

Abstract
Purpose
Genomic alterations in blood-derived circulating tumor DNA (ctDNA) from patients with colorectal cancers were correlated with clinical outcomes.
Patients and Methods
Next-generation sequencing of ctDNA (54- to 73-gene panel) was performed in 94 patients with colorectal cancer.
Results
Most patients (96%) had metastatic or recurrent disease at the time of blood draw. The median number of nonsynonymous alterations per patient was three (range, zero to 30). The most frequently aberrant genes were TP53 (52.1% of patients), KRAS (34%), and APC (28.7%). Concordance between tissue and blood next-generation sequencing ranged from 63.2% (APC) to 85.5% (BRAF). Altogether, 74 patients (79%) had one or more nonsynonymous alterations, 69 (73%) had one or more potentially actionable alterations, and 61 (65%) had an alteration actionable by a drug approved by the US Food and Drug Administration (on or off label). Lung metastases correlated with improved survival from diagnosis in univariable analysis. ctDNA of 5% or more from blood tests as well as EGFR and ERBB2 (HER2) nonsynonymous alterations correlated with worse survival (but only ERBB2 remained significant in multivariable analysis). No two patients had identical molecular portfolios. Overall, 65% versus 31% of patients treated with matched (n = 17) versus unmatched therapy (n = 18) after ctDNA testing achieved stable disease for 6 months or more, partial response, or complete response (P = .045); progression-free survival, 6.1 versus 2.3 months (P = .08); and survival not reached versus 9.4 months (P = .146; all by multivariable analysis).
Conclusion
Patients with colorectal cancer have heterogeneous ctDNA profiles, and most harbor potentially actionable ctDNA alterations. Matched therapy yielded higher rates of stable disease for 6 months or more, partial response, or complete response. ctDNA assessment may have clinical utility and merits further investigation.
INTRODUCTION
Colorectal cancer is one of the most common cancers worldwide. Globally, there were 1.4 million new cases and 693,900 deaths in 2012, with an increase in incidence and mortality rates in developing countries.1,2 At diagnosis, approximately 20% of patients have distant metastatic disease.3 For decades, systemic therapy used fluorouracil as the main active agent. Addition of irinotecan and oxaliplatin, as well as the recently developed inhibitors that target VEGF (bevacizumab, aflibercept, and regorafenib) and EGFR (cetuximab and panitumumab), have markedly improved the outcome of patients with metastatic colorectal cancer. However, prognosis remains poor (median progression-free survival [PFS], 10 months; median overall survival [OS], 19 to 28 months; 5-year survival, 10%4,5). Thus there is an unmet need to better understand the clinically relevant biology of colorectal cancer.
The molecular characteristics of colorectal cancer are better understood because of advances in next-generation sequencing (NGS) technology.6 Categorizing patients on the basis of their underlying molecular features has been proposed (ie, consensus molecular subtypes),7 and several genomic markers are now routinely used in the clinic to guide treatment. Examples of genomically guided US Food and Drug Administration (FDA)–approved therapies include anti-EGFR agents (cetuximab and panitumumab) among patients with wild-type RAS8-10 and pembrolizumab (anti-programmed cell death protein 1 antibody) for microsatellite instability-high or mismatch repair–deficient colorectal cancer.11 In additon, adding vemurafenib (BRAF inhibitor) to irinotecan and cetuximab demonstrated better clinical outcome among patients with BRAFV600–mutated colorectal cancer.12 However, despite the recent progress in targeted therapy approaches, more than 50% of patients do not respond to the aforementioned regimens, and an increased understanding of the molecular underpinnings of the disease is needed. Some of the challenges with tissue-based genomic analyses, which often are performed on archival samples, include the fact that the genomic landscape of cancer can change after therapeutic intervention,13 and the sequencing results can be confounded by intra- and intertumoral heterogeneity.14,15
To overcome the challenges of tumor heterogeneity and assess the impact of the clonal evolution that occurs with time and under therapeutic pressure, circulating tumor DNA (ctDNA) is now being actively investigated in diverse cancers.16-20 Previous studies using ctDNA analysis of colorectal cancer revealed that TP53, KRAS, and APC were the most commonly altered genes.21,22 Here, we provide an in-depth evaluation with clinical characteristics and therapeutic outcomes of patients with colorectal cancer whose ctDNA was interrogated by clinical-grade NGS.
PATIENTS AND METHODS
Patients
We reviewed the clinicopathologic and outcome data from 94 consecutively tested patients with advanced-stage colorectal cancer at the University of California San Diego Moores Cancer Center; each patient had the ctDNA test performed on their plasma (January 2015 to March 2017). The study followed the guidelines of the University of California, San Diego, Internal Review Board, the Declaration of Helsinki for the PREDICT study (NCT02478931; Profile Related Evidence Determining Individualized Cancer Therapy), and any investigational therapy for which the patients gave consent.
Sequencing, Concordance Rate, Matched Therapy, and Actionability
ctDNA sequencing was performed by Guardant Health and has been previously described (Data Supplement).16,23,24 Overall, 76 (81%) of 94 patients had NGS performed on tumor tissue using the FoundationOne assay. The methods have been previously described (Data Supplement).25 Tissue NGS and plasma ctDNA tests were compared by using the kappa statistic (Data Supplement).26 We retrospectively analyzed the treatments given after ctDNA testing and compared the clinical outcomes among patients who received matched and unmatched therapies (Data Supplement).27
Statistical Analysis
Statistical analysis was performed by M.C.S. with SPSS version 24.0 (SPSS, Chicago, IL; Data Supplement).28 The rate of stable disease (SD) for 6 months or more, partial response (PR), or complete response (CR) was compared between patients who received matched or unmatched therapy. SD, PR, and CR were determined according to an assessment by the treating physician. PFS was defined as the time from the beginning of therapy to progression or last follow-up date for patients who did not progress. OS was defined as the time from diagnosis until death or last follow-up date for patients still alive. PFS and OS were analyzed by using the Kaplan-Meier method28 and the log-rank test (univariable analysis); a Cox regression model (multivariable analysis) was used to compare variables. Patients still progression free (for PFS) or alive (for OS) at last follow-up were censored on that date.
