

Abstract
Background
18F‐florbetapir uptake by brain tissue measured by positron emission tomography (PET) is accepted by regulatory agencies like the Food and Drug Administration (FDA) and the European Medicine Agencies (EMA) for assessing amyloid load in people with dementia. Its added value is mainly demonstrated by excluding Alzheimer's pathology in an established dementia diagnosis. However, the National Institute on Aging and Alzheimer's Association (NIA‐AA) revised the diagnostic criteria for Alzheimer's disease and confidence in the diagnosis of mild cognitive impairment (MCI) due to Alzheimer's disease may be increased when using amyloid biomarkers tests like 18F‐florbetapir. These tests, added to the MCI core clinical criteria, might increase the diagnostic test accuracy (DTA) of a testing strategy. However, the DTA of 18F‐florbetapir to predict the progression from MCI to Alzheimer’s disease dementia (ADD) or other dementias has not yet been systematically evaluated.
Objectives
To determine the DTA of the 18F‐florbetapir PET scan for detecting people with MCI at time of performing the test who will clinically progress to ADD, other forms of dementia (non‐ADD), or any form of dementia at follow‐up.
Search methods
This review is current to May 2017. We searched MEDLINE (OvidSP), Embase (OvidSP), PsycINFO (OvidSP), BIOSIS Citation Index (Thomson Reuters Web of Science), Web of Science Core Collection, including the Science Citation Index (Thomson Reuters Web of Science) and the Conference Proceedings Citation Index (Thomson Reuters Web of Science), LILACS (BIREME), CINAHL (EBSCOhost), ClinicalTrials.gov (https://clinicaltrials.gov), and the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (http://www.who.int/ictrp/search/en/). We also searched ALOIS, the Cochrane Dementia & Cognitive Improvement Group’s specialised register of dementia studies (http://www.medicine.ox.ac.uk/alois/). We checked the reference lists of any relevant studies and systematic reviews, and performed citation tracking using the Science Citation Index to identify any additional relevant studies. No language or date restrictions were applied to the electronic searches.
Selection criteria
We included studies that had prospectively defined cohorts with any accepted definition of MCI at time of performing the test and the use of 18F‐florbetapir scan to evaluate the DTA of the progression from MCI to ADD or other forms of dementia. In addition, we only selected studies that applied a reference standard for Alzheimer’s dementia diagnosis, for example, National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS‐ADRDA) or Diagnostic and Statistical Manual of Mental Disorders‐IV (DSM‐IV) criteria.
Data collection and analysis
We screened all titles and abstracts identified in electronic‐database searches. Two review authors independently selected studies for inclusion and extracted data to create two‐by‐two tables, showing the binary test results cross‐classified with the binary reference standard. We used these data to calculate sensitivities, specificities, and their 95% confidence intervals. Two independent assessors performed quality assessment using the QUADAS‐2 tool plus some additional items to assess the methodological quality of the included studies.
Main results
We included three studies, two of which evaluated the progression from MCI to ADD, and one evaluated the progression from MCI to any form of dementia.
Progression from MCI to ADD was evaluated in 448 participants. The studies reported data on 401 participants with 1.6 years of follow‐up and in 47 participants with three years of follow‐up. Sixty‐one (15.2%) participants converted at 1.6 years follow‐up; nine (19.1%) participants converted at three years of follow‐up.
Progression from MCI to any form of dementia was evaluated in five participants with 1.5 years of follow‐up, with three (60%) participants converting to any form of dementia.
There were concerns regarding applicability in the reference standard in all three studies. Regarding the domain of flow and timing, two studies were considered at high risk of bias.
MCI to ADD;
Progression from MCI to ADD in those with a follow‐up between two to less than four years had a sensitivity of 67% (95% CI 30 to 93) and a specificity of 71% (95% CI 54 to 85) by visual assessment (n = 47, 1 study).
Progression from MCI to ADD in those with a follow‐up between one to less than two years had a sensitivity of 89% (95% CI 78 to 95) and a specificity of 58% (95% CI 53 to 64) by visual assessment, and a sensitivity of 87% (95% CI 76 to 94) and a specificity of 51% (95% CI 45 to 56) by quantitative assessment by the standardised uptake value ratio (SUVR)(n = 401, 1 study).
MCI to any form of dementia;
Progression from MCI to any form of dementia in those with a follow‐up between one to less than two years had a sensitivity of 67% (95% CI 9 to 99) and a specificity of 50% (95% CI 1 to 99) by visual assessment (n = 5, 1 study).
MCI to any other forms of dementia (non‐ADD);
There was no information regarding the progression from MCI to any other form of dementia (non‐ADD).
Authors' conclusions
Although sensitivity was good in one included study, considering the poor specificity and the limited data available in the literature, we cannot recommend routine use of 18F‐florbetapir PET in clinical practice to predict the progression from MCI to ADD.
Because of the poor sensitivity and specificity, limited number of included participants, and the limited data available in the literature, we cannot recommend its routine use in clinical practice to predict the progression from MCI to any form of dementia.
Because of the high financial costs of 18F‐florbetapir, clearly demonstrating the DTA and standardising the process of this modality are important prior to its wider use.
Plain language summary
18F‐florbetapir PET scan for the early diagnosis of Alzheimer's disease dementia and other dementias in people with mild cognitive impairment
Review question: In people with mild cognitive impairment (MCI), does using a 18F PET scan with florbetapir predict the progression to Alzheimer's disease dementia (ADD) and other dementias?
Background
Due to global ageing, the number of people with dementia is expected to increase dramatically in the next few decades. Diagnosing dementia at an early stage is desirable, but there is no widespread agreement on the best approach. A range of simple pen and paper tests used by healthcare professionals can assess people with poor memory or cognitive impairment. Whether or not using special PET scans that detect amyloid —one of the hallmarks of Alzheimer's disease— improves our ability to predict the progression from MCI to ADD or other forms of dementia remains unclear. Since these tests are expensive, it is important that they provide additional benefits.
Aim
We aimed to evaluate the accuracy of the 18F‐florbetapir PET scan in identifying those people with MCI who clinically progress to ADD, other types of dementia, or any form of dementia over a period of time.
Study characteristics
The evidence is current to May 2017. We found three studies including 453 participants with MCI. Two studies evaluated the progression from MCI to ADD and one study evaluated the progression from MCI to any form of dementia.
Regarding the two studies that evaluated the progression from MCI to ADD, one study had 401 participants with a follow‐up of 1.6 years and the mean age was 72 years. The other study had 47 participants with a follow‐up of three years, and the mean age was 72 years.
The other study that looked at any form of dementia included 5 participants over 90 years old.
Two of the studies were funded by the test manufacturer.
Quality of the evidence
The main limitation of this review was that our findings were based on only three studies, with insufficient detail on how the people were selected, whether the information from the scan was assessed separately from the final diagnosis. The studies were considered to be at high risk of bias due to potential conflicts of interest detected.
Key findings
In this review, we found the following results based on the three studies.
At a follow‐up of 1.6 years, using visual assessment, the scan correctly classified 89% of the participants who progressed to ADD but only 58% of the participants who did not progress to ADD. This means that in a group of 100 people with MCI, 15% of whom will develop ADD, we would expect 13 of 15 people to have a positive result and the other 2 participants to be falsely negative. Also 49 people who will not develop ADD would have a negative result, but 36 people who will not develop ADD would have a positive result (false positives).
In the study that followed up people for three years and used visual assessment, the scan correctly classified 67% of people who progressed to ADD and 71% who did not progress to ADD. This means that in a group of 100 people with MCI, 19 of whom will develop ADD, we would expect 13 people to have a positive result of the scan and 6 people to have a falsely negative result. In addition, 58 of 81 participants who will not progress to ADD would have a negative result, but 23 people who will not develop ADD would have a positive result (false positives). The small number of participants evaluated at three years lowered our confidence on these estimates of accuracy.
Regarding progression to any form of dementia, the extremely small number of participants meant that we were unable to provide meaningful estimates of accuracy.
We conclude that 18F‐florbetapir PET scans cannot be recommended for routine use in clinical practice to predict the progression from MCI to ADD or any form of dementia based on the currently available data. More studies are needed to demonstrate its usefulness.
Summary of findings
Summary of findings'. 'Diagnostic test accuracy of 18F‐florbetapir to predict the progression to ADD, any other form of dementia (non‐ADD) or any form of dementia in people with MCI.
| What is the diagnostic accuracy of 18F‐florbetapir PET amyloid biomarker for predict progression to ADD, any other form of dementia (non‐ADD) or any form of dementia in people with MCI? | |||||||
| Descriptive | |||||||
| Patient population | Participants diagnosed with MCI at time of performing the test using any of the Petersen criteria or Winblad criteria or CDR = 0.5 or any 16 definitions included by Matthews (Matthews 2008). | ||||||
| Sources of referral | Not reported (n = 2) Mixed (memory clinics, newspaper ads, radio, and other public media campaigns) (n = 1) |
||||||
| MCI criteria | ADNI criteria, CDR 0.5 criterion was included (n = 2) CIND (cognitive impairment not dementia) (Matthews 2008) (n = 1) |
||||||
| Sampling procedure | Unclear (n = 3) | ||||||
| Prior testing | The only testing prior to performing the 18F‐florbetapir PET amyloid biomarker was the application of diagnostic criteria for identifying participants with MCI | ||||||
| Settings | Community and institutionalised (n = 1) Not reported (n = 2) |
||||||
| Index test | 18F‐florbetapir PET | ||||||
| Threshold prespecified at baseline | Yes (n = 3) | ||||||
| Threshold interpretation | Visual (n = 3) Quantitative (n = 1) |
||||||
| Threshold | Visual:
SUVR (Standardised Uptake Volume ratio):
|
||||||
| 18F‐florbetapir retention region | Global cortex (n = 1) | ||||||
| Reference Standard | Alzheimer’s disease dementia: NINCDS‐ADRDA (n = 1) Unclear (n = 1) Any form of dementia: DSM‐IV criteria for dementia (n = 1) |
||||||
| Target condition | Progression from MCI to Alzheimer’s disease dementia or any other forms of dementia (non‐ADD) or any form of dementia | ||||||
| Included studies | Prospectively well‐defined cohorts with any accepted definition of MCI (as above). Three studies (N = 458 participants) were included. Number of participants included in analysis: 453. | ||||||
| Quality concerns | The participant selection and reference standard QUADAS‐2 domain: unclear risk of bias. The index test domain: low risk of bias in all three included studies. The flow and timing domain: high risk of bias in the two included studies. Unclear concerns about applicability in the reference standard domain in all three included studies. |
||||||
| Limitations | Limited investigation of heterogeneity and sensitivity analysis due to insufficient number of studies. We were unable to evaluate progression from MCI to any other form of dementia (non‐ADD) due to lack of included studies. |
||||||
| Test | Studies | Cases/Participants | Sensitivity | Specificity | Consequences in a cohort of 100 | ||
| Proportion converting1 | Missed cases2 | Overdiagnosed2 | |||||
| Alzheimer's disease dementia | |||||||
|
18F‐florbetapir by visual assessment from one to less than two years of follow‐up (Schreiber 2015) |
1 | 61/401 | 89% (95% CI 78% to 95%) | 58% (95% CI 53% to 64%) | 15 | 2 | 36 |
|
18F‐florbetapir by quantitative assessment from one to less than two years of follow‐up (Schreiber 2015) |
1 | 61/401 | 87% (95% CI 76% to 94%) | 51% (95% CI 45% to 56%) | 15 | 2 | 42 |
|
18F‐florbetapir by visual assessment from two to less than four years of follow‐up (Doraiswamy 2014) |
1 | 9/47 | 67% (95% CI 30% to 93%) | 71% (95% CI 54% to 85%) | 19 | 6 | 23 |
| Any form of dementia | |||||||
|
18F‐florbetapir by visual assessment from one to less than two years of follow‐up (Kawas 2013) |
1 | 3/5 | 67% (95% CI 9% to 99%) | 50% (95% CI 1% to 99%) | 60 | 20 | 20 |
| Investigation of heterogeneity and sensitivity analysis: The planned investigations were not possible due to the limited number of studies available for each analysis. | |||||||
| Conclusions:18F‐florbetapir PET scan is not an accurate test for detecting progression from MCI to Alzheimer’s disease dementia or any form of dementia. The strength of the evidence was weak because of considerable variation in study methods, unclear methodological quality due to poor reporting, and high risk of bias due to possible conflict of interest. There is a need for conducting studies using standardised 18F‐florbetapir PET scan methodology in larger populations. | |||||||
1. Proportion converting to ADD or any form of dementia in each included study.
2. Missed and overdiagnosed numbers were computed using the proportion converting to the target condition. ADD: Alzheimer's disease dementia ADNI: Alzheimer's Disease Neuroimaging Initiative CDR: Clinical dementia rating CIND: Cognitive impairment not dementia DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders (4th ed.) MCI: Mild cognitive impairment NINCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association QUADAS‐2: Quality Assessment of Diagnostic Accuracy Studies SUVR: Standardised uptake value ratio
Background
Dementia is a syndrome due to a brain disease — usually of a chronic or progressive nature — in which there is disturbance of multiple higher cortical functions, including memory, thinking, orientation, comprehension, calculation, learning capacity, language, and judgement. However, consciousness remains unaffected. See the glossary in Appendix 1. The impairments of cognitive function are commonly accompanied, and occasionally preceded, by a deterioration in emotional control, social behaviour, motivation, and the impairment is sufficient to interfere with everyday activities. Dementia is a collection of different subtypes distinguished by the underlying pathology. Alzheimer's disease dementia (ADD) is the most common form of dementia and other important pathologies associated with dementia are vascular disease, Lewy bodies, and frontotemporal pathology (WHO 2012).
Dementia is a serious worldwide public health problem, with a prevalence of 4.7% in adults older than 60 years (6.2% and 6.5% in Europe and the Americas, respectively). Due to its prevalence in older people, it is expected that the number of people with dementia will increase dramatically. Consequently, in the year 2050, an expected number of 115 million people will have dementia. This will result in a considerable economic burden, which currently stands at 1% of the world's Gross National Product (GNP) in direct and indirect costs (WHO 2012). These financial costs are in addition to the devastating personal and social consequences of the condition.
The definition of MCI applies to people without evidence of significant deterioration in activities of daily living, but with subjective memory complaints and cognitive impairment detected by standardised tests. MCI often precedes clinical dementia, but there is no consensus regarding how to operationalise the MCI diagnosis. There are several clinical criteria to define which people have MCI, including the Petersen criteria or Petersen Revised Criteria (Petersen 1999; Petersen 2004; Winblad 2004), Clinical Dementia Rating Scale (CDR = 0.5) (Morris 1993), or 16 other different classifications of MCI (Matthews 2008).
A diagnosis of MCI reputedly allows testing of preventive interventions that would slow the progression of MCI to dementia. If the progression of MCI to dementia could be deferred by five years, the prevalence of dementia would decrease by 43% in 2050 (Alzheimer's Association 2010). MCI has an annual progression rate to ADD from 5% to 15%. However, not every person with MCI develops dementia, and a significant number of people recover or stabilise. Therefore, future research should try to clarify which people with MCI develop dementia in order to be able to focus specifically on people who are at high risk of developing dementia. This may possibly explain the failure of therapy to alter the progression to dementia in people with MCI. Other aspects that may contribute to this failure are the disparity in diagnostic criteria and different settings of the studied participants: community, primary, secondary, and research centres (Bruscoli 2004; Mattsson 2009; Petersen 1999; Petersen 2009).
The definition of Alzheimer's disease pathology is over 100 years old. This pathology includes neuritic plaques that contain deposits of amyloid beta (Aβ) and neurofibrillary tangles (Goedert 2006). This pathology is present in approximately 84% of all people with dementia (Schneider 2007). Furthermore, Alzheimer's disease pathology is found in 88% of people diagnosed with probable ADD (Schneider 2009). Despite this, Alzheimer's disease pathology may be found concomitantly at autopsy in people thought to have other forms of dementia, such as vascular dementia, Lewy body dementia, or frontotemporal dementia (FTD) (Jellinger 2006). Furthermore, at least five common pathologies have been found in the brains of people who died and were thought to have ADD prior to death (White 2009). Also, Alzheimer's disease pathology was found in 42% of community‐dwelling older people without dementia (Schneider 2007). This has generated controversy about the importance of the presence of Alzheimer's disease pathology. The pathology can be associated with ageing per se, and, for older people, the relationship between amyloid plaque burden and cognitive impairment diminishes as age progresses (Savva 2009). Thus, this pathology could be an epiphenomenon associated with the presence of dementia, e.g. a by‐product of repair mechanisms for vascular damage (De la Torre 2004; Garcia‐Alloza 2011). On the other hand, this controversy could be because our clinical diagnostic criteria have not enough accuracy to diagnose Alzheimer's disease that is detected by histopathology in postmortem studies (Hyman 2012). In addition, other researchers think that there is no real controversy about the amyloid hypothesis, because the amyloid cascade and the Aβ deposition have a primary role in Alzheimer's disease (Selkoe 2016).
More recently, the development of Aβ pathology biomarkers in vivo has been suggested as an important advance as a diagnostic tool in the field of Alzheimer's disease, and has promoted the creation of new diagnostic criteria for people without symptoms (preclinical stages), people with MCI, and people with ADD, based on the presence of biomarkers of Alzheimer's disease. These have included Aβ tracers by positron emission tomography (PET) (Albert 2011; Dubois 2014; McKhann 2011; Sperling 2011). However, uncertainties regarding the usability of biomarkers in the diagnosis of dementia still exist, mainly due to variation between biomarker types, criteria for positivity, and differences in methodology (Noel‐Storr 2013). This prompted an important initiative, the Standards for Reporting of Diagnostic Accuracy Studies in dementia studies (STARDdem) statement (Noel‐Storr 2014). Consequently, clinical properties of dementia biomarkers should not be assumed, and formal systematic evaluations of sensitivity, specificity, and other properties of biomarkers should be performed (Davis 2013).
PET is an imaging technique using compounds labelled with short‐lived positron‐emitting radionuclides. The use of Aβ ligands permits the in vivo detection of amyloid deposition in the brain. 18F‐florbetapir is a stilbene derivative and demonstrates a high binding affinity to Aβ aggregates. 18F‐florbetapir has good uptake by brain tissue and washout kinetics in mice and monkeys (Choi 2009) and in vitro binding of Aβ plaques in postmortem ADD brain samples (Choi 2009; Lin 2010). In 2010, it was evaluated for the first time in people with ADD and healthy people without ADD (Lin 2010; Wong 2010). 18F‐florbetapir could eventually be used to differentiate between different dementia types, specifically between FTD and ADD (Kobylecki 2015).
The Food and Drug Administration (FDA) and the European Medicines Agency (EMA) approved 18F‐florbetapir for Aβ binding. These agencies have stated that a negative scan indicates sparse or no plaques, which is inconsistent with a diagnosis of ADD, thus effectively excluding this diagnosis. A positive 18F‐florbetapir scan indicates moderate to frequent or amyloid neuritic plaques. However, this might also occur in people with other neurological conditions (e.g. Lewy body dementia, Parkinson’s disease dementia) and in older adults with normal cognition. Therefore, a positive result of an 18F‐florbetapir scan does not establish the diagnosis of ADD or any other cognitive disorder definitely, and it should be combined with other diagnostic evaluations or instruments. Additionally, the effectiveness and safety of the tests have not been established by predicting development of dementia or other neurological conditions, or by monitoring responses to therapies (EMA 2013; FDA 2013).
