

Abstract
Recent work has established that IL-1β plays a central role in the inflammation and connective tissue destruction observed in both rheumatoid arthritis and osteoarthritis. These processes result from the ability of this inflammatory cytokine to activate expression of genes for neutral proteases, such as the matrix metalloproteinases. While IL-1β activates matrix metalloproteinase genes within several hours, it also activates immediate early genes, which are required for the later expression of matrix metalloproteinases and other arthritis-perpetuating genes, are also activated. To identify putative immediate early genes involved in IL-1β-mediated arthritic disease, a chondrocytic cell line (SW1353) was stimulated with this cytokine for 2 hours, total RNA was isolated, and expressed genes were identified by microarray analysis. This analysis identified alterations in the expression of multiple transcription factors, cytokines, growth factors and their receptors, adhesion molecules, proteases, and signaling intermediates that may contribute to inflammation and cartilage destruction in arthritis. Interestingly, confirmation of the expression of activating protein-1 family members by reverse transcriptase polymerase chain reaction revealed a preferential increase in junB, a known transcriptional antagonist of c-jun. The failure to observe induction of early growth response gene-1, which was detected by reverse transcriptase polymerase chain reaction to be substantially and transiently induced by 1 hour of IL-1 treatment, may be explained by the known instability of the message after early induction. However, this analysis has identified numerous IL-1β-responsive genes that warrant further investigation as mediators of disease in arthritis.
Keywords: chondrocytes, interleukin-1, matrix metalloproteinases, signal transduction, transcription factors
Introduction
IL-1β is a proinflammataory cytokine that has myriad effects on cells, increasing proliferation, activating inflammatory responses, and inducing matrix remodeling through the production of neutral proteases. Since most cells can both express and respond to IL-1β, complex signaling programs are likely required to modulate the timing and magnitude of responses to this cytokine [1]. It is well documented that elevated expression of IL-1β in the joint results in the activation of inflammatory and degradative programs in synovial cells, contributing to the progression of rheumatoid arthritis [2]. Recent work by Hasty and coworkers [3] has demonstrated that local expression of IL-1β by articular chondrocytes activates autocrine gene expression, which contributes to the pathogenesis of osteoarthritis (OA). Furthermore, IL-1-receptor antagonists are effective inhibitors of inflammation and connective tissue destruction in animal models of arthritis [2,4,5]. Together, these studies implicate IL-1β as a central mediator of pathogenesis in arthritis.
Activation of this pathologic process is complex, most likely involving the activation or repression of numerous signal/transduction pathways, with subsequent effects on many genes. Our previous work has focused on the pathways by which IL-1β induces several members of the family of matrix metalloproteinases (MMPs), enzymes that are active at neutral pH and that collectively degrade the various components of the extracellular matrix. Specifically, we have focused on MMP-1 and MMP-13, interstitial collagenases that have the unique ability to initiate cleavage of the triple helix comprising the stromal collagens types I, II, and III. Elevated expression of MMP-1 and MMP-13 by stromal cells and chondrocytes in response to IL-1β contributes to the pathogenesis of connective tissue disorders [6,7,8,9,10,11]. Studies from this laboratory and others showed that IL-1 activates MMP-1 and MMP-13 gene expression through common signaling pathways [11,12,13,14,15]. Specifically, nuclear translocation of NF-κB and mitogen-activated protein kinase (MAPK) stimulation of activator protein-1 (AP-1) are required for transcription of cytokine-induced MMP-1 and MMP-13. Furthermore, cycloheximide blocks mRNA expression of both collagenases [11,16], indicating that expression of immediate early genes is required for MMP-1 and MMP-13 gene expression. Thus, IL-1β activation of collagenase gene expression requires the increased expression of a panel of essential transcription factors and signaling intermediates, as well as the initiation of signaling cascades that activate these factors.
IL-1 also contributes to the pathogenesis of arthritis by increasing the proliferation of mesenchymal cells and by enhancing the expression of other inflammatory mediators [17,18,19]. This complex alteration in cellular phenotype probably involves the coordinated increase and decrease of genes for cytokines and growth factors, adhesion molecules, and signaling intermediates. The recent availability of microarray technology has made it possible to perform an extensive analysis of IL-1 modulation of genes in cells that are central players in arthritic disease.
We therefore carried out a microarray analysis of IL-1β-stimulated chondrocytic cells in order to identify immediate early genes that could contribute to phenotypic changes relevant to OA. In these studies, we used the human chondrosarcoma cell line SW1353, for three important reasons. First, we, and others, have found that when treated with IL-1β, SW1353 cells serve as an appropriate model for primary chondrocytes in OA [11,20,21,22]. Second, the amount of total RNA required for microarray analysis precludes the use of primary human chondrocytes for these studies. Third, the inherent genetic variation in primary human chondrocyte cultures could bias the gene expression profile and lead to erroneous conclusions. We found increased expression of a large cohort of transcription factors, cytokines, growth factors, and signaling intermediate genes that are potential regulators of metalloproteinase gene expression, proliferation, and sustained inflammation. Alternatively, this was accompanied by a decrease in a modest number of genes also belonging to these categories, suggesting that IL-1β substantially reprograms gene expression in chondrocytes. A subset of these IL-1β-stimulated genes was then confirmed by semiquantitative reverse transcriptase polymerase chain reaction (RT-PCR). This characterization of IL-1β-induced immediate early genes in chondrocytic cells identifies candidate mediators of the expression of colla-genase genes in arthritis.
