

Abstract
Follicular fluid (FF), which is the fluid that envelops the developing oocyte (egg cell) in the ovary, can be analyzed to assess trace element content as well as to determine potential exposure to toxic elements in women seeking in vitro fertilization (IVF) treatment. Such measurements may be useful in establishing associations with potential adverse effects on oocyte viability and subsequent pregnancy outcomes. The principal goal of this study was to leverage the next generation of inorganic mass spectrometry based on ICP-MS/MS to address the numerous analytical challenges of (ultra-)trace element analysis of human FF specimens. Ultra-trace element measurements are defined by the Clinical Laboratory Standards Institute as fluid concentrations below 10 μg L−1 or tissue mass fractions below 1 μg g−1. Stringent pre-analytical procedures were developed to minimize exogenous contamination during FF specimen collection and storage in a prospective study of 56 women seeking IVF treatment. ICP-MS/MS instrumental parameters were carefully optimized, and the method validated for 11 biologically important elements that included 4 at trace levels (Cu, Se, Sr, and Zn) and 7 at ultra-trace levels (As, Cd, Co, Mo, Mn, Hg, and Pb). Method limits of detection (LODs) for ultra-trace elements varied from 5.6 ng L−1 for Cd to 0.11 μg L−1 for Mo. A total of 197 human FF specimens were analyzed using the proposed ICP-MS/MS method with 84% of specimens detectable for Pb and 100% detectable for Co, Cu, Mn, Mo, Sr, and Zn. The method based on ICP-MS/MS was compared to a previous method developed for FF using SF-ICP-MS.
Keywords: human follicular fluid, trace elements, ultra-trace elements, contamination, QQQ-ICP-MS, ICP-MS/MS
Graphical Abstract
1. Introduction
Human follicular fluid (FF) represents the developing oocyte’s (i.e., the egg cell’s) first exposure to environmental contaminants, as well as being its source of essential nutrients. Studying these early environmental exposures became more practical with the introduction of in vitro fertilization (IVF) techniques, beginning in the 1970s. Human FF is a blood-plasma ultrafiltrate that envelops oocytes in the ovarian follicle and which requires an invasive procedure to collect. During the IVF treatment, oocytes (and the surrounding FF) are obtained via transvaginal needle – the FF is separated from the oocyte(s) during processing, at which time it can be harvested for research studies. As such, investigations of FF have increased as IVF has become more common.
The first investigation into the physiological origin of human FF by characterizing its elemental composition appeared more than forty years ago – wherein Na and K (in addition to other parameters) were measured in FF samples obtained from women undergoing hysterectomies.1 In the subsequent two decades after this initial report, four other investigations expanded the panel of essential elements measured to include Ca, P, S, Mg, Cu, Zn, Br and Se.2–5 The potential use of FF in biomonitoring studies was first described in 1995 when Zenzes et al. reported that Cd in FF samples was higher in smokers compared to non-smokers.6 Since then, twelve additional reports describe analyzing human FF for up to 24 element(s), principally the key toxic metals (e.g., Pb, Cd) and/or essential elements (e.g. Se, Zn).7-18 Three previous studies reported that Cu, Se, and/or Zn in FF were associated with improved pregnancy rates among study participants.16-18 Conversely, toxic elements, especially Cd and Pb, in FF may adversely affect oocyte fertilization,19 and lead to lower pregnancy rates.10-15 Interestingly, some studies have suggested Cd levels in FF are associated with higher pregnancy rates among women undergoing IVF treatment.9-11
The analysis of human FF for (ultra-)trace element content presents a major analytical challenge. The distinction between trace and ultra-trace analyses for clinical samples is defined by the Clinical Laboratory Standards Institute (CLSI) as fluid concentrations between 10 μg L−1 and 100 μg L−1 (or 0.1 to 1 μg g−1 for tissues) for the former, and below 10 μg L−1 (or < 0.1 μg g−1 for tissues) for the latter.20 With concentrations expected below 1 μg L-1 for As, Cd, Hg, and Pb,14 stringent contamination control is critical to achieving accurate and reliable results for these key toxic elements. Minimizing background contamination is mandatory to achieving the low limits of detection (LODs) that are necessary for this analysis. It is important to reduce and resolve sources of inaccuracy and imprecision that may result from spectral interferences or signal enhancement/suppression from the FF matrix and from drift over long analytical runs. While sources of bias may be minor for ‘trace’ analysis they may play a much larger role at the ultra-trace level. Accuracy assessment and method validation for FF is further diminished by the lack of standard/certified reference materials appropriate for this specific biological fluid.
Despite interest in using FF samples for biomonitoring of women undergoing IVF and their potential offspring, there is a gap in the literature to address many of the analytical challenges described. Of the eighteen studies that reported trace or ultra-trace element measurements in human FF: only eight reported an LOD or a limit of quantitation (LOQ),6, 7,9,11,14-17 plus another that noted, “concentrations <0.2 ppb were considered below the detection limit”;12 three described using FF sample recovery spikes to assess accuracy, 6, 11,14 one also utilized serum- and aqueous-based SRMs and RMs to establish accuracy;14 and another reported data for between-run repeatability.6 A previous report from our laboratory proposed a method to determine 24 trace elements in human FF using sector field (SF-)ICP-MS with a simple dilution procedure.14 Method development for that work included the preparation of a FF-based QC material, as well as a study of sample preparation procedures, investigations of aqueous vs FF-based calibration, various front-end configurations, a comparison of Q-ICP-MS with SF-ICP-MS, and validation against aqueous-based SRMs and archived serum proficiency testing materials.
The present study expands further on the analytical challenges and limitations with trace and ultra-trace element analysis of human FF by leveraging new, state-of-the-art ICP-MS/MS (i.e., QQQ) technology, comparing it to our previous SF-ICP-MS method. We also address the issue of pre-analytical contamination in more detail; and we expand on subtle matrix effects in inorganic mass spectrometry by comparing human FF and serum matrices. In contrast to our previous study, we focus here on the four key toxic elements As, Cd, Hg, and Pb; the essential elements Co, Cu, Mn, Mo, Se, and Zn; and Sr, which is potentially therapeutic.
Experimental
2.1. Instrumentation and equipment
An Agilent 8900 ICP-MS/MS (Agilent Technologies, Santa Clara, CA USA) equipped with an octopole reaction system (ORS) and axial acceleration technology was used for this study. An ultra-low particulate arrester (UPLA) air filter (Elemental Scientific, Omaha, NE USA) was installed on an SPS4 autosampler (Agilent Technologies, Santa Clara, CA USA) to minimize potential airborne contamination during analysis. Universal instrumental settings, ORS gas settings, and element-specific information is summarized in Table 1. The energy discrimination, deflect lens, plate bias, gas flow rates, ORS bias, and axial acceleration voltages were optimized to maximize signal intensity and reaction efficiency (for reaction modes) and minimize polyatomic interferences. The settings for the two gas modes used in the final method are shown in Table 1. Other lens voltages were optimized daily using Agilent’s ICP-MS tuning solution (Agilent Technologies, Santa Clara, CA USA).
