

Abstract
The failure to predict kidney toxicity of new chemical entities early in the development process before they reach humans remains a critical issue. Here, we used primary human kidney cells and applied a systems biology approach that combines multidimensional datasets and machine learning to identify biomarkers that not only predict nephrotoxic compounds but also provide hints toward their mechanism of toxicity. Gene expression and high-content imaging-derived phenotypical data from 46 diverse kidney toxicants were analyzed using Random Forest machine learning. Imaging features capturing changes in cell morphology and nucleus texture along with mRNA levels of HMOX1 and SQSTM1 were identified as the most powerful predictors of toxicity. These biomarkers were validated by their ability to accurately predict kidney toxicity of four out of six candidate therapeutics that exhibited toxicity only in late stage preclinical/clinical studies. Network analysis of similarities in toxic phenotypes was performed based on live-cell high-content image analysis at seven time points. Using compounds with known mechanism as reference, we could infer potential mechanisms of toxicity of candidate therapeutics. In summary, we report an approach to generate a multidimensional biomarker panel for mechanistic de-risking and prediction of kidney toxicity in in vitro for new therapeutic candidates and chemical entities.
Keywords: kidney toxicity, prediction, systems toxicology, in vitro, mechanism
The kidneys are the main site for the elimination of drugs and chemicals from the body. This critical role renders kidney epithelial cells uniquely susceptible to damage induced by xenobiotics and their metabolites/intermediates. It is estimated that 19%−33% of acute kidney injury (AKI) cases in the hospital are attributed to drug nephrotoxicity (Choudhury and Ahmed, 2006; Kleinknecht et al., 1987). The most frequently cited kidney toxic drugs include aminoglycoside antibiotics, analgesics, contrast media, chemotherapeutic agents, and immunosuppressants. However, exposure to environmental contaminants such as cadmium, mercuric chloride, and aristolochic acid can also lead to AKI (Vaidya et al., 2008). Kidney toxicity seen in animal toxicology studies is a major factor in the failure of drug candidates and is responsible for 8% and 9% of drug development failures in preclinical as well as clinical stages, respectively (Cook et al., 2014). Therefore, accurate methods for predicting kidney toxicity as early as possible are critical for the development of safe drugs and the risk assessment of chemicals, to allow the management of the health and environmental hazards posed by these compounds.
Currently, the main approaches for predicting potential kidney toxicity in humans include animal testing, calculation of quantitative structure-activity relationships (QSAR), and in vitro cell-based assays. Animal toxicity studies are a regulatory requirement and are therefore performed routinely. However, they are usually expensive, low-throughput, and time-consuming (Krewski et al., 2010). Furthermore, societal concern regarding excessive animal experimentation, coupled with guidance form regulatory authorities, has made reduction, refinement, and replacement of the use of animals a primary goal in toxicology (National Research Council, 2007). Computational modeling of structure-activity relationship is much better in handling a large number of compounds in a very short period of time. However, although QSAR works well for compounds with specific or well-understood chemical structures or mechanisms (Cherkasov et al., 2014; Myshkin et al., 2012), the models often do not take the biological contexts into account and thus have limited applications in predicting complex biological responses.
In vitro assays with renal epithelial cells could be used as a bridge between throughput and physiological relevance. Studies have shown the applicability and reproducibility of primary human proximal tubule cells in identifying predictors of toxicity (Su et al., 2016). However, all of the currently proposed in vitro biomarkers of kidney toxicity were only used in libraries of compounds with well-known effects and not put to the test in compounds that are in their early stages of development. Furthermore, in addition to the testing of large number of compounds, the current interest in alternatives to animal testing is driven by the requirement for the identification of injury mechanisms, which cannot be achieved by the analysis of a small number of injury biomarkers.
Therefore, the objective of this study was to develop a combined approach to not only identify biomarkers that allow classification of compounds as kidney toxic, but also to offer insight into the potential mechanisms of toxicity in vitro. We conducted a comprehensive study that integrates cellular perturbation signals (transcriptomics) along with cellular phenotypic changes (imaging-based structure, function, and behavior) to identify a sensitive and specific biomarker panel to predict toxicity. Additionally, phenotypic imaging data were used for network analyses to identify similarities between compounds with known and unknown mechanisms of toxicity. We then applied this approach to predict kidney toxicity in a new set of compounds that were discontinued in pharmaceutical development due to kidney-related findings, identified only in late stages of in vivo and clinical testing.
MATERIAL AND METHODS
Cell culture
Preparation and culture of primary human proximal tubule epithelial cells (HPTEC) were performed as described previously (Adler et al., 2016; Ramm et al., 2016). Briefly, HPTECs were purchased from Biopredic International (Rennes, France) and were cultured in supplemented DMEM/Ham’s-F12 with GlutaMAX medium on Poly-d-lysine coated black clear bottom 384-well plate (Corning Life Science) for high-content imaging (HCI) experiments and on collagen VI-coated black clear bottom 96-well plates for cell viability assays and L1000 gene expression. Cells were not used after passage 4. Human liver epithelial cells (HepG2) were obtained from ATCC (Wesel, Germany) and cultured in Dulbecco’s modified Eagle medium (DMEM, low glucose) with 10% fetal calf serum (FCS), 2 mM l-glutamine and penicillin/streptomycin (Sigma Aldrich).
Compounds
For development of the predictive models, a compound library was custom made. Most compounds were purchased from Sigma-Aldrich (St. Louis, Missouri), Enzo Life Sciences Inc. (Farmingdale, New York), and TRC Inc. (Toronto, Canada) and three synthesized compounds (GSH and CYS conjugates of halogenated alkenes) were kindly provided by Edward A. Lock (Liverpool) (Figure 2). The 46 library compounds were dissolved in DMSO or water at 100 or 200 mM, depending on solubility.
Figure 2.
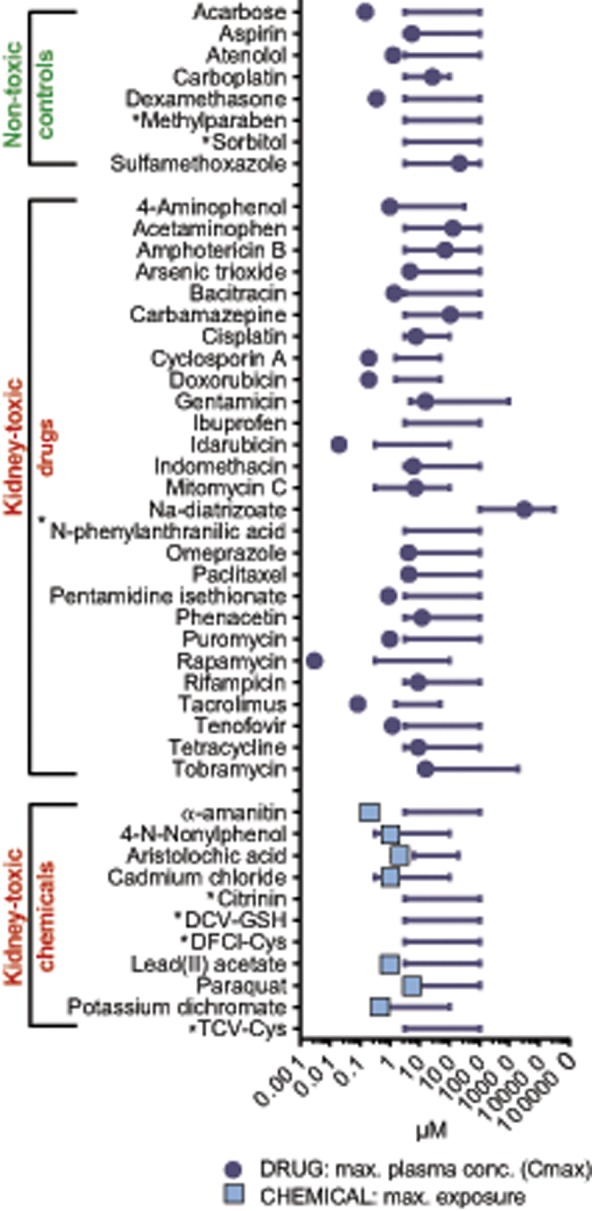
Library of 46 nontoxic and kidney toxic drugs and chemicals. The concentration range used in high-content imaging, gene expression, and CellTiter glo analyses are displayed as lines. Corresponding peak plasma concentrations of drugs are indicated as circles, exposure levels of chemicals and contaminants as squares. Asterisk (*) indicates that exposure data were not available.
