

Abstract
According to the World Health Organization, more than 1 billion people are at risk of or are affected by neglected tropical diseases. Examples of such diseases include trypanosomiasis, which causes sleeping sickness; leishmaniasis; and Chagas disease, all of which are prevalent in Africa, South America, and India. Our aim within the New Medicines for Trypanosomatidic Infections project was to use (1) synthetic and natural product libraries, (2) screening, and (3) a preclinical absorption, distribution, metabolism, and excretion–toxicity (ADME-Tox) profiling platform to identify compounds that can enter the trypanosomatidic drug discovery value chain. The synthetic compound libraries originated from multiple scaffolds with known antiparasitic activity and natural products from the Hypha Discovery MycoDiverse natural products library. Our focus was first to employ target-based screening to identify inhibitors of the protozoan Trypanosoma brucei pteridine reductase 1 (TbPTR1) and second to use a Trypanosoma brucei phenotypic assay that made use of the T. brucei brucei parasite to identify compounds that inhibited cell growth and caused death. Some of the compounds underwent structure-activity relationship expansion and, when appropriate, were evaluated in a preclinical ADME-Tox assay panel. This preclinical platform has led to the identification of lead-like compounds as well as validated hits in the trypanosomatidic drug discovery value chain.
Keywords: anti-infective drugs, cell-based assays, enzyme assays or enzyme kinetics, liquid handling, compound repositories
Introduction
Neglected tropical diseases (NTDs) form a group of diverse communicable diseases, mainly occurring in the tropical and subtropical regions and affecting people living in poverty without access to basic health services, safe water, and sanitisation.1–3 The diseases harm more than 1 billion people and lead to an economic burden of billions of dollars that contributes to a further increase in poverty of the affected people. In 2012, the World Health Organization published a Roadmap for NTDs describing the vision to control, eliminate, and eradicate NTDs. It put together recommendations for preventive chemotherapy, specialized disease management, vector and intermediate host control, accessibility to safe water, sanitation and hygiene, as well as veterinary health control, especially in areas with human-animal interaction.4
Human African trypanosomiasis (HAT), also known as sleeping sickness, is a vector-borne disease caused by the flagellated protozoan parasite Trypanosoma brucei that affects more than 10,000 people annually and threatens more than 65 million people in 36 sub-Saharan African countries.5 There are a few drugs available for the treatment of HAT, namely, suramin, pentamidine, and melarsoprol, which were described before the 1950s, and eflornithine, which was approved in the 1990s.6,7 During the past years, nifurtimox-eflornithine combination therapy has been introduced to treat HAT in several countries, with improved efficacy and tolerability in comparison with eflornithine.8,9 All of these therapies are associated with toxicity to some degree, and the urgency to discover new compounds with trypanocidal activity for the development of HAT treatment remains.
To address some of the challenges associated with NTD drug discovery, the multidisciplinary New Medicines for Trypanosomatidic Infections (NMTrypI) project was funded by the European Union and aimed at identifying chemical starting points for drugs to treat trypanosomatidic diseases. We developed a workflow (Fig. 1) that first made use of synthetic compound libraries based on scaffolds that were known from prior work to exhibit antiparasitic or antiparasite protein target activity. This workflow comprised 187 compounds from the triazole-linked privileged structure-based conjugates, aryl thiosemicarbazones, the 2-amino- 1,3,4-thiadiazole scaffold, chroman-4-one derivatives, methoxylated 2′-hydroxychalcones, flavonol derivatives, and miltefosine analogs, some of which have been progressed to lead-like compounds. Second, we have screened the MycoDiverse natural products library, which is composed of 10,049 extracts and fractions from fermentations of higher fungi (basidiomycetes and ascomycetes), complemented by 1040 extracts and fractions from entomogenous fungi, and have identified validated hits. These samples are mixtures of compounds present at unknown concentration, and a process of assay-guided purification was employed to identify and evaluate the bioactive component(s) present in a screening hit, which may be present at low or high concentration, so their potency can be established only after purification. Sample complexity ranged from those containing one or two major components to those containing 100 or more components distinguishable by chromatographic analysis. The bioactive compounds identified previously from the MycoDiverse natural products library fall into a range of classes, predominantly terpenoids but also including peptides, polyketides, nucleosides, and meroterpenoids; their molecular weights are generally low, with 84% below 500 Da.
Figure 1.

Overall workflow of the screening of synthetic compounds and natural products in the TbPTR1 target-based assay and Trypanosoma brucei phenotypic assay. Synthetic compounds (see Table 1 for details) and natural products (MycoDiverse natural products library) were screened in the TbPTR1 target-based assay and T. brucei phenotypic assay. The most promising compounds were subsequently evaluated in an absorption, distribution, metabolism, and excretion–toxicity assay panel. The synthetic compound libraries yielded multiple compound series that met the lead criteria. The MycoDiverse natural products screen yielded 40 hits from the T. brucei phenotypic assay and seven hits from the TbPTR1 target-based assay.
The higher fungi have evolved defense strategies to protect themselves from bacterial and fungal competitors, and the production of antibiotics indicates that these include chemical strategies, as exemplified by the commercially important pleuromutilins and strobilurins, respectively. Higher fungi are also subject to attack by fungivorous nematodes and protozoa and might therefore be expected to protect themselves via the production of anthelmintic and antiprotozoal chemicals that could provide valuable lead-like compounds for the development of drugs useful for the treatment of NTDs.10 There have been a number of reports of basidiomycete and ascomycete metabolites with antiprotozoal properties. Examples include striatins A and B from Cyathus striatus,11 aureobasidins from Aureobasidium pullulans,12 and palmarumycins from the ascomycete Edenia sp.13 The extra samples derived from fermentations of entomogenous (insect-associated or -pathogenic) fungi were included to access bioactive chemical diversity complementary to that from the higher fungi. In addition to the above bioactive natural products, nature has provided established antiparasitic drugs such as the antimalarial plant products quinine from Cinchona succiruba and artemisinin from Artemisia annua, the antileishmanial antibiotics amphotericin B and paromomycin, and the macrocyclic lactones (avermectins, milbemycins) from Streptomyces spp., which were breakthrough antibiotics as they were effective against different groups of parasites including helminths and ectoparasites.14 Natural products sometimes have unorthodox and often unanticipated chemical structures that offer novel routes to clinically useful drugs. The myriad structurally diverse compounds found in nature confer an important role regarding their unique potential for drug discovery. This can be crucial in meeting the challenge of drug resistance associated with structural mutations in the drug targets, as in the case of malaria. Despite their diversity, many natural products remain largely unexplored and offer rich potential as sources of drugs.15,16 The synthetic compounds and natural products were provided by various project partners and were stored centrally at the Fraunhofer Institute for Molecular Biology and Applied Ecology–ScreeningPort (Fraunhofer IME-SP) as DMSO solutions and, when required, were distributed to partner sites for T. brucei phenotypic screening through to supplying milligram quantities for in vivo studies.
The synthetic compounds and natural products were screened against a T. brucei pteridine reductase 1 (TbPTR1) target-based assay and T. brucei phenotypic assay, which were developed as 384-well microtiter plate–based assays. The key operational assay parameters being characterized included sensitivity, scalability, reproducibility, signal stability, robustness (Z′), DMSO tolerance, and pharmacological response to standard inhibitors. From the screens that made use of the synthetic compound libraries, compounds from each of the representative scaffolds underwent structure-activity relationship (SAR) expansion to improve potency and, when appropriate, were also evaluated in an absorption, distribution, metabolism, and excretion–toxicity (ADME-Tox) assay panel comprising physicochemical characterization, solubility (in water, buffer, simulated gastric fluid [SGF], fed state simulated intestinal fluids [FeSSIF] and fasted state simulated intestinal fluids [FaSSIF]), in vitro off-target liability enzyme panel (kinase and histone deacetylase [HDAC] enzymes), in vitro cytotoxicity assay panel (human A549 and WI-38 cell lines), mitochondrial toxicity (human 786-0 cell line), cytochrome (CYP) P450 inhibition (1A2, 2C9, 2C19, 2D6, and 3A4 isoforms), cardiotoxicity (hERG binding assay), and pharmacokinetics in mice. In the case of synthetic libraries, the SAR expansion yielded lead-like compounds.17–20 Because of the complex nature of the MycoDiverse natural products library, the most promising hits underwent assay-guided fractionation. Seven hits in the TbPTR1 and 40 phenotypic T. brucei assay hits underwent assay-guided purification, after which the focus was on the latter group of phenotypic assay hits because of the greater assay potency of their active fractions. This resulted in the identification of the novel active components, which were progressed to hit validation in secondary parasitological assays.
