

Abstract
The process of cell differentiation in a developing embryo is influenced by numerous factors, including various biological molecules whose presentation varies dramatically over space and time. These morphogens regulate cell fate based on concentration profiles, thus creating discrete populations of cells and ultimately generating large, complex tissues and organs. Recently, several in vitro platforms have attempted to recapitulate the complex presentation of extrinsic signals found in nature. However, it has been a challenge to design versatile platforms that can dynamically control morphogen gradients over extended periods of time. To address some of these issues, we introduce a platform using channels patterned in hydrogels to deliver multiple morphogens to cells in a 3D scaffold, thus creating a spectrum of cell phenotypes based on the resultant morphogen gradients. The diffusion coefficient of a common small molecule morphogen, retinoic acid (RA), was measured within our hydrogel platform using Raman spectroscopy and its diffusion in our platform’s geometry was modeled using finite element analysis. The predictive model of spatial gradients was validated in a cell-free hydrogel, and temporal control of morphogen gradients was then demonstrated using a reporter cell line that expresses green fluorescent protein in the presence of RA. Finally, the utility of this approach for regulating cell phenotype was demonstrated by generating opposing morphogen gradients to create a spectrum of mesenchymal stem cell differentiation states.
Graphical Abstract
Introduction
Biological development relies upon highly regulated local concentrations of proteins and/or small molecules. These so-called morphogens can be interpreted by cells based on concentration (spatial dependence), as well as the amount of time cells are exposed to each signal (temporal dependence). The integration of these cues determines cell fate and the formation of large, complex tissues and organs. One example of morphogen-guided embryonic development is the spatial patterning of the anteroposterior axis, which is simultaneously differentiated with the dorsoventral axis based on opposing gradients of retinoic acid (RA) and fibroblast growth factors (FGF). This signaling gradient induces gene expression in a position-dependent manner, ultimately producing spatially organized domains of the neural tube1,2. Another example can be found in the differentiation of the mesoderm layer into cardiac tissue in response to transforming growth factor β (TGFβ) signaling3, where the cells sub-differentiate into atrial and ventricular cardiac cells to form the atrium and ventricles of the heart4–6. Moreover, cell differentiation is not limited to embryonic development. In the adult intestine, mitotically active, undifferentiated stem cells repopulate the epithelial lining of the intestine to renew, repair and effectively absorb food while maintaining a barrier against potentially harmful microorganisms. In response to opposing gradients of Wnt and bone morphogenic protein (BMP) signaling, spatially organized domains of the epithelial layer arise to form the crypts and villi of the intestinal wall7–9.
Nearly every organ system in the body relies on morphogen signaling for proper differentiation and organization. Recreation of these developmental patterning events in human stem cells, from both embryonic and adult sources, is highly desirable for applications in disease modeling and regenerative medicine. Although patterning events involving temporal dependence have been reproduced in static well-plate cultures3,10,11, those involving spatial morphogen gradients require more intricate engineering solutions12–15.
Few platforms have been able to produce morphogen gradients over extended periods of time and across a large scale (on the order of centimeters) to guide differentiation and eventual proliferation and self-organization3,16. One early approach to producing spatially-varying differentiation employed microspheres loaded with various concentrations of BMP-2 and insulin-like growth factor 1 (IGF-1). These microspheres were embedded in alginate hydrogels, where one variety of microsphere (poly(lactic-co-glycolic acid)) released a large quantity of growth factor over a short period of time, and the other (silk) provided a sustained release over an extended period of time (weeks)17. Additionally, the researchers were able to include several different morphogens in each variety of microsphere, thereby creating gradients of multiple species within a single scaffold. Many other follow-up studies have successfully used this approach to create morphogen gradients with various growth factors for tissue regeneration18–20. However, due to eventual depletion of the morphogens, this approach is unable to sustain a heterogenous morphogen distribution for extended periods of time, as the crosslinked hydrogel cannot be reloaded with fresh microspheres without compromising the structural integrity of the matrix. In addition, because all parameters regarding the release rate of morphogens from the microspheres were preprogrammed prior to the experiment, it is difficult to produce more complex gradients (e.g. generate periodic concentration waves) after the microspheres have been loaded into the hydrogel.
Microfluidic systems provide an alternative means of delivering morphogens to cells in a scaffold and can be set up to maintain long term concentration profiles. In the past decade, several such platforms have been developed to study intracellular pathways involved in stem cell differentiation21. The use of microfluidics provides researchers with unprecedented control over the delivery and removal of soluble compounds to three-dimensional (3D), cell-laden scaffolds, and has the potential to create complex gradients of morphogens22. As a result, these systems have enabled physiologically relevant models for high throughput drug screening and a better understanding of the pathophysiology associated with several diseases23–26. One early approach utilized hydrogels with embedded microfluidic channels, wherein soluble factors diffused through the hydrogel to cells seeded on the surface. This configuration was used to deliver RA to a flat surface, creating gradients to study the differentiation of embryonic stem cells (ESCs) into neurons22. A more complex platform, consisting of four microfluidic channels adjacent to the top, bottom, left, and right sides of a cell-laden gel, has also been used to create four separate gradients (RA, Sonic hedgehog, BMP, and FGF) to study neural tube patterning2. Overall, these creative approaches have made significant contributions to the field by demonstrating the importance of controlled multifaceted morphogen presentation; however, they remain small in scale. This size limitation is due to the fact that most current platforms designed to set up morphogen gradients in 3D cell-laden hydrogels utilize gels formed inside microfluidic bioreactors,2,27 thereby limiting the size (thickness, in particular) of the cell-laden gels used.
Herein, we sought to develop an experimental platform that provides spatial and temporal control of morphogen delivery in a hydrogel over long time frames (greater than a month), and that is large enough to accommodate stem cell differentiation for patterning larger tissue structures (and, eventually, to accommodate embedded organoids, which can be on the several hundred micron scale or larger). Thus, instead of forming the gel within a microfluidic bioreactor, we form the fluidic channels within the gel. This system provides the ability to dynamically deliver and replenish morphogens via fluidic channels to a large cell-laden hydrogel, visualize cellular responses due to morphogen gradients, and run experiments over extended time periods. Moreover, the large hydrogel can easily be cut into discrete sections, each yielding a large number of cells for further analysis (Western blot, etc.). To validate this approach, we first measured the diffusion coefficient of RA with Raman spectroscopy and that of fluorescently-labeled dextran with fluorescence microscopy, modeled the diffusion patterns using COMSOL, and verified the accuracy of the models by measuring RA and dextran concentrations in samples extracted from various locations in separate hydrogels. Next, we assessed control of RA delivery in space and time using an RA-sensitive reporter cell line. Finally, using mesenchymal stem cells (MSCs) as a model system, we produced a spectrum of differentiation states by delivering two different morphogens from two parallel channels, thereby creating two opposing gradients. By changing the concentration of the morphogens in the fluidic channels, we demonstrate altered distributions of cell phenotype within the gel. Overall, these proof-of-principle studies lay the groundwork for more complex patterning applications involving stem cells in larger volumes.
