

Abstract
Liver X receptors are recognized as important regulators of cholesterol, fatty acid metabolism, inflammatory responses, and glucose homeostasis. The antineoplastic properties of synthetic liver X receptor (LXR) agonists (T0901317 and GW3965) have been reported in human carcinomas. Epidermal growth factor tyrosine kinase inhibitor (EGFR-TKI) is a first-line treatment for non-small-cell lung cancer patients with EGFR mutations. We used scratch and transwell assays to analyze cell migration and invasion. We evaluated tumor migration and invasion in vitro using a fluorescent orthotopic lung cancer model. An MMP9 (mouse) enzyme-linked immunosorbent assay kit was used to measure serum MMP9 concentrations. Protein expression was identified by western blot assays. In this study, we determined the effects of T0901317 and/or an EGFR-TKI on the lung cancer cell lines A549 and HCC827-8-1 in vitro and in vivo. We confirmed that the combination of the LXR agonist T0901317 and gefitinib can inhibit the migration and invasion of lung cancer both in vivo and in vitro, and this effect was possibly achieved by the inhibition of the ERK/MAPK signaling pathway. Our study showed that the combination of the LXR agonist T0901317 and gefitinib can inhibit the migration and invasion of lung cancer both in vivo and in vitro.
Keywords: epidermal growth factor tyrosine kinase inhibitor, invasion, liver X receptors, migration, non-small-cell lung cancer
Introduction
Lung cancer is the world’s leading cause of cancer deaths, and this disease has a poor prognosis and severely threatens human life; lung cancer includes non-small-cell lung cancer (NSCLC), which accounts for ~85% of cases 1–4. Currently, the most commonly mutated gene in NSCLC patients is epidermal growth factor receptor (EGFR) [116/400 (29%)] 3–5. Epidermal growth factor tyrosine kinase inhibitors (EGFR-TKIs) are a first-line treatment for NSCLC patients carrying EGFR mutations 3,6. However, with persistent use of TKI molecular targeted therapy alone, NSCLC patients eventually develop TKI resistance, leading to treatment failure 2,7–9.
Liver X receptors (LXRs) belong to the nuclear receptor superfamily and are ligand-activated transcriptional factors 10–13. LXRs are recognized as important regulators of cholesterol, fatty acid metabolism, inflammatory responses, and glucose homeostasis 14–16. Two isoforms have been described: LXRα and LXRβ. LXRα is highly expressed in the liver and at lower levels in the lungs, adrenal glands, intestine, adipose, macrophages, and kidneys, whereas LXRβ is ubiquitously expressed 11,17. Natural and synthetic ligands have already been developed and have effective therapeutic properties in murine models, where they have been used for the treatment of diabetes, atherosclerosis, and Alzheimer’s disease 18,19. Over recent years, the antineoplastic properties of synthetic LXR agonists (T0901317 and GW3965) have been reported in human carcinomas, such as breast and prostate 10,17,20,21. T0901317 can also induce G1 cell cycle arrest in many cancer cell lines, including H1299 cells 21. We believe that LXR agonists have a beneficial antineoplastic effect that could be useful for treating NSCLC.
We further determined whether these drugs have an effect on cell migration and invasiveness. In a previous study, we proved that the LXR agonist T0901317 could reverse EGFR-TKI treatment resistance in A549 and HCC827-8-1 human lung cancer cells by inhibiting the activation of AKT 11. Alioui and colleagues confirmed that when LXRs were genetically ablated, carcinogenic invasiveness was highly promoted in prostate cancer 13. A well-established finding that supported tumor aggressiveness was the overexpression of matrix metalloproteinases (MMPs), which allow cells to efficiently degrade the surrounding matrix and migrate 13. The authors found enhanced expression of Mmp1, Mmp2, Mmp7, and Mmp9, and marked accumulation of the MMP9 protein in Ptenpc−/−LXRαβ−/− prostate samples. Accordingly, we believe that T0901317 has therapeutic potential in lung cancer cells. We aimed to determine the effects of T0901317 and/or an EGFR-TKI on the lung cancer cell lines A549 and HCC827-8-1 in vitro and in vivo.
Materials and methods
Materials
T0901317 was purchased from Cayman Chemical (Ann Arbor, Michigan, USA). Gefitinib (Iressa) was purchased from AstraZeneca UK Limited (Macclesfield, UK). All drugs were suspended in DMSO and stored at −20°C. Antibodies against MMPs, E-cadherin, and β-actin were obtained from Cell Signaling Technology (Danvers, Massachusetts, USA).
