

Abstract
Background
Oral zuclopenthixol dihydrochloride (Clopixol) is an anti‐psychotic treatment for people with psychotic symptoms, especially those with schizophrenia. It is associated with neuroleptic malignant syndrome, a prolongation of the QTc interval, extra‐pyramidal reactions, venous thromboembolism and may modify insulin and glucose responses.
Objectives
To determine the effects of zuclopenthixol dihydrochloride for treatment of schizophrenia.
Search methods
We searched the Cochrane Schizophrenia Group's Trials Register (latest search 09 June 2015). There were no language, date, document type, or publication status limitations for inclusion of records in the register.
Selection criteria
All randomised controlled trials (RCTs) focusing on zuclopenthixol dihydrochloride for schizophrenia. We included trials meeting our inclusion criteria and reporting useable data.
Data collection and analysis
We extracted data independently. For binary outcomes, we calculated risk ratio (RR) and its 95% confidence interval (CI), on an intention‐to‐treat basis. For continuous data, we estimated the mean difference (MD) between groups and its 95% CI. We employed a random‐effect model for analyses. We assessed risk of bias for included studies and created 'Summary of findings' tables using GRADE.
Main results
We included 20 trials, randomising 1850 participants. Data were reported for 12 comparisons, predominantly for the short term (up to 12 weeks) and inpatient populations. Overall risk of bias for included studies was low to unclear.
Data were unavailable for many of our pre‐stated outcomes of interest. No data were available, across all comparisons, for death, duration of stay in hospital and general functioning.
Zuclopenthixol dihydrochloride versus:
1. placebo
Movement disorders (EPSEs) were similar between groups (1 RCT, n = 28, RR 6.07 95% CI 0.86 to 43.04 very low‐quality evidence). There was no clear difference in numbers leaving the study early (2 RCTs, n = 100, RR 0.29, 95% CI 0.01 to 6.60, very low‐quality evidence).
2. chlorpromazine
No clear differences were found for the outcomes of global state (average CGI‐SI endpoint score) (1 RCT, n = 60, MD 0.00, 95% CI ‐0.49 to 0.49) or movement disorders (EPSEs) (3 RCTs, n = 199, RR 0.94, 95% CI 0.61 to 1.45), both very low‐quality evidence. More people left the study early for any reason from the zuclopenthixol group (6 RCTs, n = 766, RR 0.54, 95% CI 0.36 to 0.81, low‐quality evidence).
3. chlorprothixene
There was no clear difference in numbers leaving the study early for any reason (1 RCT, n = 20, RR 1.00, 95% CI 0.34 to 2.93, very low‐quality evidence).
4. clozapine
No useable data were presented.
5. haloperidol
No clear differences between treatment groups were found for the outcomes global state score (average CGI endpoint score) (1 RCT, n = 49, MD 0.13, 95% CI ‐0.30 to 0.55) or leaving the study early (2 RCTs, n = 141, RR 0.99, 95% CI 0.72 to 1.35), both very low‐quality evidence.
6. perphenazine
Those receiving zuclopenthixol were more likely to require medication in the short term for EPSEs than perphenazine (1 RCT, n = 50, RR 1.90, 95% CI 1.12 to 3.22, very low‐quality evidence). Similar numbers left the study early (2 RCTs, n = 104, RR 0.63, 95% CI 0.27 to 1.47, very low‐quality evidence).
7. risperidone
Those receiving zuclopenthixol were more likely to require medications for EPSEs than risperidone (1 RCT, n = 98,RR 1.92, 95% CI 1.12 to 3.28, very low quality evidence). There was no clear difference in numbers leaving the study early ( 3 RCTs, n = 154, RR 1.30, 95% CI 0.84 to 2.02) or in mental state (average PANSS total endpoint score) (1 RCT, n = 25, MD ‐3.20, 95% CI ‐7.71 to 1.31), both very low‐quality evidence).
8. sulpiride
No clear differences were found for global state (average CGI endpoint score) ( 1 RCT, n = 61, RR 1.18, 95% CI 0.49 to 2.85, very low‐quality evidence), requiring hypnotics/sedatives (1 RCT, n = 61, RR 0.60, 95% CI 0.27 to 1.32, very low‐quality evidence) or leaving the study early (1 RCT, n = 61, RR 2.07 95% CI 0.97 to 4.40, very low‐quality evidence).
9. thiothixene
No clear differences were found for the outcomes of 'global state (average CGI endpoint score) (1 RCT, n = 20, RR 0.50, 95% CI 0.17 to 1.46) or leaving the study early (1 RCT, n = 20, RR 0.57, 95% CI 0.24 to 1.35), both very low‐quality evidence).
10. trifluoperazine
No useable data were presented.
11. zuclopenthixol depot
There was no clear difference in numbers leaving the study early (1 RCT, n = 46, RR 1.95, 95% CI 0.36 to 10.58, very low‐quality evidence).
12. Zuclopenthixol dihydrochloride (cis z isomer) versus zuclopenthixol (cis z/trans e isomer)
There were no clear differences in reported side‐effects ( 1 RCT, n = 57, RR 1.34, 95% CI 0.82 to 2.18, very low‐quality evidence) and in numbers leaving the study early (4 RCTs, n = 140, RR 2.15, 95% CI 0.49 to 9.41, very low‐quality evidence).
Authors' conclusions
Zuclopenthixol dihydrochloride appears to cause more EPSEs than clozapine, risperidone or perphenazine, but there was no difference in EPSEs when compared to placebo or chlorpromazine. Similar numbers required hypnotics/sedatives when zuclopenthixol dihydrochloride was compared to sulpiride, and similar numbers of reported side‐effects were found when its isomers were compared. The other comparisons did not report adverse‐effect data.
Reported data indicate zuclopenthixol dihydrochloride demonstrates no difference in mental or global states compared to placebo, chlorpromazine, chlorprothixene, clozapine, haloperidol, perphenazine, sulpiride, thiothixene, trifluoperazine, depot and isomers. Zuclopenthixol dihydrochloride, when compared with risperidone, is favoured when assessed using the PANSS in the short term, but not in the medium term.
The data extracted from the included studies are mostly equivocal, and very low to low quality, making it difficult to draw firm conclusions. Prescribing practice is unlikely to change based on this meta‐analysis. Recommending any particular course of action about side‐effect medication other than monitoring, using rating scales and clinical assessment, and prescriptions on a case‐by‐case basis, is also not possible.
There is a need for further studies covering this topic with more antipsychotic comparisons for currently relevant outcomes.
Plain language summary
Zuclopenthixol dihydrochloride for schizophrenia
Background
Schizophrenia is a serious mental illness where people experience delusions (strange thoughts/ideas) and/or hallucinations (hearing or seeing things that are not real). These are often known as positive or acute symptoms. People also experience negative or chronic symptoms which usually follow acute symptoms. These can include withdrawing from social contact, lack of interest in everyday activities, depression as well as problems with memory and thought processing.
Treatment usually involves a package of care involving medications known as antipsychotics, and if necessary, additional therapies such as Cognitive Behavioural Therapy, Psychoeducation or Occupational therapy.
There are many different antipsychotics available; knowing how effective each one is compared with no treatment or placebo (dummy treatment) and with other antipsychotics is important. This review compares the oral form of the antipsychotic zuclopenthixol dihydrochloride with other antipsychotics and placebo.
Searching for evidence
An electronic search was run on 9 June 2015, searching for trials that randomised people with schizophrenia into treatment groups that received either zuclopenthixol dihydrochloride or placebo or another antipsychotic.
Evidence found
The review authors found 20 trials with 1850 participants. Most of these participants were patients in psychiatric hospitals.
The trials compared oral zuclopenthixol dihydrochloride to placebo and nine other oral antipsychotics (chlorpromazine, chlorprothixene, clozapine, haloperidol, perphenazine, risperidone, sulpiride, thiothixene, and trifluoperazine). There were also trials that compared oral zuclopenthixol dihydrochloride with an injection of zuclopenthixol dihydrochloride and some that compared two different versions of zuclopenthixol dihydrochloride.
The review authors were interested in finding evidence for zuclopenthixol dihydrochloride's effect on seven main outcomes: global state, mental state, adverse effects, death, duration of stay in hospital, leaving the study early and general functioning. Unfortunately many data provided by the trials were unusable, data were available only for global state, mental state, leaving the study early and the adverse effect of movement disorders. The trials comparing zuclopenthixol dihydrochloride with sulpiride and trifluoperazine did not provide any useable data for any of these main outcomes.
Overall results suggest zuclopenthixol dihydrochloride's effect on global and mental state, and number of participants leaving the study early is similar to the other anti‐psychotics listed above.
Zuclopenthixol dihydrochloride may cause more movement disorders than clozapine, risperidone or perphenazine, but there was no difference for the other drug comparisons or placebo.
Conclusions
The evidence currently available is of very low to low quality and the meaning is therefore unclear. Data for many important outcomes are not available making conclusions about overall effectiveness of zuclopenthixol dihydrochloride difficult.
Evidence available suggest that zuclopenthixol dihydrochloride is not any worse than other antipsychotics in treating the symptoms of schizophrenia, however more trials providing good‐quality data are needed before firm conclusions can be made.
Summary of findings
Background
Description of the condition
Schizophrenia afflicts about 1% of the world's population across geographical, racial and gender barriers (Jablensky 1992).
It is characterised by distortions in thinking and perception in the context of inappropriate and/or blunted affects. It often includes the following psychopathological phenomena:
thought echo, thought insertion/withdrawal, thought broadcasting;
delusional perception and delusions of control, influence or passivity;
hallucinations (often auditory);
thought disorders;
negative symptoms.
The course of the illness can be continuous or episodic and outcomes are polymorphic (WHO 2010).
Description of the intervention
There is one oral preparation (marketed as Cisordinol, Clopixol) and two depot forms of zuclopenthixol dihydrochloride: zuclopenthixol acetate (Cisordinol‐Acutard, Clopixol‐Acuphase, Clopixol‐Acutard), and zuclopenthixol decanoate (Cisordinol depot, Clopixol depot, Clopixol Inj.). Zuclopenthixol dihydrochloride (Clopixol tablets) was manufactured by Lundbeck Pharmaceuticals, licensed in 1982 and is currently approved for use in 71 countries.
How the intervention might work
Zuclopenthixol belongs to the class of thioxanthene derivatives. Zuclopenthixol is the cis (Z)‐isomer of clopenthixoland has a high affinity for both dopamine D1 and D2 receptors. It is rapidly absorbed from the gastrointestinal tract and maximum serum concentration occurs after about four hours following a single oral dose. The elimination half‐life is approximately 20 hours (range 12 to 28 hours). It has a moderately high pre‐systemic metabolism and about 40% of the drug is eliminated this way (Dollery 1999).
Why it is important to do this review
Antipsychotic medication remains a key means of treating people with schizophrenia (Awad 1997; Dally 1967). Since the introduction of chlorpromazine in the early 1950's numerous antipsychotic drugs have been developed. These include haloperidol, thioridazine, trifluoperazine and zuclopenthixol and they are now inexpensive and accessible to millions. Zuclopenthixol has been licensed since the early 1980's and is used in at least 71 countries including many European countries, Australia, New Zealand and Canada.
A newer generation of so called 'atypical' antipsychotics have been developed and are increasingly prevalent for treating schizophrenia, especially in high‐income countries (Taylor 2000). This increase in the use of newer drugs is mirrored by a decline in the use of the older generation antipsychotics such as zuclopenthixol dihydrochloride although they are still used for treating schizophrenia and related psychotic conditions (Kozyrev 2003; Mace 2005; Percudani 2005). However, we have found it difficult to identify reviews on the effects of the oral preparation and we found no relevant meta‐analyses. This Cochrane review is the third review relevant to this compound and concentrates on the oral form of zuclopenthixol dihydrochloride. The first review investigated the effects of zuclopenthixol acetate for rapid tranquillisation of disturbed people with serious mental illnesses (Jayakody 2012), and the second concentrates on the decanoate preparation of zuclopenthixol as this is used as a long‐acting depot preparation (da Silva Freire Coutinho 1999).
Objectives
To determine the effects of zuclopenthixol dihydrochloride for the treatment of schizophrenia.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials. Where a trial was described as 'double‐blind' but it was implied that the study was randomised, we included these trials in a sensitivity analysis. If there was no substantive difference within primary outcomes (see Types of outcome measures) when these 'implied randomisation' studies were added, then we included these in the final analysis. If there was a substantive difference, only clearly randomised trials were utilised and the results of the sensitivity analysis described in the text. We excluded quasi‐randomised studies such as those allocating by using alternate days of the week.
Types of participants
We included people with schizophrenia and schizophrenia‐like disorders such as schizophreniform disorder, delusional disorder or schizoaffective disorder, diagnosed by any criteria. We also included those with 'serious/chronic mental illness' or 'psychotic illness'. Where possible we excluded people with dementia, depression and problems primarily associated with substance misuse.
Types of interventions
1. Zuclopenthixol dihydrochloride administered in oral form, any dose.
2. Placebo or no treatment.
3. Other antipsychotic drugs: any dose, administered in depot form.
4. Other antipsychotic drugs: any dose, administered in oral form.
Types of outcome measures
All outcomes were reported for the short term (up to 12 weeks), medium term (13 to 26 weeks), and long term (more than 26 weeks). None of the included studies went beyond one year.
Primary outcomes
1. Global state: average endpoint global state score
2. Adverse effects: clinically important general adverse effect
Secondary outcomes
1. Death ‐ suicide and natural causes
2. Global state
2. 1 Relapse 2. 2 Time to relapse 2. 3 Clinically important change in global state 2. 4 Any change in global state 2. 5 Average change in global state scores
3. Service outcomes
3. 1 Hospitalisation 3. 2 Time to hospitalisation 3. 3 Duration of stay in hospital
4. Mental state
4. 1 Clinically important change in general mental state 4. 2 Any change in general mental state 4. 3 Average endpoint general mental state score 4. 4 Average change in general mental state scores 4. 5 Clinically important change in specific symptoms 4. 6 Any change in specific symptoms 4. 7 Average endpoint specific symptom score 4. 8 Average change in specific symptom scores
5. Leaving the study early
5. 1 For specific reasons 5. 2 For general reasons
6. General functioning
6. 1 Clinically important change in general functioning 6. 2 Any change in general functioning 6. 3 Average endpoint general functioning score 6. 4 Average change in general functioning scores 6. 5 Clinically important change in specific aspects of functioning, such as social or life skills 6. 6 Any change in specific aspects of functioning, such as social or life skills 6. 7 Average endpoint specific aspects of functioning, such as social or life skills 6. 8 Average change in specific aspects of functioning, such as social or life skills
7. Behaviour
7. 1 Clinically important change in general behaviour 7. 2 Any change in general behaviour 7. 3 Average endpoint general behaviour score 7. 4 Average change in general behaviour scores 7. 5 Clinically important change in specific aspects of behaviour 7. 6 Any change in specific aspects of behaviour 7. 7 Average endpoint specific aspects of behaviour 7. 8 Average change in specific aspects of behaviour
8. Adverse effects
8. 1 Any general adverse effects 8. 2 Average endpoint general adverse effect score 8. 3 Average change in general adverse effect scores 8. 4 Clinically important change in specific adverse effects 8. 5 Any change in specific adverse effects 8. 6 Average endpoint specific adverse effects 8. 7 Average change in specific adverse effects
9. Engagement with services
9. 1 Clinically important engagement 9. 2 Not any engagement 9. 3 Average endpoint engagement score 9. 4 Average change in engagement scores
10. Satisfaction with treatment
10. 1 Recipient of care satisfied with treatment 10. 2 Recipient of care average satisfaction score 10. 3 Recipient of care average change in satisfaction scores 10. 4 Carer satisfied with treatment 10. 5 Carer average satisfaction score 10. 6 Carer average change in satisfaction scores
11. Quality of life
11. 1 No clinically important change in quality of life 11. 2 Not any change in quality of life 11. 3 Average endpoint quality of life score 11. 4 Average change in quality of life scores 11. 5 No clinically important change in specific aspects of quality of life 11. 6 Not any change in specific aspects of quality of life 11. 7 Average endpoint specific aspects of quality of life 11. 8 Average change in specific aspects of quality of life
12. Economic outcomes
12. 1 Direct costs 12. 2 Indirect costs
Summary of findings
We used the GRADE approach to interpret findings (Schünemann 2011) and used the GRADE profiler to import data from Review Manager (RevMan) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making.
We selected the following main outcomes for inclusion in the 12 'Summary of findings' tables.
Global state: Average endpoint global state score (clinically improved) [primary outcome]
Adverse effects: Clinically important general adverse effect [primary outcome]
Death: Suicide and natural causes [secondary outcome]
Service outcome: Duration of stay in hospital [secondary outcome]
Mental state: Average endpoint general mental state score (clinically improved) [secondary outcome]
Leaving the study early: any reason [secondary outcome]
General functioning: Average endpoint general functioning score (clinically improved) [secondary outcome]
Search methods for identification of studies
Electronic searches
Cochrane Schizophrenia Group’s Trials Register
On June 09, 2015, the Trials Search Co‐ordinator (TSC) searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials using the following search strategy which has been developed based on literature review and consulting with the authors of the review:
*Zuclopenthixol* in Intervention of STUDY
In such a study‐based register, searching the major concept retrieves all the relevant keywords and studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
Where necessary, we contacted the first author of each included study for information regarding unpublished trials. We noted the outcome of this contact in the included or awaiting assessment studies tables.
Data collection and analysis
For previous data collection and analysis see Appendix 2.
Selection of studies
For this update, review author EJB independently inspected citations from the searches and identified relevant abstracts. A random 20% sample was independently re‐inspected by Dr Marie Ann Purcell (MAP) to ensure reliability. Where disputes arose, the full report was acquired for more detailed scrutiny. Full reports of the abstracts meeting the review criteria were obtained and inspected by EJB. Again, a random 20% of full reports were‐inspected by MAP in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
For this update, review author EJB extracted data from all included studies. In addition, to ensure reliability, JX (see Acknowledgements) independently extracted data from a random sample of these studies, comprising 10% of the total. Again, any disagreements were discussed, decisions documented and, if necessary, authors of studies contacted for clarification. With remaining problems MAP helped clarify issues and these final decisions were documented. Data presented only in graphs and figures were to be extracted whenever possible, but included only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary. If studies were multi‐centre, where possible, we extracted data relevant to each component centre separately.
2. Management
2.1 Forms
We extracted data onto simple standard forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a) the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b) the measuring instrument has not been written or modified by one of the trialists for that particular trial. Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, in Description of studies we noted if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint) which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. Endpoint and change data were combined in the analysis as we used mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion:
Please note, we entered data from studies of at least 200 participants, for example, in the analysis irrespective of the following rules, because skewed data pose less of a problem in large studies. We also entered change data as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We presented and entered change data into the statistical analyses.
For endpoint data:
(a) when a scale starts from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation (SD). If this value was lower than 1, it strongly suggests a skew and the study was excluded. If this ratio was higher than 1 but below 2, there is suggestion of skew. We entered the study and tested whether its inclusion or exclusion changed the results substantially. Finally, if the ratio was larger than 2, the study was included, because skew is less likely (Altman 1996; Higgins 2011).
b) if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS) (Kay 1987), which can have values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for Zuclopenthixol dihydrochloride. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives, we reported data where the left of the line indicates an unfavourable outcome. This is noted in the relevant graphs.
Assessment of risk of bias in included studies
Again, review author EJB worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting. A random 20% was screened for validity independently by MAP.
If the raters disagreed, the final rating was made by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted the authors of the studies in order to obtain further information. We reported non‐concurrence in quality assessment , but if disputes arose as to which category a trial was to be allocated, again, we resolved this by discussion.
We noted the level of risk of bias in both the text of the review and in the 'Summary of findings' tables.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The number needed to treat for an additional beneficial outcome/number needed to treat for an additional harmful outcome (NNTB/NNTH) statistic with its confidence intervals is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' tableS, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference (SMD)). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and we calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering had been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies was possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, the additional treatment arms were presented in the comparisons. If data were binary, we simply added and combined them within the two‐by‐two table. If data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Where the additional treatment arms were not relevant, we did not use these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within the analyses. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we addressed this within the 'Summary of findings' tables by down‐rating quality. Finally, we also downgraded quality within the 'Summary of findings' tables should loss be 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat (ITT) analysis). Those leaving the study early were all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes, the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ were used for those who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when data only from people who completed the study to that point were compared to the ITT analysis using the above assumptions.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported, we reproduced these.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals available for group means, and either 'P' value or 't' value available for differences in mean, we could calculate them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the SE is reported, SDs are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recently methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups is often the core problem in randomised schizophrenia trials. We therefore did not exclude studies based on the statistical approach used. However, preferably we used the more sophisticated approaches. For example, MMRM or multiple‐imputation were preferred to LOCF and completer analyses were only presented if some kind of ITT data were not available at all. Moreover, we addressed this issue in the item "incomplete outcome data" of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, we fully discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, we fully discussed these.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 'P' value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. 'P' value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic, was interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We attempted to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol and in the published report. If the protocol was not available, we compared outcomes listed in the methods section of the trial report with actually reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). Again, these are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots were possible, we planned to seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies, even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose a random‐effects model for all analyses. The reader is, however, able to choose to inspect the data using the fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
No subgroup analysis was expected.
1.2 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of zuclopenthixol dihydrochloride for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage and with similar problems. This was not done in this update.
2. Investigation of heterogeneity
If inconsistency was high, this was reported. First, we investigated whether data had been entered correctly. Second, if data were correct, the graph was visually inspected and outlying studies were successively removed to see if homogeneity was restored. For this review we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, data were presented. If not, data were not pooled and issues discussed. We know of no supporting research for this 10% cut‐off but are investigating the use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious we simply stated hypotheses regarding these for future reviews or versions of this review. We do not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes, we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then all data were employed from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. If there was a substantial difference, we reported results and discussed them but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. A sensitivity analysis was undertaken to test how prone results were to change when completer‐only data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported results and discussed them but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then data from these trials were included in the analysis.
4. Imputed values
We also undertook a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
We synthesised all data using a random‐effects model, however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether this altered the significance of the results.
Results
Description of studies
Accross all comparisons there was a significant lack of data for the primary outcomes and the secondary outcomes. The majority of data were for the short term only, for inpatient populations and focused on adverse effects. Most studies were small with the exception of Fischer‐Cornelssen 1976.
Results of the search
Details of the search results are illustrated in the PRISMA tableFigure 1
1.
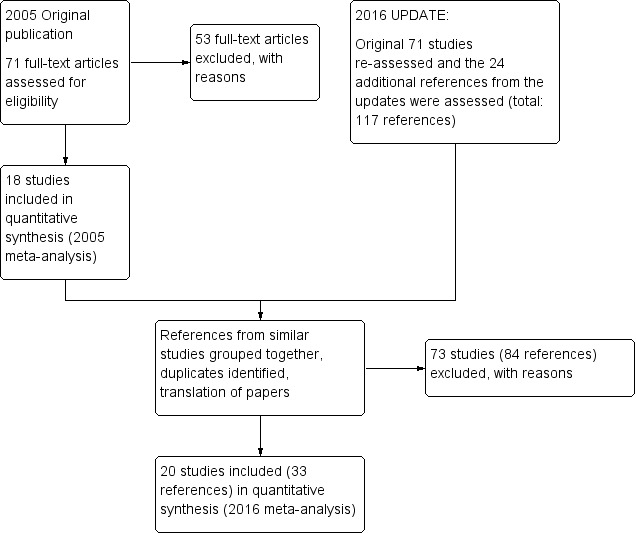
Study flow diagram ‐ update 2016.
After obtaining the initial results of the search, removing duplicates and clearly irrelevant material, the original review authors inspected 79 abstracts. Out of these 79 papers, they selected 71 full‐text papers to be assessed for eligibility. The original authors then grouped these into 'studies' where several of the reports referred to the same trial. Fifty‐three articles were excluded with reasons and 18 studies were included in the 2005 meta‐analysis.
During the update we identified 117 potential reports for the review. We had to exclude 73 (84 reports) of these studies. So, at the end of this review update we have identified 33 reports of 20 trials for inclusion in the 2016 meta‐analysis. At the time of writing zero studies are awaiting assessment and we know of no ongoing studies at the time of writing.
Included studies
We identified 20 studies (from 33 references) spanning the period 1968 to 2008 covering 12 comparisons for inclusion in this update. Fifteen were described as randomised and in the remaining five, randomisation was implied. One trial was a cross‐over trial and one was an open‐label trial. Two authors need to be contacted for additional information at the next update (Arango 2006 and Fagerlund 2004). Ban 1975 data has been updated to reflect the two‐week washout period not originally included in the initial publication.
The trial authors were not always explicit in their descriptions of randomisation or blinding and an assumption of either or both was made based on implication and context of the discussions and/or data. The majority of the data were short term and from inpatient samples. The studies used relatively small cohorts with the exception of Fischer‐Cornelssen 1976.
Where authors published research from the same cohort in several different reports the data were pooled and used only once in the meta‐analysis. Some degree of interpretation on our part was required during this process.
Further information on the included studies can be obtained in the Characteristics of included studies table.
Excluded studies
We excluded 73 studies: two had healthy volunteers, 42 used the wrong intervention (predominantly depot), four used the wrong type of participant, eight were not randomised, 16 did not have any useable data (either data that could be extracted for meta‐analysis or no data provided), and one duplicated data from elsewhere. Galdersi 1994 was originally included but excluded in this update because it was not a randomised trial.
Further information on the excluded studies can be obtained in theCharacteristics of excluded studies table.
Risk of bias in included studies
Most information is from studies at low or unclear risk of bias. It is difficult to provide a generalised assessment of the risk of bias given the low number of studies in this update. We would advise reviewing the 'Risk of bias' tables in the Characteristics of included studies table of the review.
Risk of bias overall: Unclear.
Allocation
Three studies were graded as low risk, 16 as unclear risk and one as high risk. All included studies were randomised or had implied randomisation. Most of the included studies did not explicitly describe the method by which randomisation was done.The majority of patients came from inpatient samples and there was a degree of diagnostic purity to the samples. Review of demographic tables (where published) indicated a roughly equal spread between different arms within the different studies.
Overall risk of selection bias: unclear.
Blinding
Thirteen studies were graded as low risk, four as unclear risk and three as high risk. Two of the studies were open‐label and all included studies used some form of established rating scale or modified rating scale to measure changes in global state, adverse effects and mental state. Several of the papers were vague on their methods for blinding, but it was generally implied in the majority. Only a few papers explicitly stated that blinding was conducted and this was usually mentioned in the title and/or abstract.
Overall risk of performance/detection bias: low risk.
Incomplete outcome data
Nine studies were graded as low risk, five as unclear risk and six as high risk. Reasons for loss to follow‐up are well‐reported and we have recorded these in the outcomes. Most studies, however, have not clearly described how they used data for people who were lost to follow‐up and we found no reporting of attempts to validate any assumptions by following up those who dropped out early. Per‐protocol analysis was used in eight studies.
Overall risk of attrition bias: low risk.
Selective reporting
Five studies were graded as low risk, five as unclear risk and 10 as high risk. Overall, due to poor reporting, we were unable to use a lot of the data (missing data were detected in nine of the studies). Findings presented as graphs, whether as percentiles or as inexact P values, are often of little use to a reviewer. Many studies failed to provide standard deviations when reporting mean changes. Three of the studies published the same data in multiple locations. One study presented its data inaccurately requiring EJB to make a professional judgement on what the authors intended.
Overall risk of reporting bias: high risk.
Other potential sources of bias
Two studies were graded as low risk, two as unclear risk and 16 as high risk. The predominant biases in this category are: response bias, instrument bias, interviewer bias and the Hawthorne effect. The authors of the included studies did not discuss these biases in any detail, if at all.
