

Abstract
The receptor activator of nuclear factor κB ligand (RANKL) is an important factor for osteoclastogenesis and contributes to the pathology of rheumatoid arthritis (RA); thus, the anti-RANKL antibody (Ab) has been expected to protect joint destruction in RA patients. IL-8 also has osteoclastogenic activity; however, the role of IL-8 in the bone pathology of RA as well as the relation between IL-8 and RANKL remain unclear. In the present study, clinical observation revealed serum IL-8 levels of 611 pg ml−1 in RA patients with anti-RANKL Ab and 266 pg ml−1 in the same patients without anti-RANKL Ab. In vitro assay showed that anti-RANKL Ab induced production of IL-8 from pre-osteoclast-like cells (OCLs), and IL-8 promoted the formation of OCLs from peripheral monocytes even without RANKL activity. We further showed that treatment with FK506 (tacrolimus) possibly inhibits the increase in IL-8 levels in RA patients with anti-RANKL Ab, and in vitro assay confirmed that FK506 suppressed IL-8 production in pre-OCLs. These results suggest that inhibition of RANKL induces the change in osteoclastogenesis-promoting factor from RANKL to IL-8, and FK506 may be a valuable combination drug to support the use of anti-RANKL Ab in treatment of RA.
Keywords: denosumab, FK506, osteoclast-like cells, osteoimmunology RANKL
A therapeutic anti-RANKL Ab increases IL-8 production in RA
Introduction
Homeostasis in bone metabolism is maintained by a balance between the functions of osteoblasts and osteoclasts. Osteoclasts play an important role in osteolysis and bone restructuring. Activation of these cells is associated with some diseases, including osteoporosis, bone metastasis and rheumatoid arthritis (RA). Receptor activator of nuclear factor κB ligand (RANKL) has been shown to promote the differentiation and activation of osteoclasts, receiving attention as a therapeutic target for these diseases (1–5).
In the pathology of RA, chronic autoimmune inflammation of the synovial tissue causes bone erosion, resulting in joint destruction. The inflammatory cytokines, tumor necrosis factor (TNF)-α and IL-6, produced by synovial macrophages, induce RANKL expression in synovial fibroblasts, resulting in osteoclast activation (6). Anti-TNF-α antibody (Ab) and anti-IL-6 receptor Ab have proven effective as treatments for RA patients (7, 8). Furthermore, denosumab (anti-RANKL Ab) has been reported to improve the bone erosion Sharp score of RA patients (9). Although anti-RANKL Ab may prove effective as a novel treatment for RA, one limitation is its inability to reduce joint inflammation directly (9).
IL-8 (also known as CXCL8) was the first chemokine identified to be a neutrophil chemotactic factor and is a well-known inflammatory cytokine. IL-8 levels are elevated in the serum of patients with inflammatory diseases, such as palmoplantar pustulosis, inflammatory bowel disease and those with malignant tumors (10, 11). These levels are also elevated in the serum and synovial fluid of RA patients; these levels are related to the severity of inflammation, measured as Disease Activity Score of 28 joints (DAS28) (12–14). Additionally, IL-8 promotes osteoclastogenesis during bone metastasis in breast cancer (15). Recently, IL-8 production in the synovial tissue of RA patients was found to be induced by anti-citrullinated protein antibody, resulting in osteoclast activation (16). Furthermore, IL-8 was reported to induce the generation of osteoclast-like cells (OCLs) from peripheral blood mononuclear cells (PBMCs) (17). However, the association between IL-8 and bone metabolism in RA has not been sufficiently explored in previous studies. In addition, the influence of IL-8 on osteoclastogenesis in RA during treatment with anti-RANKL Ab has not been evaluated. In this study, we evaluated the production and function of IL-8 in RA osteoclastogenesis during anti-RANKL Ab treatment.
Methods
Patient samples
Blood samples from healthy volunteers and RA patients were obtained from Osaka University Hospital. Synovial tissues were obtained from RA patients who had undergone surgery at the National Hospital Organization (NHO), Osaka Minami Medical Center. RA patients were diagnosed according to the 1987 criteria of the American College of Rheumatology (18). In addition, leukocyte removal filters [Japanese Red Cross Society (JRCS)] were used to collect PBMCs from healthy individuals. Background (e.g. age and sex) of the healthy donors from JRCS was not disclosed to protect their personal information. All donors provided written informed consent in accordance with the Declaration of Helsinki. This research was approved by the local ethics committees of Osaka University Hospital (13273-4), NHO Osaka Minami Medical Center and JRCS (280024).
Cytokine Plex
Twenty RA patients who had not previously undergone bisphosphonate treatment were selected and administered denosumab for the first time. Serum samples were obtained from these patients prior to and 1 month after denosumab treatment and were cryopreserved at −30°C. Levels of 17 cytokines and chemokines [IL-1, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, IL-17, G-CSF, macrophage colony-stimulating factor (M-CSF), IFN-γ, TNF-α, CCL2 and CCL4] in the sera were measured using Bio-Plex-Pro™ Assays (Bio-Rad, Tokyo, Japan) and a Bio-Plex™ suspension array system (Bio-Rad). Data analysis was performed using Bio-Plex Manager, version 6.1 (Bio-Rad). Bio-Plex Pro™ Human Cytokine Standard 27-Plex, Group I (Bio-Rad) was used as the reference sample for this analysis.