The selection of kidney toxic compounds was performed to provide a library as broad as possible to avoid selection bias. Therefore, the library contains drugs and chemicals with (1) different pharmacological targets, (2) different exposure levels, and importantly (3) different degrees of evidence for kidney toxicity in humans. For some compounds, robust evidence exists of their ability to damage the kidney in experimental settings and in patients (cyclosporine A, cisplatin, gentamicin), whereas others were included with case reports of toxicity only in patients with pre-existing disease states (eg, rapamycin). Finally, we also included acetaminophen as a parent compound that would require liver metabolism to be toxic in vivo, as well as toxic hepatic metabolites (4-aminophenol, halogenated vinyl cysteines and glutathione).
Pharmacological information associated with the library compounds was derived from the DrugMatrix database (https://ntp.niehs.nih.gov/results/dbsearch/index.html#DrugMatrix-and-ToxFx-Databases) in which a curation team extracted all relevant information on the compounds from the literature, the Physicians’ Desk Reference, package inserts, and other relevant sources (Supplementary Table 1). Physicochemical information associated with the library compounds was derived from PubChem (Supplementary Table 1). Pharmaceuticals and chemicals were used at six concentrations, selected to either cover or slightly exceed the human maximum plasma concentration (Cmax) or human maximal exposure (Figure 2, Supplementary Table 1).
Concentrations are nominal, without further assessment of bound and unbound compound concentrations, plastic binding, or cellular concentrations.
Antimycin A (2.5–80 µM), carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (0.16–5 µM), chloroquine (1–316 µM), oligomycin (1.25–40 µM), thapsigargin (0.13–4 µM), tunicamycin (0.31–10 µg/ml), and hydrogen peroxide (0.16–5 µM) were included in the high-content imaging experiments as controls with defined mechanism of toxicity (Supplementary Table 1). Final DMSO concentration of all treatments was 0.5%, which also served as control condition for all normalizations. Although DMSO is not generally given in vivo, the relatively high concentration of 0.5% DMSO is commonly used in vitro to ensure solubility of the toxic compounds. To ensure that DMSO itself had no significant effect on any of the readouts, medium without DMSO was included as a condition in all analyses and served as internal control.
For validation of the predictive models, six pharmaceutical compounds were selected by and donated by AstraZeneca (Figure 5A) (AZD6906, AZD6610, AZD5985, AZD8075, AZD7507, and AZD4282). These compounds were discontinued during development due to their preclinical or clinical kidney toxicity and were not marketed drugs. Additionally, six not previously used nontoxic control compounds were selected from the literature (Lin and Will, 2012) and purchased from Sigma-Aldrich and Enzo (antipyrine, atorvastatin, nadolol, candesartan, MK-801, and ramatroban). These 12 validation compounds were dissolved at 100 mM in DMSO or water, depending on solubility. Compound concentrations were selected based on each compound’s peak plasma concentration (Cmax), with 12 concentrations spanning 30× Cmax as the highest concentration and 0.06× Cmax as the lowest (Figure 5B).
Figure 5.

Validation of most predictive genes and imaging features in a set of 12 new compounds. (A) Six AstraZeneca (AZ) compounds were selected due to kidney toxic signals during development. Additional six compounds were chosen with similar pharmacological targets but no reports of kidney damage as nontoxic controls. (B) Concentration ranges (lines) of new nontoxic and AZ compounds used for following analyses with corresponding peak plasma concentrations (Cmax) (circle), spanning 12 concentrations from 30× to 0.006× Cmax. (C) Image analysis of most predictive imaging features (“Nucleus Profile 4/5 SER-Saddle_24h” and “Texture Nucleus SER-Saddle_24h”) at 24 h showed dose-dependent increase in z-score for five of the six AZ compounds. Data represent mean of four biological replicates performed in technical duplicates. Dotted line represents threshold for significant z-scores (−1.96 to 1.96), indicating two SD away from the mean of the control. (D) qRT-PCR results of HMOX1 (black) and SQSTM1 (gray) after treatment with the 12 new compounds for 24 h in low (1.3-fold Cmax), medium (6.2-fold Cmax), and high dose (30× Cmax). Data represent fold change to the average of the untreated control (dotted line) and is displayed as mean ± SD (n = 3). Multiple group comparison was conducted by one-way ANOVA followed by Dunnett’s post hoc test. p < .001 (***). (E) CellTiter Glo analysis of 12 validation compounds at 24 h showed dose-dependent decrease cell viability of two of the six AZ compounds. Data represent mean of three biological replicates performed in technical duplicates. (F) Scatter plot of z-score of Nucleus Profile 4/5 SER-Saddle against fold change of HMOX-1. Data represent maximal upregulation across all concentrations at 24 h. Dotted lines represent threshold for positive signal based on cut-offs derived from full compound library.
Cell viability measurements
Cell viability based on ATP concentrations was measured using the CellTiter-Glo (CTG) Luminescent Cell Viability Assay (Promega, Madison, Wisconsin). Cells were cultivated in 96-well plates for 3 days and then incubated with either a vehicle control (0.5% DMSO) or 46 toxic and nontoxic library compounds in six concentrations for 3, 6, 12, and 24 h (n = 3).
Association analyses
Binary logistic regression models were used to evaluate associations of the physicochemical properties, Cmax, and CTG results per time point with the odds of toxicity. Data were nontransformed and analyses were performed using Stata 13.0 (StataCorp, College Station, Texas).
High-content live-cell imaging
HPTECs were cultivated in 384-well plates for 3 days and then incubated with 30 µl of either a structural panel or a functional panel mixture of dyes in phenol red-free medium for 45 min. The structural panel consisted of MitoTracker Deep Red (0.5 µM) for mitochondria, ER-Tracker (0.5 µM) for endoplasmic reticulum (ER), LysoTracker (20 nM) for lysosomes, and Hoechst (0.5 µM) for nuclei. The functional panel consisted of TOTO-3 iodine (0.4 µM) for dead cells, tetramethylrhodamine (TMRM) (0.1 µM) for mitochondrial membrane potential (MMP), CellROX Green (4.5 µM) for reactive oxygen species (ROS), and Hoechst (0.5 µM) for nuclei. All dyes were purchased from Molecular Probes Inc. (Eugene, Oregon). After the staining, cells were washed twice with 50 µl phenol red-free medium and treated with 30 µl of either a vehicle control (0.5% DMSO), medium-only control, or toxic and nontoxic compounds diluted in phenol red-free DMEM cell culture medium in six concentrations.
Automated live-cell, multicolor image acquisition was performed on an Operetta High-Content Imaging System (Harmony software) with attached plate handler and live-cell incubator (Perkin Elmer, Waltham, Massachusetts) using a 20× objective with high numerical aperture (NA), in a single focal plane across each plate. The fluorescence images were captured according to the optimal excitation and emission wavelengths of each probe. Additionally, brightfield images were captured in both panels:
Structural plateMitoTracker Deep Red: 620–640/650–700 nm, exposure time 15 ms, focal plane −4 µM LysoTracker: 560–580/590–640 nm, exposure time 20 ms, focal plane −6 µM ER-Tracker: 460–490/500–550 nm, exposure time 20 ms, focal plane −6 µM Hoechst: 360–400/410–480 nm, exposure time 25 ms, focal plane −8 µM Brightfield: Transmission/650–700 nm, exposure time 35 ms, focal plane −6 µM
Functional plate TOTO-3 Iodine: 620–640/650–700 nm, exposure time 60 ms, focal plane −6 µM TMRM: 560–580/590–640 nm, exposure time 25 ms, focal plane −8 µM CellROX: 490–510/530–590 nm, exposure time 50 ms, focal plane −8 µM Hoechst: 360–400/410–480 nm, exposure time 25 ms, focal plane −8 µM Brightfield: Transmission/650–700 nm, exposure time 35 ms, focal plane −6 µM
Images were captured at 7 time points every 4 h, starting right after the treatment (0 h) to 24 h. Although it would have been ideal to also conduct chronic exposure to kidney toxicants, cell density of HPTECs was too high after 3 days to perform accurate image analysis. Two fields of view were imaged per well, capturing on average 1000 cells at 0 h and 1500 cells after 24 h. Quantitative image analysis was performed after automatic upload of the images to the Columbus 2.4.2 Software (Perkin Elmer). Based on the images obtained for each drug in the brightfield, nuclei, mitochondria, lysosomes, ER, ROS, TMRM, and dead cell channels, 629 numerical imaging features were analyzed, including nuclei count, area, and roundness, mitochondria intensity, standard deviation, texture, and contrast, ROS and ROS spots intensity, TMRM intensity, and texture. In brief, the Operetta algorithm “B” was used for nuclear segmentation based on Hoechst nucleus staining and algorithm “A” for detection of cytoplasmic area based on the respective mitochondria dyes in both staining panels (MitoTracker and CellROX). Second and third level parameters (eg, area, threshold, split factor, and contrast) were kept at the default setting of the software. Furthermore, the STAR method was selected to calculate a large and diverse set of morphology properties for phenotype classification and quantification of morphology changes. According to the software, “morphology” includes the outer shape of objects but also the distribution of intensity inside the objects as secondary papameters, eg, symmetry, profile, and threshold compactness. As third level parameters, we selected the Texture SER feature option. Detailed descriptions of the algorithms underlying the individual features can be found in the Columbus imaging software manual.