Cell lines
The A549 cell line was purchased from the Shanghai Institute for Biological Sciences, Chinese Academy of Cell Resource Center (Shanghai, China). A549 cells are an NSCLC line carrying the KRAS mutation. HCC827-8-1 cells were generated from HCC827 (gefitinib-sensitive) cells and maintained at the Affiliated Cancer Hospital of Nanjing Medical University. Cells were cultured at 37°C with 5% CO2 in RPMI-1640 supplemented with 10% heat-inactivated FBS, 100 units/ml penicillin, and 100 units/ml streptomycin.
Cell migration and invasion assays
Cell migration was examined by scratch assays. Cells were treated with gefitinib (5 μmol/l) and T0901317 (5 μmol/l) alone or in combination. Three days later, aliquots of the cells were seeded in six-well plates and covered fully with media. The rest of the cells were resuspended (2×105 cells/well) in 200 μl of serum-free media and placed in the upper compartments of transwell chambers (COSTAR, Corning, New York, USA). Subsequently, a 200-μl pipette tip was utilized to scratch the surface of the plates. Cell migration was observed at 0 and 24 h under an optical microscope with a magnification of ×100. All experiments were repeated in triplicate. Transwell migration chambers were used to assess A549 cell invasion ability. As described above, the remaining cells were resuspended (2×105 cells/well) in 200 μl of serum-free media and placed in the upper compartments of transwell chambers. The lower chambers contained 500 μl of RPMI-1640 and 20% fetal bovine serum. The cells in the upper chambers were wiped away using cotton wool after 48 h, and the migrated cells in the lower chamber were fixed with 4% paraformaldehyde and stained with a crystal violet solution. Then, the fixed cells were counted under a light microscope at a magnification of ×100. All experiments were repeated three times.
Western blot analysis
Cells were lysed in ice-cold RIPA buffer (Beyotime Biotechnology, Jiangsu, China). The lysates were centrifuged at 4°C for 20 min at 14 000g. The supernatants were retained for the subsequent procedures. The protein concentrations were determined using a BCA protein assay kit (Beyotime, Shanghai, China). The samples were separated on NuPAGE 10% Bis-Tris gels (Life Technologies, Beijing, China) and transferred onto polyvinylidene difluoride membranes (Millipore Corporation, Billerica, Massachusetts, USA). Then, the membranes were incubated overnight at 4°C with a primary antibody and incubated with an HRP-conjugated secondary antibody for 1 h. Then, the membranes were visualized using an ECL Plus Kit (Beyotime Biotechnology).
Mouse cancer models
Animal studies were carried out according to a protocol approved by the Institute of Animal Use and Care Committee at Washington State University. Male and female BALB/c nude mice (Nanjing Yuanduan Biotechnology Co. Ltd, Nanjing, China) were raised and tested in an SPF barrier system (Suzhou Fengshi Laboratory Animal Equipment Co., Ltd., Suzhou, China). The nude mice were implanted subcutaneously with tumor cells as follows: primary tumors were completely detached and divided into 1 mm×1 mm×1 mm tumor tissue blocks in culture medium. The tumor tissue blocks were transplanted into the lobes of 40 nude mice. The mice were divided randomly into four groups. The first group was injected with soybean oil. The second group was administered gefitinib (50 mg/kg) intraperitoneally. The third group was injected intraperitoneally with T0901317 (10 mg/kg). The last group was administered gefitinib and T0901317 (50+10 mg/kg) intraperitoneally for 4 weeks. After blood was collected, all experimental animals were killed. Fluorescence imaging of the open animals was performed to examine primary tumors and metastases.
Assay of mouse serum MMP9 concentration
We used an MMP9 (mouse) enzyme-linked immunosorbent assay kit (Abnova, Jiangsu, China) to determine the differences in the MMP9 concentration in different mouse models. Serum samples were diluted 1 : 100 and used.
Data analysis
Every experiment was conducted at least three times. All values in the figures are expressed as the mean±SD. We performed unpaired, two-sided Student’s t-tests with Microsoft Excel 2013 (Microsoft, Redmond, Washington, USA), and P values of less than 0.05 were considered to be significant.
Results
T0901317 and gefitinib reduce the migration and invasion of A549 and HCC827-8-1 cells
Cell migration is known to contribute toward tumor metastasis 22. To determine the effect of T0901317 and gefitinib on the invasive behavior of A549 cells, we used scratch and transwell assays. The results shown in Fig. 1a and b indicate that gefitinib (5 μmol/l) and T0901317 (5 μmol/l) in combination can significantly delay scratch healing in A549 and HCC827-8-1 cells. The transwell invasion chamber assays also confirmed significant differences in cell invasion among the cell groups treated with gefitinib (5 μmol/l) and T0901317 (5 μmol/l) alone and in combination (Fig. 1c).