Materials and methods
Cell culture
SW1353 chondrosarcoma cells were grown to confluence in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), penicillin/streptomycin, and L-glutamine (Cellgro, Mediatech, Herndon, VA, USA). At the beginning of each experiment, cells were washed three times with Hank's buffered sterile saline (Cellgro) to remove traces of serum and were placed in Dulbecco's modified Eagle's medium containing 0.2% lactalbumin hydrolysate (Gibco/BRL, Rockville, MD, USA). Then cells were either left unstimu-lated or treated with 10 ng/ml recombinant IL-1β (Promega, Madison, WI, USA). This concentration of IL-1 was found to be optimal for activation of MMP-13 gene expression in these cells (data not shown).
Harvest and microarray analysis of mRNA
For microarray analysis, two confluent 150-mm plates of SW1353 cells were cultured in lactalbumin hydrolysate, or lactalbumin hydrolysate plus 10 ng/ml IL-1β, for 2 hours. Cells were then washed with Hank's buffered sterile saline solution and homogenized in 2 ml Triazol Reagent (Gibco/BRL), and RNA was purified in accordance with the manufacturer's instructions. The RNA pellets were stored in 70% ethanol and shipped on dry ice to Clontech Laboratories (Palo Alto, CA, USA) via express courier. Clontech analyzed the RNA using their Atlas Human Cancer 1.2 K array (1176 unique genes) and AtlasImage 1.0 software. We chose this data baseline because it represents a large number of genes that may be expressed during development and again during disease pathology. We further classified genes that increased or decreased by twofold or more, and sorted them using Microsoft Excel.
Reverse transcriptase polymerase chain reaction
A subset of genes whose expression was increased by IL-1β was then confirmed using a radioactive RT-PCR assay. Briefly, RNA was treated with DNAse I and then 2 μg was reverse transcribed with Moloney murine leukemia virus reverse transcriptase and oligodT primers. One tenth of the reverse transcriptase reaction was amplified using gene-specific primers, Platinum Taq DNA polymerase and 33PdATP. Single-band products were resolved on native acrylamide gels and visualized by autoradiography. Table 1 shows the gene-specific primers that were used.
Table 1.
PCR primers and conditions
| Annealing | Product | ||||
| Gene | Primers | temperature | Cycles | length* | |
| MMP-13 | CCAGTGGTGGTGATGAAGATG | Sense | 52°C | 25 | 143 |
| TTCTCGGAGCCTCTCAGTC | Antisense | ||||
| c-Jun | GGAAACGACCTTCTATGACGATGCCCTCAA | Sense | 63°C | 22 | 316 |
| GAACCCCTCCTGCTCATCTGTCACGTTCTT | Antisense | ||||
| JunD | GAGTGTTCGATTCTGCCCTA | Sense | 53°C | 22 | 214 |
| TGTGGACTCGTAGCAAAACA | Antisense | ||||
| JunB | CCAGTCCTTCCACCTCGACGTTTACAAG | Sense | 58°C | 22 | 257 |
| GACTAAGTGCGTGTTTCTTTTCCACAGTAC | Antisense | ||||
| egr-1 | TCCCGGCTCCTCGACCTAC | Sense | 60°C | 25 | 177 |
| AAGGTTGCTGTCATGTCCGAAAG | Antisense |
GAPDH was amplified using Clontech's human GPDH amplimer set with an annealing temperature of 58°C and 25 cycles of amplification. MMP-13 = matrix metalloproteinase-13; egr-1 = early growth response gene-1. *Measured in base pairs.
Results and discussion
Table 2 shows the results of an analysis using Clontech's Human Cancer 1.2 K Array and total RNA from SW1353 cells treated with IL-1β (10 ng/ml) for 2 hours. This analysis revealed substantial increases or decreases in the expression of several known IL-1β-responsive genes. These include transcription factors (panel A), cytokines, growth factors, and their receptors (panel B), proteases, matrix proteins, and adhesion molecules (panel C), as well as signaling intermediates and tumor suppressors (panel D).
Table 2.
IL-1-responsive genes in chondrocytic cells
| Fold | Fold | ||
| ↑ | ↓ | Gene description | Genbank# |
| Panel A: | Transcription Factors/RNA-binding Proteins | ||
| 18.0 | I-rel (RELB) | M83221 | |
| 17.0 | nuclear factor NF-kappa-B p100 subunit; nuclear factor NF-kappa-B p52 subunit; H2TF1; oncogene lyt-10 | X61498 | |
| 15.7 | interferon regulatory factor 1 (IRF1) | X14454 | |
| 13.7 | major histocompatibility complex enhancer-binding protein MAD3 | M69043 | |
| 8.1 | nuclear factor kappa-B DNA binding subunit (NF-kappaB; NFKB) | M58603 | |
| 5.5 | octamer-binding transcription factor 2 (oct-2; OTF2); lymphoid-restricted immunoglobulin octamer | M36542 | |
| binding protein NF-A2; POU2F2 | |||
| 4.7 | IkappaB epsilon (IKBE) | U91616 | |
| 3.0 | c-rel proto-oncogene protein | X75042 | |
| 2.8 | fos-related antigen (FRA1) | X16707 | |
| 2.4 | STAT-induced STAT inhibitor 3 | AB004904 | |
