

SUMMARY
Alveolar macrophages (AMs), upon sensing pathogens, trigger host defense by activating toll-like receptor 4 (TLR4), but the counterbalancing mechanisms that deactivate AM inflammatory signaling and prevent lethal edema, the hallmark of acute lung injury (ALI), remain unknown. Here, we demonstrate the essential role of AM protease-activating receptor 2 (PAR2) in rapidly suppressing inflammation to prevent long-lasting injury. We show that thrombin, released during TLR4-induced lung injury, directly activates PAR2 to generate cAMP, which abolishes Ca2+ entry through the TRPV4 channel. Deletion of PAR2 and thus the accompanying cAMP generation augments Ca2+ entry via TRPV4, causing sustained activation of the transcription factor NFAT to produce long-lasting TLR4-mediated inflammatory lung injury. Rescuing thrombin-sensitive PAR2 expression or blocking TRPV4 activity in PAR2-null AMs restores their capacity to resolve inflammation and reverse lung injury. Thus, activation of the thrombin-induced PAR2-cAMP cascade in AMs suppresses TLR4 inflammatory signaling to reinstate tissue integrity.
Graphical Abstract
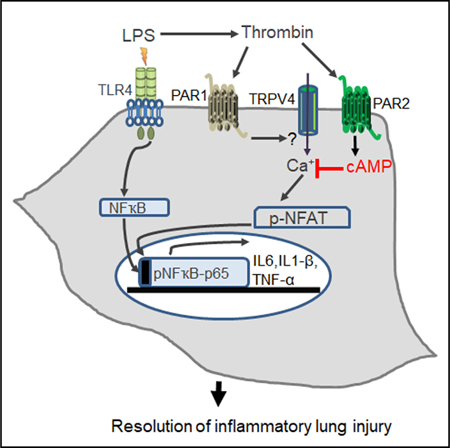
In Brief
Alveolar macrophages trigger inflammatory signaling upon TLR4 activation by pathogens, and this signaling, if unchecked, can lead to pulmonary edema, the hallmark of acute lung injury. Rayees et al. demonstrate that thrombin ligation of the alveolar macrophage protease-activating receptor 2 post-injury suppresses inflammatory lung injury via a cAMP blockade of the TRPV4 channel.
INTRODUCTION
Alveolar macrophages (AMs), the sentinel immune cells in the lung, have a pivotal function in triggering host defense (Herold et al., 2011; Rubins, 2003; Westphalen et al., 2014). Upon sensing the pathogen-associated molecular patterns, for example, lipopolysaccharide (LPS) (a cell wall component of Gram-negative bacteria), TLR4 expressed on AMs is activated with the primary purpose of inducing the production of pro-inflammatory cytokines and neutrophil transmigration into the lung via activation of nuclear factor kB (NF-ҡB) and other transcription factors (Ballinger and Christman, 2016; Herold et al., 2011). However, unchecked AM inflammatory signaling can impair the tissue repair process leading to development of acute lung injury (ALI). ALI frequently develops following sepsis, trauma, or pneumonia and, if unresolved, can progress to acute respiratory distress syndrome (ARDS) leading to high mortality and morbidity (Hook et al., 2018; Leaver and Evans, 2007). The counterbalancing mechanisms that rapidly suppress inappropriate AM inflammatory signaling and thereby facilitate induction of tissue repair to resolve ALI remain elusive.
While the inflammation and coagulation pathways are studied separately, a tight linkage between these two fundamental biological processes is required to optimize the organism’s response to injury and invasion by pathogens (Coughlin, 2005; Kaneider et al., 2007). Inflammation-induced tissue injury leads to release of several proteases such as trypsin, thrombin, elastases, FVIIa, and FXa (Coughlin, 2005; Kaneider et al., 2007). These proteases can in turn activate receptors on AMs, endothelial cells, and epithelial cells (Moraes et al., 2008; van den Boogaard et al., 2018). Protease-activating receptor 2 (PAR2), a G-protein-coupled receptor belonging to the PAR family, is strongly expressed in the lung cell types mentioned above and can be activated by several of these proteases directly or through transactivation (Churg et al., 2007; Mihara et al., 2016; O’Brien et al., 2000). Thrombin is a well-known inducer of lung vascular permeability rise by activating the endothelial PAR1 (Chava et al., 2012; Kaneider et al., 2007; Stief, 2006). AMs express both PAR1 and PAR2 (Churg et al., 2007; Jesmin et al., 2007), and thrombin activates PAR1 with higher affinity than PAR2 (Mihara et al., 2016), suggesting that transactivation of PAR2 on AMs can occur with thrombin. How PAR2, activated upon tissue injury, modifies TLR4-mediated inflammatory signaling and thereby ALI and whether it is through AM-PAR2 remain unclear. A few studies have in fact suggested an anti-inflammatory role of PAR2 but in a cellular context-dependent manner. For example, loss of PAR2 augmented lung inflammation induced by Pseudomonas aeruginosa primarily by activating macrophages, and a low dose of a PAR2 receptor agonist reduced neutrophil influx in a mouse model of ARDS (Moraes et al., 2008; White et al., 2018).
Here, we systematically evaluated the role of PAR2 in regulating TLR4 inflammatory signaling post-tissue injury. Using bone marrow transplantation, adoptive transfer of PAR2 or wild-type (WT) macrophages, and liposome-mediated delivery of thrombin-sensitive versus -insensitive PAR2 cDNA in AMs, we demonstrate that AM-PAR2 is activated by thrombin following tissue injury secondary to TLR4-mediated inflammatory signaling. Loss of AM-PAR2 thereby leads to exacerbated inflammatory signaling and tissue damage, thus impairing resolution of long-lasting tissue edema. We show that PAR2-induced cAMP generation in AMs suppresses activation of the transcription factor NFAT, secondary to Ca2+ entry through the TRPV4 channel, which prevents both cytokine generation and neutrophil influx to promote resolution of lung injury.
RESULTS
PAR2 Expressed on AMs Is Required to Resolve Lung Inflammatory Vascular Injury
PAR2 is expressed in several lung cell types and may modulate inflammatory vascular injury in response to invading microorganisms (Moraes et al., 2008; van den Boogaard et al., 2018). Thus, we exposed PAR2-null mice to nebulized LPS and assessed its role in regulating vascular injury and neutrophil infiltration into the lung. Because PAR1 can transactivate PAR2 (Kaneider et al., 2007; O’Brien et al., 2000), we also used PAR1-null mice to compare the injury-repair responses observed in PAR2-null mice (Figure S1A). In control mice, LPS increased inflammatory vascular injury within 4 h as indicated by the observed increase in lung wet-dry weight ratio as well as trans-endothelial albumin and neutrophil accumulation in the lung, which returned to near basal levels in the next 24 h (Figures 1A–1C and S1B). LPS induced a similar level of inflammatory vascular injury in PAR2-null mice as in WT mice (Figures 1A–1C and S1B). Intriguingly, lung injury failed to resolve in PAR2-null lungs (Figures 1A–1C and S1B). In contrast to PAR2-null mice, LPS failed to induce significant lung vascular injury (Figures S1C and S1D) or neutrophil accumulation in PAR1-null lungs (Figure S1E). Loss of PAR2 did not alter PAR1 expression in lung and vice versa (Figure S1A). Analysis of inflammatory cytokine protein expression in bronchoalveolar lavage (BAL) showed that LPS similarly elevated the levels of tumor necrosis factor alpha (TNF-a), interleukin-6 (IL-6), and IL1-b in WT and PAR2-null mice at 4 h (Figure 1D). At 24 h, BAL cytokines returned to near basal levels in WT mice but remained elevated in PAR2-null mice (Figure 1D). Similar findings were obtained using qPCR analysis of WT and PAR2-null BAL at 24 h (Figure S1F). In other studies, we depleted PAR2 in human macrophages to further establish the role of PAR2 in regulating LPS-induced inflammatory injury. We used the a human monocytic cell line, U-937, and, after differentiating the cells into macrophages, depleted PAR2 using small interfering RNA (siRNA). Control and PAR20depleted human macrophages were then stimulated with LPS for 4 h, and cytokine expression was determined. We observed that LPS also increased IL-6 and IL1-b levels in PAR2-depleted human macrophages more than control human macrophages (Figure 1E). These findings demonstrate the critical role of PAR2 in resolving LPS-induced lung inflammatory vascular injury.
Figure 1. Deletion of PAR2 Impairs Resolution of Lung Inflammatory Vascular Injury Induced by LPS.

(A and B) WT and Par2−/− mice were exposed to nebulized LPS (1 mg/mL) for 45 min. 30 min before sacrificing the mice at the indicated times, Evans blue-labeled albumin was injected retro-orbitally into each mouse. Lung vascular inflammatory injury was determined by measuring albumin influx (A) and lung wet-dry ratio (B). n = 6 mice/group.
(C) Neutrophil count was performed (per field) on H&E-stained WT and Par2−/− lung sections. The plot shows individual values from three independent experiments.