Despite not being approved for this purpose by the regulatory agencies, research has been conducted in people with MCI to determine whether biomarkers, such as 18F‐florbetapir for Aβ, increase the risk of developing dementia over time. The evidence for this is uncertain. For this and other reasons, the National Institute on Aging‐Alzheimer's Association (NIA‐AA) in the USA established two different criteria for MCI. Firstly, they established the Core Clinical Criteria for use in all clinical settings, without use of biomarkers, and characterised by concerns regarding a change in cognition with impairment in one or more cognitive domains with preservation of independence in functional abilities, therefore no dementia. Secondly, they established the Clinical Research Criteria, which incorporate the use of biomarkers, such as PET amyloid scans, intended for use exclusively in research settings, including academic centres and clinical trials. This will help determine whether positive scans increase the likelihood of progression from MCI to clinical dementia (Albert 2011). Lastly, it is hoped that people with MCI and positive scans will 'enrich' clinical trials, and more people who will progress to dementia in a shorter time will be included to allow more efficient studies of treatments and prevention strategies of ADD (CMS 2013).
An assumption for some researchers, and one on which this systematic review (SR) is predicated, is that if a person has both MCI and the pathology of Alzheimer's disease and develops clinical ADD subsequently, then the cause of the initial MCI and of the ADD was the Alzheimer’s pathology. Our approach is an example of assessing diagnostic test accuracy (DTA) using delayed verification of diagnosis. Instead of the reference standard being based on pathology, it is based on a clinical standard and the progression from MCI to ADD or any other form of non‐ADD or any dementia. Although, for the reasons stated above, a degree of unreliability has been introduced, defining progression has the advantage of being based on what matters most to people with MCI, their families, and clinicians involved in their care.
The 18F‐florbetapir PET scan is considered the diagnostic marker of interest and, in this SR, we assessed the DTA of 18F‐florbetapir Aβ binding in the brain and progression of the following:
From MCI to ADD.
From MCI to any other form of non‐ADD.
From MCI to any form of dementia
This SR belongs to a series of SRs regarding PET biomarkers for amyloid β, including 18F‐florbetaben and 18F‐flutemetamol (Martínez 2016).
Target condition being diagnosed
This SR assessed the following three target conditions.
ADD (progression from MCI to ADD).
Any other form of dementia (progression from MCI to any other form of non‐ADD).
Any form of dementia (progression from MCI to any form of dementia).
We compared the index test results obtained at baseline with the results of the reference standards obtained at follow‐up (delayed verification).
Index test(s)
The 18F‐florbetapir scan is an index test for the detection of Aβ deposition in the brain region of interest (ROI). The ROI is a selected brain area that physicians create for further study in various anatomical areas of the brain. 18F‐florbetapir is a molecular biomarker, described as (E)‐4‐(2‐(6‐(2‐(2‐(2[18F]fluoroethoxy)ethoxy)ethoxy)pyridine‐3‐yl)vinyl)‐N‐methylbenzamine and also referred to as 18F‐AV‐45 (Choi 2009).
Image Interpretation
Both the FDA and EMA have described the criteria for 18F‐florbetapir for Aβ positivity (EMA 2013; FDA 2013).
18F‐florbetapir diagnosis is by PET image assessment and is designated as either positive or negative by comparison of the radioactivity in cortical grey matter with activity in the adjacent white matter. This determination is made only in the cerebral cortex; the signal uptake in the cerebellum does not contribute to the scan interpretation (e.g. a positive scan may show retained cerebellar grey‐white contrast even when the cortical grey‐white contrast is lost). Specifically, a positive scan exhibited one of the following.
Two or more brain areas (each larger than a single cortical gyrus) in which there is reduced or absent grey‐white contrast. This is the most common appearance of a positive scan.
One or more areas in which grey matter radioactivity is intense and clearly exceeds radioactivity in adjacent white matter.
Readers trained in PET images with 18F‐florbetapir, should interpret the Aβ PET image made with this ligand (EMA 2013; FDA 2013).
Before the FDA and EMA described the criteria for 18F‐florbetapir PET scan positivity, the diagnosis of dementia was made using different thresholds. Therefore, we planned to use the FDA or EMA criteria applied in each included study to classify participants as either test‐positive or test‐negative, or alternatively if 18F‐florbetapir Aβ uptake and retention exceeded a certain threshold.
We considered the measurement of the 18F‐florbetapir retention (retention ratio): distribution volume ratio (DVR), standardised uptake value ratio (SUVR), or other ratios. DVR refers to the ratio of the 18F‐florbetapir distribution volume in the selected area (ROI) to the distribution volume in the reference area. SUVR is the ratio of the 18F‐florbetapir ligand standardised uptake value in the selected area (ROI) to the standardised uptake value in the reference area.
The unit of analysis of our SR was the participant. We did not include studies that analysed multiple ROIs per person.
Image analysis: not prespecified (e.g. Statistical Parametric Mapping (SPM) or other image analysis techniques).
Administration Instructions and Recommended Dosing
Time between 18F‐florbetapir injection and PET acquisition: images should be acquired in 10 minutes starting from 30 to 50 minutes after intravenous administration (EMA 2013; FDA 2013).
Injection dose: the recommended dose for 18F‐florbetapir Aβ PET is 370 MBq (10 mCi) as a single intravenous bolus in a total volume of 10 mL or less (EMA 2013; FDA 2013).
Although it is inevitable that included studies have used different imaging protocols, readers' expertise, and varied parameters, the amyloid PET data in these included studies should be technically adequate and acquired at a fully qualified and certified facility.
Clinical pathway
At this time, the clinical evaluation often has similarities between different countries (Cordella 2013; NICE 2006). It often starts with people experiencing memory complaints detected by themselves or their relatives. Frequently, general practitioners or family physicians are consulted, and they often conduct a medical evaluation using a screening test for cognitive impairment. Whenever this screening test is positive, they complete an assessment with a clinical evaluation conducted with laboratory studies that can rule out a secondary cause of cognitive impairment (e.g. hypothyroidism, renal failure, liver failure, vitamin B12 or folate deficiency, and others). In addition, these people are then referred to medical specialists in cognitive disorders (preferably a geriatrician, psychiatrist, or neurologist) in a secondary centre or directly to memory clinics where further clinical assessment, laboratory studies, and cerebral image studies are conducted to confirm the dementia diagnosis.
People with dementia, or their relatives, often directly consult these specialists or specialised memory clinics in the study of cognitive disorders. Therefore, the performance of the diagnostic tests will probably vary according to whether it is a primary consultation or a referral from primary to specialist care, or if the people have different clinical stages of the disease (MCI, mild, moderate, or severe dementia). Due to these differing pathways, the use of 18F‐florbetapir PET ligand for Aβ will be mainly used in specialist consultations and memory clinics as an addition to clinical evaluation or other tests, helping in a clinical setting to discard a diagnosis of Alzheimer's dementia with a negative scan in a person with clinical dementia and doubts about the aetiology (e.g. FTD versus ADD). Otherwise, it might be used solely in the research field in people with MCI for the enrichment of clinical trials. For example, enrolling people with MCI and a positive PET scan to study preventive interventions before people develop dementia.
However, in some memory clinics, the 18F‐florbetapir PET is used for clinical purposes in people with persistent or progressive unexplained MCI adopting the Johnson criteria (Johnson 2013), criteria without sufficient evidence. Therefore, if the 18F‐florbetapir PET is positive in a person with MCI, this positivity is considered as one of the core histopathological findings of Alzheimer's disease. The person will thus be catalogued as a patient with prodromal Alzheimer's disease or MCI due to Alzheimer's disease.
Alternative test(s)
Currently there are no standard practice tests available for the clinical diagnosis of Alzheimer's disease dementia. Below, we have listed the alternative tests that we have excluded from this SR. The Cochrane Dementia and Cognitive Improvement Group is in the process of conducting a series of DTA SRs of biomarkers and scales (see list below).
18F PET ligands for Aβ (18F‐florbetaben, 18F‐flutemetamol) (Martínez 2016).
18F‐FDG‐PET (PET F‐fluorodeoxyglucose) (Smailagic 2015).
11C‐PIB‐PET (PET‐Pittsburgh compound B) (Zhang 2014).
Cerebrospinal fluid (CSF) analysis of Aβ and tau (Kokkinou 2014; Ritchie 2013; Ritchie 2014).
Structural magnetic resonance imaging (sMRI) (Filippini 2012).
Neuropsychological tests (Mini‐Mental State Examination (MMSE); MiniCOG; Montreal Cognitive Assessment (MoCA) (Arevalo‐Rodriguez 2015; Chan 2014; Creavin 2016; Davis 2015; Fage 2015; Seitz 2014).
Informant interviews (Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE); AD8) (Harrison 2014; Hendry 2014; Lees 2014; Harrison 2015; Quinn 2014).
APOE‐ϵ4 (Elias‐Sonnenschein 2014a; Elias‐Sonnenschein 2014b; Elias‐Sonnenschein 2014c).
Single‐photon emission computed tomography (SPECT) brain imaging (Archer 2015; McCleery 2015).
Rationale
Accurate and early diagnosis of Alzheimer's disease is crucial for planning healthcare systems, because the costs of dementia are currently at least 1% of the world's GNP (WHO 2012).
18F‐florbetapir is approved for use in the clinical field mainly in people who are diagnosed clinically with dementia of uncertain aetiology, in which case diagnosis of ADD can be discarded if the test is negative. Even though 18F‐florbetapir is not approved for this purpose, this biomarker test is currently being used in the research field to search for the accurate identification of people with MCI who would progress to Alzheimer’s disease or other forms of dementia. Amyloid β tracers by PET have been included in newly diagnostic criteria in the study of people with MCI (Albert 2011; Dubois 2014). However, some uncertainties exist about the generalisability of the DTA results in clinical settings, especially in older people (Richard 2012).
It is currently believed that if the health system can identify which people are at high risk of progressing from MCI to dementia, it can focus on improving opportunities for appropriate contingency planning for them. Proper recognition of the disease may also help prevent inappropriate and potentially harmful admissions to hospital or institutional care (NAO 2007), and enable the development of new treatments designed to delay or prevent progression to more debilitating stages of the disease. Additionally, this may demonstrate a real clinical benefit for people and caregivers, and will reduce health system costs.
This SR assessed the DTA of 18F‐florbetapir Aβ PET in people with MCI.
Objectives
To determine the diagnostic test accuracy (DTA) of 18F‐florbetapir as the index test for detecting participants with mild cognitive impairment (MCI) at time of performing the test who would clinically progress to Alzheimer's disease dementia (ADD), or other forms of non‐ADD, or any form of dementia at follow‐up.
Secondary objectives
To investigate the heterogeneity of the DTA in the included studies, by evaluating the spectrum of people, referral centres, clinical criteria of MCI, 18F‐florbetapir techniques, reference standards used, duration of follow‐up, aspects of study quality, and conflicts of interest.
Methods
Criteria for considering studies for this review
Types of studies
We included longitudinal studies that had prospectively defined cohorts with any accepted definition of mild cognitive impairment (MCI), as outlined below, at time of performing the 18F‐florbetapir Aβ scan and a reference standard (see Index tests and Reference standards below). We obtained the results at the follow‐up of the studies. These studies had to employ delayed verification of progression to dementia and were sometimes labelled as 'delayed verification cross‐sectional studies' (Bossuyt 2008; Knottnerus 2002). We included case‐control studies when they incorporated a delayed verification design. This occurred in the context of a cohort study, so these studies were invariably diagnostic‐nested case‐control studies.
Participants
Participants recruited and clinically classified as having MCI at time of performing the test were eligible for inclusion. We established the diagnosis of MCI using the Petersen criteria or revised Petersen criteria (Petersen 1999; Petersen 2004; Winblad 2004), the criteria included in the Matthews study (Matthews 2008), CDR = 0.5 (CDR structured interviews collects information from both the collateral source and the subject regarding memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care, where the range of possible scores varies from none = 0 point to severe = 3 points) (Morris 1993), the National Institute on Aging‐Alzheimer's Association (NIA‐AA) core clinical criteria (Albert 2011), or a combination.
We excluded studies that included people with MCI possibly caused by any of the following:
Current or a history of alcohol or drug abuse.
Central nervous system (CNS) trauma (e.g. subdural hematoma), tumour, or infection.
Other neurological conditions (e.g. Parkinson’s or Huntington’s diseases). Regarding Parkinson's disease, many of the studies specifically excluded Parkison's disease patients from the group with mild cognitive impairment. This specific group of patients is complex in both regards to defining neuropathology and in determination of functional decline. For these reasons, this group of patients needs to be addressed in specific studies.
Index tests
The index test of this SR was the 18F‐florbetapir biomarker test. We used the criteria and cut‐off values for test positivity as reported in the included studies. We considered positivity for 18F‐florbetapir Aβ scan uptake and retention exceeding a certain threshold.
Target conditions
There were three target conditions in this SR:
Alzheimer’s disease dementia (ADD) (progression from MCI to ADD).
Any other forms of dementia (progression from MCI to any other forms of non‐ADD).
Any form of dementia (progression from MCI to any form of dementia).
Reference standards
The reference standard was the progression to the target conditions evaluated by a physician with expertise in the dementia field (preferably, a geriatrician, psychiatrist, or neurologist). For the purpose of this SR, we accepted several definitions of ADD. We included studies that applied the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS‐ADRDA) criteria (McKhann 1984), the Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria (APA 1987; APA 1994), and the International Classification of Diseases (ICD) (ICD‐10) criteria for ADD. Notably, different iterations of these standards may not be directly comparable over time (e.g. APA 1987 versus APA 1994). Moreover, the validity of the diagnoses may vary with the degree or manner in which the criteria have been operationalised (e.g. individual clinician versus algorithm versus consensus determination). We considered all these issues when we interpreted the results.
Similarly, we accepted differing clinical definitions of other dementias. For Lewy body dementia, the reference standard is the McKeith criteria (McKeith 1996; McKeith 2005); for frontotemporal dementia, the Lund criteria (Boxer 2005; Brun 1994; Neary 1998), the DSM criteria (APA 1987; APA 1994), the ICD criteria (ICD‐10), or the International Behavioural Variant FTD Criteria Consortium (Rascovsky 2011); and for vascular dementia, the National Institute of Neurological Disorders and Stroke and Association Internationale pour la Recherché et l'Enseignement en Neurosciences (NINDS‐AIREN) criteria (Román 1993), the DSM criteria (APA 1987; APA 1994), or the ICD criteria (ICD‐10).
The time interval over which progression from MCI to ADD (or other forms of dementia) occurs is very important. We used one year as the minimum period of delay in the verification of the diagnosis (the time between the assessment at which a diagnosis of MCI is made and the assessment at which the diagnosis of dementia is made).
Search methods for identification of studies
Electronic searches
We searched MEDLINE (Ovid SP) from 1946 to May 2017; Embase (Ovid SP) from 1974 to May 2017; PsycINFO (Ovid SP) from 1806 to May 2017; BIOSIS Citation Index (Thomson Reuters Web of Science) from 1922 to May 2017; Web of Science Core Collection, including the Science Citation Index (Thomson Reuters Web of Science) and the Conference Proceedings Citation Index (Thomson Reuters Web of Science) from 1946 to May 2017; LILACS (Bireme); CINAHL (EBSCOhost) from 1980 to May 2017; ClinicalTrials.gov (https://clinicaltrials.gov); and the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (http://www.who.int/ictrp/search/en/). We also searched ALOIS, the Cochrane Dementia & Cognitive Improvement Group’s specialized register of dementia studies (http://www.medicine.ox.ac.uk/alois/).
We used two approaches in designing the search. One focused solely on the specifically named index test (including a range of synonyms); the second, run in parallel, covered a more general search, linking broader terms for the index test, focused by terms describing its diagnostic use to terms for the target condition to try to capture the more difficult to locate studies of a more general nature, where these particular radioligands were included in diagnostic accuracy research but not named specifically in the parts of the electronic bibliographic record that are searchable and therefore would be missed.
See Appendix 2 for details of the sources and search strategies that we used. No language or date restrictions were applied to the electronic searches.
Searching other resources
We examined the reference lists of all relevant studies for additional studies. We also searched the Database of Abstracts of Reviews of Effects (DARE) via the Cochrane Library: www.cochranelibrary.com), the National Institute for Health Research ‐ Health Technology Assessment Database (NIHR‐HTA) (via the Cochrane Library: www.cochranelibrary.com), the Aggressive Research Intelligence Facility (ARIF) database (www.arif.bham.ac.uk) for other related systematic diagnostic accuracy reviews, and the International Federation of Clinical Chemistry and Laboratory Medicine Committee for Evidence‐based Laboratory Medicine database (C‐EBLM) (http://www.ifcc.org/ifcc‐education‐division/emd‐committees/c‐eblm/evidence‐based‐laboratory‐medicine‐c‐eblm‐base).
We checked the reference lists of any relevant studies and systematic reviews, and performed citation tracking using the Science Citation Index to identify any additional relevant studies.
Data collection and analysis
Selection of studies
Two review authors (GM, RV) independently screened the retrieved titles and abstracts for potentially eligible studies. A third review author (PF) resolved any disagreements between the two review authors. The two review authors (GM, RV) then independently assessed the full‐text articles of the selected studies with the inclusion criteria. They resolved any disagreements through discussion or, where necessary, consulted a third review author (PF) who acted as an arbitrator. When a study did not present all relevant data for creating a 2 × 2 table, we contacted the study authors directly to request further information. When more than one article presented data on the same population, we included the primary article, which was the article with the largest number of people or with the most informative data (e.g. longest time of follow‐up in the primary outcome).
Data extraction and management
We planned to extract the following data regarding the study characteristics.
-
Bibliographic details of primary paper:
author, title of study, year, and journal.
-
Basic clinical and demographic details:
number of participants;
clinical diagnosis;
MCI clinical criteria;
age;
gender;
sources of referral;
participant recruitment;
sampling procedures.
-
Details of the index test:
method of the 18F‐florbetapir test administration, including those who administered the test;
thresholds used to define positive and negative tests;
other technical aspects as seemed relevant to the review, e.g. brain areas.
-
Details of the reference standard:
definition of ADD and other dementias used in the reference standard;
duration of follow‐up from time of index test performed to defining ADD and other dementias by the reference standard: one year to less than two years; two years to less than four years; and more than four years. If participants had been followed for varied amounts of time, we recorded a mean follow‐up period for each included study. If possible, we grouped those data into minimum, maximum, and median follow‐up periods, which could then become the subject of subgroup analyses;
prevalence or proportion of population developing ADD and other dementias, with severity, if described.
We created 2 × 2 tables (cross‐relating index test results of the reference standards) as shown in Appendix 3. For each included study, we recorded the number of people lost to follow‐up. We also extracted data necessary for the assessment of quality, as defined below. Two review authors (GM, RV) independently performed data extraction. We resolved any disagreements regarding data extraction by discussion, or by consulting a third review author (PF).
