

Abstract
Kinases are crucial components in numerous cell signaling pathways. Aberrant expression and activation of protein kinases are known to be accompanied by many types of cancer, and more than 30 small-molecule kinase inhibitors have been approved by the Food and Drug Administration (FDA) for cancer chemotherapy. Biological and clinical applications of small-molecule kinase inhibitors require comprehensive characterizations about how these inhibitors modulate the protein expression and activities of kinases at the entire proteome scale. In this study, we developed a parallel-reaction monitoring (PRM)-based targeted proteomic method to monitor the alterations in protein expression of kinases in K-562 chronic myelocytic leukemia (CML) cells elicited by treatment with imatinib, an ABL kinase inhibitor approved by the FDA for CML treatment. By employing isotope-coded ATP affinity probes together with liquid chromatography-multiple-reaction monitoring (LC-MRM) analysis, we also examined the modulation of the ATP-binding affinities of kinases induced by imatinib treatment. The results revealed profound increases in protein expression levels of a large number of kinases in K-562 cells upon treatment with imatinib, which is accompanied by substantial decreases in ATP-binding capacities of many kinases. Apart from ABL kinases, we identified a number of other kinases whose ATP-binding affinities are markedly diminished upon imatinib treatment, including CHK1, a checkpoint kinase involved in DNA damage response signaling. Together, our targeted quantitative proteomic methods enabled, for the first time, dual assessments of small-molecule kinase inhibitor-induced changes in protein expression and ATP-binding affinities of kinases in live cells.
Graphical Abstract
Kinases play crucial roles in cell signaling,1 and small-molecule kinase inhibitors have been widely employed as valuable tools for delineating kinase-mediated cell signaling pathways for decades.2 In addition, aberrant activation of kinase signaling is frequently accompanied by and sometimes leads to the development of many types of cancer.3,4 As a result, kinase inhibitors have recently become a very important class of drugs in anticancer therapy, where more than 30 small-molecule kinase inhibitors have been approved by the Food and Drug Administration (FDA) for treating different types of malignancies.2
Appropriate use of small-molecule kinase inhibitors in cell signaling research and in cancer chemotherapy requires the knowledge about whether other kinases are also targeted by these inhibitors. In this respect, most kinase inhibitors are designed to disrupt, directly or indirectly, the ATP-binding capabilities of the target kinases, which possess highly conserved ATP-binding domains.2,5 Hence, despite the substantial efforts in optimizing the structures of kinase inhibitors for selective binding toward the intended kinase, the inhibitors may also bind to the ATP-binding pocket and suppress the ATP-binding capacities of other kinases, which may lead to off-target effects.2,5 In addition, cancer cells may respond to inhibitor treatment by reprogramming their kinome through altering the protein expression levels of kinases.6
The knowledge about the proteome-wide alterations in protein expression and ATP-binding affinities of kinases elicited by small-molecule inhibitors is important for understanding more completely the mechanisms underlying therapeutic efficacy, resistance, and side effects associated with the kinase inhibitor. Such knowledge is also crucial for the accurate interpretation of data when these inhibitors are used in cell signaling research. Furthermore, on the grounds that the drug safety and pharmacological properties of these FDA-approved kinase inhibitors are well-documented,7 revealing previously unrecognized kinase targets for these inhibitors may facilitate novel applications of these inhibitors in treating other human diseases. The human kinome is encoded with 518 genes,8 and many kinases are expressed at very low levels. Thus, the investigations about the alterations in protein expression and the ATP-binding capabilities of kinases in cells upon inhibitor treatment entail high-throughput and highly sensitive analytical methods.
