

Abstract
Neurodegenerative diseases are characterized by the accumulation of misfolded proteins in the brain. Insights into protein quality control mechanisms to prevent neuronal dysfunction and cell death are crucial in developing causal therapies. Here, we report that various disease‐associated protein aggregates are modified by the linear ubiquitin chain assembly complex (LUBAC). HOIP, the catalytic component of LUBAC, is recruited to misfolded Huntingtin in a p97/VCP‐dependent manner, resulting in the assembly of linear polyubiquitin. As a consequence, the interactive surface of misfolded Huntingtin species is shielded from unwanted interactions, for example with the low complexity sequence domain‐containing transcription factor Sp1, and proteasomal degradation of misfolded Huntingtin is facilitated. Notably, all three core LUBAC components are transcriptionally regulated by Sp1, linking defective LUBAC expression to Huntington's disease. In support of a protective activity of linear ubiquitination, silencing of OTULIN, a deubiquitinase with unique specificity for linear polyubiquitin, decreases proteotoxicity, whereas silencing of HOIP has the opposite effect. These findings identify linear ubiquitination as a protein quality control mechanism and hence a novel target for disease‐modifying strategies in proteinopathies.
Keywords: Huntingtin, LUBAC, OTULIN, p97, protein aggregation
Subject Categories: Neuroscience; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Quality control and stress response systems are crucial in maintaining cellular homeostasis and viability. A decreased efficiency of these pathways is associated with aging and promotes age‐related pathologies, such as neurodegenerative diseases. Several quality control systems are regulated by ubiquitination, a highly versatile post‐translational modification that can affect the function, fate, and subcellular localization of the modified substrates (rev. in Swatek & Komander, 2016; Yau & Rape, 2016). In polyubiquitin chains, the C‐terminal glycine of the donor ubiquitin is usually linked to one of the seven lysine residues of an acceptor ubiquitin via an isopeptide bond. Alternatively, the C‐terminus of ubiquitin can form a peptide bond with the N‐terminal methionine of an acceptor ubiquitin, resulting in linear or M1 ubiquitination. This type of ubiquitin linkage is exclusively generated by the linear ubiquitin chain assembly complex (LUBAC), which is composed of the two RBR (RING‐between‐RING) E3 ubiquitin ligases HOIP and HOIL‐1L and the adaptor protein SHARPIN (rev. in Hrdinka & Gyrd‐Hansen, 2017; Iwai, 2012; Rittinger and Ikeda, 2017). HOIP is the catalytically active component of LUBAC and the only known E3 ubiquitin ligase capable of assembling linear ubiquitin chains due to a unique ubiquitin‐binding domain C‐terminal to the RBR domain (Smit et al, 2012; Stieglitz et al, 2012, 2013). Notably, not only the formation of linear polyubiquitin but also its hydrolysis is a highly specific process. OTULIN was identified as an M1‐specific deubiquitinase that employs a mechanism of ubiquitin‐assisted catalysis to achieve this exceptional specificity (Keusekotten et al, 2013; Rivkin et al, 2013).
LUBAC has mostly been studied in the context of inflammation, innate immunity, and defense pathways against intracellular pathogens where it is required for NF‐κB activation (Iwai et al, 2014; Noad et al, 2017; van Wijk et al, 2017). In this study, we tested a possible impact of LUBAC on protein aggregates, which can be viewed as a special kind of “cellular pathogen”. LUBAC is recruited to different protein aggregates associated with neurodegenerative diseases, such as polyglutamine diseases (caused by mutations in Huntingtin or Ataxin‐3), amyotrophic lateral sclerosis, and frontotemporal dementia (caused by mutations in SOD1, TDP‐43, or Optineurin), and modifies proteotoxicity by affecting the fate of cellular aggregates. Thus, our study revealed a novel function of linear ubiquitination in protein quality control, which turned out to be independent of its role in NF‐κB signaling.
Results
Linear ubiquitin chains are enriched at Huntingtin aggregates
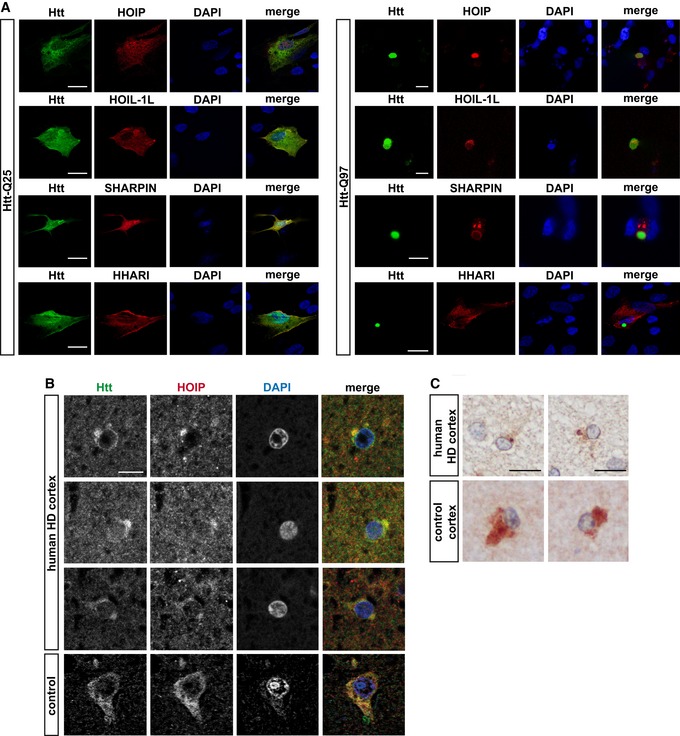
A hallmark of protein aggregates is the presence of various ubiquitin conjugates (Davies et al, 1997; Steffan et al, 2004; Bennett et al, 2007; Hipp et al, 2012; Bhat et al, 2014). We were wondering whether linear ubiquitin chains play a role in this context. As a paradigm for toxic protein aggregates, we used exon 1 of mutant Huntingtin (Htt) with an expanded polyglutamine tract (Htt‐polyQ). An increase in CAG triplet coding for glutamine in exon 1 of the Huntingtin gene beyond a critical threshold of about 35 causes Huntington's disease (HD), which is characterized by motor, cognitive, and behavioral symptoms. To address the possibility that M1‐linked ubiquitination occurs at Htt aggregates, we employed super‐resolution structured illumination microscopy (SR‐SIM) to visualize LUBAC components. In control, human neuronal SH‐SY5Y cells expressing Htt‐Q25, HOIP, HOIL‐1L, and SHARPIN were distributed throughout the cytoplasm. In contrast, these proteins were recruited to Htt aggregates in Htt‐Q97‐expressing cells (Fig 1A). The RBR E3 ubiquitin ligase HHARI (Ariadne RBR E3 ubiquitin ligase 1 or ARIH1), analyzed as a control, was not enriched at Htt‐Q97, indicating selectivity of the recruitment process. Co‐aggregation of HOIP, HOIL‐1L, and SHARPIN with Htt‐Q97 was confirmed by a filter retardation assay. This approach is based on the insolubility of Htt aggregates in SDS and their selective retention on a cellulose acetate membrane, whereas SDS‐soluble proteins are filtered through the membrane (Scherzinger et al, 1997). In lysates from cells co‐expressing Htt‐Q97 together with LUBAC components, HOIP, HOIL‐1L, and SHARPIN were retained by the membrane together with Htt‐Q97, suggesting recruitment to Htt aggregates (Fig EV1A). In addition, co‐immunoprecipitation experiments revealed an interaction between SDS‐soluble Htt‐Q97‐HA species and endogenous HOIP (Fig EV1B). Supporting a pathophysiological relevance of our observation, co‐staining of Htt‐polyQ aggregates and HOIP was also observed in human HD frontal cortex (Fig 1B and C).
Figure 1. Htt‐polyQ aggregates recruit LUBAC components.
-
AHOIP, HOIL‐1L, and SHARPIN co‐localize with Htt‐Q97 aggregates. SH‐SY5Y cells co‐expressing Htt‐Q25‐GFP or Htt‐Q97‐GFP (green) and HOIP, HOIL‐1L, SHARPIN, or HHARI (red); DAPI (blue). Scale bar, 10 μm.
-
B, CHOIP co‐localizes with Htt inclusion bodies in human HD frontal cortex. Perinuclear Htt aggregates visualized by immunofluorescence (B) and DAB immunohistochemistry (C) in human HD frontal cortex. Scale bar, 10 μm (B), 15 μm (C).
Figure EV1. HOIP, HOIL‐1L, and SHARPIN co‐aggregate with Htt‐Q97 (related to Fig 1).
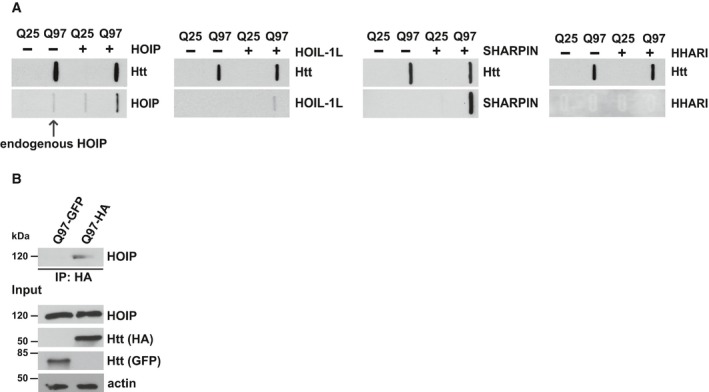
- HEK293T cells transiently co‐expressing Htt‐Q25 or Htt‐Q97 and either HOIP, HOIL‐1L, SHARPIN, or HHARI were analyzed by the filter retardation assays. Aggregated proteins retained by cellulose acetate membranes were detected by Western blotting.
- Endogenous HOIP interacts with soluble Htt‐Q97. HEK293T cells were transfected with Htt‐Q97‐HA or Htt‐Q97‐GFP (as a control for specific immunoprecipitation of HA‐tagged Htt‐Q97). Cells were lysed with 1% Triton X‐100, followed by an immunoprecipitation of Htt via the HA tag. Immunoprecipitated proteins were analyzed by Western blotting using an anti‐HOIP antibody.
Source data are available online for this figure.
Next, we tested whether the presence of LUBAC at Htt‐polyQ has functional consequences. Indeed, we found evidence for linear ubiquitin chains at Htt aggregates by utilizing confocal and super‐resolution microscopy, biochemical approaches, and mass spectrometry. M1 ubiquitin‐specific signals at both cytoplasmic and nuclear aggregates under endogenous expression of LUBAC components were detected in SH‐SY5Y cells expressing Htt‐Q97 and in cortical and striatal sections from 12‐week‐old R6/2 mice (Figs 2A and B, and EV2A and B). These mice express exon 1 of human HTT with an expanded CAG repeat under the control of the human HTT promoter and are widely used as a rodent model of HD (Mangiarini et al, 1996; Pouladi et al, 2013). Moreover, linear ubiquitin chains were present at aggregates in human HD cortical sections (Figs 2C and EV2C). In contrast, M1 ubiquitin‐positive signals at Htt aggregates were absent in HOIP CRISPR/Cas9 knockout (KO) HAP1 cells, indicating that we specifically detected linear ubiquitin chains generated by HOIP (Fig EV2D). M1‐linked ubiquitin chains were also detected by filter retardation assays using lysates from Htt‐Q97‐expressing cells under endogenous LUBAC expression. No signal was detectable after preincubation of the M1 ubiquitin antibody with recombinant M1‐linked tetra‐ubiquitin (4 × M1), whereas preincubation with recombinant K63‐linked tetra‐ubiquitin (4 × K63) was without effect, corroborating the specificity of the M1 ubiquitin antibody (Fig 2D). Increased levels of linear ubiquitin in SDS‐insoluble Htt‐Q97 fractions were confirmed by quantitative (parallel reaction monitoring) mass spectrometry (Fig EV2E and F).
Figure 2. Linear ubiquitin chains are enriched at Htt‐polyQ aggregates.
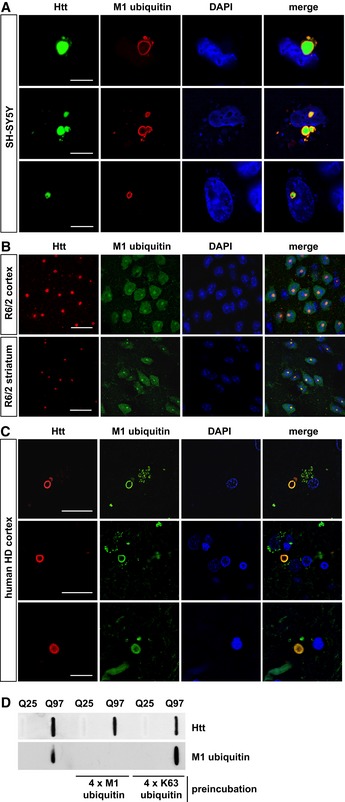
-
A–CM1‐linked ubiquitin co‐localizes with Htt‐polyQ aggregates in cultured SH‐SY5Y cells (A), R6/2 mouse cortex and striatum (B) and human HD frontal cortex (C). Autofluorescent lipofuscin appears in the green channel in human brain. Scale bar, 20 μm (A), 10 μm (B, C).
-
DHtt‐Q25‐ or Htt‐Q97‐expressing HEK293T were analyzed by filter retardation assays. As indicated, the M1 ubiquitin antibody was preincubated with recombinant tetra‐ubiquitin linked via M1 (4 × M1 ubiquitin) or K63 (4 × K63 ubiquitin).
Figure EV2. M1 ubiquitin‐specific signals occur at Htt‐polyQ aggregates and are absent in HOIP‐deficient cells (related to Fig 2).
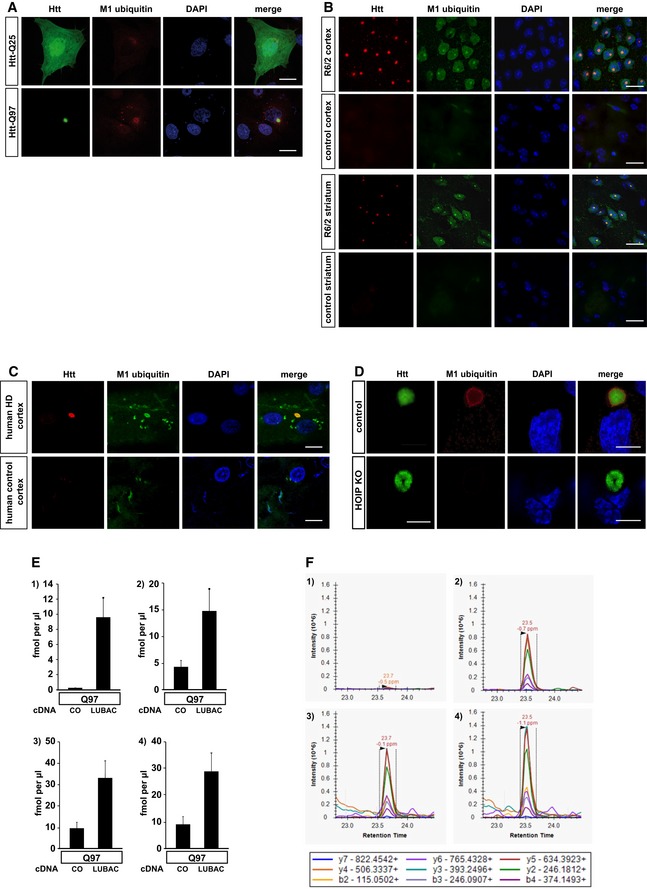
-
ASH‐SY5Y cells expressing Htt‐Q25‐GFP (upper panel) or Htt‐Q97‐GFP (lower panel) were stained for M1 ubiquitin (red) and DAPI (blue). Scale bar, 10 μm.