Assessment of methodological quality
We assessed the methodological quality of each included study using the Quality Assessment of Diagnostic Accuracy Studies 2 tool (QUADAS‐2) (Whiting 2011), as recommended by Cochrane (Davis 2013). This tool is comprised of four domains: participant selection, index test, reference standard, and participant flow. Two review authors (GM, RV), who were blinded to each other’s scores, independently performed the QUADAS‐2 assessment. We resolved any disagreements by discussion or, if necessary, consulted a third review author (PF) who acted as an arbitrator. We assessed each domain in terms of risk of bias, and also considered the first three domains in terms of applicability concerns. In Appendix 4, we have detailed the components of each of these domains and provided a rubric that shows how we made judgements concerning risk of bias. Key areas important to quality assessment are participant selection, blinding, and missing data.
We included three additional signalling questions on our checklist.
Was the PET scan interpretation done by a trained reader physician? (We included this under the ’Index test’ domain).
Was there a clear definition of a positive result? (We included this under the ’Index test’ domain).
Was the study free of commercial funding? (We included this under the ’flow and timing’ domain).
We included the item pertaining to the PET scan interpretation and the definition of positive results to take into account the subjective nature of 18F‐florbetapir Aβ scan image interpretation, which may be based on a variety of different criteria, such as extensive clinical experience, different standardised uptake values (SUV), different morphological features, or a combination of the aforementioned. We included the third additional item in order to record any potential bias resulting from commercial interest in the results due to the potential risk by the manufacturing company leading to more favourable results and conclusions than sponsorship by other sources (Lundh 2017).
We did not use QUADAS‐2 data to form a summary quality score. We produced a narrative summary that described the numbers of included studies that were at high, low, or unclear risk of bias as well as concerns regarding applicability, which we have described in Appendix 5.
Statistical analysis and data synthesis
We applied the DTA framework for the analysis of a single test and extracted the data from each included study into a 2 × 2 table, showing the binary test results cross‐classified with the binary reference standard, and we ignored any censoring that might have occurred. We acknowledge that such a reduction in the data may represent a significant oversimplification.
We used data from the 2 × 2 tables abstracted from the included studies: true positive (TP), false negative (FN), false positive (FP), true negative (TN), and entered these into Review Manager 5 (RevMan 5) (Review Manager 2014) to calculate the sensitivities, specificities, and their 95% confidence intervals. We also presented individual study results graphically by plotting estimates of sensitivities and specificities in both a forest plot and a receiver operating characteristic (ROC) space. If an individual included study published more than one threshold, we presented the graphical findings for all reported thresholds.
We planned to segment analyses into separate follow‐up mean periods for the delay in verification: one year to less than two years; two to less than four years; and greater than four years. We planned to clearly note where the same included studies contributed to the analysis for more than one reference standard follow‐up interval.
However, due to lack of data, we conducted no meta‐analyses, but we prepared a 'Summary of findings' table regardless.
Investigations of heterogeneity
We were able to include only three studies, therefore issues of heterogeneity did not arise.
Sensitivity analyses
We found insufficient data to conduct any sensitivity analyses.
Assessment of reporting bias
We did not investigate reporting bias.
Results
Results of the search
The total number of records identified for this SR was 2502. The PRISMA diagram (Figure 1) shows the selection of records through the screening and selection processes. In total, we assessed 312 studies (74 full text papers, 81 conference publications, 86 registered studies in clinicaltrials.gov, and 71 registered studies in WHO ICTRP) for eligibility in the full‐text screening. We excluded 299 studies. 115 studies were multiple publications or duplicates and 33 studies did not have extractable data for constructing 2 x 2 tables, and we received no reply when we contacted the authors. One study used an MCI definition not accepted in our protocol, and we excluded five studies with available data to extract due to duplication of participants or at high risk of duplication with the included studies (Characteristics of excluded studies). We excluded the remaining studies because they did not meet the inclusion criteria: i) not a longitudinal study (n = 44); ii) no MCI participants at time of performing the test (n = 35); iii) index test not a 18F‐florbetapir PET scan (n = 3); iv) discussion or review paper (n = 20); v) wrong design (n= 43). We included three studies and identified ten references as ongoing studies (Characteristics of ongoing studies).
1.
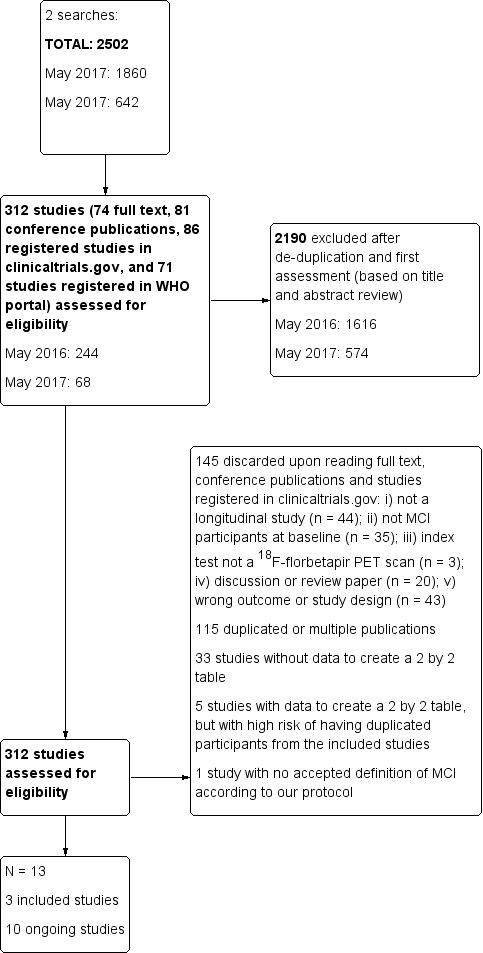
Study flow diagram.
Included studies
See Characteristics of included studies.
Doraiswamy 2014 refers to one study reported previously with ADD, MCI, and healthy control (HC) participants and a longitudinal extension at 36 months follow‐up of this cross‐sectional study with the MCI participants. Participants were recruited from 21 sites in the United States of America as part of a cross‐sectional study to determine the efficacy using both visual interpretation and quantitative interpretation to assess brain amyloid burden by 18F‐florbetapir PET scan to differentiate healthy controls (n = 79) from subjects with a clinical diagnosis of ADD (n = 45) or MCI (n = 60) study participants. The interim analysis of the longitudinal extension at 18 months reported baseline data of 51 MCI participants and the efficacy analysis included 46 participants. On the other hand, the baseline data described in Doraiswamy 2014 is of 47 MCI participants with at least one post‐baseline measurement; however, there were 52 participants at baseline planned for efficacy participants.
MCI participants had a CDR score of 0.5; complaint of memory or cognitive decline corroborated by an informant; objective evidence of cognitive impairment or marginally normal cognition with a documented history of high cognitive performance; no obvious medical cause for the impairment; subject not demented and criteria for ADD not satisfied; normal score on the Alzheimer’s Disease Clinical Studies Consortium Activities of Daily Living (ADCS ADL), 25 of 47 were female; participants with Aβ (+) by 18F‐florbetapir PET scan had a mean age of 74.47 + 7.72 years old, with 14.47 + 2.18 mean years of education, their mean MMSE was 27.29 + 2.14 points and 11 participants were APOE ϵ4 carrier positive, respectively. In those participants with Aβ (‐) by 18F‐ florbetapir PET scan, they had a mean age of 70.40 + 10.72 years old, with 15.27 + 2.42 mean years of education and their mean MMSE was 27.53 + 1.63 points. Four participants were APOE ϵ4 carrier positive, respectively. Of the 47 participants, 9 (19.1%) developed Alzheimer’s dementia. 5 participants (9.6%) had no post‐baseline measurement, therefore they were excluded from the analysis, and in those 47 with at least one post‐baseline measurement, 37 completed the study at 36 months. The Reference standard at follow‐up was not explicitly stated, although NINCDS‐ADRDA criteria for ADD (McKhann 1984) were baseline diagnostic criteria.
Potential conflicts of interest were noted. The manufacturer of 18F‐florbetapir tracer provided financial support for the study and six authors were employees.
Kawas 2013 refers to a study with participants who were 90 years old or older who either lived at home or in institutions, evaluated in the United States of America. Those included were participants of a longitudinal, population‐based study (90+ Study) and were also invited to participate in this study which was part of research to examine the relationship between measurements of brain amyloid and levels of amyloid burden measured by postmortem histopathological assessment (Clark 2011). Participants had normal cognition or cognitive or functional impairment resulting from cognition not severe enough to meet DSM‐IV diagnostic criteria and they were classified as cognitively impaired, not demented (CIND). They agreed to postmortem brain donation. The purpose of the study was to examine cross‐sectional and longitudinal associations between cognitive performance and beta amyloid load in non‐demented oldest‐old. Baseline characteristics was based on 13 non‐demented oldest‐old participants, eight were normal controls and five of them were classified as MCI at time of performing the test according to our MCI definitions (Matthews 2008), three MCI were considered as Aβ (+), and two MCI were considered as Aβ (‐). In the total group (13 participants), the mean age was 94.1 years (range 90 to 99); for those considered as Aβ (+), the mean age was 94.4 years (range 93 to 96) and 94.1 years (range 90 to 99) years old for those with Aβ (‐). Nine participants were women, two of whom were Aβ (+), and two of four men were Aβ (+) at baseline. Seven participants were reported as having been educated beyond high school age, two of them were Aβ (+) and five were Aβ (‐), and for those six having been educated up to or less than high school age, two were Aβ (+) and four were Aβ (‐), respectively. The mean MMSE was 28 (range 24 to 30); for those considered as Aβ (+), the mean MMSE was 26.5 (range 24 to 29) and 28 (range 25 to 30) for those in the Aβ (‐) group; and no data regarding APOE ϵ4 carrier were reported.
All participants were followed for a mean period of 1.5 years; three of 13 participants developed dementia during follow‐up, one in the Aβ (‐) group and two in the Aβ (+) group, and it seemed that none of the participants were lost to follow‐up. The reference standard used to classify participants at follow‐up as dementia or not was DSM‐IV (APA 1994).
Partial financial support was provided by and three authors were employees of the manufacturer of 18F‐florbetapir tracer.
Schreiber 2015 is a study with data from Alzheimer's Disease Neuroimaging Initiative (ADNI), a multicentre study, supported by the National Institute of Health, private companies, and non‐profit organisations. This ADNI database recruited participants from nearly 50 different sites. All participants were aged between 55 and 90 years, had completed at least 6 years of education, and MCI participants were single‐domain or multidomain amnestic, had subjective memory problems, had a MMSE score between 24 and 30, and had a CDR of 0.5.
The main objectives of this study were to investigate the concordance between visual and quantitative Aβ PET with the 18F‐florbetapir PET scan and if these assessments agreed or not with CSF Aβ1‐42 in MCI participants, and also to examine the prediction of progression at follow‐up according to their visual and quantitative categorization as Aβ positive or negative at baseline with the 18F‐florbetapir PET scan (ADNI‐GO; ADNI2).
The threshold used was a SUVR > 1.11 determined at baseline (Landau 2012, Landau 2013).
The study had 401 MCI participants. The mean age at baseline was 71.6 years (+ 7.5). There were 182 female participants, and 198 participants were APOE ϵ4 carrier positive. The MMSE mean was 28.1 (+ 1.7) and the mean years of education was 16.2 (+ 2.7).
No potential conflicts of interest were noted.
Excluded studies
120 studies were excluded since they did not meet the inclusion criteria for participants, index test, or target condition and 76 studies were duplicated or multiple publications. Additionally, 33 studies did not have data to create a 2 by 2 table, and three studies with data to create the 2 by 2 table were not included due to shared or high risk of shared participants with one included study (Characteristics of excluded studies).
Methodological quality of included studies
We assessed methodological quality using the QUADAS‐2 tool (Whiting 2011). Review authors’ judgements about each methodological quality item for each included study are presented in the Characteristics of included studies table and Figure 2. The overall methodological quality of the studies is summarised in Figure 2.
2.
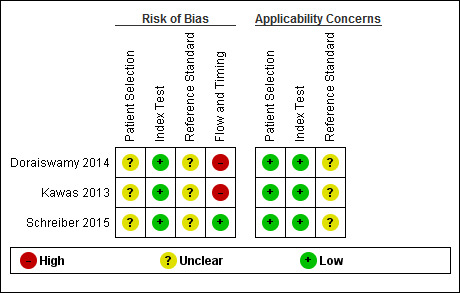
Risk of bias and applicability concerns summary: review authors' judgements about each domain for each included study
In the participant selection domain, we considered all studies (Doraiswamy 2014; Kawas 2013; Schreiber 2015) to be at unclear risk of bias due to lack of reporting on sampling procedures and exclusion criteria. We stated that the included studies avoided a case‐control design because we only considered data on performance of the index test to discriminate between people with MCI who converted to dementia and those who remained stable.
In the index test domain, we considered all three studies to have a low risk of bias (Doraiswamy 2014; Kawas 2013; Schreiber 2015). The three studies had low risk of bias because the visual assessment used in all three studies was established, the SUVR used in the Schreiber study was described previously as a SUVR > 1.11 (Landau 2012), and in all three studies, the interpretation was made blinded to the clinical data. In our two additional signalling questions, there was low risk regarding whether the index test was interpreted by a trained reader physician in all three included studies, and the positivity criteria was clearly established in two studies and, in one, it was considered as unclear.
In the reference standard domain, we considered the three studies as at unclear risk of bias (Doraiswamy 2014; Kawas 2013; Schreiber 2015). The Doraiswamy study had an unclear risk of bias because we were not able to obtain the information about which reference standard was used. Regarding the Kawas and Schreiber studies, we considered the studies as unclear risk of bias because, despite the use of DSM‐IV criteria for any form of dementia (APA 1994) and NINCDS‐ADRDA criteria (McKhann 1984) as reference standards, respectively, it was unclear if the clinician was blinded to the results of the 18F‐florbetapir PET scan to establish the dementia diagnosis.
In the flow and timing domain, we judged the Doraiswamy study to have a high risk of bias because it was unclear if it used the same criterion to diagnose the ADD at follow‐up. In our additional signalling question, there were potential conflicts of interest due to the financial support for the study and the fact that six authors were employees of the manufacturer of 18F‐Flobetapir tracer. Regarding the Kawas study, a high risk of bias was considered due to possible conflict of interest because of partial financial support and three authors were employees of the company producing 18F‐florbetapir.
For assessment of applicability, there was no concern that the included participants and setting, and the conduct and interpretation of the index test, did not match the review question. However, the target condition (as defined by the reference standard) was unclear due to lack to information about which reference standard(s) was applied (Doraiswamy 2014) and if the clinician was blinded or not to the 18F‐florbetapir PET scan result to establish the diagnosis (Kawas 2013; Schreiber 2015).
Findings
The results of the included studies are summarised in Data table 1 and Data table 2. Additionally, the summary of main results for the included studies are presented in the 'Summary of findings' table.
1. Test.

MCI to ADD by visual assessment from 2 to less than 4 years of follow‐up.
2. Test.

MCI to ADD by visual assessment from 1 to less than 2 years follow‐up.
18F‐florbetapir to predict progression from MCI to Alzheimer’s disease dementia (ADD)
Visual Assessment
Doraiswamy 2014 included data on 47 of the 52 eligible participants with MCI diagnosed with CDR = 0.5, and complaint of memory or cognitive decline corroborated by an informant; objective evidence of cognitive impairment or marginally normal cognition with a documented history of high cognitive performance; no obvious medical cause for the impairment; subject not demented and criteria for ADD not satisfied; normal score on the Alzheimer’s Disease Clinical Studies Consortium Activities of Daily Living (ADCS ADL), using a nonspecified reference standard, probably NINCDS‐ADRDA (McKhann 1984). They reported a sensitivity of 67% (95% CI 30 to 93) and a specificity of 71% (95% CI 54 to 85) to predict the progression from MCI to ADD at three years of follow up. Of the 52 participants who were given an initial clinical diagnosis of MCI, the study had data on 47 of them at the follow‐up; 6 were true positive, 11 were false positives, 3 were false negative and 27 were true negative (Figure 3).
3.
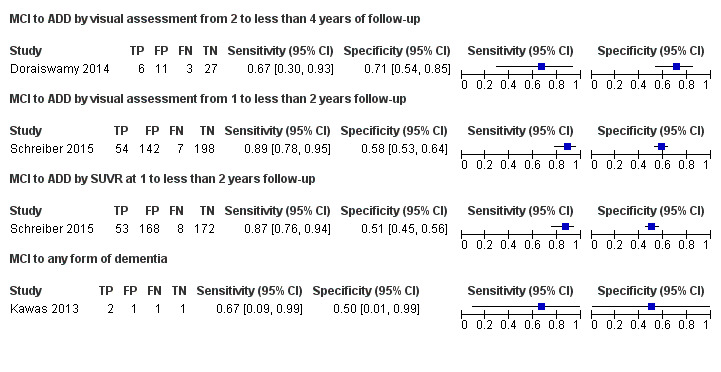
Forest plot of tests: 1 MCI to ADD by visual assessment from 2 to less than 4 years of follow‐up, 2 MCI to ADD by visual assessment from 1 to less than 2 years follow‐up, 3 MCI to ADD by SUVR at 1 to less than 2 years follow‐up, 4 MCI to any form of dementia.
Schreiber 2015 included data on 401 participants with MCI defined as cases with single‐domain or multidomain amnestic; having subjective memory concerns; MMSE score between 24 and 30 (inclusive); CDR = 0.5; Memory Box score had to be at least 0.5, and general cognition and functional performance sufficiently preserved such that a diagnosis of Alzheimer’s disease could not be made at the screening visit. They reported a sensitivity of 89% (95% CI 78 to 95) and a specificity of 58% (95% CI 53 to 64) to predict the progression from MCI to ADD at 1.6 years of follow‐up. Of the 401 participants who were given an initial clinical diagnosis of MCI, the study had data of all at follow‐up; 54 were true positive, 142 were false positives, 7 were false negative, and 198 were true negative (Figure 3).
Quantitative Assessment by SUVR > 1.1
Schreiber 2015 included data on 401 participants with MCI defined as above and reported a sensitivity of 87% (95% CI 76 to 94) and a specificity of 51% (95% CI 45 to 56) to predict the progression from MCI to ADD at 1.6 years of follow up. Of 401 participants who were given an initial clinical diagnosis of MCI, the study had data of all at follow‐up; 53 were true positive, 168 were false positives, 8 were false negative, and 172 were true negative (Figure 3).
18F‐florbetapir to predict progression from MCI to any form of dementia
Visual Assessment
Kawas 2013 included data on five participants with MCI, defined as participants with a condition known as 'cognitive impairment non‐demented (CIND)', where participants had either cognitive or functional impairment resulting from cognition not severe enough to meet DSM‐IV criteria, and it was included in Matthews 2008 MCI definitions. This study had a sensitivity of 67% (95% CI 9 to 99) and a specificity of 50% (95% CI 1 to 99) to predict the progression from MCI to any form of dementia at 1.5 years of follow‐up. Of five participants who were given an initial clinical diagnosis of MCI, the study had data on five of them at the follow‐up; two were true positive, one was false positive, one was false negative and one was true negative (Figure 3).
No data were available regarding the other target condition in this Cochrane review; progression from MCI to another form of non‐ADD.
Investigation of heterogeneity
The planned investigations were not possible due to the limited number of studies available for the analysis.
Sensitivity analyses
There were insufficient studies identified to permit any sensitivity analysis.