In the present study, we address the aforementioned analytical challenge by developing a parallel-reaction monitoring (PRM) method (Figure S1),9–11 in conjunction with stable isotope labeling by amino acids in cell culture (SILAC),12 for the proteome-wide interrogation of the protein expression levels of kinases under the same treatment conditions. We also employed the previously reported isotope-coded ATP affinity probes (Figure 1a), together with the multiple-reaction monitoring (MRM)-based targeted proteomic method,13-16 for gauging the alterations in ATP-binding affinities of kinases in K-562 cells upon a 24 h treatment with 1.0 μM imatinib. In this vein, imatinib, a small-molecule inhibitor for c-ABL kinase, was approved in 2001 by the FDA for treating chronic myelocytic leukemia (CML) in patients carrying the BCR-ABL fusion oncogene,2 and this oncogene is also present in K-562 cells.17 It is worth noting that, while liquid chromatography-multiple-reaction monitoring (LC-MRM) analysis is suitable for analyzing enriched samples like the above-described affinity-purified desthiobiotin-conjugated kinase peptides, the same analysis of kinase peptides arising from the tryptic digestion of whole cell lysate may introduce substantial background signal.18 PRM, on the other hand, allows parallel detection of all transitions in a single analysis, which enables high-throughput analysis,19 and it minimizes interferences owing to the use of mass analyzers with high resolution and high accuracy for mass detection.20 Therefore, we employ PRM for assessing kinase protein expression with the use of the tryptic digestion mixture of whole cell lysate.
Figure 1.

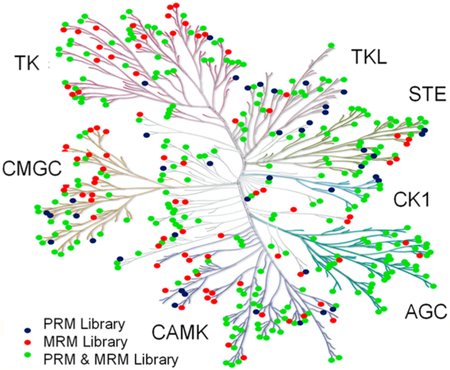
PRM- and MRM-based targeted proteomic approaches for interrogating the human kinome. (a) The chemical structures of the isotope-coded ATP affinity probes. (b) A kinome dendrogram depicting the kinases that are included in PRM- and MRM-based kinome spectral libraries. The kinome dendrogram was adapted with permission from Cell Signaling Technology (http://www.cellsignal.com). Green solid circles denote those kinases that are present in both PRM and MRM libraries, whereas blue and red solid circles designate those kinases that are included only in the PRM and MRM libraries, respectively. (c) Venn diagrams displaying the numbers of kinases (top) and protein kinases (bottom) included in the PRM- and/or MRM-based kinome libraries.
We first established a PRM kinome library based on data collected from shotgun proteomic analysis. To this end, we first collected LC-MS/MS data from shotgun proteomic analysis of the tryptic digestion mixtures of the ATP-binding proteins enriched from 8 human cell lines and the tryptic digestion mixtures of the whole cell lysates of 3 human cell lines. A Maxquant search of these data led to the identification of 11 879 protein groups.10‘11 We subsequently incorporated the tandem mass spectra and retention time information for all unmodified kinase peptides into Skyline21 for constructing the kinome PRM library. Because some kinases share highly similar sequences, we manually checked all kinase peptides and incorporated only those peptides that can be uniquely assigned to specific kinases into the library, with a maximum of four unique peptides being included for any given kinase. In doing so, the current PRM kinome library contained 1050 unique peptides representing 478 nonredundant human kinases, which encompassed 395 protein kinases, 21 lipid kinases, and 62 other kinases (Table S1). The 395 protein kinases include members from all 7 groups of the human kinase family (see kinome dendrogram in Figure 1b, Table S1). Thus, our PRM kinome library contains approximately 80% of the human kinome that includes a total of 518 protein kinases.22
To achieve high-throughput detection of kinase peptides, we adopted scheduled PRM analysis, where the mass spectrometer was programmed to acquire the MS/MS for the precursor ions of a limited number of peptides in each predefined 8 min retention time window. Therefore, accurate prediction of retention time (RT) for each kinase peptide was required for our PRM-based kinome assay. To this end, we calculated the normalized RT (iRT) value for each peptide on our target list following a previously published method with the use of a tryptic digestion mixture of bovine serum albumin (BSA) as the standard.23 The linear RT vs iRT relationship was redefined between every eight LC-PRM runs by reinjecting the tryptic digestion mixture of BSA. Since the iRT value represents an intrinsic attribute (i.e., hydrophobicity) of a peptide, we also employed it as a criterion to validate the data obtained from the PRM assay, where a marked deviation of the observed RT from the predicted RT is considered a false-positive detection.
