

Abstract
The quantification and identification of cellular phenotypes from high-content microscopy images has proven to be very useful for understanding biological activity in response to different drug treatments. The traditional approach has been to use classical image analysis to quantify changes in cell morphology, which requires several nontrivial and independent analysis steps. Recently, convolutional neural networks have emerged as a compelling alternative, offering good predictive performance and the possibility to replace traditional workflows with a single network architecture. In this study, we applied the pretrained deep convolutional neural networks ResNet50, InceptionV3, and InceptionResnetV2 to predict cell mechanisms of action in response to chemical perturbations for two cell profiling datasets from the Broad Bioimage Benchmark Collection. These networks were pretrained on ImageNet, enabling much quicker model training. We obtain higher predictive accuracy than previously reported, between 95% and 97%. The ability to quickly and accurately distinguish between different cell morphologies from a scarce amount of labeled data illustrates the combined benefit of transfer learning and deep convolutional neural networks for interrogating cell-based images.
Keywords: high-content imaging, cell phenotypes, machine learning, transfer learning, deep learning
Introduction
High-content screening (HCS) has proven to be a useful and successful technique to identify and quantify cell phenotypes.1,2 Although conventional approaches for the classification of phenotypes using cell images have shown positive results,3–7 they require several nontrivial data analysis steps. An example is Ljosa et al.7 and their pipeline workflows, which include cellular segmentation, feature extraction, profiling methods (e.g., factor analysis), and a nearest-neighbor classifier. Cell segmentation algorithms typically require manual adjustments for each new experimental setup,4 and feature extraction tends to rely on “handcrafted” features, such as those related to texture and shape (several of which are computationally expensive to measure). Principal component analysis (PCA), assuming a linear mapping, is then often used to reduce the dimensionality of these high-dimensional (>500) and highly correlated feature sets.8
Convolutional neural networks (CNNs) have recently brought about breakthroughs in computer vision and image processing—CNNs automatically discover the features needed for the classification of images based solely on the raw pixel intensity data.9 This supervised feature learning technique has been shown to be superior to using traditional handcrafted features,10,11 and the combination of segmentation and classification in a single framework12 means that image classification can be performed without the need for prior cell segmentation (a complex task that often requires careful consideration and significant computation13). A recent survey shows a rapid growth in the application of deep learning to medical image analysis,14 with several studies outperforming medical expert classification. A convenient property of CNNs is that the pipeline workflow of the traditional methods is taken care of by the network itself. Furthermore, by applying convolving filters (the weights/parameters of the network) on input layers, local connectivity and parameter sharing keeps the number of parameters relatively low, even for a deeper network.
A major bottleneck when applying supervised CNNs to cell images is the scarcity of labeled data. Importantly, studies have shown that reusing models trained on different tasks reduced these problems.15,16 Yosinski et al.17 noted that the transferability of features depends on the distance between the base task and the target task. However, the features from distant tasks may still perform better than random features. The study also illustrated that initializing the network with pretrained features improved the generalization even after considerable fine-tuning to the target dataset. Further, Zhang et al.18 showed that features trained on natural images (ImageNet19) could be transferred to biological data. Bayramoglu and Heikkilä20 used pretrained models on natural images and facial images for cell nucleus classification where the performances of transfer learning and learning from scratch were compared. All of their pretrained models had better predictive performance while requiring less training time. Phan et al.21 also successfully utilized transfer learning on bioimages, which outperformed all other methods on the mitosis detection dataset of the ICPR2012 contest.
The utility of transfer learning is in part due to the fact that the initial CNN layers capture low-level features, like edges and blobs—characteristics commonly shared between different types of images. When the number of labeled examples is small, transfer learning, like unsupervised pretraining, perhaps also helps reduce test set error through imposing a regularizing effect.22 Other factors are also likely responsible for the improved performance, such as the parameters being taken into a region of parameter space that, although better, is not accessible based only on the labeled data available.23
The Broad Bioimage Benchmark Collection (BBBC) is an important publicly available collection of microscopy images intended for validating image analysis algorithms.24 Various algorithms have been tested and validated on these datasets—ranging from traditional pipeline workflows to deep learning techniques.5,7,25 Pawlowski et al. (unpublished observations) utilized transfer learning without fine-tuning to extract features, and Ando et al. (unpublished observations) used a pretrained model on consumer images and further transformation techniques to attain the top accuracy on this benchmark dataset of 96%. However, research on transfer learning and fine-tuning of CNNs on these BBBC datasets is scarce—it is therefore important to investigate and compare this technique with the existing analysis tools that have been applied to the BBBC datasets.
