

Abstract
Glioblastoma multiforme (GBM) is an aggressive primary brain tumor, for which there is no cure. Treatment effectiveness for GBM has been limited due to tumor heterogeneity, an immunosuppressive tumor microenvironment (TME) and the presence of the blood brain barrier, which hampers the transport of chemotherapeutic compounds to the central nervous system (CNS). High-density lipoprotein (HDL)-mimicking nanodiscs hold considerable promise to achieve delivery of bioactive compounds into tumors. Herein, we tested the ability of synthetic HDL nanodiscs, to deliver chemotherapeutic agents to the GBM microenvironment and elicit tumor regression. To this end, we developed chemo-immunotherapy delivery vehicles based on sHDL nanodiscs loaded with CpG, a Toll-like receptor 9 (TLR9) agonist, together with docetaxel (DTX), a chemotherapeutic agent, for targeting GBM. Our data show that delivery of DTX-sHDL-CpG nanodiscs into the tumor mass elicited tumor regression and anti-tumor CD8+ T cell responses in the brain TME. We did not observe any overt off-target side effects. Furthermore, the combination of DTX-sHDL-CpG treatment with radiation (IR), which is the standard of care for GBM, resulted in tumor regression and long-term survival in 80% of GBM-bearing animals. Mice remained tumor free upon tumor cell rechallenge in the contralateral hemisphere, indicating the development of anti-GBM immunological memory. Collectively, these data indicate that sHDL nanodiscs constitute an effective drug delivery platform for the treatment of GBM, resulting in tumor regression, long term survival and immunological memory, when used in combination with IR. The proposed delivery platform has significant potential for clinical translation.
Keywords: HDL nanodiscs, nanoparticles, glioma, chemo-immunotherapy, local therapy, drug delivery
ACS Nano TOC Graphic
Glioblastoma multiforme (GBM) is an aggressive primary brain tumor, which bears a dismal prognosis. Current standard of care (SOC) includes surgical resection, radiation therapy and chemotherapy with temozolomide (TMZ).1 Despite advances in therapeutic approaches, the prognosis for GBM patients remains very poor, with median survival around 18–24 months’ post-diagnosis.1 Therefore, the development of effective treatment strategies to control tumor progression and improve median survival of GBM patients is urgently needed.
Radiation therapy and chemotherapy are commonly used treatment options for GBM.1 Although chemotherapeutic agents are designed to kill cancer cells, a side effect of their therapeutic activity can also elicit damage to normal cells.2–4 Since their cytotoxic effects are not selective for cancerous cells, off-target toxicity is a concern. Also, many anti-cancer drugs have poor water solubility and a short half-life in vivo.5 Reduction of possible off-target toxicity requires the development of chemotherapeutic agents with higher specificity, or highly targeted delivery platforms to achieve tumor specific killing. In addition to chemotherapy, immunotherapy is an attractive alternative strategy to overcome the immunosuppressive tumor microenvironment (TME) in GBM.6–8 CpG oligodeoxynucleotide is a TRL9 ligand expressed by most immune cells which, has been shown to trigger immune rejection and induce long-term immunity against gliomas.9 To this end, CpG loaded carbon nanotubes have been explored as effective treatment for glioma through intracranial delivery.10–12 We hypothesized, that co-delivery of chemotherapeutic drugs with CpG would achieve better tumor suppressing effects when compared to individual agents, eliciting death of tumor cells and also, anti-glioma immunity.
Ongoing research has demonstrated that nanoparticles (NPs) exhibit the ability to meet the need for targeted delivery of therapeutics and can also be used as imaging agents (theranostics) in the setting of malignant brain tumors.13–15 NPs have been previously used to co-deliver different agents, such as CpG and paclitaxel (PTX).11 However, few NPs have met regulatory approval for clinical administration. HDL is a naturally occurring nanodisc that, unlike many engineered NPs, circulates in plasma for long periods of time (t½ ~ 3–4 days).16 It also plays a major role in cholesterol transport and in the transport of other molecules, such as vitamin E, steroid hormones, signaling lipids, and micro RNAs.17–19 HDL has been utilized for the delivery of small lipophilic or amphiphilic molecules, i.e., taxol, for treating breast, prostate and ovarian cancer;20 10-hydroxycamptohecin (HCPT), for treating colon carcinoma;21 pimecrolimus and tacrolimus, for treating atopic dermatitis.22 Incorporating small molecule drugs into HDLs can improve the therapeutic efficacy by enhancing the small molecule’s solubility, circulation half-life, and distribution profile.16,20,22 Synthetic Apolipoprotein-I (ApoA-I) peptide-based sHDL nanodiscs, which are more cost-effective and easier to produce on a large scale, have been administered to humans in Phase I/II studies and were proven to be well tolerated and safe at high doses.19,23,24
Therefore, HDL is an attractive drug-delivery carrier for glioma therapeutics, capable of overcoming the current challenges encountered by traditional delivery methods, owing to their structural features, biocompatibility and intrinsic targeting ability via receptor-mediated mechanisms.19,21,25 Due to its small size, HDL NPs can diffuse through the entire solid tumor volume better than other NPs and enhance the accumulation of the cargo in tumor cells.26
To test our hypothesis, synthetic high density lipoprotein mimicking nanodiscs (sHDL) that encompass ApoAI mimetic peptide, phospholipids and CpG were developed to effectively deliver chemotherapeutic agents to the GBM TME. We assessed experimentally whether sHDL NP would target GBM in vitro and in vivo, and if sHDL-CpG loaded with chemotherapeutic agents would induce GBM tumor regression and elicit immunological memory in tumor-bearing animals.We also incorporated near-infrared fluorescent dyes and various chemotherapeutic drugs as payloads into sHDL for optical imaging of targeted drug delivery.
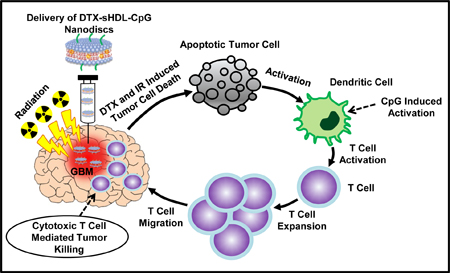
Our results demonstrate that local treatment of GBM bearing mice with HDL-mimicking nanodiscs conjugated to CpG and loaded with docetaxel (DTX), a chemotherapeutic agent, elicit tumor cells’ death with concomitant release of damage associated molecular pattern molecules (DAMPs) and tumor antigens into the TME. CpG, causes the activation of antigen presenting cells in the TME, i.e., macrophages and dendritic cells, with concomitant uptake of tumor antigens. Activated DCs, migrate to the draining lymph nodes, present tumor antigens to CD8 T cells. This elicits anti-tumor CD8+ T cell-mediated immunity. In addition, local DTX-sHDL-CpG treatment significantly improved therapeutic efficacy when tested in combination with radiation, the SOC for GBM. This resulted in tumor elimination in 80% of GBM-bearing animals and the development of long-term immunological memory against tumor rechallenge in the contralateral hemisphere, i.e., mice survived a second tumor without further treatment. This is critical, as glioma patients die of local recurrence, thus immunological memory is of paramount importance when considering the translation of therapeutic approaches to the clinical arena. In addition, the proposed therapeutic strategy did not elicit any overt off-target adverse side effects. Collectively, these data demonstrate that sHDL nanodiscs loaded with CpG and DTX are capable of inducing tumor regression long lasting anti-GBM immunological memory through a chemo-immunotherapy mediated mechanism and lend support to further developments for their implementation in a Phase 1 clinical trial for GBM patients (Figure 1).
Figure 1: Immune mediated anti-glioma mechanism of docetaxel-loaded CpG-sHDL nanodiscs.
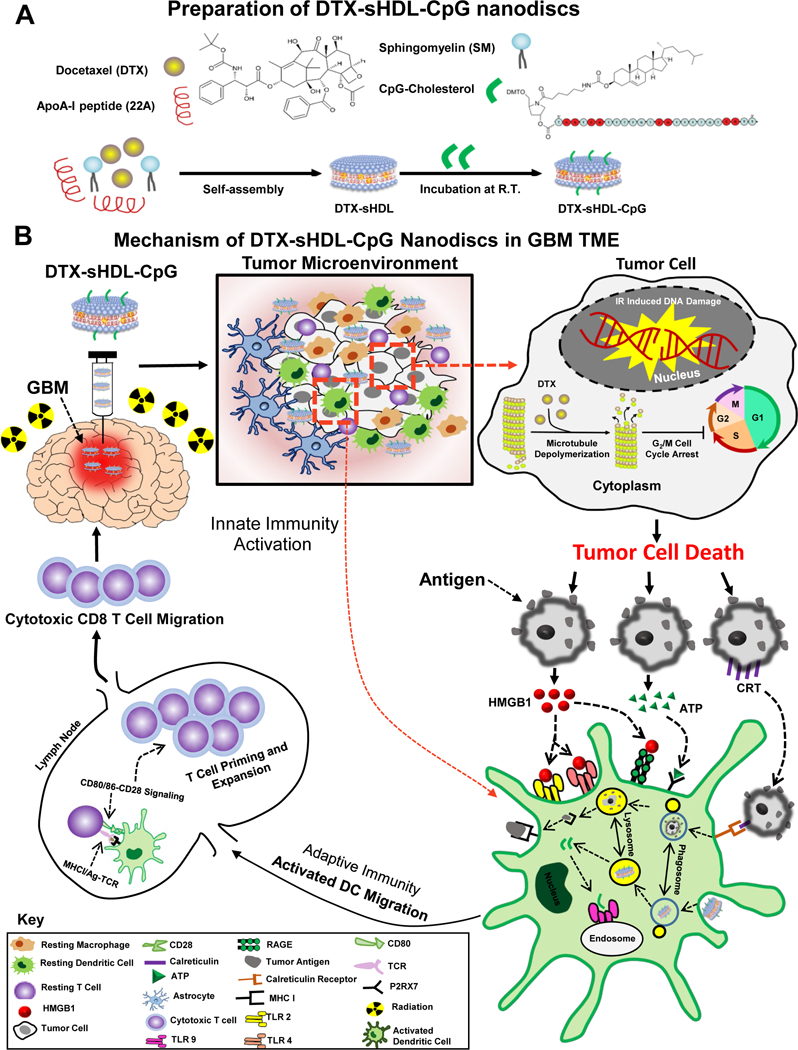
(A) DTX-sHDL-CpG was formulated by the incubation of lipid-DTX with CpG and preformed sHDL. (B) Intratumoral delivery of DTX-sHDL-CpG nanodiscs in combination with radiation results in chemoimmuno anti-glioma activity. HDL-mimicking nanodiscs deliver DTX payload to the tumor cells in the TME to suppress their microtubule depolymerization, resulting in mitotic cell cycle arrest in the G2/M phase and tumor cell death. Additionally, radiation induces double stranded DNA breaks, also leading to tumor cell death. Dying tumor cells express CRT on their surface and get engulfed by antigen presenting DCs and macrophages. The dying tumor cells also release HMGB1 and ATP, mediating DC activation. In addition, the release of CpG from the sHDL nanodiscs promotes TLR9 activation leading to further DC activation within the TME. The DCs process tumor antigens and migrate to the draining lymph node; where expansion of tumor-specific T cells takes place. Consequently, activated T cells migrate to the tumor and kill any remaining tumor cells, leading to effective anti-glioma immunity.
Results
Preparation and Characterization of sHDL Nanodiscs Loaded with Chemotherapeutic Drugs.
Chemotherapeutic agents such as paclitaxel (PTX), docetaxel (DTX) and lomustine (CCNU) have been reported to effectively induce GBM cell death in several in-vitro studies.4,27,28 However, the therapeutic efficacy of these chemotherapeutic agents in vivo is limited due to the inability of the drugs to penetrate tumor tissue and reach all the cancerous cells in the TME.29 To target the TME, we loaded chemotherapeutic drugs into HDL-mimicking nanodiscs and tested their therapeutic efficacy in glioma cells in vitro. We loaded PTX, DTX and CCNU into HDL-mimicking nanodiscs using a co-lyophilization methodology and utilized a previously reported composition of sHDL for delivering the anticancer agents, (e.g., triacetylated withaferin A, and the anti-inflammatory agent, T0901317).30,31 Dynamic light scattering (DLS) and gel permeation chromatography (GPC) were used to examine particle size, homogeneity and purity of nanodiscs. ApoA-I mimetic peptide (22A), phospholipids (1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC)) and chemotherapeutic agents were combined at a 1:1:1:0.06 weight ratio in an organic solvent, lyophilized, and hydrated with aqueous buffer. The mixture was first heated and then cooled to facilitate particle assembly. Formation of homogeneous nanodiscs’ with average size of 10–12 nm and purity of >98% was observed (Table S1, A–C). The nanodisc size determination by DLS correlated with the GPC measurement, and as the size of nanodisc increased, the retention time of GPC peak decreased. All three chemotherapeutic agents were successfully incorporated in sHDL at ~2% (w/w) loading.
To select the most potent compound for subsequent in vivo testing, the cytotoxicity of chemotherapeutic loaded HDL-mimicking nanodiscs was first evaluated in various GBM cells in vitro. Mouse GL26 cells, human HF2303 and U251 GBM cells were incubated with either free-chemotherapeutic drug, free-HDL-mimicking nanodiscs or chemotherapeutic-loaded sHDL nanodiscs at various concentrations. Half-maximal inhibitory concentration (IC50) of the different formulations were obtained from an experimentally derived dose-response curve as indicated in Figure 2 (panels A-C) and Figure S1 (panels A-B). For all three chemotherapeutic agents (DTX, PTX and CCNU) we observed comparable IC50 values between free-chemotherapeutic and chemotherapeutic-sHDL nanodiscs, implying that drug incorporation in the sHDL nanodiscs did not change their biological properties. All three chemotherapeutic agents successfully induced GBM cell death. However, free-sHDL nanodiscs did not induce tumor cell death. When chemotherapeutic agents were compared to each other, free-DTX and DTX-sHDL had the strongest cell-killing effect with the lowest IC50 values (Figure 2A–C). The IC50 value of DTX-sHDL for GL26 cells was 0.00497 μM, which is 5-fold lower than PTX-sHDL and 16,000-fold lower than CCNU-sHDL. These data indicate that DTX is the most potent agent out of the three candidates tested. Therefore, DTX was selected as the drug to be incorporated into HDL-mimicking nanodiscs for therapeutic efficacy evaluation in vivo.