In (A)–(C), the data are presented as mean ± SD. ***p < 0.001 indicates values that are significantly different from control group receiving vehicle alone. ###p < 0.001 indicates values that are significantly different from WT mice post-24 and −48 h LPS challenge.
(D) After 4 and 24 h following LPS-induced injury, bronchoalveolar lavage was obtained from WT and Par2−/− mice and the levels of the indicated cytokines were determined by ELISA. Data are represented as mean ± SD from experiments that were performed three times independently. **p < 0.01 and ***p < 0.001 indicate values that are significantly different from mice receiving vehicle alone. ##p < 0.01 and ###p < 0.001 indicate values that were significantly different from WT-mice post-4 and −24 h LPS challenge.
(E) U-937 human monocytic cells were differentiated to macrophages by exposing them to PMA (100 ng/mL) for 48 h. Differentiated macrophages were then transfected with scrambled or PAR2 siRNA. After 48 h, the cells were serum starved for 30 min followed by addition of 1 mg/mL LPS. Expression of indicated cytokines was determined using qPCR. GAPDH was used as an internal control. Data are represented as mean ± SD; experiments were performed three times individually. *p < 0.05 and **p < 0.01 indicates values that are significantly different from unstimulated scrambled siRNA transfected cells. #p < 0.05 indicates values that are significantly different from cells transfected with control siRNA post-4 h LPS challenge. In all figures, one-way ANOVA followed by paired two-tailed t test was used to assess significance between groups.
Next, we determined the contribution of myeloid cells versus parenchymal cells in mediating LPS-induced lung vascular inflammatory injury. We transplanted PAR2-null bone marrow cells into lethally irradiated WT mice (PAR2→WT chimera) or WT bone marrow into PAR2-null mice (WT→PAR2 chimera) and exposed the mice to LPS. The engraftment of donor (either WT or Par2−/−) bone marrow cells into the recipient mice was confirmed by imaging (Figure S2A). Challenging WT → Par2−/− chimeric mice with LPS induced lung edema at 4 h, which resolved at 24 h (Figure 2A). In contrast, challenging Par2−/−→ WT chimeric mice with LPS induced edema formation that persisted for 24 h (Figure 2A). Thus, PAR2 expression in hematopoietic cells was responsible for resolving the LPS-induced increase in edema formation.
Figure 2. AM PAR2 Is Required to Resolve Lung Inflammatory Vascular Injury.

(A) After bone marrow irradiation, WT mice were injected with Par2−/− bone marrow cells and vice versa. Six weeks after the transplant, the mice were challenged with nebulized LPS for 45 min, and the lung wet-dry ratio was determined at the indicated times in the chimeric mice. The plot shows individual values with mean ± SD. ***p < 0.001 indicates values significantly different from mice receiving vehicle (no LPS). n = 6 mice/group.
(B and C) Clodronate liposomes (300 mg/mouse in a volume of 60 mL) were injected i.t. into WT or PAR2-null mice, and, after 48 h, BMDMs isolated from WT or PAR2-null mice were adoptively transferred to mouse lungs. At 72 h, the mice were exposed to nebulized LPS for 45 min, and lungs and BAL were harvested at 96 h to determine lung injury (B). The plot shows individual values of lung wet-dry ratio in indicated mice post-LPS challenge (C). ***p < 0.001 indicates values that are significantly different from control group receiving vehicle alone. ###p < 0.001 indicates values that are significantly different from corresponding WT mice group post-LPS challenge. n = 5 mice/group.
(D) BAL macrophages were isolated and RNA was extracted from them to determine cytokine expression. Data are represented as mean ± SD of three independently performed experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 indicate values that are significantly different from corresponding vehicle control group. #p < 0.05, ##p < 0.01, and ###p < 0.001 indicate values that are significantly different from WT mice that received LPS.
In all figures, one-way ANOVA followed by paired two-tailed t test was used to assess significance between groups.
AMs play a critical role in dampening inflammatory injury and accelerating neutrophil clearance (Herold et al., 2011; Westphalen et al., 2014). Using fluorescence-activated cell sorting (FACS) analysis and imaging of BAL, we confirmed that loss of PAR2 did not impair AM generation (Figures S2B and S2C). So, we tested the possibility that PAR2 in AMs resolves lung injury. To this end, we depleted AMs in WT and PAR2-null mice by injecting clodronate liposomes intracheal (i.t.) and after 48 h adoptively transferred WT or PAR2-null bone-marrow-derived macrophages (BMDMs) into WT or PAR2-null mice, respectively (Di et al., 2018), and determined lung injury following LPS challenge (Figure 2B). We observed that adoptive transfer of WT-BMDMs (+/+) in PAR2-null lungs resolved lung edema and reduced BAL-inflammatory cytokines (Figures 2C and 2D). As expected, adoptive transfer of PAR2-null BMDMs (—/—) into PAR2-null lungs did not promote resolution of inflammatory lung injury (Figures 2B and 2C). Interestingly, adoptive transfer of PAR2-null BMDMs into WT lungs impaired resolution of inflammatory lung injury, mimicking our findings in PAR2-null mice (Figures 2B and 2C). These findings demonstrate that AMs expressing PAR2 play a critical role in resolving LPS-induced lung injury.
PAR2 Resolves LPS-Induced Lung Injury by Ligating Thrombin
LPS can promote localized generation of thrombin at the injury site, which can modulate vascular injury by ligating PAR1 and PAR2 (Kaneider et al., 2007; Mihara et al., 2016; Stief, 2006). To address the possibility that LPS stimulated PAR2 by generating thrombin, we used recently developed dual-tagged wildtype PAR2 (WT) which serves as a reporter for receptor activity. Also, we used the thrombin cleavage-resistant dual-tagged PAR2 alanine mutant (MT) cDNA (Mihara et al., 2016). We first transduced WT-or MT-PAR2 cDNA into PAR2-null BMDMs and stimulated these cells with thrombin to confirm thrombin activation of PAR2. We found that upon cleavage by thrombin, the WT dual receptor (yellow) separated into red and green subpopulations as the mRFP tag was released by PAR2 via thrombin cleavage (Figure 3A). However, the MT receptor remained yellow, as thrombin failed to cleave (mutated) PAR2 (Figure 3A). Next, we complexed WT-or MT-PAR2 cDNA with liposomes and delivered them i.t. into Par2−/− mice. These mice were then challenged with LPS to corroborate the role of thrombin activation of AM-PAR2 in resolving lung edema. We observed that transducing WT-PAR2 cDNA into PAR2-null mice resolved lung edema much as observed above in WT mice (Figure 3B). However, the lung injury persisted in PAR2-null mice transduced with mutated PAR2 receptor (cleavage-resistant dual-tagged MT) (Figure 3B). We next sorted CD45+/CD11c+/SiglecF+ cells from PAR2-null lungs post-liposome gene transfer using FACS analysis and confirmed that ~70% of the PAR2 expression is indeed rescued in AMs compared to WT cells (Figure 3C). To further corroborate these findings, we transfected WT-or MT-PAR2 cDNA into PAR2-null BMDMs and found that replenishing WT-PAR2 cDNA in PAR2-null BMDMs restored the expression of pro-inflammatory cytokines to the levels observed in WT-BMDMs post-LPS challenge (Figure 3D). However, enhanced cytokine generation persisted in PAR2-null BMDM-transducing MT-PAR2 cDNA (Figure 3D).
Figure 3. Thrombin Activation of PAR2 in Macrophages Suppresses LPS-Induced Inflammatory Lung Injury.

(A) PAR2-null BMDMs were transfected with WT dual tagged PAR2 cDNA (WT) or a dual tagged MT-PAR2 cDNA (resistant to thrombin cleavage [MT]) by electroporation. After 48 h, the cells were serum starved in media containing 0.1% FBS for 30 min. The cells were then stimulated with 50 U/mL thrombin, fixed with 2% paraformaldehyde for 10 min, and imaged using confocal microscope (scale bars, 5 mm). Images from a representative experiment are shown, and these experiments were performed three times independently.
(B) Liposomes complexed with either WT-PAR2 or MT-PAR2 cDNA were administered i.t into WT or Par2−/− mice, and, after 48 h, the mice were exposed to nebulized LPS for 45 min. Lung injury was assessed by determining lung wet-dry ratio, 24 h after LPS inhalation. The plot shows individual values with mean ± SD. ***p < 0.001 indicates values that are significantly different from PAR2-null mice receiving control vector. n = 5 mice/ group.
(C) Lungs from PAR2-null mice transducing WT-PAR2 or MT-PAR2 cDNA were digested with collagenase and stained with AM markers (CD11c, CD45, and Siglec-F antibodies). AMs (CD11c+/CD45+/Siglec F+) were sorted and PAR2 expression was determined using qPCR taking GAPDH as control. AMs sorted from WT lungs served as control for PAR2 expression. The plot shows mean ± SD of PAR2 expression. ***p < 0.001 indicates values that are significantly different from PAR2-null mice receiving control vector. The experiment was performed three times independently.