| 2.4 | erythroblastosis virus oncogene homolog 1 (ETS-1); p54 | J04101 | |
| 2.3 | jun-B | M29039 | |
| 2.2 | ELAV-like neuronal protein 1 (HEL-N1) + HEL-N2 | U12431 + | |
| U29943 | |||
| 2.0 | myb proto-oncogene; c-myb | M15024 | |
| 2.0 | NF-kappaB transcription factor p65 subunit; RELA; NFKB3 | L19067 | |
| 2.0 | transcription factor erf-1; AP2 gamma transcription factor | U85658 | |
| 2.0 | early growth response alpha (EGR alpha) | S81439 | |
| 6.0 | homeobox protein HOX-D3; HOX-4A | D11117 | |
| 2.0 | retinoblastoma-like protein 2 (RBL2; RB2); 130-kDa retinoblastoma-associated protein | X74594 | |
| 2.0 | mothers against dpp homolog 4 (SMAD4); MADR4; pancreatic carcinoma gene 4 (DPC4) | U44378 | |
| Panel B: | Cytokines/Growth Factors/Receptors | ||
| 21 | leukemia inhibitory factor precursor (LIF); differentiation-stimulating factor (D factor); melanoma-derived | X13967; | |
| LPL inhibitor (MLPLI); HILDA | M63420 | ||
| 8.7 | interleukin-11 (IL-11); adipogenesis inhibitory factor (AGIF) | M57765 | |
| 6.1 | interferon-gamma (IFN-gamma) receptor beta subunit precursor; IFN-gamma accessory factor 1 (AF1); | U05875 | |
| IFN-gamma transducer 1 (IFNGT1) | |||
| 6.1 | interleukin-6 precursor (IL-6); B-cell stimulatory factor 2 (BSF2); interferon beta-2 (IFNB2); | X04602; | |
| hybridoma growth factor | M14584 | ||
| 5.9 | heparin-binding EGF-like growth factor (HBEGF); diphtheria toxin receptor (DTR) | M60278 | |
| 5.0 | interferon gamma-induced protein precursor (gamma-IP10) | X02530 | |
| 4.3 | putative serine/threonine protein kinase PRK | U56998 | |
| 4.3 | CD40 receptor-associated factor 1 (CRAF1) | U21092 | |
| 3.6 | platelet-derived growth factor A subunit precursor (PDGFA; PDGF1) | X06374 | |
| 3.0 | bone morphogenetic protein 3B precursor (BMP3B); growth differentiation factor 10 (GDF10); | D49493 | |
| bone inducing protein (BIP) | |||
| 2.5 | CD40 ligand (CD40-L); tumor necrosis factor (TNF)-related activation protein (TRAP); T-cell antigen GP39 | L07414 | |
| 2.1 | interleukin-12 beta subunit precursor (IL-12B); cytotoxic lymphocyte maturation factor 40-kDa subunit | M65290 | |
| (CLMF p40); NK cell stimulatory factor subunit 2 (NKSF2) | |||
| 2.1 | glial growth factor 2 precursor (GGFHPP2); neuregulin; heregulin-beta3 + neu differentiation factor + | L12260; | |
| heregulin-alpha | L12261 + | ||
| U02326 + | |||
| M94165 | |||
| 2.0 | platelet-derived growth factor B subunit precursor (PDGFB; PDGF2); bacaplermin; c-sis | X02811; | |
| X02744; | |||
| M12783; | |||
| M16288 | |||
| 7.8 | bone morphogenetic protein 4 (BMP4) + bone morphogenetic protein 2B (BMP2B) | D30751 + | |
| M22490 | |||
| 2.3 | platelet-derived growth factor receptor alpha subunit (PDGFRA); CD140A antigen | M21574 | |
| 2.3 | platelet-derived growth factor receptor beta subunit (PDGFRB); CD140B antigen | M21616 | |
| Panel C: | Proteases/Matrix Proteins | ||
| 3.0 | matrix metalloproteinase 13 (MMP-13); collagenase 3 precursor | X75308 | |
| 2.3 | matrix metalloproteinase 7 (MMP-7); matrilysin | X07819 | |
| 2.1 | matrix metalloproteinase 12 (MMP-12); metalloelastase | L23808 | |
| 2.1 | collagen 6 alpha 2 subunit (COL6A2) | M34570 | |
| 2.1 | tissue-type plasminogen activator precursor (T-plasminogen activator; TPA) | M15518; | |
| X07393; | |||
| M18182 | |||
| 2.0 | urokinase-type plasminogen activator receptor GPI-anchored form precursor (U-PAR); monocyte activation | U08839; | |
| antigen MO3; CD87 antigen | M83246; | ||
| X51675 | |||
| 3.0 | chymotrypsin-like protease CTRL-1 precursor | X71874 | |
| 2.0 | cysteine protease | D55696 | |
| Adhesion Molecules | |||
| 3.5 | ninjurin-1 | U72661 | |
| 3.0 | desmocollin 3A/3B precursor (DSC3) + desmocollin 4 (DSC4) | X83929 + | |
| D17427 | |||
| 2.8 | tenascin precursor (TN); hexabrachion (HXB); cytotactin; neuronectin; GMEM; miotendinous antigen; | X78565; | |
| glioma-associated extracellular matrix antigen | M55618 | ||
| 2.2 | alpha-catenin related protein (CAPR); alpha 2 catenin (CTNNA2); alpha N-catenin | M94151 | |
| 4.0 | opioid-binding protein/cell adhesion molecule precursor (OBCAM); opioid-binding cell adhesion | L34774 | |
| molecule (OPCML) | |||
| 3.0 | integrin beta 4 (ITGB4); CD104 antigen | X53587; | |
| X52186; | |||
| X51841 | |||
| 2.2 | integrin alpha 1 (ITGA1); laminin & collagen receptor; VLA1; CD49A antigen | X68742 | |
| Panel D: | Signaling Intermediates | ||
| 7.0 | rho6 protein | Y07923 | |
| 2.9 | Gem; induced immediate early protein; ras family member (ras-like protein KIR) | U10550 | |
| 2.6 | regulator of G protein signaling 3 (RGS3; RGP3) | U27655 | |