Overall risk of other bias: high risk.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12
Summary of findings for the main comparison. Zuclopenthixol dihydrochloride versus placebo.
| Patient or population: people with schizophrenia Setting: Hospital Intervention: ZUCLOPENTHIXOL Comparison: PLACEBO (short term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with PLACEBO (short term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Adverse effects: Clinically important general adverse effect (extrapyramidal effects ‐ UKU side effect rating scale) | 80 per 1000 | 486 per 1000 (69 to 1000) | RR 6.07 (0.86 to 43.04) | 28 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2, 3,4, 5, 6 | Risk assumed to be moderate and rounded from 7.69% to 8%. |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Service outcomes: Duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Leaving the study early (any reason) | 40 per 1000 | 12 per 1000 (0 to 264) | RR 0.29 (0.01 to 6.60) | 100 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1, 6 | Risk assumed to be moderate and rounded from 3.85% to 4%. Low number of events in both RCTs. |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'very serious' ‐ possible selection bias, blinding may have been single, attrition bias and reporting bias.
2 Risk of bias: rated 'very serious' ‐ selection, attrition and reporting bias
3 Risk of inconsistency: rated 'not serious' ‐ suspected but not found.
4 Risk of publication bias: rated 'strongly suspected' ‐ multiple papers published with the same patient cohort.
5 Risk of large effect: rated 'very large' ‐ RR 6.07, small n number but result likely when versus placebo.
6 Risk of imprecision: rated 'very serious' ‐ low n numbers
Summary of findings 2. Zuclopenthixol dihydrochloride versus chlorpromazine.
| Patient or population: schizophrenia Setting: Outpatient and inpatient (predominantly hospitalised) Intervention: ZUCLOPENTHIXOL Comparison: CHLORPROMAZINE (short term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with CHLORPROMAZINE (short term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (CGI‐SI, high score not reported, average score = 2.2) | The mean CGI‐SI endpoint score in the intervention group (MD) was 0 (‐0.49 lower to 0.49 higher) | ‐ | 64 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 5 | Translated study. | |
| Adverse effects: Clinically important general adverse effect (EPSEs) | 300 per 1000 | 282 per 1000 (183 to 435) | RR 0.94 (0.61 to 1.45) | 199 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 2 4 5 6 | Risk control rounded to 30% and set to moderate. Mixture of inpatient and outpatient, though predominantly hospitalised patients. |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (BPRS, high score = 34.2) | The mean mental state: average endpoint score (BPRS, high score = 34.2) in the intervention group was 0.4 more (2.43 fewer to 3.23 more) | ‐ | 221 (3 RCTs) | ⊕⊕⊝⊝ LOW 2 6 | Mixture of inpatient and outpatient, though predominantly hospitalised patients. | |
| Leaving the study early (any reason) | 70 per 1000 | 38 per 1000 (25 to 57) | RR 0.54 (0.36 to 0.81) | 766 (6 RCTs) | ⊕⊕⊝⊝ LOW 3 6 | Mixture of inpatient and outpatient, though predominantly hospitalised patients. Risk control set to moderate and rounded to 7%; extreme values not likely. |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated 'serious' ‐ Selection and attrition bias likely.
2 Risk of bias: rated 'serious' ‐ Selection attrition bias.
3 Risk of bias: rated 'serious' ‐ Selection, attrition and reporting bias.
4 Risk of bias: rated 'serious' ‐ Selection, attrition and reporting bias. Risk of inconsistency: rated as 'serious' ‐ All three papers reported on differing population sizes and obtained different levels of EPSEs in the experimental and control groups.
5 Risk of imprecision: rated 'very serious' ‐ low n numbers
6 Risk of indirectness: rated 'very serious' ‐ mixed samples
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 3. Zuclopenthixol dihydrochloride versus chlorprothixene.
| Patient or population: schizophrenia Setting: Hospital Intervention: ZUCLOPENTHIXOL Comparison: CHLORPROTHIXENE (medium term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with CHLORPROTHIXENE (medium term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Adverse effects: Clinically important general adverse effect (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Leaving the study early (any reason) | 400 per 1000 | 400 per 1000 (136 to 1000) | RR 1.00 (0.34 to 2.93) | 20 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'serious' ‐ selection, reporting bias likely.
2 Risk of publication bias: rated as 'strongly suspected' ‐ several papers published using the same cohort of patients.
3 Risk of imprecision: rated 'very serious' ‐ low n numbers
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 4. Zuclopenthixol dihydrochloride versus clozapine.
| Patient or population: schizophrenia Setting: Hospital Intervention: ZUCLOPENTHIXOL Comparison: CLOZAPINE (short term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with CLOZAPINE (short term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Adverse effects: Clinically important general adverse effect (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Leaving the study early (any reason) | 0 per 1000 | 0 per 1000 (0 to 0) | not estimable | 407 (1 RCT) | ⊕⊕⊝⊝ LOW 1 2 | Multi‐centre, multi‐drug trial with disproportionate numbers of people in different arms of the study. The authors did not report that anybody left the study in the Zuclopenthixol and clozapine arms. |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'serious' ‐ selection, attrition, reporting and performance bias likely.
2 Risk of indirectness: rated 'serious' ‐ Multiple study arms
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 5. Zuclopenthixol dihydrochloride versus haloperidol.
| Patient or population: schizophrenia Setting: Hospital only. Intervention: ZUCLOPENTHIXOL Comparison: HALOPERIDOL (short term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with HALOPERIDOL (short term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (CGI, mean score = 1.25) | The mean CGI endpoint score in the intervention group (MD) was 0.13 more (‐0.3 fewer to 0.55 more) | ‐ | 49 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 3 4 | Small study, multiple scales used (NOSIE30, BPRS, CGI). Paper only reported outcomes of some of these scales. | |
| Adverse effects: Clinically important general adverse effect (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Leaving the study early (any reason) | 300 per 1000 | 297 per 1000 (216 to 405) | RR 0.99 (0.72 to 1.35) | 141 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 3 4 | Risk control rounded to 30% from 29.25% and, as extreme values unlikely, it was set to moderate. |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'very serious' ‐ likely selection, attrition, reporting and diagnostic purity bias.
2 Risk of bias: rated as 'serious' ‐ likely selection, attrition, reporting and diagnostic purity bias. Risk of inconsistency: rated as 'serious' ‐ both papers generated differing values for people leaving the study and do not appear consistent (face validity).
3 Risk of imprecision: rated 'serious' ‐ low n numbers
4 Risk of indirectness: rated 'serious' ‐ not all scales reported
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 6. Zuclopenthixol dihydrochloride versus perphenazine.
| Patient or population: schizophrenia Setting: Hospital Intervention: ZUCLOPENTHIXOL Comparison: PERPHENAZINE (short and medium term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with PERPHENAZINE (short and medium term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Adverse effects: Clinically important general adverse effect (EPSEs requiring medication ‐ medium term) | 400 per 1000 | 760 per 1000 (448 to 1000) | RR 1.90 (1.12 to 3.22) | 50 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Leaving the study early (any reason, short and medium term) | 207 per 1000 | 130 per 1000 (56 to 304) | RR 0.63 (0.27 to 1.47) | 104 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'serious' ‐ likely selection and reporting bias.
2 Risk of imprecision: rated 'very serious' ‐ low n numbers
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 7. Zuclopenthixol dihydrochloride versus risperidone.
| Patient or population: schizophrenia Setting: Mostly hospital. Huttunen group did not state Intervention: ZUCLOPENTHIXOL Comparison: RISPERIDONE (short and medium term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with RISPERIDONE (short and medium term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Adverse effects: Clinically important general adverse effect ‐ short term (EPSEs requiring medication) | 271 per 1000 | 520 per 1000 (303 to 888) | RR 1.92 (1.12 to 3.28) | 98 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2 5 6 | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (PANSS, average score = 20.5) ‐ medium term | The mean PANNS endpoint score in the intervention group (MD) was 3.2 fewer (‐7.71 fewer to 1.31 more) | ‐ | 25 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | Small study. Standard deviations may be standard errors. Open label trial. | |
| Leaving the study early (any reason, short and medium term) | 310 per 1000 | 403 per 1000 (260 to 626) | RR 1.30 (0.84 to 2.02) | 154 (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 3 4 | Risk control taken as mean of extremes: high + low / 2 (changed from 21.43% to 31%) |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'very serious' ‐ open label trial, likely selection bias.
2 Risk of imprecision: rated as 'very serious' ‐ strongly suspect reported standard deviations are standard errors, low n numbers
3 Risk of publication bias: rated as 'strongly suspected' ‐ all studies published findings across several consecutive years in different journals and at conferences.
4 Risk of bias: rated as 'very serious' ‐ study 1 (selection, reporting, diagnostic purity and attrition bias likely ‐ author emailed); study 2 (open label); study 3 (reporting and attrition bias).
5 Risk of publication bias: rated as 'strongly suspected' ‐ multiple papers over consecutive years published with the same data.
6 Risk of bias: rated as 'serious' ‐ diagnostic purity and attrition bias; likely selection and reporting bias (authors contacted)
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 8. Zuclopenthixol dihydrochloride versus sulpiride.
| Patient or population: schizophrenia Setting: Not specified in study. Intervention: ZUCLOPENTHIXOL Comparison: SULPIRIDE (short term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with SULPIRIDE (short term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (CGI ‐ unchanged/worse) | 226 per 1000 | 266 per 1000 (111 to 644) | RR 1.18 (0.49 to 2.85) | 61 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | The study did not report clinical improvement so unchanged/worse is reported for this outcome. Usually we report clinical improvement. |
| Adverse effects: Clinically important general adverse effect ‐ requiring hypnotics/sedatives | 419 per 1000 | 252 per 1000 (113 to 554) | RR 0.60 (0.27 to 1.32) | 61 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (BPRS) | The mean BPRS endpoint score in the intervention group (MD) was 1.3 fewer (‐5.08 fewer to 2.48 more) | ‐ | 61 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| Leaving the study early (any reason) | 230 per 1000 | 476 per 1000 (223 to 1000) | RR 2.07 (0.97 to 4.40) | 61 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Control of risk rounded up from 22.58% to 23%. |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'serious' ‐ diagnostic purity, attrition bias. Per‐Protocol analysis suspected.
2 Risk of imprecision: rated as ' very serious' ‐ percentages used to describe data and comparisons are made against an assumption of baseline measurements. Low n numbers.
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 9. Zuclopenthixol dihydrochloride versus thiothixene.
| Patient or population: schizophrenia Setting: hospital Intervention: ZUCLOPENTHIXOL Comparison: THIOTHIXENE (medium term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with THIOTHIXENE (medium term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (unchanged/worse ‐ CGI) | 600 per 1000 | 300 per 1000 (102 to 876) | RR 0.50 (0.17 to 1.46) | 20 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | The study did not report clinical improvement so unchanged/worse is reported for this outcome. Usually we report clinical improvement. |
| Adverse effects: Clinically important general adverse effect (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported on this outcome. | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported on this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported on this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported on this outcome. | |
| Leaving the study early (any reason) | 700 per 1000 | 399 per 1000 (168 to 945) | RR 0.57 (0.24 to 1.35) | 20 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No studies reported on this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'serious' ‐ likely selection and reporting bias.
2 Risk of publication bias: rated as 'strongly suspected' ‐ several papers published using the same cohort.
3 Risk of imprecision: rated as 'serious' ‐ low n numbers
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 10. Zuclopenthixol dihydrochloride versus trifluoperazine.
| Patient or population: schizophrenia Setting: Hospital Intervention: ZUCLOPENTHIXOL Comparison: TRIFLUOPERAZINE (short term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with TRIFLUOPERAZINE (short term) | Risk with ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Adverse effects: Clinically important general adverse effect (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Leaving the study early (any reason) | 0 per 1000 | 0 per 1000 (0 to 0) | not estimable | 72 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | Multi‐centre and multi‐drug trial with low numbers in each arm. |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'serious' ‐ selection, attrition, reporting and performance bias likely.
2 Risk of imprecision: rated 'very serious' ‐ low n numbers
3 Risk of indirectness: rated 'very serious' ‐ multiple arms, some missing
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 11. Zuclopenthixol dihydrochloride versus zuclopenthixol depot.
| Patient or population: schizophrenia Setting: Hospital Intervention: ZUCLOPENTHIXOL Comparison: ZUCLOPENTHIXOL DEPOT (long term) | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | ||
| Risk with ZUCLOPENTHIXOL DEPOT (long term) | Risk with ZUCLOPENTHIXOL | ||||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | ||
| Adverse effects: Clinically important general adverse effect (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | ||
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | ||
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | ||
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | ||
| Leaving the study early (any reason) | 80 per 1000 | 156 per 1000 (29 to 846) | RR 1.95 (0.36 to 10.58) | 46 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | Control of risk rounded from 7.69% to 8%. | |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | ||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | |||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||||
1 Risk of bias: rated as 'very serious' ‐ single researcher did randomisation, selection bias likely, open label trial and incomplete outcome data.
2 Risk of publication bias: rated as 'strongly suspected' ‐ several papers published using the same data and cohort.
3 Risk of imprecision: rated 'very serious' ‐ low n numbers
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Summary of findings 12. Zuclopenthixol dihydrochloride (Cis Z isomer) versus Zuclopenthixol (Cis Z/Trans E isomer).
| Patient or population: schizophrenia Setting: Mostly inpatient. 4 Studies included of which two did not state but implied inpatient. Intervention: CIS‐(Z) ZUCLOPENTHIXOL Comparison: CIS(Z)/TRANS(E) ZUCLOPENTHIXOL (short term) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with CIS(Z)/TRANS(E) ZUCLOPENTHIXOL (short term) | Risk with CIS‐(Z) ZUCLOPENTHIXOL | |||||
| Global state: Average endpoint global state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Adverse effects: Clinically important general adverse effect | 470 per 1000 | 630 per 1000 (385 to 1000) | RR 1.34 (0.82 to 2.18) | 57 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | Control of risk set at moderate and rounded up from 46.4% to 47%. |
| Death: Suicide and natural causes (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Service outcomes: duration of stay in hospital (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Mental state: Average endpoint general mental state score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| Leaving the study early: Any reason | 28 per 1000 | 61 per 1000 (14 to 265) | RR 2.15 (0.49 to 9.41) | 140 (4 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | |
| General functioning: Average endpoint general functioning score (clinically improved) (no data) | not pooled | not pooled | not estimable | (0 studies) | No study reported this outcome. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Risk of bias: rated as 'serious' ‐ selection bias throughout. Very difficult to ascertain from published materials if selection bias has been minimised.
2 Risk of imprecision: rated 'very serious' ‐ low n numbers
For risks rated as serious, we downgraded by 1.
For risks rated as very serious we downgraded by 2.
Studies relevant to this review fall into 12 comparisons. We identified 20 randomised trials from which it was possible to extract numerical data. These data can be compared to the original published data in Appendix 3.
1. ZUCLOPENTHIXOL versus PLACEBO ‐ all short term
1.1 Leaving the study early (any reason)
For this outcome we found two relevant studies, with a total of 100 people. There was not a clear difference between zuclopenthixol dihydrochloride and placebo (risk ratio (RR) 0.29, 95% confidence interval (CI) 0.01 to 6.60, very low‐quality evidence) (Analysis 1.1).
1.1. Analysis.
Comparison 1 ZUCLOPENTHIXOL versus PLACEBO ‐ all short term, Outcome 1 Leaving the study early (any reason).
1.2 Adverse effects: 1. Clinically important change in specific adverse effects ‐ cardiovascular ‐ orthostatic
For this outcome we found a single study (total n = 28). We did not find evidence of a clear difference between the two treatments (RR 0.29, 95% CI 0.01 to 6.60) (Analysis 1.2).
1.2. Analysis.
Comparison 1 ZUCLOPENTHIXOL versus PLACEBO ‐ all short term, Outcome 2 Adverse effects: 1. Clinically important change in specific adverse effects ‐ cardiovascular ‐ orthostatic.
1.3 Adverse effects: 2. Clinically important change in specific adverse effects ‐ central nervous system ‐ arousal state
We identified one study relevant to this outcome.
1.3.1 excitation
We found one trial to be relevant to this subgroup, which included a total of 28 participants. There was not a clear difference (RR 2.62, 95% CI 0.12 to 59.40) (Analysis 1.3).
1.3. Analysis.
Comparison 1 ZUCLOPENTHIXOL versus PLACEBO ‐ all short term, Outcome 3 Adverse effects: 2. Clinically important change in specific adverse effects ‐ central nervous system ‐ arousal state.
1.3.2 sleepiness / sedation
We found one trial to be relevant to this subgroup (total n = 28). For this outcome, within this subgroup, we did find evidence that 'zuclopenthixol' was clearly different in its effects compared with 'placebo ‐ all short term' (RR 2.89, 95% CI 1.01 to 8.30). Zuclopenthixol increases the likelihood of sleepiness/sedation when compared to placebo (Analysis 1.3).
1.4 Adverse effects: 3. Clinically important change in specific adverse effects ‐ endocrine ‐ menstruation started
We identified one study relevant to this outcome involving 36 participants. There was not a clear difference (RR 7.00, 95% CI 0.39 to 126.48) (Analysis 1.4).
1.4. Analysis.
Comparison 1 ZUCLOPENTHIXOL versus PLACEBO ‐ all short term, Outcome 4 Adverse effects: 3. Clinically important change in specific adverse effects ‐ endocrine ‐ menstruation started.
1.5 Adverse effects: 4a. Any general adverse effects ‐ movement disorders ‐ EPSEs (UKU side effect rating scale, no scores)
There is a single trial (total n = 28), we did not find evidence of a clear difference between the two treatments when measured on the UKU (Lingjaerde 1987) (RR 6.07, 95% CI 0.86 to 43.04, very low‐quality evidence ). (Analysis 1.5).
1.5. Analysis.
Comparison 1 ZUCLOPENTHIXOL versus PLACEBO ‐ all short term, Outcome 5 Adverse effects: 4a. Any general adverse effects ‐ movement disorders ‐ EPSEs (UKU side effect rating scale, no scores).
1.6 Adverse effects: 4b. Clinically important change in specific adverse effects ‐ movement disorders ‐ EPSEs
1.6.1 parkinsonism
There is a single trial in this subgroup, which included a total of 36 participants. For this subgroup, we did not find evidence of a clear difference between the two treatments (RR 5.00, 95% CI 0.26 to 97.37) (Analysis 1.6).
1.6. Analysis.
Comparison 1 ZUCLOPENTHIXOL versus PLACEBO ‐ all short term, Outcome 6 Adverse effects: 4b. Clinically important change in specific adverse effects ‐ movement disorders ‐ EPSEs.
1.6.2 oculogyric crisis
We found one trial to be relevant to this subgroup (total n = 36). For this subgroup, we did not find evidence of a clear difference between the two treatments (RR 3.00, 95% CI 0.13 to 69.09) (Analysis 1.6).
1.6.3 tremor
We found one trial to be relevant to this subgroup (total n = 36). For this subgroup, we did not find evidence of a clear difference between the two treatments (RR 5.00, 95% CI 0.26 to 97.37) (Analysis 1.6).
2. ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term
This comparison has 15 outcomes.
2.1 Global state: 1. Average endpoint global state score ‐ Unchanged/worse (CGI, scores not reported)
For this outcome we found two relevant studies (total n = 135). There was not a clear difference in the CGI (Guy 1976) (RR 0.92, 95% CI 0.75 to 1.13) (Analysis 2.1).
2.1. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 1 Global state: 1. Average endpoint global state score ‐ Unchanged/worse (CGI, scores not reported).
2.2 Global state: 2. Average endpoint global state score ‐ no Recovery
We identified one study relevant to this outcome, with a total of 64 people. We did not find evidence of a clear difference between the two treatments in this comparison (RR 1.02, 95% CI 0.89 to 1.16) (Analysis 2.2).
2.2. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 2 Global state: 2. Average endpoint global state score ‐ No Recovery.
2.3 Global state: 3a. Average endpoint global state score (GAS, high score not reported, average score = 63.4)
For this outcome we found a single study (total n = 60). We did not find evidence of a clear difference between the two treatments in this comparison (MD ‐0.60 95% CI ‐8.12 to 6.92) (Analysis 2.3).
2.3. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 3 Global state: 3a. Average endpoint global state score (GAS, high score not reported, average score = 63.4).
2.4 Global state: 3b. Average endpoint global state score (CGI‐SI, high score not reported, average score = 2.2)
We identified one study relevant to this outcome, with a total of 60 people. We did not find evidence of a clear difference (mean difference (MD) 0.00, 95% CI ‐0.49 to 0.49, very low‐quality evidence) (Analysis 2.4).
2.4. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 4 Global state: 3b. Average endpoint global state score (CGI‐SI, high score not reported, average score = 2.2).
2.5 Mental state: 1. No clinically important change in general mental state ‐ Not improved (PANSS, scores not reported)
We identified one study relevant to this outcome (total n = 120). There was not a clear difference (RR 0.98, 95% CI 0.81 to 1.18) (Analysis 2.5).
2.5. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 5 Mental state: 1. No clinically important change in general mental state ‐ Not improved (PANSS, scores not reported).
2.6 Mental state: 2. No clinically important change in general mental state ‐ No clinical response
We identified one study relevant to this outcome involving 64 participants. We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.78, 95% CI 0.25 to 2.42) (Analysis 2.6).
2.6. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 6 Mental state: 2. No clinically important change in general mental state ‐ No clinical response.
2.7 Mental state: 3. Average endpoint general mental state score (BPRS, high score = 34.2)
For this outcome we found three relevant studies, with a total of 221 people. There was not a clear difference (MD 0.40, 95% CI ‐2.43 to 3.23) (Analysis 2.7).
2.7. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 7 Mental state: 3. Average endpoint general mental state score (BPRS, high score = 34.2).
2.8 Leaving the study early (any reason)
For this outcome we found six relevant studies (total n = 766). For this outcome, we did find evidence that 'zuclopenthixol' was clearly different in its effects compared with 'chlorpromazine ‐ all short term' (RR 0.54, 95% CI 0.36 to 0.81, low‐quality evidence). Chlorpromazine was more likely to cause patients to leave a study early for any reason (Analysis 2.8).
2.8. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 8 Leaving the study early (any reason).
2.9 Adverse effects: 1. Any general adverse effects ‐ side‐effects (CGI, high score not reported)
For this outcome we found a single study involving 94 participants. For this outcome, we did find evidence that 'zuclopenthixol' was clearly different in its effects compared with 'chlorpromazine ‐ all short term' (RR 0.86, 95% CI 0.77 to 0.97). There is a lower risk of general side‐effects measured on the CGI with zuclopenthixol (Analysis 2.9).
2.9. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 9 Adverse effects: 1. Any general adverse effects ‐ side effects (CGI, high score not reported).
2.10 Adverse effects: 2. Average endpoint general adverse effect score (TESS, high score not reported, average score = 12.00)
For this outcome we found a single study, with a total of 60 people. There was not a clear difference (MD 4.48, 95% CI ‐2.38 to 11.34) (Analysis 2.10).
2.10. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 10 Adverse effects: 2. Average endpoint general adverse effect score ‐ average score (TESS, high score not reported, average score = 12.00).
2.11 Adverse effects: 3. Any change in specific adverse effects ‐ cardiovascular ‐ postural hypotension (dizziness/syncope)
For this outcome we found a single study (total n = 43). There was not a clear difference (RR 0.11, 95% CI 0.01 to 1.73) (Analysis 2.11).
2.11. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 11 Adverse effects: 3. Any change in specific adverse effects ‐ cardiovascular ‐ postural hypotension (dizziness/syncope).
2.12 Adverse effects: 4. Any change in specific adverse effects ‐ central nervous system ‐ arousal
2.12.1 excitation
There is a single trial in this subgroup (total n = 43). For this subgroup, we did not find evidence of a clear difference between the two treatments (RR 0.62, 95% CI 0.07 to 5.47) (Analysis 2.12).
2.12. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 12 Adverse effects: 4. Any change in specific adverse effects ‐ central nervous system ‐ arousal.
2.12.2 sedation
There are two relevant trials in this subgroup (total n = 163). There was not a clear difference (RR 1.11, 95% CI 0.73 to 1.70) (Analysis 2.12).
2.13 Adverse effects: 5. Any change in specific adverse effects ‐ metabolic ‐ weight change ‐ loss or gain of weight of 10 pounds
There is a single study relating to this outcome (total n = 29). We did not find evidence of a clear difference between the two treatments (RR 0.62 95% CI 0.22 to 1.75) (Analysis 2.13).
2.13. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 13 Adverse effects: 5. Any change in specific adverse effects ‐ metabolic ‐ weight change ‐ loss or gain of weight of 10 pounds.
2.14 Adverse effects: 6a. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs
For this outcome we found three relevant studies involving 199 participants. There was not a clear difference (RR 0.94, 95% CI 0.61 to 1.45, very low‐quality evidence) (Analysis 2.14).
2.14. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 14 Adverse effects: 6a. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs.
2.15 Adverse effects: 6b. Any change in specific adverse effects ‐ movement disorders ‐ additional medication use
These data were presented in other data tables because it is not quantitative. The authors of all references did not specify which patient arms received which additional medication, benzhexol, diazepam, trihexphenidyl and scopolamine if necessary (Analysis 2.15).
2.15. Analysis.
Comparison 2 ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term, Outcome 15 Adverse effects: 6b. Any change in specific adverse effects ‐ movement disorders ‐ additional medication use.
| Adverse effects: 6b. Any change in specific adverse effects ‐ movement disorders ‐ additional medication use | ||
|---|---|---|
| Study | Zuclopenthixol | Chlorpromazine |
| Kingstone 1970 | n = 5, authors do not state which additional medication. | n = 2, authors do not state which additional medication. |
| Kordas 1968 | Benzhexol and diazepam if necessary. | Benzhexol and diazepam if necessary. |
| Wang 1995a | Trihexphenidyl or scopolamine used if necessary. Authors do not report frequency of use or which group used which. | Trihexphenidyl or scopolamine used if necessary. Authors do not report frequency of use or which group used which. |
3. ZUCLOPENTHIXOL versus CHLORPROTHIXENE ‐ all medium term
In this comparison, there were two outcomes.
3.1 Global state: Average endpoint global state score ‐ unchanged/worse (CGI)
For this outcome we found a single study (n = 20) that demonstrated no difference (RR 0.38, 95% CI 0.14 to 1.02) (Analysis 3.1).
3.1. Analysis.
Comparison 3 ZUCLOPENTHIXOL versus CHLORPROTHIXENE ‐ all medium term, Outcome 1 Global state: Average endpoint global state score ‐ Unchanged/worse (CGI).
3.2 Leaving the study early (any reason)
We identified one study relevant to this outcome (total n = 20). There was not a clear difference between 'zuclopenthixol' and 'chlorprothixene ‐ all medium term' (RR 1.00 95% CI 0.34 to 2.93, very low‐quality evidence) (Analysis 3.2).
3.2. Analysis.
Comparison 3 ZUCLOPENTHIXOL versus CHLORPROTHIXENE ‐ all medium term, Outcome 2 Leaving the study early (any reason).
4. ZUCLOPENTHIXOL versus CLOZAPINE ‐ all short term
4.1 Leaving the study early (any reason)
We identified one study relevant to this outcome, with a total of 407 people. We found no evidence of a clear difference between 'zuclopenthixol' and 'clozapine ‐ all short term' (RR not estimable) (Analysis 4.1).
4.1. Analysis.
Comparison 4 ZUCLOPENTHIXOL versus CLOZAPINE ‐ all short term, Outcome 1 Leaving the study early (any reason).
4.2 Adverse effects: Any general adverse effects ‐ side‐effects ‐ frequency per day
These data were presented in other data tables because the information is a list with percentages only (Analysis 4.2). A single RCT reported the following results.