Figure 2: Optimization and characterization of DTX-sHDL-CpG nanodiscs.
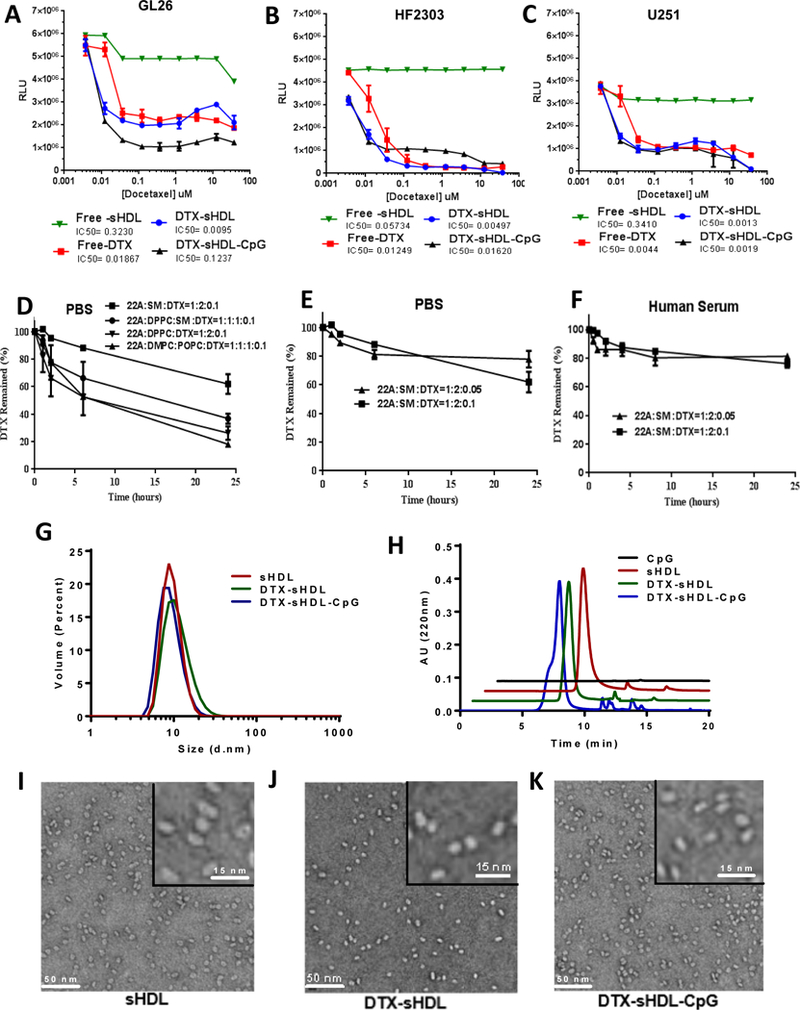
(A-C) Dose response curves for mouse (GL26, Panel A) and human (HF2303, Panel B; U251, Panel C) glioma cells treated with free-DTX; HDLs loaded with DTX; HDLs conjugated with CpG and loaded with DTX; or empty HDLs of equivalent HDL concentration to the chemotherapeutic loaded-HDLs. Cells were incubated with sHDL nanodiscs for 48 hours at indicated doses, then cell viability was evaluated. Bars represent ± SEM corresponding to three technical replicates. (D) Measurement of DTX-sHDL nanodiscs’ stability in PBS. (E) Measurement of DTX-sHDL nanodiscs’ stability with high (3.2%) or low (1.6%) drug loading in PBS. (F) Measurement of DTX-sHDL nanodiscs’ stability with high (3.2%) or low (1.6%) drug loading in human serum. For collecting stability measurements, various formulations of DTX-sHDL were suspended in PBS or human serum and incubated at 37°C with the DTX concentration of 1 mg/ml and sHDL concentration of 10 mg/mL. At different time points, 100 μl mixture of each sample was collected and filtered. The amount of drug that remained in the particles was determined by UPLC analysis. (G-H) Particle size distribution of blank sHDL, DTX-sHDL, and DTX-sHDL-CpG determined by dynamic light scattering (DLS, Panel D) and gel permeation chromatography (GPC, Panel E) at sHDL concentration of 1mg/mL. (I-K) Images demonstrating particle size distribution and morphology of blank sHDL (I), DTX-sHDL (J) and DTX-sHDL-CpG (K) taken by transmission electron microscopy (Scale bar illustration for low magnification = 50nm). Higher magnification images are shown in the upper right corner for each formulation (scale bar =15nm).
In order to optimize DTX-sHDL formulation with maximum drug retention and nanodisc stability, we prepared DTX-sHDL nanodiscs composed of lipids with varying fatty acid saturation. Purity of each generated formulation has been summarized in Table S1 (E-I). The peptide, lipid and chemotherapeutic ratio was kept at 1:2:0.1 (w/w); however, the lipid composition was varied. To select a formulation that retained maximum DTX, an in vitro drug release in PBS was conducted (Figure 1A). Nanodiscs were incubated at 37°C and the DTX retained in the nanodiscs over-time was quantified. We observed a positive correlation between the saturation of phospholipids [sphingomyelin (SM) > 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) > DMPC > POPC] and the retention of DTX in sHDL nanodiscs in PBS and plasma. Following a 24 h incubation, more than 60% of DTX remained encapsulated in SM-based sHDL, while less than 40% of the chemotherapeutic remained in the nanodiscs for all other formulations. Thus, SM was selected as the lipid component of choice to generate DTX-sHDL nanodiscs.
We next investigated the stability of HDL-mimicking nanodiscs loaded with various amounts of DTX in PBS or human serum during a 24 h incubation period. Two formulations were prepared; DTX was loaded at 1.6% and 3.2% (w/w). We observed that HDL-mimicking nanodiscs loaded with 1.6% DTX released the chemotherapeutic agent slowly in PBS and 80% of the drug remained within the nanodisc after a 24 h incubation (Figure 2E). Although, HDL-mimicking nanodiscs loaded with 3.2% DTX released the chemotherapeutic agent slowly in PBS, only 60% of the drug remained within the nanodisc after a 24 h incubation. A similar drug release trend was confirmed in human serum when evaluating the same two formulations (Figure 2F). Since DTX-sHDL nanodiscs with 22A: SM: DTX at a weight ratio of 1:1:0.05 displayed highest drug retention in PBS and human serum, they were selected as the final formulation to assess their in vivo efficacy. HDL-mimicking nanodiscs composed of same amounts of peptide and lipids without the chemotherapeutic agent were used as free-sHDL control.
We next developed the chemo-immunotherapeutic platform by incorporating CpG-cholesterol in the HDL-mimicking nanodiscs. CpG-cholesterol was incubated with DTX-sHDL in PBS at room temperature (25°C), allowing the cholesterol moiety to incorporate into the lipid bilayer of sHDL and form DTX-sHDL-CpG. DLS and GPC characterization of the three formulations have been summarized in Table S1 (D-E, J) and Figure 2 G–H. GPC curves demonstrated high purity of the sHDL, DTX-sHDL and DTX-sHDL-CpG solutions after preparation. The peak of the 22A peptide and CpG shifted to sHDL, indicating that 22A and CpG have been completely incorporated into the HDL-mimicking nanodiscs. Transmission electron microscopy (TEM) was used to examine the morphology of the nanodiscs (Figure 2 F–H). Both DLS and TEM revealed a homogeneous hydrodynamic size of ~ 10 nm and discoidal shape for blank-sHDL, DTX-sHDL and DTX-sHDL-CpG nanodiscs. This demonstrates that DTX or CpG have a negligible impact on HDL-mimicking nanodiscs formation and homogeneity.
Cellular Uptake of sHDL Nanodiscs by GBM Cells in vitro and Assessment of HDL-Mimicking Nanodiscs’ Biodistribution in vivo.
We next evaluated the uptake of sHDL by rodent and human GBM cells. To do this, we utilized 1, 1’-diocadecyl-3,3,3’,3’-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate (DiD: a fluorescent tracer with a hydrophobic anchor) labeled HDL-mimicking nanodiscs. Rodent (GL26-, CNS1) and human (HF2303) GBM cells stably transfected with mCitrine were used for imaging purposes. Cells were exposed to increasing concentrations of DiD-sHDL for 2 h. DiD-sHDL signal was imaged using scanning confocal microscopy and quantified as a percentage of total GBM cells. The cellular uptake of HDL-mimicking nanodiscs was dose dependent in both rodent and human GBM cells (Figure 3A).
Figure 3: In vitro and in vivo HDL-mimicking nanodiscs uptake.
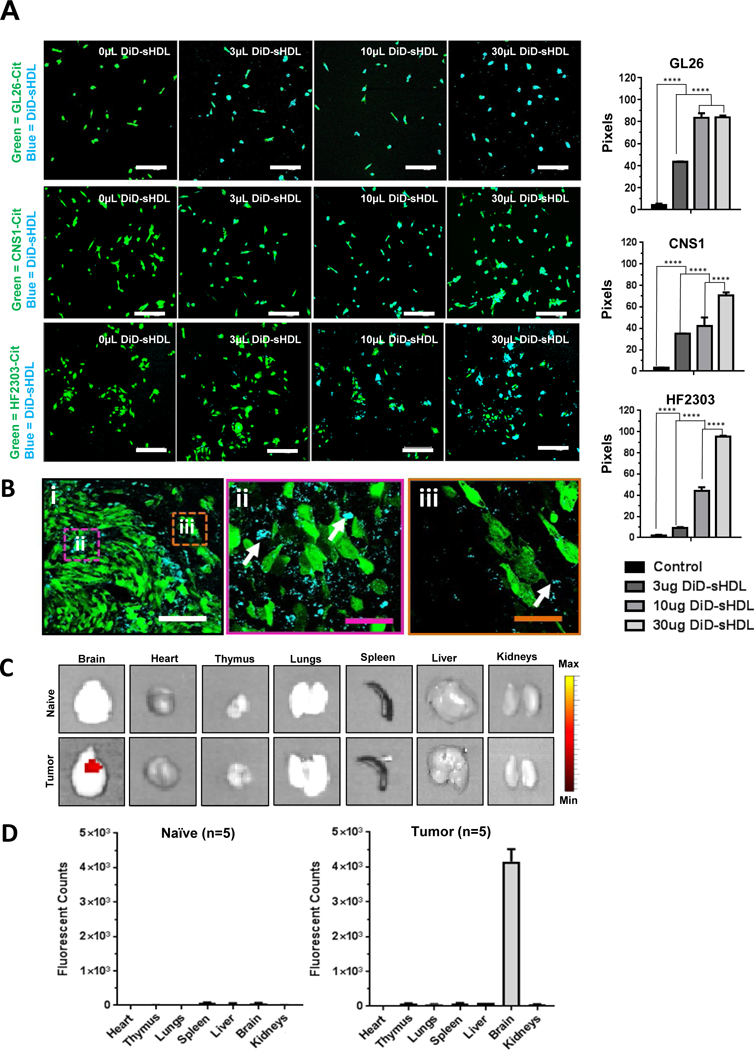
(A) In-vitro data showing intracellular localization of sHDL-nanodiscs in mouse (GL26), rat (CNS1) and human (HF2303) GBM cells 2 hrs post incubation. Imaging of the in-vitro assay shows cyan nanodiscs co-localized with citrine (green) expressing GMB cells in a dose dependent manner (white scale bar =20μm). ****p<0.0001; one-way ANNOVA test; Bars represent ± SEM corresponding to three technical replicates. (B) Mice harboring GL26-cit tumors 5 days post implantation (dpi) were intratumorally injected with 0.5mg/Kg DiD-sHDL nanodiscs. At 24hrs after the injection brains were harvested for confocal imaging. (i) DiD-sHDL nanodiscs (cyan) localized within the tumor mass (green). (ii) Higher magnification image of the tumor core delineated in panel i. (iii) Higher magnification image of the tumor border delineated in panel i. White arrows indicate DiD-sHDL nanodiscs; white scale bar = 150μm; purple and orange scale bars = 33μm. (C) Tumor naive mice (n=5) or mice harboring 21 dpi GL26-wt tumors (n=5) were intratumorally injected with 0.5mg/Kg DiR-sHDL. At 24hrs after the injection, organs were harvested for ex vivo optical imaging by IVIS and (D) fluorescence signal for each organ was quantified.
We also examined the in vivo GBM targeting efficiency of HDL-mimicking nanodiscs’ in GBM bearing animals by utilizing the GL26 syngeneic mouse glioma model, which exhibits histopathological characteristics encountered in human GBM.32–34 Mice were implanted with GL26-cit tumors in the striatum and administered with DiD-sHDL intratumorally (i.t.) 7 days post implantation (dpi). They were perfused 24 h after the injection and brains were processed for confocal imaging. Our results indicate that the DiD-sHDL signal was strongly associated with the TME (Figure 3Bi–iii). To assess the biodistribution of HDL-mimicking nanodiscs, GL26-wt cells were implanted in the striatum of C57BL6 mice. At 21 dpi, tumor bearing and naïve (non-tumor bearing) mice received i.t. 1, 1’-dioctadecyl-3,3,3’-tetramethylinodtricarbocyanie iodide (DiR, fluorescent tracer with a hydrophobic anchor). Mice were perfused at 24 h after injection, and the fluorescence intensity of several organs (heart, thymus, lungs, spleen, liver, brain and kidneys) was visualized under IVIS optical imaging system. DiR-sHDL signal was highly tumor specific; we did not observe fluorescence signal either in the contralateral hemisphere of the tumor bearing animals or within the naïve brain (Figure 5B). Furthermore, the fluorescence signal in the liver, thymus, heart, lungs, kidneys and spleens of naïve and tumor bearing animals injected i.t. with DiR-sHDL was insignificant (Figure 5C–D), indicating negligible off-site biodistribution through this delivery route. Collectively, these data demonstrate that sHDL nanodiscs can be used as a delivery platform for GBM treatment since they can reach and remain in the target tissue (brain tumor) with no accumulation in off-target organ systems.