Table 1:
Agilent 8900 instrumental conditions.
| Instrumental Settings | Octopole Reaction System Settings | |||
|---|---|---|---|---|
| RF Power | 1550 V | Gas | He | O2 |
| Carrier Gas | 1.05 L min−1 | Gas Flow Rate | 10 mL min−1 | 0.5 mL min−1 |
| Sample Uptake Rate | 0.4 mL min−1 | Energy Discrimination | 5.0 V | −7.0 V |
| Spray Chamber | 2°C | Axial Acceleration | 1.0 V | 1.5 V |
| Lens Configuration | x-lens | Octopole Bias | −100.0 V | −5.0 V |
| Replicates (sweeps/replicate) | 3 (100) | |||
| Settings for elements measured using O2 | ||||
| Analyte | Q1 m/z | Q2 m/z | Integration Time (s) |
Internal Standard |
| Mn | 55 | 55 | 1 | Ga |
| Co | 59 | 59 | 1 | Ga |
| As | 75 | 91 | 1 | Rh* |
| Mo | 98 | 130 | 1 | Y |
| Cd | 114 | 114 | 5 | Rh |
| Hg | 202 | 202 | 2 | Ir |
| Pb | ∑206, 207, 208 | ∑206, 207, 208 | 5 | Ir |
| Internal Standard | ||||
| Ga | 71 | 71 | 0.5 | |
| Y | 89 | 105 | 0.5 | |
| Rh | 103 | 103 | 0.5 | |
| Rh | 103 | 119* | 0.5 | |
| Ir | 193 | 193 | 0.5 | |
| Settings for elements measured using High-Energy He | ||||
| Analyte | Q1 m/z | Q2 m/z | Integration Time (s) |
Internal Standard |
| Cu | 65 | 65 | 0.5 | Ga |
| Zn | 66 | 66 | 0.5 | Rh |
| Se | 78 | 78 | 0.5 | Y |
| Sr | 88 | 88 | 0.5 | Y |
| Internal Standard | ||||
| Ga | 71 | 71 | 0.5 | |
| Y | 89 | 89 | 0.5 | |
| Rh | 103 | 103 | 0.5 | |
Rh monitored as RhO at m/z = 119, i.e., using a mass shift in O2 mode.
2.2. Reagents and Solutions
A multi-element stock solution containing As, Cd, Co, Cu, Mn, Mo, Pb, Se, Sr, and Zn was prepared from single-element solutions (High Purity Standards, Charleston, SC USA) and used for the duration of this study. An intermediate standard was prepared monthly by diluting the multi-element stock and a separate Hg (High Purity Standards, Charleston, SC USA) standard 1+99. Five working standards were prepared weekly from the intermediate stock solution by diluting appropriately in 0.5% (v/v) HNO3. Standard 1 contained the following concentrations of elements: 0.15 μg L−1 Mn, 0.03 μg L−1 Co, 125 μg L−1 Cu, 100 μg L−1 Zn, 0.15 μg L−1 As, 15 μg L−1 Se, 8 μg L−1 Sr, 0.25 μg L−1 Mo, 10 ng L−1 Cd, 0.08 μg L−1 Hg, and 0.1 μg L−1 Pb; each subsequent standard doubled in concentration. Following the highest standard, a check blank was included to assess carry-over issues, especially for Hg. A multi-element spike solution was also prepared weekly from the single-element stock solutions in 0.5% (v/v) HNO3; it contained 2.5 μg L−1 Mn, 0.5 μg L−1 Co, 1000 μg L−1 Cu, 500 μg L−1 Zn, 2.7 μg L−1 As, 150 μg L−1 Se, 75 μg L−1 Sr, 3.5 μg L−1 Mo, 150 ng L−1 Cd, 1.6 μg L−1 Hg, and 1.0 μg L−1 Pb.
Standard and spike solutions were prepared and stored in 100-mL Nalgene polypropylene volumetric flasks (Thermo Fisher Scientific, Rochester, NY). Instrumental rinse solution and diluent were prepared and stored in acid-washed 2-L fluorinated ethylene propylene containers (GFS Chemicals, Inc., Columbus, OH, USA); both contained 0.5% (v/v) HNO3, 1,000 μg L−1 Au (Inorganic Ventures, Christiansburg, VA), and 0.005% (v/v) Triton X-100 (Sigma Aldrich Co., St. Louis, MO USA,); 0.5 μg L−1 Ir, Y, Rh, and Ga were added to the diluent solution as internal standards (single elements stocks, High Purity Standards, Charleston, SC USA). All HNO3 used in this work was double distilled in-house from reagent grade acid using a DuoPUR Sub-boiling point Distillation System (Milestone Inc., Shelton, CT). Double de-ionized (DDI) (>18 MΩ·cm) water was produced using a NANOpure DIamond UV/UF water system (Barnstead International, Dubuque, IA).
Pooled human serum (~500 mL) was obtained from ZenBio, Inc. (Research Triangle Park, NC) and used for initial method development studies. Two QC materials were prepared from pooled FF, derived from approximately 80 de-identified 2-mL aliquots that were obtained from the University of California at San Francisco (UCSF) Center for Reproductive Health and shipped on dry ice to the Trace Elements Laboratory at the Wadsworth Center, New York State Department of Health (NYS DOH). Both pools were filtered through cheesecloth (Majestic Chef, Austin, TX) to remove suspended particulate matter. A low-level pool (QC 1) was used for matrix-matching calibration standards and also analyzed as a QC sample at the start of each run; the high-level pool (QC 2) was supplemented with Co, Cu, Zn, As, Se, Sr, Mo, Cd, Hg, and Pb and analyzed at least twice per run (start and end). The FF QC materials were also characterized via standard additions over multiple (n>8) independent runs and evaluated over the course of the study against a matrix-matched calibration curve.
2.3. Contamination control
2.3.a. Screening and certifying collection supplies used in the IVF clinic.
The prospective nature of this study facilitated the implementation of protocols designed to minimize field contamination of trace elements during FF sample collection and processing at the IVF clinic. However, some compromises were necessary – the primary goal of specimen collection and processing in the IVF clinic is to obtain viable oocytes and the research component is secondary by necessity. The established practice of acid-washing all supplies used for ultra-trace element analysis conflicts with need to maintain sterile conditions during specimen collection and processing. In this work, supplies that were deemed critical to ensuring the oocyte’s viability (e.g., butterfly needles) were pre-screened according to an in-house protocol consistent with one used by the Center for Disease Control and Prevention (CDC) for the National Health and Nutrition Examination Survey (NHANES).21 Other supplies (e.g., FF storage containers) were acid-washed by the laboratory and sent to the IVF clinic for use.
2.3.b. Screening and certifying analytical supplies/reagents.
A custom screening protocol was developed for all laboratory supplies and reagents and is briefly described here. Prior to the first use for this study, all containers were acid-washed and then filled to volume with 0.5% (v/v) HNO3, which was analyzed directly from the container. Once solutions/reagents were consumed or reached their expiration date, the containers were acid-washed using 2% (v/v) HNO3 for at least 24 hours, rinsed thoroughly in DDI H2O, and air-dried in a class 100 clean room (Terra Universal Inc., Fullerton, CA USA). Rinse, diluent, and 0.5% (v/v) HNO3 (working calibration blank) were analyzed in triplicate prior to preparing samples; the resulting signals (in cps) were monitored closely as part of the QA/QC plan for this project. Internal standards and other stock solutions were analyzed individually prior to being placed into use. Autosampler tubes were prepared as reagent blanks (150 μL DDI H20; 150 μL 0.5% HNO3; 3.4 mL diluent), allowed to sit overnight and then analyzed. Pipet tips (Axygen Inc. Union City, CA USA; Thermo Fisher Scientific, Vantaa, Finland) were also screened on a lot-to-lot basis.
Autosampler tubes examined in this study include Greiner 15-mL centrifuge tubes (Greiner Bio-One, Frickenhausen, Germany), Falcon 15-mL centrifuge tubes (Corning Inc., Reynosa, Tamaulipas, Mexico), Sarstedt 13-mL centrifuge tubes (Sarstedt Inc., Newton, NC USA), Nalgene 5-mL Cryovials (Thermo Scientific, Rochester, NY USA), and VWR Metal Free 15-mL centrifuge tubes (VWR International, Radnor, PA USA).