Discussion
Summary of main results
The volume and quality of evidence regarding the DTA of 18F‐florbetapir for early diagnosis of ADD and other dementias in participants with MCI was limited. We identified three studies in this SR. However, we were not able to construct a meta‐analysis because we planned to perform an analysis according to their follow‐up mean periods for the delay in verification: one year to less than two years; two to less than four years; and greater than four years. Neither of the two studies with the same target condition, progression from MCI to ADD, had the same follow‐up period. We did not perform sensitivity analyses and were not able to analyse heterogeneity.
Two included studies (Doraiswamy 2014; Schreiber 2015) addressed the DTA of 18F‐florbetapir analysed by visual assessment for the prediction of progression from MCI to ADD at follow‐up, and one also evaluated the progression from MCI to ADD at follow‐up analysed quantitatively with a threshold of SUVR > 1.1 (Schreiber 2015). One study addressed the DTA of 18F‐florbetapir analysed by visual assessment for the prediction of progression from MCI to any form of dementia at follow‐up (Kawas 2013). The results are summarised in the 'Summary of findings' table (Table 1). Two studies were evaluated as at high risk of bias, mainly due to the potential conflict of interest regarding financial support by the company who manufactured the 18F‐florbetapir tracer (Doraiswamy 2014; Kawas 2013). No other study had information about the progression to any other form of dementia (non‐ADD).
Regarding the objectives of our SR, to determine the DTA of the 18F‐florbetapir PET scan for detecting participants with MCI at time of performing the test who would clinically progress to ADD, or to other forms of dementia or any form of dementia at follow‐up, the results were as follows:
18F‐florbetapir PET scan for Alzheimer’s disease dementia (ADD)
Progression from MCI to ADD in those with a follow‐up between two to less than four years had a sensitivity of 67% (95% CI 30 to 93) and a specificity of 71% (95% CI 54 to 85) by visual assessment (Figure 3).
Progression from MCI to ADD in those with a follow‐up between one to less than two years had a sensitivity of 89% (95% CI 78 to 95) and a specificity of 58% (95% CI 53 to 64) by visual assessment and a sensitivity of 87% (95% CI 76 to 94) and a specificity of 51% (95% CI 45 to 56) by quantitative assessment by SUVR (Figure 3).
The DTA of18F‐florbetapir included a wide range of low‐to‐moderate and good sensitivity and low‐to‐moderate specificity for predicting progression to ADD through visual or SUVR assessment evaluation at different follow‐up. In other words, the low‐to‐moderate or good sensitivity could be affected by a high false negative rate. One hypothesis that could explain false negatives is that some people with probable ADD diagnosis may have different and multiple brain pathologies, the most common being Alzheimer's disease pathology combined with microscopic infarcts or neocortical Lewy body disease. These heterogeneous pathological findings are similar in those with MCI (Schneider 2007; Schneider 2009). In addition, the soluble Aβ oligomers are not detected by 18F‐florbetapir, and they have been playing a central role in Alzheimer's pathogenesis in the amyloid hypothesis (Heyden 2013), with the possibility of producing false negatives. Indeed, a study found two of 11 participants with an autopsy performed > one year after the 18F‐florbetapir PET scan as having a positive neuropathological diagnosis (probable or definite Alzheimer's disease), and they had a negative 18F‐florbetapir PET scan (Clark 2012).
Moreover, the presence of neurofibrillary tangles (NFTs), the other histopathologic core of Alzheimer's disease, is not detected by amyloid tracers. For example, the data from cohort studies indicated that plaques and tangles independently contributed to cognitive impairment in Alzheimer's disease pathology without any other primary neuropathologic diagnosis (Serrano‐Pozo 2013). Furthermore, NFT formation might be either unrelated to amyloid plaques formation or a temporally distinct process, or both (Royall 2014).
In addition, the low‐to moderate specificity could be affected by a high false positive rate. A positive 18F‐florbetapir PET scan for Aβ, has been found in other neurological conditions. It was positive in seven of 11 cases of dementia with Lewy bodies, in one of five Parkinson's disease participants (Siderowf 2014), and in six of eight FTD participants evaluated with a SUVR > 1.11 (Kobylecki 2015). The latter could be explained due to the presence of mixed pathology in the same participant, however, in one study with a pathology diagnosis, in three cases with non‐ADD by histopathology, the 18F‐florbetapir PET had a low likelihood of Alzheimer's disease by NIA/Reagan Institute criteria in all of them (Clark 2011). On the other hand, the false positive rate could be explained because it has affinity to amyloid in vessel walls, in particular, to cerebral amyloid angiopathy as this was shown in patients with intracerebral haemorrhage due to cerebral amyloid angiopathy (CAA)(Gurol 2016). The latter would indicate that some MCI participants have vascular MCI due to CAA. The other important option for a high false positive rate is that in many people without cognitive impairment, it is possible to find Aβ deposits at autopsy (Gelber 2012) generating doubt about the pathophysiological relevance of the Aβ hypothesis in Alzheimer's disease.
Duration of follow‐up is also important in predicting the progression of MCI to ADD, because the reported progression rate of MCI to ADD is between 8% and 16% per year (Mitchell 2009). We took it for granted that, given a long follow‐up period, a high percentage of people with MCI at time of performing the test would progress to Alzheimer’s disease, thus affecting the predictive accuracy of the 18F‐florbetapir PET scan. This was found in a systematic review with PiB PET where the data were separated into short follow‐up and longer than two years of follow‐up (Ma 2014). The authors included five studies with 102 participants in total with a variable specificity between 58% to 100%. However, in this SR, the progression rate in both included studies was relatively similar despite the follow‐up in one study being almost double the other (15.2% at 1.6 years and 19.1% at three years of follow‐up). This difference is probably explained by the setting of recruitment or other characteristics of the MCI participants and other underlying factors affecting these progression rates (Doraiswamy 2014; Schreiber 2015). In consequence, due to the lack of data, we were not able to investigate the effect of the follow‐up on the progression rate from MCI to ADD or any form of dementia.
The MCI subtypes have been studied regarding their relationship with the progression to ADD. In the largest longitudinal study to our knowledge, results from the follow‐up of 550 MCI participants indicated that the MCI subtype, presence of storage memory impairment, multiple domain condition, and presence of APOE ϵ4 allele increased the risk of progression to dementia. Multivariate survival and Kaplan‐Meier analyses showed that amnestic MCI with storage memory impairment had the most and closest risk of progression to dementia (Espinosa 2013). In our review, one study included only amnestic MCI, and this could explain the decrease in false negative rate with an increase in sensitivity. This may explain why the sensitivity was higher in this study (Schreiber 2015) than the study which included any type of MCI (Doraiswamy 2014). In addition, some ’high risk factors’ such as positive family history of dementia, presence of Abeta and tau protein in cerebrospinal fluid, and the APOE ϵ4 allele may also contribute to a faster progression rate to dementia. To support this, the Schreiber study showed different Cox proportional hazards regression models, where visual analysis adjusted by age, sex, and educational level had a higher hazard ratio to predict the progression than when analyses added APOE ϵ4 allele or 18F‐FDG‐PET as covariates (Schreiber 2015). Another study using a multimodal approach to predict the progression including MRI, 18F‐florbetapir PET, and 18F‐FDG‐PET had better predictive accuracy than the single modality (Xu 2016). In conclusion, further studies should include high‐quality research with more detailed data about the characteristics of MCI, not only to explore the underlying mechanisms but also to elucidate the causal pathways that link 18F‐florbetapir PET scan positivity to diverse MCI subtypes and disease progression.
18F‐florbetapir PET scan for any other forms of dementia (non‐Alzheimer’s disease dementia (non‐ADD))
Data for any other forms of dementia (non‐Alzheimer's disease dementia) were limited in this SR. Although 18F‐florbetapir retention is a poor predictor of subsequent progression to Alzheimer’s disease, the current available data suggested that 18F‐florbetapir may not play a role in any other forms of dementia (non‐ADD).
18F‐florbetapir PET scan for any form of dementia
Progression from MCI to any form of dementia in those with a follow‐up between one to less than two years had a sensitivity of 67% (95% CI 9 to 99) and a specificity of 50% (95% CI 1 to 99) by visual assessment (Figure 3).
Kawas 2013 had poor sensitivity and specificity to predict any form of dementia (Kawas 2013). This could be explained by the reasons stated before, specifically due to the small sample of the oldest‐old participants of this study, and the high prevalence of Alzheimer's pathology in older people without a stated dementia (Savva 2009; Gelber 2012). The DTA may be different in those studies with a younger population.
Strengths and weaknesses of the review
We conducted an extensive, comprehensive, and sensitive literature search, using eleven different electronic databases without any limitation to language. However, we were able to include only three studies with 453 eligible participants, therefore our DTA estimates were relatively imprecise. This paucity of evidence reflected the very significant challenges inherent in conducting long‐term prospective studies of well‐characterised participants, followed up to the point of progression of clinical dementia. The methodological quality assessment and data syntheses were based on the recommended methods (Davis 2013). To increase the reliability of our findings, we included only studies that fulfilled delayed verification of progression from MCI to ADD or other form of dementia (non‐ADD) or any form of dementia at follow‐up.
The included studies did have significant methodological limitations that weakened confidence in the results of this SR. First, considerable uncertainty remained concerning the clinical diagnosis of ADD; the histopathological diagnosis would be the better way to define the diagnosis, but this is not a realistic option for a clinical trial. Second, the three studies lacked information regarding the selection of participants. It was not clear if the reference standard interpretation was made without knowledge of the 18F‐florbetapir PET scan results in two studies, and a major problem in two of the studies was a potential conflict of interest with the company that produced the tracer.
The selection of participants with MCI in these studies could be another weakness, because we did not have all the necessary baseline data to perform risk stratification to detect the MCI subgroup who would progress to ADD. However, this selection of participants such as type of MCI, age, presence of APOE ϵ4 allele, structural abnormality at MRI, hypometabolism at FDG‐PET scan, and alteration in cerebrospinal fluid could help determine different subgroups of people at higher risk of developing dementia at follow‐up, and could enable stratification that could help avoid biases, and develop more efficient studies in the future (Hampel 2012, Caroli 2015, Wolz 2016). In this way, Xu 2016 described a multimodal approach with a 18F‐florbetapir PET scan and 18F‐FDG‐ PET scan, Pascoal 2017 described the combination of p‐tau levels in cerebrospinal fluid with 18F‐florbetapir PET scan status, and in Schreiber 2015, age, sex, educational level, ADAS‐cog at baseline, APOE ϵ4 allele, and 18F‐FDG‐PET scan status were included in the Cox regression model.
Finally, an important weakness of this SR was the nonresponse from the majority of the authors about their studies. This has resulted in a lack of data for analysis in this review.
Applicability of findings to the review question
Regarding the question of this SR:
Could the 18F‐florbetapir PET scan identify those MCI participants who would progress to a clinical dementia at follow up?
There were concerns regarding the applicability of the included participants and setting and in the index test domain in all three studies. In addition, in all three studies, there were concerns regarding the applicability of the reference standard due to the lack of information about the knowledge or not of the index test result to make the diagnosis. There was also lack of information regarding which reference standard was used. In two studies, there were concerns regarding applicability because of potential conflicts of interest. Therefore, due to the limited number of included studies and levels of heterogeneity with respect to the domains mentioned above, it was difficult to determine to what extent the findings from this systematic review could be applied to clinical practice.
The DTA of the 18F‐florbetapir PET scan for identifying Alzheimer’s disease pathology and identifying those people with MCI who would convert to ADD or any form of dementia could be affected by a number of factors that have not been determined so far. The most important, is the lack of a large study to evaluate this question; we included only three studies, two that addressed the progression from MCI to ADD with 448 participants and one that addressed the progression from MCI to any form of dementia with only five participants.
We have to wait for new longer‐term longitudinal studies. The 18F‐florbetapir test is expensive, therefore, we believe it is important to clearly determine its DTA prior to recommending its adoption in clinical practice, because the actual sensitivity and especially the specificity are too low to have enough accuracy to be used in clinical practice to predict the progression from MCI to ADD.
Authors' conclusions
Implications for practice.
Today, the use of 18F‐florbetapir has not been established for predicting development of Alzheimer's disease (FDA 2013, EMA 2013), and is not indicated in people with MCI except in clinical trials and research studies ( Albert 2011).
However, the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association have proposed the usage of amyloid PET in people with persistent or progressive unexplained MCI (Johnson 2013). The DTA of 18F‐florbetapir PET scans, as determined in this SR, has a variable sensitivity and low‐to‐moderate specificity to predict the progression from MCI to ADD, based on two studies with 448 participants at follow‐up, and there were only five participants to predict the progression from MCI to any form of dementia in one study.
Due to the aforementioned, and the methodological limitations of the included studies, it is not possible to recommend the routine use of 18F‐florbetapir in clinical practice. The 18F‐florbetapir biomarker is expensive, therefore it is important to clearly determine its DTA prior to it being recommended for clinical practice.
Implications for research.
The FDA and EMA have established the 18F‐florbetapir positivity criteria in order to use these in ADD patient evaluation and their use in MCI participants is accepted in research settings and clinical trials (Albert 2011). However, their use has also been proposed in clinical practice by the Nuclear Medicine Society and the Alzheimer's Association (Johnson 2013).
One problem found in the evaluation of the DTA of the 18F‐florbetapir PET scan is that many studies used different SUVR, visual assessment or both. This produces different accuracies for the tracer even in patients with ADD when compared with HC. Therefore, it is necessary to consider visual assessment as the option to interpret the 18F‐florbetapir PET scan, because this is the approach to the interpretation established by FDA and EMA (EMA 2013, FDA 2013)
On the other hand, clinical assessment in people with memory complaints is not always made with only one test; it could potentially use different tests like volumetric hyppocampal MRI, FDG‐PET, SPECT, CSF, and others. This may be sensible as neurodegenerative diseases are complex disorders with occasionally multiple and overlapping pathophysiological processes. Multitracer imaging may be helpful in combining metabolic, inflammation, or apoptosis markers with those labelling typical protein aggregations seen in the progression of MCI to Alzheimer’s disease. In future, various PET imaging modalities are needed to evaluate the usefulness of the various PET tracers as predictors of progression to Alzheimer’s disease in MCI studies with clinical follow‐up. There is a hypothesis that amyloid deposition is an early event in Alzheimer’s disease that reaches a relative plateau even at the MCI stage, while downstream biomarkers measure neuronal loss and dysfunction, and cognitive measures are more dynamic at the symptomatic disease stage (Jack 2013). Based on this hypothesis, the combination of structural imaging, functional imaging, and cognitive tests may be better predictors of when an individual will convert. However, there is a lack of studies with 18F‐florbetapir combined with other tests. The only study combining MRI, 18F‐FDG PET, and 18F‐florbetapir PET suggests a better accuracy when these tests are using a multimodal approach rather than a single modality (Xu 2016), and one study combining a positive18F‐florbetapir PET with a positive cerebrospinal fluid p‐tau showed a sensitivity of 92% and a specificity of 55% (including as a negative index test all those with discordant results) to predict the progression to ADD at two years of follow‐up (Pascoal 2017). However, when the analysis included the discordant results as a positive index test, the sensitivity increased to 97% and the specificity decreased to 18%.
Additionally, if we consider the hierarchical evidence needed for the level of efficacy of diagnostic imaging tests, we are currently in the second step of five according to Herscovitch (Herscovitch 2015): technical efficacy, diagnostic accuracy efficacy, diagnostic thinking efficacy, therapeutic impact, patient health outcomes, and, finally, societal efficacy. Therefore, we need further research about accuracy before progressing to the other steps with their specific studies before we can incorporate the 18F‐florbetapir PET scan into clinical practice.
Acknowledgements
Gabriel Martínez is a PhD candidate in Methodology of Biomedical Research and Public Health at the Department of Paediatrics, Obstetrics and Gynaecology and Preventive Medicine, Universitat Autònoma de Barcelona, Barcelona, Spain.
We are grateful to the authors of included and excluded studies who responded to our requests for additional information.
We thank the Cochrane Dementia and Cognitive Improvement Group (CDCIG) for strong support, especially Sue Marcus in finalizing the review.
We thank Anna Noel‐Storr, Information Specialist of the CDCIG, for her assistance with the design of the search strategy.
We thank Gerard Urrútia and Marta Roqué i Figuls for their contribution in the preparation of the protocol for the review (Martínez 2016)
We thank the peer reviewers for their many helpful suggestions.
Appendices
Appendix 1. Glossary
Aetiology: the cause, set of causes, or manner of causation of a disease or condition.
Amyloid beta (Aβ): an amyloid that is derived from a larger precursor protein and is the primary component of plaques characteristic of Alzheimer's disease.
Biomarker: measurable and quantifiable biological parameters (e.g., specific enzyme concentration, specific hormone concentration, presence of biological substances) which serve as indices for health‐ and physiology‐related assessments, such as disease risk, psychiatric disorders, environmental exposure and its effects, disease diagnosis; metabolic processes; etc.
Bolus: a single dose of a drug or other medicinal preparation given all at once.
Cingulate cortex: one of the convolutions on the medial surface of the cerebral hemispheres.
Cortical: the thin layer of grey matter on the surface of the cerebral hemispheres. It reaches its highest development in humans and is responsible for intellectual faculties and higher mental functions.
Epiphenomenon: A secondary effect or by‐product. A secondary symptom or pathology, occurring simultaneously with a disease or condition but not directly related to it.
Frontotemporal: relating to the frontal and the temporal cerebral lobes.
Histopathology: the study of changes in tissues caused by disease.
Hypothyroidism: a syndrome that results from abnormally low secretion of thyroid hormones from the thyroid gland.
Index test: the test under evaluation.
In vivo: (of processes) performed or taking place in a living organism.
Ligand: a molecule that binds to another molecule, used especially to refer to a small molecule that binds specifically to a larger molecule, e.g., an antigen binding to an antibody, a hormone or neurotransmitter binding to a receptor, or a substrate or allosteric effector binding to an enzyme.
Neuritic plaques: accumulations of extracellularly deposited amyloid fibrils within tissues. Is one of the hallmarks of Alzheimer's disease.
Neurofibrillary tangles: abnormal structures located in various parts of the brain and composed of dense arrays of paired helical filaments (neurofilaments and microtubules). Are aggregates of hyperphosphorylated tau protein that are most commonly known as a primary marker of Alzheimer's disease.
Parietal lobe: upper central part of the cerebral hemisphere. It is located anterior to the occipital lobe, and superior to the temporal lobes.
Positron: an extremely small piece of matter with a positive electrical charge, having the same mass as an electron.
Precuneus: is a part of the parietal lobe of the brain, lying on the medial surface of the cerebral hemisphere.
Prodromal: relating to prodrome; indicating an early stage of a disease.
Radionuclide (sometimes called a radioisotope or isotope): is a chemical which emits a type of radioactivity called gamma rays. The radioactivity can be detected by special scanners.
Reference standard: the best available method for establishing the presence or absence of the target condition.
Sensitivity: a measure of a test’s ability to correctly detect people with the disease. It is the proportion of diseased cases that are correctly identified by the test. It is calculated as follows: Sensitivity = Number with disease who have a positive test/Number with disease.
Specificity: a measure of a test’s ability to correctly identify people who do not have the disease. It is the proportion of people without the target disease who are correctly identified by the test. It is calculated as follows: Specificity = Number without disease who have a negative test/Number without disease.