Materials and Methods
Chemicals and Assay Kits
All chemical reagents, cell culture media, and standard inhibitors were of the highest quality and included penicillin G (P-11-010, PAA Laboratories GmbH, Austria), MitoTracker Red CMXRos (Thermo, Waltham, MA), SYBR Green (Invitrogen, Waltham, MA), Trichostatin A (Sigma-Aldrich, St. Louis, MO), SU6656 (A15518-10, Calbiochem, Burlington, MA), E-4031 (BML-KC158-0005, Enzo Life Sciences, Inc., Farmingdale, NY), valinomycin (V0627, Sigma-Aldrich), paclitaxel (T7191, Sigma-Aldrich), methotrexate (ALX-440-045-M100, Enzo Life Sciences, Inc.), cytochrome c (C2037, Sigma-Aldrich), dihydrobiopterin (H2B; 37272, Sigma-Aldrich), pyrimethamine (46706, Sigma-Aldrich), alpha-naphthoflavone (N5757-1G, Sigma-Aldrich), sulfaphenazole (S0758-1G, Sigma-Aldrich), troglitazone (T2573-5MG, Sigma-Aldrich) quinidine (Q3625-5G, Sigma-Aldrich), and ketoconazole (K1003, Sigma-Aldrich). Compounds were dissolved to yield stock solutions in 100% v/v DMSO (Carl Roth GmbH & Co. KG, Karlsruhe, Germany) and stored at −20 °C. Assay kits used in the ADME-Tox assay panel included cytotoxicity CellTiter-Glo (CTG) reagent (Promega Corp., Madison, WI); hERG assay (Predictor hERG, Thermo); CYP P450 1A2, 2C9, 2C19, 2D6, and 3A4 assays (P450-Glo, Promega Corp.); CYP P450 preparations as Supersomes (Corning Inc., Corning, NY); Aurora B ADP-Glo Kinase Enzyme System (Promega Corp.); and HDAC (HDAC-Glo Class I/II Kits, Promega Corp.).
Biological Reagents
T. brucei (Lister 427) reference strain was kindly provided by Professor Sergio Schenkman (UNIFESP, São Paulo, Brazil). The A549 cell line was obtained from DSMZ (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany), the WI-38 cell line was obtained from ATCC (ATCC CCL-75, Manassas, VA), and the 786-O cell line was from Cell Lines Services GmbH (Eppelheim, Germany). Human recombinant C-ter-His-FLAG-HDAC1 (50051), HDAC-3/NcoR2 (50003), and N-ter-GST-HDAC-6 (50006) were purchased from BPS Bioscience (San Diego, CA) TbPTR1 enzyme was expressed and purified as previously described.21
Synthetic and Natural Product Libraries
The composition of the designed synthetic compound libraries and the natural product libraries were as follows:
18 triazole-linked privileged structure-based conjugates, which contain a substructure characterizing various drugs22;
28 aryl thiosemicarbazones with known antiparasitic activity23;
57 derivatives of the thiadiazole scaffold that can target the folate/biopterin pathway with potential for activity against the parasite in combination with dihydrofolate reductase (DHFR) inhibitors24;
three chroman-4-one derivatives that target pteridine reductase 1 and are shown to possess antiparasitic activity25;
13 2′-hydroxy chalcones derived from chalcones that have been shown to display a broad spectrum of pharmacological activities, including antiparasitic activity26;
16 flavonol derivatives with known antitrypanosomatidic activity21;
52 analogues of miltefosine, which is an alkyl phospholipid compound exhibiting antileishmanial activity and approved for clinical use27; and
the MycoDiverse natural products library (10,049 mixtures from basidiomycetes and ascomycetes) complemented with 1040 samples from entomogenous fungi, which contains compounds produced by a diverse collection of fungi after stimulation using proprietary fermentation technologies. Strains were fermented under a variety of conditions in shaken Erlenmeyer flasks (250 mL containing 100 mL medium) and two different harvest times were used for each fermentation. Upon harvest, half of the fermentation material (50 mL) was processed by solid phase extraction (SPE) of the aqueous broth after centrifugation to generate four fractions of decreasing polarity and lyophilization and organic solvent extraction of the biomass to generate a fifth sample. The fractions and extracts were then concentrated to dryness and redissolved in 100% DMSO (v/v) at a concentration factor of 40× and stored at −20 °C in deep-well 96-well blocks and formatted into 384-well plates for screening. The remainder of the harvested fermentation material (40–50 mL fermentation broth) was stored frozen at −20 °C.
After screening, active fractions underwent fractionation from material stored at −20 °C, after thawing and extraction by SPE or biomass extraction. The resulting extracts were concentrated to dryness and redissolved in DMSO-MeOH (3:1, 1.0 mL). Aliquots (0.9 mL) were chromatographed by reversed-phase high-performance liquid chromatography (HPLC) on an Xbridge Prep Phenyl 5 µm OBD column (19 mm × 100 mm) with a guard column (19 mm × 10 mm), using gradient elution of 10% to 100% acetonitrile in water in the presence of 0.1% formic acid over 8 min, held at 100% acetonitrile for 3 min before returning to starting conditions over 1 min at a flow rate of 17 mL/min. This method was adjusted as appropriate if the components in a particular extract were either more polar or more lipophilic than usual. Twenty-four fractions were collected for each fractionation and concentrated to dryness in a Genevac HT12 centrifugal evaporator. The resulting dried fractions were redissolved in DMSO-MeOH (3:1, 0.45 mL), and aliquots (25 µL) were transferred to 96-well microtiter plates and lyophilized, followed by dissolution in DMSO (100% v/v) and formatting into 384-well plates for screening. Specific chromatographic methods were developed for each selected active primary fraction to separate and resolve its constituent components/HPLC peaks using a reversed-phase column chemistry orthogonal to that used for the primary fractionation step (usually SymmetryShield RP8). The primary fraction was then fractionated on the semipreparative scale and individual HPLC peaks collected into secondary fractions that were concentrated to dryness, redissolved, and shipped for assay as described above.
Scale-up fermentation of the hits producing the most promising activities were repeated in multiple shake flasks under exactly the same conditions as used for the initial (primary) fermentation and harvested after the same period of time that produced the target activity. Aqueous broth actives were harvested by capture using Diaion HP20 resin and elution with MeOH/acetonitrile. Biomass actives were extracted using MeOH-acetone (1:1). The target compounds were purified by preparative HPLC methods scaled up from the purification methods developed from the small-scale assay-guided purification investigations. Acquisition of spectroscopic data for natural product database searching was performed using HPLC–mass spectrometry (MS) of the active primary fractions by orthogonal reversed-phase HPLC-MS conducted on a SymmetryShield RP8 column (3.5 µm; 4.6 mm × 75 mm) eluted with a linear gradient of 10% to 95% MeCN in water, containing 10 mM ammonium formate + 0.1% formic acid, at a flow rate of 1 mL/min, held at 95% MeCN for 1.0 min before returning to initial conditions over 0.5 min; the total run time was 12 min. Ultraviolet (UV)–visible spectra were acquired using a Waters 2996 photodiode array detector, and positive and negative ion electrospray mass spectra were acquired using a Waters Acquity SQ detector. Putative molecular weights, UV-visible maxima, and producing organism taxonomic data were used to search two natural products databases to identify known compounds, namely, Antibase and the Chapman and Hall Dictionary of Natural Products. 1H and 13C nuclear magnetic resonance (NMR) spectra were acquired at 500 MHz and 125 MHz, respectively, at 298K in DMSO-d6, using a Bruker AVANCE III 500 MHz NMR spectrometer at the University of Surrey (Guildford, UK). Data sets for structure elucidation included COSY, HSQC, HMBC, and NOESY spectra, which were interpreted at Hypha Discovery using Mestrenova software.