4.2. Analysis.
Comparison 4 ZUCLOPENTHIXOL versus CLOZAPINE ‐ all short term, Outcome 2 Adverse effects: Any general adverse effects ‐ side effects ‐ frequency per day.
| Adverse effects: Any general adverse effects ‐ side effects ‐ frequency per day | ||
|---|---|---|
| Study | Zuclopenthixol (n = 36) | Clozapine (n = 38) |
| Fischer‐Cornelssen 1976 | Drowsiness (12%); Stimulation (14%); Confusion (1%); GI (1%); anticholinergic (14%); dizziness (6%); Orthostatic reaction (1%); Headache (2%); Hypersalivation (2%); hypokinesia (4%); hyperkinesia (2%); dyskinesia (0.4%); rigor (5%); tremor (5%); akathisia (3%) | Drowsiness (12%); Stimulation (21%); Confusion (3%); GI (8%); anticholinergic (8%); dizziness (15%); Orthostatic reaction (6%); Headache (3%); Hypersalivation (4%); hypokinesia (2%); hyperkinesia (4%); dyskinesia (0%); rigor (1%); tremor (3%); akathisia (3%) |
Drowsiness ‐ no difference (Zuclopenthixol 12% / Clozapine 12%)
Stimulation ‐ favours zuclopenthixol (14% / 21%)
Confusion ‐ favours zuclopenthixol (1% / 3%)
GI ‐ favours zuclopenthixol (1% / 8%)
Anticholinergic ‐ favours clozapine (14% / 8%)
Dizziness ‐ favours zuclopenthixol (6% / 15%)
Orthostatic reaction ‐ favours zuclopenthixol (1% / 6%)
Headache ‐ slightly favours zuclopenthixol (2% / 3%)
Hypersalivation ‐ slightly favours zuclopenthixol (2% / 4%)
Hypokinesia ‐ slightly favours clozapine (4% / 2%)
Hyperkinesia ‐ slightly favours zuclopenthixol(2% / 4%)
Dyskinesia ‐ no difference (0.4% / 0%)
Rigor ‐ favours clozapine (5% / 1%)
Tremor ‐ slightly favours clozapine (5% / 3%)
Akathisia ‐ no difference (3% / 3%)
5. ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term
This particular comparison has seven outcomes.
5.1 Global state: 1. Average endpoint global state score ‐ unchanged/worse (CGI)
We identified one study relevant to this outcome. No clear difference was observed (n = 63, 1 RCT, RR 0.86, 95% CI 0.60 to 1.24) (Analysis 5.1).
5.1. Analysis.
Comparison 5 ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term, Outcome 1 Global state: 1. Average endpoint global state score ‐ Unchanged/worse (CGI).
5.2 Global state: 2. Average endpoint global state score (CGI, high score = 1.25)
For this outcome we found a single study (total n = 49). We found no evidence of a clear difference (MD 0.13, 95% CI ‐0.30 to 0.55, very low‐quality evidence) (Analysis 5.2).
5.2. Analysis.
Comparison 5 ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term, Outcome 2 Global state: 2. Average endpoint global state score (CGI, mean score = 1.25).
5.3 Leaving the study early (any reason)
For this outcome we found two relevant studies involving 141 participants. We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.99, 95% CI 0.72 to 1.35, very low‐quality evidence). For this outcome heterogeneity is high (Chi2 = 0.0; df = 0.0; P = 0.0; I2 = 100%) (Analysis 5.3).
5.3. Analysis.
Comparison 5 ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term, Outcome 3 Leaving the study early (any reason).
5.4 Adverse effects: 1. Any change in specific adverse effects ‐ interference with functioning
There is a single trial in this subgroup (total n = 63). There was not a clear difference (RR 0.91 95% CI 0.64 to 1.29) (Analysis 5.4).
5.4. Analysis.
Comparison 5 ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term, Outcome 4 Adverse effects: 1. Any change in specific adverse effects ‐ interference with functioning.
5.5 Adverse effects: 2. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs ‐ requiring medication
There is a single trial (total n = 63), we did not find evidence of a clear difference between the two treatments (RR 0.83, 95% CI 0.47 to 1.45) (Analysis 5.5).
5.5. Analysis.
Comparison 5 ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term, Outcome 5 Adverse effects: 2. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs ‐ requiring medication.
5.6 Adverse effects: 3. Any change in specific adverse effects ‐ requiring additional medication
There is a single trial with a total of 63 people. We did not find evidence of a clear difference between the two treatments (RR 0.99, 95% CI 0.47 to 2.10) (Analysis 5.6).
5.6. Analysis.
Comparison 5 ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term, Outcome 6 Adverse effects: 3. Any change in specific adverse effects ‐ requiring additional medication.
5.7 Adverse effects: 4. Any change in specific adverse effects ‐ requiring hypnotics/sedatives
We found one trial to be relevant, which included a total of 63 participants. There was not a clear difference (RR 0.81, 95% CI 0.44 to 1.47) (Analysis 5.7).
5.7. Analysis.
Comparison 5 ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term, Outcome 7 Adverse effects: 4. Any change in specific adverse effects ‐ requiring hypnotics/sedatives.
6. ZUCLOPENTHIXOL versus PERPHENAZINE
In this comparison, there were four outcomes.
6.1 Global state: Average endpoint global state score ‐ unchanged/worse (global rating ‐ investigator opinion) ‐ medium term
For this outcome we found a single study (n = 50, 1 RCT, MD 1.50, 95% CI 0.48 to 4.68), which demonstrated no significant difference (Analysis 6.1).
6.1. Analysis.
Comparison 6 ZUCLOPENTHIXOL versus PERPHENAZINE, Outcome 1 Global state: Average endpoint global state score ‐ unchanged/worse (global rating ‐ investigator opinion) ‐ medium term.
6.2 Leaving the study early (any reason) ‐ short/medium term
For this outcome we found two relevant studies, with a total of 104 people. We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.63, 95% CI 0.27 to 1.47, very low‐quality evidence) (Analysis 6.2).
6.2. Analysis.
Comparison 6 ZUCLOPENTHIXOL versus PERPHENAZINE, Outcome 2 Leaving the study early (any reason) ‐ short/medium term.
6.3 Adverse effects: 1. Any change in specific adverse effects ‐ central nervous system ‐ arousal ‐ requiring medication ‐ medium term
There is a single trial with a total of 50 people. There was not a clear difference between 'zuclopenthixol' and 'perphenazine' (RR 2.00, 95% CI 0.98 to 4.10) (Analysis 6.3).
6.3. Analysis.
Comparison 6 ZUCLOPENTHIXOL versus PERPHENAZINE, Outcome 3 Adverse effects: 1. Any change in specific adverse effects ‐ central nervous system ‐ arousal ‐ requiring medication ‐ medium term.
6.4 Adverse effects: 2. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs ‐ requiring medication ‐ medium term
We found one trial to be relevant, which included a total of 50 participants. We found evidence of a clear difference between 'zuclopenthixol' and 'perphenazine' (RR 1.90, 95% CI 1.12 to 3.22, very low‐quality evidence). Zuclopenthixol is more likely to require medication in the short term for EPSEs than perphenazine (Analysis 6.4).
6.4. Analysis.
Comparison 6 ZUCLOPENTHIXOL versus PERPHENAZINE, Outcome 4 Adverse effects: 2. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs ‐ requiring medication ‐ medium term.
7. ZUCLOPENTHIXOL versus RISPERIDONE
This particular comparison has nine outcomes.
7.1 Mental State: 1. Average endpoint general mental state score (PANSS, average score = 45.8) ‐ medium term
For this outcome we found a single study (total n = 25). We did not find evidence of a clear difference between the two treatments in this comparison (MD ‐3.20, 95% CI ‐7.71 to 1.31, very low‐quality evidence) (Analysis 7.1).
7.1. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 1 Mental State: 1. Average endpoint general mental state score (PANSS, average score = 45.8) ‐ medium term.
7.2 Mental State: 2. Average endpoint general mental state score (PANSS General, average score medium term = 20.5) ‐ short/medium term
For this outcome we found two relevant studies, the data from which we divided into two subgroups in accordance with our protocol.
7.2.1 short term
There is a single trial in this subgroup, with a total of 19 people. We found evidence of a clear difference between 'zuclopenthixol' and 'risperidone' within this subgroup (MD ‐2.40, 95% CI ‐4.52 to ‐0.28). PANSS scores were more likely to improve with zuclopenthixol than with risperidone (Analysis 7.2).
7.2. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 2 Mental State: 2. Average endpoint general mental state score (PANSS General, average score medium term = 20.5) ‐ short/medium term.
7.2.2 medium term
We found one trial to be relevant to this subgroup, with a total of 25 people. For this subgroup, we did not find evidence of a clear difference between the two treatments (MD ‐0.30, 95% CI ‐2.72 to 2.12) (Analysis 7.2).
7.3 Mental State: 3. Average endpoint general mental state score (PANSS positive, average score medium term = 9.8) ‐ medium term
We identified one study relevant to this outcome involving 25 participants. For this outcome, we did not find evidence that 'zuclopenthixol' was clearly different in its effects compared with 'risperidone' (MD ‐1.00, 95% CI ‐2.69 to 0.69) (Analysis 7.3).
7.3. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 3 Mental State: 3. Average endpoint general mental state score (PANSS Positive, average score = 9.8) ‐ medium term.
7.4 Mental State: 4. Average endpoint general mental state score (PANSS negative, average score = 11.5) ‐ medium term
We identified one study relevant to this outcome (total n = 25). We did not find evidence of a clear difference between the two treatments in this comparison (MD ‐1.50, 95% CI ‐4.05 to 1.05) (Analysis 7.4).
7.4. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 4 Mental State: 4. Average endpoint general mental state score (PANSS Negative, average score 11.5) ‐ medium term.
7.5 Leaving the study early (any reason) ‐ short/medium term
For this outcome we found three relevant studies, with a total of 154 people. We did not find evidence of a clear difference between the two treatments in this comparison (RR 1.30, 95% CI 0.84 to 2.02, very low‐quality evidence) (Analysis 7.5).
7.5. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 5 Leaving the study early (any reason) ‐ short/medium term.
7.6 Adverse Effects: 1. Any general adverse effects ‐ additional medication use ‐ short/medium term
For zuclopenthixol (n = 18, 1 RCT), benzodiazepines were used on seven occasions and anticholinergics were used on 11 occasions. For risperidone (n = 15, 1 RCT), benzodiazepines were used on eight occasions and anticholinergics on seven occasions. One of the RCTs (Glenthoj 2007) did not differentiate between the two antipsychotics but reported combined totals of benzodiazepine use on 11 occasions, anticholinergic use on 10 occasions and antidepressant use on one occasion (Analysis 7.6).
7.6. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 6 Adverse Effects: 1. Any change in general adverse effects ‐ additional medication use ‐ short/medium term.
| Adverse Effects: 1. Any change in general adverse effects ‐ additional medication use ‐ short/medium term | ||
|---|---|---|
| Study | Zuclopenthixol | Risperidone |
| Fagerlund 2004 | Benzodiazepines n = 7 Anticholinergics n = 11 |
Benzodiazepines n = 8 Anticholinergics n = 7 |
| Glenthoj 2007 | Anticholinergic n = 10, Benzodiazepines n = 11, Antidepressant n = 1 | Authors do not differentiate the use of additional medication between the two groups, totals only given. |
7.7 Adverse effects: 2a. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs ‐ requiring medication ‐ short term
There is a single trial (total n = 98). We found evidence of a clear difference between 'zuclopenthixol' and 'risperidone' (RR 1.92, 95% CI 1.12 to 3.28, very low‐quality evidence). Zuclopenthixol was more likely to require medications fors EPSEs than risperidone (Analysis 7.7).
7.7. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 7 Adverse effects: 2a. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs ‐ requiring medication ‐ short term.
7.8 Adverse effects: 2b. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs (ESRS) ‐ short term
There is a single trial, which included a total of 19 participants. There was a clear difference between 'zuclopenthixol' and 'risperidone' (MD 4.50, 95% CI 0.67 to 8.33). Zuclopenthixol was more likely to cause EPSEs as measured on the ESRS (Chouinard 1980) than risperidone (Analysis 7.8).
7.8. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 8 Adverse Effects: 2b. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs (ESRS) ‐ short term.
7.9 Adverse effects: 3. Any change in specific adverse effects ‐ negative and cognitive symptoms of schizophrenia ‐ short term
We identified one study relevant to this outcome, the data from was used in one subgroup in accordance with our protocol.
7.9.1 UKU ‐ asthenia/lassitude/increased fatigability
We found one trial to be relevant to this subgroup, with a total of 98 people. There was not a clear difference between 'zuclopenthixol' and 'risperidone' within this subgroup (RR 0.82, 95% CI 0.67 to 1.01) (Analysis 7.9).
7.9. Analysis.
Comparison 7 ZUCLOPENTHIXOL versus RISPERIDONE, Outcome 9 Adverse effects: 3. Any change in specific adverse effects ‐ negative and cognitive symptoms of schizophrenia ‐ short term.
8. ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term
In this comparison, there were seven outcomes.
8.1 Global state: 1. Average endpoint global state score ‐ unchanged/worse (CGI)
We identified one study relevant to this outcome (n = 61). No clear difference was noted (RR 1.18, 95% CI 0.49 to 2.85, very low‐quality evidence) (Analysis 8.1).
8.1. Analysis.
Comparison 8 ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term, Outcome 1 Global state: 1. Average endpoint global state score ‐ Unchanged/worse (CGI).
8.2 Global State: 2. Average endpoint global state score ‐ moderately or severely ill (CGI)
We identified one study relevant to this outcome (total n = 61). We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.99, 95% CI 0.75 to 1.30). This outcome had important levels of heterogeneity (Chi2 = 0.0; df = 0.0; P = 0.0; I2 = 100%) (Analysis 8.2).
8.2. Analysis.
Comparison 8 ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term, Outcome 2 Global State: 2. Average endpoint global state score ‐ Moderately or severely ill (CGI).
8.3 Mental State: Average endpoint general mental state score (BPRS, average = 5.7)
We identified one study relevant to this outcome involving 61 participants. We did not find evidence of a clear difference between the two treatments in this comparison (MD ‐1.30, 95% CI ‐5.08 to 2.48) (Analysis 8.3).
8.3. Analysis.
Comparison 8 ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term, Outcome 3 Mental State: Average endpoint general mental state score (BPRS, average = 5.7).
8.4 Leaving the study early (any reason)
For this outcome we found a single study involving 61 participants. There was not a clear difference between 'zuclopenthixol' and 'sulpiride ‐ all short term' (RR 2.07, 95% CI 0.97 to 4.40, very low‐quality evidence; ) (Analysis 8.4).
8.4. Analysis.
Comparison 8 ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term, Outcome 4 Leaving the study early (any reason).
8.5 Adverse Effects: 1. Any general adverse effects ‐ additional medication use
See tables. Amitrypilline was used four times in each of the zuclopenthixol and sulpiride groups (Analysis 8.5).
8.5. Analysis.
Comparison 8 ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term, Outcome 5 Adverse Effects: 1. Any change in general adverse effects ‐ additional medication use.
| Adverse Effects: 1. Any change in general adverse effects ‐ additional medication use | ||
|---|---|---|
| Study | Zuclopenthixol | Sulpiride |
| Mahadevan 1991 | n = 4 Amitriptyline | n = 4 Amitryptyline |
8.6 Adverse Effects: 2. Any change in specific adverse effects ‐ metabolic ‐ weight change
We identified one study relevant to this outcome (total n = 61). There was not a clear difference between 'zuclopenthixol' and 'sulpiride ‐ all short term' (MD ‐1.60, 95% CI ‐8.35 to 5.15) (Analysis 8.6).
8.6. Analysis.
Comparison 8 ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term, Outcome 6 Adverse Effects: 2. Any change in specific adverse effects ‐ metabolic ‐ weight change.
8.7 Adverse Effects: 3. Any change in specific adverse effects ‐ requiring additional medication ‐ hypnotics/sedative
There is a single trial which included a total of 61 participants. There was not a clear difference between 'zuclopenthixol' and 'sulpiride ‐ all short term' (RR 0.60, 95% CI 0.27 to 1.32) (Analysis 8.7).
8.7. Analysis.
Comparison 8 ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term, Outcome 7 Adverse effects: 3. Any change in specific adverse effects ‐ requiring additional medication ‐ hypnotics/sedatives.
9. ZUCLOPENTHIXOL versus THIOTHIXENE ‐ all medium term
This comparison has two outcomes.
9.1 Global state: Average endpoint global state score ‐ unchanged/worse (CGI)
For this outcome we found one study (n = 20). No significant difference was found (RR 0.50, 95% CI 0.17 to 1.46, very low‐quality evidence) (Analysis 9.1).
9.1. Analysis.
Comparison 9 ZUCLOPENTHIXOL versus THIOTHIXENE ‐ all medium term, Outcome 1 Global state: Average endpoint global state score ‐ unchanged/worse (CGI).
9.2 Leaving the study early (any reason)
For this outcome we found a single study (total n = 20). We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.57, 95% CI 0.24 to 1.35, very low‐quality evidence) (Analysis 9.2).
9.2. Analysis.
Comparison 9 ZUCLOPENTHIXOL versus THIOTHIXENE ‐ all medium term, Outcome 2 Leaving the study early (any reason).
10. ZUCLOPENTHIXOL versus TRIFLUOPERAZINE ‐ all short term
This comparison has a single outcome.
10.1 Leaving the study early (any reason)
We identified one study relevant to this outcome (total n = 72). We found no evidence of a clear difference between 'zuclopenthixol' and 'trifluoperazine ‐ all short term' (RR not estimable, no events were reported by the authors) (Analysis 10.1).
10.1. Analysis.
Comparison 10 ZUCLOPENTHIXOL versus TRIFLUOPERAZINE ‐ all short term, Outcome 1 Leaving the study early (any reason).
11. ZUCLOPENTHIXOL versus ZUCLOPENTHIXOL DEPOT ‐ all long term
This comparison has four outcomes.
11.1 Leaving the study early (any reason)
We identified one study relevant to this outcome involving 46 participants. There are no subgroups in this outcome. There was not a clear difference between 'zuclopenthixol' and 'zuclopenthixol depot ‐ all long term' (RR 1.95, 95% CI 0.36 to 10.58, very low‐quality evidence) (Analysis 11.1).
11.1. Analysis.
Comparison 11 ZUCLOPENTHIXOL versus ZUCLOPENTHIXOL DEPOT ‐ all long term, Outcome 1 Leaving the study early (any reason).
11.2 Behaviour: Average change in specific aspects of behaviour ‐ Violence during follow‐up
We identified one study relevant to this outcome involving 46 participants. We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.87, 95% CI 0.44 to 1.71). This outcome had important levels of heterogeneity (Chi2 = 0.0; df = 0.0; P = 0.0; I2 = 100%) (Analysis 11.2).
11.2. Analysis.
Comparison 11 ZUCLOPENTHIXOL versus ZUCLOPENTHIXOL DEPOT ‐ all long term, Outcome 2 Behaviour: Average change in specific aspects of behaviour ‐ Violence during follow‐up.
11.3 Adverse Effects: 1a. Any general adverse effects ‐ additional medication use
One person in the zuclopenthixol group was prescribed propranolol (reasons not stated by the author). One person in the depot group was prescribed venlafaxine and one person prescribed lithium (Analysis 11.3).
11.3. Analysis.
Comparison 11 ZUCLOPENTHIXOL versus ZUCLOPENTHIXOL DEPOT ‐ all long term, Outcome 3 Adverse Effects: 1a. Any general adverse effects ‐ additional medication use.
| Adverse Effects: 1a. Any general adverse effects ‐ additional medication use | ||
|---|---|---|
| Study | Zuclopenthixol | Depot |
| Arango 2006 | n = 1 Propanolol | n = 1 venlafaxine n = 1 Lithium |
11.4 Adverse Effects: 1b. Any change in specific adverse effects ‐ additional medication use ‐ benzodiazepine use at least once
For this outcome we found a single study (total n = 46). There was not a clear difference between 'zuclopenthixol' and 'zuclopenthixol depot ‐ all long term' (RR 1.30, 95% CI 0.59 to 2.86) (Analysis 11.4).
11.4. Analysis.
Comparison 11 ZUCLOPENTHIXOL versus ZUCLOPENTHIXOL DEPOT ‐ all long term, Outcome 4 Adverse Effects: 1b. Any change in specific adverse effects ‐ additional medication use ‐ benzodiazepine use at least once.
12. CIS‐(Z) ZUCLOPENTHIXOL versus CIS(Z)/TRANS(E) ZUCLOPENTHIXOL ‐ all short term
This particular comparison has five outcomes.
12.1 Global state: Average endpoint global state score ‐ Unwell
We identified three studies relevant to this outcome involving 131 participants. We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.97, 95% CI 0.80 to 1.17) (Analysis 12.1).
12.1. Analysis.
Comparison 12 CIS‐(Z) ZUCLOPENTHIXOL versus CIS(Z)/TRANS(E) ZUCLOPENTHIXOL ‐ all short term, Outcome 1 Global state: Average endpoint global state score ‐ Unwell.
12.2 Mental state: Average endpoint general mental state score ‐ not improved
We identified one study relevant to this outcome (total n = 57). We did not find evidence of a clear difference between the two treatments in this comparison (RR 0.97, 95% CI 0.45 to 2.07) (Analysis 12.2).
12.2. Analysis.
Comparison 12 CIS‐(Z) ZUCLOPENTHIXOL versus CIS(Z)/TRANS(E) ZUCLOPENTHIXOL ‐ all short term, Outcome 2 Mental state: Average endpoint general mental state score ‐ Not improved.
12.3 Leaving the study early (any reason)
We identified four studies relevant to this outcome (total n = 140). There was not a clear difference between 'cis‐(z) zuclopenthixol' and 'cis(z)/trans(e) zuclopenthixol ‐ all short term' (RR 2.15, 95% CI 0.49 to 9.41, very low‐quality evidence) (Analysis 12.3).
12.3. Analysis.
Comparison 12 CIS‐(Z) ZUCLOPENTHIXOL versus CIS(Z)/TRANS(E) ZUCLOPENTHIXOL ‐ all short term, Outcome 3 Leaving the study early (any reason).
12.4 Adverse Effects: 1a. Any general adverse effects ‐ side‐effects reported
We identified one study relevant to this outcome (total n = 57). There was not a clear difference between 'cis‐(z) zuclopenthixol' and 'cis(z)/trans(e) zuclopenthixol ‐ all short term' (RR 1.34, 95% CI 0.82 to 2.18, very low‐quality evidence) (Analysis 12.4).
12.4. Analysis.
Comparison 12 CIS‐(Z) ZUCLOPENTHIXOL versus CIS(Z)/TRANS(E) ZUCLOPENTHIXOL ‐ all short term, Outcome 4 Adverse Effects: 1a. Any general adverse effects ‐ side effects reported.
12.5 Adverse Effects: 1b. Any change in specific adverse effects ‐ individual side‐effects
EPSEs were the most frequent adverse effect (no data) and the authors report no significant difference between the two isomers. For the Cis Z isomer: Dry mouth, Disturbance of accommodation, Disturbance of urination, Constipation, Dizziness, Headache, Increased sweating, Drowsiness, Anxiety, Parkinsonism, Akathisia, Tardive dyskinesia and others were reported. For the Cis Z/Trans E isomer: Dry mouth, Disturbance of urination, Constipation, Dizziness, Drowsiness, Parkinsonism, Akathisia, Tardive dyskinesia and others were reported (Analysis 12.5).
12.5. Analysis.
Comparison 12 CIS‐(Z) ZUCLOPENTHIXOL versus CIS(Z)/TRANS(E) ZUCLOPENTHIXOL ‐ all short term, Outcome 5 Adverse Effects: 1b. Any change in specific adverse effects ‐ individual side effects.
| Adverse Effects: 1b. Any change in specific adverse effects ‐ individual side effects | ||
|---|---|---|
| Study | Cis Z | Cis(Z)/Trans(E) |
| Gravem 1981 | EPSEs most frequent. Authors state no significant difference between two isomers. |
EPSEs most frequent. |
| Heikkila 1981 | Dry mouth, Disturbance of accommodation, Disturbance of urination, Constipation, Dizziness, Headache, Increased sweating, Drowsiness, Anxiety, Parkinsonism, Akathisia, Tardive dyskinesia and others | Dry mouth, Disturbance of urination, Constipation, Dizziness, Drowsiness, Parkinsonism, Akathisia, Tardive dyskinesia, others |
Discussion
In the original publication the authors completed the following analyses (Appendix 4).
Zuclopenthixol versus placebo (only short term)
Zuclopenthixol versus other typical antipsychotics (only short term)
Zuclopenthixol versus atypical antipsychotics (only short term)
Cis (Z) zuclopenthixol versus cis (Z)/Trans (E) zuclopenthixol (only short term)
For this update the original four analyses were modified and adapted in an attempt to reflect the evidence base for realistic clinical questions e.g. should I use zuclopenthixol instead of clozapine? It was felt that the original four comparisons were misleading to clinicians, patients and policy makers as they suggested that zuclopenthixol had been compared to all other possible antipsychotics. This was not the original authors intent.
This review update identified comparisons against nine alternative antipsychotics, only two of which were newer atypical drugs. Comparisons one and four remain and have been updated (see below).
Summary of main results
Compared to placebo
Data were lacking for this comparison and where data were available, they were only for the short term. Participants did not leave a study earlier when prescribed oral zuclopenthixol (n = 100, 2 RCTs, RR 0.29, 95% CI 0.01 to 6.60) which is surprising given that we expect the side‐effects of being on medication to be more frequent than being on no medication. This information is probably only of use to researchers and caution should be used in transferring this result into community or inpatient settings.
Extrapyramidal effects detected using the UKU side effect rating scale also did not reveal any significant difference, which is again surprising as you would expect more EPSEs on antipsychotic medication than on placebo (n = 28, 1 RCT, RR 6.07, 95% CI 0.86 to 43.04). This may be a consequence of the low number of trials and low participant numbers in these trials (see Table 1).
There was some evidence that zuclopenthixol increased the likelihood of patients experiencing more 'sleepiness/sedation' than placebo (n = 28, 1 RCT, RR 2.89 95% CI 1.01 to 8.3). There was little evidence on whether zuclopenthixol was different to non‐pharmacological treatment (e.g. psychotherapeutic methods, community care or no care at all) in terms of genuine improvements in patients' mental health.
The dose range of oral zuclopenthixol in these studies was 150 mg to 205 mg/day.
Compared to chlorpromazine
Again, despite a large number of participants in this comparison, data were lacking and only available for the short term. Chlorpromazine increases the likelihood of patients leaving a study early (n = 766, 6 RCTs, RR 0.54, 95% CI 0.36 to 0.81), though the reasons for this can only be hypothesised as being related to treatment as a whole as this information was not directly reported or commented on by the researchers. This information is probably only of use to researchers and caution should be used in transferring this result into community or inpatient settings (see Table 2).
More side‐effects were reported in patients on chlorpromazine (identified using the CGI; n = 94, 1 RCT, RR 0.86, 95% CI 0.77 to 0.97). Additional medication was used for patients on chlorpromazine and zuclopenthixol (trihexphenidyl, scopolamine, benzhexol and diazepam), but there are no data on frequency so inferring likelihood of use in clinical practice is difficult. The evidence does not help us decide to prescribe outside the usual clinical need. There was no evidence that zuclopenthixol was different to chlorpromazine with regards to improvements in patients' mental health.
The dose range of oral zuclopenthixol in these studies was 25 mg to 600 mg/day and the dose range of chlorpromazine was 100 mg to 1800 mg/day.
Compared to chlorprothixene
The evidence for this comparison is severely limited (see Table 3). More research is needed in the form of high‐quality randomised controlled trials.
The dose of oral zuclopenthixol in this single study was 50 mg to 200 mg/day and the dose range of chlorprothixene was 150 mg to 600 mg/day.
Compared to clozapine
Data were severely lacking for this comparison with no information about global state or mental state changes (see Table 4). Zuclopenthixol appears to have lower likelihood (at face value) of patients reporting 'stimulation', 'confusion', 'GI', 'dizziness', 'orthostatic reaction', 'headache', 'hypersalivation' and 'hyperkinesia' side‐effects than clozapine. Conversely, there is an increased likelihood of reporting 'anticholinergic', 'hypokinesia', 'rigor' and 'tremor' side‐effects.
The dose range of oral zuclopenthixol in this single study was 100 mg/day and the dose range of clozapine was 200 mg to 300 mg/day.
Compared to haloperidol
Table 5 demonstrates the poverty of information available for this comparison. No differences were detected for any of the outcomes when the dose range of oral zuclopenthixol was 40 mg to 205 mg/day and the dose range of haloperidol was4 mg to 12.3 mg/day. It is unlikely that there are no differences between the two medications, but we cannot infer any conclusions based on the current available evidence.
Compared to perphenazine
Patients did not leave a study any earlier whilst prescribed perphenazine or zuclopenthixol in the medium term (n = 104, 2 RCTs, RR 0.63, 95% CI 0.27 to 1.47). Caution should be used in translating this information into clinical practice, but this is useful to those carrying out research.