Stilbene: organic compounds that contain 1,2‐diphenylethylene as a functional group.
Target condition: the disease or condition that the index test is expected to detect.
Temporal lobe: lower lateral part of the cerebral hemisphere responsible for auditory, olfactory, and semantic processing. It is located inferior to the lateral fissure and anterior to the occipital lobe.
Vascular: relating to, affecting, or consisting of a vessel or vessels, especially those which carry blood.
Appendix 2. Search strategy for 18F‐florbetapir Aβ scan
| Source | Search strategy |
| MEDLINE In‐process and other non‐indexed citations and Medline® 1946 to May 2017(Ovid SP) | 1. Florbetapir.ti,ab,nm. 2. (AMYViD or amyvid*).ti,ab,nm. 3. "florbetapir‐fluorine‐18".ti,ab,nm. 4. "18F‐AV‐45" or "(18)F‐AV‐45" or "[18F]AV‐45" or "[(18)F]AV‐45".ti,ab. 5. "[18F]Florbetapir".ti,ab,nm. 6. "florbetapir‐PET".ti,ab,nm. 7. or/1‐6 8. Fluorine Radioisotopes/du 9. Aniline Compounds/du 10. Ethylene Glycols/du 11. Stilbenes/du 12. Radioligand Assay/ 13. radioligand*.ti,ab. 14. or/8‐13 15. Alzheimer Disease/ri [Radionuclide Imaging] 16. Plaque, Amyloid/ri [Radionuclide Imaging] 17. or/15‐16 18. 14 and 17 19. 7 or 18 |
| Embase 1974 to May 2017 (Ovid SP) | 1. Florbetapir.ti,ab. 2. (AMYViD or amyvid*).ti,ab. 3. "florbetapir‐fluorine‐18".ti,ab. 4. "18F‐AV‐45" or "(18)F‐AV‐45" or "[18F]AV‐45" or "[(18)F]AV‐45".ti,ab. 5. "[18F]Florbetapir".ti,ab. 6. "florbetapir‐PET".ti,ab. 7. exp florbetapir f 18/ 8. or/1‐7 9. exp *radioligand/ 10. Alzheimer disease/ 11. Alzheimer*.ti,ab. 12. amyloid plaque/di [Diagnosis] 13. mild cognitive impairment/ 14. or/10‐13 15. 9 and 14 16. 8 or 15 |
| PsycINFO 1806 to May 2017 (Ovid SP) | 1. Florbetapir.ti,ab. 2. (AMYViD or amyvid*).ti,ab. 3. "florbetapir‐fluorine‐18".ti,ab. 4. "18F‐AV‐45" or "(18)F‐AV‐45" or "[18F]AV‐45" or "[(18)F]AV‐45".ti,ab. 5. "[18F]Florbetapir".ti,ab. 6. "florbetapir‐PET".ti,ab. 7. or/1‐6 |
| BIOSIS Citation Index (Thomson Reuters Web of Science) (1922 to May 2017) | Topic=(Florbetapir OR AMYViD OR amyvid* OR "florbetapir‐fluorine‐18" OR "18F‐AV‐45" OR "[18F]Florbetapir" OR "florbetapir‐PET") Timespan=All years. Databases=BCI |
| Web of Science Core Collection, including the Science Citation Index and the Conference Proceedings Citation Index (Thomson Reuters Web of Science) (1946 to May 2017) |
Topic=(Florbetapir OR AMYViD OR amyvid* OR "florbetapir‐fluorine‐18" OR "18F‐AV‐45" OR "[18F]Florbetapir" OR "florbetapir‐PET") Timespan=All years. Databases=SCI‐EXPANDED, SSCI, A&HCI, CPCI‐S, CPCI‐SSH, BKCI‐S, BKCI‐SSH, CCR‐EXPANDED, IC. |
| LILACS (BIREME) | Florbetapir OR AMYViD OR amyvid* OR "florbetapir‐fluorine‐18" OR "18F‐AV‐45" OR "[18F]Florbetapir" OR "florbetapir‐PET" [Words] |
| CINAHL (EBSCOhost) (1980 to May 2017) | S1 TX Florbetapir S2 TX AMYViD S3 TX amyvid* S4 TX "florbetapir‐fluorine‐18" S5 TX "18F‐AV‐45" S6 TX "[18F]Florbetapir" S7 TX "florbetapir‐PET" S8 S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S7 |
| ClinicalTrials.gov (www.clinicaltrials.gov) | Florbetapir OR AMYViD OR amyvid* OR "florbetapir‐fluorine‐18" OR "18F‐AV‐45" OR "[18F]Florbetapir" OR "florbetapir‐PET" |
| World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (http://apps.who.int/trialsearch) | Florbetapir OR AMYViD OR amyvid OR "florbetapir‐fluorine‐18" OR "18F‐AV‐45" OR "[18F]Florbetapir" OR "florbetapir‐PET" |
| ALOIS, the Cochrane Dementia & Cognitive Improvement Group’s specialized register of dementia studies (http://www.medicine.ox.ac.uk/alois/). | Imaging AND PET |
Appendix 3. Tables (2 × 2) cross‐relating index test results of the reference standards
Table 1. Progression from mild cognitive impairment (MCI) to Alzheimer’s disease dementia (ADD)
| Index test information | References standard information | |
| ADD present | ADD absent | |
| Index test‐positive | 18F‐florbetapir PET ligand for Aβ (+) who progress to ADD (TP) | 18F‐florbetapir PET ligand for Aβ (+) who remain MCI (FP) and 18F‐florbetapir PET ligand Aβ (+) who progress to non‐ADD (FP) |
| Index test‐negative | 18F‐florbetapir PET ligand for Aβ (‐) who progress to ADD (FN) | 18F‐florbetapir PET ligand for Aβ (‐) who remain MCI (TN) and 18F‐florbetapir PET ligand for Aβ (‐) who progress to non‐ADD (TN) |
ADD: Alzheimer's disease dementia FN: False negative FP: False positive
MCI: Mild cognitive impairment
PET: Positron emission tomography TN: True negative TP: True positive
Table 2. Progression from mild cognitive impairment (MCI) to non‐Alzheimer’s disease dementia (non‐ADD)
| Index test information | References standard information | |
| Non‐ADD present | Non‐ADD absent | |
| Index test‐positive | 18F‐florbetapir PET ligand for Aβ (+) who progress to non‐ADD (TP) |
18F‐florbetapir PET ligand for Aβ (+) who remain MCI (FP) and 18F‐florbetapir PET ligand for Aβ (+) who progress to ADD (FP) |
| Index test‐negative | 18F‐florbetapir PET ligand for Aβ (‐) who progress to non‐ADD (FN) |
18F‐florbetapir PET ligand for Aβ (‐) who remain MCI (TN) and 18F‐florbetapir PET ligand for Aβ (‐) who progress to ADD (TN) |
ADD: Alzheimer's disease dementia FN: False negative FP: False positive MCI: Mild cognitive impairment
PET: Positron emission tomography TN: True negative TP: True positive
Table 3. Progression from mild cognitive impairment (MCI) to any form of dementia
| Index test information | References standard information | |
| Any forms of dementia present | Dementia absent | |
| Index test‐positive | 18F‐florbetapir PET ligand for Aβ (+) who progress to any form of dementia (TP) | 18F‐florbetapir PET ligand for Aβ (+) who remain MCI (FP) |
| Index test‐negative | 18F‐florbetapir PET ligand for Aβ (‐) who progress to any form of dementia (FN) | 18F‐florbetapir PET ligand for Aβ (‐) who remain MCI (TN) |
FN: False negative FP: False positive
MCI: Mild cognitive impairment
PET: Positron emission tomography TN: True negative TP: True positive
Appendix 4. Assessment of methodological quality table: Quality Assessment of Diagnostic Accuracy Studies 2 (QUADAS‐2) tool
| Domain | Patient selection | Index test | Reference standard | Flow and timing |
| Description | Describe methods of patient selection: describe included participants (prior testing, presentation, intended use of index test and setting) | Describe the index test and how it was conducted and interpreted |
Describe the reference standard and how it was conducted and interpreted | Describe any patient who did not receive the index test(s) or reference standard, or both, or who were excluded from the 2 × 2 table (refer to flow diagram): describe the time interval and any interventions between index test(s) and reference standard |
| Signalling questions (yes/no/unclear) | Was a consecutive or random sample of patients enrolled? | Were the index test results interpreted without knowledge of the results of the reference standard? | Is the reference standard likely to correctly classify the target condition? | Was there an appropriate interval between index test(s) and reference standard? |
| Was a case‐control design avoided? | If a threshold was used, was it prespecified? | Were the reference standard results interpreted without knowledge of the results of the index test? | Did all patients receive a reference standard? | |
| Did the study avoid inappropriate exclusions? | Did all patients receive the same reference standard? | |||
| Were all patients included in the analysis? | ||||
| Risk of bias (high/low/unclear) | Could the selection of patients have introduced bias? | Could the conduct or interpretation of the index test have introduced bias? | Could the reference standard, its conduct, or its interpretation have introduced bias? | Could the patient flow have introduced bias? |
| Concerns regarding applicability (high/low/unclear) | Are there concerns that the included participants did not match the review question? | Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Are there concerns that the target condition as defined by the reference standard does not match the review question? |
Appendix 5. Anchoring statements for quality assessment of 18F‐florbetapir Aβ scan diagnostic studies
Table 4. Review question and inclusion criteria
| Category | Review question | Inclusion criteria |
| Patients | Participants with mild cognitive impairment (MCI), no dementia | Participants that fulfil the criteria for the clinical diagnosis of MCI at baseline |
| Index test | 18F‐florbetapir PET ligand for Aβ biomarker | 18F‐florbetapir PET ligand for Aβ biomarker |
| Target condition | Alzheimer’s disease dementia (ADD) (progression from MCI to ADD) Any other forms of dementia (progression from MCI to any other forms of dementia | ADD (progression from MCI to ADD) Any other forms of dementia (progression from MCI to any other forms of dementia) |
| Reference standard | NINCDS‐ADRDA; DSM; ICD; McKeith criteria; Lund criteria; International Behavioural Variant FTD Criteria Consortium; NINDS‐ARIEN criteria | NINCDS‐ADRDA; DSM; ICD; McKeith criteria; Lund criteria; International Behavioural Variant FTD Criteria Consortium; NINDS‐ARIEN criteria |
| Outcome | N/A | Data to construct a 2 × 2 table |
| Study design | N/A | Longitudinal cohort studies and nested case‐control studies if they incorporate a delayed verification design (case‐control nested in cohort studies) |
ADD: Alzheimer's disease dementia DSM: Diagnostic and Statistical Manual of Mental Disorders FTD: Frontotemporal dementia ICD: International Classification of Diseases MCI: Mild cognitive impairment NINCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association NINDS‐AIREN: National Institute of Neurological Disorders and Stroke and Association Internationale pour la Recherché et l'Enseignement en Neurosciences PET: Positron emission tomography
Anchoring statements for quality assessment 18F‐florbetapir PET ligand for Aβ diagnostic studies
We have provided some core anchoring statements for quality assessment in the diagnostic test accuracy (DTA) review of the 18F‐florbetapir PET ligand for Aβ biomarker in dementia. These statements are designed for use with the Quality Assessment of Diagnostic Accuracy Studies 2 (QUADAS‐2) tool and are based on the guidance for quality assessment of DTA reviews of Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) in dementia (Quinn 2014). In assessing individual items, the score of unclear should only be given if there is genuine uncertainty. In these situations, we contacted the relevant study teams for additional information. Whenever we scored one question as high risk of bias, we considered the study as having a high risk of bias.
Table 5. Anchoring statements to assist with the 'Risk of bias' assessment
| Question | Response and weighting | Explanation |
| Patient selection | ||
| Was the sampling method appropriate? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | Where sampling is used, the designs least likely to cause bias are consecutive sampling or random sampling. Sampling that is based on volunteers or selecting subjects from a clinic or research resource is prone to bias. |
| Was a case‐control or similar design avoided? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | Designs similar to case‐control that may introduce bias are those designs where the study team deliberately increase or decrease the proportion of subjects with the target condition, which may not be representative. Some case‐control methods may already be excluded if they mix subjects from various settings. |
| Are exclusion criteria described and appropriate? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | We automatically graded the study as unclear if the study authors did not detail exclusions (pending contact with study authors). Where a study details exclusions, we graded the study as 'low risk' if we considered exclusions to be appropriate. Certain exclusions common to many studies of dementia are: medical instability; terminal disease; alcohol/substance misuse; concomitant psychiatric diagnosis; other neurodegenerative conditions. Exclusions are not appropriate if they comprise ‘difficult to diagnose’ patients. We labelled post‐hoc and inappropriate exclusions as at 'high risk' of bias. |
| Index test | ||
| Was the 18F‐florbetapir PET ligand for Aβ biomarker's assessment/interpretation performed without knowledge of clinical dementia diagnosis? |
No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | Terms such as 'blinded' or 'independently and without knowledge of' are sufficient and full details of the blinding procedure are not required. Interpretation of the results of the index test may be influenced by knowledge of the results of the reference standard. If the index test is always interpreted prior to the reference standard, then the person interpreting the index test cannot be aware of the results of the reference standard and so this item could be rated as ‘yes’. For certain index tests, the result is objective and knowledge of the reference standard should not influence the result, e.g. level of protein in cerebrospinal fluid; in this instance, the quality assessment may be 'low risk' even if blinding was not achieved. |
| Was the 18F‐florbetapir PET ligand for Aβ biomarker's threshold prespecified? |
No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | For scales and biomarkers, there is often a reference point (in units or categories) above which subjects are classified as 'test‐positive'; this may be referred to as the threshold, clinical cut‐off, or dichotomisation point. A study is classified at high risk of bias if the study authors define the optimal cut‐off post‐hoc based on their own study data because selecting the threshold to maximise sensitivity and/or specificity may lead to overoptimistic measures of test performance. Certain papers may use an alternative methodology for analysis that does not use thresholds and these papers should be classified as not applicable. |
| Was the 18F‐florbetapir PET ligand for Aβ scan interpretation done by a trained reader physician? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | If a trained reader physician performed the scan interpretation, we scored this item as ’yes’. If no definition of trained reader was done, we scored this item as ’unclear’. If a nontrained reader physician performed the scan interpretation, we scored this item as ’no’. |
| Did the study provide a clear definition of what was considered to be a 18F‐florbetapir PET ligand for Aβ biomarker’s positive result? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | If the study clearly stated the definition of a positive result (e.g. SUV), we scored this item as ’yes’. If the study did not give a definition of what it considered a positive result or the definition of a positive result varied between the participants, we scored this item as ’no’. If the study gave insufficient information to permit judgement, we scored the item as ’unclear’. |
| Reference standard | ||
| Is the assessment used for clinical diagnosis of dementia acceptable? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | Commonly used international criteria to assist with clinical diagnosis of dementia included those detailed in DSM‐IV and ICD‐10. Criteria specific to dementia subtypes included but were not limited to NINCDS‐ADRDA criteria for Alzheimer’s dementia; McKeith criteria for Lewy body dementia; Lund criteria and International Behavioural Variant FTD Criteria Consortium for frontotemporal dementia; and the NINDS‐AIREN criteria for vascular dementia. Where the criteria used for assessment were not familiar to the review authors or the Cochrane Dementia and Cognitive Improvement group (‘unclear’), we classified this item as 'high risk of bias'. |
| Were clinical assessments for dementia performed without knowledge of the 18F‐florbetapir PET ligand for Aβ biomarker? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | Terms such as 'blinded' or 'independently and without knowledge of' were sufficient and full details of the blinding procedure were not required. Interpretation of the results of the reference standard may be influenced by knowledge of the results of the index test. |
| Patient flow | ||
| Was there an appropriate interval between 18F‐florbetapir PET ligand for Aβ biomarker and clinical dementia assessment? |
No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | As we test the accuracy of the 18F‐florbetapir PET ligand for Aβ biomarker for MCI progression to dementia, there will always be a delay between the index test and the reference standard assessments. The time between the reference standard and the index test will influence the accuracy (Geslani 2005; Okello 2007; Visser 2006), and therefore we noted time as a separate variable (both within and between studies) and will test its influence on the diagnostic accuracy. We have set a minimum mean time to follow‐up assessment of 1 year. If more than 16% of subjects have assessment for MCI progression before nine months, this item was scored ‘no’. |
| Did all subjects get the same assessment for dementia regardless 18F‐florbetapir PET ligand for Aβ biomarker? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | There may be scenarios where participants who score 'test‐positive' on the index test have a more detailed assessment. Where dementia assessment differs between participants, this should be classified as high risk of bias. |
| Were all patients who received 18F‐florbetapir PET ligand for Aβ biomarker’s assessment included in the final analysis? |
No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | If the number of patients enrolled differs from the number of patients included in the 2 × 2 table, then there is the potential for bias. If patients lost to dropouts differ systematically from those who remain, then estimates of test performance may differ. If there are dropouts, these should be accounted for; a maximum proportion of dropouts for a study to remain at low risk of bias has been specified as 20%. |
| Were missing 18F‐florbetapir PET ligand for Aβ biomarker's results reported? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | Where missing or uninterpretable results are reported, and if there is substantial attrition (we have set an arbitrary value of 50% missing data), we will score this as ‘no’. If the study did not report these results, we scored this as ‘unclear’ and we contacted the study authors. |
| Was the study with 18F‐florbetapir PET ligand for Aβ biomarker free of commercial funding? | No = high risk of bias Yes = low risk of bias Unclear = unclear risk of bias | If the funding source is clearly stated and is not commercial, this should be scored as ‘no’. If the funding source is clearly stated and is commercial, this should be scored as ’yes ’. If not enough information is given to assess whether the funding source is commercial, the scored is ’unclear’. |
| Anchoring statements to assist with assessment for applicability | ||
| Question | Explanation | |
| Were included patients representative of the general population of interest? | The included patients should match the intended population as described in the review question. The review authors should consider population in terms of symptoms; pretesting; potential disease prevalence; setting. If there is a clear ground for suspecting an unrepresentative spectrum, the item should be rated poor applicability. | |
| Index test | ||
| Were sufficient data on 18F‐florbetapir PET ligand for Aβ biomarker’s application given for the test to be repeated in an independent study? | Variation in technology, test execution, and test interpretation may affect estimate of accuracy. In addition, the background, and training/expertise of the assessor should be reported and taken in consideration. If 18F‐florbetapir PET ligand for Aβ biomarker was not performed consistently, this item should be rated poor applicability. | |
| Reference standard | ||
| Was clinical diagnosis of dementia made in a manner similar to current clinical practice? | For many reviews, inclusion criteria and 'Risk of bias' assessments will already have assessed the dementia diagnosis. For certain reviews, an applicability statement relating to the reference standard may not be applicable. There is the possibility that a form of dementia assessment, although valid, may diagnose a far larger proportion of people with disease than usual clinical practice. In this instance, the item should be rated poor applicability. | |
DSM: Diagnostic and Statistical Manual of Mental Disorders FTD: Frontotemporal dementia ICD: International Classification of Diseases MCI: Mild cognitive impairment NINCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association NINDS‐AIREN: National Institute of Neurological Disorders and Stroke and Association Internationale pour la Recherché et l'Enseignement en Neurosciences PET: Positron emission tomography
Data
Presented below are all the data for all of the tests entered into the review.