TbPTR1 Target-Based Assay
The in vitro TbPTR1 target-based assay was based on that of Shanks et al.28 As PTR1 enzymes use H2B as a substrate and also require NADPH for the reaction, the reduction of H2B to 5,6,7,8-tetrahydrobiopterin (H4B) by PTR1 in this assay is nonenzymatically linked with the reduction of cytochrome c (cyt c Fe3+ → cyt c Fe2+), which is detected at 550 nm. TbPTR1 activity was assayed in a buffer containing 20 mM sodium citrate (pH 6.0). The final reaction mixture (50 µL with 1% v/v DMSO) contained test compound at 10 µM or natural product mixture at an unknown concentration and TbPTR1 (10 nM), H2B (3 µM), cyt c (100 µM), and NADPH (500 µM). Compound screening was performed by addition of compound (in 100% v/v DMSO) to assay plates followed by addition of 45 µL reaction mix (enzyme, H2B, and cyt c in 20 mM sodium citrate buffer). A preread (0 min) was made at 550 nm using an EnVision Multilabel 2103 Reader followed by incubation of the assay plates at 30 °C for 10 min. The reaction was initiated by the addition of 5 µL NADPH (5 mM in ultrapure water) followed by kinetically reading the assay plates at 550 nm using the EnVision Multilabel 2103 Reader at 10, 20, 30, 40, and 50 min (typically, the linear range was between 10 and 40 min). Based on the rate of reaction (ΔAbsorbance 550 nm/min), the data were normalized to the positive control methotrexate for TbPTR1 (1 µM yielding 100% inhibition) and negative controls (NCs; 1% v/v DMSO, yielding 0% inhibition), and the percentage inhibition was calculated for all samples. The preread (0 min) measurement was used to flag optically interfering samples.
T. brucei Phenotypic Assay
T. brucei brucei parasite cells were cultured in HMI-9 media supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco Life Science, Waltham, MA) at 37 °C in 5% CO2 in a humidified incubator. Trypanosomes were routinely passaged and maintained at an exponential growth phase, ranging from 5 × 104 to 1 × 106 trypanosomes/mL. For screening, parasite cells were cultured in HMI-9 media (composed of 2× Iscove’s Modified Dulbecco’s Media, 1 mM hypoxanthine, 0.16 mM thymidine, 50 µM bathocuproine disulphonic acid, 1.5 mM L-cysteine, 1 mM sodium pyruvate, and 0.0014% v/v β-mercaptoethanol; all reagents from Sigma-Aldrich), with the pH adjusted to 7.3 and the media sterilized by filtration and stored at 4 °C.
In the screening campaign, the Janus MDT liquid handler equipped with a 384 head was used to dilute and dispense compounds. In confirmatory screening, compound transfer was performed manually with a 16-channel pipette. Intermediate plates were prepared by dispensing 19.4 µL phosphate-buffered saline (PBS)/well in a plate, followed by the addition of controls and/or test compound/natural product mixture. Prior to the transfer, the contents in the stock plate were mixed thrice by aspirating/dispensing 5 µL, and then 0.6 µL were dispensed into the intermediate plate, yielding a 33.3-fold dilution. Ten microliters from each well of the intermediate plates were transferred to assay plates. Compound plating was immediately followed by parasite plating: exponentially growing T. brucei were harvested, counted in a Neubauer hemocytometer, and resuspended at 8000 parasites/mL in complete HMI-9 media. The parasite suspension was kept stirring at low speed, and 50 µL/well were dispensed in assay plates with the aid of the WellMate, except for column 24, which was left blank for manual addition of 50 µL/well of media. The final volume was 60 µL/well, containing 0.5% v/v DMSO, with test compound/natural product mixture diluted 200× in relation to the concentration in stock plates. The positive controls contained 120 nM pentamidine. The plates were maintained at 37 °C and 5% CO2 in a humidified incubator for 72 h. Plates were developed by adding 15 µL of lysis buffer (containing 30 mM Tris HCl pH 7.4, 7.5 mM EDTA, 0.012% of Saponin, and 0.12% Triton X-100 [all reagents from Sigma-Aldrich] and SYBR Green diluted 1:2000) per well followed by agitation at 1500 rpm for 45 s in the MixMate plate mixer and incubation for 1 h in the dark at room temperature (RT). Plates were read using the EnVision Multilabel 2103 Reader with excitation at 485 nm and emission at 535 nm. Plates with Z′29 <0.4 were failed and repeated. The pentamidine IC50 was determined using Prism GraphPad software (version 6) using a four-parameter logistic fit. Compounds/natural product mixtures that reduced parasite growth by >60%, in comparison with intraplate controls, were categorized as hits. For the MycoDiverse natural products library screen, the selected hits were fractionated guided by the assay activity. This first stage of assay-guided fractionation entailed reextracting stored frozen fermentation material and fractionating the resulting fresh extracts using a standard reversed-phase HPLC method to generate 24 fractions for each hit. Aliquots of these were tested at three concentrations (three points of 1:5 dilutions), with the top concentration for testing at 200-fold dilution, using the same screening methodology described above.
Compound Solubility Studies
The solubility of compounds under various conditions (in triplicate) was determined after 4 h incubation at 37 °C in SGF (containing pepsin) and FaSSIF and FeSSIF. Assays were performed in 384-well plates in 100 µL volume in the presence of a maximum of 5% v/v DMSO. Kaolin was used as the insoluble control. Compounds at varying concentrations in each liquid were prepared and mixed using an orbital shaker (e.g., MixMate; Eppendorf AG, Hamburg, Germany) for 10 min at RT, sealed, and stored in the dark at RT for 6 h. Measurements were performed using a NEPHELOstar Galaxy (BMG Labtech, Ortenberg, Germany). Compounds were tested at increasing concentrations until the relative nephelometry units (RNU) significantly increased compared with the background, which is indicative of compound precipitation. Compounds were considered to be soluble at the highest concentration when the RNU did not significantly increase compared with the background.
Cytotoxicity Assay
A549 cells and WI-38 cells were grown on surface-modified T175 cell culture flasks in Dulbecco’s Modified Eagle Medium with 10% fetal calf serum (FCS), streptomycin (100 µg/mL), and 100 U/mL penicillin G. At about 80% confluency, cells were washed, trypsinized, resuspended, and counted in RPMI-1640 medium before seeding (in triplicate) into white 384-well microtiter plates (20 µL) at 500 cells/well and incubated at 37 °C in the presence of 5% CO2. At 24 h postseeding, baseline growth was assessed using a control plate and CTG reagent (Promega Corp.). A total of 20 µL/well of CTG detection mix was added, and plates were read using an EnVision Multilabel 2103 Reader after a 10-min incubation in the dark. A set of parallel assay plates were dosed with compounds as 11-point dose-response curves. Each test compound (200 nL of 10 mM top concentration in 100% v/v DMSO) was added to cells seeded in polystyrene 384-well cell culture microtiter plates using the Echo 550 Liquid Handler and read after 48 h of incubation at 37 °C in the presence of 5% CO2 using CTG as described above (24 h postseeding). The compounds/positive control (paclitaxel with final concentration of 10 µM and 1% v/v DMSO) and NC (final 1% v/v DMSO) were added into the 384-well plates (200 nL/well; 1% v/v DMSO) using the Echo 550 Liquid Handler. The raw data were normalized to percentage of cell growth by using the baseline growth and the corresponding NC containing only 1% v/v DMSO. The luminescence signal of each sample (S) was converted into percentage of cell growth compared with the average signal of the baseline control (BC). In case of a sample signal being higher than the average baseline, the following formula was used: % effect = (S – BC)/(NC – BC) × 100. In case of a sample signal being lower than the average baseline, the following formula was used: % effect = (S – BC)/BC × 100. The relative growth was used to calculate cell viability and proliferation parameters (GI50, TGI, and LC50) as described elsewhere.30 The GI50 value corresponded to the compound concentration where growth was reduced to 50%, the TGI corresponded to full growth inhibition (100% inhibition), and the LC50 corresponded to 50% cell death compared with the BC measurement.