RESULTS
Patient Characteristics
We evaluated 94 patients with colorectal cancer who had NGS of ctDNA. Median age at diagnosis was 50 years (range, 25 to 84 years). The majority of patients had metastasis or recurrence when blood was drawn for ctDNA testing (n = 90 [96%]) and had one or more nonsynonymous alterations (n = 74 [79%]; Table 1; Data Supplement).
Table 1.
Characteristics of Patients With Colorectal Cancer (N = 94)

ctDNA Sequencing Results
The median time from diagnosis to ctDNA analysis was 19.2 months (95% CI, 15.6 to 29.2 months). The median number of nonsynonymous alterations per patient was three (range, zero to 30). The most frequent nonsynonymous alterations were in TP53 (52.1%), KRAS (34%), APC (28.7%), BRAF (19.1%), PIK3CA (17%), and EGFR (16%) genes (Fig 1A). Location of the primary colorectal cancer (right or left side) did not influence the frequency of these alterations.
Fig 1.
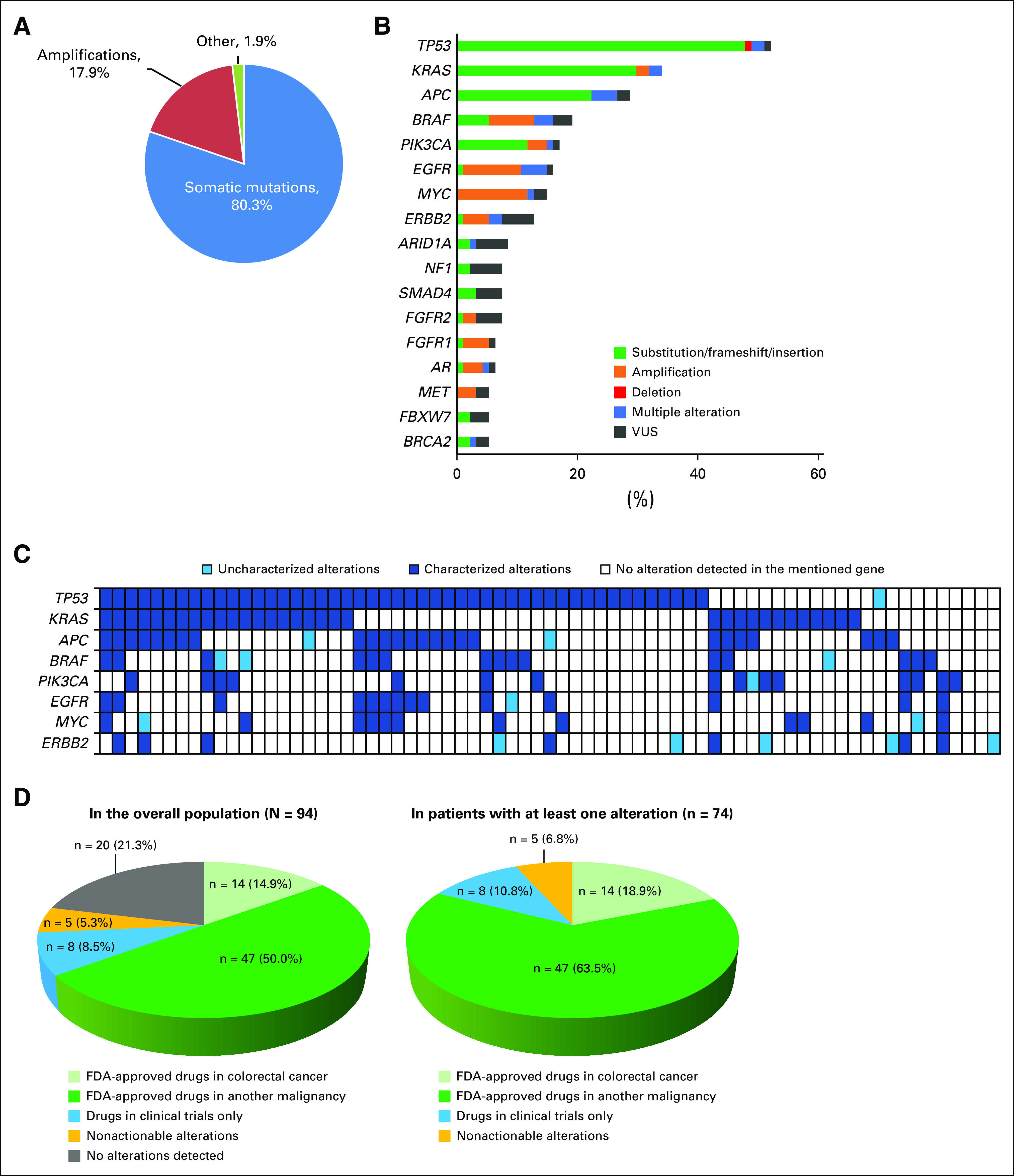
(A) Pie chart showing the types of alterations in the overall population (N = 94). In total, 375 nonsynonymous alterations were detected in 94 patients (74 patients had at least one alteration). Other alterations included three deletions, three insertions, and one fusion.. Frequencies are percent of alterations. (B) Frequency of the most common circulating tumor DNA (ctDNA) alterations. Only genes that were altered in five or more patients are displayed in the bar graph. The other altered genes in our population (fewer than five patients had the alteration) were ABL1, AKT1, ALK, ARAF, ATM, BRCA1, CCND1, CCND2, CCNE1, CDH1, CDK6, CDKN2A, CTNNB1, ESR1, FGFR3, GNAQ, GNAS, HRAS, JAK3, KIT, MAP2K1, MLH1, MTOR, NOTCH1, NRAS, PTEN, RAF1, RB1, RET, RHOA, RIT1, ROS1, SDK6, SMO, STK11, VHL. This analysis included only nonsynonymous alterations. Alterations of unknown significance (variant of unknown significance; VUSs) versus characterized mutations (indels, amplifications, fusions, and single nucleotide variant (SNV) point mutations) were considered at the variant level. Multiple alteration indicates that different alterations were found in same gene (eg, alterations in both BRAF amplification and SNVs found in same patient). Frequencies are percent of patients. (C) Oncoprint of the most frequent alterations. Only alterations identified in more than 10 patients were represented. Each row represents the mentioned alteration; each column represents one patient. Only patients with at least one alteration in one of these genes—TP53, KRAS, APC, BRAF, PIK3CA, EGFR, MYC, or ERBB2—are displayed (n = 71 patients; the other 23 patients had no alterations in the represented genes and their corresponding columns would have been empty or white). (D) Pie charts representing the potential actionability of the detected alterations in the overall population (N = 94; left) and in patients with at least one nonsynonymous alteration (n = 74; right). Percentages are percent of patients. FDA, US Food and Drug Administration.
