

Abstract
Background
Sodium‐glucose cotransporter (SGLT) 2 inhibitors were recently approved as glucose‐lowering interventions in people with type 2 diabetes mellitus (T2DM). Potential beneficial or harmful effects of SGLT 2 inhibitors in people at risk for the development of T2DM are unknown.
Objectives
To assess the effects of SGLT 2 inhibitors focusing on the prevention or delay of T2DM and its associated complications in people with impaired glucose tolerance, impaired fasting blood glucose or moderately elevated glycosylated haemoglobin A1c (HbA1c) or any combination of these.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, PubMed, EMBASE, ClinicalTrials.gov, the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) and reference lists of systematic reviews, articles and health technology assessment reports. We asked investigators of ongoing for information about additional trials. The date of the last search of all databases was January 2016.
Selection criteria
Randomised controlled trials (RCTs) of any duration comparing SGLT 2 inhibitors with any glucose‐lowering intervention, behaviour‐changing intervention, placebo or no intervention in people with impaired fasting glucose, impaired glucose tolerance, moderately elevated HbA1c or combinations of these.
Data collection and analysis
Two review authors read all abstracts, assessed quality and extracted data independently. We resolved discrepancies by consensus or the involvement of a third author.
Main results
We could not include any RCT in this systematic review. One trial was published in two abstracts, but did not provide separate information of the participants with impaired glucose tolerance, impaired fasting glucose or both. We identified two ongoing trials, both evaluating the effects of dapagliflozin (and metformin) in people at risk for the development of type 2 diabetes and a follow‐up of 24 to 26 weeks. Both trials will mainly report on surrogate outcome measures with some data on adverse effects and health‐related quality of life.
Authors' conclusions
Due to lack of data it is not possible to conclude whether SGLT 2 inhibitors prevent or delay the diagnosis of T2DM and its associated complications.
Plain language summary
Sodium‐glucose cotransporter (SGLT) 2 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus
Review question
What are the effects of the group of glucose‐lowering drugs called 'sodium‐glucose cotransporter (SGLT) 2 inhibitors' for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus?
Background
The SGLT 2 inhibitors (such as canagliflozin, dapagliflozin and empagliflozin) are glucose‐lowering drugs that reduce blood glucose levels by increasing the secretion of glucose from the kidneys to the urine. SGLT 2 inhibitors were recently approved for the treatment of diabetes in people with type 2 diabetes mellitus. It is currently not known whether SGLT 2 inhibitors should be prescribed for people with raised blood glucose levels who do not meet the criteria for having type 2 diabetes. We wanted to find out whether these drugs would prevent or only delay the development of type 2 diabetes. Furthermore, we wanted to analyse the effects of SGLT 2 inhibitors on patient‐important outcomes such as complications of diabetes (for example kidney and eye disease, heart attacks, strokes), death from any cause, health‐related quality of life and side effects of the medications.
Study characteristics
We searched the medical literature and registers of ongoing trials for randomised controlled trials (clinical studies where people are randomly put into one of two or more treatment groups) of SGLT 2 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications. We wanted to synthesise the findings of several studies to answer our review question. Unfortunately, we did not find such a study.
This evidence is up to date as of January 2016.
Key results
We could not include any study in our systematic review. However, we identified two ongoing studies, both evaluating the effects of dapagliflozin in people at risk for the development of type 2 diabetes and a follow‐up of 24 to 26 weeks. Both rather short‐term studies will mainly report on laboratory outcome measures with some data on side effects and health‐related quality of life.
Quality of the evidence
Because we could not include any study we were not able to investigate the quality of the evidence. In both ongoing studies the participants know what kind of medication they are taking which may create problems with the measurement of some outcomes such as health‐related quality of life and side effects.
Background
Description of the condition
'Prediabetes', 'borderline diabetes', the 'prediabetic stage', 'high risk of diabetes', 'dysglycaemia' or 'intermediate hyperglycaemia' are often characterised by various measurements of elevated blood glucose concentrations (such as isolated impaired fasting glucose (IFG), isolated impaired glucose tolerance (IGT), isolated elevated glycosylated haemoglobin A1c (HbA1c) or combinations thereof) (WHO/IDF 2006). These elevated blood glucose levels that indicate hyperglycaemia are too high to be considered normal but are below the diagnostic threshold for type 2 diabetes mellitus (T2DM). Therefore, due to the continuous glycaemic spectrum from the normal to the diabetic stage, a sound evidence base is needed to define glycaemic thresholds for people at high risk of diabetes. The different terms used to describe various stages of hyperglycaemia may cause people to have different emotional reactions. For example, the term 'prediabetes' may imply (at least for lay people) that the disease diabetes is unavoidable, whereas (high) risk of diabetes has the positive connotation to possibly being able to avoid the disease altogether. In addition to the disputable construct of intermediate health states termed "prediseases" (Viera 2011), the label 'prediabetes' may be associated by many people with dire consequences. Alternatively, any diagnosis of 'prediabetes' may be an opportunity to review, for example, eating habits and physical activity levels, thus enabling 'affected' individuals to actively change their way of life.
The American Diabetes Association (ADA) and the World Health Organization (WHO) established the most commonly used criteria to define people with a high risk of developing T2DM. IGT was the first glycaemic measurement used by the US National Diabetes Data Group to define the prediabetic stage (NDDG 1979). It is based on the measurement of plasma glucose two hours after ingestion of 75 g of glucose. The dysglycaemic range is defined as a plasma glucose level of between 7.8 to 11.1 mmol/L (140 to 200 mg/dL) two hours after the glucose load. Studies indicate that IGT is caused by insulin resistance and defective insulin secretion (Abdul‐Ghani 2006; Jensen 2002). In 1997, the ADA and later the WHO introduced the IFG concept to define 'prediabetes' and intermediate hyperglycaemia (ADA 1997; WHO 1999). The initial definition of IFG was 6.1 to 6.9 mmol/L (110 to 125 mg/dL). Later, the ADA reduced the lower threshold for defining IFG to 5.6 mmol/L (100 mg/dL) (ADA 2003). However, the WHO did not endorse this lower cut‐off point for IFG to define 'prediabetes' (WHO/IDF 2006). IFG seems to be associated with β‐cell dysfunction (impaired insulin secretion) and an increase in the hepatic glucose output (DeFronzo 1989). More recently, HbA1c has been introduced to identify people at high risk of developing T2DM. In 2009, the International Expert Committee (IEC) suggested certain HbA1c ranges to identify people with a high risk of T2DM. People with HbA1c measurements of between 6.0% to 6.4% fulfilled this criterion (IEC 2009). Shortly afterwards, the ADA re‐defined this HbA1c level as 5.7% to 6.4% to identify people with a high risk of developing T2DM (ADA 2010). Unlike IFG and IGT, HbA1c reflects longer‐term glycaemic control, i.e. how a person's blood glucose levels have been during the preceding two to three months (Inzucchi 2012).
In 2010, the International Diabetes Federation (IDF) estimated the prevalence of IGT to be 343 million people, and this is predicted to increase to 471 million people by 2035 (IDF 2013). Studies have shown poor correlations between HbA1c and IFG/IGT (Gosmanov 2014; Selvin 2011). Notably, the various glycaemic tests do not seem to identify the same people as there is an imperfect overlap among the glycaemic modalities available to define dysglycaemia (Gosmanov 2014; Selvin 2011). The risk of progression from people at risk to T2DM depends on the diagnostic criteria used to identify the risk. Some people with dysglycaemia will never develop T2DM, and some people will return to normoglycaemia. IGT is often accepted as the best glycaemic variable for risk to predict progression to T2DM. However, studies indicate that less than half of the people defined as 'prediabetic' by means of IGT will develop T2DM in the following 10 years. Both IFG and HbA1c are thought to predict a different risk spectrum for developing T2DM (Cheng 2006; Morris 2013). Most importantly, dysglycaemia is commonly an asymptomatic condition, and naturally often remains 'undiagnosed' (CDC 2015).
It has not been clarified whether or not any particular intervention, especially glucose‐lowering drugs, should be recommended for people at risk for T2DM (Yudkin 2014). Trials have indicated that the progression to T2DM is reduced, or possibly just delayed, with behavioural interventions (increased physical activity, dietary changes or both) (Diabetes Prevention Program 2002; Diabetes Prevention Program FU 2009; Finnish Diabetes Prevention Study Group 2001). A recent meta‐analysis of 22 trials with interventions that changed behaviour in people at high risk of T2DM concluded that the effect of these interventions on longer‐term diabetes prevention is not clarified(Dunkley 2014). Therefore, more research is needed to establish optimal strategies for reducing T2DM with behavioral approaches (Dunkley 2014).
International diabetes associations and clinicians do not generally accept the prescription of pharmacological glucose‐lowering interventions for the prevention of T2DM. Several groups of pharmacological glucose‐lowering interventions have been investigated in people at risk of T2DM. Some findings indicate that the progression to T2DM is reduced or may be just delayed (Diabetes Prevention Program 2002; Diabetes Prevention Program FU 2009). However, the ADA recommends metformin for people at risk for T2DM and with a body mass index of over 35 kg/m², aged less than 60 years and women with prior gestational diabetes mellitus (ADA 2015).
Description of the intervention
Recently, several members of a new class of glucose‐lowering interventions, sodium‐glucose co‐transporter (SGLT) 2 inhibitors, have been approved to treat people with T2DM. In 2012, dapagliflozin was introduced as the first SGLT 2 inhibitor to the European market (EMA 2012). In 2014, the Food and Drug Administration (FDA) approved dapagliflozin (FDA 2014a) and empagliflozin (FDA 2014b). Canagliflozin was the first SGLT 2 inhibitor to be approved by the FDA in 2013 (FDA 2013). Currently, three different SGLT 2 inhibitors are available for people with T2DM in the USA and Europe: canagliflozin, dapagliflozin and empagliflozin.
For people with T2DM, SGLT 2 inhibitors can be prescribed as a monotherapy, usually if diet and exercise alone are insufficient in controlling T2DM or if there are metformin intolerances or contraindications; SGLT 2 inhibitors can also be applied as combination therapy with existing pharmacological glucose‐lowering interventions (ADA 2015). Recently, a large scale randomised controlled trial indicated a beneficial effect on all‐cause mortality in people with T2DM and established cardiovascular disease when they added empagliflozin to existing glucose‐lowering medications (approximately 50% of the participants were on dual background glucose‐lowering therapy and approximately 74% received metformin treatment) compared with placebo (EMPA‐REG 2015).
The doses of the approved SGLT 2 inhibitors for people with T2DM differ. All approved SGLT 2 inhibitors are orally administered. For canagliflozin, the recommended starting dose is 100 mg once daily, which can be increased to 300 mg/day (Janssen Pharmaceuticals 2013). For dapagliflozin, the recommended starting dose is 5 mg/day or 10 mg/day (AstraZeneca Pharmaceuticals 2014). For empagliflozin, the recommended starting dose is 10 mg/day, which can be increased to 25 mg/day (Boehringer Ingelheim Pharmaceuticals 2014). All FDA‐approved SGLT 2 inhibitors are rapidly absorbed after oral administration and have a half‐life that allows once‐daily administration (Scheen 2015).
Adverse effects of the intervention
The most frequently reported adverse effects of SGLT 2 inhibitors are urogenital tract infections (AstraZeneca Pharmaceuticals 2014; Boehringer Ingelheim Pharmaceuticals 2014; EMPA‐REG 2015; Janssen Pharmaceuticals 2013). A new report from FDA warns that there may be an increased risk of ketoacidosis associated with SGLT 2 inhibitors (FDA 2015).
