

Abstract
This review describes catalytic asymmetric cycloaddition reactions of silyl-protected enoldiazo compounds for the construction of highly functionalized carbo- and heterocycles which possess one or more chiral center(s). The enoldiazo compound or its derivative, donor-acceptor cyclopropene, form electrophilic vinylogous metal carbene intermediates that combine stepwise with nucleophilic dipolar reactants to form products from [3+1]-, [3+2]-, [3+3]-, [3+4]-, and [3+5]-cycloaddition, generally in high yield and with exceptional stereocontrol and regioselectivity.
TOC Graphic
Review of recent advances in asymmetric catalytic cycloaddition reactions of silyl-protected enoldiazo compounds.
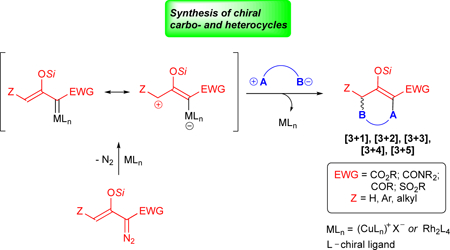
1. Introduction
A reaction in which two or more unsaturated molecules (or parts of the same molecule) combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity is defined (IUPAC) as cycloaddition.1 Occurring as concerted or stepwise processes, cycloaddition reactions are among the most useful synthetic constructions in organic chemistry.2 Of these transformations the concerted thermal [4+2]-cycloaddition – the Diels-Alder reaction – is, by far, the best known and most widely applied,2,3 and other concerted cycloaddition processes, especially Huisgen [3+2]-dipolar cycloadditions4 and photochemical [2+2]-cycloaddition,5 further expand the synthetic utility of this ring-forming methodology. However, recognition of the limitations of the symmetry-allowed processes has prompted alternative stepwise approaches to cycloaddition, the most significant of which have been organocatalytic and transition metal mediated approaches to cyclization.
Access to heterocyclic compounds and enantiocontrol have been principal limitations of concerted cycloaddition methodologies. Organocatalysis, especially as applied to [3+3]- and [3+2]-cycloaddition processes,6 and commonly based on vinyliminium ion chemistry, has afforded convenient stereocontrolled approaches to many heterocyclic compounds (Scheme 1). These stepwise reactions occur under mild conditions; and although intermolecular cyclization reactions are facile and can give high stereocontrol, greater stereocontrol is generally achieved in intramolecular examples. However, only one heteroatom is generally introduced, and relatively large amounts of the catalyst (20–30 mol% or even more) are often required.6a
Scheme 1.
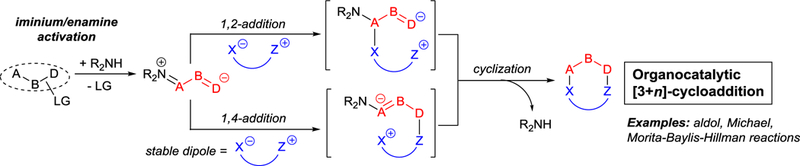
Organocatalytic [3+n]-cycloaddition.
Metal catalysts are widely recognized to provide new opportunities for highly selective cycloaddition reactions since complexation with the metal modifies reactivity and can improve selectivity.7 The carbene-based cyclopropanation reactions ([2+1]-cycloaddition) exemplify the enormity of the control in both reactivity and stereoselectivity that can be achieved.8 Transition metal catalyzed reactions that activate cyclopropanes, employ trimethylenemethane (TMM), or involve vinylcarbenes in dipolar cycloaddition processes have been reported,7 but in comparison with concerted cycloadditions or those promoted by organocatalysts, this approach is still in its infancy. Its perceived advantages are low catalyst loading, potential for high stereocontrol, and access to heterocyclic compounds having one or more heteroatoms.
Recently the transition metal catalyzed vinylcarbene approach to cycloaddition has shown considerable promise for highly chemo-, regio-, and enantio-selective cycloaddition reactions. Examples of [3+4]-,9 [3+3]-,10–14 [3+2]-,14–20 and even a [3+1]-21 and [3+5]-22 cycloaddition reactions have been reported using dipolar metallo-vinylcarbenes, generally in high yields with low catalyst loadings. The transformations of silyl-protected enoldiazo compounds (Scheme 2) involve initial nucleophilic attachment to the electrophilic vinylogous position of the metallo-vinylcarbene followed by electrophilic addition to the original carbenic center with displacement of the catalyst. High stereocontrol has been achieved with silyl-protected enoldiazo-acetates, -acetamides, -ketones, and -sulfones in most of these cycloaddition reactions, and also with styryldiazoacetates in [3+2]-cycloaddition reactions. Our earlier review of cycloaddition reactions involving silyl-protected enoldiazo compounds12 reported the breadth of these transformations mainly with diazoacetates and with dirhodium catalysts. This report documents the extensive growth in these reactions since 2016, their mechanistic implications for future expansion, and the applicability of chiral copper catalysts to compliment and improve previously documented performance with chiral dirhodium catalysts.
Scheme 2.
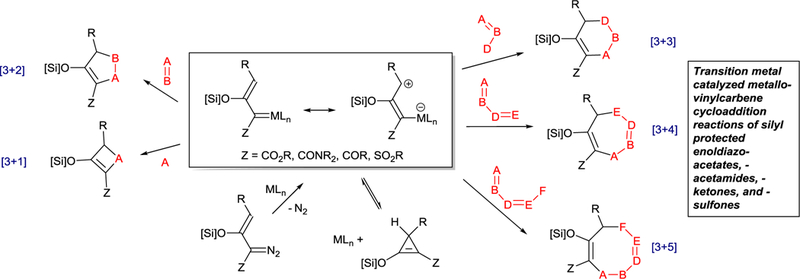
Transition metal catalyzed cycloaddition reactions of silyl-protected enol-diazo-acetates, -acetamides, -ketones, and -sulfones yield hetero- and carbocycles.
2. Asymmetric [3+1]-cycloaddition
The robust and efficient synthesis of cyclobutane and cyclobutene rings is important because this structural unit is present in many natural products and drugs,23 but it is commonly difficult to achieve because of ring strain. In addition to the well-known [2+2]-cycloaddition5,24 a methodology of [3+1]-cycloaddition that leads to the structurally diverse four-membered carbo- and hetero-cycles has recently been reported.25 Formal [3+1]-cycloaddition of diazo compounds that involves ring-expansion of α-diazocyclopropane compounds has found its application in the synthesis of cyclobutenes,26 azetines,27 and azetidines.28 However, the first enantioselective [3+1]-cycloaddition has recently been reported in which chiral copper(I)-catalyzed cycloaddition reactions of enoldiazoacetates 1 with sulfur ylides 2 (Scheme 3) form highly functionalized cyclobutenes in good yields and high stereocontrol.21 The initial step is the formation of a metal carbene from enoldiazoacetate 1 using CuOTf1/2∙Tol with chiral side-armed bisoxazoline (sabox) ligand 3 that facilitates dinitrogen extrusion. The nucleophilic addition of a sulfur ylide to the vinylogous position of the copper carbene intermediate, followed by the intramolecular cyclization with displacement of the sulfide (R3)2S and elimination of the copper(I) catalyst, results in tri- and tetrasubstituted cyclobutenes 4 with high stereocontrol. The nucleophilic displacement of the sulfide in the ring-forming reaction is the first example of an SN2-like process in an ylide transformation. Cyclobutenes 4 are also formed by cycloaddition with donor-acceptor (D-A) cyclopropenes 5 that are formed from and returned to the metal enol-carbene.21 Further examples of this novel transformation are expected to reveal their applicability to general carbene/ylide capture.
Scheme 3.
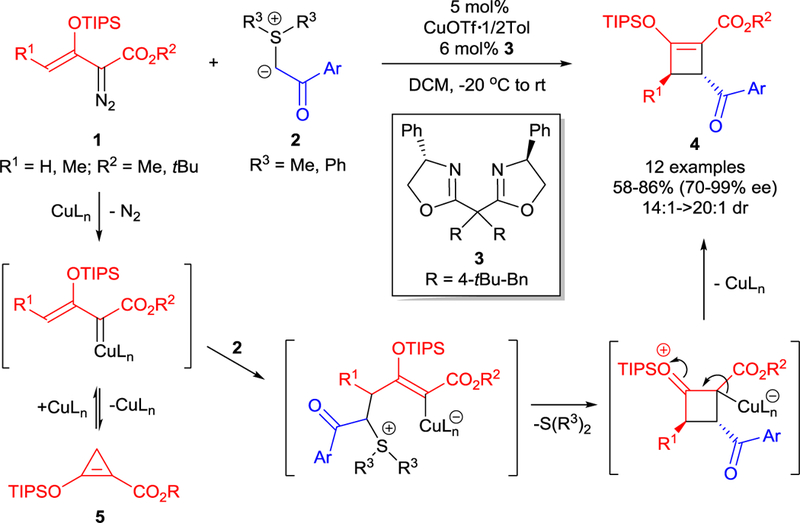
Copper(I)-catalyzed asymmetric [3+1]-cycloaddition of enoldiazoacetates 1 with sulfur ylides 2.