Distinctiveness of genomic portfolios.
Among the 74 patients with one or more nonsynonymous ctDNA alterations, 59 (79.7%) had distinct gene alteration portfolios; 15 patients (20.3%) had similar genomic alteration portfolios (when alterations were considered at the gene level only, irrespective of the specific variant). This was the case only when patients had a small number of alterations (three or fewer) and included mostly the three genes with the most frequent alterations: TP53, KRAS, and APC (Fig 1B). Of note, all 15 patients had distinct genomic alterations when gene variants were considered. Thus, no two patients had identical molecular profiles (considering both genes and their loci). Among the 74 patients with one or more detectable nonsynonymous ctDNA alterations, 93.2% (69 of 74 patients) had one or more characterized anomalies that were potentially actionable by FDA-approved drugs (82.4%) or by experimental drugs (10.8%) in a clinical trial if an FDA-approved drug was not available (Fig 1C-D).
Comparison of ctDNA and tissue NGS testing.
Overall, 76 patients (80.6%) had tissue NGS (Foundation Medicine; see Patients and Methods). The median time interval between the tissue biopsy and ctDNA testing was 5.8 months (range, 0.03 to 81 months; 95% CI, 3.2 to 8.3 months). We found statistically significant correlations between driver alterations detected in ctDNA (TP53, KRAS, APC, and BRAF) when compared with those detected in tissue, with concordance rates ranging from 63.2% to 85.5%, depending on the genes (Appendix Fig A1; Data Supplement). We did not observe a difference in the concordance rate when the time interval between the blood draw and tissue biopsy were taken into consideration (a cutoff of 6 months was used because it was the median time interval). Although APC alterations seemed to be detected more frequently in the tissue than in the plasma (22 patients were positive in both tests, 27 patients were positive in tissue only, and one patient was positive in plasma only), BRAF alterations seemed to be detected more frequently in the ctDNA test (eight patients were positive in both tests, two were positive in tissue only, and nine were positive in plasma only). Of note, there was 100% concordance between ctDNA and tissue DNA testing specifically for BRAFV600E mutation (n = 6). In contrast, BRAF amplification was seen only with ctDNA testing (n = 8).
OS Analysis
First, we analyzed the impact of several clinical variables on OS calculated from the time of diagnosis (Table 2). In univariable analysis, the presence of nonsynonymous alterations in APC, BRAF, EGFR, MYC, and ERBB2 genes from ctDNA and the presence of one or more gene alterations with ctDNA of 5% or more correlated with a poorer survival (all P < .05). Of interest, patients with metastases localized in their lungs had a statistically significantly improved survival in univariable analysis. After the multivariable analysis, patients in whom an alteration was detected with ctDNA of 5% or more still had statistically significant survival (Fig 2A).
Table 2.
OS Analyses (N = 94)

Fig 2.

Kaplan-Meier curves for overall survival analysis from (A) the time of diagnosis and (B) the time of blood draw used for the ctDNA testing. The variables that were significant in the multivariable analyses (Table 2) are represented. P values are from the log-rank test.
Among 76 patients who had tissue NGS analysis, patients with BRAF alterations had significantly worse OS, and patients with lung metastases had better OS (statistically significant after the multivariable analysis). EGFR, MYC, and ERBB2 alterations in tissue DNA were not assessed because of the small number of patients harboring these alterations (fewer than 10 patients in each group; Data Supplement).
When survival was calculated from the time of blood draw (Table 2), the presence of nonsynonymous alterations in APC, BRAF, PIK3CA, EGFR, MYC, and ERBB2 genes as well as alterations with ctDNA of 5% or more were associated with a shorter median OS (all P < .05). However, only ERBB2 alterations remained significant in the multivariable model (Fig 2B).
Analysis of Outcomes for Patients With Matched Therapy Versus Patients With Unmatched Therapy
Outcomes of the patients who were treated with matched therapy after their ctDNA testing (n = 17) versus those who were given unmatched therapy (n = 18) were evaluated (Figure 3A; Data Supplement). Patients had a median of one prior therapy in the metastatic setting. Overall, we observed improved outcomes for patients with matched therapy (Fig 3). Altogether, 65% of patients in the matched therapy group attained SD for 6 months or more, PR, or CR versus 31% of patients in the unmatched therapy group (P = .060 in univariable analysis; P = .045 in multivariable analysis [multivariable analysis was performed using line of therapy as a confounding variable]). In addition, patients in the matched therapy group had a median PFS of 6.1 months compared with 2.3 months for patients in the unmatched therapy group (P = .143 for univariable analysis; P = .079 for multivariable analysis). Finally, patients who received matched therapy survived longer than unmatched therapy group, with a median OS (calculated from the treatment start date) not reached (at 11.1 months) compared to 9.4 months, respectively (albeit not statistically significant; Data Supplement).
Fig 3.