In this study, we present state-of-the-art deep CNNs pretrained on natural images, with minor image preprocessing and without segmentation. These models were used to predict mechanisms of action (MoAs) and nucleus translocation, based only on pixel intensities that automatically pass through the network to give the final predictions. We used two different BBBC datasets, BBBC021v1 (hereafter referred to as the MoA dataset)24 and BBBC014v1 (hereafter referred to as the translocation dataset),24 to evaluate the models’ predictive performance as well as to visualize the filter output activations (feature maps) throughout the network. This visualization was done to understand the different levels of abstraction processed and the transferability of the networks. After the parameter values were transferred, the networks were fine-tuned to fit the data—the transferred parameters can thus be thought of as good initial parameter values. An extensive comparison with randomized initialization of parameter values was done, and we hypothesized that the pretrained parameters would significantly improve performance in terms of both accuracy and learning time relative to this baseline comparison.
Methods
Data
Datasets
The MoA dataset contains MCF-7 breast cancer cell images available from the BBBC (https://data.broadinstitute.org/bbbc/BBBC021/).24 The MoA dataset is a subset of the BBBC021v1 dataset that had been identified as clearly having 1 out of the 12 primary MoAs, and thus was annotated with a label. The 12 different MoAs represent a wide cross section of morphological phenotypes ( Fig. 1a ) that had been either identified visually or based on the literature (when not visually identifiable).24 The BBBC21v1 dataset contains 13,200 fields of view (imaged in three channels), which were generated from a total of 55 microtiter plates, each having 60 wells (6 of them being DMSO control treatments) and 4 fields of view per well. These plates contained MCF-7 breast cancer cells that had been treated for 24 h with a collection of 113 small molecules (compounds) at eight concentrations (each well treated with 1 compound at one concentration). The wells had been fixed; labeled for DNA, F-actin, and B-tubulin; and imaged by fluorescence microscopy as described by Caie et al.26 We followed the same strategy as used in previous studies on the same dataset5,7,25 as well as previously unpublished studies (Ando et al., Pawlowski et al.). We extracted a total of 38 compounds and 103 treatments (compound–concentration pairs) that had been labeled with 1 out of the 12 different MoAs ( Table 1 ), summing up to 1208 images out of the 13,200 images that the BBBC021v1 dataset offered.
Figure 1.

(a) The different MoAs in the MoA dataset. Images were cropped and processed from the original training images. Act = actin disruption; MD = microtubule destabilization; Aur = aurora kinase inhibition; DR = DNA replication; Eg5 = Eg5 inhibition; PD = protein degradation; Ch = cholesterol lowering; DD = DNA damage; Epi = epithelial; KI = kinase inhibition; PS = protein synthesis; MS = microtubule stabilization. (b) The two different classes (positive and negative) in the translocation dataset. Positive means translocation; negative means no translocation. Images were cropped and processed from the original training images.
Table 1.
Summary Table of the MoA Dataset.
| Compound | Number of Concentrations | Mechanism of Action (MoA) | Replicates | Number of Images |
|---|---|---|---|---|
| Cytochalasin B | 2 | Actin disruption | 3 | 24 |
| Cytochalasin D | 1 | Actin disruption | 3 | 12 |
| Latrunculin B | 2 | Actin disruption | 3 | 24 |
| AZ-A | 6 | Aurora kinase inhibition | 3 | 72 |
| AZ258 | 3 | Aurora kinase inhibition | 3 | 36 |
| AZ841 | 3 | Aurora kinase inhibition | 3 | 36 |
| Mevinolin/lovastatin | 3 | Cholesterol lowering | 3 | 36 |
| Simvastatin | 3 | Cholesterol lowering | 3 | 36 |
| Chlorambucil | 1 | DNA damage | 3 | 12 |
| Cisplatin | 1 | DNA damage | 3 | 12 |
| Etoposide | 3 | DNA damage | 3 | 36 |
| Mitomycin C | 4 | DNA damage | 3 | 48 |
| Camptothecin | 3 | DNA replication | 3 | 36 |
| Floxuridine | 2 | DNA replication | 3 | 24 |
| Methotrexate | 1 | DNA replication | 3 | 12 |
| Mitoxantrone | 2 | DNA replication | 3 | 24 |
| AZ-C | 7 | Eg5 inhibition | 3 | 84 |
| AZ138 | 5 | Eg5 inhibition | 3 | 60 |
| AZ-J | 3 | Epithelial | 3 | 36 |
| AZ-U | 3 | Epithelial | 3 | 36 |
| PP-2 | 2 | Epithelial | 2 | 16 |
| Alsterpaullone | 2 | Kinase inhibition | 2 | 16 |
| Bryostatin | 1 | Kinase inhibition | 2 | 8 |
| PD-169316 | 2 | Kinase inhibition | 2 | 16 |
| Colchicine | 1 | Microtubule destabilization | 3 | 12 |
| Demecolcine | 4 | Microtubule destabilization | 3 | 48 |
| Nocodazole | 2 | Microtubule destabilization | 3 | 24 |
| Vincristine | 7 | Microtubule destabilization | 3 | 84 |
| Docetaxel | 3 | Microtubule stabilization | 3 | 36 |
| Epothilone B | 3 | Microtubule stabilization | 3 | 36 |
| Taxol | 3 | Microtubule stabilization | 3 | 36 |
| ALLN | 2 | Protein degradation | 3 | 24 |
| lactacystin | 1 | Protein degradation | 3 | 12 |
| MG-132 | 2 | Protein degradation | 3 | 24 |
| Proteasome inhibitor I | 2 | Protein degradation | 3 | 24 |
| Anisomycin | 2 | Protein synthesis | 3 | 24 |
| Cyclohexamide | 3 | Protein synthesis | 3 | 36 |
| Emetine | 3 | Protein synthesis | 3 | 36 |
The “Number of Images” column represents the number of images before augmentation (through cropping).