Tests. Data tables by test.
| Test | No. of studies | No. of participants |
|---|---|---|
| 1 MCI to ADD by visual assessment from 2 to less than 4 years of follow‐up | 1 | 47 |
| 2 MCI to ADD by visual assessment from 1 to less than 2 years follow‐up | 1 | 401 |
| 3 MCI to ADD by SUVR at 1 to less than 2 years follow‐up | 1 | 401 |
| 4 MCI to any form of dementia | 1 | 5 |
3. Test.

MCI to ADD by SUVR at 1 to less than 2 years follow‐up.
4. Test.

MCI to any form of dementia.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Doraiswamy 2014.
| Study characteristics | |||
| Patient sampling |
|
||
| Patient characteristics and setting |
|
||
| Index tests |
After a training session, three nuclear medicine physicians with no access to clinical information, independently rated each PET image for amyloid burden based on successive levels of florbetapir retention from 0 to 4 as follows: (0) None: predominantly white matter tracer retention with no appreciable cortical gray matter retention above cerebellar grey matter levels; (1) Low: evidence of increased tracer retention above cerebellar grey levels in 1 or 2 cortical grey regions; (2) Low‐moderate: either (a) predominantly white matter pattern, but at least 2 cortical regions with increased retention relative to cerebellar grey, or (b) predominantly a cortical gray matter pattern, with most cortical areas mildly positive relative to cerebellum; (3) Moderate‐high: specific cortical retention generally greater than or equal to white matter retention and at least one cortical area with greatly increased retention relative to cerebellar grey; (4) High: Specific cortical uptake greater than or equal to white matter background and multiple cortical areas with greatly increased retention relative to cerebellar grey.
The visual reads were used to classify each data set as either visually positive for Aβ or visually negative for Aβ Visual rating scores of 2 to 4 were considered positive and 0 to 1 were considered negative.
|
||
| Target condition and reference standard(s) |
|
||
| Flow and timing |
|
||
| Comparative | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Unclear | Low | ||
| DOMAIN 2: Index Test All tests | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Was the PET scan interpretation done by a trained reader physician? | Yes | ||
| Was there a clear definition of a positive result? | Yes | ||
| Low | Low | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Unclear | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Unclear | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Unclear | ||
| Were all patients included in the analysis? | Yes | ||
| Was the study free of commercial funding? | No | ||
| High | |||
Kawas 2013.
| Study characteristics | |||
| Patient sampling |
|
||
| Patient characteristics and setting |
Nine of the participants were women, two of them were Aβ (+), and two of four men were Aβ (+) at baseline. APOE ϵ4 carrier: not reported MMSE: the mean MMSE was 28 (range 24 to 30); for those considered as in the Aβ (+) group, the mean was 26.5 (range 24 to 29) and 28 (range 25 to 30) for those in the Aβ (‐) group. Years of education: seven participants were reported having studied after high school: two of them were Aβ (+) and five were Aβ (‐); for those six having studied at high school or with less education, two were Aβ (+) and four were Aβ (‐), respectively.
|
||
| Index tests |
After a training session, three nuclear medicine physicians with no access to clinical information, independently rated each PET image for amyloid burden based on successive levels of florbetapir retention from from 0 (no amyloid) to 4 (high levels of cortical amyloid). The median of the three visual scores was used to dichotomize participants into Aβ (‐) (score, 0 to 1 point) and Aβ (+) (score, 2 to 4 points). |
||
| Target condition and reference standard(s) |
|
||
| Flow and timing |
|
||
| Comparative | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Unclear | Low | ||
| DOMAIN 2: Index Test All tests | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Was the PET scan interpretation done by a trained reader physician? | Yes | ||
| Was there a clear definition of a positive result? | Unclear | ||
| Low | Low | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Unclear | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Was the study free of commercial funding? | No | ||
| High | |||
Schreiber 2015.
| Study characteristics | |||
| Patient sampling |
|
||
| Patient characteristics and setting |
|
||
| Index tests |
The reader was trained using an online electronic training tool produced by the company who produced the tracer, and the reader was blinded to all clinical data and any other imaging test of each participant.
The threshold used was a SUVR > 1.11 determined at baseline.(Landau 2012, Landau 2013). |
||
| Target condition and reference standard(s) |
Unclear whether clinicians conducting follow‐up were aware of the ¹⁸F‐florbetapir PET scan results. |
||
| Flow and timing |
Number included in analysis: MCI
|
||
| Comparative | |||
| Notes | Dr Schreiber kindly sent the ADNI identification code for each MCI participant (mail received 04/07/2017). | ||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Unclear | Low | ||
| DOMAIN 2: Index Test All tests | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Was the PET scan interpretation done by a trained reader physician? | Yes | ||
| Was there a clear definition of a positive result? | Yes | ||
| Low | Low | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Unclear | Unclear | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Was the study free of commercial funding? | Yes | ||
| Low | |||
Aβ: Amyloid Beta ADD: Alzheimer's disease dementia ADNI: Alzheimer's Disease Neuroimaging Initiative APOE ϵ4: Apolipoprotein E4 CDR: Clinical dementia rating CIND: Cognitive impairment not dementia CT: Computed tomography DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders (4th ed.) FN: False negative FP: False positive MBq: Megabecquerel MCI: Mild cognitive impairment mCi: Millicurie MMSE: Mini‐mental state examination MPRAGE: Magnetization‐Prepared Rapid Gradient‐Echo NINCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association PET: Positron emission tomography ROI: Region of interest SUVR: Standardised uptake value ratio
T: Tesla TN: True negative TP: True positive
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Altomare 2016 | MCI diagnosis at baseline was not made with any of our accepted definitions by protocol for MCI participants. Dr Altomare kindly responded to some questions regarding the method of his study (mail received 16/06/2017). |
| Apostolova 2016 | Not having data for constructing a 2 x 2 table. The study was focused on the development of neuropsychiatric symptoms and not on Alzheimer's disease or dementia progression. |
| Brendel 2014 | Not having data for constructing a 2 x 2 table. The study was focused on longitudinal quantitative analyses of 18F‐florbetapir PET and their association with progression of dementia. |
| Brendel 2015 | Not having data for constructing a 2 x 2 table. The study was focused on testing the effects of different reference regions and atrophy‐based partial volume effects on the discriminatory power and longitudinal performance of amyloid PET. |
| Cheewakriengkrai 2014 | Not having data for constructing a 2 x 2 table. The study was focused on the relationship between regional distributions of brain fibrillar amyloid deposition, neurodegenerative biomarkers in CSF (CSF Aβ 1‐42, t‐tau, p‐tau) and cognitive function (ADAS‐cog) at 24 months follow‐up. |
| Chen 2015a | Not having data for constructing a 2 x 2 table. The study compared the power of template‐based cerebellar, pontine, and cerebral white matter reference regions to track 24‐month florbetapir standardized uptake value (SUV) ratio (SUVR) changes; and to relate those changes to 24‐month clinical declines |
| Chen 2015b | Not having data for constructing a 2 x 2 table. The study was focused in the diagnostic potential of FDG PET, florbetapir, PiB and CSF biomarkers in monitoring the progression from mild cognitive impairment (MCI) to Alzheimer’s disease (ADD) and cognitively normal (NC) to MCI in a longitudinal study |
| Chincarini 2015 | Not having data for constructing a 2 x 2 table. The study was focused on examining different approaches to amyloid‐PET quantification and a longitudinal analyses of Aβ deposition. |
| Chincarini 2016 | The study focused on the evaluation of brain amyloidosis (ELBA) with a new method on imaging of the 18F‐florbetapir PET scan. We did not include this study because we preferred to include the Schreiber study for the following reasons:
|
| Durkanova 2015 | Not having data for constructing a 2 x 2 table. The study was focused in evaluate five different test strategies for integrating use of florbetapir and FDG PET information to predict rates of cognitive and functional decline over 2 years. |
| Fan 2015 | Not having data for constructing a 2 x 2 table. The study was focused on investigating whether different translocator protein genotypes influenced cognitive function, amyloid load, and disease progression over time. |
| Greenia 2014 | Not having data for constructing a 2 x 2 table. The study was focused on evaluating the 18F‐florbetapir PET and the relationship with cognitive decline in the oldest‐old. |
| Hochstetler 2014 | Not having data for constructing a 2 x 2 table. The study was focused on trying to define trajectories of cognitive and functional decline, and characteristics associated with distinct trajectories, using Growth Mixture Modeling. |
| Joshi 2014 | Not having data for constructing a 2 x 2 table. The study was focused on the estimation of longitudinal change in Aβ burden over 2 years. |
| Klein 2015 | Not having data for constructing a 2 x 2 table. The study was focused on the evaluation of native space compared to SPM template methods and a variety of possible SUVR reference regions with highest longitudinal change in the SUVR at 24 months. |
| Landau 2014 | Not having data for constructing a 2 x 2 table. The study was focused on the 18F‐florbetapir PET longitudinal evaluation in cognitively normal, MCI, and ADD participants, examining characteristics of normal individuals with subthreshold florbetapir retention and the influence of reference region selection on estimated trajectories across the entire range of amyloid measurements. |
| Landau 2016 | This study was focused on comparing participants with amyloid beta negative MCI and participants with ADD enrolled in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) with their Aβ amyloid positive counterparts on a number of clinical, neuropsychological, and biomarker characteristics with an average available follow‐up time for longitudinal cognitive measurements of 1.4 + 0.8 years. The conversion rate in those MCI participants with PET negative was 11% and the conversion in those with PET positive was 45%. We did not include this study, and we preferred Schreiber 2015 to be included for the following reasons:
|
| Lee 2015 | Not having data for constructing a 2 x 2 table. This study was focused in the correlation between florbetapir and FDG PET and cognition measured by MMSE at follow‐up. |
| Lim 2014 | Not having data for constructing a 2 x 2 table. This study was focused on evaluating the florbetapir status at baseline and different cognitive composite measures at 36 months. |
| Manitsirikul 2015 | Not having data for constructing a 2 x 2 table. The study was focused on the relationship between regional distributions of brain fibrillar amyloid deposition, neurodegenerative biomarkers in brain (FDG) and CSF (tau), brain structural change, and cognitive function at 24‐month follow‐up. |
| Margolin 2013 | Not having data for constructing a 2 x 2 table. The study was focused on evaluating the 18F‐florbetapir PET and the relationship with cognitive decline at follow‐up. |
| Mathotaarachchi 2015 | Not having data for constructing a 2 x 2 table. The study was focused on the regional effects of amyloid retention measured by the 18F‐florbetapir PET scan on the rate of hypometabolism measured by FDG PET scan over the follow‐up. |
| Mattsson 2014a | This study was focused on comparing the diagnostic test accuracy with CSF Aβ42 and the 18F‐florbetapir PET scan in three different groups, healthy controls, Alzheimer's disease dementia, and MCI (progressive vs stable MCI) participants. We did not include this study, as we preferred Schreiber 2015 to be included for the following reasons:
|
| Mattsson 2014b | Not having data for constructing a 2 x 2 table. This study was focused in determine the extent to which CSF and 18F‐florbetapir PET contribute independent diagnostic information in AD studies, and to determine the nature and degree of pathology in discordantly classified individuals in healthy controls, ADD patients, and MCI participants. |
| Mattsson 2015a | Not having data for constructing a 2 x 2 table. The study was focused on testing if CSF and amyloid beta PET scan biomarkers were independently related to other Alzheimer's disease markers, and to examine individuals who were discordantly classified by these two biomarker modalities with a follow‐up for up to three years. |
| Mattsson 2015b | Not having data for constructing a 2 x 2 table. The study was focused on relationships in a large number of brain regions in MCI participants with cognitive evaluations for up to three years with Logical Memory delayed recall and Rey Auditory Verbal Learning Test delayed recall. |
| Ming 2015 | Not having data for constructing a 2 x 2 table. The study was focused on MCI participants and 18F‐florbetapir at baseline and follow‐up for up to three years with cognitive evaluations with MMSE, ADAS11 and CDR sum of boxes. |
| Mohades 2014 | Not having data for constructing a 2 x 2 table. The study was focused on comparing neurodegeneration in 18F‐florbetapir accumulators and nonaccumulators based on a 24‐month assessment. |
| Morbelli 2015 | Not having data for constructing a 2 x 2 table. The study was focused on MCI participants that had longitudinal evaluation with the 18F‐florbetapir PET scan over two years and different methods to establish the PET positivity. |
| Pascoal 2016 | Not having data for constructing a 2 x 2 table. The study was focused on neuropsychological and clinical decline in participants with MCI and if they were associated with brain amyloid‐beta deposition and tau hyperphosphorylation. |
| Pascoal 2017 | The study was focused on amnestic MCI individuals and whether the synergism between Aβ aggregation and tau hyperphosphorylation could determine the progression from amnestic MCI to ADD dementia. We did not include this study because we preferred the Schreiber study to be included for the following reasons:
Dr Pascoal kindly responded to some questions regarding the method of his study and provided the ADNI identification code of the participants (mail received 16/06/2017). |
| Pontecorvo 2011 | Not having data for constructing a 2 x 2 table. The study was focused on the evaluation of the correlation of florbetapir SUVR with cognitive change from baseline to month 24 in MCI and cognitively normal participants, PET PiB, and CSF amyloid and tau levels. |
| Risacher 2014 | Not having data for constructing a 2 x 2 table. The study was focused on the comparative assessment of two‐year change in amyloid deposition, glucose metabolism, and hippocampal atrophy in healthy controls, MCI and ADD participants. |
| Shokouhi 2016 | Not having data for constructing a 2 x 2 table. The study was focused on evaluating the effect of reference tissue normalization in a test–retest 18F‐florbetapir SUVR study using different reference regions and evaluating the correlation between 18F‐florbetapir PET and concurrent CSF Aβ1–42 levels in a MCI cohort over the course of 2 years. |
| Siderowf 2013 | Not having data for constructing a 2 x 2 table. The study was focused on evaluating cognitive decline measured by ADAS‐cog in participants with negative and positive 18F‐florbetapir PET scan imaging with a clinical follow‐up of 18 months. |
| Teipel 2015 | Not having data for constructing a 2 x 2 table. The study was focused on comparing penalized regression analysis, with more classical unregularised regression models in respect to predicting conversion from MCI to ADD in 127 MCI subjects who had a clinical follow‐up between 6 and 31 months. |
| Toledo 2015 | Not having data for constructing a 2 x 2 table. The study was focused on determining the association between CSF and PET amyloid biomarkers (cross‐sectional and longitudinal measures) and comparing the cut‐offs for these measures. |
| Wisse 2015 | Not having data for constructing a 2 x 2 table. The study was focused on characterising MCI participants separated into four groups according to their abnormal amyloid‐beta 42 levels and abnormal hippocampal volume or hypometabolism using fluorodeoxyglucose PET and the conversion rate at 24 months. |
| Xu 2016 | The study was focused on exploring the contribution of different neuroimaging modalities in their predictive power and characterised the sensitive biomarkers from each modality. We did not include this study, as we preferred the Schreiber study to be included for the following reasons:
|
Aβ: Amyloid Beta ADAS11: Alzheimer's disease assessment scale‐11 ADAScog: Alzheimer's Disease Assessment Scale‐Cognitive subscale ADD: Alzheimer's disease dementia ADNI: Alzheimer's Disease Neuroimaging Initiative CDR: Clinical dementia rating CSF: Cerebrospinal fluid ELBA: Evaluation of brain amyloidosis FDG: Fluorodeoxyglucose MCI: Mild cognitive impairment MMSE: Mini‐mental state examination PET: Positron emission tomography PiB: Pittsburgh compound B SPM: statistical parametric mapping SUV: Standardised uptake value SUVR: Standardised uptake value ratio
Characteristics of ongoing studies [ordered by study ID]
JPRN‐UMIN000019926.
| Trial name or title | Clinical and neuroimaging study on preclinical Alzheimer's disease |
| Target condition and reference standard(s) | Estimation of progression rate at 36 months of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir, PET PiB, 18F‐flutemetamol |
| Starting date | 2016 |
| Contact information | Hiroshi Mori mori@med.osaka‐cu.ac.jp |
| Notes |
NCT01325259.
| Trial name or title | FluoroAv45 Imaging Research‐in Alzheimer's Disease (FAIR‐AD) |
| Target condition and reference standard(s) | Cogitive decline after 2 years of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir |
| Starting date | 2009 |
| Contact information | vincent.camus@univ‐tours.fr |
| Notes |
NCT01554202.
| Trial name or title | Multi‐modal Neuroimaging in Alzheimer's Disease (IMAP) |
| Target condition and reference standard(s) | Cognitive decline over three years of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir |
| Starting date | 2008 |
| Contact information | Vincent de La Sayette, University Hospital, Caen |
| Notes |
NCT01638949.
| Trial name or title | Multi‐modal Neuroimaging in Alzheimer's Disease (IMAP+) |
| Target condition and reference standard(s) | Cognitive decline over three years of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir |
| Starting date | 2012 |
| Contact information | Vincent de La Sayette, University Hospital, Caen |
| Notes |
NCT01687153.
| Trial name or title | A Study of Brain Aging in Vietnam War Veterans (DOD‐ADNI) |
| Target condition and reference standard(s) | Cognitive decline over one year of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir |
| Starting date | 2012 |
| Contact information | Michael W. Weiner, University of California, San Francisco Paul Aisen, USC Alzheimer's Therapeutic Research Institute (ATRI) Ronald Petersen, Mayo Clinic |
| Notes |
NCT01746706.
| Trial name or title | Can the Assessment of the Subhippocampal Region Contribute to the Detection of Early Diagnosis of Alzheimer's Disease? A Validation Study Using PET With florbetapir (AV‐45) |
| Target condition and reference standard(s) | Cognitive decline over two years of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir |
| Starting date | 2011 |
| Contact information | Bernard Belaiguesa, Assistance Publique Hopitaux De Marseille |
| Notes |
NCT02164643.
| Trial name or title | Longitudinal Study of Brain Amyloid imaGing in MEMENTO (MEMENTOAmyGing) |
| Target condition and reference standard(s) | Cognitive decline over two years of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir and 18F‐flutemetamol |
| Starting date | 2014 |
| Contact information | Genevieve Chene, CIC‐EC7 ‐ ISPED ‐ CHU de Bodeaux |
| Notes |
NCT02330510.
| Trial name or title | Amyloid and Glucose PET Imaging in Alzheimer and Vascular Cognitive Impairment Patients With Significant White Matter Disease (MITNEC C6) |
| Target condition and reference standard(s) | Cognitive decline over two years of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir |
| Starting date | 2014 |
| Contact information | Maryam Niapour, maryam.niapour@sunnybrook.ca Christopher JM Scott, christopher.scott@sri.utoronto.ca |
| Notes |
NCT02343757.
| Trial name or title | Alzheimer's Disease Imaging With PET/MRI ‐ Beta‐amyloid |
| Target condition and reference standard(s) | Assessing the diagnosis of a participant at one year of follow‐up, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir |
| Starting date | 2014 |
| Contact information | James O'Donnell, Jamesk.ODonnell@UHhospitals.org |
| Notes |
NCT02854033.
| Trial name or title | Alzheimer's Disease Neuroimaging Initiative 3 (ADNI3) Protocol |
| Target condition and reference standard(s) | Rate of progression to MCI or dementia due to ADD, reference standard not specified |
| Index and comparator tests | 18F‐florbetapir and 18F‐florbetaben |
| Starting date | 2016 |
| Contact information | Paul Aisen, Director, Alzheimer's Therapeutic Research Institute, University of Southern California |
| Notes |
ADD:Alzheimer's disease dementia MCI: Mild cognitive impairment PET: Positron emission tomography PiB: Pittsburgh Compound B
Contributions of authors
Gabriel Martínez, Robin WM Vernooij, and Paulina Fuentes Padilla: contributed to the conception, design, and draft of the protocol; overall responsibility of study selection; data extraction; contact of the authors; draft of discussion; and authors’ conclusion sections.