| 2.5 | dual-specificity protein phosphatase 8; HVH5 | U27193 | |
| 2.3 | death receptor 5 (DR5); cytotoxic TRAIL receptor 2 (TRICK2A) | AF016268 | |
| 2.1 | protein kinase DYRK3 | Y12735 | |
| 2.1 | jagged 1 (HJ1) | AF028593 | |
| 6.0 | C-fgr proto-oncogene (p55-FGR); SRC2 | M19722 | |
| 4.0 | rho7 protein | X95456 | |
| 3.0 | GTP-binding protein ras associated with diabetes (RAD1) | L24564 | |
| 2.7 | rho-associated coiled-coil containing protein kinase p160ROCK | U43195 | |
| 2.5 | frizzled | L37882 | |
| 2.3 | cyclin-dependent kinase 4 inhibitor D (CDKN2D); p19-INK4D | U40343; | |
| U20498 | |||
| 2.3 | phosphotyrosyl phosphatase activator | X73478 | |
| 2.3 | protein kinase C-binding protein RACK7 | U48251 | |
| 2.2 | A-raf proto-oncogene serine/threonine-protein kinase; PKS2 | L24038 | |
| 2.2 | STE20-like kinase; MST2 | U26424 | |
| 2.1 | phosphorylase B kinase beta regulatory subunit | X84908 | |
| 2.0 | protein phosphatase 2A B56-alpha | L42373 | |
| Tumor Suppressors | |||
| 11.0 | melanoma differentiation-associated protein 7 precursor (MDA7) | U16261 | |
| 2.3 | tumor suppressor protein DCC precursor; colorectal cancer suppressor | X76132 |
We focused on genes for transcription factors because they have been reported as IL-1-responsive immediate early genes in chondrocytes [23]. Members of the NF-κB, AP-1, and ets families of transcription factors substantially increased in the Human Cancer 1.2 K array (see Table 2, panel A). To confirm the effect of IL-1β on AP-1 family members, we assayed Jun family members by RT-PCR (Fig. 1). We found that the c-Jun and JunB genes increased transiently within 1 hour of IL-1 treatment, before the induction of the MMP-13 gene. The expression profiles for c-Jun and JunB were similar to the profile for egr-1, another immediate early gene in IL-1-stimulated chondrocytes [23]. In contrast, the JunD gene was constitutively expressed, and IL-1β increased this expression modestly. These data show that in IL-1β-stimulated chondrocytes, all three Jun family members are expressed and can contribute to MMP gene expression. Furthermore, RT-PCR analysis of the JunB gene (Fig. 1) validated the gene expression profile derived from the Human Cancer 1.2 K array (Table 2, panel A).
Figure 1.
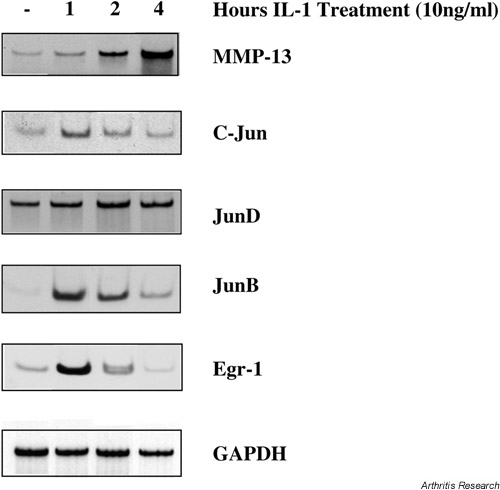
Early gene expression in IL-1-treated SW1353 chondrocytes. SW1353 cells were stimulated for the indicated times with IL-1β (10 ng/ml) and total RNA was isolated. RNA was treated with DNAse I and then reverse transcribed with Moloney murine leukemia virus reverse transcriptase and oligodT primers. A tenth of the reverse transcriptase reaction was amplified using gene-specific primers, Platinum Taq DNA polymerase, and 33PdATP. Single-band products were resolved on native acrylamide gels and visualized by autoradiography.
Transcription factor genes that were modulated by IL-1β included members from the NF-κB family (relB, p100/p52, p105/p50, IκB-ε, c-rel, and p65), the AP-1 family (fra-1 and junB), and the ETS family (ets-1). While it is well established that NF-κB proteins translocate to the nucleus upon IL-1 stimulation [24], we were surprised to find so many family members expressed simultaneously. This suggests that in IL-1-stimulated chondrocytes, NF-κB family members may compete for dimerization partners and for binding sites in responsive genes.
Members of the AP-1 family of transcription factors are essential transcriptional activators for both the MMP-1 and MMP-13 genes [25,26,27], and these transcription factors are activated by extracellular stimuli through MAPK pathways [28]. JunB was identified as an IL-1-inducible gene in chondrocytes by the Human Cancer 1.2 K array (Table 1) and this finding was confirmed by RT-PCR (Fig. 1). Levels of JunB mRNA peaked within 1 hour of IL-1 stimulation and began to decline after 2 hours. This was somewhat surprising, since MAPK pathways do not activate JunB [29] and in some systems JunB antagonizes transcriptional activation by c-Jun [30]. Since c-Jun, JunB, and JunD are all expressed in IL-1-treated SW1353 chondrocytic cells, it is not clear which family member is responsible for transcriptional activation of collagenase genes. Perhaps the functional transcriptional complex is determined by induced expression and activation of AP-1 accessory proteins, such as the Fos family member Fra-1, or the ets family member ets-1 [31]. This cohort of AP-1, Fos, and ets family members may also preferentially bind to different AP-1 sites in MMP promoters, with subtle effects on gene expression [32].