How the intervention might work
In healthy individuals, glucose is filtered and reabsorbed in the kidneys, so that the amount of excreted glucose in the urine is almost zero. The main protein in the kidneys that is responsible for the reabsorption of the glucose in the urine is the sodium–glucose cotransporter 2 (Mather 2011). In people with T2DM, the reabsorption of glucose in the kidneys is increased, which contributes to the development of hyperglycaemia. Moreover, sodium–glucose cotransporter 2 expression seems to be increased (Rahmoune 2005). The sodium–glucose cotransporter 2 proteins are placed in the proximal tubuli (Rahmoune 2005). The action of SGLT 2 inhibitors does not depend on either the secretion of insulin or on the insulin sensitivity in peripheral tissues. SGLT 2 inhibitors reduce fasting plasma glucose, post‐prandial glucose levels and HbA1c in people with T2DM (List 2011).
Why it is important to do this review
There has been an increased focus on the prevention or delay of diabetes with non‐pharmacological interventions and glucose‐lowering medications. Currently, several trials are ongoing to clarify whether the progression from an at‐risk status to T2DM can be stopped or postponed with glucose‐lowering compounds (ClinicalTrials.gov). However, a more important issue for people with dysglycaemia is whether or not these interventions reduce the risk of death and complications — especially cardiovascular disease — related to T2DM. The present systematic review focuses on one the benefits and harms of SGLT 2 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for development of type 2 diabetes mellitus.
Objectives
To assess the effects of sodium‐glucose cotransporter (SGLT) 2 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus.
Secondary objectives were to analyse the strength and quality of the body of evidence for the associations between various definitions of risk (impaired glucose tolerance, impaired fasting glucose, HbA1c thresholds) and the therapeutic consequences of SGLT 2 inhibition.
Methods
Criteria for considering studies for this review
Types of studies
We considered all randomised controlled trials (RCTs) of any design eligible for this review, if inclusion criteria were fulfilled. We would have included published and unpublished trials in all languages.
Types of participants
We planned to include nondiabetic individuals who are at an increased risk of type 2 diabetes mellitus (T2DM) development.
Diagnostic criteria for people at risk of T2DM development
To be consistent with changes to the classification of, and diagnostic criteria for, dysglycaemia (impaired fasting glucose (IFG), impaired glucose tolerance (IGT) and elevated glycosylated haemoglobin A1c (HbA1c)) over the years, the diagnosis should have been established using the standard criteria valid at the trial start (e.g. ADA 1997; ADA 2010; NDDG 1979; WHO 1999). Ideally, the diagnostic criteria should have been described. If necessary, we planned to use the trial authors' definition of risk but we would have contacted trial authors for additional information. Differences in the glycaemic measurements used to define risk may introduce substantial heterogeneity. Therefore we planned to subject the diagnostic criteria to a subgroup analysis.
Types of interventions
We planned to investigate the following comparisons of sodium–glucose cotransporter (SGLT) 2 inhibitors versus all pharmacological glucose‐lowering interventions, behaviour‐changing interventions, placebo or no intervention.
Intervention
SGLT 2 inhibitors.
Comparator
Any pharmacological glucose‐lowering intervention (e.g. acarbose, metformin, sulphonylurea).
Behaviour‐changing interventions (e.g. diet, exercise, diet and exercise).
Placebo.
No intervention.
The included trials had to have identical concomitant interventions in both the intervention and comparator groups to enable us to establish fair comparisons.
Minimum duration of intervention
We wanted to include trials irrespective of the duration of the intervention.
Exclusion criteria
We planned to exclude trials of people diagnosed with the 'metabolic syndrome' because this is a special population which is not representative of people with just intermediate hyperglycaemia. Also, the composite of risk indicators such as elevated blood lipids, insulin resistance, obesity and high blood pressure which is termed metabolic syndrome is of doubtful clinical usefulness and uncertain distinct disease entity. However, if we had identified any trials investigating participants with any definition of the metabolic syndrome, we would have summarised some basic trial information in an additional table.
We would have excluded trials evaluating participants with intermediate hyperglycaemia in combination with another condition, e.g. cystic fibrosis, acute myocardial infarction and stroke, if such trials were identified.
We planned to include trials that explicitly described that a fraction of the included participants had intermediate hyperglycaemia. We contacted the investigators in order to get separate data on the group with intermediate hyperglycaemia and wanted to include these in the systematic review and meta‐analyses.
We planned to include trials on obese people or participants with previous gestational diabetes, if trial investigators had described that the participants had intermediate hyperglycaemia.
We planned to include a trial even if it did not report one or more of our primary or secondary outcome measures in the publication. If we identified a trial that did not report any of our primary or secondary outcomes, we would have contacted the corresponding trial author for supplementary data. If no additional data were available, the trial would have been presented in a supplementary table.
Types of outcome measures
Primary outcomes
All‐cause mortality.
Incidence of T2DM.
Serious adverse events.
Secondary outcomes
Cardiovascular mortality.
Non‐fatal myocardial infarction.
Congestive heart failure.
Non‐fatal stroke.
Amputation of lower extremity.
Blindness or severe vision loss.
End‐stage renal disease.
Non‐serious adverse events.
Hypoglycaemia.
Health‐related quality of life.
Time to progression to T2DM.
Measures of blood glucose control.
Socioeconomic effects.
Method and timing of outcome measurement
All‐cause mortality: defined as death from any cause and measured at the end of the intervention and the end of follow‐up.
Incidence of T2DM and time to progression to T2DM: defined according to the diagnostic criteria valid at the time the diagnosis was established using the standard criteria valid at the time of the trial commencing (e.g. ADA 2008; WHO 1998) and measured at the end of the intervention and the longest reported end of follow‐up. If necessary, we would use the trial authors' definition of T2DM.
Serious adverse events: defined according to the International Conference on Harmonization Guidelines as any event that lead to death, was life‐threatening, required in‐patient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability, or any important medical event that had jeopardised the participant or required intervention to prevent it (ICH‐GCP 1997) or as reported in trials and measured at any time of the intervention and during follow‐up. Cardiovascular mortality, non‐fatal myocardial infarction, non‐fatal stroke, amputation of lower extremity, blindness or severe vision loss, hypoglycaemia (mild, moderate, severe/serious): defined as reported in trials. Measured at the end of the intervention and at the end of follow‐up.
End‐stage renal disease: defined as dialysis, renal transplantation or death due to renal disease and measured at the end of the intervention and at the end of follow‐up.
Non‐serious adverse events: defined as number of participants with any untoward medical occurrence not necessarily having a causal relationship with the intervention and measured at the end of the intervention and at the end of follow‐up.
Health‐related quality of life: defined as mental and physical health‐related quality of life as separate and combined, evaluated by a validated instrument such as Short‐Form 36 and measured at the end of the intervention and at the end of follow‐up.
Measures of blood glucose control: fasting blood glucose, blood glucose two hours after ingestion of 75 g glucose and HbA1c measurements and measured at the end of the intervention and at the end of follow‐up.
Socioeconomic effects: defined as costs of the intervention, absence from work and medication consumption and measured at the end of the intervention and at the end of follow‐up.
Specification of key prognostic variables
Age.
Gender.
Equity issues (access to health care, social determinants).
Ethnicity.
Hypertension.
Cardiovascular disease.
Obesity.
Previous gestational diabetes.
Summary of findings
We presented a 'Summary of finding' table and reported the following outcomes, listed according to priority.
All‐cause mortality.
Incidence of T2DM.
Serious adverse events.
Cardiovascular mortality.
Non‐fatal myocardial infarction/stroke.
Health‐related quality of life.
Socioeconomic effects.
Search methods for identification of studies
Electronic searches
We searched the following sources from inception of each database to the specified date and placed no restrictions on the language of publication.
Cochrane Central Register of Controlled Trials (CENTRAL) (21 January 2016).
Ovid MEDLINE(R) In‐Process & Other Non‐Indexed Citations and Ovid MEDLINE(R) <1946 to Present> (21 January 2016).
PubMed (subsets not available on Ovid) (21 January 2016).
EMBASE <1974 to 2016 January 20> (21 January 2016).
ClinicalTrials.gov (21 January 2016).
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) Search Portal (http://apps.who.int/trialsearch/) (21 January 2016).
We continuously applied a MEDLINE (via Ovid SP) email alert service to identify newly published trials using the same search strategy as described for MEDLINE (for details on search strategies, see Appendix 1). If we had identified new trials for inclusion, we would have evaluated these, and incorporated the findings into our review and resubmit another Cochrane review draft (Beller 2013).
If additional key words of relevance had been detected during any of the electronic or other searches, we would have modified the electronic search strategies to incorporate these terms.
Searching other resources
We planned to identify other potentially eligible trials or ancillary publications by searching the reference lists of included trials, systematic reviews, meta‐analyses and health technology assessment reports. However, no included trials in full publication, systematic reviews, meta‐analyses and health technology assessment reports were identified. In addition we planned to contact authors of included trials, to get clarified if we missed any potentially relevant trial. However, we could not include any trial. We identified one abstract which fulfilled the inclusion criteria, as the trial included obese people with some of the participants having IFG and/or IGT (Sarich 2010). When searching for contact information of the investigators, we identified another abstract reporting the same trial (Sarich 2010a). We contacted the primary and secondary authors of the abstracts. The primary author responded that he was unable to provide additional information. We contacted the investigators of the identified ongoing trials who replied that they were not aware of any other existing or ongoing trial.
As none of the existing SGLT 2 inhibitors is approved for people at risk for T2DM, we did not search the databases of the regulatory agencies (i.e. the European Medicines Agency (EMA) and the US Food and Drugs Administration (FDA)).
Data collection and analysis
Selection of studies
Two review authors (BH, JK) independently scanned the abstract or title, or both, of every record retrieved, to determine which trials to be assessed further. Differences between review authors were resolved by discussion and involvement of a third author (BR). We prepared a flow diagram of the number of trials identified and excluded at each stage in accordance with the PRISMA (Preferred reporting items for systematic reviews and meta‐analyses) flow chart of study selection (Liberati 2009).
Data extraction and management
If we identified trials that fulfilled inclusion criteria, two review authors (BH, JK) would have independently extracted key participant and intervention characteristics. Data would have been reported on efficacy outcomes and adverse events using standardised data extraction sheets from the CMED Group. We planned to dissolve disagreements by discussion or, if required, by involvement of a third review author (BR).
We provide information about potentially relevant ongoing trial including the trial identifier in the 'Characteristics of ongoing studies' table and planned to also report data in a joint appendix 'Matrix of trial endpoint (publications and trial documents)'. We wanted to find the protocol of each included trial and planned to report primary, secondary and other outcomes in comparison with data in publications in this joint appendix.
We planned to email all authors of the included trials to enquire whether they would be willing to answer questions regarding their trials. We planned to present the results of this survey in an appendix. Thereafter we would have sought relevant missing information on the trial from the primary trial author(s), if required.
Dealing with duplicate and companion publications
In the event of duplicate publications, companion documents or multiple reports of a primary trial, we planned to maximise the information yield by collecting all available data and using the most complete data set aggregated across all known publications. Duplicate publications, companion documents or multiple reports of a primary trial would have been listed as secondary references under the primary reference of the included or excluded trial.
Assessment of risk of bias in included studies
We planned to assess the risk of bias independently by two review authors (BH, JK) of each included trials. We would have resolved any disagreements by consensus, or by consultation with a third review author (BR). If adequate information was unavailable from the trial publication, trial protocol or both, we would have contacted the trial authors for missing data on risk of bias items.
We planned to use the Cochrane 'Risk of bias' assessment tool (Higgins 2011a; Higgins 2011b) and would have judged risk of bias criteria as at either low, high, or unclear risk. We planned to evaluate individual bias items as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) where any of the specified criteria for a judgement on low, unclear or high risk of bias justifies the associated categorisation.