Zuclopenthixol has a greater likelihood of EPSEs that require medication (n = 50, 1 RCT, RR 1.90, 95% CI 1.12 to 3.22). The number needed to treat for an additional harmful outcome (NNTH) is 2.8, thus on average, 2.8 patients would have to receive zuclopenthixol treatment (instead of perphenazine treatment) for one additional patient to require medication for EPSEs. Any patients prescribed zuclopenthixol should be advised that this is the case and appropriate clinical caution should be taken (asking patients about EPSEs on each contact and/or consideration to EPSE rating scales). There is no evidence on how frequently this should be done.
The dose range of oral zuclopenthixol in this single study was 10 mg to 250 mg/day and the dose range of perphenazine was 8 mg to 72 mg/day. See Table 6.
Compared to risperidone
The baseline average PANSS general score of 26.4 SD 5.5 reduced to an average score of 19.3 SD 1.8 for patients on zuclopenthixol. The patients in the risperidone arm scored higher baseline averages (32.4 SD 6.2) and experienced a reduction in average scores at follow‐up (21.7 SD 2.9) that were roughly equivalent to zuclopenthixol. Despite the disparity in the patient baseline mental states (worse mental states in the risperidone arm), in the short term, there was a clear difference in favour of zuclopenthixol in the PANSS General score (n = 19, 1 RCT, MD ‐2.40, 95% CI ‐4.52 to ‐0.28).
The baseline average PANSS positive score of 19.1 SD 3.0 reduced to an average score of 9.8 SD 1.9 for patients on zuclopenthixol. The patients in the risperidone arm had similar baseline averages (20.9 SD 4.4) and experienced a similar reduction in average scores at follow‐up (10.8 SD 2.4). In the medium term there was no difference seen (n = 25, 1 RCT, MD ‐1.00, 95% CI ‐2.69 to 0.69).
Additional medication use (benzodiazepines and anticholinergics) in the short and medium term was associated with both antipsychotics, but frequency of use was the only data reported by the RCTs. No clear difference is observed at face value.
In the short term, zuclopenthixol has a greater likelihood of EPSEs requiring medication than risperidone (n = 98, 1 RCT RR 1.92, 95% CI 1.12 to 3.28, NNTH = 4). Any patients prescribed zuclopenthixol should be advised that this is the case and appropriate clinical caution should be taken (asking patients about EPSEs on each contact and/or consideration to EPSE rating scales). There is no evidence on how frequently this should be done.
For the dose ranges (zuclopenthixol 3.8 mg to 38 mg/day, risperidone 1.9 mg to 8 mg/day) in the included trials, the evidence seems to suggest some clinical benefit for using zuclopenthixol over risperidone, in terms of mental state measured using the PANNS, both in the short and medium term. There is some indication in the short term at these doses that zuclopenthixol will lead to more EPSEs that need medication than risperidone, but both are associated with additional medication use, specifically benzodiazepines and anticholinergics (see Table 7).
Compared to sulpiride
For the outcomes in the protocol, very limited evidence exists for this comparison. In the short term, no clear differences were detected causing patients to leave the study early for any reason, in the requirement of hypnotics and/or sedatives, in the global state measured using the CGI and in the mental state measured using the BPRS. Amitriptyline was used four times in the zuclopenthixol group and four times in the sulpiride group, suggesting that the occurrence of mood symptoms could be similar for patients prescribed both antipsychotics (see Table 8).
More research is needed in the form of high‐quality randomised controlled trials.
The dose of oral zuclopenthixol in this single study was 25 mg to 150 mg/day and the dose range of sulpiride was 200 mg to 1200 mg/day.
Compared to thiothixene
Extremely limited evidence is available for this comparison and only in the medium term. No clear difference was detected causing patients to leave the study early for any reason (n = 20, 1 RCT, RR 0.57, 95% CI 0.24 to 1.35) or in global state scores using the CGI (n = 20, 1 RCT, RR 0.50, 95% CI 0.17 to 1.46). More research is needed in the form of high‐quality randomised controlled trials.
The dose of oral zuclopenthixol in this single study was 50 mg to 200 mg/day and the dose range of thiothixene was 10 mg to 40 mg/day (see Table 9).
Compared to trifluoperazine
We did not identify any significant findings for this comparison (see Table 10) for any of the protocol outcomes. More research is needed in the form of high‐quality randomised controlled trials.
The dose of oral zuclopenthixol in this single study was 100 mg/day and the dose range of trifluoperazine was 20 mg to 30 mg/day.
Compared to zuclopenthixol depot
Evidence for this comparison was limited only to the longer term, where there was no clear difference detected causing patients to leave the study early for any reason (n = 46, 1 RCT, RR 1.95, 95% CI 0.36 to 10.58). More research is needed in the form of high quality‐randomised controlled trials.
The dose of oral zuclopenthixol in this single study was 35 mg/day and the dose of depot was 233 mg every 14 days (see Table 11).
Compared to zuclopenthixol isomer
In the short term, there was no clear difference detected causing patients to leave the study early for any reason (n = 140, 4 RCTs, RR 2.15, 95% CI 0.49 to 9.41), and no clear difference in general adverse effects (n = 57, 1 RCT, RR 1.34, 95% CI 0.82 to 2.18).
Dry mouth, disturbance of accommodation, disturbance of urination, constipation, dizziness, headache, increased sweating, drowsiness, anxiety, parkinsonism, akathisia, tardive dyskinesia and 'others' were reported for both isomers but no data were presented. This is perhaps useful only in as far as informing what side‐effects could develop in patients prescribed zuclopenthixol, but with no information on frequency, it is not possible to advise on how common these effects are.
More research is needed in the form of high quality‐randomised controlled trials.
The dose range of oral cis (Z) zuclopenthixol in these studies was 10 mg to 200 mg/day and the dose range of cis(Z)/trans(E) zuclopenthixol was 20 mg to 200 mg/day (see Table 12).
Overall completeness and applicability of evidence
1. Completeness
1.1 Relevance of the evidence to the review question
Zuclopenthixol dihydrochloride is generally used for patients with a psychotic illness and predominantly those with schizophrenia‐spectrum diagnoses. The primary and secondary outcomes of this review (death, service outcomes, mental state, leaving the study early, general functioning, behaviour, adverse effects, engagement with services, satisfaction with treatment, quality of life and economic) were not generally the primary focus of the included evidence. The extracted data were predominantly around mental state, leaving the study early, general functioning and adverse effects. Much of the data were obtained from rating scales which are not always used in clinical practice but are heavily used in research.
The included papers focused on (1) behaviour and therapeutic effect (versus placebo); (2) efficacy, dosage, tolerance and safety (versus chlorpromazine); (3) efficacy, dosage, tolerance and safety (versus clozapine); (4) efficacy, dosage, tolerance and safety (versus haloperidol); (5) efficacy, dosage, tolerance and safety (versus trifluoperazine); (6) clinical profile (versus perphenazine); (7) efficacy, tolerability, baseline and follow‐up basal ganglia volumes, executive functions, selective attention and reaction times (versus risperidone); (8) no clear focus (versus thiothixene and chlorprothixene); (9) violence reduction (versus depot zuclopenthixol) and (10) serum levels and clinical effect (versus zuclopenthixol isomers).
The main interventions of the included studies were antipsychotics and the outcomes were generally related to this. None of the included studies covered other psycho‐social aspects.
1.2 Overall judgement of the external validity of the review
Data were obtained predominantly for the short term (up to 12 weeks) and for inpatient populations. Some data were obtained for the medium term (13 to 26 weeks). Longer‐term data (> 26 weeks) were obtained only for depot zuclopenthixol. The studies were conducted either in Europe or North America (though this update does add some Chinese research).
The majority of trials involved inpatient participants with little in the way of physical and psychiatric co‐morbidity and with well‐defined schizophrenia or schizoaffective disorder. Such people are a minority in everyday care, where it is the norm to find people (not in a hospital setting) who suffer from less well‐defined illnesses combined with problems such as depression, personality disorder and substance abuse/misuse. Much of the data were obtained from rating scales which are not always used in clinical practice but are heavily used in research.
The results of this study can be generalised in a limited capacity in the short term (up to 12 weeks) to inpatient populations based mainly in European and American healthcare systems that are similar to those during the period 1968 to 2008.
2. Applicability of findings
2.1 The results and current practice
Current practice is unlikely to be significantly changed in Europe or the USA by the results of this meta‐analysis. The results do continue to support the importance of reviewing patients for EPSEs when they are prescribed oral zuclopenthixol, but advice on frequency of monitoring cannot be drawn from the data included. Much of the data focus on older antipsychotics, which tend to be used as second‐ or third‐line agents currently (including zuclopenthixol), though the data on superiority/inferiority were lacking and limited to only nine comparisons.
Small but significant improvements in average endpoint mental states are seen with zuclopenthixol when compared to the newer risperidone, but at the cost of an increased chance of EPSEs requiring further medication in the form of anticholinergics and benzodiazepines. Again, of the newer agents, evidence was only found for risperidone and clozapine, thus not allowing us to comment on the wide range of other newer anti‐psychotics.
The evidence is primarily generated from inpatient samples and caution should be given to using the results for patients in the community, general hospital settings or outpatient departments. The dose range of oral zuclopenthixol dihydrochloride across all of the included studies was 10 mg to 600 mg/day.
2.2 The results and current international practice
The authors of this paper are not familiar with all areas of current international clinical practice and advise caution interpreting and generalising the findings of this update to areas outside of Europe and the USA where healthcare systems could be very different from those covered in the included studies.
Quality of the evidence
1. Possibility of a robust conclusion
The evidence identified in this review is only for 12 comparisons (n = 1850, 20 RCTs, 1968 to 2008), and most of these comparisons are for older antipsychotics and/or antipsychotics that are not used commonly in clinical practice currently.
The largest study had 723 participants but the majority of studies were small. Data were obtained for patients with a psychotic illness and predominantly those with schizophrenia‐spectrum diagnoses. The evidence was mainly for the short term (up to 12 weeks) and for inpatient populations. Some data were obtained for the medium term (13‐26 weeks) and longer‐term data (> 26 weeks) were obtained only for depot zuclopenthixol.
The included studies were all RCTs of varying degrees of quality though no significant key methodological flaws were detected. Inconsistency of results between RCTs was difficult to assess given the small number of included studies and the wide number of non‐equal comparisons.
This makes drawing robust conclusions about the use of zuclopenthixol dihydrochloride for schizophrenia in the broader sense more difficult.
2. Overall quality of the evidence
The overall quality of the evidence contributing to the findings of this review ranged from very low to medium, with much of the evidence falling into either very low to low quality using GRADE. The reasons for the down grading of the included evidence was mostly because of biases (reporting, selection, Berkson, publication and attrition), and these reasons are clarified in the footnote section of each 'Summary of findings' table.
Potential biases in the review process
1. Preventing biases: strengths
The Cochrane systematic review process is designed to prevent and minimise biases introduced by authors of reviews. A single review author (EJB), independently of the original co‐authors (AK and DS), and at a different point in time, re‐appraised the original papers for inclusion in the review update in addition to appraising the suitability of new evidence for inclusion.
EJB utilised a strict system of appraisal of evidence as guided by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and attempted to contact the original authors where possible/appropriate. At several stages in the review process, a 10% sample of appraised papers, included papers and extracted data was assessed by review author MAP to ensure consensus was reached.
EJB has no known conflicts of interest.
2. Preventing biases: limitations
A systematic review is only ever as good at preventing bias in the findings as the original evidence used in determining the findings. For example, the review data are predominantly from inpatient samples, so the review findings are predominantly focused on this patient population. The review cannot correct for many of the biases of the original evidence. There may also be biases introduced through the systematic approach that cannot be controlled/minimised. The methods used to identify the evidence could have introduced biases at different stages e.g. searching, study selection, data collection, analysis.
3. Missing relevant studies
Judging by the preponderance of small positive studies included in this review update, it is likely that not all evidence has been identified. Several of the papers included had results published that could not be extracted for meta‐analysis, even though they may have been relevant to the review question.
Agreements and disagreements with other studies or reviews
The previous incarnation of this review suggested oral zuclopenthixol dihydrochloride caused movement disorders, perhaps more so than newer antipsychotics, but no more frequently than other older antipsychotics. This update agrees partially with these conclusions (it does cause movement disorders), but restricts their breadth as data were only obtained for two newer agents (clozapine and risperidone) and only for seven older agents. The only two comparisons that suggested oral zuclopenthixol dihydrochloride caused more EPSEs was with perphenazine and risperidone. There was no significant evidence of more EPSEs being reported when compared to placebo. More side‐effects were reported with chlorpromazine than with zuclopenthixol.
Additionally, the previous version of this review indicated that oral zuclopenthixol dihydrochloride may offer a clinical advantage in terms of global state over other older alternatives in the short term, but would need to be taken with additional medication to temper the sequelae of EPSEs. The results of this update do not support this statement, at least for the comparisons where data were obtained.
This update suggests zuclopenthixol dihydrochloride only has a modest clinical advantage with regards to mental state (measured on the PANSS) when compared to risperidone only (short term).
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
There is evidence that zuclopenthixol causes movement disorders that require additional medication, but no more so than other agents (excepting perphenazine and risperidone where it caused more, and chlorpromazine where it caused less). There is some suggestion from this review update that oral zuclopenthixol may have some clinical advantage with regards to improvements in mental state compared to risperidone in the short term. There was no evidence of a disadvantage or advantage when compared to the other agents in this review.
No significant data were obtained for the following outcomes: death, global state, general functioning, quality of life or satisfaction with treatment. This leaves patients with schizophrenia with a difficult decision to make when deciding on whether to take zuclopenthixol dihydrochloride versus another agent. It should still remain an option for treatment.
If an older drug is going to be used though, zuclopenthixol dihydrochloride is a viable option, but may be best taken with additional medication to offset movement disorders.
2. For family/care‐givers of people with schizophrenia
There is no relevant data from the included research that pertain to this important dimension of care in the update. There is evidence that zuclopenthixol dihydrochloride causes movement disorders, thus it is important that care‐givers/family are aware of the relevant symptoms/signs, that they have clear and straightforward ways to seek help and that they know what to do in the event of an emergency.
3. For clinicians
There is no evidence that zuclopenthixol dihydrochloride is any worse than other compounds and some indications that it is in fact better. Any difference in the isomers is speculative, but there are some data to suggest a real advantage in improvements in mental state scores in comparison with risperidone in the short term only.
As with other older generation drugs, the use of zuclopenthixol dihydrochloride is tainted by the need for drugs to counter movement disorders. All patients commenced on zuclopenthixol should be advised of the potential for movement disorders and the likely need for additional medication. The patient on zuclopenthixol dihydrochloride should have regular reviews for such side‐effects as this may affect adherence to treatment and engagement with services, though evidence for this is lacking.
There are no significant data for death, global states, service outcomes, general functioning, behaviour, engagement with services, satisfaction with treatment or quality of life, which leaves clinicians without a definitive answer when deciding whether or not to suggest zuclopenthixol dihydrochloride in the treatment of schizophrenia. What data are available are mostly for inpatient samples in Western healthcare systems.
The evidence does suggest that zuclopenthixol dihydrochloride should remain an option in the treatment of schizophrenia where indicated, though the clinical effects may be more pronounced in inpatient samples than for other populations.
4. For managers/policy makers
There are no data on service outcomes and no medium‐ or long‐term data. In the context of finite resources, the lack of good quality data leaves managers and policy makers with difficult decisions to make. Zuclopenthixol dihydrochloride should remain a choice in the treatment of people for whom older generation drugs are indicated.
Implications for research.
1. General
If the recommendations of the CONSORT statement (Moher 2001) had been anticipated by trialists much more data would have been available. Allocation concealment is essential for the result of a trial to be considered valid and gives the assurance that selection bias is kept to the minimum. Well‐described and tested blinding could have encouraged confidence in the control of performance and detection bias. It is also important to know how many, and from which groups, people were withdrawn, in order to evaluate exclusion bias. It would have been helpful if authors had presented data in a useful manner that reflects association between intervention and outcome, for example, relative risk, odds‐ratio, risk or mean differences, as well as raw numbers. Binary outcomes should be calculated in preference to continuous results, as they are easier to interpret. If P values are used, the exact value should be reported.
2. Specific
2.1 Reviews
From the excluded studies there are several reviews that are suggested. This review, although far from complete, will not need updating again in the future unless any significant, large multi‐centre double‐blind randomised controlled trials (RCTs) are published. Each of the comparisons in this review would potentially serve as a review in its own right.
2.2 Trials
We do think that more trials are indicated. These should not only be large and long, but should also adhere to a pragmatic design in order to increase applicability. Methods should be strict and involve good concealment of allocation and follow‐up. Participants should be people recognisable in everyday life and not those who are so strictly diagnosed as to render them unrecognisable to routine care.
Interventions should involve standard doses of zuclopenthixol and a control drug that is a real choice in the region of the study. This could equally be a new generation drugs such as risperidone, or an older medication such as chlorpromazine. Outcomes should be measured over months rather than weeks as this is the usual period a person would be asked to take the drug. We suggest that if scales are to be used, validated and clinically meaningful outcomes are pre‐defined.
Routine outcomes such as relapse, employment, housing status, satisfaction with care, serious or troubling adverse effects can all be easily recorded without the use of scales and we would suggest that these are included in the design of the study. Quality of life measures were generally missing from the trials and we would advocate for more studies including this outcome.
What's new
| Date | Event | Description |
|---|---|---|
| 25 October 2017 | New citation required but conclusions have not changed | New data do not substantively alter the overall conclusions of this review. |
| 3 May 2017 | New search has been performed | Results from 2012 and 2015 searches added to review. Two new studies included, 20 new studies excluded. Review now includes 20 trials, reporting data for 12 comparisons. |
History
Protocol first published: Issue 4, 2005 Review first published: Issue 4, 2005
| Date | Event | Description |
|---|---|---|
| 9 June 2015 | Amended | Search updated and 16 References (14 Studies) were added to 'Studies awaiting classification' section of the review. |
| 5 June 2015 | Amended | July 2012 update included in analyses. Risk of bias tables updated. Original included and excluded studies reviewed. Text revised. Structure revised. |
| 23 July 2012 | Amended | Update search of Cochrane Schizophrenia Group's Trial Register (see Search methods for identification of studies), 8 studies added to Studies awaiting classification. |
| 23 April 2008 | Amended | Converted to new review format. |
| 10 July 2005 | New citation required and conclusions have changed | Substantive amendment |
Notes
No notes to be published.
Acknowledgements
With thanks to Judy Wright, Gill Rizzello, Tessa Grant, John Rathbone and Clive Adams of the Cochrane Schizophrenia Group, University of Leeds, Leeds, UK. Thank you also to Mahesh Jayaram, Specialist Registrar, Leeds, UK. Additional thanks to Dr Marie Ann Purcell (Consultant General Practitioner, Market Surgery, Wath‐upon‐Dearne, Rotherham, UK). We would like to thank Daniel Strech for his contribution to the protocol and previous version of this review. We would also like to thank Amna Bibi, Michael Albert and Valerie Taylor for peer reviewing this version of the review.
Jun Xia helped us with data extraction for the update version by carrying out the reliability checks.
Parts of this review were generated using RevMan HAL v 4.2. You can find more information about RevMan here.
'Summary of findings' tables were generated by the online software GradPro. You can find more information about GradePro here.
Appendices
Appendix 1. Previous searches
1. Electronic searches: We searched the Cochrane Schizophrenia Group's study‐based Register (December 2004) using the phrase:[ ( (**zuclopenthixol* or *ciatyl* or *cisordinol* or *clopenthixol* or *clopixol* or *sordinol*) in REFERENCE) and ( (clopenthixol* or 0‐108* or cisordinol* or clopixol* or zuclopenthix*) in STUDY)]
This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group Module).
1.2. Reference searching We inspected references of all identified studies for more studies.
1.3. Personal contact We contacted the first author of each included study for more information regarding unpublished trials.
1.4. Drug companies We contacted the Lundbeck Limited for further data.
2. Cochrane Schizophrenia Group Trials Register (July 2012)
The Trials Search Co‐ordinator searched the Cochrane Schizophrenia Group’s Trials Register (10 July 2012).
1.1 Intervention search
The ‘Intervention’ field will be searched using the phrase:
((*zuclopenthix* or *ciatyl* or *cisordinol* or *clopenthixol* or *clopixol* or *sordinol*) AND *placebo*) The Cochrane Schizophrenia Group’s Trials Register is compiled by systematic searches of major databases, handsearches of relevant journals and conference proceedings (see group module).
Trials identified through the searching activities are each assigned to awaiting classification of relevant review titles.
Appendix 2. Previous methods
1. Selection of trials We independently inspected the citations identified from the search. We identified potentially relevant abstracts and ordered full papers and reassessed these for inclusion and methodological quality. We discussed and reported any disagreement.
2. Quality assessment We allocated trials to three quality categories, as described in the Cochrane Collaboration Handbook (Higgins 2011). When disputes arose as to which category a trial was allocated, again, we attempted resolution by discussion. When this was not possible and further information was necessary to clarify into which category to allocate the trial, we did not enter data but allocated the trial to the list of those awaiting assessment. We only included trials in Category A or B in the review.
3. Data management
3.1 Data extraction We independently extracted data from selected trials. When disputes arose, we attempted resolution by discussion. When this was not possible and further information was necessary to resolve the dilemma, we did not enter data but added this outcome of the trial to the list of those awaiting assessment.
3.2 Intention to treat analysis We excluded data from studies where more than 50% of participants in any group were lost to follow up, except for the outcome of 'leaving the study early'. In studies with less than 50% dropout rate, everyone allocated to the intervention was counted whether or not they completed follow up. We considered those leaving early to have had the negative outcome, except for the event of death and adverse effects.
Where attrition rates were high (25‐50%), we analysed the impact of including this type of data in a sensitivity analysis. If inclusion of high attrition data resulted in a substantive change in the estimate of effect, then we did not pool this data, but presented the data separately.
4. Data analysis
4.1 Binary data For binary outcomes we calculated a standard estimate of the relative risk (RR) and its 95% confidence intervals (CI) (fixed effect). Where possible, we estimated the number needed to treat (NNT) using an on‐line calculator (http://www.nntonline.net/). If heterogeneity was found (see section 5) we used a random‐effects model.
4.2 Continuous data 4.2.1 Intention‐to‐treat analyses versus analyses that only take into account those who completed the study: in the case of continuous data, it was supposed that in many cases an intention‐to‐treat analysis would not be available, so an analysis was presented on those who completed the study.
4.2.2 Rating scales: A wide range of instruments are available to measure mental health outcomes. These instruments vary in quality and it has been shown that the use of rating scales which have not been described in a peer‐reviewed journal (Marshall 2000) are associated with bias, or may not be valid, or even ad hoc. Therefore, some minimum standards were set: (a) the psychometric properties of the instrument should have been described in a peer‐reviewed journal; (b) the instrument should either be a self‐report, or completed by an independent rater or relative (not the therapist); and (c) the instrument should be a global assessment of an area of functioning.
4.2.3 Normal distribution of data: mental health continuous data are often not normally distributed. Most statistics assume a normal distribution. To avoid including non‐normally distributed data in the statistical analysis we applied the following criteria to all data before inclusion:
a. Standard deviations and means were reported or derivable from data in the paper, or were obtainable from the authors. b. When a scale started from zero, the standard deviation, when multiplied by two, was less than the mean (as otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution (Altman 1996)). Endpoint scores on scales often have a finite start and end point and this rule can be applied to them. c. When continuous data are presented on a scale which includes a possibility of negative values (such as change on a scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not. It is thus preferable to use scale end point data, which typically cannot have negative values. If end point data were not available, we chose to use change data, because the statistics used in Metaview are rather robust towards skew. d. If a scale starts from a positive value (such as PANSS, which can have values from 30‐210) the calculation described above in (b) should be modified to take the scale starting point into account. In these cases skew is present if 2SD> (S‐Smin), where S is the mean score and Smin is the minimum score.
4.2.4 Endpoint versus change data: where possible, we presented endpoint data and if both endpoint and change data were available for the same outcomes then we only reported the former in this review.
4.2.5 Summary statistic: For continuous outcomes we calculated weighted mean differences (WMD) and respective 95% CI (fixed effect). If heterogeneity was found (see section 5) we used a random effects model.
4.3 Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997, Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
Where cluster studies were appropriately analysed taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies was possible using the generic inverse variance technique.
5. Test for inconsistency Firstly, consideration of all the included studies within any comparison was undertaken to estimate clinical heterogeneity. Then visual inspection of graphs was used to investigate the possibility of statistical heterogeneity. This was supplemented employing, primarily, the I‐squared statistic. This provides an estimate of the percentage of inconsistency thought to be due to chance. Where the I‐squared estimate included 75% this was interpreted as evidence of high levels of heterogeneity (Higgins 2003). Data were then re‐analysed using a random effects model to see if this made a substantial difference. If it did, and results became more consistent, falling below 75% in the estimate, the studies were added to the main body trials. If using the random effects model did not make a difference and inconsistency remained high, data were not summated, but were presented separately and reasons for heterogeneity investigated.
6. Addressing publication bias We entered all data from the included studies into a funnel graph (trial effect against trial size) in an attempt to investigate the likelihood of overt publication bias (Egger 1997).
7. Sensitivity analyses The effect of including studies with high attrition rates was analysed in a sensitivity analysis.