3. Asymmetric [3+2]-cycloaddition
A convenient way to construct five-membered rings is to use [3+2]-dipolar cycloaddition, and a growing list of these reactions use vinyldiazo compounds. The most common 1,2-dipoles used in these reactions are polarized alkenes activated by electron-donating groups, and examples with enol ethers29 (asymmetric [3+2]-cycloaddition with vinyldiazoacetates) and enamides30 have been reported. Catalytic [3+2]-cycloaddition reactions of enodiazo compounds 6 with polarized alkenes 7 occur stepwise via initial nucleophilic attack of the polarized alkene at the vinylogous position of the electrophilic metallo-enolcarbene followed by the intramolecular cyclization to form five-membered carbocycles 8 with elimination of the metal catalyst (Scheme 4). Reactions exemplified by this transformation15,16,20,31–38 have recently been reviewed.12
Scheme 4.
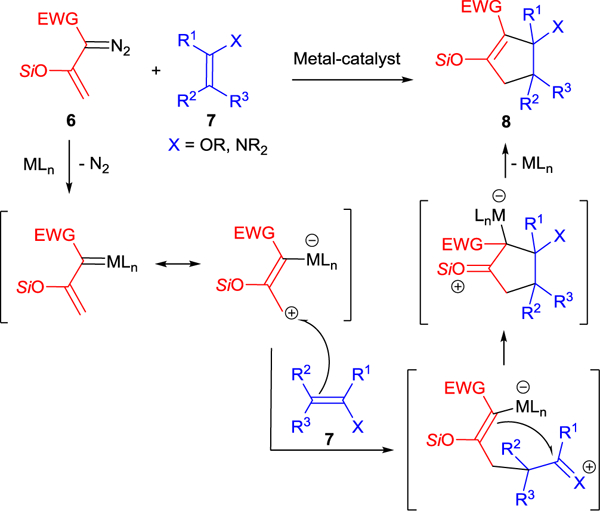
Metal-catalyzed [3+2]-cycloaddition of enoldiazo compounds 6 with polarized alkenes 7.
4. Asymmetric [3+3]-cycloaddition
4.1. Nitrones
The copper(I)-catalyzed [3+3]-cycloaddition of nitrones 10 using silyl-protected enoldiazo-acetates,39 -acetamides,39,40 -ketones39 and -sulfones41 (9) (Scheme 5) illustrates the exceptional selectivity that can be achieved in these transformations as well as their inherent complexity. Because of their inherent stability, nitrones are popular for catalytic dipolar cycloaddition transformations.4b,10–13,42 In previous investigations of nitrone cycloaddition reactions with enoldiazoacetates, copper(I) catalysis was not as effective or selective as catalysis by chiral rhodium(II) carboxylates43 or silver(I)/t-BuBOX.44 However, reinvestigation of this process revealed that enoldiazocarbonyl compounds and enoldiazosulfones offered consistently higher yields and enantioselectivities under copper(I) catalysis39–41 than did the corresponding enoldiazoacetates under rhodium(II) catalysis.43 The key factor in this discovery was the modification of the chiral box ligands on copper, and the greater electrophilic reactivity of these copper catalysts over chiral dirhodium carboxylate catalysts. The greater electrophilic reactivity of the copper catalysts also made possible the facile return of the donor-acceptor cyclopropene intermediate 12 (Scheme 5) to the reactive copper carbene.
Scheme 5.
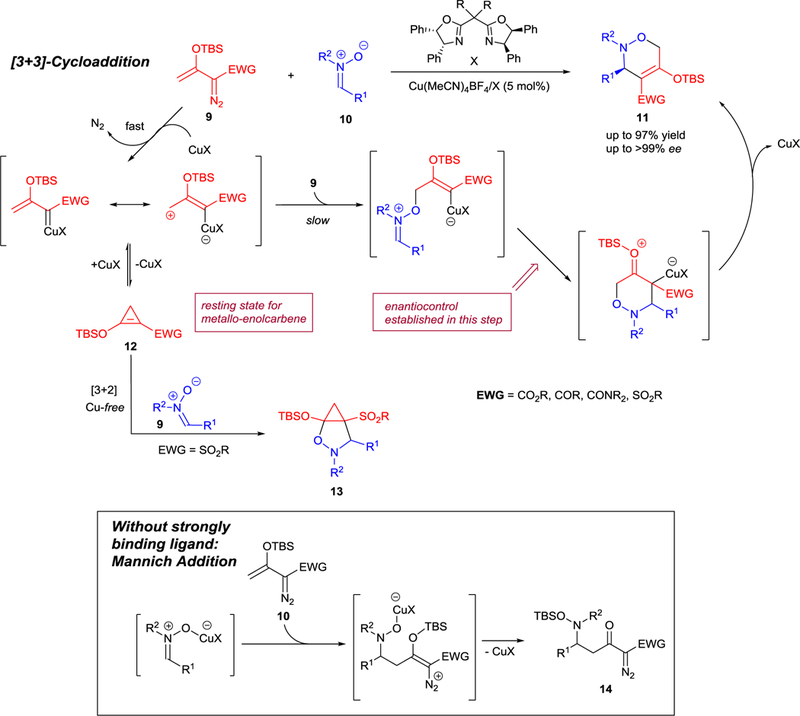
Complexity in the reactants and catalyst in the [3+3]-cycloaddition reaction with nitrones 10.
In its mechanism of action copper(I) tetrafluoroborate with chiral bisoxazoline ligands facilitates the rapid extrusion of dinitrogen from the enoldiazo compound to form the metallo-enolcarbene, which is in equilibrium with its donor-acceptor (D-A) cyclopropene 12 (Scheme 5). The metallo-enolcarbene has electrophilic centers at both the carbenic and its vinylogous position, but the vinylogous position is favored for nucleophilic attack. Nitrone addition produces a vinylcopper intermediate that is subject to electrophilic addition which occurs intramolecularly. Stabilization of the electrophilic addition product is facilitated by the electron-donating siloxy group resulting in the subsequent extrusion of the copper catalyst to produce the [3+3]-cycloaddition product 11.40
The use of 4,4’,5,5’-tetraphenylsubstituted chiral sabox ligands in copper(I) catalyzed [3+3]-cycloaddition of nitrones 10 with any enoldiazocarbonyl compound 15−17 provides high yields and exceptional enantiocontrol of oxazines 18−20 at room temperature. In contrast, the dirhodium(II) catalyst, Rh2(S-PTA)4, forms the same product (18 from enoldiazoacetates 15) with opposite chirality but with lower enantiocontrol and yields in reactions performed at low temperatures (Scheme 6A).43 Sterically encumbered sabox ligand 21 was the most effective in reactions of enoldiazo-ketones 17 (Scheme 6C), as well as -acetates 15, and -acetamides 16.39 However, the use of sabox ligand 22 also provides very high yields and enantioselectivities in the formation of oxazines 19 from reactions with enoldiazoacetamides 16 (Scheme 6B).40
Scheme 6.
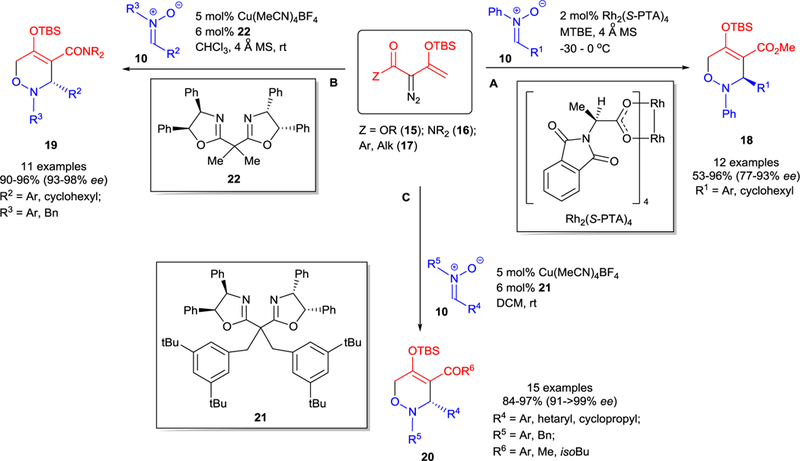
Metal-catalyzed asymmetric [3+3]-cycloaddition of enoldiazocarbonyl compounds 15−17 with nitrones 10.
There are very few reports on the cycloaddition reactions of diazosulfones. Examples of gold(I)-catalyzed [3+2]-cycloaddition of vinyldiazosulfones45 and [4+2]-cycloaddition of D-A cyclopropene generated from TBS-protected enoldiazosulfone using dirhodium catalysis46 were reported. The first example of highly enantioselective [3+3]-cycloaddition of enoldiazosulfones 23 with nitrones 10 to form chiral sulfonyloxazines 24 was recently reported using exclusively copper(I) catalysis with 4,4’-diphenyl 25, 26 or 4,4’,5,5’-tetraphenyl-sabox 27 ligands (Scheme 7).41 The use of 1.5 equiv. of enoldiazosulfone 23 was required due to the facile TBS transfer to nitrone 10 from the corresponding D-A cyclopropene 28. Furthermore, an extensive metal catalyst screening showed product discrimination between [3+3]-, [3+2]-cycloaddition, and the Mannich addition (Scheme 5).
Scheme 7.

Copper(I)-catalyzed asymmetric [3+3]-cycloaddition of enoldiazosulfones 23 with nitrones 10.