The second dataset used in this study was the translocation dataset ( Fig. 1b ) provided by Ilya Ravkin, and also available from the BBBC (https://data.broadinstitute.org/bbbc/BBBC014/).24 The images are Human U2OS cells of cytoplasm to nucleus translocation of the MCF-7 and A549 (human alveolar basal epithelial) in response to tumor necrosis factor alpha (TNFα) concentrations. For each well, there was one field of view and two imaging channels, one for the nuclear counterstain (DAPI) and the other for the signal stain (FITC). A total 96 wells contained 12 concentration points and 4 replicates for each cell type. In this study, the four highest (labeled positive) and four lowest (including 0 concentration; labeled negative) concentrations were used for each cell type (i.e., 64 out of the total 96 wells), resulting in 64 (two-channeled) images (32 images per cell type).
Image Preprocessing
For the MoA dataset, the three different 16-bit range channels (labeled for DNA, F-actin, and B-tubulin), with a resolution of 1280 × 1024 pixels, were stacked into a three-channel image. The images were then normalized plate-wise, by subtracting the mean pixel intensities of DMSO images (the control samples) and then dividing by the standard deviation of their pixel intensities. After the normalization, an Anscombe transformation27 was performed, followed by a mapping to an 8-bit range. Similarly, for the translocation dataset, the two 8-bit channels (DAPI and FITC) of each sample, with a resolution of 1360 × 1024 pixels, were stacked with an addition of a zero matrix to create a three-channel input. These images were in the 8-bit range and were variance stabilized by the Anscombe transformation, and then mapped back to an 8-bit range. The resulting images for both datasets were cropped into 16 images (translocation dataset) and 4 images (MoA dataset) to increase the number of training samples, resulting in a total of 4832 images (680 × 512 pixels; 1–40 cells per image) and 512 images (320 × 256 pixels; ca. 30 cells per image), respectively.
CNN Architectures
Three different state-of-the-art architectures were implemented in TensorFlow via Keras:28 ResNet50,29 InceptionV3,30 and InceptionResnetV2.31 They were all pretrained on the ImageNet dataset, containing 13 million natural images.19
Residual Network
Utilizing a very deep CNN can have a negative effect on model performance—arising from the difficulty in finding suitable parameters for the deeper layers. Adding further layers to a suitably deep model can lead to higher training error not caused by overfitting.29,32,33 Residual networks use residual mapping H(x) = F(x) + x, where x is the original feature vector (identity mapping) added to the deeper version of the network F(x) (output of the stacked layers). Importantly, if the mappings are optimal, it is easier for the network to push the residuals to zero than fit an identity mapping with stacks of nonlinear layers.29 The implication of this is that although F(x) is not learning anything, the output will simply be an identity mapping x. Thus, in the worst-case scenario the output equals the input, and in the best-case scenario some important features are learned. Residual mappings therefore assist in avoiding the degradation problem that occurs for very deep CNNs. Another important aspect of residual networks is the intermediate normalization layers (also called batch normalization), which help to solve the problem of vanishing and exploding gradients.
The residual network used in this study had 50 layers (49 convolutional layers and a final fully connected classification layer), based on ResNet50 from the paper “Deep Residual Learning for Image Recognition.”29
Inception Network
It is often difficult to determine the best network filter sizes and whether or not to use pooling layers. To overcome this, inception architectures use many different filter sizes and pooling layers in parallel (an inception block), the outputs of which are concatenated and inputted to the next block. In this way, the network chooses which filter sizes or combinations thereof to use. To solve the problem of a large increase in computational cost, the Inception networks utilize 1 × 1 convolutions to shrink the volume of the next layer. This network architecture was introduced by Szegedy et al.34 to make a network deeper and wider, hence more powerful, while keeping the computational cost low. The Inception network can thus go very deep and, like ResNet50, utilizes intermediate normalization layers to avoid vanishing and exploding gradients.