8. General Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for zuclopenthixol dihydrochloride.
Appendix 3. Previous effects of interventions
1. The search We found a total of 85 citations using the search strategy. Out of these 85 only 26 fulfilled the criteria for our review. 2. COMPARISON 1. ZUCLOPENTHIXOL versus PLACEBO This comparison included two studies (total n=74). Neither study reported global or mental state outcomes. 2.1 Adverse effects Serafetinides 1972 showed that zuclopenthixol is associated with less orthostatic adverse effects than placebo but not to conventional levels of statistical significance (n=28, 1 RCT, RR 0.29, CI 0.01 to 6.60). The two studies, Serafetinides 1972 and Kordas 1968, reported different extrapyramidal adverse effects but overall zuclopenthixol did increase a persons risk of having any of these symptoms compared with placebo (n=64, RR 5.37, CI 1.12 to 29.34 NNH 2 CI 2 to 31). Other adverse effects reported by Serafetinides 1972 were excitation, sleepiness/sedation and weight change. Excitation, sleepiness/sedation were more prominent with zuclopenthixol than placebo (n=28, 1 RCT, RR 2.89, CI 1.01 to 8.30 NNH 3 CI 2 to 435). However weight gain/loss of ten pounds showed that people in the placebo group were no more prone to lose/gain weight than those in the zuclopenthixol group (n=28, 1 RCT, RR 0.43 CI 0.17 to 1.11). 2.2 Leaving the study early Serafetinides 1972 reported leaving the study early due to adverse effects and we found fewer people allocated zuclopenthixol left in the short term compared with those given placebo. However these did not meet conventional levels of statistical significance (n=28, RR 0.29 CI 0.01 to 6.6). 3. COMPARISON 2. ZUCLOPENTHIXOL versus OTHER TYPICAL ANTIPSYCHOTICS (only short term) This comparison included ten studies (total n=478). 3. 1 Global state Seven studies presented categorical data on global state and reported these in different ways. We were most frequently able to extract data on the short term outcome of unchanged or worse. Being allocated to the various control drugs was significantly associated with being unchanged or worse, compared with being given zuclopenthixol (n=357, 7 RCTs, RR 0.72 CI 0.53 to 0.98, NNT 10 CI 6 to 131). 3.2 Mental state There appeared to be no significant difference between about 122 mg/day zuclopenthixol and about 435 mg/day chlorpromazine, on BPRS as a continuous outcome measure (n=41, 1 RCT, WMD ‐2.66 CI ‐9.09 to 3.77). Other BPRS and CPRS data that were too skewed to present graphically also did not point to any difference between groups. 3.3 Adverse effects Several studies report general adverse effects. None of the findings suggest any clear difference between zuclopenthixol and other typical antipsychotics across a whole range of effects, including movement disorders (n=280, 6 RCTs, RR needing additional antiparkinsonian medication 1.07 CI 0.86 to 1.33) and general agitation (n=162, 3 RCTs, RR needing treatment with hypnotic/sedative drugs 1.09 CI 0.76 to 1.56). Zuclopenthixol did not clearly cause more adverse effects than other typical antipsychotics. 3.4 Leaving the study early Although two studies did report the reasons for leaving the study early, most simply stated that people had left and did not specify causes. Fewer people allocated zuclopenthixol left in the short term compared with those given other typical antipsychotics (n=424, 22% vs 30%, 8 RCTs, RR 0.70 CI 0.51 to 0.95, NNT 12 CI 7 to 67). 4. COMPARISON 3. ZUCLOPENTHIXOL versus ATYPICAL ANTIPSYCHOTICS (only short term) This comparison included three studies (total n=233). 4.1 Global state Only Huttunen 1995 presented categorical data on global state. We were able to extract data on the short term outcome of unchanged or worse. There was no clear difference between zuclopenthixol and risperidone for the outcome of being 'unchanged or worse' (n=98, 1 RCT, RR 1.30 CI 0.80 to 2.11). 4.2 Mental state There appeared to be no significant difference between zuclopenthixol and risperidone on 'no clinical response' (n=98, 1 RCT, RR not achieving at least 20% reduction in PANSS total score 1.39 CI 0.92 to 2.10). Other, skewed data, for BPRS also did not show any difference between groups. 4.3 Adverse effects Three studies comparing zuclopenthixol with atypical antipsychotics reported different adverse effects. Huttunen 1995 reported general adverse effects (n=98) but showed no difference between the two groups. Fischer 1976 reported different specific autonomic adverse effects (n=74): gastrointestinal adverse effects, anticholinergic adverse effects, orthostatic reaction, headache, hypersalivation and dizziness. Again we found no significant differences between those allocated zuclopenthixol and those given atypical antipsychotics. Huttunen 1995 and Fischer 1976 recorded the different aspects of extrapyramidal adverse effects: needing antiparkinsonian medication, hypokinesia, hyperkinesia, rigor, tremor and akathisia. Huttunen 1995 showed that the zuclopenthixol group had been prescribed with antiparkinsonian medication more frequently compared to people treated with risperidone (n=98, 1 RCT, RR 1.92 CI 1.12 to 3.28, NNH 3 CI 3 to 17). Psychic adverse effects (drowsiness, stimulation, confusion) reported by Fischer 1976, were not different between zuclopenthixol and clozapine (n=74). Mahadevan 1991 reported weight change as continuous data. We found no significant difference between people allocated zuclopenthixol and those given sulpiride (n=61, 1 RCT, WMD 1.60 CI 8.35 to 5.15). 4.4 Leaving the study early Huttunen 1995 and Mahadevan 1991 reported the outcome leaving the study early (45% vs 30%) and we found that fewer people allocated atypical antipsychotics left in the short term compared with those given zuclopenthixol but not to conventional levels of statistical significance (n=159, 2 RCTs, RR 1.48 CI 0.98 to 2.22). 5. COMPARISON 4. CIS‐ (Z) ZUCLOPENTHIXOL versus CIS (Z)+TRANS (E) FORM OF ZUCLOPENTHIXOL (only short term) This comparison included four studies (total n=140). 5.1 Global state Three studies Gravem 1978, Gravem 1981, Heikkila 1981a reported the outcome unchanged or worse in the category of global state but found no difference between the groups (n=131, RR 1.08 CI 0.76 to 1.52). 5.2 Adverse effects General adverse effects were reported by the same studies. None of the findings suggest any clear difference between cis (z) clopenthixol and cis (z)+ trans (e) clopenthixol (n=131, 3 RCTs, RR interfering with functioning/outweighing therapeutic effect 0.75 CI 0.46 to 1.22). Gravem 1981 (n=20) found no difference for the frequency of treatment of extrapyramidal adverse effects between the isomers (n=20, RR 1.40, CI 0.67 to 2.94). Gravem 1978 (n=57) provided data for the outcome 'sedation'. Again these researchers found no clear differences between the isomers (RR 0.19 CI 0.02 to 1.55). 5.3 Leaving the study early Aaes‐Jorgensen 1981b and Gravem 1981 did not show any significant difference for the outcome leaving the study early by only one week (n=29, 2 RCTs, RR 5.0 CI 0.27 to 92.62).
Appendix 4. Previous discussion
1. The studies Zuclopenthixol dihydrochloride has been widely proposed as a product specifically designed for the management of schizophrenia for inpatients and outpatients (Bhattacharya 1987). Low frequency of adverse effects and good tolerability has also been stressed by open clinical studies and materials produced for marketing purposes (Gravem 1981). In this systematic search for controlled clinical trials we found a small number of studies, some presenting important methodological flaws. 1.1 Applicability of findings The studies were conducted either in Europe or North America. The majority of trials involved inpatient participants with little in the way of physical and psychiatric co‐morbidity and with well‐defined schizophrenia or schizoaffective disorder. Such people are a minority in everyday care, where it is the norm to find people (not in a hospital setting) who suffer from less well defined illnesses combined with problems such as depression and substance abuse. 1.2 Limited data, confusing data The collection and quality of the data reported was very variable. All included studies reported data only for the short term (less than 12 weeks). To further undermine the value of the studies, many reported mean figures without giving the standard deviation and therefore these averages were meaningless. Among the 12 groups of defined outcomes, only five were addressed by the studies. We found no data on hospital and services outcomes, engagement with services, satisfaction with treatment and economic outcomes. There was a lack of information on outcomes that are clinically important such as death, general functioning, behaviour, treatment and hospitalisation. For such a widely used drug there are surprisingly few data. Outcomes were commonly reported using graphs and p‐values instead of tables and confidence intervals. The excessive use of graphs did not allow us to acquire sufficient numbers to calculate many measures of effectiveness. 1.3 Quality of studies We appreciate that studies in this population group bring unique difficulties. There were however important methodological difficulties with the trials and therefore any conclusions must be viewed with caution. There is a danger of inclusion of at least a moderate risk of bias in these results (Higgins 2011). 2. ZUCLOPENTHIXOL versus PLACEBO (only short term) This comparison included two short, small studies (total n=74) but it is a pity that neither reported global or mental state outcomes. Even from the few data there is indication that zuclopenthixol increases a persons risk of having extrapyramidal adverse effects compared with placebo (NNH 2 CI 2 to 31). Zuclopenthixol is also sedating (NNH 3 CI 2 to 435) but does not definitely cause weight gain in the short term. These may not be unexpected results to clinicians who frequently administer this drug, but as far as we are aware, this is the first time that these effects have been quantified from the best available data. 3. ZUCLOPENTHIXOL versus OTHER TYPICAL ANTIPSYCHOTICS (only short term) 3.1 Global state It has often been stated that there is little to choose between one antipsychotic and another. The older drugs are now often placed in one large category but this review does suggest that the compounds may have different levels of efficacy and zuclopenthixol is statistically significantly better than other older drugs in terms of a broad global outcome (n=357, 7 RCTs, RR unchanged or worse 0.72 CI 0.53 to 0.98, NNT 10 CI 6 to 131). However this result comes from small short trials and needs to be replicated. Real differences between drugs could point to subtle effects that can inform future drug design. It is obvious that, like many others, this is an under researched drug and we would be able to ascertain much more concrete evidence regarding the effects of this compound from larger, longer trials. 3.2 Mental state Data relevant to mental state are few and there is insufficient information on this outcome for us to use in this review. 3.3 Adverse effects Zuclopenthixol did not clearly cause greater or lesser adverse effects than other typical antipsychotics. At these moderately high doses however, over half of both the treatment and control groups required additional drugs to offset movement disorders. In comparison to newer treatment regimens and more modern drugs, this must be seen as disadvantage of using oral zuclopenthixol, but no more so than for other older drugs given in similar doses. 3.4 Leaving the study early Many people left these short studies early (22% zuclopenthixol vs 30% control) and data were lost. Although there was less attrition from the zuclopenthixol groups (NNT 12 CI 7 to 67) we would hope, with more modern trial designs, such low follow up could be avoided in the future. It might well be that despite its disadvantages zuclopenthixol is more acceptable than other older drugs which would be an important finding in need of replication. 4. ZUCLOPENTHIXOL versus ATYPICAL ANTIPSYCHOTICS (only short term) We were surprised to find that zuclopenthixol had been compared to the newer generation of drugs. The new generation are most commonly compared to themselves or older drugs such as haloperidol or chlorpromazine. However the studies were too small and short to be truly informative. 4.1 Global and mental state With a study size of more than 98, conducted over a longer period of time, we may have been able to ascertain more definite results in the comparison zuclopenthixol versus risperidone. However, at the present time, in terms of global measures and mental state scores given in the trials we uncovered, zuclopenthixol compares favourably with risperidone. 4.2 Adverse effects If there had been greater standardisation of studies the three relevant trials would not have all reported different adverse effects. With the exception of the movement disorders in terms of general and specific autonomic effects (gastrointestinal, anticholinergic, orthostatic, headache, hypersalivation, dizziness and weight changes), zuclopenthixol was not clearly different from the newer generation of drugs. People did need more additional antiparkinsonian medication however compared to those treated with risperidone (n=98, 1 RCT, RR 1.92 CI 1.12 to 3.28, NNH 3 CI 3 to 17). This is a real practical disadvantage of using zuclopenthixol dihydrochloride, but no more so than for the other older generation drugs. 4.3 Leaving the study early Normal clinical care would not expect loss to follow up of about 38% across six to ten weeks so it is likely that overall study design promotes attrition. Two studies however did suggest that taking zuclopenthixol may be less acceptable than taking either risperidone or sulpiride (45% vs 30% attrition) and that this did not reach conventional levels of statistical significance (n=159, 2 RCTs, RR 1.48 CI 0.98 to 2.22) may be a function of study size and duration. 5. CIS‐ (Z) ZUCLOPENTHIXOL versus CIS (Z)+TRANS (E) FORM OF ZUCLOPENTHIXOL (only short term) Three studies attempted to highlight differences between the isomers of zuclopenthixol hydrochloride. Studies were small and short and found no clear differences in terms of global state, adverse effects or attrition. Any differences were always likely to be small and therefore studies would have had to be very large, probably with thousands of participants, to highlight clear disparity in effect. In current circumstances, whether or not there really is a clinically meaningful difference between the two isomers is likely to remain unanswered. 6. Missing data There are no data on outcomes relating to service use (e.g. hospitalisation), relapse, satisfaction with care, behaviour and social function. It would be ideal if newer generation drugs were not widely circulated without such data, but we are sceptical that this is the case considering that three of the studies included in this review are trials of zuclopenthixol versus new compounds. Note: the 8 citations in the awaiting classification section of the review may alter the conclusions of the review once assessed.
Appendix 5. Previous conclusions
Implications for practice 1. For people with schizophrenia There is evidence that zuclopenthixol causes movement disorders, perhaps more so than newer drugs, but no more frequently than the other older generation antipsychotics. There is some suggestion from this review that oral zuclopenthixol may have some clinical advantage over other older drugs in terms of global state, at least in the short term. If an older drug is going to be used, zuclopenthixol dihydrochloride is a viable option but may be best taken with additional medication to offset movement disorders that occur in about half the people taking this drug. If the clinical advantage is real, zuclopenthixol does come in a depot form, and these data support those in the review of the depot zuclopenthixol (Coutinho 2004) suggesting a moderate advantage over other similar compounds. 2. For clinicians There is no evidence that zuclopenthixol dihydrochloride is any worse than other compounds and some indications that it is in fact better. Any difference in the isomers is speculative, but there are some data to suggest a real advantage in comparison with other older drugs. As with other older generation drugs, the use of zuclopenthixol dihydrochloride is tainted by the need for drugs to counter movement disorders. If the advantage, in terms of global effect, over its fellow older generation drugs is real, then zuclopenthixol could be a preferred choice of these older generation drugs. However in light of just these few trials, there does appear to be an advantage for the newer generation of drugs in terms of extrapyramidal adverse effects. 3. For managers/policy makers There are no data on service outcomes and no medium or long term data. In the context of finite resources, the lack of good quality data leaves managers and policy makers with difficult decisions to make. However, the short term data do favour zuclopenthixol dihydrochloride over several of the older drugs and therefore it should remain a choice in the treatment of people for whom older generation drugs are indicated. Implications for research 1. General If the recommendations of the CONSORT statement (Moher 2001) had been anticipated by trialists much more data would have been available. Allocation concealment is essential for the result of a trial to be considered valid and gives the assurance that selection bias is kept to the minimum. Well‐described and tested blinding could have encouraged confidence in the control of performance and detection bias. It is also important to know how many, and from which groups, people were withdrawn, in order to evaluate exclusion bias. It would have been helpful if authors had presented data in a useful manner which reflects association between intervention and outcome, for example, relative risk, odds‐ratio, risk or mean differences, as well as raw numbers. Binary outcomes should be calculated in preference to continuous results, as they are easier to interpret. If p‐values are used, the exact value should be reported. 2. Specific We do think that more trials are indicated. These should not only be large and long but should also adhere to a pragmatic design in order to increase applicability. Methods should be strict and involve good concealment of allocation and follow up. Participants should be people recognisable in everyday life and not those who are so strictly diagnosed as to render them unrecognisable to routine care. Interventions should involve standard doses of zuclopenthixol and a control drug that is a real choice in the region of the study. This could equally be a new generation drugs such as risperidone, or an older medication such as chlorpromazine. Outcomes should be measured over months rather than weeks as this is the usual period a person would be asked to take the drug. We suggest that if scales are to be used, validated and clinically meaningful outcomes are pre‐defined. Routine outcomes such as relapse, employment, housing status, satisfaction with care, serious or troubling adverse effects can all be easily recorded without the use of scales and we would suggest that these are included in the design of the study.
Data and analyses
Comparison 1. ZUCLOPENTHIXOL versus PLACEBO ‐ all short term.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early (any reason) | 2 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.01, 6.60] |
| 2 Adverse effects: 1. Clinically important change in specific adverse effects ‐ cardiovascular ‐ orthostatic | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.01, 6.60] |
| 3 Adverse effects: 2. Clinically important change in specific adverse effects ‐ central nervous system ‐ arousal state | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 excitation | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 2.62 [0.12, 59.40] |
| 3.2 sleepiness / sedation | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 2.89 [1.01, 8.30] |
| 4 Adverse effects: 3. Clinically important change in specific adverse effects ‐ endocrine ‐ menstruation started | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.39, 126.48] |
| 5 Adverse effects: 4a. Any general adverse effects ‐ movement disorders ‐ EPSEs (UKU side effect rating scale, no scores) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 6.07 [0.86, 43.04] |
| 6 Adverse effects: 4b. Clinically important change in specific adverse effects ‐ movement disorders ‐ EPSEs | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 parkinsonism | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.26, 97.37] |
| 6.2 oculogyric crisis | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.09] |
| 6.3 tremor | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.26, 97.37] |
Comparison 2. ZUCLOPENTHIXOL versus CHLORPROMAZINE ‐ all short term.
Comparison 3. ZUCLOPENTHIXOL versus CHLORPROTHIXENE ‐ all medium term.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: Average endpoint global state score ‐ Unchanged/worse (CGI) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Leaving the study early (any reason) | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.34, 2.93] |
Comparison 4. ZUCLOPENTHIXOL versus CLOZAPINE ‐ all short term.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early (any reason) | 1 | 407 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Adverse effects: Any general adverse effects ‐ side effects ‐ frequency per day | Other data | No numeric data |
Comparison 5. ZUCLOPENTHIXOL versus HALOPERIDOL ‐ all short term.
Comparison 6. ZUCLOPENTHIXOL versus PERPHENAZINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: Average endpoint global state score ‐ unchanged/worse (global rating ‐ investigator opinion) ‐ medium term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Leaving the study early (any reason) ‐ short/medium term | 2 | 104 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.27, 1.47] |
| 3 Adverse effects: 1. Any change in specific adverse effects ‐ central nervous system ‐ arousal ‐ requiring medication ‐ medium term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Adverse effects: 2. Any change in specific adverse effects ‐ movement disorders ‐ EPSEs ‐ requiring medication ‐ medium term | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 7. ZUCLOPENTHIXOL versus RISPERIDONE.
Comparison 8. ZUCLOPENTHIXOL versus SULPIRIDE ‐ all short term.
Comparison 9. ZUCLOPENTHIXOL versus THIOTHIXENE ‐ all medium term.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: Average endpoint global state score ‐ unchanged/worse (CGI) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Leaving the study early (any reason) | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.24, 1.35] |
Comparison 10. ZUCLOPENTHIXOL versus TRIFLUOPERAZINE ‐ all short term.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early (any reason) | 1 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 11. ZUCLOPENTHIXOL versus ZUCLOPENTHIXOL DEPOT ‐ all long term.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early (any reason) | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 1.95 [0.36, 10.58] |
| 2 Behaviour: Average change in specific aspects of behaviour ‐ Violence during follow‐up | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.44, 1.71] |
| 3 Adverse Effects: 1a. Any general adverse effects ‐ additional medication use | Other data | No numeric data | ||
| 4 Adverse Effects: 1b. Any change in specific adverse effects ‐ additional medication use ‐ benzodiazepine use at least once | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 1.3 [0.59, 2.86] |
Comparison 12. CIS‐(Z) ZUCLOPENTHIXOL versus CIS(Z)/TRANS(E) ZUCLOPENTHIXOL ‐ all short term.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: Average endpoint global state score ‐ Unwell | 3 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.80, 1.17] |
| 2 Mental state: Average endpoint general mental state score ‐ Not improved | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.45, 2.07] |
| 3 Leaving the study early (any reason) | 4 | 140 | Risk Ratio (M‐H, Random, 95% CI) | 2.15 [0.49, 9.41] |
| 4 Adverse Effects: 1a. Any general adverse effects ‐ side effects reported | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.82, 2.18] |
| 5 Adverse Effects: 1b. Any change in specific adverse effects ‐ individual side effects | Other data | No numeric data |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aaes‐Jorgensen 1981.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 1 week before cross‐over.
Design: cross‐over.
Country: Denmark. Funding: Not mentioned. |
|
| Participants | Diagnosis: paranoid schizophrenia.
History: not clear in article but states duration of illness 2 months ‐ 2 years.
N = 9.
Age: mean 58 years, range 39‐81.
Sex: all male.
Inclusion criteria: all patients who had been previously treated with cis(Z)/trans(E)‐clopenthixol (from 2 months to several years). Exclusion: not mentioned. Setting: not reported. |
|
| Interventions | 1. Cis(Z)‐clopenthixol hydrochloride: dose range 5 mg to 50 mg/day. Taken twice daily. N = 4. 2. Clopenthixol hydrochloride (cis(Z) and trans(E) forms): dose 10 mg to 100 mg/day. Taken twice daily. N = 5. | |
| Outcomes | 5.2 Leaving the study early for General Reasons (zero people left the study early). Unable to use: Physiological ‐ serum concentrations of Cis(Z)‐clopenthixol and clopenthixol. Non‐clinical data all reported post‐cross‐over. |
|
| Notes | Cross‐over trial. Patient 4 excluded during statistical analysis (also received multiple drugs). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Done "in a randomised way". Authors do not state how. Attempted to contact authors but Dikemark Hospital no longer exists. |
| Allocation concealment (selection bias) | Unclear risk | Authors do not comment. Attempted to contact authors but Dikemark Hospital no longer exists. |
| Blinding | Low risk | Authors state "Double blindly". |
| Incomplete outcome data (attrition bias) None | Low risk | None detected. |
| Selective reporting (reporting bias) | Low risk | All data appear to have been reported that was sought by the study's aims. |
| Other bias | High risk | Selection bias (n = 9) ‐ diagnostic purity bias. |
Arango 2006.
| Methods | Allocation: randomised Blindness: none ‐ open‐label Duration: 1 year Funding: Theodore and Vada Stanley Foundation Grant Setting: 3 inpatient units |
|
| Participants | Diagnosis: schizophrenia N = 46 Age: mean ˜34 years, range 24‐44 Sex: 38 male, 8 female Inclusion: DSM IV criteria for schizophrenia, a violent episode in the previous year with a score of greater than or equal to 3 on the physical aggression sub‐scale or the MOAS. A family member living with the patient and willing to collaborate with the researchers. Informed consent signed by the patient and the informant. Exclusion: Any other axis I disorders, including alcohol and/or drug abuse/dependence, mental retardation. |
|
| Interventions | 1. Zuclopenthixol (oral) 35 mg/day n = 20 2. Zuclopenthixol (depot) 233 mg every 14 days (˜16.6 mg/day) n = 26 Additional medication: All received biperiden, other psychotropics were permitted. |
|
| Outcomes | Useable: 4. Mental State ‐ PANSS positive only 5. Leaving the study Early 7. Behaviour ‐ number of violent episodes 8. Adverse Effects ‐ additional medication Unable to use: 7. Behaviour ‐ MOAS, baseline data only 8. Adverse effects ‐ UKU ‐ no data provided by group |
|
| Notes | Madrid Inclusion criteria do not match included data and significant selective reporting. Authors will need to be contacted at next update to obtain clarification. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | A researcher at one centre did the randomisation. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding | High risk | Open‐label. |
| Incomplete outcome data (attrition bias) None | High risk | Missing UKU, missing PANSS. |
| Selective reporting (reporting bias) | High risk | MOAS not reported other than at baseline. |
| Other bias | High risk | Selection bias ‐ Berkson |
Balasubramanian 1991.
| Methods | Allocation: randomised. Blindness: double. Duration: 10 weeks. Design: outpatient and Inpatient Country: UK. | |
| Participants | Diagnosis: acute functional psychosis (schizophrenia n = 68, schizoaffective n = 7, hypomania n = 4, unknown n = 4 and unreported n = 11. History: acutely ill / exacerbation of chronic illness, 0‐20 episodes. N = 94. Age: mean ˜ 36.9 years, range 18‐65. Age of one patient not recorded. Sex: 48 male, 34 female, 13 unspecified (12 patients not analysed by authors and sex not reported in 1 patient). Lundbeck UK contacted to request information. Inclusion criteria: BPRS 15 or more. Exclusion criteria: previous intolerance to neuroleptics, additional serious psychiatric or neurological disorder, serious physical illness, pregnant or lactating women and those of child‐bearing potential who intend to become pregnant, addiction to drugs or alcohol, depot neuroleptic at an unstable dose or dosing frequency. Setting: hospital and community. | |
| Interventions | 1. Zuclopenthixol: dose range 25 mg to 150 mg/day. N = 50.
2. Chlorpromazine: dose range 100 mg to 600 mg/day. N = 44. Additional medication: amitriptyline, temazepam, procyclidine. |
|
| Outcomes | 2. Global state: CGI. Table 1 5. Leaving the study early. Zuclo n = 32 completed study, Chlor n = 21 completed the study. 8. Adverse effects. Table 1 Unable to use: Mental state: BPRS (> 50% loss to follow‐up in one group). Adverse effects: UKU side effects rating scale (data not reported by group). |
|
| Notes | Zuclopenthixol group previous episodes 0‐17. Chlorpromazine group previous episodes 0‐20. Most common zuclopenthixol dose 75 mg/day. Most common chlorpromazine dose 600 mg/day |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Authors do not mention. Lundbeck UK contacted to request information regarding potential biases 5th January 2016. |
| Allocation concealment (selection bias) | Unclear risk | Authors do not mention. |
| Blinding | Low risk | Clearly stated in the paper as double‐blind design. |
| Incomplete outcome data (attrition bias) None | High risk | 11 patients were excluded and not analysed. Missing outcome data. |
| Selective reporting (reporting bias) | Low risk | All data appear to have been reported on. |
| Other bias | High risk | Attrition bias ‐ per protocol analysis. |
Ban 1975.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 12 weeks.
Design: single‐centre.
Country: Canada. Funding: Public health service research grant ‐ Pfizer |
|
| Participants | Diagnosis: schizophrenia.
N = 30.
Age: mean 37 years, range 18‐58.
Sex: 13 male, 17 female.
Inclusion: diagnosis of schizophrenia Exclusion criteria: not reported. Setting: hospital. |
|
| Interventions | 1. Clopenthixol: dose range 50 mg to 200 mg/day. Average 150 mg/day. N = 10. 2. Thiothixene: dose range 10 mg to 40 mg/day. average 30 mg/day. N = 10. 3. Chlorprothixene: dose range 150 mg to 600 mg/day. Average 450 mg/day. N = 10. | |
| Outcomes | Useable: 2. Global state: CGI. 5. Leaving the study early 8. Adverse effects: number of adverse effects in every group, partial TESS Unable to use: 4. Mental state: BPRS (data not reported by group). 7. Behaviour: NOSIE (no data available). 8. Adverse effects: TESS (no data available). |
|
| Notes | 15 chronic and 15 new admissions. Original Cochrane authors did not include the two‐week washout period in the duration of the study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Suggested but not reported by authors. "10 week double blind." |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned. |
| Blinding | Low risk | "10 week double blind." |
| Incomplete outcome data (attrition bias) None | Low risk | None obvious and none mentioned by authors. |
| Selective reporting (reporting bias) | High risk | Multiple areas where data not reported. Several papers produced with same data. |
| Other bias | High risk | Selection bias ‐ sampling ‐ Berkson |
CSzG Study ID 9828 2005.
| Methods | Allocation: inpatient Blindness: randomised by specially assigned person Duraton:: 6 weeks |
|
| Participants | Diagnosis: CCMD ‐ 3 N = 120 Age: mean ˜33 years, range 24‐42 Exclusion: severe physical diseases, alcohol and substance dependence |
|
| Interventions | 1. Clopenthixol 40 mg to 70 mg/day. N = 60 2. Chlorpromazine 300 mg to 550 mg/day. N = 60 Other antipsychotics were not allowed during the treatment. Benzhexol and diazepam were used if necessary. |
|
| Outcomes | Able to use: 4. Mental state: BPRS, PANSS 8. Adverse Effects: EPSEs and excessive sedation Unable to use: 4. Mentals State: SANS ‐ no useable data |
|
| Notes | Paper kindly translated and data extracted by Jun Xia. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Not mentioned. No detailed information was described in the study. No Concerns raised by the data extractor. |
| Allocation concealment (selection bias) | Low risk | Not mentioned. No detailed information was described in the study. No Concerns raised by the data extractor. |
| Blinding | Low risk | Randomised. No detailed information was described in the study. No Concerns raised by the data extractor. |
| Incomplete outcome data (attrition bias) None | Low risk | All outcomes reported. |
| Selective reporting (reporting bias) | Low risk | None found. |
| Other bias | Low risk | None obvious. |
Dehnel 1968.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 13 weeks.
Design: single centre.
Country: USA. Funding: Not reported. |
|
| Participants | Diagnosis: chronic schizophrenia.
History: hospitalised patients with schizophrenia with duration of illness 2 months‐to 3 years.
N = 50.
Age: mean ˜ 43 years, range 25‐59.
Sex: all male.
Inclusion criteria: patients under 60 years of age and free of significant physical illness such as liver disease or severe cardiovascular disease. Exclusion criteria: not discussed. Setting: not reported. |
|
| Interventions | 1. Clopenthixol: dose 132.5 mg/day, range 25 mg to 250 mg/day. N = 25. 2. Perphenazine: dose 51.2 mg/day, range 8 mg to 80 mg/day. N = 25. | |
| Outcomes | Useable: 2. Global state: CGI. 5. Leaving the study early. 8. Adverse effects: requiring additional medication ‐ benztropine methanesulphate; other medications. Unable to use: 4. Mental state: BPRS (no SD), PIP (no data by group). 8. Adverse Effects ‐ table 3 ‐ week 12 missing |
|
| Notes | All but two patients were taking 1 or more psychoactive drugs (mostly phenothiazines) before starting the study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported, suggested but not clear. |
| Allocation concealment (selection bias) | Unclear risk | Not reported, suggested but not clear. |
| Blinding | Low risk | Reasonable methods employed for blinding. |
| Incomplete outcome data (attrition bias) None | Low risk | Reasons given by authors. |
| Selective reporting (reporting bias) | Low risk | Not detected. |
| Other bias | High risk | Per‐protocol analysis. |
Fagerlund 2004.
| Methods | Allocation: Random Blindness: open‐label Duration: 13 weeks Funding: Danish Medical Counscil, H:S Research council, University of Copenhagen Faculty of Humanities Location: Copenhagen |
|
| Participants | Diagnosis: ICD10 F20 Schizophrenia N = 31 Age: 27.3 years (+/‐5.9), range 19‐37 Sex: not discussed History: untreated psychosis 4‐78 months Inclusion: anti‐psychotic naive admitted for treatment and written informed consent Exclusion: known retardation, need acute medication, compulsorily hospitalised |
|
| Interventions | 1. Zuclopenthixol 6 mg to 26 mg average 9.6 mg. N = 10 2. Risperidone 2 mg to 7 mg average 3.6 mg. N = 15 3: Healthy controls N = 25 Additional Medication: Benzodiazepiens and anticholinergics but not on the day of examination |
|
| Outcomes | Usable: 4. Mental State: PANSS ‐ table 2 5. Leaving the study Early Not Useable: 6. General Functioning: Cognitive Functions (CANTAB, WCST, verbal fluency, Figural fluency, trail making tests A & B, DART) ‐ no useable data 8. Adverse Effects: ESRS ‐ no data |
|
| Notes | Not all data published by authors. When review next updated the authors should be contacted to obtain this information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated but not elaborated on. |
| Allocation concealment (selection bias) | High risk | Open‐label. |
| Blinding | High risk | Open‐label. |
| Incomplete outcome data (attrition bias) None | Unclear risk | Authors state they will publish other data elsewhere though not found in searches to date. |
| Selective reporting (reporting bias) | Unclear risk | Authors state they will publish other data elsewhere though not found in searches to date. |
| Other bias | High risk | Selection bias, Berkson |
Fischer‐Cornelssen 1976.
| Methods | Allocation: randomised not clear.