Cytochrome (CYP) P450 Inhibition Assay
These assays (in triplicate) made use of microsomal preparations of CYP450 (1A2, 2C9, 2C19, 2D6, and 3A4) from baculovirus-infected insect cells (Corning Inc.) and cytochrome c reductase (and cytochrome b5 for CYP450 3A4). For detection of CYP450 activity, the luminescence-based P450-Glo (Promega Corp.) assay system was used that contained a luminogenic CYP450 substrate, lyophilized luciferin detection reagent, and reconstitution buffer. The substrates were luciferin derivatives of CYP450-specific substrates that produce (4S)-4,5-dihydro-2-(6-hydroxy-benzothiazolyl)-4-thiazolecarboxylic acid (D-luciferin) after cleavage by CYP450 (CYP450 3A4, luciferin-IPA; CYP450 2C19, luciferin-H EGE; CYP450 2C9, luciferin-H; CYP450 2D6, luciferin-ME EGE; CYP450 1A2, luciferin-1A2). CYP450 reactions were initiated by addition of the NADPH regeneration system to the enzyme-substrate mixture with the luciferin detection reagent stopping the reaction and the D-luciferin being converted to oxyluciferin under production of light being proportional to the CYP450 activity. The CYP450 assays were performed using the Tecan Fluent liquid-handling automation platform (Tecan Group Ltd, Männedorf, Switzerland) in 384-well assay format. Compounds were added into an empty 384-well plate (100 nL/well in 1% v/v DMSO) using the Echo 550 Liquid Handler followed by addition of 5 µL/well of CYP450/substrate mixture and incubation for 30 min at 37 °C, after which the reaction was initiated by addition of 5 µL/well NADPH regeneration system. After a further 30-min incubation at 37 °C, the CYP450 reaction was stopped, and the luciferase reaction was simultaneously initiated by addition of 10 µL/well of luciferin detection reagent, followed by an additional 30-min incubation at 37 °C. The luminescence signal was detected using an Infinite M1000 PRO plate reader. The NCs yielded 0% inhibition (1% v/v DMSO), and standard CYP450 specific inhibitors were used as positive controls, yielding 100% inhibition (CYP450 1A2, alpha-naphthoflavone; CYP450 2C9, sulfaphenazole; CYP450 2C19, troglitazone; CYP450 2D6, quinidine; CYP450 3A4, ketoconazole).
HDAC Assay
Inhibition of HDAC enzymes (in triplicate) was determined using the homogeneous, single-addition, bioluminogenic HDAC-Glo I/II assay (Promega Corp.). The kit contains a proluminogenic substrate with an acetylated lysine peptide sequence derived from histone 4 conjugated to aminoluciferin. HDAC-mediated deacetylation of the lysine residue facilitates luminogenic substrate susceptibility to specific proteolytic cleavage by the enzyme in the developer reagent. The aminoluciferin product of that cleavage acts as a substrate for luciferase, and the amount of light produced in this reaction is proportional to HDAC enzyme activity. Human recombinant HDAC enzymes were purchased from BPS Bioscience (San Diego, CA), and standard inhibitor trichostatin A (Sigma-Aldrich) was dissolved to a yield stock solution in 100% v/v DMSO and stored at −20 °C. Plate handling was performed using a Cell Explorer HTS platform equipped with an Echo 550 Liquid Handler and Multidrop liquid-handling systems with luminescence measurements taken using an EnVision Multilabel 2103 Reader. The compounds/positive control (trichostatin A with final concentration of 1 µM and 1% v/v DMSO) and high control (final 1% v/v DMSO) were added into the 384-well plates (100 nL/well; 1% v/v DMSO) using the Echo 550 Liquid Handler. The HDAC-Glo I/II assay reagent was prepared by (1) rehydration of lyophilized HDAC-Glo I/II substrate (with an acetylated peptide concentration of 100 µM) in 10 mL HDAC-Glo I/II assay buffer and (2) addition of 10 µL of developer reagent (containing trypsin). The microtiter plates were mixed briefly by orbital shaking (500–700 rpm), and luminescence was measured at steady-state signal: background, which was achieved after 20 min.
Aurora B Kinase Assay
Inhibition of Aurora B kinase was determined (in triplicate) using the ADP-Glo Kinase Enzyme System (Promega Corp.). The positive control was SU6656 at a final concentration of 1 µM, with the NC being DMSO at the same concentration (v/v). An enzyme master mix containing of 1× buffer, 50 µM DTT, and 17.5 ng/µL (35 µL/well) Aurora B (all reagents provided in the kit) was prepared. A substrate master mix containing 1× buffer, 36 µM adenosine triphosphate (ATP), and 7.5 ng/µL (15 ng/well) myelin basic protein (MBP) as substrate (buffer and MBP were provided in the Aurora B Kinase Enzyme System; ultrapure ATP was provided in the ADP-Glo Kinase Assay System) was prepared. Two microliters of the enzyme master mix and 2 µL of the substrate master mix were added to each well of a 384-well low-volume plate. The plate was sealed using Thermowell sealing tape (Corning Inc.) and incubated for 45 min at RT. The enzymatic reaction was stopped by adding 4 µL ADP-Glo reagent (provided in the ADP-Glo Kinase Assay System) and the plate sealed using Thermowell sealing tape and incubated for 40 min at RT. Following this, 8 µL detection reagent was added to each well, and the plate was sealed again and incubated for 45 min at RT, with the luminescence measured using the EnVision Multilabel 2103 Reader. DMSO concentration was tolerated up to 2% v/v final.
hERG Cardiotoxicity Assay
The Predictor hERG fluorescence polarization assay (Thermo) was used to test compounds for potential cardiotoxicity (in triplicate). To each well of an assay plate, 100 nL of the test/control compound was added followed by addition of 5 µL homogenized membrane solution (undiluted) and 5 µL of tracer (1 nM final concentration in assay). The plates were incubated for 2 h at 25 °C in a humidity-controlled incubator, and the fluorescence polarization was measured using an EnVision Multilabel 2103 Reader. The NCs (0% inhibition) and positive controls with E-4031, a blocker of hERG-type potassium channels (yielding 100% inhibition), were used to normalize the raw data.
Mitochondrial Toxicity Assay
This assay made use of the MitoTracker Red CMXRos dye (Thermo), which stains mitochondria in live cells and its accumulation is dependent on the presence of a membrane potential. The renal carcinoma 786-0 cell line was used for mitochondrial toxicity screening. Cells were harvested from a 75 cm2 flask at 80% confluency by washing once with 5 mL RT PBS and incubating with 1 mL 0.05% v/v trypsin/0.02% v/v EDTA for 3 min. Cells were suspended in 10 mL prewarmed cell culture media (RPMI-1 640 supplemented with 10% v/v FCS, 100 U/mL penicillin, and 100 µg/mL streptomycin) and counted using a Scepter (Merck Millipore, Darmstadt, Germany). The 786-0 cells were diluted to 75,000 cells/mL, and 20 µL of this suspension added to each well of a 384-well plate (in triplicate). Cells were incubated for 36 h at 37 °C and 5% CO2, and compounds were added using a predilution plate. The positive control was valinomycin at a final concentration of 1 µM, with the NC being DMSO at the same concentration. Ten microliters of compounds and controls were added to cells and incubated for 6 h at 37 °C and 5% CO2 in a humidity-controlled atmosphere. After incubation, 10 µL of a 200 nM solution of MitoTracker Red CMXRos in prewarmed cell culture media was added to each well, and the 786-0 cells were incubated for an additional 45 min at 37 °C and 5% CO2. MitoTracker Red CMXRos uptake was measured using an Opera Imaging System. To facilitate automatic image analysis, the layout containing the compound area as well as the valinomycin and DMSO control areas were created and stored. A sublayout of five evenly dispersed fields per well were used. These settings also included a measurement height of 1 µm, which was stored in an exposure file format. By using the stored settings and files, an automated run was repeatedly created and executed. The images were transferred to a file server and uploaded into Columbus 2.4.0 using the built-in helper function and analyzed therein.