The enhanced stability of the D-A cyclopropene with sulfonyl group uncovered a [3+2]-cycloaddition process not previously reported in catalytic reactions of enoldiazocarbonyl compounds that form carbonyl D-A cyclopropenes (12) as reaction intermediates. [3+2]-Cycloaddition of 12 with the nitrone occurs with Rh(II)-catalysts or under metal-free conditions to form bicyclic product 13 (Scheme 5) that is stable to subsequent ring opening. In contrast, Cu(I)-catalysts also form 12 but are able to open the cyclopropene ring, regenerate metal carbene, and facilitate [3+3]-cycloaddition,41 demonstrating the greater reactivity of these copper(I) catalysts towards 12, and the more limited amount of 12 available at any one time for [3+2]-cycloaddition.
The Lewis acidity of the transition metal is an important consideration in the selection of a catalyst appropriate for the transformation. In the absence of stabilizing ligands on, for example, copper(I) activates the nitrone for electrophilic addition to the vinylogous center of the vinyldiazo compound, causing subsequent TBS transfer and formation of the Mannich product 14 (Scheme 5).39–41
Formal asymmetric [3+3]-cycloaddition of nitrones 10 with D-A cyclopropene 29 has been reported using Ag(I)-catalysis (Scheme 8).44 The parent γ-phenyl-enoldiazoacetate 1b does not undergo dirhodium(II) catalyzed cycloaddition with nitrones, but chiral AgSbF6/(S)-tBuBOX (30) catalysis facilitated formal [3+3]-cycloaddition of 29 at low temperature (−78 °C); and a one-pot TBS removal afforded cis-diarylsubstituted 3,6-dihydro-1,2H-oxazines 31, which exist exclusively in the enol form, in high yields and with exceptional stereocontrol. The cause of the lack of reactivity of dirhodium(II) catalysts may be steric in origin, but this requires further study.
Scheme 8.
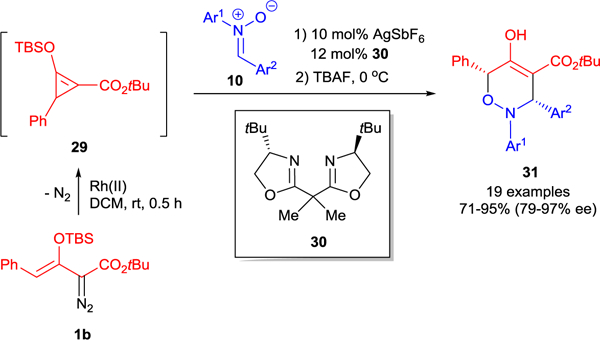
Silver-catalyzed asymmetric formal [3+3]-cycloaddition of donor-acceptor cyclopropene 29 with nitrones 10.
4.2. N-Acyliminopyridinium/quinolinium ylides
Asymmetric synthesis of chiral diazines via cycloaddition reactions is less explored47 than that of the corresponding oxazines. The first example of the catalytic synthesis of chiral bi- or tricyclic diazines was reported using chiral dirhodium(II) catalysis in the dearomatizing [3+3]-cycloaddition of acylimino-pyridinium or -quinolinium ylides 32 with enoldiazoacetates 15 (Scheme 9).12,48 Variations between Rh2(S-PTTL)4 or Rh2(S-PTAD)4 chiral catalysts, and toluene or fluorobenzene as solvents provided high yields and enantioselectivities of bicyclic diazines 33 in [3+3]-cycloaddition reactions performed at 0 °C. Recent studies of this reaction using copper(I) catalysis39 identified a general catalyst, Cu(MeCN)4BF4 with chiral sabox ligand 34, that provided high yields and excellent enantiocontrol for the synthesis of bicyclic diazines 33, 35, 36 in [3+3]-cycloaddition of acylimino-pyridinium and -quinolinium ylides 32 with enoldiazoketones 17 as well as with -acetamides 16 and -acetates 15 at room temperature (Scheme 9).
Scheme 9.
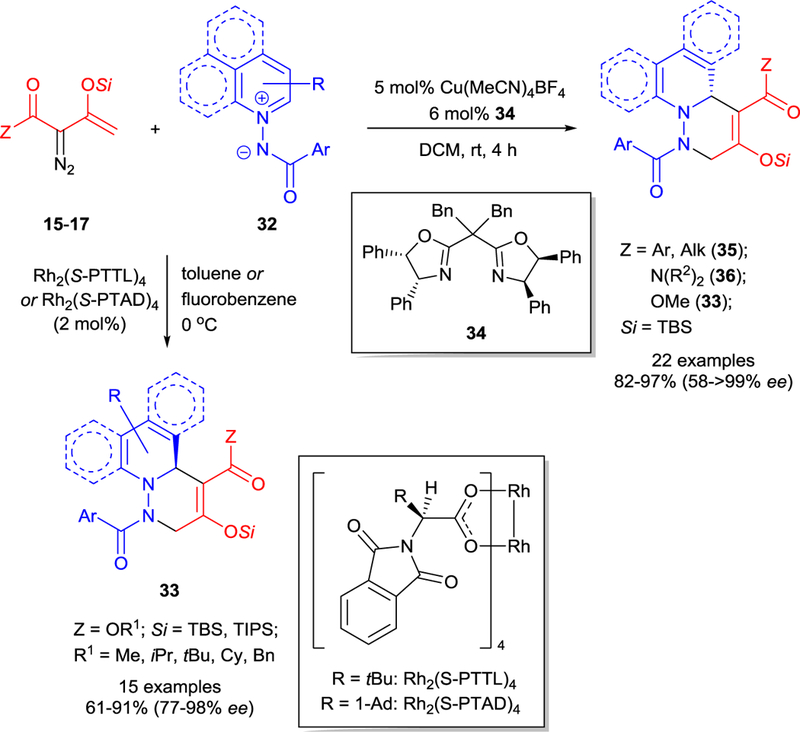
Metal-catalyzed asymmetric [3+3]-cycloaddition of enoldiazocarbonyl compounds 15−17 with N-acyliminopyridinium/quinolinium ylides 32.
4.3. Pyridinium/isoquinolinium methylides
Similar to acyliminopyridinium ylides 32, pyridinium and isoquinolinium dicyanomethylides underwent rhodium(II)-catalyzed [3+3]-cycloaddition with enoldiazoacetates forming chiral bi- or tricyclic azines.12,49 A complexity of this transformation arose from the competition between [3+3]- (with the metal carbene intermediate) versus [3+2]-cycloaddition (with the D-A cyclopropene as an intermediate) when 1 mol% of catalyst was used. Higher catalyst loadings (3 mol%) provided exclusive [3+3]-cycloaddition and initiated the discovery that the D-A cyclopropene generated from the metal carbene also formed the metal carbene intermediate. The D-A cyclopropene was both formed from the metal carbene intermediate and formed the metal carbene intermediate. Increasing the amount of the Lewis base shifted the reaction towards [3+2]-cycloaddition.49 High yields, diastereoselectivity, and enantiocontrol for the formation of chiral bi- or tricyclic azines were achieved in [3+3]-cycloaddition of enoldiazoacetates. Products of [3+2]-cycloaddition were obtained in moderate yields via direct catalyst-free treatment of a D-A cyclopropene with a methylide. The experimental data was consistent with a recent theoretical study50 that confirmed the [3+3]-cycloaddition as the major reaction pathway with Rh2(S-PTIL)4 catalyst, and that preferential [3+2]-cycloaddition occurred in reactions catalyzed by electron-deficient Rh2(CF3COO)4 and the chiral rhodium(II) catalyst Rh2(S-TCPTTL)4 which has greater steric repulsions from its ligands compared to Rh2(S-PTIL)4.50
4.4. Phenylhydrazones
Beyond dipolar species used in catalytic asymmetric [3+3]-cycloaddition of enoldiazo compounds, phenylhydrazones, as previously reviewed,12 were also utilized in formal [3+3]-cycloaddition reactions of enoldiazoacetates: sequential chiral dirhodium(II) catalyzed vinylogous N−H insertion followed by a Lewis acid-catalyzed Mannich type cyclization to chiral diazines.51 The use of Rh2(R-PTL)4 as the catalyst in toluene at low temperatures provided high enantiocontrol (up to 97% ee) in the N−H insertion step. Treatment of the N−H insertion intermediate with 5 mol% Sc(OTf)3 resulted in a highly diastereoselective one-pot cyclization, and TIPS removal afforded cis-3,6-disubstituted 1,2,3,6-tetrahydropyridazines in moderate to high yields.
5. Asymmetric [3+4]-cycloaddition
[3+4]-Cycloaddition, although commonly a cascade cyclopropanation/Cope rearrangement, is a versatile tool for the construction of seven-membered carbo- and heterocycles, which are common structural units of many natural products52 including tropane alkaloids.53 Stereocontrol is delivered in the cyclopropanation step; the Cope rearrangement is stereospecific.
5.1. Dienes
Although asymmetric [3+4]-cycloaddition of vinyldizoacetates with dienes via cyclopropanation/Cope rearrangement has been known since 1993,54 enoldiazoacetates were used in this transformation for the first time in 2009.55 The use of Rh2(S-PTAD)4 or Rh2(S-DOSP)4 catalysts in the reaction of enoldiazoacetate 1a with dienes 37 led to divinylcyclopropane intermediate 38 that underwent stereocontrolled Cope rearrangement to cycloheptadienes 39 (Scheme 10); and the results from Rh2(S-PTAD)4 catalysis were superior of those using Rh2(S-DOSP)4.55 This methodology was successfully used to form cycloheptadiene 41 (65%, 90% ee), which was converted in 8 steps to 5-epi-vibsanin E (42). Notably, the analogous reaction of a,β-unsubstituted vinyldiazoacetate with triene 40 catalyzed by Rh2(S-PTAD)4 showed diminished enantiocontrol (up to 57% ee).55
Scheme 10.