-
B, CM1‐linked ubiquitin co‐localizes with Htt‐polyQ aggregates in R6/2 mouse cortex and striatum (B) and human HD frontal cortex (C). Autofluorescent lipofuscin appears in the green channel in human brain. Scale bar, 20 μm (B), 10 μm (C).
-
DHAP1 WT (control) or HOIP KO HAP1 cells expressing Htt‐Q97‐GPF (green) were stained for M1 ubiquitin (red) and DAPI (blue). Scale bar, 10 μm.
-
EQuantification of ubiquitin chains associated with SDS‐insoluble Htt‐Q97 from HEK293T cells by mass spectrometry (1: M1, 2: K11, 3: K48, and 4: K63). The standard error is shown with error bars (n = 3).
-
FExtracted chromatograms from the M1 ubiquitin measurements. Upper panels show the quantification of the ubiquitin chain peptide, while the lower panels show the spiked in reference peptide used for quantification. Panels 1 and 3 show an example of Htt‐Q97, and panels 2 and 4 show an example of Htt‐Q97 + LUBAC. Above each peak, the retention time and mass error are shown. Below the panels a legend identifying the product ions observed and used for quantification is shown.
HOIP is recruited to Htt‐polyQ via the PUB domain in a p97/VCP‐dependent manner
To address the mechanism of HOIP recruitment to Htt‐polyQ, we performed a mutational analysis (Fig 3A). Deletion of the NZF domains, the UBA domain, the C‐terminal RBR domain, or mutation of the catalytic cysteine (C885A) did not result in a major impairment of HOIP binding to Htt‐Q97 (Fig 3B and C). In contrast, deletion of the N‐terminal PUB domain markedly interfered with HOIP recruitment. The PUB domain of HOIP has been reported to interact with the PIM domain of the triple‐A ATPase p97/VCP (Elliott et al, 2014; Schaeffer et al, 2014), a versatile quality control enzyme that uses ATP hydrolysis to extract ubiquitinated proteins from macromolecular complexes or lipid membranes. The ability of p97/VCP to bind to protein aggregates and to deliver misfolded species to the proteasome explains its crucial role in proteotoxic stress (rev. in van den Boom & Meyer, 2018). Hence, its dysfunction has been associated with neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). We tested whether p97/VCP plays a role in recruiting HOIP to Htt aggregates by knocking down p97/VCP expression. Of note, p97/VCP silencing by RNA interference was toxic in Htt‐polyQ‐expressing cells; therefore, we were not able to decrease p97/VCP expression in Htt‐polyQ‐expressing cells by more than 50–60% (Fig EV3A). Yet, partial silencing of p97/VCP significantly decreased HOIP recruitment to Htt‐Q97. Binding of HOIP to Htt‐Q97 was restored by WT p97/VCP but not by p97/VCP∆PIM lacking the C‐terminal PIM motif (Fig 3D). Vice versa, binding of p97/VCP to Htt‐Q97 was decreased in HOIP KO HAP1 cells and restored by WT HOIP but not HOIP∆PUB (Fig 3E). HOIP‐dependent binding of p97/VCP to SDS‐soluble Htt‐polyQ species was observed by affinity purification of Htt followed by immunoblotting (Fig 3F). These data indicate a feed‐forward loop of HOIP and p97/VCP binding to misfolded Htt, promoting efficient recruitment of quality control components.
Figure 3. HOIP is recruited to Htt‐polyQ via the PUB domain in a p97/VCP‐dependent manner.
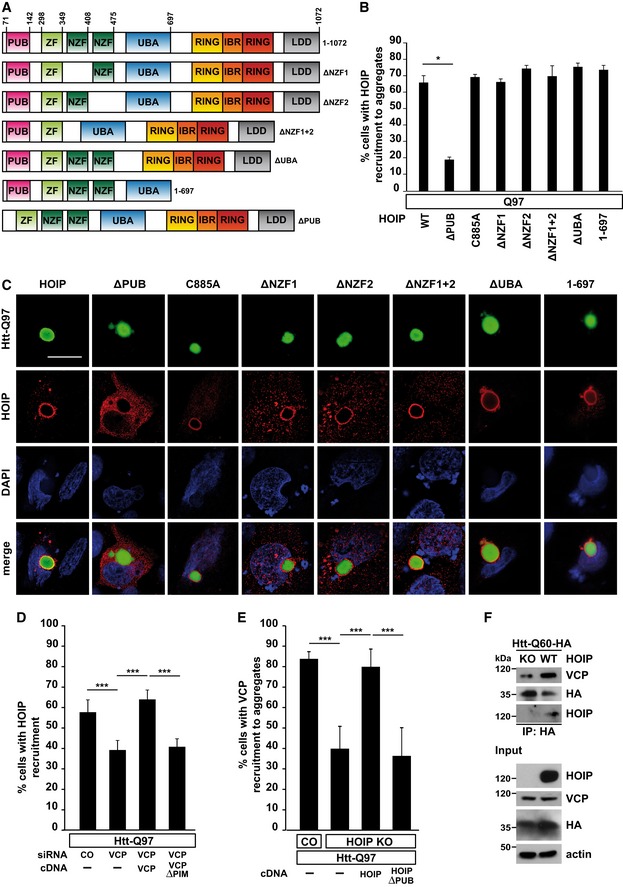
- Scheme of WT HOIP and HOIP mutants.
- Quantification of the co‐localization of WT HOIP or HOIP mutants and Htt‐Q97 aggregates. Data are displayed as mean ± SD and were analyzed by Mann–Whitney U‐test, n = 3.
- Htt‐Q97‐GFP (green) was co‐expressed with HA‐tagged WT or HOIP mutants (red) in SH‐SY5Y cells and analyzed by immunocytochemistry using an antibody against HA. DAPI (blue). Scale bar, 10 μm.
- Recruitment of HOIP to Htt‐Q97 is decreased in p97/VCP‐deficient cells. p97/VCP‐deficient SH‐SY5Y cells co‐expressing Htt‐Q97 and HA‐HOIP were reconstituted with either WT VCP or VCP∆PIM and analyzed by immunocytochemistry. Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test, n = 9.
- Recruitment of p97/VCP to Htt‐Q97 is decreased in HOIP KO cells. WT HAP1 cells (control) or HOIP KO HAP1 cells reconstituted with either WT HOIP or HOIP∆PUB were analyzed by immunocytochemistry. Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Bonferroni's multiple comparison test, n = 5.
- The interaction of soluble Htt‐Q60 with p97/VCP is dependent on HOIP. WT HAP1 cells or HOIP KO HAP1 cells were transfected with Htt‐Q60‐HA. 48 h after transfection, cells were lysed with 1% Triton X‐100, followed by an immunoprecipitation of Htt‐Q60 via the HA tag. Immunopurified proteins were detected by Western blotting using an anti‐VCP and anti‐HOIP antibody.
Figure EV3. OTULIN silencing protects neuronal cells from apoptotic cell death (related to Fig 4).
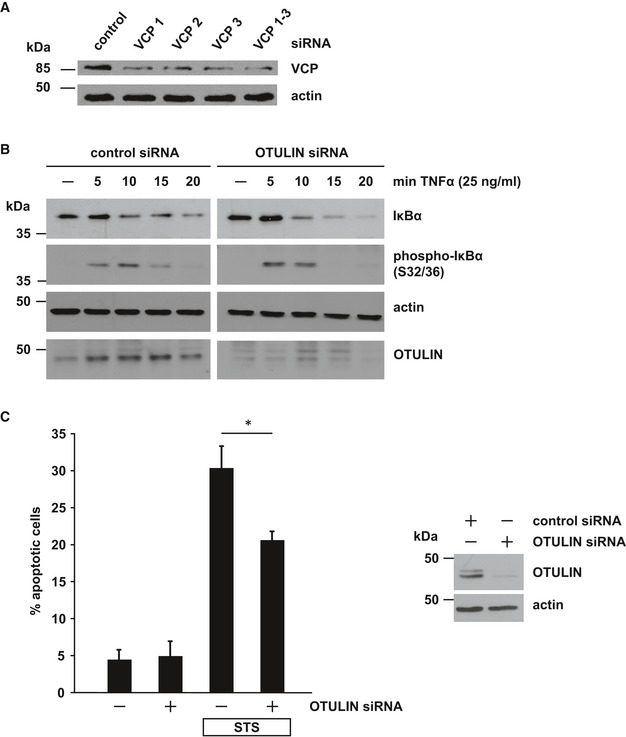
- Partial silencing of VCP/p97 in SH‐SY5Y cells by three different siRNAs. Cells were harvested 48 h after transfection of control or VCP/p97‐specific siRNAs and analyzed by immunoblotting.
- Decreased OTULIN expression promotes NF‐κB activation. Degradation of IκBα or phospho‐IκBα after TNF‐α treatment (25 ng/ml) of SH‐SY5Y cells transfected with control siRNA or OTULIN siRNA was visualized by immunoblotting using the indicated antibodies.
- OTULIN RNA interference decreases staurosporine‐induced cell death. SH‐SY5Y cells were treated with staurosporine (STS, 1 μM, 3 h) 2 days after transfection with control siRNA or OTULIN siRNA. Apoptotic cell death was quantified by immunocytochemistry using an antibody against active caspase‐3. Data are displayed as mean ± SD and were analyzed by a Mann–Whitney U‐test (*P ≤ 0.05), n = 4.
Source data are available online for this figure.
Linear ubiquitination decreases Htt‐polyQ‐induced proteotoxicity independently of NF‐κB activation
To study a possible impact of linear ubiquitination on Htt aggregate toxicity, we quantified nuclear phospho‐c‐Jun in primary neurons expressing Htt‐Q97 or Htt‐Q25 as a control, based on considerable evidence for a role of the JNK/c‐Jun pathway in mediating Htt‐induced toxicity (Jordan, 1991; Liu, 1998; Merienne et al, 2003; Garcia et al, 2004; Scappini et al, 2007; Perrin et al, 2009; Taylor et al, 2013; Su et al, 2016). Nuclear phospho‐c‐Jun was detected in 8% of Htt‐Q25‐expressing neurons and in 67% of Htt‐Q97‐expressing neurons, whereas co‐expression of HOIP reduced nuclear c‐Jun phosphorylation to control levels (Fig 4A). In line with this observation, silencing of HOIP in Htt‐Q97‐expressing cells by RNA interference significantly increased nuclear phosphorylation of c‐Jun, which was rescued by restoring HOIP expression (Fig 4B). Linear ubiquitination catalyzed by HOIP is antagonized by OTULIN, a deubiquitinase with unique specificity for M1‐linked polyubiquitin (Keusekotten et al, 2013; Rivkin et al, 2013). A recent study reported that the expression of catalytically inactive OTULIN (C129A) results in a decrease in NF‐κB signaling and strong induction of cell death, suggesting that OTULIN promotes rather than antagonizes LUBAC activity (Heger et al, 2018). However, in our neuronal cellular models silencing of OTULIN protected against apoptotic cell death and it increased NF‐κB activation (Fig EV3B and C), confirming previous studies showing antagonistic effects of OTULIN on LUBAC (Fiil et al, 2013; Keusekotten et al, 2013; Takiuchi et al, 2014; Damgaard et al, 2016). In support of a protective effect mediated by increased linear ubiquitination, silencing of OTULIN markedly reduced Htt‐Q97‐induced toxicity, reflected by a decrease in both c‐Jun phosphorylation and caspase‐3 activation (Fig 4C and D).
Figure 4. Linear ubiquitination occurs at SDS‐soluble and SDS‐insoluble Htt‐polyQ species and decreases Htt‐polyQ toxicity.
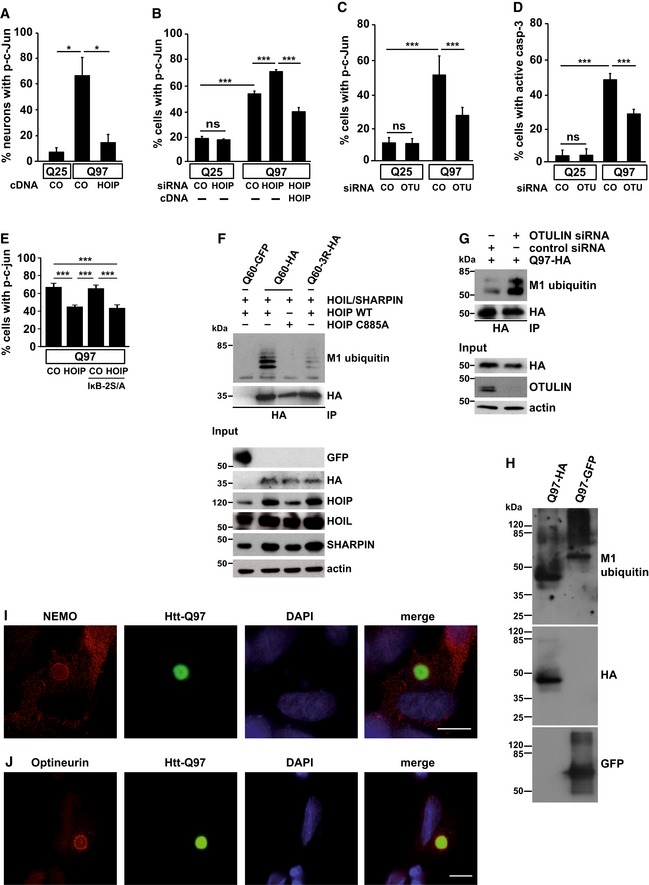
-
AIncreased expression of HOIP decreases nuclear phospho‐c‐Jun in primary neurons. Primary mouse hippocampal neurons expressing Htt‐Q25 or Htt‐Q97 ± HOIP were analyzed by immunocytochemistry using an antibody against phospho‐c‐Jun. Data are displayed as mean ± SD and were analyzed by two independent Mann–Whitney U‐tests, n = 3.