Materials and methods
Bioreactor system fabrication
In order to ensure reproducibility of the bioreactor construction, namely channel and port locations, a jig was designed using Creo Parametric and 3D printed from ABS (Acrylonitrile butadiene styrene, Stratasys F170) (Fig. 1). This jig was used to guide the drilling of two 1/4” holes on each end of the polystyrene box with 11 mm separation between the holes on each face. The drilling was completed using a mill and a plastic cutting drill bit (McMaster-Carr, 27465A84, specialized drill bit designed to avoid cracking plastic). The barbed end of female Luer bulkheads was cut off, and bulkheads were mounted to each of the four holes in the polystyrene box using locking nuts. A 1” piece of square stock was placed in the middle of the polystyrene box in contact with the four bulkheads. PDMS (10:1 PDMS:initiator) was then poured into the space between the square stock and the polystyrene box. After cross-linking the PDMS “walls” in an oven at 60C for 2 hours, the square stock was removed and 1.5 mL of PDMS was added to the bottom of the box and crosslinked to completely coat the inside of the polystyrene box with PDMS. The PDMS was used to act as a frame for the gelatin to adhere and serve as a gasket to prevent leaking. Finally, the bulkheads were removed and the resulting PDMS frame and polystyrene box were sterilized in bleach for 30 minutes then ethanol for 1 hour. The box and PDMS were then transferred to the biohood and were placed in a DI water bath for 1 hour. This was repeated 3 times. The devices were then dried on a heated surface (37C). After drying, the PDMS frame was placed back into the polystyrene box and new, sterile, female Luer bulkheads (with barbed end intact) were screwed into the four embedded locking nuts. The embedded locking nuts ensure that the integrity of the ports is maintained for several weeks of perfusion. Additionally, the hydrogel itself sticks to the PDMS walls, so the gelatin does not pull away from the ports and the channels do not leak when perfused. Sterile 1/16” silicon tubing (Cole Palmer) was inserted through each pair of bulkheads to create a mold for the two parallel channels (9 mm apart) to be formed during hydrogel fabrication. For experiments, the PDMS-lined polystyrene box was placed in a larger polystyrene box to further protect the hydrogel from contamination. The larger box also contained the two media reservoirs (50 mL conical tubes with two holes drilled into the cap for tubing) for continuous, nonmixing, recirculation of media. Individual bioreactors were fabricated for each experiment; the polystyrene box, PDMS frame, and tubing were not re-used.
Figure 1:

Bioreactor fabrication and assembly process. First, a 3D-printed jig is used to guide drilling of holes for the perfusion ports, which are then fitted with Luer bulkheads and locking nuts. One-inch square stock is placed within the outer box in contact with the Luer bulkheads to provide a temporary mold for a PDMS layer in which the locking nuts are embedded. The bottom of the box is also coated with PDMS to complete the inner frame. After the PDMS frame has crosslinked, silicon tubing is inserted to form a mold for two parallel perfusion channels located 9 mm apart. Finally, a hydrogel solution (with or without cells) is poured into the PDMS frame, the hydrogel is crosslinked, and the silicone tubing pieces are removed to leave embedded, parallel channels within the hydrogel. The overall hydrogel dimensions are 25 × 25 × 25 mm.
Cell culture and transfection
RFP-expressing fibroblasts and human bone marrow-derived MSCs were purchased from Angio-Proteomie (Boston, MA) and Lonza (Allendale, NJ), respectively. The cells were maintained in a humidified 5% CO2 environment at 37°C. MSC maintenance media consisted of DMEM supplemented with 20% v/v fetal bovine serum (FBS), 50 U/mL penicillin, and 50 μg/mL streptomycin, all procured from Life Technologies (Carlsbad, CA). Red fluorescent protein (RFP)-expressing fibroblasts were transfected with the pGreenFire1-RARE Lentivector virus28–31 (System Bioscience, Palo Alto, CA). RARE-RFP-fibroblasts were seeded at 1 million cells/ml in a 6-well plate, where a multiplicity of infection of 5 was used to infect the cells. After 48 hours, cells were treated with 10 nM of RA (20 mg/ml RA in DMSO stock solution) and imaged for compromised green fluorescent protein (GFP; 2-hour half-life) expression to verify successful transduction.
Hydrogel scaffold fabrication for perfusion
Hydrogels were formed inside the PDMS frame as described previously, with the modification of using two isolated embedded perfusion channels32. Briefly, gelatin powder (Sigma Aldrich, St. Louis, MO) was dissolved in MSC maintenance media at a final concentration of 10% w/v. After 1 h of sonication at 37°C, the gelatin solution was sterile-filtered (0.2 μm, Thermo Fisher, Waltham, MA) and maintained at 37°C in a water bath. A 20% w/v solution of microbial transglutaminase (mTG, MooGloo, Eliot, ME) in PBS was prepared separately and sterile-filtered. The sterile mTG and gelatin solutions were then added to a conical tube containing a pellet of trypsinized RARE-RFP-fibroblasts (3 million cells/mL) or MSCs (3 million cells/mL) in a 1:9 mTG to gel ratio for a total volume of 20 mL. The cells were resuspended by manual pipetting, and the solution was immediately poured over the silicone tubing in the PDMS frame. The bioreactor was incubated at 37°C for 30 minutes to crosslink the hydrogel. The silicone tubing pieces were then withdrawn from the hydrogel, leaving two parallel channels aligned with the Luer bulkheads (Fig. 2A). Autoclaved Tygon (E-3603) lab tubing (1/16 in. ID × 1/8 in. OD) (Cole Palmer, Vernon Hills, IL) was then washed 3 times with DI water and connected to the Luer bulkheads, and the bioreactor was perfused using a peristaltic pump perfusion system (Fig. 2B)32.
Figure 2:

A closed-circuit microfluidic hydrogel platform allows for dual delivery of osteogenic and chondrogenic media. (A) An illustration of the cross-section between the two parallel channels. The area was divided into three regions, each with a width of 3mm (regions A-C), for the Western blot study and divided into five regions, each with a width of 1.8mm (regions 1–5), for the immunofluorescence study. (B) An illustration of the perfusion setup shows how the media is drawn from a conical tube reservoir, around a pumphead, through the gel, and back into the reservoir.