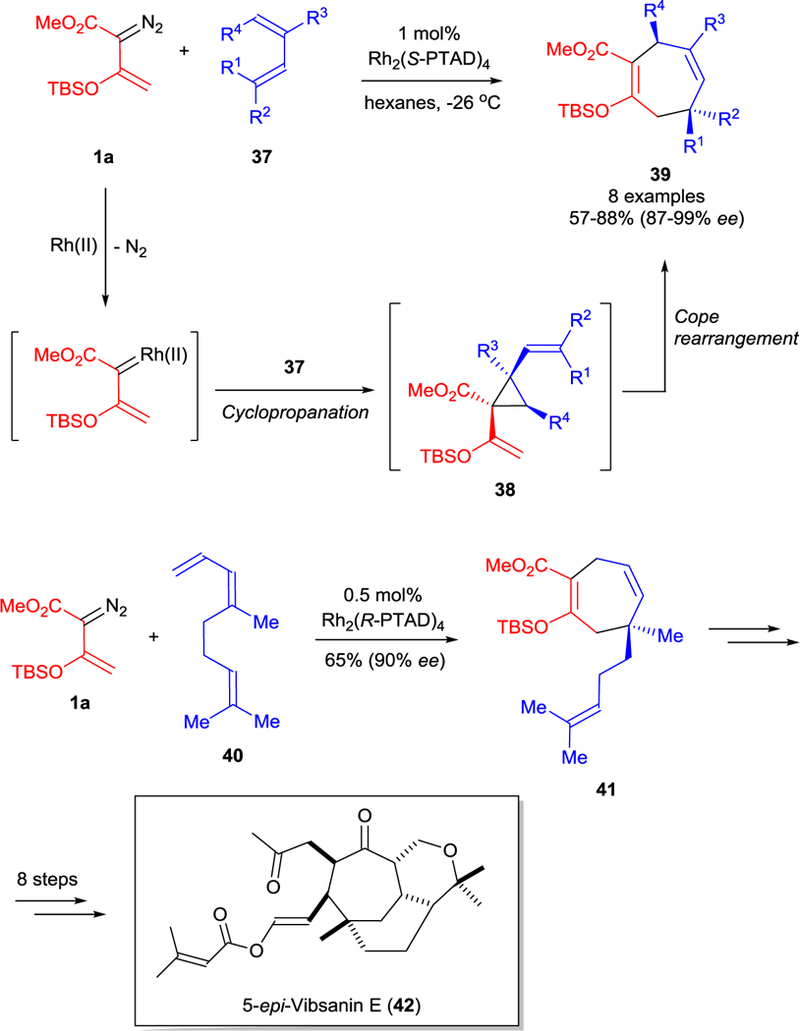
Rhodium(II)-catalyzed asymmetric formal [3+4]-cycloaddition of enoldiazoacetate 1a with dienes 37. Application in the synthesis of 5-epi-vibsanin E (42).
Asymmetric [3+4]-cycloaddition using chiral dirhodium(II) catalysis was later applied in total synthesis of (+)-barekoxide and (─)-barekol.56 The reaction of 1a with diene 43 (containing a tetrasubstituted alkene unit) with Rh2(R-PTAD)4 occurred only in refluxing hexane presumably due to the reduced reactivity of the diene, and afforded tricyclic compound 44 in 47% yield (Scheme 11). Nevertheless, this precursor provided (+)-barekoxide (45) in only 2 steps, which was easily converted to (─)-barekol (46) via acid-catalyzed hydrolysis-elimination.
Scheme 11.
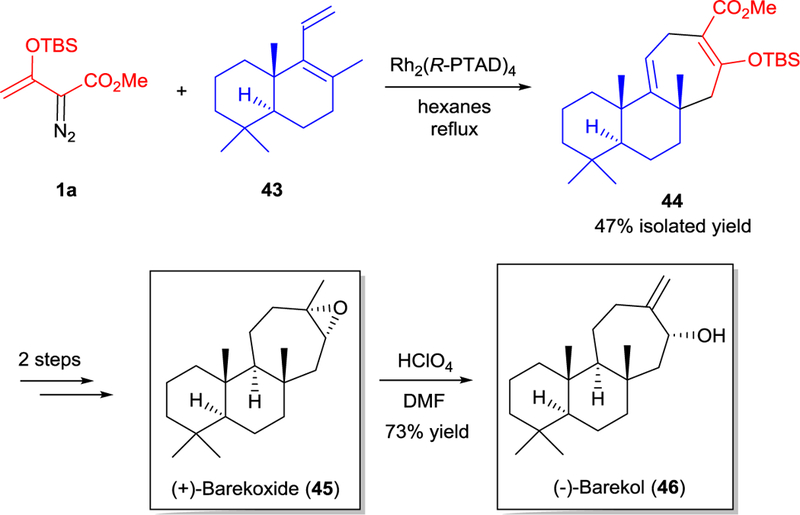
Asymmetric formal [3+4]-cycloaddition in total synthesis of (+)-barekoxide (45) and (─)-barekol (46).
Another pathway has been disclosed for the [3+4]-cycloaddition of enoldiazoacetates: instead of addition at the rhodium carbene center, nucleophilic attack of an activated diene occurs at the vinylogous position of rhodium-enolcarbenes.9a However, the “vinylogous attack” pathway is highly catalyst [using sterically encumbered tetrakis(triarylcyclopropanecarboxylate) dirhodium(II)] and substrate (using EDG-activated dienes) dependent.
5.2. Pyrroles
Pyrroles are particularly interesting “diene” substrates for asymmetric [3+4]-cycloaddition with enoldiazo compounds because the products of this reaction contain the tropane unit constructed in only one step. In the first asymmetric example of [3+4]-cycloaddition of pyrroles with enoldiazoacetates57 only enoldiazoacetate 1a provided high yields (up to 86%) and asymmetric induction (up to 98% ee) of [3,2,1]-bicyclic products 48 under Rh2(S-PTAD)4 catalysis with substituted pyrroles 47 (Scheme 12). In contrast, cycloadditions with β-unsubstituted vinyldiazoacetates occurred in 58% yield and 65% ee as the highest.56,58 The use of this methodology was applied in total synthesis of isostemofoline (50) from bicyclic compound 49 (Scheme 18).59
Scheme 12.
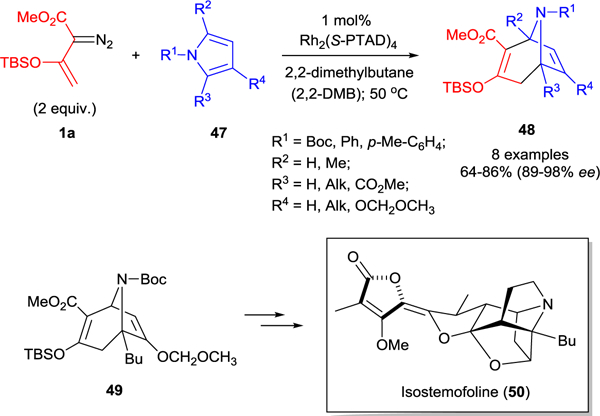
Rh2(S-PTAD)4-catalyzed asymmetric [3+4]-cycloaddition of enoldiazoacetate 1a with substituted pyrroles 47: application in total synthesis of isostemofoline (50).
Scheme 18.
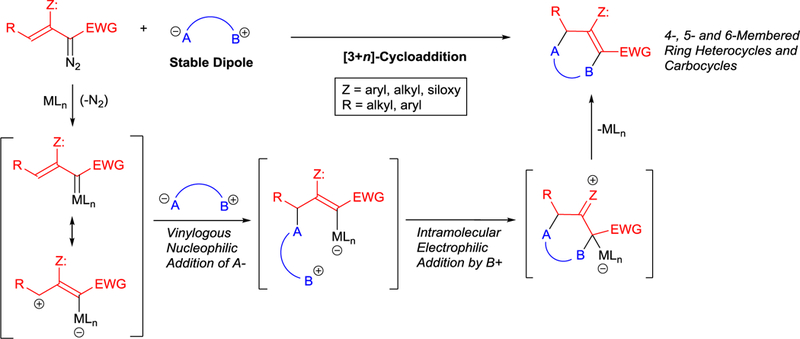
Electronic control of cycloaddition by the 3-silyloxy substituent.
Analogous [3+4]-cycloaddition of pyrrole 47a with enoldiazoacetate 1c having a chiral appendage was used in the total synthesis of (+)-batzelladine B (52).60 The bicyclic “tropane” derivative 51 was obtained in high yield and excellent diasterecontrol using Rh2(S-PTTL)4 catalyst (Scheme 13). The efficient construction of the tropane unit by this methodology also made possible the total syntheses of (+)-batzelladine E, (+)-batzelladine K, and (−)-dehydrobatzelladine C.61
Scheme 13.
