

Summary
Alternative splicing is a prevalent mechanism of gene regulation that is modulated in response to a wide range of extracellular stimuli. Stress-activated protein kinases (SAPKs) play a key role in controlling several steps of mRNA biogenesis. Here, we show that osmostress has an impact on the regulation of alternative splicing (AS), which is partly mediated through the action of p38 SAPK. Splicing network analysis revealed a functional connection between p38 and the spliceosome component SKIIP, whose depletion abolished a significant fraction of p38-mediated AS changes. Importantly, p38 interacted with and directly phosphorylated SKIIP, thereby altering its activity. SKIIP phosphorylation regulated AS of GADD45α, the upstream activator of the p38 pathway, uncovering a negative feedback loop involving AS regulation. Our data reveal mechanisms and targets of SAPK function in stress adaptation through the regulation of AS.
Keywords: cell signaling, p38 SAPK, alternative splicing, stress responses, GADD45
Graphical Abstract
Highlights
-
•
The p38 SAPK regulates stress-mediated alternative splicing
-
•
Splicing network analysis reveals a functional connection between p38 and SKIIP
-
•
p38 mediates stress-dependent alternative splicing by targeting SKIIP
-
•
SKIIP phosphorylation by p38 induces GADD45α splicing to modulate SAPK signaling
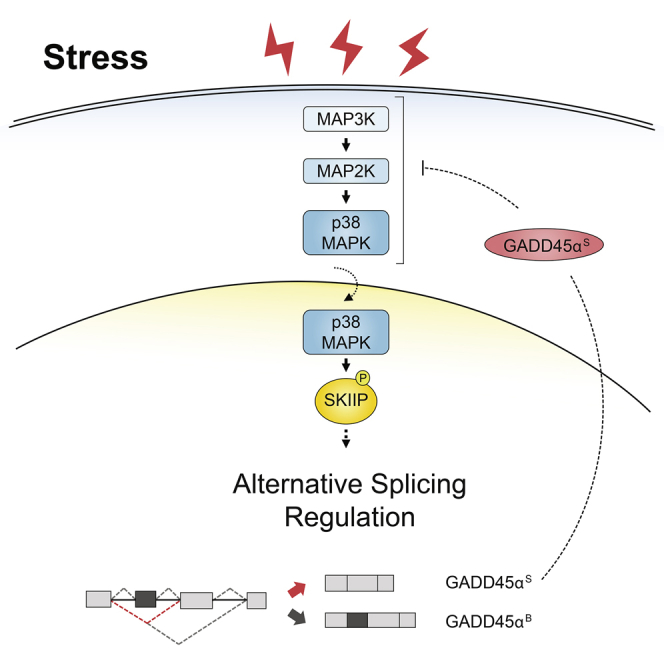
Carbonell et al. reports that p38 SAPK regulates alternative splicing to generate protein diversity in response to osmostress. The authors also show that SKIIP mediates splicing by p38. This mechanism, in turn, allows control of p38 activity through alternative splicing of the upstream regulator of the p38 pathway GADD45α.
Introduction
Cells respond and adapt to environmental changes through activation of stress-activated protein kinases (SAPKs). Activation of the p38 SAPK has a pivotal role in the regulation of cellular responses to diverse kinds of stresses, thereby controlling proliferation, differentiation, and development of specific cell types (Kyriakis and Avruch, 2012). Interest in the p38 pathway has increased since its deregulation has been implicated in the development of many pathological conditions such as cancer, inflammation-related diseases, and cardiovascular dysfunction (for review, see Corre et al., 2017, Gupta and Nebreda, 2015, Yokota and Wang, 2016). Activation of p38 coordinates essential physiological functions such as cell-cycle progression and gene expression (de Nadal and Posas, 2015, Gubern et al., 2016, Joaquin et al., 2012). A comprehensive program of gene expression is required for cell adaptation to stress (Amat et al., 2019, Ferreiro et al., 2010b, Nadal-Ribelles et al., 2012), and p38 regulates multiple steps of the mRNA biogenesis and stability (Borisova et al., 2018, Degese et al., 2015). Upon stress, p38 is recruited to chromatin via its interaction with specific transcription factors, allowing recruitment of the RNA Pol II machinery (Chow and Davis, 2006, Edmunds and Mahadevan, 2004, Ferreiro et al., 2010a, Simone et al., 2004). Moreover, the p38 SAPK phosphorylates several transcription and chromatin remodeling factors, thereby modulating their stability and localization, as well as their interaction with DNA and other regulatory proteins (Cuadrado and Nebreda, 2010, de Nadal and Posas, 2015, Forcales et al., 2012, Rampalli et al., 2007, Yang et al., 2013).
Alternative splicing (AS) is modulated in response to a wide range of extracellular stimuli, including oxidative stress, heat shock, osmostress, and ultraviolet radiation (Biamonti and Caceres, 2009, Gonçalves et al., 2017, Kim Guisbert and Guisbert, 2017, Shalgi et al., 2014, Shkreta and Chabot, 2015). Examples of modulation of the activity of proteins involved in splicing through the activation of p38 have been reported. Thus, upon osmostress and UV irradiation, p38 phosphorylates the heterogeneous nuclear ribonucleoprotein hnRNPA1, a regulator of RNA splicing, leading to its relocalization into the cytoplasm where it concentrates into stress granules (Guil et al., 2006, van der Houven van Oordt et al., 2000). Cell exposure to UV leads to phosphorylation of the splicing factor SPF45 by ERK and p38 (Al-Ayoubi et al., 2012). Another example of an RNA binding protein targeted by p38 is the Hu antigen R (HuR), which induces changes in Tra2β alternative splicing profiles in response to oxidative stress (Akaike et al., 2014). Although the above examples indicate that stress adaptive responses lead to the regulation of splicing by SAPKs, the effects and mechanisms of p38 activation on AS modulation remain poorly understood. Here, we examined the genome-wide contribution of p38 activation to AS regulation upon osmostress. Moreover, a functional splicing network analysis revealed connections between p38 and the splicing machinery, through p38-mediated regulation of the SKIIP protein. Finally, we describe a molecular mechanism underlying AS regulation upon osmostress and the functional consequences of differential isoform expression under stress conditions.
Results
p38 SAPK Regulates Stress-Mediated Alternative Splicing
We initially investigated the potential role of p38 in osmostress-induced splicing regulation in mammals using a custom splicing-sensitive microarray platform that contained exon and exon-junction probes covering 1,920 AS events in 491 genes encoding splicing factors and cancer-related proteins (Muñoz et al., 2009). The comparison of RNAs from non-treated, stressed (100 mM NaCl, 2 h), and stressed cells pre-treated with the SB203580 p38 inhibitor resulted in the identification of 9 genes containing p38-dependent alternative splicing events (Table S1; see STAR Methods for details). To validate these results, a number of p38-induced splicing isoforms were assessed by semiquantitative RT-PCR followed by capillary electrophoresis using primers hybridizing to the flanking constitutive exons, as well as by qPCR using oligonucleotides covering exons and exon junctions (see Table S3). A representative example is the regulation of an alternative 5′ splice site (5′ss) event on the exon 10 of the dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) (Figure S1A). While the upstream 5′ss was preferentially selected in basal conditions (DYRK1AB), a switch in 5′ss usage to the downstream site was observed under stress (DYRK1AS). The regulation of this event was dependent on p38 activity, as it was reduced by the addition of SB203580. Interestingly, stress effects were recapitulated using minigenes corresponding to the alternatively spliced region of DYRK1A (Figure S1B, left), suggesting that regulation of DYRK1A AS by p38 depends on its primary sequence. To confirm that the modulation of DYRK1A AS was directly mediated by p38 activation, we co-transfected the DYRK1A minigene with constructs expressing p38 and a constitutively active form of MKK6, the upstream activator of p38 (MKK6DD). The constitutively active MKK6 mutant MKK6DD, where the Ser207 and Thr211 has been replaced with Asp codons, is commonly used to specifically activate the p38 MAPK irrespectively of stress (Takekawa et al., 1998). AS regulation of DYRK1A minigene upon genetic activation of p38 was similar to that observed upon stress (Figure S1B; right). In summary, these initial results revealed a role for p38 SAPK in stress-induced AS and prompted us to further investigate the global impact of p38 SAPK in alternative splicing regulation upon stress.
We therefore examined splicing transcriptome-wide by RNA sequencing (RNA-seq) analysis of untreated and stressed (NaCl 100 mM) cells in the presence or the absence of SB203580 (Figure 1; Table S2). Stress-induced changes in 793 AS events were detected (|ΔPSI| ≥ 15), of which 70% corresponded to negative ΔPSI and 30% to positive ΔPSI values, distributed as follows: cassette exons (58%), alternative 3′ss (7%), alternative 5′ss (8%), and intron retention (27%) (Figure 1A, striped bars). Approximately half of the stress-dependent splicing events were attenuated in the presence of the SB203580 inhibitor (Figures 1A, filled-bars, and 1B), indicating a prevalent role for the p38 SAPK in splicing regulation upon stress. Remarkably, all the changes affected by SB203580 reduced the effect of stress on splice site selection, with no changes exacerbated by the inhibitor.
Figure 1.

Transcriptome-wide Analysis of Stress-Induced p38-Mediated Splicing Events
(A) Barplot representing stress-induced AS events (NaCl-induced, 100 mM NaCl for 3 h) and those that are reverted in the presence of the p38 inhibitor SB203502 (p38-dependent). On the y axis, the number of events displaying higher PSI upon osmostress (DPSI ≥ +15%, top), or lower PSI after NaCl treatment (DPSI ≤ −15%, bottom) are indicated. On the x axis, different categories of events are shown. Striped bars represent NaCl-induced events, filled bars represent p38-dependent events.
(B) Heatmap representation of p38-dependent events. The color gradient indicates PSI values of each event in each duplicate sample, ranging from 0 to 100. Events are separated into negative and positive ΔPSI changes relative to the basal splicing state. n = number of events detected in each splicing category. Detailed information for each event can be found in Table S2.
(C) Examples of validation of p38-dependent AS changes identified by the RNA-seq analysis. HeLa cells were treated as in (A) and AS changes were assessed by RT-PCR followed by capillary electrophoresis. Schematic diagrams show constitutive exons (gray boxes) and alternative exons (colored boxes) for FST (HsaALTA0003478), FBOXO32 (HsaEX0025293), CBX4 (HsaEX0012775), and LATS2 (HsaEX0035466). Percentage of stress-induced isoforms (S) was calculated over the total of stress (S) and basal (B) isoforms.