Leon Flicker: contributed to the conception, and designed and reviewed the draft protocol and final manuscript.
Xavier Bonfill Cosp: reviewed the draft protocol and final manuscript.
Javier Zamora: designed and drafted the protocol, performed statistical analyses, updated the statistical methods section and final manuscript.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research (NIHR), UK.
This review was supported by the NIHR, via Cochrane Infrastructure funding to the Cochrane Dementia and Cognitive Improvement group. The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, the NIHR, the NHS, or the Department of Health
Declarations of interest
Gabriel Martínez has no known conflicts of interest. Leon Flicker has no known conflicts of interest. Robin WM Vernooij has no known conflicts of interest. Paulina Fuentes Padilla has no known conflicts of interest. Javier Zamora has no known conflicts of interest. Xavier Bonfill Cosp has no known conflicts of interest.
New
References
References to studies included in this review
Doraiswamy 2014 {published data only}
- Doraiswamy PM, Clark C, Sperling R, Reiman E, Pontecorvo M, Sabbagh M, et al. Prognostic significance of florbetapir F18 PET imaging in MCI and mormal elderly: final results from a longitudinal multicenter trial. Alzheimer's & Dementia 2011;7 Suppl(4):S108. [Google Scholar]
- Doraiswamy PM, Sperling RA, Coleman RE, Johnson KA, Reiman EM, Davis MD, et al. Amyloid‐β assessed by florbetapir F 18 PET and 18‐month cognitive decline: a multicenter study. Neurology 2012;79(16):1636‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doraiswamy PM, Sperling RA, Johnson K, Reiman EM, Wong TZ, Sabbagh MN, et al. Florbetapir F 18 amyloid PET and 36‐month cognitive decline: a prospective multicenter study. Molecular Psychiatry 2014;19(9):1044‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Sperling RA, Gidicsin CM, Carmasin JS, Maye JE, Coleman RE, et al. Florbetapir (F18‐AV‐45) PET to assess amyloid burden in Alzheimer's disease dementia, mild cognitive impairment, and normal aging. Alzheimer's & Dementia 2013;9 Suppl(5):S72‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00857506. Observational study of cognitive outcomes for subjects who have had prior PET amyloid imaging With Florbetapir F 18 (18F‐AV‐45). https://clinicaltrials.gov/show/NCT00857506 (first received 6 March 2009).
Kawas 2013 {published data only}
- Kawas CH, Greenia DE, Bullain SS, Clark CM, Pontecorvo MJ, Joshi AD, et al. Amyloid imaging and cognitive decline in nondemented oldest‐old: the 90+ study. Alzheimer's & Dementia 2013;9(2):199‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schreiber 2015 {published data only}
- ADNI 2 PET Technical Procedures Manual AV‐45 (Florbetapir F 18) & FDG. adni.loni.usc.edu/wp‐content/uploads/2010/05/ADNI2_PET_Tech_Manual_0142011.pdf (accessed prior to 12 October 2017).
- ADNI‐GOPET Technical Procedures Manual AV‐45 & FDG. adni.loni.usc.edu/wp‐content/uploads/2010/05/ADNIGO_PET_Tech_Manual_01142011.pdf (accessed prior to 12 October 2017).
- Alzheimer’s Disease Neuroimaging Initiative 2 (ADNI2). www.adni‐info.org/Scientists/doc/ADNI2_Procedures_Manual_20130624.pdf (accessed prior to 12 October 2017).
- Alzheimer’s Disease Neuroimaging Initiative Grand Opportunity (ADNI‐GO). www.adni‐info.org/Scientists/doc/ADNI_GO_Procedures_Manual_06102011.pdf (accessed prior to 12 October 2017).
- NCT01078636. Alzheimer's disease neuroimaging initiative grand opportunity (ADNI‐GO). clinicaltrials.gov/show/NCT01078636 (first received 2 March 2010).
- NCT01231971. Alzheimer's disease neuroimaging initiative 2 (ADNI2). clinicaltrials.gov/show/NCT01231971 (first received 1 November 2010).
- Schreiber S, Landau SM, Fero A, Schreiber F, Jagust WJ, Alzheimer’s Disease Neuroimaging Initiative. Comparison of visual and quantitative Florbetapir F 18 positron emission tomography analysis in predicting mild cognitive impairment outcomes. JAMA Neurology 2015;72(10):1183‐90. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Altomare 2016 {published data only}
- Altomare D, Festari C, Ferrari C, Muscio C, Padovani A, Frisoni GB, et al. Brain amyloidosis and cognitive decline in MCI: 12‐month follow‐up. Alzheimer's & Dementia 2016;12(7 Supplement):P16‐P17. [Google Scholar]
Apostolova 2016 {published data only}
- Apostolova L, Goukasian N, Do T, Grotts J, Ringman J, Elashoff D. Effect of brain amyloidosis on the emergence of neuropsychiatric behaviors in MCI over time. Neurology 2016;86 Suppl 16:P2.232. [Google Scholar]
Brendel 2014 {published data only}
- Brendel M, Hoegenauer M, Delker A, Bartenstein P, Rominger A. Longitudinal amyloid PET in mild cognitive impaired patients. Journal of Nuclear Medicine 2014;55 Suppl 1:193. [Google Scholar]
Brendel 2015 {published data only}
- Brendel M, Högenauer M, Delker A, Sauerbeck J, Bartenstein P, Seibyl J, et al. Improved longitudinal [(18)F]‐AV45 amyloid PET by white matter reference and VOI‐based partial volume effect correction. NeuroImage 2015;108:450‐9. [DOI] [PubMed] [Google Scholar]
Cheewakriengkrai 2014 {published data only}
- Cheewakriengkrai L, Manitsirikul S, Mohades S, Wang S, Shin M, Benedet AL, et al. Neurodegeneration associated with longitudinal changes of abeta1‐42 and fibrillary amyloid. Alzheimer's & Dementia 2014;10 Suppl(4):839‐40. [Google Scholar]
Chen 2015a {published data only}
- Chen K, Roontiva A, Thiyyagura P, Lee W, Liu X, Ayutyanont N, et al. Improved power for characterizing longitudinal amyloid‐beta PET changes and evaluating amyloid‐modifying treatments with a cerebral white matter reference region. Journal of Nuclear Medicine 2015;56(4):560‐66. [DOI] [PubMed] [Google Scholar]
Chen 2015b {published data only}
- Chen X, Wang R, Gao R, Cao H, Wong D, Zhou Y. Evaluation of the diagnostic value of FDG and amyloid PET imaging with CSF biomarkers in monitoring the progression in Alzheimer's disease. Journal of Nuclear Medicine 2015;56 Suppl 3:1569. [Google Scholar]
Chincarini 2015 {published data only}
- Chincarini A, Sensi F, Guerra UP, Morbelli S, Bossert I, Rei L, et al. Amyloid‐PET quantification: methods and rationale. Clinical and Translational Imaging 2015;3 Suppl(1):S20. [Google Scholar]
Chincarini 2016 {published data only}
- Chincarini A, Sensi F, Rei L, Bossert I, Morbelli S, Guerra UP, et al. Standardized uptake value ratio‐independent evaluation of brain amyloidosis. Journal of Alzheimer's Disease 2016;54(4):1437‐57. [DOI] [PubMed] [Google Scholar]
Durkanova 2015 {published data only}
- Durcanova B, Diaz‐Aguilar D, Parker E, Lee J, Yi L, Silverman D. Optimal strategies for using amyloid imaging and FDG PET in prognostic evaluation of mild cognitive impairment (MCI). Journal of Nuclear Medicine 2015;56 Suppl 3:192. [Google Scholar]
Fan 2015 {published data only}
- Fan Z, Harold D, Pasqualetti G, Williams J, Brooks DJ, Edison P. Can studies of neuroinflammation in a TSPO genetic subgroup (HAB or MAB) be applied to the entire AD cohort?. Journal of Nuclear Medicine 2015;56(5):707‐13. [DOI] [PubMed] [Google Scholar]
Greenia 2014 {published data only}
- Greenia D, Kawas C, Caunca M, Bullain S, Corrada M. PET amyloid imaging with florbetapir predicts cognitive decline in the oldest‐old. Neurology 2014;82 Suppl 10:P4.011. [Google Scholar]
Hochstetler 2014 {published data only}
- Hochstetler H, Wang S, Yu P, Trzepacz PT, Case M, Henley D, et al. Empirically defining trajectories of late‐life cognitive and functional decline. Alzheimer's & Dementia 2014;10 Suppl(4):687‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Joshi 2014 {published data only}
- Joshi A, Pontecorvo M, Navitsky MA, Kennedy IA, Mintun M, Devous MD. Measuring change in beta‐amyloid burden over time using florbetapir‐PET and a subcortical white matter reference region. Alzheimer's & Dementia 2014;10 Suppl(4):902. [Google Scholar]
Klein 2015 {published data only}
- Klein G, Sampat M, Staewen D, Scott D, Suhy J. Comparison of SUVR methods and reference regions in amyloid PET. Journal of Nuclear Medicine 2015;56 Suppl 3:1741. [Google Scholar]
Landau 2014 {published data only}
- Landau S, Fero A, Baker S, Jagust W. Modeling longitudian Florbetapir change across the disease spectrum. Alzheimer's & Dementia 2014;10 Suppl(4):P7. [Google Scholar]
Landau 2016 {published data only}
- Landau SM, Horng A, Fero A, Jagust WJ. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology 2016;86(15):1377‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2015 {published data only}
- Lee J, Torosyan N, Dahlbom M, Silverman D. Amyloid imaging and FDG PET as predictors of subsequent cognitive decline in MCI subpopulation. Journal of Nuclear Medicine 2015;56 Suppl 3:190. [Google Scholar]
Lim 2014 {published data only}
- Lim YY, Maruff P, Pietrzak RH, Ames D, Ellis KA, Harrington K, et al. Effect of amyloid on memory and non‐memory decline from preclinical to clinical Alzheimer's disease. Brain 2014;137(1):221‐31. [DOI] [PubMed] [Google Scholar]
Manitsirikul 2015 {published data only}
- Manitsirikul S, Mathotaarachchi SS, Mohamedes S, Gauthier S, Beaudry T, Rosa‐Neto P. How to follow up and cluster subjects by longitudinal changes of fibrillary amyloid imaging and CSF biomarkers? A 24‐month follow up. Alzheimer's & Dementia 2015;11 Suppl(7):P19‐21. [Google Scholar]
Margolin 2013 {published data only}
- Margolin RA, Andrews RD, Lukic AS, Zhao X, Tudor IC, Salloway S, et al. Biomarkers and cognition in amyloid positive and amyloid‐negative ADNI‐2 MCI subjects: implications for AD therapeutic trials. Journal of Nutrition, Health & Aging 2013;17(9):795‐96. [Google Scholar]
Mathotaarachchi 2015 {published data only}
- Mathotaarachchi SS, Mohades S, Shin M, Beaudry T, Benedet AL, Pascoal TA, et al. Should a global or a regional measure of amyloidosis be used in a longitudinal study?. Alzheimer's & Dementia 2015;11 Suppl(7):P19. [Google Scholar]
Mattsson 2014a {published data only}
- Mattsson N, Insel PS, Landau S, Jagust W, Donohue M, Shaw LM, et al. Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer's disease. Annals of Clinical and Translational Neurology 2014;1(8):534‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mattsson 2014b {published data only}
- Mattsson N, Insel P, Landau S, Jagust W, Shaw L, Trojanowski JQ, et al. Combining CSF AB42 and PET florbetapir to predict diagnosis, tau, atrophy, and cognition. Alzheimer's & Dementia 2014;10 Suppl(4):P174. [Google Scholar]
Mattsson 2015a {published data only}
- Mattsson N, Insel PS, Donohue M, Landau S, Jagust WJ, Shaw LM, et al. Independent information from cerebrospinal fluid amyloid‐β and florbetapir imaging in Alzheimer's disease. Brain 2015;138(3):772‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mattsson 2015b {published data only}
- Mattsson N, Insel PS, Aisen PS, Jagust W, Mackin S, Weiner M, et al. Brain structure and function as mediators of the effects of amyloid on memory. Neurology 2015;84(11):1136‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ming 2015 {published data only}
- Lu M, Pontecorvo MJ, Siderowf A, Joshi AD, Devous MD, Mintun MA, et al. Prognostic value of 18F‐Florbetapir scan: a 36‐month follow up analysis using ADNI data. Clinical and Translational Imaging 2015;3 Suppl 1:S126. [Google Scholar]
Mohades 2014 {published data only}
- Mohades S, Mathotaarachchi SS, Parent M, Shin M, Wang S, Benedet AL, et al. Neurodegeneration and cortical atrophy in [18f] florbetapir accumulators and non‐accumulators. Alzheimer's & Dementia 2014;10 Suppl(4):P26‐7. [Google Scholar]
Morbelli 2015 {published data only}
- Morbelli S, Nobili F, Sensi F, Guerra U, Rei L, Bossert I, et al. SUVratio (SUVr)‐independent semiquantification of brain amyloidosis: a software‐aided integration of visual and quantitative analyses. European Journal of Nuclear Medicine and Molecular Imaging 2015;42 Suppl 1:S547. [Google Scholar]
Pascoal 2016 {published data only}
- Pascoal T, Benedet A, Mathotaarachchi S, Soucy JP, Beaudry T, Gauthier S, et al. Amyloidbeta and hyperphosphorylated tau synergy drives clinical progression in individuals with mild cognitive impairment. Neurology 2016;86 Suppl(16):P2.228. [Google Scholar]
Pascoal 2017 {published data only}
- Pascoal TA, Mathotaarachchi S, Shin M, Benedet AL, Mohades S, Wang S, et al. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimer's & Dementia 2017;13:644‐53. [DOI] [PubMed] [Google Scholar]
Pontecorvo 2011 {published data only}
- Pontecorvo MJ, Joshi A, Skovronsky D, Clark C, Mintun M. Florbetapir PET correlates with cognitive decline, PiB PET and CSF markers in the ADNI database. Alzheimer's & Dementia 2011;7 Suppl(4):S697. [Google Scholar]
Risacher 2014 {published data only}
- Risacher SL, Kim S, Nho KT, West JD, Petersen RC, Aisen PS, et al. Two‐year longitudinal change in amyloid deposition, glucose metabolism, and hippocampal atrophy in ADNI‐2 participants: relation to genetic risk. Alzheimer's & Dementia 2014;10 Suppl(4):P211‐12. [Google Scholar]
Shokouhi 2016 {published data only}
- Shokouhi S, Mckay JW, Baker SL, Kang H, Brill AB, Gwirtsman HE, et al. Reference tissue normalization in longitudinal (18)F‐florbetapir positron emission tomography of late mild cognitive impairment. Alzheimer's Research & Therapy 2016;8(1):article no 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Siderowf 2013 {published data only}
- Siderowf A, Joshi A, Lu M, Mintun M, Pontecorvo M. Lack of substantial progression of cognitive deficits in patients with negative amyloid imaging: implications for clinical trials. Neurology 2013;80 Suppl(7):P01.016. [Google Scholar]
Teipel 2015 {published data only}
- Teipel SJ, Kurth J, Krause B, Grothe MJ, Alzheimer's Disease Neuroimaging Initiative. The relative importance of imaging markers for the prediction of Alzheimer's disease dementia in mild cognitive impairment ‐ beyond classical regression. NeuroImage: Clinical 2015;8:583‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Toledo 2015 {published data only}
- Toledo JB, Bjerke M, Da X, Landau SM, Foster NL, Jagust W, et al. Nonlinear association between cerebrospinal fluid and florbetapir F‐18 beta‐amyloid measures across the spectrum of Alzheimer disease. JAMA Neurology 2015;72(5):571‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wisse 2015 {published data only}
- Wisse LE, Butala N, Das SR, Davatzikos C, Dickerson BC, Vaishnavi SN, et al. Suspected non‐AD pathology in mild cognitive impairment. Neurobiology of Aging 2015;36(12):3152‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xu 2016 {published data only}
- Xu L, Wu X, Li R, Chen K, Long Z, Zhang J, et al. Prediction of progressive mild cognitive impairment by multi‐modal neuroimaging biomarkers. Journal of Alzheimer's Disease 2016;51(4):1045‐56. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
JPRN‐UMIN000019926 {unpublished data only}
- JPRN‐UMIN000019926. Clinical and neuroimaging study on preclinical Alzheimer's disease. apps.who.int/trialsearch/Trial2.aspx?TrialID=JPRN‐UMIN000019926 (first received 1 December 2015).
NCT01325259 {unpublished data only}
- NCT01325259. FluoroAv45 imaging research‐in Alzheimer's disease (FAIR‐AD). clinicaltrials.gov/show/NCT01325259 (first received 29 March 2011).
NCT01554202 {unpublished data only}
- NCT01554202. Multi‐modal neuroimaging in Alzheimer's disease (IMAP). clinicaltrials.gov/show/NCT01554202 (first received 14 March 2012).
NCT01638949 {unpublished data only}
- NCT01638949. Multi‐modal neuroimaging in Alzheimer's disease (IMAP+). clinicaltrials.gov/show/NCT01638949 (first received 12 July 2012).
NCT01687153 {unpublished data only}
- NCT01687153. A study of brain aging in Vietnam war veterans (DOD‐ADNI). clinicaltrials.gov/show/NCT01687153 (first received 18 September 2012).
NCT01746706 {unpublished data only}
- NCT01746706. Can the assessment of the subhippocampal region contribute to the detection of early diagnosis of Alzheimer's disease? A validation study using PET with florbetapir (AV‐45). https://clinicaltrials.gov/show/NCT01746706 (first received 11 December 2012).
NCT02164643 {unpublished data only}
- NCT02164643. Longitudinal study of brain amyloid imaging in MEMENTO (MEMENTOAmyGing). clinicaltrials.gov/show/NCT02164643 (first received 16 June 2014).
NCT02330510 {unpublished data only}
- NCT02330510. Amyloid and glucose PET imaging in Alzheimer and vascular cognitive impairment patients with significant white matter disease (MITNEC C6). clinicaltrials.gov/show/NCT02330510 (first received 5 January 2015).
NCT02343757 {unpublished data only}
- NCT02343757. Alzheimer's disease imaging with PET/MRI ‐ beta‐amyloid. clinicaltrials.gov/show/NCT02343757 (first received 22 January 2015).
NCT02854033 {unpublished data only}
- NCT02854033. Alzheimer's disease neuroimaging initiative 3 (ADNI3) protocol. clinicaltrials.gov/show/NCT02854033 (first received 3 August 2016).
Additional references
Albert 2011
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia 2011;7(3):270‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Alzheimer's Association 2010
- Alzheimer's Association. Changing the trajectory of Alzheimer's disease: a national imperative. http://www.alzheimersreadingroom.com/2010/05/changing‐trajectory‐of‐alzheimers.html (first accessed prior to 12 October 2017).