Four biological replicates were measured with technical duplicates per plate. Features were exported as mean of 2 fields/well and the data were normalized via calculating the z-scores to the average and standard deviation of the 8 vehicle (0.5% DMSO) treated control wells on each plate for each time point. Therefore, all the vehicle-treated wells in a 384-well plate had an average value of 0 in all imaging outputs and each drug-treated response is represented in positive or negative values relative to 0.
Gene expression profiling using L1000 platform
HPTECs were cultured in 96-well plates for 3 days and then treated with toxic and nontoxic compounds for 6 and 24 h. Cells were lysed with 100 µl of TCL buffer (Qiagen) and cell lysates were added to a TurboCapture 384 plate to perform mRNA isolation and cDNA synthesis in the same well (TurboCapture 384 mRNA kit, Qiagen). High-throughput gene expression analysis was performed based on the measurement of 1000 transcripts as described previously (Adler et al., 2016; Peck et al., 2006; Subramanian et al., 2017). The method involves ligation-mediated amplification (LMA) using locus-specific probes engineered to contain unique molecular barcodes, universal biotinylated primers, and 5.6-micron optically addressed polystyrene microspheres coupled to capture probes complementary to the barcode sequences (Duan et al., 2014). Specific probes were annealed to HPTEC cDNA, ligated by a Taq ligase, amplified, and then hybridized to barcoded Luminex beads. Beads are analyzed using Luminex FLEXMAP 3D instrument (Thermo Fisher) by identifying specific bead region and measuring the density of the hybridized probes on each bead with laser beams. Data were calibrated and normalized first to 80 invariantly expressed transcripts in each well and then to the controls treated with 0.5% DMSO in each plate.
Real-time PCR
HPTECs were cultured in collagen VI-coated 48-well plates for 3 days and then treated with toxic and nontoxic compounds for 24 h. Total RNA was isolated using RNeasy Mini Kit (Qiagen) and transcribed into cDNA with the QuantiTect Reverse Transcription Kit (Qiagen). Real-time PCR (qPCR) was carried out on QuantStudio 7 (Thermo Fisher) using QuantiFast SYBR Green PCR Kit (Qiagen). All samples were measured in duplicates and normalized to GAPDH. Changes in the mRNA expression were calculated using the ΔΔCt method relative to 0.5% DMSO control. Following primers were used: GAPDH_forward GAA GGT GAA GGT CGG AGT, GAPDH_reverse GAA GAT GGT GAT GGG ATT TC, HMOX1_forward AAG ACT GCG TTC CTG CTC AA, HMOX1_reverse GGG GGC AGA ATC TTG CAC TT, SQSTM1_forward AAG CCG GGT GGG AAT GTTG, and SQSTM1_reverse CCT GAA CAG TTA TCC GAC TCC AT.
Preparation of imaging and L1000 gene expression data for Random Forest machine learning
Random Forest (RF) machine-learning algorithms were used for the systematic identification of imaging features and genes with the highest predictive power. Clinical observations of kidney toxicity in humans (Supplementary Table 1) were used as the anchor (target variable) for the RF training and testing for the kidney toxicity classification. Class labels as nontoxic = 0 (10 instances, including 8 compounds, DMSO, and medium controls) or toxic = 1 (38 compounds) to the kidney were used. To account for the imbalance in the number of nontoxic versus toxic conditions, we applied balancing weights to the class labels during the training of each RF tree. The datasets were subjected to the following steps of clean-up. First, median values for every feature across all replicates of each compound × concentration combination were picked. Second, to capture the responses with the greatest magnitude for each compound, maximum vectors of the imaging and gene expression datasets were constructed for use with RF classifiers. For imaging data, z-scores with the greatest magnitude, maximum or minimum, for every feature across all doses of a compound were selected. For gene expression data, the maximum log2(fold changes) for every gene across all doses of a particular compound were selected. This produced a maximum vector for each time point with dimensions of n rows and f columns, corresponding to the number of compounds and features, respectively. Third, to remove noise from the data, gene expression maximum vectors were filtered such that log2(fold change) values between −1 and 1 were set to zero, and imaging maximum vectors were filtered such that z-score values between −1 and 1 were set to zero.
Finally, we chose early (4 h for imaging and 6 h for gene expression) and late (24 h for imaging and gene expression) time points from each dataset and concatenated the max vectors in order to produce two matrices with shapes of (n rows and 2f columns), where rows correspond to compounds and columns correspond to measured features at a particular time point.
Random Forest-based identification of predictive imaging features and genes
To score and select features for subsequent steps, the scikit-learn Python package was used (Pedregosa et al., 2011). Recursive feature elimination (RFE) was performed using a RF classifier, trained to predict whether compounds were toxic to proximal tubule cells or not. Maximum vectors were supplied to a k-fold cross-validation scheme, in which the maximum vector was split into two parts where a fraction of compounds was used to train the classifier, and the remainder of compounds were used to test and assess the performance of the classifier. This 9-fold cross-validation was repeated until the classifier had returned predictions for every compound in the dataset after training on the other compounds. One hundred different combinations of cross-validation splits were predefined, such that the same splits could be performed multiple times on imaging and gene expression data. For each of these cross-validation, split combinations RFE would be performed twice: once using the true class labels for compounds (toxic or not) and once using shuffled class labels, to compare how the model performs with real and randomized class labels. During each round of RFE, an RF model with 100 trees was fit, where each tree was trained on a balanced subsample of the data (ie, the class labels were weighed to account for the imbalanced number of nephrotoxic vs nonnephrotoxic compounds). The scikitlearn implementation of RF produces a variable importance score for each feature based on Gini impurity (Menze et al., 2009). The variables with the lowest scores were recursively cut from the dataset until only one variable remained. At each step, the importance for all variables and the predictions made by the model via cross-validation were recorded. Ultimately, this produced 100 different runs of the RFE trained on data with true and randomized class labels.
Performance for each run was computed as the area under the receiver operator characteristic (AUROC) curve. Statistical and visual comparisons were carried out via the seaborn Python package to determine that the performance of the model was better than random with true as opposed to randomized/scrambled class labels. Inspection of AUROC versus number of features plots depicting the decay in performance as features decreased. Here, this was at roughly 20 genes and imaging features. At this stage, the features were ranked in importance, and a cut-off was set around rank 20. The number of times each feature occurred at or below this rank cut-off was counted and used to sort the features for selection and use in further applications.
Network analysis of imaging data and hierarchical clustering
To generate a network that allows distinguishing between different mechanisms of toxicity, we selected 18 compounds with known mechanism of toxicity to create a “background network.” They consisted of 7 compounds with well-defined mechanism of action (CCCP, antimycin A, tunicamycin, chloroquine, hydrogen peroxide, oligomycin A, and thapsigargin) and additionally, 11 compounds from the library of kidney toxic compounds (Figure 6).
Figure 6.

Overview of the pipeline for classification of mechanisms of toxicity based on imaging data using network overlays. Selection of 18 background compounds with well-defined mechanism of toxicity were used to generate a “background network.” Using the complete high-content imaging dataset (six concentrations, seven time points) of these 18 compounds, a network was generated by displaying the dataset in two dimensions via embedding it into a k-nearest neighbors graph. Each compound or mechanistic group can be located separately in the network. The addition of high-content imaging data for one new compound, not part of the initial background, allows overlay with the existing, labeled network and thereby decoding of the potential mechanisms of toxicity.
The networks were created using the Ayasdi software platform (ayasdi.com, Ayasdi Inc., Menlo Park, California). This software employs a technique called Topological Data Analysis (TDA) to compare multidimensional features within a highly complex dataset and cluster based on similarities of the data, successfully used in a variety of scientific questions, eg, breast cancer subgroups (Nicolau et al., 2011). This approach displays high dimensional data (such as the imaging dataset with 629 numerical features for each of the 18 compounds at seven time points and six concentrations) as a 3D network in which each “node” comprises conditions similar to each other (eg, different concentrations of the same drug) in multiple dimensions. Lines or “edges” are drawn between nodes that contain similar and slightly overlapping data points. This method combines features of standard clustering methodologies and also provides a geometric representation of the data (Lum et al., 2013).