Fig. 1.
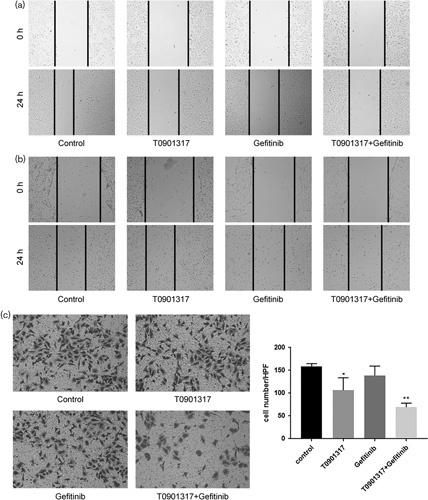
The combination of T0901317 and gefitinib inhibits the migration and invasion of A549 cells. (a) The migration capability of A549 cells after treatment with the combination of T0901317 and gefitinib was determined by wound-healing assays. (b) Transwell assays showed that the combination of T0901317 and gefitinib decreased A549 cell invasion, **P<0.01 compared with the control. (c) The migration capability of HCC827-8-1 cells after treatment with the combination of T0901317 and gefitinib was determined by wound-healing assays. *P<0.05.
T0901317 and gefitinib reduce the migration and invasion of A549 cells, possibly by reducing MMP9 expression
We identified that MMP9 expression decreased after cells were cultivated in gefitinib (5 μmol/l) and T0901317 (5 μmol/l) in combination for 3 days (Fig. 2a). It has been reported that high MMP9 expression tended to enhance the metastasis of NSCLC 23. These results indicate that the combination treatment of T0901317 and gefitinib could reduce the expression of MMP9 in A549 cells.
Fig. 2.

Western blot results showed that the combination of T0901317 and gefitinib regulated the expression of MMP9 and E-cadherin. (a) The combination of T0901317 and gefitinib reduced the expression of MMP9 in A549 cells. (b) The combination of T0901317 and gefitinib induced the expression of E-cadherin in HCC827-8-1 cells.
T0901317 and gefitinib reduce the migration of HCC827-8-1 cells, possibly by reducing E-cadherin expression
Reduced E-cadherin levels in cancer have been discovered in the progression and metastasis of many malignancies 24,25. The expression of E-cadherin increased after cells were cultivated in gefitinib (5 μmol/l) and T0901317 (5 μmol/l) in combination for 3 days (Fig. 2b). The results showed that the combination of T0901317 and gefitinib could increase the expression of E-cadherin in HCC827-8-1 cells.
T0901317 and gefitinib reduce tumor metastasis
A fluorescence orthotopic lung cancer model in nude mice showed that the group treated with the combination of T0901317 and gefitinib showed a slower metastasis rate than the other groups (Fig. 3a). The metastasis rate of this group was significantly lower than that of the control group, and statistically significant differences were noted (Fig. 3b and Table 1).
Fig. 3.
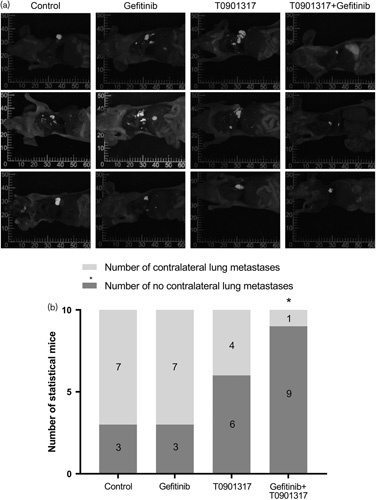
A fluorescent orthotopic lung cancer model showed that the combination of T0901317 and gefitinib inhibited the metastasis rate in nude mice. (a) The combination of T0901317 and gefitinib inhibited metastasis in nude mice. (b) The group treated with the combination of T0901317 and gefitinib showed a slower metastasis rate than the other groups. *P<0.05.
Table 1.
Statistical results of the orthotopic transplantation tumor model of nude mice with fluorescent lung cancer

T0901317 and gefitinib reduce serum MMP9 concentrations in mice
An MMP9 (mouse) enzyme-linked immunosorbent assay kit was used to measure the concentration of MMP9 in the serum of different treatment groups. It was found that the concentration of MMP9 in the T0901317 and gefitinib combination group was significantly lower than that in the control group (Fig. 4).