-
BHtt‐Q97‐induced toxicity is increased in cells silenced for HOIP. SH‐SY5Y cells transiently transfected with Htt‐Q25 or Htt‐Q97 and either control siRNA (CO) or HOIP siRNA were analyzed by immunocytochemistry using an antibody against phospho‐c‐Jun. For rescue experiments, HOIP was co‐expressed 24 h after silencing. Quantification of Htt‐expressing cells with nuclear phospho‐c‐Jun is based on Student's t‐test from three independent experiments. Data are displayed as mean ± SD unless otherwise stated.
-
C, DOTULIN silencing decreases the toxicity of Htt‐Q97. SH‐SY5Y cells transiently transfected with either control siRNA (CO) or OTULIN siRNA and Htt‐Q25 or Htt‐Q97 were analyzed by immunocytochemistry using antibodies against phospho‐c‐Jun (C) or cleaved caspase‐3 (D). Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test, n = 9–11.
-
EThe protective effect of HOIP is independent of NF‐κB signaling. SH‐SY5Y cells transiently transfected with Htt‐Q97‐GFP and either control vector (CO) or HOIP ± the NF‐κB super‐repressor IκB‐2S/A were analyzed by immunocytochemistry using an antibody against phospho‐c‐Jun. Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test, n = 9.
-
FSDS‐soluble Htt‐polyQ species are modified by linear ubiquitin chains. HEK293T cells were co‐transfected with Htt‐Q60‐GFP (as a control for specific immunoprecipitation of HA‐tagged Htt‐Q60), Htt‐Q60‐HA, or Htt‐Q60‐3R‐HA (three lysine residues replaced by arginines) and the plasmids indicated. Cells were lysed under denaturing conditions (1% SDS), followed by immunoprecipitation of Htt via the HA tag (1% Triton X‐100, 0.1% SDS). Immunopurified proteins were detected by Western blotting using the M1 ubiquitin‐specific antibody 1E3 and anti‐HA.
-
GSilencing of OTULIN increases linear ubiquitination of Htt‐Q97. A linear ubiquitination assay was performed as described in (F) with HEK293T cells transfected with Htt‐Q97‐HA and control siRNA or OTULIN siRNA.
-
HSDS‐insoluble Htt‐polyQ species are modified by linear ubiquitin chains. HEK293T cells expressing Htt‐Q97‐HA or Htt‐Q97‐GFP were lysed under denaturing conditions in 1.5% SDS. After centrifugation, the pellets containing the SDS‐insoluble aggregates (SDS‐insoluble fraction) were dissolved in formic acid. Formic acid‐dissolved aggregates were analyzed by immunoblotting using the M1 ubiquitin‐specific 1F11/3F5/Y102L antibody.
-
IEndogenous NEMO co‐localizes with Htt‐Q97 aggregates. NEMO (red) and Htt‐Q97‐GFP (green) were analyzed by immunocytochemistry in SH‐SY5Y cells. DAPI (blue). Scale bar, 10 μm.
-
JEndogenous Optineurin co‐localizes with Htt‐Q97 aggregates. Optineurin (red) and Htt‐Q97‐GFP (green) were analyzed by immunocytochemistry in SH‐SY5Y cells. DAPI (blue). Scale bar, 10 μm.
Linear ubiquitination is essential for canonical NF‐κB activation in response to various stimuli (Haas et al, 2009; Rahighi et al, 2009; Tokunaga et al, 2009; Niu et al, 2011; Lafont et al, 2017). To address the possibility that linear ubiquitination decreased Htt‐Q97 toxicity by activating NF‐κB‐mediated gene transcription, we used the NF‐κB super‐repressor IκB‐2S/A (serines 32 and 36 replaced by alanines), which blocked nuclear translocation of the NF‐κB subunit p65 (Sun et al, 1996). The protective activity of HOIP in cells co‐expressing Htt‐Q97 and IκB‐2S/A was not impaired (Fig 4E), revealing that the effects of linear ubiquitination on aggregate toxicity cannot be explained by a transcriptional activity of NF‐κB.
The Htt exon 1 fragment used to study Htt aggregation consists of an N‐terminal region of 17 amino acids (N17), followed by the polyQ tract and a proline‐rich region. The N17 domain contains three lysine residues (K6, K9, and K15) that can be modified by ubiquitin (Steffan et al, 2004; Bhat et al, 2014; Juenemann et al, 2015). To test whether Htt is a direct substrate of LUBAC, we mutated K6, K9, and K15 to arginines (Htt‐Q60‐3R). To avoid misinterpretation due to ubiquitination of GFP, we used HA as a tag, which lacks lysine residues. Cells expressing LUBAC and either Htt‐Q60 or Htt‐Q60‐3R were harvested under denaturing conditions, and Htt was immunoprecipitated via the HA tag. The immunopellet was analyzed by immunoblotting using the 1E3 antibody specific for M1‐linked ubiquitin or anti‐HA to assess immunoprecipitated Htt. We detected M1‐ubiquitin‐positive signals for SDS‐soluble Htt‐Q60 in the presence of catalytically active HOIP but not when the C885A HOIP mutant was expressed (Fig 4F). Signal intensity was strongly reduced for the Htt‐Q60‐3R mutant. Increased linear ubiquitination of SDS‐soluble Htt‐Q97 was also observed after OTULIN silencing (Fig 4G). In addition, we analyzed SDS‐insoluble, formic acid‐soluble Htt under endogenous expression of LUBAC components, which confirmed the presence of M1‐linked ubiquitin at Htt‐Q97 (Fig 4H).
The presence of M1‐linked ubiquitin at misfolded Htt‐polyQ prompted us to test whether proteins specifically binding to linear ubiquitin chains are also recruited to Htt‐polyQ. NEMO (NF‐κB essential modifier), the regulatory component of the IκB kinase complex, and Optineurin, an autophagy receptor and NF‐κB regulator, bind M1 ubiquitin through their UBAN domains (ubiquitin binding in ABIN and NEMO; Laplantine et al, 2009; Lo et al, 2009; Rahighi et al, 2009). Indeed, we observed enrichment of both endogenous NEMO and Optineurin at Htt‐Q97 by immunocytochemistry (Figs 4I and J, and EV4A and B) and filter retardation assays (Fig EV4C and D). NEMO binding to Htt‐Q97 did not occur in HOIP KO cells but was restored after HOIP expression, indicating HOIP‐dependent recruitment of NEMO to Htt (Fig EV4E). In conclusion, recruitment of LUBAC components to Htt‐polyQ remodels the surface of misfolded Htt species with linear ubiquitin chains and proteins specifically binding to M1‐linked ubiquitin. Htt‐polyQ oligomers seem to be the major targets for LUBAC‐mediated quality control, since not only SDS‐insoluble Htt‐polyQ aggregates but also SDS‐soluble aggregate precursors were found to be modified with M1‐linked ubiquitin.
Figure EV4. Endogenous NEMO and Optineurin are recruited to Htt‐Q97 (related to Fig 4).
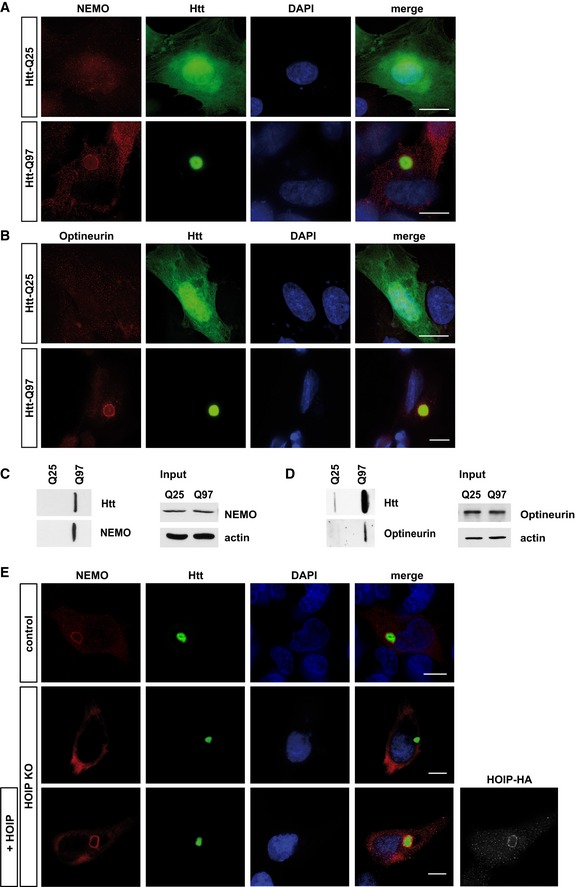
-
A, BSH‐SY5Y cells expressing Htt‐Q25‐GFP (upper panel) or Htt‐Q97‐GFP (lower panel) were stained for (A) NEMO (red) or (B) Optineurin (red) and DAPI (blue). Scale bar, 10 μm.
-
C, DHtt‐Q25‐ or Htt‐Q97‐expressing HEK293T were analyzed by filter retardation assays. Both endogenous NEMO (C) and Optineurin (D) are retained on a cellulose acetate membrane together with SDS‐insoluble Htt‐Q97.
-
EHOIP is required to recruit NEMO to Htt‐Q97. NEMO (red) and Htt‐Q97‐GFP (green) were analyzed by immunocytochemistry in WT HAP1 cells (control), HOIP KO HAP1 cells, or HOIP KO HAP1 cells reconstituted with HOIP. DAPI (blue). Scale bar, 10 μm.
Source data are available online for this figure.
Sequestration of the transcription factor Sp1 to Htt‐polyQ is modulated by linear ubiquitination
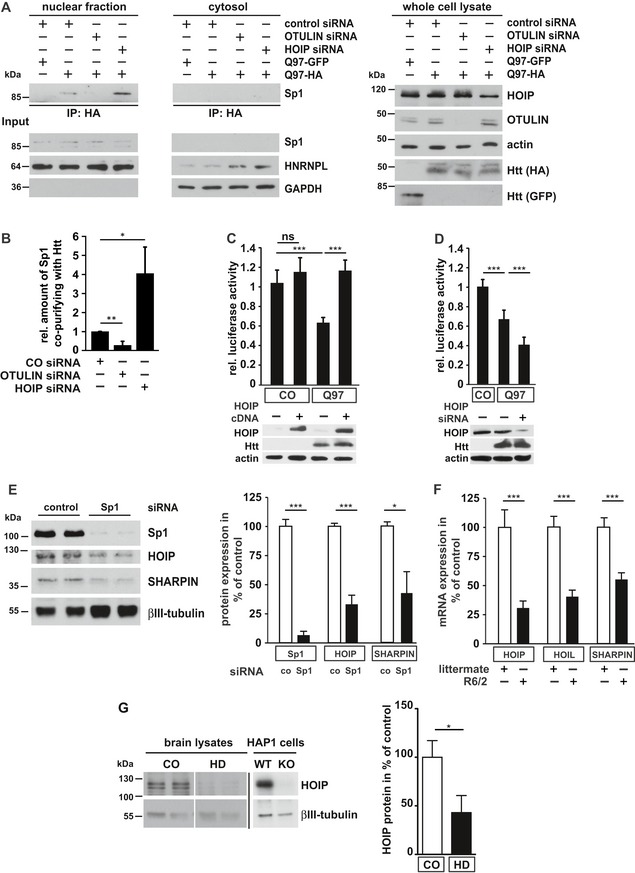
Next, we analyzed how the interactive surface of Htt‐polyQ is affected by linear ubiquitination. A major pathogenic mechanism contributing to toxic effects of mutant Htt is transcriptional dysregulation caused by the interaction of the polyglutamine tract with low complexity sequence domains often found in transcription factors (Kim et al, 2016; Li et al, 2016). To test whether modification of Htt‐Q97 by linear ubiquitin chains may shield Htt from unfavorable interactions, we chose the transcription factor Sp1 that has been reported to interact with soluble species of mutant Htt, causing disruption of Sp1 transcriptional activity (Dunah et al, 2002; Li et al, 2002; Zhai et al, 2005; Paul et al, 2014). Co‐immunoprecipitation experiments confirmed binding of endogenous Sp1 to SDS‐soluble Htt‐Q97 (Fig 5A, lane 2). Importantly, increased linear ubiquitination upon OTULIN silencing significantly decreased sequestration of Sp1 by Htt‐Q97, whereas decreased linear ubiquitination upon HOIP silencing increased the amount of Sp1 bound to Htt‐Q97 (Fig 5A and B). Moreover, luciferase reporter assays indicated that Sp1‐mediated transcription was reduced in Htt‐Q97‐expressing cells (Fig 5C). Impaired Sp1 function could be restored by increasing linear ubiquitination (Fig 5C), whereas HOIP silencing further decreased Sp1‐mediated transcription in Htt‐Q97‐expressing cells (Fig 5D).
Figure 5. Linear ubiquitination of Htt‐Q97 prevents sequestration of the transcription factor Sp1.
-
AThe interaction between Htt‐Q97 and Sp1 is decreased by linear ubiquitination. HEK293T cells were co‐transfected with Htt‐Q97‐HA and the siRNAs indicated. Htt‐Q97‐GFP was transfected as a control for specific immunoprecipitation of HA‐Htt‐Q97. Nuclear (left panel) and cytosolic fractions (middle panel) were subjected to an anti‐HA immunoprecipitation followed by Western blotting using an anti‐Sp1 antibody. Right panel: input controls (whole cell lysates).
-
BQuantification of the interaction between Htt‐Q97 and Sp1 in nuclear fractions. Shown is the relative amount of Sp1 co‐purifying with Htt‐Q97. Signal intensities were normalized to nuclear Sp1. Data are displayed as mean ± SD and were analyzed by two independent Mann–Whitney U‐tests, n = 4.
-
C, DImpairment of Sp1‐mediated transcription by Htt‐Q97 is modified by HOIP. (C) Shown is the relative luciferase activity in HEK293T cells expressing Htt‐Q97‐HA, a Sp1 luciferase reporter construct, and HOIP cDNA. Data are displayed as mean ± SD and were analyzed by Kruskal–Wallis test followed by Dunn's multiple comparison test, n = 9–11. (D) Shown is the relative luciferase activity in HEK293T cells expressing Htt‐Q97‐HA, a Sp1 luciferase reporter construct, and HOIP siRNA. Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test, n = 9.
-
ESp1 silencing decreases HOIP and SHARPIN protein expression. SH‐SY5Y cells were transfected with control or Sp1‐specific siRNAs and analyzed for protein expression 2 days after transfection by immunoblotting (left panel). Quantification of normalized Sp1, HOIP, and SHARPIN protein expression levels (right panel). Data are displayed as mean ± SEM with n = 5 from unpaired two‐tailed Student's t‐tests.
