substrates through the protein‐conducting SecY channel (Economou & Wickner, 1994). How any of these ATPases perform mechanical work is poorly understood.
SecA is a multi‐domain protein (Fig EV1A and B) with two nucleotide‐binding, RecA‐like domains (NBD1 and NBD2), which bind the nucleotide at their interface and move relative to one another during the ATP hydrolysis cycle (Hunt et al, 2002). A two‐helix finger, consisting of two helices connected by a loop, inserts into the cytoplasmic opening of the SecY channel (Fig EV1A and B; Zimmer & Rapoport, 2009; Li et al, 2016). A conserved Tyr residue within the loop contacts the translocating polypeptide chain (Erlandson et al, 2008a; Bauer et al, 2014), which is positioned above the SecY channel by a clamp formed by rotation of the polypeptide‐crosslinking domain (PPXD) toward NBD2 (Fig EV1B and C).
Figure EV1. Structure of the SecA‐SecY complex.
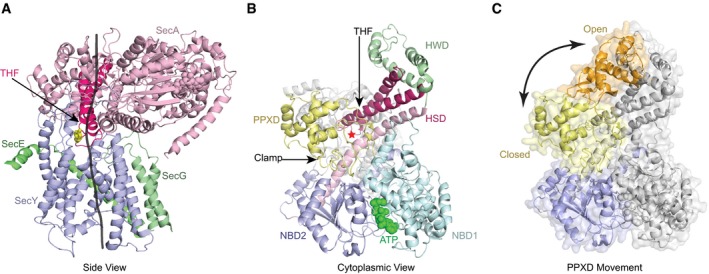
- Side view with a translocating polypeptide (black line) modeled into the crystal structure (PDB 3DIN). SecA is in pink, SecY in blue, and SecG and SecE in green. The lateral gate of the SecY channel is in the front. The two‐helix finger (THF) of SecA is highlighted in red and a conserved Tyr residue at its tip shown in yellow in stick presentation.
- View from the cytosol. SecA's domains are shown in different colors, and ATP is shown in its binding site. The red star indicates the position of the translocating polypeptide.
- Clamp conformational change. The PPXD from the crystal structure of SecA with an open clamp conformation (PDB 1M74) aligned to SecA in the closed conformation. NBD2 is in blue. The closed PPXD is in yellow and the open conformation is in orange. The arrow indicates the PPXD movement.
The SecY channel is formed from three polypeptide chains (SecY, SecE, and SecG). The large SecY subunit consists of N‐ and C‐terminal halves and forms an hourglass‐shaped pore. The cytoplasmic cavity is empty, while the extracellular cavity is filled with a plug domain. At the constriction in the middle of the membrane is a pore ring of amino acids. During translocation, the plug is displaced (Li et al, 2016; Fessl et al, 2018), and the polypeptide chain moves through the pore ring across the membrane.
Several models have been proposed to explain SecA function. In a ratcheting model (Allen et al, 2016; Corey et al, 2019), the finger serves as a sensor for bulky amino acid residues or short α‐helical stretches of the substrate. When such a residue is encountered, SecA converts from the ADP‐bound to the ATP‐bound state and the SecY channel opens, allowing the residue to diffuse through the pore. Following ATP hydrolysis, the channel closes, trapping the bulky residue on the other side of the membrane. In this model, the finger does not move relative to the channel. By contrast, in a power‐stroke model (Bauer et al, 2014), ATP binding at the NBDs would cause the two‐helix finger to interact with the polypeptide chain and push it into the channel; following ATP hydrolysis, the finger would disengage and allow free diffusion of the chain in either direction. Here, the finger would undergo large movements toward and away from the channel. In one extreme version of this model, the “plunging model”, large domains of SecA would reach entirely through membrane to deliver the substrate to the other side (Economou & Wickner, 1994; Banerjee et al, 2017). As proposed, however, the power‐stroke models fail to explain how a SecA domain would return to its starting position without erasing the work done during its power stroke. One possibility is that the clamp holds the polypeptide chain when the two‐helix finger resets (Zimmer et al, 2008), but this model seems to be in contradiction with the observation that the polypeptide chain can slide back and forth through the SecA‐SecY complex (Erlandson et al, 2008b; Bauer et al, 2014). Recent single‐molecule experiments support the idea that the clamp of SecA undergoes nucleotide‐dependent conformational changes (Chada et al, 2018; Ernst et al, 2018; Vandenberk et al, 2018), but it remains unclear whether they occur during translocation, as the studies were performed in the absence of SecY and translocation substrate.
Here, we use single‐molecule Fӧrster resonance energy transfer (FRET) experiments to follow conformational changes of SecA during protein translocation. Single‐molecule experiments are required because the ATP hydrolysis cycles of all SecA molecules cannot be synchronized in traditional biochemical assays. Our results show that, upon ATP binding to SecA, the two‐helix finger undergoes a large conformational change that pushes the polypeptide into the SecY channel. When the finger resets, the clamp tightens around the polypeptide, thus preserving the progress of translocation. Passive sliding of the polypeptide chain occurs after ATP hydrolysis, when the clamp opens. Our results lead to a comprehensive model for SecA function, which may also be applicable to hexameric ATPases.
Results
Experimental design
We used single‐molecule FRET in combination with a reconstituted translocation system (Fig 1A). A translocation intermediate was generated, using purified SecA, SecYEG, and substrate. SecA and SecY were labeled with different fluorophores, and the translocation complex was immobilized on a glass surface via the substrate. This strategy ensured that all components were present in each observed complex. In contrast, if two different dyes are placed into the same protein (Allen et al, 2016; Ernst et al, 2018; Fessl et al, 2018; Vandenberk et al, 2018), one cannot exclude that the unlabeled components are missing and that FRET changes are caused by the dissociation or association of the complex, rather than by conformational changes within the complex. It should be noted that attaching complexes to a glass surface via the SecY channel or lipids yielded very few FRET traces, likely because substrate was absent from many complexes. Also, complexes assembled in vivo required ADP•BeFx during purification, which could not be substituted with other nucleotides in the FRET experiments.
Figure 1. SecA's two‐helix finger makes large movements during the ATP hydrolysis cycle.
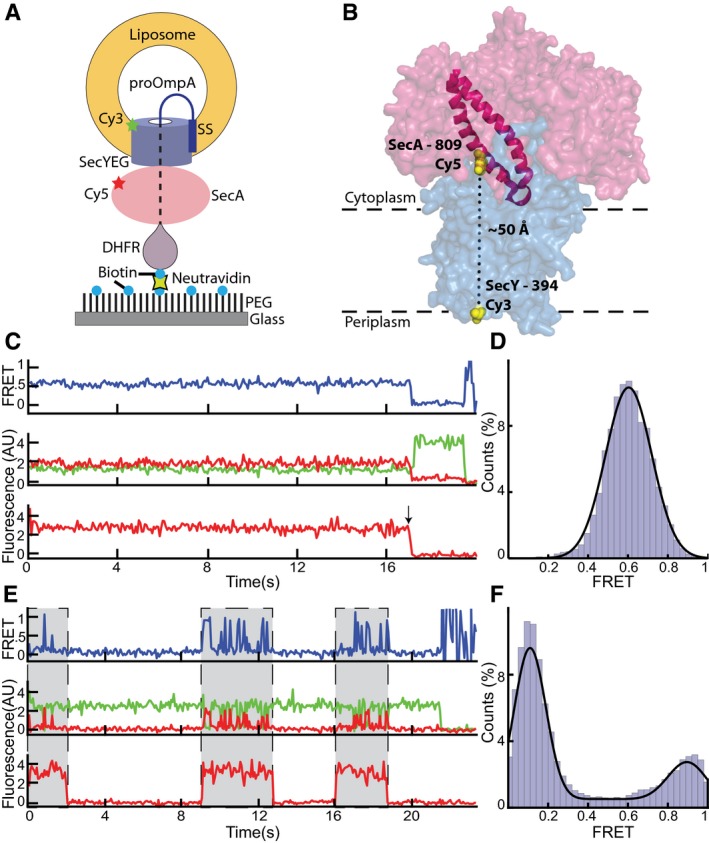
- Experimental setup to measure single‐molecule FRET in translocation complexes immobilized on a surface.
- Cy5 and Cy3 fluorophores were introduced into the two‐helix finger of SecA (PDB 3DIN; red space filling model; helices highlighted) at position 809 and into SecY (blue) at position 394, respectively.
- Representative traces obtained with ADP•BeFx. The upper FRET trace was calculated from the middle traces obtained by exciting the donor fluorophore and measuring both donor (green) and acceptor (red) fluorescence. The lowest trace was obtained by exciting the acceptor fluorophore directly. The arrow indicates a bleaching event.
- Distribution of FRET values determined from 97 traces as in (C) fit with a Gaussian model (black curve).
- As in (C), but in the presence of ATP. Periods in which a fluorescently labeled SecA molecule is bound are indicated by gray shading.
- As in (D), but with ATP (257 traces).
Source data are available online for this figure.
In our specific experimental setup, we introduced single cysteines at different positions into cysteine‐lacking Escherichia coli SecA and labeled them with the acceptor fluorophore Cyanine 5 (Cy5). The donor fluorophore (Cy3) was attached to a single cysteine introduced at different positions into cysteine‐free E. coli SecY. All SecA and SecY mutants retained translocation and ATPase activity after labeling (Appendix Fig S1 and S2). Proteoliposomes were then reconstituted with labeled SecYEG complex and mixed with labeled SecA, ATP, and substrate. The substrate consisted of a fusion of the first 175 amino acids of proOmpA, including the N‐terminal signal sequence (SS), a dihydrofolate reductase (DHFR) domain, and a biotinylation tag. The proteoliposomes were then attached to a coverslip by neutravidin, which interacted with both the biotinylated C‐terminus of the substrate and biotinylated polyethylene glycol (PEG) molecules at the surface.