Random sequence generation (selection bias due to inadequate generation of a randomised sequence) ‐ assessment at trial level
For each included trial we planned to describe the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
Low risk of bias: the trial authors achieved sequence generation using computer random number generation or a random number table. Drawing of lots, tossing a coin, shuffling cards or envelopes, and throwing dice are adequate if an independent person performed this who was not otherwise involved in the trial. We considered the use of the minimisation technique as equivalent to being random.
Unclear risk of bias: there was insufficient information about the sequence generation process.
High risk of bias: the sequence generation method was non‐random (e.g. sequence generated by odd or even date of birth; sequence generated by some rule based on date (or day) of admission; sequence generated by some rule based on hospital or clinic record number; allocation by judgement of the clinician; allocation by preference of the participant; allocation based on the results of a laboratory test or a series of tests; or allocation by availability of the intervention). We excluded such trials.
Allocation concealment (selection bias due to inadequate concealment of allocations prior to assignment) ‐ assessment at trial level
We planned to describe for each included trial the method used to conceal allocation to interventions prior to assignment and we wanted to assess whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
Low risk of bias: central allocation (including telephone, interactive voice‐recorder, web‐based and pharmacy‐controlled randomisation); sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes.
Unclear risk of bias: insufficient information about the allocation concealment.
High risk of bias: using an open random allocation schedule (e.g. a list of random numbers); the trial used assignment envelopes without appropriate safeguards; alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. We excluded such trials.
We also planned to evaluate trial baseline data to incorporate assessment of baseline imbalance into the risk of bias judgement for selection bias (Corbett 2014; Egbewale 2014; Riley 2013). Chance imbalances may also affect judgements on the risk of attrition bias. In case of unadjusted analyses we would have distinguished between trials we rate at low risk of bias on the basis of both randomisation methods and baseline similarity, and trials we rated at low risk of bias on the basis of baseline similarity alone (Corbett 2014). We planned to re‐classify judgements of unclear, low or high risk of selection bias as specified in Appendix 2.
Blinding of participants and study personnel (performance bias due to knowledge of the allocated interventions by participants and personnel during the trial) ‐ assessment at outcome level
We planned to evaluate the risk of detection bias separately for each outcome (Hróbjartsson 2013). We wanted to note whether outcomes were self reported, investigator‐assessed or adjudicated outcome measures (see below).
Low risk of bias: blinding of participants and key study personnel is ensured, and it is unlikely that the blinding could have been broken; no blinding or incomplete blinding, but we judge that the outcome is unlikely to have been influenced by lack of blinding.
Unclear risk of bias: insufficient information about the blinding of participants and study personnel; the trial does not address this outcome.
High risk of bias: no blinding or incomplete blinding, and the outcome is likely to have been influenced by lack of blinding; blinding of trial participants and key personnel attempted, but likely that the blinding could have been broken, and the outcome is likely to be influenced by lack of blinding.
Blinding of outcome assessment (detection bias due to knowledge of the allocated interventions by outcome assessment) ‐ assessment at outcome level
We planned to evaluate the risk of detection bias separately for each outcome (Hróbjartsson 2013). We wanted to note whether outcomes were self reported, investigator‐assessed or adjudicated outcome measures (see below).
Low risk of bias: blinding of outcome assessment was ensured, and it was unlikely that the blinding could have been broken; no blinding of outcome assessment, but we judged that the outcome measurement is unlikely to have been influenced by lack of blinding.
Unclear risk of bias: insufficient information about the blinding of outcome assessors; the trial did not address this outcome.
High risk of bias: no blinding of outcome assessment, and the outcome measurement was likely to have been influenced by lack of blinding; blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement was likely to be influenced by lack of blinding.
Incomplete outcome data (attrition bias due to amount, nature or handling of incomplete outcome data) ‐ assessment at outcome level
For each included trial and for each outcome, we planned to describe the completeness of data, including attrition and exclusions from the analyses. We would have stated whether the trial reported attrition and exclusions and the number of participants included in the analysis at each stage (compared with the number of randomised participants per intervention/comparator groups). We also wanted to note if the trial reported the reasons for attrition or exclusion, and whether missing data were balanced across groups or were related to outcomes. We planned to consider the implications of missing outcome data per outcome such as high drop‐out rates (e.g. above 15%) or disparate attrition rates (e.g. difference of 10% or more between trial arms).
Low risk of bias: no missing outcome data; reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk was not enough to have a clinically‐ relevant impact on the intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes was not enough to have a clinically relevant impact on observed effect size; appropriate methods, such as multiple imputation, were used to handle missing data.
Unclear risk of bias: insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to induce bias; the trial did not address this outcome.
High risk of bias: reason for missing outcome data was likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk enough to induce clinically‐relevant bias in intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes enough to induce clinically relevant bias in observed effect size; ‘as‐treated’ or similar analysis done with substantial departure of the intervention received from that assigned at randomisation; potentially inappropriate application of simple imputation.
Selective reporting (reporting bias due to selective outcome reporting) ‐ assessment at trial level
We planned to assess outcome reporting bias by integrating the results of the appendix 'Matrix of trial endpoints (publications and trial documents)' (Boutron 2014; Mathieu 2009), with those of the appendix 'High risk of outcome reporting bias according to Outcome Reporting Bias In Trials (ORBIT) classification' (Kirkham 2010). This analysis would have formed the basis for the judgement of selective reporting.
Low risk of bias: the trial protocol was available and all of the trial’s pre‐specified (primary and secondary) outcomes that are of interest in the Cochrane review were reported in the pre‐specified way; the study protocol was unavailable but it was clear that the published reports included all expected outcomes (ORBIT classification).
Unclear risk of bias: insufficient information about selective reporting.
High risk of bias: not all of the trial’s pre‐specified primary outcomes were reported; one or more primary outcomes are reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not pre‐specified; one or more reported primary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect); one or more outcomes of interest in the Cochrane review are reported incompletely so that we cannot enter them in a meta‐analysis; the trial report failed to include results for a key outcome that we would expect to have been reported for such a trial (ORBIT classification).
Other bias (bias due to problems not covered elsewhere) ‐ assessment at trial level
Other risk of bias reflects other circumstances that may threaten the validity of the trials, e.g. funding bias and academic bias (Lundh 2012).
Low risk of bias: the trial appeared to be free of other sources of bias.
Unclear risk of bias: there was insufficient information to assess whether an important risk of bias existed; insufficient rationale or evidence that an identified problem introduced bias.
High risk of bias: the trial had a potential source of bias related to the specific trial design used; the trial has been claimed to have been fraudulent; or the trial had some other serious problem.
We planned to present a 'Risk of bias' graph and a 'Risk of bias' summary figure.
For 'Risk of bias' evaluation we wanted to group outcome measures as follows.
Health‐related quality of life.
Incidence of T2DM.
Macrovascular complications: non‐fatal myocardial infarction, non‐fatal stroke.
Measures of blood glucose control.
Microvascular complications: amputation of lower extremity, blindness/severe vision loss, end‐stage renal disease.
Mortality: all‐cause mortality, cardiovascular mortality.
Non‐serious adverse events (including hypoglycaemic episodes, depending on measurement).
Serious hypoglycaemic episodes (including hypoglycaemic episodes, depending on measurement).
Socioeconomic effects.
Time to progression to T2DM.
Furthermore, we wanted to distinguish between self reported, investigator‐assessed and adjudicated outcome measures.
We defined the following outcomes as self reported.
Non‐serious adverse events.
Hypoglycaemia, if reported by participant(s).
Health‐related quality of life.
Blood glucose control, if measured by trial participant(s).
We defined the following outcomes as investigator‐assessed.
All‐cause mortality.
Incidence of T2DM.
Time to progression to T2DM.
Serious adverse events.
Cardiovascular mortality.
Non‐fatal myocardial infarction.
Non‐fatal stroke.
Amputation of lower extremity.
Blindness or severe vision loss.
End‐stage renal disease.
Hypoglycaemia, if measured by trial personnel.
Blood glucose control, if measured by trial personnel.
Socioeconomic effects.
Summary assessment of risk of bias
Risk of bias for a trial across outcomes
Some risk of bias domains, like selection bias (sequence generation and allocation sequence concealment), affect the risk of bias across all outcome measures in a trial. Otherwise, we would not have performed a summary assessment of the risk of bias across all outcomes for a trial. In case of high risk of selection bias, we planned to exclude the trial.
Risk of bias for an outcome within a trial and across domains
We planned to assess the risk of bias for an outcome measure by including all entries relevant to that outcome, i.e. both trial‐level entries and outcome specific entries. 'Low' risk of bias was defined as low risk of bias for all key domains, 'unclear' risk of bias as unclear risk of bias for one or more key domains and 'high' risk of bias as high risk of bias for one or more key domains.
Risk of bias for an outcome across trials and across domains
These are our main summary assessments that we would have incorporated into our judgements about the quality of evidence in the 'Summary of finding' table(s). 'Low' risk of bias is defined as most information coming from trials at low risk of bias, 'unclear' risk of bias as most information coming from trials at low or unclear risk of bias and 'high' risk of bias as sufficient proportion of information coming from trials at high risk of bias.
Measures of treatment effect
When at least two included trials were available for a comparison of a given outcome, we would have expressed dichotomous data as a risk ratio (RR) with 95% confidence intervals (CIs) and with Trial Sequential Analysis (TSA)‐adjusted CIs if the diversity‐adjusted required information size was not reached. We planned to express continuous data that were reported on the same scale as mean difference (MD) with 95% CIs and with TSA‐adjusted CIs if the diversity‐adjusted required information size was not reached. For trials that address the same outcome but use different outcome measure scales, we planned to use standardised mean differences (SMD) with 95% CIs. We wanted to calculate time‐to‐event data as hazard ratios (HRs) with 95% CIs with the generic inverse variance method. We preferentially would have used unadjusted HRs as adjustment may differ among the included trials. For outcomes meta‐analysed as SMD and the generic inverse variance method, we are presently unable to conduct TSA and adjust the 95% CIs.
The scales measuring health‐related quality of life may go in different directions. Some scales increase in values with improved health‐related quality of life, whereas other scales decrease in values with improved health‐related quality of life. To adjust for the different directions of the scales, we wanted to multiply by –1 the scales that report better health‐related quality of life with decreasing values.
Unit of analysis issues
We planned to take into account the level at which randomisation occurred, such as cross‐over trials, cluster‐randomised trials and multiple observations for the same outcome. If more than one comparison from the same trial was eligible for inclusion in the same meta‐analysis, we would have either combined groups to create a single pair‐wise comparison or appropriately reduced the sample size so that the same participants do not contribute multiply (splitting the 'shared' group into two or more groups). While the latter approach offers some solution to adjusting the precision of the comparison, it does not account for correlation arising from the same set of participants being in multiple comparisons (Higgins 2011a).
We planned to reanalyse cluster‐randomised trials that did not appropriately adjust for potential clustering of participants within clusters in their analyses. The variance of the intervention effects was planned to be inflated by a design effect (DEFF). Calculation of a DEFF involves estimation of an intra‐cluster correlation (ICC). We planned to obtain estimates of ICCs through contact with authors, or impute them using estimates from other included studies that report ICCs, or using external estimates from empirical research (e.g. Bell 2013). We planned to examine the impact of clustering using sensitivity analyses.
Dealing with missing data
If possible, we would have obtained missing data from the authors of the included trials. We planned to carefully evaluate important numerical data such as screened, randomly assigned participants as well as intention‐to‐treat (ITT), and as‐treated and per‐protocol populations.
We planned to investigate attrition rates (e.g. drop‐outs, losses to follow‐up, withdrawals), and wanted to critically appraise issues concerning missing data and imputation methods (e.g. last observation carried forward (LOCF)).
Where included trials did not report means and standard deviations (SDs) for outcomes and we did not receive the needed information from trial authors, we would have imputed these values by assuming the SDs of the missing outcome to be the average of the SDs from the trials that reported this information.