Cell preparations
PBMCs were separated from whole blood samples and leukocyte removal filters using Ficoll-Paque PLUS (GE Healthcare BioSciences, Tokyo, Japan). Synovial cells were collected by cutting and grinding synovial tissues. These cells were cryopreserved using CELLBANKER1 (Nippon Zenyaku Kogyo, Tokyo, Japan) in liquid nitrogen. CD14+ cells were isolated from PBMCs using CD14 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany).
Induction and culture of OCLs
CD14+ cells were cultured for 10 days in 96-well flat-bottom plates under the following conditions: (i) 50 ng ml−1 M-CSF (BioLegend, San Diego, CA, USA) and 125 ng ml−1 RANKL (BioLegend); (ii) 50 ng ml−1 M-CSF and 5 µg ml−1 anti-RANKL Ab (R&D Systems, Minneapolis, MN, USA) with or without 10 ng ml−1 IL-8 (PeproTech, Rocky Hill, New Jersey, USA); and (iii) 50 ng ml−1 M-CSF, 125 ng ml−1 RANKL and 50 ng ml−1 TNF-α (BioLegend) with or without 3 ng ml−1 FK506. After culture, the number of OCLs per well was counted for each culture condition. Ten days after culture, cells were washed with phosphate-buffered saline (PBS), supplied with fresh media and cultured again overnight under the following conditions: (i) no medication, (ii) anti-RANKL Ab (5 µg ml−1) and (iii) anti-RANKL Ab (5 µg ml−1) and FK506 (3 ng ml−1). Media were collected from overnight cultures and cryopreserved at −30°C.
Tartrate-resistant acid phosphatase staining
OCLs induced from CD14+ cells were fixed for 5 min using a 10% formalin solution and stained for 1 h at 37°C using a tartrate-resistant acid phosphatase (TRAP) staining kit (Cosmo Bio Co. LTD, Tokyo, Japan). TRAP+ multinuclear cells were counted as OCLs. On the other hands, TRAP+ mononuclear cells were regarded as pre-OCLs.
Immunofluorescence staining
OCLs and pre-OCLs induced from peripheral monocytes were stimulated using 1 ng ml−1 lipopolysaccharide (LPS) (Sigma-Aldrich, St. Louis, MO, USA), 5 μg ml−1 anti-RANKL Ab (R&D Systems) or 5 μg ml−1 anti-Mouse IgG2B κ isotype control Ab (BD BioSciences, San Diego, CA, USA), with GolgiStop protein transport inhibitor (BD BioSciences), and incubated for 5 h at 37°C. Cells were then stained for 15 min at 4°C with fluorescent antibody RANK-Alexa Fluor (AF)-488 (Novus, Littleton, CO, USA). Next, cells were fixed for 30 min at 26°C using Cytofix/Cytoperm™ fixation kit (BD) and stained for 30 min at 4°C with fluorescent antibodies: IL-8-PE (BioLegend), PE Mouse IgG2b κ isotype control (BD BioSciences) and cathepsin K-PE (Abcam, Tokyo, Japan). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Finally, cells were photographed using a BZ-X710-All-in-One Fluorescence Microscope (Keyence, Osaka, Japan).
Measurement of IL-8 levels in culture media
IL-8 levels in culture media collected from each culture were measured with a flow cytometer (BD FACS Canto II) using a Cytometric Bead Array Kit (BD).
Culture of synovial cells from RA patients
Synovial cells from RA patients were cultured for 5 days in 96-well flat-bottom plates in the presence of 50 ng ml−1 M-CSF and 125 ng ml−1 RANKL, either with or without 3 ng ml−1 tacrolimus (FK506) (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan). M-CSF and RANKL were included to create an osteoclastogenesis-promoting environment. After culture, cells were washed with PBS, provided with fresh media and cultured overnight under the following conditions: (i) no medication, (ii) anti-RANKL Ab (5 µg ml−1) and (iii) anti-RANKL Ab (5 µg ml−1) with FK506 (3 ng ml−1). Media were collected from overnight cultures and cryopreserved at −30°C.
Evaluation of cell signaling
CD14+ cells were cultured with M-CSF and RANKL. Ten days after culture, the culture medium was changed and cells were incubated with the following: (i) M-CSF only, (ii) M-CSF and RANKL and (iii) M-CSF and RANKL with anti-RANKL Ab, at 37°C for 1 h. Cells were fixed for 30 min at 26°C using Cytofix/Cytoperm™ (BD) and then permeabilized for 30 min at 4°C using BD Phosflow™ Perm Buffer III (BD). Finally, cells were stained for 30 min at 4°C with fluorescent antibodies: extracellular signal-regulated kinase (ERK)1/2(pT202/pY204)-AF488 (BD, 20A), nuclear factor κB (NF-κB) p65(pS529)-PE (BD, K10-895, 12, 50) and Src(pY418)-AF 647 (BD, K98-37). The mean fluorescence intensities (MFIs) of phosphorylated ERK, NF-κB and Src in cells were measured using a flow cytometer (BD Biosciences).