Three treatment replicates were analyzed for each condition. Error bars represent SD from the mean value. Statistical significance refers to the results of two-sided t test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
We next validated a set of selected p38-mediated AS events by RT-PCR (Figure 1C). Follistatin (FST), which plays a protective role under a variety of stresses (Zhang et al., 2018), displays alternative 3′ splice sites in exon 6. In basal conditions, the mRNA that encodes the long FST 315 protein isoform (FSTB) was preferentially expressed. However, the production of FSTS, which encodes the FSTB 288 protein lacking the acidic C-terminal tail (Shimasaki et al., 1988), was induced upon stress depending on p38 activation. We also observed a p38-dependent change in the inclusion of cassette exon 5 in FBXO32 (also known as MAFbx or Atrogin-1), originally identified as a muscle-specific gene involved in muscle atrophy (Bodine et al., 2001, Gomes et al., 2001). Skipping of FBXO32 exon 5 upon stress generates a shorter mRNA isoform (FBXO32S) containing an in-frame premature termination codon predicted to reduce total mRNA transcript levels by nonsense-mediated mRNA decay (NMD). Osmostress also induced skipping of exon 4 of chromobox 4 (CBX4), a member of the Polycomb group (PcG) that functions as a regulator of cell proliferation and apoptosis (Luis et al., 2011, Wang et al., 2016, Zheng et al., 2015). The p38-dependent isoform of CBX4 (CBX4S) also contains a premature stop codon. Last, expression of a p38-dependent isoform of the large tumor suppressor kinase 2 (LATS2), a serine or threonine kinase involved in cell-cycle regulation (Furth and Aylon, 2017), was observed upon stress. In this case, skipping of exon 7 (LATS2S) encoded a shorter protein isoform lacking the AGC-C-terminal domain. Collectively, microarray and RNA-seq data validations provided multiple examples of p38-induced, in-frame and out-of-frame, alternatively spliced isoforms.
In order to assess the global impact on the coding sequence (CDS) of p38-mediated AS, we compared NaCl-induced and p38-dependent events to a background set of AS events that were not affected by osmostress (|ΔPSI| NaCl-Ctrl < 15%) and had an intermediate PSI in the dataset (30 < PSI < 70 across all samples) (Figures 2A and S2A). Remarkably, NaCl-induced events, as well as p38-dependent events, showed a higher proportion of CDS-disrupting events compared to control, suggesting a significant impact on protein expression through AS regulation. AS events with higher PSI in stressed cells also showed a higher proportion of exons mapping in the 5′ untranslated regions (UTR) of genes, which might be relevant for translational regulation of the mRNAs.
Figure 2.

Analysis of Transcript Features Associated with NaCl and p38-Induced AS Events
(A) Proportion of events disrupting the transcript open reading frame (CDS-disrupting), preserving it (CDS-preserving), mapping in 3′/5′ untranslated regions (UTR) or in non-coding RNA, classified as in Irimia et al. (2014). Numbers of events are shown for each category: background alternative events, NaCl-induced events (ALL) and subsets of NaCl-induced events with ΔPSI ≥ 15 or ΔPSI ≤ −15.
(B) Number of transcripts per gene containing background, NaCl-induced and p38-dependent AS events. Top and bottom represent exons displaying higher inclusion or skipping upon NaCl treatment, respectively.
(C) Distribution of length and ratio of intron/exon length in NaCl-induced and p38-dependent exons. Top and bottom represent exons displaying higher inclusion or skipping upon NaCl treatment, respectively. Boxplots corresponding to each feature represent the inter-quantile range (IQR) with median values shown by the black line. Outliers were discarded. Significant changes were calculated by Mann-Whitney-Wilcoxon test comparing each set of exons versus the background set (∗p < 0.05).
See also Figure S2.
To gain insight on potential regulatory AS mechanisms induced by osmostress, we examined using MATT (Gohr and Irimia, 2019) the distribution of sequence features on NaCl-induced cassette exons and their flanking introns, compared with the background described above. NaCl-induced cassette exons were typically part of simpler transcriptional units compared to background alternative exons (Figure 2B). Introns surrounding NaCl-dependent exons tended to be shorter, whereas exons included after NaCl treatment showed a tendency toward being longer than the control set (Figure 2C). Exons included after osmostress typically had lower GC content, while the upstream introns tended to display higher GC content. On the contrary, exons skipped after NaCl treatment featured lower GC content in the upstream and downstream introns (Figure S2B). Furthermore, NaCl-dependent exons were typically flanked by weaker splice sites compared to control alternative exons, with the exception of stronger branch points in exons included upon NaCl and stronger 5′ splice sites in skipped exons (Figures S2C and S2D). Moreover, p38-dependent AS events showed a higher SF1-BBP binding score (an approximation of branch point strength) and a higher number of predicted branch point sequences (Figure S2C, bottom), pointing to a special dependence on efficient branch point recognition.
Splicing Network Analysis Revealed a Functional Connection between p38 and the Prp19-Related Splicing Factor SKIIP
p38 SAPK phosphorylates hnRNPA1 and SPF45 splicing factors, with consequences in certain AS decisions (Al-Ayoubi et al., 2012, van der Houven van Oordt et al., 2000). We therefore asked whether the stress-induced, p38-mediated AS events described above are regulated through these spliceosomal components. To test this hypothesis, we subjected hnRNPA1- and SPF45-depleted cells to osmostress and assayed p38-dependent isoforms by RT-PCR (Figure S3A). Although relative expression of stress and basal isoforms was affected by hnRNPA1 and SPF45 knockdown in some cases, there was a significant induction of FST, FBXO32, CBX4, and LATS2 stress isoforms in hnRNPA1- and SPF45-depleted cells (Figure S3B). These results suggest that additional spliceosomal components are involved in p38-mediated AS upon stress.
To identify functional targets of p38 SAPK in the splicing machinery, we took advantage of an unbiased functional splicing network platform in which connections between 270 spliceosomal components and splicing regulators are established based upon the comparison between the profile of effects of their individual knockdown on 36 AS events (Papasaikas et al., 2015). Functional links between any perturbation that affects these events and its potential targets within the splicing machinery can thus be established by comparing the profile of AS changes. To define only specific p38-mediated events, we compared the splicing profiles in the network with those induced in response to p38 activation by the constitutively active MKK6DD. As referred above, MKK6DD is commonly used to specifically activate p38 independently of stress conditions (Takekawa et al., 1998). HeLa cells were co-transfected with plasmids expressing p38 and MKK6DD and exon inclusion was compared to that of GFP-transfected control cells by RT-PCR. The most pronounced AS change induced by MKK6DD and p38 overexpression within the set of 36 events monitored in this assay occurred in the growth arrest and DNA damage-inducible 45 (GADD45α) gene. GADD45α has been described as an upstream activator of the p38 SAPK pathway, because it interacts and activates the MAP3K MTK1 (Takekawa and Saito, 1998) (Figure 3A). Variations in GADD45α alternative exons 2 and 3 were assessed by RT-PCR using primers complementary to exons 1 and 4 (Figure 3B). In cells transfected with a control vector, the full-length mRNA isoform was preferentially expressed (GADD45αB) whereas in cells overexpressing MKK6DD, two splicing variants were generated either by skipping of exon 2 (GADD45S) or skipping of both exons 2 and 3 (GADD45αS1). The expression of both isoforms was p38-dependent, as their accumulation was reduced in the presence of SB203580 (Figure 3B). Importantly, both isoforms were induced upon NaCl treatment (events HsaEX6031508 and HsaEX0026819, Table S2). Skipping of exon 2 maintains the reading frame and codes for an isoform with a 34-amino acid deletion (Zhang et al., 2009), while skipping of exons 2 and 3 triggers the inclusion of a premature termination codon.
Figure 3.

The Profile of Splicing Changes upon Activation of p38 by MKK6DD Reveals a Functional Association with the Splicing Factor SKIIP
(A) Perturbation profile of AS induced by activation of p38 by MKK6DD. Positive and negative scaled Z scores indicate increased exon inclusion or skipping, respectively.
(B) Validation of GADD45α AS was assessed by RT-PCR followed by capillary electrophoresis. Percentage of exon 2 (S) and exon 2 and 3 (S1) skipping was calculated over the total of stress (S and S1) and basal (B) isoforms. Data are shown as means ± SD of three biological replicates. Error bars represent SD from the mean value. Statistical significance refers to the results of two-sided t test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
(C) Network connectivity. Lines connecting RNA processing factors and MKK6DD expression indicate similar effects on the profile of AS changes by the indicated protein knockdown or induction of p38. Inset table: effects of the indicated protein knockdowns on GADD45α AS. The percentage of exon 2 inclusion (% inc), p values, and Z score values are indicated.
(D and E) SKIIP is essential for p38-dependent GADD45αS induction. HeLa cells were treated with scrambled or SKIIP stealth siRNA (D) and subsequently transfected with an empty vector or with MKK6DD and p38 expression vectors (left) or were treated for 2 h with 100 mM NaCl (right) (E). GADD45αS AS was assessed by RT-PCR and qPCR. Levels of GADD45α stress-induced isoform (S) and basal isoform (B) were determined by qPCR using primers targeting exon-junctions. Relative expression of stress isoform was quantified as fold change over non-treated cells. Data are shown as means ± SD of at least four independent experiments. Significant changes were calculated by Student’s t test comparing transfected or treated versus non-transfected or non-treated samples (∗p < 0.05). Western blot analysis of HeLa cells expressing control or SKIIP siRNA is shown.
See also Figure S3.
Assessment of the perturbation profiles of splicing caused by MKK6DD expression revealed functional links between p38 and several splicing factors involved in different steps of pre-mRNA splicing, including the RNA lariat debranching enzyme (DBR1), C22ORF19 (THOC5, a component of the THO/TREX complex), the related-U5snRNP components SYNCRIP (heterogeneous nuclear riboprotein Q), and SKIIP (SNW domain-containing protein 1) (Figure 3C). Of note, depletion of SYNCRIP and SKIIP specifically promoted GADD45α AS, while depletion of DBR1 and C22ORF19 had more modest effects (Figure 3C, inset table). To validate the functional link between SYNCRIP and SKIIP upon p38 activation, we assessed GADD45α AS in small interfering RNA (siRNA)-mediated SYNCRIP or SKIIP knockdown cells (Figures 3D and S4A). While SYNCRIP and SKIIP depletion did not significantly affect the AS pattern of GADD45α under basal conditions, they efficiently suppressed induction of GADD45α exon 2 skipping (GADD45αS isoform) by genetically activated p38 (Figures 3E, left, and S4B). Similarly, GADD45αS isoform expression upon osmostress was also prevented in siRNA-mediated SKIIP knockdown cells (Figure 3E, right). These data support the notion that SYNCRIP and SKIIP are essential mediators of p38-dependent GADD45α AS. Of note, both splicing factors interacted with each other by immunoprecipitation experiments (Figure S4C).