We planned to investigate the impact of imputation on meta‐analyses by performing sensitivity analyses.
Assessment of heterogeneity
In the event of substantial clinical or methodological heterogeneity, we planned not to report trial results as the pooled effect estimate in a meta‐analysis.
We would have identified heterogeneity (inconsistency) by visually inspecting the forest plots and by using a standard Chi² test with a significance level of α = 0.1. In view of the low power of this test, we also wanted to consider the I² statistic, which quantifies inconsistency across trials to assess the impact of heterogeneity on the meta‐analysis (Higgins 2002; Higgins 2003); where an I² statistic ≥ 75% indicates a considerable level of heterogeneity (Higgins 2011a).
Assessment of reporting biases
If we included 10 or more trials that investigate a particular outcome, we would have used funnel plots to assess small‐trial effects. Several explanations may account for funnel plot asymmetry, including true heterogeneity of effect with respect to trial size, poor methodological design (and hence bias of small trials) and publication bias. Therefore, we planned to interpret results carefully (Sterne 2011).
Data synthesis
Unless effects appear homogeneous across trials, we primarily planned to summarise low risk of bias data using a random‐effects model (Wood 2008). We wanted to interpret random‐effects meta‐analyses with due consideration of the whole distribution of effects, ideally by presenting a prediction interval (Higgins 2009). A prediction interval specifies a predicted range for the true treatment effect in an individual trial (Riley 2011). In addition, we also planned to perform statistical analyses according to the statistical guidelines contained in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
Trial Sequential Analyses
In a single trial, sparse data and interim analyses increase the risk of type I and type II errors. To avoid type I errors, group sequential monitoring boundaries are applied to decide whether a trial could be terminated early because of a sufficiently small P value; that is, the cumulative Z‐curve crosses the monitoring boundaries (Lan 1983). Likewise, before reaching the planned sample size of a trial, the trial authors may stop the trial due to futility if the cumulative Z‐score crosses the futility monitoring boundaries (Higgins 2011a). Sequential monitoring boundaries for benefit, harm, or futility can be applied to meta‐analyses as well, called trial sequential monitoring boundaries (Higgins 2010; Wetterslev 2008). In Trial Sequential Analysis (TSA), the addition of each trial in a cumulative meta‐analysis is regarded as an interim meta‐analysis and helps to clarify if significance is reached or futility is reached or whether additional trials are needed (Wetterslev 2008).
TSA combines a calculation of the diversity‐adjusted required information size (cumulated meta‐analysis sample size to detect or reject a specific relative intervention effect) for meta‐analysis with the threshold of data associated with statistics. We planned to perform TSA on all outcomes included in the 'Summary of findings' table (Brok 2009; Pogue 1997; Wetterslev 2008).
The idea in TSA is that if the cumulative Z‐curve crosses the boundary for benefit or harm before a diversity‐adjusted required information size is reached, a sufficient level of evidence for the anticipated intervention effect has been reached with the assumed type I error and no further trials may be needed. If the cumulative Z‐curve crosses the boundary for futility before a diversity‐adjusted required information size is reached, the assumed intervention effect can be rejected with the assumed type II error and no further trials may be needed. If the Z‐curve does not cross any boundary, then there is insufficient evidence to reach a conclusion. To construct the trial sequential monitoring boundaries, the required information size is needed and is calculated as the least number of participants needed in a well‐powered single trial and subsequently adjusted for diversity among the included trials in the meta‐analysis (Brok 2009; Wetterslev 2008). We planned to apply TSA as it decreases the risk of type I and II errors due to sparse data and multiple updating in a cumulative meta‐analysis, and it provides us with important information in order to estimate the risks of imprecision when the required information size is not reached. Additionally, TSA provides important information regarding the need for additional trials and the required information size of such trials (Wetterslev 2008).
We wanted to apply trial sequential monitoring boundaries according to an estimated clinical important effect. We would have based the required information size on an a priori effect corresponding to a 10% relative risk reduction for beneficial effects of the intervention and a 30% relative risk increase for harmful effects of the interventions.
We planned to perform TSA for continuous outcomes with mean difference values, by using the trials applying the same scale to calculate the required sample size. For the continuous outcomes we planned to test the evidence for the achieved differences in the cumulative meta‐analyses.
For the heterogeneity adjustment of the required information size we wanted to use the diversity (D²) estimated in the meta‐analyses of included trials. If diversity was zero in a meta‐analysis, we would have performed a sensitivity analysis with an assumed diversity of 20% when future trials are included possibly changing future heterogeneity among trials. Otherwise, we would have been at risk of underestimating the required information size.
Quality of evidence
We planned to present the overall quality of the evidence for each outcome according to the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, which takes into account issues related not only to internal validity (risk of bias, inconsistency, imprecision, publication bias) but also to external validity, such as directness of results. Two review authors (BH, JK) planned to independently rate the quality of evidence for each outcome. We wanted to present a summary of the evidence in a 'Summary of findings' table. This provide key information about the best estimate of the magnitude of the effect, in relative terms and as absolute differences, for each relevant comparison of alternative management strategies, numbers of participants and trials addressing each important outcome and rating of overall confidence in effect estimates for each outcome. We would have created the 'Summary of findings' table based on the methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) by means of Review Manager (RevMan)'s table editor (RevMan 2014). We planned to include an appendix named 'Checklist to aid consistency and reproducibility of GRADE assessments' (Meader 2014) to help with standardisation of the 'Summary of findings' tables. Alternatively, we would have used the GRADEpro Guideline Development Tool (GDT) software (GRADEpro GDT 2015) and presented evidence profile tables as an appendix. We planned to present results for the outcomes as described in the 'Types of outcome measures' section. If meta‐analysis was not possible, we would have presented the results in a narrative format in the 'Summary of findings' table. We planned to justify all decisions to downgrade the quality using footnotes, and we wanted to make comments to aid the reader's understanding of the Cochrane review where necessary.
Subgroup analysis and investigation of heterogeneity
We expected the following characteristics to introduce clinical heterogeneity, and planned to perform the following subgroup analyses and an investigation of interactions.
Trials of long duration (two years or longer) versus trials of short duration (less than 2 years).
Diagnostic criteria (IFG, IGT, HbA1c).
Age, depending on data.
Sex.
Ethnicity, depending on the data.
Comorbid conditions, such as hypertension, obesity or both.
Participants with previous gestational diabetes mellitus.
Sensitivity analysis
We planned to perform sensitivity analyses to explore the influence of the following factors (when applicable) on effect sizes by restricting analysis to the following.
Published trials.
Taking into account risk of bias, as specified in the Assessment of risk of bias in included studies section.
Trials using the following filters: imputation, language of publication, source of funding (industry versus other) or country.
We also wanted to test the robustness of results by repeating the analysis using different measures of effect size (RR, odds ratio, etc) and different statistical models (fixed‐effect and random‐effects models).
Results
Description of studies
For a detailed description of studies, see the sections Characteristics of excluded studies, Ongoing studies, and Table 1.
1. Potentially relevant trial published in abstracts.
| Study ID (type of publication) | Sarich 2010; Sarich 2010a (abstracts) |
| Methods | Type of trial: interventional Allocation: randomised Intervention model: parallel assignment Masking: double blind Duration of the intervention: 14 days |
| Participants |
Condition: obese participants, including some with impaired fasting glucose or impaired glucose tolerance Enrollment: 80 |
| Interventions |
Intervention 1: diet + canagliflozin 30 mg daily (n = 12) Intervention 2: diet + canagliflozin 100 mg daily (n = 12) Intervention 3: diet + canagliflozin 300 mg daily (n = 12) Intervention 4: diet + canagliflozin 600 mg daily (n=12) Intervention 5: diet + canagliflozin 300 mg twice a day (n = 12) Comparator: diet + placebo (n = 20) |
| Outcomes |
Primary and secondary outcomes: not specified. Outcomes reported in abstract: 24‐hour urinary glucose excretion, fasting plasma glucose, mean 24‐hour plasma glucose, insulin levels, weight, renal threshold for glucose excretion, hypoglycaemia, adverse events, self‐reported appetite and satiety measures, urine volume or frequency, vital signs, urine volume and frequency, electrocardiograms and laboratory tests |
| Conclusion | "CANA was well tolerated and increased 24‐h UGE, decreased RTG, and reduced body weight in obese healthy subjects. In addition, the data suggest that CANA is effective in reducing MPG in patients with IFG and/or IGT." |
| Notes | We contacted primary and secondary authors through email. The primary author (Sarich) responded that he was not aware whether additional information regarding this trial would be available. We asked the primary author Sarich if he could find a person who could provide us with additional information regarding this trial. Unknown how many participants were normoglycaemic or had IFG or IGT in each of the randomised groups. |
CANA: canagliflozin; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; MPG: mean plasma glucose; RTG: renal threshold for glucose excretion; UGE: urinary glucose excretion
Results of the search
The initial search of the databases identified 874 records after duplicates were removed. We excluded most of the references on the basis of their titles and abstracts because they clearly did not meet the inclusion criteria (Figure 1). We evaluated five references further. One reference of potential interest was only published as two abstracts (Sarich 2010; Sarich 2010a). Three of the retrieved references referred to ongoing trials (EudraCT 2015‐001552‐30; NCT02338193; NCT01248364). However, we excluded one of these records because it was not a randomised controlled trial (NCT01248364).
1.
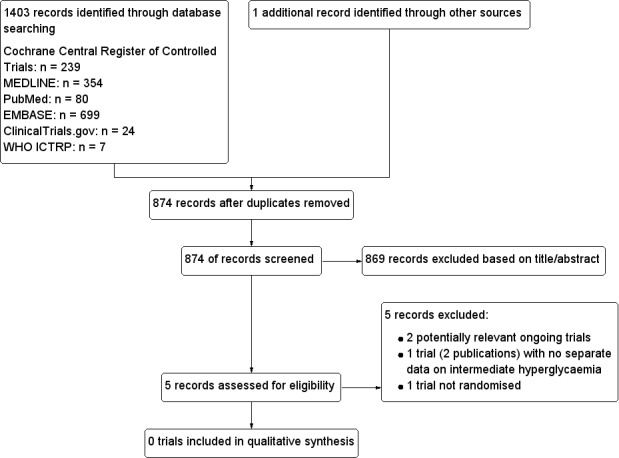
Study flow diagram.
We identified one abstract which fulfilled the inclusion criteria, as the trial included obese people with some of the participants having impaired fasting glucose (IFG), impaired glucose tolerance (IGT) or both (Sarich 2010). When searching for contact information on the trial authors of this abstract, we identified a reference to another abstract reporting the same trial (Sarich 2010a). We contacted the authors of the abstracts but none of the authors was able to provide additional information. Therefore, no separate information on the participants with impaired fasting glucose (IFG), impaired glucose tolerance (IGT) or both was available and we could not include the trial. More information regarding this trial is available in Table 1.
We contacted the investigators of the two identified ongoing trials in order to get additional information. Furthermore, we asked if they were aware of any other existing or ongoing trial of relevance for this review, but they could not name any specific trial. We did not discover a health technology assessment report, a systematic review or a meta‐analysis evaluating the effects of SGLT 2 inhibitors in participants with intermediate hyperglycaemia. Moreover, we contacted Prof. Silvio Inzucchi, senior author of a large scale trial of empagliflozin in people with type 2 diabetes mellitus, to ask if he was aware of any existing or ongoing trials investigating the effects of SGLT 2 inhibitors in participants with intermediate hyperglycaemia (EMPA‐REG 2015). In reply, Prof. Inzucchi was not aware of any ongoing or completed trial on this topic.
Included studies
We could not identify any trial addressing our review objectives and providing data on persons at risk for the development of type 2 diabetes mellitus.