The Inception network used in this study was InceptionV3 from the paper “Rethinking the Inception Architecture for Computer Vision,”30 excluding the auxiliary classifiers. This network had 95 layers in total, a number much larger than ResNet50 due to the width of each inception block.
Inception-Residual Network
Szegedy et al.31 evaluated a network combining inception blocks and residuals (similar to the ResNet50 residuals). They showed an improvement in training speed after introducing these residuals, making it possible to implement even deeper networks at a reasonable cost.
In this study, we implemented an Inception-ResNet architecture based on InceptionResnetV2 from the paper “Inception-v4, Inception-ResNet and the Impact of Residual Connections on Learning.”31 This network is even deeper than ResNet50 and InceptionV3 combined—totaling 245 layers.
Fine-Tuning of the Pretrained Networks
As mentioned before, our networks were all pretrained on the ImageNet dataset. Concretely, instead of randomly initializing the parameters (e.g., using Xavier initialization), the parameters of our networks used parameters that had been learned from the ImageNet dataset. Our networks, with their pretrained parameters, were then fine-tuned to better fit the MoA and translocation datasets.
Downsampling and Data Augmentation
Before the MoA images were inputted into the network, they were downsampled to have the dimensions of 224 × 224 × 3 for ResNet50 and 299 × 299 × 3 for InceptionV3 and InceptionResnetV2. For the translocation dataset, all images were downsampled to have dimensions of 256 × 256 × 3. To increase the number of training examples for MoA, the input images were randomly rotated and mirrored. Further, jitter, blur, and Gaussian noise were then randomly applied to both prevent the network from identifying noise as important features and augment the data further.
Model Evaluation and Deep Visualization
Model Evaluation
To evaluate the models of the MoA dataset, we used a “leave-one-compound-out” cross-validation—resulting in a 38-fold cross-validation. In each fold, predictions were made for all the treatments of the excluded compound. An element-wise median over the replicates was first calculated to obtain a prediction vector for each well. These vectors were then used to calculate the element-wise median over the wells, to obtain prediction vectors for each treatment. Finally, the highest values in the resulting 12-dimensional prediction vectors, containing the MoA predictions for the treatments, decided the models’ final predictions for the treatments. This procedure was repeated for all cross-validation folds, with a reset to the pretrained parameter values after each fold, resulting in a total of 103 final predictions. For the translocation dataset, we used a twofold cross-validation where the images of one cell line were “left out” as the test set, while the remaining images for the other cell line were used for training. At test time, the trained model made predictions of whether translocation had occurred for each of the left-out images—resulting in 32 predictions for each fold.
Activation Maximization
To compare the models before and after fine-tuning (fit to our MoA data), we investigated the ImageNet ResNet50 model (i.e., our initial model before fine-tuning) and a fine-tuned ResNet50 model (trained on all images for 10 epochs) and contrasted a selection of their filters. We used the high-level Keras visualization toolkit keras-vis35 to do this and applied an activation maximization function to generate an input image that maximizes certain filter output activations. This allows us to understand the input patterns that activate certain filters. The network filters of the deeper layers learn more abstract representations of the data, and tend to produce more intricately structured visualizations.36
Results and Discussion
To evaluate the models on the MoA dataset, we predicted the MoA for each treatment of the left-out compound for each fold in the cross-validation—hence testing our deep CNN models on unseen compounds and their treatments 103 times. We illustrate the accuracies of these predictions by plotting confusion matrices for all MoAs ( Fig. 2 ). ResNet50, InceptionV3, and InceptionResnetV2 attained mean accuracies of 97%, 97%, and 95%, respectively (and per image accuracies of 88%, 88%, and 86%, respectively)—thus comparing well with previous state-of-the-art algorithms in terms of accuracy (Suppl. Table S1), where our ResNet50 and InceptionV3 applications reached greater accuracy than any model yet reported based on the BBBC.23 Furthermore, there were only two treatments that were misclassified by the three models: colchicine, 0.03 µmol/L (microtubule destabilizer) and latrunculin B, 1.0 µmol/L (actin disruptor). Supplemental Figure S3 illustrates the near-imperceptible features of their MoAs, which explains why the models had difficulties correctly classifying these two treatments. However, several of the correctly predicted compounds had a treatment soft prediction of less than 0.5 (Suppl. Tables S2–S4), which means that although our model correctly predicted the MoA for these treatments, there were strong uncertainties in several of the predictions.