Of SKIIP and SYNCRIP, only SKIIP contains putative sites of p38 phosphorylation (S/T-P). We therefore selected SKIIP for further analysis as a potential direct target of p38. To globally assess the role of SKIIP in p38-dependent stress-induced AS events, we performed RNA-seq experiments comparing RNAs from SKIIP knockdown versus control cells under basal or stressed (100 mM NaCl) conditions. As described before, stress induced 793 splicing events, approximately half of which were mediated by p38 (i.e., affected by the SB203580 p38 inhibitor) (Figure 1; Table S2). Of those, a total of 155 events (43%) showed involvement of SKIIP before and/or after NaCl addition and were thus defined as SKIIP-dependent (Figure 4). Specifically, 103 of the 793 events changed after SKIIP knockdown, while 79 osmostress-induced changes could not be observed in the SKIIP knockdown cells treated with NaCl (Table S2). These data support a prominent role for SKIIP in p38-mediated splicing upon stress.
Figure 4.

SKIIP Mediates a Large Proportion of p38-Dependent A Changes
Heatmap representation of p38-dependent splicing events whose modulation is affected by SKIIP knockdown, either in basal condition or in stressed cells (100 mM NaCl for 3 h). The color gradient indicates PSI values of each event in each sample, ranging from 0 to 100. Events are separated into negative and positive ΔPSI changes relative to the basal splicing state. n = number of events detected in each splicing category.
Detailed information for each event can be found in Table S2.
p38 Interacts with and Phosphorylates Thr180, Ser224, and Ser232 of SKIIP to Mediate Alternative Splicing
Most SAPKs directly interact with their substrates. We therefore assessed whether p38 could interact with SKIIP by performing immunoprecipitation experiments using extracts from HeLa cells expressing flag-tagged p38 and HA-tagged SKIIP. FLAG-p38 co-immunoprecipitated when HA-SKIIP was pulled down using anti-HA antibodies (Figure 5A), indicating that both proteins indeed interact with each other.
Figure 5.

Regulation of GADD45α AS upon Stress Depends on p38 Phosphorylation of SKIIP
(A) SKIIP interacts with p38 in vivo. HA-SKIIP and Flag-p38α were expressed in HeLa cells, immunoprecipitated with anti-HA coupled Sepharose beads, and analyzed by western blotting with anti-HA and anti-Flag antibodies.
(B) p38 phosphorylates the SNW1 domain of SKIIP. Wild type GST-SKIIP (WT) and GST-SKIIP-3A (Thr180, Ser224, and Ser232 mutated to Ala) (3A) were purified from E. coli and subjected to an in vitro kinase assay using activated p38α.
(C) HeLa cells stably carrying siRNA-resistant pLVX-TetOne T7-SKIIP WT or pLVX-TetOne T7-SKIIP-3A were incubated with doxycycline (500 ng/mL) when indicated and transfected with SKIIP siRNA or scrambled siRNA (20 nM) to deplete endogenous SKIIP. Left: T7-SKIIP and endogenous SKIIP levels were analyzed by western blotting with anti-T7 and anti-SKIIP antibodies. Right: After 72 h, cells were stimulated for 2 h with 100 mM NaCl, and the relative expression of GADD45αS was analyzed by qPCR. Statistical significance refers to the results of two-sided t test comparisons between relative expression of GADD45αS values of NaCl-treated versus non treated cells. ∗∗p < 0.001. n.s., non-significant (p > 0.05). A minimum of three biological replicates was used for each condition. Error bars represent SD from the mean value.
(D) Regulation of CBX4 and LATS2 upon stress also depends on SKIIP phosphorylation. HeLa cells stably carrying siRNA-resistant pLVX-TetOne T7-SKIIP WT or pLVX-TetOne T7-SKIIP-3A were incubated with doxycycline (500 ng/mL) when indicated and transfected with SKIIP siRNA or scrambled siRNA (20 nM). After 72 h, cells were stimulated for 2 h with 100 mM NaCl, and splicing changes compared to basal state were assessed by RT-PCR as in Figure 1C. Data are shown as means ± SD of at least three biological replicates. Significant changes were calculated by Student’s t test comparing NaCl treated versus non-treated samples (∗p < 0.05; ∗∗p < 0.01).
(E) Induction of SKIIP expression is sufficient to rescue the effect of SKIIP knockdown on AS. HeLa cells stably expressing siRNA were incubated with doxycycline and transfected as in (D). Relative levels of AACS exon 17 skipping were analyzed by qPCR. Data are shown as means ± SD from three independent experiments. Significant changes were calculated by Student’s t test comparing transfected or treated with doxycycline versus non-transfected or non-treated samples (∗∗p < 0.01).
See also Figure S3.
To assess whether SKIIP was a direct target for p38 SAPK, we analyzed the phosphorylation of SKIIP in an in vitro kinase assay using purified proteins. p38α activated by MKK6DD was found to phosphorylate SKIIP (Figure 5B, left). The SKIIP protein contains three putative MAPK consensus sites, all of them located in the SNW1 domain. The potential phosphosites Thr180, Ser224, and Ser232 were mutated to alanine (SKIIP-3A) and SKIIP phosphorylation by p38 was then assayed in vitro. Combined mutation of Thr180, Ser224, and Ser232 strongly reduced p38 phosphorylation of SKIIP (Figure 5B, right).
SKIIP localizes into the nucleus in basal conditions (Abankwa et al., 2013, Brès et al., 2009, Makarova et al., 2004, Zhang et al., 2003). To assess whether p38 phosphorylation induced changes in SKIIP localization upon stress, we performed immunofluorescence assays of HeLa cells stably expressing doxycycline-inducible T7 tag wild type and SKIIP-3A non-phosphorylatable mutant. Both protein were nuclear in basal and stress conditions, indicating that p38-mediated phosphorylation of SKIIP does not affect its subcellular localization (Figure S5).
To assess whether SKIIP phosphorylation by p38 altered SKIIP activity, we stably expressed, under the control of the Tet promoter, siRNA-insensitive forms of wild type (WT) SKIIP or containing mutations in the three phosphorylation sites (3A) in HeLa cells depleted of endogenous SKIIP by means of an siRNA or in cells treated with scrambled siRNAs as control (Figure 5C, left). Transfected SKIIP was induced by doxycycline, and the cells were subjected to osmostress as before. Expression of the GADD45αS stress-dependent isoform was measured by qPCR. As previously shown, induction of GADD45α exon 2 skipping upon osmostress was strongly impaired in SKIIP knockdown cells. Notably, when endogenous SKIIP was depleted, expression of wild-type SKIIP was sufficient to significantly restore GADD45αS induction upon stress. In contrast, induction of the stress-dependent GADD45αS isoform was impaired upon expression of the non-phosphorylatable SKIIP-3A mutant under the same conditions (Figure 5C, right). Moreover, other p38-dependent splicing changes mediated by SKIIP that were identified in the RNA-seq analysis, such as the splicing of CBX4 and LATS2, were also dependent on SKIIP phosphorylation (Figure 5D). Of note, induction of the SKIIP-3A mutant (as well as the WT version) was sufficient to rescue the effect of the SKIIP knockdown in non-stress responsive SKIIP-dependent alternative splicing events such as exon 17 skipping in the AACS gene (van der Lelij et al., 2014) (Figure 5E), indicating that the SKIIP-3A mutant showed effects specifically in stress-dependent AS events. Therefore, our data confirm that phosphorylation of SKIIP by p38 mediates alternative splicing of several genes in response to stress.
Expression of the GADD45αS Isoform upon Stress Impairs SAPK Activation
In addition to being induced upon stress, GADD45 proteins have been implicated in activation of the p38 SAPK pathway by interaction with the N-terminal region of MTK1 MAP3K, an upstream activator of p38. GADD45-like proteins activate MTK1 kinase activity and induce apoptotic cell death (Takekawa and Saito, 1998). We therefore reasoned that stress-induced GADD45 AS might have an impact on the activation of the MTK1 upstream kinase. To elucidate the biological relevance of the induction of the new GADD45α isoform upon stress, we assessed SAPK signaling dynamics in the presence of the wild-type and stress-induced GADD45α splicing isoforms (GADD45αB or GADD45αS). For this purpose, we used a highly sensitive system based on a kinase translocation reporter (KTR) that measures kinase activity in single cells (Regot et al., 2014). KTR reporters change their nucleocytoplasmic shuttling upon phosphorylation, which can be followed by fluorescence microscopy, allowing quantitative and simultaneous measurement of kinase activity at single-cell resolution. 3T3 cell lines stably expressing a p38 KTR were transiently transfected with either GADD45αB- or GADD45αS-expressing isoforms (Figure 6, top), and kinase activity was monitored upon osmostress by fluorescence time-lapse microscopy. Osmostress induced similar stress-activation of p38 in control and GADD45αB cells, while cells expressing the stress-induced GADD45αS isoform showed more limited kinase activation upon NaCl treatment (Figure 6, bottom). These data suggest that the activation of p38 can be tuned down by induction of the stress-mediated GADD45αS isoform upon stress, thereby establishing a negative feed-back loop of p38 activity regulated by the expression of a GADD45 splicing isoform.
Figure 6.

Activation of the p38 Pathway upon Stress Is Tuned Down by Expression of the Stress-Induced GADD45αS Isoform
NIH 3T3 mouse fibroblasts constitutively expressing a p38 kinase translocation reporter (KTR) were transiently transfected with pcDNA3 (mock), pcDNA3 GADD45αB, or pcDNA3 GADD45αS. Two days post-transfection, the cells were seeded into 12-well glass-bottom plates coated with fibronectin. The following day the cells were subjected to time-lapse microscopy as described in the STAR Methods.