Excluded studies
We retrieved one reference describing an ongoing non‐randomised trial (NCT01248364) which is listed in 'Excluded studies'.
Risk of bias in included studies
Because of lack of included trials we could not assess risk of bias and did not establish a 'Risk of bias' graph and a 'Risk of bias' summary figure.
Effects of interventions
We could not include any trial in this review either due to lack of trials fulfilling inclusion criteria or because separate data on participants with intermediate hyperglycaemia were not available. However, in the abstracts it was reported that no participant experienced hypoglycaemia (Sarich 2010; Sarich 2010a).
Discussion
Summary of main results
Our extensive search for randomised controlled trials (RCTs) evaluating participants with intermediate hyperglycaemia randomised to sodium–glucose cotransporter (SGLT) 2 inhibitors did not reveal any trial to be included. However, one trial published in two abstracts included some participants of interest. Unfortunately, no additional information was available (Sarich 2010; Sarich 2010a). The only outcome we could get information about through the abstract was hypoglycaemia, and no participant reported this outcome. We identified two ongoing trials, both evaluating the effects of dapagliflozin in people at risk for the development of type 2 diabetes mellitus (T2DM) and a follow‐up of 24 to 26 weeks. Both trials will mainly report on surrogate outcome measures with some data on adverse effects and health‐related quality of life.
Overall completeness and applicability of evidence
We performed an extensive search in major databases, conference proceedings and trials registers. We did not apply any language restrictions. Despite, we did not discover any published trial. We contacted investigators of the ongoing trials about further information of other ongoing or completed trials of relevance, but the investigators could not provide us with information about additional trials.
Quality of the evidence
We could not investigate how the Grading of Recommendations Assessment, Development and Evaluation (GRADE) considerations would impact on the findings of our key outcome results because we could not include any trial.
Potential biases in the review process
We searched several databases, conference proceedings and contacted investigators in order to obtain unpublished trials. However, even with this effort it was not possible to retrieve any trial. A strength of this systematic review was the fact that all investigators of the ongoing trials kindly provided additional information about these trials. We know that one completed trial included participants of our interest (Sarich 2010; Sarich 2010a). However, the full report of this short‐term trial of two weeks, if it happened, is unlikely to have a major impact on the overall conclusion of our review.
Agreements and disagreements with other studies or reviews
No other systematic review, meta‐analysis or health‐technology assessment report is currently available to compare our results with.
SGLT 2 inhibitors are a relatively new class of glucose‐lowering interventions in people with T2DM. Recently, a large scale indicated RCT beneficial effects of SGLT 2 inhibitors regarding patient important outcomes in participants with T2DM (EMPA‐REG 2015). Whether the same beneficial effects exist in people with intermediate hyperglycaemia remains to be proven. Currently, two RCTs in people with intermediate hyperglycaemia are ongoing, but none of these are investigating the effects of the SGLT 2 inhibitors on micro‐ or macrovascular complications associated with T2DM.
Presently, the American Food and Drug Administration has not approved any glucose‐lowering intervention for people with intermediate hyperglycaemia. However, the American Diabetes Association recommends metformin in certain people with intermediate hyperglycaemia.
The best way to clarify whether SGLT 2 inhibitors could have a therapeutic role in people with intermediate hyperglycaemia is through risk of bias minimising RCTs. Observational studies and non‐randomised trials can only detect associations between an intervention and an outcome. Causality cannot be reliably appraised with observational studies or non‐randomised trials as the influence of undetectable factors like confounding factors and generally risk of bias is unknown or difficult to appraise.
More RCTs investigating the potential beneficial effects of SGLT 2 inhibitors on people with intermediate hyperglycaemia might be initiated in the near future. Future RCTs investigating the effect of SGLT 2 inhibitors in people with T2DM should be performed with minimisation of systematic errors, meaning low risk of bias on all risk of bias domains ('Assessment of risk of bias in included studies'). As no glucose‐lowering interventions are approved for people with intermediate hyperglycaemia, the most appropriate comparisons of a RCT would be to compare SGLT 2 inhibitors with placebo. All intervention groups should be treated identically regarding other aspects, e.g. treatment of hypertension and dietary advice. Outcomes of interest for future trials in people with intermediate hyperglycaemia should focus on patient‐important outcome measures, like diabetes related complications (e.g. myocardial infarction, end‐stage renal disease). Combination of outcomes, so called composite outcomes, is often problematic, especially if mortality is included as one of the components (Cordoba 2010). Health‐related quality of life measured with validated scales as well as socioeconomic effects of the interventions should also be included. Moreover, it is of major importance to investigate whether possible adverse effects exist. Assessment of surrogate markers, e.g. cholesterol levels, weight changes, glycaemic measures and even incidence of T2DM is of limited value because changes in these outcomes may substantially differ over time and people may even oscillate between intermediate hyperglycaemia, T2DM and normoglycaemia over various points in time (Yudkin 2011; Yudkin 2014). To detect differences between interventions with patient‐important outcomes that occur relatively rare will require a large sample size and long intervention and follow‐up periods. It is impossible to judge how long an intervention period should be in order to assess patient‐important outcomes. Several of the patient‐important outcomes might take years to evolve. An intervention period of at least two years would probably be a good start. Regarding a potential reduction in the incidence of T2DM, it should be kept in mind, that a long follow‐up period, also after the end of the intervention, is required in order to establish whether T2DM is really prevented, or just delayed. Non‐inferiority designs should be avoided (Kaji 2015).
Millions of people worldwide are said to have intermediate hyperglycaemia, depending on definition of intermediate hyperglycaemia, and it is predicted that the prevalence will increase (IDF 2013). A pharmacological prevention of T2DM and its associated complications is of huge interest for the pharmacological industry. It will potentially open up a complete new market targeting a non‐diseased population, as long as the concept of 'predisease' is not generally accepted. The interest in sponsoring future clinical trials of SGLT 2 inhibitors in people with intermediate hyperglycaemia would therefore primarily, if not solely, be initiated by the pharmaceutical industry. However, data have shown that industry sponsored studies lead to more favourable results and conclusions than sponsorship by other sources, which should be kept in mind (Lundh 2012). Finally, any intervention interfering with the life of non‐diseased persons has to demonstrate very firm beneficial effects on patient‐important outcomes because even rare but severe adverse effects when translated to population wide settings could result in deleterious consequences impacting huge numbers of people.
Authors' conclusions
Implications for practice.
There is no evidence to show whether sodium–glucose cotransporter (SGLT) 2 inhibitors influence the risk for developing type 2 diabetes or its associated complications in people with intermediate hyperglycaemia. No data are available for patient‐important outcomes such as macrovascular and microvascular complications.
Implications for research.
No randomised controlled trials have investigated the effects of sodium–glucose cotransporter (SGLT) 2 inhibitors for the prevention or delay of type 2 diabetes mellitus. Future randomised controlled trials should focus on patient‐important outcomes to clarify if people with intermediate hyperglycaemia could benefit of this glucose‐lowering intervention. We identified two ongoing trials, both evaluating the effects of dapagliflozin in people at risk for the development of type 2 diabetes and a follow‐up of 24 to 26 weeks. Both trials will mainly report on surrogate outcome measures with some data on adverse effects and health‐related quality of life.
Notes
We have based parts of the 'Methods' and 'Appendix 1' sections of this Cochrane Review on a standard template established by the CMED Group.
Congestive heart failure was added as an outcome.
Acknowledgements
The authors would like to thank Kristine Færch and Karen Elkind‐Hirsch for answering our request for information on the ongoing trials.
Appendices
Appendix 1. Search strategies
| MEDLINE (OvidSP) |
|
Block 1: 'Prediabetes' 1. Prediabetic state/ 2. Glucose Intolerance/ 3. (prediabet* or pre diabet*).tw. 4. intermediate hyperglyc?emi*.tw. 5. ((impaired fasting adj2 glucose) or IFG or impaired FPG).tw. 6. glucose intolerance.tw. 7. ((impaired glucose adj (tolerance or metabolism)) or IGT).tw. 8. ("HbA(1c)" or HbA1 or HbA1c or "HbA 1c" or ((glycosylated or glycated) adj h?emoglobin)).tw. 9. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) adj4 (diabetes or T2D* or NIDDM or "type 2" or "type II")).tw. 10. or/1‐9 Block 2: SGLT 2 inhibitors 11. Sodium‐Glucose Transporter 2/ai [Antagonists & Inhibitors] 12. gliflozin?.tw. 13. ((SGLT2 or SGLT 2) adj2 inhibitor?).tw. 14. (sodium adj2 glucose adj (cotransporter or co transporter) adj 2 inhibitor?).tw. 15. atigliflozin.tw. 16. (bexagliflozin or EGT 1442 OR EGT0001442 or THR 1442).tw. 17. (canagliflozin or invokana or JNJ 24831754 or JNJ 24831754AAA or JNJ 24831754ZAE or TA 7284).tw. 18. (dapagliflozin or BMS‐512148 or farxiga or forxiga).tw. 19. (empagliflozin or BI 10773 or BI10773 or jardiance).tw. 20. (ertugliflozin or PF 04971729).tw. 21. (ipragliflozin or ASP1941 or ASP 1941 or suglat).tw. 22. (luseogliflozin or TS 071 OR TS 71).tw. 23. (sergliflozin or remogliflozin).tw. 24. (sotagliflozin or LX 4211 OR LX4211 or LP 802034).tw. 25. (tofogliflozin or CSG452 or CSG 452 or R 7201).tw. 26. BI 44847.tw. 27. or/11‐26 Block 1 and block 2 28. 10 and 27 29. exp animals/ not humans/ 30. 28 not 29 |