(D and E) WT and Par2−/− BMDMs were transfected with WT-PAR2 cDNA or MT-PAR2 cDNA. After 48 h of transfection, the cells were stimulated with LPS for 4 h. RNA was isolated and the expression of indicated pro-inflammatory genes (D) and PAR2 (E) was determined by qPCR. The data are represented as mean ± SD of three independent experiments. *p < 0.05 and **p < 0.01 indicate a significant increase in expression levels in comparison to unstimulated cells. #p < 0.05 and ##p < 0.01 indicate significant increase in expression of cytokines in comparison to WT-BMDMs post-LPS challenge.
(F) WT mice were exposed to nebulized LPS for 45 min and after 1 h PAR2-AP (SLIGRL-NH2) or control peptide (LRGILS) was delivered i.t. (1 mg/kg of body weight). Lung wet-dry ratio was determined at 4 h after LPS exposure. The plot shows individual values with mean ± SD n = 6 mice /group. ***p < 0.001 indicates values significantly different from control mice. ###p < 0.001 indicates values that are significantly different from mice receiving PAR2-AP with LPS.
In all figures, one-way ANOVA followed by paired two-tailed t test was used to assess significance between groups.
Next, we investigated whether activation of PAR2 directly modulates LPS-induced injury using a PAR2-activating peptide (PAR2-AP). We instilled PAR2-AP i.t. into WT mice and after 1 h challenged these mice with LPS followed by assessment of lung vascular injury at 4 h. PAR2-AP significantly reduced LPS-induced lung vascular injury at 4 h (Figure 3E). These findings indicate that following lung injury by LPS the ligand thrombin activates PAR2 in AMs to promote their pro-resolution role following ALI.
PAR2 Suppresses a Ca2+-Dependent Increase in NFAT Activity and Cytokine Generation
PAR2 activates multiple G protein-linked signaling pathways, including intracellular Ca2+ signaling, which can then regulate cytokine generation in a Ca2+-dependent manner (Jairaman et al., 2015; Ramachandran et al., 2009). Thus, we surmised that PAR2 switches off inflammatory signaling by regulating cytosolic Ca2+ levels. The thrombininduced rise in intracellular Ca2+ occurs in two phases. The first phase is due to mobilization of Ca2+ from endoplasmic reticulum stores (ERs) followed by Ca2+ entry through plasmalemmal channels (Yazbeck et al., 2017). We stimulated WT and PAR2-null BMDMs with thrombin in Ca2+-free media to deplete ER Ca2+ stores followed by add-back of 2 mM Ca2+ to determine the role of PAR2 in regulating cytosolic Ca2+ increase. We observed that, under Ca2+-free bath conditions, PAR2 deletion had no effect on thrombin-induced release of Ca2+ from ER stores (Figures 4A and 4B). However, loss of PAR2 significantly increased Ca2+ entry upon Ca2+ repletion (Figures 4A and 4B).
Figure 4. Loss of PAR2 Augments Ca2+Entry and Cytokine Generation via NFAT Activity.

(A and B) WT and Par2−/− BMDMs were loaded with Fura2 for 15 min following which they were stimulated with thrombin in Ca2+-free media to deplete intracellular stores. Ca2+ entry was invoked by repleting 2 mM Ca2+ at ~360 s. A representative trace is shown in (A), while (B) shows individual data points along with mean ± SD of 12–15 cells. Results show data from experiments that were repeated three times. ***p < 0.001 indicates values that were significantly different from WT-BMDMs.
(C) WT and Par2−/− BMDMs were stimulated with LPS (1 mg/mL) for indicted times and lysed. Phosphorylation of p65 subunit of NF-ҡB and NFAT was determined by western blotting using NFAT or NF-ҡB phosphor-specific antibodies. Immunoblotting with anti-actin antibody served as a loading control. A representative immunoblot is shown from experiments that were independently repeated three times.
(D) BMDMs were left unstimulated or stimulated with LPS for 4 h and RNA was isolated. The expression of indicated pro-inflammatory genes was determined by qPCR. Data are represented as mean ± SD from three independent experiments. *p < 0.05 and **p < 0.01 indicate values that were significantly different from unstimulated BMDMs. #p < 0.05 and ##p < 0.01 indicate values that were significantly different from WT-BMDMs post-LPS challenge.
(E) WT and Par2−/− BMDMs were transfected with scrambled or NFATc1 siRNA, and after 48 h the cells were stimulated with LPS for 4 h. The RNA was isolated and the expression of indicated pro-inflammatory genes was determined by qPCR. Data are represented as mean ± SD from three independent experiments. *p < 0.05 and **p < 0.01 indicate values significantly different from untreated BMDMs. #p < 0.05 and ##p < 0.01 indicate values that were significantly different from WT-BMDMs post-LPS challenge.
(F) DNA fragments obtained from transfected and control WT and Par2−/− BMDMs were purified. qPCR was performed using primers specific for TNF-a, IL-6, and IL1-b. Data are represented as mean ± SD of three independent experiments. *p < 0.05 and **p < 0.01 indicate values significantly different from untreated BMDMs. #p < 0.05 indicates values that were significantly different from WT-BMDMs post-LPS challenge.
In all figures, one-way ANOVA followed by paired two-tailed t test was used to assess significance between groups.
Ca2+ entry induces the activities of both NF-ҡB as well as NFAT but in a cell-context-dependent manner (Bair et al., 2009; Rao, 2009). While it is known that LPS via TLR4 induces pro-inflammatory cytokine generation through NF-ҡB (Ballinger and Christman, 2016; Herold et al., 2011), the role of NFAT activity in inducing gene transcription by LPS in macrophages is only suspected. NFAT is basally phosphorylated. NFAT dephosphorylation by calcineurin, a Ca2+-dependent phosphatase, is required to increase NFAT activity (Rao, 2009). So, we determined the phosphorylation state of NFAT and NF-ҡB to assess the possibility that PAR2 suppresses inflammatory signaling by inhibiting NFAT activity as well. Intriguingly, we found that, in PAR2-null BMDMs, LPS induced the dephosphorylation of NFAT (i.e., activation of NFAT) at 4 h, which was sustained for the next 8 h. However, LPS modestly and transiently altered NFAT phosphorylation in WT-BMDMs. We next measured the phosphorylation of p65-NFҡB, a readout of NF-ҡB activity (Ballinger and Christman, 2016; Herold et al., 2011), and, as expected, LPS increased the phosphorylation of p65-NF-ҡB in WT-BMDMs at 2 h, which increased further at 4 h. At 8 h, the phosphorylation declined to the 2 h level following LPS stimulation. However, LPS-induced p65-NF-ҡB phosphorylation was markedly elevated at 2 h and persisted up to 8 h in PAR2-null BMDMs as compared to WT BMDMs (Figure 4C). Consistent with this observation, LPS-induced TNF-a, IL-6, and IL1-b expression was ~2-to 3-fold higher in PAR2-null BMDMs as compared to WT-BMDMs (Figure 4D).
To determine whether PAR2 via NFAT modulates inflammatory cytokine gene expression, we depleted NFATc1 in BMDMs using siRNA followed by their stimulation with LPS and measured TNF-a, IL-6, and IL-1b mRNA expression. Knockdown of NFATc1 reduced the expression of these cytokines by ~30% in WT-BMDMs (Figure 4E). Interestingly, NFATc1 depletion in PAR2-null BMDMs reduced LPS-induced elevation of TNF-a, IL-6, and IL-1b expression to similar levels seen in WT-BMDMs (Figure 4E). Given that NF-ҡB activity was higher in PAR2-null BMDMs than WT-BMDMs, while depletion of NFAT blunted the LPS-induced elevation in cytokine expression in PAR2-null BMDMs, we examined whether NFAT modulated NF-ҡB binding to the TNF-a, IL-6, and IL-1b promoters, thereby enhancing their expression in response to LPS. Hence, we analyzed LPS-induced NF-ҡB binding to the indicated promoters in WT-BMDM and Par2−/− BMDM without or with depletion of NFATc1 using chromatin immunoprecipitation (ChIP) assays. Interestingly, we found that LPS increased NF-ҡB binding to the TNF-a, IL-6, and IL-1b promoters in Par2−/− BMDMs by 3-fold compared to WT-BMDMs. Depletion of NFATc1 reduced the increase in NF-ҡB binding to the promoters of these cytokines in Par2−/− BMDMs. (Figure 4F). These findings indicate that loss of PAR2 results in activation of NFAT, which in turn promotes NF-ҡB binding to cytokine promoters leading to increased inflammatory cytokine generation and thereby impairs resolution of inflammation.