Fig. 4.
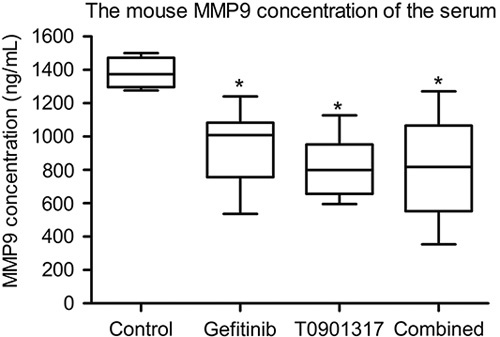
The concentration of MMP9 was significantly decreased in the T0901317 and gefitinib combination group. *P<0.05.
Discussion
NSCLC has such a poor prognosis that the development of effective agents is very important. Gefitinib has been used to treat lung cancer patients for a long time. The antineoplastic properties of LXR agonists have also been reported continuously over recent years. Our study shows a possible therapeutic role for T0901317 combined with gefitinib; this combination has antineoplastic properties that suppress migration and invasion in vivo and in vitro.
T0901317 and gefitinib affect cell proliferation 3 days after dosing according to our previous study 11. We chose to conduct the experiment within 3 days after adding the drugs. The cell scratch assay showed that the combination of gefitinib and T0901317 can significantly reduce the migration of A549 and HCC827-8-1 cells in vitro. Meanwhile, the transwell assay indicates that the combination of gefitinib and T0901317 can significantly reduce the invasion of A549 cells. The lung cancer in-situ nude mouse model showed only 10% metastases in the combination group and 70% metastases in the control group. The serum MMP9 concentration in the mice was lower in the combination group than in the other groups. Consistent with this observation, we confirmed that the protein expression of MMP9 was significantly decreased after treatment with T0901317 and gefitinib in combination.
In our previous study, we found that phospho-ERK levels were lower in the combined treatment group than in the control group or the single drug group. The ERK/MAPK pathway is often aberrantly activated in human cancers, and this activation can lead to enhanced cell invasiveness. The fact that MMPs play a vital role downstream of the ERK/MAPK signaling pathway is widely acknowledged 23,26. These results indicate that T0901317 and gefitinib can inhibit the metastasis of A549 cells by suppressing the ERK/MAPK signaling pathway. LXR agonists and EGFR-TKIs have a potential synergistic effect in inhibiting the activation of the ERK/MAPK pathway. The migration of HCC827-8-1 cells may be inhibited by the reduction of E-cadherin.
Conclusion
Our study showed that the combination of the LXR agonist T0901317 and gefitinib can inhibit the migration and invasion of lung cancer both in vivo and in vitro, and this effect was possibly achieved by the inhibition of the ERK/MAPK signaling pathway. We will explore more details about the antineoplastic effect of the LXR agonist and attempt to identify the potential mechanism in a future study.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81372396) and the Natural Science Foundation of Jiangsu Province (BK20141016).
Authors’ contributions: J.F. and H.C. conceived and supervised the experiment and modified the manuscript. R.L. and S.D. participated in the design of this study, conducted the western blot experiment, and drafted the manuscript. C.S. and X.X. conducted the cell and RT-PCR experiments. R.M. and J.W. carried out the statistical analysis. All authors read and approved the final manuscript.
Conflicts of interest
There are no conflicts of interest.
Footnotes
*Rui Lou and Haixia Cao contributed equally to the writing of this article.