| EMBASE (OvidSP) |
|
Block 1: 'Prediabetes' 1. impaired glucose tolerance/ 2. (prediabet* or pre diabet*).tw. 3. intermediate hyperglyc?emi*.tw. 4. ((impaired fasting adj2 glucose) or IFG or impaired FPG).tw. 5. glucose intolerance.tw. 6. ((impaired glucose adj (tolerance or metabolism)) or IGT).tw. 7. ("HbA(1c)" or HbA1 or HbA1c or "HbA 1c" or ((glycosylated or glycated) adj h?emoglobin)).tw. 8. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) adj4 (diabetes or T2D* or NIDDM or "type 2" or "type II")).tw. 9. or/1‐8 Block 2: SGLT 2 inhibitors 10. exp sodium glucose cotransporter 2 inhibitor/ 11. gliflozin?.tw. 12. ((SGLT2 or SGLT 2) adj2 inhibitor?).tw. 13. (sodium adj2 glucose adj (cotransporter or co transporter) adj 2 inhibitor?).tw. 14. atigliflozin.tw. 15. (bexagliflozin or EGT 1442 OR EGT0001442 or THR 1442).tw. 16. (canagliflozin or invokana or JNJ 24831754 or JNJ 24831754AAA or JNJ 24831754ZAE or TA 7284).tw. 17. (dapagliflozin or BMS‐512148 or farxiga or forxiga).tw. 18. (empagliflozin or BI 10773 or BI10773 or jardiance).tw. 19. (ertugliflozin or PF 04971729).tw. 20. (ipragliflozin or ASP1941 or ASP 1941 or suglat).tw. 21. (luseogliflozin or TS 071 OR TS 71).tw. 22. (sergliflozin or remogliflozin).tw. 23. (sotagliflozin or LX 4211 OR LX4211 or LP 802034).tw. 24. (tofogliflozin or CSG452 or CSG 452 or R 7201).tw. 25. BI 44847.tw. 26. or/10‐25 Block 1 and block 2 27. 9 and 26 [28:Wong 2006"sound treatment studies" filter – BS version] 28. random*.tw. or clinical trial*.mp. or exp health care quality/ 29. 27 and 28 |
| Cochrane Central Register of Controlled Trials (Cochrane Register of Studies Online) |
| 1. MESH DESCRIPTOR Prediabetic state 2. MESH DESCRIPTOR Glucose Intolerance 3. (prediabet* or pre diabet*):TI,AB,KY 4. (intermediate hyperglyc?emi*):TI,AB,KY 5. ((impaired fasting ADJ2 glucose) or IFG or impaired FPG):TI,AB,KY 6. glucose intolerance:TI,AB,KY 7. ((impaired glucose ADJ (tolerance or metabolism)) or IGT):TI,AB,KY 8. ("HbA(1c)" or HbA1 or HbA1c or "HbA 1c" or ((glycosylated or glycated) ADJ h?emoglobin)):TI,AB,KY 9. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) ADJ4 (diabetes or T2D* or NIDDM or "type 2" or "type II")):TI,AB,KY 10. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 11. MESH DESCRIPTOR Sodium‐Glucose Transporter 2 WITH QUALIFIERS AI 12. gliflozin?:TI,AB,KY 13. ((SGLT2 or SGLT 2) ADJ2 inhibitor?):TI,AB,KY 14. (sodium ADJ2 glucose ADJ (cotransporter or co transporter) ADJ2 inhibitor?):TI,AB,KY 15. atigliflozin:TI,AB,KY 16. (bexagliflozin or EGT 1442 OR EGT0001442 or THR 1442):TI,AB,KY 17. (canagliflozin or invokana or JNJ 24831754 or JNJ 24831754AAA or JNJ 24831754ZAE or TA 7284):TI,AB,KY 18. (dapagliflozin or BMS‐512148 or farxiga or forxiga):TI,AB,KY 19. (empagliflozin or BI 10773 or BI10773 or jardiance):TI,AB,KY 20. (ertugliflozin or PF 04971729):TI,AB,KY 21. (ipragliflozin or ASP1941 or ASP 1941 or suglat):TI,AB,KY 22. (luseogliflozin or TS 071 OR TS 71):TI,AB,KY 23. (sergliflozin or remogliflozin):TI,AB,KY 24. (sotagliflozin or LX 4211 OR LX4211 or LP 802034):TI,AB,KY 25. (tofogliflozin or CSG452 or CSG 452 or R 7201):TI,AB,KY 26. BI 44847:TI,AB,KY 27. #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 28. 10 AND 27 |
| PubMed (subsets unavailable on Ovid) |
| 1. ((prediabet*[tiab] OR pre diabet*[tiab] OR hyperglyc*[tiab] OR ("impaired fasting"[tiab] AND glucose[tiab]) OR IFG[tiab] OR "impaired FPG"[tiab] OR "glucose intolerance"[tiab] OR ("impaired glucose"[tiab] AND (tolerance[tiab] OR metabolism[tiab])) OR IGT[tiab] OR "HbA(1c)"[tiab] OR HbA1[tiab] OR HbA1c[tiab] OR "HbA 1c"[tiab] OR "glycosylated hemoglobin"[tiab] OR "glycosylated haemoglobin"[tiab] OR "glycated hemoglobin"[tiab] OR "glycated haemoglobin"[tiab] OR ((risk[tiab] OR progress*[tiab] OR prevent*[tiab] OR inciden*[tiab] OR conversion[tiab] OR develop*[tiab] OR delay*[tiab]) AND (diabetes[tiab] OR T2D*[tiab] OR NIDDM[tiab] OR "type 2"[tiab] OR "type II"[tiab])))) 2. (("sodium glucose"[tiab] OR "sodium dependent glucose"[tiab] OR SGLT2[tiab] OR "SGLT 2"[tiab] OR gliflozin*[tiab] OR atigliflozin[tiab] OR bexagliflozin[tiab] OR "EGT 1442"[tiab] OR EGT0001442[tiab] OR "THR 1442"[tiab] OR canagliflozin[tiab] OR invokana[tiab] OR "JNJ 24831754"[tiab] OR "JNJ 24831754AAA"[tiab] OR "JNJ 24831754ZAE"[tiab] OR "TA 7284"[tiab] OR dapagliflozin[tiab] OR "BMS 512148"[tiab] OR farxiga[tiab] OR forxiga[tiab] OR empagliflozin[tiab] OR "BI 10773"[tiab] OR BI10773[tiab] OR jardiance[tiab] OR ertugliflozin[tiab] OR "PF 04971729"[tiab] OR ipragliflozin[tiab] OR ASP1941[tiab] OR "ASP 1941"[tiab] OR suglat[tiab] OR luseogliflozin[tiab] OR "TS 071"[tiab] OR "TS 71"[tiab] OR sergliflozin[tiab] OR remogliflozin[tiab] OR sotagliflozin[tiab] OR "LX 4211"[tiab] OR LX4211[tiab] OR "LP 802034"[tiab] OR tofogliflozin[tiab] OR CSG452[tiab] OR "CSG 452"[tiab] OR "R 7201"[tiab] OR "BI 44847"[tiab])) 3. #1 AND #2 4. pubstatusaheadofprint OR publisher[sb] OR pubmednotmedline[sb] 5. #3 AND #4 6. (random*[tiab] OR placebo[tiab] OR trial[tiab] OR groups[tiab]) OR (meta analysis[tiab] OR review[tiab] OR search*[tiab]) 7. #5 AND #6 |
| ClinicalTrials.gov (advanced search) |
|
Search terms: (prediabetes OR prediabetic OR "pre diabetes" OR "pre diabetic" OR hyperglycemia OR hyperglycaemia OR hyperglycemic OR hyperglycaemic OR "impaired glucose tolerance" OR "impaired fasting glucose" OR "glucose intolerance" OR IGT OR IFG OR ((diabetes OR "type 2" OR "type II" OR T2D OR T2DM) AND (risk OR progress OR progression OR progressed OR incident OR incidence OR conversion OR developed OR development OR develop OR delay OR delayed OR prevention OR prevent OR prevented))) AND ("sodium glucose" OR "sodium dependent glucose" OR SGLT2 OR "SGLT 2" OR gliflozin OR gliflozins OR atigliflozin OR bexagliflozin OR "EGT 1442" OR EGT0001442 OR "THR 1442" OR canagliflozin OR invokana OR "JNJ 24831754" OR "JNJ 24831754AAA" OR "JNJ 24831754ZAE" OR "TA 7284" OR dapagliflozin OR "BMS 512148" OR farxiga OR forxiga OR empagliflozin OR "BI 10773" OR BI10773 OR jardiance OR ertugliflozin OR "PF 04971729" OR ipragliflozin OR ASP1941 OR "ASP 1941" OR suglat OR luseogliflozin OR "TS 071" OR "TS 71" OR sergliflozin OR remogliflozin OR sotagliflozin OR "LX 4211" OR LX4211 OR "LP 802034" OR tofogliflozin OR CSG452 OR "CSG 452" OR "R 7201" OR "BI 44847") Study Type: Interventional |
| WHO ICTRP Search Portal (standard search) |
| prediabetes AND dapagliflozin OR prediabetes AND canagliflozin OR prediabetes AND empagliflozin OR prediabetes AND SGLT OR prediabetes AND SGLT2 OR pre diabetes AND dapagliflozin OR pre diabetes AND canagliflozin OR pre diabetes AND empagliflozin OR pre diabetes AND SGLT OR pre diabetes AND SGLT2 OR impaired glucose tolerance AND dapagliflozin OR impaired glucose tolerance AND canagliflozin OR impaired glucose tolerance AND empagliflozin OR impaired glucose tolerance AND SGLT OR impaired glucose tolerance AND SGLT2 OR impaired fasting glucose AND dapagliflozin OR impaired fasting glucose AND canagliflozin OR impaired fasting glucose AND empagliflozin OR impaired fasting glucose AND SGLT OR impaired fasting glucose AND SGLT2 OR glucose intolerance AND dapagliflozin OR glucose intolerance AND canagliflozin OR glucose intolerance AND empagliflozin OR glucose intolerance AND SGLT OR glucose intolerance AND SGLT2 OR diabetes AND risk AND dapagliflozin OR diabetes AND risk AND canagliflozin OR diabetes AND risk AND empagliflozin OR diabetes AND risk AND SGLT OR diabetes AND risk AND SGLT2 diabetes AND prevent* AND dapagliflozin OR diabetes AND prevent* AND canagliflozin OR diabetes AND prevent* AND empagliflozin OR diabetes AND prevent* AND SGLT OR diabetes AND prevent* AND SGLT2 |
Appendix 2. Selection bias decisions
| Selection bias decisions for trials reporting unadjusted analyses: comparison of results obtained using method details alone with results using method details and trial baseline informationa | |||
| Reported randomisation and allocation concealment methods | Risk of bias judgement using methods reporting | Information gained from study characteristics data | Risk of bias using baseline information and methods reporting |
| Unclear methods | Unclear risk | Baseline imbalances present for important prognostic variable(s) | High risk |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited or no baseline details | Unclear risk | ||
| Would generate a truly random sample, with robust allocation concealment | Low risk | Baseline imbalances present for important prognostic variable(s) | Unclear riskc |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited baseline details, showing balance in some important prognostic variablesb | Low risk | ||
| No baseline details | Unclear risk | ||
| Sequence is not truly randomised, or allocation concealment is inadequate | High risk | Baseline imbalances present for important prognostic variable(s) | High risk |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited baseline details, showing balance in some important prognostic variablesb | Unclear risk | ||
| No baseline details | High risk | ||
| aTaken from Corbett 2014; judgements highlighted in grey indicate situations in which the addition of baseline assessments would change the judgement about risk of selection bias, compared with using methods reporting alone. bDetails for the remaining important prognostic variables are not reported. cImbalance identified that appears likely to be due to chance. | |||
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| NCT01248364 | Not a randomised controlled trial. Trial is completed, but only data for the participants with type 2 diabetes are published. |
Characteristics of ongoing studies [ordered by study ID]
EudraCT 2015‐001552‐30.