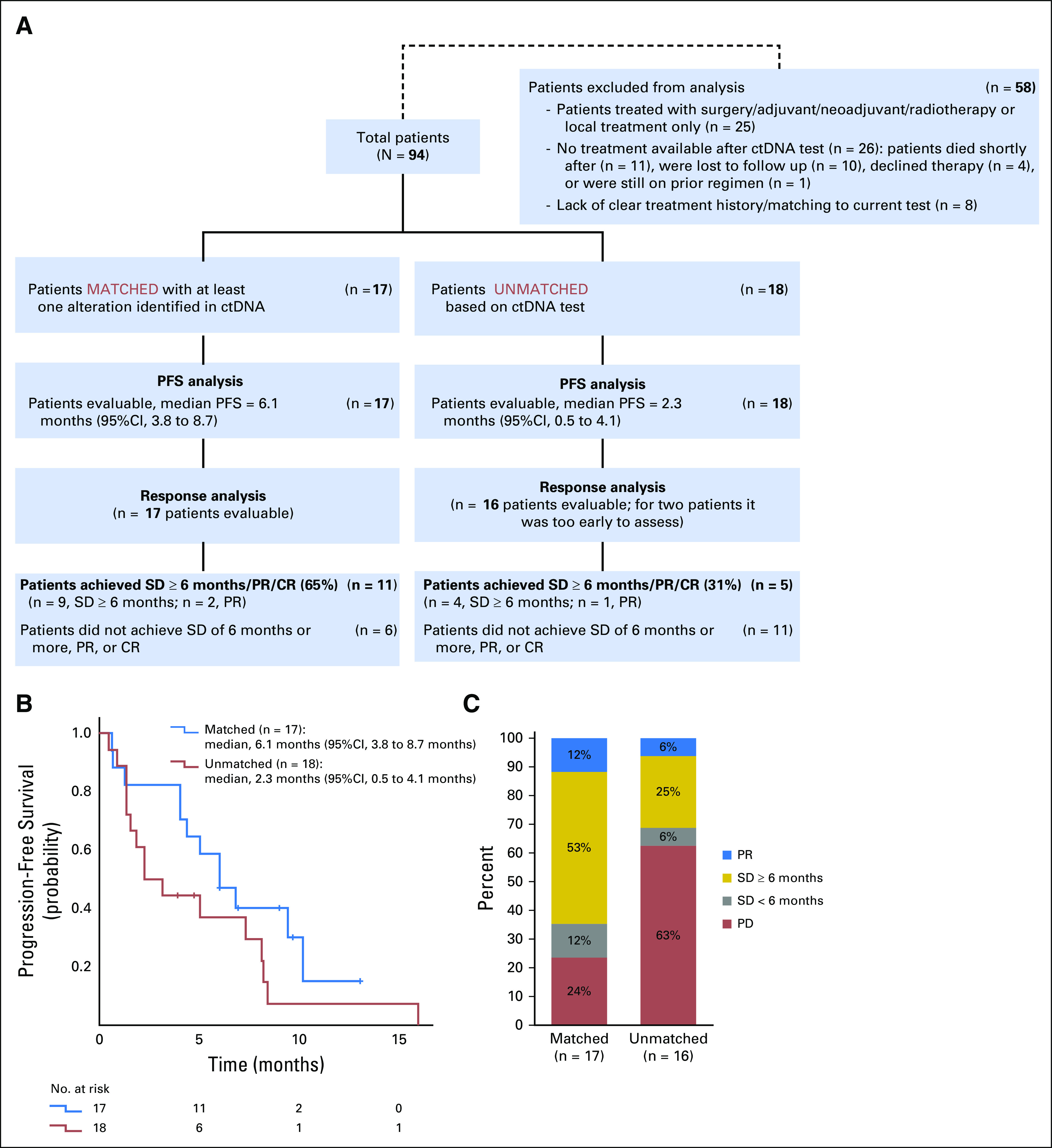
Treatment outcome analyses. (A) Diagram representing the treatment analyses and comparison of patients with matched treatment v patients with unmatched treatment. (B) Kaplan-Meier curves comparing the progression-free survival (PFS) for the patients with matched treatment (n = 17) v patients with unmatched treatment (n = 18). Univariable analysis (log-rank test) P = .143; multivariable analysis (Cox regression) P = .079. (C) Comparison of response outcomes for evaluable patients with matched treatment (n = 17) v those with unmatched treatment (n = 16). Univariable analysis (logistic regression) P = .060; multivariable analysis (multiple logistic regression) P = .045. CR, complete response; ctDNA, circulating tumor DNA; PD, progressive disease; PR, partial response; SD, stable disease.
Illustrative Case: Serial ctDNA Testing in a Patient Who Progressed While Receiving Anti-EGFR–Based Therapy
A 49-year-old-man with metastatic rectal adenocarcinoma was started on fourth-line therapy with irinotecan plus cetuximab. The patient’s baseline ctDNA when therapy was initiated showed alterations including APC E422* (2.2%) and TP53 S127F (1.9%) (Fig 4A). Tumor regression was initially seen, but upon progression that included new lung metastases and lymphangitic spread (Fig 4B), ctDNA among previously observed alterations increased approximately 20-fold (33.8% for APC E422*; 39% for TP53 S127F), and emerging alterations included APC I1307fs and EGFR amplification (Fig 4A).
Fig 4.

Patients who had serial circulating tumor DNA (ctDNA) testing and were progressing on anti-EGFR–based therapy. A 49-year-old-man with metastatic adenocarcinoma of the rectum had a history of previous treatment with (1) capecitabine plus oxaliplatin and (2) fluorouracil plus irinotecan plus bevacizumab; (3) treatment on a clinical trial with anti-CD73 included fourth-line therapy with irinotecan plus cetuximab. (A) Patient’s baseline ctDNA at the start of therapy showing alterations (amount in percent). (B) The patient showed initial tumor regression, but at 9 months, the tumor progressed with new lung metastases and lymphangitic spread (red arrow). ctDNA among previously observed alterations increased approximately 20-fold (33.8% for APC E422*; 39% for TP53 S127F) along with emerging alterations, including MTOR E162V, APC I1307fs, and EGFR amplification. Among the ctDNA alterations observed in this patient, the following were characterized alterations: TP53 S127F, APC E422*, APC I1307fs, and EGFR amplification. MTOR E162V was a variant of uncertain significance; ARID1A K1808K was a synonymous substitution. (†) Only levels of ctDNA mutations were quantified using %ctDNA and represented; EGFR amplification was detected at progression but not quantified.
DISCUSSION
Despite the expansion of our recent understanding of the molecular biology of colorectal cancer6,7 and the development of salutary systemic therapies for advanced-stage disease, overall prognosis remains poor.4,5 Thus, there is an urgent need for innovative treatment approaches for patients with metastatic colorectal cancer. Here we present the biologic features and clinical correlates of genomic alterations among 94 patients with mostly advanced-stage colorectal cancer by using targeted NGS that assessed ctDNA.