-
FHOIP‐, HOIL‐1L‐, and SHARPIN‐specific mRNAs are decreased in R6/2 striatum. Quantification of mRNA from the striatum of 9‐ to 12‐week‐old R6/2 mice and non‐transgenic littermates. Data are displayed as mean ± SD with n = 6–9 biological replicates from unpaired two‐tailed Student's t‐tests.
-
GHOIP protein levels are decreased in human HD brain. Total protein lysates from human brain (anterior cingulate cortex) were analyzed by immunoblotting using antibodies against HOIP and βIII‐tubulin. HOIP‐positive signals were normalized to βIII‐tubulin. To verify the specificity of the HOIP antibody, WT and HOIP KO HAP1 cells were analyzed in parallel on a different gel. Data are displayed as mean ± SEM with n = 5 for control and n = 7 for HD patients from unpaired two‐tailed Student's t‐tests.
Since linear ubiquitination decreased Htt proteotoxicity, we wondered whether this protective modification is dysregulated in HD. A bioinformatical promoter analysis of HOIP, HOIL‐1L, and SHARPIN revealed that all three LUBAC components share the presence of abundant and highly conserved Sp1 binding sites (Fig EV5A and B). In the human promoter sequences, 19 (HOIP), 27 (HOIL‐1L), and 19 (SHARPIN) Sp1 binding sites are significantly overrepresented showing Z scores of 6.75, 10.81, and 6.13, respectively, indicative of co‐regulation of LUBAC genes by Sp1. To test the functional relevance of Sp1 binding sites in LUBAC promoters, we silenced Sp1 expression in SH‐SY5Y cells and quantified expression of LUBAC components. Commercially available antibodies against HOIL‐1L were not suited to detect endogenous HOIL‐1L; we therefore concentrated on HOIP and SHARPIN. In Sp1 knockdown cells, expression levels of HOIP and SHARPIN were significantly reduced to about 32 and 44% of expression levels in control cells, respectively (Fig 5E). In addition, HOIP‐, HOIL‐1L‐, and SHARPIN‐specific mRNAs isolated from the striatum of 9‐ to 12‐week‐old R6/2 mice were markedly decreased in comparison with gender‐matched non‐transgenic littermates (Fig 5F). Moreover, HOIP protein levels were significantly reduced in human HD cortices compared to non‐HD control cortices analyzed by immunoblotting (Fig 5G), suggesting that expression of LUBAC components is dysregulated in HD.
Figure EV5. Presence of highly conserved SP1 binding sites in LUBAC component genes (related to Figs 5 and 6).
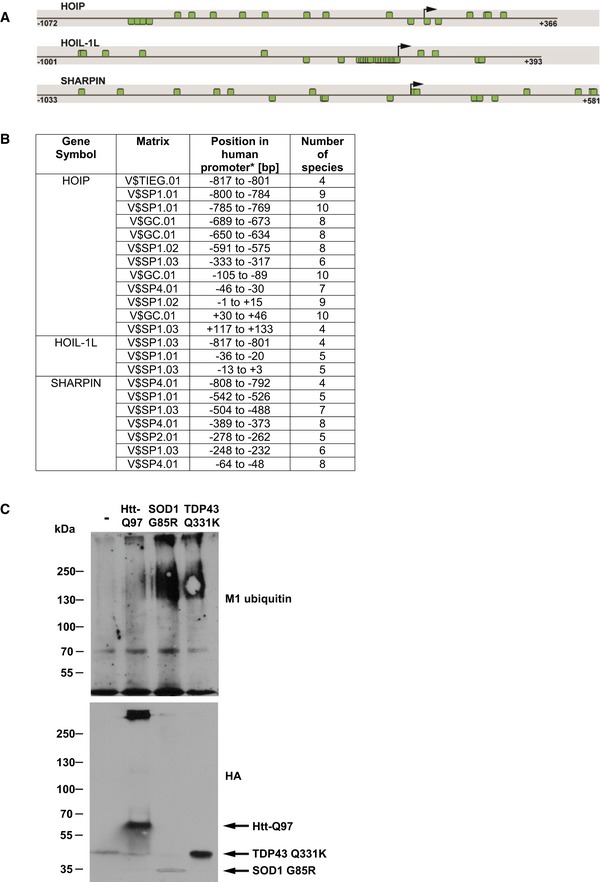
- In silico promoter analysis of HOIP, HOIL‐1L, and SHARPIN. Promoter sequence of human HOIP, HOIL‐1L, and SHARPIN showing SP1 binding sites. The black arrow indicates the transcription start site (TSS), and the positions are denoted relative to the TSS. Predicted SP1 binding sites are highlighted by green boxes. Binding sites above each line are located on the plus strand, whereas binding sites below the line are on the minus strand.
- Species conservation of V$SP1F binding sites in the promoter sequences of HOIL, HOIL‐1L, and SHARPIN (*relative to the transcriptional start site).
- SDS‐insoluble SOD1‐G85R, TDP‐43‐Q331K, and Htt‐Q97‐HA are modified by linear ubiquitin chains. HEK293T cells expressing Htt‐Q97‐HA, SOD1‐G85R‐HA, or TDP‐43‐Q331K‐HA were lysed under denaturing conditions in 1.5% SDS. After centrifugation, the pellets containing the SDS‐insoluble aggregates (SDS‐insoluble fraction) were dissolved in formic acid. Formic acid‐dissolved aggregates were analyzed by immunoblotting using the M1 ubiquitin‐specific 1F11/3F5/Y102L antibody.
Source data are available online for this figure.
Linear ubiquitination occurs at various protein aggregates and promotes removal of misfolded species via the proteasome
Based on the fact that HOIP is recruited to Htt‐polyQ in a p97/VCP‐dependent manner, we wondered whether HOIP binding to protein aggregates is a general phenomenon. Recruitment of HOIP and linear ubiquitination under endogenous LUBAC expression was also observed at other protein aggregates associated with neurodegenerative diseases, such as mutant Ataxin‐3, TDP‐43, SOD1, and Optineurin in SH‐SY5Y cells (Fig 6A and B) and in primary neurons (Fig 6C). Linear ubiquitination assays with SOD1 G85R and TDP‐43 Q331K revealed that similar to Htt‐polyQ, SDS‐soluble aggregate precursors are modified by linear ubiquitin chains upon increased expression of LUBAC or OTULIN silencing (Fig 6D). M1 ubiquitin‐positive signals were also observed for SDS‐insoluble SOD1 G85R and TDP‐43 Q331K species under endogenous LUBAC expression (Fig EV5C).
Figure 6. Linear ubiquitin chains are found at multiple protein aggregates.
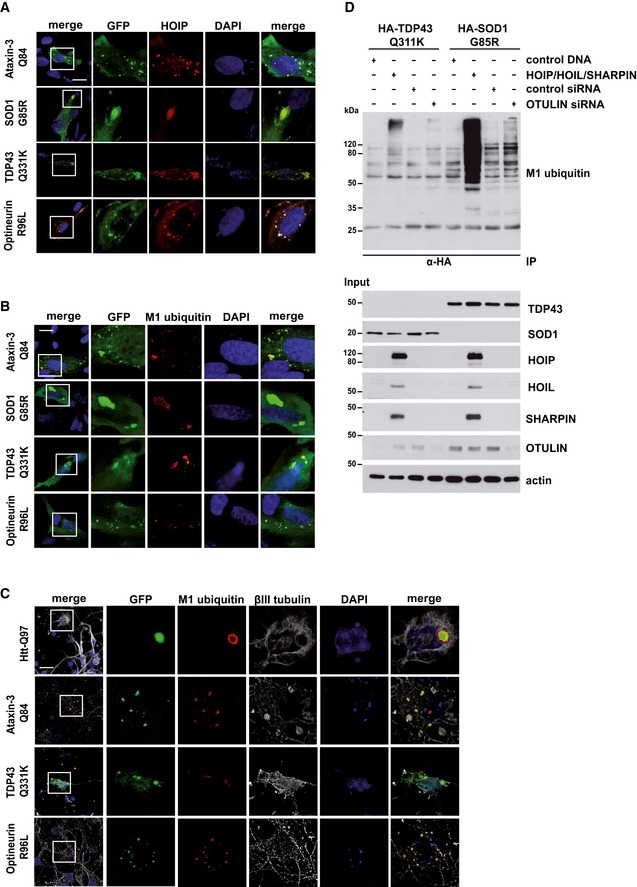
-
AHOIP is recruited to disease‐associated protein aggregates formed by Ataxin‐3‐Q84, SOD1 G85R, TDP‐43 Q331K, or Optineurin R96L. SH‐SY5Y cells co‐expressing disease‐associated proteins (green) and HOIP (red) were analyzed by immunocytochemistry; DAPI (blue). Scale bar, 20 μm.
-
B, CLinear ubiquitin accumulates at disease‐associated protein aggregates under endogenous LUBAC expression. M1‐linked ubiquitin (red) and disease‐associated proteins (green) were analyzed by immunocytochemistry in SH‐SY5Y cells using the 1E3 antibody (Millipore) (B) or primary striatal neurons using the 1F11/3F5/Y102L antibody (Genentech) (C). DAPI (blue). Scale bar, 20 μm.
-
DSDS‐soluble SOD G85R and TDP‐43 Q331K species are modified by linear ubiquitin chains. HEK293T cells were co‐transfected with disease‐associated proteins and either with the plasmids indicated or with control siRNA or OTULIN siRNA. Forty‐eight hours after transfection, cells were lysed under denaturing conditions (1% SDS), followed by immunoprecipitation of SOD G85R and TDP‐43 Q331K via the HA tag (1% Triton X‐100, 0.1% SDS). Immunopurified proteins were detected by Western blotting using the M1 ubiquitin‐specific antibody 1E3.
Next, we tested whether linear ubiquitination affects the cellular fate of misfolded protein species. HOIP silencing increased the percentage of transfected SH‐SY5Y cells with Htt‐Q97 aggregates detected by immunocytochemistry, whereas OTULIN silencing or increased expression of WT HOIP but not HOIP C885A had the opposite effect (Fig 7A and B). In the presence of the NF‐κB super‐repressor IκB‐2S/A, the effect of HOIP on Htt‐Q97 aggregates was not affected, arguing for an NF‐κB‐independent activity (Fig 7C). To assess the impact of autophagosomal degradation on this effect, we used mouse embryonic fibroblasts (MEFs) from either WT or ATG5 KO mice that are defective in autophagy (Kuma et al, 2004). Increased expression of WT HOIP but not catalytically inactive HOIP C885A decreased the number of cells with Htt‐Q97 aggregates in both WT and ATG5 KO MEFs, indicating an autophagy‐independent effect of HOIP (Fig 7D). However, HOIP was not effective in WT or ATG5 KO MEFs treated with the proteasomal inhibitor MG‐132, suggesting that linear ubiquitination promotes proteasomal degradation of Htt‐Q97 (Fig 7D). Consistent with this notion, we observed a similar effect in SH‐SY5Y cells expressing TDP‐43 Q331K. HOIP silencing increased and OTULIN silencing decreased the percentage of transfected SH‐SY5Y cells with visible TDP‐43 inclusions (Fig 7E). Vice versa, increased expression of WT HOIP but not catalytically inactive HOIP C885A decreased the fraction of cells with aggregates (Fig 7F). The effect of WT HOIP was abrogated in the presence of the proteasomal inhibitor MG‐132 or the p97/VCP inhibitor NSM‐873 (Fig 7F). HOIP C885A in all conditions significantly increased the number of cells with TDP‐43 aggregates, indicative of a dominant negative effect, most probably by competing with endogenous HOIP (Fig 7F). The combination of MG‐132 and NSM‐873 did not result in an additive effect, suggesting that p97/VCP and the proteasome function in the same pathway in removing misfolded TDP‐43 species (Fig 7G).
Figure 7. Linear ubiquitination decreases the number of cells with aggregates in a proteasome‐dependent manner.
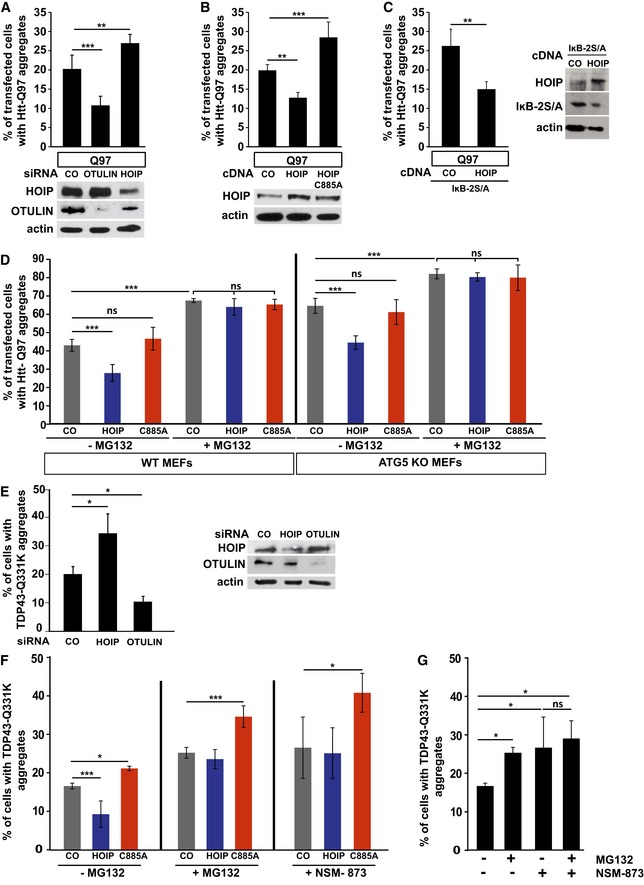
- Linear ubiquitination reduces the fraction of cells with Htt‐Q97 aggregates. SH‐SY5Y cells were transfected with Htt‐Q97 and either control siRNA, HOIP siRNA, or OTULIN siRNA. Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test, n = 9. Expression levels of HOIP and OTULIN were analyzed by Western blotting.
- Linear ubiquitination reduces the fraction of cells with Htt‐Q97 aggregates. SH‐SY5Y cells were transfected with Htt‐Q97 and either WT HOIP or the catalytically inactive HOIP mutant C885A. Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test, n = 5. Expression levels of WT and mutant HOIP and OTULIN were analyzed by Western blotting.
- The effect of HOIP on Htt‐Q97 aggregation is independent of NF‐κB signaling. SH‐SY5Y cells transiently transfected with Htt‐Q97 and either control vector (CO) or WT HOIP plus the NF‐κB super‐repressor IκB‐2S/A were analyzed by immunocytochemistry. Data are mean ± SD with n = 5 from an unpaired two‐tailed Student's t‐test. Expression levels of WT HOIP and IκB‐2S/A were analyzed by Western blotting.