Measuring the diffusion coefficients of small and large compounds
For measurement of the diffusion coefficient of RA (Sigma Aldrich, St. Louis, MO) in a gelatin hydrogel, 64×27×1.2mm glass slides were used as a mold. First, 5 glass slides were glued and stacked on top of each other and allowed to cure at 60C. The stack of slides was then placed in the center of a 100mm petri dish, where PDMS was poured over the glass slides and filled until the slides were thoroughly covered. This was allowed to crosslink for 1 hour at 60C, after which the crosslinked PDMS was cut out of the petri dish. The glass slides were removed to create a well for the gel. On one of the short ends of the PDMS mold, two holes were created using a 1mm biopsy punch, and silicon tubing was placed to serve as ports for recirculating RA in DI water at a final concentration of 2μM. The same stack of glass slides was then placed vertically into the PDMS chamber against the face where the biopsy punches were made. A 1:9 solution of mTG to gelatin was mixed in DI water and poured into the mold. After 30 minutes of crosslinking at 37C, the glass slides were removed, leaving space for an RA reservoir on one end of the hydrogel.
For the RA diffusion experiment, the PDMS mold containing a crosslinked hydrogel was placed on the confocal Raman microscope stage (Renishaw) and the laser was focused below the surface of the hydrogel at a location 0.2cm from the gel/reservoir interface. An external reservoir of 2μM RA in DI water was connected to the reservoir in the PDMS frame via a custom-built peristaltic pump described previously25. The internal reservoir was filled with 2μM RA and Raman spectra acquisition began immediately (Fig. 3A). The peristaltic pump recirculated RA from the large external reservoir, and Raman spectra were recorded every 30 minutes for 5 hours (532nm laser, 50% power, 5X magnification, 5 second acquisition time, grating 2400 l/mm). The peak Raman intensity between 1590–1595cm−1, corresponding to the RA C=C bond stretching Raman signal in our hydrogel, was plotted as a function of time after background subtraction (Fig. 3B). The data from 5 independent experiments were combined and analyzed with a modified equation developed for the diffusion of semi-infinite media by Crank33,34. At time equal to 0, the RA concentration throughout the gel is 0 and the gel/reservoir interface concentration of RA was maintained at C0. As RA molecules diffuse into the gel, the concentration dependence on the distance in the gel from the gel/reservoir interface, defined as x = 0, is given by the equation below:
where C is the concentration at distance x from the interface at an elapsed time t, and Dt is the mutual-diffusion coefficient. An additional constant term, B, was added to account for a small offset in the y-intercept, due to imperfect background subtraction. The data were fit using a nonlinear least-squares solver (lsqcurvefit function) in Matlab (Mathworks, Natick, MA).
Figure 3:

The diffusion coefficient of a model small compound (RA) in the hydrogel scaffold was measured using Raman spectroscopy. (A) A schematic of the experiment shows the reservoir of RA maintained at a concentration of 2 μM adjacent to a hydrogel contained within a PDMS frame. Confocal Raman microscopy measurements were collected at a focal point below the surface of the gel at a location 0.2 cm from the reservoir-gel interface. (B) The intensity of the RA-associated Raman peak (1590–1595cm−1) increased over time as RA diffused into the hydrogel. (C) The maximum intensity of the Raman peak for RA was plotted as a function of time (n=5 independent experiments), and the data were fit with a modified equation developed for the diffusion of semi-infinite media in order to determine the diffusion coefficient of RA. (D) COMSOL model of RA diffusion in a hydrogel with two fluidic channels. Delivery of RA from the left channel is simulated and the RA concentration gradient is shown at 24 hours of perfusion. (E) Values of RA from the COMSOL model were validated experimentally at distances from 1–3 mm.
Diffusion of a larger compound, fluorescently-labeled 10 kDa dextran (Thermo Fisher, Waltham, MA) was measured in a hydrogel with two perfusion channels fabricated as described in Fig. 1 without inclusion of cells. Syringe pumps were used to perfuse the left channel with 1 mM Texas red 10 kDa dextran and the right channel with 1 mM FITC 10 kDa dextran at 300 μl/minute (Fig. 4A). The fluorescence intensity of the Texas red dextran was measured 0.2 cm from the left channel every minute for 60 minutes using a confocal microscope (Zeiss LSM 710). To determine a diffusion coefficient, the resulting data were then fit using the above diffusion equation. Additionally, the entire length between the red and green channels was imaged after 60 minutes to map out the extent to which each variety of dextran diffused.
Figure 4:

The diffusion coefficient of a model large compound (fluorescently-labeled 10 kDa dextran) in the hydrogel scaffold was measured using confocal microscopy. (A) A fluorescent image of the experiment shows the left perfusion channel (red) and the right perfusion channel (green) under perfusion with a syringe pump. Fluorescence intensity of Texas red dextran at a spot 0.2 cm from the left channel was measured over time to determine the diffusion coefficient. (B-C) After 60 minutes of perfusion, opposing gradients of both Texas red- and FITC-dextran molecules were apparent. (D-E) Using the acquired diffusion coefficient, a COMSOL model was created to simulate Texas red dextran diffusion from the left port and FITC dextran diffusion from the right port. (F) Local concentrations of both dextrans obtained from the COMSOL simulation were validated experimentally at positions corresponding to x=5, 7, and 8 mm.
COMSOL model of diffusion and experimental validation
Diffusion of RA was modeled using the Transport of Diluted Species physics interface of the Chemical Reaction Engineering module included in the COMSOL Multiphysics 5.3 modeling suite (COMSOL AB, Stockholm, Sweden) with the experimentally-measured diffusion coefficient as an input. A model of diffusion in the hydrogel was generated using 8 mM RA in a channel, and the simulation was run to a 24-hour timepoint. At the 24-hour mark, the RA concentrations in the gel at 1 mm, 2 mm, and 3 mm from gel/reservoir interface were obtained. In order to experimentally verify the simulation, a gel was cast in the same geometry (using PBS instead of complete media for simplicity). The reservoir was then filled with recirculating 8 mM RA. After 24 hours, the gel was cross-sectionally cut into 10 equal sections. A 0.5 mm biopsy punch was used to punch out gel at distances of 1 mm, 2 mm, and 3 mm from the gel/reservoir interface. The punches were then placed into a sterile 0.45 μm pore centrifuge tube filter (Spin-X Centrifuge Tube Filters, Corning, New York, NY) and spun at 10,000xG to extract the liquid, and the RA concentration in the liquid phase was determined using a microplate reader (Synergy H1 Multi-mode Microplate Reader, BioTek) to measure absorbance at 350 nm.
The diffusion of 10 kDa dextran from two parallel channels in a hydrogel was modeled using the same COMSOL settings described for RA with the experimentally-measured diffusion coefficient as an input, and the simulation was run for 60 minutes. At the 60-minute mark, the dextran concentrations in the gel at 1 mm, 2 mm and 4 mm distances from the right channel (10 kDa FITC dextran) were obtained. In order to experimentally verify the simulation, a parallel perfusion channel hydrogel was cast (using PBS instead of complete media for simplicity). Each parallel channel was then connected to a syringe containing either 1mM of 10 kDa Texas red dextran or 10 kDa FITC dextran. The hydrogel was then perfused at a rate of 300 μl/minute for 60 minutes. A 0.5 mm biopsy punch was used to punch out gel at distances of 1 mm, 2 mm, and 4mm from the right channel (FITC dextran side). The punches were then placed into a sterile 0.45 μm pore centrifuge tube filter (Spin-X Centrifuge Tube Filters, Corning, New York, NY) and spun at 10,000xG to extract the liquid, and the dextran concentrations in the liquid phase were determined using a microplate reader (Synergy H1 Multi-mode Microplate Reader, BioTek) to measure fluorescence using the FITC filter and the Texas red filter.