In the presence of methotrexate, the DHFR domain of the substrate is tightly folded and too large to move through the SecA‐SecY complex, therefore preventing complete translocation of the fusion protein (Bauer & Rapoport, 2009). Essentially, all channels were occupied with translocation intermediate (Appendix Fig S3). In the presence of ATP, the substrate is constantly sliding out of the proteoliposomes and is then pushed back into the SecY channel (Bauer et al, 2014). Thus, despite the fact that, on average, the DHFR domain is abutting the channel, the polypeptide chain is undergoing continuous translocation.
FRET was monitored in a flow chamber with wide field total internal reflection fluorescence (TIRF) microscopy. As expected from our setup, fluorescent spots were only detected on the surface in the presence of all components (Appendix Fig S4). Alternating excitation of Cy3 and Cy5 allowed for measurement of both FRET between SecY and SecA, as well as direct detection of SecA. Our experimental design ensured that both partners are present and allowed single SecA molecules to be monitored through many hydrolysis cycles over a period as long as 30 s, i.e., observation times far longer than those allowed by solution FRET experiments. Although the time resolution was limited to 33 ms per frame, this is about 20 times faster than the duration of an ATP hydrolysis cycle measured in bulk (Appendix Fig S2).
FRET traces were either obtained in the presence of ATP or the nucleotide was exchanged in the flow chamber to either ADP•BeFx, which mimicks the transition state of ATP hydrolysis, or ATPγS, a slowly hydrolyzing ATP analog. Complexes could not be imaged in the presence of ADP alone, as SecA binds only weakly to SecY in the presence of this nucleotide (Bauer et al, 2014). While the fluorescent spots were stable with nucleotide analogs, they rapidly disappeared in the presence of ATP, likely because SecA dissociates in its ADP‐bound state, allowing the substrate to slide backwards in the SecY pore until the entire proteoliposome is released from the glass surface. We therefore added unlabeled SecA when imaging with ATP, keeping the total concentration below the Kd of SecA dimerization (Woodbury et al, 2002) to ensure that only active SecA monomers are observed. The increased concentration allowed SecA to rebind abandoned complexes before they dissociated. Although most rebinding SecA molecules were unlabeled, some were labeled and allowed the observation of FRET over extended time periods.
Movement of the two‐helix finger of SecA during protein translocation
We first analyzed movements of the two‐helix finger of SecA. To this end, the donor fluorophore was placed into a periplasmic loop of E. coli SecY (position 394) and the acceptor fluorophore into position 809 of the two‐helix finger (Fig 1B). The probes are predicted to be about 50 Å apart according to crystal structures obtained in the presence of the transition state analog ADP•BeFx (Zimmer et al, 2008; Li et al, 2016). Consistent with the observation that ADP•BeFx allows stable binding of SecA to the SecY channel, a static FRET signal between SecA and SecY was observed in all traces (Fig 1C; top trace). Direct excitation of the SecA‐bound fluorophore showed that SecA remained bound to the channel (bottom trace). At the end of a trace, the acceptor fluorescence bleached in one step, and the donor fluorescence was de‐quenched (see arrow), as expected for a FRET signal. In no case did acceptor fluorescence return, confirming that, in the presence of ADP•BeFx, SecY‐bound SecA is not exchanged with SecA in bulk solution. The analysis of many traces showed that the FRET ratios had a Gaussian distribution with a mean value of 0.60 ± 0.12 (Fig 1D). While distance estimates based on FRET probes in a proteinaceous environment are unreliable due to orientation restrictions of the fluorophores, a naïve estimate using the standard FRET equation gives a distance of 51 Å, in close agreement with the structural prediction.
In the presence of ATP, SecA repeatedly bound and dissociated from the SecY channel, as demonstrated by direct excitation of the acceptor fluorophore (Fig 1E; bottom trace). While bleaching and dissociation cannot be distinguished a priori in individual traces, the imaging lifetime of individual SecA molecules in the presence of ATP was generally shorter than in the presence of ADP•BeFx (Appendix Fig S5), suggesting that SecA does indeed dissociate in these traces. The FRET signal was highly dynamic when SecA was bound (top trace), alternating between high and low states with mean FRET ratios of 0.90 ± 0.09 and 0.11 ± 0.08 (Fig 1F; additional examples of traces are shown in Appendix Fig S6). The high and low FRET states likely correspond to states in which the two‐helix finger is either inserted into or withdrawn from the SecY channel. The large FRET difference indicates that the finger undergoes a substantial conformational change, although its precise movement cannot be deduced from the FRET values. The low FRET state shows a significantly higher occupancy than the high FRET state (Fig 1F). When the donor fluorophore was placed at a different position in SecY (position 103), a markedly similar behavior was observed (Fig EV2A–C). Again, a constant FRET level was observed in the presence of ADP•BeFx, which matched well the estimated inter‐fluorophore distance derived from the crystal structures. As before, in the presence of ATP, the FRET traces were dynamic during SecA‐bound periods (Fig EV2D). Histograms derived from these traces also showed two populations at low and high FRET levels, with a higher occupancy in the low FRET state (Fig EV2E).
Figure EV2. SecA's two‐helix finger movements observed by alternative fluorophore positions.
- Cy5 and Cy3 fluorophores were introduced into the two‐helix finger of SecA (PDB 3dIN; red space filling model; helices highlighted) at position 809 and into SecY (blue) at position 103, respectively.
- Representative traces obtained with ADP•BeFx. The upper FRET trace was calculated from the middle traces obtained by exciting the donor fluorophore and measuring both donor (green) and acceptor (red) fluorescence. The lowest trace was obtained by exciting the acceptor fluorophore directly. The arrow indicates a bleaching event.
- Distribution of FRET values determined from 106 traces as in (B), fit with a Gaussian model (black curve).
- As in (B), but in the presence of ATP. Periods in which a fluorescently labeled SecA molecule is bound are indicated by gray shading.
- As in (C), but with ATP (200 traces).
- Traces obtained in the presence of different nucleotides were used to determine the number of states best fit by the Markov model.
- Transition density plot of idealized ATP FRET states obtained in (F).
- The distribution of dwell times of the low FRET states observed in ATP was fit with exponentials (1,172 low FRET states). The inset shows average dwell time and error, defined as the standard error based on the number of traces.
- As in (H), but with high FRET (1,349 high FRET states).
Source data are available online for this figure.
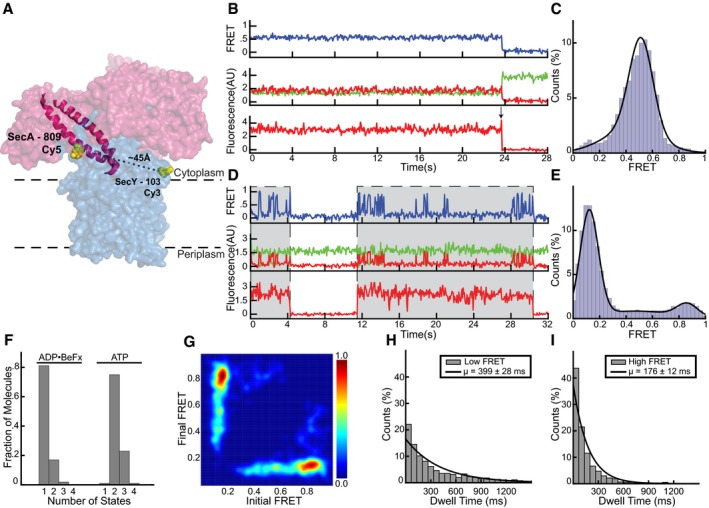
To connect these FRET changes to the ATP hydrolysis cycle of SecA, we fit the FRET traces with a hidden Markov model (McKinney et al, 2006; Bronson et al, 2009; van de Meent et al, 2014; Fig 2A). These models employ a maximum evidence approach to find the most likely number of structural conformations that underlie the observed data. Individual traces were fit with an increasing complexity of models, which were scored positively for the closeness of their fit to the data and negatively for the number of discrete FRET states included. In this way, the most parsimonious model was selected that reproduces the data without evoking extraneous conformations (Bronson et al, 2009). This analysis confirmed that in the presence of ADP•BeFx only one conformational state exists, while in the presence of ATP, and with both donor positions, the majority of traces showed two states (Figs 2B and EV2F). Transitions between these idealized FRET states can also be plotted as transition density plots (TDPs) to show how these FRET states connect to each other (McKinney et al, 2006). Transition density plots of idealized FRET states obtained in the presence of ATP showed symmetry across the principal diagonal, indicating cycling between only two FRET states (Figs 2C and EV2G). Thus, the high and low FRET states simply interchange with each other. The distribution of dwell times for the two FRET states observed with ATP could each be fit with a single exponential and demonstrated that the mean lifetime for the low FRET state is about twice as long as that of the high FRET state (Figs 2D and E, and EV2H and I). The low and high FRET states likely correspond to ADP‐ and ATP‐bound states, respectively, as previous experiments showed that SecA spends most of its time during the ATP hydrolysis cycle in the ADP‐bound state (Robson et al, 2009). This assumption is consistent with the relatively high intermediate FRET signal observed with the transition state mimic ADP•BeFx (Fig 1C and D). Furthermore, the sum of the high and low FRET lifetimes gives an estimate of the overall ATP hydrolysis rate that agrees with bulk measurements performed at the same temperature (Appendix Fig S2). Finally, FRET experiments with ATPγS, a slowly hydrolyzing analog, also showed two conformational states (Fig 2B), but the high FRET state now lasted as long as the low FRET state (Fig 2F and G). Interestingly, the high FRET value was close to that measured in the presence of ADP•BeFx (0.67 ± 0.11 vs. 0.6 ± 0.11), suggesting that ATPγS extends the duration of the transition state of ATP hydrolysis.