Pit assay for OCLs
Peripheral monocytes were cultured with M-CSF and RANKL, or M-CSF and IL-8 with anti-RANKL Ab, in osteoassay plates coated with calcium phosphate crystals (Corning International, Inc., New York, USA). Ten days after culture, OCLs were removed using 0.25% trypsin (Sigma-Aldrich) and gentle agitation by pipette. Pit formation was observed and photographed by microscopy (Keyence). Resorption pit area per a focus (×200) in microscopy was also measured by software BZ-X Analyser (Keyence).
Statistical analysis
Statistical analyses were performed using R version 3.12 (R project for Statistical Computing), and EZR version 1.29 (19). Statistical significance was evaluated using the Wilcoxon signed-rank test and paired Student’s t-test. One-way analysis of variance (ANOVA) with Tukey’s post hoc test was performed for multiple comparisons. Data were expressed as mean ± SD. P values ≤0.05 were considered statistically significant.
Results
Denosumab-induced increase of serum IL-8 levels in RA patients
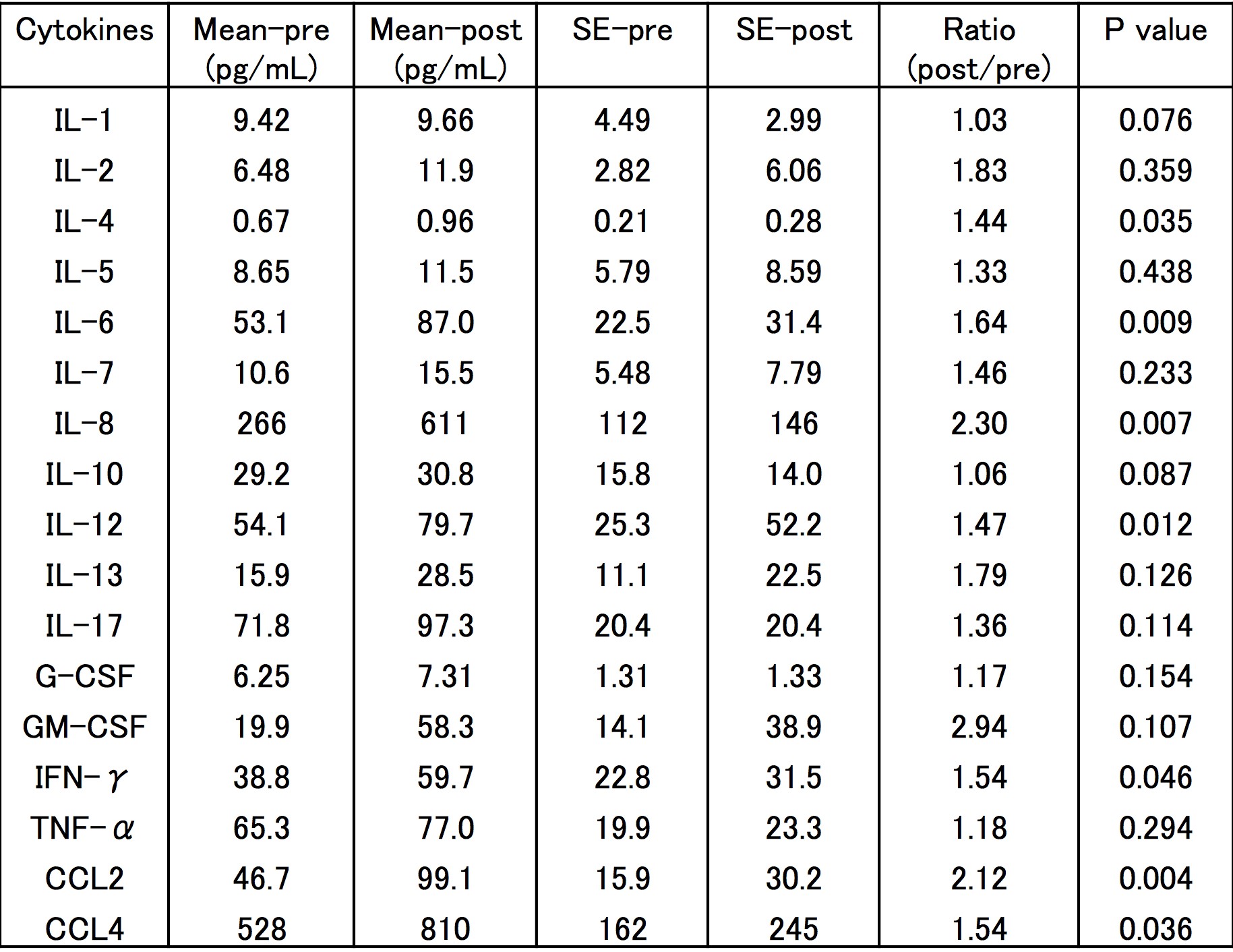
To investigate the production of IL-8 and other cytokines in RA patients during RANKL inhibition, serum levels of 17 cytokines, including IL-8, were measured in RA patients prior to and 1 month after denosumab treatment. Clinical backgrounds of the RA patients included in the study are shown in Table 1. Levels of some cytokines such as IL-6 slightly increased before and after denosumab treatment; serum IL-8 levels, in particular, increased apparently and significantly (P = 0.007) (Fig. 1 and Supplementary Figure 1). To evaluate the influence on inflammation of increased IL-8 levels after denosumab treatment, clinical information of RA patients was also evaluated. Inflammatory markers such as C-reactive protein (CRP) and neutrophil percentages in white blood cells did not change following denosumab treatment (Supplementary Figure 2A). In bone metabolism of RA following denosumab treatment, levels of osteocalcin, a marker of bone formation, in the sera of RA patients did not change. In contrast, TRAP-5b, a marker of bone erosion, significantly decreased after denosumab treatment (P = 0.001) (Supplementary Figure 2B).