| Trial name or title |
Title: Effect of dapagliflozin, metformin and physical activity on glucose variability, body composition and cardiovascular risk in pre‐diabetes (The PRE‐D Trial) ‐ A randomised, parallel, open‐label, intervention study Acronym: PRE‐D Trial |
| Methods |
Type of trial: interventional
Allocation: randomised
Intervention model: parallel assignment
Masking: none (open‐label) Duration of the intervention: 13 weeks (4 weeks of titration metformin, thereafter full dose) |
| Participants |
Condition: intermediate hyperglycaemia Enrollment: 160 participants Inclusion criteria: HbA1c 5.7%‐6.4% (39‐47 mmol/mol), age 30‐70 years, BMI ≥ 25 kg/m2 Exclusion criteria: uncontrolled medical issues including but not limited to cardiovascular pulmonary, rheumatologic, haematologic, oncologic, infectious, gastrointestinal or psychiatric disease; diabetes or other endocrine disease; immunosuppression; treatment with hormones which affect glucose metabolism; treatment with loop diuretics or thiazolidinediones; treatment with beta blockers or peroral steroids; bariatric surgery within the past 2 years; impaired renal function defined as an estimated GFR < 60 mL/min/1.73m2; neurogenic bladder disorders; alcohol/drug abuse or in treatment with disulfiram at time of inclusion; pregnant or lactating women; fertile women not using birth control agents including oral contraceptives, gestagen injection, subdermal implantation hormonal vaginal ring, transdermal application, or intra‐uterine devices; allergic to one or more of the medications used in the study; treatment with peroral steroids; concomitant participation in other intervention study; unable to understand the informed consent and the study procedures |
| Interventions |
Intervention: dapagliflozin (10 mg daily) Comparator 1: metformin (850 mg twice daily) Comparator 2: physical activity (interval training 5 days a week, 30 min per session) Comparator 3: lifestyle advice (healthy lifestyle and weight loss according to the national recommendations from the Danish Health and Medicines Authority) |
| Outcomes |
Primary end point(s): reduction in mean amplitude of glycaemic excursions from baseline to end‐of treatment. Secondary end point(s): reduction in HbA1c; reduction in intra‐day glycaemic variability; reduction in daily time spent > 6.1 mmol/L, > 7.0 mmol/L, > 7.8 mmol/L and > 11.1 mmol/L; reduction in fasting and 2‐hour glucose concentrations during OGTT; improvement in insulin secretion, insulin sensitivity and disposition index; reduction of mean amplitude of glycaemic excursions and intra‐day glycaemic variability in subgroups of individuals with different baseline characteristics; reduction in body weight and change in body fat distribution; changes in basal and physical activity energy expenditure and substrate oxidation; changes in time spent sedentary and in moderate‐to‐vigorous physical activity intensity; reduction in blood pressure and lipids; changes in biomarkers of metabolic functions; number of adverse events and side effects; changes in self‐rated health and quality of life; adherence to the different interventions. All secondary outcomes measured from baseline to end‐of‐treatment (13 weeks) and at the end of follow‐up (26 weeks) |
| Starting date |
Trial start date: February 2016; inclusion of first participant Trial completion date: June 2017; last participant, last visit |
| Contact information |
Responsible party/principal investigator: professor, chief physician Marit Eika Jørgensen, MD PhD, Steno Diabetes Center, Niels Steensens Vej 2, DK‐2820 Gentofte, Denmark. E‐mail: maej@steno.dk. Phone: +45 3075 6008. Contact: sponsor, senior researcher Kristine Færch, MSc PhD, Steno Diabetes Center, Niels Steensens Vej 2, DK‐2820 Gentofte, Denmark. E‐mail: krif@steno.dk. Phone: +45 3079 1461. |
| Notes |
Trial record: EudraCT 2015‐001552‐30. The sponsor provided us with the full protocol for internal use. |
NCT02338193.
| Trial name or title |
Title: a randomized study evaluating dapagliflozin and metformin, alone and in combination, in overweight women with a recent history of gestational diabetes mellitus: effects on anthropometric measurements and cardiometabolic abnormalities Acronym: DAPA‐GDM |
| Methods |
Type of trial: interventional
Allocation: randomised
Intervention model: parallel assignment
Masking: single blind (investigator) Duration of the intervention: 24 weeks |
| Participants |
Condition: impaired fasting glucose, impaired glucose tolerance or both post partum. Enrollment: 72 Inclusion criteria: overweight/obese (BMI >25 kg/m2) females 18 years to 45 years of age, who experienced gestational diabetes during recent (within 12 months) pregnancy, postpartum metabolic abnormalities determined by a 75 gm OGTT (inclusive of prior gestational diabetes women with impaired fasting glucose, impaired glucose tolerance, or both postpartum), completed lactation, using adequate contraception during study period unless sterilized, written consent for participation in the trial Exclusion criteria: cholestasis during the past pregnancy; any hepatic diseases in the past; gallstones; abnormal liver function tests or renal impairment; presence of significant systemic disease; heart problems including congestive heart failure; history of pancreatitis, or diabetes mellitus (type 1 or 2); renal impairment; significantly elevated triglyceride levels; untreated or poorly controlled hypertension; prior history of a malignant disease requiring chemotherapy; known hypersensitivity or contraindications to use of insulin sensitizer such as metformin or thiazolidinediones; history of hypersensitivity reaction to dapagliflozin or other SGLT 2 inhibitors; current use of metformin, thiazolidinediones, GLP‐1 receptor agonists, DPP‐4 inhibitors, SGLT 2 inhibitors or weight loss medications; uncontrolled thyroid disease (documented normal TSH) or hyperprolactinaemia; liver enzymes levels exceeding more than twice normal lab values; use of drugs known to exacerbate glucose tolerance; history of diabetes or prior use of medications to treat diabetes except gestational diabetes; currently lactating; eating disorders or gastrointestinal disorders; suspected pregnancy; desiring pregnancy in next 6 months; breastfeeding, or known pregnancy in last 2 months; active or prior history of substance abuse or significant intake of alcohol or history of alcoholism; patient not willing to use adequate contraception during study period and up to 4 weeks after last dose of study drug (unless sterilized); debilitating psychiatric disorder such as psychosis or neurological condition that might confound outcome variables; inability or refusal to comply with protocol; not currently participating or having participated in an experimental drug study in previous three months |
| Interventions |
Intervention 1: 10 mg dapagliflozin once daily Intervention 2: 5 mg dapagliflozin/1000 mg metformin twice a day Comparator: 1000 mg metformin twice a day |
| Outcomes |
Primary outcome(s): percent change in body weight, change in percent body weight with combination therapy compared to monotherapy, both from baseline to week 24 Secondary outcome(s): measures of insulin sensitivity and secretion, surrogate measures of insulin action derived from OGTT, plasma lipid fractions, creatinine and calculated eGFR, glycaemic control, blood pressure, waist circumference, waist‐to‐height ratio, body mass index, liver enzymes, all measured as change from baseline to 24 weeks Other outcome(s): none |
| Starting date |
Trial start date: August 2015 Trial completion date: estimated study completion date: March 2018 |
| Contact information |
Responsible party/principal investigator: Karen E Elkind‐Hirsch, PhD 225 231‐5278 karen.elkind‐hirsch@womans.org
Contact: Martha Paterson, MD 225‐924‐8247 martha.paterson@womans.org United States, Louisiana Woman's Hospital Recruiting Baton Rouge, Louisiana, United States, 70815 Contact: Karen Elkind‐Hirsch, PhD. 225‐231‐5278 Contact: Ericka Seidemann, MS 225‐231‐5275 ericka.seidemann@womans.org Principal Investigator: Karen Elkind‐Hirsch, Ph.D. Sub‐Investigator: Martha Paterson, M.D |
| Notes |
Trial record: NCT02338193 We contacted Karen Elkind‐Hirsch through email. She informed us, that the trial has started in December 2015, and it will take at least two years before the data are published (personal communication). |
BMI: body mass index; DDP‐4: dipeptidyl peptidase‐4; eGFR: estimated glomerular filtration rate; GLP‐1: glucagon like peptide‐1; HbA1c: glycosylated haemoglobin A1c; OGTT: oral glucose tolerance test; SGLT: sodium‐glucose cotransporter; TSH: thyroid stimulating hormone
Contributions of authors
All review authors read and approved the final review draft.
Bianca Hemmingsen (BH): review draft, search strategy development, acquisition of trial reports, trial selection, data extraction, data analysis, data interpretation, contacting additional sources, and future review updates.
Jesper Krogh (JK): trial selection, data extraction, and future review updates.
Maria‐Inti Metzendorf (MIM): search strategy development, performed the searches, and future review updates.
Bernd Richter (BR): review draft, search strategy development, and future review updates.
Sources of support
Internal sources
No sources of support supplied
External sources
-
World Health Organization (WHO), Other.
This review is part of a series of reviews funded by the WHO on interventions for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus.
Declarations of interest
BH: this review is part of a series of reviews on interventions for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus which is funded by the WHO.
JK: none known.
MIM: none known.
BR: none known.
New
References
References to studies excluded from this review
NCT01248364 {published data only}
- A Study to Determine Acute (After First Dose) and Chronic (After 28 Days) Effects of Empagliflozin (BI 10773) on Pre and Postprandial Glucose Homeostasis in Patients With Impaired Glucose Tolerance and Type 2 Diabetes Mellitus and Healthy Subjects. https://clinicaltrials.gov/ct2/show/study/NCT01248364 (accessed 8 March 2016).
References to ongoing studies
EudraCT 2015‐001552‐30 {published and unpublished data}
- Effect of dapagliflozin, metformin and physical activity on glucose variability, body composition and cardiovascular risk in pre‐diabetes (The PRE‐D Trial) ‐ a randomised, parallel, open‐label, intervention study. https://www.clinicaltrialsregister.eu/ctr‐search/trial/2015‐001552‐30/DK/ (accessed 8 March 2016).
NCT02338193 {published data only}
- Dapagliflozin and metformin, alone and in combination, in overweight/obese prior GDM women (DAPA‐GDM). https://clinicaltrials.gov/ct2/show/NCT02338193 (accessed 5 March 2016).
Additional references
Abdul‐Ghani 2006
- Abdul‐Ghani MA, Jenkinson CP, Richardson DK, Tripathy D, DeFronzo RA. Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: results from the Veterans Administration Genetic Epidemiology Study. Diabetes 2006;55(5):1430‐5. [PUBMED: 16644701] [DOI] [PubMed] [Google Scholar]
ADA 1997
- The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 1997;20(7):1183‐97. [DOI] [PubMed] [Google Scholar]
ADA 2003
- Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 2003;26(Suppl 1):S5‐20. [DOI] [PubMed] [Google Scholar]
ADA 2008
- American Diabetes Association. Standards of medical care in diabetes‐2008. Diabetes Care 2008;31(Suppl 1):S12‐54. [PUBMED: 18165335] [DOI] [PubMed] [Google Scholar]
ADA 2010
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010;33(Suppl 1):S62‐9. [PUBMED: 20042775] [DOI] [PMC free article] [PubMed] [Google Scholar]
ADA 2015
- American Diabetes Association. Standards of medical care in diabetes‐2015. Diabetes Care 2015;38(Suppl 1):S1‐93. [PUBMED: 24357209] [PubMed] [Google Scholar]
AstraZeneca Pharmaceuticals 2014
- AstraZeneca Pharmaceuticals. Farxiga product information. http://www.azpicentral.com/farxiga/pi_farxiga.pdf#page=1 (accessed 15th October 2015).
Bell 2013
- Bell ML, McKenzie JE. Designing psycho‐oncology randomised trials and cluster randomised trials: variance components and intra‐cluster correlation of commonly used psychosocial measures. Psychooncology 2013;22(8):1738‐47. [DOI] [PubMed] [Google Scholar]
Beller 2013
- Beller EM, Chen JK, Wang UL, Glasziou PP. Are systematic reviews up‐to‐date at the time of publication?. Systematic Reviews 2013;2:36. [DOI: 10.1186/2046-4053-2-36] [DOI] [PMC free article] [PubMed] [Google Scholar]
Boehringer Ingelheim Pharmaceuticals 2014
- Boehringer Ingelheim Pharmaceuticals Inc. JARDIANCE® product information. http://docs.boehringer‐ingelheim.com/Prescribing%20Information/PIs/Jardiance/jardiance.pdf (accessed 15th October 2015).
Boutron 2014
- Boutron I, Altman DG, Hopewell S, Vera‐Badillo F, Tannock I, Ravaud P. Impact of spin in the abstracts of articles reporting results of randomized controlled trials in the field of cancer: the SPIIN randomized controlled trial. Journal of Clinical Oncology 2014;32(36):4120‐6. [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive‐‐Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [PUBMED: 18824466] [DOI] [PubMed] [Google Scholar]
CDC 2015
- Centers for Disease Control and Prevention. 2014 National Diabetes Statistics Report. http://www.cdc.gov/diabetes/data/statistics/2014statisticsreport.html (accessed 15 th October 2015).
Cheng 2006
- Cheng C, Kushner H, Falkner BE. The utility of fasting glucose for detection of prediabetes. Metabolism: Clinical and Experimental 2006;55(4):434‐8. [PUBMED: 16546472] [DOI] [PubMed] [Google Scholar]
ClinicalTrials.gov
- https://clinicaltrials.gov/ct2/results?term=prediabetes+drugs&Search=Search. Accessed 12th February 2016.