PAR2 Attenuates NFAT Activity by Generating cAMP
PAR2 is also known to induce cAMP levels by coupling to Gas (Driesbaugh et al., 2015; Scott et al., 2003). cAMP opposes the rise in intracellular Ca2+ (Chabardè s et al., 1999; Chow and Davis, 2000). Thus, we assessed cAMP concentration in WT-or PAR2-null-BAL post-LPS challenge. We found that LPS increased cAMP in WT-BAL but not in Par2−/−-BAL (Figure 5A). Next, we determined whether loss of PAR2 impaired cAMP generation by thrombin. We found that thrombin significantly increased cyclic AMP in WT-BMDMs, but this response was not observed in PAR2-null BMDMs (Figure 5B). We stimulated adenylate cyclase directly using forskolin to determine whether it rescues cAMP generation post-thrombin stimulation in PAR2-null BMDMs. Additionally, we used rolipram, an inhibitor of phosphodiesterase 4, which cleaves cAMP back to AMP (Xiao et al., 2011). We observed that forskolin induced a similar increase in cAMP in both WT and PAR2-null BMDMs. Interestingly, combining thrombin with forskolin failed to rescue cAMP levels in PAR2-null BMDMs (Figure 5B). Further, rolipram alone or in combination with thrombin did not induce any significant increase in intracellular cAMP in WT or PAR2-null BMDMs (Figure 5B). These findings indicate that thrombin ligation of PAR2 is necessary for cAMP generation. To further corroborate this notion, we compared cAMP generation and Ca2+ entry in PAR2-null BMDMs following transfection with WT-PAR2 cDNA versus MT-PAR2 cDNA. We observed that transduction of WT-PAR2 into PAR2-null BMDMs restored thrombin-induced cAMP generation and Ca2+ entry to levels similar to WT-BMDMs, whereas transduction of MT-PAR2 cDNA into PAR2-null BMDMs did not (Figures 5C–5E).
Figure 5. PAR2 Suppresses Ca2+Entry and NFAT-Dependent Cytokine Generation by Generating cAMP.

(A) BAL-AM from control or LPS challenged mice were obtained after 30 min, and intracellular cAMP was measured using ELISA kit. Data are represented as mean ± SD of three independently performed experiments. ***p < 0.001 indicates a significant increase in cAMP concentration in comparison to control cells. ##p < 0.01 indicates a significant increase in cAMP as compared to the Par2−/− plus LPS group.
(B) WT and Par2−/− BMDMs were stimulated with indicated agonists and cAMP levels were determined using an ELISA kit. Data represent mean ± SD of three experiments. ***p < 0.001 indicates values significantly different from corresponding Par2−/− BMDMs.
(C) BMDMs were transfected with WT-PAR2 or MT-PAR2 cDNA. After 48 h of transfection, the cells were stimulated with thrombin for 30 min (50 U/mL) and intracellular cAMP was estimated using the ELISA kit. The data are represented as mean ± SD of three experiments. **p < 0.01 and ***p < 0.001 indicate a significant increase in cAMP levels in comparison to unstimulated or MT-PAR2 transfected cells post thrombin stimulation.
(D and E) BMDMs were transfected with WT-PAR2 or MT-PAR2 cDNA or control vector. The cells were stimulated with thrombin in Ca2+-free media followed by repletion of extracellular Ca2+ (2 mM) at ~300 s. A representative trace is shown in (D) from three independent experiments, while (E) shows individual data points along with mean ± SD of 12–15 cells. ***p < 0.001 indicates values that were significantly different from WT BMDMs. ###p < 0.001 indicates values that were significantly different from Par2−/−-null BMDMs transfected with WT-PAR2 after thrombin stimulation.
(F and G) BMDMs were stimulated with thrombin in Ca2+-free media followed by stimulation with 250 mM 8-Br-cAMP at ~180 s and subsequent repletion of extracellular Ca2+ (2 mM) at ~360 s. A representative trace is shown in (F), while (G) shows individual data points along with mean ± SD of 12–15 cells. Results show data from experiments that were repeated three times. ***p < 0.001 indicates values that were significantly different WT BMDMs treated with thrombin only. ###p < 0.001 indicates values that were significantly different from BMDMs were treated with 8-Br-cAMP.
(H) BMDMs were pre-treated with 250 mM 8-Br-cAMP for 15 min followed by LPS stimulation for 4 h. The phosphorylation of p65-NF-ҡB and NFAT was estimated by western blotting. A representative immunoblot is shown from independent experiments that were repeated 3 times.
(I) In a separate experiment under the same experimental conditions (i.e., 8-Br-cAMP pre-treatment and LPS stimulation of BMDMs), expression of the indicated pro-inflammatory genes was measured by qPCR. Data are represented as mean ± SD of three independent experiments. *p < 0.05 and **p < 0.05 indicate values that were significantly different from untreated BMDMs. #p < 0.05 and ###p < 0.001 indicate values that were significantly different from WT-BMDMs post-LPS challenge.
In all figures, one-way ANOVA followed by paired two-tailed t test was used to assess significance between groups.
To establish that impaired cAMP generation was responsible for Ca2+ entry and NFAT-dependent inflammatory signaling in PAR2-null BMDMs, we used the cell-permeable analog, 8-Br-cAMP (Wang and Adjaye, 2011). We found that treatment of PAR2-null BMDMs with 8-Br-cAMP inhibited thrombin-induced Ca2+ entry (Figures 5F and 5G) and diminished LPS-induced NFAT and NF-ҡB activities as well as pro-inflammatory cytokine generation (Figures 5H and 5I).
PAR2 Blocks Ca2+ Entry and Downstream Signaling by Inhibiting TRPV4 Activity
Stromal interacting molecule 1 (STIM1) plays a critical role in inducing Ca2+ entry upon depletion of the ER Ca2+ store (Yazbeck et al., 2017). Thus, we knocked down STIM1 in WT and PAR2-null BMDMs and assessed whether depletion of STIM1 blocked cytokine expression. We found that depletion of STIM1 had no effect on LPS-induced cytokine expression (Figure S4A). Since PAR2 has been shown to interact with the TRPV4 channel (Grant et al., 2007; Zhao et al., 2015), we depleted TRPV4 using siRNA or inhibited TRPV4 using a TRPV4-specific antagonist (GSK 2193874) (Balakrishna et al., 2014; Thorneloe et al., 2012) to address the possibility that the PAR2-cAMP cascade functioned by blocking TRPV4 activity. We found that blocking TRPV4 with GSK 2193874 (GSK-I) rescued the induction of cytokine expression by LPS to the level observed in WT-BMDMs (Figure 6A). In other studies, we showed that GSK-I treatment or TRPV4 depletion in PAR2-null BMDMs blocked Ca2+ entry following thrombin stimulation (Figures 6B–6E). We also found that TRPV4 depletion markedly reduced the phosphorylation of NF-ҡB and NFAT (Figure 6F). Also, direct activation of TRPV4 with its agonist (GSK1016790A) (Vincent and Duncton, 2011) enhanced Ca2+ entry in PAR2-null BMDMs more than in WT-BMDMs (Figures 6G and 6H). Addition of cAMP inhibited TRPV4 induction by GSK1016790A (GSK101). These results together with above findings identify the PAR2-cAMP cascade as a suppressor of TRPV4 activity and NFAT-mediated cytokine generation (Figures 6G and 6H).
Figure 6. TRPV4 Induces Ca2+ Entry and Promotes NFAT-Dependent Inflammatory Cytokine Generation.

(A) WT and Par2−/− BMDMs were pre-treated with 100 nM GSK-I for 5 min followed by addition of 1 mg/mL of LPS. The expression of indicated pro-inflammatory genes was determined after 4 h by qPCR. Data are represented as mean ± SD of three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 indicate values that were significantly different from unstimulated BMDMs. ##p < 0.01 indicates values that were significantly different from WT-BMDMs post-LPS challenge.
(B–E) BMDMs were left un-transfected (B and C) or transfected with control or TRPV4 siRNA (D and E). These cells were stimulated with thrombin in Ca2+-free media followed by repletion of extracellular Ca2+ (2 mM). GSK-I (100 nM) was then added at the peak phase of Ca2+ entry (arrow) in un-transfected cells to assess TRPV4 activity (B and C). A representative trace is shown in (B) and (D), while (C) and (E) show individual data points along with mean ± SD of 8–15 cells. Results show data from experiments that were repeated three times. ***p < 0.001 indicates values that were significantly different from WT BMDMs. ###p < 0.001 indicates values that were significantly different from TRPV4-depleted BMDMs or BMDMs treated with GSK-I.
Blocking TRPV4 Activity in Par2−/− Mice Resolves Lung Edema
To address whether TRPV4 serves as a critical signal impairing resolution of lung injury post-LPS challenge in PAR2-null mice, we injected GSK-I (0.5 mg/kg body weight) i.t. 1 h after LPS inhalation. Interestingly, blockage of TRPV4 in PAR2-null mice promoted resolution of edema formation and reduced neutrophil accumulation more than in PAR2-null mice receiving vehicle alone (Figures 7A, 7B, and S4B). Thus, PAR2 plays a key role in suppressing TRPV4 signaling and thereby suppresses inflammatory lung injury.
Figure 7. Inhibition of TRPV4 Resolves ALI.

(A) WT and Par2−/− mice were challenged with LPS for 45 min followed by i.t. administration of TRPV4 antagonist, GSK-I (0.5 mg/kg, body weight), or vehicle post-30 min of lung injury. After 4 and 24 h, lung edema and neutrophil accumulation in the lungs were determined. Plot shows individual values of lung edema with mean ± SD. ***p < 0.001 indicates values that are significantly different from their respective control groups. n = 5 mice/group.