2.4. Sample Collection and Preparation
After undergoing a baseline evaluation, female study subjects were treated with an antagonist, flare, or Lupron down regulated ovarian hyperstimulation protocol according to the UCSF Center for Reproductive Health clinical guidelines. Ultrasounds and daily serum estradiol measures were performed regularly during a two-week period to assess and confirm uterine lining and ovarian follicle maturation. After the lead follicle reached 17mm or larger in diameter, with the majority >13 mm, human chorionic gonadotropin was administered subcutaneously for follicle maturation and oocyte retrieval was performed 36 hours later. A transvaginal ultrasound-guided oocyte retrieval was performed using a 17 gauge (1.4 × 350 mm with 900 mm tubing) oocyte aspiration needle (Vitrolife Sweden AB, Vastra Frolunda, Sweden), via a needle guide mounted on the transvaginal ultrasound probe. Between 3.5 and 5.0 mL of FF was aspirated from the two largest follicles in each ovary, for a maximum of four specimens per subject. Each follicle was evacuated completely. The aspiration needle was rinsed with culture media and the clinician ensured that the aspiration tubing was completely empty between follicle collections. FF specimens were individually labelled, tracked, and centrifuged for 10 minutes at 1,500 x g before being aliquoted into 1.8-mL polypropylene cryovials, and stored at −80°C until shipment to the laboratory at Wadsworth. Additional FF routinely collected during aspiration procedures at UCSF was pooled before being processed and stored at −80°C until shipment.
Analysis of these de-identified human FF specimens for trace elements was approved by the NYS DOH Institutional Review Board (IRB) (Study #15–019). FF samples from study participants were shipped on dry ice to the Trace Elements Laboratory at the Wadsworth Center, NYS DOH. Upon receipt, samples were visually examined and classified according to a previously established protocol 22 into one of five categories: clean, lightly cloudy, cloudy, lightly blood contaminated or blood contaminated. Samples were stored at −80°C pending analysis. The FF samples and QC materials were removed from the freezer on the day of analysis and mixed thoroughly on a rocker (Boekel Scientific, Feasterville, PA USA) for 1–2 hours. Samples, standards, and QC materials were diluted 1 + 24 into the autosampler tube for analysis by ICP-MS/MS. All ICP-MS/MS data were processed using the Agilent MassHunter software with internal standardization, except in Figures 1 and 3b where the raw signals are shown for the purpose of visualization. Prism (7.0, GraphPad Software) was used for all graphs and statistical analyses.
Figure 1.
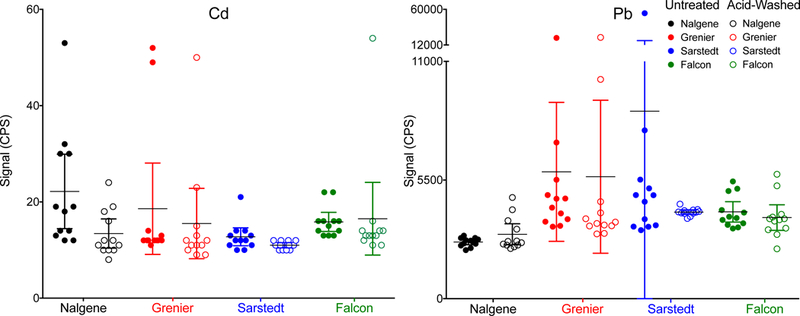
ICP-MS/MS signal (cps) for Cd and Pb in untreated (filled circles) and acid-washed (open circles) autosampler tubes from four manufactures: Nalgene (black), Grenier (red), Sarstedt (blue), and Falcon (green). For each group, the individual data points are shown along with the mean ± 95% CI.
Figure 3.
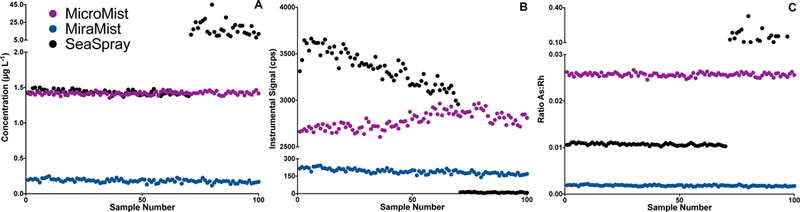
Within-run signal stability for As in a base serum sample injected 100 times (~22 hours) using three nebulizers: MicoMist (purple), MiraMist (blue), and SeaSpray (black). The stability is expressed in panels A, B, and C as As concentration, instrument signal (cps), and the ratio of As:Rh (analyte cps to internal standard cps).
3. Results and Discussion
3.1. Pre-analytical issues and contamination studies
Initially, four brands of autosampler tubes were evaluated: Greiner, Falcon, Sarstedt, and Nalgene. For each of the four brands, acid-washed tubes were compared with untreated tubes. Each experimental group consisted of 12 autosampler tubes, selected at random from each lot, which were prepared as described previously (section 2.3.b) and analyzed in a randomized order on the ICP-MS/MS. Of the 11 elements reported in these FF samples, contamination issues were particularly troublesome for Pb and Cd; thus these two elements were the focus of efforts to minimize background levels. Background contamination data, expressed as the signal in counts per second (cps), in the four tube brands (acid-washed and untreated) evaluated are shown for Cd and Pb in Figure 1. Acid-washing reduced Cd contamination in the Nalgene, Grenier, and Sarstedt tubes, with the latter achieving the lowest background levels. The effectiveness of acid-washing on Pb for the Grenier and Falcon tubes was unclear while acid-washing Nalgene tubes appeared to make Pb contamination slightly worse, which was counter intuitive.
The mixed results obtained by acid-washing four brands of autosampler vials in the initial experiment prompted a second study comparing acid-washed Sarstedt tubes with VWR “Metal Free” Centrifuge Tubes (VWR tubes). In this second study, untreated and acid-washed VWR tubes were evaluated (n=20) with special focus on Pb and Cd contamination (Figure 2). The average background concentration of Cd and Pb in the untreated VWR tubes was 2 ng L−1 and 0.6 μg L−1, respectively after leaching in 3.7 mL of reagent blank. Acid-washing the VWR tubes resulted in reductions of 0.5 μg L−1 and 4 ng L−1 for Pb and Cd, respectively. Of the 20 acid-washed VWR tubes, three had measurable amounts of Pb similar to that found in the untreated group, indicating that acid-washing was less than 100% effective. Based on these data, acid-washed Sarstedt tubes were selected as fit for our purposes.
Figure 2.
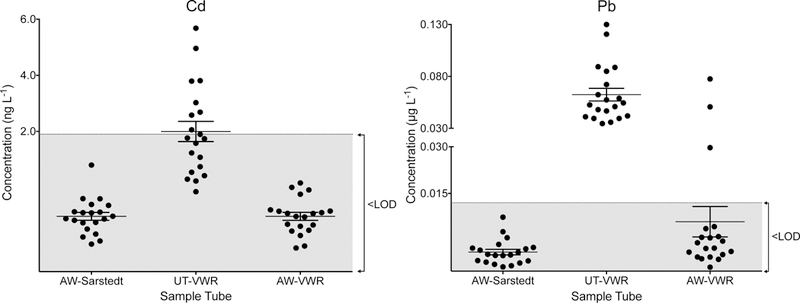
Concentrations of Cd and Pb are plotted in 1) acid-washed Sarstedt autosampler vials (AW-Sarstedt); 2) untreated VWR tubes (UT-VWR); and 3) acid-washed VWR tubes (AW-VWR). For each group, the individual data points (filled circles) are shown along with the mean ± 95% CI.