(A) Western blot displaying the expression of pcDNAD3, pcDNA3 GADD45αB, and pcDNA3 GADD45αS isoforms.
(B) Time-lapse microscopy. The arrow indicates the time point when 100 mM NaCl was added. Data represent the median of the cytoplasm/nucleus fluorescent ratio over time. The colored shadows represent SEM from more than 30 independently tracked individual cells.
Discussion
The p38 SAPK plays a major role in the regulation of gene expression upon stress (de Nadal and Posas, 2015, Klein et al., 2013, Yang et al., 2013). However, although some splicing events are known to be regulated by p38 (Al-Ayoubi et al., 2012, Robbins et al., 2008), a comprehensive picture of the impact of this SAPK on the modulation of AS in human cells is lacking. Here, we assessed the transcriptome-wide AS profile mediated by p38 activation. In response to stress, changes in more than 700 splicing events, involving all types of AS patterns, are induced. Approximately half of these events are dependent on p38 SAPK activity, indicating a major role for this kinase in splicing regulation upon stress. NaCl-dependent exons, embedded in simpler transcription units, showed specific characteristics in length and GC content compared to control alternative exons. These length and GC content features may reflect the previously described different architectures of exon and intron definition (Amit et al., 2012). We also found that AS events induced by osmostress, as well as p38-dependent events, are predicted to disrupt their ORF in significantly higher proportions, compared to a control set of alternative splicing events (Fisher’s exact test p values < 0.001). This suggests that SAPK-dependent alternative splicing regulation might decrease the coding potential of stressed cells to increase translation efficiency of genes indispensable for stress-adaptation and survival.
To understand how p38 regulates splicing in response to stress, we systematically correlated the AS changes induced by p38 activation with those promoted by depletion of splicing factors by means of a network analysis (Papasaikas et al., 2015). Using this approach, we functionally linked p38 SAPK signaling with the Prp19-related splicing factor SKIIP (Bessonov et al., 2008) and demonstrated that p38 regulates AS through SKIIP phosphorylation. SKIIP is known to affect the splicing of specific introns in a subset of cell cycle- and apoptosis-related genes (Chen et al., 2011, van der Lelij et al., 2014) and its ortholog is required for AS and mRNA maturation of several salt-tolerance genes in Arabidopsis (Feng et al., 2015). Indeed, under salt-stress conditions in plants, SKIIP controls proper splicing of these genes, while skip-1 mutants accumulate aberrantly spliced transcripts as well as mRNAs harboring open reading frame (ORF)-disrupting intron retention events (Feng et al., 2015). It will be of interest to assess whether the p38-like SAPK in Arabidopsis also regulates salt-tolerance genes through phosphorylation of SKIIP. Although plants deficient in SKIIP are hypersensitive to both salt and other osmotic stresses (Feng et al., 2015), we were not able to detect clear defects on cell survival upon osmostress in human cells deficient in SKIIP under the conditions of our experimental set up (data not shown). The changes induced by SKIIP on stress-splicing might be not as critical for cell fate and be more relevant for fine tuning of the adaptive response to stress. Another possible explanation could be redundancy in gene regulation by different splicing factors, as suggested by the transcriptome-wide data showing that half of the AS events induced by p38 in response to stress are independent of SKIIP. Thus, although the p38-SKIIP axis contributed to the stress-adaptive response, it may not by itself be enough to make a difference for cell survival.
Environmental insults can alter splicing decisions through multiple molecular mechanisms, including the activation of post-translational modifications, the induction of subcellular redistribution of splicing factors, or modulating the interplay between the splicing and transcription machineries (Dutertre et al., 2011, Shang et al., 2017). For instance, p38 regulates the nucleo-cytoplasmic transport of hnRNPA1, a well-known AS regulator (van der Houven van Oordt et al., 2000), and the splicing factor SF3B1 acts as a regulator of the heat shock response by modulating both the concentration and the activity of the HSF1 heat shock transcription factor (Kim Guisbert and Guisbert, 2017). Phosphorylation represents an important regulatory process for both core splicing components and auxiliary splicing factors (Naro and Sette, 2013). Here, we showed that the p38 kinase directly phosphorylates SKIIP in response to osmostress and revealed the relevance of such phosphorylation to AS. p38 phosphorylates SKIIP at the Thr180, Ser224, and Ser232, which are located within the SNW1 domain. The SNW1 domain contains the highly conserved motif SNWKN that is essential for its interaction with several transcriptional regulators and with the splicing factors Brr2 and Snu114 (Folk et al., 2004, Sato et al., 2015). Although neither Brr2 nor Snu114 alter their association with SKIIP in response to stress based on co-immunoprecipitation experiments (data not shown), SKIIP phosphorylation could modulate its association with other spliceosomal components, favoring the selection of specific splice sites. Indeed, SKIIP associates with SYNCRIP and the effect of depletion of SYNCRIP is similar to that of depletion of SKIIP in GADD45 splicing events. SKIIP has been proposed to modulate expression and splicing of the cell-cycle regulator p21Cip1 upon stress, by recruiting the 3′ splice site-recognizing factor U2AF65 to p21Cip1 gene and transcripts (Chen et al., 2011). Other SKIIP-associated factors, including Prp19, are also selectively required for p21Cip1 expression under stress (Chen et al., 2011). Thus, despite the Prp19 (TNC) complex being incorporated at late stages of spliceosome assembly, right before splicing catalysis, changes in the levels and/or activity (e.g., through phosphorylation) of its components can modulate splice site selection, consistent with the concept that AS can be regulated at many steps of the spliceosome cycle (Papasaikas et al., 2015, Tejedor et al., 2015) through a large number of potential interactions. It is also important to consider that both SKIIP and Prp19 have been implicated in other processes, including transcription, DNA damage response, mRNA stability, and even signaling events, which can also contribute to their roles in the stress response. For instance, Prp19 cooperates with the transforming growth factor β (TGF-β)-activated kinase 1 (TAK1) to promote the activation of p38 in hepatocellular carcinoma (Yin et al., 2016) which, in turn, can regulate the activity of SKIIP (this manuscript), another component of the Prp19 complex, to exert effects in AS and protein isoform expression that also modulate p38 signaling, establishing an intricate regulatory circuit.
Our results demonstrate that phosphorylation of SKIIP by p38 is essential for stress-dependent skipping of GADD45α exon 2 (GADD45αs). Previous studies showed that expression of the GADD45α isoform lacking exon 2 differentially regulates cell proliferation and cell-cycle transition in response to arsenic treatment (Zhang et al., 2009). However, the role of the GADD45αS isoform in additional biological processes was not defined. GADD45 proteins have been implicated in mediating regulation of the p38 and JNK SAPK pathways. GADD45β interacts directly with MTK1 (MEKK4), a MAP3K upstream of p38 and JNK, resulting in apoptosis through activation of the JNK and p38 SAPKs (Mita et al., 2002, Miyake et al., 2007, Takekawa and Saito, 1998, Tamura et al., 2012). Here, we showed that expression of the stress-mediated GADD45αS isoform partially impaired the activation of p38 upon osmostress, suggesting that this splicing isoform could be acting as a negative regulator of MTK1 activation. This mechanism could establish a negative feed-back loop to downregulate the activity of the SAPK signaling pathways through AS of a downstream target. Of note, other studies have pointed out that activation of signaling pathways can be modulated through AS of genes encoding signaling proteins; for instance, AS regulation of MNK2 influences the p38 pathway (Maimon et al., 2014), and expression of different isoforms of MKK7 modulates JNK signaling responses (Martinez et al., 2015). Furthermore, AS can provide molecular mechanisms for crosstalk between insulin and wingless signaling in Drosophila (Hartmann et al., 2009).