Figure 5: Anti-tumor efficacy and T cell immunity exhibited by intratumoral sHDL-CpG-DTX treatment.
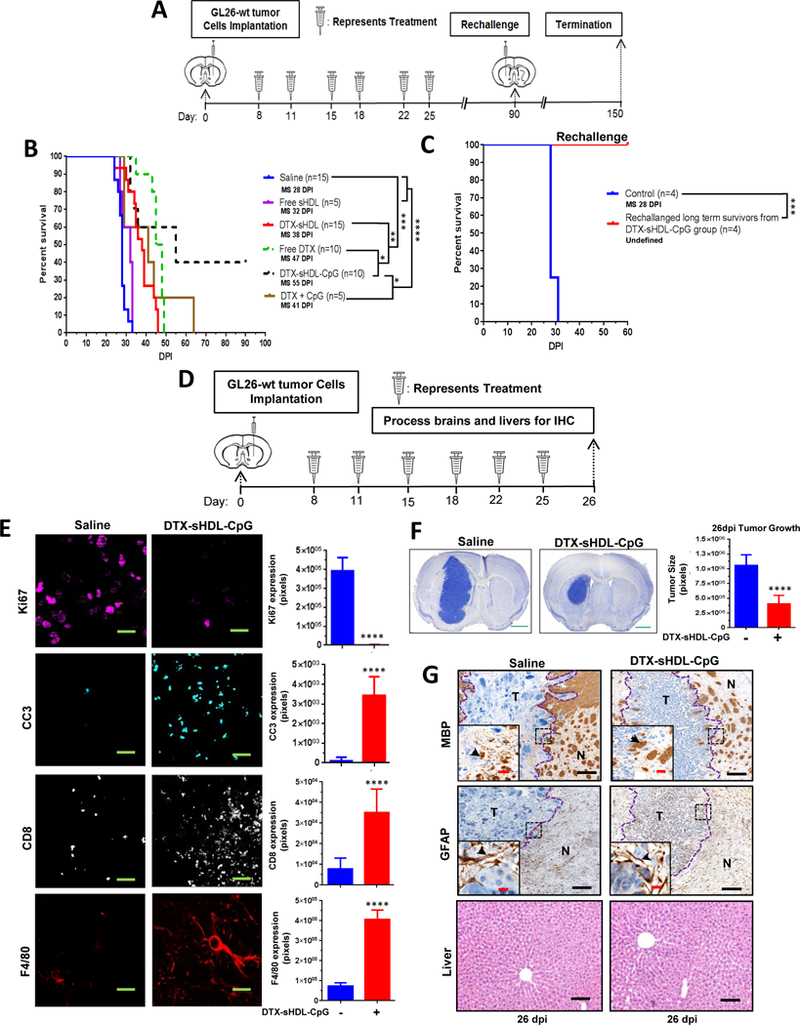
(A) GL26-wt tumors were implanted stereotactically into the right striatum of C57BL/6 mice that were treated intratumorally with saline, 0.5mg/Kg free-DTX, CpG-DTX, free-sHDL, DTX-sHDL, or DTX-sHDL-CpG loaded nanodiscs at 8, 11, 15, 18, 22 and 25 days post tumor implantation (dpi). Long-term survivors from the DTX-sHDL-CpG treatment group were rechallenged in the contralateral hemisphere with GL26-wt cells. For the control group, GL26-wt cells were implanted in naive mice, which did not receive further treatment. (B) Kaplan-Meier survival analysis of saline (n=15), free sHDL (n=5), DTX-sHDL (n=15), free DTX (n=10), DTX-sHDL-CpG (n=10), or CpG +DTX (n=5) treated mice. (C) Kaplan-Meier survival plot for rechallenged long-term survivors from (B) sHDL-CpG-DTX treatment group (n=4) and control mice (n=4). Data were analyzed using the log-rank (Mantel-Cox) test. *p < 0.05;**p < 0.001;****p <0.0001; MS=median survival. (D) C57BL/6 mice bearing GL26-wt tumors were treated with saline (n=3) or sHDL-CpG-DTX (n=3) nanodiscs at 8, 11, 15, 18, 22 and 25 days post tumor implantation (dpi). At the end of the treatment, brains and livers were harvested for histopathology. (E) Immunohistochemistry staining for Ki67, cleaved caspase-3 (CC3), CD8 and F4/80 on 50μm vibratome tumor sections (green scale bar = 10μm). Immunofluorescence staining in n=3 different tumors in each treatment group was quantified by Image J. Bar graphs represent total number of positive cells for Ki67, CC3, CD8 and F4/80 in saline and sHDL-CpG-DTX groups. ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n=3 biological replicates). (F) Nissl staining of 50μm brain sections from saline (26 dpi) and DTX-sHDL-CpG (26 dpi) GL26 tumor bearing mice. The bar graph represents tumor size quantification by ImageJ of 26 dpi GL26 tumor brain sections were stained with nissl after saline or DTX-sHDL-CpG treatment from n= 3 different tumors. ****p <0.001; unpaired t-test (green scale bar = 1mm). (G) Immunohistochemistry staining for myelin basic protein (MBP), and glial fibrillary acidic protein (GFAP) in 5μm paraffin embedded tumor sections. Low magnification panels show normal brain (N) and tumor (T) tissue (black scale bar = 100 μm). Black arrows in the high magnification panels (red scale bar = 20 μm) indicate positive staining for the areas delineated from the low magnification panels. H&E staining of 5μm paraffin embedded liver sections from saline (26 dpi) and DTX-sHDL-CpG (26 dpi) treated mice (black scale bar =200μm).
GBM Tumor Progression in the Clinic is due to Local Reoccurrence of the Tumor after Standard of Care (SOC) Treatment.
In humans, GBM carries a poor prognosis, even with maximal safe surgical resection and chemoradiation.1 Neuropathological diagnosis is performed at the time of surgery (Figure 4A). A surgical biopsy is obtained, and after cryosectioning, hematoxylin and eosin staining of the tumor sections is performed (Figure 4Bi). GBMs exhibit typical features including hypercellularity pseudopalisading necrosis, microvascular proliferation, and internal hemorrhage (Figure 4Bii). Despite treatment, patients invariably develop disease progression and tumor recurrence.1 Progression free survival is defined as the interval between surgery and the appearance of new tumor recurrence on magnetic resonance imaging (MRI). Tumor recurrence may occur locally, or adjacent to the tumor resection cavity. Conversely, tumor recurrence occasionally appears distant from the initial site of disease. Local drug delivery at the time of surgery has the advantage of treating residual disease and preventing or prolonging the time to local recurrence. This is important because recurrence most often occurs locally after surgery. In order to validate this statement, we retrospectively analyzed 50 consecutive cases of patients who underwent surgery for GBM at our institution. The results demonstrate that 45/50 (90%) of patients developed local recurrence at the 3-year follow up (Figure 4C, 4E), while just 3/50 (6%) had distant recurrence (>2cm away from the surgical resection cavity) (Figure 4D, 4E). Two patients had no evidence of disease recurrence at the time of most recent follow-up. GBMs are diffuse; however, these results demonstrate that macroscopic tumor recurrence occurs most often at the at the initial site of disease. Local drug delivery has the ability to provide the maximum concentration of drug directly to the site where disease progression or relapse is most commonly observed.
Figure 4: Patients with high grade GBM display local tumor recurrence.
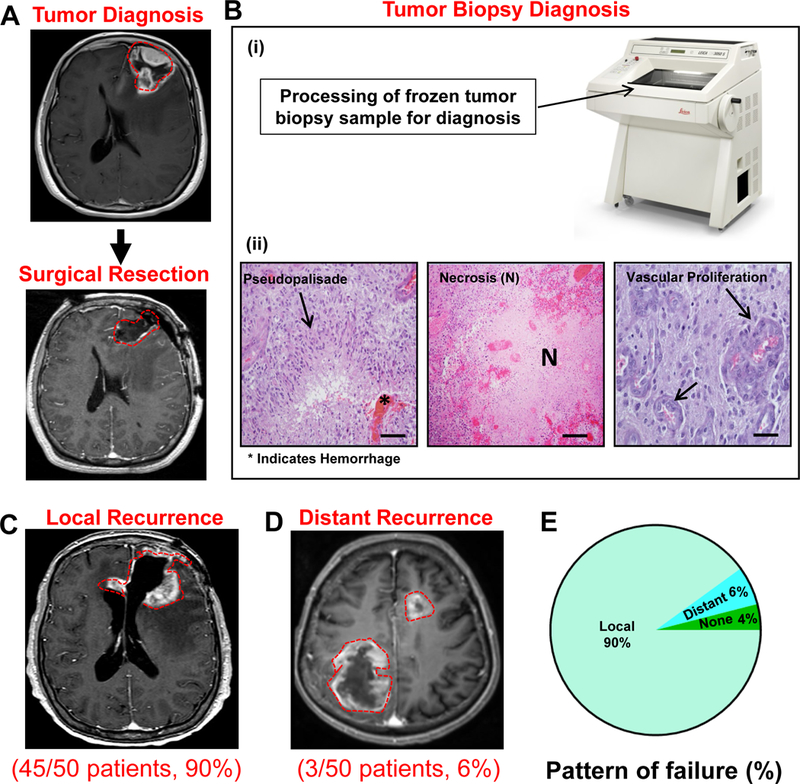
A retrospective analysis monitoring for local versus distant (>2 cm away from the surgical resection cavity) disease recurrence in 50 patients with GBM who underwent surgery at the University of Michigan Health System. (A) Upper panel: Representative MRI displaying a newly diagnosed left frontal brain tumor. Lower panel: Representative MRI displaying a complete resection of the tumor with no residual disease. (B) Processing of the tumor surgical biopsy for neuropathological diagnosis. (i) Surgical biopsy of the tumor was frozen and cryosectioned. (ii) H&E staining of the tumor sections display pseudopalisading necrosis, hemorrhage, and microvascular proliferation (scale bar = 100 μm). At the time of recurrence, 46/50 (90%) of patients primarily displayed local disease recurrence adjacent to the resection cavity (C), whereas only 3/50 (6%) patients had distant disease recurrence (D). Three years after tumor surgical resection, 2/50 (4%) patients did not have disease recurrence. (E) Pie Chart displaying retrospective analysis of 50 GBM patients that underwent surgical resection of the tumor.
Therapeutic Efficacy of DTX-sHDL Nanodiscs in an Intracranial GBM Model.
We next investigated the potential of the DTX-sHDL-CpG nanodisc formulation of eliciting tumor regression in an intracranial GBM model. Mice bearing GL26-wt tumors were treated with saline, free-sHDL, free-DTX, DTX-CpG, DTX-sHDL, or DTX-sHDL-CpG at the indicated doses and treatment schedule (Figure 5A). We observed a ~1.2 fold (p<0.05) increase in median survival (MS) of mice in the free-DTX treated group (MS: 47 dpi), a ~1.4 fold (p<0.05) increase in MS of mice in DTX-sHDL treatment group (MS: 38 dpi), and ~1.5 fold (p<0.05) increase in MS of mice in CpG + DTX treatment group (MS: 41 dpi) when compared to the control mice in the saline treated group (MS: 28 dpi). DTX-sHDL-CpG (MS: 55 dpi) treated group displayed the highest survival advantage of ~2 fold increase (p<0.001) when compared to all other treatment groups. Also, 40% of the tumor bearing mice treated with DTX-sHDL-CpG survived long term (90+ dpi) and remained tumor free (Figure 5B). When the long term survivors from the DTX-sHDL-CpG group were rechallenged with GL26-wt tumors in the contralateral hemisphere (Figure 5A), they remained tumor free without further treatment compared to control mice implanted with tumors, which succumbed due to tumor burden (MS: 28 days) (p<0.0001). These results suggest the development of immunological memory in the tumor bearing animals treated with DTX-sHDL-CpG nanodiscs (Figure 5C).
To determine whether DTX-sHDL-CpG nanodiscs’ efficacy is mediated by the immune system of the host, CD8-knockout mice were implanted with GL26-wt cells and treated with saline or DTX-sHDL-CpG nanodiscs at the indicated dose and treatment schedule (Figure S2A). There was no statistically significant difference in MS between the saline or DTX-sHDL-CpG WT in CD8 KO treated mice, indicating the critical role played by CD8+ T cells in mediating therapeutic response in this treatment group (Figure S2B).
Subsequently, we analyzed the in vivo expression of Ki67 (proliferation marker), Cleaved Caspase 3 (CC3; apoptosis marker), CD8 (T cell marker) and F4/80 (macrophage marker) in GL26 tumors, one day after the end of saline or DTX-sHDL-CpG i.t treatment (Figure 5D). DTX-sHDL-CpG group showed a significant decrease of Ki67 expression (p<0.00001) and increased CC3 expression (p<0.00001) compared to the saline treated group (Figure 5E). We also observed increased infiltration of F4/80+ macrophages (p<0.0001) and CD8+ T cells (p<0.0001) in DTX-sHDL-CpG treated tumors when compared to the saline treated group. Additionally, we quantified the size of the tumor burden one day after the final DTX-sHDL-CpG or saline treatment (26 dpi). We observed a ~3-fold (p<0.0001) decrease in tumor size in the DTX-sHDL-CpG treated mice (Figure 5F).