Corbett 2014
- Corbett MS, Higgins JP, Woolacott NF. Assessing baseline imbalance in randomised trials: implications for the Cochrane risk of bias tool. Research Synthesis Methods 2014;5(1):79‐85. [DOI] [PubMed] [Google Scholar]
Cordoba 2010
- Cordoba G, Schwartz L, Woloshin S, Bae H, Gotzsche PC. Definition, reporting, and interpretation of composite outcomes in clinical trials: systematic review. BMJ (Clinical research ed.) 2010;341:c3920. [PUBMED: 20719825] [DOI] [PMC free article] [PubMed] [Google Scholar]
DeFronzo 1989
- DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non‐insulin‐dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism: Clinical and Experimental 1989;38(4):387‐95. [PUBMED: 2657323] [DOI] [PubMed] [Google Scholar]
Diabetes Prevention Program 2002
- Knowler WC, Barrett‐Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. The New England Journal of Medicine 2002;346(6):393‐403. [PUBMED: 11832527] [DOI] [PMC free article] [PubMed] [Google Scholar]
Diabetes Prevention Program FU 2009
- Diabetes Prevention Program Research Group, Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, et al. 10‐year follow‐up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009;374(9702):1677‐86. [DOI: 10.1016/S0140-6736(09)61457-4; PUBMED: 19878986] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dunkley 2014
- Dunkley AJ, Bodicoat DH, Greaves CJ, Russell C, Yates T, Davies MJ, et al. Diabetes prevention in the real world: effectiveness of pragmatic lifestyle interventions for the prevention of type 2 diabetes and of the impact of adherence to guideline recommendations: a systematic review and meta‐analysis. Diabetes Care 2014;37(4):922‐33. [PUBMED: 24652723] [DOI] [PubMed] [Google Scholar]
Egbewale 2014
- Egbewale BE, Lewis M, Sim J. Bias, precision and statistical power of analysis of covariance in the analysis of randomized trials with baseline imbalance: a simulation study. BMC Medical Research Methodology 2014;14:49. [DOI: 10.1186/1471-2288-14-49] [DOI] [PMC free article] [PubMed] [Google Scholar]
EMA 2012
- European Medicines Agency. Summary of the European public assessment report (EPAR) for Forxiga. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002322/human_med_001546.jsp&mid=WC0b01ac058001d124 (accessed 15 th October 2015).
EMPA‐REG 2015
- Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. The New England Journal of Medicine 2015;373(22):2117‐28. [PUBMED: 26378978] [DOI] [PubMed] [Google Scholar]
FDA 2013
- U.S. Food, Drug Administration. FDA news release: FDA approves Invokana to treat type 2 diabetes. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm345848.htm (accessed 15th October 2015).
FDA 2014a
- U.S. Food, Drug Administration. FDA news release: FDA approves Farxiga to treat type 2 diabetes. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm380829.htm (accessed 15th October 2015).
FDA 2014b
- U.S. Food, Drug Administration. FDA news release: FDA approves Jardiance to treat type 2 diabetes. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm407637.htm (accessed 15th October 2015).
FDA 2015
- U.S. Food, Drug Administration. FDA Drug Safety Communication: FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm (accessed 15th October 2015).
Finnish Diabetes Prevention Study Group 2001
- Tuomilehto J, Lindström J, Eriksson JG, Valle TT, Hämäläinen H, Ilanne‐Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. The New England Journal of Medicine 2001;344(18):1343‐50. [PUBMED: 11333990] [DOI] [PubMed] [Google Scholar]
Gosmanov 2014
- Gosmanov AR, Wan J. Low positive predictive value of hemoglobin A1c for diagnosis of prediabetes in clinical practice. The American Journal of the Medical Sciences 2014;348(3):191‐4. [PUBMED: 24556928] [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime, Inc.). GRADEpro GDT: GRADEpro Guideline Development Tool. Available from www.gradepro.org. Hamilton: McMaster University (developed by Evidence Prime, Inc.), 2015.
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21(11):1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analysis. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JPT, Thompson SG, Spiegelhalter DJ. A re‐evaluation of random‐effects meta‐analysis. Journal of the Royal Statistical Society: Series A (Statistics in Society) 2009;172(1):137‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2010
- Higgins JP, Whitehead A, Simmonds M. Sequential methods for random‐effects meta‐analysis. Statistics in Medicine 2011;30(9):903‐21. [PUBMED: 21190240] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011;343:d5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hróbjartsson 2013
- Hróbjartsson A, Thomsen AS, Emanuelsson F, Tendal B, Hilden J, Boutron I, et al. Observer bias in randomized clinical trials with measurement scale outcomes: a systematic review of trials with both blinded and nonblinded assessors. Canadian Medical Association Journal 2013;185(4):E201‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice CFR & ICH Guidelines. USA: Barnett International/PAREXEL, 1997; Vol. 1.
IDF 2013
- International Diabetes Federation. International Diabetes Federation. IDF Diabetes Atlas. 6th Edition. Brussels: International Diabetes Federation, 2013. [Google Scholar]
IEC 2009
- International Expert Committee. International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 2009;32(7):1327‐34. [PUBMED: 19502545] [DOI] [PMC free article] [PubMed] [Google Scholar]
Inzucchi 2012
- Inzucchi SE. Clinical practice. Diagnosis of diabetes. The New England Journal of Medicine 2012;367(6):542‐50. [PUBMED: 22873534] [DOI] [PubMed] [Google Scholar]
Janssen Pharmaceuticals 2013
- Janssen Pharmaceuticals. INVOKANA® prescribing information sheet. Published 2013. https://www.invokanahcp.com/prescribing‐information.pdf (accessed 15th October 2015).
Jensen 2002
- Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE. Beta‐cell function is a major contributor to oral glucose tolerance in high‐risk relatives of four ethnic groups in the U.S. Diabetes 2002;51(7):2170‐8. [PUBMED: 12086947] [DOI] [PubMed] [Google Scholar]
Kaji 2015
- Kaji AH, Lewis RJ. Noninferiority trials: is a new treatment almost as effective as another?. JAMA 2015;313(23):2371‐2. [PUBMED: 26080342] [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI: 10.1136/bmj.c365] [DOI] [PubMed] [Google Scholar]
Lan 1983
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika 1983;70:659‐63. [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic and meta‐analyses of studies that evaluate interventions: explanation and elaboration. PLoS Medicine 2009;6(7):e1000100. [DOI: 10.1371/journal.pmed.1000100] [DOI] [PMC free article] [PubMed] [Google Scholar]
List 2011
- List JF, Whaley JM. Glucose dynamics and mechanistic implications of SGLT2 inhibitors in animals and humans. Kidney International. Supplement 2011;79(Suppl 120):S20‐7. [DOI] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
Mather 2011
- Mather A, Pollock C. Glucose handling by the kidney. Kidney International. Supplement 2011;120:S1‐6. [PUBMED: 21358696] [DOI] [PubMed] [Google Scholar]
Mathieu 2009
- Mathieu S, Boutron I, Moher D, Altman DG, Ravaud P. Comparison of registered and published primary outcomes in randomized controlled trials. JAMA 2009;302(9):977‐84. [DOI] [PubMed] [Google Scholar]
Meader 2014
- Meader N, King K, Llewellyn A, Norman G, Brown J, Rodgers M, et al. A checklist designed to aid consistency and reproducibility of GRADE assessments: development and pilot validation. Systematic Reviews 2014;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Morris 2013
- Morris DH, Khunti K, Achana F, Srinivasan B, Gray LJ, Davies MJ, et al. Progression rates from HbA1c 6.0‐6.4% and other prediabetes definitions to type 2 diabetes: a meta‐analysis. Diabetologia 2013;56(7):1489‐93. [PUBMED: 23584433] [DOI] [PubMed] [Google Scholar]
NDDG 1979
- National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. National Diabetes Data Group. Diabetes 1979;28(12):1039‐57. [PUBMED: 510803] [DOI] [PubMed] [Google Scholar]
Pogue 1997
- Pogue JM, Yusuf S. Cumulating evidence from randomized trials: utilizing sequential monitoring boundaries for cumulative meta‐analysis. Controlled Clinical Trials 1997;18(6):580‐93; discussion 661‐6. [PUBMED: 9408720] [DOI] [PubMed] [Google Scholar]
Rahmoune 2005
- Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non‐insulin‐dependent diabetes. Diabetes 2005;54(12):3427‐34. [PUBMED: 16306358] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Riley 2011
- Riley RD, Higgins JP, Deeks JJ. Interpretation of random effects meta‐analyses. BMJ 2011;342:d549. [DOI] [PubMed] [Google Scholar]
Riley 2013
- Riley RD, Kauser I, Bland M, Thijs L, Staessen JA, Wang J, et al. Meta‐analysis of randomised trials with a continuous outcome according to baseline imbalance and availability of individual participant data. Statistics in Medicine 2013;32(16):2747‐66. [DOI] [PubMed] [Google Scholar]
Sarich 2010
- Sarich T, Devineni D, Ghosh A, Wexler D, Shalayda K, Blake J, et al. Canagliflozin, a novel inhibitor of sodium glucose co‐transporter 2, increases 24‐hour urinary glucose excretion and reduces body weight in obese subjects over 2 weeks of treatment. Diabetologia. 2010; Vol. 53, Issue 0:pp. S349‐S350.
Sarich 2010a
- Sarich T, Devineni D, Ghosh A, Wexler D, Shalayda K, Blake J, et al. Canagliflozin, a novel inhibitor of sodium glucose co‐transporter 2 (SGLT2), increases 24‐h urinary glucose excretion and decreases body weight in obese subjects. Diabetes. 2010; Vol. 59(Suppl 1):567‐P.
Scheen 2015
- Scheen, A. Pharmacodynamics, efficacy and safety of sodium–glucose co‐transporter type 2 (SGLT2) inhibitors for the treatment of type 2 diabetes mellitus. Drugs 2015;75(1):33‐59. [DOI] [PubMed] [Google Scholar]
Selvin 2011
- Selvin E, Steffes MW, Ballantyne CM, Hoogeveen RC, Coresh J, Brancati FL. Racial differences in glycemic markers: a cross‐sectional analysis of community‐based data. Annals of Internal Medicine 2011;154(5):303‐9. [PUBMED: 21357907] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta‐analyses of randomised controlled trials. BMJ 2011;343:d4002. [DOI] [PubMed] [Google Scholar]
Viera 2011
- Viera AJ. Predisease: when does it make sense?. Epidemiologic Reviews 2011;33:122‐34. [DOI] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [PUBMED: 18083463] [DOI] [PubMed] [Google Scholar]
WHO 1998
- Alberti KM, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part I: diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabetic Medicine 1998;15(7):539‐53. [DOI] [PubMed] [Google Scholar]
WHO 1999
- World Health Organization. Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Part 1. Diagnosis and classification of diabetes mellitus. Geneva: World Health Organization, 1999. [Google Scholar]
WHO/IDF 2006
- World Health Organization/International Diabetes Federation. Definition and diagnosis of diabetes mellitus and intermediate hyperglycaemia. Report of a WHO/IDF consultation. 2006. http://www.idf.org/webdata/docs/WHO_IDF_definition_diagnosis_of_diabetes.pdf (accessed 15th October 2015).
Wong 2006
- Wong SSL, Wilczynski NL, Haynes RB. Developing optimal search strategies for detecting clinically sound treatment studies in EMBASE. Journal of the Medical Library Association 2006;94(1):41‐7. [PMC free article] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yudkin 2011
- Yudkin JS, Lipska KJ, Montori VM. The idolatry of the surrogate. BMJ (Clinical research ed.) 2011;343:d7995. [PUBMED: 22205706] [DOI] [PubMed] [Google Scholar]
Yudkin 2014
- Yudkin JS, Montori VM. The epidemic of pre‐diabetes: the medicine and the politics. BMJ 2014;349:g4485. [DOI] [PMC free article] [PubMed] [Google Scholar]