Table 1.
Background of RA patients before denosumab treatment
| Clinical data | Before denosumab treatment |
|---|---|
| Number | 20 |
| Age (year) | 59.7 ± 11.3 |
| Female (%) | 100 |
| Disease duration (year) | 12.8 ± 11.1 |
| WBC (µl−1) | 5534 ± 2069 |
| Neutrophil (%) | 60.5 ± 11.0 |
| CRP (mg dl−1) | 0.21 ± 0.38 |
| ESR (mm h−1) | 27.4 ± 20.6 |
| MMP-3 (ng ml−1) | 69.1 ± 66.9 |
| SAA (µg ml−1) | 14.6 ± 27.5 |
| DAS28 ESR | 1.91 ± 1.27 |
| RF (mg dl−1) | 52.9 ± 49.1 |
| Osteocalcin (ng ml−1) | 0.41 ± 0.50 |
| TRAP-5b (ng ml−1) | 429 ± 157 |
| CTX (µg mmol−1) | 3.8 ± 1.12 |
| Treatments | TCZ (5), TCZ + FK506 (2) |
| ETN (2), ETN + MTX (1) | |
| MTX (6), MTX + PSL (4) |
Values are mean ± SD. CTX, collagen type 1 cross-linked C-telopeptide; ESR, erythrocyte sedimentation rate; ETN, etanercept; MMP-3, matrix metalloproteinase-3; PSL, prednisolone; RF, rheumatoid factor; SAA, serum amyloid A protein; TCZ, tocilizumab; WBC, white blood cell.
Fig. 1.

Serum IL-8 levels in RA patients increased after denosumab treatment. Serum cytokine or chemokine levels in RA patients who had not previously undergone bisphosphonate treatment were measured prior to and 1 month after denosumab treatment. Statistical significance was evaluated using Wilcoxon signed-rank test. CCL, chemokine ligand.
Anti-RANKL Ab-induced increase of IL-8 production in OCLs and synovial cells
To resolve the mechanism of IL-8 increase following anti-RANKL Ab treatment, in vitro assays using OCLs and synovial cells were performed. OCLs were induced from peripheral monocytes of healthy donors using M-CSF and RANKL. OCLs were observed as TRAP+ multinuclear cells following TRAP staining (Fig. 2A). TRAP+ cells were also observed to express RANK (Fig. 2B). In these culture cells, IL-8 production was observed by immunofluorescence staining. OCLs were found to produce IL-8 following LPS stimulation. Conversely, small mononuclear cells (pre-OCLs) produced IL-8 when exposed to anti-RANKL Ab or control Ab (Fig. 2C). IL-8 levels in culture medium increased significantly (P = 0.031) after overnight incubation with anti-RANKL Ab, compared with those obtained after incubation with control Ab (Fig. 2D). Interestingly, IL-8 levels in culture medium decreased significantly after overnight incubation with combined M-CSF and RANKL compared with those obtained after overnight incubation with M-CSF alone (P = 0.004) (Fig. 2D). In a similar assay using synovial cells, IL-8 levels in culture medium increased significantly after overnight incubation with anti-RANKL Ab compared with those obtained after overnight incubation without anti-RANKL Ab (P = 0.033) (Fig. 2E). Additionally, IL-8 production after anti-RANKL Ab treatment was amplified by TNF-α (Fig. 2F).
Fig. 2.