Figure 2.

Confusion matrices for hard predictions of compound–concentration pairs of the MoA dataset, with a mean accuracy of 97%, 97%, and 95% for ResNet50, InceptionV3, and InceptionResnetV2, respectively. Zeros are excluded for better visualization.
Transfer learning—the transfer of knowledge between tasks—is often beneficial when a limited amount of annotated data is available, such as in image cytometry, where manual annotations are time-consuming to acquire and require a high level of expertise to make. Furthermore, CNNs trained on biomedical images, captured under specific experimental condition and imaging setups, can have poor generalizability. To overcome these limitations, large annotated datasets, like ImageNet, can be used to pretrain state-of-the-art CNNs (such as the ResNet and Inception architectures). The transferred parameter values—providing good initial values for gradient descent—can be fine-tuned to fit the target data, as we have done in this paper. As we show in Figures 4 and 5, relative to training from scratch (using Xavier initializations) transfer learning allows the fitting of deeper networks based on fewer task-specific annotated images. It also gives faster convergence (i.e., fewer training epochs are required) and improved classification performance and generalizability. In Figure 3 , for the MoA dataset, the Xavier initialized models took many more epochs to converge (reaching mean prediction accuracies of 70%, 84%, and 91% for ResNet50, InceptionV3, and InceptionResnetV2, respectively). Note that for the deepest architecture we explored, InceptionResnetV2, the difference between the final prediction accuracies was much less pronounced than for either of the architectures in isolation (Figs. 4 and 5). For these two architectures, there was also more variability in the predictive performance across replicate runs based on the pretrained initializations, even when run for 20 epochs ( Fig. 4 ). From a practical perspective, one may wish to run multiple replicates and report the best performance. However, with a more skewed distribution, running many replicates and reporting the best one may give an exaggerated measure of predictive performance. For the MoA dataset, we show the mean and SEM (standard error of the mean) across three replicate runs of each model during the training epochs ( Fig. 3 ). The variability across replicates is likely a consequence of parameter uncertainty and confounding, the stochastic nature of gradient descent, and the fact that different local minima may be found for each run.
Figure 4.

A comparison of test set accuracy between pretrained applications and Xavier initialized applications of the same architectures and the same hyperparameter settings for the translocation dataset. Each bar plot (nine in total) represents 30 replicate models, all trained with identical hyperparameter values.
Figure 5.

A comparison between the pretrained model and the fine-tuned model showing a subset of images that maximize certain filter output activations in the different layers of ResNet50 (attained using the keras-vis toolkit). The fine-tuned model was trained on the MoA dataset for 10 epochs.
Figure 3.
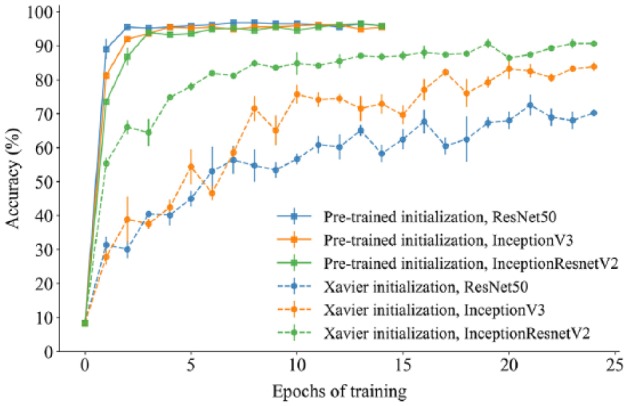
A comparison of test set accuracy between pretrained applications and Xavier initialized applications of the same architectures and the same hyperparameter settings for the MoA dataset. The plot illustrates how pretrained applications greatly improve learning, where the pretrained ResNet50 application attained near 90% accuracy after just a single epoch of training.
An alternative and often used mode of transfer learning is to cut away the first layers of the trained network and freeze their parameters—capturing generic image representations or “off-the-shelf” features—while training from scratch the parameters in the final added layer(s).36 Pawlowski et al. (2016) utilized this alternative approach, together with InceptionV3, to obtain a prediction accuracy of 91% on the same MoA dataset as used in this study. A hybrid approach, between freezing and fine-tuning, where the earlier layers are given a slower learning rate than the later layers, has also been proposed.20 However, in a thorough comparison of transfer learning strategies for digital pathology—across seven architectures (including the three explored in this paper), eight image classification datasets, and various transfer methods (including the aforementioned fine-tuning and freezing of layers)—Mormont et al.37 found that fine-tuning outperformed all other methods regardless of the network architecture applied. Furthermore, this outperformance was especially pronounced for multiclass datasets. As we had 12 MoA classes to predict, we are confident that the fine-tuning transfer learning strategy was the best option for this dataset.