(B) Neutrophil count was performed as described in Figure 1C. The plot shows individual values of neutrophils from three independent experiments with mean ± SD. ***p < 0.001 indicates a significant increase in neutrophil count as compared to the respective vehicle control group.
In all figures, one-way ANOVA followed by paired two-tailed t test was used to assess significance between groups.
(F) BMDMs were transfected with control or TRPV4 siRNA. Cells were then stimulated with 1 mg/mL LPS for 4 h. The phosphorylation of NFAT and NF-ҡB was estimated by western blotting. A representative immunoblot is shown from experiments that were independently repeated 3 times.
(G and H) BMDMss were incubated in 2 mM Ca2+ containing media followed by stimulation with 50 nM of a TRPV4 agonist (GSK-101). Subsequently, 8-Br-cAMP was added at the peak phase of Ca2+ entry. A representative trace is shown in (G), and (H) shows individual data points along with mean ± SD of 12–15 cells. Results show data from experiments that were repeated three times. ***p < 0.001 indicates values that were significantly different from WT BMDMs. ###p < 0.001 indicates values that were significantly different from BMDMs treated with 8-Br-cAMP.
DISCUSSION
Our results identified PAR2 in AMs as a checkpoint for suppressing TLR4-mediated inflammatory signaling induced by LPS to resolve pulmonary edema and thereby reinstate tissue integrity. We specifically demonstrated that, following lung injury, thrombin activates PAR2, which controls cAMP rise. cAMP antagonizes the TRPV4-dependent increase in cytosolic Ca2+ limiting the ability of NFAT to promote NF-ҡB-mediated generation of pro-inflammatory cytokines, leading to resolution of ALI.
Infectious agents such as endotoxin LPS (cell wall component of Gram-negative bacteria), in addition to inducing inflammatory signaling, can activate the coagulation pathway, thus stimulating generation of proteases such as thrombin, which can activate PAR½ directly or indirectly (Coughlin, 2005; Kaneider et al., 2007; Mihara et al., 2016). Since PAR2 is expressed in several lung cell types (O’Brien et al., 2000; Ramachandran et al., 2009), an important unanswered question is whether PAR2 modulates lung injury by regulating inflammatory signaling in parenchymal cells, such as endothelial cells, or whether hematopoietic cells, such as macrophages, are involved. Our results identified an anti-inflammatory role of PAR2 in AMs, which proved to be critical for suppressing LPS-induced lung injury. In contrast to the present findings, many studies conducted thus far in nonhematopoietic cells have described a pro-inflammatory role of PAR2 (Ostrowska et al., 2007; van den Boogaard et al., 2018). Intriguingly, bone marrow cell transplantation studies demonstrated that PAR2 in hematopoietic cells plays a key role in resolving lung injury, because transplantation of PAR2-null bone marrow into lethally irradiated WT mice impaired resolution of LPS-induced lung edema formation in the chimeric WT mice.
The function of AM-TLR4 is to initiate inflammatory signaling upon sensing pathogens for the purpose of host defense. This signaling has to be terminated in a timely fashion to prevent tissue damage and thus limit ALI (Herold et al., 2011; Rubins, 2003). We showed that adoptive transfer of WT-BMDMs in AM-depleted PAR2-null mice resolved lung injury similar to that observed in WT mice demonstrating that PAR2 in AMs is required for resolving lung injury. Importantly, we showed that AM-depleted WT lungs receiving PAR2-null BMDMs failed to resolve LPS-induced lung injury, supporting the above finding of long-lasting inflammatory signaling by PAR2-null AMs, which thereby impaired the tissue repair process. These findings also indicate that PAR1 in AMs induced observed inflammatory lung injury following LPS in PAR2-null mice. Overall, our data unequivocally demonstrate that PAR2 expression in AMs (as opposed to parenchymal cells) is responsible for the resolution of lung injury induced by LPS.
PAR2 can be ligated by several proteases produced during inflammatory injury including elastases and trypsin (Kaneider et al., 2007; Mihara et al., 2016). Thrombin can activate its high-affinity receptor PAR-1 on AMs (Mercer et al., 2014; Roche et al., 2003). PAR1 can also transactivate PAR2 (Kaneider et al., 2007; O’Brien et al., 2000). Thus, PAR2 may indirectly contribute to lung injury resolution following TLR4 activity. Strikingly, we showed that rescuing thrombin-cleavage sensitive PAR2 (WT-PAR2) expression in AMs resolved lung edema in Par2−/−mice, while restoration of a mutated PAR2 receptor lacking the thrombin cleavage site failed to do so. Our findings rule out PAR1 transactivation of PAR2 as a mechanism of PAR2 activation and instead demonstrate that activation of PAR2 by thrombin is both necessary and sufficient for resolution of lung injury. This conclusion is supported by recent findings showing that PAR2 can ligate thrombin directly (Mihara et al., 2016).
PAR2 regulates the increase in cAMP and intracellular Ca2+ via heterotrimeric G-proteins, which in turn trigger various signaling cascades (Jairaman et al., 2015; Ramachandran et al., 2009; Scott et al., 2003; Zhao et al., 2015). Thus, to address the mechanisms of AM-expressed PAR2 in promoting resolution of lung inflammation, we focused on whether loss of PAR2 leads to long-lasting inflammatory signaling in AMs secondary to generation of these second messengers. We showed that in PAR2-null AMs, thrombin failed to generate cAMP, while it augmented Ca2+ entry. We also showed that LPS failed to induce lung injury in PAR1-null mice. PAR1 and PAR2 are known to play opposing roles in thrombin-induced signaling in several cell types (Kahn et al., 1999; Sidhu et al., 2014; Soh et al., 2010). We therefore speculate from these findings that thrombin-mediated activation of PAR1 in AM-induced Ca2+ entry through the plasmalemmal channel. Our findings that depletion of TRPV4 as well as use of a TRPV4 antagonist inhibited both Ca2+ entry into PAR2-null BMDMs and pulmonary edema support such a contention. Thus, we have identified thrombin-activated PAR2 in AMs as a suppressor of TRPV4 activity and resultant lung injury. Consistently, TRPV4 has been shown to induce an increase in lung vascular permeability and inflammation (Balakrishna et al., 2014; Yin et al., 2016).
Gas is known to induce cAMP by activating adenylate cyclase (Godinho et al., 2015). Gai can contribute in inducing cytosolic Ca2+ and in suppressing cAMP levels (Chow and Davis, 2000; Lee et al., 2007; Scott et al., 2003). We showed that PAR2 deletion did not alter cAMP generation by forskolin, a direct activator of adenylate cyclase (Faruqi et al., 2000). However, thrombin alone or in combination with forskolin failed to rescue cAMP generation in PAR2-null BMDMs to the levels seen in WT-BMDMs. Evidence indicates that Gas mutants deficient in the capacity to bind adenylate cyclase compromised cAMP generation in the presence of forskolin (Vortherms et al., 2004; Zheng et al., 2010). We therefore speculate that inefficient coupling between Gas and adenylate cyclase in PAR2-null AMs impaired cAMP generation by forskolin. Indeed, we showed that rescuing thrombin-sensitive PAR2 in PAR2-null BMDMs restored cAMP generation, thus supporting the concept that PAR2 ligation by thrombin induced cAMP.
How does loss of PAR2 increase AM TLR4 inflammatory signaling? AM TLR4 is known to induce the immune response by increasing NF-ҡB activity (Ballinger and Christman, 2016; Herold et al., 2011). A rise in intracellular Ca2+ can increase the transcriptional activities of both NF-ҡB and NFAT and thereby inflammatory signaling albeit in a cell-context-dependent manner (Bair et al., 2009; Chow and Davis, 2000; Rao, 2009). Findings from the current study showed that loss of PAR2 augmented TRPV4 activity in macrophages leading to activation of NF-ҡB and NFAT. Importantly, we showed that knockdown of NFATc1 suppressed cytokine generation in PAR2-null AMs. Furthermore, depletion of NFATc1 reduced NF-ҡB binding to the cytokine promoters in PAR2-null macrophages. These findings indicate that activated NFAT enhances cytokine generation by promoting the binding of NF-ҡB to the TNF-a, IL-6, and IL1-b promoters. However, it remains to be seen whether NFAT influences NF-ҡB binding to cytokine promoters directly or by modulating NF-ҡB phosphorylation and nuclear transport. We also showed that a cell-permeable cAMP analog blocked TRPV4-induced Ca2+ entry and thereby NFAT and NF-ҡB activation as well as cytokine generation, indicating that cAMP was responsible for blocking TRPV4 activity and its consequences. Additionally, a blockade of TRPV4 inhibited cytokine expression and lung vascular permeability increase in PAR2-null mice. Thus, impaired cAMP generation resulting in sustained TRPV4 activity in AMs could explain the observed persistent increase in inflammatory signaling seen after PAR2 deletion.