- Catalytically active HOIP decreases the number of cells with Htt‐Q97 aggregates independently of autophagy. WT and ATG5 KO MEFs were transiently transfected with Htt‐Q97‐GFP and either control vector (CO), HOIP, or HOIP C885A. Cells were treated 24 h after transfection with the proteasomal inhibitor MG132 (1 μM, 16 h). Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Tukey's multiple comparison test, n = 7.
- Linear ubiquitination reduces the fraction of cells with TDP‐43 Q331K aggregates. SH‐SY5Y cells were transfected with TDP‐43 Q331K and either control siRNA, HOIP siRNA, or OTULIN siRNA. Data are displayed as mean ± SD and were analyzed by Kruskal–Wallis test, followed by Dunn's multiple comparison test, n = 7. Expression levels of HOIP and OTULIN were analyzed by Western blotting.
- Catalytically active HOIP decreases the number of cells with TDP‐43 aggregates dependent on proteasomal degradation. SH‐SY5Y cells were transfected with TDP‐43 Q331K and either control vector (CO), HOIP, or HOIP C885A. Cells were treated 48 h after transfection with the proteasomal inhibitor MG132 (1 μM) or the p97/VCP inhibitor NSM‐873 (1 μM) for 3 h. Data are displayed as mean ± SD and were analyzed by one‐way ANOVA followed by Dunnett's multiple comparison test, n = 5.
- Inhibition of the proteasome and p97/VCP does not have additive effects on the degradation of misfolded TDP‐43 species. SH‐SY5Y cells expressing TDP‐43 Q331K were treated 48 h after transfection with MG132 (1 μM) and/or NSM‐873 (1 μM) for 3 h. All data are displayed as mean ± SD, and were analyzed by one‐way ANOVA followed by Bonferroni's multiple comparison test, n = 5.
Discussion
In neurodegenerative diseases, the accumulation of misfolded proteins perturbs protein homeostasis, resulting in progressive neuronal dysfunction. An emerging concept in the regulation of protein quality control pathways is the complexity of ubiquitin signaling based on the multitude of possible ubiquitin polymers, including heterotypic (mixed or branched) chains. Our study identified linear ubiquitination mediated by LUBAC as a potent protein quality control mechanism that decreases the toxic potential of misfolded protein species and promotes their removal. In support of our findings, it has been reported that linear ubiquitination mediated by LUBEL, the Drosophila orthologue of HOIP, protects flies against toxicity induced by heat shock (Asaoka et al, 2016). Notably, LUBAC assembles M1‐linked ubiquitin preferentially on pre‐existing K63‐linked ubiquitin and presumably other linkages (Emmerich et al, 2013; Fiil et al, 2013; Hrdinka et al, 2016). Our data support the notion that M1‐linked ubiquitin is added to ubiquitination sites in the N‐terminal domain of Htt. However, ubiquitination of proteins recruited to Htt or LUBAC, such as NEMO, seems to contribute to the ubiquitin coating of protein aggregates. Linear ubiquitin chains are not only present at Htt aggregates but also on oligomeric aggregate precursors; therefore, it is conceivable that LUBAC also affects the aggregation process.
The presence of LUBAC, linear polyubiquitin, and proteins interacting with LUBAC components and/or M1‐linked ubiquitin, such as NEMO, remodels the interactive surface of misfolded protein species, thereby preventing unfavorable interactions. As an example, endogenous expression levels of HOIP or OTULIN conversely affected the sequestration of Sp1 by Htt‐polyQ, a transcription factor that has been reported to be dysregulated in HD (Dunah et al, 2002; Li et al, 2002; Zhai et al, 2005). Since Sp1 transcriptionally regulates HOIP, HOIL‐1L, and SHARPIN, Sp1 dysfunction decreases LUBAC expression, presumably impairing efficient linear ubiquitination of misfolded Htt in HD.
LUBAC is required for the activation of NF‐κB, which is implicated in immune and inflammatory signaling as well as cell death regulation (rev. in Kupka et al, 2016; Rittinger & Ikeda, 2017). Strikingly, the effects of HOIP on Htt‐polyQ‐induced proteotoxicity and aggregation were unaffected by the presence of the NF‐κB super‐repressor mutant IκB that efficiently blocks NF‐κB transcriptional activity. These results indicate that LUBAC has a function in protein quality control that is NF‐κB‐independent, although LUBAC‐mediated effects on NF‐κB may also play a role in HD.
It has recently been discovered that LUBAC synthesizes linear ubiquitin chains at the surface of intracellular, cytosol‐invading bacteria, such as Salmonella Typhimurium. As a consequence, the pathogen–host interface is modified to allow local activation of NF‐κB and recruitment of autophagy receptors to promote clearance of bacteria by xenophagy, thereby restricting bacterial proliferation (Noad et al, 2017; van Wijk et al, 2017). Linear ubiquitination at misfolded cytoplasmic protein assemblies obviously pursues a similar purpose, yet via different mechanisms. First, prerequisites for the recruitment of HOIP to bacteria are the NFZ domains and the catalytic activity of HOIP (Noad et al, 2017). Binding of HOIP to misfolded Htt is independent of the NZF domains and catalytic activity, but requires the N‐terminal PUB domain that interacts with the PIM domains of p97/VCP or OTULIN (Elliott et al, 2014; Schaeffer et al, 2014). Indeed, we observed that the presence of p97/VCP at misfolded Htt species favors HOIP recruitment and vice versa, indicating a feed‐forward mechanism. Second, local activation of NF‐κB at protein aggregates may occur, since binding of p65 to mutant Htt has been described; however, NF‐κB transcriptional activity is not required for the beneficial effects of HOIP on Htt‐polyQ‐induced toxicity and removal of misfolded species. Third, although the autophagy adaptor protein Optineurin is recruited to linear polyubiquitin at mutant Htt, HOIP preferentially promotes degradation of misfolded protein species via the proteasome. Inhibition of either the proteasome or p97/VCP blocks the effects of HOIP on mutant Htt or TDP‐43 removal, whereas HOIP is still effective in autophagy‐deficient ATG5 KO cells.
In conclusion, our study revealed that cytoplasmic protein assemblies are sensed as a special kind of “cellular pathogens” or danger‐associated molecular patterns, resulting in the recruitment and activation of LUBAC as part of a quality control pathway related to innate immunity. It will be interesting to see whether linear ubiquitination also limits spreading of misfolded protein species to neighboring cells, as it does with intracellular bacteria. Our findings emphasize common features of protein quality control in diverse disease entities and thus can help to unveil potential future therapeutic avenues. Since linear ubiquitination is a highly specific process, pharmacological modulation of this pathway seems feasible.
Materials and Methods
Reagents and Tools table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti‐β‐actin | Sigma‐Aldrich | Cat#A5316, RRID: AB_476743 |
| Rabbit monoclonal anti‐cleaved caspase‐3 | Cell Signaling Technology | Cat#9664, RRID: AB_2070042 |
| Mouse monoclonal anti‐GAPDH | Thermo Fisher Scientific | Cat#AM4300, RRID: AB_2536381 |
| Mouse monoclonal anti‐GFP | Thermo Fisher Scientific | Cat#14‐6674, RRID: AB_2572899 |
| Rabbit polyclonal anti‐HA | Sigma‐Aldrich | Cat#H6908, RRID: AB_260070 |
| Mouse monoclonal anti‐HA | Covance Research Products Inc | Cat#NMS‐101R, RIDD: N/A |
| Mouse monoclonal anti‐HHARI | Santa Cruz Biotechnology | Cat#sc‐390763, RRID: N/A |
| Rabbit polyclonal anti‐HNRNPL | Sigma‐Aldrich | Cat#HPA052661, RRID: N/A |
| Mouse monoclonal anti‐HOIL‐1L | Santa Cruz Biotechnology | Cat#sc‐365523, RRID: AB_10841591 |
| Goat polyclonal anti‐HOIP | Acris | Cat#AP16062PU‐N, RRID: AB_1928030 |
| Rabbit polyclonal anti‐HOIP | Bethyl | Cat#A303‐560A, RRID: AB_10949139 |
| Mouse monoclonal anti‐Huntingtin | Millipore | Cat#MAB2166, RRID: AB_11213141 |
| Mouse monoclonal anti‐Huntingtin | Millipore | Cat#MAB5374, RRID: AB_10055116 |
| Rabbit polyclonal anti‐IKKβ | Cell Signaling Technology | Cat#2370, RRID: AB_2122154 |
| Rabbit monoclonal anti‐M1 ubiquitin | Millipore | Cat#MABS199, RRID: AB_2576212 |
| Mouse monoclonal anti‐NEMO | Santa Cruz Biotechnology | Cat#sc‐8032, RRID: AB_627786 |
| Rabbit polyclonal anti‐NEMO | Santa Cruz Biotechnology | Cat#sc‐8330, RRID: AB_2124846 |
| Rabbit polyclonal anti‐NF kappa B p65 | Santa Cruz Biotechnology | Cat#sc‐372, RRID: AB_632037 |
| Rabbit polyclonal anti‐OTULIN | Cell Signaling Technology | Cat#14127, RRID: AB_2576213 |
| Rabbit polyclonal anti‐Optineurin | Sigma‐Aldrich | Cat#HPA003279, RRID: AB_1079527 |
| Rabbit monoclonal anti‐phospho‐c‐jun | Cell Signaling Technology | Cat#3270, RRID: AB_2129575 |
| Rabbit polyclonal anti‐Sp1 | Santa Cruz Biotechnology | Cat#sc‐59, RRID: AB_2171050 |
| Mouse monoclonal anti‐VCP | Abcam | Cat#ab11433, RRID: AB_298039 |
| Biological samples | ||
| Mouse: C57BL/6J cortex/hippocampus | Charles River | Cat#000664 |
| Mouse: R6/2: B6CBA‐Tg(HDexon1)62Gpb/3J striatum/cortex | The Jackson Laboratory | JAX: 006494 |
| Chemical, peptides and recombinant proteins | ||
| Human TNF‐α | Peprotech | Cat#300‐01A |
| MG‐132 | Sigma‐Aldrich | Cat#M8699 |
| NSM‐873 | Sigma‐Aldrich | Cat#SML1128 |
| Experimental models: cell lines | ||
| SH‐SY5Y | Leibniz‐Institut DSMZ‐Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH https://www.dsmz.de/home.html | DSMZ no.: ACC 209 |
| HEK293T | ATCC https://www.lgcstandards-atcc.org/ | ATCC® CRL‐1573™ |
| HAP1 WT and HAP1 RNF31 KO | Horizon | https://www.horizondiscovery.com |
| Oligonucleotides | ||
| HOIP siRNA human | ||
| GGUACUGGCGUGGUGUCAAGUUUAA | Invitrogen | HSS123836 |
| GAGAUGUGCUGCGAUUAUAUGGCUA | HSS123837 | |
| CACCACCCUCGAGACUGCCUCUUCU | HSS182838 | |
| OTULIN siRNA human | ||
| GGAAGAAUGAGGACCUGGUUGAUAA | Invitrogen | HSS131920 |
| GCGGAGGAAUAUAGCCUCUAUGAAG | HSS131921 | |
| UCUCCAAGUACAACACGGAAGAAUU | HSS131922 | |
| VCP siRNA human | ||
| CCCAAGAUGGAUGAAUUGCAGUUGU | Invitrogen | HSS111263 |
| GGAGUUCAAAGUGGUGGAAACAGAU | HSS111264 | |
| CACCUUCCAAGGGAGUUCUGUUCUA | HSS187663 | |
| RT–PCR primer mouse HOIP | ||
| Fwd: CCCAGTGTCACCAGACCTTC | Sigma‐Aldrich | N/A |
| Rev: CCTCACAACTCCGTCCTCTG | ||
| RT–PCR primer mouse HOIL‐1L | ||
| Fwd: TCTCCCCAACACAGGACATC | Sigma‐Aldrich | N/A |
| Rev: AAATGGTGACGGTGTGCAT | ||
| RT–PCR primer mouse SHARPIN | ||
| Fwd: TCCAGTTCCAGTCTCCCAAG | Sigma‐Aldrich | N/A |
| Rev: GCGGTTTGAGGCATTGAT | ||
| RT–PCR primer mouse EIF4A2 | ||
| Fwd: CGATCTACCTACCAATCGTGAA | Sigma‐Aldrich | N/A |
| Rev: ACCTTTCCTCCCAAATCGAC | ||
Methods and Protocols
Cell lines
HEK293T cells
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 100 IU/ml penicillin–100 μg/ml streptomycin sulfate.
SH‐SY5Y cells
Cells were cultured in Dulbecco's modified Eagle's medium F‐12 (DMEM/F12) supplemented with 15% (v/v) fetal bovine serum (FBS), 100 IU/ml penicillin–100 μg/ml streptomycin sulfate, and 1% non‐essential amino acids.
HAP1 WT and HAP1 HOIP (RNF31) KO
Cells were cultured in Iscove's modified Dulbecco's medium (IMDM) supplemented with 10% (v/v) FBS, 100 IU/ml penicillin–100 μg/ml streptomycin sulfate, and 8 mM L‐glutamine.
Mouse embryonic fibroblasts (MEFs)
MEFs derived from wild‐type or ATG5 KO mice (Kuma et al, 2004) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 100 IU/ml penicillin–100 μg/ml streptomycin sulfate.
Primary hippocampal or striatal neuron cultures
Mouse hippocampus or striatum from embryonic day 18 (E18) embryos was dissected into ice‐cold HBSS and dissociated in acetylated trypsin (Sigma) for 20 min at 37°C. Subsequently, tissues were rinsed with HBSS and treated with DNase I to break down DNA and triturated with pipette tips. The cell suspension was resuspended in Neurobasal medium (Invitrogen) and plated in 24‐well plates on coverslips coated with poly‐L‐lysine (Sigma) and laminin (Sigma) at a concentration of 100,000 neurons/well for immunocytochemistry. Forty‐five minutes after seeding, medium was removed and replaced by fresh one. Primary neurons were cultured in Neurobasal medium with B27 (Invitrogen) and glutamine (Invitrogen) in a humidified incubator (37°C, 5% CO2). At day 5, in vitro striatal neurons were transfected using 2 μl of Lipofectamine 2000 per well. One day after transfection, primary neurons were fixed in 4% paraformaldehyde/4% glucose in PBS for 10 min, permeabilized in 0.1% (v/v) Triton X‐100 in PBS and subjected to immunocytochemistry. Animal protocols were performed in compliance with institutional and governmental regulations.