IL-8 production in OCL cultures induced from peripheral monocytes. (A) CD14+ cells from PBMCs of healthy donors were cultured with M-CSF (50 ng ml−1) and RANKL (125 ng ml−1). Ten days after culture, TRAP staining was performed. (B) Expression of RANKL in culture cells was evaluated by immunofluorescence staining (RANK-AF488 and DAPI). (C) IL-8 production in culture cells containing OCLs and pre-OCLs after LPS (1 ng ml−1) stimulation, anti-RANKL Ab (5 µg ml−1) treatment and control Ab (5 µg ml−1) treatment was evaluated by immunofluorescence staining (IL-8-PE, isotype control Ab-PE). (D) Ten days after culture of CD14+ cells with M-CSF and RANKL, the medium was changed, and cultured cells were incubated overnight in the following conditions: M-CSF only, M-CSF and RANKL, M-CSF and RANKL with anti-RANKL Ab (5 µg ml−1), and M-CSF and RANKL with control Ab (5 µg ml−1). After incubation, IL-8 levels in culture supernatant were measured (n = 5). (E) Synovial cells were cultured with M-CSF and RANKL. Five days after culture, medium was changed. Culture cells were incubated overnight with or without anti-RANKL Ab. After incubation, IL-8 levels in culture supernatant were measured (n = 5). (F) IL-8 levels in the culture supernatant of OCLs with M-CSF and RANKL [with or without TNF-α (50 ng ml−1)] after anti-RANKL Ab treatment were evaluated (n = 3). Representative images (A–C) from five healthy donors are shown. Statistical significance was evaluated using paired Student’s t-test (C and D). aRANKL Ab, anti-RANKL antibody.
Recovery of phosphorylation levels of ERK, Src and NF-κB by anti-RANKL Ab
To evaluate intracellular signaling pathways associated with RANK-RANKL signaling during anti-RANKL Ab treatment, phosphorylation levels of ERK, Src and NF-κB were measured in cells cultured for 10 days with M-CSF and RANKL 1 h after stimulation with M-CSF only, both M-CSF and RANKL, or either group with anti-RANKL Ab. Flow cytometry was used for this assay to allow easy, quantitative evaluation of differences in phosphorylation levels between samples. The median MFIs for phosphorylated (p) ERK, Src and NF-κB were decreased by RANKL and recovered by anti-RANKL Ab (Fig. 3).
Fig. 3.

The intracellular pathways associated with RANK-RANKL signaling and IL-8 production were evaluated with and without anti-RANKL Ab. Peripheral monocytes cultured with M-CSF and RANKL for 10 days had their media changed and were then incubated with M-CSF only, M-CSF and RANKL, and M-CSF and RANKL with anti-RANKL Ab at 37°C for 1 h. The MFIs of phosphorylated (p) ERK, NF-κB and Src were measured using a flow cytometer. Representative images from five RA patients are shown. Red, blue, green and gray lines represent M-CSF and RANKL with anti-RANKL Ab, M-CSF only, M-CSF and RANKL, and unstained, respectively. Additionally, the median MFIs for pERK, pNF-κB and pSrc were also evaluated (n = 5). Error bars indicate SD. Statistical significance was evaluated using paired Student’s t-test.
Induction of OCLs by IL-8
To evaluate the role of IL-8 in the formation of OCLs, peripheral monocytes from healthy donors were cultured for 10 days with M-CSF and IL-8 with anti-RANKL Ab. Although OCLs were not induced by M-CSF with anti-RANKL Ab, exogenous addition of IL-8 was capable of inducing OCLs despite RANKL inhibition by anti-RANKL Ab (Fig. 4A and B). Furthermore, it was observed that OCLs induced by IL-8 were capable of eroding the bone in pit assays (Fig. 4C and D) and expressed cathepsin K (Fig. 4E).
Fig. 4.

IL-8 promotes the formation of OCLs during RANKL inhibition with anti-RANKL Ab. (A) CD14+ cells from PBMCs of healthy donors were cultured under the following conditions: M-CSF (50 ng ml−1) and RANKL (125 ng ml−1), M-CSF with anti-RANKL Ab (5 µg ml−1) with or without IL-8 (10 ng ml−1). Ten days after culture, TRAP staining was performed on each culture. Representative images (A) from five healthy donors are shown. (B) The number of OCLs per well in each culture condition was counted (n = 5). (C, D) Pit assays were performed and resorption pit area per a focus was measured on the same cultures. (E) Expression of cathepsin K was evaluated by immunofluorescence staining in the same cultures (cathepsin K-PE and DAPI). Statistical significance was evaluated using one-way ANOVA with Tukey’s post hoc test (B) and paired Student’s t-test (D). aRANKL Ab, anti-RANKL antibody.
Suppression of IL-8 production by FK506
In our results from a clinical retrospective survey (Fig. 1), serum IL-8 levels after denosumab treatment did not increase in two RA patients treated with FK506; however, this was not replicated by methotrexate (MTX) treatment (Supplementary Figure 3). Therefore, we focused on FK506 as a potential agent to suppress IL-8 production during anti-RANKL Ab treatment. To evaluate the function of FK506, an in vitro assay using OCLs was performed. In this in vitro assay, TNF-α was added to the OCL culture to mimic the inflammatory conditions of RA. Peripheral monocytes were cultured with M-CSF, RANKL and TNF-α, with or without FK506. After 10 days of culture, culture media were changed and cells were incubated with or without anti-RANKL Ab. After overnight incubation, IL-8 production in culture media was measured. Pretreatment of FK506 suppressed IL-8 production during anti-RANKL Ab treatment (Fig. 5A). This phenomenon was also observed in in vitro assays using synovial cells from RA patients (Fig. 5B).