Therefore, the combined data suggest that splicing modulation upon stress can be an additional control mechanism that complements the regulation of gene induction to maximize cell survival upon environmental insults. Although several regulatory elements are likely to mediate such changes in AS, the p38 SAPK plays a key role by targeting different components of the central core of the splicing machinery such as SKIIP. While SKIIP regulation cannot explain all the effects of p38 observed in transcriptome-wide analyses, it does illustrate how SAPK can have a direct impact on the function of spliceosome components to mediate splicing changes upon p38-mediated stress. Collectively, our data indicate that control of the splicing machinery by post-translational modifications can provide a fast mechanism for the generation of new isoforms upon external insults and establish regulatory circuits that modulate stress responses.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | Cat. No. S9026 |
| mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | Cat. No. sc-32233, RRID:AB_627679 |
| mouse monoclonal anti-hnRNPA1 | Santa Cruz Biotechnology | Cat. No. sc-32301, RRID:AB_627729 |
| mouse monoclonal anti-Flag | Sigma-Aldrich | Cat. No. S2220 |
| mouse monoclonal anti-HA | House-made from the 12CA5 hybridoma | N/A |
| rabbit polyclonal anti-SKIIP | Santa Cruz Biotechnology | Cat. No. sc-30139, RRID:AB_2302187 |
| rabbit monoclonal anti-p38α SAPK | Santa Cruz Biotechnology | Cat. No. sc-535, RRID:AB_632138 |
| rabbit polyclonal anti-T7 epitope tag antibody | Novus | Cat. No. NB 600-372, RRID:AB_527035 |
| rabbit polyclonal anti-GADD45α | Santa Cruz Biotechnology | Cat. No. sc-792, RRID:AB_2108185 |
| anti-rabbit IgG-Alexa488 | Life technologies | A11008 |
| rabbit polyclonal anti-SPF45 | Provided by Dr. Valcárcel, Centre for Genomic Regulation, Barcelona. | N/A |
| rabbit polyclonal anti-SYNCRIP | Cell Signaling | Cat. No. 8588 |
| Chemicals, Peptides and Recombinant Proteins | ||
| SB203580 | Calbiochem | Cat. No. 559389 |
| Lipofectamine RNAiMAX | ThermoFisher | Cat. No. 13778030 |
| FuGENE 6 | Promega Transfection Reagent | Cat. No. E2691 |
| Hoechst 33342 | Sigma | 14533-100MG |
| GST-MKK6DD | Gubern et al., 2016 | N/A |
| GST-p38α | Gubern et al., 2016 | N/A |
| GST-SKIIP | This paper | N/A |
| GST-SKIIP-3A | This paper | N/A |
| Deposited Data | ||
| RNA seq data | GSE117699 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117699 |
| Splicing-sensitive microarray | GSE117996 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117996 |
| Experimental Models: Cell Lines | ||
| HeLa CCL-2 | ATCC | Cat. No. CCL-2, RRID:CVCL_0030 |
| HEK293 | ATCC | Cat. No. CRL-1573, RRID:CVCL_0045 |
| 3T3 cell lines stably expressing KTRs | Provided by Dr. Covert, Shriram Center for Bioengineering and Chemical Engineering, Stanford. | N/A |
| HeLa cells stably expressing doxycycline-inducible T7-SKIIP | This paper | N/A |
| Oligonucleotides | ||
| Primers for construct generation, see Table S3 | This paper | N/A |
| Primers for semiquantitative PCR analysis, see Table S3 | This paper | N/A |
| Primers for qPCR analysis, see Table S3 | This paper | N/A |
| Primers for semiquantitative PCR analysis of network alternative splicing events, see Table S3 | Papasaikas et al., 2015 | N/A |
| ON-TARGETplus Non-targeting siRNA #2 | Dharmacon | Cat. No. D-001810-02-05 |
| SMARTpool: ON-TARGETplus SNW1 siRNA (SKIIP) | Dharmacon | Cat. No. L-012446-00-0005 |
| ON-TARGETplus siRNA - Human HNRPA1 ORF | Dharmacon | Cat. No. J-008221-11 |
| stealth SKIIP 774: caggtctctccaaagtgaatgaact | Invitrogen | Custom |
| stealth scrambled 774 | Invitrogen | Custom |
| Recombinant DNA | ||
| pcDNA3 | Invitrogen | Cat. No. V79020 |
| pEGFP-C1 | Clontech | N/A |
| pGEX-6P-1 | GE Healthcare | Cat. No. 28-9546-48 |
| pEFmlink-MKK6DD | Alonso et al., 2000 | N/A |
| pcDNA3-3HA-p38α | Provided by Dr. Engelberg, The Hebrew University of Israel, Jerusalem. | N/A |
| pcDNA3-flag-p38α | Enslen et al., 1998 | N/A |
| pcGT7 | Provided by Dr. Valcárcel; Centre for Genomic Regulation, Barcelona. | N/A |
| Lenti-X Tet-One Puro Inducible Expression System | Clonetech | Cat. No. 631847 |
| pMDG2 envelope vector | Provided by Dr. Moffat, University of Toronto, Toronto. | N/A |
| Psp2 envelope vector | Provided by Dr. Moffat, University of Toronto, Toronto. | N/A |
| pGEX2T1-MKK6DD | Gubern et al., 2016 | N/A |
| pGEX5x3-p38α | Gubern et al., 2016 | N/A |
| pGEX-SKIIP | This paper | N/A |
| pGEX-SKIIP-3A | This paper | N/A |
| HA-SKIIP | This paper | N/A |
| T7-SKIIP | This paper | N/A |
| T7-SKIIP-3A | This paper | N/A |
| pLVX-TetOne T7-SKIIP | This paper | N/A |
| pLVX-TetOne T7-SKIIP-3A | This paper | N/A |
| DYRK1A minigene | This paper | N/A |
| pCDNA3-GADD45αB | This paper | N/A |
| pCDNA3-GADD45αS | This paper | N/A |
| Software and Algorithms | ||
| VAST-TOOLS, version 2.0.0 | Irimia et al., 2014, Tapial et al., 2017 | https://github.com/vastgroup/vast-tools |
| MATT | Gohr and Irimia, 2019 | N/A |
| MATLAB | THE MATHWOKS INC | N/A |
| NIS elements AR software | NIKON INSTRUMENTS INC | N/A |
| Other | ||
| rabbit polyclonal T7-Tag® Antibody Agarose | Millipore | Cat. No. 69026-3, RRID:AB_10947861 |
| Splicing sensitive microarray platform | Muñoz et al., 2009 | N/A |
| FluoroBrite DMEM | GIBCO | Cat. No. A1896701 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Francesc Posas (francesc.posas@irbbarcelona.org).
Experimental Model and Subject Details
Cells were maintained in Dulbecco’s modified Eagle’s medium (Biological Industries) containing 10% fetal calf serum (Sigma) and supplemented with 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin (GibCO)and were cultured in a 5% CO2 humidified incubator at 37°C.
HeLa (human, cervical epithelial, female) and HEK293 (human, embryonic kidney epitelial, female) cells were purchased from the American Type Culture Collection (ATCC). NIH 3T3 (mouse, embryonic, male) cell lines stably expressing JNK and p38 KTRs were provided by Dr. Covert’s lab. HeLa cells stably expressing doxycycline-inducible T7-SKIIP proteins were constructed using the Lenti-X Tet-One Inducible Expression System. Briefly, lentiviral particles were generated by co-transfecting HEK293T cells with the lentiviral vectors pLVX-TetOne T7-SKIIP or pLVX-TetOne T7-SKIIP-3A along with the lentiviral packaging and envelope vectors pMDG2 and Psp2 and were incubated for 72 hours before harvesting the media. After a brief centrifugation to remove cell debris, virus-containing supernatant was used to infect HeLa cells in the presence of 8 μg/ml polybrene. Infected cells were selected with puromycin (2 μg/mL) for 48 hours.
Cells were transiently transfected with the indicated plasmids or with small interference RNAs (siRNAs) using the FuGENE 6 transfection reagent or Lipofectamine RNAiMAX respectively, according to the manufacturer’s instructions.
When specified, cells were treated with 100 mM NaCl and with 10 μM SB203580 for 30 minutes prior to the treatments.
Method Details
Splicing-sensitive microarray analysis
HeLa cells were not treated (c1), treated for 2 hours with 100 mM NaCl (c2) or pre-treated for 30 minutes with SB203580 before stress (c3). RNA was isolated, reverse transcribed, labeled with Cy5 or Cy3 fluorochromes and hybridized to custom a splicing sensitive microarray platform (Muñoz et al., 2009). A reciprocal experiment using switching of fluorochromes was also carried out. Two independent differential hybridization microarray assays (μa) were performed: Osmostress μa (combining c1 with c2) and SB μa (combining c2 with c3). Exon-junction probes with log2 (ratio) changes higher than 0.4, and changes in gene expression higher than 1.3, p value < 0.01, z-score > 3 were considered.
Minigene assays
HeLa cells (150000 cells/well) were seeded in 6-well plates and transfected with 0.5 μg of minigene/well. After 24 hours, cells were treated as indicated in legend figures, RNA was extracted, reverse transcribed and minigene splicing patterns were analyzed by qPCR (primers in Table S3).
RNA-seq analysis
100000 HeLa cells/well were grown and 20 nM SKIIP 774 stealth scrambled siRNA was used. 72 hours after siRNA transfection cells were treated with NaCl (100 mM) in the presence or not of SB203502 (10 μM). Stranded mRNA-seq libraries (TruSeq) were prepared from 0.5 ng RNA. Libraries were pooled and sequenced on two Illumina HiSeq 2500 lanes using 125 bp paired-end reads, yielding around 150 million read pairs per sample. Duplicates were sequenced for each condition. Library preparation and sequencing was done in the CRG Genomics Unit. Splicing analyses of RNA-Seq data were performed using Vertebrate Alternative Splicing and Transcription Tools (VAST-TOOLS, version 2.0.0) (https://github.com/vastgroup/vast-tools) (Irimia et al., 2014, Tapial et al., 2017). For all events, a minimum read coverage was required as described in Irimia et al. (2014): only events with a minimum of 10 actual reads in each sample were considered. PSI values for single replicates were quantified for all types of alternative events, including single and complex cassette exons (S, C1, C2, C3), microexons (MIC) alternative 5′ and 3′ splice sites (Alt5, Alt3) and retained introns (IR-S, IR-C). For cassette exon events, PSI values of all annotated exons was also quantified with VAST-TOOLS Annotation module (ANN). To detect differentially spliced events upon NaCl treatment, we required a ΔPSI of at least 15% between the merged replicates of untreated and stressed cells. To define the AS events that were p38-dependent, we required an AS change after treatment with NaCl + SB203502 inhibitor to be at least 25% lower than the change caused by the salt treatment (ΔPSI NaCl-NaCl SB ≥ (ΔPSI NaCl-Mock) / 4 for events with higher PSI in NaCl-treated cells, ΔPSI NaCl-NaCl SB ≤ (ΔPSI NaCl-Mock) / 4 for events with lower PSI in NaCl-treated cells). To define AS events affected by SKIIP knockdown, we required a ΔPSI of at least 15% between the merged replicates of control and SKIIP-knockdown, in both conditions of untreated (abs (ΔPSI SKIIPctrl-SCRctrl) ≥ 15%) and osmostress (abs (ΔPSI SKIIPNaCl-SCRNaCl) ≥ 15%). Heatmaps were obtained using the pheatmap R package.
In the following analyses, NaCl-induced events and p38-dependent sets described above were compared to a background set of alternative events (n = 1421) that were not affected by osmostress (ΔPSI NaCl-Ctrl < 15%) and had an intermediate PSI in the dataset (30 < PSI < 70 across all samples). ORF-disrupting potential was predicted as described in Irimia et al. (2014): briefly, exons were mapped on the coding sequence (CDS) or 5′ / 3′ untranslated regions (UTR) of genes. CDS-mapped events were subsequently classified as ORF-disrupting events if predicted to cause frameshift upon exclusion or inclusion or if containing a premature stop codon. Alternative sequences inserting an in-frame stop that would generate a truncated protein at least 300 amino acids shorter than the reference protein were also considered as ORF-disrupting events. Sequence feature analysis was performed with MATT (Gohr and Irimia, 2019), comparing NaCl-induced and p38-dependent cassette exons to background exons (n = 581). Splice site strength were determined according to Yeo and Burge (2004). Branch point features were calculated according to Corvelo et al. (2010) on the 150 nucleotides at the 3′end of each intron, excluding the first 20 nucleotides at their 5′end. SF1 binding motif, as a prediction of branch point strength, was obtained from Corioni et al. (2011).