Blindness: double.
Duration: 7 weeks.
Design: multicentre.
Country: Switzerland. Funding: not stated |
|
| Participants | Diagnosis: acute paranoid schizophrenia. History: moderate to severe hospitalised acute patients with schizophrenia. N = 723. Age: not reported. Sex: not reported. Inclusion criteria: moderate to severe acute patients with schizophrenia. Setting: hospital. | |
| Interventions | 1. Clopenthixol: dose 100 mg/day. N = 36. 2. Clozapine: dose 200 mg to 300 mg/day. N = 371. 3. Chlorpromazine: dose 200 mg to 350 mg/day. N = 212. 4. Haloperidol: dose 4 mg to 8 mg/day. N = 78. 5. Trifluoperazine: dose 20 mg to 30 mg/day. N = 36. | |
| Outcomes | Useable: 8. Adverse effects: drowsiness, stimulation, confusion, gastrointestinal side‐effects, anticholinergic side‐effects, dizziness, orthostatic reaction, headache, hypersalivation, hypokinesia, hyperkinesia, dyskinesia, rigor, tremor, akathisia. Unable to use: 2. Global state: number of patients completing the study not provided. 4. Mental state: BPRS (no clear data). |
|
| Notes | All of the higher dose ranges are medians. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported. Sandoz UK contacted by online form to clarify bases and seek additional data 5th January 2016. |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding | Unclear risk | Authors state double‐blind but do not say how. |
| Incomplete outcome data (attrition bias) None | Unclear risk | Total figures are not provided. |
| Selective reporting (reporting bias) | High risk | Sandoz side‐effect check list not reported. |
| Other bias | High risk | Selection bias, performance bias, unclear if intention‐to‐treat analysis was used. |
Glenthoj 2007.
| Methods | Allocation: random Blindness: not reported Duration: 12 weeks Funding: committee on medicine and science ‐ personal grant and an undisclosed non‐restricted grant |
|
| Participants | Diagnosis: F20 schizophrenia n = 45 Age: ˜21‐33 years Sex: 25 male, 13 female, 7 unknown Included: drug naive first episode patients with schizophrenia, F20 ICD10 criteria for schizophrenia Excluded: mental retardation, severe somatic diseases, severe head trauma, pregnancy, presence of MRI contraindications, acute need of medication, compulsory hospitalisation in patients, presence of psychotic illness in controls or their first‐degree relatives. |
|
| Interventions | 1. Zuclopenthixol (n = 8) 10.3 mg/day average (3.8 mg to 16.8 mg) 2. Risperidone (n = 11) 3.4 mg/day average (1.9 mg to 4.9 mg) 3. Control (n = 20) Of the original n = 45 it is unclear as to which groups the remaining n = 6 were attributed. Other medication: Anticholinergic n = 1‐, antidepressant n = 1, benzodiazepines n = 11 |
|
| Outcomes | Usable: 4. Mental State: PANSS ‐ table 2 (PANSS general, mean and SD baseline and follow‐up). 5. Leaving the study early 8: Adverse effects ‐ ESRS ‐ table 2 |
|
| Notes | 3 patients minimally medicated prior to the start of the study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Not reported. DRCMR research group contacted 5th January 2016 to clarify these biases and seek any missing information. |
| Allocation concealment (selection bias) | High risk | Not reported. |
| Blinding | High risk | Not reported. |
| Incomplete outcome data (attrition bias) None | High risk | Conflicting reports from several papers using same population cohort. |
| Selective reporting (reporting bias) | High risk | Supplementary publications highlight dropout and inconsistencies in the randomisation. |
| Other bias | High risk | Berkson bias |
Gravem 1978.
| Methods | Allocation: unclear. "In the investigation patients ... were allocated." Implied when table 1 reviewed. Blindness: double. Duration: 4 weeks Design: RCT Country: Norway | |
| Participants | Diagnosis: acute and chronic psychoses, mainly schizophrenia
N = 20
Age: 20‐66 years (mean 36)
Sex: 13 male, 7 female
Inclusion criteria: acute psychoses and chronic exacerbations. Exclusion: below 15 years age, serious somatic disease, pathological laboratory findings, pregnant patients. Setting: Inpatient |
|
| Interventions | 1. Cis(Z)‐clopenthixol 48mg/day range 10 mg to 150 mg/day. N = 10. 2. Cis(Z)/trans(E) ‐ clopenthixol 88 mg/day range 10 mg to 200 mg/day. N = 10. | |
| Outcomes | 2. 6 Average Endpoint in global state scores Table 7, CGI 5. Leaving the study early n = 2, (one for refusal due to side‐effects, oculogyric crisis and one for non‐compliance) Unable to use: Physiological: serum concentrations of Cis(Z)‐clopenthixol and clopenthixol (non‐clinical data all reported post‐cross‐over). 4. 4 Average endpoint general mental state score Table 3 and 4 ‐ CGI time 0, 2 weeks and 4 weeks no SD 4. 4 Average change in general mental state scores Table 5 ‐ BPRS ‐ total score time 0, 2 weeks and 4 weeks. no SD 4. 8 Average change in specific symptom scores Thinking disturbance: Table 5 ‐ BPRS ‐ time 0, 2 weeks and 4 weeks. no SD 4. 8 Average change in specific symptom scores Withdrawal ‐ retardation: Table 5 ‐ BPRS ‐ time 0, 2 weeks and 4 weeks. noS.D 4. 8 Average change in specific symptom scores Hostile‐suspiciousness: Table 5 ‐ BPRS ‐ time 0, 2 weeks and 4 weeks. no SD 4. 8 Average change in specific symptom scores Anxious‐depression: Table 5 ‐ BPRS ‐ time 0, 2 weeks and 4 weeks. no SD 8. 1 Clinically important general adverse effects Table 8, individual side‐effects and totals. Frequencies not patient numbers. |
|
| Notes | none | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported but implied; "A double blind clinical investigation". |
| Allocation concealment (selection bias) | Unclear risk | patients were "allocated" Attempted to contact authors but Dikemark Hospital no longer exists. Google search did not reveal any contact details of the authors. |
| Blinding | Low risk | Authors state double‐blinding but the details are not clear in the paper. |
| Incomplete outcome data (attrition bias) None | Low risk | The two excluded patients had reasons given by the authors. |
| Selective reporting (reporting bias) | Low risk | Reported all outcomes stated in the study aims. |
| Other bias | Unclear risk | Berkson and diagnostic purity bias. |
Gravem 1981.
| Methods | Allocation: patients are allocated to each group and it is unclear if a process of randomisation occurred. A review of table one suggests an element of randomisation ‐ implied randomisation.
Blindness: double.
Duration: 8 weeks.
Design: cross‐over.
Country: Denmark. Funding: not declared. |
|
| Participants | Diagnosis: chronic schizophrenia.
History: illness duration 5‐20 years
N = 57
Age: mean ˜50 years, range 24‐79
Sex: 53 male and 4 female
Inclusion criteria: chronic patients already in neuroleptic treatment, preferably clopenthixol Exclusion: not mentioned Setting: inpatient |
|
| Interventions | 1. Cis(Z)‐clopenthixol hydrochloride, n = 29 average dose 47.6 mg/day
2. Clopenthixol hydrochloride (cis(Z) and trans(E) forms), n = 28 average dose 150 mg/day Table 2 dosing reported by authors is unclear. |
|
| Outcomes | 2.6 Global State ‐ Average change in global state scores (Table 1) 4.3 Average endpoint general mental state score 5.2 Leaving the study early (for general reasons) ‐ zero people left the study 6.3 General Functioning ‐ average endpoint general functioning scores (Table 4) Unable to use: Physiological ‐ serum concentrations of Cis(Z)‐clopenthixol and clopenthixol (non‐clinical data all reported post‐cross‐over). |
|
| Notes | none. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported but implied; "A double blind clinical investigation". |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding | Low risk | Stated in paper. "The drugs were given double blindly in a randomised way." |
| Incomplete outcome data (attrition bias) None | Low risk | No incomplete outcome data. |
| Selective reporting (reporting bias) | Low risk | None noted. |
| Other bias | Unclear risk | Berkson and diagnostic purity bias. |
Heikkila 1981.
| Methods | Allocation: "The patients were allocated..." Implied randomisation.
Blindness: double.
Duration: 8 weeks
Design: RCT
Country: Finland Funding: not stated |
|
| Participants | Diagnosis: 40 patients with schizophrenia and 14 patients with oligophrenia. History: vast majority ill or hospitalised more than 5 years. N = 54 Age: Mean ˜45 years Sex: 28 male, 26 female Inclusion/exclusion: not reported Setting: not reported. | |
| Interventions | 1. Cis(Z)‐clopenthixol 73 mg/day (10 mg to 150 mg/day). N = 26 2. Cis(Z)/Trans(E)‐clopenthixol 114 mg/day (20 mg to 300 mg/day). N = 28 | |
| Outcomes | 2. 5 Average endpoint global state score Table 3 and 5: Severity of illness, scale unclear 5. Leaving the study early Table 3 and 5: n = 5 left, reasons not stated in the article. 8. 3 Average endpoint general adverse effect score Table 7: Single side‐effects week 8 total Unable to use: 8. 4 Average change in general adverse effect scores Table 6: Side‐effects interfering with patient's functioning ‐ CGI. No SD. 8. 8 Average change in specific adverse effects Table 7: Single side‐effects. No SD. Physiological: serum concentrations of Cis(Z)‐clopenthixol and clopenthixol (non‐clinical data all reported post‐cross‐over). |
|
| Notes | none | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported ‐ Finland Central Hospital contacted by email 5th January 2016. |
| Allocation concealment (selection bias) | Unclear risk | Not reported ‐ Finland Central Hospital contacted by email 5th January 2016. |
| Blinding | Unclear risk | Reported but not detailed |
| Incomplete outcome data (attrition bias) None | High risk | At week 8, five patients were not included in the outcome data and not accounted for by the authors. |
| Selective reporting (reporting bias) | Unclear risk | Not enough information to make a clear decision. |
| Other bias | Low risk | Per protocol analysis, attrition bias |
Heikkila 1981a.
| Methods | Allocation: not stated
Blindness: double, implied randomisation
Duration: 12 weeks.
Design: multicentre.
Country: Finland. Funding: Not stated |
|
| Participants | Diagnosis: chronic schizophrenia (n = 58) or other psychotic (n = 5, paranoic state, depressive/PD) History: n = 40 duration of illness > 10 years and n = 11 for > five years. N = 63. Age: mean ˜ 43 years. Sex: 41 male, 22 female. Inclusion criteria: chronic schizophrenic inpatients or other psychotic inpatients who might benefit from either of these drugs or already receiving such treatment. Exclusion criteria: patients under 15 years of age, with serious concomitant somatic illness or pathological laboratory findings, and pregnant patients. Setting: hospitalised. | |
| Interventions | 1. Cis(Z)‐zuclopenthixol: dose 40 mg/day. N = 30. 2. Haloperidol: dose 10 mg/day. N = 33. | |
| Outcomes | Useable: 2. Global state: CGI. 5. Leaving the study early [n = 27 left (13 Zuclo and 14 Halo) ‐ only 13 accounted for: n = 1 discharge, n = 9 insufficient effect and side‐effects, n = 3 reasons unrelated to treatment] 8. Adverse effects: use of other therapeutic drugs. CGI, frequency only. Unable to use ‐ 2. Global State: NOSIE 30 (unable to use data). CGI ‐ percentages only 4. Mental state: BPRS (no SD). |
|
| Notes | Previous neuroleptic medication: 14 different neuroleptics. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. Hospital District of South West Finland contacted to seek further information on biases 5th January 2016. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding | Low risk | Double‐blind. |
| Incomplete outcome data (attrition bias) None | High risk | 27 patients dropped out and only 12 accounted for by the authors. |
| Selective reporting (reporting bias) | Unclear risk | Not able to ascertain from paper. |
| Other bias | High risk | Berkson bias, diagnostic purity bias |
Huttunen1995.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 6 weeks.
Design: multicentre.
Country: Finland. Funding: not stated |
|
| Participants | Diagnosis: chronic schizophrenia or schizophreniform disorder with an acute exacerbation (DSM‐III‐R).
History: community patients with acute exacerbation of chronic schizophrenia. N = 98. Age: mean ˜ 47 years, range 18‐65. Sex: 47 male, 51 female. Inclusion criteria: acute exacerbations. 18‐65y, acute psychotic symptoms necessitating antipsychotic treatment. Diagnosis of chronic or sub‐chronic schizophrenia or schizophreniform disorder according to DSM III‐R Exclusion criteria: patients with clinically significant organic or neurological disorder, serious psychotic disorder other than schizophrenia or schizophreniform disorder, clinically relevant abnormalities in laboratory tests, patients likely to be noncompliant, as well as pregnant or lactating women and those of reproductive age without adequate contraception. Setting: community. |
|
| Interventions | 1. Zuclopenthixol: dose 38 mg/day, range 10 mg to 100 mg/day. N = 50.
2. Risperidone: dose 8 mg/day, range 2 mg to 20 mg/day. N = 48. Authors aimed to avoid anti‐parkinsonism medication and benzodiazepines. |
|
| Outcomes | Useable: 5. Leaving the study early (n = 98 to n = 40) 8. Adverse effects: use of antiparkinsonian medication. 8. Adverse effects: UKU side effect rating scale Unable to use 1. Death: n = 1 patient on risperidone. died 1 month after the trial. This patient was only on the drug for one week and the pathology diagnosis was viral myocarditis. The authors deemed this to not be related to risperidone. 4. Mental state: PANSS score, BPRS (originally included but upon review data is unusable) Vital signs, body weight, laboratory screening (no data by group). 8. Adverse effects: extrapyramidal symptom rating scale (data not useable) |
|
| Notes | Discontinuation of anti‐parkinsonism and other psychotropic medication. Author contacted to seek clarification of some data: no reply at the time of writing. Information from multiple papers. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised but not stated how. E‐mailed M.O Huttunen Summer 2015 with no response to date. |
| Allocation concealment (selection bias) | Unclear risk | Randomised but not stated how. E‐mailed M.O Huttunen Summer 2015 with no response to date. |
| Blinding | Unclear risk | Mentioned but not clear how. E‐mailed M.O Huttunen Summer 2015 with no response to date. |
| Incomplete outcome data (attrition bias) None | Low risk | Intention‐to‐treat analysis |
| Selective reporting (reporting bias) | High risk | Report mean for non‐normal data instead of median. Only reported CGI data on comparison to previous neuroleptic treatment. |
| Other bias | High risk | Diagnostic purity and attrition biases. |
Kingstone 1970.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 3 weeks. Funding: Ayerst, McKenna and Harrison LTD |
|
| Participants | Diagnosis: schizophrenic reaction (different subtypes) plus one extreme incapacitating anxiety in a schizoid personality. N = 41 Age: 18‐65 years, mean ˜31 years Sex: 16 male, 25 female History: Duration of illness 0.4 ‐ 5.3 years Included: acute psychotic symptomatology Excluded: not mentioned. |
|
| Interventions | 1. Clopenthixol (N = 20) 122 mg/day (75 mg to 600 mg), n = 5 additional medication 2. Chlorpromazine (N = 21) 435 mg/day (150 mg to 1800 mg), n = 2 additional medication All patients had previous treatment. |
|
| Outcomes | Used: 2. Global state ‐ Global assessment ‐ table 2 4. Mental state ‐ BPRS ‐ table 1 5. Leaving the study early n = 4 (Chlorpromazine, 1 exhausted tablets and 3 due to side‐effects). n = 1 (zuclopenthixol, side‐effects). 8. Adverse effects ‐ table 3 and additional medication n = 7 (authors do not mention which additional medication) Unable to use: 5. Leaving the study early "Several patients ... diagnosis changed ... to a non‐psychotic disorder or to a brain syndrome were dropped from the study." No data. |
|
| Notes | none. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Implied randomisation. Response: Patients assigned a number belonging to a bottle of medications. Research unit at the Royal Victoria Hospital in Quebec contacted for more information on biases and missing patient information 5th January 2016. |
| Allocation concealment (selection bias) | Low risk | Contents of bottles not known to anyone in the study until the end. |
| Blinding | Low risk | Study states "double blind". |
| Incomplete outcome data (attrition bias) None | High risk | Several patients not included in the study because of a change in diagnosis. The numbers were not given and not included in the analysis. |
| Selective reporting (reporting bias) | High risk | Missing data, areas not reported but alluded to in discussion. |
| Other bias | High risk | Berkson bias, attrition bias, per protocol analysis |
Kordas 1968.
| Methods | Allocation: "Patients were subsequently allocated" ‐ randomisation implied Blindness: single (patient) Duration: 6 weeks. Design: single centre. Country: Greece. | |
| Participants | Diagnosis: chronic schizophrenia.
History: duration of illness 6 years and over.
N = 54.
Age: mean ˜43 years, range 26‐64.
Sex: 29 male, 25 female.
Inclusion: established diagnosis of schizophrenia in hospital records Exclusion criteria: not reported. setting: hospital. |
|
| Interventions | 1. Clopenthixol: dose 150 mg/day. N = 18.
2. Chlorpromazine: dose 300 mg/day. N = 18.
3. Placebo. N = 18. Additional Medication: Artane and Phenergan. |
|
| Outcomes | Used: 5. Leaving the study early n = 0 8. Adverse effects: number of patients experiencing side‐effects. (all on clopenthixol, n = 3 restarted menstruation, n = 2 parkinsonism, n = 1 oculogyric crisis Not used: 7. Behaviour: behavioural scale, Aof V (table III) ‐ no usable data |
|
| Notes | none | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not mentioned. Contacted Dromokaītion Hospital Athens to obtain further information on bias 5th January 2016. |
| Allocation concealment (selection bias) | Unclear risk | "...patients were subsequently allocated into three groups..." Unclear how. |
| Blinding | Unclear risk | "He was the only person knowing the identity of the tablets and to which group the patients belonged." Unclear if single‐ or double‐blind. |
| Incomplete outcome data (attrition bias) None | Unclear risk | First rating deleted from analysis as was considered to be training by the researchers. |
| Selective reporting (reporting bias) | High risk | See above |
| Other bias | High risk | Hospitalised patients used ‐ Berkson bias. |
Mahadevan 1991.
| Methods | Allocation: randomised.
Blindness: single.
Duration: 10 weeks.
Design: multicentre.
Country: UK. Funding: not declared. |
|
| Participants | Diagnosis: acute schizophrenia (RDC).
N = 61.
Age: mean ˜ 39 years, range 18‐65.
Sex: 26 male, 35 female. Inclusion criteria: aged 18‐65 years inclusive, were experiencing an acute schizophrenic episode according to RDC and minimum BPRS score of 15. Exclusion criteria: concurrent serious physical illness, patients who had previously demonstrated intolerance to neuroleptics, received a depot neuroleptic in the previous 2 weeks, or were suffering from a serious additional psychiatric or neurological disorder, patients who were pregnant or lactating; dependence or addiction to drugs or alcohol. Setting: hospital. |
|
| Interventions | 1. Zuclopenthixol dihydrochloride: dose range 25 mg to 150 mg/day. N = 30.
2. Sulpiride: dose 200 mg to 1200 mg/day. N = 31. Additional medication: temazepam (insomnia), procyclidine (EPSEs), amitriptyline (depression). |
|
| Outcomes | usable: 2. Global state: CGI. 4. Mental state: BPRS. 5. Leaving the study Early 8. Adverse effects: change in mean weight. |
|
| Notes | Multicentre | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised but not elaborated on by authors. Bolton General Hospital contacted 5th January 2016 to seek clarification from the first author regarding biases. |
| Allocation concealment (selection bias) | Low risk | Randomly allocated but not elaborated on. |
| Blinding | Low risk | Clearly stated. |
| Incomplete outcome data (attrition bias) None | Low risk | Authors discuss |
| Selective reporting (reporting bias) | High risk | Not all data was reported in the paper. |
| Other bias | High risk | Diagnostic purity bias, attrition bias, per‐protocol analysis |
Remvig 1987.
| Methods | Randomised, double‐blind, 12 weeks | |
| Participants | Acute psychosis N = 54 Age: 20‐60 years (mean 38) Sex: 25 female, 15 male, 14 unspecified Inclusion: hospitalised in the preceding 7 days, acute psychosis, exacerbation of chronic psychosis, neuroleptic treatment was anticipated to be at least 3 weeks, informed consent. Excluded: mania/depression, serious somatic disease, organic brain damage, pregnant, history of abuse, previous good response to neuroleptic |
|
| Interventions | 1. Zuclopenthixol 37 mg/day (10 mg to 20 mg) n = 27 2. Perphenazine 30 mg/day (8 mg to 72 mg) n = 27 Additional medication: benzodiazepines, methotrimeprazine, anti‐parkinsonism medication |
|
| Outcomes | 5. Leaving the study early Unable to use: 2. Global state: CGI 4. Mental state: CPRS 8 Adverse effects |
|
| Notes | Multicentre | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. Glostrup hospital contacted 5th January 2016 to obtain further information to clarify biases and seek additional outcome data if available. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding | Low risk | Double‐blind stated. |
| Incomplete outcome data (attrition bias) None | Low risk | Not detected. |
| Selective reporting (reporting bias) | High risk | Multiple data not reported and often unclear. |
| Other bias | High risk | Berkson bias evident. |
Serafetinides 1972.
| Methods | Allocation: randomised.
Blindness: double.
Duration: 24 weeks.
Design: single centre.
Country: USA. Funding: United states Public health service grant |
|
| Participants | Diagnosis: chronic schizophrenia.
History: duration of illness 2 years or longer.
N = 57.
Age: mean ˜ 42 years, range 21‐61 years.
Sex: 25 male, 32 female.
Inclusion criteria: no evidence of organic disease, duration of illness 2 years or longer, chronic schizophrenia, permission from a family member Exclusion: no complicating organic illness or known brain damage Setting: hospital. |
|
| Interventions | 1. Clopenthixol: dose 205 mg/day. N = 15.
2. Haloperidol: dose 12.3 mg/day. N = 14.
3. Chlorpromazine: dose 830 mg/day. N = 14.
4. Placebo. N = 13. Additional medication: antiparkinsonian medication, sedative. |
|
| Outcomes | Useable: 5. Leaving the study early: n = 4 (behavioural deterioration n = 2, 1 Chlorpromazine and 1 placebo, Intestinal obstruction secondary faecal impaction n = 2, both chlorpromazine) 8. Adverse effects ‐ table 10 and 11 Unable to use ‐ 2. Global state: CGI (no SD) 4. Mental state: BPRS (no SD). Short‐term data not reported (first 12 weeks medication free) |
|
| Notes | Originally reported as a 12‐week study in previous review. Combination of several papers (6 in total) all reporting on the same cohort. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "In a double blind placebo controlled trial..." Implied but not clear. |
| Allocation concealment (selection bias) | High risk | Not specifically discussed by the authors but "subjects were housed together on a special two‐wing ward, males in one wing and females in the other" so it is unlikely that allocation concealment was maintained. |
| Blinding | Low risk | Authors state double‐blind. Not discussed how. |
| Incomplete outcome data (attrition bias) None | Unclear risk | Incomplete data described by the authors but not used in outcome data. |
| Selective reporting (reporting bias) | High risk | Multiple papers, same patient cohort. |
| Other bias | High risk | Selection bias (Berkson), Suspected data mining, likely per‐protocol analysis. |
Wang 1995a.
| Methods | Allocation: Quote” Patients were randomly divided into two groups ”(p.4)
Blinding: Double‐blind Duration: Quote”8 weeks”(p. 4) Location: Quote ”Inpatients, WuHan, China” (p.4) |
|
| Participants | Schizophrenia, CCMD‐3;
Sex: male 31 female 29
Age:mean˜34.1 years, SD ˜12.5
Total N = 64
Inclusion and exclusion criteria: Patients with schizophrenia fulfilled CCMD‐3 were included; Patients with severe heart, liver, kidney diseases were excluded. |
|
| Interventions | 1. Zuclopenthixol Group: (n = 33)
Management: newly hospitalised patients had to stop the previous drugs for three days and then were randomly divided into Zuclopenthixol Group or Chlorpromazine Group.
Chlorpromazine or Zuclopenthixol was enclosed in the same colour, style capsule by pharmacist.
In the first week of treatment, the dosage of Zuclopenthixol was 20 mg/day and from the second week, it was increased 10 mg/day every week.
No other antipsychotic drugs were allowed during the treatment.
Trihexyphenidyl or scopolamine was used when necessary.
MECT was used two times per week for 12 times. 2. Chlorpromazine Group: (n = 27) management: Newly hospitalised patients had to stop the previous drugs for three days and then were randomly divided into Zuclopenthixol Group or Chlorpromazine Group. Chlorpromazine or Zuclopenthixol was enclosed in the same colour, style capsule by pharmacist. In the first week of treatment, the dosage of Chlorpromazine was 200 mg/day and from the second week, it was increased 50 mg/day every week. No other antipsychotic drugs were allowed during the treatment; Trihexyphenidyl or scopolamine was used when necessary; |
|
| Outcomes | 2. Global State ‐ Clinical response
4. Mental State ‐ BPRS score
8. Adverse effects ‐ Adverse events, TESS; Unable to use: electrocardiogram; electroencephalogram; liver function; blood routine examination. |
|
| Notes | We have noticed that the N number does not always add up in this study, for example, the total sample size randomised was claimed to be 64, and 61 completed, but the number randomised to each group was reported as 33 and 27. Author to be contacted at next update. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Authors state patients randomised, not how |
| Allocation concealment (selection bias) | Unclear risk | Not reported after translation and data extraction |
| Blinding | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) None | Unclear risk | N number unclear |
| Selective reporting (reporting bias) | Unclear risk | Not reported after translation and data extraction |
| Other bias | High risk | Berkson bias |
BPRS ‐ Brief Psychiatric Rating Scale, CCMD‐3 ‐ Chinese classification of mental disorders,CGI ‐ Clinical Global Impression, CPRS (Montgomery 1979) ‐ Comprehensive Psychopathological Rating Scale, DSM ‐ Diagnostic and Statistical Manual of Mental Disorders, EPSEs ‐ extrapyramidal side‐effects, ESRS ‐ Extrapyramidal Symptom Rating Scale, ICD 10 ‐ International Classification of Diseases, RCT ‐ randomised controlled trial, MRI ‐ magnetic resonance imaging, NOSIE ‐ Nourses' Observation Scale for Inpatient Evaluation, PANSS ‐ Positive and Negative Syndrome Scale, PIP ‐ Psychotic Inpatient Profile, RCT ‐ randomised controlled trial, RDC ‐ Research Diagnostic Criteria, SANS ‐ Scale for the Assessment of Negative Symptoms, SD ‐ standard deviation, TESS ‐ Treatment Emergent Symptom Scale, UKU ‐ side effects scale.