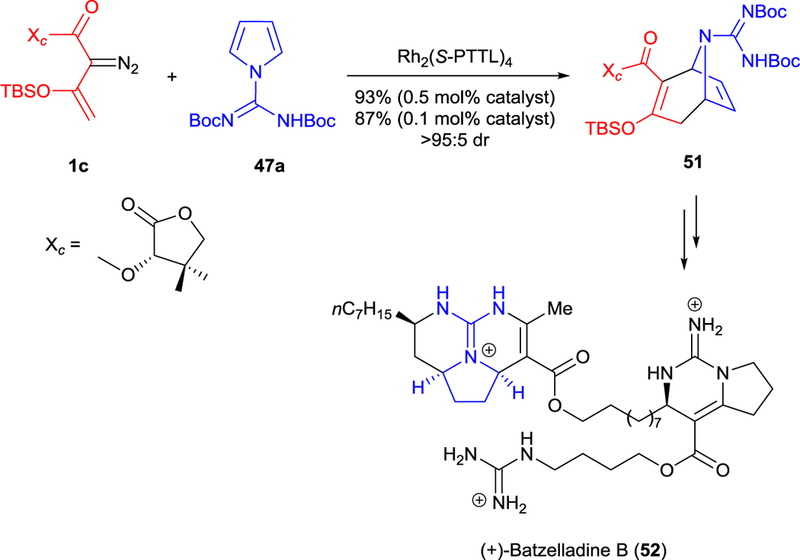
Rh2(S-PTTL)4-catalyzed asymmetric [3+4]-cycloaddition of enoldiazoacetate 1c with N-substituted pyrrole 47a for the total synthesis of (+)-batzelladine B (52).
6. Asymmetric [3+5]-cycloaddition
[3+5]-Cycloaddition is a transformation that requires the use of 1,5-dipolar species.12 Pyridinium zwitterions as 1,5-dipoles have found an application in [5+2]-cycloaddition with activated alkynes for the construction of 1,4-diazepines,62 and dirhodium(II) catalyzed [3+5]-cycloaddition of enoldiazoacetates with pyridinium zwitterions was subsequently reported.12,22a
Further studies of the synthetic utility of pyridinium zwitterion 53 uncovered substrate dependent regiodivergent [3+5]- and [4+2]-dipolar cycloadditions that afforded two different types of cyclic products that are formed in competition with excellent regiocontrol (Scheme 14).22b The origin of the regioselectivity and the reaction mechanism have been investigated in detail in combined experimental and computational studies.22b Zwitterion 53 undergoes preferential [3+5]-cycloaddition with an electrophilic reaction partner generated from 1a to form 55, however the reaction with a Pd-bound nucleophile initiates the attack at the electron-deficient C4-position of pyridine ring with subsequent cyclization to tricyclic compound 54 − a product of formal [4+2]-cycloaddition (Scheme 14).
Scheme 14.
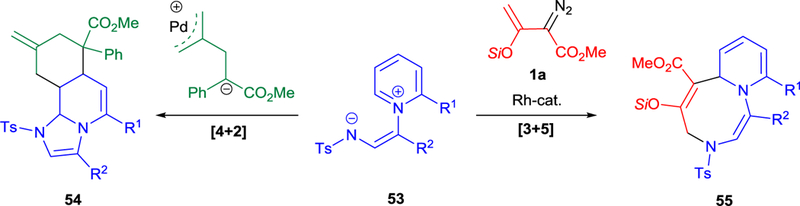
Divergent outcomes in metal-catalyzed cycloaddition of zwitterion 53 with electrophilic and nucleophilic reaction partners.
7. Cycloaddition reactions of chiral substrates
There are several examples of cycloaddition processes of enoldiazo compounds in which one of the reactants is a single enantiomer. In such cases chiral ligands for the transition metal catalyst are not required.
7.1. Azomethine imines
Azomethine imines are attractive dipoles for the construction of bicyclic pyrazolines. A number of examples on organo- and metal-catalyzed (copper, silver, magnesium, nickel) asymmetric [3+2]-cycloadditions has been reported.63 However, there are few reports of their [3+3]-cycloadditions, one of which is the efficient synthesis of bicyclic pyrazolines from azomethine imine with high diastereocontrol.64 The first report of the cycloaddition of enoldiazoacetates 15 with azomethine imines 56 uncovered two catalyst-divergent pathways using Cu(OTf)2 ([3+2]-cycloaddition to form 57 via a Lewis acid catalyzed pathway) and Rh2(OAc)4 catalysis ([3+3]-cycloaddition that occurs through a metallo-enolcarbene to form 58) (Scheme 15).65 The origin of the high diastereocontrol in the formation of the cis-stereoisomer is in the relative stabilities of the intermediates formed in the nucleophilic attack at the vinylogous position of the metallo-enolcarbene (favored versus disfavored), and this interpretation is consistent with a recent computational study of this particular transformation.66 The theoretical study also eliminated the possibility of [3+2]-cycloaddition between azomethine imine 56 and D-A cyclopropene 12a to form 59 – the product that was not observed in the experimental studies (Scheme 15).65
Scheme 15.
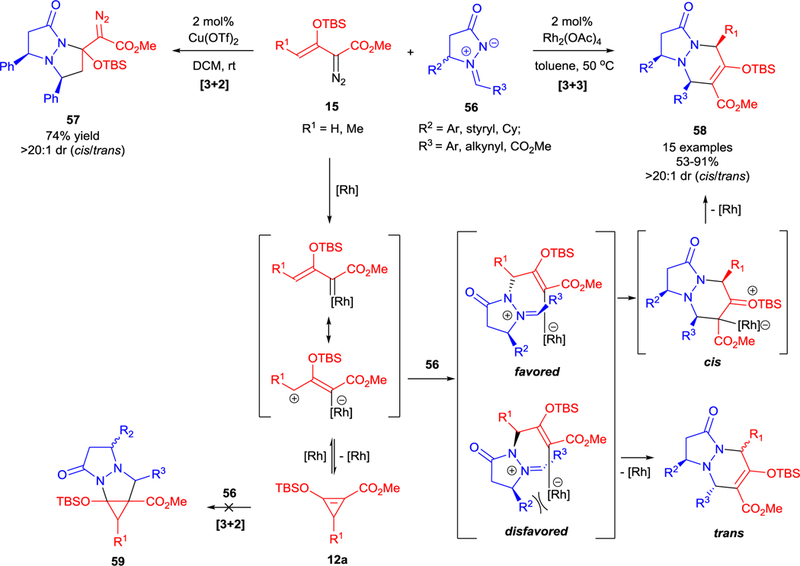
Diverse pathways in the reaction of enoldiazoacetates 15 with azomethine imines 56. The origin of diastereselectivity in [3+3]-cycloaddition.
7.2. Carbonyl ylides
Regioselectivity and access to structurally different products from the same starting materials by variations of catalysts are among the most challenging tasks in catalytic synthetic chemistry. A strategy of discrimination between two diazo compounds in metal-catalyzed cycloaddition reactions is known26,67 but underexplored due to its complexity. Recently reported regiodivergent copper(I)- and rhodium(II)-catalyzed [3+3]- and [3+2]-cycloadditions of enoldiazoacetates 15 with α-diazocarboximides 60 are new examples of discrimination between two diazo compounds, and they have allowed easy access to novel tricyclic azepine (62) and oxazole (64, 65) derivatives with excellent diastereoselectivities (Scheme 16).68 α-Diazocarboximides 60 are precursors to cyclic carbonyl ylides 6169 the generation of which is facilitated by a metal catalyst. Intermediate 61 undergoes the [3+3]-cycloaddition reaction with enoldiazoacetate 15 using copper(I) catalysis to produce 62 with excellent diastereocontrol or, using dirhodium(II) catalysis, 1,4-proton transfer to generate intermediate 63 that further undergoes [3+2]-cycloaddition. Interestingly, the ligand on the dirhodium core determines regioselectivity in the [3+2]-cycloaddition: with catalysis by rhodium perfluorobutyrate, Rh2(pfb)4, cycloaddition affords regioisomer 64, but using the sterically encumbered Rh2(esp)2, bis[rhodium(α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid)], the preferred regioisomer is 65 (Scheme 16).
Scheme 16.
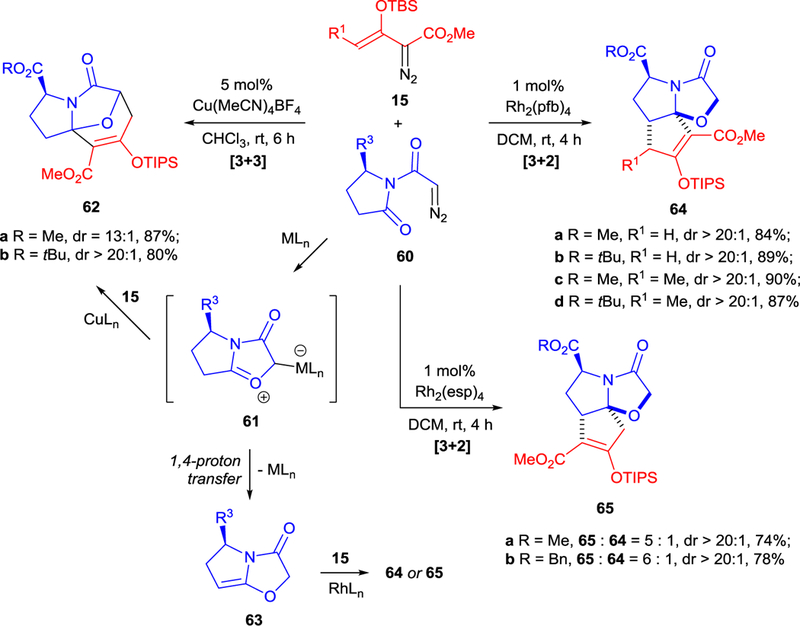
Catalytic regiodivergent diastereoselective [3+3]- and [3+2]-cycloaddition of enoldiazoacetates 15 with carbonyl ylides 61. Synthesis of chiral tricyclic heterocycles.