Next, we assessed the potential inflammatory response and disruption of the surrounding brain architecture due to DTX-sHDL-CpG nanodisc therapy. Brain architecture of mice treated with saline or DTX-sHDL-CpG nanodiscs was evaluated by immunohistochemistry for myelin basic protein (MBP), as an index of oligodendrocyte integrity and glial fibrillary acid protein (GFAP), as an index of astrocyte integrity. Brain structure was preserved, with no apparent reduction in MBP or GFAP expression, demyleinization or overt inflammation due to the DTX-sHDL-CpG treatment compared to the saline control group (Figure 5G). Furthermore, liver tissue sections from both treatment groups showed no signs of necrosis, inflammation, or changes in cellular structures (Figure 5G). These data demonstrate that DTX-sHDL-CpG treatment does not induce overt toxicity or adverse side effects in the brain or the liver.
Expansion of Anti-Tumor Cytotoxic T Cells in Response to Chemo-Immunotherapy.
Cytotoxic T lymphocytes are crucial for mediating tumor specific adaptive immunity; however, their activity is suppressed in the presence of GBM.35 Previously, DTX has been shown to enhance antitumor T cell responses against lung cancer by eliciting immunogenic cell death (ICD).36 We examined whether the tumor regression caused by DTX-sHDL-CpG treatment (Figure 5B–C) was due to the release of danger signals (e.g., CRT and HMGB1) associated with ICD. Briefly, mice bearing GL26 tumors harboring a surrogate tumor antigen, ovalbumin (OVA), were treated with saline or DTX-sHDL-CpG nanodiscs at the indicated doses and schedule (Figure 6A); mice were euthanized one day after the end of the treatment and brains were processed for flow cytometry analysis. Analysis of GL26-OVA tumors at day 26 revealed ~ 2.3 fold (p<0.0001) higher expression levels of calreticulin (CRT) on the surface of tumor cells treated with DTX-sHDL-CpG compared to the saline treated group (Figure 6B). We also observed a ~1.9 fold (p<0.0001) increase of intracellular HMGB1 expression within the tumor cells in the DTX-sHDL-CpG treatment group (Figure 6B). These data demonstrate that DTX-sHDL-CpG treatment triggers ICD-associated danger signals in the GBM TME.
Figure 6: Chemo-immunotherapy induces tumor-specific CD8 T cell responses within GBM TME.
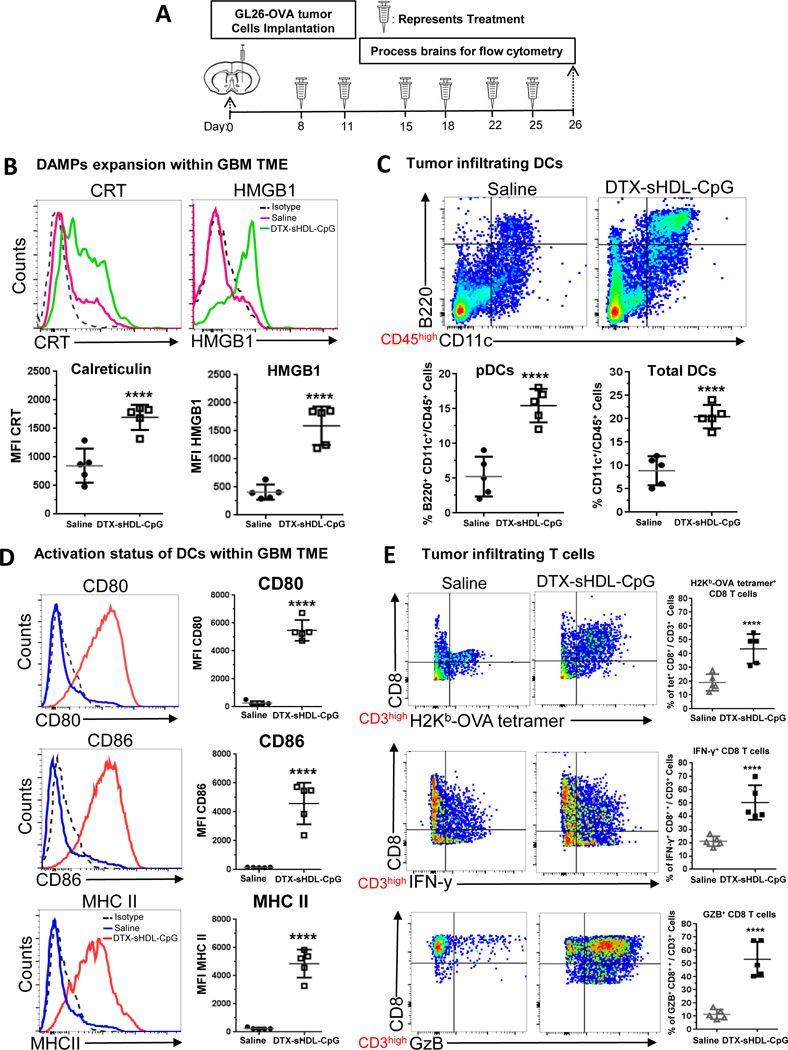
(A) Mice bearing GL26-OVA tumors were treated with sHDL-CpG-DTX nanodiscs or saline (n=5 mice /treatment group) at 8, 11, 15, 18, 22 and 25 days post tumor implantation (dpi), at end of the treatment brains were harvested for flowcytometry analysis. Levels of ICD markers, CRT (B) and HMGBI, within saline and DTX-sHDL-CpG TME of GL26 tumor-bearing were determined at 26 dpi. Representative histograms display each marker’s expression levels (solid lines: pink= saline, green= DTX-sHDL-CpG) compared to isotype controls (black dotted line). MFI= mean fluorescence intensity. ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates). (C) The percent of pDCs (CD11c+/B220+) and pan DCs (CD11c+) within the CD45+ cell population in the TME of saline and sHDL-CpG-DTX treated mice was assessed at 26dpi. Representative flow plots for each group are displayed. ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates). (D) Activation status of CD11c+ DCs in the tumor was compared between saline and sHDL-CpG-DTX treatment groups at 26dpi. Activation status of DCs was assessed by the expression levels of CD80, CD86, and MHC II. Representative histograms display each marker’s expression levels (solid lines: blue= saline, red= DTX-sHDL-CpG) compared to isotype control (black dotted line). ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates). (E) Tumor-specific CD8+ T cells within the TME of GL26-OVA tumors were analyzed by staining for the SIINFEKL-Kb tetramer. Activation status of CD8+ T cells within the TME was analyzed by staining for granzyme B (Gzb) and IFNγ after stimulation with the tumor lysate. Representative flow plots for each group are displayed. ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates).
Additionally, we assessed whether DTX-sHDL-CpG treatment recruits antigen presenting cells to the tumor milieu. Since CpG, a potent TLR9 agonist, directly stimulates plasmacytoid dendritic cells (pDCs),9 we aimed to elucidate their role in the induction of immune mediated anti-GBM activity. We observed an increase in the percentage of tumor infiltrating pDCs (CD45+/CD11c+ / B220+) and pan DCs (CD45+/CD11c+), ~2.9 fold (p<0.0001) and ~2.5 fold (p<0.0001) respectively in the TME of DTX-sHDL-CpG treated mice when compared to saline treated mice (Figure 6C). We also identified a ~1.5 fold (p<0.0001) increase in macrophages (CD45+/F4/80+) in the TME of DTX-sHDL-CpG treated mice (Figure S3). When DCs recognize danger signals (e.g., CRT and HMGB1) or CpG, they express elevated levels of molecules involved in antigen presentation such as MHC II and co-stimulatory molecules, CD80 and CD86.9 To examine the effect of DTX-sHDL-CpG treatment on the DC activation status, we assessed the expression levels of CD80, CD86 and MHC II. We observed an increase in the frequency of CD45+/CD11c+ /CD80+ (~5 fold, p<0.0001), CD45+/CD11c+ /CD86+ (~8.4 fold, p<0.0001), and CD45+/CD11c+ /MHC II+ (~8.3 fold, p<0.0001) DCs in the TME of DTX-sHDL-CpG treated mice when compared to saline treated control mice (Figure 6D). These data suggest that the DTX and CpG loaded sHDL nanodiscs trigger innate and adaptive immune responses within the TME by activating tumor antigen presenting DCs and macrophages, which then prime tumor specific T cells.
Given that DTX-sHDL-CpG treatment resulted in tumor regression (Figure B–C), we sought to identify whether therapeutic efficacy is mediated by a CD8+ T cells’ tumor antigen specific anti-tumor response. We examined tumor-specific T cells in the TME of saline and DTX-sHDL-CpG treated mice. Tumor-specific T cells were identified using the SIINFEKL-H2Kb tetramer, the OVA cognate antigen (tumor antigen-specific T cells: CD3+/ CD8+/ SIINFEKL-H2Kb tetramer+). We observed a ~1.3 fold (p<0.0001) increase in tumor antigen-specific CD8+ T cells in the TME of mice treated with DTX-sHDL-CpG (Figure 6F). We also assessed the impact of DTX-sHDL-CpG nanodiscs on the activation status of CD3+/CD8+ T cells in the TME, by assessing their interferon-γ (IFNγ) and granzyme B (Gzb) expression levels. In CD8+ T cells isolated from the TME of mice treated with DTX-sHDL-CpG, IFNg levels were ~1.4 fold higher (p<0.00130) and GZb levels were ~1.7 fold higher (p<0.0100), when compared to levels encountered in CD8+ T cells isolated from control mice treated with saline (Figure 6E).
Next, we sought to evaluate the activation status of antigen presenting dendritic cells and CD8+ T cells within the draining lymph nodes (dLNs) of DTX-sHDL-CpG treated mice. Briefly, mice bearing GL26 tumors harboring surrogate tumor antigen, ovalbumin (OVA) were treated with saline or DTX-sHDL-CpG nanodiscs at indicated doses and schedule (Figure 7A). Mice were euthanized one day after the end of the treatment and dLNs were processed for flow cytometry. We observed an increase in the percentage of pDCs (CD45+/CD11c+ / B220+) and pan DCs (CD45+/CD11c+), ~1.5 fold (p<0.0001) and ~1.8 fold (p<0.0001) respectively in the dLNs of DTX-sHDL-CpG treated mice compared to saline treated mice (Figure 7B). To examine the effect of DTX-sHDL-CpG treatment on DC activation in the dLNs, the expression of CD80, CD86 and MHC II was assessed. We observed an increase in the frequency of CD45+/CD11c+ /CD80+ (~1.8 fold, p<0.0001), CD45+/CD11c+ /CD86+ (~3.5 fold, p<0.0001), and CD45+/CD11c+ /MHC II+ (~4.5 fold, p<0.0001) DCs in the TME of DTX-sHDL-CpG treated mice in compared to saline treated mice (Figure 7C). These data suggest that DTX-sHDL-CpG treatment induces the activation of DCs in the dLNs by enhancing the expression of molecules involved in antigen presentation.
Figure 7: Chemo-immunotherapy induces activation of CD8+ T cells in the draining lymph nodes and splenocytes.
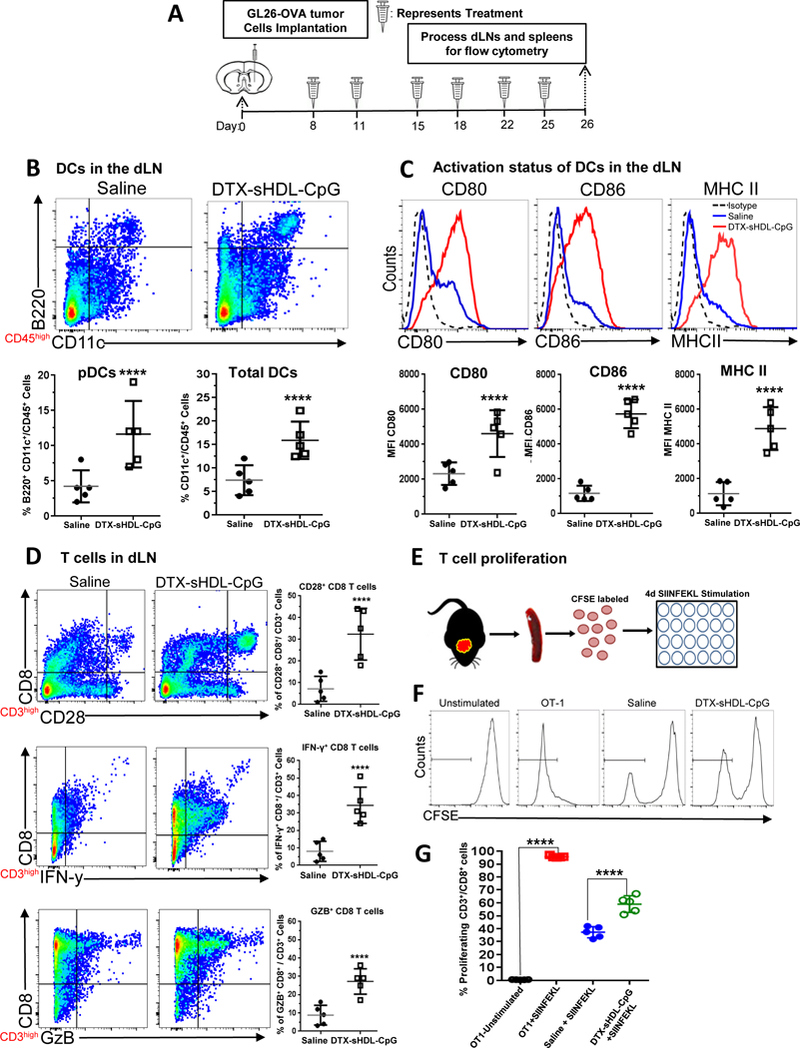
(A) Mice bearing GL26-OVA tumors were treated with sHDL-CpG-DTX nanodiscs or saline (n=5 mice / treatment group) at 8, 11, 15, 18, 22 and 25 days post tumor implantation (dpi), at end of the treatment draining lymph nodes (dLN) and spleens were harvested for flowcytometry analysis. (B) The percent of pDCs (CD11c+/B220+) and pan DCs (CD11c+) within the CD45+ cell population in the draining lymph node of saline and sHDL-CpG-DTX treated mice was assessed at 26dpi. Representative flow plots for each group are displayed. ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates). (C) Activation status of CD11c+ DCs in the draining lymph nodes was compared between saline and sHDL-CpG-DTX treatment groups at 26dpi. Activation status on DCs was analyzed for the expression levels of CD80, CD86, and MHC II. Representative histograms display each marker’s expression levels (solid lines: blue= saline, red= DTX-sHDL-CpG) compared to isotype control (black dotted line). ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates). (D) Activation status of CD8+ T cells within the dLNs of GL26-OVA tumor bearing mice was assessed by staining for CD28. Also, activation status of CD8+ T cells within the dLNs was analyzed by staining for granzyme B (Gzb) and IFNγ after stimulation with the tumor lysate. Representative flow plots for each group are displayed. ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates). (E) Experimental design showing splenocytes from saline or sHDL-CpG-DTX treated GL26 tumor bearing mice labeled with CFSE and then stimulated with 100nM of SIINFEKL peptide during 4 days in culture to assess CD8+ T cell proliferation. (F) Histograms show representative CFSE staining from unstimulated splenocytes (negative control), OT-1 splenocytes undergoing rapid proliferation in response to SIINFEKL (positive control), and the effect of SIINFEKL-induced T cell proliferation on splenocytes from saline or DTX-sHDL-CpG treated GL26 tumor bearing mice. (G) Quantification of splenocytes undergoing T cell proliferation. ****p < 0.0001; unpaired t-test. Bars represent mean ± SEM (n= 5 biological replicates).