Here, TDA was performed using variance normalized Euclidean (VNE) distance as a metric to normalize the z-score data, considering that each column in the dataset could have significantly different variance. Subsequently, TDA used two filter functions, called “Neighborhood Lenses 1 and 2.” These filters generate an embedding of the high-dimensional VNE distance data into two dimensions by embedding a k-nearest neighbors graph of the data. A k-nearest neighbors graph is generated by connecting each point to its nearest neighbors. This graph is embedded in two dimensions using Ayasdi’s proprietary graph layout algorithm used in their visualizations. These filters work to emphasize the structure of the data.
Statistical methods and software
Unless otherwise indicated, data are presented as mean ± SD. Statistical difference (p < .05) as calculated by Student’s t test. Multiple group comparison was conducted by ANOVA followed by Dunnett’s post hoc test. p < .05 was considered significant and represented by * as compared with corresponding controls. All graphs were generated using GraphPad Prism (GraphPad, Inc., La Jolla, California) or JMP Pro 12.0.1 (SAS, Cary, North Carolina). Hierarchical clustering was performed using GeneE software (software.broadinstitute.org/GENE-E). Logistic regression analyses were performed using Stata 13.0 (StataCorp, College Station, Texas). RF analyses pipelines, report generation, and figure visualization were performed using Python.
Data and code availability
The gene expression and high-content imaging datasets generated during and/or analyzed during the current study, as well as Source code and Python scripts for RF analyses are available in the Dryad repository (https://doi.org/10.5061/dryad.646v2r1).
RESULTS
Overview of the Experimental Design
We generated a library of 46 drugs and chemicals, classified to cause drug-induced kidney injury (DIKI) (38) or nontoxic controls (8) (Figs. 1 and 2, Supplementary Table 1). Previously characterized human proximal tubular epithelial cells (HPTECs) (Adler et al., 2016) were challenged with six concentrations of each compound, selected to span or exceed the human maximal exposure to achieve robust toxic responses (Figure 2).
Figure 1.

Overview of data generation and prediction model construction.
To identify biomarkers that predict compound toxicity in a sensitive and specific manner, we generated two main datasets with the compound library:
Gene expression analysis was conducted in six concentrations after 6 and 24 h of treatment by measuring 978 transcripts (plus 22 housekeeping genes for normalization). These transcripts, called L1000, represent a selection of landmark genes characteristic of the variability of the transcriptome (Adler et al., 2016; Lamb, 2007). Due to high noise and only 80% confidence, we did not infer the L1000 data to the whole transcriptome. All 978 transcripts were used for RF feature reduction.
Automated high-content live-cell imaging was performed with the library compounds at six concentrations and seven time points (0–24 h, every 4h). We quantified and extracted 629 cellular imaging features, including changes in the structure, intensity, texture, and shape of the nucleus, mitochondria, ER, and lysosomes. We also measured three functional changes relevant to cell viability: MMP, ROS, and plasma membrane integrity (Figure 1). All 629 features were used for RF feature reduction and for mechanistic network analyses. In addition to the 46 library compounds, we imaged seven compounds that served as mechanistic controls, such as tunicamycin for ER-stress or antimycin A for mitochondrial damage (Supplementary Table 1).
Random Forest Machine Learning to Identify Genes and Imaging Features Predictive for Toxicity
Performance of the Random Forest model
The first aim of our study was to identify a panel of biomarkers across both gene expression and imaging datasets that are most accurate in distinguishing between kidney toxic and nontoxic compounds (Figure 1). We decided to apply a RF machine-learning algorithm for the systematic identification of imaging features and genes with the highest predictive power. RF produces highly accurate classifiers, it runs efficiently on large databases, and although it can handle thousands of input variables without variable deletion, it provides estimates of what variables are important in the classification (Breiman, 2001; Shi and Horvath, 2006). In a RFE step, the gene/imaging feature with the lowest importance score was removed and area under the receiver operating characteristic (AUROC) curve was monitored as performance parameter across the elimination cycles until only one gene/imaging feature was left (Figs. 3A and 3B). To assure that associations of selected genes/features with kidney toxicity were statistically significant and not due to chance, we randomized/scrambled the compound labels and ran the RF again (Figs. 3A and 3B). As expected, the scrambled run type generated on average AUROC values around 0.5, indicating no predictivity. Feature reduction of 978 genes at two time points (= 1956 total variables) yielded on average AUROC values of 0.8 (Figure 3A). This performance started to drop slightly when the elimination reached around 20 genes and went down to an AUROC of 0.6 with just one gene left. RF-based reduction of 629 imaging features at two time points (= 1258 total variables) performed better than the genes with average AUROC values of >0.9 (Figure 3B). Additionally, this good predictivity was maintained longer, and started dropping at around 3 imaging features until it reached an AUROC of 0.8 with just one feature left.
Figure 3.

Performance of Random Forest-based predictive model. Recursive feature elimination of 978 genes (A) and 629 imaging read-outs (B) at two time points. Results at each round of feature reduction are derived from the performance of 100 trees and are shown as average area under the receiver operating characteristic (AUROC) curve as lines with standard deviation as band. Identification of most predictive genes and imaging read-outs. Bar graphs display ranking of most predictive genes (C) and imaging features (D) based on the number of appearances among the last 20 features during recursive reduction shown as counts.
Identification of most predictive genes and imaging features
To identify genes and imaging features that were the least likely to get eliminated during the RF, the number of times each variable occurred at or below the last 20 RF cycles was counted and used to sort the features based on their counts (Figs. 3C and 3D).
Among all 978 genes across two time points (= 1956 total variables), heme oxygenase-1 (HMOX1 at 6 h and 24 h) and sequestosome-1 (SQSTM1 at 24h) appeared the most times among the most predictive 20 genes (Figure 3C). Both genes belong to the Nrf2 pathway and are relevant signaling hubs for diverse cellular events such as oxidative stress response (Agarwal and Bolisetty, 2013; Katsuragi et al., 2015). Overall, genes measured at 24 h performed better (33 of the most important 50 genes) than genes measured at the earlier 6 h time point (17/50). However, in addition to HMOX1, several genes appear on the list of final genes at both time points, including CDKN1A (Cyclin Dependent Kinase Inhibitor 1A), LYN (Lck/Yes novel tyrosine kinase), GADD45A (Growth arrest and DNA-damage-inducible protein alpha), and ARPP19 (cAMP-regulated phosphoprotein 19). Further genes that yielded high counts during the last 20 RF cycles include PMAIP1 (Phorbol-12-myristate-13-acetate-induced protein 1) and MMP1 (Matrix metalloproteinase-1) (Figure 3C).
Of all 629 imaging features at two time points (= 1258 total variables), Cell Radial Deviation SER-Dark, based on the mitochondrial dye and describing cell shape, ranked number one among the last most important features (Figure 3D). Additional features that are calculated based on changes in cell shape appeared 20× among the 50 highest ranked. Other important groups of features describe nucleus shape (7/50, eg, Nucleus Profile 4/4 SER-Saddle), nucleus texture (6/50, eg, Texture Nucleus SER-Saddle), and different mathematical ways to describe nucleus intensity (4/50), such as max, min, quantile, and mean). Additional features that seemed of high predictive importance include changes in the dye for ROS (both in “Spots,” labeling mitochondrial DNA, and throughout the “Cell”), and changes in MMP. Similar to the genes, the list of most relevant imaging features contained mostly features at the later time point at 24 h (41/50) rather than very early at 4 h (9/50) (Figure 3D).
Comparison of Most Predictive Genes and Imaging Features to Standard Toxicity Assays and Physicochemical Properties
Next, we wanted to directly compare how standard toxicity and cell viability assays perform in classifying the library of 46 kidney toxic and nontoxic compounds versus the genes and imaging features identified by the RF. Therefore, two new datasets were generated by analyzing all 46 library compounds for their ability to reduce cell viability (CellTiter Glo ATP assay) (Supplementary Data 2) and to increase the number of dead cells (uptake of TOTO-3 iodine dye). Similar to the gene expression and imaging dataset, a max vector (maximal response across each six-point dose-response curve) was build.
Using GraphPad Prism Software, we calculated ROC curves at 24 h for the two standard assays (Cell Viability, Cell Death), two highest ranked genes (HMOX1, SQSTM1), and three highest ranked imaging features (Cell Radial Deviation SER-Dark = “Cell Shape,” Texture Nucleus SER-Saddle = “Nucleus Texture,” and Nucleus Profile 4/4 SER-Saddle = “Nucleus Profile”) based on all 46 compounds (Figure 4A). The maximal achievable Youden Index (YI = Sensitivity + Specificity − 1) of each ROC curve was used to determine a cut-off for each of the seven readouts at which the accuracy of the prediction reached a maximum (Figure 4A).
Figure 4.