Our PRM- and MRM-based kinome spectral libraries commonly contain 350 nonredundant kinases (Figure 1c,d). A combination of the PRM and MRM data, therefore, permits independent assessments about how the expression levels and ATP-binding capacities of kinases are modulated by imatinib.
The PRM-based targeted proteomic method led to the quantification of 295 unique kinases in K-562 cells with or without imatinib treatment, including ~250 protein kinases, which cover approximately 50% of the human kinome (Figure 2a and Table S2). Similarly, the MRM-based targeted proteomic method resulted in the quantification of the ATP-binding affinities of 332 unique kinases (Figure 2b and Table S2). All transitions from PRM (4–6 transitions) and MRM (3 transitions) used for quantification of each kinase peptide displayed the same retention time with the dot product (dotp) value being >0.7 (Figure S2).24 Furthermore, over 80% of the kinases quantified with the PRM approach and all the kinases quantified with the MRM method appeared in both forward and reverse labeling experiments (Figure S3a). The ratios of the quantified peptides obtained from forward and reverse experiments exhibited a linear fit (Figure S3b), which confirms the excellent reproducibility of the targeted quantitative proteomic methods.
Figure 2.

Differential protein expression (a) and ATP-binding affinities (b) of kinase proteins in K-562 cells induced by imatinib treatment. The kinase protein expression data represent the mean of results obtained from three forward and two reverse SILAC labeling experiments, and the ATP-binding affinity results reflect five forward and six reverse ATP probe labeling experiments. (The ratios obtained from individual measurements are listed in Table S2.) Blue, red, and gray bars represent those kinases with ratios (in imatinib-treated/control cells) that are <0.67, >1.5, and between 0.67 and 1.5, respectively.
Our quantification results showed that exposure of K-562 cells to imatinib led to pronounced diminutions in the ATP-binding affinities of a large number of kinases while concomitantly stimulating the protein expression levels of many kinases (Figure 2, Table S2). For instance, substantially more kinases display diminished ATP-binding capabilities than those with heightened ATP-binding affinities (65 vs 8), whereas a much larger number of kinases exhibit elevated protein expression than those with reduced expression upon imatinib treatment (48 vs 9, Figure 2, Table S2).
We also examined the accuracy of the PRM method by monitoring the expression levels of three representative kinases (AK1, CCND3, and SCYL3) in K-562 cells with and without imatinib treatment. The Western blot data showed that the expression levels of these kinases are elevated after imatinib treatment, which is consistent with the PRM results (Figure 3). It is worth noting that our LC-PRM method, which quantifies proteins based on unique peptides, permits the independent measurements of different isoforms of kinases. For instance, for the two isoforms of ABL kinases, the expression of ABL1 protein was clearly elevated, whereas that of ABL2 protein kept unchanged upon a 24 h treatment with imatinib (Figure 4a, Table S2). In this vein, imatinib was shown to bind to the kinase domains of ABL1 and ABL2;25,27 however, imatinib administration leads to the differential reprogramming of the two isoforms of ABL. The detailed mechanism(s) contributing to the elevated expression of ABL1, but not ABL2, is not clear and awaits further investigation.
Figure 3.

Western blot analyses for validating the protein expression levels of kinases in K-562 cells with or without imatinib treatment. (a) Images from Western blot analyses of the expression levels of select kinases in K-562 cells with or without imatinib treatment. (b) Representative PRM traces for monitoring the expression levels of the same kinases as shown in (a). (c) The quantification results for the ratios of kinase proteins in imatinib-treated cells over untreated cells. Error bars represent standard deviations.
Figure 4.