In trace element analysis, the practice of acid-washing sample collection supplies, storage devices, and other lab materials has been well established for decades.20 While acid washing such materials and/or using supplies marketed specifically as “metal-free” may seem intuitively the best practice, the data reported here suggest that this may be insufficient for ultra-trace element analysis, at least for some key elements. In this study, acid-washing with 2% (v/v) HNO3 prepared using DDI and ultra-pure re-distilled acid produced using a Milestone sub-boiling point quartz still did not reduce background contamination in at least one or more of the five brands of autosampler tubes tested here for Mn, Co, Zn, As, Sr, Cd, and Pb. The data further suggest that the source of contamination might be inherent to the materials used to manufacture the polypropylene polymer and/or the production processes, such that acid-washing may increase contamination by mobilizing some these elements, particularly Pb. Although efforts were taken to minimize contamination, sporadic Pb signals were still evident, albeit at “ultratrace” levels below 0.1 μg L−1 (i.e., below 4 ng L−1 in the autosampler tube).
3.2. ICP-MS/MS method optimization.
Several optimizations of instrumental hardware and settings were conducted using a pooled human sera so as to conserve the limited amount of human FF available for validating the finalized method. Human serum is a similar biological fluid and was judged an acceptable alternative for method optimization purposes. For optimization studies, the signal to noise ratio (S/N) was utilized to select the most favorable dilution factor, analyte integration time (or dwell time), and the ORS cell gas conditions for the final method. Two dilution factors (1 + 24 and 1 + 49) and six integration times (0.1s to 10s) were compared. An approximate 2-fold improvement in detection limits for As, Cd, and Hg led us to select a 1 + 24 dilution factor over 1 + 49 while tolerating a two-fold increase (i.e., 75 μL to 150 μL) in FF sample consumed. The integration time selected for each element (Table 1) represented the best compromise to achieve the LOD to support ultra-trace element analysis, while holding the total data acquisition time to ~4 mins, thereby limiting FF specimen consumption to just 150 μL.
Up to five different ORS cell gas modes were evaluated for each element: no gas; He with Kinetic Energy Discrimination (KED) only; High Energy (HE-) He, i.e., KED + Collision Induced Dissociation (CID); O2; and NH3. For the two reaction modes, appropriate mass shifts were evaluated for select elements.23 The following parameters were optimized for each gas mode as described in section 2.1: energy discrimination, deflect lens, plate bias, gas flow rate, ORS bias, and axial acceleration. Each gas mode was evaluated based on: 1) triplicate analysis of 20 archived human serum samples obtained from the New York State Biomonitoring Proficiency Testing (PT) Program for Trace Elements (n = 3 days); 2) analysis of NIST SRM 1640 Trace Elements in Natural Water, SRM 1643e Trace Elements in Water, and SRM 1598a Inorganic Constituents in Animal Serum, which were analyzed in duplicate over three days (n=6); and 3) an estimated LOD for each gas mode which was based on analysis of the base serum (n=6). These data were analyzed using a 2-way ANOVA with Tukey’s test for multiple comparisons for As, Cd, Pb, Hg, Cu, Mn, Co, Mo, and Sr (and Sidak’s test for multiple comparisons for Se and Zn) to identify statistically significant differences between gas modes. While some differences between modes did reach statistical significance (p<0.05), they were minor (<10% difference) and judged as unimportant given the known uncertainty associated with the target/assigned value for each material. For the finalized FF method, O2 was selected as the cell gas because it yielded better LODs for As, Cd, Hg, and Se; additionally, use of O2 resulted in slightly better (lower) or equivalent LODs for Pb, Mn, and Co compared to other gas modes.
The ability to leverage O2 as a cell gas to mass shift the analyte or interfering ion(s), while limiting those ions entering the reaction cell to a single m/z, is a particular advantage of MS/MS technology when analyzing complex biological matrices.24 In this study, As and Mo were “mass shifted” and analyzed as 75As16O+ and 98Mo16O2+, respectively. In contrast, O2 gas was used to mass shift 98Mo16O+ (away from 114Cd+) and 186W16O+ (away from 202Hg+) to achieve interference-free analysis. Since there is an order of magnitude more Mo than Cd in FF, this interference is quite significant. While FF samples are expected to contain little endogenous W, significant amounts of W contamination have been observed, even after acid-washing (data not shown). A second gas mode was deemed necessary for Cu and Zn because, during the initial studies, use of O2 resulted in count rates exceeding 106 counts-per-second such that detection was consistently forced into analog mode; therefore, HEHe was selected as the sensitivity was better optimized for Cu in FF samples. Although a single “universal” gas mode would have been ideal to minimize sample uptake (i.e., volume), use of HEHe gas mode avoided potential detector saturation, while adding only ~30s of data acquisition time.
System stability was assessed by analyzing a serum sample over a 22-hour run by comparing three nebulizers: MicroMist, MiraMist and SeaSpray. The MicroMist is the standard nebulizer for the Agilent 8900 system; however previous experience with biological matrices led us to investigate two “high-matrix” nebulizers. Data for As are shown in Figure 3 (A-C) as representative for all elements; the data are presented in three ways over the time period: (a) concentration in μg L−1, (b) signal in cps, and (c) As:Rh ratio (cps As:cps Rh). The MicroMist (purple) and SeaSpray (black) nebulizers yielded similar concentration data until replicate 70 (~15 hours). The SeaSpray nebulizer clogged, resulting in the erratic pattern observed for the concentration and As:Rh panels, while the MicroMist proved more robust at tolerating the diluted FF matrix over the course of the analytical run. The MiraMist nebulizer, was operated at a sample uptake rate of 0.4 mL min−1, consistent with that used for the other two nebulizers. This resulted in considerably less signal compared to either the MicroMist or SeaSpray, probably due to decreased nebulization (and/or transport) efficiency when this nebulizer is operated at such low flow rates. At a sample uptake rate of 1.0 mL min−1, the sensitivity of the MiraMist is comparable to that of the MicroMist; however this higher flow rate increases the volume of sample required, which is detrimental when analyzing low-volume samples such as FF. Consequently, the MicroMist was selected based on the lower sample volume consumption, and acceptable within-run precision (defined as as a relative standard deviation (RSD) of <3%) for the 11 analytes measured. Following the 22-hour run, some minor carbon build-up was evident on the sample/skimmer cones but the orifices were not blocked and no significant discoloration of the first extraction lens was observed.
Matrix effects were studied with calibration standards in three matrices: aqueous, serum, and FF, each containing the appropriate internal standards. Calibration slopes were compared using a method equivalent to an ANCOVA in the Prism software.25 Compared to a simple aqueous matrix, FF suppressed the signals for Cd, Cu, Zn, Co, and Sr from 8% for Co to 18% for Zn and enhanced the signal for As and Se by 10% and 15%, respectively. The serum matrix behaved similarly to FF in that signal suppression was found for Cd, Cu, Zn, Co, and Sr compared to the aqueous standards. Signal enhancement in serum was more prominent than in FF for As and Se at 26% and 40%, respectively. Additionally, a 12% signal enhancement for Hg was present in serum that was not observed in FF. Matrix-matching the calibration curve is a common practice in clinical ICP-MS,26 however, use of aqueous- or surrogate matrix-based (e.g., serum) calibration standards may be appropriate if matrix effects are negligible. In this work, aqueous standards were clearly inappropriate for FF-based samples based on the differences noted above for Cd, Cu, Zn, Se, Co, Sr and As. While serum was a better surrogate matrix for Cd, Cu, Zn, Co, and Sr it was still unacceptable for As, Se, and Hg. The option of using a digestion step, compared to a “dilute-and-shoot” approach as adopted here, was previously explored and reported on for FF.14 In that report, the disadvantages of digestion included poorer LODs given small sample volumes involved and the potential for additional contamination especially at ultra-trace levels, while advantages include the relative freedom from some types of matrix effects.