Where possible, contact has been attempted with authors and/or institutions to seek clarification of bias and to obtain missing and/or unclear outcome data.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aaes‐Jorgensen 1981a | Allocation: randomised. Participants: healthy volunteers. |
| Ahlfors 1980 | Allocation: randomised. Participants: people with chronic schizophrenia. Interventions: clopenthixol decanoate versus perphenazine enanthate, not oral form of zuclopenthixol. |
| Al Haddad 1996 | Allocation: randomised. Participants: people with acute psychosis. Interventions: zuclopenthixol acetate versus haloperidol, not oral form of zuclopenthixol. |
| Arango 2002 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral ziclopenthixol versus depot zuclopenthixol. outcomes: frequency of violent episodes (no data reported by group). |
| Baastrup 1993 | Allocation: randomised. Participants: people with acute psychosis, mania. |
| Bereen 1987 | Allocation: not randomised, case‐series. |
| Bhattacharya 1987 | Allocation: not randomised, case series. |
| Bobon 1989 | Allocation: randomised. Participants: people with acute psychosis. Interventions: zuclopenthixol acetate versus haloperidol, not oral form of zuclopenthixol. |
| Bourdouxhe 1987 | Allocation: randomised. Participants: people with acute psychosis. Interventions: zuclopenthixol acetate versus haloperidol, not oral form of zuclopenthixol. |
| Brook 1998 | Allocation: randomised. Participants: people with acute psychosis. Interventions: zuclopenthixol acetate versus haloperidol, not oral form of zuclopenthixol. |
| Burke 2002 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone versus sulpiride, chlorpromazine, trifluoperazine, haloperidol, flupenthixol, zuclopenthixol. Outcomes: SAPS and SANS scale, unable to use data. |
| Chin 1998 | Allocation: randomised. Participants: people with acute schizophrenia. Interventions: zuclopenthixol acetate versus haloperidol, not oral form of zuclopenthixol. |
| Chouinard 1991 | Allocation: randomised. Participants: people with acute schizophrenia. Interventions: zuclopenthixol acetate versus liquid haloperidol, not oral form of zuclopenthixol. |
| Clark 1969 | Allocation: not clear, double‐blind. Participants: people with chronic schizophrenia. Interventions: sordinol versus chlorpromazine versus placebo (unable to use data) |
| CTRI‐2014‐07‐004712 | Wrong intervention. |
| Den 2000 | Allocation: not clear, double‐blind. Participants: people with schizophrenia. Interventions: ritanserin versus placebo with other neuroleptics one of which was zuclopenthixol. Outcomes: PANSS, CGI, ESRS, no usable date. |
| Dencker 1980 | Allocation: randomised. Participants: people with schizophrenia. Interventions: clopenthixol decanoate vesus flupenthixol palmitate, not oral form of zuclopenthixol. |
| Dom 1978 | Allocation: randomised. Participants: people with chronic schizophrenia. Interventions: variable doses versus constant dose of zuclopenthixol decanoate, not oral form of zuclopenthixol. |
| Fagerlund 2003 | Allocation: randomised, blindness not clear. Participants: people with first‐episode schizophrenia. Interventions: zuclopenthixol versus risperidone. Outcomes: PANSS, cognitive function, no usable data. |
| Fan 1999 | Not clear if oral or depot. Authors need to be contacted at next update. |
| Fensbo 1990 | Allocation: randomised, double‐blind. Participants: people with acute psychosis, exacerbation of chronic psychosis and mania. Interventions: zuclopenthixol versus haloperidol. Outcomes: leaving the study early, CGI, BPRS, no usable data. |
| Fricchione 2010 | Depot not oral intervention. |
| Galdersi 1994 | Originally included but excluded at this update. It is not an RCT and blindness is not mentioned. |
| Glenthoj 2000 | Allocation: randomised. Participants: people with first‐episode schizophrenia. Interventions: risperidone versus zuclopenthixol. Outcomes: PANSS, PPI, cognitive functions, no data provided. |
| Gravem 1990 | Allocation: randomised. Participants: people with schizophrenia, manic‐depressive illness, psychosis. Interventions: zuclopenthixol acetate and zuclopenthixol decanoate, not oral form of zuclopenthixol. |
| Hicklin 1967 | Allocation: not randomised. |
| Hovens 2003 | Allocation: randomised. Participants: people with acute psychosis. Interventions: risperidone + lorazepam versus zuclopenthixol + lorazepam; zuclopenthixol and lorazepam administered either in oral/injection form, not stated specifically for individual group. |
| Hovens 2005 | Not randomised. Not blinded. |
| Huang 2001 | Allocation: randomised. Participants: people with schizophrenia. Interventions: zuclopenthixol decanoate versus clorpromazine, not oral form of zuclopenthixol. |
| Karsten 1981 | Allocation: randomised. Participants: people with oligophrenia. |
| Knegtering 2002a | Allocation: randomised. Participants: people with psychosis. Interventions: risperidone versus quetiapine, not oral form of zuclopenthixol. |
| Koskinen 1991 | Allocation: randomised. Participants: people with chronic schizophrenia. Interventions: zuclopenthixol decanoate versus haloperidol decanoate, not oral form of zuclopenthixol. |
| Kristiansen 2001 | Allocation: randomised. Participants: healthy individuals. |
| Kristiansen 2003 | No data. |
| Lamure 2003 | Not oral form. |
| Lemmens 1994 | Study duplicates data presented by authors already included in review. |
| Liu 1997b | injection not oral. |
| Liu 1998a | Injection not oral. |
| Loza 2001 | Allocation: randomised. Participants: people with first‐episode paranoid schizophrenia. Interventions: typical antipsychotics (zuclopenthixol, perphenazine, haloperidol, perazine) versus atypical antipsychotics (risperidone, olanzapine, quetiapine). Outcomes: PANSS, DL, WCST, no usable data. |
| Lublin 1991 | Allocation: randomised. Participants: Psychotic psychiatric in‐patients with tardive dyskinesia. Interventions: concomitant use of zuclopenthixol and haloperidol. Outcomes: CGI, BPRS, UKU side effects rating scale, no usable data. |
| Mackeprang 2001 | Allocation: randomised. Participants: people with first‐episode schizophrenia. Interventions: zuclopenthixol versus risperidone. Outcomes: SPECT, MRI, cognitive functions test, rapid visual information processing test. E‐mailed author for outcomes. Deceased. Colleague replied that the data had not yet been published. Unable to use data. |
| Malt 1995 | Allocation: randomised. Participants: people with learning disability. |
| Mann 1985 | Allocation: not randomised, case series. |
| Martyns 1993 | Allocation: randomised. Participants: people with chronic schizophrenia. Interventions: clopenthixol decanoate versus flupenthixol decanoate, not oral form of zuclopenthixol. |
| Mazurek 2003 | Allocation: randomised. Participants: people with paranoid schizophrenia. Interventions: typical antipsychotics (zuclopenthixol, perphenazine, haloperidol, perazine, levomepromazine) vs atypical antipsychotics (risperidone, olanzapine, clozapine). Information about number of patients using each drug is not provided. |
| Meyers 1972 | Allocation: not randomised. |
| Mosolov 2000 | Allocation: randomised. Participants: people with acute schizophrenia. Interventions: after 2 day wash‐out period for group 1. clopixol acuphase i.m. for 7 days and then oral clopixol for 14 days. Group 2. halopeidol i.m. for 7 days and then oral form for 14 days (no usable data). |
| NCT00206960 | No data. |
| Pagsberg 2004 | Wrong intervention, no data. |
| Ropert 1988 | Allocation: randomised. Participants: people with psychosis, mania, chronic schizophrenia. Interventions: zuclopenthixol acetate versus chlorpromazine, not oral form of zuclopenthixol. |
| Rubio 2005 | Not oral form. |
| Rubio 2006 | Zuclopenthixol depot. |
| Rubio 2009 | Not oral form. |
| Rubioz 2006 | Not oral form. |
| Saxena 1990b | Depot zuclopenthixol. |
| Saxena 1996 | Allocation: not clear, double ‐blind. Participants: people with schizophrenia. interventions: zuclopenthixol decanoate versus fluphenazine decanoate, not oral form of zuclopenthixol. |
| Schooler 1993 | Allocation: randomised. Participants: people with schizophrenia. Interventions: antipsychotics (not mentioned) and family management. |
| Shelton 1969 | Allocation: randomisation not clear. Participants: people with schizophrenia. Interventions: zuclopenthixol versus trifluoperazine. Outcome: measurement of achilles reflex, no other data reported by group, unable to use data. |
| Singh 1992 | Allocation: randomised. Participants: mentally handicapped in‐patients with behavioural disorder with or without a psychiatric diagnosis. |
| Sofronov 2013 | Depot zuclopenthixol. |
| Svestka 1986 | Allocation: randomised. Participants: people with schizophrenia. Interventions: clopenthixol decanoate versus oxyprothepine decanoate, not oral form of zuclopenthixol. |
| Syvalahti 1997 | Allocation: randomised. Participants: people with schizophrenia. Interventions: citalopram versus placebo with other neuroleptics i.e. haloperidol, chlorpromazine, levomepromazine, zuclopenthixol, thioridazine and perphenazine. Outcomes: plasma concentrations of drugs, no usable data. |
| Taymeeyapradit 2002 | Allocation: randomised. Participants: people with acute psychosis. Interventions: zuclopenthixol acetate versus haloperidol, not oral form of zuclopenthixol. |
| Tegeler 1985 | Allocation: randomised. Participants: people with schizophrenia. Interventions: clopenthixol decanoate versus fluphenazine decanoate, not oral form of zuclopenthixol. |
| Uys 1996 | Allocation: randomised. Participants: people with acute psychosis. Interventions: zuclopenthixol acetate versus clothiapine, not oral form of zuclopenthixol. |
| Venkateswarlu 1990 | Allocation: not randomised, case series. |
| Viala 1988 | Allocation: randomised. Participants: people with chronic psychosis. Interventions: zuclopenthixol decanoate versus fluphenazine decanoate, not oral form of zuclopenthixol. |
| Walker 1983 | Allocation: randomised. Participants: people with chronic schizophrenia. Interventions: clopenthixol decanoate versus fluphenazine decanoate, not oral form of zuclopenthixol. |
| Weiser 1975 | Allocation: randomised. Participants: people with acute schizophrenia. Interventions: droperidol versus clopenthixol i. m. versus clozapine i.m., not oral form of zuclopenthixol. |
| Wisted 1990 | Depot zuclopenthixol. |
| Wistedt 1991 | Allocation: randomised. Participants: people with chronic schizophrenia. Interventions: zuclopenthixol decanoate versus haloperidol decanoate, not oral form of zuclopenthixol. |
| Xiaanao 1999 | Allocation: randomised. Participants: people with schizophrenia. Interventions: concomitant use of clopenthixol and other psychotropics. |
| Youssef 1991 | Allocation: not clear. Participants: people with psychosis. Interventions: haloperidol decanoate versus other neuroleptics (fluphenazine decanoate, flupenthixol decanoate, clopenthixol decanoate, oral thioridazine, oral haloperidol, oral pimozide), not oral form of zuclopenthixol. |
BPRS ‐ Brief Psychiatric Rating Scale, CGI ‐ Clinical Global Impression, ESRS ‐ Extrapyramidal Symptom Rating Scale, i.m. ‐ intramuscular, MRI ‐ magnetic resonance imaging, PANSS ‐ Positive and Negative Syndrome Scale, RCT ‐ randomised controlled trial, SANS ‐ Scale for the Assessment of Negative Symptoms, SAPS ‐ Scale for the Assessment of Positive Symptoms, UKU ‐ side effects scale.
Differences between protocol and review
Differences have previously been clarified in the methods section. The main difference is in the comparisons (four in the original review and 12 in the update).
In the original publication the authors completed the following analyses (Appendix 3).
Zuclopenthixol versus placebo (only short term)
Zuclopenthixol versus other typical antipsychotics (only short term)
Zuclopenthixol versus atypical antipsychotics (only short term)
Cis (Z) zuclopenthixol versus cis (Z)/Trans (E) zuclopenthixol (only short term)
For this update the original four analyses were modified and adapted in an attempt to reflect the evidence base for realistic clinical questions e.g. Should I use zuclopenthixol instead of clozapine? It was felt that the original four comparisons were misleading to clinicians, patients and policy makers as they suggested that zuclopenthixol had been compared to all other possible antipsychotics. This was not the original authors intent.
This review update identified comparisons against nine alternative antipsychotics, only two of which were newer atypicals. Comparisons one and four remain and have been updated (see below).
Contributions of authors
Edward Bryan ‐ protocol revisions, review update and re‐write.
Marie Purcell ‐ Peer review and proof reading.
Ajit Kumar ‐ protocol, review development and writing.
Sources of support
Internal sources
-
Sheffield Health and Social Care, Sheffield NHS Foundation Trust, UK.
Employs lead author Edward J Bryan.
-
Market Surgery, Rotherham, UK.
Employs review author Marie Ann Purcell.
-
Leeds Community Healthcare NHS Trust, Leeds, UK.
Employs review author Ajit Kumar.
External sources
No sources of support supplied
Declarations of interest
Edward Bryan: none known.
Marie Purcell: none known.
Ajit Kumar: none known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Aaes‐Jorgensen 1981 {published data only}
- Aaes‐Jorgensen T, Gravem A, Jorgensen A. Serum levels of the isomers of clopenthixol in patients given cis(Z)‐clopenthixol or cis(Z)‐trans(E)‐clopenthixol. Acta Psychiatrica Scandinavica Supplementum 1981;294:70‐7. [MEDLINE: ; PMID 6951396] [PubMed] [Google Scholar]
Arango 2006 {published data only}
- Arango C, Bombin I, Gonzalez‐Salvador T, Garcia‐Cabeza I, Bobes J. Randomised clinical trial comparing oral versus depot formulations of zuclopenthixol in patients with schizophrenia and previous violence. European Psychiatry 2006;21(1):34‐40. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Bombin I, Arango C. Antipsychotic treatment adherence and violence in schizophrenia outpatients: a randomized prospective trial. European Neuropsychopharmacology 2004;14(Suppl 3):S289. [Google Scholar]
Balasubramanian 1991 {published data only}
- Balasubramanian K, Baloch N, Briscoe MH, Chattree S, Cooper CJ, Durani SK, et al. A double blind multicentre comparison of oral zuclopenthixol and oral chlorpromazine in the treatment of acute psychosis. British Journal of Clinical Research 1991;n/a:149‐56. [MEDLINE: ; PMID 5600894]5600894 [Google Scholar]
Ban 1975 {published data only}
- Ban TA, Lehmann HE, Sterlin C, Climan M. Comprehensive clinical studies with thiothixene. Diseases of the Nervous System 1975;36(9):473‐7. [MEDLINE: ; PMID 240651] [PubMed] [Google Scholar]
- Lehmann HE, Ban TA. Thiothixene versus chlorprothixene versus clopenthixol. Psychopharmacology Bulletin 1970;6(4):118‐20. [13027] [Google Scholar]
- Sterlin C, Ban TA, Jarrold L. The place of thiothixene among the thioxanthenes. Current Therapeutic Research Clinical and Experimental 1972;14(4):205‐14. [MEDLINE: ; PMID 4623599] [PubMed] [Google Scholar]
CSzG Study ID 9828 2005 {published data only}
- 刘启珍, 王慧芳, 陈洪来, 刘翠珍. [Title only available in Chinese characters] [氯噻吨与氯丙嗪治疗精神分裂症的对照研究]. Shandong Archives of Psychiatry [山东精神医学] 2005;18(3):175‐6. [Google Scholar]
Dehnel 1968 {published data only}
- Dehnel LL, Vestre ND, Schiele BC. A controlled comparison of clopenthixol and perphenazine in a chronic schizophrenic population. Current Therapeutic Research Clinical and Experimental 1968;10:169‐76. [MEDLINE: ; PMID 4967871] [PubMed] [Google Scholar]
Fagerlund 2004 {published data only}
Fischer‐Cornelssen 1976 {published data only}
- Fischer‐Cornelssen KA, Ferner UJ. An example of European multicentre trials: multispectral analysis of clozapine. Psychopharmacology Bulletin 1976;12:34‐9. [PubMed] [Google Scholar]
Glenthoj 2007 {published data only}
- Glenthoj A, Glenthoj BY, Mackeprang T, Pagsberg AK, Hemmingsen RP, Jernigan TL, et al. Basal ganglia volumes in drug‐naive first‐episode schizophrenia patients before and after short‐term treatment with either a typical or an atypical antipsychotic drug. Psychiatry Research 2007;154(3):199‐208. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Glenthoj B, Fagerlund B, Rasmussen H, Pinborg L, Svarer C, Friberg L, et al. Extrastriatal dopamine D2/D3‐receptors in drug‐naïve first‐episode schizophrenic patients: relation to psychopathology, cognition and treatment outcome. International Journal of Neuropsychopharmacology 2008;11(Suppl 1):6‐7. [Google Scholar]
- Glenthoj B, Glenthoj A, Jagersma E, Pagsberg AK, Hemmingsen RP, Svarer C, et al. Effects of typical and atypical antipsychotic medication on caudate nucleus volume in first‐episode schizophrenic patients: relation to dopamine d2/3 receptor occupancy psychopathology and extrapyramidal side‐effects. Proceedings of the 8th World Congress of Psychiatry; 2005 Sep 10‐15; Cairo, Egypt. 2005.
Gravem 1978 {published data only}
- Gravem A, Engstrand E, Guleng RJ. Cis(Z)‐clopenthixol and clopenthixol (Sordinol) in chronic psychotic patients. A double‐blind clinical investigation. Acta Psychiatrica Scandinavica 1978;58(5):384‐8. [MEDLINE: ; PMID 362830] [DOI] [PubMed] [Google Scholar]
Gravem 1981 {published data only}
- Gravem A, Bugge A. Cis(Z)‐clopenthixol and clopenthixol in the treatment of acute psychoses and exacerbations of chronic psychoses. A double‐blind clinical investigation. Acta Psychiatrica Scandinavica Supplementum 1981;294:13‐24. [MEDLINE: ; PMID 7041515] [DOI] [PubMed] [Google Scholar]
Heikkila 1981 {published data only}
- Heikkila L, Karsten D, Valli K. A double‐blind clinical investigation of cis(Z)‐clopenthixol and clopenthixol in chronic schizophrenic patients. Acta Psychiatrica Scandinavica Supplementum 1981;294:25‐9. [MEDLINE: ; PMID 7041516] [PubMed] [Google Scholar]
Heikkila 1981a {published data only}
- Heikkila L, Eliander H, Vartiainen H, Turunen M, Pedersen V. Zuclopenthixol and haloperidol in patients with acute psychotic states. A double‐blind, multicentre study. Current Medical Research and Opinion 1992;12(9):594‐603. [EMBASE 1992128093] [DOI] [PubMed] [Google Scholar]
- Heikkila L, Laitinen J, Vartiainen H. Cis(Z)‐clopenthixol and haloperidol in chronic schizophrenic patients ‐ a double‐blind clinical multicentre investigation. Acta Psychiatrica Scandinavica Supplementum 1981;294:30‐8. [MEDLINE: ; PMID 7041517] [PubMed] [Google Scholar]
Huttunen1995 {published data only}
- Huttunen MO, Piepponen T, Rantanen H, Larmo I, Nyholm R, Raitasuo V. Risperidone versus zuclopenthixol in the treatment of acute schizophrenic episodes: a double‐blind parallel‐group trial. Acta Psychiatrica Scandinavica 1995;91(4):271‐7. [DOI] [PubMed] [Google Scholar]
- Huttunen MO, Piepponen T. Risperidone versus zuclopenthixol in the treatment of acute schizophrenic episodes. European Psychiatry 1994;9(Suppl 1):85. [DOI] [PubMed] [Google Scholar]
- Moller HJ, Huttunen MO. The prevalence of extrapyramidal symptoms during short and long term treatment with risperidone. 7th Biennial European Winter Workshop on Schizophrenia; 1994 Jan 23‐28; Les Diablerets, Switzerland 1994;11(2):106. [MEDLINE: ; PMID 8104468]8104468 [Google Scholar]
Kingstone 1970 {published data only}
- Kingstone E, Kolivakis T, Kossatz I. Double blind study of clopenthixol and chlorpromazine in acute hospitalized schizophrenics. Internationale Zeitschrift Fur Klinische Pharmakologie Therapie und Toxikologie 1970;3(1):41‐5. [MEDLINE: ; PMID 4909377] [PubMed] [Google Scholar]
Kordas 1968 {published data only}
- Kordas SK, Kazamias NG, Georgas JG, Papadokostakis JG. Clopenthixol: a controlled trial in chronic hospitalized schizophrenic patients. British Journal of Psychiatry 1968;114(512):833‐6. [MEDLINE: ; PMID 4874163] [DOI] [PubMed] [Google Scholar]
Mahadevan 1991 {published data only}
- Mahadevan K, Gadhvi HM, Suri AK, Hussain MF, Sharma VK, Sharma SK, et al. A multicentre comparison of oral zuclopenthixol dihydrochloride and oral sulpiride in the treatment of acute schizophrenia. British Journal of Clinical Research 1991;2:13‐20. [Cochrane Library CN‐00282616] [Google Scholar]
Remvig 1987 {published data only}
- Remvig J, Larsen H, Rask P, Skausig OB, Skov S, Stromgren LS. Zuclopenthixol and perphenazine in patients with acute psychotic states. A double‐blind multicentre study. Pharmacopsychiatry 1987;20(4):147‐54. [MEDLINE: ; PMID 3615572] [DOI] [PubMed] [Google Scholar]
Serafetinides 1972 {published data only}
- Serafetinides EA. Consistency and similarity of drug EEG responses in chronic schizophrenic patients. International Pharmacopsychiatry 1973;8(4):214‐6. [MEDLINE: ; PsycINFO 52‐05954; PMID 4589640] [DOI] [PubMed] [Google Scholar]
- Serafetinides EA. Voltage laterality in the EEG of psychiatric patients. Diseases of the Nervous System 1973;34(3):190‐1. [MEDLINE: ; PsycINFO 1973‐29665‐001; PMID 4715636] [PubMed] [Google Scholar]
- Serafetinides EA, Clark ML. Psychological effects of single dose antipsychotic medication. Biological Psychiatry 1973;7(3):263‐7. [MEDLINE: ; PsycINFO 52‐03694; PMID 4586833] [PubMed] [Google Scholar]
- Serafetinides EA, Collins S, Clark ML. Haloperidol, clopenthixol, and chlorpromazine in chronic schizophrenia. Chemically unrelated antipsychotics as therapeutic alternatives. Journal of Nervous and Mental Disease 1972;154(1):31‐42. [MEDLINE: ; PMID 4550211] [DOI] [PubMed] [Google Scholar]
- Serafetinides EA, Willis D. A method of quantifying EEG for psychopharmacological research. International Pharmacopsychiatry 1973;8(4):245‐7. [DOI] [PubMed] [Google Scholar]
- Serafetinides EA, Willis D, Clark ML. Haloperidol, clopenthixol, and chlorpromazine in chronic schizophrenia. Journal of Nervous and Mental Disease 1972;155(5):366‐9. [MEDLINE: ; PMID 4563078] [DOI] [PubMed] [Google Scholar]
Wang 1995a {published data only}
- Wang R. Double blind study of clopenthixsol treating schizophrenia. Medical Journal of Chinese Civil Administration 1995; Vol. 7, issue 1:4‐5, 7.
References to studies excluded from this review
Aaes‐Jorgensen 1981a {published data only}
- Aaes‐Jorgensen T. Serum concentrations of cis(Z)‐ and trans(E)‐clopenthixol after administration of cis(Z)‐clopenthixol and clopenthixol to human volunteers. Acta Psychiatrica Scandinavica Supplementum 1981;294:64‐9. [MEDLINE: ; PMID 6951395] [PubMed] [Google Scholar]
Ahlfors 1980 {published data only}
- Ahlfors UG, Dencker SJ, Gravem A, Remvig J. Clopenthixol decanoate and perphenazine enanthate in schizophrenic patients. A double‐blind Nordic multicentre trial. Acta Psychiatrica Scandinavica Supplementum 1980;279:77‐91. [MEDLINE: ; PMID 6996426] [DOI] [PubMed] [Google Scholar]
Al Haddad 1996 {published data only}
- Al Haddad MK, Kamel C, Mawgood MA, Sequeria RP. Zuclopenthixol versus haloperidol in the initial treatment of schizophrenic psychoses,affective psychoses and paranoid states: a controlled clinical trial. Arab Journal of Psychiatry 1996;7(1):44‐54. [PsycINFO 2001‐16512‐004] [Google Scholar]
Arango 2002 {published data only}
- Arango C, Bombýn I, Bobes´J, Gonzalez‐, Cabeza Garcýa‐, Salvador MT. Prediction and prevention of violence in schizophrenia outpatients: preliminary report. Schizophrenia Research 2002;53(3 Suppl.1):233. [MEDLINE: ] [Google Scholar]
Baastrup 1993 {published data only}
- Baastrup PC, Alhfors UG, Bjerkenstedt L, Dencker SJ, Fensbo C, Gravem A, et al. A controlled Nordic multicentre study of zuclopenthixol acetate in oil solution, haloperidol and zuclopenthixol in the treatment of acute psychosis. Acta Psychiatrica Scandinavica 1993;37(1):48‐58. [MEDLINE: ; PMID 8093824] [DOI] [PubMed] [Google Scholar]
Bereen 1987 {published data only}
- Bereen FJ, Harte FB, Maguire J, Singh AN. The use of oral zuclopenthixol in the treatment of functional psychotic illness. Pharmatherapeutica 1987;5(1):61‐8. [PubMed] [Google Scholar]
Bhattacharya 1987 {published data only}
- Bhattacharya SN, Ghoshal J, Sharma SK, Halstead N, John B, Launer MA, et al. Acute functional psychoses:treatment with zuclopenthixol dihydrochloride ('Clopixol') tablets. Pharmatherapeutica 1987;5(1):1‐8. [PubMed] [Google Scholar]
Bobon 1989 {published data only}
- Bobon D, Bleeker E. Zuclopenthixol acetate and haloperidol in acute psychotic patients ‐ a randomized multicentre study. 2nd Congress of the European College of Neuropsychopharmacology; 1989 May 24‐26; Gothenburg, Germany. 1989. [MEDLINE: ; PMID 240651]240651
- Bobon D, Bleeker E. Zuclopenthixol acetate and haloperidol in acute psychotic patients ‐ a randomized multicentre study. New Strategies in the Treatment of Aggressive, Acutely Psychotic Patients. Proceedings of the 8th World Congress of Psychiatry; 1989 October 13‐19, Athens, Greece. Excerpta Medica, 1989:47‐59. [MEDLINE: ; PMID 240651]240651
Bourdouxhe 1987 {published data only}
- Bourdouxhe S, Mirel J, Denys W, Bobon D. Zuclopenthixol acetate and haloperidol in acute psychoses ‐ results of the AMDP subgroup of a Belgian multicenter study [L'acetate de zuclopenthixol et l'haloperidol dans la psychose aigue ‐ Resultats du sous groupe AMDP d'une etude multicentrique belge]. Acta Psychiatrica Belgica 1987;87(2):236‐44. [MEDLINE: ; PsycINFO 26‐73137; PMID 3618274] [PubMed] [Google Scholar]
Brook 1998 {published data only}
- Brook S, Berk M, Selemani S, Kolloori J, Nzo I. A randomised controlled double blind study of zuclopenthixol acetate compared to haloperidol in acute psychosis. 10th European College of Neuropsychopharmacology Congress; 1997 Sep 13‐17; Vienna, Austria. 1997. [MEDLINE: ; PMID 7542829]7542829
- Brook S, Berk M, Selemani S, Kolloori J, Nzo I. A randomised controlled double blind study of zuclopenthixol acetate compared to haloperidol in acute psychosis. Unpublished Report submitted to Human Psychopharmacology Clinical and Experimental 1996. [MEDLINE: ; PMID 7542829]7542829
- Brook S, Berk M, Selemani S, Kolloori J, Nzo I. A randomized controlled double blind study of zuclopenthixol acetate compared to haloperidol in acute psychosis. Human Psychopharmacology 1998;13(1):17‐20. [EMBASE 1998035700] [Google Scholar]
Burke 2002 {published data only}
- Burke JG, Reveley MA. Improved antisaccade performance with risperidone in schizophrenia. Journal of Neurology, Neurosurgery, and Psychiatry 2002;72:449‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chin 1998 {published data only}
- Chin CN, Hamid AR, Philip G, Ramlee T, Mahmud M, Zulkifli G, et al. A double blind comparison of zuclopenthixol acetate with haloperidol in the management of acutely disturbed schizophrenics. Medical Journal of Malaysia 1998;53(4):365‐71. [MEDLINE: ; PsycINFO 52‐05954; PMID 4589640] [PubMed] [Google Scholar]
Chouinard 1991 {published data only}
- Chouinard G, Safadi G, Beauclair L. A double‐blind controlled study of intramuscular zuclopenthixol acetate and liquid oral haloperidol in the treatment of schizophrenic patients with acute exacerbation. Journal of Clinical Psychopharmacology 1994;14(6):377‐84. [MEDLINE: ; PMID 7884017] [PubMed] [Google Scholar]
- Chouinard G, Safadi G, Beauclair L. The management of acutely schizophrenic patients newly admitted from the emergency room: a double‐blind clinical trial comparing zuclopenthixol acetate and liquid haloperidol. Modern Trends in the Treatment of Chronic Schizophrenia. Proceedings of a Symposium; 1991 June 10, Florence, Italy. Excerpta Medica, 1991:35‐43. [MEDLINE: ; PMID 7884017]7884017
Clark 1969 {published data only}
- Clark. Sordinol versus chlorpromazine versus placebo. Psychopharmacology Bulletin 1969;3:54‐6. [01003] [Google Scholar]
CTRI‐2014‐07‐004712 {published data only}
- CTRI‐2014‐07‐004712. Rapid tranquillization of violent or agitated patients in a psychiatric emergency setting: Pragmatic, randomized, allocation concealed, participant and assessor blinded trial of intramuscular zuclopenthixol acetate versus intramuscular haloperidol plus promethazine. Http://apps.who.int/trialsearch/Trial.aspx?TrialID=CTRI/2014/07/004712 2014.