Validation of temporal control of morphogen delivery
Temporal control of a morphogen gradient was validated using the RARE-RFP-fibroblasts. These RA reporter cells were embedded in hydrogels in the dual port perfusion bioreactor for real-time visualization of the cellular response to the diffusion of RA. First, the left port of the gel was perfused with media containing 2 μM RA, while the right port was perfused with standard growth media. The hydrogel was perfused for 1 hour to allow for RA diffusion before the perfusion reservoir was changed to standard growth media. The RA-responsive GFP expression and the constitutively active RFP expression in the RARE-RFP-fibroblasts were then visualized using confocal microscopy. After 24 hours of perfusion with standard growth media in both channels, the right port was perfused with 2 μM RA in media while the left port received standard growth media. GFP and RFP expression were visualized again after 1 hour of RA diffusion from the right channel. Then, both channels were perfused with 2 μM RA after another 24-hour period of standard media perfusion and confocal microscopy was performed a third time after 1 hour of RA diffusion from both channels. Finally, after 24 hours of perfusion with standard growth media, GFP and RFP were visualized to confirm no GFP was being expressed. Survival of cells located in the entire region between the perfusion channels was verified by perfusing an RFP-fibroblast-laden hydrogel with standard media for 14 days, then staining the cells with Calcein (ThermoFisher) and Sytox blue (ThermoFisher) to identify live and dead cells, respectively. The Calcein-AM and Sytox Blue images were analyzed in ImageJ to assess cell viability ([Calcein-AM count] / [Calcein-AM + Sytox Blue count]) on day 14.
Formation of gradients of MSC differentiation
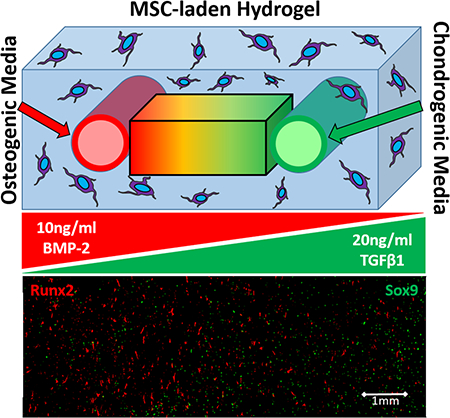
To demonstrate heterogeneous morphogen presentation, one of the two fluidic channels were perfused with osteogenic media and the other was perfused with chondrogenic media. Osteogenic media consisted of high-glucose DMEM, 10 ng/ml BMP-2, 0.1 μM dexamethasone (Sigma Aldrich), 10 mM β-glycerol phosphate (Sigma Aldrich), 0.2 mM ascorbic acid (Sigma Aldrich), 20% FBS (ThermoFisher), and 1% Pen/Strep35 (ThermoFisher)36. Chondrogenic media consisted of high-glucose DMEM with sodium pyruvate (Sigma Aldrich), 10% ITS Premix (ThermoFisher), 0.1 μM dexamethasone, 1 μM ascorbic acid, 4% Proline (Sigma Aldrich), 20 ng/ml or 10 ng/ml TGFβ−1 (Peprotech, Rocky Hill, NJ), 20% FBS, and 1% Pen/Strep37. After the cell laden hydrogels were crosslinked, the perfusion began immediately at a rate of 300 μl/minute. The osteogenic and chondrogenic media in the conical tube reservoirs were replaced with fresh differentiation media every 7 days, and the hydrogels were perfused continuously for 35 days.
On day 35, hydrogels were divided into two pieces by slicing the gel in the center, perpendicular to the two perfusion channels, so that each piece contained both osteogenic and chondrogenic regions. One half of the hydrogel was further subdivided into 5 sections using a razor to make cuts in the same direction as the initial cut (perpendicular to the channels). The intact half of the hydrogel was removed from the PDMS frame and immediately placed in a bath of 4% paraformaldehyde on a rocker for fixation prior to immunofluorescence staining. The 5 thin sections of the other half of the hydrogel were removed from the PDMS frame and a 3 mm biopsy punch was used to collect three samples from each section. The samples were located 1) adjacent to the osteogenic channel, 2) at the midpoint between the two channels, and 3) adjacent to the chondrogenic channel. This resulted in 5 samples for each of the 3 locations, and the samples from a single hydrogel were combined for use in a Western blot (i.e. 5 biopsy samples collected for each location from 10 hydrogels resulted in n=10).
Predefined sections of the hydrogel designated for Western blot were cut and dissolved in Collagenase Type IV (2% W/V) for 1 hour. The resulting dissolved gel-cell mixture was centrifuged at 400*g to pellet the cells. The pellet was washed 3 times with DPBS (without magnesium or calcium) with centrifugation. The pellet was then resuspended in RIPA buffer (Sigma Aldrich) with 1:100 v/v protease and phosphatase inhibitors (Sigma Aldrich) for 5–10 minutes on ice followed by centrifugation at 12,000*g for 15 minutes at 4C. The supernatant was then removed and frozen at −20C for future use.
Protein concentrations were determined using the Pierce BCA Protein Assay Kit (ThermoFisher Scientific). 10 μg of protein were pipetted from each protein sample, and the appropriate amount of RIPA buffer, Laemmli buffer (BioRAD), and betamercapto ethanol (Sigma Aldrich) were added to yield a total volume of 25 μl. These samples were boiled at 95C for 5 minutes, and then cooled on ice. 4–20% Criterion TGX Precast Midi Protein Gels (BioRAD) were pre-run at 70V for 20 minutes in running buffer (Corning) during this boiling and cooling period. Boiled protein samples and appropriate loading ladder were added to the gel and was then run at 80V until the protein samples had run down the majority of the gels. Gels were then transferred to nitrocellulose membranes on an iBlot 2 dry blotting system (ThermoFisher Scientific) according to the manufacturer’s protocol.
The membrane was blocked with TBS-based Odyssey blocking buffer (Li-COR Biosciences) for 30 minutes at room temperature on a shaker plate. Primary antibodies were dissolved in the Odyssey blocking buffer with 0.05% Tween 20 (Sigma Aldrich) at the following concentrations: RUNX2 (1:1000, Abcam ab23981), SOX9 (1:1000, Cell Signaling Technology, 82630S), and GAPDH (1:5000, Cell Signaling Technology, 14C10). Primary antibody incubation was performed at 4C overnight on a shaker plate. Membranes were then washed with TBS (Corning) containing 0.05% Tween 20 (3 washes for 5 minutes each on a shaker plate). Appropriate Li-COR 800CW secondary antibodies were diluted in TBST at 1:15,000, and then incubated on the membranes for 2 hours at room temperature on a shaker plate. Membranes were again washed with TBST (3 washes for 5 minutes each on a shaker plate) before being imaged on a Li-COR Odyssey scanner, and imaging processing was performed in the manufacturer’s software.