Concerning the translocation dataset, the three models attained accuracies of up to 100% after just single epochs of training ( Fig. 4 )—an accuracy depending heavily on the stochastic process of mini-batch gradient descent and due to the small training set. The greatest difference between pretrained initialization and Xavier initialization can be seen when only training with one epoch. However, pretrained initialization outperforms Xavier initialization even after 20 epochs of training. The quick learning is arguably a strong indication of transferability of the pretrained parameters. These results also suggest that deep CNN models can be successfully applied to smaller datasets of high-content imaging. Interestingly, InceptionResnetV3 seems to be the most robust model for five epochs and beyond, with good accuracy without pretraining.
Neural networks have often been thought of as “black boxes” due to the difficult problem of understanding what they have learned and how they have learned it. For very deep neural networks, such as those studied in this paper, the problem is even more acute due to the multitude of nonlinear interacting components.17 We applied “deep visualisations”17 to gain some insights into the workings of our neural networks and the transferability of the pretrained parameters ( Fig. 5 ). Notably, the early layers of the two models showed similar patterns of activation between the pretrained and fine-tuned images, whereas the deeper layers, activated by higher-level abstractions, were more dissimilar. Furthermore, the fine-tuned images of the deeper layers showed less elaborate patterns than the layers prior to fine-tuning. We speculate that this could be due to the initial networks’ ability to capture a greater variety of object types. As our images are only of cells, it may be that although the pretrained networks provide reasonable initial values for the weight parameters, for the deeper layers many of these weights will be shrunk toward zero to accommodate for this reduction in object variety.
Conclusion and Potential Future Work
Transfer learning and deep CNNs, when used in combination, produce highly accurate classification of MoAs. These models were able to quickly distinguish the different cell phenotypes despite a limited quantity of labeled data. However, although the MoA dataset is one of the very few good benchmarking datasets publicly available, it no longer presents significant challenges for many of the current state-of-the-art models, many of which have already reported accuracies of 90% and above. It would therefore be interesting to evaluate these models on more difficult classification tasks of MoAs, and evaluate them further in the field of high-content imaging.
Finally, as mentioned earlier, there were strong uncertainties in many of our predictions, whereby the probability for the selected MoA class was less than 0.5 (Suppl. Tables S2–S4). Furthermore, these probabilities were simply point estimates without any information on their variability. Indeed, as we show in Figure 4 , replicate runs of the models, finding different local minima in the loss function, will output different point estimates. For medical image data in general, there also often exists a high degree of uncertainty in the annotated labels.38 Accounting for these various forms of uncertainty is invaluable. Deep learning methods that assign confidence to predictions will also be better received by clinicians. Perhaps the most promising means of accounting for uncertainty will come with the fusion of Bayesian modeling and deep learning, thus permitting the incorporation of parameter, model, and observational uncertainty in a natural probabilistic manner. However, due to their high computational expense and the requirement to specify prior distributions on all the network weights, such fully Bayesian approaches are currently infeasible and approximate solutions are required. The simplest means of doing this, as proposed by Gal and Ghahramani,39 is to use dropout between all the network layers and run the model multiple times during testing, which results in an approximate Bayesian posterior distribution for the predicted probabilities. However, such approximate Bayesian methods40 are based on rather limited distributional assumptions and are prone to underestimate uncertainty. The Bayesian hypernetworks of Krueger et al. (unpublished observations), combining Bayesian methods, deep learning, and generative modeling ideas, provide one means of overcoming this uncertainty underestimation problem. As an alternative and less computationally expensive approach, one can use a method known as conformal prediction,41 which works atop machine learning algorithms to enable assessments of uncertainty and reliability. Conformal prediction can be readily applied to deep learning applications at no additional cost42 and can also be utilized in a manner that accounts for uncertainty in the local minima achieved by the gradient descent algorithm.43 Furthermore, conformal prediction does not make any distributional assumptions and circumvents the need to specify priors on the parameters, and interestingly can provide stronger guarantees of validity than Bayesian methods, even when based on the true probability distribution of the data.41 In future work, we plan to explore and contrast these various methods for uncertainty quantification together with transfer learning and neural network architectures applied to the MoA and translocation datasets.
Supplemental Material
Supplemental material, Supplementary_Material_for_Transfer_Learning_with_Deep_Convolutional_Neural_Networks_for_Classifying_Cellular_Morphological_Changes_by_kensert_et_al for Transfer Learning with Deep Convolutional Neural Networks for Classifying Cellular Morphological Changes by Alexander Kensert, Philip J. Harrison and Ola Spjuth in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Availability: The code for the analysis is available at https://github.com/pharmbio/kensert_CNN. Due to stochastic procedures, like randomly dividing the datasets into mini-batches, results will differ somewhat from session to session.