We also examined the activation status CD8+ T cells within the dLN of GBM bearing mice in response to DTX-sHDL-CpG treatment. Since T cell activation is mediated by the interaction of costimulatory ligands (i.e., CD80 or CD86) with CD28 on the surface of T cells,37,38 we aimed to test whether CD8-T cells in the dLNs of tumor bearing mice treated with DTX-sHDL-CpG express CD28. We observed a ~3.8 fold (p<0.0001) increase in CD3+/CD8+/ CD28+ cells in the dLNs after DTX-sHDL-CpG (Figure 7D). We also tested the impact of DTX-sHDL-CpG treatment on the activation status of CD3+/ CD8+ T cells in the dLNs, by assessing their IFNγ and Gzb expression levels. CD8+ T cells isolated from the dLNs of mice treated with DTX-sHDL-CpG exhibited ~3.7 fold higher levels of IFNγ (p<0.00130) and ~3.1 fold higher levels of Gzb (p<0.0100), when compared to mice treated with saline (Figure 7D). This indicates that there is an expansion of activated CD8+ T cells in the dLNs of mice treated with DTX-sHDL-CpG nanodiscs.
Since the success of immunotherapy relies on the activation and expansion of tumor specific CD8+ T cells, we examined whether DTX-sHDL-CpG treatment would trigger the proliferation of splenic CD8+ T cells isolated from GL26-OVA bearing mice one day after the end of the treatment (Figure 7E). Isolated T cells were labeled with 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE), a fluorescent dye that gets incorporated into dividing daughter cells, and then stimulated with OVA cognate peptide SIINFEKL for 4 days. T cells from OT-1 mice were used as a positive control for the assay, which are engineered to recognize the SIINFEKL peptide, and almost 100% of the T cells underwent cell division in response to SIINFEKL stimulation (Figure 6E–G). we observed a ~1.5 fold (p<0.0001) increase in splenic CD8+ T cell expansion in mice treated with DTX-sHDL-CpG as opposed to the saline treated group (Figure 6E–G). Overall, our data show that DTX-sHDL-CpG nanodiscs mediate robust immune responses through tumor-specific CD8+ T cell activation and expansion.
Therapeutic Efficacy and Anti-Tumor Immunological Memory in Glioma Bearing Mice Treated with DTX-sHDL-CpG Nanodiscs in Combination with Radiation.
Since radiation therapy (IR) is the SOC for GBM patients,1 we sought out to determine if the anti-tumor efficacy elicited by DTX-sHDL-CpG monotherapy could be enhanced by combining it with radiation. Mice implanted with GL26-wt tumors in the striatum were treated with saline or DTX-sHDL-CpG in combination with IR at the indicated doses and treatment schedule (Figure 8A). We observed a ~1.5 fold (p<0.007) increase in the MS of mice after IR (MS: 43 dpi), and a ~ 2.0 fold (p<0.001) increase in the MS of mice after DTX-sHDL-CpG (MS: 55 dpi) treatment compared to the saline treated group (MS: 28 dpi) group; whereas mice in the DTX-sHDL-CpG + IR did not reach MS (Figure 8B). As described previously (Figure 5B), DTX-sHDL-CpG treatment resulted in tumor regression in 40% of the mice. Notably, DTX-sHDL-CpG in combination with IR effectively inhibited tumor growth resulting in tumor regression in 80% of the mice (Figure 8B). When the long term survivors from the DTX-sHDL-CpG and DTX-sHDL-CpG+ IR groups were rechallenged with GL26-wt tumors in the contralateral hemisphere (Figure 8A), they remained tumor free without further treatment compared to control mice implanted with tumors, which succumbed due to tumor burden (MS= 28 days) (p<0.0001). These data confirm development of robust immunological memory against tumor relapse (Figure 8C).
Figure 8: Intratumoral DTX-sHDL-CpG treatment in combination with radiation enhances survival of GBM-bearing mice.
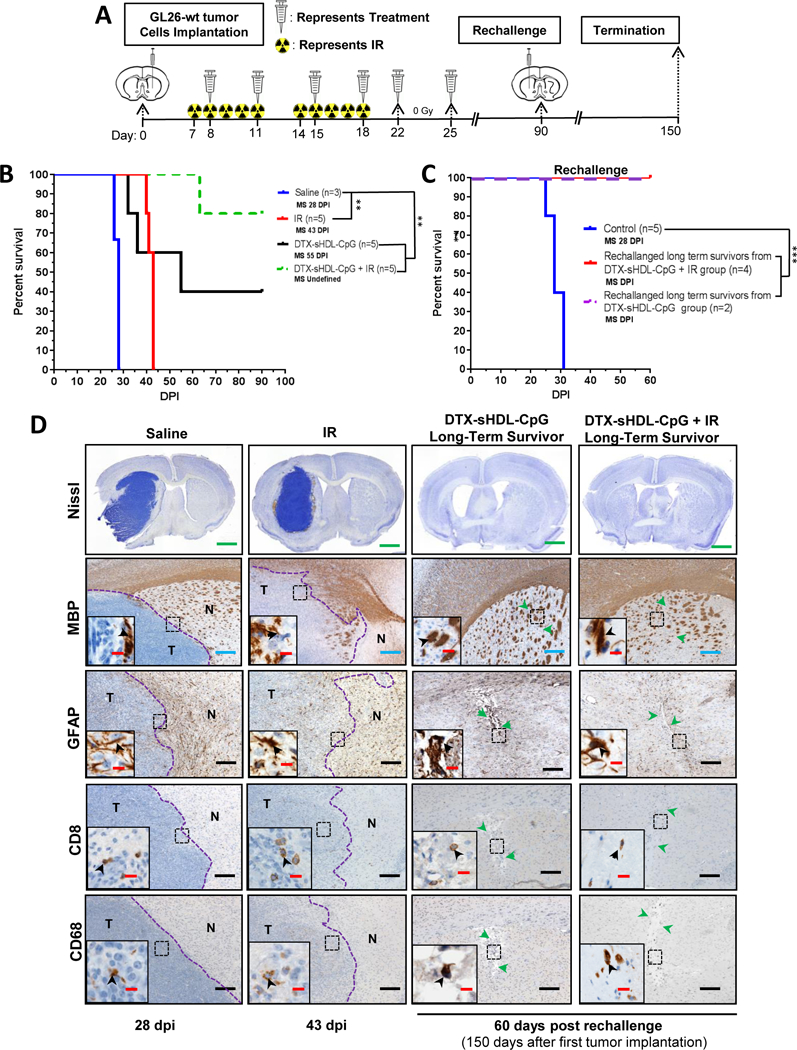
(A) Experimental design showing mice with GL26-wt tumors treated intratumorally with DTX-sHDL-CpG nanodiscs at 8d, 11d, 15d, 18d, 22d and 25d post tumor implantation along with 2 Gy/d IR for 10 days. Long-term survivors from the sHDL-CpG-DTX+ IR treatment group were rechallenged in the contralateral hemisphere with GL26-wt cells. For the control group, GL26-wt cells were implanted in naïve mice, and did not receive further treatment. (B) Kaplan-Meier survival analysis for saline (n=5), DTX-sHDL-CpG (n=5), or DTX-sHDL-CpG + IR (n=5) treated mice. (C) Kaplan-Meier survival analysis of rechallanged longterm survivors from (B) DTX-sHDL-CpG (n=2) and DTX-sHDL-CpG + IR (n=4) treatment groups and control mice (n=5). Data were analyzed using the log-rank (Mantel-Cox) test. ****p <0.0001; **p < 0.005. MS=median survival. (D) Nissl staining of 50μm brain sections from saline (28 dpi), IR (43 dpi), and long term survivors from DTX-sHDL-CpG and DTX-sHDL-CpG + IR treatment groups (60dpi after rechallenge with GL26 cells) (scale bar = 1 mm). Paraffin embedded 5μm brain sections for each treatment group were stained for myelin basic protein (MBP), glial fibrillary acidic protein (GFAP), CD8 or CD68. Low magnification panels show normal brain (N) and tumor (T) tissue (blue scale bar = 200 μm; black scale bar = 100 μm). Green arrows in the low magnifications panels indicate scar tissue. Black arrows in the high magnification panels (red scale bar = 20 μm) indicate positive staining for the areas delineated in the low magnification panels.
We also assessed the potential inflammatory response and disruption of the surrounding brain tissue due to DTX-sHDL-CpG + IR therapy. Brain architecture of mice treated with either saline, IR, DTX-sHDL-CpG or DTX-sHDL-CpG + IR was evaluated by immunohistochemistry using MBP (myelin sheaths), GFAP (astrocytes), CD8 (T cells) and CD68 (macrophages) antibodies. We observed IBA1+ microglia within the tumor and the surrounding brain parenchyma in the saline treated controls, IR alone, and around the injection site of the DTX-sHDL-CpG and DTX-sHDL-CpG + IR treated animals (Figure S4A). We also observed CD8+ T cells and CD68+ macrophages within the tumor and the surrounding brain parenchyma in saline treated controls, IR alone, and around the injection site in the DTX-sHDL-CpG and DTX-sHDL-CpG + IR long-term survivors (Figure 8D). There was no increase in immune cellular infiltrates due to the DTX-sHDL-CpG treatment. These data indicate the absence of inflammatory responses in the brain due to DTX-sHDL-CpG + IR treatment. Also, the brain architecture was preserved and there was no apparent reduction in MBP or GFAP expression, demyleinization or overt inflammation due to the DTX-sHDL-CpG, IR, or DTX-sHDL-CpG + IR treatment compared to the saline treated group, indicating the absence of neuropathological side effects (Figure 8D). Additionally, tissue sections of liver stained with H&E showed no signs of necrosis, inflammation, or changes in cellular structures due DTX-sHDL-CpG + IR treatment (Figure S4B). Enhanced therapeutic efficacy observed in this study suggests that DTX-sHDL-CpG nanodiscs in combination with IR is capable of eliciting anti-tumor mechanisms that inhibit tumor progression leading to long term survival and immunological memory.