After 7 minutes in the paraformaldehyde bath, the half of each hydrogel designated for immunofluorescence was further subdivided into 5 equal sections and washed 3 times with PBS. The slices were then placed in a 5% normal goat serum for blocking and membrane permeabilization. After 1 hour of blocking, 10μl of Runx2 conjugated Alexa fluor 647, Sox9 conjugated Alexa fluor 488, Rhodamine Phalloidin (actin), and Hoechst 33342 (nuclear stain) were added to the blocking solution and allowed to bind overnight where they were then visualized with confocal microscopy (Zeiss LSM 710). Separate slices were immunostained with 10μl of Osteocalcin conjugated Alexa fluor 680, Collagen II conjugated Alexa fluor 488, Rhodamine Phalloidin (actin), and Hoechst 33342 (nuclear stain) and allowed to bind overnight where they were then visualized with confocal microscopy. Confocal z-stacks were acquired for the 9-mm area located between the two perfusion channels in each hydrogel section. The two nuclear markers (Runx2 and Sox9) were used for quantification of cell differentiation fate. Five 1.8-mm regions were defined in 2D projection images of the z-stacks for quantification of differentiation status as a function of location relative to each channel. The images were thresholded using ImageJ software, and the number of positive cells (Runx2 and Sox9) were counted and normalized to the total cell number (Hoechst) in each region. The actin marker provided visualization of cell morphology.
Results and discussion
The process of stem cell differentiation is challenging to model in vitro on a biologically relevant scale with currently available technology. Therefore, we developed a platform in which large, 3D tissue constructs (on the order of centimeters) could be provided with time-varying morphogen gradients over long periods of time (on the order of weeks to months).
Modeling small and large molecule diffusion gradients
After fabricating the bioreactor and hydrogel perfusion system, we sought to develop a predictive model for designing gradients for stem cell differentiation using small and large compounds. To do so, we used RA as a model small biomolecule as its strong Raman signal allows for sensitive, label-free detection with non-destructive imaging in the hydrogel scaffolds38,39. In addition to its strong Raman signal, RA is also relevant for modeling diffusion in our platform because it is an important morphogen in biological development (e.g. spatial organization of the neural tube)1,2. First, confocal Raman microscopy was used to measure RA concentration in the hydrogel over time in order to calculate a diffusion coefficient. We set up our system such that RA could diffuse into the hydrogel from a reservoir as shown in Fig. 3A. Raman measurements collected at a distance of 0.2 cm from the gel/reservoir interface showed increases in the intensity of the RA peak over time38,39 (Fig. 3B). The peak intensity of the RA signal (maximum intensity between 1590–1595 cm−1) as a function of time was fit using the equation for diffusion in semi-infinite media34,40, resulting in a diffusion coefficient of 3.4 ± 0.3 × 10−7 cm2/s (Fig. 3C). Although the diffusion coefficient of RA in gelatin hydrogels has not been previously reported, this result is in the expected range based on published diffusion coefficients for other molecules tested in a collagen film.41 Dodson et al. measured diffusion coefficients of 2.4 to 3.0 × 10−6 cm2/s for various retinoids in a solution of CD3OD and D2O (1:1) and 2.3 × 10−6 cm2/s for all-trans-retinoic acid in a bi-phase assay (agarose matrix and a toluene-based fluid)42. The difference between the diffusion coefficient measured in our study and that previously reported is likely due to differences in hydrogel density and composition (i.e. 10% gelatin vs. 5% agarose).
The diffusion of RA through the hydrogel was then modeled using COMSOL with the Raman-derived diffusion coefficient applied as an input parameter. A representative concentration gradient after 24 hours is shown in Fig. 3D. The resulting values were further validated experimentally by perfusing RA through channels in a gelatin hydrogel for 24 hours and subsequently quantifying the RA concentration at different locations using a microplate reader (Fig. 3.E). The experimental and model data show good agreement, suggesting the COMSOL model is a good predictor of small molecule diffusion.
To model diffusion gradients of larger molecules, hydrogel channels were perfused with 10 kDa dextran conjugated to Texas red and 10 kDa dextran conjugated to FITC. The fluorescence intensity of 10 kDa Texas red dextran in the hydrogel was measured over time at a single location and fit with the diffusion equation (Fig. 4A). Fluorescence intensity measurements collected at a distance of 0.2 cm from the channel showed increases in intensity over time, resulting in a calculated diffusion coefficient of 3.6 ± 0.18 × 10−6 cm2/s. This result closely agrees with previously published diffusion coefficients for 10 kDa dextran43,44. After 60 minutes of perfusion, opposing gradients of both fluorescent molecules were apparent as shown in an image of the entire length between the red and green channels (Fig. 4B) and a plot of the intensity profiles of the two varieties of dextran as a function of distance from the respective channels (Fig. 4C). Although the crossover point for the two profiles is dependent on the relative gain settings for each color channel during imaging, the opposing diffusion gradients are clearly present, and the profiles intersect near the center of the hydrogel.
The diffusion of Texas red-conjugated 10 kDa dextran and FITC-conjugated 10 kDa dextran from two parallel channels in a perfused hydrogel was modeled using COMSOL (Fig. 4D-E). The simulated diffusion gradients were validated experimentally by perfusing a hydrogel for 60 minutes with the two dextran molecules in isolated channels (Fig. 4B) and subsequently quantifying the concentrations of both dextrans at distances of 1 mm, 2 mm, and 4 mm from the right (FITC-perfused) channel (x=8, 7, and 5 mm in COMSOL simulation) with a microplate reader (Fig. 4F). The experimental and model data show good agreement, suggesting the COMSOL model is a good predictor of large molecule diffusion.
Demonstration of temporal control of morphogen delivery
RFP-expressing fibroblasts were transduced with a lentivirus to report RA signaling via GFP expression. These RA reporter cells were embedded in the dual port perfusion bioreactor for real-time, dynamic visualization of cellular response to the diffusion of RA. The cell-laden hydrogel was imaged with confocal microscopy to visualize constitutively active RFP and the reporter GFP expression after 4 conditions: 1) perfusion of the left channel with 2 μM RA for 1 hour, 2) perfusion of the right channel with 2 μM RA for 1 hour, 3) perfusion of both channels with 2 μM RA for 1 hour, and 4) perfusion with standard media for 24 hours. Standard media was perfused for 24 hours between each RA condition. As shown in Fig. 5 A-D, the GFP signal was localized to the region of the gel near the channels(s) perfused with RA. This signal was reversible as shown by conditions 2–4 and that this on/off experiment demonstrates both spatial and temporal control of morphogen delivery to the hydrogel. Because the cells near the center of the hydrogel were not exposed to RA and did not express GFP in this experiment, an additional hydrogel containing RFP-expressing fibroblasts was perfused with standard media for 14 days and stained with Calcein and Sytox blue to confirm cell viability throughout the hydrogel region between the two channels (Fig. 5E). This experiment confirmed that 97% of the cells in the region between the two channels were alive at 14 days.