Author Contributions: A.K. and O.S. conceived the project; A.K. performed the data analysis; A.K., P.J.H., and O.S. contributed to interpretation, discussion, and manuscript preparation.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The computations were partly performed on resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). This project received financial support from the Swedish Foundation for Strategic Research (SSF) as part of the HASTE project under the call “Big Data and Computational Science.”
ORCID iD: Alexander Kensert  https://orcid.org/0000-0002-5295-010X
https://orcid.org/0000-0002-5295-010X
References
- 1. Carpenter A. E., Jones T. R., Lamprecht M., et al. Cellprofiler: Image Analysis Software for Identifying and Quantifying Cell Phenotypes. Genome Biol. 2006, 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liberali P., Snijder B., Pelkmans L. Single-Cell and Multivariate Approaches in Genetic Perturbation Screens. Nat. Rev. Genet. 2015, 16, 18–32. [DOI] [PubMed] [Google Scholar]
- 3. Sommer C., Gerlich D. W. Machine Learning in Cell Biology—Teaching Computers to Recognize Phenotypes. J. Cell Sci. 2013, 126, 5529–5539. [DOI] [PubMed] [Google Scholar]
- 4. Caicedo J., Cooper S., Heigwer F., et al. Data-Analysis Strategies for Image-Based Cell Profiling. Nat. Methods 2017, 14, 849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh S., Bray M., Jones T., et al. Pipeline for Illumination Correction of Images for High-Throughput Microscopy. J. Microsc. 2014, 256, 231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uhlmann V., Singh S., Carpenter A. E. CP-CHARM: Segmentation-Free Image Classification Made Accessible. BMC Bioinformatics 2016, 17, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ljosa V., Caie P. D., Ter Horst R., et al. Comparison of Methods for Image-Based Profiling of Cellular Morphological Responses to Small-Molecule Treatment. J. Biomol. Screen. 2013, 18, 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kraus O. Z., Frey B. J. Computer Vision for High Content Screening. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 102–109. [DOI] [PubMed] [Google Scholar]
- 9. LeCun Y., Bengio Y., Hinton G. Deep Learning. Nature 2015, 521, 436EP. [DOI] [PubMed] [Google Scholar]
- 10. Xu Y., Mo T., Feng Q., et al. Deep Learning of Feature Representation with Multiple Instance Learning for Medical Image Analysis. In 2014 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Philadelphia, 2014, pp 1626–1630. [Google Scholar]
- 11. Pärnamaa T., Parts L. Accurate Classification of Protein Subcellular Localization from High-Throughput Microscopy Images Using Deep Learning. G3 2017, 7, 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kraus O. Z., Ba J. L., Frey B. J. Classifying and Segmenting Microscopy Images with Deep Multiple Instance Learning. Bioinformatics 2016, 32, i52–i59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arbelle A., Reyes J., Chen J.-Y., et al. A Probabilistic Approach to Joint Cell Tracking and Segmentation in High-Throughput Microscopy Videos. Med. Image Anal. 2018, 47, 140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Litjens G., Kooi T., Bejnordi B. E., et al. A Survey on Deep Learning in Medical Image Analysis. Med. Image Anal. 2017, 42, 60–88. [DOI] [PubMed] [Google Scholar]
- 15. Razavian A. S., Azizpour H., Sullivan J., et al. CNN Features Off-the-Shelf: An Astounding Baseline for Recognition. CoRR 2014, abs/1403.6382. [Google Scholar]
- 16. Donahue J., Jia Y., Vinyals O., et al. DeCAF: A Deep Convolutional Activation Feature for Generic Visual Recognition. CoRR 2013, abs/1310.1531. [Google Scholar]
- 17. Yosinski J., Clune J., Bengio Y., et al. How Transferable Are Features in Deep Neural Networks? CoRR 2014, abs/1411.1792.15. [Google Scholar]
- 18. Zhang W., Li R., Zeng T., et al. Deep Model Based Transfer and Multi-Task Learning for Biological Image Analysis. In Proceedings of the 21st ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD ’15), Paris, 2015, pp 1475–1484. [Google Scholar]
- 19. Russakovsky O., Deng J., Su H., et al. Imagenet Large Scale Visual Recognition Challenge. Int. J. Comput. Vis. 2015, 115, 211–252. [Google Scholar]