egr-1 is an IL-1-inducible immediate early gene in chondrocytes [23] and has been implicated in the elevated MMP-1 gene expression observed in rheumatoid synoviocytes [33,34]. Surprisingly, egr-1 expression was increased only 1.3-fold in this Human Cancer 1.2 K array (data not shown). However, when this gene was analyzed by RT-PCR, we found a robust increase in egr-1 mRNA within 1 hour of IL-1 stimulation, which declined significantly by 2 hours (Fig. 1). Thus, the relatively short half-life of egr-1 mRNA resulted in an underestimation of IL-1 induction of this gene in the microarray. It is therefore probable that an array using RNA from cells treated for 1 hour with IL-1 would identify additional important genes.
IL-1β repressed three transcription factor genes, including HOX-4A, the retinoblastoma-like protein 2, and mothers against dpp homologue 4 (SMAD4). SMAD4 contributes to transforming growth factor-β (TGF-β)-dependent transcription of the alpha 2(I)-collagen (COL1A2) [35] and aggrecan [36] genes. Furthermore, our microarray data demonstrated that IL-1 inhibited Col2A1 expression by 1.6-fold in SW1353 cells (data not shown). It has been reported that IL-1 inhibition of Col2A1 gene expression requires the p38 MAPK pathway [37]. Perhaps inhibition of SMAD4 synthesis is another component of IL-1-dependent Col2A1 gene repression. While IL-1 promotes cartilage degradation through MMP synthesis and inhibits matrix deposition by reducing Col2A1 gene expression, TGF-β has the opposite effect on chondrocytes [3]. Thus, it is tempting to speculate that IL-1 antagonizes the effects of TGF-β by blocking expression of essential signaling intermediates, such as SMAD4.
Cytokines and growth factors and their receptors were a major group of IL-1-modulated genes in chondrocytes (Table 2, panel B). For instance, leukemia inhibitor factor precursor (LIF) was substantially induced by IL-1β in SW1353 cells, and this is consistent with previous reports of LIF as an IL-1-responsive gene in chondrocytes [38,39]. Recently, LIF was reported to induce MMP-13 gene expression [40], and this may contribute to the role of this cytokine in arthritis pathology [38]. IL-6 expression was increased in IL-1-treated cells, and while this may affect a variety of cellular functions in chondrocytes, this cytokine does not seem to be required for IL-1-induced suppression of proteoglycan synthesis in cartilage [41]. Interestingly, there was a concurrent increase in expression of platelet-derived growth factor (PDGF) subunits, and a decrease in PDGF receptor subunits. Perhaps this is a mechanism of ensuring paracrine, but not autocrine, effects of PDGF in cytokine-stimulated chondrocytes. The role of growth factors in IL-1-dependent effects is not clear cut, since IL-1 can both induce and inhibit proliferation of chondrocytes [17]. However, in the present model of SW1353 cells under serum-free conditions, we did not observe a mitogenic effect of IL-1 (data not shown).
Bone morphogenetic protein 4 (BMP4) is a member of the TGF-β superfamily of proteins and is required for chondrocyte differentiation and cartilage maintenance [42,43]. The observed suppression of its expression in chondrocytes by IL-1β (Table 2, panel B) is consistent with a previous report in osteoblastic cells [44] and suggests that IL-1β may enhance cartilage degradation by blocking an essential autocrine signal for chondrocyte maturation and function.
IL-1 induction of several MMP family members is seen, including collagenase-3 (MMP-13), matrilysin (MMP-7), and metalloelastase (MMP-12) (Table 2, panel C). The collagenase-1 (MMP-1) gene was not significantly induced in this array, and this is consistent with our previous findings that MMP-1 is expressed at lower levels than MMP-13 in SW1353 cells [11]. Furthermore, the magnitude of increase observed for these MMP genes was modest, despite the fact that MMP genes are strongly induced in response to IL-1β. However, the most likely explanation for the low level of induction seen here is the fact that MMPs are not 'early response genes'. Rather, they are the downstream targets of the signal/transduction pathways and transcription factors that do represent early responses. Indeed, RT-PCR confirmed that MMP-13 mRNA continues to increase 4 hours after IL-1 treatment (Fig. 1). This is consistent with our previously published finding that steady-state MMP-13 mRNA levels peak 12 hours after IL-1 treatment of chondrocytes [11].
Tenascin, an adhesion molecule that is expressed during cartilage development [45], was induced by IL-1β in SW1353 cells (Table 2, panel C). Interestingly, the tenascin gene has been reported to be IL-1-inducible in synovial cells and chondrocytes, and its expression is enhanced in rheumatoid arthritis and OA [46,47]. A possible physiologic consequence of IL-1β-induced tenascin expression is that it may promote chondrocyte migration towards the sites of lesions in OA [47]. IL-1β coordinately reduced expression of integrin a1, a cell-adhesion molecule that chondrocytes use to bind to type II collagen [48]. Thus, IL-1β treatment may modify adhesion molecule expression, so that matrix adhesion is compromised.