Fig. 5.

FK506 suppresses IL-8 production during anti-RANKL Ab treatment. (A) CD14+ cells from PBMCs of healthy donors were cultured with M-CSF (50 ng ml−1), RANKL (125 ng ml−1) and TNF-α (50 ng ml−1) with or without FK506 (3 ng ml−1). Ten days after culture, the culture medium was changed and cells were incubated overnight with anti-RANKL Ab or anti-RANKL Ab and FK506. After incubation, IL-8 levels in the supernatant were measured (n = 5). (B) Synovial cells were cultured with M-CSF and RANKL with or without FK506. Five days after culture, the culture medium was changed and cells were incubated overnight with anti-RANKL Ab or anti-RANKL Ab and FK506. After incubation, IL-8 levels in supernatant were measured (n = 5). Statistical significance was evaluated using paired Student’s t-test. aRANKL Ab, anti-RANKL antibody.
Discussion
IL-8 is a well-known inflammatory cytokine associated with neutrophil responses; however, it has not been known to promote osteoclastogenesis during bone metabolism (20). In our clinical data, serum IL-8 levels were found to increase significantly after denosumab treatment. Despite this increase, inflammatory marker levels such as CRP and neutrophil percentages in white blood cells did not change. These results implied that increased serum IL-8 levels are not associated with inflammation in RA. Therefore, we focused on the relationship between IL-8 and osteoclastogenesis in RA. Our results suggested that IL-8 promotes the formation of OCLs in environments with anti-RANKL Ab. This phenomenon indicated that IL-8 might compensate for RANKL function in the induction of OCLs in RANKL-low environments. Furthermore, IL-8 production was promoted by TNF-α. Therefore, this promotion of osteoclastogenesis by IL-8 may be amplified by TNF-α-rich environments, such as those seen in RA. In the case that anti-RANKL Ab is used to treat RA patients, IL-8 inhibition may also be an important treatment.
Our results indicated that RANKL suppresses IL-8 production induced by M-CSF and that neutralization of RANKL by anti-RANKL Ab recovers IL-8 production. Interestingly, previous studies have reported that RANKL suppresses the production of inflammatory cytokines. According to a report on osteoprotegerin (OPG), a decoy receptor against RANKL with a similar function to anti-RANKL Ab, OPG stimulation induces the release of IL-8 and IL-6 in vitro (21). Additionally, OPG knockout reduces IL-6 and inflammatory cytokines in an ischemic mouse brain, and RANKL treatment also reduces these cytokines in the ischemic mouse brain (22). This report also suggested that RANK-RANKL signaling inhibits TLR-4 signaling and thus the production of inflammatory cytokines, such as IL-6, in activated microglia. In our clinical findings, the IL-6 production also increased slightly after denosumab treatment (Fig. 1). According to these reports and our results, RANKL may suppress the production of inflammatory cytokines such as IL-6 or IL-8. Reversible signals of RANKL, stimulated by RANK-Fc, can reportedly induce IL-8 production in B lymphocyte leukemia cells (23). Although an association between IL-8 production and this reversible signal was suspected, the reversible signal was not considered to influence an increase in IL-8 levels in this study because IL-8 production with M-CSF and anti-RANKL Ab did not increase compared with that under M-CSF only.
From our data, the expression levels of pERK, pSrc and pNF-κB decreased when cultured with RANKL and recovered by anti-RANKL Ab treatment. Generally, RANKL can enhance the phosphorylation of ERK, Src and NF-κB. Most previous studies have evaluated intracellular signals such as the ERK pathway immediately after stimulation by RANKL in mouse bone marrow cells, human cell lines such as RAW264.7 cells or human PBMCs treated by M-CSF. In our assay, PBMCs treated by both M-CSF and RANKL for a long time were used to investigate the influence of RANKL inhibition on promoted osteoclastogenesis, such as the pathology of RA, and this experimental condition was different from that in a previous study. A previous study indicated that repeated stimulations of RANKL reduced the phosphorylation of intracellular signals such as ERK compared with a single stimulation of RANKL (24). Our results are surmised to have been caused by the sustainable treatment with RANKL on osteoclast precursors (pOC) as well as the results of repeated stimulations by RANKL than previously. Although RANKL stimulation can promote phosphorylation of ERK, Src and NF-κB (25), continuous stimulations of RANKL may regulate their phosphorylation according to the differentiation stage of pOC. Expression of pERK, pSrc and pNF-κB is known to promote IL-8 production (26–28). Recovering the expression levels of pERK, pSrc and pNF-κB by anti-RANKL Ab treatment may enhance IL-8 production.