3.3. Accuracy, precision, and performance parameters
Method accuracy was assessed in two ways: 1) by comparing values assigned using the method of standard additions for the two FF QC materials with values obtained from external FF matrix- matched calibration; and 2) by spike recovery studies on 20 human FF samples selected at random, and representing 10% of the total number analyzed. In all of these studies, the calibration strategies included the appropriate internal standards as listed in Table 1. Mean values (± the expanded uncertainty, U) obtained by standard additions and matrix- matched calibration are compared for each element in the two QC materials (Fig 4). Results show excellent agreement, i.e., defined as an overlapping ±U, for 9 of the 11 elements (As, Pb, Hg, Cu, Zn, Se, Co, Mo, and Sr), with a small negative bias observed for Cd (<10%). The negative bias observed for Mn (18%) for QC 2 was judged acceptable (±20%), while noting that both QC 1 and the spike recoveries were within ±10%. Spike recoveries ranged from 96% for Mo to 109% for As, on average. Ideally, method accuracy is best established using an appropriate matrix-based CRM.27 The FF matrix effects reported here precluded using a serum or aqueous CRM, yet FF-based CRMs are not available. A multi-laboratory characterization may also be an option, but is not feasible here since few labs have validated methods for human FF. Absent this, the dual approach selected here (standard additions and spike recoveries) represents a reasonable alternative.
Figure 4.
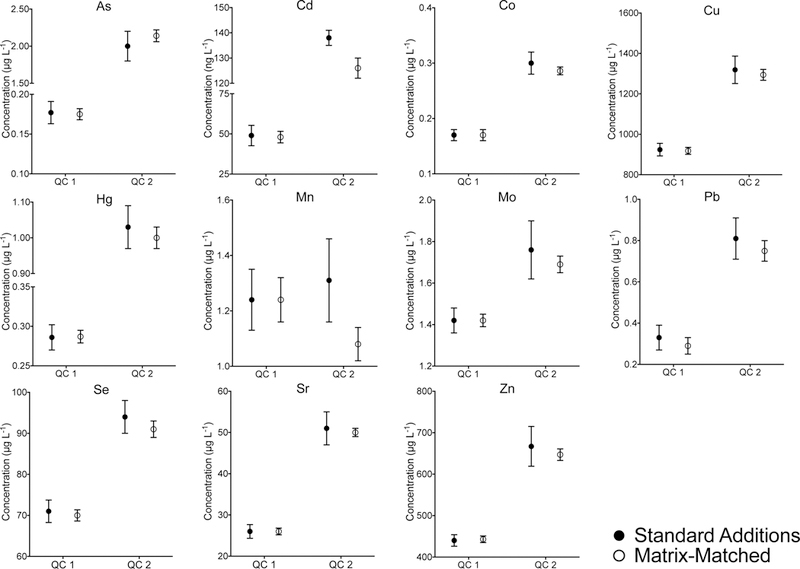
Comparison of values obtained for two human FF-based QC materials for 1) material characterization using standard additions as a reference method (filled in circles) and 2) analyses against an external matrix-matched calibration as part of the method validation and sample batches (open circles). The error bars represent the expanded uncertainty, U, calculated as
Method repeatability (between-run %RSD) was assessed using the FF QC data collected across the study period (~2.5 months), which included 15 analytical runs. Repeatability ranged from 3% for Cu, Zn, and Se, to 22% for Pb in QC 1 (n=27) and from 4% for Cu, Zn, Sr, and Mo to 12% for Pb in QC 2 (n=32). Since the Pb concentration in QC 1 approaches the LOQ this may explain in part the inflated value for the %RSD. Duplicate analyses were performed on a 10% random selection of samples; repeat analysis was performed on up to an additional 30% of samples based on internal lab QC policies. Duplicate values were typically within ±15% of the initial value, except for Pb where less stringent criteria (±0.08 μg L−1) were applied since most of the duplicate samples were below the method LOQ. Based on the duplicate data for Pb, it is possible that FF specimen heterogeneity may have contributed to poorer than expected precision for actual patient specimens, given turbidity, and possible proteinaceous debris observed for some. Over 15 analytical runs, calibration curve slopes varied (RSD) from 5% for Zn, Se, and Sr to 15% for Mn, while the curve fit (r2) ranged from 0.9984 for Mn to 0.9997 for Zn, Sr, and Hg. Within-run signal drift was monitored using QC 2, which was analyzed at the start and end of each analytical run, with no problems observed. Method LODs calculated according to the ISO/IUPAC harmonized guidelines are shown in Table 2. 27 Method LOQs were calculated as 10 SD.
Table 2.
Trace elements in 197 FF samples analyzed by ICP-MS/MS. Data are in μg L−1 except where noted.
| As | Cd | Co | Cu | Hg | Mn | Mo | Pb | Se | Sr | Zn | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Geometric Mean | 0.329 | 18.4* | 0.17 | 965 | 0.276 | 0.68 | 1.04 | 0.06 | 66 | 28.7 | 480 |
| 95th percentile | 2.285 | 50.5* | 0.37 | 1398 | 1.110 | 1.61 | 1.97 | 0.20 | 90 | 47.0 | 653 |
| % Detectable | 97 | 97 | 100 | 100 | 92 | 100 | 100 | 84 | 99 | 100 | 100 |
| LOD | 0.042 | 5.6* | 0.01 | 53 | 0.026 | 0.05 | 0.11 | 0.03 | 7 | 0.6 | 21 |
ng L−1
3.4. Comparison between ICP-MS/MS and SF-ICP-MS FF data.
In a previous report from our group, we compared Q-ICP-MS and SF-ICP-MS for the analysis of 67 FF samples for 10 elements.14 In that pilot study, it was concluded that both instrumental methods could be used to determine Cu, Sr, and Zn, but the other 7 elements required SF-ICP-MS. The original FF samples from the previous pilot study were archived at –80 °C for future research studies. A total of n=60 (90%) had sufficient volume remaining to be re-analyzed using the proposed ICP-MS/MS method. Method comparisons proved difficult for those elements (As, Cd, Co, Hg, Mn, Mo, and Pb) where a majority of values were below the LOQ for one method. One metric for comparing results close to the LOD is to set an absolute range that is considered fit-for-purpose given both the biological variation and analytical uncertainty in the LOD-LOQ range. Since little is known about trace element variation within human follicles, or between follicles from the same subject, or even between subjects, we propose acceptability criteria based on analytical uncertainty alone. These acceptability criteria are defined as ±15% or ± a fixed value, whichever is greater. The latter are element-specific: ±0.5 μg L−1 for As, Hg, Mn, and Pb; ±50 ng L−1for Cd and Co; and ±1 μg L−1 for Mo. A comparison between ICP-MS/MS and SF-ICP-MS for the analysis of 60 common FF specimens is shown in Figure 5 for 7 elements. The data in Fig 5 are graphed as a Bland-Altman difference plot, i.e., where the difference between two methods is plotted as a function of the mean value for both methods. The respective method LOQs are shown as red lines, which helps remind us of the relative uncertainty approaching that region. More than 90% of the data for the common samples are within the quality criteria proposed for As, Cd, Co, Hg, Mn, Mo, and Pb. Figure 6 shows a scatterplot comparison between the two methods for Cu, Se, Sr, and Zn, where there is a broad range of concentrations that are well above the LOQ. The data show good agreement between the methods and within ±15% as shown by the dashed lines.