Figure 5:

Live-cell imaging of the response to RA in RFP-expressing fibroblasts that report RA signaling via GFP expression. The dual perfusion channel, cell-laden hydrogel was imaged to visualize constitutively active RFP and the reporter GFP expression after exposing the device sequentially to the following conditions: (A) perfusion of the left channel with 2 μM RA for 1 hour, (B) perfusion of the right channel with 2 μM RA for 1 hour, (C) perfusion of both channels with 2 μM RA for 1 hour, and (D) perfusion with standard media for 24 hours. Standard media was perfused for 24 hours between each RA condition. The GFP signal was localized to the region of the gel near the channels(s) perfused with RA, and the signal was reversible. (E) To confirm the cells remain viable in the entire region between the perfusion channels, RFP-expressing fibroblasts embedded in a hydrogel perfused with standard media for 14 days were stained with Calcein and Sytox blue (97% live cells).
One difficult aspect of replicating in vivo developmental progression is the inability to control and manipulate temporal changes in morphogen concentrations in large 3D scaffolds. Traditionally, stem cell differentiation protocols have been developed and optimized in 2D cultures. This approach focuses on the direct delivery of soluble morphogens to a monolayer of cells, resulting in a large yield of differentiated cells. However, these results are not indicative of the complex stem cell niche that arises when cells react to the endogenous cues of 3D physical environments, including morphogen gradients, cell-to-scaffold, and cell-to-cell interactions. These concerns have prompted the development of systems to better control morphogen delivery in 3D culture. Our device, as demonstrated using the RARE reporter cells, allows researchers to introduce a morphogen such as RA to the cells through diffusion and monitor real-time responses to morphogen exposure. In addition, this platform enables users to turn off the morphogen signal by simply changing the perfusion media.
Spatially tuned control of MSC fate with morphogen gradients
MSCs were uniformly embedded in 3D hydrogels and the two parallel channels were perfused with osteogenic (containing 10ng/ml BMP-2) or chondrogenic (containing 20 ng/ml TGFβ1) media provided by two separate conical tube reservoirs (Fig. 2A-B). Stem cell differentiation was characterized using immunofluorescence and Western blot after 35 days of perfusion. High-magnification images of nuclear (Runx2 and Sox9) and extracellular matrix (Osteocalcin and Collagen II) markers of osteogenic and chondrogenic differentiated MSCs confirm that both cell fates were achieved in the hydrogels (Fig. 6). Immunofluorescence of Runx2 (a marker of osteocytes) and Sox9 (a marker of chondrocytes) was first used to visualize (Fig. 7A) and quantify (Fig. 7C-D) MSC differentiation in response to each morphogen. The number of Runx2-positive cells (osteocytes, normalized to total cell count (Fig. 7B)) was maximized within a 3.6 mm distance from the channel (Fig. 7C, Regions 1 and 2) and decreased significantly at 3.6–7.2 mm (Regions 3 and 4, p<0.05). The region of the gel furthest from the channel (7.2–9 mm, Region 5) had an additional significant reduction in differentiation of MSCs into osteocytes (Fig. 7C, p<0.05). The Sox9 immunofluorescence showed a steeper gradient (Fig. 7D), with a significant decrease in the percentage of positive cells in Regions 3 and 4 relative to Region 5 (1.8–5.4 mm from channel). The differentiation of cells into chondrocytes further decreased significantly at distances 5.4–7.2 mm and 7.2–9 mm from the channel (Fig. 7D, p<0.05). Protein expression for Runx2 and Sox9 was also confirmed and quantified using Western blot (p<0.05, Fig. 7E-F) and showed significant decreases in protein expression at each 3 mm increment in distance from the channel perfused with morphogens.
Figure 6:

Osteogenic and chondrogenic MSC fates were examined using immunofluorescence after 35 days of hydrogel perfusion with differentiation media. Cells in the osteogenic region expressed Runx2 in the nucleus and osteocalcin in the extracellular matrix. Cells in the chondrogenic regions expressed Sox9 in the nucleus and Collagen II in the extracellular matrix. Hoechst and actin stains were used to visualize cell nuclei and cell morphology, respectively.
Figure 7:

Opposing gradients of osteogenic and chondrogenic differentiation can be generated by delivery of soluble morphogens localized to isolated channels. (A) MSCs demonstrated gradient patterns of differentiation with respect to osteogenic and chondrogenic media. This representative immunofluorescence image shows spatially opposing gradients of osteogenic and chondrogenic differentiation of MSCs, with the highest level of Runx2 expression (red, marker of osteogenic cells) located near the osteogenic media channel (region 1) and more prominent Sox9 expression (green, marker of chondrogenic cells) located closer to the chondrogenic media channel (region 5). (B) A Hoechst nuclear counterstain was used to visualize the whole-cell population for normalization of the Runx2 and Sox9 positive cell counts. (C) Osteogenic differentiation induced by 10 ng/ml BMP-2 had the highest immunofluorescence expression of the differentiation protein Runx2 in region 1 and significantly decreased as distance increased from the source channel (regions 2–5). (D) Similarly, chondrogenic differentiation induced by 20ng/mL of TGFβ1 had the highest immunofluorescence expression of the differentiation protein Sox9 in the region closest to the chondrogenic media channel (region 5) and significantly decreased further away from the source channel (regions 1–4). (C-D) The immunofluorescence results were further validated by protein expression measured by Western blot. The highest expression of Runx2 (E) occurred nearest to the osteogenic media channel (region A), and significantly decreased as distance increased (regions B and C). The highest expression of Sox9 (F) was measured nearest to the chondrogenic media channel (region C), and significantly decreased further away from the source channel (regions A and B). *p<0.05, n=10 hydrogels.
Together, these immunofluorescence and Western blot results confirm that spatial gradients of stem cell differentiation were achieved, resulting in both bone and cartilage regions within a single, 3D hydrogel. The differentiation of MSCs into bone and cartilage using these morphogens has been well-characterized in previous 2D and 3D studies35,45,46. Thus, we used these differentiation protocols as a proof-of-concept validation of our stem cell differentiation platform and observed the expected behavior of MSC differentiation. Additionally, we addressed an aspect of in vitro stem cell differentiation that has been underdeveloped: the ability to control morphogen gradients presented to cells for extended periods of time in a 3D construct.