PRM and MRM results showing the differential expression and ATP-binding affinities of kinases in K-562 cells induced by imatinib treatment. (a) Representative PRM and MRM traces for the quantification of ABL kinases. (b) Venn diagram depicting the overlap in the numbers of kinases that were quantified by the PRM- and MRM-based kinome profiling methods for lysates of K-562 cells after a 24 h treatment with 1 μM of imatinib or DMSO. (c) A scatter plot depicting the lack of correlation between the ratios of kinases in imatinib-treated cells over control DMSO-treated cells obtained from the PRM and MRM methods (plotted in logarithm scale).
The two methods facilitated the quantification of approximately 160 common kinases (Figure 4b). A comparison of kinase ratios (with/without kinase inhibitor treatment) obtained from PRM and MRM analyses revealed the lack of apparent correlation between the inhibitor-induced alterations in kinase protein expression and ATP-binding capability (Figure 4c), underscoring that the alterations in overall ATP-binding capacities of most kinases are not attributable to changes in protein expression levels of these kinases.
Despite the fact that the two isoforms of ABL kinase exhibit differential reprogramming after imatinib treatment, the ATP-binding affinities of both isoforms were substantially attenuated (Figure 4a), which is in keeping with a previous finding.28 In addition, our results showed the decreased ATP-binding capacities of several other kinases, including FGR, GAK, IRAK, and MELK (Figures 2b and S4, Table S2). These kinases were previously shown to bind directly to imatinib.29,30 Hence, our isotope-coded ATP affinity probe, together with LC-MRM analysis, allows for the revelation of candidate kinases that are targeted by imatinib.
Apart from previously reported kinase targets of imatinib, we identified a number of kinases with reduced ATP-binding affinities but not with diminished protein expression in K-562 cells after imatinib exposure, including YES1 and CHK1. In this context, our results showed that the expression levels of YES1 and CHK1 proteins were not changed in K-562 cells upon imatinib treatment, whereas their ATP-binding affinities were decreased by approximately 2-fold (Table S2 and Figure 5), supporting that imatinib can also compromise the ATP-binding capabilities of YES1 and CHK1. Along this line, it was previously observed that imatinib-resistant cells displayed elevated expression of YES1.31 In addition, our Western blot data showed that imatinib treatment led to a marked diminution in the kinase activity of CHK1 (as manifested by reduced autophosphorylation at Ser296), albeit with no appreciable change in its protein expression level (Figure 5a,b).
Figure 5.

Imatinib inhibits the activity of CHK1. (a) Representative MRM and PRM traces for the quantifications of CHK1. (b) Western blot for the validation of the protein expression and activity of CHK1 in K-562 cells with and without imatinib treatment. (c) Clonogenic survival assay results showing the effect of imatinib on sensitizing MDA-MB-231 cells toward NCS. Error bars represent standard deviation. *, 0.01 < p < 0.05; **, 0.001 < p < 0.01; #, no significant difference. The p-values were calculated against the control using the two-tailed, unpaired Student’s t-test.
In response to DNA double-strand break (DSB) formation in cells, CHK1 coordinates DNA damage response signaling and cell cycle checkpoint control.32 Therefore, we also asked whether the cellular sensitivity toward neocarzinostatin (NCS), a radiomimetic agent that induces DNA DSBs, could be modulated by cotreatment with imatinib. The result from the clonogenic survival assay indeed showed that imatinib rendered MDA-MB-231 cells more sensitive toward NCS (Figure 5c).
In summary, we made significant advances in quantitative analysis of kinome in human cells. By developing a PRM-based method for quantifying the protein expression of kinases and combining it with our previously reported MRM-based targeted proteomic approach,14 we are able to assess independently the protein expression and ATP-binding affinities of kinases. In particular, our PRM and MRM kinome libraries each encompass approximately 80% of the human kinome, with 350 kinases being commonly included in both libraries (Figure 1c). Previously published methods for profiling the kinase inhibitor-induced alterations of the human kinome in cells rely on the use of affinity resin immobilized with multiple kinase inhibitors.6,33 However, the binding of kinases to inhibitor-immobilized affinity resin, similar to the ATP affinity probe method employed here,13,14,16 can be influenced by the protein expression level and sometimes by activity-induced conformational changes of protein kinases.6 To the best of our knowledge, our PRM-based targeted proteomic approach facilitated, for the first time, the assessment of the alterations in protein expression levels of kinases upon small-molecule inhibitor treatment. Our results unveiled pronounced increases in expression levels of a large number of kinases in cells upon treatment with imatinib.