7.3. Sulfur ylides
The copper(I)-catalyzed reaction of enol-substituted carbonyl ylides (electrophile) and sulfur ylides (nucleophile) generated in situ from enoldiazoimides 66 and sulfonium salts 67, respectively, provides convenient access to substituted indolizidinones 68 which are not available by other synthetic methods (Scheme 17).70 The introduction of an asymmetric center to enoldiazoimides leads to chiral indolizidinones, and one representative example was reported. The reaction is considered as a [4+2]-cycloaddition that involves the following steps: C−O bond cleavage, silyl group migration, acetyl group migration, and C−S bond cleavage.70 The use of acetyldiazoimides did not lead to a cycloaddition product; and this observation indicates the important role of silyl-protected enoldiazoimide 66 in this complex transformation.
Scheme 17.
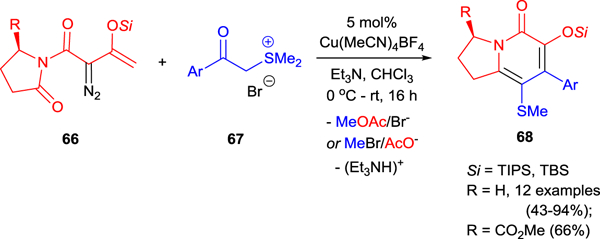
Copper(I)-catalyzed formal [4+2]-cycloaddition of enoldiazoimides 66 with sulfur ylides.
8. Influence of substituents at the 3-position
As is evident throughout this review, the siloxy substituent at the 3-position of vinyldiazoesters appears to have an overwhelming influence on the associated metal carbene to undergo [3+n]-cycloaddition reactions. The extended conjugation of vinyldiazo compounds provides the electronic framework for vinylogous nucleophilic addition reactions of their corresponding metal carbenes. The siloxy substituent (Z: = [Si]O) at the internal vinyl position of vinyldiazo compounds lowers the energy for ring closure of the metal-associated intermediate from vinylogous nucleophilic addition to the metal carbene (Scheme 18) and provides a mechanistic advantage for cycloaddition. However, although numerous processes are now available in which silyl protected enoldiazo compounds have been used effectively with high yields and selectivity, there are few comparable substituents that have been employed. 2-Diazo-3-phenyl-3-butenoate, where phenyl is at the same position as [Si]O in enoldiazo compounds, has been reported in cycloaddition reactions with vinyl ethers71 and imines.72 [3+2]-Cycloaddition occurred with n-BuOCH=CH2 (45% yield), but not with PhCH=NAr, although a methyl substituent at the 3-position, instead of phenyl, imparted reactivity with both vinyl ethers and imines.
Because of the significant difference in the outcome of cycloaddition reactions from reactions of enoldiazoacetates and styryldiazoacetates, the role of the 3-silyloxy substituent as a directive influence in cycloaddition reactions has been interpreted as being due to both steric and electronic influences.73 The “steric” influence resides in the conformation of the intermediate metal carbene resulting from 3- versus 4-substituents (A and B in Scheme 19). The prediction is that other substituents at the 3-position would also favor [3+3]-cycloaddition reactions with the same dipolar species that are effective for enoldiazo compounds. Unfortunately, this comparison has not been made, and the results published thus far do not provide direct comparison.
Scheme 19.
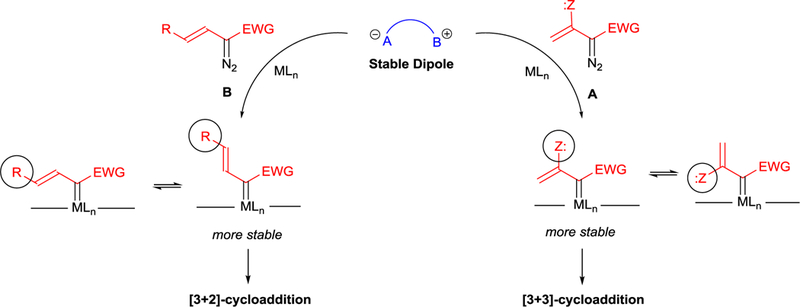
Steric influence on cycloaddition of vinyldiazo compounds.
9. Conclusions
Asymmetric cycloaddition reactions of enoldiazo compounds have broad scope with enormous structural variations for the synthesis of highly functionalized chiral carbo- and heterocycles. Generally, silyl-protected enoldiazo compounds have demonstrated higher stereocontrol than analogous vinyl or styryldiazo compounds in comparable cycloaddition reactions, and they are more stable. The unique feature of these silyl enoldiazo compounds for cycloaddition is not only their ability to form metallo-enolcarbenes that undergo nucleophilic attack at the electrophilic vinylogous position, but also the enhanced nucleophilicity of the original metal carbenic carbon that favors cyclization. This mechanistic pathway has established the platform for [3+n]-cycloaddition reactions that occur with high stereocontrol. Chiral dirhodium(II) and, more recently, copper(I) compounds are particularly suitable catalysts with which to achieve high stereocontrol in cycloaddition of enoldiazo compounds and, because of their different ligand configurations, the stereochemical outcome from the use of chiral dirhodium(II) catalysts can be complimentary to that from chiral copper(I) catalysts. However, although chiral rhodium(II) catalysis has a longer history in cycloaddition reactions of enoldiazo compounds, it is most effective for reactions with enoldiazoacetates. Copper(I) catalysis using box ligands have demonstrated advantages over rhodium(II) catalysis in [3+n]-cycloaddition reactions of all classes of enoldiazo compounds, and further development of asymmetric cycloaddition reactions using chiral copper(I) catalysis should be expected in the near future. The major breakthrough in understanding the effectiveness of these reactions is the discovery of D-A cyclopropenes (Scheme 2), which are formed from the intermediate metallo-enolcarbene but also react with the catalyst to reform the metallo-enolcarbene and serve as a resting state for the carbene. Unexpectedly stable at room temperature, D-A cyclopropenes are also reactive with dipolar compounds forming products from [3+2]-cycloaddition. Knowing the nature and stability of D-A cyclopropenes is important in understanding their role in cycloaddition reactions. Enoldiazo-acetamides and -acetates, which are stable at room temperature, undergo dinitrogen extrusion at 50 °C and higher to form D-A cyclopropenes quantitatively.74 The enhanced stability of enoldiazoacetamides and their D-A cyclopropenes compared to -acetates makes rhodium(II) catalysis less suitable for cycloaddition. Enoldiazoketones are the most reactive of the enoldiazocarbonyl compounds, undergoing catalyst-free dinitrogen extrusion even at room temperature. Their D-A cyclopropenes are also reactive, and they readily undergo rearrangement to furan derivatives under dirhodium(II) catalysis75 which, because of this, complicates cycloaddition with this undesirable competing reaction.39 Enoldiazosulfones and their D-A cyclopropenes are thermally the most stable in this series; however, its silyl group is unstable to hydrolysis. These features make enoldiazosulfones challenging for cycloaddition through metallo-enolcarbenes, and only copper(I) catalysis has been able to overcome these limitations.41 Future investigations on regiodivergent catalytic asymmetric cycloaddition of silyl-protected enoldiazo compounds and possibly new vinyldiazo compounds with other electron-donating groups, as well as the exploration of new dipolar reagents are expected to become a new wave in the synthesis of chiral carbo- and hetero-cycles.
Acknowledgements
The research described in this review was supported by grants from the U. S. National Science Foundation (CHE-1464690 and −1763168) and the National Institutes of Health (GM465030).
Photos and Biographies:
 Kostiantyn O. Marichev received his B.Sc. and M.Sc. degrees in Chemistry from Donetsk National University and his Ph.D. degree in Organic Chemistry from the L. M. Litvinenko Institute of Physical Organic & Coal Chemistry NAS of Ukraine (2013) under the supervision of Prof. Nikolai Korotkikh focused on chemistry and catalytic applications of N-heterocyclic carbenes (NHC). In 2013 Dr. Marichev joined the University of Nebraska-Lincoln (UNL) as a postdoctoral research associate with Prof. James M. Takacs where his research dealt with supramolecular ligand systems for site-selective catalytic asymmetric hydroboration of polyalkenes and ruthenium-catalyzed aminations of alcohols using borrowing-hydrogen methodology. Currently, Dr. Marichev is a postdoctoral research associate at the University of Texas at San Antonio (UTSA) with Prof. Michael P. Doyle where his research is focused on metal carbene and diazo chemistry, including catalytic asymmetric cycloaddition reactions of enoldiazo compounds.