Previous microfluidic platforms have been designed for tight control of the spatial presentation of morphogens through simple Fickian diffusion principles,2,21,47,48 over relatively short culture times and in small volumes. In the current work, we explored the ability to further tune the MSC differentiation gradient over a 35-day period by modulating the morphogen concentration in the chondrogenic differentiation media (10 ng/mL TGFβ1 compared to 20 ng/mL TGFβ1, Fig. 8). The lower concentration of TGFβ1 produced a steeper gradient of Sox9 expression as shown by both immunofluorescence (Fig. 8A) and Western blot (Fig. 8B). While the percentage of positive cells was equivalent for both concentrations of TGFβ1 within the first 1.8 mm region next to the channel as detected with immunofluorescence of Sox9, perfusion with the lower morphogen concentration resulted in significantly fewer differentiated cells within each of the other 4 regions relative to the higher TGFβ1 concentration (†p<0.001, Fig. 8A). Western blot analysis showed that protein expression differed significantly between the two concentrations of TGFβ1 within all three 3-mm regions (*p<0.05, Fig. 8B). These results indicate that the spectrum of differentiation states can be further modulated by varying the concentration of a morphogen provided to the hydrogel via the perfusion channel17,22,23,49. Because our platform is designed with external reservoirs that can be replenished or replaced, and perfusion circuits that could employ valves to automatically switch between reservoirs, we have an additional level of control for designing complex morphogen presentation gradients in space and time.
Figure 8:

MSC differentiation gradients generated by TGFβ1 can be tuned based on source channel concentration. (A) Quantitative analysis of Sox9 immunofluorescence reveals a decrease in the percentage of differentiated cells when the concentration of TGFβ1 is decreased from 20ng/mL (solid bars) is decreased to 10ng/mL (striped bars). Regions 1–4 showed a statistical difference (†p<0.001) between the 20ng/mL and 10ng/mL TGFβ1 conditions. (B) Quantitative analysis of protein expression of Sox9 showed a decrease in chondrogenic differentiation with a decrease in TGFβ1 concentration from 20ng/mL (solid bars) to 10ng/mL (striped bars). Regions A-C showed a statistical difference (*p<0.05) between the two TGFβ1 conditions. n=10 hydrogels per TGFβ1 concentration.
As stem cell studies increase in complexity, so must the versatility of the platforms needed to perform experiments. Several novel approaches have been developed to study the precise mechanisms by which individual components regulate stem cell differentiation in 3D. For example, vertebral neural development50, kidney development51, liver development52, and cardiac development53 have been studied using 3D, multicellular masses known as organoids54 to mimic in vivo architectures and complex functions. These structures can grow to millimeters in size over the course of months and exhibit layered differentiation. However, to achieve proper organization, organoids derived from stem cells require complex, region-specific dynamic cues to differentiate into a multifaceted, functional organoid. One approach to growing more complex organoids is to manually fuse separate, differentiated organoids such as individual brain regions55; however, human intervention may impart heterogeneity and fusion of more than two regions remains challenging. Our perfused scaffold-based platform represents an alternative approach for generating large, complex architectures, as it allows for differentiation of stem cells into multiple tissue types within a single, large-scale hydrogel without the need for joining separate constructs. However, our large-scale scaffold still presents finite boundaries to the cells, which is a common limitation for in vitro models of developmental processes, regardless of the size of the construct. An advantage of using a large, 3D hydrogel is the ability to dissect and analyze spatially discrete regions using several common readouts (e.g. immunofluorescence and Western blot) that would be difficult to obtain in microfluidic system. Another limitation in this study is the absence of cellular metabolic activity and consumption of morphogens in the COMSOL model. While simulated diffusion gradients at early timepoints can be validated experimentally, the model does not account for the complex cellular binding and uptake of morphogens which is beyond the scope of the current work. Together with the diffusion model, our hydrogel perfusion platform, with external reservoirs that can be turned on/off or replenished as desired, would allow users to predict and tune spatial and temporal morphogen gradients to achieve control over a targeted range of stem cell lineages/commitments localized to specific regions of a scaffold.
Conclusion
Recreation of the spatial and temporal morphogen signaling gradients that occur in nearly every organ system in the body remains a significant, unmet need for disease modeling and regenerative medicine research. Currently available strategies for directing stem cell differentiation in vitro have advanced our knowledge of stem cell biology, but recapitulation of developmental patterning events has been limited by the longevity of differentiation experiments and a lack of dynamic control over morphogen gradients in space and time. In this work, we developed a perfusion bioreactor platform for simultaneous differentiation of stem cells to multiple fates in a 3D scaffold and demonstrated dynamic, temporal control of morphogen delivery. These results establish this perfused hydrogel system and the accompanying computational diffusion model as a new toolset for the design of complex differentiation protocols in both basic science and translational research.
Acknowledgments
This research was funded and supported by NSF BMAT 1506717 (LMB), NIH R00EB013630 (LMB) and a NARSAD Young Investigator Award from the Brain and Behavior Research Foundation (ESL). EKN was supported through NSF Graduate Research Fellowship DGE-1445197. YCO and RB were supported through CMMI-1634856. DAB was partially by NIH T32GM007347.