Human brain sections
Huntington disease (HD) and control brain tissues were provided by the Neurobiobank Munich, Ludwig‐Maximilians‐University (LMU) Munich, and the Institute of Anatomy, Ruhr University Bochum (RUB), Germany, according to the guidelines of the local ethical committees (LMU, Reg. No. 345‐13; RUB, Reg. No. 17‐5939). Available demographic and clinical data are listed in the following table.
| Case No. | Age at death | Sex | Number of CAG repeats | Postmortem delay (in h) |
|---|---|---|---|---|
| HD01 | 38 | Male | No information yet | 10 |
| HD02 | 74 | Male | 40 | 34–38 |
| HD03 | 72 | Male | > 40 | 15.0 |
| HD04 | 57 | Male | 45 | 48 |
| HD05 | 70 | Male | 42 | 56 |
| HD06 | 66 | Male | 47 | < 15 |
| HD07 | 45 | Female | 54 | 8 |
| CO01 | 37 | Female | Not applicable | 27 |
| CO02 | 76 | Female | Not applicable | 26 |
| CO03 | 70 | Male | Not applicable | 39 |
| CO04 | 53 | Male | Not applicable | 29 |
| CO06 | 64 | Female | Not applicable | 32 |
Transfection and siRNA knockdown
Transient transfection was performed with Lipofectamine and Plus reagent (Invitrogen) according to the manufacturer's instructions. For RNA interference, cells were transfected with stealth siRNA oligos (Invitrogen) using Lipofectamine RNAiMAX (Invitrogen) or Lipofectamine 2000 (Invitrogen) for co‐transfection of siRNA and DNA plasmids. Per 3.5 cm dish, 1.5 μg Htt plasmid and 0.2 μg HOIP or OTULIN plasmid were co‐transfected.
Preparation of brain lysates
Frozen tissue (anterior cingulate cortex) was homogenized in lysis buffer containing 50 mM HEPES pH 7.5, 300 mM NaCl, 250 mM sucrose, 1% NP‐40, 0.2% sarkosyl, 1 mM PMSF, and 2 × Complete Protease Inhibitor (Roche) at 10% (w/v) with 20 strokes in a Glass‐Teflon douncer. Subsequently, samples were spun (20,000 × g for 10 min at 4°C), the supernatant was collected, and SDS loading buffer was added prior to SDS–PAGE and immunoblotting onto 0.2‐μm nitrocellulose membrane.
Treatment of cells with inhibitors
For the induction of linear ubiquitin chains, cells were stressed with TNF‐α (Peprotech, Cat#300‐01A) for 15 min with 25 ng/ml. Proteasomal inhibition was conducted by treatment of the cells with 1 μM MG132 (Sigma‐Aldrich, Cat#M8699). Transfected cells were either stressed for 16 h with 1 μM MG132 24 h post‐transfection or with 1 μM MG132 48 h post‐transfection for 3 h. Inhibition of p97/VCP was obtained by treatment for 3 h with 1 μM NSM‐873 (Sigma‐Aldrich, Cat#SML1128) 48 h post‐transfection.
Immunoblotting
SDS–PAGE and Western blotting were described previously (Winklhofer et al, 2003). Antigens were detected with the enhanced chemiluminescence (ECL) detection system (Promega).
DNA constructs
The following constructs were described previously: wild‐type (WT) human HOIP, HOIL‐1L, SHARPIN, HA‐HHARI (Muller‐Rischart et al, 2013), WT p97/VCP (Ritz et al, 2011). The HOIP mutant C885A was generated by site‐directed mutagenesis using the following primers: HOIP‐C885A‐fwd 5′‐CCATGTCACTTTCACGTACCGCGGAGGAGCCCGGTCC‐3′ and HOIP‐C885A‐rev 5′‐CCTGGCCCGAGGAGGCGCCATGCACTTTCACTGTACCCTG‐3′. The amplified fragments were digested with EcoRI and NotI and cloned into the pcDNA3.1+ vector (Invitrogen). HA‐HOIP 1‐697 was generated using the following primers: HOIP 1‐697‐fwd 5′‐GGAGACCCAAGCTGGCTAGC‐3′ and HOIP 1‐697‐rev 5′‐ATATTCTAGATTACTGGGCAAGCAAGCGGCGCAG‐3′. The amplified fragment was digested with EcoRI and XbaI and cloned into pc.DNA3.1‐N‐HA. HA‐HOIP ΔNZF1, HA‐HOIP ΔNZF2, HA‐HOIP ΔNZF1 + 2, HA‐HOIP ΔUBA, and HA‐HOIP ΔPUB were generated by overlap extension PCR. The amplified fragments were generated using the following primers: HOIP ΔUBA‐fwd: 5′‐ATATGCGGCCGCATGTACCCATACGATGTTCCAGATTAC‐3′, HOIP ΔUBA‐rev: 5′‐ATATGAATTCCTTCCGCCTGCGGGGGATAC‐3′, HOIP ΔUBA‐left arm‐rev: 5′‐GCGCGCTCTAGACTACTTCCGCCTGCGGGGGATACTC‐3′, HOIP ΔUBA‐right‐arm‐fwd: 5′‐GCAGCAGCAGGCTCGGATCCACTAGTCCAGTGTGGTG‐3′; HOIP ΔNZF1‐fwd: 5′‐GATCCACTAGTCCAGTGTGGTGG‐3′, HOIP Δ NZF1‐rev: 5′‐ CAGCGGGTTTAAACGGGCCCTC‐3′, HOIP Δ NZF1‐left arm‐rev: 5′‐GCTGGGAGGCTGAAGATCAGGTTCTAGGCCTCCAGTTC, HOIP Δ NZF1‐right arm‐fwd: 5′‐GAACCTGATCTTCAGCCTCCCAGCTTGGTGGTG‐3′; HOIP Δ NZF2‐fwd: 5′‐GATCCACTAGTCCAGTGTGGTGG‐3′, HOIP Δ NZF2‐rev: 5′‐CAGCGGGTTTAAACGGGCCCTC‐3′, HOIP Δ NZF2‐left arm‐rev: 5′‐TTGTGCTGGAATGGCAGAGGCCAGCAAAGCATCC‐3′, HOIP Δ NZF2‐right arm‐fwd: 5′‐CTGGCCTCTGCCATTCCAGCACAACATGCCCCCCG; HOIP ΔPUB‐fwd: 5′‐GATCCACTAGTCCAGTGTGGTGG‐3′, HOIP ΔPUB‐rev: 5′‐CAGCGGGTTTAAACGGGCCCTC‐3′, HOIP ΔPUB‐left arm‐rev: CTGCAACGCTTTCCCCGAAGGGCAGGAGGAG, HOIP ΔPUB‐right arm‐fwd: CTTCGGGGAAAGCGTTGCAGCGGACAAGGC. The amplified fragments were digested with EcoRI and BamHI and cloned into pcDNA3.1‐N‐HA. VCP ΔPIM was generated using the following primers: VCPΔPIM‐fwd 5′‐GTGGATCCATGGCCTCTGGAGCCG‐3′ and VCP ΔPIM‐rev 5′‐GTCTCGAGTTAATCGTCATTGTCTTCTGTGTACACATTGCC‐3′. The amplified fragment was digested with BamHI and XhoI and cloned into pcDNA3.1+ vector (Invitrogen). WT p97/VCP‐V5 and VCP ΔPIM‐V5 were cloned into pcDNA3.1‐N‐V5 by amplification with the primers p97/VCP‐V5‐fwd 5′‐GGAGACCCAAGCTGGCTAGC‐3′, WT p97/VCP‐V5‐rev 5′‐GGCTGATCAGCGGGTTTAAACG‐3′, and p97/VCP ΔPIM‐V‐rev 5′‐ATATCTCGAGTTAATCGTCATTGTCTTCTGTGTACACATTG‐3′ and subsequent digestion of the amplified fragments with BamHI and XhoI. GFP‐Htt‐Q25 and GFP‐Htt‐Q97 were cloned into the pRC/CMV vector (Invitrogen). HA‐Htt‐Q97 was generated by PCR using the primers HA‐Htt‐Q97‐fwd 5′‐CCGAATTCGCGACCCTGGAAAAGCTGATG‐3′ and HA‐HttQ97‐rev 5′‐CAACTCGAGTCACGGTCGGTGCAGCGG‐3′. The amplified fragment was digested with EcoRI and XhoI and cloned into pcDNA3.1‐N‐HA. HA‐Htt‐Q60 and HA‐Htt‐Q60‐3R were generated by gene synthesis (Eurofins Genomics) and subcloned from pEX‐K4 into pcDNA3.1+ by digestion with KpnI and NotI. GFP‐tagged Htt‐Q60 and Htt‐Q60‐3R were generated by PCR using the primers GFP‐Htt‐Q60‐fwd 5′‐GTGAATTCATGGCGACACTCGAGAAGTTGATGAAAGCGTTCGA‐3′, GFP‐Htt‐Q60 and Q60‐3R‐rev 5′‐GTGGATCCCCTGGCCTGTGTAAGG‐3′, and GFP‐Htt‐Q60‐3R‐fwd 5′‐GTGAATTCATGGCGACACTCGAGCGGTTGATGCGAGCGTTCGA‐3′. The amplified fragments were digested with EcoRI and BamHI and cloned into the pEGFP‐N1 vector (Clontech). Human TDP‐43 (TARDBP) cDNA was amplified from cDNA preparation from HEK293 cells (TARDBP_for: ATGTCTGAATATATTCGGGTAACC and TARDBP_rev: CTACATTCCCCAGCCAGAAGA). The Q331K mutations were generated by PCR using mutant‐specific primers (Q331k_for: CACTAAAGAGCAGTTGGGGTAT and Q331K_rev: ATACCCCAACTGCTCTTTAGTG). TARDBP wild‐type and mutant DNAs were inserted in the pEGFP‐N1 vector (Clontech) allowing the expression of C‐terminally tagged TDP‐43. HA‐TDP‐43 Q331K was generated by PCR using the primers HA‐TDP‐fwd 5′‐ATATGGATCCTCTGAATATATTCGGGTAACCG‐3′ and HA‐TDP‐43‐rev 5′‐ATATGCGGCCGCCTACATTCCCCAGCCAGAAGAC‐3′. The amplified fragment was digested with BamHI and NotI and cloned into pcDNA3.1‐N‐HA vector. pEGFP‐C1‐Ataxin‐3‐Q84 was ordered from Addgene (#22123; Chai et al, 2002). SOD G85R‐GFP was described before (Witan et al, 2009). SOD1 G85R‐HA was generated by PCR using the primers SOD1 G85R‐HA‐fwd 5′‐AAAAGAATTCATGGCCACGAAGGCCGTG‐3′ and SOD1 G85R‐HA‐rev 5′‐AAAAGCGGCCGCTTGGGCGATCCCAATTACACC‐3′. The amplified fragment was digested with EcoRI and NotI and cloned into pcDNA3.1‐C‐HA vector. Optineurin R96L was ordered from Addgene (#68846; Turturro et al, 2014). IκB‐2S/A was generated by overlap extension PCR using the following primers: mut‐IκB‐2S‐fwd 5′‐CCACGACGCCGGCCTGGACGCCATGAAAG‐3′, mut‐IκB‐2S ‐rev 5′‐CGTCTTTCATGGCGTCCAGGCCGGCGTCG‐3′, BamHI‐IκB2S ‐fwd 5′‐ATATGGATCCTTCCAGGCGGCCGAGCGCCCCCAGGAG‐3′, and IκB2S‐NotI‐rev 5′‐ATATGCGGCCGCCTATAACGTCAGACGCTGGCCTCCAAACACACAGTC‐3′. The amplified fragments were digested with BamHI and NotI and cloned into the pcDNA3.1‐N‐HA vector.
Immunocytochemistry
SY‐SY5Y, HAP1, MEFs, and neuronal cells were cultivated on glass coverslips (Laboratory Glassware Marienfeld). 24–72 h after transfection, cells were fixed for 15 min with 4% paraformaldehyde in PBS pH 7.4 and permeabilized with 0.2% (v/v) Triton X‐100 in PBS for 10 min or 0.5% saponin, 1% BSA in PBS for 45 min at room temperature. After blocking with 1% BSA or 5% goat/donkey serum, cells were stained with primary antibodies at a dilution of 1:100 to 1:1,000 in PBS or 0.5% saponin, 1% BSA in PBS at 4°C overnight, washed with PBS, and incubated with fluorescent dye‐conjugated secondary antibodies Alexa Fluor 488, 555, or 647 (Thermo Scientific), at a dilution of 1:1,000 for 1 h at room temperature. Cells were mounted in Fluoroshield with DAPI (Sigma) or Prolong Gold (Thermo Fisher Scientific).
Microscopy
Fluorescence microscopy was performed using a Zeiss ELYRA PS.1 equipped with an LSM880 (Carl Zeiss, Jena) or C2+ system (Nikon) with a 63× oil or 100× oil immersion objective. Super‐resolution images were generated by the Structured Illumination Microscopy (SIM) technology from five phases and three to five rotations of the SIM grid and acquisition times of 100–150 ms. All channels were acquired independent and subsequently. Raw confocal SIM images were processed and generated using the ZEN software (ZEN Black 2.1; Carl Zeiss, Jena).
Immunohistochemistry of human brain sections
For immunofluorescence histochemistry, paraffin‐embedded sections (5 μm) of the middle frontal gyrus were deparaffinized with xylene and ethanol and briefly washed with deionized water. After antigen retrieval through microwaving in 100 mM citrate buffer pH 6.0 and blocking with 2% FBS, the primary antibody was added and incubation at 4°C overnight followed. After washing with 0.02% Brij35 (Thermo Fisher, Germany) in PBS, sections were incubated with the secondary Alexa‐conjugated antibody. For nuclear staining, DAPI was used. Images were taken with the confocal microscope LSM710 (Zeiss, Jena).
For peroxidase immunohistochemistry sections, retrieval procedure was performed as described above followed by blocking of endogenous peroxidase with 5% H2O2 in methanol. Then, sections were transferred in PBS with 0.02% Brij35 and blocked with 2% FBS in PBS. Incubation with the primary antibody was performed overnight at 4°C. After rinsing with 0.02% Brij35 in PBS, antibody binding was detected and enhanced by DCS Super Vision 2 HRP‐Polymer‐Kit (DCS, Germany) using the chromogen DAB. Counterstaining with hematoxylin for cellular structures was performed. Microscopic images were obtained with a BX50 microscope and Cell‐D software (Olympus).
For M1 ubiquitin 1E3‐ and Htt (mEM48) immunofluorescence histochemistry, 4% paraformaldehyde‐fixed blocks of human HD frontal cortices were frozen and incubated as described (Petrasch‐Parwez et al, 2004). Cryosections (12 μm) were mounted on Superfrost slides (Thermo Fisher Scientific, Germany), dried at 40°C for 30 min, blocked for 1 h in 10% NGS with 0.3% Triton X‐100 in PBS, and incubated with the M1 ubiquitin antibody (1:100) in the blocking solution for 48 h at 4°C. Then, sections were washed with PBS, blocked with 2% BSA in PBS for 1 h, and incubated with the mEM48 (1:500) in 2% BSA in PBS for 2 h at room temperature. Finally, sections were washed and mounted with Prolong Gold with DAPI (Thermo Fisher Scientific). Confocal images were obtained using a Zeiss ELYRA PS.1 equipped with an LSM880 (Zeiss). Super‐resolution and confocal images were processed using the ZEN2.1 software (Zeiss).