Performance of standard toxicity assays compared with RF-derived most predictive genes and imaging features. (A) Receiver operating characteristic curve (ROC) for (1) standard toxicity assays (cell viability: Celltiter Glo ATP assay; cell death: number of dead cells based on TOTO-3 iodine dye), (2) predictive genes, identified by RF feature reduction (HMOX1 and SQSTM1), and (3) imaging features identified by RF feature reduction (Cell Radial Deviation SER-Dark, Texture Nucleus SER-Saddle, Nucleus Profile 4/5 SER-Saddle). Data represent maximal upregulation of 38 kidney toxic compounds and 8 nontoxic controls across all concentrations at 24 h. Area under the ROC curve (AUROC) was calculated using GraphPad Prism software. Cut-off was calculated using the maximum YI (Youden Index = Sensitivity +Specificity − 1). (B) ROC-derived cut-offs were used as a threshold to calculate number of False Positive (FP), True Positive (TP), False Negative (FN), and True Negative (TN) compounds. Sensitivity or True Positive Rates (TPR) were calculated as #True Positive/All Positive. Specificity or True Negative Rates (TNR) were calculated as #True Negative/All Negative. Accuracy was calculated as (TP + TN)/(TP + FP + FN + TN).
In a second step, the ROC curve-derived cut-offs were applied to the max values of each compound to determine the number of compounds correctly or incorrectly classified (Figure 4B). Four of the seven assays misclassified one of the eight nontoxic compounds as toxic, whereas Number of Dead Cells, Nucleus Texture and Nucleus Profile showed 100% Specificity. All assays failed to identify a certain number of toxic compounds correctly, ranging from a maximum of 12 false negative compounds using Cell Viability and Number of Dead Cells, to a minimum of only 6 false negative compounds using Nucleus Profile (Figure 4B). Overall, the standard assays performed worst with AUROCs of 0.829 for Cell Viability and 0.791 for number of dead cells, whereas Nucleus Profile and Nucleus texture performed best with AUROCs of 0.92 (Figure 4A).
The only toxic compound misclassified by all seven assays was acetaminophen, consistent with previous observations in HPTEC cells showing a lack of necessary drug metabolizing enzymes, thereby preventing this drug from exhibiting nephrotoxicity in vitro (Adler et al., 2016). This was confirmed by the correct identification of 4-aminophenol, an acetaminophen metabolite responsible for toxicity, by all seven assays (Figure 4B).
In addition to standard toxicity assays, we were interested whether physicochemical properties of our library compounds could serve as predictors of toxicity. Therefore, a set of 10 standard physicochemical properties, such as lipophilicity, polar surface area, molecular weight, acidity, and maximal plasma concentration (Cmax) was collected for all 46 compounds. Yet, logistic regression modeling showed that none of the parameters was associated with increased odds for a compound to be nephrotoxic in humans (Supplementary Data 3a).
Finally, because many of the extracted imaging features were derived via algorithms specific to the Columbus analysis software, we calculated the predictive performance of an imaging feature that can easily be translated to any imaging software. “Nucleus Intensity Maximum,” is calculated based on the intensity histogram in the nucleus mask and describes the maximum intensity in the Hoechst channel as an average per well. Nucleus Intensity Max ranked number 16 of 629 in the list of most important features (Figure 3D), but it generated a Sensitivity of 86.8% and Specificity of 75% (Supplementary Data 3b). The AUROC of 0.913 was very competitive to Nucleus Profile and texture with 0.92.
Validation of RF-Derived Genes and Imaging Features by Predicting Toxicity in a Set of 12 New Compounds
To further test the RF-derived genes and imaging features that seem to perform better than standard assays in a library of 46 drugs and chemicals, we performed external validation by measuring the two highest ranked genes and two best performing imaging features in a set of 12 new compounds at 24 h. Six of the compounds had previously been discontinued by AstraZeneca (AZ), either in clinical or preclinical development, due to kidney-related findings that were not anticipated based on early toxicological in vitro testing (Figure 5A). Additional six compounds were selected because they had no reports on kidney toxicity in patients and some have overlapping targets with the AZ candidates: MA-801/Dizocilpine is an NMDA receptor antagonist like AZD4282, Ramatroban is a prostaglandin D2 receptor CRTH2 antagonist like AZD8057 and AZD5985. All concentration ranges were based on the respective plasma Cmax of the compounds, with 30× Cmax as the highest concentration (Figure 5B). Figure 5C shows a dose-dependent significant upregulation of the two most predictive imaging features (Nucleus Profile and Nucleus Texture) after treatment with four of the six AZ compounds, ranging from a mean z-score increase of 2.0–13.4. Notably, none of the six nontoxic controls crossed the line of 1.96 for a significant z-score. Of the two most predictive genes, HMOX1 showed a significant increase after exposure to all of the six toxic AZ compounds, varying in extend from a mean upregulation of 1.8–96-fold compared with vehicle control (Figure 5D). SQSTM1 was significantly upregulated for three of the six toxic AZ compounds, ranging from a mean upregulation of 5.6–20.4-fold.
To allow benchmarking against the currently used standard method of toxicity prediction, we also measured the 12 new compounds at the same concentrations using the ATP Cell Viability assay (CellTiter Glo) after 24 h of exposure. Again, none of the six nontoxic controls showed a decrease in cell viability up to 30× Cmax (Figure 5E). However, only two of the six toxic AZ compounds significantly decreased viability of the cells, and this loss in cell viability was only detectable at concentrations much higher than required for HMOX1 or Nucleus imaging feature upregulation (Figure 5E). Finally, integration of the two types of readouts by plotting the maximum upregulation of the Nucleus Profile imaging feature against HMOX1 allowed clear separation of AZD4282, AZD8075, AZD6906, and AZD5985 from the nontoxic control compounds (Figure 5F) by applying the same ROC-derived cut-offs that were determined previously with the 46 compound library (Figure 4B).
Organ Specificity of Nucleus Texture Imaging Feature
A direct comparison of a subset of compounds in both liver (HepG2) and kidney (HPTEC) cells showed that the Nucleus Texture feature successfully detected the liver toxicant (acetaminophen) and the kidney toxicant (gentamicin) in the correct cell type while general toxicants (aflatoxin B1 and cadmium chloride) induced upregulation of Nucleus Texture in both cell types (Supplemental Data 3c).
Topological Data Analysis to Identify Compound-Induced Mechanisms of Toxicity
The second aim of this study was to classify compounds by their potential mechanism of toxicity with the help of network analysis. To this end, we selected 18 compounds with well-described mechanism of toxicity and used them to generate a “Background Network” in the Ayasdi software that applies TDA to build networks from multidimensional data (Figure 6). The complete high-content imaging dataset, consisting of 629 imaging features for seven time points and six concentrations of the selected 18 compounds and nontoxic conditions (0.5% DMSO control, aspirin, and sulfamethoxazole), was uploaded to the software and embedded in a 3D network based on their k-nearest neighbor similarity. Of the 12 mechanistic groups making up the “Background Network,” seven were represented by one compound, such as oligomycin A for Mitochondrial ATP-synthase inhibition (Symersky et al., 2012), tunicamycin for ER-stress via protein accumulation (Oslowski and Urano, 2011), cyclosporine A for Immunomodulation (Yocum, 1996), and phenacetin for Analgesic nephropathy (Murray, 1972). The remaining six assigned mechanisms were represented by two compounds each, such as cadmium chloride and hydrogen peroxide for ROS and Apoptosis (Khojastehfar et al., 2015), chloroquine & gentamicin for Lysosomal disorders (Anderson and Borlak, 2006), and cisplatin & doxorubicin for DNA intercalation (Cheung-Ong et al., 2013) (Figure 6).
Locating the 12 groups in the network showed that individual mechanistic clusters exhibited little overlap and presented distinct flares in the “Background Network” (Figure 7A). Only groups with mechanisms in which a certain degree of similarity would be expected, as in Mitochondrial ATP-synthase inhibition & ER-stress via Ca2+ ATPase inhibition, showed overlap of their location in the network (Figure 7A).
Figure 7.

Network analysis of imaging data to identify mechanistic clusters. (A) Network of 18 background compounds with separation of 12 mechanistic groups based on complete high-content imaging dataset (six concentrations, seven time points). Nodes in the network represent clusters of compounds with similar imaging profiles and edges connect nodes that contain samples in common. Coloring represents concentration of the mechanistic category in each node as the −log10(p value) and ranks from 0.2 (blue) to 67 (red). (B) Overlay of new compounds on the background network to infer mechanistic information. Examples displayed here: Tacrolimus only overlaps with Immunomodulation (= cyclosporine A), Tobramycin overlaps with lysosomal disorders (= gentamicin, chloroquine), necrosis (= p-aminophenol, amphotericin), and mitochondrial respiration (= CCCP, antimycin A). (C) Two-way hierarchical clustering of kidney toxic compounds based on their overlap (blue label) with one or more of the 12 network-based groups shows formation of clusters of compounds with similar mechanism.