Overall, 79% of patients (74 of 94) had one or more nonsynonymous ctDNA alterations (Table 1). The most frequently characterized alterations were in TP53 (51% of patients) followed by KRAS (34%), APC (27%), BRAF (16%), PIK3CA (16%), and EGFR (15%) genes (Fig 1). Although the frequency of APC alterations detected in this study is less than what has been previously reported, frequencies of other genomic alterations are consistent with those in previous publications.21,22 Concordance was statistically significant between ctDNA and tissue DNA among the driver or truncal gene alterations (overall concordance: TP53, 68%; KRAS, 74%; APC, 63%; BRAF, 86% [all P < .05]). Hong et al29 previously reported that the ctDNA NGS had 100% sensitivity for tissue-detected as well as digital droplet polymerase chain reaction–based plasma-detected BRAFV600E mutation among patients with colorectal cancer, which is consistent with our data. Considering that BRAFV600E is targetable with a combination of anti-EGFR and anti-BRAF therapies, testing for this alteration is important.12 It should be noted that some of the high overall concordance was driven by negative concordance. Sensitivity of ctDNA was variable for detection of mutations found in tissue. For instance, sensitivity of ctDNA for detection of patients with tissue APC positivity was 44.9%; sensitivity of ctDNA for detection of those with tissue BRAF positivity was 80% (this analysis was not restricted to BRAFV600E). The apparent low capacity for detecting APC alterations could have implications for plasma monitoring of this alteration.
We did not observe differences in the concordance rate of driver alterations when we compared patients with a time interval between the blood draw and tissue biopsy of 6 months or less versus more than 6 months (Data Supplement). This observation differs from previous reports, which showed that the longer the time interval between the two tests, the lower the rate of concordance.20,30 The small number of patients in each of our subgroups may have confounded our ability to discern such differences.
Importantly, certain clinical and ctDNA factors were associated with survival outcome. When survival was evaluated from the time of blood draw to the last follow-up, the presence of APC, BRAF, PIK3CA, EGFR, MYC, and ERBB2 nonsynonymous alterations (including single-nucleotide variations and amplifications) were all associated with poor overall survival (all P < .05 by univariable analysis), and ERBB2 alterations remained significant after the multivariable analysis (P < .001) (Table 2; Fig 2). Many of these poor prognostic alterations are potentially targetable with either FDA-approved drugs (on- or off-label use) or with investigational agents currently in clinical development. Examples include using an EGFR inhibitor (eg, cetuximab) plus a BRAF inhibitor (eg, vemurafenib) for patients with BRAF alterations12 and anti-HER2 agents (eg, trastuzumab and/or lapatinib) for ERBB2 alterations.31
Accumulating evidence suggests that a biomarker-based approach to selecting targeted therapies, especially if they are based on genomic markers, is associated with improved outcomes.32-36 Despite the relatively small numbers of patients in our study, we observed improvement in disease parameters in patients who received matched therapies (n = 17) compared with patients who received unmatched therapies (n = 18; SD was 6 months or more, PR, or CR [65% v 31%; P = .045]; median PFS, 6.1 v 2.3 months [P = .079]; median OS, not reached v 9.4 months [P = .146] using multivariable analysis; Figs 3-4; Data Supplement). The change in PFS and OS represented a nonsignificant trend, suggesting the need for larger studies. However, other factors may be operative and could have confounded our results. For instance, patients often received concomitant chemotherapy, and the putative interaction between TP53 and bevacizumab could have inflated the results.37-39 These considerations are important because some matched therapies have not proved to be effective in colorectal cancers.40,41
Although the precision medicine approach of matching patients with genomically or immunotargeted treatments is potentially promising, there are challenges for implementing this strategy (eg, tumor heterogeneity and genomic complexity among patients). In this study, we observed a median of three alterations per patient (range, zero to 30 alterations), and among 74 patients with one or more alterations, there were no two patients with identical molecular profiles. These findings suggest that matched monotherapy may not be optimal. Rather, customized combinations are most likely required for each individual. Preliminary results from the I-PREDICT (NCT02534675; Investigation of Profile-Related Evidence Determining Individualized Cancer Therapy) study of patients with aggressive malignancies showed a statistically significant improvement in time-to-treatment failure among patients receiving individualized combinations of matched therapies on the basis of genomic alterations when compared with patients receiving unmatched therapies.42 Expansion of this trial is ongoing.
We have also observed that patients who had high mutation allele frequency (MAF) and ctDNA of 5% or more had significantly worse survival (Table 2), which is consistent with previous reports.20,30 This observation is not surprising because ctDNA levels are reflective of the underlying tumor burden and can undergo dynamic changes after therapy.43,44 Moreover, Tie et al45 reported that, among patients with early-stage colon cancer who had surgical resection, detection of postsurgical ctDNA was strongly associated with recurrent disease, which indicates that ctDNA can be a very sensitive biomarker for residual disease and treatment response. Along with this notion, we have also presented a patient with metastatic rectal adenocarcinoma whose ctDNA level increased by 20-fold when his tumor showed progression of lung metastases and lymphangitic spread while he was receiving treatment with irinotecan and cetuximab (Fig 4). It is unclear at this time whether increases in ctDNA level warrant a change in treatment strategy. Similarly, it is uncertain whether addition of new targeted therapy agents upon progression is warranted. In this study, EGFR amplification surfaced along with tumor progression while the patient was receiving anti-EGFR therapy (cetuximab; Fig 4). Emergence of alterations similar to those in ctDNA and other anomalies such as the appearance of KRAS alterations have been implicated in the evolution of resistance in patients with colorectal cancer who were treated with EGFR inhibitors.46
There were several limitations to our study. First, it was performed retrospectively with a relatively small sample size at a single institution. Thus, our clinical findings require further validation with prospective studies. Second, multiple comparisons could result in overestimating the implications of P values; however, we have included a Bonferroni correction to attenuate this limitation. Third, it would be interesting to determine the impact of microsatellite stability status on the ctDNA alteration profile; this analysis could not be performed in this study because only 26 of our patients had microsatellite testing and 25 of them were microsatellite stable. Finally, because of the limited number of patients and the variable duration of time between tissue and ctDNA sampling, it was not feasible to analyze the effect of MAF for different alterations or the correlation between tissue and ctDNA MAF. Despite these limitations, our study provided an in-depth investigation of the clinical utility of ctDNA testing among patients with colorectal cancer.