Figure 2. The two‐helix finger switches between two states.
- A representative FRET trace (blue line) was obtained as in Fig 1 and fit with a hidden Markov model (black dashed line).
- Traces as in (A) obtained in the presence of different nucleotides were used to determine the number of states best fit by the Markov model.
- Transition density plot of idealized ATP FRET states obtained in (B).
- The distributions of dwell times of the low FRET states observed in ATP were fit with a single exponential (1,500 low FRET states). The inset shows average dwell time and error, defined as the standard error based on the number of traces.
- As in (D), but with high FRET (1,656 high FRET states).
- Representative traces obtained with ATPγS. The upper FRET trace was calculated from the middle traces obtained by exciting the donor fluorophore and measuring both donor (green) and acceptor (red) fluorescence. The lowest trace was obtained by exciting the acceptor fluorophore directly. The arrow indicates a bleaching event.
- Distribution of FRET values determined from 168 traces as in (D) fit with a Gaussian model (black curve).
Source data are available online for this figure.
Given that the average FRET efficiency observed for the two‐helix finger is different in the transition state of ATP hydrolysis (ADP•BeFx) and the ADP‐bound state (0.6 vs. 0.1), we asked whether the transition between them happens before or after Pi release. We therefore measured FRET in the presence of ADP and Pi, as well as ADP and vanadate (Vi), a phosphate analog that binds more stably. In both conditions, the two‐helix finger was primarily in the low FRET state (Fig EV3A–D), indicating that it withdraws before Pi release. The two‐helix finger was more dynamic in the presence of ADP•Pi and ADP•Vi than with ADP•BeFx, either because of increased conformational flexibility or frequent dissociation of Pi/Vi.
Figure EV3. Representative FRET data for SecA conformations before Pi release.
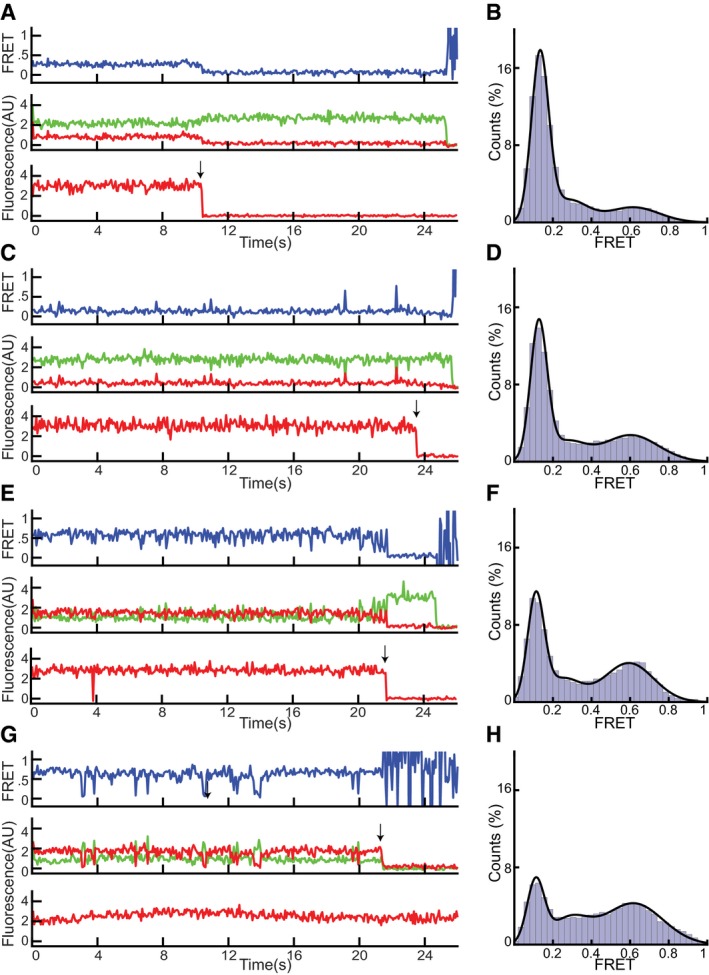
- Representative traces for the two‐helix finger (positions 809 in SecA and 394 in SecY) obtained with ADP•Pi. The upper FRET trace was calculated from the middle traces obtained by exciting the donor fluorophore and measuring both donor (green) and acceptor (red) fluorescence. The lowest trace was obtained by exciting the acceptor fluorophore directly. The arrow indicates a bleaching event.
- Distribution of FRET values determined from 227 traces as in (A) fit with a Gaussian model (black curve).
- As in (A), but with ADP•Vi.
- As in (B), but with ADP•Vi (202 traces).
- As in (A), but for the clamp (positions 233 in SecA and 103 in SecY).
- As in (B), but for the clamp (274 traces).
- As in (A), but for the clamp with ADP•Vi.
- As in (B), but for the clamp with ADP•Vi (157 traces).
Source data are available online for this figure.
Taken together, these results show that, during protein translocation, the two‐helix finger of SecA undergoes a large conformational change. It alternates between two conformations during ATP hydrolysis: In the short‐lived ATP‐bound state, the finger inserts deeply into the SecY channel and gives a high FRET signal, and in the longer ADP‐bound state, it withdraws from the pore and produces low FRET. In the transition state, mimicked by ADP•BeFx, the finger is in an intermediate position, but it retracts completely following completion of ATP hydrolysis, before Pi release. Movement of the two‐helix finger into the channel would push the polypeptide forward, and movement away would reset the finger for the next cycle.
Movement of the clamp of SecA
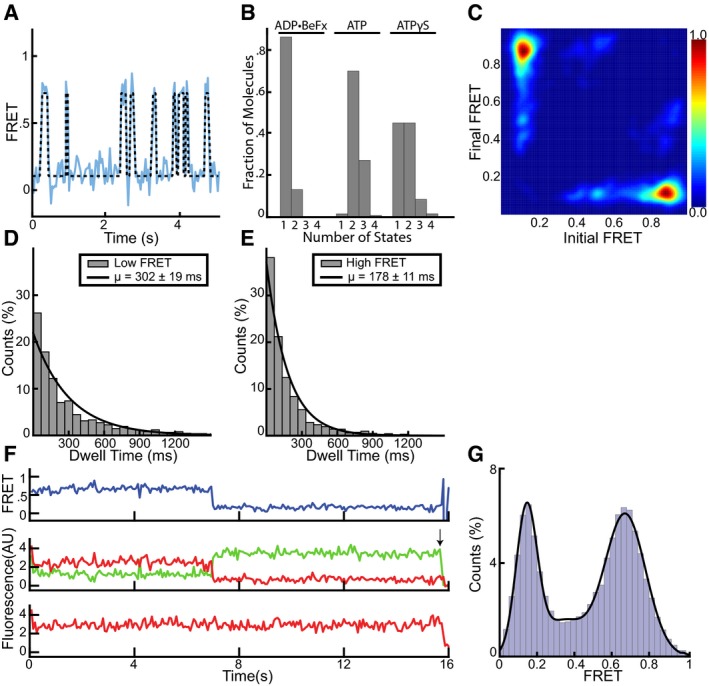
The observation of only two states of the two‐helix finger during ATP hydrolysis requires a mechanism that prevents the finger from dragging the polypeptide backwards when the finger moves away from the channel. A likely candidate for holding the polypeptide during finger resetting is the clamp, a groove formed by the rotation of the PPXD toward NBD2 (Fig EV1B and C; Zimmer et al, 2008). Rotation of the PPXD can be inferred from crystal structures of soluble SecA that show this domain at different distances from NBD2 (Hunt et al, 2002; Osborne et al, 2004; Chen et al, 2015), and movement of the translocating polypeptide chain through the clamp is indicated by crosslinking experiments (Bauer & Rapoport, 2009). However, it remained unclear whether the clamp simply forms a conduit for the translocating polypeptide chain or cyclically binds and releases it during ATP hydrolysis. To test whether the clamp undergoes nucleotide‐dependent movements, we placed the acceptor fluorophore into the PPXD (position 233) and the donor fluorophore at position 103 in the N‐terminal half of SecY (Fig 3A and B). In the presence of ADP•BeFx, a static FRET signal of 0.60 ± 0.08 was observed (Fig 3C and D). Again, the distance estimated with the standard FRET equation agreed well with those measured in crystal structures (Zimmer et al, 2008; Li et al, 2016). In the presence of ATP, we once again observed exchange of SecA molecules on the SecY channel, and changes between two conformations when SecA was bound to the channel (Figs 3E and F, and 4A and B; additional examples of traces are shown in Appendix Fig S7). Similar results were obtained when the donor fluorophore was moved to position 336 in the C‐terminal half of SecY (Fig EV4A–H), demonstrating that FRET changes are due to conformational changes of the SecA clamp, rather than the channel. Experiments with the slowly hydrolyzing ATP analog ATPγS showed an increase in the occupancy of the high FRET state (Fig 4C and D), consistent with the clamp movements being linked to ATP hydrolysis. The predominance of the high FRET state in ATPγS indicates that clamp is closed during ATP hydrolysis, though it is unclear whether the initial closure occurs during ATP binding or hydrolysis.