In a next step, the remaining 25 library compounds were individually overlaid on the background landscape by creating one new network analysis per compound. For this reason, each of the 25 new networks looked slightly different than the original Background Network as it had the additional dataset of one more compound to consider. The overlap with the 12 previously defined mechanistic groups allowed inference of information about the new molecule (Supplementary Data 4). Figure 7B shows the overlay of two example compounds on the Background Network. Tobramycin nodes overlapped with the clusters associated with Lysosomal disorders, Necrosis, and Mitochondrial respiration, which is consistent with effects of tobramycin seen in preclinical and clinical studies (Mingeot-Leclercq and Tulkens, 1999). Tacrolimus, as a second example, only overlapped with the cluster labeled Immunosuppression. The background compound underlying this cluster was cyclosporine A, a compound that is structurally unrelated to tacrolimus and binds to different cytosolic proteins in target cells (Randhawa et al., 1997), yet, both act as immunosuppressive drugs and both have been reported to induce kidney toxicity. Therefore, although the molecular basis of nephrotoxicity is not as well understood for these two compounds, using high-content imaging data and the comparative network analysis, we were able to confirm their overlap in toxicity profile (Randhawa et al., 1997).
In a last step, all overlaps with one or more of the 12 mechanistic groups for the 46 kidney-toxic drugs and chemicals plus the seven additional mechanistic compounds were submitted as a binary matrix to two-way hierarchical clustering (Figure 7C). Despite integrating concentrations and time points into the initial network analysis at which no apparent cytotoxicity was detectable, the analysis was able to cluster about 50% of compounds into groups with distinct labels. These groups include for example a cluster with six compounds that are all known to induce DNA damage (aristolochic acid, carboplatin, cisplatin, doxorubicin, idarubicin, and mitomycin C). Another cluster includes four different analgesics (ibuprofen, phenacetin, acetaminophen, and N-phenylanthranilic acid) and a third cluster includes three antibiotics of the library (gentamicin, tobramycin, and bacitracin) that are all inducing lysosomal damage. The remaining compounds showed overlap with a mixture of different mechanisms (eg, citrinin overlapped both with Apoptosis/ROS and with Necrosis) and were therefore harder to capture under one term. However, these mixed results are both a reflection of the multifaceted behavior of toxic chemicals in the cells and also capture the time- and dose-dependency of the imaging data across six doses and seven time-points.
Overlap of AZ Compounds with Imaging Network to Infer Mechanism of Toxicity
In addition to using the individual genes and imaging features to predict the potential for drug-induced kidney toxicity (Figure 5), we also used the complete imaging dataset (629 features, 7 time points, 12 concentrations) of the six AZ compounds to perform network analysis for identification of potential mechanisms of toxicity. To that end, we overlaid each AZ compound onto a network based on all of the library compounds, instead of just the 18 background compounds.
Figure 8 shows the six networks that were formed by adding the AZ compounds one at a time. Circles indicate the location of nodes where the respective AZ compound clusters and they were used to find overlaps with other drugs in the network.
Figure 8.

Overlap of AZ compounds with the network of kidney toxic library compounds. Coloring represents concentration in the respective drug in each node as the −log10(p value) and ranks from 0.2 (blue) to 67 (red). Circles indicate the location of nodes where an AZ compound clusters and were used to find overlaps in the network of new compounds with known library compounds to infer mechanistic information.
AZD6906, a GABAB receptor agonist, showed distinct overlap with calcineurin inhibitors cyclosporine A and tacrolimus. The imaging feature responsible for the formation of this cluster was an increase in MMP compared with the rest of the network. AZD4282, a NMDA receptor antagonist, partly overlapped with paclitaxel and cadmium chloride. AZD8075, a Prostaglandin D2 receptor (CrTh2) antagonist, showed overlap with four DNA-damaging compounds, which was a very different profile than the second compound with the same pharmacological target, AZD5985, which overlapped with compounds related to analgesic nephropathy and inhibition of ATP synthesis (N-phenylanthranilic acid, indomethacin, and rapamycin). The last two compounds, AZD7507 and AZD6610, were not identified as toxic in the predictive analyses, however, in the network analysis, they still formed separate clusters and did not overlap with the nontoxic controls. Instead, AZD6610 showed overlap with compounds like potassium dichromate and lead (IV) acetate, known to induce cell stress and apoptosis. Therefore, imaging-based network analysis could add sensitivity to a pure biomarker-based prediction.
Because these six compounds were discontinued in their development and no details on their mechanism of kidney toxicity are available, we could not confirm whether the overlaps we identified in the network analysis are accurate. However, these overlaps and similarities with known toxicants could be used to help identify potential interactions of unknown compounds with cells and thereby support toxicity mitigation strategies in early drug development.
DISCUSSION
The etiology and pathophysiology of AKI are complex and often multifaceted, (Makris and Spanou, 2016) and it is this complexity that underlies the challenge of developing new treatments for AKI (Gallagher et al., 2017). Notably, the complex physiology of the kidney also renders it susceptible to a wide range of toxic insults, an important consideration in early development of drugs and chemicals. Currently, there is no well-established in vitro method that screens large number of compounds for their potential to induce kidney toxicity. Therefore, this study aimed to develop an unbiased approach that combines multidimensional datasets and machine learning to identify biomarkers that not only predict nephrotoxic compounds but also provide hints toward their mechanisms of toxicity.
Machine Learning to Identify Biomarkers
To avoid the pitfalls associated with either studying a small number of compounds (limiting the generalizability of results to compounds with disparate mechanisms) or employing a restricted set of analytic readouts (Loo and Zink, 2017; Wilmes et al., 2013), we accepted a diverse range of compounds’ dose to response based on Cmax, time to response, mechanism of action, and even degrees of evidence for kidney toxicity in humans.
RF identified two genes, HMOX1 and SQSTM1, as effective discriminators between toxic and nontoxic compounds. Both genes belong to the Nrf2 pathway and serve as signaling hubs for diverse cellular events such as oxidative stress response (Agarwal and Bolisetty, 2013; Katsuragi et al., 2015). Notably, both genes and their products have previously been implicated in renal injury response. Upregulated HMOX1 and SQSTM1 expression has been observed in vitro as a feature of cellular stress responses to toxic chemicals (Adler et al., 2016; Alegre et al., 2018), and greater HMOX1 expression has also been observed in the setting of clinical AKI and inflammatory renal disease (Yokoyama et al., 2011; Zager et al., 2012). Furthermore, HMOX1 upregulation is seen as part of the renal response to ischemia reperfusion injury across several species (Grigoryev et al., 2013), and SQSTM1 has been implicated in preventing oxidative stress-induced apoptosis in kidneys of patients receiving cisplatin (Xia et al., 2014). In addition, numerous other genes ranked by the RF as being associated with nephrotoxicity (eg, PMAIP1, CDKN1A, MMP1, GADD45A) have all previously been reported as important components of cellular stress responses including apoptosis, DNA damage response, and remodeling of the extracellular matrix (Lenz et al., 2000; Oda et al., 2000).
The highest ranked imaging features identified by the RF did not include those readouts expected to be most tightly associated with cellular toxicity, such dead cell number, ROS induction, or loss of MMP. This might be because these commonly assessed endpoints are often too selective and only perform well for a subset of toxicants with a particular target (McKim, 2010). Rather, the unbiased feature selection identified more general readouts as highly predictive of toxicity, including cellular shape and nuclear texture (Figure 3D). This finding is consistent with previous reports that changes in chromatin and actin cytoskeleton are highly predictive of the nephrotoxicity of xenobiotics (Su et al., 2016). Changes to nucleus texture can occur for a wide variety of reasons, including DNA damage or simply apoptosis (Ziegler and Groscurth, 2004). In our work, such changes were predictive of the toxicity of compounds affording a broad range of insults, including DNA damage, activation of oxidative stress pathways, and lysosomal damage.
The strength of detecting a broad range of toxic cellular insults can also be a disadvantage because it can limit organ specificity of the prediction. Using biomarkers of general cell stress rather than pathways specific for kidney cell damage could lead to an increased number of false-positive hits as every cytotoxic compound could be identified as positive. However, a direct comparison of a subset of compounds in both liver (HepG2) and kidney (HPTEC) cells showed that the Nucleus Texture feature successfully detected the liver toxicant (acetaminophen) and the kidney toxicant (gentamicin) in the correct cell type while general toxicants (aflatoxin B1 and cadmium chloride) induced upregulation of Nucleus Texture in both cell types (Supplementary Data 3c). These data suggest that our Nucleus Texture imaging biomarker could potentially be used in liver cells for the detection of hepatotoxicity, which also poses a significant problem with candidate compounds in drug development.