Kostiantyn O. Marichev received his B.Sc. and M.Sc. degrees in Chemistry from Donetsk National University and his Ph.D. degree in Organic Chemistry from the L. M. Litvinenko Institute of Physical Organic & Coal Chemistry NAS of Ukraine (2013) under the supervision of Prof. Nikolai Korotkikh focused on chemistry and catalytic applications of N-heterocyclic carbenes (NHC). In 2013 Dr. Marichev joined the University of Nebraska-Lincoln (UNL) as a postdoctoral research associate with Prof. James M. Takacs where his research dealt with supramolecular ligand systems for site-selective catalytic asymmetric hydroboration of polyalkenes and ruthenium-catalyzed aminations of alcohols using borrowing-hydrogen methodology. Currently, Dr. Marichev is a postdoctoral research associate at the University of Texas at San Antonio (UTSA) with Prof. Michael P. Doyle where his research is focused on metal carbene and diazo chemistry, including catalytic asymmetric cycloaddition reactions of enoldiazo compounds.
 Michael P. (Mike) Doyle is the Rita and John Feik Distinguished University Chair in Medicinal Chemistry at the University of Texas at San Antonio. He is a graduate of the College of St. Thomas and Iowa State University, has had academic appointments at undergraduate institutions (Hope College and Trinity University) and graduate universities (University of Arizona and University of Maryland), as well as being Vice President, then President, of a science foundation (Research Corporation) before taking his current position. Doyle is a Fellow of the American Chemical Society, the American Association for the Advancement of Science, and the Royal Society of Chemistry, and he is widely recognized for his research in catalytic methods for metal carbene transformations.
Michael P. (Mike) Doyle is the Rita and John Feik Distinguished University Chair in Medicinal Chemistry at the University of Texas at San Antonio. He is a graduate of the College of St. Thomas and Iowa State University, has had academic appointments at undergraduate institutions (Hope College and Trinity University) and graduate universities (University of Arizona and University of Maryland), as well as being Vice President, then President, of a science foundation (Research Corporation) before taking his current position. Doyle is a Fellow of the American Chemical Society, the American Association for the Advancement of Science, and the Royal Society of Chemistry, and he is widely recognized for his research in catalytic methods for metal carbene transformations.
References
- 1.IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by McNaught AD and Wilkinson A. Blackwell Scientific Publications, Oxford: (1997). XML on-line corrected version: http://goldbook.iupac.org (2006-) created by Nic M, Jirat J, Kosata B ; updates compiled by Jenkins A. ISBN 0–9678550-9–8. 10.1351/goldbook. [DOI] [Google Scholar]
- 2.(a) Carruthers W, Cycloaddition reactions in organic synthesis, Pergamon Press, Oxford, England, 1990; [Google Scholar]; (b) Kobayashi. Kobayashi S and Jorgensen K, Cycloaddition reactions in organic synthesis, Wiley VCH, Weinheim, 2003. [Google Scholar]
- 3.(a) Hamer J, 1,4-Cycloaddition reactions, Academic Press, New York, 1967; [Google Scholar]; (b) Nicolaou KC, Snyder SA, Montagnon T and Vassilikogiannakis G, Angew. Chem., Int. Ed, 2002, 41, 1668; [DOI] [PubMed] [Google Scholar]; (c) Fringuelli F and Taticchi A, The Diels-Alder Reaction, Wiley, New York, 2003; [Google Scholar]; (d) Chirkin E and Porée F, Curr. Org. Chem, 2016, 20, 2284; [Google Scholar]; (e) Constantino A, Francisco C, Cubides-Roman D and Lacerda V Jr., Curr. Org. Synth., 2018, 15, 84. [Google Scholar]
- 4.(a) Huisgen R, Angew. Chem., Int. Ed, 1963, 2, 565; [Google Scholar]; (b) Hashimoto T and Maruoka K, Chem. Rev, 2015, 115, 5366. [DOI] [PubMed] [Google Scholar]
- 5.Poplata S, Tröster A, Zou Y-Q and Bach T, Chem. Rev, 2016, 116, 9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Moyano A and Rios R, Chem. Rev, 2011, 111, 4703; [DOI] [PubMed] [Google Scholar]; (b) Buchanan GS, Feltenberger JB and Hsung RP, Curr. Org. Synth, 2010, 7, 363; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Deng J, Wang X-N and Hsung RP, “A formal [3+3] cycloaddition approach to natural product synthesis” In Methods and applications of cycloaddition reactions in organic syntheses, Nishiwaki N, Ed. Wiley-VCH; 2013, Ch. 12; [Google Scholar]; (d) Narayan R, Potowski M, Jia Z-J, Antonchick AP and Waldmann H, Acc. Chem. Res, 2014, 47, 1296; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Adrio J and Carretero JC, Chem. Commun, 2014, 50, 12434. [DOI] [PubMed] [Google Scholar]
- 7.(a) Lautens M, Klute W and Tam W, Chem. Rev, 1996, 96, 49; [DOI] [PubMed] [Google Scholar]; (b) Frühauf H-W, Chem. Rev, 1997, 97, 523; [DOI] [PubMed] [Google Scholar]; (c) Gerasyuto AI, Ma Z-X, Buchanan GS and Hsung RP, Beil. J. Org Chem, 2013, 9, 1170; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gao Y, Fu X-F and Yu Z-X, Top. Curr. Chem, 2014, 346, 195. [DOI] [PubMed] [Google Scholar]
- 8.(a) Doyle MP, Angew. Chem., Int. Ed, 2009, 48, 850; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ford A, Miel H, Ring A, Slattery CN, Maguire AR and McKervey MA, Chem. Rev, 2015, 115, 9981. [DOI] [PubMed] [Google Scholar]
- 9.(a) Guzmán PE, Lian Y and Davies HML, Angew. Chem., Int. Ed, 2014, 53, 13083; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krainz T, Chow S, Korica N, Bernhardt PV, Boyle GM, Parsons PG, Davies HML and Williams CM, Eur. J. Org. Chem, 2016, 41. [Google Scholar]
- 10.Xu X and Doyle MP, Acc. Chem. Res, 2014, 47, 1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng Y, Cheng Q-Q and Doyle MP, Synlett, 2017, 1695. [Google Scholar]
- 12.Cheng Q-Q, Deng Y, Lankelma M and Doyle MP, Chem. Soc. Rev, 2017, 46, 5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng Q-Q, Yu Y, Yedoyan J and Doyle MP, ChemCatChem, 2018, 10, 488. [Google Scholar]
- 14.Navarro C, Shapiro ND, Bernasconi M, Horibe T and Toste FD, Tetrahedron, 2015, 71, 5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng Y, Yglesias MV, Arman H and Doyle MP, Angew. Chem., Int. Ed, 2016, 55, 10108. [DOI] [PubMed] [Google Scholar]
- 16.Jing C, Cheng Q-Q, Deng Y, Arman H and Doyle MP, Org. Lett, 2016, 18, 4550. [DOI] [PubMed] [Google Scholar]
- 17.Padwa A, Helv. Chim. Acta, 2005, 88, 1357. [Google Scholar]
- 18.Spangler JE and Davies HML, J. Am. Chem. Soc, 2013, 135, 6802. [DOI] [PubMed] [Google Scholar]
- 19.Briones JF and Davies HML, J. Am. Chem. Soc, 2013, 135, 13314. [DOI] [PubMed] [Google Scholar]
- 20.Smith AG and Davies HML, J. Am. Chem. Soc, 2012, 134, 18241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng Y, Massey LA, Zavalij PY and Doyle MP, Angew. Chem., Int. Ed, 2017, 56, 7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Lee DJ, Ko D and Yoo EJ, Angew. Chem., Int. Ed, 2015, 54, 13715; [DOI] [PubMed] [Google Scholar]; (b) Baek S, Lee JY, Ko D, Baik M-H and Yoo EJ, ACS Catal, 2018, 8, 6353. [Google Scholar]
- 23.(a) Dembitsky VM, J. Nat. Med, 2008, 62, 1; [DOI] [PubMed] [Google Scholar]; (b) Misale A, Niyomchon S and Maulide N, Acc. Chem. Res, 2016, 49, 2444; [DOI] [PubMed] [Google Scholar]; (c) Fan Y-Y, Gao X-H and Yu J-M, Sci. China Chem, 2016, 59, 1126. [Google Scholar]
- 24.(a) Alcaide B, Almendros P and Aragoncillo C, Chem. Soc. Rev, 2010, 39, 783; [DOI] [PubMed] [Google Scholar]; (b) Xu Y, Conner ML and Brown MK, Angew. Chem., Int. Ed, 2015, 54, 11918. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Jiang K, Ouyang Q, Liu T-Y and Chen Y-C, Org. Lett, 2016, 18, 2738. [DOI] [PubMed] [Google Scholar]