In conclusion, we have interrogated tumors from 94 patients with mostly advanced-stage colorectal cancer who had clinical-grade NGS performed on blood-derived ctDNA. The median number of nonsynonymous alterations per patient was three and, importantly, each patient harbored a unique molecular profile. Concordance with common alterations in tissue ranged from 63% to 86%; differences between ctDNA and tissue could reflect dynamic changes in ctDNA after treatment. As was demonstrated by an illustrative patient, ctDNA may be shed from multiple metastatic sites or there could be differences in sensitivity between tissue and ctDNA sequencing. The presence of ctDNA of less than 5% independently correlated with longer survival, whereas ERBB2 ctDNA nonsynonymous alterations were associated with shorter survival. Although the number of patients receiving targeted therapy in our study was modest, this is the first study, to our knowledge, to demonstrate objective evidence of the clinical utility of ctDNA NGS in metastatic colorectal cancer across multiple biomarkers beyond BRAFV600E. Further clinical investigations using ctDNA to guide therapy in patients with colorectal cancer are needed.47
Appendix
Methods
Digital sequencing of circulating tumor DNA.
Circulating tumor DNA (ctDNA sequencing was performed by Guardant Health, which was previously described (Guardant360, Redwood City, CA; http://www.guardanthealth.com/guardant360/). Guardant360 is a clinical laboratory certified by the Clinical Laboratory Improvement Amendment, accredited by the College of American Pathologists, and approved by the New York State Department of Health. The analytical and clinical validation of Guardant360 was conducted in conformance with evidentiary standards established by the Standards for Reporting of Diagnostic Accuracy, Reporting of Tumor Marker Studies, Evaluation of Genomic Applications in Practice and Prevention, recent next-generation sequencing (NGS), and Standardization of Clinical Testing Biomarker Guidelines.16
The sequencing uses molecular barcoding and hybrid capture followed by NGS of the critical exons in a panel of 54 to 73 genes and reports all four major types of genomic alterations (point mutations, insertions-deletions [indels], fusions, and copy number amplifications)16,23 See the Data Supplement for detailed gene panels. The analytic sensitivity reaches detection levels of one to two single mutant fragments from a 10-mL blood sample (0.1% limit of detection), and analytic specificity is greater than 99.9999%. The fractional concentration or mutation allele frequency (MAF) for a given somatic mutation is calculated as the fraction of ctDNA harboring that mutation in a background of wild-type ctDNA fragments at the same nucleotide position. The lower limit of detection of ctDNA was 0.04% for single nucleotide variants and fusions and 0.02% for indels.16,24 Only nonsynonymous alterations that include characterized alterations and variants of unknown significance (VUSs) were included in our analysis. When characterized alterations are referred to, VUSs were excluded in the analysis.
NGS of tissue.
Overall, 76 (81%) of 94 patients who had ctDNA results also had NGS performed by agencies accredited by Clinical Laboratory Improvement Amendment and the College of American Pathologists on tumor tissue using the FoundationOne assay (http://www.foundationone.com/; hybrid capture–based NGS; 182, 236, or 315 genes, depending on the time period). At > 250× median depth of coverage, > 99% of base substitutions present at MAF ≥ 10% were successfully detected. For indels at MAF ≥ 20%, 98% were detected. The methods have been previously described.25
Concordance Rate
For the 76 patients who had both types of tests (tissue NGS and plasma ctDNA testing that covered the same genes and alteration types revealed in the tissue NGS), we assessed the concordance for the most frequent alterations and the corresponding kappa statistic, which is a conservative measurement of relative agreement that takes into account agreement by chance. Kappa values range from 1 (perfect agreement) to 0 (no agreement other than what would be expected by chance).26
Matched Therapy and Actionability
We retrospectively analyzed the treatments given after ctDNA testing and compared the clinical outcomes among patients who received matched and unmatched therapies. A therapy was considered matched if at least one agent in the treatment regimen targeted at least one abnormality or pathway component aberrant in a patient’s ctDNA molecular profile. Patients were evaluable if therapy was administered for more than 10 days.
Actionability implies that the protein product of a genomic abnormality can be affected by a specific targeted drug.27 A potentially actionable alteration was defined as an alteration that was either the direct target (such as an EGFR inhibitor targeting an EGFR mutation) or a pathway component (such as an mTOR inhibitor for a PIK3CA mutation [because mTOR is downstream of PIK3CA]) that could be targeted by at least one US Food and Drug (FDA)–approved or investigational drug in a clinical trial. Actionability was considered at the variant level; VUSs (functional consequences and clinical significance of these gene variants are not established, as opposed to characterized alterations) were considered nonactionable.
Statistical Analysis
Medians and 95% CIs or ranges were reported. Fisher’s exact tests were used to compare categorical variables, and the nonparametric Mann-Whitney U test was used to compare two groups on one continuous variable. Binary logistic regressions were performed for categorical end points, and multiple linear regressions were performed for continuous variables. The rate of stable disease of 6 months or more, partial response, or complete response was compared between patients with matched and unmatched therapy. Stable disease, partial response, or complete response was determined per assessment of the treating physician. Progression-free survival was defined as the time from the beginning of therapy to progression or last follow-up date for patients who did not progress. Overall survival was defined as the time from diagnosis until death or last follow-up date for patients still alive. Progression-free survival and overall survival were analyzed by the Kaplan-Meier method,28 and the log-rank test (univariable analysis) or Cox regression model (multivariable analysis) was used to compare variables. When appropriate, Bonferroni correction was used for multiple comparisons. Statistical analysis was performed by M.C.S. with SPSS version 24.0 (SPSS, Chicago, IL).
Figure A1.