This result demonstrates that unbiased phenotypic profiling, not geared toward prespecified mechanisms or narrow endpoints, can identify predictors of toxicity that have broader applicability across a diverse chemical space.
Understanding mechanisms for de-risking
Here we employed network comparison based on TDA as a machine-learning framework to classify compounds by mechanism of action. This approach accurately clustered many of the compounds into distinct groups with overlapping toxicity, such as DNA damaging agents, aminoglycoside antibiotics, analgesics, and immunosuppressants. Additionally, even for compounds that did not fall into a specific cluster, we could make important mechanistic predictions. As an example, diatrizoic acid, a radiocontrast medication, clustered with drugs inducing DNA/RNA damage and lysosomal pathology, consistent with previous reports that (1) this hypertonic agent induces DNA fragmentation in renal tubular cells (Hizoh et al., 1998); and (2) that radioconstrast agents can induce reversible lysosomal alterations in rat kidneys representing first signs of cellular injury (Tervahartiala et al., 1991).
Another advantage of mechanistic clustering is that it might complement quantitative readouts for toxicity prediction to improve assessment of potentially misclassified drugs. For example, acetaminophen was not detected in the toxicity panel due to a lack of hepatic biotransformation, which is a limitation of an in vitro system with kidney cells only. However, in the mechanistic clustering based on all high-content imaging data, acetaminophen still overlapped with other analgesics, demonstrating the superiority of examining both toxicity prediction and mechanism. A second toxic and yet negatively classified compound was tenofovir, likely due to a low activity of its uptake transporter OAT1 in our cells (Adler et al., 2016; Kohler et al., 2011). However, based on our imaging-based network analysis, we could again identify changes in the mitochondria and similarities to analgesic-induced tubulointerstitial nephritis. This is consistent with observations in patients, where tenofovir toxicity presents with the morphologic abnormalities in proximal tubule mitochondria and renal Fanconi-Syndrome, a milder case of tubulointerstitial nephritis (Hall et al., 2011).
A potential caveat in this analysis is that our method is based on overlaps with a network of known compounds. This renders the approach highly flexible but the mechanistic insight gained is only as good as the mechanistic coverage of the library compounds. Second, rather than getting specific activated or deactivated pathways as readouts, our imaging panel yields information on organelle changes, ROS, and loss of MMP. That said, the main advantage lies in the depth of data obtained providing unique ability to pick up both early changes such as development of oxidative stress and lysosomal alterations, as well as late damage such as necrosis. Furthermore, by manually assessing the overlap of new compounds with the background network, new compounds need not be matched to only one mechanistic category. Rather, it is acknowledged that one compound can cluster with several different mechanisms of cellular response—partly because of multifactorial mechanisms of toxicity, and partly because of changes in cell response to injury over time.
Applying Biomarkers and Mechanistic De-risking Strategy to Therapeutic Candidates
In our validation study, using one nucleus texture imaging feature and HMOX1 gene expression, we correctly classified four of six toxic compounds whose adverse effects were previously only detected in preclinical and clinical studies. The performance of newly identified biomarkers was significantly better than standard cell viability readouts, which could only identify toxicity when using the highest concentration of two of the six compounds.
It can be speculated that the false negative classifications may be due to limitations in transporters or biotransformation in the kidney cell system employed. For example, AZD6610 is metabolized in vivo through hepatic CYP450 and UDP-glucuronosyltransferase (UGT) enzymes (Darnell et al., 2012), which could have led to a lack of toxic metabolites in our in vitro system. Further, the chemical structure of the second compound, AZD7507, contains an amino group that might, in its cationic form (), bind to the megalin transporter located at the brush-border membrane and expressed in HPTECs (Adler et al., 2016). Subsequent internalization of the megalin-bound compound by endocytosis could achieve 100–1000 times higher drug concentrations in the proximal tubule cells that observed in serum (Mingeot-Leclercq and Tulkens, 1999), such that our Cmax-based concentration range would be underestimating the actual intracellular concentrations. The mechanistic network analysis of all of the six drugs showed at least a partial overlap with some of the kidney toxic library compounds. Because these six compounds were discontinued in their development and no details on their mechanism of kidney toxicity were available, we could not confirm whether the identified overlaps in the network are accurate.
In summary, using RF feature selection and comparative network analysis across two large omics datasets, we developed an integrative systems toxicology approach that enabled two goals: (1) A strict feature reduction, supported by highly accurate machine learning algorithms, allowed the identification of HMOX1 gene expression in combination with nucleus texture imaging feature as robust biomarkers to predict drug toxicity. (2) Measurement of as many readouts as possible, while using a limited, yet diverse set of compounds, allowed insight into the potential mechanisms of toxicity. Compounds flagged by this in vitro screening can then be triaged for further testing in lower throughput repeat-dose in vitro studies, 3D microphysiological systems, or preclinical in vivo assays.
CONFLICT OF INTEREST
SR and JTM are full-time employees of AstraZeneca, MP is a full-time employee of Bayer, VSV is a full-time employee of Pfizer, HS is a full-time employee of Merck, and WWC is a full-time employee of Biogen.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Marc Hafner for helpful discussions and advice on cross-validation methods. Furthermore, the authors thank Shom Goel for reviewing and providing feedback on the manuscript. The authors also acknowledge a collaborative research agreement with Ayasdi, who kindly provided software, training, and support for TDA analyses.
FUNDING
Work in the Vaidya laboratory is supported by Outstanding New Environmental Sciences (ONES) award from NIH/NIEHS (ES017543) and Innovation in Regulatory Science Award from Burroughs Wellcome Fund (BWF-1012518). Further funding was provided by a Regulatory Science Ignition Award from Harvard Program in Therapeutic Sciences at Harvard Medical School, National Institutes of Health (NIH) (UH3-TR000504), and Merrimack Pharmaceuticals. The work was also supported by a collaborative research agreement with AstraZeneca and resources made available through the NIH LINCS (U54-HG006097 to V.S.V.); and Sao Paulo Research Foundation (Fapesp 2016/04935-2 to M.B.M.).