Analysis of kinase protein expression and ATP-binding affinity data together led to the discovery of a number of kinases exhibiting diminutions in ATP-binding affinity but not in protein expression in K-562 cells upon imatinib administration, underscoring that imatinib disrupts the ATP-binding activities of many kinases. Among these kinases, we validated that the activity of CHK1 kinase could be inhibited upon imatinib treatment, which is further substantiated by the observation that imatinib could sensitize cancer cells toward NCS, a radiomimetic agent capable of inducing DNA doublestrand breaks. We envision that our methods will be generally applicable for investigating how the protein expression levels and ATP-binding affinities of the human kinome are modulated by other kinase inhibitors in live cells. Such knowledge will be valuable for the appropriate use of kinase inhibitors in cell signaling research and in therapeutic interventions of human diseases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 CA210072). The authors would like to thank Prof. Jian-Jian Li for kindly providing the MDA-MB-231 cells used in this study.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.anal-chem.9b00289.
Detailed experimental procedures, representative PRM quantification data, and kinome map for the quantified kinases (PDF)
List of kinases included in the Skyline human PRM kinome library (XLSX)
Expression levels and ATP probe labeling efficiency of kinases after imatinib treatment (XLSX)
Notes
The authors declare no competing financial interest.
All the raw files for LC-PRM and LC-MRM analyses of kinases were deposited into PeptideAtlas with the identifier number of PASS01178 (http://www.peptideatlas.org/PASS/PASS01178).
REFERENCES
- (1).Lemmon MA; Schlessinger J Cell 2010, 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wu P; Nielsen TE; Clausen MH Trends Pharmacol. Sci 2015, 36, 422–439. [DOI] [PubMed] [Google Scholar]
- (3).Druker BJ Cancer Cell 2002, 1, 31–36. [DOI] [PubMed] [Google Scholar]
- (4).Cui Y; Steagall WK; Lamattina AM; Pacheco-Rodriguez G; Stylianou M; Kidambi P; Stump B; Golzarri F; Rosas IO; Priolo C; Henske EP; Moss J; El-Chemaly S Cancer Res. 2017, 77, 1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dancey J; Sausville EA Nat. Rev. Drug Discovery 2003, 2, 296–313. [DOI] [PubMed] [Google Scholar]
- (6).Duncan JS; Whittle MC; Nakamura K; Abell AN; Midland AA; Zawistowski JS; Johnson NL; Granger DA; Jordan NV; Darr DB; Usary J; Kuan PF; Smalley DM; Major B; He X; Hoadley KA; Zhou B; Sharpless NE; Perou CM; Kim WY; Gomez SM; Chen X; Jin J; Frye SV; Earp GS; Graves LM; Johnson GL Cell 2012, 149, 307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chong CR; Sullivan DJ Jr. Nature 2007, 448, 645–6. [DOI] [PubMed] [Google Scholar]
- (8).Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S Science 2002, 298, 1912–34. [DOI] [PubMed] [Google Scholar]
- (9).Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ Mol. Cell. Proteomics 2012, 11, 1475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Miao W; Li L; Wang Y Anal. Chem 2018, 90, 6835–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Miao W; Li L; Wang Y Anal. Chem 2018, 90, 11751–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (13).Patricelli MP; Nomanbhoy TK; Wu J; Brown H; Zhou D; Zhang J; Jagannathan S; Aban A; Okerberg E; Herring C; Nordin B; Weissig H; Yang Q; Lee JD; Gray NS; Kozarich JW Chem. Biol 2011, 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Miao W; Xiao Y; Guo L; Jiang X; Huang M; Wang Y Anal. Chem 2016, 88, 9773–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Patricelli MP; Szardenings AK; Liyanage M; Nomanbhoy TK; Wu M; Weissig H; Aban A; Chun D; Tanner S; Kozarich JW Biochemistry 2007, 46, 350–8. [DOI] [PubMed] [Google Scholar]