In vitro p38 kinase assay
GST-p38α was activated in vitro in a small volume (15 μl/assay) by mixing with constitutively active GST-MKK6DD (MKK6Ser207D Thr211D is a constitutively activated form of MKK6 (Takekawa et al., 1998)) in 1x kinase assay buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 2 mM DTT) with 100 μM cold ATP in the presence or absence of 10 μM SB203580 for 20 minutes at 30°C. This activated GST-p38α was used to phosphorylate in vitro either eluted GST-fused proteins. The reactions were carried out in 1x kinase assay buffer in the presence of 1 μCi/assay of radiolabeled 32P-γ-ATP (3000 Ci/mmol, Perkin-Elmer) in a final volume of 40 μl/assay for 20 minutes at 30°C. Reactions were stopped by adding SB 5x (250 mM Tris-HCl pH 6.8, 0.5 M DTT, 10% SDS, 20% glycerol, 0.5% Bromophenol Blue) and boiling at 100°C for 5 minutes. Phosphorylated proteins were subjected to SDS-PAGE and were Coomassie blue-stained or were transfer-blotted onto a PVDF membrane and exposed to KODAK BIOMAX XAR films or a phosphorimager.
Network analysis
To integrate the effects of p38 activation in the functional splicing network, HeLa cells were seeded in 6-well plates (200000 cells/well) and were either transfected with a control expression vector (pEGFP-C1; 1ug) or co-transfected with vectors encoding the constitutively active mutant MKK6 (pEFmlink-MKK6DD; 0,5 ug) and p38 (pcDNA3-3HA-p38α; 0,5 ug). After 24 hours, total RNA was isolated by an automated procedure using oligo-dT-coated 96-well plates (mRNA catcher PLUS, Life Technologies) following the manufacturrer’s recommendations and reverse transcribed to cDNA in 96-well plates using Superscript III (Life Technologies) in teh poresence of oligo-dT (Sigma-Aldrich) and random primers (Life Technologies). PCR reactions for the 36 alternative splicing events included in the splicing network (Papasaikas et al., 2015) were carried out using the primer pairs listed in Table S3 and reagents provided in the GoTaq DNA polymerase kit (Promega) in a Zephyr compact liquid handling workstation (Perkin Elmer), and alternative isoform ratios were determined by high throughput capillary electrophoresis using a HT DNA High Sensitivity LabChip chip (Perkin Elmer) as previously described (Tejedor et al., 2015).
Immunocytochemistry
HeLa cells were grown in 8-chamber glass slides. The expression of T7-epitope tagged SKIIP and SKIIP 3A was induced upon the addition of doxyciclin to the cell culture media at at 12.5 μg/ml concentration for 24 hours. Cells were then stressed with 100 mM NaCl for 60 minutes. After treatment, HeLa cells were fixed with BD CytofixTM Fixation Buffer and permeabilized with BD PhosflowTM Perm Buffer III (BD Biosciences) following the manufacturer’s indications. Fixed cells were then blocked with 3% BSA/TBS for 30 minutes. Rabbit anti T7 antibody (Novus) was incubated at a 1/500 dilution in blocking buffer overnight. The secondary anti-rabbit IgG-Alexa488 antibody (Life technologies) was incubated at a1/1000 dilution in blocking buffer for 1 hour in the presence of 8 μM Hoechst 33342 (Sigma) to stain the nuclei. Images were taken on an epifluorescense Nikon Eclipse Ti miscroscope.
Time-lapse microscopy
NIH 3T3 mouse fibroblasts constitutively expressing the KTRs were transiently transfected with pcDNAD3 (mock), pCDNA3 GADD45αB or pCDNA3 GADD45αS. Two days post-transfection the cells were seeded (100000 cells/well) into 12-well glass-bottom plates previously coated with fibronectin. The following day the cells were washed twice with 1x PBS and cultured with 1 mL of FluoroBrite DMEM supplemented with 1% fetal calf serum (Sigma) for 1 hour prior to imaging. Cells were imaged with a Nikon Eclipse Ti fluorescence microscope controlled by the NIS elements AR software. Temperature (37°C), CO2 (5%), and humidity were held constant during the entire time-lapsed experiment. Cells were imaged for a period of four hours with captures every 3 minutes. The fluorescent channels for the cerulean p38KTR and iRFP-Histone H2B were acquired. Four independent background fields, taken from a well containing media only, were imaged for every fluorescent channel and used for the flat fielding of the time lapse images.
Plasmids and constructs
pGEX-SKIIP was obtained by cloning the SKIIP open reading frame into the BamHI/XhoI sites of the pGEX-6P-1 vector in-frame with the GST N-terminal tag. pGEX-SKIIP-3A (where T180, S224 and S232 were mutated to A) was generated by sequential site-directed mutagenesis of pGEX-SKIIP. T7-SKIIP and T7-SKIIP-3A were generated by PCR amplification using pGEX-SKIIP and pGEX-SKIIP-3A as a template, respectively. The obtained PCR product was cloned into the XbaI/BamHI restriction sites of the pcGT7 expression vector. HA-SKIIP was generated by subcloning SKIIP from pGEX-SKIIP into the BamHI/XhoI sites of the pCDNA3-3HA pcDNA vector. For construction of the T7-tagged SKIIP inducible lentiviral vectors (pLVX-TetOne T7-SKIIP and pLVX-TetOne T7-SKIIP-3A) cDNAs were amplified by PCR from the corresponding T7-SKIIP vectors. These fragments were cloned into the pLVX-TetOne-Puro Vector between the AgeI/BamHI restriction sites. GADD45αB and GADD45αS constructs were generated by PCR amplification using cDNA from HeLa cells subjected to stress as a template and were cloned into the EcoRV/XhoI sites of the pcDNA3 vector. For minigene constructs, DYRK1A and MIB1 minigenes were obtained by overlap PCR and were cloned into the EcoRI/XhoI restriction sites of pcDNA3. The GADD45α minigene was constructed by PCR amplification of the indicated region between exon 1 and exon 4 that was inserted into the pcDNA3 EcoRV/XhoI restriction sites. Primers used for construct generation are listed in Table S3.
RNA Extraction and Reverse Transcription
Total RNA was isolated using the QIAGEN RNeasy mini kit (Cat. No. 74104) and was resuspended in RNase-free water (Ambion). First strand cDNA synthesis was performed with 500 ng of RNA, 50 pmol of oligo-dT (Sigma-Aldrich), 75 ng of random primers (Life Technologies), and superscript III reverse transcriptase (Life Technologies) in a 20 μL final volume, following the manufacturer’s instructions.
PCR and qPCR
GoTaq DNA Polymerase (Promega) was used for semiquantitative PCR amplification according to the manufacturer’s instructions. After 30 cycles of 15’’ at 95°C, 30” at 57°C and 1’ at 72°C the PCR products were resolved by capillary electrophoresis using the Labchip GX Caliper workstation (Caliper, Perkin Elmer) and HT DNA 5K LabChip chip (Perkin Elmer) or analyzed on 2% agarose gels.
Inclusion/skipping levels of specific alternative exons were validated by quantitative PCR using exon-exon junction and exon specific primers (Table S3). qPCR amplification was carried out using 1 μL of 1:20 diluted cDNA with 5 μL of 2X SYBR Green Master Mix (Roche) and 4 pmol of specific primer pairs in a final volume of 10 μL in 384 well-white microtiter plates (Roche). qPCR mixes were analyzed in triplicates in a Light Cycler 480 system (Roche).
Western blotting and immunoprecipitation assays
After the indicated treatments, cells were washed with ice-cold PBS and scraped into 500 μL of IP/lysis buffer (10 mM Tris HCL pH 7.5, 1% NP40, 2 mM EDTA, 50 mM NaF, 50 mM b-glycerophosphate, 1 mM Sodium Vanadate, supplemented with the protease inhibitors 1 mM PMSF, 1 mM Benzamidine, 200 μg/ml Leupeptin and 200 μg/ml Pepstatin). The lysates were cleared by centrifugation. For immunoprecipitation assays, 10% of the total lysate was retained as input. Remaining lysates were subjected to immunoprecipitation with either 50 μL anti-T7-Tag Antibody Agarose or 50 μL Sepharose-protein A beads (GE Healthcare, 50% slurry equilibrated in IP buffer) coupled to specific antibodies by mixing overnight at 4°C. Immune complexes were collected by brief centrifugation and were rapidly washed five times with IP buffer. Immunoprecipitates, input samples or total lysates were subjected to SDS-PAGE and western blotting.
Bacterial expression and purification of recombinant proteins
E. coli cells transformed with pGEX2T1-MKK6DD, pGEX5x3-p38α, pGEX-SKIIP or pGEX-SKIIP-3A constructs were grown at 37°C until they reached an OD600 of 0.5 absorbance units. At this point, GST-tagged proteins were induced for 4 hours by adding 1 mM IPTG and switching the culture temperature to 25°C. After induction, cells were collected by centrifugation and resuspended in 1/50 volume of STET 1X buffer (100 mM NaCl, 10 mM Tris-HCl pH 8.0, 10 mM EDTA pH8.0, 5% Triton X-100 supplemented with 2 mM DTT, 1 mM PMSF, 1 mM Benzamidine, 200 mg/ml Leupeptin and 200 mg/ml Pepstatin). Cells were lysed by ice-cold brief sonication and cleared by high speed centrifugation. GST-fused proteins were pulled down from supernatants with 300 μl of glutathione-Sepharose beads (GE Healthcare, 50% slurry equilibrated with STET) by mixing for 45 minutes at 4°C. The glutathione-Sepharose beads were collected by brief centrifugation and were washed four times in STET buffer and twice in 50 mM Tris-HCl pH 8.0 buffer supplemented with 2 mM DTT. The GST-fused proteins were then eluted in 200 μl of 50 mM Tris-HCl pH 8.0 buffer supplemented with 2 mM DTT and 10 mM reduced glutathione (Sigma) by mixing for 45 minutes at 4°C and stored at 80°C.
Quantification and Statistical Analysis
PCR and qPCR analysis
Results are expressed as the mean ± SD unless otherwise specified. P values were calculated using a Student’s two-tailed t test with Welch’s correction (GraphPad) or using mean ± standard error, t test 2 tails (Excel); ∗, P value between 0.01 and 0.05; ∗∗, P value between 0.001 and 0.01; ∗∗∗, P value less than 0.001. Statistical details of experiments can be found in Figure legends and Method Details section.
Splicing-sensitive microarray analysis
Exon-junction probes with log2 (ratio) changes higher than 0.4, and changes in gene expression higher than 1.3, p value < 0.01, z-score > 3 were considered.
RNaseq analysis
Splicing analyses of RNA-Seq data were performed using Vertebrate Alternative Splicing and Transcription Tools (VAST-TOOLS, version 2.0.0) (https://github.com/vastgroup/vast-tools) (Irimia et al., 2014, Tapial et al., 2017). See RNA-seq analysis in METHOD DETAILS for detailed information.