REFERENCES
- Adler M., Ramm S., Hafner M., Muhlich J. L., Gottwald E. M., Weber E., Jaklic A., Ajay A. K., Svoboda D., Auerbach S. (2016). A quantitative approach to screen for nephrotoxic compounds in vitro. J. Am. Soc. Nephrol. 27, 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A., Bolisetty S. (2013). Adaptive responses to tissue injury: Role of heme oxygenase-1. Trans. Am. Clin. Climatol. Assoc. 124, 111–122. [PMC free article] [PubMed] [Google Scholar]
- Alegre F., Moragrega A. B., Polo M., Marti-Rodrigo A., Esplugues J. V., Blas-Garcia A., Apostolova N. (2018). Role of p62/SQSTM1 beyond autophagy: A lesson learned from drug-induced toxicity in vitro. Br. J. Pharmacol. 175, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N., Borlak J. (2006). Drug-induced phospholipidosis. FEBS Lett. 580, 5533–5540. [DOI] [PubMed] [Google Scholar]
- Breiman L. (2001). Random Forests. Mach. Learn. 45, 5–32. [Google Scholar]
- Cherkasov A., Muratov E. N., Fourches D., Varnek A., Baskin I. I., Cronin M., Dearden J., Gramatica P., Martin Y. C., Todeschini R. (2014). QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 57, 4977–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Ong K., Giaever G., Nislow C. (2013). DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 20, 648–659. [DOI] [PubMed] [Google Scholar]
- Choudhury D., Ahmed Z. (2006). Drug-associated renal dysfunction and injury. Nat. Clin. Pract. Nephrol. 2, 80–91. [DOI] [PubMed] [Google Scholar]
- Cook D., Brown D., Alexander R., March R., Morgan P., Satterthwaite G., Pangalos M. N. (2014). Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. 13, 419–431. [DOI] [PubMed] [Google Scholar]
- Darnell M., Ulvestad M., Ellis E., Weidolf L., Andersson T. B. (2012). In vitro evaluation of major in vivo drug metabolic pathways using primary human hepatocytes and HepaRG cells in suspension and a dynamic three-dimensional bioreactor system. J. Pharmacol. Exp. Ther. 343, 134–144. [DOI] [PubMed] [Google Scholar]
- Duan Q., Flynn C., Niepel M., Hafner M., Muhlich J. L., Fernandez N. F., Rouillard A. D., Tan C. M., Chen E. Y., Golub T. R., et al. (2014). LINCS canvas browser: Interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 42, W449–W460. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher K. M., O’neill S., Harrison E. M., Ross J. A., Wigmore S. J., Hughes J. (2017). Recent early clinical drug development for acute kidney injury. Expert Opin. Investig. Drugs 26, 141–154. [DOI] [PubMed] [Google Scholar]
- Grigoryev D. N., Cheranova D. I., Heruth D. P., Huang P., Zhang L. Q., Rabb H., Ye S. Q. (2013). Meta-analysis of molecular response of kidney to ischemia reperfusion injury for the identification of new candidate genes. BMC Nephrol. 14, 231.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. M., Hendry B. M., Nitsch D., Connolly J. O. (2011). Tenofovir-associated kidney toxicity in HIV-infected patients: A review of the evidence. Am. J. Kidney Dis. 57, 773–780. [DOI] [PubMed] [Google Scholar]
- Hizoh I., Strater J., Schick C. S., Kubler W., Haller C. (1998). Radiocontrast-induced DNA fragmentation of renal tubular cells in vitro: Role of hypertonicity. Nephrol. Dial. Transpl. 13, 911–918. [DOI] [PubMed] [Google Scholar]
- Katsuragi Y., Ichimura Y., Komatsu M. (2015). p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282, 4672–4678. [DOI] [PubMed] [Google Scholar]
- Khojastehfar A., Aghaei M., Gharagozloo M., Panjehpour M. (2015). Cadmium induces reactive oxygen species-dependent apoptosis in MCF-7 human breast cancer cell line. Toxicol. Mech. Methods 25, 48–55. [DOI] [PubMed] [Google Scholar]
- Kleinknecht D., Landais P., Goldfarb B. (1987). Drug-associated acute renal failure. A prospective collaborative study of 81 biopsied patients. Adv. Exp. Med. Biol. 212, 125–128. [DOI] [PubMed] [Google Scholar]
- Kohler J. J., Hosseini S. H., Green E., Abuin A., Ludaway T., Russ R., Santoianni R., Lewis W. (2011). Tenofovir renal proximal tubular toxicity is regulated by OAT1 and MRP4 transporters. Lab. Investig. 91, 852–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewski D., Acosta D. Jr, Andersen M., Anderson H., Bailar J. C. 3rd, Boekelheide K., Brent R., Charnley G., Cheung V. G., Green S. Jr (2010). Toxicity testing in the 21st century: A vision and a strategy. J. Toxicol. Environ. Health B Crit. Rev. 13, 51–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J. (2007). The connectivity map: A new tool for biomedical research. Nat. Rev. 7, 54–60. [DOI] [PubMed] [Google Scholar]
- Lenz O., Elliot S. J., Stetler-Stevenson W. G. (2000). Matrix metalloproteinases in renal development and disease. J. Am. Soc. Nephrol. 11, 574–581. [DOI] [PubMed] [Google Scholar]
- Lin Z., Will Y. (2012). Evaluation of drugs with specific organ toxicities in organ-specific cell lines. Toxicol. Sci. 126, 114–127. [DOI] [PubMed] [Google Scholar]
- Loo L. H., Zink D. (2017). High-throughput prediction of nephrotoxicity in humans. Altern. Lab. Anim. 45, 241–252. [DOI] [PubMed] [Google Scholar]
- Lum P. Y., Singh G., Lehman A., Ishkanov T., Vejdemo-Johansson M., Alagappan M., Carlsson J., Carlsson G. (2013). Extracting insights from the shape of complex data using topology. Sci. Rep. 3, 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris K., Spanou L. (2016). Acute kidney injury: Definition, pathophysiology and clinical phenotypes. Clin. Biochem. Rev. 37, 85–98. [PMC free article] [PubMed] [Google Scholar]
- McKim J. M., Jr (2010). Building a tiered approach to in vitro predictive toxicity screening: A focus on assays with in vivo relevance. Comb. Chem. High Throughput Screen. 13, 188–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menze B. H., Kelm B. M., Masuch R., Himmelreich U., Bachert P., Petrich W., Hamprecht F. A. (2009). A comparison of Random Forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinformatics 10, 213.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P., Tulkens P. M. (1999). Aminoglycosides: Nephrotoxicity. Antimicrob. Agents Chemother. 43, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray R. M. (1972). Analgesic nephropathy: Removal of phenacetin from proprietary analgesics. Br. Med. J. 4, 131–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myshkin E., Brennan R., Khasanova T., Sitnik T., Serebriyskaya T., Litvinova E., Guryanov A., Nikolsky Y., Nikolskaya T., Bureeva S. (2012). Prediction of organ toxicity endpoints by QSAR modeling based on precise chemical-histopathology annotations. Chem. Biol. Drug Des. 80, 406–416. [DOI] [PubMed] [Google Scholar]
- Nicolau M., Levine A. J., Carlsson G. (2011). Topology based data analysis identifies a subgroup of breast cancers with a unique mutational profile and excellent survival. Proc. Natl. Acad. Sci. U.S.A. 108, 7265–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council. (2007). Toxicity Testing in the 21st Century: A Vision and a Strategy. The National Academic Press, Washington, D.C. [Google Scholar]
- Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. (2000). Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058. [DOI] [PubMed] [Google Scholar]
- Oslowski C. M., Urano F. (2011). Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck D., Crawford E. D., Ross K. N., Stegmaier K., Golub T. R., Lamb J. (2006). A method for high-throughput gene expression signature analysis. Genome Biol. 7, R61.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F., Varoquaux G., Alexandre G., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., et al. (2011). Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- Ramm S., Adler M., Vaidya V. S. (2016). A high-throughput screening assay to identify kidney toxic compounds. Curr. Protoc. Toxicol. 69, 9 10 1–9 10 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa P. S., Starzl T. E., Demetris A. J. (1997). Tacrolimus (FK506)-associated renal pathology. Adv. Anat. Pathol. 4, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T., Horvath S. (2006). Unsupervised learning with Random Forest predictors. J. Comput. Graph. Statist. 15, 118–138. [Google Scholar]
- Su R., Xiong S., Zink D., Loo L. H. (2016). High-throughput imaging-based nephrotoxicity prediction for xenobiotics with diverse chemical structures. Arch. Toxicol. 90, 2793–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Narayan R., Corsello S. M., Peck D. D., Natoli T. E., Lu X., Gould J., Davis J. F., Tubelli A. A., Asiedu J. K., et al. (2017). A next generation connectivity map: l 1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symersky J., Osowski D., Walters D. E., Mueller D. M. (2012). Oligomycin frames a common drug-binding site in the ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 109, 13961–13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervahartiala P., Kivisaari L., Kivisaari R., Virtanen I., Standertskjöld-Nordenstam C. G. (1991). Contrast media-induced renal tubular vacuolization. A light and electron microscopic study on rat kidneys. Invest. Radiol. 26, 882–887. [DOI] [PubMed] [Google Scholar]
- Vaidya V. S., Ferguson M. A., Bonventre J. V. (2008). Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 48, 463–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes A., Limonciel A., Aschauer L., Moenks K., Bielow C., Leonard M. O., Hamon J., Carpi D., Ruzek S., Handler A., et al. (2013). Application of integrated transcriptomic, proteomic and metabolomic profiling for the delineation of mechanisms of drug induced cell stress. J. Proteom. 79, 180–194. [DOI] [PubMed] [Google Scholar]
- Xia M., Yu H., Gu S., Xu Y., Su J., Li H., Kang J., Cui M. (2014). p62/SQSTM1 is involved in cisplatin resistance in human ovarian cancer cells via the Keap1-Nrf2-ARE system. Int. J. Oncol. 45, 2341–2348. [DOI] [PubMed] [Google Scholar]
- Yocum D. E. (1996). Cyclosporine, FK-506, rapamycin, and other immunomodulators. Rheum. Dis. Clin. North Am. 22, 133–154. [DOI] [PubMed] [Google Scholar]
- Yokoyama T., Shimizu M., Ohta K., Yuno T., Okajima M., Wada T., Toma T., Koizumi S., Yachie A. (2011). Urinary heme oxygenase-1 as a sensitive indicator of tubulointerstitial inflammatory damage in various renal diseases. Am. J. Nephrol. 33, 414–420. [DOI] [PubMed] [Google Scholar]
- Zager R. A., Johnson A. C., Becker K. (2012). Plasma and urinary heme oxygenase-1 in AKI. J. Am. Soc. Nephrol. 23, 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler U., Groscurth P. (2004). Morphological features of cell death. News Physiol. Sci. 19, 124–128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene expression and high-content imaging datasets generated during and/or analyzed during the current study, as well as Source code and Python scripts for RF analyses are available in the Dryad repository (https://doi.org/10.5061/dryad.646v2r1).