Figure 5.
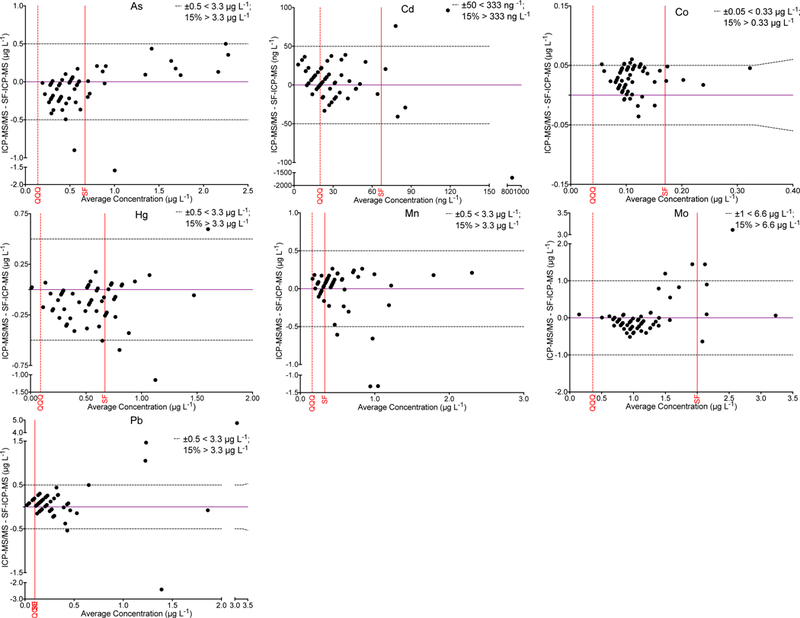
The difference between values obtained by ICP-MS/MS (QQQ) and SF-ICP-MS is shown as a function of the mean concentration of As, Cd, Co, Hg, Mn, Mo, and Pb in 60 common samples. For these elements a large proportion of the data were below the method LOQ for either the ICP-MS/MS method or the SF-ICP-MS method. Thus, acceptability criteria (black dashed lines) are proposed as ± an absolute criterion (shown per element) or ±15%, whichever is greater. Method LOQs are shown for the QQQ method (red dashed line) and SF method (solid red line).
Figure 6.
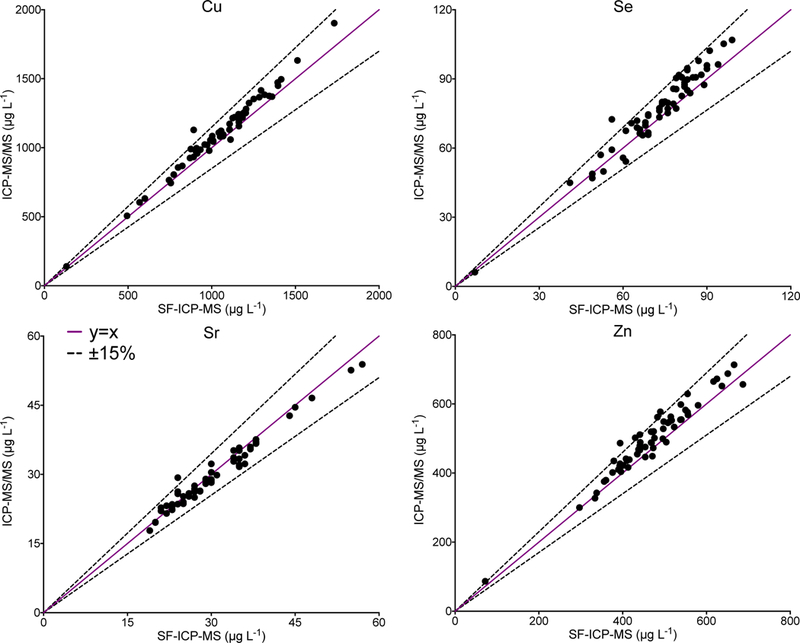
Values obtained by ICP-MS/MS are shown as a function of those reported by SF-ICP-MS for Cu, Se, Sr, and Zn in 60 common samples. The solid purple line dashed black lines represent y=x ±15%.
3.5. Distribution of trace elements in human FF
The proposed method was used to analyze 197 FF samples for 11 elements. Descriptive data, including the geometric mean, 95th percentile, and percent detected are shown for each element in Table 2. The proposed ICP-MS/MS method achieved >90% detectability for 10 of the 11 elements measured, which is comparable to the performance of the SF-ICP-MS method in the pilot study. The exception was Pb for which 84% were detectable in this study compared to 90% in the pilot study. Another notable difference was that Cd was detected in 97% of FF specimens in this study compared to 52% in the pilot study using SF-ICP-MS (LOD= 20 ng L−1), which reflects a lower LOD by ICP-MS/MS (6 ng L−1). The proposed ICP-MS/MS method has a 2–8 fold lower LOD for As, Cd, Co, Hg, Mn, and Mo compared to the LOD by SF-ICP-MS. The LODs calculated for Sr, Se, Cu and Zn by ICP-MS/MS are naturally inflated because of the relatively large endogenous levels in human FF, and are similar to those reported using the SF-ICP-MS method.
A review of the literature on the elemental composition of human FF was conducted to provide a comparison to the data reported here. These studies are summarized in Table 3 using a style similar to that adopted by Atomic Spectrometry Updates for the analysis of clinical and biological materials.28 The geometric mean values (n=197) for Cu, Sr and Zn reported in this study were similar to those reported by most other studies. Values for Se found here were most similar to those reported by us previously using SF-ICP-MS14 along with data reported by Nanto et al.,4 and Ozkaya et al.,.13 It is evident from Table 3 that for FF Se there is a clear lack of consensus among the 8 studies listed. The lowest values obtained for the four key toxic elements in human FF, As, Cd, Hg, and Pb, were reported in this study by ICP-MS/MS. This could be the result of lower exposures for this population of women, or a combination of analytical bias, exogenous environmental contamination, and/or contamination of FF by blood.
Table 3.
Literature review of (ultra-)trace element determination in human FF using methods based on atomic spectrometry. Data are reported in μg L−1.