- (16).Xiao Y; Guo L; Wang Y Mol. Cell. Proteomics 2014, 13, 1065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Milani G; Lana T; Bresolin S; Aveic S; Pasto A; Frasson C; Te Kronnie G Mol. Cancer Res 2017, 15, 683–695. [DOI] [PubMed] [Google Scholar]
- (18).Reiter L; Rinner O; Picotti P; Hüttenhain R; Beck M; Brusniak M-Y; Hengartner MO; Aebersold R Nat. Methods 2011, 8, 430. [DOI] [PubMed] [Google Scholar]
- (19).Doerr A Nat. Methods 2012, 9, 950. [DOI] [PubMed] [Google Scholar]
- (20).Rauniyar N Int. J. Mol. Sci 2015, 16, 28566–28581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S Science 2002, 298, 1912. [DOI] [PubMed] [Google Scholar]
- (23).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Proteomics 2012, 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).de Graaf EL; Altelaar AF; van Breukelen B; Mohammed S; Heck AJ J. Proteome Res. 2011, 10, 4334–4341. [DOI] [PubMed] [Google Scholar]
- (25).Young MA; Shah NP; Chao LH; Seeliger M; Milanov ZV; Biggs WH; Treiber DK; Patel HK; Zarrinkar PP; Lockhart DJ; Sawyers CL; Kuriyan J Cancer Res. 2006, 66, 1007. [DOI] [PubMed] [Google Scholar]
- (26).Procaccia V; Nakayama H; Shimizu A; Klagsbrun M Biochem. Biophys. Res. Commun 2014, 448, 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Virgili A; Koptyra M; Dasgupta Y; Glodkowska-Mrowka E; Stoklosa T; Nacheva EP; Skorski T Cancer Res. 2011, 71, 5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Giles FJ; Cortes JE; Kantarjian HM Curr. Mol. Med 2005, 5, 615–623. [DOI] [PubMed] [Google Scholar]
- (29).Davis MI; Hunt JP; Herrgard S; Ciceri P; Wodicka LM; Pallares G; Hocker M; Treiber DK; Zarrinkar PP Nat. Biotechnol 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- (30).Fabian MA; Biggs WH 3rd; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai AG; Lelias JM; Mehta SA; Milanov ZV; Velasco AM; Wodicka LM; Patel HK; Zarrinkar PP; Lockhart DJ Nat. Biotechnol 2005, 23, 329–36. [DOI] [PubMed] [Google Scholar]
- (31).Kim TM; Ha SA; Kim HK; Yoo J; Kim S; Yim SH; Jung SH; Kim DW; Chung YJ; Kim JW Blood Cancer J. 2011, 1, No. e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu Q; Guntuku S; Cui XS; Matsuoka S; Cortez D; Tamai K; Luo G; Carattini-Rivera S; DeMayo F; Bradley A; Donehower LA; Elledge SJ Genes Dev. 2000, 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- (33).Klaeger S; Heinzlmeir S; Wilhelm M; Polzer H; Vick B; Koenig P-A; Reinecke M; Ruprecht B; Petzoldt S; Meng C; Zecha J; Reiter K; Qiao H; Helm D; Koch H; Schoof M; Canevari G; Casale E; Depaolini SR; Feuchtinger A; Wu Z; Schmidt T; Rueckert L; Becker W; Huenges J; Garz A-K; Gohlke B-O; Zolg DP; Kayser G; Vooder T; Preissner R; Hahne H; Tõnisson N; Kramer K; Götze K; Bassermann F; Schlegl J; Ehrlich H-C; Aiche S; Walch A; Greif PA; Schneider S; Felder ER; Ruland J; Médard G; Jeremias I; Spiekermann K; Kuster B Science 2017, 358, No. eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.