We found that IL-1β changed the expression levels for several signaling proteins and tumor suppressor genes (Table 2, panel D), but the significance of these changes is not clear. Proteins associated with G-protein signaling were both increased (rho6, Gem, and regulator of G protein signaling) and decreased (rho7, RAD1, and p160ROCK). Jagged, which is a transmembrane ligand for the Notch signaling pathway [49], is induced by IL-1β. Interestingly, jagged-1 is important for epithelial–mesenchymal cell interaction in development [50], and mutations in the jagged gene lead to Alagille syndrome, a congenital connective tissue disorder whose hallmarks include craniofacial and vertebral deformities [51]. Zhao and colleagues [52] identified frizzled (denoted FZD2) as the human homologue of the Drosophila polarity-determining gene. Of potential importance is the fact that frizzled family members have recently been implicated by Carson and colleagues in rheumatoid arthritis [53]. A new frizzled family member that is expressed in chondrocytes and is involved in skeletal morphogenesis has been described [54]. Perhaps down-regulation of frizzled family members by IL-1β is deleterious to chondrocyte function and can contribute to OA.
Conclusions
Along with confirming changes in gene expression already known to be associated with IL-1β stimulation, chondrocyte biology, and MMP gene regulation, this microarray identified several other induced and repressed genes whose roles in chondrocyte biology are yet to be defined. While the significance of these findings in terms of understanding IL-1 effects on chondrocytes is still uncertain, the documentation of these changes in gene expression may provide the basis for future studies on the molecular effects of IL-1 on chondrocytes and on other cell types as well.
Abbreviations
AP-1 = activator protein-1; COL2A1 = procollagen 2 alpha 1; egr-1 = early growth response gene-1; ets = erythroblastosis gene twenty-six; IL-1β = interleukin-1β; LH = lactalbumin hydrolysate; LIF = leukemia inhibitory factor; MAPK= mitogen-activated protein kinase; MMP = matrix metalloproteinase; NF-κB = nuclear factor-κB; OA = osteoarthritis; PDGF = platelet-derived growth factor; RT-PCR = reverse transcriptase polymerase chain reaction; SMAD4 = mothers against dpp homolog 4; TGF-β = transforming growth factor-β.
Acknowledgments
Acknowledgements
The authors would like to acknowledge the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grants AR-46977 and AR-02024 to MPV; AR-26599 to CEB), the National Cancer Institute (grant CA-77267 to CEB) and the RGK Foundation, Austin Texas (grant to CEB) for funding of this research.
References
- Dinarello CA. Interleukin-1 and tumor necrosis factor: effector cytokines in autoimmune diseases. Semin Immunol. 1992;4:133–145. [PubMed] [Google Scholar]
- Arend WP, Dayer JM. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum. 1995;38:151–160. doi: 10.1002/art.1780380202. [DOI] [PubMed] [Google Scholar]
- Shlopov BV, Gumanovskaya ML, Hasty KA. Autocrine regulation of collagenase 3 (matrix metalloproteinase 13) during osteoarthritis. Arthritis Rheum. 2000;43:195–205. doi: 10.1002/1529-0131(200001)43:1<195::AID-ANR24>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Gabay C, Arend WP. Treatment of rheumatoid arthritis with IL-1 inhibitors. Springer Semin Immunopathol. 1998;20:229–246. doi: 10.1007/BF00832009. [DOI] [PubMed] [Google Scholar]
- van den Berg WB, Joosten LA, van de Loo FA. TNF alpha and IL-1 beta are separate targets in chronic arthritis. Clin Exp Rheumatol. 1999;17:S105–214. [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, Lee O, Brinckerhoff CE. Regulation of collagenase gene expression by IL-1 beta requires transcriptional and post-transcriptional mechanisms. Nucleic Acids Res. 1994;22:4818–4827. doi: 10.1093/nar/22.22.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, White LA, Barchowsky A, Brinckerhoff CE. src-related tyrosine kinases regulate transcriptional activation of the interstitial collagenase gene, MMP-1, in interleukin-1-stimulated synovial fibroblasts. Arthritis Rheum. 1996;39:574–582. doi: 10.1002/art.1780390406. [DOI] [PubMed] [Google Scholar]
- Vincenti MP, Schroen DJ, Coon CI, Brinckerhoff CE. v-src activation of the collagenase-1 (matrix metalloproteinase-1) promoter through PEA3 and STAT: requirement of extracellular signal-regulated kinases and inhibition by retinoic acid receptors. Mol Carcinog. 1998;21:194–204. [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, Mengshol JA, Yocum S, Mitchell P, Brinckerhoff CE. Cloning of the gene for interstitial collagenase-3 (matrix metalloproteinase-13) from rabbit synovial fibroblasts: differential expression with collagenase-1 (matrix metalloproteinase-1). Biochem J. 1998;331:341–346. doi: 10.1042/bj3310341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum. 1998;41:1987–1994. doi: 10.1002/1529-0131(199811)41:11<1987::AID-ART14>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43:801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Frleta D, Vincenti MP. Integration of the NF-kappaB and mitogen-activated protein kinase/AP-1 pathways at the collagenase-1 promoter: divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts. Cytokine. 2000;12:1469–1479. doi: 10.1006/cyto.2000.0743. [DOI] [PubMed] [Google Scholar]