Pharmacokinetics Using LC-MS
BALB/c mice were treated with compound alone, 1 mg/kg intravenously or 20 mg/kg by mouth, based on the toxicity and solubility of the compound as described in the literature. Plasma samples were analyzed by LC-MS. Chromatographic separation was carried out using a Shimadzu LC system consisting of two pumps, column oven, degasser, and autosampler. Attached to this system was an analytical column (C18, Gemini 5 mm 110 A Phenomenex, 150 mm × 2 mm). The HPLC system was connected to a triplequadrupole mass spectrometer equipped with a turbo-ion spray source operated with unit resolution in the positive ion mode (ESIQQQMS, Shimadzu LCMS-30). Plasma samples of mice (NMRI and BALB/c) were obtained by serial sampling from submandibular or tail vein and stored at −20 °C until analyzed.
Hardware
Screening of synthetic compounds and MycoDiverse natural products library was performed in 384-well microtiter plates. The Cell Explorer HTS platform (PerkinElmer, Waltham, MA) equipped with an Echo 550 Liquid Handler (Labcyte, Sunnyvale, CA) and Multidrop (Thermo) liquid-handling systems were used for the TbPTR1 target-based screen. Assay measurements were made using an EnVision Multilabel 2103 Reader (PerkinElmer). The T. brucei phenotypic screen was performed using a Janus MDT (PerkinElmer), equipped with a 384-head, WellMate (Thermo) and MixMate plate mixer (Eppendorf AG). Assay measurements were taken using the EnVision Multilabel 2103 Reader (PerkinElmer) or Infinite M1000 PRO plate reader (Tecan Group Ltd). Images from the mitochondrial toxicity assay were taken using an Opera automated microscope (PerkinElmer). The CYP450 assay was performed using the Tecan Fluent liquid-handling automation platform (Tecan Group Ltd).
Data Analysis
The screening data were analyzed using ActivityBase (IDBS, Guildford, UK), and outlier elimination in the control wells was performed using the 3-sigma method. Unless stated, dose-response experiments were performed in 11-point format with the IC50 value, Hill slope, minimum signal, and maximum signal for each dose-response curve obtained using a four-parameter logistic fit in the XE module of ActivityBase (IDBS).
Results
Design, Synthesis, and Characterization of Synthetic Compound Libraries and Composition of the Hypha MycoDiverse Natural Products Library
A total of 187 synthetic compounds from the following scaffolds were designed and synthesized (see Table 1 for details), namely, (1) triazole-linked privileged structure-based conjugates22; (2) aryl thiosemicarbazones23; (3) thiadiazole-based compounds that can target the folate/biopterin pathway with potential for activity against the parasite in combination with DHFR inhibitors24; (4) chroman-4-one derivatives that target PTR1 showing anti-parasitic activity25; (5) 2′-hydroxy chalcone derivatives, which displayed a broad spectrum of pharmacological activities, including antiparasitic activity26; (6) flavonol derivatives21; and (7) analogues of miltefosine, an alkyl phospholipid compound that exhibits antileishmanial activity and has been approved for clinical use.27 Most of the compounds belonging to these classes have been shown to exhibit antiparasitic activity.21–27 Our focus was to enhance the core scaffold of each class of compound aiming to improve the potency against TbPTR1 and various properties such as their antiparasitic activity (inhibition in the T. brucei phenotypic assay) or ADME-Tox properties that would increase their significance in the drug discovery value chain. Suitably active compounds were evaluated in the ADME-Tox assays initially at 10 µM and, when necessary, were subsequently screened in dose-response format to determine their overall liabilities.
Table 1.
Summary of the Output of the Progression of Synthetic Compound Libraries and the MycoDiverse Natural Products Library for Trypanosomatidic Activity.
| Compound Class | Quantity | Hit Validation | Hit-to-Lead | Reference |
|---|---|---|---|---|
| Triazole-linked privileged conjugates | 18 compounds | ✓ | [22] | |
| Aryl thiosemicarbazones | 28 compounds | ✓ | ✓ | [23] |
| Thiadiazole derivatives | 57 compounds | ✓ | ✓ | [24] |
| Chroman-4-one derivatives | 3 compounds | ✓ | [25] | |
| Chalcone derivatives | 13 compounds | ✓ | [26] | |
| Flavonol derivatives | 16 compounds | ✓ | [21] | |
| Miltefosine analogues | 52 compounds | ✓ | ✓ | [27] |
| MycoDiverse natural products library | 11,089 mixtures | ✓ | — |
The MycoDiverse natural products library (10,049 mixtures from basidiomycetes and ascomycetes and 1040 entomogenous fungi-derived samples) can represent a valuable source of drugs and compounds for pharmaceutical development in many therapeutic areas. A proprietary stimulatory fermentation technique is used to unlock the biosynthetic potential of this underexploited group of microorganisms, resulting in bioactive chemistry with a high degree of structural novelty, which offers an opportunity to prospect this underexploited group of organisms to identify and develop new antiparasitic agents.
Screening Using the TbPTR1 Target-Based Assay
A TbPTR1 target-based assay was successfully developed and validated to meet the criteria for screening. The key parameters investigated for the assay included enzyme and substrate titrations, assay buffer composition, incubation time and temperature, Km for NADPH (Fig. 2A), Z′, DMSO tolerance (Fig. 2B), and reproducibility of the potencies of methotrexate as the reference compound (Fig. 2C). As this assay made use of a kinetic readout, it was possible to identify compounds that optically interfere from the outset.
Figure 2.

(A) Determination of the Km for the NADPH for TbPTR1 enzyme (Km 229.1 ± 25.4 µM). The duplicate data from these experiments were fitted using the four-parameter logistic equation using GraphPad Prism (version 5.02, GraphPad Software, Inc.), and standard deviations are within ±10% of the values of the rates. (B) DMSO tolerance. (C) Compound concentration-response curve for TbPTR1 target-based assay against methotrexate (IC50 2.62 ± 0.12 nM; Hill slope 0.72 ± 0.04), and standard deviations are within ±10% of the values. (D) Summary of compounds evaluated (see Table 1 for details) were shown to inhibit T. brucei growth at 50 µM. See the Materials and Methods section for full experimental details.
As expected, many of the compounds from the synthetic libraries were active (IC50 <1 µM) in the TbPTR1 target-based assay as these scaffolds were known to be active against this enzyme.21–27 In addition, we have now established that these compounds also inhibit T. brucei growth at 50 µM (Fig. 2D).
With regard to screening of the MycoDiverse natural products library against TbPTR1, the performance of the screen was exceptional, as shown by the Z′ >0.85 for each assay plate, which contained multiple single concentrations of miltefosine, methotrexate, and DMSO to allow the quality control of each assay plate to be performed (Fig. 3A). As a number of TbPTR1 inhibitors are known, each assay plate also contained eight-point dose responses of F0046, methotrexate, and pyrimethamine; the average IC50 values across all 34 assay plates for each reference compound were for methotrexate 8.6 ± 2.3 nM, F0046 73 ± 27 nM, and pyrimethamine 16.7 ± 5.0 nM (Fig. 3B). Although miltefosine was included as a control, it is not an inhibitor of TbPTR1 and, as expected, did not act in this manner. Based on test wells that yielded ≥50% inhibition, seven apparent hits were identified (hit rate of 0.069%), which were evaluated in conjunction with the hits from the T. brucei phenotypic assay and underwent assay-guided fractionation. These were subsequently deprioritized to focus on the phenotypic assay hits, which showed more promising active fraction potency.
Figure 3.

(A) Layout of samples and controls in assay plates for the TbPTR1 target-based screen against the MycoDiverse natural products library. (B) Dose-response curves (DRC) and average IC50 values across all 34 assay plates for each reference compound were for methotrexate 8.6 ± 2.3 nM, F0046 73 ± 27 nM, and pyrimethamine 16.7 ± 5.0 nM. Standard deviations are within ±10% of the values.
Screening Using the T. brucei Phenotypic Assay
The T. brucei phenotypic assay relied on the indirect determination of parasite population viability by quantification of total DNA present in a well by the SYBR Green I DNA fluorescent dye. The assay consisted of incubating the bloodstream forms of T. brucei brucei in the presence of compounds for 72 h, followed by cell lysis and addition of the DNA-binding SYBR Green I dye.31 This assay was successfully miniaturized into a 384-well microtiter plate and met the criteria for suitability in a screening campaign. The parameters investigated included concentrations of cells, assay media composition, incubation time and temperature, Z′, DMSO tolerance, and reproducibility of the potencies of the reference compound pentamidine (3.17 ± 0.69 nM).