The basis of cAMP inhibition of TRPV4 activity downstream of PAR2 is not clear. TRPV4 activity is regulated in part by phosphorylation of the channel (Cao et al., 2018) Additionally, altered channel conformation is thought to regulate activation of TRPV4 channels because TRPV4 agonist-mediated Ca2+ entry was preserved in cells expressing a phosphodefective TRPV4 mutant (Cao et al., 2018). Alignment of TRPV4 protein sequence with cAMP-binding proteins such as EPAC and NLRP3 (Lee et al., 2012; Ster et al., 2007) identified the shared ‘‘LM/FNRP’’ sequence within TRPV4. Thus, a likely scenario may be that cAMP downregulates TRPV4 activity, directly by affecting channel conformation (Arniges et al., 2006). This notion is supported by studies in which cAMP has been shown to alter the conformation of HCN-cation channels (Lee and MacKinnon, 2017).
In summary, the present study has identified PAR2-cAMP signaling in AMs as a key mechanism in tightly controlling TLR4 signaling, thereby dampening inflammatory injury and hence leading to resolution of lung injury. We showed that TLR4, while activating the canonical NF-ҡB-driven immune response in AMs, also enabled thrombin activation of PAR2. PAR2 induces an increase in cAMP, which in turn prevents TRPV4 activity (perhaps downstream of PAR1 in AMs) and thereby NFAT-driven sustained inflammatory injury, thus resolving lung edema. Because of this pivotal role of PAR2, we suggest that activation of PAR2 in AMs is potentially a useful target in preventing ALI.
STAR★METHODS
CONTACTS FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dolly Mehta (dmehta@uic.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Illinois. Par1−/−, Par2−/− and C57BL/6J mice breeding pairs were initially obtained from Jackson Laboratory (Farmington, CT, USA) and bred at the University of Illinois at Chicago. Their colonies were maintained in a pathogen-free housing facility at the University. C57B6 mice were used as wild-type (WT). Male and female mice between 6–8 weeks old were used in the study.
Bone-Marrow-Derived Macrophages
Bone-marrow-derived macrophages were isolated from both male and female mice by flushing the femur and tibia with RPMI containing 1% antibiotic/antimycotic, 10% FBS and 25 ng/ml M-CSF. The cells were incubated for 5 days (first 3 days with M-CSF) at 37 ○C in a 5% CO2 incubator and media changed every 3 days. Cells were serum starved in 0.1% FBS containing media for 30 min for every experiment followed by addition of LPS (1 mg/ml) or thrombin (50 U/ml) in M-CSF free media containing 0.1% FBS.
Cell Line
U-937, a male human monocytic cell line was used in the study. Before use, the cells were differentiated to macrophages by exposing them to PMA (100 ng/ml) for 48 h.
METHOD DETAILS
Induction of Lung Injury
Lung injury was induced using nebulized LPS (45 min at 1 mg/ml LPS). The mice were euthanized, and edema was assessed by measuring lung wet-dry weight ratio 4, 24 and 48 h after LPS challenge. Loss of alveolar-capillary barrier function was determined by measuring extravasation of Evans blue–conjugated albumin (EBA) (20 mg/kg) as described previously (Tauseef et al., 2012). Briefly, EBA was injected retro-orbitally 30 min before sacrificing the mice. Lungs were homogenized. Lung homogenate and the plasma were incubated with formamide for 18 h at 55○C and then centrifuged to isolate supernatant. The optical density was measured spectrophotometrically at 620 nm (Evans blue) and 740 nm (hemoglobin correction). Evans blue-albumin extravasation was calculated by transendothelial albumin influx in lungs versus plasma. Further, left lobe of lungs from mice were excised after LPS induced injury, weighed, and dried at 55○C for 24 h and lung wet–dry ratio calculated as a measure of lung edema as described previously (Tauseef et al., 2012).
Bronchoalveolar Lavage
After 4 and 24 h of LPS inhalation, each mouse was sacrificed, bronchoalveolar lavage (BAL) was performed using an 18-gauge blunt needle. Briefly, the tracheotomy was performed, 1 mL of cold PBS was slowly injected into the lungs, aspirated back and repeated 3 more times. BAL fluid was collected for further processing or layered onto culture dishes for alveolar macrophage isolation. Briefly, the BAL fluid was poured on dishes, supplied with 10% serum and alveolar macrophages allowed to adhere for 60 mins. The non-adherent cells were removed by washing with PBS (Bang et al., 2011). Total RNA was isolated from macrophages using a QIAGEN RNA isolation kit (QIAGEN).
Bone Marrow Transplantation
Par2−/− and WT mice were exposed to lethal irradiation with 1,000 rads (10 Gy) (Weber et al., 2015). 2–3 h after irradiation, the mice were anaesthetized by intraperitoneal injections of ketamine (100 mg/kg) and xylazine (15 mg/kg) followed by retro-orbitally injecting 0.2 mL 3 × 106 donor bone marrow cells (PAR2 null or WT) using a 27-gauge needle. Experiments were performed 6 weeks after the day of transplantation (Tauseef et al., 2012)
Adoptive Transfer
AM depletion was achieved by i.t. instillation of commercially available clodronate liposomes (Encapsula Nanosciences). The depletion was followed by reconstitution, at 48 h, using 1×106 BMDM for each mouse. The cells were injected by non-invasive intratracheal instillation method (Di et al., 2018). Briefly, the mouse was suspended in a semi-recumbent position on a solid support. The tongue was grasped and positioned in an upward-leftward direction to pour the cell suspension in the back of oral cavity, where from it is aspirated by the anaesthetized mouse. The LPS inhalation of mice was done at 72 h followed by their sacrifice at 96 h. The lungs were harvested for measurement of edema. Bronchoalveolar lavage was collected, and the macrophages were isolated for qPCR and imaging.
Measurement of Gene Expression
The RNA was extracted from BMDM using TRIzol (Invitrogen) or QIAGEN RNA isolation kit for alveolar macrophages, as per manufacturer’s instructions. The quantity and integrity of RNA were measured using a Nanodrop spectrophotometer (Thermo Fischer). Equal quantities of RNA were used for cDNA synthesis and gene expression was determined by Syber green-based real-time q-PCR using ViiA7 (Applied Biosystem, Foster City, CA) (Tauseef et al., 2012). The primer sequences used in the study areNFATc1 forward: 5′-AATAACATGCGAGCCATCATC-3′, reverse: 5′-TCACCCTGGTGTTCTTCCTC-3′; TRPV4 forward: 5′-TCACCTTCGTGCTCCTGTTG-3′, reverse: 5′-AGATGTGCTTGCTCTCCTTG-3′; PAR2 forward: 5′-TGGCCATTGGAGTCTTCCT GTT-3′, reverse: 5′-TAGCCCTCTGCCTTTTCTTCTC-3′; PAR1 forward: 5′՛-GTTGATCGTTTCCACGGTCT-3′, reverse: 5′-ACGCA GAGGAGGTAAGCAAA-3′; IL6 forward: 5′-AGTCCGGAGAGGAGACTTCA-3′, reverse: 5′-TTGCCATTGCACAACTCTTT-3′; TNF-a forward: 5′-CCCCAAAGGGATGAGAAGTT-3′, reverse: 5′-ACTTGGTGGTTTGCTACGA-3′; IL1b forward: 5′-GGGCTGCTTCCAAA CCTTTG-3′, reverse: 5′-TGATACTGCCTGCCTGAAGCTC-3′; GAPDH forward: 5′-GTGAAGGTCGGTGTGAACGG-3′; reverse: 5′-TCATGAGCCCTTCCACAATG-3′; Human IL1b forward: 5′-AAATACCTGTGGCCTTGGGC-3′; reverse: 5′-TTGGGATCTACACTCT CCAGCT-3′; Il-6 forward: 5′-GTAGCCGCCCCACACAGA-3′; reverse: 5′-CATGTCTCCTTTCTCAGGGCTG-3′; PAR2 forward: 5′-CTGCATCTGTCCTCACTGGA-3′; reverse: 5′-ACAGAGAGGAGGTCAGCCAA-3′; GAPDH forward: 5′-GTCTCCTCTGACTTCAA CAGCG-3′; reverse: 5′-ACCACCCTGTTGCTGTAGCCAA.
Cytosolic Ca2+ Measurement
BMDM were cultured on 35 mm glass bottom dishes for Ca2+ estimation. An increase in intracellular Ca2+ in macrophages was measured using Fura 2-AM dye. Cells were incubated with Fura 2-AM dye for 15 mins, washed with HBSS (without Ca2+) and stimulated with thrombin (50 nM) to deplete intracellular Ca2+ levels. Subsequently, 2 mM Ca2+ was added followed by GSK-I or cAMP treatment to determine Ca2+ entry (Yazbeck et al., 2017).
Immunoblotting
At the time of termination of experiment(s), BMDM were washed with cold PBS and lysed in RIPA buffer/Laemmli buffer. The lysates were heated at 100○C for 7 min. Protein samples were loaded in SDS gels and standard western blotting procedure was followed to detect desired proteins. b-actin was used as an experimental control.