Discussion
Devising effective treatments for patients with GBM remains a challenge in clinical neuro-oncology.39 Current standard of care (SOC) for GBM patients includes surgical resection of the tumor mass in combination with IR and adjuvant TMZ. Given the invasive nature of gliomas and their proximity to structures with important function, complete resection is not always possible.39 Aggressive tumor behavior or the presence of residual disease are risk factors for early recurrence. Furthermore, surgery and chemo-radiation confers only a modest improvement in overall survival, highlighting the need for the development of more effective and safe therapies. Potent chemotherapeutic agents historically have been unsuccessful in treating GBM. Several key factors that contribute to these agents’ ineffectiveness include poor aqueous solubility, rapid clearance, and short half-life.14
Ongoing research shows that nanoparticles (NPs) hold considerable promise for delivering active compounds to the GBM TME.40,41 NP delivery systems are capable of increasing the bioavailability of chemotherapeutic agents within the brain tissue for a prolonged time; however, their optimal physicochemical properties such as stability, biocompatibility, size, chemical composition, and off-target toxicity remains unclear. Only a few NP formulations have met regulatory approval for clinical translation.42 Furthermore, moving nano-medicine from concept to clinical approval can be extremely costly due to drug development, toxicology testing and rigorous pre-approval of the clinical trial requirements. To ameliorate some of these challenges, we used sHDL-mimicking nanodiscs for our study, which have already been developed and tested in phase I and II human clinical trials for treatment of acute coronary syndrome.19,24,43 The HDL-mimicking nanodiscs’ delivery platform is attractive for translation because of the ease of synthesis, established large-scale manufacturing, proven human safety, and non-immunogenicity after in vivo delivery.19
Other groups have used sHDL nanodiscs for drug delivery,44–46 however the biomimetic HDL used in these studies was prepared with full-length ApoA-I protein purified from plasma or produced recombinantly and combined with lipid based drugs to form HDL nanodiscs. The resulting nanodiscs were heterogeneous in size distribution and required several technically complex purification steps for removing impurities such as endotoxins, surfactants and particulates prior to in vivo administration.44–46 To avoid these technical issues, we utilized a fully synthetic ApoA-I-mimetic peptide, 22A, to prepare highly homogeneous HDL-mimicking nanodiscs with an average diameter of 8–12 nm (Figure 2). The ultra-small size of the nanodiscs could facilitate the intratumoral delivery of the drug cargo to the GBM TME, especially since the tumor cells utilize large amounts of lipids and cholesterol to proliferate.26,47
Unlike many engineered NPs, HDL-mimicking nanodiscs are capable of naturally circulating for long periods of time (~1–3 days),19 increasing the potential for drug distribution and retention in the GBM TME. Many chemotherapeutic drugs have poor aqueous solubility, extensive tissue binding and short half-lives. Therefore, incorporation of drugs into sHDL nanodiscs could improve their tumor penetration without affecting the drug efficacy.21 In order to select the most potent compound for this study, we evaluated the cytotoxicity of the chemotherapeutic drugs loaded onto HDL-mimicking nanodiscs, in various rodent and human GBM cells in vitro. Out of all the chemotherapeutic compounds tested, docetaxel (DTX) contributed to the highest GBM cell death with an IC50 = 0.0095μM (mouse glioma cells; GL26); 0.00497μM (human glioma cells; HF20303); and 0.0013μM (human glioma cells; U251) < PTX = 0.0214μM (mouse glioma cells; GL26); 0.0406μM (human glioma cells; HF2303); and 0.0046μM (human glioma cells; U251) < CCNU = 79.83μM (mouse glioma cells; GL26); 81.49μM (human glioma cells; HF2303), and 56.97μM (human glioma cells; U251). Therefore, DTX was chosen as the chemotherapeutic agent for this study (Figure 2). Mechanistically, DTX prevents mitotic cell division of tumor cells by inhibiting microtubule depolymerization.36 Furthermore, DTX has been shown to enhance antitumor T cell responses against lung cancer through eliciting immunogenic cell death (ICD).36 Under this mechanism, dying tumor cells that express calreticulin (CRT) on their surface are engulfed by DCs and macrophages.36 It has been previously demonstrated that GBM cells undergoing cell death, both in vitro and in vivo, release an endogenous damage associated molecular pattern molecule (DAMP), e.g., high-mobility group boxl (HMGB1)6 and ATP.48 HMGB1 binds to TLR2/4 and RAGE, while ATP binds to PTRX7 to elicit DC activation required for effective adaptive immune response.6,49,50 Concurrently, dying tumor cells release tumor antigens in situ into the TME. The activated DCs phagocytose the tumor antigens and migrate to the draining lymph node, where they present the antigens to immature T cells through MCH class I (Figure 1). This response triggers expansion of tumor antigen specific CD8+ T cells, which directly elicit tumor cell death.6,49,50
Immune-mediated therapies have the potential to be a powerful treatment modality for GBM.6–8 Since the GBM TME is highly immunosuppressive, various approaches have been explored to reverse this. Particularly, an oligonucleotide containing 5’-C-phospate-G-3’ (CpG), a potent TLR9 agonist, has emerged has a powerful immune stimulator for the development of long lasting tumor specific immunity by direct stimulation of DCs and macrophages in the TME.9 Also, CpG has been tested to treat patients with recurrent GBM in a phase II clinical trial. This study demonstrated that treatment with CpG resulted in a partial tumor response only in a few patients.51 The ineffectiveness of the treatment could have been due to CpG’s transportation away from the site of injection, thus interacting with immune cells located at distant sites in relation to the tumor, limiting the therapeutic efficacy of the treatment.52 The shortcoming faced by free CpG delivery has been successfully addressed by nanosystems in different tumor models.52 These nanosystems have provided the means to protect free-CpG from degradation and directly deliver it to the target immune cells within the tumor milieu, thus potentiating anti-tumor immune response.52 Therefore, in order to activate the antigen presenting cells within the GBM TME, we loaded HDL-mimicking nanodiscs with CpG.
Systemic chemotherapy treatments for GBM have been reported to promote severe immunosuppressive effects and systemic toxicity.53,54 Evidence suggests that this mode of treatment damages the bone marrow and consequently affects the proliferation and activation status of resident immune cells.55 The depletion of lymphocytes due to chemotherapy has been shown to decrease the effectiveness of immunotherapeutic agents.56,57 This remains as a major hurdle when combining multiple treatment modalities to achieve maximum clinical benefit for GBM.58 Recently, efforts have been made to identify strategies for integrating chemotherapy and immunotherapy to augment antitumor effects. Local chemotherapy in combination with immunotherapy resulted in a survival benefit with recruitment of tumor infiltrating immune cells and memory T cells into the GBM TME, which protected the animals from tumor rechallenge.8,58 Furthermore, a recent genotype-targeted molecular based treatment study demonstrated that local delivery of NPs loaded with chemotherapeutic agent at the tumor margins after surgical resection reduced the chances of tumor relapse.59 This study also revealed that local delivery of chemotherapeutic agent results in sustained release of the drug formulation at the tumor site treating residual tumor cells while avoiding adverse local or systemic toxicity.59
The results reported herein demonstrate a promising therapeutic potential for the use of HDL-mimicking nanodiscs loaded with DTX and CpG to treat GBM. Intratumoral delivery of DTX-sHDL-CpG (MS: 55 dpi) displayed improved efficacy compared to DTX-CpG (MS: 41 dpi). Additionally, combining DTX-sHDL-CpG nanodisc treatment with IR resulted in further increased therapeutic efficacy (MS: not reached). We observed 80% long term survival in the DTX-sHDL-CpG + IR treatment group. In addition, despite no further treatment, long term survivors did not succumb to tumor rechallenge (Figure 8C). Chemoimmunotherapy in combination with radiation has been shown to increase danger signals (DAMPs) associated with ICD within the tumor and broaden epitope recognition by tumor-associated immune cells resulting in a robust anti-tumor T cell response.60,61 Our results indicate that local tumor delivery of DTX-sHDL-CpG nanodiscs in combination with IR elicits effective anti-tumor response and promotes lasting immunological memory to prevent GBM recurrence.
The role of memory T cells in promoting immunological memory after immunotherapy is critical for preventing tumor recurrence. Our proposed strategy consisting of targeting the GBM TME with local delivery of DTX-sHDL-CpG nanodiscs elicits tumor cell death through immune-chemotherapy, and also elicits immunological memory against tumor relapse (Figures 5,7). Our findings have direct implications on the development of an effective adjuvant treatment for GBM.
The work presented here is, to the best of our knowledge, the first report based on sHDL chemo-immunotherapy to elicit immunogenic GBM cell death resulting in long term survival and anti-GBM specific immunological memory in an intracranial syngeneic mouse glioma model. Our strategy could be readily applied to other chemotherapeutic agents known to induce ICD.62–64 There is strong interest in improving GBM patients’ response rate and therapeutic efficacy of immunotherapy combined with chemotherapy or radiation. The strategy presented in this study may have a wide-ranging impact in the field of drug delivery, nanotechnology, and brain cancer chemo-immunotherapy. It also has strong potential for translation to Phase I clinical trials for GBM.
Conclusions
We have optimized the use of HDL-mimicking nanodiscs loaded with DTX, a chemotherapeutic agent known to be active against GBM cells. We demonstrate that intratumoral delivery of the DTX-sHDL-CpG nanodiscs results in a significant increase in median survival with no overt off target systemic toxicity. In addition, we report that modifying HDL-mimicking nanodiscs with ligands that stimulate immune responses, such as CpG, results in immune mediated anti-GBM activity. We also demonstrate that the anti-tumor efficacy elicited by DTX-sHDL-CpG nanodiscs loaded with a chemotherapeutic agent can be enhanced in combination with radiation. Of note, DTX-sHDL-CpG nanodiscs elicited anti-tumor immunological memory that prevents tumor recurrence. Taken together, our results demonstrate that DTX-sHDL-CpG has the potential for effective clinical translation as a treatment option for patients with GBM.
Materials and Methods
Reagents
Paclitaxel (PTX), docetaxel (DTX) and lomustine (CCNU) were purchased from Sigma-Aldrich with purity over 99% (St. Louis, MO). ApoA-I mimetic peptides 22A (PVLDLFRELLNELLEALKQKLK) were synthesized by Genscript Inc. (Piscataway, NJ). The purity of peptide was determined to be over 95% by the reverse phase HPLC. 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and egg sphingomyelin (SM) were purchased from Avanti Polar Lipids (Alabaster, AL). Fluorescent dyes (DiO and DiR) were purchased from Invitrogen (Carlsbad, CA). Additional reagents used were of analytical grade and obtained from commercial suppliers.
Preparation and Characterization of Drug-Loaded sHDL and Dye-Loaded sHDL
The drug loaded synthetic HDL (sHDL) was prepared by lyophilization method. Briefly, 22A peptide, lipids and anti-cancer drugs with various weight ratios were dissolved and well mixed in glacial acetic acid, which was then removed by freeze-drying method. Lyophilized product was hydrated using 1M phosphate-buffered saline (PBS, pH 7.4) followed by thermal cycling varying from 50 °C to 20 °C for a minimum of 3 heat and cool cycles (10 min each) applying gentle shaking to obtain homogenous HDL-mimicking nanodiscs. Fluorescent dye (DiD or DiR) loaded sHDL was prepared by the same process. DTX-sHDL-CpG was prepared by incubating CpG-cholesterol with DTX-sHDL solution at room temperature for 4 h after DTX-sHDL preparation.
Gel permeation chromatography (GPC) was used to separate particles based on their sizes. The purity and homogeneity of prepared sHDL was calculated by dividing area under the curve of sHDL to the total chromatography peaks’ area using a Shimadzu HPLC system equipped with a TSKgel G2000SWxl column (7.8 mm ID × 30 cm, Tosoh Bioscience LLC) and the detection wavelengths were set at 220 nm for quantification of 22A peptide. The particle size of drug-sHDL was measured by dynamic light scattering (DLS) on a Malvern Zetasizer (Westborough, MA). The sHDL morphology was assessed by transmission electron microscopy (TEM) after proper dilution of the original samples. The diluted sample solution was deposited on a carbon film-coated 400 mesh copper grid (Electron Microscopy Sciences) and dried for 1 min. Samples were then negatively stained with 1% (w/v) uranyl formate, and the grid was dried before TEM observation. All specimens were imaged on a 100kV Morgagni TEM equipped with a Gatan Orius CCD.
Stability Study
In vitro study was performed to quantify the stability of DTX-sHDL particles for formulation screening purpose. Briefly, different formulations of DTX-sHDL were suspended in PBS or human serum and incubated at 37°C with the DTX concentration of 1 mg/ml. At 0, 0.5, 1,2, 4, 8, 24 h after incubation, 100 μl mixture of each sample was collected and filtered through 0.22 μm membrane to separate precipitated drug. After filtration, 50 μl of each sample was mixed with 450 μl acetonitrile to dissolve the all component of the nanodiscs and precipitate proteins. After centrifuge, the drug content incorporated in sHDL was determined by UPLC analysis.
Cell Line and Cell Culture Conditions
Mouse, GL26-WT, GL26-Cit, GL26-OVA, rat CNS-1, and human HF2303, U251 glioblastoma cells were grown in Dulbecco’s modified eagle (DMEM) media supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 0.3 mg/mL L-glutamine. For mCitrne or OVA selection, medium was additionally supplemented with 6pg/mL G418. Cells were maintained in a humidified incubator at 95% air/5% CO2 at 37°C and passaged every 2–4 days.
Animal Strains
Six to eight week old female C57BL/6 and CD8 knockout mice were purchased from Jackson Laboratory (Bar Harbor, ME) and were housed in pathogen free conditions at the University of Michigan. All experimental studies were performed in compliance with Institutional Animal Care & Use Committee (IACUC).
Intracranial GBM Models
Syngeneic tumors were established in C57BL/6 mice by stereotactically injecting 20,000 GL26-WT, GL26-Cit or 60,000 GL26-OVA cells into the right striatum using a 22-gauge Hamilton syringe (1 μL over 1 minute) with the following coordinates: +1.00 mm anterior, 2.5 mm lateral, and 3.00 mm deep.
Nanodisc Uptake Assay
To determine the cellular uptake of sHDL by GBM cells, 50,000 GL26-Cit, CNS1-Cit or HF2303-Cit cells per well were plated onto glass cover slips coated with poly-L lysine in a 24-well plate 12 hours before treatment. Cells were then treated with 0ug, 3ug, 10ug and 30ug of DiD-HDL in 0.4mL of fully supplemented DMEM for 2 hours. Cells were washed three times with PBS and fixed in 4% Paraformaldehyde (PFA) for 30 minutes and mounted onto slides with ProLong® Gold anti-fade (Thermo Fisher Scientific, Life technologies™, P36930). DiD-sHDL uptake was imaged with confocal microscopy (Carl Zeiss: MIC System) at 63× with oil-immersion lens and quantified using image J software.
Cytotoxicity Assay
Human, and mouse GBM cells were plated at 1,000 cells per well in 96-well plate 24 h prior to treatment. Cells were then incubated with increasing concentrations (0.01uM-300uM) of free-sHDL, free-CCNU, PTX, DTX; HDLs loaded with CCNU, PTX, and DTX for 48 h Cell viability was determined with CellTiter-Glo viability assay following manufacture’s protocol. IC50 values for each chemotherapeutic reagent were calculated from dose-response curves generated using graphpad prism.
Biodistribution
In order to qualitatively evaluate the biodistribution of HDL-mimicking nanodiscs in vivo, fluorescent dye DiR was loaded into the HDL-mimicking nanodiscs, which were administered intratumorally (i.t.) into normal or tumor bearing mice. DiR-loaded sHDL was prepared with methods mentioned above. DiR-sHDL was diluted with PBS (pH=7.4) to 20 μg/ml of DiR before injection. Twenty one days post GL26-wt tumor or saline implantation, mice (n=5/ group) injected with 0.5 mg/Kg DiR-sHDL in 5 μL volume. From each group, mice were transcardially perfused at 24 h, and heart, thymus, lungs, spleen, liver, brain, and kidneys were harvested. Fluorescent signal within each organ was measured with IVIS spectrum analysis.