The purpose of the T. brucei phenotypic screen against the Hypha MycoDiverse natural products library (10,049 extracts and fractions from fermentations of higher fungi complemented with 1040 entomogenous fungi-derived samples) was to identify samples with inhibitory activity of T. brucei growth (antiparasitic activity). The MycoDiverse set was screened at a 200-fold dilution in 0.5% v/v DMSO, yielding a mean Z′ across the screen of 0.79 for compound plates. Samples with antiparasitic activity ≥60% were selected as hits, totaling 1645 hits and an overall primary hit rate of 14.8%. It was also clear that most of the hits were caused by biomass extracts, which tend to contain significantly more concentrated material than the SPE fractions generated from the aqueous fermentation broths. Because of the large number of samples to progress, the number of hits was reduced for confirmation assays using the previously known Hypha Discovery antitumor assay data to deselect samples toxic to human tumor cells. Thus, any sample showing ≥50% cytotoxicity against one out of three or four cell lines (A549, A375, MCF7, and HT29) was deselected (data not shown). In addition, the Hypha Discovery collections of fungi have a high degree of taxonomic characterization, enabling further filtering of particular species or genera that appeared to be overrepresented. In this way, 287 hits were selected for confirmation in dose-response experiments (seven-point dose-response curves with 1:3 dilution series). The confirmation rate was 83%, with the quality control yielding a mean Z′ of 0.58.
After analyzing the maximum activity and dose-response curve fitting of the Hypha MycoDiverse confirmed hits, 40 samples were selected for reversed-phase HPLC fractionation. The 24 fractions for each hit were tested at three concentrations (three points of 1:5 dilutions), with the top concentration for testing at 200-fold dilution using the same screening methodology described above. The mean Z′ was 0.54, and the pentamidine-positive control yielded a mean IC50 of 1.51 ± 0.66 nM. All 40 fractionations were successful in that the fresh secondary tube extracts were all active, yielding at least one fraction with T. brucei growth inhibitory activity at the top concentration tested, whereas many of them yielded multiple fractions active at 1:5 or 1:25 dilutions, suggesting the presence of potent active components. There was no evidence for any loss of activity due to the separation of two or more components combining synergistically to cause activity in the unfractionated samples. None of the 40 hits had TbPTR1 inhibitory activity, so the fractions were not tested in this assay.
As the same chromatographic method was used for each fractionation, the active fraction numbers were used as an initial guide to the active component retention times, and the frequent occurrence of the same active fraction numbers in fractionations from different hits suggested the presence of common active components. All active fractions were analyzed by reversed-phase HPLC with diode-array UV, electrospray ionization MS (positive and negative ionization modes), and evaporative light-scattering detection (HPLC-UV-MS-ELSD). The resulting data were inspected for the presence of known compounds or indications of the presence of the same or similar active compounds in active fractions from fractionations of different hits, with searching of putative physicochemical data in natural products databases (Antibase and the Chapman and Hall Dictionary of Natural Products) where possible.
The most promising active fractions were then advanced to a further fractionation step (secondary fractionation): new chromatographic methods were developed to separate the components present in each fraction, and the resulting secondary fractions were tested for T. brucei growth inhibitory activity to unequivocally link activity to chromatographic peak(s) to allow further physicochemical characterization of the active components and estimation of the potency of their antiparasitic activity. Specific examples of validated hits from the MycoDiverse natural products library screen include E03124-A4 (an extract from fermentation of an organism classified as Stilbella erythrocephala). This was found to contain two major components with molecular weights of 1670 and 1656 that were subsequently identified as the known peptaibol metabolites, antiamoebins I and III, respectively, which have been reported to have antimalarial activity with low micromolar potency.32 Figure 4A shows pairs of ELSD and diode array UV-visible chromatograms for the crude extract of E03124 (top), purified antiamoebin I (middle), and purified antiamoebin III (bottom), while Figure 4B shows the mass spectra of the two components. ELSD is useful for detecting compounds with little or no UV detection, which is the case for the antiamoebins. Purified antiamoebins I and III had IC50 values of 49.0 ± 4.0 nM and 1.65 ± 0.08 µM, respectively. E03139-A4 (from a Tolypocladium sp.) was found to contain components with putative molecular weights of 1620, 1634, and 1648, corresponding to the known Tolypocladium spp. metabolites efrapeptins. These are linear peptides that are chemically distinct from the antiamoebins but have also been reported to have antimalarial activity32; they were not further purified. E03172-A4 (from Verticillium griseum) was found to contain a major active component with molecular weight 197 and UV maxima at 220 and 277 nm that was identified as the mycotoxin tenuazonic acid, which is produced by a range of fungi and does not appear to have been reported to have antiparasitic properties, but it is a protein synthesis inhibitor and is most frequently reported as having phytotoxic effects.33 When tested as a pure compound, tenuazonic acid showed only weak activity in the phenotypic assay, with an IC50 of 2.7 µM, so it was obviously present in the crude extract at a very high concentration. The structures of these compounds are shown in Figure 5 . The identification of these compounds at this stage validates the capability of the MycoDiverse natural products library assay-guided fractionation process to isolate and characterize compounds with potent bioactivities. Further investigation of a number of higher fungal fermentations led to the identification of several novel compounds belonging to various sesquiterpene families and a novel polyketide. These compounds had antitrypanosomal potency in the mid-low micromolar range and will be reported separately.
Figure 4.
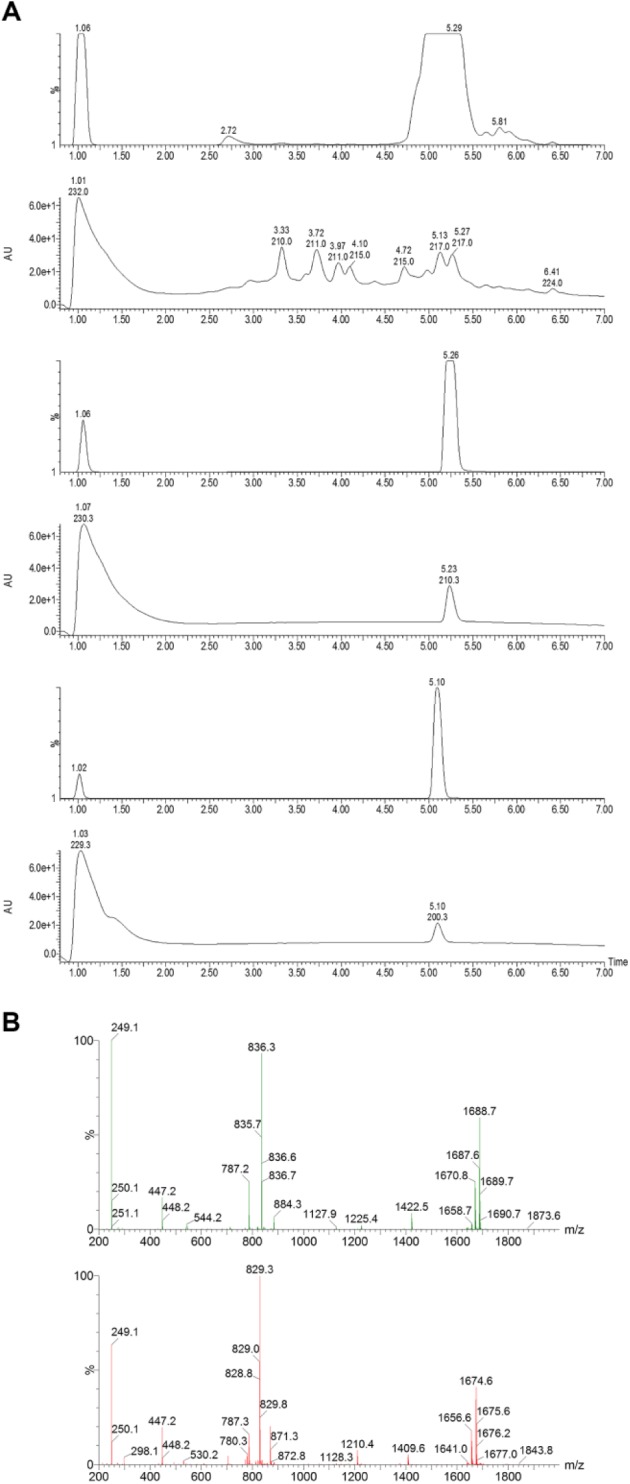
(A) Evaporative light-scattering detection (upper) and diode array ultraviolet-visible (lower) chromatograms of (top) MycoDiverse natural products library sample E03124-A4, a solid-phase extraction fraction derived from fermentation of Stibella erythrocephala; (middle) purified compound antiamoebin I; (bottom) purified compound antiamoebin III. The high-performance liquid chromatography–mass spectrometry (HPLC-MS) analyses were conducted on a SymmetryShield RP8 column as detailed in the Materials and Methods section. (B) Mass spectra of active compounds purified from MycoDiverse natural products library sample E03124-A4: (top) antiamoebin I; (bottom): antiamebin III. The HPLC-MS analyses were conducted on a SymmetryShield RP8 column as detailed in the Materials and Methods section.