- 20. Bayramoglu N., Heikkilä J. Transfer Learning for Cell Nuclei Classification in Histopathology Images. In Computer Vision—ECCV 2016 Workshops, Amsterdam, 2016, pp 532–539. [Google Scholar]
- 21. Phan H. T. H., Kumar A., Kim J., et al. Transfer Learning of a Convolutional Neural Network for hep-2 Cell Image Classification. In 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI), Prague, 2016, pp 1208–1211. [Google Scholar]
- 22. Goodfellow I., Bengio Y., Courville A. Deep Learning. MIT Press: Cambridge, MA, 2016. [Google Scholar]
- 23. Erhan D., Bengio Y., Courville A., et al. Why Does Unsupervised Pre-Training Help Deep Learning? J. Mach. Learn. Res. 2010, 11, 625–660. [Google Scholar]
- 24. Vebjorn L., Katherine S. L., Carpenter A. E. Annotated High-Throughput Microscopy Image Sets for Validation. Nat. Methods 2012, 9, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Godinez W. J., Hossain I., Lazic S. E., et al. A Multi-Scale Convolutional Neural Network for Phenotyping High-Content Cellular Images. Bioinformatics 2017, 33, 2010–2019. [DOI] [PubMed] [Google Scholar]
- 26. Caie P. D., Walls R. E., Ingleston-Orme A., et al. High-Content Phenotypic Profiling of Drug Response Signatures across Distinct Cancer Cells. Mol. Cancer Ther. 2010, 9, 1913–1926. [DOI] [PubMed] [Google Scholar]
- 27. Anscombe F. J. The Transformation of Poisson, Binomial and Negative-Binomial Data. Biometrika 1948, 35, 246–254. [Google Scholar]
- 28. Chollet F. Keras. 2015. https://keras.io.
- 29. He K., Zhang X., Ren S., et al. Deep Residual Learning for Image Recognition. In 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, 2016, pp 770–778. [Google Scholar]
- 30. Szegedy C., Vanhoucke V., Ioffe S., et al. Rethinking the Inception Architecture for Computer Vision. CoRR 2015, abs/1512.00567. [Google Scholar]
- 31. Szegedy C., Ioffe S., Vanhoucke V. Inception-v4, Inception-Resnet and the Impact of Residual Connections on Learning. CoRR 2016, abs/1602.07261. [Google Scholar]
- 32. He K., Sun J. Convolutional Neural Networks at Constrained Time Cost. CoRR 2014, abs/1412.1710. [Google Scholar]
- 33. Srivastava R. K., Greff K., Schmidhuber J. Highway Networks. CoRR 2015, abs/1505.00387. [Google Scholar]
- 34. Szegedy C., Liu W., Jia Y., et al. Going Deeper with Convolutions. In 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston, 2015, pp 1–9. [Google Scholar]
- 35. Kotikalapudi R. keras-vis. 2017. https://github.com/raghakot/keras-vis.
- 36. Yosinski J., Clune J., Nguyen A. M., et al. Understanding Neural Networks through Deep Visualization. CoRR 2015, abs/1506.06579. [Google Scholar]
- 37. Mormont R., Geurts P., Marée R. Comparison of Deep Transfer Learning Strategies for Digital pathology. In 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), Salt Lake City, UT, 2018, pp 2262–2271. [Google Scholar]
- 38. Armato S. G., Roberts R. Y., Kocherginsky M., et al. Assessment of Radiologist Performance in the Detection of Lung Nodules. Acad. Radiol. 2009, 16, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gal Y., Ghahramani Z. Dropout as a Bayesian Approximation: Representing Model Uncertainty in Deep Learning. In Proceedings of the 33rd International Conference on Machine Learning, New York, 2016, pp 1050–1059. [Google Scholar]
- 40. Blei D. M., Kucukelbir A., McAuliffe J. D. Variational Inference: A Review for Statisticians. J. Am. Stat. Assoc. 2017, 112, 859–877. [Google Scholar]
- 41. Vovk V., Gammerman A., Shafer G. Algorithmic Learning in a Random World, 1st Ed Springer-Verlag: Berlin, 2005. [Google Scholar]
- 42. Papadopoulos H. Tools in Artificial Intelligence. IntechOpen: Rijeka, 2008, pp 315–330. [Google Scholar]
- 43. Cortes-Ciriano I., Bender A. Deep Confidence: A Computationally Efficient Framework for Calculating Reliable Errors for Deep Neural Networks. J. Chem. Inform. Model. 2018. DOI: 10.1021/acs.jcim.8b00542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplementary_Material_for_Transfer_Learning_with_Deep_Convolutional_Neural_Networks_for_Classifying_Cellular_Morphological_Changes_by_kensert_et_al for Transfer Learning with Deep Convolutional Neural Networks for Classifying Cellular Morphological Changes by Alexander Kensert, Philip J. Harrison and Ola Spjuth in SLAS Discovery