Figure 3. SecA's clamp opens and closes during the ATPase cycle.
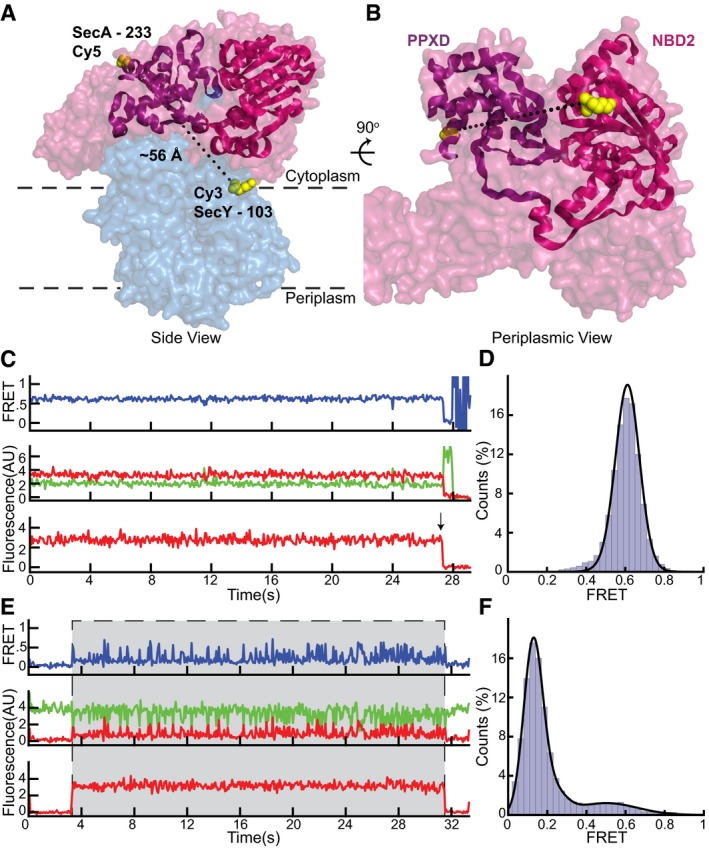
- Cy5 and Cy3 fluorophores were introduced into the clamp of SecA (PDB 3DIN; red space filling model) at position 233 and into the N‐terminal half of SecY (blue) at position 103, respectively. The PPXD and NBD2 making up the clamp are shown as a violet and magenta ribbon models, respectively.
- Rotated view of (A) with SecY masked except for the labeled residue 103.
- Representative traces obtained with ADP•BeFx. The upper FRET trace was calculated from the middle traces obtained by exciting the donor fluorophore and measuring both donor (green) and acceptor (red) fluorescence. The lowest trace was obtained by exciting the acceptor fluorophore directly. The arrow indicates a bleaching event.
- Distribution of FRET values determined from 197 traces as in (B) fit with a Gaussian model (black curve).
- As in (C), but with ATP. Periods in which a fluorescently labeled SecA molecule is bound are indicated by gray shading.
- As in (D), but with ATP (228 traces).
Source data are available online for this figure.
Figure 4. Clamp dynamics and comparison with the two‐helix finger.
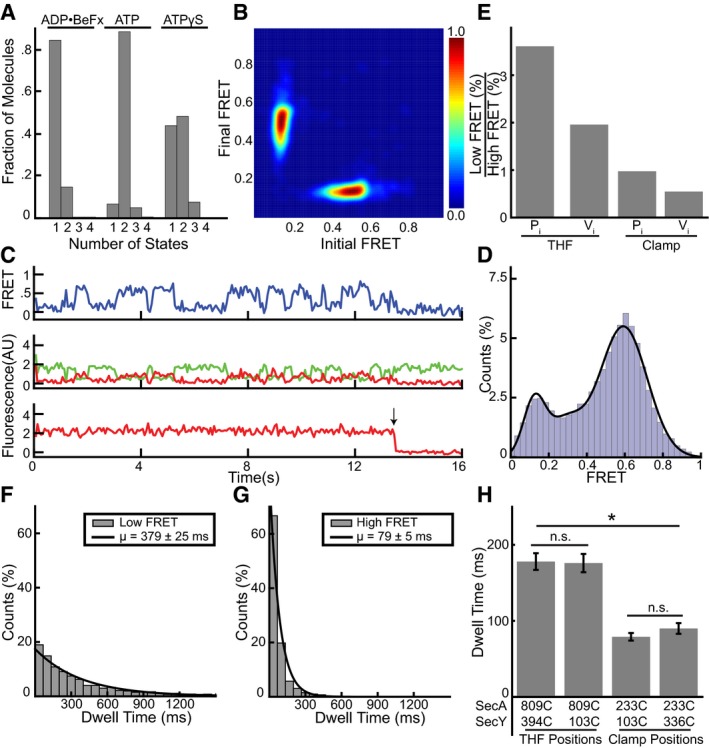
- FRET traces of clamp movements in the presence of different nucleotides were used to determine the number of states best fit by a Markov model.
- Transition density plot of idealized ATP FRET states obtained in (A).
- Representative traces obtained with ATPγS. The upper FRET trace was calculated from the middle traces obtained by exciting the donor fluorophore and measuring both donor (green) and acceptor (red) fluorescence. The lowest trace was obtained by exciting the acceptor fluorophore directly. The arrow indicates a bleaching event.
- Distribution of FRET values determined from 315 traces as in (C) fit with a Gaussian model (black curve).
- Comparison of high and low FRET state occupancy in ADP•Pi and ADP•Vi for the clamp and THF.
- The distributions of dwell times of the low FRET states observed in ATP were fit with a single exponential (1,539 low FRET states). The inset shows average dwell time and error, defined as the standard error based on the number of traces.
- As in (F), but with high FRET (1,773 high FRET states).
- Comparison of dwell times of the high FRET states for the two‐helix finger (THF) and clamp for different fluorophore positions. Errors as in (G) with significance based on two‐sample Kolmogorov–Smirnov tests with a 1% threshold. n.s. P = 0.012 (left), n.s. P = 0.681 (right); *P < 1*10−35 (between each pair of THF/clamp mutants). 809C/394C: 1,656 high FRET states; 809C/103C: 1,349 states; 233C/103C: 1,773 states; 233C/336C: 1,778 states.
Source data are available online for this figure.
Figure EV4. Clamp movements observed by alternative fluorophore positions.
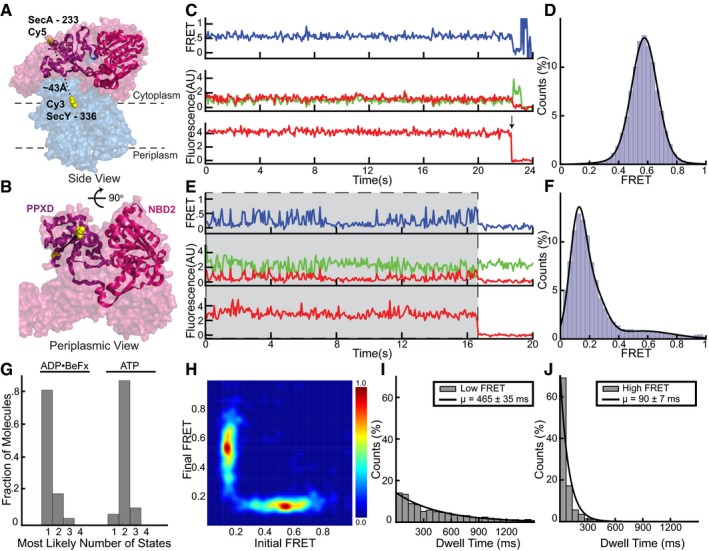
- Cy5 and Cy3 fluorophores were introduced into the clamp of SecA (PDB 3DIN; red space filling model) at position 233 and into the C‐terminal half of SecY (blue) at position 336, respectively. The PPXD and NBD2 making up the clamp are shown as a violet and magenta ribbon models, respectively.
- Rotated view of (A) with SecY masked except for the labeled residue 336.
- Representative traces obtained with ADP•BeFx. The upper FRET trace was calculated from the middle traces obtained by exciting the donor fluorophore and measuring both donor (green) and acceptor (red) fluorescence. The lowest trace was obtained by exciting the acceptor fluorophore directly. The arrow indicates a bleaching event.
- Distribution of FRET values determined from 163 traces as in (B), fit with a Gaussian model (black curve).
- As in (C), but in the presence of ATP. Periods in which a fluorescently labeled SecA molecule is bound are indicated by gray shading.
- As in (D), but with ATP (178 traces).
- Traces obtained in the presence of different nucleotides were used to determine the number of states best fit by the Markov model.
- Transition density plot of idealized ATP FRET states obtained in (F).
- The distribution of dwell times of the low FRET states observed in ATP was fit with an exponential (1,550 low FRET states). The inset shows average dwell time and error, defined as the standard error based on the number of traces.
- As in (I), but with high FRET (1,778 high FRET states).
Source data are available online for this figure.
Interestingly, whereas the two‐helix finger adopted different conformations in the ATP‐bound state and in the transition state of ATP hydrolysis, the clamp did not change much, as the high FRET signal for the clamp was similar in ATP and ADP•BeFx (0.49 ± 0.09 and 0.60 ± 0.08). Data taken in the presence of ADP and either Pi or Vi show that the clamp remains closed even when ATP hydrolysis is completed, as there was a clear bias toward the higher FRET state in both conditions (Fig EV3E–H). The clamp remains closed while the two‐helix finger undergoes a transition to the low FRET state, as the ratio of low to high FRET occupancy was significantly higher for the finger than for the clamp, both with ADP•Pi and with ADP•Vi (Fig 4E). Thus, it seems that the two‐helix finger starts moving away from the channel during ATP hydrolysis, while the clamp remains closed and only opens after Pi is released.