Figure 5.

The structures of known compounds isolated from the MycoDiverse natural products library by assay-guided fractionation using the phenotypic T. brucei assay: (A) tenuazonic acid; (B) antiamoebin I; (C) antiamoebin III; (D) efrapeptin D; (E) efrapeptin E.
Discussion
The Need to Identify New Drugs to Treat Neglected Parasitic Diseases
With the goal of providing chemical starting points for the discovery and development of new drugs for HAT chemotherapy, we have attempted to discover such suitable compounds using a combination of target and phenotypic approaches. This has enabled the bringing together of various classes of synthetic compounds and natural products and screening them in (1) a target-based assay for TbPTR1, a good target for trypanocidal activity,34 and (2) a T. brucei phenotypic screening approach to identify compounds with trypanocidal activity from a target agnostic perspective. Most of the synthetic compound libraries were known to include inhibitors of TbPTR1. Our aim was to understand their ADME-Tox properties in a panel of assays, which included their physicochemical characterization, solubility (in water, buffer, SGF, FeSSIF, and FaSSIF), in vitro off-target liability enzyme panel (kinase and HDAC enzymes), in vitro cytotoxicity assay panel (human A549 and WI-38 cell lines), mitochondrial toxicity (human 786-0 cell line), cytochrome P450 inhibition (1A2, 2C9, 2C19, 2D6, and 3A4 isoforms), cardiotoxicity (hERG binding assay), and rodent pharmacokinetics. Some of the classes of synthetic molecules delivered compounds that can be progressed in the drug discovery value chain to lead-like compounds.
Natural compounds are a proven source of molecules that can serve as chemical starting points for drug development, and in some cases, they may have properties enabling their use without further chemical modification. We made use of the MycoDiverse natural products library derived from fermentations of higher (basidiomycetes and ascomycetes) and entomogenous fungi, which is composed of 11,089 samples that are mixtures of compounds at unknown concentrations. In this case, the active samples underwent purification, leading to the physicochemical and biological characterization of the active component. This necessitated a process of assay-guided purification, supported, where necessary, by larger-scale fermentations to produce sufficient purified material, before the true development potential of these compounds could be assessed.
In the past, natural products have been shown to be a good starting point for drug development or even provided established antiparasitic drugs such as the antimalarial plant products quinine from Cinchona succiruba12 and artemisinin from Artemisia annua. The MycoDiverse natural products library originates from fermentations of higher fungi (basidiomycetes and ascomycetes), which have been shown to have defense strategies for protection against bacterial and fungal competitors and are expected to produce antihelmintic and antiprotozoal chemicals as they were shown to be attacked by fungivorous nematodes and protozoa.8
TbPTR1 Target-Based and T. brucei Phenotypic Screening Activities
In general, target-based assays and associated screening campaigns can be performed more easily than cell-based assays for reasons such as complexity, incubation periods, and multiparameter analysis of data. The TbPTR1 target-based assay that was developed performed robustly and enabled a typical screening batch to be processed in a single day. The hit rate for the MycoDiverse natural products library was relatively low (at 0.069%), and after elimination of samples causing apparent activity by optical interference, 14 hits were selected for the first stage of assay-guided purification. Twelve of these also showed strong growth inhibition in the T. brucei phenotypic assay.
In contrast to the TbPTR1 target-based screen, each T. brucei phenotypic screening batch required approximately 1 wk to be completed because of its complex nature and was therefore associated with a lower overall robustness. Despite these challenges, the T. brucei phenotypic assay performance was consistent toward the reference compound pentamidine, yielding an IC50 of 3.17 ± 0.69 nM, which is in good agreement with the independently reported value of 5.57 ± 1.06 nM.31 Based on the criteria for selection of hits and the ≥60% decrease in parasite growth, the overall hit rate was high (14.8%), and a strategy was adopted to reduce the number of samples for further study. These data further demonstrate that phenotypic screening can be useful in the discovery of new anti-infective drugs as most or all potential targets can be exposed to compounds in this strategy, as has been argued.33
Because of the nature of the natural product libraries that include mixtures of potentially valuable compounds, the initial hit selection was facilitated based on the MycoDiverse natural products library database information (including producing organism identities and toxicity data against human tumor cell lines), which led to a manageable number of samples selected for hit confirmation and dose-response studies. The TbPTR1 target-based screen against the libraries facilitated the triaging of actives for assay-guided purification investigations of the most promising hits. As each fungus fermented is represented by multiple samples (SPE fractions, biomass extracts, different fermentation harvest times, fermentations under different conditions) in these libraries, one hit per organism for confirmation and dose-response profiling was selected, favoring the less complex SPE fractions and shorter fermentation times and filtering species and genera that were overrepresented. This also reduced the number of hits for confirmation to 287, with most being confirmed. The Hypha Discovery database information was used to filter hits, with some hits selected for progression to assay-guided fractionation based on potency and producing organism diversity. Overall, 40 active primary sample hits were progressed to fractionation. This entailed reextracting stored frozen fermentation material and fractionating the resulting fresh extracts using a standard reversed-phase HPLC method to generate 24 fractions for each hit. The most promising active fractions were then advanced to secondary fractionation: new chromatographic methods were developed to separate the components present in each fraction, and the resulting secondary fractions were tested for T. brucei growth inhibitory activity to unequivocally link activity to chromatographic peak(s) to allow further physicochemical characterization of the active components and estimation of the potency of their antiparasitic activity.
This assay-guided natural product isolation campaign necessarily employed a reductionist approach to focus on identifying new compounds with promising antiparasitic properties given the complexity of the MycoDiverse natural products library samples and the time frame of the NMTrypI project (3 y, with most of the hit follow-up work conducted in the second half of the project). Nevertheless, there was no evidence for loss of activity on fractionation of the selected hits that might indicate the presence of synergistic component combinations in the unfractionated samples. The capability of this screening and assay-guided purification approach to identify compounds with promising properties was initially validated by the identification of known compounds with antitrypanosomal activity, including antiamoebins I and III, the efrapeptins, and tenuazonic acid. Although the antiamoebins had promising antiparasitic potencies, tenuazonic acid was only weakly active when pure, illustrating the potential for some hits to be caused by the presence of compounds with low potency activity at high concentration in the screening samples.
In summary, we have identified multiple synthetic compound series, some of which are associated with profiles that meet the generally accepted lead-like criteria, as well as characterized natural compounds that are validated hits. These outputs can undergo further optimization with the expectation that some of these scaffolds can be further exploited for antiparasitic drug discovery purposes.
Acknowledgments
The Authors acknowledge the collaboration of the COST ACTION 1307 for the positive and stimulating scientific environment that contributed to generating the idea and disseminating our drug discovery work in a unified high impact manuscript. I.P., W.M., J.P.-H. and R.C.W. gratefully acknowledge the support of the Klaus Tschira Foundation. C.B. and A.Q. gratefully acknowledge the School of doctorate of CME (Clinical and Experimental Medicine) at the University of Modena for favouring their PhD student research work.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has received funding from the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agreement No. 603240 (NMTrypI, New Medicines for Trypanosomatidic Infections).