- Bondeson J, Foxwell B, Brennan F, Feldmann M. Defining therapeutic targets by using adenovirus: Blocking NF-kappaB inhibits both inflammatory and destructive mechanisms in rheumatoid synovium but spares anti-inflammatory mediators. Proc Natl Acad Sci U S A. 1999;96:5668–5673. doi: 10.1073/pnas.96.10.5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Aupperle KR, Bennett B, Manning AM, Firestein GS. Jun N-terminal kinase in rheumatoid arthritis. J Pharmacol Exp Ther. 1999;291:124–130. [PubMed] [Google Scholar]
- Reunanen N, Westermarck J, Hakkinen L, Holmstrom TH, Elo I, Eriksson JE, Kahari VM. Enhancement of fibroblast collagenase (matrix metalloproteinase-1) gene expression by ceramide is mediated by extracellular signal-regulated and stress-activated protein kinase pathways. J Biol Chem. 1998;273:5137–5145. doi: 10.1074/jbc.273.9.5137. [DOI] [PubMed] [Google Scholar]
- McCachren SS, Greer PK, Niedel JE. Regulation of human synovial fibroblast collagenase messenger RNA by interleukin-1. Arthritis Rheum. 1989;32:1539–1545. doi: 10.1002/anr.1780321207. [DOI] [PubMed] [Google Scholar]
- Blanco FJ, Geng Y, Lotz M. Differentiation-dependent effects of IL-1 and TGF-beta on human articular chondrocyte proliferation are related to inducible nitric oxide synthase expression. J Immunol. 1995;154:4018–4026. [PubMed] [Google Scholar]
- Blanco FJ, Lotz M. IL-1-induced nitric oxide inhibits chondrocyte proliferation via PGE2. Exp Cell Res. 1995;218:319–325. doi: 10.1006/excr.1995.1161. [DOI] [PubMed] [Google Scholar]
- Alvaro-Gracia JM, Zvaifler NJ, Firestein GS. Cytokines in chronic inflammatory arthritis. V. Mutual antagonism between inter-feron-gamma and tumor necrosis factor-alpha on HLA-DR expression, proliferation, collagenase production, and granulocyte macrophage colony-stimulating factor production by rheumatoid arthritis synoviocytes. J Clin Invest. 1990;86:1790–1798. doi: 10.1172/JCI114908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RA, Schena M, Chai A, Shalon D, Bedilion T, Gilmore J, Woolley DE, Davis RW. Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci U S A. 1997;94:2150–2155. doi: 10.1073/pnas.94.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden P, Solymar D, Sucharczuk A, Lindman B, Cannon P, Heller RA. Cytokine control of interstitial collagenase and collagenase-3 gene expression in human chondrocytes. J Biol Chem. 1996;271:23577–23581. doi: 10.1074/jbc.271.38.23577. [DOI] [PubMed] [Google Scholar]
- Mix KS, Mengshol JA, Benbow U, Vincenti MP, Sporn MB, Brinck-erhoff CE. A synthetic triterpenoid selectively inhibits the induction of matrix metalloproteinases 1 and 13 by inflammatory cytokines. Arthritis Rheum. 2001;44:1096–1104. doi: 10.1002/1529-0131(200105)44:5<1096::AID-ANR190>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S, Glowacki J, Arbiser JL, Apperley JF. Interleukin-1 beta-modulated gene expression in immortalized human chondrocytes. J Clin Invest. 1994;94:2307–2316. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Porte D, Tuckermann J, Becker M, Baumann B, Teurich S, Higgins T, Owen MJ, Schorpp-Kistner M, Angel P. Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene. 1999;18:667–678. doi: 10.1038/sj.onc.1202333. [DOI] [PubMed] [Google Scholar]
- Auble DT, Brinckerhoff CE. The AP-1 sequence is necessary but not sufficient for phorbol induction of collagenase in fibroblasts. Biochemistry. 1991;30:4629–4635. doi: 10.1021/bi00232a039. [DOI] [PubMed] [Google Scholar]
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis elecent recognized by a TPA-modulated transacting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Szabowski A, Maas-Szabowski N, Andrecht S, Kolbus A, Schorpp-Kistner M, Fusenig NE, Angel P. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell. 2000;103:745–755. doi: 10.1016/s0092-8674(00)00178-1. [DOI] [PubMed] [Google Scholar]
- Buttice G, Duterque-Coquillaud M, Basuyaux JP, Carrere S, Kurki-nen M, Stehelin D. Erg, an Ets-family member, differentially regulates human collagenase1 (MMP1) and stromelysin1 (MMP3) gene expression by physically interacting with the Fos/Jun complex. Oncogene. 1996;13:2297–2306. [PubMed] [Google Scholar]
- White LA, Brinckerhoff CE. Two activator protein-1 elements in the matrix metalloproteinase-1 promoter have different effects on transcription and bind Jun D, c-Fos, and Fra-2. Matrix Biol. 1995;14:715–725. doi: 10.1016/s0945-053x(05)80014-9. [DOI] [PubMed] [Google Scholar]
- Trabandt A, Aicher WK, Gay RE, Sukhatme VP, Fassbender HG, Gay S. Spontaneous expression of immediately-early response genes c-fos and egr-1 in collagenase-producing rheumatoid synovial fibroblasts. Rheumatol Int. 1992;12:53–59. doi: 10.1007/BF00300977. [DOI] [PubMed] [Google Scholar]
- Trabandt A, Gay RE, Birkedal-Hansen H, Gay S. Expression of collagenase and potential transcriptional factors in the MRL/l mouse arthropathy. Semin Arthritis Rheum. 1992;21:246–251. doi: 10.1016/0049-0172(92)90055-i. [DOI] [PubMed] [Google Scholar]