To assess HDL-mimicking nanodiscs’ accumulation within the GBM tumor microenvironment, fluorescent dye DiD was loaded into the HDL-mimicking nanodiscs, which were administered it into GBM bearing mice. DiD-loaded sHDL was prepared with methods mentioned above. DiD-sHDL was diluted with PBS (pH=7.4) to 20 μg/ml of DiD before injection. Seven days post GL26-cit tumor implantation, mice (n=3/group) were it injected with 0.5 mg/Kg DiD-sHDL in 5 μL volume. From each group, mice were transcardially perfused at 24 h, and brains were processed for imaging. DiD-sHDL accumulation within the TME was imaged with confocal microscopy (Carl Zeiss: MIC System) at 63× with oil-immersion lens.
Therapeutic Study in Tumor Bearing Animals
To evaluate the therapeutic efficacy of HDL-mimicking nanodiscs loaded with DTX and CpG, saline, 0.5 mg/kg of free-DTX, CpG-DTX, free-sHDL, DTX-sHDL, or sHDL-CpG-DTX loaded HDL-mimicking nanodiscs were administered in a 5 μL volume intratumorally into GL26 tumor bearing mice on 8, 11, 15, 18, 22 and 25 days post tumor implantation. Each treatment group consisted of at least n=5 mice. When mice displayed signs of neurological deficits, they were transcardiacly perfused with Tyrode’s solution and 4% PFA.
Radiotherapy
Eight days post GL26-wt tumor cells’ implantation, a dose of 2 Gy Irradiation (IR) was administered to mice 5 days a week for two weeks.
Immunohistochemistry
Some of the PFA-fixed brains were serially sectioned 50 μm thick using the vibratome system and placed consecutively into six wells (in a 12-well tissue culture plate containing 2 mL of PBS with 0.01% sodium azide), where each well contained sections representing the whole brain. Some PFA-fixed brains were paraffin embedded and serially sectioned 5 μm thick using a microtome. Sections from each mouse were permeabilized with TBS-0.5% Triton-X (TBS-Tx) for 20 min. This was followed by antigen retrieval at 96 °C with 10 mM sodium citrate (pH 6) for an additional 20 min. Then, the sections were cooled at room temperature (RT) and washed 5 times with TBS-Tx (5 min per wash) and blocked with 10% goat serum in TBS-Tx for 1 h at RT. Brain sections were incubated in primary antibody Ki-67 (Abcam, ab15580, 1:1000), cleaved caspase 3 (Cell Signaling, 9661, 1:400), GFAP (Millipore, AB5541, 1:1000), MBP (Millipore, MAB386, 1:500), IBA1 (Abcam, ab178846 1:2000), CD8 (Cedarlane, 361003, 1:2000), or CD68 (Abcam, ab125212m 1:200) diluted in 1% goat serum TBS-Tx overnight at RT. The next day sections were washed with TBS-Tx 5 times. Brain sections labeled with Ki67 or CC3 were incubated in fluorescent-dye conjugated secondary antibody, while brain sections labeled with GFAP, MBP, CD8 or CD68 were incubated with HRP secondary antibody, which were diluted in 1% goat serum TBS-Tx in the dark for 4 h. Fluorescently labeled sections were washed in PBS 3 times and mounted onto microspore slides and coverslipped with ProLong Gold. HRP labeled sections were subjected to 3, 3′-diaminobenzidine (DAB) (Biocare Medical) with nickel sulfate precipitation. The reaction was quenched with 10% sodium azide; sections were washed 3 times in 0.1 M sodium acetate followed by dehydration in xylene, and coverslipped with DePeX Mounting Medium (Electron Microscopy Sciences). High magnification images at 63X were obtained using confocal microscopy (Carl Zeiss: MIC-System) and stains were quantified using ImageJ software.
For tumor size quantification, one well per mouse was stained with Nissl as described previously.65 Sections comprising of tumor (approximately 10–12 sections per mouse) were imaged using the brightfield (Olympus BX53) setting and tumor size was quantified using ImageJ’s Otsu threshold to determine the tumor size in pixels.
For histological assessment, livers were embedded in paraffin, sectioned 5μm thick using the microtome system and H&E stained as described by us previously.65 Brightfield images were obtained using Olympus MA BX53 microscope.
Flow Cytometry
For flow cytometry analysis of the cells within the TME and draining lymph nodes of GL26 tumor bearing mice, one day post DTX-sHDL-CpG or saline treatment, mice were euthanized and brains were extracted. Tumor mass within the brain and draining lymph nodes were carefully dissected and homogenized using Tenbroeck (Corning) homogenizer in DMEM media containing 10% FBS. Then, tumor infiltrating immune cells was enriched with 30%70% Percoll (GE Lifesciences) density gradient. These cells were resuspended in PBS containing 2% FBS (flow buffer) and non-specific antibody binding was blocked with CD16/CD32. Dendritic cells were labeled with CD45, CD11c, CD80, CD86, MHC II and B220 antibodies. Macrophages were labeled with CD45, F4/80 and CD206 antibodies. Tumor specific T cells were labeled with CD45, CD3, CD8 and SIINFEKL-H2Kb-tetramer. Activated T cells in the draining lymph node were labeled with CD45, CD3, CD8 and CD28 antibodies. For identifying DAMPs in the tumor microenvironment, tumor mass was dissociated to single cell suspension and CD45 cells were labeled with magnetic beads (Miltenyi) using the manufactures’ instructions at 4 °C. Purified cells were washed and passed through a preconditioned MS column placed in the magnetic field of a MACS separator. Cells that were negative for CD45 were collected, resuspeneded in flow buffer and labeled with CRT and HMGB1 antibodies for flow cytometry analysis. Live/dead staining was carried out using fixable viability dye (eBioscience). Intracellular Granzyme B and IFNγ were stained using BD intracellular staining kit using the manufacturer’s instructions. All stains were carried out for 30min at 4°C with 3X flow buffer washes between live/dead staining, blocking, surface staining, cell fixation, intracellular staining and data acquisition. For T cell functional analysis, purified immune cells from the TME were stimulated with 100 μg/mL of GL26-OVA lysate for 24 h in DMEM media containing 10% FBS followed by 6 hs incubation with Brefeldin and monensin. Flow data has been measured with FACSAria flow cytometer (BD Bioscience) and analyzed using Flow Jo version 10 (Treestar).
T Cell Proliferation Analysis
Splenocytes from 26 dpi GL26-wt tumor bearing mice treated with saline or DTX-sHDL-CpG were CFSE labeled and cultured with 100 nM SIINFEKL peptide (Anaspec) for 4 days. Unstimulated splenocytes were used as negative control and splenocytes from Rag2 knockout/transgenic OT-I T cell receptor mice (Taconic) stimulated with SIINFEKL were used as a positive control. Then cells were stained with CD3 and CD8 antibodies in flow buffer as detailed above, and T cell proliferation was assessed based on CFSE dye dilution.
Analysis of Local and Distant GBM Recurrence in Patients
In order to assess the incidence of local versus distant disease progression after surgery for GBM, a retrospective analysis of patients’ medical records was performed. Fifty consecutive patients who underwent surgery for solitary GBM at the University of Michigan were selected for inclusion. The most recently treated patients with 3 years of complete follow up were included for analysis. Following surgery, serial MRIs were performed to monitor for disease recurrence. If and when disease progression occurred, it was noted to be local if gadolinium contrast enhancing disease was located immediately adjacent to the surgical resection cavity or distant if it was located more than 2 cm away with normal appearing, non-enhancing brain parenchyma between. The percentage of patients with local versus distant disease progression was calculated accordingly. The University of Michigan Institutional Review Board approved the study protocol.
Statistical Analysis
Sample sizes were chosen based on preliminary data from pilot experiments and previously published results in the literature. All animal studies were performed after randomization. Data were analyzed by one-or two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons post-test or log rank (Mantel-Cox) test with Prism 6.0 (GraphPad Software). Data were normally distributed and variance between groups was similar. P values less than 0.05 were considered statistically significant. All values are reported as means ± SD with the indicated sample size. No samples were excluded from analysis.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) Grants R21-NS091555 to M.G.C., A.S. and PRL; R37NS094804, and R01NS074387 to M.G.C.; R01NS076991, R01NS082311, and R01NS096756 to P.R.L; National Institutes of Health/National Cancer Institute (NIH/NIC) Grant T32–0CA009676 to M.G.C; National institutes of Health/National Institute of Biomedical Imaging and Bioengineering (NIH/NIBIB) Grant R01-EB022563 to J.J.M., P.R.L, and M.G.C.;
American Heart Association (AHA) Pre-doctoral Fellowship 15PRE25090050 to R.K.; Broomfield International Student Fellowship to R.K. and D.L.; Barbour Fellowship to D.L.; University of Michigan M-Cube; the Center for RNA Biomedicine; the Department of Neurosurgery; the University of Michigan Rogel Comprehensive Cancer Center; Leah’s Happy Hearts Foundation; and the Biointerfaces Institute at the University of Michigan.
Footnotes
Associated Content
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.
Table S1: Characterization summary of different 22A sHDL-mimicking nanodiscs, Figure S1: Cytotoxicity of chemotherapeutic loaded HDL-mimicking nanodiscs, Figure S2: Intratumoral DTX-sHDL-CpG treatment in CD8 knockout mice, Figure S3: Chemo-immunotherapy enhances macrophage responses within GBM TME, and Figure S4:mo-immunotherapy as described in the text (PDF)
References
- (1).Alifieris C; Trafalis DT Glioblastoma Multiforme: Pathogenesis and Treatment. Pharmacol. Ther. 2015, 152, 63–82. [DOI] [PubMed] [Google Scholar]
- (2).Forsyth P; Cairncross G; Stewart D; Goodyear M; Wainman N; Eisenhauer E Phase II Trial of Docetaxel in Patients with Recurrent Malignant Glioma: A Study of the National Cancer Institute of Canada Clinical Trials Group. Invest. New Drugs 1996, 14, 203–206. [DOI] [PubMed] [Google Scholar]
- (3).Prados MD; Schold SC; Spence AM; Berger MS; McAllister LD; Mehta MP; Gilbert MR; Fulton D; Kuhn J; Lamborn K; Rector DJ; Chang SM Phase II Study of Paclitaxel in Patients with Recurrent Malignant Glioma. J. Clin. Oncol. 1996, 14, 2316–2321. [DOI] [PubMed] [Google Scholar]
- (4).Jakobsen JN; Urup T; Grunnet K; Toft A; Johansen MD; Poulsen SH; Christensen IJ; Muhic A; Poulsen HS Toxicity and Efficacy of Lomustine and Bevacizumab in Recurrent Glioblastoma Patients. J. Neurooncol. 2018, 137, 439–446. [DOI] [PubMed] [Google Scholar]
- (5).Kalepu S; Nekkanti V Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta. Pharm. Sin. B 2015, 5, 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Curtin JF; Liu N; Candolfi M; Xiong W; Assi H; Yagiz K; Edwards MR; Michelsen KS; Kroeger KM; Liu C; Muhammad AK; Clark MC; Arditi M; Comin-Anduix B; Ribas A; Lowenstein PR; Castro MG HMGB1 Mediates Endogenous TLR2 Activation and Brain Tumor Regression. PLoSMed. 2009, 6, 83–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kamran N; Kadiyala P; Saxena M; Candolfi M; Li Y; Moreno-Ayala MA; Raja N; Shah D; Lowenstein PR; Castro MG Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Mol. Ther. 2017, 25, 232–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Candolfi M; Yagiz K; Wibowo M; Ahlzadeh GE; Puntel M; Ghiasi H; Kamran N; Paran C; Lowenstein PR; Castro MG Temozolomide Does Not Impair Gene Therapy-Mediated Antitumor Immunity in Syngeneic Brain Tumor Models. Clin. Cancer Res. 2014, 20, 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Carpentier AF; Auf G; Delattre JY CpG-Oligonucleotides for Cancer Immunotherapy : Review of the Literature and Potential Applications in Malignant Glioma. Front. Biosci. 2003, 8, 115–127. [DOI] [PubMed] [Google Scholar]
- (10).Fan H; Zhang I; Chen X; Zhang L; Wang H; Da Fonseca A; Manuel ER; Diamond DJ; Raubitschek A; Badie B Intracerebral CpG Immunotherapy with Carbon Nanotubes Abrogates Growth of Subcutaneous Melanomas in Mice. Clin. Cancer Res. 2012, 18, 5628–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lollo G; Vincent M; Ullio-Gamboa G; Lemaire L; Franconi F; Couez D; Benoit JP Development of Multifunctional Lipid Nanocapsules for the Co-Delivery of Paclitaxel and CpG-ODN in the Treatment of Glioblastoma. Int. J. Pharm. 2015, 495, 972–980. [DOI] [PubMed] [Google Scholar]