- Mythri SV, Tharyan P, Sunder S, Kattula D, Kirubakaran R, Adams CE. Rapid tranquillization of violent or agitated patients in a psychiatric emergency setting: Pragmatic, randomized, allocation concealed, participant and assessor blinded trial of intramuscular zuclopenthixol acetate versus intramuscular haloperidol plus promethazine [A comparative study of the effects of an intramuscular injection of zuclopenthixol acetate versus an intramuscular injection of a combination of haloperidol plus promethazine in people with violence or agitation presenting to a psychiatric hospital as an emergency]. Study Protocol 2013.
Den 2000 {published data only}
- Boer JA, Vahlne JO, Post P, Heck AH, Daubenton F, Olbrich R. Ritanserin as add on medication to neuroleptic therapy for patients with chronic or subchronic schizophrenia. Human Psychopharmacology 2000;15(3):179‐89. [EMBASE 2000170004] [DOI] [PubMed] [Google Scholar]
Dencker 1980 {published data only}
- Dencker SJ, Lepp M, Malm U. Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. I. A one year double‐blind study of clopenthixol decanoate and flupenthixol palmitate. Acta Psychiatrica Scandinavica Supplementum 1980;279:10‐28. [MEDLINE: ; PMID 6931470] [DOI] [PubMed] [Google Scholar]
- Dencker SJ, Malm U, Jorgensen A, Overo KF. Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. IV. Serum levels and clinical outcome. Acta Psychiatrica Scandinavica Supplementum 1980;279:55‐63. [MEDLINE: ; PMID 6931473] [DOI] [PubMed] [Google Scholar]
Dom 1978 {published data only}
- Dom R, Mesmaecker L, Broucke M, Hest T, Baro F. Maintenance treatment of chronic schizophrenic patients. A study with the long‐acting thioxanthene derivative, cis(Z)‐clopenthixol decanoate‐sordinol depot. Acta Psychiatrica Scandinavica 1978;57(4):299‐304. [MEDLINE: ; PMID 352094] [DOI] [PubMed] [Google Scholar]
Fagerlund 2003 {published data only}
- Fagerlund B, Mackeprang T, Gade A, Glenthoj BY. Effects of risperidone and zuclopenthixol on cognitive deficits in drug‐naive first episode schizophrenic patients. Schizophrenia Research 2003;60:133‐4. [CN‐00394142] [Google Scholar]
Fan 1999 {published data only}
- Fan S, Sun ZG, Li J. The comparison of effects of clopenthixal and clozapine in the treatment of schizophrenia [氯噻噸與氯氮平治療精神分裂癥的效果比較]. Acta Acadimae Medicinae Qingdao 1999; Vol. 35, issue 2:125‐7.
Fensbo 1990 {published data only}
- Fensbo C. Zuclopenthixol acetate, haloperidol, and zuclopenthixol in the treatment of acutely psychotic patients ‐ A controlled multicenter study. Meeting of the Scandinavian Society for Psychopharmacology (1989, Copenhagen, Denmark) [Zuclopenthixol acetat, haloperidol og zuclopenthixol i behandling af akut psykotiske patienter. En kontrolleret multicenterundersogelse]. Nordisk Psykiatrisk Tidsskrift 1990;44(3):295‐7. [PsycINFO 28‐73892] [Google Scholar]
Fricchione 2010 {published data only}
- Fricchione Parise V, Balletta G, Manna G. Risperidone depot versus zuclopenthixol depot for schizophrenia and schizophrenialike psychoses: Real world outcomes. European Neuropsychopharmacology 2010; Vol. 1:472‐3.
Galdersi 1994 {published data only}
- Galderisi S, Maj M, Mucci A, Bucci P, Kemali D. QEEG alpha1 changes after a single dose of high‐potency neuroleptics as a predictor of short‐term response to treatment in schizophrenic patients. Biological Psychiatry 1994;35(6):367‐74. [MEDLINE: ; EMBASE 1994114037; PMID 8018782] [DOI] [PubMed] [Google Scholar]
Glenthoj 2000 {published data only}
- Glenthoj BY, Mackeprang T, Fagerlund B, Hemmingsen R. Effects of antipsychotics on information‐processing and extrastriatal dopamine d2/d3 receptors in first‐episode drug‐naive schizophrenic patients. 39th Annual Meeting of the American College of Neuropsychopharmacology; 2000; Dec 10‐14; San Juan; Puerto Rico. 2000:191. [MEDLINE: ; PMID 7756183]7756183
- Glenth¢j BY, Mackeprang T, Fagerlund B, Hemmingsen RP. Effects of antipsychotic treatment on prepulse inhibition of the startle response (ppi) and cognition in first episode drug‐naive schizophrenic patients. Schizophrenia Research 2001;49(1,2):133. [PsycINFO 2001‐16512‐004] [Google Scholar]
Gravem 1990 {published data only}
- Gravem A, Elgen K. Cis(Z) clopenthixol. The neuroleptically active isomer of clopenthixol. A presentation of five double blind clinical investigations and other studies with cis(Z) clopenthixol (Cisordinol(R), Clopixol(R)). Acta Psychiatrica Scandinavica, Supplementum 1981;64(294):5‐12. [MEDLINE: ; PMID 6122337] [PubMed] [Google Scholar]
- Gravem Arne, Tove AJ. Co‐injection of zuclopenthixol acetate and zuclopenthixol decanoate. Nordisk Psykiatrisk Tidsskrift 1990;44(4):403‐5. [PsycINFO 78‐07960] [Google Scholar]
Hicklin 1967 {published data only}
- Hicklin A, Angst J. Retrospective and prospective studies on the clinical effect of clopenthixol (sordinol) in endogenous phychoses [Retrospektive und prospektive Studie uber die klinische Wirkung von Clopenthixol (Sordinol) bei endogenen Psychosen]. Schweizerische Medizinische Wochenschrift. Journal Suisse De Medecine 1967;97(19):615‐21. [MEDLINE: ; PMID 5600894] [PubMed] [Google Scholar]
Hovens 2003 {published data only}
- Hovens JEJM, Beijeman S, Tollenar J, Dries PJT, Loonen AJM. Risperidone versus zuclopenthixol in acute psychosis in emergency psychiatry: a prospective naturalistic study. Journal of the European College of Neuropsychopharmacology 2003;13(4):S293. [Google Scholar]
Hovens 2005 {published data only}
- Hovens JE, Dries PJT, Melman CTM, Wapenaar RJC, Loonen AJM. Oral risperidone with lorazepam versus oral zuclopenthixol with lorazepam in the treatment of acute psychosis in emergency psychiatry: a prospective, comparative, open‐label study. Journal of Psychopharmacology 2005; Vol. 19, issue 1:51‐7. [MEDLINE: ] [DOI] [PubMed]
Huang 2001 {published data only}
- Huang SH, Pan YH, Wu FY, Qiu YP. Comparison of clopenthixol decanoate to chlorpromazine in the treatment of schizophrenia. Chinese General Practice 2001;4(10):775‐6. [CN‐00393723] [Google Scholar]
Karsten 1981 {published data only}
- Karsten D, Kivimaki T, Linna SL, Pollari L, Turunen S. Neuroleptic treatment of oligophrenic patients. A double‐blind clinical multicentre trial of cis(Z)‐clopenthixol and haloperidol. Acta Psychiatrica Scandinavica Supplementum 1981;294(64):39‐45. [MEDLINE: ; PMID 7041518] [PubMed] [Google Scholar]
Knegtering 2002a {published data only}
- Knegtering H, Castelein S, Linde J, Bous J. Sexual dysfunctions and serum prolactin levels in patients using risperidone or quetiapine: a randomised trial. Schizophrenia Research 2002;53(3 Suppl 1):163. [Google Scholar]
Koskinen 1991 {published data only}
- Koskinen T, Wistedt B, Thelander S. Zuclopenthixol decanoate vs haloperidol decanoate: a double‐blind comparative trial. 5th World Congress of Biological Psychiatry; 1991 Jun 9‐14; Florence, Italy. 1991:563‐5. [MEDLINE: ; PMID 7756183]7756183
Kristiansen 2001 {published data only}
- Kristiansen KT, Mackeprang T, Fink‐Jensen A, Hemmingsen RP, Glenth¢j BY. Effects of antipsychotics and other agents on prepulse inhibition of the starlte response(ppi). Schizophrenia Research 2001;49(1,2):216. [PsycINFO 2001‐16512‐004] [Google Scholar]
Kristiansen 2003 {published data only}
- Kristiansen KT, Mackeprang T, Glenthoj BY. Acute effects of antipsychotics and diazepam on prepulse inhibition of the startle response. Schizophrenia Research 2003; Vol. 60, issue 1 Suppl 1:100.
Lamure 2003 {published data only}
- Lamure M, Toumi M, Chabannes J‐P, Dansette G‐Y, Benyaya J, Hansen K. Zuclopenthixol versus haloperidol: an observational randomised pharmacoeconomic evaluation of patients with chronic schizophrenia exhibiting acute psychosis. International Journal of Psychiatry in Clinical Practice 2003;7(3):177‐85. [Google Scholar]
Lemmens 1994 {published data only}
- Lemmens P, Smedt G, Gheuens J, Tritsmans L. Efficacy of risperidone in the treatment of schizophrenia. Proceedings of the 7th Biennial Winter Workshop on Schizophrenia; 1994 Jan 23‐28; Les Diablerets, Switzerland 1994; Vol. 11, issue 2:106.
Liu 1997b {published data only}
- Liu P, Bian H, Chen H. Observation of clinical effect of clopixol‐acuphase injection for acute psychosis. Chinese Journal of Pharmaco Epidemiology [药物流行病学杂志] 1997; Vol. 6, issue 4:202‐4.
Liu 1998a {published data only}
- Liu S. A follow‐up of effects of sustain treatment of clopixol and cpz in schizophrenic patients. Sichuan Mental Health [四川精神卫生] 1998; Vol. 11, issue 3:162‐3.
Loza 2001 {published data only}
- Loza B, Kucharska‐Pietura K, Debowska G. Atypical versus typical antipsychotic treatment prognosis in first‐episode paranoid schizophrenia based on wcst and dichotic listening scores. European Neuropsychopharmacology (Abstracts of the 14th Congress of the European College of Neuropsychopharmacology; 2001 Oct 13‐17; Istanbul, Turkey) 2001;11(3):285. [14th Congress of the European College of Neuropsychopharmacology [Congress Information System]: Conifer, Excerpta Medica Medical Communications BV, 2001 P2103#] [Google Scholar]
Lublin 1991 {published data only}
- Lublin H, Gerlach J, Hagert U, Meidahl B, Molbjerg C, Pedersen V, et al. Zuclopenthixol, a combined dopamine D1/D2 antagonist, versus haloperidol, a dopamine D2 antagonist, in tardive dyskinesia. European Neuropsychopharmacology 1991;1(4):541‐8. [EMBASE 1998035700] [DOI] [PubMed] [Google Scholar]
Mackeprang 2001 {published data only}
- Mackeprang T, Fagerlund B, Videbaek C, Hemmingsen RP, Glenthoj BY. Extrastriatal dopamine(d2/d3)‐receptor occupancy and cognition in first episode schizophrenic patients. Schizophrenia Research 2001;49(Suppl.1,2):194. [LMD163341] [Google Scholar]
Malt 1995 {published data only}
- Malt UF, Nystad R, Bache T, Noren O, Sjaastad M, Solberg KO, et al. Effectiveness of zuclopenthixol compared with haloperidol in the treatment of behavioural disturbances in learning disabled patients. British Journal of Psychiatry 1995;166(3):374‐7. [MEDLINE: ; PMID 7788130] [DOI] [PubMed] [Google Scholar]
Mann 1985 {published data only}
- Mann BS, Moslehuddin KS, Owen RT, Clayton AR, Rohatgi KK, Sud P, et al. A clinical assessment of zuclopenthixol dihydrochloride ('Clopixol Tablets') in the treatment of psychotic illness. Pharmatherapeutica 1985;4(6):386392. [PubMed] [Google Scholar]
Martyns 1993 {published data only}
- Martyns Yellowe IS. The decanoates of flupenthixol and clopenthixol in the treatment of chronic schizophrenic in‐patients. Implications for community psychiatry. West African Journal of Medicine 1993;12(2):110‐3. [MEDLINE: ; PMID 8104468] [PubMed] [Google Scholar]
- Martyns‐Yellowe IS. The positive and negative symptoms of schizophrenia: patterns of response to depot neuroleptic treatment. West African Journal of Medicine 1994;13(4):200‐3. [MEDLINE: ; PMID 7756183] [PubMed] [Google Scholar]
Mazurek 2003 {published data only}
- Mazurek I, Loza B, Lecyk A. Atypical versus typical antipsychotic treatment prognosis based on veps and wcst scores in paranoid schizophrenia. Journal of the European College of Neuropsychopharmacology 2003;13(4):S347. [P.2.156] [Google Scholar]
Meyers 1972 {published data only}
- Meyers C, Lhoas JP, Huvelle R. Rediscovery of a neuroleptic drug in a new form: clopenthixol [Redecouverte d'un neuroleptique sous une autre presentation: le clopenthixol]. Schweizerische Rundschau fur Medizin Praxis 1972;61(26):869‐71. [Cochrane Library CN‐00282616] [PubMed] [Google Scholar]
Mosolov 2000 {published data only}
- Mosolov SN, Molodetskikh AV, Eryomin AV, Graikov GM, Nourislamov SV. Effect and tolerability of Clopixol and haloperidol in patients with acute symptoms of schizophrenia: comparative randomized investigation. Sotsial'Naia I KlinIcheskaia Psikhiatria 2000;10(2):47‐52. [MEDLINE: ; PMID 4967871]4967871 [Google Scholar]
NCT00206960 {published data only}
- NCT00206960. Effects of classical and atypical antipsychotics on dopamine receptor binding of 123i‐epidepride, cognition, startle response and extrapyramidal side‐effects in drug‐naive first‐episode schizophrenic patients. http://www.clinicaltrials.gov 2005.
Pagsberg 2004 {published data only}
- Pagsberg A, Jagersma E, Baare W, Mackeprang T, Glenthoej by. Change in caudate nucleus volume after three‐month treatment in drug‐ naive first‐episode schizophrenia patients. Schizophrenia Research 2004;67(1):99. [Google Scholar]
Ropert 1988 {published data only}
- Ropert R, Hoepfner Petersen HE, Benyay J. Where zuclopenthixol acetate stands amid the 'modified release' neuroleptics [Place de l'acetate de zuclopenthixol au sein des neuroleptiques a liberation modifiee]. Congres de Psychiatrie et de Neurologie de Langue Francaise, LXXXVIth Session; 1988 Jun 13‐17; Chambery, France. 1988:350‐62. [MEDLINE: ; PMID 3615572]3615572
Rubio 2005 {published data only}
- Rubio G, Martínez‐Gras I, Ponce G, López‐Muñoz F, Alamo C, Jimenez‐Arriero MA, et al. Risperidone versus zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity: a long‐term controlled study. European Neuropsychopharmacology 2005;15(Suppl 3):S476. [Google Scholar]
Rubio 2006 {published data only}
- Rubio G, Martinez I, Ponce G, Jimenez‐Arriero MA, Lopez‐Munoz F, Alamo C. Long‐acting injectable risperidone compared with zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity. Canadian Journal of Psychiatry [Revue Canadienne de Psychiatrie]. Canada, 2006; Vol. 51, issue 8:531‐9. [MEDLINE: ] [DOI] [PubMed]
- Rubio G, Martinez‐Gras I, Ponce G, Jiménez‐Arriero MA, López‐Muñoz F, Alamo C. Long‐acting injectable risperidone versus zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity. Proceedings of the 159th Annual Meeting of the American Psychiatric Association; 2006 May 20‐25; Toronto, Canada 2006.
Rubio 2009 {published data only}
- Rubio G, Rodriguez‐Jimenez R, Martinez‐Gras I, Jimenez‐Arriero MA, Palomo T. Prepulse inhibition of the startle response in amisulpride‐treated patients: comparison with typical antipsychotics. Proceedings of the 9th World Congress of Biological Psychiatry; 2009 Jun 28‐Jul 2 ;Paris, France. 2009:394.
Rubioz 2006 {published data only}
- Rubioz G, Martine I, Recio A, Ponce G, Lopez‐Munoz F, Alamo C, et al. Risperidone versus zuclopenthixol in the treatment of schizophrenia with substance abuse comorbidity: a long‐term randomized, controlled, crossover study. European Journal of Psychiatry 2006;20(3):133‐46. [Google Scholar]
Saxena 1990b {published data only}
- Saxena B, MacCrimmon D, Busse E, Turner P, Cooper L, Kenny J, et al. I.M clopixol v/s modecate in chronic schizophrenia – a double‐blind study. Proceedings of the 17th Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1990 Sep 10‐14; Kyoto, Japan 1990:247.
Saxena 1996 {published data only}
- Saxena B. The value of depot neuroleptic injections in the treatment of chronic schizophrenia. Schizophrenia 1996 ‐ Breaking down the Barriers. Proceedings of the 4th International Conference; 1996 Oct 6‐9; Vancouver, Canada. 1996. [MEDLINE: ; PsycINFO 1973‐29665‐001; PMID 4715636]4715636
Schooler 1993 {published data only}
- Schooler NR, Keith SJ, Severe JB, Matthews SM. Treatment strategies in schizophrenia: effects of dosage reduction and family management on outcome. International Congress on Schizophrenia Research 1993;9:260. [Google Scholar]
Shelton 1969 {published data only}
- Shelton WH, Bishop MP, Gallant DM. The Achilles reflex and antipsychotic drugs. Current Therapeutic Research Clinical and Experimental 1969;11(1):22‐6. [MEDLINE: ; PMID 4973850] [PubMed] [Google Scholar]
Singh 1992 {published data only}
- Singh I, Owino WJE. A double‐blind comparison of zuclopenthixol tablets with placebo in the treatment of mentally handicapped in‐patients with associated behavioural disorders. Journal of Intellectual Disability Research 1992;36:541‐9. [DOI] [PubMed] [Google Scholar]
Sofronov 2013 {published data only}
- Sofronov A, Spikina A, Savelyev A. The role of modern antipsychotics in correction of neurocognitive deficit in schizophrenia patients. European Psychiatry 2013; Vol. 28:1247.
Svestka 1986 {published data only}
- Svestka J, Nahunek K, Ceskova E. Controlled cross‐over comparison of oxyprothepine and clopenthixol decanoate in the prophylaxis of schizophrenic psychoses. Activitas Nervosa Superior 1986;28(1):35‐6. [PsycINFO 75‐08452] [Google Scholar]
Syvalahti 1997 {published data only}
- Syvalahti EK, Taiminen T, Saarijarvi S, Lehto H, Niemi H, Ahola V, ET AL. Citalopram causes no significant alterations in plasma neuroleptic levels in schizophrenic patients. Journal of International Medical Research 1997;25(1):24‐32. [MEDLINE: ; PMID 9027670] [DOI] [PubMed] [Google Scholar]
Taymeeyapradit 2002 {published data only}
- Taymeeyapradit U, Kuasirikul S. Comparative study of the effectiveness of zuclopenthixol acetate and haloperidol in acutely disturbed psychotic patients. Journal of the Medical Association of Thailand 2002;85(12):1301‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Tegeler 1985 {published data only}
- Tegeler J. A comparative trial of Cis(Z)‐clopenthixol‐decanoate and fluphenazine‐decanoate. Pharmacopsychiatry 1985;18(1):78‐9. [MEDLINE: ; PMID 9027670]9027670 [Google Scholar]
Uys 1996 {published data only}
- Uys H, Berk M. A controlled double blind study of zuclopenthixol acetate compared to clothiapine in acute psychosis including mania and exacerbation of chronic psychosis. Unpublished Report 1996. [MEDLINE: ]
- Uys H, Berk M. A controlled double blind study of zuclopenthixol acetate compared with clothiapine in acute psychosis including mania and exacerbation of chronic psychosis. 20th Congress of the Collegium Internationale Neuro‐psychopharmacologicum; 1996 Jun 23‐27; Melbourne, Australia. 1996. [MEDLINE: ; PsycINFO 52‐05954; PMID 4589640]4589640
Venkateswarlu 1990 {published data only}
- Venkateswarlu T, Staley CJ, Neal CD. Zuclopenthixol dihydrochloride in the management of behavioural disorders in elderly demented patients:a dose‐ranging study. International Journal of Geriatric Psychiatry 1990;5(4):265‐70. [Google Scholar]
Viala 1988 {published data only}
- Viala A, Ba B, Durand A, Gouezo F, Hou N, Jorgensen A. Comparative study of the pharmacokinetics of zuclopenthixol decanoate and fluphenazine decanoate. Psychopharmacology 1988;94(3):293‐7. [MEDLINE: ; PMID 2895936] [DOI] [PubMed] [Google Scholar]
Walker 1983 {published data only}
- Walker CA. A double‐blind comparative trial of the decanoates of clopenthixol and fluphenazine in the treatment of chronic schizophrenic out‐ patients. Pharmatherapeutica 1983;3(5):289‐93. [MEDLINE: ; PMID 6133287] [PubMed] [Google Scholar]
Weiser 1975 {published data only}
- Weiser G, Tahedl A, Reisecker F, Meyer H. Advantages of the initial therapy of acute schizophrenia with large doses of droperidol/A comparative study (author's transl) [Vorteile der Initialbehandlung akuter Schizophrenien mit hochdosiertem Droperidol. Eine Vergleichsstudie]. Arzneimittel Forschung 1975;25(11):1845‐8. [MEDLINE: ; PMID 1243093] [PubMed] [Google Scholar]
Wisted 1990 {published data only}
Wistedt 1991 {published data only}
- Wistedt B, Koskinen T, Thelander S, Nerdrum T, Pedersen V, Molbjerg C. Zuclopenthixol decanoate and haloperidol decanoate in chronic schizophrenia: a double‐blind multicentre study. Acta Psychiatrica Scandinavica 1991;84(1):14‐21. [MEDLINE: ; PMID 1681680] [DOI] [PubMed] [Google Scholar]
Xiaanao 1999 {published data only}
- Xiaanoao G, Jiaju X, Mingshen W. Efficacy of clopenthixol in treating delusions and halucinations in schizophrenia. Journal of Clinical Psychology 1999;9(4):202‐3. [Google Scholar]
Youssef 1991 {published data only}
- Youssef HA. Duration of neuroleptic treatment and relapse rate: a 5‐year follow‐up study with haloperidol decanoate. Clinical Neuropharmacology 1991;14(suppl.2):S16‐S23. [PubMed] [Google Scholar]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Awad 1997
- Awad GA, Voruganti LNP, Heslegrave RJ. Measuring quality of life in patients with schizophrenia. Pharmacoeconomics 1997;11(1):32‐47. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Chouinard 1980
- Chouinard G, Ross‐Chouinard A, Annable L, Jones BD. The Extrapyramidal Symptom Rating Scale. Canadian Journal of Neurological Science 1980;7:233. [Google Scholar]
da Silva Freire Coutinho 1999
- Silva Freire Coutinho E, Fenton M, Quraishi SN. Zuclopenthixol decanoate for schizophrenia and other serious mental illnesses. Cochrane Database of Systematic Reviews 1999, Issue 3. [DOI: 10.1002/14651858.CD001164] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dally 1967
- Dally P. Chemotherapy of Psychiatric Disorders. London: Logos Press Limited, 1967. [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Dollery 1999
- Dollery. Drug discovery and development in the molecular era. British Journal of Clinical Pharmacology January 1999;47(1):5‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Zellweger‐Zähner T, Schneider M, Junker C, Lengeler C, Antes G. Language bias in randomised controlled trials published in English and German. Lancet 1997;350:326‐9. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy U. ECDEU Assessment Manual for Psychopharmacology. Revised. Rockville: National Institute of Mental Health, 1976. [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Jablensky 1992
- Jablensky A, Sartorius N, Ernberg G, Anker M, Korten A, Cooper JE, et al. Schizophrenia: manifestations, incidence and course in different cultures. A World Health Organization ten‐country study. Psychological Medicine 1992;20(Monograph Supplement):1‐97. [DOI] [PubMed] [Google Scholar]
Jayakody 2012
- Jayakody K, Gibson RC, Kumar A, Gunadasa S. Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD000525.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13(2):261‐76. [DOI] [PubMed] [Google Scholar]
Kozyrev 2003
- Kozyrev VN, Smulevich AB, Drobizhev MIu, Kraeva GK, Kubrakov MA. Psychotropic drugs used in a psychiatric hospital (pharmaco‐epidemiologic aspects).. Zhurnal Nevrologii Psikhiatrii Im S S Korsakova 2003;103(11):25‐32. [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Lingjaerde 1987
- Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross‐sectional study of side effects in neuroleptic‐treated patients. Acta Psychiatrica Scandinavica Supplementum 1987;76(334):100. [DOI] [PubMed] [Google Scholar]
Mace 2005
- Mace S, Taylor D. A prescription survey of antipsychotic use in England and Wales following the introduction of NICE guidance. International Journal of Psychiatry in Clinical Practice 2005;9(2):124‐9. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Adams C, Bradley C, Joy C, Fenton M. Unpublished rating scales ‐ a major source of bias in randomised controlled trials of treatments for schizophrenia?. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schultz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel group randomised trials. Lancet 2001;357:1191‐4. [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Percudani 2005
- Percudani M. Barbui C. Fortino I. Petrovich L. Epidemiology of first‐ and second‐generation antipsychotic agents in Lombardy, Italy. Pharmacopsychiatry 2005;38(3):128‐31. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. on behalf of the Cochrane Applicability and Recommendations Methods Group. Chapter 12: Interpreting results and drawing conclusions. In Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Taylor 2000
- Taylor D, Kerwin R. Prescribing in schizophrenia. Evaluating the effect of introducing a new treatment protocol. Psychiatric Bulletin 2000;24:106‐8. [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):iii‐92. [MEDLINE: ] [PubMed] [Google Scholar]
WHO 2010
- Multiple. Pocket Guide to the ICD‐10 Classification of Mental and Behavioural Disorders. 16. Elsevier, 2010. [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Kumar 2005
- Kumar A, Strech D. Zuclopenthixol dihydrochloride for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD005474] [DOI] [PubMed] [Google Scholar]