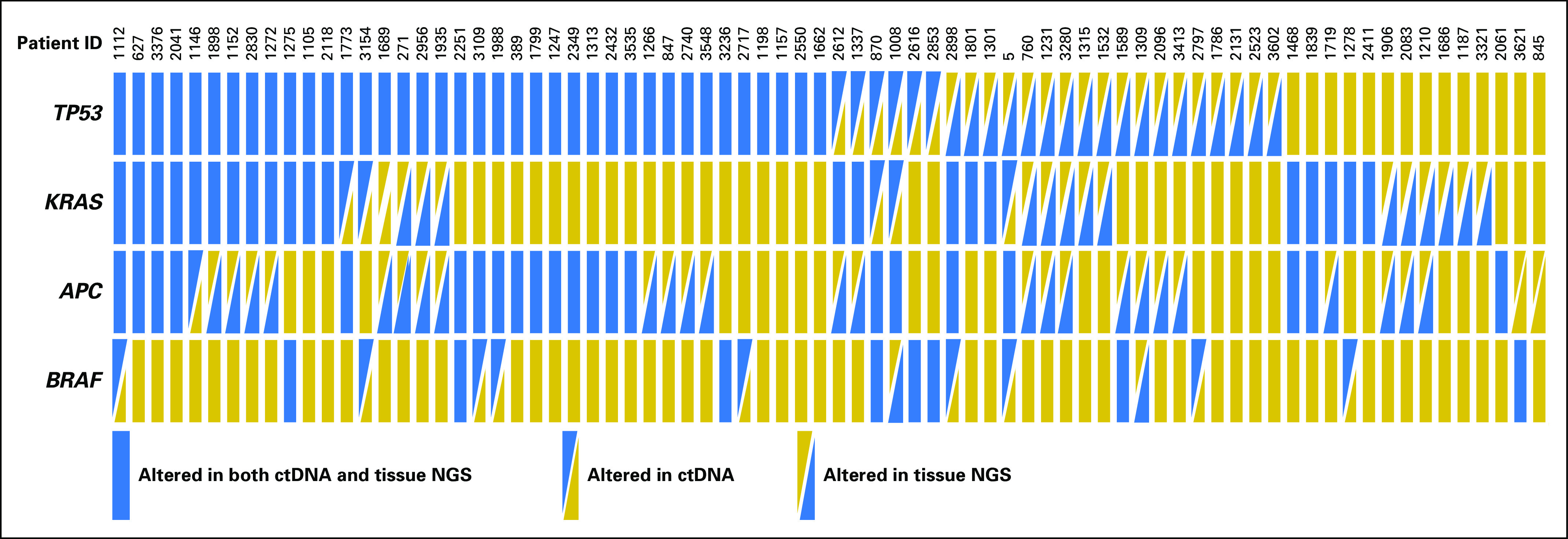
Concordance between circulating tumor DNA (ctDNA) and tissue DNA analyses among commonly altered genomic alterations. NGS, next-generation sequencing.
Footnotes
Supported by Grant No. P30 CA023100 (R.K.) from the National Cancer Institute and funded in part by the Joan and Irwin Jacobs Fund philanthropic fund.
AUTHOR CONTRIBUTIONS
Conception and design: Shumei Kato, Maria C. Schwaederlé, Scott M. Lippman, AmirAli Talasaz, Razelle Kurzrock
Administrative support: Scott M. Lippman
Provision of study materials or patients: Paul T. Fanta, Victoria M. Raymond
Collection and assembly of data: Shumei Kato, Maria C. Schwaederlé, Paul T. Fanta, Richard B. Lanman, Victoria M. Raymond, AmirAli Talasaz
Data analysis and interpretation: Shumei Kato, Maria C. Schwaederlé, Paul T. Fanta, Ryosuke Okamura, Lawrence Leichman, Scott M. Lippman, Victoria M. Raymond, AmirAli Talasaz, Razelle Kurzrock
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Shumei Kato
No relationship to disclose
Maria C. Schwaederlé
Employment: Merck (I)
Stock and Other Ownership Interests: Merck (I)
Travel, Accommodations, Expenses: Merck (I)
Paul T. Fanta
No relationship to disclose
Ryosuke Okamura
No relationship to disclose
Lawrence Leichman
No relationship to disclose
Scott M. Lippman
No relationship to disclose
Richard B. Lanman
Employment: Guardant Health, Veracyte
Leadership: Guardant Health, BIOLASE
Stock and Other Ownership Interests: Guardant Health, BIOLASE
Research Funding: Guardant Health
Victoria M. Raymond
Employment: Trovagene, Guardant Health
Stock and Other Ownership Interests: Trovagene, Guardant Health
AmirAli Talasaz
Employment: Guardant Health
Leadership: Guardant Health
Stock and Other Ownership Interests: Guardant Health
Research Funding: Guardant Health
Patents, Royalties, Other Intellectual Property: Guardant Health
Travel, Accommodations, Expenses: Guardant Health
Razelle Kurzrock
Leadership: CureMatch
Stock and Other Ownership Interests: CureMatch, IDbyDNA
Consulting or Advisory Role: Actuate Therapeutics, Loxo Oncology, XBiotech, Neo-Med, Roche
Speakers’ Bureau: Roche
Research Funding: Guardant Health (Inst), Sequenom (Inst), Merck Serono (Inst), Genentech (Inst), Pfizer (Inst), Foundation Medicine (Inst), Incyte (Inst), Konica Minolta (Inst)
REFERENCES
- 1.Arnold M, Sierra MS, Laversanne M, et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–691. doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.van der Geest LG, Lam-Boer J, Koopman M, et al. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin Exp Metastasis. 2015;32:457–465. doi: 10.1007/s10585-015-9719-0. [DOI] [PubMed] [Google Scholar]
- 4.Cassidy J, Clarke S, Díaz-Rubio E, et al. XELOX vs FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br J Cancer. 2011;105:58–64. doi: 10.1038/bjc.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–1075. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 9.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 10.Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 11.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 3722015:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopetz S, McDonough SL, Morris VK, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG 1406) J Clin Oncol. 2017;35 doi: 10.1200/JCO.20.01994. suppl; abstr 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russo M, Siravegna G, Blaszkowsky LS, et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016;6:147–153. doi: 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10:e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwaederle M, Chattopadhyay R, Kato S, et al. Genomic alterations in circulating tumor DNA from diverse cancer patients identified by next-generation sequencing. Cancer Res. 2017;77:5419–5427. doi: 10.1158/0008-5472.CAN-17-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato S, Krishnamurthy N, Banks KC, et al. Utility of genomic analysis in circulating tumor DNA from patients with carcinoma of unknown primary. Cancer Res. 2017;77:4238–4246. doi: 10.1158/0008-5472.CAN-17-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato S, Janku F. Cell-free DNA as a novel marker in cancer therapy. Biomarkers Med. 2015;9:703–712. doi: 10.2217/bmm.15.38. [DOI] [PubMed] [Google Scholar]
- 20.Schwaederlé MC, Patel SP, Husain H, et al. Utility of genomic assessment of blood-derived circulating tumor DNA (ctDNA) in patients with advanced lung adenocarcinoma. Clin Cancer Res. 2017;23:5101–5111. doi: 10.1158/1078-0432.CCR-16-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strickler JH, Loree JM, Ahronian LG, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 2018;8:164–173. doi: 10.1158/2159-8290.CD-17-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thierry AR, El Messaoudi S, Mollevi C, et al. Clinical utility of circulating DNA analysis for rapid detection of actionable mutations to select metastatic colorectal patients for anti-EGFR treatment. Ann Oncol. 2017;28:2149–2159. doi: 10.1093/annonc/mdx330. [DOI] [PubMed] [Google Scholar]