ORCID iDs: Cecilia Pozzi  https://orcid.org/0000-0003-2574-3911
https://orcid.org/0000-0003-2574-3911
Lucio H. Freitas-Junior
https://orcid.org/0000-0002-8904-7897
Chiara Borsari
https://orcid.org/0000-0002-4688-8362
References
- 1. Molyneux D. H., Savioli L., Engels D. Neglected Tropical Diseases: Progress towards Addressing the Chronic Pandemic. Lancet 2017, 389, 312–325. [DOI] [PubMed] [Google Scholar]
- 2. Field M. C., Horn D., Fairlamb A. H., et al. Anti-Trypanosomatid Drug Discovery: An Ongoing Challenge and a Continuing Need. Nat. Rev. Microbiol. 2017, 15, 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nii-Trebi N. I. Emerging and Neglected Infectious Diseases: Insights, Advances, and Challenges. Biomed. Res. Int. 2017, 2017, 5245021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization. Integrating Neglected Tropical Diseases in Global Health and Development. Fourth WHO Report on Neglected Tropical Diseases. http://www.who.int/neglected_diseases/resources/9789241565448/en/ (accessed Dec 13, 2018).
- 5. Lejon V., Bentivoglio M., Franco J. R. Human African Trypanosomiasis. Handb. Clin. Neurol. 2013, 114, 169–181. [DOI] [PubMed] [Google Scholar]
- 6. Docampo R., Moreno S. N. Current Chemotherapy of Human African Trypanosomiasis. Parasitol. Res. 2003, 90, S10–S13. [DOI] [PubMed] [Google Scholar]
- 7. Kennedy P. G. Clinical Features, Diagnosis, and Treatment of Human African Trypanosomiasis (Sleeping Sickness). Lancet Neurol. 2013, 12, 186–194. [DOI] [PubMed] [Google Scholar]
- 8. Priotto G., Kasparian S., Mutombo W., et al. Nifurtimox-Eflornithine Combination Therapy for Second-Stage African Trypanosoma brucei Gambiense Trypanosomiasis: A Multicentre, Randomised, Phase III, Non-Inferiority Trial. Lancet. 2009, 374, 56–64. [DOI] [PubMed] [Google Scholar]
- 9. Alirol E., Schrumpf D., Amici Heradi J., et al. Nifurtimox-Eflornithine Combination Therapy for Second-Stage Gambiense Human African Trypanosomiasis: Médecins Sans Frontières Experience in the Democratic Republic of the Congo. Clin. Infect. Dis. 2013, 56, 195–203. [DOI] [PubMed] [Google Scholar]
- 10. Rohlfs M., Albert M., Keller N. P., et al. Secondary Chemicals Protect Mould From Fungivory. Biol. Lett. 2007, 3, 523–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Inchausti A., Yaluff G., Rojas de, Arias A., et al. Leishmanicidal and Trypanocidal Activity of Extracts and Secondary Metabolites from Basidiomycetes. Phytother. Res. 1997, 11, 193–197. [Google Scholar]
- 12. Tanaka A. K., Valero V. B., Takahashi H. K., et al. Inhibition of Leishmania amazonensis Growth and Infectivity by Aureobasidin A. J. Antimicrob. Chemother. 2007, 59, 487–492. [DOI] [PubMed] [Google Scholar]
- 13. Martinez-Luis S., Della-Togna G., Coley P. D., et al. Antileishmanial Constituents of the Panamanian endophyte Fungus Edenia sp. J. Nat. Prod. 2008, 71, 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kayser O., Kiderlen A., Croft S. L. Natural Products as Potential Antiparasitic Drugs. St. Nat. Prod. Chem. 2002, 26, 779–848. [Google Scholar]
- 15. Harvey A. L., Edrada-Ebel R., Quinn R. J. The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [DOI] [PubMed] [Google Scholar]
- 16. Kinghorn A. D., Pan L., Fletcher J. N., et al. The Relevance of Higher Plants in Lead Compound Discovery Programs. J. Nat. Prod. 2011, 74, 1539–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hefti F. F. Requirements for a Lead Compound to Become a Clinical Candidate. BMC Neurosci. 2008, 9(Suppl. 3), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roy R. Early Probe and Drug Discovery in Academia: A Minireview. High Throughput 2018, 7, E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rademacher C., Seeberger P. H. High-Throughput Synthesis of Diverse Compound Collections for Lead Discovery and Optimization. Handb. Exp. Pharmacol. 2016, 232, 73–89. [DOI] [PubMed] [Google Scholar]
- 20. Zhao H., Dietrich J. Privileged Scaffolds in Lead Generation. Expert Opin. Drug Discov. 2015, 10, 781–790. [DOI] [PubMed] [Google Scholar]
- 21. Borsari C., Luciani R., Pozzi C., et al. Profiling of Flavonol Derivatives for the Development of Anti-Trypanosomatidic Drugs. J. Med. Chem. 2016, 59, 7598–7616. [DOI] [PubMed] [Google Scholar]
- 22. Uliassi E., Piazzi L., Belluti F., et al. Development of a Focused Library of Triazole-Linked Privileged Structure-Based Conjugates Leading to the Discovery of Novel Phenotypic Hits against Protozoan Parasitic Infections. Chem. Med. Chem. 2018, 13, 678–683. [DOI] [PubMed] [Google Scholar]
- 23. Linciano P., Moraes C. B., Alcantara L. M., et al. Aryl Thiosemicarbazones for the Treatment of Trypanosomatidic Infections. Eur. J. Med. Chem. 2018, 146, 423–434. [DOI] [PubMed] [Google Scholar]
- 24. Linciano P., Dawson A., Pöhner I., et al. Exploiting the 2-Amino-1,3,4-Thiadiazole Scaffold to Inhibit Trypanosoma brucei Pteridine Reductase in Support of Early-Stage Drug Discovery. ACS Omega 2017, 2, 5666–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Pisa F., Landi G., Dello Iacono L., et al. Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity. Molecules 2017, 22, E426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borsari C., Santarem N., Torrado J., et al. Methoxylated 2′-Hydroxychalcones as Antiparasitic Hit Compounds. Eur. J. Med. Chem. 2017, 126, 1129–1135. [DOI] [PubMed] [Google Scholar]
- 27. Pachioni J. A., Magalhães J. G., Lima E. J., et al. Alkylphospholipids—A Promising Class of Chemotherapeutic Agents with a Broad Pharmacological Spectrum. J. Pharm. Pharm. Sci. 2013, 16, 742–759. [DOI] [PubMed] [Google Scholar]
- 28. Shanks E. J., Ong H. B., Robinson D. A., et al. Development and Validation of a Cytochrome c-Coupled Assay for Pteridine Reductase 1 and Dihydrofolate Reductase. Anal. Biochem. 2010, 2, 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang J. H., Chung T. D. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 30. Ellinger B., Silber J., Prashar A., et al. A Phenotypic Screening Approach to Identify Anticancer Compounds Derived from Marine Fungi. Assay Drug Dev. Technol. 2014, 12, 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faria J., Moraes C. B., Song R., et al. Drug Discovery for human African Trypanosomiasis: Identification of Novel Scaffolds by the Newly Developed HTS SYBR Green Assay for Trypanosoma brucei. J. Biomol. Screen. 2015, 20, 70–81. [DOI] [PubMed] [Google Scholar]
- 32. Nagaraj G, Uma M. V., Shivayogi M. S., et al. Antimalarial Activities of Peptide Antibiotics Isolated from Fungi. Antimicrog. Ag. Chemother. 2001, 45, 145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cole R. J., Cox R. H. Handbook of Toxic Fungal Metabolites; Academic Press, Waltham, MA, 1981. [Google Scholar]
- 34. Sienkiewicz N., Ong H. B., Fairlamb A. H. Trypanosoma brucei Pteridine Reductase 1 Is Essential for Survival In Vitro and for Virulence in Mice. Mol. Microbiol. 2010, 77, 658–671. [DOI] [PMC free article] [PubMed] [Google Scholar]