Network analysis
Robust estimates of isoform ratios upon p38 activation were obtained using the median PSI values of biological triplicates for p38 and MKK6DD-transfected cells relative to control GPF-transfected cells. Computational data analysis to integrate these results into the splicing network was performed as described in Papasaikas et al. (2015).
Time-lapse microscopy analysis
Image analysis comprising flat fielding, image registration, object identification and cell tracking were performed using custom software as described in Regot et al. (2014). Quantification and graphics were performed using custom MATLAB scripts.
Data and Software Availability
The accession number for the RNA seq reported in this paper is GEO: GSE117699
The accession number for the Splicing-sensitive microarray platform reported in this paper is GEO: GSE117996
Vertebrate Alternative Splicing and Transcription Tools (VAST-TOOLS, version 2.0.0): https://github.com/vastgroup/vast-tools
Acknowledgments
We thank L. Subirana and A. Fernández for technical assistance, members of our laboratories for critical feedback, L. Vigevani and S. Bannal for experimental assistance, Dr. Covert for providing strains, and Dr. S. de la Luna and Dr. Regot for discussion. C.C. was supported by an FPI predoctoral fellowship from the Spanish Government. Support from the CRG Genomics Facility is also acknowledged. This work was supported by grants from the Spanish Ministry of Economy and Competitiveness (BFU2015-64437-P and FEDER, PGC2018-094136-B-I00 and FEDER, BFU2014-52125-REDT, and BFU2014-51672-REDC to F.P.; BFU2017-85152-P and FEDER to E.d.N.; BFU2014-005153 to J.V.), the Catalan Government (2017 SGR 799), the European Research Council (AdvG 670146 to J.V.), the Fundación Botín and the Banco Santander through its Santander Universities Global Division (to F.P. and J.V.), the Unidad de Excelencia Maria de Maeztu (MDM-2014-0370), and the Centre of Excellence Severo Ochoa. F.P. is a recipient of an ICREA Acadèmia (Generalitat de Catalunya).
Author Contributions
C.C. was instrumental in all phases of the study. C.C., A.U., M.J., and J.R.T. designed and conducted the experiments. P.P., C.V., R.B., and B.M. performed the bioinformatic analysis. C.C. and M.J. performed the experiments required for the revision of the manuscript. C.C., J.V., E.d.N., and F.P. designed the experiments and wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: April 16, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.060.
Contributor Information
Eulàlia de Nadal, Email: eulalia.nadal@irbbarcelona.org.
Juan Valcárcel, Email: juan.valcarcel@crg.eu.
Francesc Posas, Email: francesc.posas@irbbarcelona.org.
Supplemental Information
References
- Abankwa D., Millard S.M., Martel N., Choong C.S., Yang M., Butler L.M., Buchanan G., Tilley W.D., Ueki N., Hayman M.J., Leong G.M. Ski-interacting protein (SKIP) interacts with androgen receptor in the nucleus and modulates androgen-dependent transcription. BMC Biochem. 2013;14:10. doi: 10.1186/1471-2091-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike Y., Masuda K., Kuwano Y., Nishida K., Kajita K., Kurokawa K., Satake Y., Shoda K., Imoto I., Rokutan K. HuR regulates alternative splicing of the TRA2β gene in human colon cancer cells under oxidative stress. Mol. Cell. Biol. 2014;34:2857–2873. doi: 10.1128/MCB.00333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ayoubi A.M., Zheng H., Liu Y., Bai T., Eblen S.T. Mitogen-activated protein kinase phosphorylation of splicing factor 45 (SPF45) regulates SPF45 alternative splicing site utilization, proliferation, and cell adhesion. Mol. Cell. Biol. 2012;32:2880–2893. doi: 10.1128/MCB.06327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso G., Ambrosino C., Jones M., Nebreda A.R. Differential activation of p38 mitogen-activated protein kinase isoforms depending on signal strength. J. Biol. Chem. 2000;275:40641–40648. doi: 10.1074/jbc.M007835200. [DOI] [PubMed] [Google Scholar]
- Amat R., Böttcher R., Le Dily F., Vidal E., Quilez J., Cuartero Y., Beato M., de Nadal E., Posas F. Rapid reversible changes in compartments and local chromatin organization revealed by hyperosmotic shock. Genome Res. 2019;29:18–28. doi: 10.1101/gr.238527.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit M., Donyo M., Hollander D., Goren A., Kim E., Gelfman S., Lev-Maor G., Burstein D., Schwartz S., Postolsky B. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 2012;1:543–556. doi: 10.1016/j.celrep.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Bessonov S., Anokhina M., Will C.L., Urlaub H., Lührmann R. Isolation of an active step I spliceosome and composition of its RNP core. Nature. 2008;452:846–850. doi: 10.1038/nature06842. [DOI] [PubMed] [Google Scholar]
- Biamonti G., Caceres J.F. Cellular stress and RNA splicing. Trends Biochem. Sci. 2009;34:146–153. doi: 10.1016/j.tibs.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Borisova M.E., Voigt A., Tollenaere M.A.X., Sahu S.K., Juretschke T., Kreim N., Mailand N., Choudhary C., Bekker-Jensen S., Akutsu M. p38-MK2 signaling axis regulates RNA metabolism after UV-light-induced DNA damage. Nat. Commun. 2018;9:1017. doi: 10.1038/s41467-018-03417-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brès V., Yoshida T., Pickle L., Jones K.A. SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol. Cell. 2009;36:75–87. doi: 10.1016/j.molcel.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Zhang L., Jones K.A. SKIP counteracts p53-mediated apoptosis via selective regulation of p21Cip1 mRNA splicing. Genes Dev. 2011;25:701–716. doi: 10.1101/gad.2002611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C.W., Davis R.J. Proteins kinases: chromatin-associated enzymes? Cell. 2006;127:887–890. doi: 10.1016/j.cell.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Corioni M., Antih N., Tanackovic G., Zavolan M., Krämer A. Analysis of in situ pre-mRNA targets of human splicing factor SF1 reveals a function in alternative splicing. Nucleic Acids Res. 2011;39:1868–1879. doi: 10.1093/nar/gkq1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre I., Paris F., Huot J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget. 2017;8:55684–55714. doi: 10.18632/oncotarget.18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvelo A., Hallegger M., Smith C.W., Eyras E. Genome-wide association between branch point properties and alternative splicing. PLoS Comput. Biol. 2010;6:e1001016. doi: 10.1371/journal.pcbi.1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A., Nebreda A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- de Nadal E., Posas F. Osmostress-induced gene expression--a model to understand how stress-activated protein kinases (SAPKs) regulate transcription. FEBS J. 2015;282:3275–3285. doi: 10.1111/febs.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degese M.S., Tanos T., Naipauer J., Gingerich T., Chiappe D., Echeverria P., LaMarre J., Gutkind J.S., Coso O.A. An interplay between the p38 MAPK pathway and AUBPs regulates c-fos mRNA stability during mitogenic stimulation. Biochem. J. 2015;467:77–90. doi: 10.1042/BJ20141100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre M., Sanchez G., Barbier J., Corcos L., Auboeuf D. The emerging role of pre-messenger RNA splicing in stress responses: sending alternative messages and silent messengers. RNA Biol. 2011;8:740–747. doi: 10.4161/rna.8.5.16016. [DOI] [PubMed] [Google Scholar]
- Edmunds J.W., Mahadevan L.C. MAP kinases as structural adaptors and enzymatic activators in transcription complexes. J. Cell Sci. 2004;117:3715–3723. doi: 10.1242/jcs.01346. [DOI] [PubMed] [Google Scholar]
- Enslen H., Raingeaud J., Davis R.J. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- Feng J., Li J., Gao Z., Lu Y., Yu J., Zheng Q., Yan S., Zhang W., He H., Ma L., Zhu Z. SKIP Confers Osmotic Tolerance during Salt Stress by Controlling Alternative Gene Splicing in Arabidopsis. Mol. Plant. 2015;8:1038–1052. doi: 10.1016/j.molp.2015.01.011. [DOI] [PubMed] [Google Scholar]
- Ferreiro I., Barragan M., Gubern A., Ballestar E., Joaquin M., Posas F. The p38 SAPK is recruited to chromatin via its interaction with transcription factors. J. Biol. Chem. 2010;285:31819–31828. doi: 10.1074/jbc.M110.155846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro I., Joaquin M., Islam A., Gomez-Lopez G., Barragan M., Lombardía L., Domínguez O., Pisano D.G., Lopez-Bigas N., Nebreda A.R., Posas F. Whole genome analysis of p38 SAPK-mediated gene expression upon stress. BMC Genomics. 2010;11:144. doi: 10.1186/1471-2164-11-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folk P., Půta F., Skruzný M. Transcriptional coregulator SNW/SKIP: the concealed tie of dissimilar pathways. Cell. Mol. Life Sci. 2004;61:629–640. doi: 10.1007/s00018-003-3215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcales S.V., Albini S., Giordani L., Malecova B., Cignolo L., Chernov A., Coutinho P., Saccone V., Consalvi S., Williams R. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 2012;31:301–316. doi: 10.1038/emboj.2011.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth N., Aylon Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 2017;24:1488–1501. doi: 10.1038/cdd.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohr A., Irimia M. Matt: Unix tools for alternative splicing analysis. Bioinformatics. 2019;35:130–132. doi: 10.1093/bioinformatics/bty606. [DOI] [PubMed] [Google Scholar]