Tacrolimus (FK506) is an immunosuppressive drug that is also a well-known calcineurin inhibitor. Calcineurin is activated by calmodulin binding and stimulated by T-cell receptor signaling. Activated calcineurin then promotes the activation of the nuclear factor of activated T cells (NFAT), which induces the gene expression of several cytokines, including IL-2. FK506 suppresses the function of the calcineurin complex, blocking cytokine production and T-cell activation (29). Therefore, FK506 is often used to prevent graft rejection in patients undergoing organ transplantation (30). In Japan, it is also considered an effective treatment for RA (31); FK506 has been reported to improve ACR20, ACR50 and ACR70 levels to 48.5, 27.2 and 11.7%, respectively (32). FK506 has also been found to improve the Sharp scores of patients (33), directly suppress the differentiation of osteoclasts (by blocking the expression of NFATc1 activated by RANK-RANKL signaling) (34), and suppress IL-8 production by regulating the function of NF-κB (35). In our study, FK506 decreased IL-8 production in the culture media of OCLs. Although the most important function of FK506 is suppression of T cells and osteoclasts directly in the pathology of RA, this decrease in IL-8 production may be another interesting function of FK506 for the treatment of RA.
There are some limitations in our study. Unfortunately, IL-8 is not present in mice; therefore, we could not corroborate our findings in any mouse models of arthritis. The evaluation of RA patients treated with denosumab was retrospective, and the sample size was limited. Therefore, we could not evaluate, in detail, the clinical data of RA patients who underwent FK506 treatment with denosumab. Moreover, we were not able to investigate the effect of IL-8 on bone metabolism in RA between patients who exhibited increased IL-8 levels and those who exhibited a decrease in IL-8 levels after denosumab treatment because there were only a small number of patients with decreased IL-8 levels. Finally, the duration of observation in this retrospective study was only 1 month, making it impossible to perform long-term observations of serum IL-8 levels, clinical data (i.e. CRP, ESR, DAS28 and TRACP-5b levels), and bone density of RA patients after denosumab treatment. Therefore, further research is needed to reveal the full role of IL-8 during bone metabolism in RA.
Neutralizing RANKL function with anti-RANKL Ab treatment is expected to suppress bone erosion in RA. However, this suppression may be incomplete because treatment induces the production of osteoclastogenesis-promoting IL-8, particularly in inflammatory environments. To support anti-RANKL Ab treatment in RA patients, FK506 may be a favorable combination drug owing to its ability to suppress IL-8 production.
Funding
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (JP18H05282 to AK), the Japan Agency for Medical Research and Development (AMED) (JP18cm016335 and JP18cm059042 to AK).
Conflicts of interest statement: Hideki Tsuboi and Y. Shima received the fee for lectures from Daiichi Sankyo Company. The remaining authors declared no conflicts of interest.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
T.M. and Y. Shima designed the study. Y. Shima conducted the study. A.K. supervised the project. T.M., Y. Shima, H.T., M.N., and A.O. collected clinical samples. T.M. analyzed the data. K.F. assisted some experiments and advised on data interpretation. All authors interpreted the data. T.M. and Y. Shima drafted manuscript. T.M., M.N., and Y. Shima revised manuscript content. All authors approved final version of manuscript. Y. Shima takes responsibility for the integrity of data analysis.
References
- 1. Manolagas, S. C. 2000. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 21:115. [DOI] [PubMed] [Google Scholar]
- 2. Mundy, G. R. 2002. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2:584. [DOI] [PubMed] [Google Scholar]
- 3. Cummings, S. R., San Martin, J., McClung, M. R.et al. ; FREEDOM Trial. 2009. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 361:756. [DOI] [PubMed] [Google Scholar]
- 4. Steger, G. G. and Bartsch, R. 2011. Denosumab for the treatment of bone metastases in breast cancer: evidence and opinion. Ther. Adv. Med. Oncol. 3:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanaka, S. 2013. Regulation of bone destruction in rheumatoid arthritis through RANKL-RANK pathways. World J. Orthop. 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okamoto, K. and Takayanagi, H. 2011. Osteoclasts in arthritis and Th17 cell development. Int. Immunopharmacol. 11:543. [DOI] [PubMed] [Google Scholar]
- 7. Feldmann, M. 2002. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2:364. [DOI] [PubMed] [Google Scholar]
- 8. Nishimoto, N., Kishimoto, T. and Yoshizaki, K. 2000. Anti-interleukin 6 receptor antibody treatment in rheumatic disease. Ann. Rheum. Dis. 59 (Suppl. 1):i21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen, S. B., Dore, R. K., Lane, N. E.et al. ; Denosumab Rheumatoid Arthritis Study Group. 2008. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 58:1299. [DOI] [PubMed] [Google Scholar]