Support for this model comes from comparing the kinetics of the conformational changes of the clamp and two‐helix finger. The sums of the lifetimes of the high and low FRET states observed for each domain were about the same (~500 ms), suggesting that they are measurements of the same hydrolysis cycle. However, the division of this cycle between the high and low FRET states was different. The high FRET state of the clamp lasted about one‐fifth as long as the low FRET state (Figs 4F and G, and EV4I and J) and only half as long as the high FRET state of the two‐helix finger (Fig 4H), supporting the idea that the two‐helix finger and clamp move during different stages of the ATP hydrolysis cycle (Fig 5A). Our data not only suggest that the clamp remains closed when the finger starts moving away from the channel, but also that the clamp does not close until the finger has moved all the way into the channel. The latter conclusion is based on the fact that the clamp has a shorter high FRET lifetime than the two‐helix finger and that it transitions to the low FRET state later, so that the period the clamp spends in the high FRET state is shifted relative to that of the two‐helix finger (Fig 5A). Taken together, these data lead to a model in which the clamp is open during the power stroke of the two‐helix finger, but then closes to hold the polypeptide chain while the finger resets. However, the exact point of clamp closure remains uncertain; the data cannot discern between closure during ATP binding or hydrolysis.
Figure 5. Power‐stroke model for polypeptide translocation by SecA.
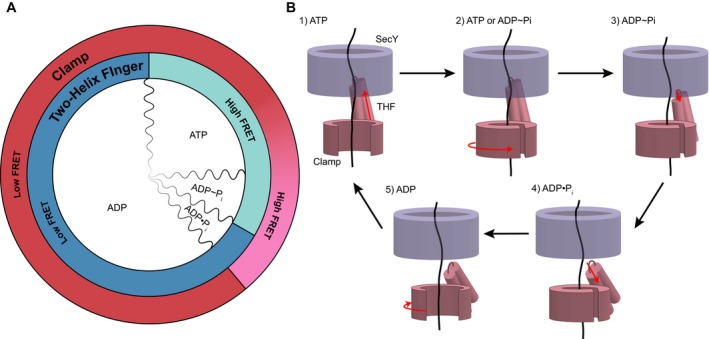
- Alignment of observed FRET states with the ATP hydrolysis cycle. Two‐helix FRET states are depicted in shades of blue and clamp states in shades of red. The red gradient from low to high FRET indicates uncertainty as to the precise point of clamp closure. The subdivisions of ATP hydrolysis (wavy lines) are meant to indicate qualitative, rather than quantitative, durations of the hydrolysis steps. ADP˜Pi indicates the transition state of ATP hydrolysis.
- SecY is shown in blue and SecA domains in pink (THF, two‐helix finger). Domain movements are indicated by red arrows. Clamp closure occurs either during ATP binding or hydrolysis. See text for details.
Discussion
Our results lead to a new model for how the SecA ATPase moves polypeptides into the SecY channel. The model combines features of the previously proposed “push‐and‐slide” mechanism (Bauer et al, 2014) with an essential role for the clamp to hold the polypeptide chain during resetting of the finger. Without this additional mechanism, the bidirectional movement of the two‐helix finger toward and away from the channel would result in no net translocation, as the finger would drag the polypeptide with it when it retracts. Thus, clamp tightening is essential to preserve progress of the substrate when the finger moves away from the channel.
Our single‐molecule FRET results, together with previous bulk solution experiments (Robson et al, 2009; Bauer et al, 2014), lead to a model for the coupling of ATP hydrolysis by SecA with polypeptide translocation (Fig 5B). When ATP binds to SecA, the two‐helix finger inserts deeply into the SecY channel, pushing the polypeptide substrate toward the extracellular side of the membrane (stage 1). Next, the clamp closes around the polypeptide chain, with SecA's PPXD contacting the NBD2 through the conserved C‐loop segment (Zimmer et al, 2008; Chen et al, 2015). Contact between the C‐loop and NBD2 could trigger ATP hydrolysis, i.e., clamp closure would occur before ATP hydrolysis (stage 2). Alternatively, the clamp could close only during ATP hydrolysis. In the transition state of ATP hydrolysis (stage 3), the two‐helix finger has started to retract, while the clamp is closed. After ATP hydrolysis, but before Pi release, the two‐helix finger has retracted all the way, while the clamp remains closed (stage 4). This allows the two‐helix finger to reset without dragging the polypeptide chain backwards. After Pi release, the clamp reopens, allowing the passive sliding of the polypeptide in either direction (stage 5). Our model is based on independent observations of the movements of the two‐helix finger and clamp, aligned by using complexes trapped in the transition state of ATP hydrolysis (ADP•BeFx) or after ATP hydrolysis, but before Pi release (ADP•Pi or ADP•Vi). Linking the FRET changes of the two domains was also facilitated by measuring the kinetics during ATP hydrolysis and by performing experiments in ATPγS, which biases the system toward an ATP‐bound or ATP hydrolysis intermediate state. However, future experiments with three‐color FRET will be required to simultaneously follow the movements of both domains.
Passive sliding of the polypeptide chain remains a major part of the translocation mechanism (Erlandson et al, 2008b; Bauer et al, 2014), as SecA spends most of its time in the ADP‐bound state (stage 5), in which the two‐helix finger is disengaged and the clamp is open. Indeed, ADP release is rate‐limiting in the ATP hydrolysis cycle and the apo‐state is exceedingly transient (Robson et al, 2009). Furthermore, in the ATP‐bound state, the two‐helix finger does not interact strongly with all amino acids encountered (Bauer et al, 2014), so that a power stroke does not always result in active pushing. But, passive sliding in the ADP‐bound state allows translocation of any polypeptide segment encountered by SecA. Although we employed a stalled translocation intermediate in our study, the polypeptide chain is continuously sliding backwards and must be re‐inserted by SecA into the SecY channel, thus mimicking real translocation, even if the C‐terminus of the polypeptide does not enter the channel. Backsliding in vivo might be reduced by the membrane potential across the inner membrane, folding of the polypeptide in the periplasm, or by binding of the polypeptide to periplasmic proteins.
Our results argue against a proposed ratcheting model, in which the two‐helix finger makes only small movements relative to the channel and the polypeptide chain is free to slide in the ATP‐bound state (Allen et al, 2016; Corey et al, 2019). The FRET data indicate that the finger makes in fact very large movements, alternating between a withdrawn conformation and one in which it inserts into the channel. The large movements of the two‐helix finger must originate from much smaller conformational changes at the interface between the NBDs, which are propagated and amplified through a long linker helix (Hunt et al, 2002). Whether the two‐helix finger, or any other SecA domain, moves all the way through the membrane, as previously proposed and supported by recent crosslinking experiments (Economou & Wickner, 1994; Banerjee et al, 2017) requires further investigation, as it is difficult to see from the available crystal structures how the finger could pass the SecY pore constriction. Our in vitro experiments did not include SecDFYajC, components that facilitate protein secretion in vivo (Pogliano & Beckwith, 1993). Although these components are not essential, they might modify the movements of SecA during translocation (Economou et al, 1995). Surprisingly, the two‐helix finger of SecA can be crosslinked to a cytosolic loop in SecY without abolishing translocation activity (Whitehouse et al, 2012). However, this loop is the longest and most flexible on the cytosolic face of SecY and might therefore not arrest the finger. Crosslinking to a more rigid position in SecY's cytosolic cavity does in fact eliminate translocation activity (Whitehouse et al, 2012).
Hexameric ATPases that move polypeptides, such as the 19S subunit of the proteasome, the Cdc48 ATPase, and the Clp proteins, may use a similar mechanism as SecA. In this case, each of the six subunits has a loop analogous to the two‐helix finger of SecA, which pushes the polypeptide chain through the central pore (Hinnerwisch et al, 2005; Martin et al, 2008; Han et al, 2017; Puchades et al, 2017; Ho et al, 2018). Because it is difficult to separate the movements of the six loops during the ATP hydrolysis cycles, even with single‐molecule experiments (Aubin‐Tam et al, 2011; Sen et al, 2013; Olivares et al, 2014), monomeric SecA provides a unique, tractable model to determine the mechanism by which ATPases move polypeptides.
In fact, recent structures of the Plasmodium translocon of exported proteins (PTEX) suggest an analogous model for the Hsp101 ATPase (Ho et al, 2018). Here, a set of three pore loops undergo similar movements as the two‐helix finger of SecA during the ATPase cycle, pushing the polypeptide chain forward. When the loops release the substrate, other loops assume the role of SecA's clamp, holding the polypeptide in place and thus preventing its backward movement. It is therefore possible that the proposed SecA mechanism is generally employed by polypeptide‐moving ATPases.