- Gomes M.D., Lecker S.H., Jagoe R.T., Navon A., Goldberg A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves V., Pereira J.F.S., Jordan P. Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes (Basel) 2017;9:9. doi: 10.3390/genes9010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubern A., Joaquin M., Marquès M., Maseres P., Garcia-Garcia J., Amat R., González-Nuñez D., Oliva B., Real F.X., de Nadal E., Posas F. The N-Terminal Phosphorylation of RB by p38 Bypasses Its Inactivation by CDKs and Prevents Proliferation in Cancer Cells. Mol. Cell. 2016;64:25–36. doi: 10.1016/j.molcel.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Guil S., Long J.C., Cáceres J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 2006;26:5744–5758. doi: 10.1128/MCB.00224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta J., Nebreda A.R. Roles of p38α mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 2015;282:1841–1857. doi: 10.1111/febs.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann B., Castelo R., Blanchette M., Boue S., Rio D.C., Valcárcel J. Global analysis of alternative splicing regulation by insulin and wingless signaling in Drosophila cells. Genome Biol. 2009;10:R11. doi: 10.1186/gb-2009-10-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M., Weatheritt R.J., Ellis J.D., Parikshak N.N., Gonatopoulos-Pournatzis T., Babor M., Quesnel-Vallières M., Tapial J., Raj B., O’Hanlon D. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159:1511–1523. doi: 10.1016/j.cell.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joaquin M., Gubern A., González-Nuñez D., Josué Ruiz E., Ferreiro I., de Nadal E., Nebreda A.R., Posas F. The p57 CDKi integrates stress signals into cell-cycle progression to promote cell survival upon stress. EMBO J. 2012;31:2952–2964. doi: 10.1038/emboj.2012.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Guisbert K.S., Guisbert E. SF3B1 is a stress-sensitive splicing factor that regulates both HSF1 concentration and activity. PLoS ONE. 2017;12:e0176382. doi: 10.1371/journal.pone.0176382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A.M., Zaganjor E., Cobb M.H. Chromatin-tethered MAPKs. Curr. Opin. Cell Biol. 2013;25:272–277. doi: 10.1016/j.ceb.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis J.M., Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- Luis N.M., Morey L., Mejetta S., Pascual G., Janich P., Kuebler B., Cozutto L., Roma G., Nascimento E., Frye M. Regulation of human epidermal stem cell proliferation and senescence requires polycomb- dependent and -independent functions of Cbx4. Cell Stem Cell. 2011;9:233–246. doi: 10.1016/j.stem.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Maimon A., Mogilevsky M., Shilo A., Golan-Gerstl R., Obiedat A., Ben-Hur V., Lebenthal-Loinger I., Stein I., Reich R., Beenstock J. Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep. 2014;7:501–513. doi: 10.1016/j.celrep.2014.03.041. [DOI] [PubMed] [Google Scholar]
- Makarova O.V., Makarov E.M., Urlaub H., Will C.L., Gentzel M., Wilm M., Lührmann R. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 2004;23:2381–2391. doi: 10.1038/sj.emboj.7600241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez N.M., Agosto L., Qiu J., Mallory M.J., Gazzara M.R., Barash Y., Fu X.D., Lynch K.W. Widespread JNK-dependent alternative splicing induces a positive feedback loop through CELF2-mediated regulation of MKK7 during T-cell activation. Genes Dev. 2015;29:2054–2066. doi: 10.1101/gad.267245.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita H., Tsutsui J., Takekawa M., Witten E.A., Saito H. Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol. Cell. Biol. 2002;22:4544–4555. doi: 10.1128/MCB.22.13.4544-4555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Z., Takekawa M., Ge Q., Saito H. Activation of MTK1/MEKK4 by GADD45 through induced N-C dissociation and dimerization-mediated trans autophosphorylation of the MTK1 kinase domain. Mol. Cell. Biol. 2007;27:2765–2776. doi: 10.1128/MCB.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz M.J., Pérez Santangelo M.S., Paronetto M.P., de la Mata M., Pelisch F., Boireau S., Glover-Cutter K., Ben-Dov C., Blaustein M., Lozano J.J. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell. 2009;137:708–720. doi: 10.1016/j.cell.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Nadal-Ribelles M., Conde N., Flores O., González-Vallinas J., Eyras E., Orozco M., de Nadal E., Posas F. Hog1 bypasses stress-mediated down-regulation of transcription by RNA polymerase II redistribution and chromatin remodeling. Genome Biol. 2012;13:R106. doi: 10.1186/gb-2012-13-11-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naro C., Sette C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013;2013:151839. doi: 10.1155/2013/151839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasaikas P., Tejedor J.R., Vigevani L., Valcárcel J. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell. 2015;57:7–22. doi: 10.1016/j.molcel.2014.10.030. [DOI] [PubMed] [Google Scholar]
- Rampalli S., Li L., Mak E., Ge K., Brand M., Tapscott S.J., Dilworth F.J. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007;14:1150–1156. doi: 10.1038/nsmb1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S., Hughey J.J., Bajar B.T., Carrasco S., Covert M.W. High-sensitivity measurements of multiple kinase activities in live single cells. Cell. 2014;157:1724–1734. doi: 10.1016/j.cell.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins E.W., Travanty E.A., Yang K., Iczkowski K.A. MAP kinase pathways and calcitonin influence CD44 alternate isoform expression in prostate cancer cells. BMC Cancer. 2008;8:260. doi: 10.1186/1471-2407-8-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N., Maeda M., Sugiyama M., Ito S., Hyodo T., Masuda A., Tsunoda N., Kokuryo T., Hamaguchi M., Nagino M., Senga T. Inhibition of SNW1 association with spliceosomal proteins promotes apoptosis in breast cancer cells. Cancer Med. 2015;4:268–277. doi: 10.1002/cam4.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalgi R., Hurt J.A., Lindquist S., Burge C.B. Widespread inhibition of posttranscriptional splicing shapes the cellular transcriptome following heat shock. Cell Rep. 2014;7:1362–1370. doi: 10.1016/j.celrep.2014.04.044. [DOI] [PubMed] [Google Scholar]
- Shang X., Cao Y., Ma L. Alternative Splicing in Plant Genes: A Means of Regulating the Environmental Fitness of Plants. Int. J. Mol. Sci. 2017;18:432. doi: 10.3390/ijms18020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimasaki S., Koga M., Esch F., Mercado M., Cooksey K., Koba A., Ling N. Porcine follistatin gene structure supports two forms of mature follistatin produced by alternative splicing. Biochem. Biophys. Res. Commun. 1988;152:717–723. doi: 10.1016/s0006-291x(88)80097-4. [DOI] [PubMed] [Google Scholar]
- Shkreta L., Chabot B. The RNA Splicing Response to DNA Damage. Biomolecules. 2015;5:2935–2977. doi: 10.3390/biom5042935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone C., Forcales S.V., Hill D.A., Imbalzano A.N., Latella L., Puri P.L. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 2004;36:738–743. doi: 10.1038/ng1378. [DOI] [PubMed] [Google Scholar]
- Takekawa M., Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- Takekawa M., Maeda T., Saito H. Protein phosphatase 2Calpha inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J. 1998;17:4744–4752. doi: 10.1093/emboj/17.16.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura R.E., de Vasconcellos J.F., Sarkar D., Libermann T.A., Fisher P.B., Zerbini L.F. GADD45 proteins: central players in tumorigenesis. Curr. Mol. Med. 2012;12:634–651. doi: 10.2174/156652412800619978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapial J., Ha K.C.H., Sterne-Weiler T., Gohr A., Braunschweig U., Hermoso-Pulido A., Quesnel-Vallières M., Permanyer J., Sodaei R., Marquez Y. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017;27:1759–1768. doi: 10.1101/gr.220962.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejedor J.R., Papasaikas P., Valcárcel J. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol. Cell. 2015;57:23–38. doi: 10.1016/j.molcel.2014.10.029. [DOI] [PubMed] [Google Scholar]
- van der Houven van Oordt W., Diaz-Meco M.T., Lozano J., Krainer A.R., Moscat J., Cáceres J.F. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J. Cell Biol. 2000;149:307–316. doi: 10.1083/jcb.149.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lelij P., Stocsits R.R., Ladurner R., Petzold G., Kreidl E., Koch B., Schmitz J., Neumann B., Ellenberg J., Peters J.M. SNW1 enables sister chromatid cohesion by mediating the splicing of sororin and APC2 pre-mRNAs. EMBO J. 2014;33:2643–2658. doi: 10.15252/embj.201488202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Li L., Wu Y., Zhang R., Zhang M., Liao D., Wang G., Qin G., Xu R.H., Kang T. CBX4 Suppresses Metastasis via Recruitment of HDAC3 to the Runx2 Promoter in Colorectal Carcinoma. Cancer Res. 2016;76:7277–7289. doi: 10.1158/0008-5472.CAN-16-2100. [DOI] [PubMed] [Google Scholar]
- Yang S.H., Sharrocks A.D., Whitmarsh A.J. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013;513:1–13. doi: 10.1016/j.gene.2012.10.033. [DOI] [PubMed] [Google Scholar]
- Yeo G., Burge C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- Yin J., Wang L., Zhu J.M., Yu Q., Xue R.Y., Fang Y., Zhang Y.A., Chen Y.J., Liu T.T., Dong L., Shen X.Z. Prp19 facilitates invasion of hepatocellular carcinoma via p38 mitogen-activated protein kinase/twist1 pathway. Oncotarget. 2016;7:21939–21951. doi: 10.18632/oncotarget.7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota T., Wang Y. p38 MAP kinases in the heart. Gene. 2016;575:369–376. doi: 10.1016/j.gene.2015.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Dowd D.R., Staal A., Gu C., Lian J.B., van Wijnen A.J., Stein G.S., MacDonald P.N. Nuclear coactivator-62 kDa/Ski-interacting protein is a nuclear matrix-associated coactivator that may couple vitamin D receptor-mediated transcription and RNA splicing. J. Biol. Chem. 2003;278:35325–35336. doi: 10.1074/jbc.M305191200. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Beezhold K., Castranova V., Shi X., Chen F. Characterization of an alternatively spliced GADD45alpha, GADD45alpha1 isoform, in arsenic-treated epithelial cells. Mol. Carcinog. 2009;48:454–464. doi: 10.1002/mc.20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Liu K., Han B., Xu Z., Gao X. The emerging role of follistatin under stresses and its implications in diseases. Gene. 2018;639:111–116. doi: 10.1016/j.gene.2017.10.017. [DOI] [PubMed] [Google Scholar]
- Zheng C., Li J., Wang Q., Liu W., Zhou J., Liu R., Zeng Q., Peng X., Huang C., Cao P., Cao K. MicroRNA-195 functions as a tumor suppressor by inhibiting CBX4 in hepatocellular carcinoma. Oncol. Rep. 2015;33:1115–1122. doi: 10.3892/or.2015.3734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA seq reported in this paper is GEO: GSE117699
The accession number for the Splicing-sensitive microarray platform reported in this paper is GEO: GSE117996
Vertebrate Alternative Splicing and Transcription Tools (VAST-TOOLS, version 2.0.0): https://github.com/vastgroup/vast-tools