- 10. Yoshimura, T., Matsushima, K., Tanaka, S.et al. 1987. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc. Natl Acad. Sci. USA 84:9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Russo, R. C., Garcia, C. C., Teixeira, M. M. and Amaral, F. A. 2014. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 10:593. [DOI] [PubMed] [Google Scholar]
- 12. Osiri, M., Wongpiyabovorn, J., Sattayasomboon, Y. and Thammacharoenrach, N. 2016. Inflammatory cytokine levels, disease activity, and function of patients with rheumatoid arthritis treated with combined conventional disease-modifying antirheumatic drugs or biologics. Clin. Rheumatol. 35:1673. [DOI] [PubMed] [Google Scholar]
- 13. Klimiuk, P. A., Sierakowski, S., Domyslawska, I. and Chwiecko, J. 2011. Serum chemokines in patients with rheumatoid arthritis treated with etanercept. Rheumatol. Int. 31:457. [DOI] [PubMed] [Google Scholar]
- 14. Hidaka, T., Suzuki, K., Kawakami, M.et al. 2001. Dynamic changes in cytokine levels in serum and synovial fluid following filtration leukocytapheresis therapy in patients with rheumatoid arthritis. J. Clin. Apher. 16:74. [DOI] [PubMed] [Google Scholar]
- 15. Kamalakar, A., Bendre, M. S., Washam, C. L.et al. 2014. Circulating interleukin-8 levels explain breast cancer osteolysis in mice and humans. Bone 61:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krishnamurthy, A., Joshua, V., Haj Hensvold, A.et al. 2016. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 75:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bendre, M. S., Montague, D. C., Peery, T., Akel, N. S., Gaddy, D. and Suva, L. J. 2003. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone 33:28. [DOI] [PubMed] [Google Scholar]
- 18. Arnett, C. F., Edworthy, M. S., Bloch, A. D.et al. 1988. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 31:315. [DOI] [PubMed] [Google Scholar]
- 19. Kanda, Y. 2013. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant 48:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mukaida, N. 2003. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 284:L566. [DOI] [PubMed] [Google Scholar]
- 21. Perez de Ciriza, C., Lawrie, A. and Varo, N. 2015. OPG expression on endothelial cells and modulation by IL-1B, PDGF, insulin, and glucose. Biochem. Physiol. 4:179. [Google Scholar]
- 22. Shimamura, M., Nakagami, H., Osako, M. K.et al. 2014. OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc. Natl Acad. Sci. USA 111:8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Secchiero, P., Corallini, F., Barbarotto, E.et al. 2006. Role of the RANKL/RANK system in the induction of interleukin-8 (IL-8) in B chronic lymphocytic leukemia (B-CLL) cells. J. Cell. Physiol. 207:158. [DOI] [PubMed] [Google Scholar]
- 24. Lee, K., Chung, Y. H., Ahn, H., Kim, H., Rho, J. and Jeong, D. 2016. Selective regulation of MAPK signaling mediates RANKL-dependent osteoclast differentiation. Int. J. Biol. Sci. 12:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wada, T., Nakashima, T., Hiroshi, N. and Penninger, J. M. 2006. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 12:17. [DOI] [PubMed] [Google Scholar]
- 26. Namba, S., Nakano, R., Kitanaka, T., Kitanaka, N., Nakayama, T. and Sugiya, H. 2017. ERK2 and JNK1 contribute to TNF-α-induced IL-8 expression in synovial fibroblasts. PLoS ONE 12:e0182923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trevino, J. G., Summy, J. M., Gray, M. J.et al. 2005. Expression and activity of SRC regulate interleukin-8 expression in pancreatic adenocarcinoma cells: implications for angiogenesis. Cancer Res. 65:7214. [DOI] [PubMed] [Google Scholar]
- 28. Mukaida, N., Mahe, Y. and Matsushima, K. 1990. Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J. Biol. Chem. 265:21128. [PubMed] [Google Scholar]
- 29. Dutta, S. and Ahmad, Y. 2011. The efficacy and safety of tacrolimus in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 3:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klintmalm, G. 1994. A review of FK506: a new immunosuppressant agent for the prevention and rescure of graft rejection. Transplantation Rev. 8:53. [Google Scholar]
- 31. Miyasaka, N. 2011. Treatment trends of rheumatoid arthritis in Japan: changes toward globalization and its unique innovation. Inflammation Regenerant 31:25. Available at: https://doi.org/10.2492/inflammregen.31.25" xlink:type="simple" xmlns:xlink="http://www.w3.org/1999/xlink">https://doi.org/10.2492/inflammregen.31.25. [Google Scholar]
- 32. Kawai, S., Hashimoto, H., Kondo, H., Murayama, T., Kiuchi, T. and Abe, T. 2006. Comparison of tacrolimus and mizoribine in a randomized, double-blind controlled study in patients with rheumatoid arthritis. J. Rheumatol. 33:2153. [PubMed] [Google Scholar]
- 33. Tanaka, Y., Kawai, S., Tamamoto, K. and Miyasaka, N. 2013. Prevention of joint destruction by tacrolimus in patients with early rheumatoid arthritis: a post hoc analysis of a double-blind, randomized, placebo-controlled study. Modern Rheum. 24:1045. [DOI] [PubMed] [Google Scholar]
- 34. Miyazaki, M., Fujikawa, Y., Takita, C. and Tsumura, H. 2007. Tacrolimus and cyclosporine A inhibit human osteoclast formation via targeting the calcineurin-dependent NFAT pathway and an activation pathway for c-Jun or MITF in rheumatoid arthritis. Clin. Rheumatol. 26:231. [DOI] [PubMed] [Google Scholar]
- 35. Mukaida, N., Okamoto, S., Ishikawa, Y. and Matsushima, K. 1994. Molecular mechanism of interleukin-8 gene expression. J. Leukoc. Biol. 56:554. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.