| Element | Mean | n | LOD | Technique | Year | Ref. | |
|---|---|---|---|---|---|---|---|
| As | 4 | 35 | - | ICP-MS | 2006 | 10 | |
| As | 0.6 | 67 | 0.2 | SF-ICP-MS | 2012 | 14 | |
| As | 1.65 | 101 | 0.17 | HG-ETAAS | 2016 | 16 | |
| As | 0.329 | 197 | 0.042 | ICP-MS/MS | 2019 | * | |
| Cd | 0.94b | 221 | - | ETAAS | 2018 | 18 | |
| Cd | 6.73 | 10 | 0.1 | ETAAS | 1995 | 6 | |
| Cd | 2.44 | 21 | 1.1 | ETAAS | 2002 | 9 | |
| Cd | <0.2 | 33 | 0.2 | ICP-MS | 2009 | 12 | |
| Cd | 0.03 | 67 | 0.02 | SF-ICP-MS | 2012 | 14 | |
| Cd | 0.74 | 101 | 0.33 | ETAAS | 2016 | 16 | |
| Cd | 0.0184 | 197 | 0.0056 | ICP-MS/MS | 2019 | * | |
| Cd | 0.34 | 220 | 0.1 | ETAAS | 2001 | 7 | |
| Cd | 0.61 | 340 | 0.001a | ETAAS | 2013 | 15 | |
| Cd | 0.33 | 614 | 0.25 | ETAAS | 2008 | 11 | |
| Co | 0.09 | 67 | 0.05 | SF-ICP-MS | 2012 | 14 | |
| Co | 0.17 | 194 | 0.011 | ICP-MS/MS | 2019 | * | |
| Cu | 3.4b | 221 | - | ETAAS | 2018 | 18 | |
| Cu | 407.9 | 30 | - | ICP-AES | 2011 | 13 | |
| Cu | 1160 | 33 | - | FAAS | 1987 | 3 | |
| Cu | 1000 | 35 | - | ICP-MS | 2006 | 10 | |
| Cu | 1035 | 67 | 57 | SF-ICP-MS | 2012 | 14 | |
| Cu | 1030 | 73 | - | PIXE | 1991 | 4 | |
| Cu | 960 | 101 | 230 | FAAS | 2016 | 16 | |
| Cu | 965 | 197 | 53 | ICP-MS/MS | 2019 | * | |
| Cu | 368 | 340 | 0.041a | ETAAS | 2013 | 15 | |
| Cu | 560 | 53 | 10 | FAAS | 2017 | 17 | |
| Hg | 0.5 | 67 | 0.2 | SF-ICP-MS | 2012 | 14 | |
| Hg | 2.03 | 101 | 0.1 | HG-ETAAS | 2016 | 16 | |
| Hg | 0.276 | 197 | 0.026 | ICP-MS/MS | 2019 | * | |
| Hg | 2.48 | 612 | 0.28 | HG-ETAAS | 2008 | 11 | |
| Mn | 1 | 35 | - | ICP-MS | 2006 | 10 | |
| Mn | 0.4 | 67 | 0.1 | SF-ICP-MS | 2012 | 14 | |
| Mn | 0.68 | 197 | 0.05 | ICP-MS/MS | 2019 | * | |
| Mo | 2 | 35 | - | ICP-MS | 2006 | 10 | |
| Mo | 1.1 | 67 | 0.6 | SF-ICP-MS | 2012 | 14 | |
| Mo | 1.04 | 197 | 0.11 | ICP-MS/MS | 2019 | * | |
| Pb | 18.0 | 221 | - | ETAAS | 2018 | 18 | |
| Pb | 11.29 | 23 | - | ETAAS | 2001 | 8 | |
| Pb | 3 | 35 | - | ICP-MS | 2006 | 10 | |
| Pb | 0.15 | 67 | 0.03 | SF-ICP-MS | 2012 | 14 | |
| Pb | 28.8 | 101 | 12.2 | ETAAS | 2016 | 16 | |
| Pb | 0.06 | 194 | 0.03 | ICP-MS/MS | 2019 | * | |
| Pb | 1.41 | 340 | 0.015a | ETAAS | 2013 | 15 | |
| Pb | 18.2 | 615 | 0.66 | ETAAS | 2008 | 11 | |
| Se | 9.8 | 221 | - | ETAAS | 2018 | 18 | |
| Se | 50.3 | 30 | - | HG-ETAAS | 2011 | 13 | |
| Se | 18 | 35 | - | ICP-MS | 2006 | 10 | |
| Se | 71 | 67 | 3 | SF-ICP-MS | 2012 | 14 | |
| Se | 62.2 | 73 | - | ETAAS | 1991 | 4 | |
| Se | 38.69 | 135 | - | ETAAS | 1995 | 5 | |
| Se | 66 | 197 | 7 | ICP-MS/MS | 2019 | * | |
| Se | 0.547 | 340 | 0.023a | HG-ETAAS | 2013 | 15 | |
| Sr | 23 | 35 | - | ICP-MS | 2006 | 10 | |
| Sr | 29 | 67 | 0.8 | SF-ICP-MS | 2012 | 14 | |
| Sr | 28.7 | 197 | 0.6 | ICP-MS/MS | 2019 | * | |
| Zn | 461c | 221 | — | ETAAS | 2018 | 18 | |
| Zn | 307.6 | 30 | - | ICP-AES | 2011 | 13 | |
| Zn | 720 | 33 | - | FAAS | 1987 | 3 | |
| Zn | 400 | 35 | - | ICP-MS | 2006 | 10 | |
| Zn | 454 | 67 | 21 | SF-ICP-MS | 2012 | 14 | |
| Zn | 450 | 73 | - | PIXE | 1991 | 4 | |
| Zn | 760 | 101 | 180 | FAAS | 2016 | 16 | |
| Zn | 480 | 197 | 21.00 | ICP-MS/MS | 2019 | * | |
| Zn | 502 | 340 | 0.09a | ETAAS | 2013 | 15 | |
| Zn | 668 | 53 | 40 | FAAS | 2017 | 17 | |
Reported LOQ.
This work.
Estimated median value based on graphs of results.
units in μg L−1.
In this study, human FF from two populations of women undergoing IVF treatment was analyzed for trace elements using a new analytical method based on ICP-MS/MS. One population (“pilot study”) was drawn from 60 archived FF specimens analyzed previously for 24 elements using SF-ICP-MS, and re-analyzed for 11 elements by ICP-MS/MS. A second population of 197 FF specimens were collected prospectively using an updated protocol that included more stringent contamination controls procedures. The two population groups were compared using a two-tailed Mann-Whitney U test limited to the four key toxic elements (As, Cd, Hg, and Pb). All data below the methods LOD were included without any truncation or transformation, i.e., as reported by the Agilent MassHunter software, to avoid potentially biasing the test to the null hypothesis. The p-values reported by the statistical software program (Prism GraphPad) indicated both Cd and Pb were significantly lower (p<0.0001) in the current study population compared to the pilot study population, where FF specimens were collected without any pre-analytical considerations. The population data for As, Cd, Hg, and Pb are shown as scatter plots on a semi-log scale in Figure 7. The two populations appear side-by-side with the pilot (open circles) and prospective (filled in circles) shown for the four key elements. The error bars shown Figure 7 represent the geometric mean ± geometric standard deviation. It is plausible that the extensive efforts to minimize pre-analytical background contamination as described in section 3.1 may explain the observed shift for Cd and Pb to lower values.
Figure 7.
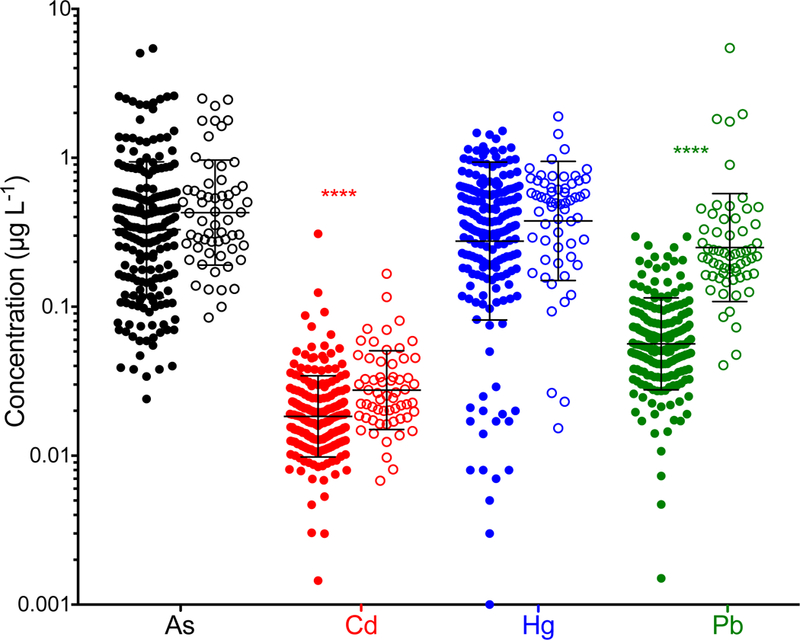
Values obtained by ICP-MS/MS for As (black), Cd (red), Hg (blue), and Pb (green) in the 197 FF samples measured as part of the present study (filled in circles) compared to those measured in 60 of the FF samples that were originally measured by SF-ICP-MS (open circles). The geometric mean ± geometric standard deviation are shown for each group.