- Zhang W, Ou J, Inagaki Y, Greenwel P, Ramirez F. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor beta1 stimulation of alpha 2(I)-collagen (COL1A2) transcription. J Biol Chem. 2000;275:39237–39245. doi: 10.1074/jbc.M003339200. [DOI] [PubMed] [Google Scholar]
- Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–14473. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- Robbins JR, Thomas B, Tan L, Choy B, Arbiser JL, Berenbaum F, Goldring MB. Immortalized human adult articular chondrocytes maintain cartilage-specific phenotype and responses to interleukin-1beta. Arthritis Rheum. 2000;43:2189–2201. doi: 10.1002/1529-0131(200010)43:10<2189::AID-ANR6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Lotz M, Moats T, Villiger PM. Leukemia inhibitory factor is expressed in cartilage and synovium and can contribute to the pathogenesis of arthritis. J Clin Inves. 1992;90:888–896. doi: 10.1172/JCI115964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IK, Waring P, Novak U, Hamilton JA. Production of leukemia inhibitory factor by human articular chondrocytes and cartilage in response to interleukin-1 and tumor necrosis factor alpha. Arthritis Rheum. 1993;36:790–794. doi: 10.1002/art.1780360608. [DOI] [PubMed] [Google Scholar]
- Varghese S, Yu K, Canalis E. Leukemia inhibitory factor and oncostatin M stimulate collagenase-3 expression in osteoblasts. Am J Physiol. 1999;276:E465–471. doi: 10.1152/ajpendo.1999.276.3.E465. [DOI] [PubMed] [Google Scholar]
- Van de Loo FA, Arntz OJ, Van den Berg WB. Effect of inter-leukin 1 and leukaemia inhibitory factor on chondrocyte metabolism in articular cartilage from normal and interleukin-6-deficient mice: role of nitric oxide and IL-6 in the suppression of proteoglycan synthesis. Cytokine. 1997;9:453–462. doi: 10.1006/cyto.1997.0188. [DOI] [PubMed] [Google Scholar]
- Luyten FP, Chen P, Paralkar V, Reddi AH. Recombinant bone morphogenetic protein-4, transforming growth factor-beta 1, and activin A enhance the cartilage phenotype of articular chondrocytes in vitro. Exp Cell Res. 1994;210:224–229. doi: 10.1006/excr.1994.1033. [DOI] [PubMed] [Google Scholar]
- Shukunami C, Akiyama H, Nakamura T, Hiraki Y. Requirement of autocrine signaling by bone morphogenetic protein-4 for chondrogenic differentiation of ATDC5 cells. FEBS Lett. 2000;469:83–87. doi: 10.1016/s0014-5793(00)01251-5. [DOI] [PubMed] [Google Scholar]
- Virdi AS, Cook LJ, Oreffo RO, Triffitt JT. Modulation of bone morphogenetic protein-2 and bone morphogenetic protein-4 gene expression in osteoblastic cell lines. Cell Mol Biol (Noisy-le-grand) 1998;44:1237–1246. [PubMed] [Google Scholar]
- Pacifici M, Iwamoto M, Golden EB, Leatherman JL, Lee YS, Chuong CM. Tenascin is associated with articular cartilage development. Dev Dyn. 1993;198:123–134. doi: 10.1002/aja.1001980206. [DOI] [PubMed] [Google Scholar]
- McCachren SS, Lightner VA. Expression of human tenascin in synovitis and its regulation by interleukin-1. Arthritis Rheum. 1992;35:1185–1196. doi: 10.1002/art.1780351011. [DOI] [PubMed] [Google Scholar]
- Chevalier X, Claudepierre P, Groult N, Godeau GJ. Influence of interleukin 1 beta on tenascin distribution in human normal and osteoarthritic cartilage: a quantitative immunohistochem-ical study. Ann Rheum Dis. 1996;55:772–775. doi: 10.1136/ard.55.10.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser RF, Sadiev S, Tan L, Goldring MB. Integrin expression by primary and immortalized human chondrocytes: evidence of a differential role for alpha1beta1 and alpha2beta1 integrins in mediating chondrocyte adhesion to types II and VI collagen. Osteoarthritis Cartilage. 2000;8:96–105. doi: 10.1053/joca.1999.0277. [DOI] [PubMed] [Google Scholar]
- Lindsell CE, Shawber CJ, Boulter J, Weinmaster G. Jagged: a mammalian ligand that activates Notch1. Cell. 1995;80:909–917. doi: 10.1016/0092-8674(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Mitsiadis TA, Henrique D, Thesleff I, Lendahl U. Mouse Serrate-1 (Jagged-1): expression in the developing tooth is regulated by epithelial-mesenchymal interactions and fibroblast growth factor-4. Development. 1997;124:1473–1483. doi: 10.1242/dev.124.8.1473. [DOI] [PubMed] [Google Scholar]
- Pilia G, Uda M, Macis D, Frau F, Crisponi L, Balli F, Barbera C, Colombo C, Frediani T, Gatti R, Iorio R, Marazzi MG, Marcellini M, Musumeci S, Nebbia G, Vajro P, Ruffa G, Zancan L, Cao A, DeVirgilis S. Jagged-1 mutation analysis in Italian Alagille syndrome patients. Hum Mutat. 1999;14:394–400. doi: 10.1002/(SICI)1098-1004(199911)14:5<394::AID-HUMU5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Lee CC, Baldini A, Caskey CT. A human homologue of the Drosophila polarity gene frizzled has been identified and mapped to 17q21.1. Genomics. 1995;27:370–373. doi: 10.1006/geno.1995.1060. [DOI] [PubMed] [Google Scholar]
- Sen M, Lauterbach K, El-Gabalawy H, Firestein GS, Corr M, Carson DA. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2000;97:2791–2796. doi: 10.1073/pnas.050574297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang B, Moos M, Jr, Vukicevic S, Luyten FP. Primary structure and tissue distribution of FRZB, a novel protein related to Drosophila frizzled, suggest a role in skeletal morphogenesis. J Biol Chem. 1996;271:26131–26137. doi: 10.1074/jbc.271.42.26131. [DOI] [PubMed] [Google Scholar]