- 26.Barluenga J, Riesgo L, López LA, Rubio E and Tomás M, Angew. Chem., Int. Ed, 2009, 48, 7569. [DOI] [PubMed] [Google Scholar]
- 27.Barluenga J, Riesgo L, Lonzi G, Tomás M and López LA, Chem. – Eur. J, 2012, 18, 9221. [DOI] [PubMed] [Google Scholar]
- 28.Schmid SC, Guzei IA and Schomaker JM, Angew. Chem., Int. Ed, 2017, 56, 12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Davies HML, Xiang B, Kong N and Stafford DG, J. Am.Chem. Soc, 2001, 123, 7461; [DOI] [PubMed] [Google Scholar]; (b) Briones JF and Davies HML, J. Am. Chem. Soc, 2013, 135, 13314. [DOI] [PubMed] [Google Scholar]
- 30.(a) Carbery DR, Org. Biomol. Chem, 2008, 6, 3455; [DOI] [PubMed] [Google Scholar]; (b) Gopalaiah K and Kagan HB, Chem. Rev, 2011, 111, 4599. [DOI] [PubMed] [Google Scholar]
- 31.(a) Kiss L, Forró E and Fülöp F in Amino Acids, Peptides and Proteins in Organic Chemistry, Wiley-VCH, Weinheim, 2009, p.367; [Google Scholar]; (b) Kiss L and Fülöp F, Chem. Rev, 2014, 114, 1116. [DOI] [PubMed] [Google Scholar]
- 32.a) Kuhl A, Hahn MG, Dumić M and Mittendorf J, Amino Acids, 2005, 29, 89; [DOI] [PubMed] [Google Scholar]; b) Cabrele C, Martinek TA, Reiser O and Berlicki L, J. Med. Chem, 2014, 57, 9718. [DOI] [PubMed] [Google Scholar]
- 33.a) Mittendorf J, Kunisch F, Matzke M, Militzer H-C, Schmidt A and Schönfeld W, Bioorg. Med. Chem. Lett, 2003, 13, 433; [DOI] [PubMed] [Google Scholar]; b) Petraitiene R, Petraitis V, Kelaher AM, Sarafandi AA, Mickiene D, Groll AH, Sein T, Bacher J and Walsh TJ, Antimicrob. Agents Chemother, 2005, 49, 2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laursen JS, Engel-Andreasen J and Olsen CA, Acc. Chem. Res, 2015, 48, 2696. [DOI] [PubMed] [Google Scholar]
- 35.Herndon JW, “Metal carbene cycloadditions” In Comprehensive Organic Synthesis 2nd Ed., Knochel P and Molander GA, Eds. Elsevier Ltd; 2014, Vol. 5, p 1179. [Google Scholar]
- 36.Barluenga J, Tudela E, Ballesteros A and Tomás M, J. Am. Chem. Soc, 2009, 131, 2096. [DOI] [PubMed] [Google Scholar]
- 37.Lian Y and Davies HML, J. Am. Chem. Soc, 2010, 132, 440. [DOI] [PubMed] [Google Scholar]
- 38.(a) Xiong H, Xu H, Liao S, Xie Z and Tang Y, J. Am. Chem. Soc, 2013, 135, 7851; [DOI] [PubMed] [Google Scholar]; (b) Zhu J, Liang Y, Wang L, Zheng Z-B, Houk KN and Tang Y, J. Am. Chem. Soc, 2014, 136, 6900. [DOI] [PubMed] [Google Scholar]
- 39.Marichev KO, Adly FG, Carranco AM, Garcia EC, Arman H and Doyle MP, ACS Catal, 2018, 8, 10392. [Google Scholar]
- 40.Cheng Q-Q, Yedoyan J, Arman H and Doyle MP, J. Am. Chem. Soc, 2016, 138, 44. [DOI] [PubMed] [Google Scholar]
- 41.Adly FG, Marichev KO, Jensen JA, Arman H and Doyle MP, Org. Lett, 2019, 21, 40. [DOI] [PubMed] [Google Scholar]
- 42.(a) Hamer J and Macaluso A, Chem. Rev, 1964, 64, 473; [Google Scholar]; (b) Gothelf KV and Jørgensen KA, Chem. Rev, 1998, 98, 863; [DOI] [PubMed] [Google Scholar]; (c) Anderson LL, Asian J Org. Chem, 2016, 5, 9. [Google Scholar]
- 43.Wang X, Xu X, Zavalij PY and Doyle MP, J. Am. Chem. Soc, 2011, 133, 16402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu X, Zavalij PJ and Doyle MP, Chem. Commun, 2013, 49, 10287. [DOI] [PubMed] [Google Scholar]
- 45.Lonzi G and Lopez LA, Adv. Synth. Catal, 2013, 355, 1948. [Google Scholar]
- 46.Davies HML, Houser JH and Thornley C, J. Org. Chem, 1995, 60, 7529. [Google Scholar]
- 47.(a) Chrzanowska M and Rozwadowska MD, Chem. Rev, 2004, 104, 3341; [DOI] [PubMed] [Google Scholar]; (b) Perreault C, Goudreau SR, Zimmer LE and Charette AB, Org. Lett, 2008, 10, 689; [DOI] [PubMed] [Google Scholar]; (c) Zhang O, Yang L and Tong X, J. Am. Chem. Soc, 2010, 132, 2550. [DOI] [PubMed] [Google Scholar]; (d) Zhou Y-Y, Li J, Ling L, Liao S-H, Sun X-L, Li Y-X, Wang L-J and Tang Y, Angew. Chem., Int. Ed, 2013, 52, 1452; [DOI] [PubMed] [Google Scholar]; (e) Guo C, Fleige M, Janssen-Müller D, Daniliuc CG and Glorius F, Nat. Chem, 2015, 7, 842. [DOI] [PubMed] [Google Scholar]
- 48.Xu X, Zavalij PJ and Doyle MP, Angew. Chem., Int. Ed, 2013, 52, 12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu X, Zavalij PY and Doyle MP, J. Am. Chem. Soc, 2013, 135, 12439. [DOI] [PubMed] [Google Scholar]
- 50.Li S-J and Fang D-C, Organometallics, 2018, 37, 1373. [Google Scholar]
- 51.Xu X, Zavalij PJ and Doyle MP, Angew. Chem., Int. Ed, 2012, 51, 9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin Z, He Y and Chiu P, Chem. Soc. Rev, 2018, 47, 8881. [DOI] [PubMed] [Google Scholar]
- 53.(a) Fodor G and Dharanipragada R, Nat. Prod. Rep, 1986, 3, 181; [DOI] [PubMed] [Google Scholar]; (b) Gadzikowska M and Grynkiewicz G, Acta Pol. Pharm, 2002, 59, 149; [PubMed] [Google Scholar]; (c) Grynkiewicz G and Gadzikowska M, Pharmacol. Rep, 2008, 60, 439. [PubMed] [Google Scholar]
- 54.(a) Davies HML, Tetrahedron, 1993, 49, 5203; [Google Scholar]; (b) Davies HML, Peng Z-Q and Houser JH, Tetrahedron Lett, 1994, 35, 8939. [Google Scholar]
- 55.Schwartz BD, Denton JR, Lian Y, Davies HML and Williams CM, J. Am. Chem. Soc, 2009, 131, 8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lian Y, Miller LC, Born S, Sarpong R and Davies HML, J. Am. Chem. Soc, 2010, 132, 12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy RP and Davies HML, J. Am. Chem. Soc, 2007, 129, 10312. [DOI] [PubMed] [Google Scholar]
- 58.Davies HML, Matasi JJ, Hodges LM, Huby NJS, Thornley C, Kong N and Houser JH, J. Org. Chem, 1997, 62, 1095. [Google Scholar]
- 59.Kende AS, Smalley TL Jr. and Huang H, J. Am. Chem. Soc, 1999, 121, 7431. [Google Scholar]
- 60.Parr BT, Economou C and Herzon SB, Nature, 2015, 525, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Economou C, Romaire JP, Scott TZ, Parr BT and Herzon SB, Tetrahedron, 2018, 74, 3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee DJ, Han HS, Shin J and Yoo EJ, J. Am. Chem. Soc, 2014, 136, 11606. [DOI] [PubMed] [Google Scholar]
- 63.Požgan F, Mamari HA, Grošelj U, Svete J and Štefane B, Molecules, 2018, 23, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.(a) Shintani R and Hayashi T, J. Am. Chem. Soc, 2006, 128, 6330; [DOI] [PubMed] [Google Scholar]; (b) Shapiro ND, Shi Y and Toste FD, J. Am. Chem. Soc, 2009, 131, 11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qian Y, Zavalij PY, Hu W and Doyle MP, Org. Lett, 2013, 15, 1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Yang Y, Zhao J and Xue Y, Eur. J. Org. Chem, 2018, 3086. [Google Scholar]
- 67.(a) Xu G, Zhu C, Gu W, Li J and Sun J, Angew. Chem., Int. Ed, 2015, 54, 883; [DOI] [PubMed] [Google Scholar]; b) Cheng Q-Q, .Yedoyan J, Arman H and Doyle MP, Angew. Chem., Int. Ed, 2016, 55, 5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deng Y, Massey LA, Rodriguez Núñez YA, Arman H and Doyle MP, Angew. Chem., Int. Ed, 2017, 56, 12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.For a review on cyclic carbonyl ylides, see: Padwa A, Chem. Soc. Rev, 2009, 38, 3072. [DOI] [PubMed] [Google Scholar]
- 70.Cheng Q-Q, Massey LA, Willett BS, Deng Y, Arman H and Doyle MP, Angew. Chem., Int. Ed, 2018, 57, 10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davies HML, Hu B, Saikali E and Bruzinski PR, J. Org. Chem, 1994, 59, 4535. [Google Scholar]
- 72.Doyle MP, Yan M, Hu W and Gronenberg LS, J. Am. Chem. Soc, 2003, 125, 4692. [DOI] [PubMed] [Google Scholar]
- 73.Qin C and Davies HML, J. Am. Chem. Soc, 2013, 135, 14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deng Y, Jing C and Doyle MP, Chem. Commun, 2015, 51, 12924. [DOI] [PubMed] [Google Scholar]
- 75.Marichev KO, Wang Y, Carranco AM, Garcia EC, Yu Z-X and Doyle MP, Chem. Commun, 2018, 54, 9513. [DOI] [PubMed] [Google Scholar]