- 23.Vowles J, Odegaard J, Mortimer S, et al. Vol. 77. AACR; 2017. Analytical validation of Guardant360 v2. 10. suppl; abstr 5705. [Google Scholar]
- 24.Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24:3539–3549. doi: 10.1158/1078-0432.CCR-17-3831. [DOI] [PubMed] [Google Scholar]
- 25.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McHugh ML. Interrater reliability: The kappa statistic. Biochem Med (Zagreb) 2012;22:276–282. [PMC free article] [PubMed] [Google Scholar]
- 27.Vidwans SJ, Turski ML, Janku F, et al. A framework for genomic biomarker actionability and its use in clinical decision making. Oncoscience. 2014;1:614–623. doi: 10.18632/oncoscience.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goel MK, Khanna P, Kishore J. Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res. 2010;1:274–278. doi: 10.4103/0974-7788.76794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong DS, Morris VK, El Osta B, et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 2016;6:1352–1365. doi: 10.1158/2159-8290.CD-16-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwaederle M, Husain H, Fanta PT, et al. Use of liquid biopsies in clinical oncology: Pilot experience in 168 patients. Clin Cancer Res. 2016;22:5497–5505. doi: 10.1158/1078-0432.CCR-16-0318. [DOI] [PubMed] [Google Scholar]
- 31.Sartore-Bianchi A, Trusolino L, Martino C, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:738–746. doi: 10.1016/S1470-2045(16)00150-9. [DOI] [PubMed] [Google Scholar]
- 32.Jardim DL, Schwaederle M, Wei C, et al. Impact of a biomarker-based strategy on oncology drug development: A meta-analysis of clinical trials leading to FDA approval. J Natl Cancer Inst. 2015;107:djv253. doi: 10.1093/jnci/djv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwaederle M, Zhao M, Lee JJ, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: A meta-analysis. JAMA Oncol. 2016;2:1452–1459. doi: 10.1001/jamaoncol.2016.2129. [DOI] [PubMed] [Google Scholar]
- 34.Schwaederle M, Parker BA, Schwab RB, et al. Precision oncology: The UC San Diego Moores Cancer Center PREDICT experience. Mol Cancer Ther. 2016;15:743–752. doi: 10.1158/1535-7163.MCT-15-0795. [DOI] [PubMed] [Google Scholar]
- 35.Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18:6373–6383. doi: 10.1158/1078-0432.CCR-12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wheler JJ, Janku F, Naing A, et al. Cancer therapy directed by comprehensive genomic profiling: A single center study. Cancer Res. 2016;76:3690–3701. doi: 10.1158/0008-5472.CAN-15-3043. [DOI] [PubMed] [Google Scholar]
- 37.Said R, Hong DS, Warneke CL, et al. P53 mutations in advanced cancers: Clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget. 2013;4:705–714. doi: 10.18632/oncotarget.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwaederlé M, Lazar V, Validire P, et al. VEGF-A expression correlates with TP53 mutations in non-small cell lung cancer: Implications for antiangiogenesis therapy. Cancer Res. 2015;75:1187–1190. doi: 10.1158/0008-5472.CAN-14-2305. [DOI] [PubMed] [Google Scholar]
- 39.Wheler JJ, Janku F, Naing A, et al. TP53 alterations correlate with response to VEGF/VEGFR inhibitors: Implications for targeted therapeutics. Mol Cancer Ther. 2016;15:2475–2485. doi: 10.1158/1535-7163.MCT-16-0196. [DOI] [PubMed] [Google Scholar]
- 40.Hochster HS, Uboha N, Messersmith W, et al. Phase II study of selumetinib (AZD6244, ARRY-142886) plus irinotecan as second-line therapy in patients with K-RAS mutated colorectal cancer. Cancer Chemother Pharmacol. 2015;75:17–23. doi: 10.1007/s00280-014-2609-3. [DOI] [PubMed] [Google Scholar]
- 41.Kim ST, Lee J, Park SH, et al. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer. 2017;17:211. doi: 10.1186/s12885-017-3196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sicklick JK, Leyland-Jones B, Kato S, et al. Personalized, molecularly matched combination therapies for treatment-na. J Clin Oncol. 2017;35 suppl; abstr 2512. [Google Scholar]
- 43.Janku F, Huang HJ, Claes B, et al. BRAF mutation testing in cell-free DNA from the plasma of patients with advanced cancers using a rapid, automated molecular diagnostics system. Mol Cancer Ther. 2016;15:1397–1404. doi: 10.1158/1535-7163.MCT-15-0712. [DOI] [PubMed] [Google Scholar]
- 44.Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 45.Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwaederle M, Chattopadhyay R, Kato S, et al. Genomic alterations in circulating tumor DNA from diverse cancer patients identified by next-generation sequencing. Cancer Res. 2017;77:5419–5427. doi: 10.1158/0008-5472.CAN-17-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reference deleted. [Google Scholar]