In Vivo Gene Delivery
Liposomes were prepared using a mixture of chloroform, Dimethyl Dioctadecyl Ammonium Bromide and cholesterol as described previously (Tauseef et al., 2012; Yazbeck et al., 2017). The chloroform was evaporated from the mixture using a rotavapor system at a speed of 105 rpm for 15–20 min at 37○C to make a lipid layer. Lipid layer was extracted by sonicating the solution for 1 h at 42○C in presence of 5% glucose. The liposomes were filtered through a 0.45-micron filter and subsequently cDNA was added. The cDNA loaded liposomes were administered intratracheally into the mouse lungs followed by LPS inhalation after 48h. The lungs were harvested for edema measurement after 24 h of LPS challenge, and confirmation of cDNA expression in alveolar macrophages was done by qPCR.
Lung Histology and Confocal Imaging
Lungs were fixed in formalin and sections (4 mm) were either co-immunostained with PE Siglec-F and anti-PAR2 antibodies along with appropriate secondary antibodies or stained with hematoxylin and eosin to assess inflammatory changes as described earlier (Tauseef et al., 2012; Yazbeck et al., 2017). Lung sections were observed under an LSM880 confocal microscope (Carl Zeiss). After transfection, BMDM were seeded either on 35 mm glass bottom dishes or coverslips and after 48 h, cells were washed with PBS and starved for 30 min in 0.1% FBS containing media and subsequently stimulated with thrombin for the indicated time. The cells were washed, fixed with 2% para-formaldehyde and imaging was done using an LSM880 confocal microscope under 63x oil objective.
Transfection
BMDM or U937 cells were transfected with indicated cDNA or siRNA using Amaxa Nucleofactor electroporation system (Lonza) as described (Mihara et al., 2016; Yazbeck et al., 2017). Briefly, the cells were detached from the plates using accutase, spun down with complete media, and cDNA/siRNA applied to cells in a transfection reagent and electroporated. The experimental controls were transfected with control siRNA or vector plasmids.
ELISA Analysis
The levels of TNF-a, IL-6 and IL1-b were estimated from bronchoalveolar lavage fluid of mice by using commercially available ELISA kits. The assays were performed as per the manufacturer’s instructions (Signosis). The absorbance was measured at 450nm. The concentration of cytokines in samples was calculated from absorbance using standard curve.
Chromatin Immunoprecipitation (ChIP) Assay
WT or Par2−/− BMDM transfected with scrambled or NFATc1 siRNA were stimulated with LPS. Formaldehyde crosslinked protein-DNA complexes (100–125 mg) were immunoprecipitated with the anti-NFҡB antibody or IgG. DNA fragments were purified using phenol-chloroform-isoamyl alcohol extraction, followed by ethanol precipitation, and then resuspended in 18–20 mL nuclease water for using as a starting material in PCR amplification. Promoter regions of TNF-a, IL-6 and IL1-b that correspond to a 149 bp, 179 bp and 182 bp fragment respectively, containing NFҡB binding sites were quantified by real-time q-PCR. Normal rabbit IgG was used as negative antibody control and DNA from the input (30–40 mg protein-DNA complexes) was used as an internal control (Chava et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
All values are given as mean ± SD. Statistical analysis was performed using Graph Pad Prism version 7.0 (Graph Pad Software, La Jolla, CA). Multiple groups were compared using One-way ANOVA followed by a paired Student’s t test to assess significance between two groups. The statistics details can be found in the individual figure legends.
Supplementary Material
KEY RESOURCES TABLE
| Reagent or Resource | Source | Identifier |
|---|---|---|
|
Antibodies | ||
| Phospho-NFATc1 | Invitrogen | Cat# PA5–38301; RRID: AB_2554902 |
| Phospho-P65-NFҡB | Santa Cruz Biotechnology | Cat# sc-33039; RRID: AB_2238379 |
| b-actin | Santa Cruz Biotechnology | Cat# sc-47778; RRID: AB_2714189 |
| TRPV4 | Abcam | Cat# 39260; RRID: AB_1143677 |
|
Chemicals, peptides and recombinant proteins | ||
| Lipopolysaccharide | Sigma Aldrich | Cat# 2880 |
| Cat# 2630 | ||
| 8-Bromoadenosine 3′,5′-cyclic monophosphate | Sigma Aldrich | Cat# B5386 |
| Trpv4 antagonist GSK2193874 | Sigma Aldrich | Cat# SML0942 |
| Trpv4 agonist GSK1016790A | Sigma Aldrich | Cat# G0798 |
| Vectashield Antifade Mounting Medium with DAPI | Vector Laboratories Inc | Cat# H-1200 |
| Rolipram | EMD Millipore | Cat# 557330 |
| Fura 2-AM | Invitrogen | Cat# F1221 |
| PE-Siglec-F | eBioscience | Cat# 12–1702-82; RRID: AB_2637129 |
| PE-CD11c | eBioscience | Cat# 12–0114-82; RRID: AB_465552 |
| EF450 Ly-6G | eBioscience | Cat# 48–5931-82; RRID: AB_1548788 |
| APC-CD11b | eBioscience | Cat# 17–0112-82; RRID: AB_469343 |
| PE-Cy7 CD45 | eBioscience | Cat# 25–0451-82; RRID: AB_2734986 |
| cAMP ELISA kit | Cayman Chemical | Cat# 501040 |
| M-CSF | Sigma Aldrich | Cat# M6518 |
|
Experimental models: Cell lines | ||
| U-937 | N/A | Gifted by Dr. Kostandin Pajcini, University of Illinois at Chicago, USA |
|
Experimental models: Organisms/Strains | ||
| C57BL/6J wildtype | (Tauseef et al., 2012) | N/A |
| Par1−/− | Jackson laboratories | #002862 |
| Par2−/− | Jackson laboratories | #004993 |
|
Oligonucleotides | ||
| siRNA Nfatc1 | Thermo Fisher Scientific | Cat# MSS275981 |
| siRNA TRPV4 | Dharmacon | Cat# M-040742–01-0005 |
| siRNA Par2 | QIAGEN | Cat# SI02757419 |
| siRNA Scramble (Non targeting siRNA) | Dharmacon Inc | Cat# D-001810–01-05 |
|
Recombinant DNA | ||
| eYFP-PAR2-mRFP (WT PAR2) | (Mihara et al., 2016) | N/A |
| eYFP-PAR2-mRFP (MT PAR2) | (Mihara et al., 2016) | N/A |
| pCDNA3.1 (Vector) | (Mihara et al., 2016) | N/A |
|
Software and Algorithms | ||
| Zen Lite | Zeiss Inc | https://www.zeiss.com/microscopy/us/software-cameras.html |
| Graph Pad Prism Version 7.0 | Graph pad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| Summit 4.0 | Beckman coulter | https://www.beckman.com/assets/training/flowcytometry/summitsoftware/index.html |
Highlights.