References
- 1.Sadler TW, in American Journal of Medical Genetics - Seminars in Medical Genetics, 2005, vol. 135 C, pp. 2–8. [DOI] [PubMed] [Google Scholar]
- 2.Demers CJ, Soundararajan P, Chennampally P, Cox GA, Briscoe J, Collins SD and Smith RL, Development, 2016, 143, 1884–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kinney MA and McDevitt TC, Trends Biotechnol, 2013, 31, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckingham M, Meilhac S and Zaffran S, Nat. Rev. Genet, 2005, 6, 826–835. [DOI] [PubMed] [Google Scholar]
- 5.Birket MJ, Ribeiro MC, Verkerk AO, Ward D, Leitoguinho AR, Den Hartogh SC, Orlova VV, Devalla HD, Schwach V, Bellin M, Passier R and Mummery CL, Nat. Biotechnol, 2015, 33, 970–979. [DOI] [PubMed] [Google Scholar]
- 6.Schneider MD, Cell Stem Cell, 2017, 21, 151–152. [DOI] [PubMed] [Google Scholar]
- 7.Beaulieu J-F, Cell Sci J, 1992, 102, 427–36. [DOI] [PubMed] [Google Scholar]
- 8.Barker N, Nat. Rev. Mol. Cell Biol, 2014, 15, 19–33. [DOI] [PubMed] [Google Scholar]
- 9.Lane SW, Williams DA and Watt FM, Nat. Biotechnol, 2014, 32, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lippmann ES, williams CE, Ruhl DA, Estevez-Silva MC, Chapman ER, Coon JJ and Ashton RS, Stem Cell Reports, 2015, 4, 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gouti M, Tsakiridis A, Wymeersch FJ, Huang Y, Kleinjung J, Wilson V and Briscoe J, PLoS Biol,, DOI: 10.1371/journal.pbio.1001937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer TN, Schwesinger C, Bush KT, Stuart RO, Rose DW, Shah MM, Vaughn DA, Steer DL and Nigam SK, Dev. Biol, 2004, 275, 44–67. [DOI] [PubMed] [Google Scholar]
- 13.Dhawan U, Pan HA, Shie MJ, Chu YH, Huang GS, Chen PC and Chen WL, Nanoscale Res. Lett, 2017, 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barati D, Shariati SRP, Moeinzadeh S, Melero-Martin JM, Khademhosseini A and Jabbari E, J. Control. Release, 2016, 223, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown TE and Anseth KS, Chem. Soc. Rev, 2017, 46, 6532–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thakur R, Zhang Y, Amin A and Wereley S, Microfluid. Nanofluidics, 2015, 18, 1425–1431. [Google Scholar]
- 17.Wang X, Wenk E, Zhang X, Meinel L, Vunjak-Novakovic G and Kaplan DL, J. Control. Release, 2009, 134, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen FM, Zhang M and Wu ZF, Biomaterials, 2010, 31, 6279–6308. [DOI] [PubMed] [Google Scholar]
- 19.Vo TN, Kasper FK and Mikos AG, Adv. Drug Deliv. Rev, 2012, 64, 1292–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sant S, Hancock MJ, Donnelly JP, Iyer D and Khademhosseini A, Can. J. Chem. Eng, 2010, 88, 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Der Meer AD and Van Den Berg A, Integr. Biol, 2012, 4, 461–470. [DOI] [PubMed] [Google Scholar]
- 22.Cosson S and Lutolf MP, Sci. Rep, 2014, 4, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakaguchi K, Shimizu T, Horaguchi S, Sekine H, Yamato M, Umezu M and Okano T, Sci. Rep,, DOI: 10.1038/srep01316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu M-H, Huang S-B and Lee G-B, Lab Chip, 2010, 10, 939. [DOI] [PubMed] [Google Scholar]
- 25.van der Helm MW, van der Meer AD, Eijkel JCT, van den Berg A and Segerink LI, Tissue Barriers, 2016, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S and Takayama S, Kidney Res. Clin. Pract, 2015, 34, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uzel SGM, Amadi OC, Pearl TM, Lee RT, So PTC and Kamm RD, Small,, DOI: 10.1002/smll.201501905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cunningham TJ and Duester G, Nat. Rev. Mol. Cell Biol, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uzkudun M, Marcon L and Sharpe J, Mol. Syst. Biol,, DOI: 10.15252/msb.20145882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rankin SA, Han L, McCracken KW, Kenny AP, Anglin CT, Grigg EA, Crawford CM, Wells JM, Shannon JM and Zorn AM, Cell Rep,, DOI: 10.1016/j.celrep.2016.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rankin SA, McCracken KW, Luedeke DM, Han L, Wells JM, Shannon JM and Zorn AM, Dev. Biol,, DOI: 10.1016/j.ydbio.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Grady B, Wang J, Faley S, Balikov D, Lippmann E and Bellan LM, SLAS Technol, 2018, 247263031877505. [DOI] [PubMed] [Google Scholar]
- 33.Crank J, in The Mathematics of Diffusion, 1975, pp. 1–10. [Google Scholar]
- 34.Kwak S and Lafleur M, Appl. Spectrosc, 2003, 57, 768–773. [DOI] [PubMed] [Google Scholar]
- 35.Jaiswal N, Haynesworth SE, Caplan AI and Bruder SP, J. Cell. Biochem, 1997, 64, 295–312. [PubMed] [Google Scholar]
- 36.Yang J, Zhang YS, Yue K and Khademhosseini A, Acta Biomater, 2017, 57, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barry F, Boynton RE, Liu B and Murphy JM, Exp. Cell Res, 2001, 268, 189–200. [DOI] [PubMed] [Google Scholar]
- 38.Freudiger CW, Min W, Saar BG, Lu S, Holtom GR, He C, Tsai JC, Kang JX and Xie XS, Science (80-. )., 2008, 322, 1857–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freudiger CW, Min W, Saar BG and Xie XS, Opt. Photonics News, 2009, 20, 30. [Google Scholar]
- 40.Westwater JW and Drickamer HG, J. Am. Chem. Soc, 1957, 79, 1267–1268. [Google Scholar]
- 41.Ho HO, Lin CW and Sheu MT, J. Control. Release, 2001, 77, 97–105. [DOI] [PubMed] [Google Scholar]
- 42.Dodson CS, Peresypkin AV, Rengarajan K, Wu SX and Nickerson JM, Curr. Eye Res, 2002, 24, 66–74. [DOI] [PubMed] [Google Scholar]
- 43.Al-Baradi AM, Mears M, Jones RAL and Geoghegan M, J. Polym. Sci. Part B Polym. Phys,, DOI: 10.1002/polb.23120. [DOI] [Google Scholar]
- 44.Kihara T, Ito J and Miyake J, PLoS One,, DOI: 10.1371/journal.pone.0082382. [DOI] [Google Scholar]
- 45.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S and Marshak DR, Science (80-. )., 1999, 284, 143–147. [DOI] [PubMed] [Google Scholar]
- 46.Mackay AM, Beck SC, Murphy JM, Barry FP, Chichester CO and Pittenger MF, Tissue Eng, 1998, 4, 415–428. [DOI] [PubMed] [Google Scholar]
- 47.Smith RL, Demers CJ and Collins SD, Microfluid. Nanofluidics, 2010, 9, 613–622. [Google Scholar]
- 48.Esch EW, Bahinski A and Huh D, Nat. Rev. Drug Discov, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hübner J, Lindner M, Drewell C, Bauer S, Thomas A, Sambo NS, Sonntag F, Lauster R and Marx U, Lab Chip, 2015, 15, 2688–2699. [DOI] [PubMed] [Google Scholar]
- 50.Kelava I and Lancaster MA, Dev. Biol, 2016, 420, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morizane R and Bonventre JV, Trends Mol. Med, 2017, 23, 246–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huch M, Gehart H, Van Boxtel R, Hamer K, Blokzijl F, Verstegen MMA, Ellis E, Van Wenum M, Fuchs SA, De Ligt J, Van De Wetering M, Sasaki N, Boers SJ, Kemperman H, De Jonge J, Ijzermans JNM, Nieuwenhuis EES, Hoekstra R, Strom S, Vries RRG, Van Der Laan LJW, Cuppen E and Clevers H, Cell, 2015, 160, 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoang P, Wang J, Conklin BR, Healy KE and Ma Z, Nat. Protoc, 2018, 13, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murrow LM, Weber RJ and Gartner ZJ, Development, 2017, 144, 998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bagley JA, Reumann D, Bian S, Lévi-Strauss J and Knoblich JA, Nat. Methods, 2017, 14, 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paguirigan AL and Beebe DJ, BioEssays, 2008, 30, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kshitiz D. H. Kim, Beebe DJ and Levchenko A, Trends Biotechnol, 2011, 29, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]