Immunohistochemistry of R6/2 mouse brain sections
Twelve‐week‐old R6/2 male mice were anesthetized [25% (v/v) ketamine, 5% (v/v) xylazine, and 2.5% (v/v) vetranquil in NaCl 0.9% (w/v)] and transcardially perfused with PBS and 4% (w/v) paraformaldehyde sequentially. Brains were harvested immediately, post‐fixed in 4% (w/v) paraformaldehyde for 24 h followed by cryoprotection in 30% (w/v) sucrose solution, and embedded in Tissue‐Tek O.C.T (Sakura Finetek, the Netherlands). For M1 ubiquitin and Htt immunofluorescent staining, coronal brain sections (10 μm) (Bregma 1.10–0.14 mm) were cooked in 100 mM citrate buffer pH 6.0 for 30 min, washed in PBS, and blocked with 10% (v/v) goat serum and 0.5% (v/v) Triton X‐100 in PGBA [0.1% (w/v) gelatin in phosphate buffer 0.1 M] for 1 h at room temperature. Brain sections were stained with primary antibodies 1E3 (1:100) and mEM48 (1:750) in blocking solution for 48 h at 4°C, washed with PBS, and incubated with fluorescent dye‐conjugated secondary antibodies (1:1,000) in blocking solution for 1 h at room temperature. After washing in PBS, brain sections were mounted in Fluoromount with DAPI (SouthernBiotech, USA). Images were obtained using a Zeiss ELYRA PS.1 equipped with an LSM880. Super‐resolution and confocal images were processed using the ZEN2.1 software.
Analysis of proteotoxic stress
SH‐SY5Y or neuronal cells were transfected with the indicated constructs for 72 h. Afterward, cells were fixed and immunostained with anti‐phospho‐c‐Jun antibody and analyzed by fluorescence microscopy (Nikon Eclipse E400).
Filter retardation assay
To detect Htt aggregates, transfected HEK293T cells were lysed in 1% (v/v) Triton X‐100, 50 mM MgCl2, and 0.2 mg/ml DNase I in PBS. After centrifugation (180,000 × g for 30 min at 4°C), the pellet was resuspended in 2% SDS in 100 mM Tris (pH 7.0). After 1‐h incubation at room temperature, the homogenates were diluted 1:5 in 100 mM Tris (pH 7.0) and filtered through a cellulose acetate membrane with 0.2 μm pore size (GE) using a Slot Blot Blotting Manifold (Hoeffer).
Analysis of SDS‐insoluble proteins
The method was performed as previously described by Juenemann et al (2015). In brief, HEK293T cells expressing the proteins of interest were grown on 10‐cm dishes and lysed under denaturing conditions in TEX buffer [70 mM Tris–HCL pH 6.8, 1.5% SDS (w/v), 20% glycerol (v/v)] 3 days after transfection. After vortexing for 10 s, the samples were heated up to 99°C and DNA was sheared by passing the samples 15 times through a 23‐Gauge needle. DTT was added to the samples at a final concentration of 50 mM and boiled for 10 min at 99°C. Afterward, the samples were centrifuged for 60 min (20,000 rcf, room temperature). The SDS‐insoluble pellets were dissolved in 70 μl 100% formic acid by incubation of the samples for 40 min at 37°C while shaking at 1,000 rpm. Formic acid was evaporated overnight at 30°C using a Speedvac system (Eppendorf). The remaining protein pellets were solved in Laemmli sample buffer and boiled for 10 min at 95°C. Formic acid‐dissolved aggregates were analyzed by immunoblotting using the M1 ubiquitin‐specific 1F11/3F5/Y102L antibody.
Linear ubiquitination assays
HEK293T cells were transfected with the indicated constructs and harvested in cold PBS. Subsequently, cells were lysed in 1% (w/v) SDS in PBS and boiled for 30 min at 95°C. The lysates were cleared by centrifugation at 20,000 × g. 1% (v/v) Triton X‐100 in PBS was added to the lysates in order to reduce the SDS concentration to 0.1%. Samples were incubated overnight with anti‐HA agarose beads at 4°C with gentle rotation. Beads were washed three times with 1% (v/v) Triton X‐100 in PBS. Immunopurified proteins were eluted by adding Laemmli sample buffer and boiling for 10 min and then analyzed by Western blotting using the M1 ubiquitin‐specific antibody 1E3.
Co‐immunoprecipitation
HEK293T or HAP1 cells transfected with the indicated constructs were harvested in cold PBS and subsequently lysed in 1% (v/v) Triton X‐100 in PBS supplemented with Protease Inhibitor Cocktail (Roche), N‐Ethylmaleimide, and sodium orthovanadate. The lysates were cleared by centrifugation at 20,000 × g and incubated overnight with anti‐HA agarose beads at 4°C with gentle rotation. Beads were washed three times with lysis buffer. Immunopurified proteins were eluted by adding Laemmli sample buffer and boiling for 10 min.
Luciferase reporter assays
HEK293T cells were transiently transfected with Htt‐Q97 or control plasmid and the Cignal Sp1 reporter construct (Qiagen) for 72 h. Cells were harvested and lysed in 1× reporter lysis buffer (Promega). Lysates were cleared by centrifugation, and luciferase activity was determined luminometrically (LB96V, Berthold Technologies) by the luciferase assay system (Promega). Measured values were analyzed with WinGlow software (Berthold Technologies) and normalized to protein levels. Quantifications are based on at least three independent experiments performed in triplicates.
Quantitative RT–PCR
mRNA of 9‐ to 12‐week‐old R6/2 male mice and their wild‐type littermates from in‐house colonies was prepared from fresh‐frozen striatal brain tissue. RNA was prepared from ~70 mg of brain tissue, homogenized using QIAzol lysis reagent (Qiagen). Subsequently, RNA was purified using RNeasy Lipid Tissue Mini Kit (Qiagen) following the manufacturer's instructions. 1,000 ng RNA was reverse‐transcribed into cDNA using the iScript™ cDNA Synthesis Kit (Bio‐RAD, CA, USA). cDNA was diluted 1:5 in PCR‐grade dH2O, and 1.5 μl of the cDNA‐mix was used per 20 μl real‐time PCR with 0.25 μM of each primer and 2 × FastStart Essential DNA Green Master Mix (Roche). For each target and reference gene, samples were run in triplicates on a LightCycler 96 (Roche). On each plate, relevant negative controls were run. Melt curves were studied for all assays, with the Tm checked to be within known specifications for each assay. Standard curves were run for each primer pair. Relative quantification of HOIP, HOIL‐1L, and SHARPIN was performed using the method.
In silico promoter analysis
All sequences analyzed are from the promoter sequence retrieval database ElDorado 12‐2016 (Genomatix, Germany), which is based on NCBI build GRCh38. The following Genomatix/Entrez Gene identifiers were used: GXP_294687/55072 (RNF31), GXP_153638/10616 (RBCK1), GXP_51601/81858 (SHARPIN). Promoter sequences of RNF31 (HOIP), RBCK1 (HOIL‐1L), and SHARPIN were aligned using the DiAlign TF program (Cartharius et al, 2005) of the Genomatix software suite GEMS Launcher in order to evaluate overall promoter similarity and to predict common (conserved) binding sites of the V$SP1F and further matrix families. The corresponding position weight matrices were derived from the Matrix Family Library Version 10.0 (October 2016), which contains 210 matrix families for vertebrates. The V$SP1F family comprises the following matrices (in the IUPAC notation) due to their similar binding profiles and functionality of the corresponding transcription factors: GC box elements V$GC.01 (NRGGGGCGGGGCNK); Sp1 transcription factor V$SP1.01 (SGGGGCGGGGN), V$SP1.02 (NVKGGGCGGRGYBN), V$SP1.03 (NNGGGGCGGGGNN); Sp2 transcription factor V$SP2.01 (TGGGCGGGACTA); Sp4 transcription factor V$SP4.01 (NNAGGGGGCGKNNN), V$SP4.02 (NNWAGGCGTGNC); TGF beta‐inducible early gene V$TIEG.01 (GGGGTGTGT).
The promoter sequence of each human gene (HOIP, HOIL‐1L, and SHARPIN) was aligned separately to the homologous promoter sequences of different species in order to determine evolutionarily conserved binding sites of the V$SP1F family. From the ElDorado 12‐2016 database, the promoter sequences of HOIP, HOIL‐1L, and SHARPIN for 14 different species were derived, i.e., Homo sapiens (human), Macaca mulatta (rhesus monkey), Pan troglodytes (chimpanzee), Mus musculus (mouse), Rattus norvegicus (rat), Oryctolagus cuniculus (rabbit), Equus caballus (horse), Bos taurus (cow), Sus scrofa (pig), Monodelphis domestica (opossum), Canis lupus (dog), Xenopus tropicalis (frog), Danio rerio (zebrafish), and Gallus gallus (chicken). If V$SP1F binding sites are aligned in the same region of promoter sequences from different species with help the DiAlign TF program, they were defined as conserved.
In order to calculate statistical overrepresentation of transcription factor binding sites (TFBSs), the program Overrepresented TFBS (Genomatix) was used. A Z‐score of a binomial distribution comparing the rate of occurrence of TFBS (V$SP1F) in the input set to the expected rate estimated from the background set divided by the standard deviation was determined (Ho Sui et al, 2005). For computation of the Z‐score, the number of nucleotides in all human promoters (i.e., the background sequences), the number of detected TFBSs in the background sequences, the number of the nucleotides of the input promoter sequence, and the number of detected TFBSs in the input sequence were derived from MatBase (Matrix Library 10.0, December 2016) and the ElDorado 12‐2016 database (Genomatix).
Mass spectrometry analysis of ubiquitination topologies
Samples prepared for mass spectrometry analysis were digested using a gel‐assisted sample preparation protocol on a Digest‐O‐R (Axel Semrau, Germany; Kanashova et al, 2015). Briefly, samples were loaded into the top of an SDS–PAGE gel before running for 10 min at 80 V to concentrate proteins into the gel. The small band was then removed and shredded by centrifugation through a Spin‐X centrifuge tube. The shredded gel was loaded into a filter plate before washing with 10 cycles of acetonitrile (ACN) and 50 mM ammonium bicarbonate (ABC) each with 5‐min incubation before vacuum removal through the plate filter. Gel pieces were treated with 10 mM dithiothreitol for 30 min before removing and treatment with 55 mM iodoacetamide for 20 min. A mix of lys‐C and trypsin was then added at a 1:50 ratio to the sample and incubated for 10 h. After digest, peptides were eluted from the gel pieces by alternating buffer washes of ACN and 50 mM ABC with centrifuging at 100 g (Hettich Zentrifugen, Germany) for 5 min between each buffer addition to collect elute.
Eluted peptides were centrifuged in a SpeedVac (Eppendorf, Belgium) till dry before resuspension with 1% ACN 0.05% trifluoroacetic acid (TFA) and the addition of heavy labeled standard peptides. Clean‐up was performed using Sep‐Pak tC18 96‐well μElution Plate (Waters, Belgium) following the manufacturer's protocols. Eluted peptides from the C18 clean‐up were centrifuged in a SpeedVac (Eppendorf, Belgium) till dry before resuspension in 1% ACN 0.05% TFA.
Samples were analyzed on an UltiMate 3000 RSLCnano coupled to a Q Exactive™ Plus Hybrid Quadrupole‐Orbitrap™ Mass Spectrometer (Thermo Fisher Scientific). A gradient was formed on an Acclaim PepMap™ RSLC 75 μm × 15 cm C18 Column with buffer A (100% H2O, 0.1% FA) exchanged for buffer B (100% ACN, 0.1% FA) from 2 to 35% over 33 min and a flow rate of 300 nl per min.
A scheduled parallel reaction monitoring (sPRM) method was used. The MS cycle was started with a full scan at 35,000 resolution followed by PRM scans acquired at a resolving power of 70,000 with a normalized collision energy of 25. The quadrupole isolation window for the PRM events was set to 1 m/z unit with a 3‐min scheduling window. PRM scheduling and subsequent analysis were performed in Skyline (skyline.ms). Statistical analysis was subsequently performed in R (R‐project.org).
Quantification and statistical analysis
Data represent the mean ± SD or SEM (indicated in the figure legends) from n ≥ 3 biological replicates. For stable cell lines, 100–900 cells were assessed for each biological replicate and for primary cell culture of at least seven individual neurons (significance levels: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). All statistical analyses were performed by using GraphPad Prism (version 5; San Diego, CA, USA). To analyze the distribution of datasets (parametric/non‐parametric), Kolmogorov–Smirnov, D'Agostino–Pearson, and Shapiro–Wilk normality test were performed. Based on the outcome of these tests, appropriate parametric and non‐parametric tests were chosen. For the comparison of two independent parametric datasets, Student's t‐test was used. For the comparison of more than two parametric datasets, one‐way ANOVA was used. To correct for α‐error inflation resulting from multiple comparisons, ANOVA was followed by Bonferroni's, Dunnett's, or Tukey's post hoc multiple comparison tests. For the direct comparison of two non‐parametric datasets, Wilcoxon–Mann–Whitney (U‐test) was used, and for the comparison of more than two datasets, Kruskal–Wallis one‐way analysis of variance was performed. To correct for α‐error inflation resulting from multiple comparisons, Kruskal–Wallis test was followed by Dunn's post hoc multiple comparison test.
Author contributions
Conceptualization: KFW, JT; Methodology: KFW, JT, GD, EMW, VB, AS‐V, JM, NF, MP, CSc, CSh, EP‐P, AB, DT, TA; Investigation: EMW, VB, AS‐V, JM, NF, MP, CSc, AB, JL, CSh, PG, KM, DT, ACW, DAS, AS‐V, LA, LAB; Formal analysis: EMW, MP, VB, JM, CSc, CSh; Resources: EP‐P, TA, RG, GE, CSa, AMC, CB; Writing—original draft: KFW, JT; Writing—review and editing: KFW, JT, FUH, MSH; Visualization: EMW, VB, AS‐V, JM, MP, CSc, CSh, DT, DAS, JL, LA, LAB; Supervision: KFW, JT, FUH, MSH, GD, RG, GE, CSa, WW, CH; Project administration: KFW, JT; Funding acquisition: KFW.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Drs. A. Voigt for Htt‐polyQ plasmids, H. Meyer for the p97/VCP plasmid, N. Mizushima for ATG5 KO MEFs, L. Lanier for providing videos of striatal neuron preparation, and Genentech for providing the 1F11/3F5/Y102L antibody. We wish to thank the patients and their families for brain samples. The research leading to these results has received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (TA 167/6‐1, WI 2111/4‐1, WU 164/5‐1, SFB1177 to C.B.), the Munich Cluster for Systems Neurology (C.H., F.U.H., K.F.W., M.S.H., W.W.), Germany's Excellence Strategy—EXC‐2033—Projektnummer 390677874 (J.T., K.F.W.), the Hans and Ilse Breuer Foundation (M.P.), the German Federal Ministry of Education and Research through the Integrated Network MitoPD and HIT‐Tau (grants 031A430E and 01EK1605C to W.W.), and the Medical Faculty of the Ruhr University Bochum (FoRUM F832R‐2014 to K.F.W.). SR‐SIM microscopy was funded by the German Research Foundation and the State Government of North Rhine‐Westphalia (INST 213/840‐1 FUGG).