References
- 1.Yang J, Lin J, Liu T, Chen T, Pan S, Huang W, Li S. Analysis of lncRNA expression profiles in non-small cell lung cancers (NSCLC) and their clinical subtypes. Lung Cancer 2014; 85:10–15. [DOI] [PubMed] [Google Scholar]
- 2.Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res 2014; 4:411–435. [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi K, Hagiwara K. Epidermal growth factor receptor (EGFR) mutation and personalized therapy in advanced nonsmall cell lung cancer (NSCLC). Target Oncol 2013; 8:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Mello, Madureira P, Carvalho LS, Araújo A, O'Brien M, Popat S. EGFR and KRAS mutations, and ALK fusions: current developments and personalized therapies for patients with advanced non-small-cell lung cancer. Pharmacogenomics 2013; 14:1765–1777. [DOI] [PubMed] [Google Scholar]
- 5.Das BR, Bhaumik S, Ahmad F, Mandsaurwala A, Satam H. Molecular spectrum of somatic EGFR and KRAS gene mutations in non small cell lung carcinoma: determination of frequency, distribution pattern and identification of novel variations in Indian patients. Pathol Oncol Res 2015; 21:675–687. [DOI] [PubMed] [Google Scholar]
- 6.Kogure Y, Saka H, Oki M, Saito TI, Ahmed SN, Kitagawa C, et al. Post-progression survival after EGFR-TKI for advanced non-small cell lung cancer harboring EGFR mutations. PLoS One 2015; 10:0135393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tetsu O, Phuchareon J, Eisele DW, Hangauer MJ, McCormick F. AKT inactivation causes persistent drug tolerance to EGFR inhibitors. Pharmacol Res 2015; 102:132–137. [DOI] [PubMed] [Google Scholar]
- 8.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010; 362:2380–2388. [DOI] [PubMed] [Google Scholar]
- 9.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010; 11:121–128. [DOI] [PubMed] [Google Scholar]
- 10.Rough JJ, Monroy MA, Yerrum S, Daly JM. Anti-proliferative effect of LXR agonist T0901317 in ovarian carcinoma cells. J Ovarian Res 2010; 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao H, Yu S, Chen D, Jing C, Wang Z, Ma R, et al. Liver X receptor agonist T0901317 reverses resistance of A549 human lung cancer cells to EGFR-TKI treatment. FEBS Open Bio 2017; 7:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alioui A, Dufour J, Leoni V, Loregger A, Moeton M, Iuliano L, et al. Liver X receptors constrain tumor development and metastasis dissemination in PTEN-deficient prostate cancer. Nat Commun 2017; 8:445. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Wu Y, Yu DD, Hu Y, Cao HX, Yu SR, Liu SW, et al. LXR ligands sensitize EGFR-TKI-resistant human lung cancer cells in vitro by inhibiting Akt activation. Biochem Biophys Res Commun 2015; 467:900–905. [DOI] [PubMed] [Google Scholar]
- 14.Savic D, Ramaker RC, Roberts BS, Dean EC, Burwell TC, Meadows SK, et al. Distinct gene regulatory programs define the inhibitory effects of liver X receptors and PPARG on cancer cell proliferation. Genome Med 2016; 8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J Biol Chem 2006; 281:27816–27826. [DOI] [PubMed] [Google Scholar]
- 16.Xiao L, Wang J, Jiang M, Xie W, Zhai Y. The emerging role of constitutive androstane receptor and its cross talk with liver X receptors and peroxisome proliferator-activated receptor A in lipid metabolism. Vitam Horm 2013; 91:243–258. [DOI] [PubMed] [Google Scholar]
- 17.Candelaria NR, Addanki S, Zheng J, Nguyen-Vu T, Karaboga H, Dey P, et al. Antiproliferative effects and mechanisms of liver x receptor ligands in pancreatic ductal adenocarcinoma cells. PLoS One 2014; 9:e106289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noghero A, Perino A, Seano G, Saglio E, Lo Sasso G, Veglio F, et al. Liver X receptor activation reduces angiogenesis by impairing lipid raft localization and signaling of vascular endothelial growth factor receptor-2. Arterioscler Thromb Vasc Biol 2012; 32:2280–2288. [DOI] [PubMed] [Google Scholar]
- 19.Castrillo A, Tontonoz P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu Rev Cell Dev Biol 2004; 20:455–480. [DOI] [PubMed] [Google Scholar]
- 20.Komati R, Spadoni D, Zheng S, Sridhar J, Riley KE, Wang G. Ligands of therapeutic utility for the liver X receptors. Molecules 2017; 22:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chuu CP, Lin HP. Antiproliferative effect of LXR agonists T0901317 and 22(R)-hydroxycholesterol on multiple human cancer cell lines. Anticancer Res 2010; 30:3643–3648. [PubMed] [Google Scholar]
- 22.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65:87–108. [DOI] [PubMed] [Google Scholar]
- 23.Yadav L, Puri N, Rastogi V, Satpute P, Ahmad R, Kaur G. Matrix metalloproteinases and cancer: roles in threat and therapy. Asian Pac J Cancer Prev 2014; 15:1085–1091. [DOI] [PubMed] [Google Scholar]
- 24.Yang YL, Chen MW, Xian L. Prognostic and clinicopathological significance of downregulated E-cadherin expression in patients with non-small cell lung cancer (NSCLC): a meta-analysis. PLoS One 2014; 9:99763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ, et al. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis 2014; 35:867–876. [DOI] [PubMed] [Google Scholar]
- 26.Brown GT, Murray GI. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J Pathol 2015; 237:273–281. [DOI] [PubMed] [Google Scholar]