Post-injury, thrombin activation of AM PAR2 suppresses TLR4-mediated inflammation
PAR2-generated cAMP abolishes Ca2+ entry through TRPV4
Ca2+ entry causes NFAT-dependent long-lasting inflammatory signaling in Par2−/− AMs
Rescuing thrombin-sensitive PAR2 or TRPV4 blockade in Par2−/− AMs resolves injury
ACKNOWLEDGMENTS
This work was supported by NIH United States grants P01-HL077806, PO1-HL060678, and HL137179-01. We acknowledge technical help from Drs. Md-Ruhul Amin and Koichiro Mihara.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.053.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arniges M, Fernández-Fernández JM, Albrecht N, Schaefer M, and Valverde MA (2006). Human TRPV4 channel splice variants revealed a key role of ankyrin domains in multimerization and trafficking. J. Biol. Chem 281, 1580–1586. [DOI] [PubMed] [Google Scholar]
- Bair AM, Thippegowda PB, Freichel M, Cheng N, Ye RD, Vogel SM, Yu Y, Flockerzi V, Malik AB, and Tiruppathi C (2009). Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-kappaB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cdelta. J. Biol. Chem 284, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, et al. (2014). TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol 307, L158–L172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger MN, and Christman JW (2016). Pulmonary Macrophages: Overlooked and Underappreciated. Am. J. Respir. Cell Mol. Biol 54, 1–2. [DOI] [PubMed] [Google Scholar]
- Bang BR, Chun E, Shim EJ, Lee HS, Lee SY, Cho SH, Min KU, Kim YY, and Park HW (2011). Alveolar macrophages modulate allergic inflammation in a murine model of asthma. Exp. Mol. Med 43, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Anishkin A, Zinkevich NS, Nishijima Y, Korishettar A, Wang Z, Fang J, Wilcox DA, and Zhang DX (2018). Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation. J. Biol. Chem 293, 5307–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabardès D, Imbert-Teboul M, and Elalouf JM(1999). Functional properties of Ca2+-inhibitable type 5 and type 6 adenylyl cyclases and role of Ca2+ increase in the inhibition of intracellular cAMP content. Cell. Signal 11, 651–663. [DOI] [PubMed] [Google Scholar]
- Chava KR, Tauseef M, Sharma T, and Mehta D (2012). Cyclic AMP response element-binding protein prevents endothelial permeability increase through transcriptional controlling p190RhoGAP expression. Blood 119, 308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CW, and Davis RJ (2000). Integration of calcium and cyclic AMP signaling pathways by 14–3-3. Mol. Cell. Biol 20, 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg A, Wang X, Wang RD, Meixner SC, Pryzdial EL, and Wright JL (2007). Alpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophages. Am. J. Respir. Cell Mol. Biol 37, 144–151. [DOI] [PubMed] [Google Scholar]
- Coughlin SR (2005). Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost 3, 1800–1814. [DOI] [PubMed] [Google Scholar]
- Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, Mittal M, Hong Z, Kanneganti TD, Rehman J, and Malik AB (2018). The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasomeinduced inflammation.. Immunity 49, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driesbaugh KH, Buzza MS, Martin EW, Conway GD, Kao JP, and Antalis TM (2015). Proteolytic activation of the protease-activated receptor (PAR)-2 by the glycosylphosphatidylinositol-anchored serine protease testisin. J. Biol. Chem 290, 3529–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, and Coughlin SR (2000). Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J. Biol. Chem 275, 19728–19734. [DOI] [PubMed] [Google Scholar]
- Godinho RO, Duarte T, and Pacini ES (2015). New perspectives in signaling mediated by receptors coupled to stimulatory G protein: the emerging significance of cAMP efflux and extracellular cAMP-adenosine pathway. Front. Pharmacol 6, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, et al. (2007). Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J. Physiol 578, 715–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Mayer K, and Lohmeyer J (2011). Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front. Immunol 2, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook JL, Islam MN, Parker D, Prince AS, Bhattacharya S, and Bhattacharya J (2018). Disruption of staphylococcal aggregation protects against lethal lung injury. J. Clin. Invest 128, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jairaman A, Yamashita M, Schleimer RP, and Prakriya M (2015). Store-Operated Ca2+ Release-Activated Ca2+ Channels Regulate PAR2-Activated Ca2+ Signaling and Cytokine Production in Airway Epithelial Cells. J. Immunol 195, 2122–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesmin S, Gando S, Zaedi S, and Sakuraya F (2007). Differential expression, time course and distribution of four PARs in rats with endotoxin-induced acute lung injury. Inflammation 30, 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, and Coughlin SR (1999). Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest 103, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, and Kuliopulos A (2007). ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat. Immunol 8, 1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver SK, and Evans TW (2007). Acute respiratory distress syndrome. BMJ 335, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, and MacKinnon R (2017). Structures of the human HCN1 hyperpolarization-activated channel. Cell 168, 111–120.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Rivera R, Dubin AE, and Chun J (2007). LPA(4)/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing G(s)-, G(q)/G(i)-mediated calcium signaling and G(12/13)-mediated Rho activation. J. Biol. Chem 282, 4310–4317. [DOI] [PubMed] [Google Scholar]
- Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, and Chae JJ (2012). The calciumsensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer PF, Williams AE, Scotton CJ, José RJ, Sulikowski M, Moffatt JD, Murray LA, and Chambers RC (2014). Proteinase-activated receptor-1, CCL2, and CCL7 regulate acute neutrophilic lung inflammation. Am. J. Respir. Cell Mol. Biol 50, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara K, Ramachandran R, Saifeddine M, Hansen KK, Renaux B, Polley D, Gibson S, Vanderboor C, and Hollenberg MD (2016). Thrombin-Mediated Direct Activation of Proteinase-Activated Receptor-2: Another Target for Thrombin Signaling. Mol. Pharmacol 89, 606–614. [DOI] [PubMed] [Google Scholar]
- Moraes TJ, Martin R, Plumb JD, Vachon E, Cameron CM, Danesh A, Kelvin DJ, Ruf W, and Downey GP (2008). Role of PAR2 in murine pulmonary pseudomonal infection. Am. J. Physiol. Lung Cell. Mol. Physiol 294, L368–L377. [DOI] [PubMed] [Google Scholar]
- O’Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, and Brass LF (2000). Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J. Biol. Chem 275, 13502–13509. [DOI] [PubMed] [Google Scholar]
- Ostrowska E, Sokolova E, and Reiser G (2007). PAR-2 activation and LPS synergistically enhance inflammatory signaling in airway epithelial cells by raising PAR expression level and interleukin-8 release. Am. J. Physiol. Lung Cell. Mol. Physiol 293, L1208–L1218. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, and Hollenberg MD (2009). Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol. Pharmacol 76, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A (2009). Signaling to gene expression: calcium, calcineurin and NFAT. Nat. Immunol 10, 3–5. [DOI] [PubMed] [Google Scholar]
- Roche N, Stirling RG, Lim S, Oliver BG, Oates T, Jazrawi E, Caramori G, and Chung KF (2003). Effect of acute and chronic inflammatory stimuli on expression of protease-activated receptors 1 and 2 in alveolar macrophages. J. Allergy Clin. Immunol 111, 367–373. [DOI] [PubMed] [Google Scholar]
- Rubins JB (2003). Alveolar macrophages: wielding the double-edged sword of inflammation. Am. J. Respir. Crit. Care Med 167, 103–104. [DOI] [PubMed] [Google Scholar]
- Scott G, Leopardi S, Parker L, Babiarz L, Seiberg M, and Han R (2003). The proteinase-activated receptor-2 mediates phagocytosis in a Rho-dependent manner in human keratinocytes. J. Invest. Dermatol 121, 529–541. [DOI] [PubMed] [Google Scholar]
- Sidhu TS, French SL, and Hamilton JR (2014). Differential signaling by protease-activated receptors: implications for therapeutic targeting. Int. J. Mol. Sci 15, 6169–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh UJ, Dores MR, Chen B, and Trejo J (2010). Signal transduction by protease-activated receptors. Br. J. Pharmacol 160, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ster J, De Bock F, Guèrineau NC, Janossy A, Barrère-Lemaire S, Bos JL, Bockaert J, and Fagni L (2007). Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc. Natl. Acad. Sci. USA 104, 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stief TW (2006). Thrombin generation by exposure of blood to endotoxin: a simple model to study disseminated intravascular coagulation. Clin. Appl. Thromb. Hemost 12, 137–161. [DOI] [PubMed] [Google Scholar]
- Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE, Dietrich A, et al. (2012). TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxininduced lung vascular permeability and inflammation. J. Exp. Med 209, 1953–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, et al. (2012). An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci. Transl. Med 4, 159ra148. [DOI] [PubMed] [Google Scholar]
- van den Boogaard FE, Brands X, Duitman J, de Stoppelaar SF, Borensztajn KS, Roelofs JJTH, Hollenberg MD, Spek CA, Schultz MJ, van ‘t Veer C, and van der Poll T (2018). Protease-Activated Receptor 2 Facilitates Bacterial Dissemination in Pneumococcal Pneumonia. J. Infect. Dis 217, 1462–1471. [DOI] [PubMed] [Google Scholar]
- Vincent F, and Duncton MA (2011). TRPV4 agonists and antagonists. Curr. Top. Med. Chem 11, 2216–2226. [DOI] [PubMed] [Google Scholar]
- Vortherms TA, Nguyen CH, Berlot CH, and Watts VJ (2004). Using molecular tools to dissect the role of Galphas in sensitization of AC1. Mol. Pharmacol 66, 1617–1624. [DOI] [PubMed] [Google Scholar]
- Wang Y, and Adjaye J (2011). A cyclic AMP analog, 8-Br-cAMP, enhances the induction of pluripotency in human fibroblast cells. Stem Cell Rev 7, 331–341. [DOI] [PubMed] [Google Scholar]
- Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, and Muller WA (2015). TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J. Exp. Med 212, 1883–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, and Bhattacharya J (2014). Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJV, Chinea LE, Pilling D, and Gomer RH (2018). Protease activated-receptor 2 is necessary for neutrophil chemorepulsion induced by trypsin, tryptase, or dipeptidyl peptidase IV. J. Leukoc. Biol 103, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, O’Callaghan JP, and O’Donnell JM (2011). Effects of repeated treatment with phosphodiesterase-4 inhibitors on cAMP signaling, hippocampal cell proliferation, and behavior in the forced-swim test. J. Pharmacol. Exp. Ther 338, 641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazbeck P, Tauseef M, Kruse K, Amin MR, Sheikh R, Feske S, Komarova Y, and Mehta D (2017). STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca2+ Entry. Sci. Rep 7, 42758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Michalick L, Tang C, Tabuchi A, Goldenberg N, Dan Q, Awwad K, Wang L, Erfinanda L, Nouailles G, et al. (2016). Role of Transient Receptor Potential Vanilloid 4 in Neutrophil Activation and Acute Lung Injury. Am. J. Respir. Cell Mol. Biol 54, 370–383. [DOI] [PubMed] [Google Scholar]
- Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, Liedtke W, Jimenez-Vargas NN, Vanner SJ, and Bunnett NW (2015). Neutrophil Elastase Activates Protease-activated Receptor-2 (PAR2) and Transient Receptor Potential Vanilloid 4 (TRPV4) to Cause Inflammation and Pain. J. Biol. Chem 290, 13875–13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shen H, Xiong Y, Yang X, and He J (2010). The beta1-adrenergic receptor mediates extracellular signal-regulated kinase activation via Galphas. Amino Acids 38, 75–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.