The EMBO Journal (2019) 38: e100730
See also: RL Wiseman (May 2019)
References
- Asaoka T, Almagro J, Ehrhardt C, Tsai I, Schleiffer A, Deszcz L, Junttila S, Ringrose L, Mechtler K, Kavirayani A, Gyenesei A, Hofmann K, Duchek P, Rittinger K, Ikeda F (2016) Linear ubiquitination by LUBEL has a role in Drosophila heat stress response. EMBO Rep 17: 1624–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR (2007) Global changes to the ubiquitin system in Huntington's disease. Nature 448: 704–708 [DOI] [PubMed] [Google Scholar]
- Bhat KP, Yan S, Wang CE, Li S, Li XJ (2014) Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin‐protein ligase E3A. Proc Natl Acad Sci USA 111: 5706–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, Meyer H (2018) VCP/p97‐mediated unfolding as a principle in protein homeostasis and signaling. Mol Cell 69: 182–194 [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21: 2933–2942 [DOI] [PubMed] [Google Scholar]
- Chai Y, Shao J, Miller VM, Williams A, Paulson HL (2002) Live‐cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci USA 99: 9310–9315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard RB, Walker JA, Marco‐Casanova P, Morgan NV, Titheradge HL, Elliott PR, McHale D, Maher ER, McKenzie ANJ, Komander D (2016) The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 166: 1215–1230 e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537–548 [DOI] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296: 2238–2243 [DOI] [PubMed] [Google Scholar]
- Elliott PR, Nielsen SV, Marco‐Casanova P, Fiil BK, Keusekotten K, Mailand N, Freund SM, Gyrd‐Hansen M, Komander D (2014) Molecular basis and regulation of OTULIN‐LUBAC interaction. Mol Cell 54: 335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, Cohen P (2013) Activation of the canonical IKK complex by K63/M1‐linked hybrid ubiquitin chains. Proc Natl Acad Sci USA 110: 15247–15252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiil BK, Damgaard RB, Wagner SA, Keusekotten K, Fritsch M, Bekker‐Jensen S, Mailand N, Choudhary C, Komander D, Gyrd‐Hansen M (2013) OTULIN restricts Met1‐linked ubiquitination to control innate immune signaling. Mol Cell 50: 818–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M, Charvin D, Caboche J (2004) Expanded huntingtin activates the c‐Jun terminal kinase/c‐Jun pathway prior to aggregate formation in striatal neurons in culture. Neuroscience 127: 859–870 [DOI] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H (2009) Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF‐R1 signaling complex and is required for TNF‐mediated gene induction. Mol Cell 36: 831–844 [DOI] [PubMed] [Google Scholar]
- Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, Maltzman A, de Sousa EMF, Hung J, Zeng Y, Verschueren E, Kirkpatrick DS, Vucic D, Lee WP, Roose‐Girma M, Newman RJ, Warming S, Hsiao YC, Komuves LG, Webster JD et al (2018) OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559: 120–124 [DOI] [PubMed] [Google Scholar]
- Hipp MS, Patel CN, Bersuker K, Riley BE, Kaiser SE, Shaler TA, Brandeis M, Kopito RR (2012) Indirect inhibition of 26S proteasome activity in a cellular model of Huntington's disease. J Cell Biol 196: 573–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW (2005) oPOSSUM: identification of over‐represented transcription factor binding sites in co‐expressed genes. Nucleic Acids Res 33: 3154–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdinka M, Fiil BK, Zucca M, Leske D, Bagola K, Yabal M, Elliott PR, Damgaard RB, Komander D, Jost PJ, Gyrd‐Hansen M (2016) CYLD limits Lys63‐ and Met1‐linked ubiquitin at receptor complexes to regulate innate immune signaling. Cell Rep 14: 2846–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdinka M, Gyrd‐Hansen M (2017) The Met1‐linked ubiquitin machinery: emerging themes of (De)regulation. Mol Cell 68: 265–280 [DOI] [PubMed] [Google Scholar]
- Iwai K (2012) Diverse ubiquitin signaling in NF‐kappaB activation. Trends Cell Biol 22: 355–364 [DOI] [PubMed] [Google Scholar]
- Iwai K, Fujita H, Sasaki Y (2014) Linear ubiquitin chains: NF‐kappaB signalling, cell death and beyond. Nat Rev Mol Cell Biol 15: 503–508 [DOI] [PubMed] [Google Scholar]
- Jordan IK (1991) Ethical issues in the genetic study of deafness. Ann N Y Acad Sci 630: 236–239 [DOI] [PubMed] [Google Scholar]
- Juenemann K, Wiemhoefer A, Reits EA (2015) Detection of ubiquitinated huntingtin species in intracellular aggregates. Front Mol Neurosci 8: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanashova T, Popp O, Orasche J, Karg E, Harndorf H, Stengel B, Sklorz M, Streibel T, Zimmermann R, Dittmar G (2015) Differential proteomic analysis of mouse macrophages exposed to adsorbate‐loaded heavy fuel oil derived combustion particles using an automated sample‐preparation workflow. Anal Bioanal Chem 407: 5965–5976 [DOI] [PubMed] [Google Scholar]
- Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, Wauer T, Hospenthal MK, Gyrd‐Hansen M, Krappmann D, Hofmann K, Komander D (2013) OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1‐linked polyubiquitin. Cell 153: 1312–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hosp F, Frottin F, Ge H, Mann M, Hayer‐Hartl M, Hartl FU (2016) Soluble oligomers of PolyQ‐expanded Huntingtin target a multiplicity of key cellular factors. Mol Cell 63: 951–964 [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Kupka S, Reichert M, Draber P, Walczak H (2016) Formation and removal of poly‐ubiquitin chains in the regulation of tumor necrosis factor‐induced gene activation and cell death. FEBS J 283: 2626–2639 [DOI] [PubMed] [Google Scholar]
- Lafont E, Kantari‐Mimoun C, Draber P, De Miguel D, Hartwig T, Reichert M, Kupka S, Shimizu Y, Taraborrelli L, Spit M, Sprick MR, Walczak H (2017) The linear ubiquitin chain assembly complex regulates TRAIL‐induced gene activation and cell death. EMBO J 36: 1147–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Veron M, Agou F, Israel A (2009) NEMO specifically recognizes K63‐linked poly‐ubiquitin chains through a new bipartite ubiquitin‐binding domain. EMBO J 28: 2885–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ (2002) Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 22: 1277–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu H, Dong P, Li D, Legant WR, Grimm JB, Lavis LD, Betzig E, Tjian R, Liu Z (2016) Real‐time imaging of Huntingtin aggregates diverting target search and gene transcription. eLife 5: e17056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YF (1998) Expression of polyglutamine‐expanded Huntingtin activates the SEK1‐JNK pathway and induces apoptosis in a hippocampal neuronal cell line. J Biol Chem 273: 28873–28877 [DOI] [PubMed] [Google Scholar]
- Lo YC, Lin SC, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, Eliezer D, Wu H (2009) Structural basis for recognition of diubiquitins by NEMO. Mol Cell 33: 602–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493–506 [DOI] [PubMed] [Google Scholar]
- Merienne K, Helmlinger D, Perkin GR, Devys D, Trottier Y (2003) Polyglutamine expansion induces a protein‐damaging stress connecting heat shock protein 70 to the JNK pathway. J Biol Chem 278: 16957–16967 [DOI] [PubMed] [Google Scholar]
- Muller‐Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, Lichtenthaler SF, Motori E, Hrelia S, Wurst W, Trumbach D, Langer T, Krappmann D, Dittmar G, Tatzelt J, Winklhofer KF (2013) The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell 49: 908–921 [DOI] [PubMed] [Google Scholar]
- Niu J, Shi Y, Iwai K, Wu ZH (2011) LUBAC regulates NF‐kappaB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J 30: 3741–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, Randow F (2017) LUBAC‐synthesized linear ubiquitin chains restrict cytosol‐invading bacteria by activating autophagy and NF‐kappaB. Nat Microbiol 2: 17063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM, Snyder SH (2014) Cystathionine gamma‐lyase deficiency mediates neurodegeneration in Huntington's disease. Nature 509: 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin V, Dufour N, Raoul C, Hassig R, Brouillet E, Aebischer P, Luthi‐Carter R, Deglon N (2009) Implication of the JNK pathway in a rat model of Huntington's disease. Exp Neurol 215: 191–200 [DOI] [PubMed] [Google Scholar]
- Petrasch‐Parwez E, Habbes HW, Weickert S, Lobbecke‐Schumacher M, Striedinger K, Wieczorek S, Dermietzel R, Epplen JT (2004) Fine‐structural analysis and connexin expression in the retina of a transgenic model of Huntington's disease. J Comp Neurol 479: 181–197 [DOI] [PubMed] [Google Scholar]
- Pouladi MA, Morton AJ, Hayden MR (2013) Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci 14: 708–721 [DOI] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I (2009) Specific recognition of linear ubiquitin chains by NEMO is important for NF‐kappaB activation. Cell 136: 1098–1109 [DOI] [PubMed] [Google Scholar]
- Rittinger K, Ikeda F (2017) Linear ubiquitin chains: enzymes, mechanisms and biology. Open Biol 7: 170026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz D, Vuk M, Kirchner P, Bug M, Schutz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, Glatter T, Gstaiger M, Aebersold R, Weihl CC, Meyer H (2011) Endolysosomal sorting of ubiquitylated caveolin‐1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol 13: 1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivkin E, Almeida SM, Ceccarelli DF, Juang YC, MacLean TA, Srikumar T, Huang H, Dunham WH, Fukumura R, Xie G, Gondo Y, Raught B, Gingras AC, Sicheri F, Cordes SP (2013) The linear ubiquitin‐specific deubiquitinase gumby regulates angiogenesis. Nature 498: 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scappini E, Koh TW, Martin NP, O'Bryan JP (2007) Intersectin enhances huntingtin aggregation and neurodegeneration through activation of c‐Jun‐NH2‐terminal kinase. Hum Mol Genet 16: 1862–1871 [DOI] [PubMed] [Google Scholar]
- Schaeffer V, Akutsu M, Olma MH, Gomes LC, Kawasaki M, Dikic I (2014) Binding of OTULIN to the PUB domain of HOIP controls NF‐kappaB signaling. Mol Cell 54: 349–361 [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE (1997) Huntingtin‐encoded polyglutamine expansions form amyloid‐like protein aggregates in vitro and in vivo . Cell 90: 549–558 [DOI] [PubMed] [Google Scholar]
- Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING‐IBR‐RING domain and the unique LDD extension. EMBO J 31: 3833–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL (2004) SUMO modification of Huntingtin and Huntington's disease pathology. Science 304: 100–104 [DOI] [PubMed] [Google Scholar]
- Stieglitz B, Morris‐Davies AC, Koliopoulos MG, Christodoulou E, Rittinger K (2012) LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep 13: 840–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz B, Rana RR, Koliopoulos MG, Morris‐Davies AC, Schaeffer V, Christodoulou E, Howell S, Brown NR, Dikic I, Rittinger K (2013) Structural basis for ligase‐specific conjugation of linear ubiquitin chains by HOIP. Nature 503: 422–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su KH, Cao J, Tang Z, Dai S, He Y, Sampson SB, Benjamin IJ, Dai C (2016) HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat Cell Biol 18: 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Elwood J, Greene WC (1996) Both amino‐ and carboxyl‐terminal sequences within I kappa B alpha regulate its inducible degradation. Mol Cell Biol 16: 1058–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Komander D (2016) Ubiquitin modifications. Cell Res 26: 399–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiuchi T, Nakagawa T, Tamiya H, Fujita H, Sasaki Y, Saeki Y, Takeda H, Sawasaki T, Buchberger A, Kimura T, Iwai K (2014) Suppression of LUBAC‐mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells 19: 254–272 [DOI] [PubMed] [Google Scholar]
- Taylor DM, Moser R, Regulier E, Breuillaud L, Dixon M, Beesen AA, Elliston L, Silva Santos Mde F, Kim J, Jones L, Goldstein DR, Ferrante RJ, Luthi‐Carter R (2013) MAP kinase phosphatase 1 (MKP‐1/DUSP1) is neuroprotective in Huntington's disease via additive effects of JNK and p38 inhibition. J Neurosci 33: 2313–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K (2009) Involvement of linear polyubiquitylation of NEMO in NF‐kappaB activation. Nat Cell Biol 11: 123–132 [DOI] [PubMed] [Google Scholar]
- Turturro S, Shen X, Shyam R, Yue BY, Ying H (2014) Effects of mutations and deletions in the human optineurin gene. Springerplus 3: 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJL, Fricke F, Herhaus L, Gupta J, Hotte K, Pampaloni F, Grumati P, Kaulich M, Sou YS, Komatsu M, Greten FR, Fulda S, Heilemann M, Dikic I (2017) Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF‐kappaB and restricts bacterial proliferation. Nat Microbiol 2: 17066 [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Henn IH, Kay‐Jackson PC, Heller U, Tatzelt J (2003) Inactivation of parkin by oxidative stress and C‐terminal truncations: a protective role of molecular chaperones. J Biol Chem 278: 47199–47208 [DOI] [PubMed] [Google Scholar]
- Witan H, Gorlovoy P, Kaya AM, Koziollek‐Drechsler I, Neumann H, Behl C, Clement AM (2009) Wild‐type Cu/Zn superoxide dismutase (SOD1) does not facilitate, but impedes the formation of protein aggregates of amyotrophic lateral sclerosis causing mutant SOD1. Neurobiol Dis 36: 331–342 [DOI] [PubMed] [Google Scholar]
- Yau R, Rape M (2016) The increasing complexity of the ubiquitin code. Nat Cell Biol 18: 579–586 [DOI] [PubMed] [Google Scholar]
- Zhai W, Jeong H, Cui L, Krainc D, Tjian R (2005) In vitro analysis of huntingtin‐mediated transcriptional repression reveals multiple transcription factor targets. Cell 123: 1241–1253 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7