4. Conclusions
The principal goal of this research was to leverage the next generation of inorganic mass spectrometry (ICP-MS/MS) coupled with more stringent pre-analytical procedures to achieve improved measurements limited to 11 key elements in human FF obtained as part of a prospective study of women undergoing IVF treatments. According to the CLSI, most of the elements measured here can be classified as being present in human FF at ultra-trace concentrations. The analytical challenges achieving the low LODs reported were quite significant. The data suggest that implementing customized protocols designed to minimize pre-analytical contamination from supplies used in specimen collection, processing, and storage, are important. Our experience also suggests that routine acid-washing plasticware may be insufficient to eliminate background contamination for some key elements at ultra-trace levels, and may even complicate matters by mobilizing others. The shift in Cd and Pb to lower levels reported here compared to the previous “pilot” study data may be due to the improved pre-analytical protocols and contamination control. The results of this study add further to the literature on the trace and ultra-trace element content of human FF and help to provide the basis for a future reference range.
Acknowledgements
The authors would like to thank the staff of the UCSF Center for Reproductive Health for implementing the sample collection and processing protocols established as part of this project. This work was funded in part by the National Institute of Environmental Health Sciences (NIEHS), grant number 1R56 ES023886–01 (MSB) and by the Office of the Director, National Institutes of Health (OD), and the National Institute of Environmental Health Sciences (NIEHS), grant number 1U2CES026542–01 to the Wadsworth Center (PJP).
References
- 1.Shalgi R, Soferman N and Kraicer PF, J. Reprod. Fertil, 1972, 28, 335–340. [DOI] [PubMed] [Google Scholar]
- 2.Chong AP, Taymor ML and Lechene CP, Am J Obstet Gynecol, 1977, 128, 209–211. [DOI] [PubMed] [Google Scholar]
- 3.Ng SC, Karunanithy R, Edirisinghe WR, Roy AC, Wong PC and Ratnam SS, Gynecol. Obstet. Invest, 1987, 23, 129–132. [DOI] [PubMed] [Google Scholar]
- 4.Nanto V, Kiilholma P, Nikkanen V, Pakarinen P, Hyora H, Rosengard U and Brenner R, Acta Radiol. Suppl, 1991, 376, 176–178. [PubMed] [Google Scholar]
- 5.Paszkowski T, Traub AI, Robinson SY and McMaster D, Clin. Chim. Acta, 1995, 236, 173–180. [DOI] [PubMed] [Google Scholar]
- 6.Zenzes MT, Krishnan S, Krishnan B, Zhang HM and Casper RF, Fertil. Steril, 1995, 64, 599–603. [DOI] [PubMed] [Google Scholar]
- 7.Fiala J, Hruba D, Crha I, Rezl P and Totusek J, Int. J. Occup. Med. Environ. Health, 2001, 14, 185–188. [PubMed] [Google Scholar]
- 8.Paksy K, Gati I, Naray M and Rajczy K, Toxicol J Environ. Health Part A, 2001, 62, 359–366. [DOI] [PubMed] [Google Scholar]
- 9.Younglai EV, Foster WG, Hughes EG, Trim K and Jarrell JF, Arch. Environ. Contam. Toxicol, 2002, 43, 121–126. [DOI] [PubMed] [Google Scholar]
- 10.Silberstein T, Saphier O, Paz-Tal O, Trimarchi JR, Gonzalez L and Keefe DL, J Trace Elem Med Biol, 2006, 20, 205–207. [DOI] [PubMed] [Google Scholar]
- 11.Al-Saleh I, Coskun S, Mashhour A, Shinwari N, El-Doush I, Billedo G, Jaroudi K, Al-Shahrani A, Al-Kabra M and Mohamed GED, Int. J. Hyg. Environ. Health, 2008, 211, 560–579. [DOI] [PubMed] [Google Scholar]
- 12.Silberstein T, Saphier O, Paz-Tal O, Gonzalez L, Keefe DL and Trimarchi JR, Fertil. Steril, 2009, 91, 1771–1774. [DOI] [PubMed] [Google Scholar]
- 13.Ozkaya MO, Naziroglu M, Barak C and Berkkanoglu M, Biol. Trace Elem. Res, 2011, 139, 1–9. [DOI] [PubMed] [Google Scholar]
- 14.Kruger PC, Bloom MS, Arnason JG, Palmer CD, Fujimoto VY and Parsons PJ, J. Anal. At. Spectrom, 2012, 27, 1245–1253. [Google Scholar]
- 15.Singh AK, Chattopadhyay R, Chakravarty B and Chaudhury K, Reprod. Toxicol, 2013, 42, 116–124. [DOI] [PubMed] [Google Scholar]
- 16.Tolunay HE, Sukur YE, Ozkavukcu S, Seval MM, Ates C, Turksoy VA, Ecemis T, Atabekoglu CS, Ozmen B, Berker B and Sonmezer M, Eur. J. Obstet. Gynecol. Reprod. Biol, 2016, 198, 73–77. [DOI] [PubMed] [Google Scholar]
- 17.Sun Y, Lin Y, Niu M, Kang YF, Du SR and Zheng BH, Int. J. Clin. Exp. Med, 2017, 10, 3547–3553. [Google Scholar]
- 18.Wdowiak A, Wdowiak E and Bojar I, Ann. Agr. Env. Med, 2018, 25, 213–218. [DOI] [PubMed] [Google Scholar]
- 19.Bloom MS, Kim K, Kruger PC, Parsons PJ, Arnason JG, Steuerwald AJ and Fujimoto VY, J. Assist. Reprod. Genet, 2012, 29, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.CLSI, Control of Preanalytical Variation in Trace Element Determinations; Approved Guideline CLSI document C38-A., Clinical and Laboratory Standards Institute, Wayne, PA, 1997. [Google Scholar]
- 21.CDC, CDC Screening (“Lot Testing”) Guidelines for Elemental Analysis of Biological Matrices, Atlanta, GA, Center for Disease Control and Prevention, 2005. [Google Scholar]
- 22.Levay PF, Huyser C, Fourie FLR and Rossouw DJ, J. Assist. Reprod. Genet, 1997, 14, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugiyama N and Nakano K, Reaction Data for 70 elements using O2, NH3, and H2 gases with the Agilent 8800 Triple Quadrupole ICP-MS, Agilent Technologies, 2014. [Google Scholar]
- 24.Balcaen L, Bolea-Fernandez E, Resano M and Vanhaecke F, Analytica Chimica Acta, 2015, 894, 7–19. [DOI] [PubMed] [Google Scholar]
- 25.GraphPad, Comparing slopes and intercepts, https://www.graphpad.com/guides/prism/7/curve-fitting/index.htm?reg_comparingslopesandintercepts.htm, (accessed October 12, 2018).
- 26.Palmer CD, Lewis ME Jr., Geraghty CM, Barbosa F Jr. and Parsons PJ, Spectrochim. Acta Part B At. Spectrosc., 2006, 61, 980–990. [Google Scholar]
- 27.Thompson M, Ellison Stephen LR and Wood R, Pure and Applied Chemistry, 2002, 74, 835. [Google Scholar]
- 28.Taylor A, Barlow N, Day MP, Hill S, Martin N and Patriarca M, J. Anal. At. Spectrom, 2018, 33, 338–382. [Google Scholar]