Materials and Methods
Protein expression and purification
SecA and SecY were expressed and purified as previously described (Bauer et al, 2014). Cysteine‐free SecA N95 (lacking the non‐essential C‐terminus; Matsuyama et al, 1990) with a C‐terminal His‐6 tag and 3C protease cleavage site was cloned into a pET30b (EMD Millipore, Burlington, Massachusetts) vector and expressed in BL21(DE3) E. coli (New England Biolabs, Ipswich, Massachusetts) for 4 h at 37°C after induction at OD600 0.8 with 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG). Cells were collected by centrifugation for 10 min at 4,000 g, resuspended in buffer A (50 mM HEPES/KOH pH 7.5, 300 mM NaCl, 15 mM imidazole, 20 mM b‐mercaptoethanol [BME]) and lysed by two passes through an EmulsiFlex‐C3 (Avestin, Ottawa, Canada) at 20,000 psi. Soluble components were separated from the membrane fraction by centrifugation at 110,000 g for 45 min. The supernatant was bound to 2 ml Ni2+ resin, washed with 50 ml buffer B (50 mM HEPES/KOH pH 7.5, 100 mM NaCl), and incubated overnight in 5 ml buffer B at 4°C with 5 μM 3C protease. The flow‐through was then collected and subjected to anion exchange chromatography (HiTrap Q FF, GE Healthcare Life Sciences, Marlborough, Massachusetts) followed by size exclusion chromatography (Superdex 200 10/300GL, GE Healthcare Life Sciences) in buffer C (50 mM HEPES/KOH pH 7.5, 50 mM KCl).
The three E. coli SecYEG protein components, with an N‐terminal His‐6 tag on SecE, were cloned into a pBAD22 vector (ATCC, Manassas, Virginia) under a single L‐arabinose‐inducible promoter. The cells were grown to OD600 0.6 and induced for 4 h at 37°C by addition of 10 ml 20% L‐arabinose. The cells were collected, lysed, and fractionated in the same manner as for SecA. The membrane fraction was solubilized for 90 min in buffer D (buffer A, 10% glycerol) with 1% n‐dodecyl β‐D‐maltoside (DDM, Anatrace Inc., Maumee, Ohio). The extract was subjected to high‐speed centrifugation at 110,000 g for 45 min. Subsequent steps were carried out in buffers containing 0.03% DDM. The protein was bound to 1 ml Ni2+ resin in buffer D, washed with 50 ml buffer D, and eluted in 5 ml buffer E (buffer B, 10% glycerol) with 250 mM imidazole. The eluate was then subjected to cation exchange chromatography (HiTrap SP FF, GE Healthcare Life Sciences) and size exclusion chromatography (Superdex 200 10/300, GE Healthcare Life Sciences) in buffer F (buffer C, 10% glycerol).
proOmpA(1‐175)‐DHFR‐Avitag (proOmpA‐DHFR) was cloned into a pET30b vector and co‐expressed with the biotin ligase BirA encoded on the pBirAcm plasmid (Avidity, LLC., Aurora, Colorado). Both vectors were transformed into BL21(DE3) E. coli and the cells grown to OD600 0.6. Expression was induced by the addition of 1 mM IPTG and the media was further supplemented with 20 μM D‐biotin. After 2 h at 37°C, cells were collected by centrifugation and lysed by two passes through an EmulisFlex‐C3 at 20,000 psi in Buffer A. The lysate was centrifuged for 45 min at 110,000 g, and the insoluble pellet containing proOmpA was collected. The pellet was incubated in buffer A with 6 M urea for 1 h at 23°C and then centrifuged again at 110,000 g for 45 min. The supernatant was collected and diluted with buffer A to 2 M urea before being mixed with 1 ml High Capacity Streptavidin Agarose (Thermo Fisher Scientific, Waltham, Massachusetts) for 1 h at 4°C. The beads were then washed with 50 ml buffer C with 2 M urea before the protein was eluted by addition of 10 ml buffer C with 6 M urea.
Protein labeling
Both SecA and SecY were labeled using the same protocol. 500 μl of 10 μM purified protein was incubated with 40 μM of tris(2‐carboxyethyl)phosphine (TCEP) for 20 min on ice. 100 μM maleimide‐conjugated Cyanine 3 (Cy3) or Cyanine 5 (Cy5) (Lumiprobe, Hunt Valley, Maryland) was added to the SecY and SecA, respectively, from 10 mM stock in dimethyl sulfoxide (DMSO) and rotated overnight at 4°C. Labeling was quenched by the addition of 10 mM 1,4‐dithiothreitol (DTT). Dye excess was then removed via gel filtration through a 30 cm column packed with Superfine G‐50 Sephadex (GE Healthcare Life Sciences) equilibrated in buffer C for SecA or buffer F with 0.03% DDM for SecY. The first visible dye peak was collected and further purified by size exclusion chromatography (Superdex 200 10/300, GE Healthcare Life Sciences). Labeling efficiencies were generally around 80% for SecA mutants and 60% for SecY mutants.
Liposome preparation and membrane protein reconstitution
Liposomes were prepared from E. coli Polar Lipid Extract (Avanti Polar Lipids, Alabaster, Alabama). 2 mg of lipids from 25 mg/ml chloroform stock was dried under nitrogen stream, resuspended in 500 μl diethyl ether, dried again, and stored under vacuum overnight to remove all solvent traces. The resulting lipid film was hydrated in 500 μl of buffer C by vortexing, followed by shaking for 1 h at 750 rpm at 23°C. This suspension was then sonicated in a bath sonicator (Branson Ultrasonics, Danbury, Connecticut) for 30 min and subjected to 5 freeze/thaw cycles. Finally, the liposomes were passed 21 times through a 50‐nm polycarbonate filter (Avestin) in a Mini‐Extruder (Avanti Polar Lipids).
To reconstitute SecYEG into these liposomes, 50 μl of 4 mg/ml liposomes was mixed with 0.4% Triton X‐100 and 20 pmol (single‐molecule experiments) or 200 pmol (bulk experiments) of purified protein. The reconstitution volume was brought to 100 μl by addition of buffer C, and the mixture was incubated for 30 min at 4°C. Detergent was then removed by 4 sequential batches of SM‐2 Bio‐Beads (Bio‐Rad Laboratories, Hercules, California) for 1, 4, 12, and 2 h. The final proteoliposomes were centrifuged for 5 min at 14,000 g to remove any insoluble material before use.
Microscope setup
The microscope used was a through‐objective TIRF microscope built on an inverted microscope body (Olympus IX71, Olympus, Tokyo, Japan) as described elsewhere (Graham et al, 2016). Samples were illuminated by 532 nm and 641 nm lasers (Coherent Sapphire 532, and Cube 641, Coherent, Santa Clara, California). The beams were expanded and focused through the rear window into the back aperture of an oil immersion objective (Olympus UPlanSApo, 100×, NA 1.40). A suspended lens in front of the back microscope port allowed for adjustment of the TIRF angle. Cy3 and Cy5 emission was separated by a dichroic mirror (Chroma) and projected onto two halves of an EM‐CCD camera (Hamamatsu, ImageEM 9100‐13, Hamamatsu Photonics, Hamamatsu City, Japan). The field of view was manipulated by a digitally controlled, automated microstage (Mad City Labs Inc., Madison, Wisconsin), and the microscope focus was manually adjusted.
Flow chamber preparation
Cover glasses (22 × 60 mm No. 1.5; Fisher Scientific, Hampton, New Hampshire) were cleaned by four alternating 30‐min washes in ethanol and KOH, with intervening rinses in deionized water, followed by a 10‐min sonication in acetone. The glasses were silanized by a 2‐min treatment with 2% (3‐aminopropyl)triethoxysilane (APTES, Sigma‐Aldrich, St. Louis, Missouri) in acetone, thoroughly washed with deionized water, and heated for 30 min at 110°C. 150 mg 5 kDa methoxypolyethylene glycol–succinimidyl valerate (mPEG‐SVA) and 2.5 mg 5 kDa biotin‐PEG‐SVA (both Laysan Bio, Arab, Alabama) were dissolved in 1 ml of 100 mM NaHCO3 pH 8.2. 100 μl of the PEG solution was sandwiched between pairs of cleaned cover glasses and incubated at room temperature for 4 h. Glasses were then separated, washed thoroughly with deionized water, and stored under vacuum.
Flow chambers were constructed by sandwiching rectangles of double‐sided Kapton tape (Bertech, Torrance, California) with 5 × 15 mm slits cut into them between cleaned cover glasses and 2.5 × 2.5 × 0.5 cm quartz glass blocks (Quartz Scientific Inc., Fairport Harbor, Ohio). To allow perfusion of the chambers, tubing (PFTE #24; Cole‐Parmer, Vernon Hills, Illinois) was inserted through holes drilled 10 mm apart in the quartz and sealed by epoxy. Before use, flow chambers were flushed with 500 μl of buffer G (50 mM HEPES/KOH pH 7.5, 50 mM KCl, 5 mM MgCl2, 8 mM protocatechuic acid [PCA, Santa Cruz Biotechnology, Dallas, Texas], 200 nM protocatechuate 3,4‐dioxygenase [PCD, Sigma‐Aldrich], 20 mM BME, 0.2 mg/ml bovine serum albumin [BSA, New England Biolabs], 10 mM 6‐hydroxy‐2,5,7,8‐tetramethylchroman‐2‐carboxylic acid [Trolox, Sigma‐Aldrich]), and then incubated with 0.5 mg/ml neutravidin (Thermo Fisher Scientific) in buffer G for 30 min. Unbound neutravidin was removed by a second wash with 500 μl of buffer G.