- (12).Zhao D; Alizadeh D; Zhang L; Liu W; Farrukh O; Manuel E; Diamond DJ; Badie B Carbon Nanotubes Enhance CpG Uptake and Potentiate Antiglioma Immunity. Clin. Cancer Res. 2011, 17, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Parrish KE; Sarkaria JN; Elmquist WF Improving Drug Delivery to Primary and Metastatic Brain Tumors: Strategies to Overcome the Blood-Brain Barrier. Clin. Pharmacol. Ther. 2015, 97, 336–346. [DOI] [PubMed] [Google Scholar]
- (14).van Tellingen O; Yetkin-Arik B; de Gooijer MC; Wesseling P; Wurdinger T; de Vries HE Overcoming the Blood-Brain Tumor Barrier for Effective Glioblastoma Treatment. Drug Resist. Updat. 2015, 19, 1–12. [DOI] [PubMed] [Google Scholar]
- (15).Zhang F; Xu CL; Liu CM Drug Delivery Strategies to Enhance the Permeability of the Blood-Brain Barrier for Treatment of Glioma. Drug Des. Devel. Ther. 2015, 9, 2089–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gille A; Easton R; D’Andrea D; Wright SD; Shear CL CSL112 Enhances Biomarkers of Reverse Cholesterol Transport after Single and Multiple Infusions in Healthy Subjects. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2106–2114. [DOI] [PubMed] [Google Scholar]
- (17).Vickers KC; Palmisano BT; Shoucri BM; Shamburek RD; Remaley AT MicroRNAs Are Transported in Plasma and Delivered to Recipient Cells by High-Density Lipoproteins. Nat. Cell. Biol. 2011, 13, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang M; Briggs MR HDL: The Metabolism, Function, and Therapeutic Importance. Chem. Rev. 2004, 104, 119–137. [DOI] [PubMed] [Google Scholar]
- (19).Kuai R; Li D; Chen YE; Moon JJ; Schwendeman A High-Density Lipoproteins: Nature’s Multifunctional Nanoparticles. ACS Nano 2016, 10, 3015–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Weiss HM; Fresneau M; Moenius T; Stuetz A; Billich A Binding of Pimecrolimus and Tacrolimus to Skin and Plasma Proteins: Implications for Systemic Exposure after Topical Application. DrugMetab. Dispos. 2008, 36, 1812–1818. [DOI] [PubMed] [Google Scholar]
- (21).Yuan Y; Wen J; Tang J; Kan Q; Ackermann R; Olsen K; Schwendeman A Synthetic High-Density Lipoproteins for Delivery of 10-Hydroxycamptothecin. Int. J. Nanomedicine 2016, 11, 6229–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sykes E; Woodburn K; Decker D; Kessel D Effects of Cremophor EL on Distribution of Taxol to Serum Lipoproteins. Br. J. Cancer 1994, 70, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Krause BR; Remaley AT Reconstituted HDL for the Acute Treatment of Acute Coronary Syndrome. Curr. Opin. Lipidol. 2013, 24, 480–486. [DOI] [PubMed] [Google Scholar]
- (24).Li D; Gordon S; Schwendeman A; Remaley AT Apolipoprotein Mimetic Peptides for Stimulating Cholesterol Efflux In Apolipoprotein Mimetics in the Management of Human Disease, Anantharamaiah GM; Goldberg D, Eds. Springer International Publishing: Cham, 2015; pp 29–42. [Google Scholar]
- (25).Glickson JD; Lund-Katz S; Zhou R; Choi H; Chen IW; Li H; Corbin I; Popov AV; Cao W; Song L; Qi C; Marotta D; Nelson DS; Chen J; Chance B;Zheng G Lipoprotein Nanoplatform for Targeted Delivery of Diagnostic and Therapeutic Agents. Molecular Imaging 2008, 7, 101–110. [PubMed] [Google Scholar]
- (26).Tang J; Kuai R; Yuan W; Drake L; Moon JJ; Schwendeman A Effect of Size and Pegylation of Liposomes and Peptide-Based Synthetic Lipoproteins on Tumor Targeting. Nanomedicine 2017, 13, 1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zou L; Tao Y; Payne G; Do L; Thomas T; Rodriguez J; Dou H Targeted Delivery of Nano-PTX to the Brain Tumor-Associated Macrophages. Oncotarget 2017, 8, 6564–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li AJ; Zheng YH; Liu GD; Liu WS; Cao PC; Bu ZF Efficient Delivery of Docetaxel for the Treatment of Brain Tumors by Cyclic RGD-Tagged Polymeric Micelles. Mol. Med. Rep. 2015, 11, 3078–3086. [DOI] [PubMed] [Google Scholar]
- (29).Banks WA From Blood-Brain Barrier to Blood-Brain Interface: New Opportunities for CNS Drug Delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [DOI] [PubMed] [Google Scholar]
- (30).Guo Y; Yuan W; Yu B; Kuai R; Hu W; Morin EE; Garcia-Barrio MT; Zhang J; Moon JJ; Schwendeman A; Eugene Chen Y Synthetic High-Density Lipoprotein-Mediated Targeted Delivery of Liver X Receptors Agonist Promotes Atherosclerosis Regression. EBioMedicine 2018, 28, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kuai R; Subramanian C; White P; N Timmermann B; Moon J; Cohen M; Schwendeman A Synthetic High-Density Lipoprotein Nanodisks for Targeted Withalongolide Delivery to Adrenocortical Carcinoma. Int. J. Nanomedicine 2017, 12, 6581–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Candolfi M; Curtin JF; Nichols WS; Muhammad AG; King GD; Pluhar GE; McNiel EA; Ohlfest JR; Freese AB; Moore PF; Lerner J; Lowenstein PR; Castro MG Intracranial Glioblastoma Models in Preclinical Neuro-Oncology: Neuropathological Characterization and Tumor Progression. J. Neurooncol. 2007, 85, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Persaud-Sharma D; Burns J; Trangle J; Moulik S Disparities in Brain Cancer in the United States: A Literature Review of Gliomas. MedSci (Basel) 2017, 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Urbanska K; Sokolowska J; Szmidt M; Sysa P Glioblastoma Multiforme-an Overview. Contemp. Oncol. (Pozn) 2014, 18, 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nduom EK; Weller M; Heimberger AB Immunosuppressive Mechanisms in Glioblastoma. Neuro. Oncol. 2015, 17, vii9–vii14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ishii H; Chikamatsu K; Igarashi S; Takahashi H; Sakamoto K; Higuchi H; Tanaka S; Matsuoka T; Masuyama K Establishment of Synergistic Chemoimmunotherapy for Head and Neck Cancer Using Peritumoral Immature Dendritic Cell Injections and Low-Dose Chemotherapies. Transl. Oncol. 2018, 11, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rudd CE; Taylor A; Schneider H CD28 and CTLA-4 Coreceptor Expression and Signal Transduction. Immunol. Rev. 2009, 229, 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lenschow DJ; Walunas TL; Bluestone JA CD28/B7 System of T Cell Costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [DOI] [PubMed] [Google Scholar]
- (39).Mallick S; Benson R; Hakim A; Rath GK Management of Glioblastoma after Recurrence: A Changing Paradigm. Journal of the Egyptian National Cancer Institute 2016, 28, 199–210. [DOI] [PubMed] [Google Scholar]
- (40).Jain KK Use of Nanoparticles for Drug Delivery in Glioblastoma Multiforme. Expert Rev. Neurother. 2007, 7, 363–372. [DOI] [PubMed] [Google Scholar]
- (41).Wegscheid ML; Morshed RA; Cheng Y; Lesniak MS The Art of Attraction: Applications of Multifunctional Magnetic Nanomaterials for Malignant Glioma. Expert Opin. DrugDeliv. 2014, 11, 957–975. [DOI] [PubMed] [Google Scholar]
- (42).Bobo D; Robinson KJ; Islam J; Thurecht KJ; Corrie SR Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [DOI] [PubMed] [Google Scholar]
- (43).Kingwell BA; Chapman MJ; Kontush A; Miller NE HDL-Targeted Therapies: Progress, Failures and Future. Nat. Rev. DrugDiscov. 2014, 13, 445–464. [DOI] [PubMed] [Google Scholar]
- (44).Duivenvoorden R; Tang J; Cormode DP; Mieszawska AJ; Izquierdo-Garcia D; Ozcan C; Otten MJ; Zaidi N; Lobatto ME; van Rijs SM; Priem B; Kuan EL; Martel C; Hewing B; Sager H; Nahrendorf M; Randolph GJ; Stroes ES; Fuster V; Fisher EA; et al. A Statin-Loaded Reconstituted High-Density Lipoprotein Nanoparticle Inhibits Atherosclerotic Plaque Inflammation. Nat. Commun. 2014, 5, 3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang X; Chen B Recombinant High Density Lipoprotein Reconstituted with Apolipoprotein AI Cysteine Mutants as Delivery Vehicles for 10-Hydroxycamptothecin. Cancer Lett. 2010, 298, 26–33. [DOI] [PubMed] [Google Scholar]
- (46).Mooberry LK; Nair M; Paranjape S; McConathy WJ; Lacko AG Receptor Mediated Uptake of Paclitaxel from a Synthetic High Density Lipoprotein Nanocarrier. J. Drug Target 2010, 18, 53–58. [DOI] [PubMed] [Google Scholar]
- (47).Guo D; Bell EH; Chakravarti A Lipid Metabolism Emerges As a Promising Target for Malignant Glioma Therapy. CNS Oncol. 2013, 2, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Vénéreau E; Ceriotti C; Bianchi M Damps from Cell Death to New Life. Front. Immunol. 2015, 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yamazaki T; Hannani D; Poirier-Colame V; Ladoire S; Locher C; Sistigu A; Prada N; Adjemian S; Catani JP; Freudenberg M; Galanos C; André F; Kroemer G; Zitvogel L Defective immunogenic cell death of HMGBl-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ. 2014, 21, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dumitriu IE; Bianchi ME; Bacci M; Manfredi AA; Rovere-Querini P The Secretion of HMGB1 is Required for the Migration of Maturing Dendritic Cells. J. Leukoc. Biol. 2007, 81, 84–91. [DOI] [PubMed] [Google Scholar]
- (51).Carpentier A; Metellus P; Ursu R; Zohar S; Lafitte F; Barrie M; Meng Y; Richard M; Parizot C; Laigle-Donadey F; Gorochov G; Psimaras D; Sanson M; Tibi A; Chinot O; Carpentier AF Intracerebral Administration of CpG Oligonucleotide for Patients with Recurrent Glioblastoma: A Phase II Study. Neuro. Oncol. 2010, 12, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Badie B; Berlin JM The Future of CpG Immunotherapy in Cancer. Immunotherapy 2013, 5, 1–3. [DOI] [PubMed] [Google Scholar]
- (53).Brooks WH; Netsky MG; Normansell DE; Horwitz DA Depressed Cell-Mediated Immunity in Patients with Primary Intracranial Tumors. Characterization of a Humoral Immunosuppressive Factor. J. Exp. Med. 1972, 136, 1631–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Grossman SA; Ye X; Lesser G; Sloan A; Carraway H; Desideri S; Piantadosi S Immunosuppression in Patients with High-Grade Gliomas Treated with Radiation and Temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lombardi G; Rumiato E; Bertorelle R; Saggioro D; Farina P; Della Puppa A; Zustovich F; Berti F; Sacchetto V; Marcato R; Amadori A; Zagonel V Clinical and Genetic Factors Associated with Severe Hematological Toxicity in Glioblastoma Patients during Radiation plus Temozolomide Treatment: A Prospective Study. Am. J. Clin. Oncol. 2015, 38, 514–519. [DOI] [PubMed] [Google Scholar]
- (56).van der Most RG; Robinson BW; Lake RA Combining Immunotherapy with Chemotherapy to Treat Cancer. Discov. Med. 2005, 5, 265–270. [PubMed] [Google Scholar]
- (57).Jackson C; Ruzevick J; Brem H; Lim M Vaccine Strategies for Glioblastoma: Progress and Future Directions. Immunotherapy 2013, 5, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mathios D; Kim JE; Mangraviti A; Phallen J; Park CK; Jackson CM; Garzon-Muvdi T; Kim E; Theodros D; Polanczyk M; Martin AM; Suk I; Ye X; Tyler B; Bettegowda C; Brem H; Pardoll DM; Lim M Anti-PD-1 Antitumor Immunity is Enhanced by Local and Abrogated by Systemic Chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Shankar GM; Kirtane AR; Miller JJ; Mazdiyasni H; Rogner J; Tai T; Williams EA; Higuchi F; Juratli TA; Tateishi K; Koerner MVA; Tummala SS; Fink AL; Penson T; Schmidt SP; Wojtkiewicz GR; Baig A; Francis JM; Rinne ML; Batten JM; et al. Genotype-Targeted Local Therapy of Glioma. Proc. Natl. Acad. Sci. USA 2018. 115, E8388–E8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Mann J; Ramakrishna R; Magge R; Wernicke AG Advances in Radiotherapy for Glioblastoma. Frontiers in Neurology 2018, 8, 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Wu Q; Allouch A; Martins I; Brenner C; Modjtahedi N; Deutsch E; Perfettini J-L Modulating Both Tumor Cell Death and Innate Immunity is Essential for Improving Radiation Therapy Effectiveness. Frontiers in Immunology 2017, 8, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kroemer G; Galluzzi L; Kepp O; Zitvogel L Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [DOI] [PubMed] [Google Scholar]
- (63).Obeid M; Tesniere A; Ghiringhelli F; Fimia GM; Apetoh L; Perfettini JL; Castedo M; Mignot G; Panaretakis T; Casares N; Metivier D; Larochette N; van Endert P; Ciccosanti F; Piacentini M; Zitvogel L; Kroemer G Nat. Med. 2007, 13, 54–61. [DOI] [PubMed] [Google Scholar]
- (64).Apetoh L; Ghiringhelli F; Tesniere A; Obeid M; Ortiz C; Criollo A; Mignot G; Maiuri MC; Ullrich E; Saulnier P; Yang H; Amigorena S; Ryffel B; Barrat FJ; Saftig P; Levi F; Lidereau R; Nogues C; Mira JP; Chompret A; et al. Nat. Med 2007, 13, 1050–1059. [DOI] [PubMed] [Google Scholar]
- (65).Calinescu AA; Yadav VN; Carballo E; Kadiyala P; Tran D; Zamler DB; Doherty R; Srikanth M; Lowenstein PR; Castro MG Survival and Proliferation of Neural Progenitor-Derived Glioblastomas under Hypoxic Stress is Controlled by a CXCL12/CXCR4 Autocrine-Positive Feedback Mechanism. Clin. Cancer Res. 2017, 23, 1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.