Single‐molecule FRET complex assembly and surface tethering
Translocation intermediate complexes were assembled in bulk by mixing 20 nM Cy3‐labeled SecYEG proteoliposomes, 20 nM Cy5‐labeled SecA, 1 μM proOmpA(1‐175)‐DHFR‐biotin with 5 mM ATP, and 50 μM methotrexate (MTX, Sigma‐Aldrich) in 50 μl buffer H (50 mM HEPES/KOH pH 7.5, 50 mM KCl, 5 mM MgCl2, 20 mM BME, 0.02 mg/ml BSA). Reactions were incubated for 10 min at 37°C while shaking at 650 rpm in a Thermomixer R (Eppendorf, Germany). When indicated, 1 mM ADP•BeFx, 1 mM ADP and Vi (sodium orthovanadate, New England Biolabs), 5 mM ATPγS (Jena Bioscience, Jena, Germany), or 1 mM ADP and Pi were added after the initial 10 min, along with 1 U of hexokinase (Roche Applied Science, Germany) and 20 mM glucose, and incubated for an additional 5 min. Assembled complexes were diluted 1:20 in buffer G supplemented with 50 μM MTX and 5 mM ATP, 1 mM ADP•BeFx, 1 mM ADP•Vi, 5 mM ADP•Pi, or 5 mM ATPγS. 20 nM unlabeled SecA was added at this point during ATP experiments. The diluted reactions were added to flow cells and incubated for 5 min at 23°C. Unbound protein was then washed out with 500 μl of buffer G containing 50 pM Cy5‐labeled SecA, 50 μM MTX, and 5 mM ATP, 1 mM ADP•BeFx, 1 mM ADP•Vi, 5 mM ADP•Pi, or 5 mM ATPγS. ATP reactions also contained 1 nM unlabeled SecA and an ATP regeneration system consisting of 8 mM phosphocreatine and 50 ng/ml creatine kinase (Sigma‐Aldrich).
Single‐molecule imaging
The 532 nm and 641 nm lasers were set to 45 W/cm2 and 35 W/cm2 surface density, respectively. The camera EM gain was set to its maximal setting. The camera integration time was set to 33 ms, and the illumination was switched between the two lasers by Uniblitz VS14 shutters (Vincent Associates, Rochester, New York) synchronized to the camera. Alternating excitation (AlEx) illumination was used, where single frames switched between direct excitation of the Cy3 molecules and direct excitation of the Cy5 molecules. 1,000 frames were recorded for each video, consisting of 33 total seconds. The stage was translated and refocused between each video to find previously unexposed, unbleached regions of the cover glass. Data for each mutant/nucleotide combination were aggregated from multiple videos taken from at least four separate immobilizations collected over at least two different days.
Extraction of FRET data from videos
Initial analysis was performed in the iSMS single‐molecule FRET software suite (Preus et al, 2015) in MATLAB (MathWorks, Natick, Massachusetts). The Cy3 and Cy5 channels of each video were aligned by maximizing the co‐localization of fluorescence spots projected on each half of the camera field of view. Raw fluorescence traces were extracted from co‐localized spots by fitting a five‐pixel‐diameter circular aperture function to each spot and integrating the intensity within the circle. A mean background signal calculated from a ring of pixels with a 10 pixel diameter centered on each fit spot was subtracted from the integrated intensity. These raw traces were filtered to select complexes with 1:1 SecA:SecY stoichiometry as evidenced by single‐step photobleaching, as well as clear anti‐correlated FRET behavior either within a trace or in response to acceptor bleaching. In general, each video had roughly 25 monomeric SecY spots, about 15 of which co‐localized with a monomeric SecA. Of these co‐localized spots, about 10–20% exhibited anti‐correlated FRET activity (Appendix Table S1). Traces were manually segmented to account for multiple SecA molecules binding and dissociating within one video. Raw fluorescence was then corrected for Cy3 bleed‐through into the Cy5 channel as well as direct excitation of Cy5 by the 532 nm laser. These correction factors were calculated independently for each imaging session based on the measured fluorescence in the Cy5 channel after Cy5 and Cy3 photobleaching, respectively. A γ‐factor correction was also calculated for each session based on total fluorescence in both channels before and after acceptor bleaching. FRET was then calculated from these corrected fluorescence traces (Lee et al, 2005).
Estimation of inter‐fluorophore distances
FRET histograms were compiled by a non‐weighted aggregation of FRET values calculated from each frame prior to photobleaching/dissociation of all traces for a given mutant/nucleotide combination. These histograms were fit with Gaussian mixture models using the fitgmdist() function in MATLAB. The mean and standard deviation of Gaussian components aligned to prominent peaks were used to calculate the estimated distances between fluorophores, using 55 Å as a value for the Fӧrster radius between Cy3 and Cy5 (Yu et al, 1994). Calculated values were compared to inter‐Cα distances measured between corresponding amino acids in published structures of the SecA/SecY complex (PDBs 3DIN, 5EUL) using PyMol (Schrӧdinger LLC., New York, New York). The number of traces included in each histogram is indicated in the corresponding figure legends.
Hidden Markov analysis
Hidden Markov model analysis of individual FRET traces was performed in the vbFRET software suite in MATLAB (Bronson et al, 2009). This program finds the idealized parameters, including the number, value, and transition probabilities, of FRET states for each trace using a maximum evidence approach. The program was run with up to 5 discrete FRET states allowed per trace using default parameters, including 10 fitting attempts per trace, a maximum of 100 iterations per VBEM, and a convergence threshold of 10−5. The most likely number of states represented in each FRET trace was reported. The number of traces analyzed in this manner is the same as the number reported for the corresponding FRET histograms. Transition density plots were constructed from these idealized states for ATP data by plotting the FRET value fit by vbFRET one frame before each predicted transition against the FRET value one frame after each prediction (McKinney et al, 2006). Transition density plots for data in the presence of ADP•BeFx could not be plotted as the model predicted no transitions in most traces.
Dwell time analysis
FRET states from individual traces were then grouped into consensus high and low FRET states using ebFRET (van de Meent et al, 2014), again using default parameters except for a precision of 10−5. The mean dwell times of these high and low FRET states were found by fitting single exponential functions to the ensembles of individual states grouped into each FRET. Since events that terminate within the integration time of a given frame often get attributed the duration of the full frame, each data point was treated as left‐censored by up to one frame while fitting. Also, the first and last FRET state of each trace was excluded as the state was not necessarily observed for its full duration. A small population of long‐lived, non‐exponentially distributed, low FRET states was truncated by limiting the analysis to states with durations <2 s. Error on these mean lifetimes was the standard error of the mean (SEM) based on the number of individual traces fit by ebFRET for the given mutant/nucleotide combination. The number of FRET states from which the dwell times were measured is indicated in the corresponding figure legends.
Translocation assay
Bulk translocation assays were performed on wild type and labeled SecA and SecYEG purified in the same manner as for single‐molecule experiments. 35S‐Met‐labeled proOmpA was generated by in vitro translation. mRNA was transcribed from linearized template with an SP6 promoter followed by the Kozak consensus ribosome binding site directly 5′ to the proOmpA gene start codon using a RiboMax SP6 in vitro transcription kit (Promega, Madison, Wisconsin). 2 μg of transcription product RNA was mixed with 35 μl of nuclease‐treated rabbit reticulocyte lysate (Promega), 1 μl of 1 mM amino acid mixture minus methionine (Promega), and 2 μl (.022 μCi) of EasyTag Express S35 Labeling Mix (PerkinElmer, Waltham, Massachusetts). Translation product was precipitated by the addition of 150 μl saturated ammonium sulfate and resuspended in 50 μl 6 M urea pH 6.8.
Reactions were assembled in buffer H. 100 nM SecA, and 100 nM SecYEG proteoliposomes were mixed with 1 μl of the in vitro translation products in a 50 μl total volume. Reactions were initiated by the addition of 5 mM ATP and incubated for 10 min at 37°C while shaking at 650 rpm. For the channel saturation experiments, 1 μl of purified proOmpA‐DHFR was added at varying concentrations along with 50 μM MTX before the addition of the in vitro translation product and incubated for an additional 10 min at 37°C. Reactions were terminated by transfer to ice and addition of 0.4 mg/ml proteinase K, and, where indicated, 0.2% Triton X‐100. Digests were continued for 45 min and quenched with 2 mM phenylmethylsulfonyl fluoride (PMSF). Reactions were then precipitated in 10% trichloroacetic acid (TCA) and resuspended in 1X Laemmli buffer with 300 mM Tris base. Samples were then analyzed by SDS–PAGE. The gels were vacuum‐dried, exposed to autoradiography film overnight, and imaged by a Personal Molecular Imager (Bio‐Rad).
ATPase assays
ATP activity was measured with an EnzChek Phosphate Assay Kit (Thermo Fisher Scientific). Activity was measured with both wild‐type proteins and fluorophore‐labeled mutants. 100 μl reactions were prepared in 1× EnzChek assay buffer (Thermo Fisher Scientific) with 200 μM 2‐amino‐6‐mercapto‐7‐methylpurine riboside (MESG), 100 nM SecA, 100 nM SecYEG proteoliposomes, 0.02 mg/ml BSA, 20 mM BME, 50 μM MTX, and 1 μM proOmpA‐DHFR. 0.1 U of purine nucleoside phosphorylase (Thermo Fisher Scientific) was added, and the reactions were incubated for 30 min at 23°C. 5 mM ATP was then added, and the change in absorbance of 360 nm light (OD360) was monitored over 90 min at 15‐s intervals in a M5 plate reader (Molecular Devices, San Jose, California). Reactions were run in quadruplicate together with a control reaction lacking ATP. The dependence on SecA concentration was measured similarly with 0, 1, 5, 10, 25, 50, 75, 100, 150, or 200 nM SecA. A standard curve was generated by measuring the absorbance of Pi standards (Thermo Fisher Scientific). Average ATP consumption rates were converted to a per molecule SecA rates using the SecA concentrations and ATPase rates measured from the SecA titration experiment.
Author contributions
MAC performed all experiments. Initial tests of FRET constructs were performed by BWB. JJL provided expertise with data acquisition and analysis. MAC and TAR designed the experiments and wrote the paper. TAR supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank T. Graham for the construction and maintenance of the microscope, as well as training. We thank the Nikon imaging center at Harvard Medical School for help with early FRET experiments. We thank R. Powers and N. Sever for reading of the manuscript. M.A.C. is supported by National Institutes of Health/National Institute of General Medical Sciences T32 GM008313 training grant. T.A.R. and J.J.L. are supported by R01 NIH grants GM052586 and GM115487, respectively. T.A.R. is a Howard Hughes Medical Institute Investigator.
The EMBO Journal (2019) 38: e101140
Data availability
Individual FRET traces for all reported experiments are included as Source Data.
Raw video data are available as.cxd files upon request.
References
- Allen WJ, Corey RA, Oatley P, Sessions RB, Baldwin SA, Radford SE, Tuma R, Collinson I (2016) Two‐way communication between SecY and SecA suggests a Brownian ratchet mechanism for protein translocation. Elife 5: e15598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin‐Tam M‐E, Olivares AO, Sauer RT, Baker TA, Lang MJ (2011) Single‐molecule protein unfolding and translocation by an ATP‐fueled proteolytic machine. Cell 145: 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee T, Zheng Z, Abolafia J, Harper S, Oliver DB (2017) The SecA protein deeply penetrates into the SecYEG channel during insertion, contacting most channel transmembrane helices and periplasmic regions. J Biol Chem 292: 19693–19707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer BW, Rapoport TA (2009) Mapping polypeptide interactions of the SecA ATPase during translocation. Proc Natl Acad Sci USA 106: 20800–20805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer BW, Shemesh T, Chen Y, Rapoport TA (2014) A “Push and Slide” mechanism allows sequence‐insensitive translocation of secretory proteins by the SecA ATPase. Cell 157: 1416–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar N, Rapoport T (2017) Toward an understanding of the Cdc48/p97 ATPase. F1000Research 6: 1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson JE, Fei J, Hofman JM, Gonzalez RL, Wiggins CH (2009) Learning rates and states from biophysical time series: a Bayesian approach to model selection and single‐molecule FRET data. Biophys J 97: 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada N, Chattrakun K, Marsh BP, Mao C, Bariya P, King GM (2018) Single‐molecule observation of nucleotide induced conformational changes in basal SecA‐ATP hydrolysis. Sci Adv 4: eaat8797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bauer BW, Rapoport TA, Gumbart JC (2015) Conformational changes of the clamp of the protein translocation ATPase SecA. J Mol Biol 427: 2348–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey RA, Allen WJ, Collinson I (2016) Protein translocation: what's the problem? Biochem Soc Trans 44: 753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey RA, Ahdash Z, Shah A, Pyle E, Allen WJ, Fessl T, Lovett JE, Politis A, Collinson I (2019) ATP‐induced asymmetric pre‐protein folding as a driver of protein translocation through the Sec machinery. Elife 8: e41803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranford‐Smith T, Huber D (2018) The way is the goal: how SecA transports proteins across the cytoplasmic membrane in bacteria. FEMS Microbiol Lett 365: fny093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economou A, Wickner W (1994) SecA promotes preprotein translocation by undergoing ATP‐driven cycles of membrane insertion and deinsertion. Cell 78: 835–843 [DOI] [PubMed] [Google Scholar]
- Economou A, Pogliano JA, Beckwith J, Oliver DB, Wickner W (1995) SecA membrane cycling at SecYEG is driven by distinct ATP binding and hydrolysis events and is regulated by SecD and SecF. Cell 83: 1171–1181 [DOI] [PubMed] [Google Scholar]
- Erlandson KJ, Miller SBM, Nam Y, Osborne AR, Zimmer J, Rapoport TA (2008a) A role for the two‐helix finger of the SecA ATPase in protein translocation. Nature 455: 984–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandson KJ, Or E, Osborne AR, Rapoport TA (2008b) Analysis of polypeptide movement in the SecY channel during SecA‐mediated protein translocation. J Biol Chem 283: 15709–15715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst I, Haase M, Ernst S, Yuan S, Kuhn A, Leptihn S (2018) Large conformational changes of a highly dynamic pre‐protein binding domain in SecA. Commun Biol 1: 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessl T, Watkins D, Oatley P, Allen WJ, Corey RA, Horne J, Baldwin SA, Radford SE, Collinson I, Tuma R (2018) Dynamic action of the Sec machinery during initiation, protein translocation and termination. Elife 7: e35112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TGW, Walter JC, Loparo JJ (2016) Two‐stage synapsis of DNA ends during non‐homologous end joining. Mol Cell 61: 850–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Monroe N, Sundquist WI, Shen PS, Hill CP (2017) The AAA ATPase Vps4 binds ESCRT‐III substrates through a repeating array of dipeptide‐binding pockets. Elife 6: e31324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW, Horwich AL (2005) Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell 121: 1029–1041 [DOI] [PubMed] [Google Scholar]
- Ho C‐M, Beck JR, Lai M, Cui Y, Goldberg DE, Egea PF, Zhou ZH (2018) Malaria parasite translocon structure and mechanism of effector export. Nature 561: 70–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J (2002) Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science 297: 2018–2026 [DOI] [PubMed] [Google Scholar]
- Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, Weiss S (2005) Accurate FRET measurements within single diffusing biomolecules using alternating‐laser excitation. Biophys J 88: 2939–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Park E, Ling J, Ingram J, Ploegh H, Rapoport TA (2016) Crystal structure of a substrate‐engaged SecY protein‐translocation channel. Nature 531: 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT (2008) Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor‐dependent recognition of ssrA‐tagged substrates. Mol Cell 29: 441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Kimura E, Mizushima S (1990) Complementation of two overlapping fragments of SecA, a protein translocation ATPase of Escherichia coli, allows ATP binding to its amino‐terminal region. J Biol Chem 265: 8760–8765 [PubMed] [Google Scholar]
- McKinney SA, Joo C, Ha T (2006) Analysis of single‐molecule FRET trajectories using hidden Markov modeling. Biophys J 91: 1941–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Meent J‐W, Bronson JE, Wiggins CH, Gonzalez RL (2014) Empirical Bayes methods enable advanced population‐level analyses of single‐molecule FRET experiments. Biophys J 106: 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA (2014) Mechanochemical basis of protein degradation by a double‐ring AAA+ machine. Nat Struct Mol Biol 21: 871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Or E, Boyd D, Gon S, Beckwith J, Rapoport T (2005) The bacterial ATPase SecA functions as a monomer in protein translocation. J Biol Chem 280: 9097–9105 [DOI] [PubMed] [Google Scholar]
- Osborne AR, Clemons WM, Rapoport TA (2004) A large conformational change of the translocation ATPase SecA. Proc Natl Acad Sci USA 101: 10937–10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogliano KJ, Beckwith J (1993) The Cs sec mutants of Escherichia coli reflect the cold sensitivity of protein export itself. Genetics 133: 763–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preus S, Noer SL, Hildebrandt LL, Gudnason D, Birkedal V (2015) iSMS: single‐molecule FRET microscopy software. Nat Methods 12: 593–594 [DOI] [PubMed] [Google Scholar]
- Puchades C, Rampello AJ, Shin M, Giuliano CJ, Wiseman RL, Glynn SE, Lander GC (2017) Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358: eaao0464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport TA, Li L, Park E (2017) Structural and mechanistic insights into protein translocation. Annu Rev Cell Dev Biol 33: 369–390 [DOI] [PubMed] [Google Scholar]
- Robson A, Gold VAM, Hodson S, Clarke AR, Collinson I (2009) Energy transduction in protein transport and the ATP hydrolytic cycle of SecA. Proc Natl Acad Sci USA 106: 5111–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen M, Maillard RA, Nyquist K, Rodriguez‐Aliaga P, Pressé S, Martin A, Bustamante C (2013) The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell 155: 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberk N, Karamanou S, Portaliou AG, Zorzini V, Hofkens J, Hendrix J, Economou A (2018) The preprotein binding domain of SecA displays intrinsic rotational dynamics. Structure 27: 90–101 [DOI] [PubMed] [Google Scholar]
- Whitehouse S, Gold VAM, Robson A, Allen WJ, Sessions RB, Collinson I (2012) Mobility of the SecA 2‐helix‐finger is not essential for polypeptide translocation via the SecYEG complex. J Cell Biol 199: 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury RL, Hardy SJS, Randall LL (2002) Complex behavior in solution of homodimeric SecA. Protein Sci 11: 875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Tang WK, Zhang T, Xia D (2017) A mighty “Protein Extractor” of the cell: structure and function of the p97/CDC48 ATPase. Front Mol Biosci 4: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedidi RS, Wendler P, Enenkel C (2017) AAA‐ATPases in protein degradation. Front Mol Biosci 4: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Chao J, Patek D, Mujumdar R, Mujumdar S, Waggoner AS (1994) Cyanine dye dUTP analogs for enzymatic labeling of DNA probes. Nucleic Acids Res 22: 3226–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Slevin JT, Whiteheart SW (2007) Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett 581: 2140–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer J, Nam Y, Rapoport TA (2008) Structure of a complex of the ATPase SecA and the protein‐translocation channel. Nature 455: 936–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer J, Rapoport TA (2009) Conformational flexibility and peptide interaction of the translocation ATPase SecA. J Mol Biol 394: 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
Individual FRET traces for all reported experiments are included as Source Data.
Raw video data are available as.cxd files upon request.