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd Edition. Washington, DC: American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th Edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
Archer 2015
- Archer HA, Smailagic N, John C, Holmes RB, Takwoingi Y, Coulthard EJ, et al. Regional Cerebral Blood Flow Single Photon Emission Computed Tomography for detection of Frontotemporal dementia in people with suspected dementia. Cochrane Database of Systematic Reviews 2015, Issue 6. [DOI: 10.1002/14651858.CD010896.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Arevalo‐Rodriguez 2015
- Arevalo‐Rodriguez I, Smailagic N, Figuls MRI, Ciapponi A, Sanchez‐Perez E, Giannakou A, et al. Mini‐Mental State Examination (MMSE) for the detection of Alzheimer's disease and other dementias in people with mild cognitive impairment (MCI). Cochrane Database of Systematic Reviews 2015, Issue 3. [DOI: 10.1002/14651858.CD010783.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bossuyt 2008
- Bossuyt PM, Leeflang MM, editor(s). Chapter 6: Developing criteria for including studies. In: Deeks JJ, Bossuyt PM, Gatsonis C, editor(s). Cochrane Handbook for Systematic Reviews of Diagnostic Test Accuracy Version 0.4 (updated September 2008). The Cochrane Collaboration, 2008. Available from srdta.cochrane.org.
Boxer 2005
- Boxer AL, Miller BL. Clinical features of frontotemporal dementia. Alzheimer Disease and Associated Disorders 2005;19(Suppl 1):S3‐6. [DOI] [PubMed] [Google Scholar]
Brun 1994
- Brun A, Englund B, Gustafson L, Passant U, Mann DMA, Neary D, et al. Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. Journal of Neurology, Neurosurgery, and Psychiatry 1994;57(4):416‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bruscoli 2004
- Bruscoli M, Lovestone S. Is MCI really just early dementia? A systematic review of conversion studies. International Psychogeriatrics 2004;16(2):129‐40. [DOI] [PubMed] [Google Scholar]
Caroli 2015
- Caroli A, Prestia A, Wade S, Chen K, Ayutyanont N, Landau SM, et al. Alzheimer disease biomarkers as outcome measures for clinical trials in MCI. Alzheimer Disease and Associated Disorders 2015;29(2):101‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chan 2014
- Chan CCH, Fage BA, Smailagic N, Gill SS, Herrmann N, Nikolaou V, et al. Mini‐Cog for the diagnosis of Alzheimer’s disease dementia and other dementias within a secondary care setting. Cochrane Database of Systematic Reviews 2014, Issue 12. [DOI: 10.1002/14651858.CD011414] [DOI] [PMC free article] [PubMed] [Google Scholar]
Choi 2009
- Choi SR, Golding G, Zhuang Z, Zhang W, Lim N, Hefti F, et al. Preclinical properties of 18F‐AV‐45: a PET agent for Abeta plaques in the brain. Journal of Nuclear Medicine 2009;50(11):1887‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Clark 2011
- Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir‐PET for imaging beta‐amyloid pathology. JAMA 2011;305(3):275‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Clark 2012
- Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐β plaques: a prospective cohort study. Lancet Neurology 2012;11(8):669‐78. [DOI] [PubMed] [Google Scholar]
CMS 2013
- Centers for Medicare & Medicaid Services. Decision memo for beta amyloid positron emission tomography in dementia and neurodegenerative disease (CAG‐00431N). www.cms.gov/medicare‐coverage‐database/details/nca‐decision‐memo.aspx?NCAId=265 (accessed 08/10/2015).
Cordella 2013
- Cordella CB, Borson S, Boustani M, Chodosh J, Reuben D, Verghese J, et al. Alzheimer's Association recommendations for operationalizing the detection of cognitive impairment during the Medicare Annual Wellness Visit in a primary care setting. Alzheimer's & Dementia 2013;9(2):141‐50. [DOI] [PubMed] [Google Scholar]
Creavin 2016
- Creavin S, Wisniewski S, Noel‐Storr A, Trevelyan C, Hampton T, Rayment D, et al. Mini‐Mental State Examination (MMSE) for the detection of dementia in clinically unevaluated people aged 65 and over in community and primary care populations. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD011145.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Davis 2013
- Davis DHJ, Creavin ST, Noel‐Storr A, Quinn TJ, Smailagic N, Hyde C, et al. Neuropsychological tests for the diagnosis of Alzheimer’s disease dementia and other dementias: a generic protocol for cross‐sectional and delayed‐verification studies. Cochrane Database of Systematic Reviews 2013, Issue 3. [DOI: 10.1002/14651858.CD010460] [DOI] [PMC free article] [PubMed] [Google Scholar]
Davis 2015
- Davis DHJ, Creavin ST, Yip JLY, Noel‐Storr AH, Brayne C, Cullum S. Montreal Cognitive Assessment for the diagnosis of Alzheimer’s disease and other dementias. Cochrane Database of Systematic Reviews 2015, Issue 10. [DOI: 10.1002/14651858.CD010775.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
De la Torre 2004
- Torre JC. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurology 2004;3(3):184‐90. [DOI] [PubMed] [Google Scholar]
Dubois 2014
- Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria. Lancet Neurology 2014;13(6):614‐29. [DOI] [PubMed] [Google Scholar]
Elias‐Sonnenschein 2014a
- Elias‐Sonnenschein LS, Viechtbauer W, Ramakers I, Ukoumunne O, Verhey FRJ, Visser PJ. APOE‐ϵ4 allele for the diagnosis of Alzheimer's and other dementia disorders in people with mild cognitive impairment in a community setting. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD010948] [DOI] [Google Scholar]
Elias‐Sonnenschein 2014b
- Elias‐Sonnenschein LS, Viechtbauer W, Ramakers I, Ukoumunne O, Verhey FRJ, Visser PJ. APOE‐ϵ4 allele for the diagnosis of Alzheimer's and other dementia disorders in people with mild cognitive impairment in a primary care setting. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD010949] [DOI] [Google Scholar]
Elias‐Sonnenschein 2014c
- Elias‐Sonnenschein LS, Viechtbauer W, Ramakers I, Ukoumunne O, Verhey FRJ, Visser PJ. APOE‐ϵ4 allele for the diagnosis of Alzheimer's and other dementia disorders in people with mild cognitive impairment in a secondary care setting. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD010950] [DOI] [Google Scholar]
EMA 2013
- European Medicines Agency. Assessment report. Amyvid. International non‐proprietary name: florbetapir (18F) Procedure No. EMEA/H/C/002422. www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002422/WC500137634.pdf (accessed 8th April 2015).
Espinosa 2013
- Espinosa A, Alegret M, Valero S, Vinyes‐Junque G, Hernandez I, Mauleon A, et al. A longitudinal follow‐up of 550 mild cognitive impairment patients: evidence for large conversion to dementia rates and detection of major risk factors involved. Journal of Alzheimer’s Disease 2013;34(3):769‐80. [DOI] [PubMed] [Google Scholar]
Fage 2015
- Fage BA, Chan CCH, Gill SS, Noel‐Storr AH, Herrmann N, Smailagic N, et al. Mini‐Cog for the diagnosis of Alzheimer's disease dementia and other dementias within a community setting. Cochrane Database of Systematic Reviews 2015, Issue 2. [DOI: 10.1002/14651858.CD010860.pub2] [DOI] [PubMed] [Google Scholar]
FDA 2013
- Food, Drug Administration. Amyvid. www.accessdata.fda.gov/drugsatfda_docs/label/2013/202008s020lbl.pdf (accessed 8 April 2015).
Filippini 2012
- Filippini G, Casazza G, Bellatorre AG, Lista C, Duca P, Beecher D, et al. The role of MRI in the early diagnosis of Alzheimer’s disease or other dementias in persons with mild cognitive impairment (MCI). Cochrane Database of Systematic Reviews 2012, Issue 2. [DOI: 10.1002/14651858.CD009628] [DOI] [Google Scholar]
Garcia‐Alloza 2011
- Garcia‐Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, et al. Cerebrovascular lesions induce transient β‐amyloid deposition. Brain 2011;134(12):3697‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gelber 2012
- Gelber RP, Launer LJ, White LR. The Honolulu‐Asia Aging Study: epidemiologic and neuropathologic research on cognitive impairment. Current Alzheimer Research 2012;9(6):664‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Geslani 2005
- Geslani DM, Tierney MC, Herrmann N, Szalai JP. Mild cognitive impairment: an operational definition and its conversion rate to Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders 2005;19(5‐6):383‐9. [DOI] [PubMed] [Google Scholar]
Goedert 2006
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science 2006;314(5800):777‐81. [DOI] [PubMed] [Google Scholar]
Gurol 2016
- Gurol ME, Becker JA, Fotiadis P, Riley G, Schwab K, Johnson KA, et al. Florbetapir‐PET to diagnose cerebral amyloid angiopathy: a prospective study. Neurology 2016;87(19):2043‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hampel 2012
- Hampel H, Lista S, Khachaturian Z. Development of biomarkers to chart all Alzheimer’s disease stages: the royal road to cutting the therapeutic Gordian Knot. Alzheimer's & Dementia 2012;8(4):312‐36. [DOI] [PubMed] [Google Scholar]
Harrison 2014
- Harrison JK, Fearon P, Noel‐Storr AH, McShane R, Stott DJ, Quinn TJ. Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) for the diagnosis of dementia within a general practice (primary care) setting. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD010771.pub2] [DOI] [PubMed] [Google Scholar]
Harrison 2015
- Harrison JK, Fearon P, Noel‐Storr AH, McShane R, Stott DJ, Quinn TJ. Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) for the diagnosis of dementia within a secondary care setting. Cochrane Database of Systematic Reviews 2015, Issue 3. [DOI: 10.1002/14651858.CD010772.pub2] [DOI] [PubMed] [Google Scholar]
Hendry 2014
- Hendry K, Lees RA, McShane R, Noel‐Storr AH, Stott DJ, Quinn TJ. AD‐8 for diagnosis of dementia across a variety of healthcare settings. Cochrane Database of Systematic Reviews 2014, Issue 5. [DOI: 10.1002/14651858.CD011121] [DOI] [PMC free article] [PubMed] [Google Scholar]
Herscovitch 2015
- Herscovitch P. Regulatory approval and insurance reimbursement: the final steps in clinical translation of amyloid brain imaging. Clinical and Translational Imaging 2015;3:75‐7. [Google Scholar]
Heyden 2013
- Hayden EY, Teplow DB. Amyloid β‐protein oligomers and Alzheimer’s disease. Alzheimer's Research & Therapy 2013;5(5):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hyman 2012
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's & Dementia 2012;8(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
ICD‐10
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems (ICD‐10 Version: 2010). apps.who.int/classifications/icd10/browse/2010/en (accessed 29 January 2015).
Jack 2013
- Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurology 2013;12(2):207‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jellinger 2006
- Jellinger K. Clinicopathological analysis of dementia disorders in the elderly ‐ update. Journal of Alzheimer's Disease 2006;9(Supplement 3):61‐70. [DOI] [PubMed] [Google Scholar]
Johnson 2013
- Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer's Association. Journal of Nuclear Medicine 2013;54(3):476‐90. [DOI] [PubMed] [Google Scholar]
Knottnerus 2002
- Knottnerus JA, Weel C, Muris JW. Evaluation of diagnostic procedures. BMJ 2002;324(7335):477‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kobylecki 2015
- Kobylecki C, Langheinrich T, Hinz R, Vardy ER, Brown G, Martino ME, et al. 18F‐Florbetapir PET in patients with frontotemporal dementia and Alzheimer disease. Journal of Nuclear Medicine 2015;56(3):386‐91. [DOI] [PubMed] [Google Scholar]
Kokkinou 2014
- Kokkinou M, Smailagic N, Noel‐Storr AH, Hyde C, Ukoumunne O, Worrall RE, et al. Plasma and Cerebrospinal fluid (CSF) Abeta42 for the differential diagnosis of Alzheimer's disease dementia in participants diagnosed with any dementia subtype in a specialist care setting. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD010945] [DOI] [PMC free article] [PubMed] [Google Scholar]
Landau 2012
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Annals of Neurology 2012;72(4):578‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Landau 2013
- Landau SM, Lu M, Joshi AD, Pontecorvo M, Mintun MA, Trojanowski JQ, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β‐amyloid. Annal of Neurology 2013;74(6):826‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lees 2014
- Lees RA, Stott DJ, McShane R, Noel‐Storr AH, Quinn TJ. Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) for the early diagnosis of dementia across a variety of healthcare settings. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD011333] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lin 2010
- Lin KJ, Hsu WC, Hsiao IT, Wey SP, Jin LW, Skovronsky D, et al. Whole‐body biodistribution and brain PET imaging with [18F]AV‐45, a novel amyloid imaging agent ‐ a pilot study. Nuclear Medicine and Biology 2010;37(4):497‐508. [DOI] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ma 2014
- Ma Y, Zhang S, Li J, Zheng DM, Guo Y, Feng J, et al. Predictive accuracy of amyloid imaging for progression from mild cognitive impairment to Alzheimer disease with different lengths of follow‐up: a meta‐analysis. Medicine 2014;93(27):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Martínez 2016
- Martínez G, Flicker L, Vernooij RWM, Fuentes Padilla P, Zamora J, Figuls MRI, et al. 18F PET ligands for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD012216] [DOI] [Google Scholar]
Matthews 2008
- Matthews FE, Stephan BC, McKeith IG, Bond J, Brayne C, Medical Research Council Cognitive Function and Ageing Study. Two‐year progression from mild cognitive impairment to dementia: to what extent do different definitions agree?. Journal of the American Geriatrics Society 2008;56(8):1424‐33. [DOI] [PubMed] [Google Scholar]
Mattsson 2009
- Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009;302(4):385‐93. [DOI] [PubMed] [Google Scholar]
McCleery 2015
- McCleery J, Morgan S, Bradley KM, Noel‐Storr AH, Ansorge O, Hyde C. Dopamine transporter imaging for the diagnosis of dementia with Lewy bodies. Cochrane Database of Systematic Reviews 2015, Issue 1. [DOI: 10.1002/14651858.CD010633.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
McKeith 1996
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996;47(5):1113‐24. [DOI] [PubMed] [Google Scholar]
McKeith 2005
- McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 2005;65(12):1863‐72. [DOI] [PubMed] [Google Scholar]
McKhann 1984
- McKhann GM, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34(7):939‐44. [DOI] [PubMed] [Google Scholar]
McKhann 2011
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia 2011;7(3):263‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mitchell 2009
- Mitchell AJ, Shiri‐Feshki M. Rate of progression of mild cognitive impairment to dementia ‐ meta‐analysis of 41 robust inception cohort studies. Acta Psychiatrica Scandinavica 2009;119(4):252‐65. [DOI] [PubMed] [Google Scholar]
Morris 1993
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43(11):2412‐4. [DOI] [PubMed] [Google Scholar]
NAO 2007
- National Audit Office. Improving services and support for people with dementia. Report by the Comptroller and Auditor General. HC 604 Session General 2006‐2007. 4 July 2007. www.nao.org.uk/wp‐content/uploads/2007/07/0607604.pdf (accessed 25th March 2015).
Neary 1998
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51(6):1546‐54. [DOI] [PubMed] [Google Scholar]
NICE 2006
- National Institute for Health Care Excellence. Dementia: supporting people with dementia and their carers in health and social care. NICE guidelines [CG42]. www.nice.org.uk/guidance/cg42 (accessed 17th April 2015).
Noel‐Storr 2013
- Noel‐Storr AH, Flicker L, Ritchie CW, Nguyen GH, Gupta T, Wood P, et al. Systematic review of the body of evidence for the use of biomarkers in the diagnosis of dementia. Alzheimer's & Dementia 2013;9(3):e96‐105. [DOI] [PubMed] [Google Scholar]
Noel‐Storr 2014
- Noel‐Storr AH, McCleery JM, Richard E, Ritchie CW, Flicker L, Cullum SJ, et al. Reporting standards for studies of diagnostic test accuracy in dementia: The STARDdem Initiative. Neurology 2014;83(4):364‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Okello 2007
- Okello A, Edison P, Archer H, Hinz R, Fox N, Kennedy AM, et al. Amyloid deposition and cerebral glucose metabolism in mild cognitive impairment: a longitudinal 11C‐PIB and 18F‐FDG PET study. Journal of Neurology, Neurosurgery, and Psychiatry 2007;78(2):219‐20. [Google Scholar]
Petersen 1999
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Archives of Neurology 1999;56(3):303‐8. [DOI] [PubMed] [Google Scholar]
Petersen 2004
- Petersen RC. Mild cognitive impairment as a diagnostic entity. Journal of Internal Medicine 2004;256(3):183‐94. [DOI] [PubMed] [Google Scholar]
Petersen 2009
- Petersen RC, Roberts RO, Knopman DS, Boeve BF, Geda YE, Ivnik RJ, et al. Mild cognitive impairment: ten years later. Archives of Neurology 2009;66(12):1447‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Quinn 2014
- Quinn TJ, Fearon P, Noel‐Storr AH, Young C, McShane R, Stott DJ. Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) for the diagnosis of dementia within community dwelling populations. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD010079.pub2] [DOI] [PubMed] [Google Scholar]
Rascovsky 2011
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134(Pt 9):2456‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager. Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Richard 2012
- Richard E, Schmand B, Eikelenboom P, Westendorp RG, Gool WA. The Alzheimer myth and biomarker research in dementia. Journal of Alzheimer's Disease: JAD 2012;31(Suppl 3):S203‐9. [DOI] [PubMed] [Google Scholar]
Ritchie 2013
- Ritchie C, Smailagic N, Ladds EC, Noel‐Storr AH, Ukoumunne O, Martin S. CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer's disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database of Systematic Reviews 2013, Issue 10. [DOI: 10.1002/14651858.CD010803] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ritchie 2014
- Ritchie C, Smailagic N, Noel‐Storr AH, Takwoingi Y, Flicker L, Mason SE, et al. Plasma and cerebrospinal fluid amyloid beta for the diagnosis of Alzheimer's disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database of Systematic Reviews 2014, Issue 6. [DOI: 10.1002/14651858.CD008782.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Román 1993
- Román GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS‐AIREN International Workshop. Neurology 1993;43(2):250‐60. [DOI] [PubMed] [Google Scholar]
Royall 2014
- Royall DR, Palmer RF. The temporospatial evolution of neuritic plaque‐related and independent tauopathies: implications for dementia staging. Journal of Alzheimer’s Disease 2014;40(3):541‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Savva 2009
- Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C, Medical Research Council Cognitive Function and Ageing Study. Age, neuropathology, and dementia. The New England Journal of Medicine 2009;360(22):2302‐9. [DOI] [PubMed] [Google Scholar]
Schneider 2007
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology 2007;69(24):2197‐204. [DOI] [PubMed] [Google Scholar]
Schneider 2009
- Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Annals of neurology 2009;66(2):200‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Seitz 2014
- Seitz DP, Fage BA, Chan CCH, Gill SS, Herrmann N, Smailagic N, et al. Mini‐Cog for the diagnosis of Alzheimer’s disease dementia and other dementias within a primary care setting. Cochrane Database of Systematic Reviews 2014, Issue 12. [DOI: 10.1002/14651858.CD011415] [DOI] [PMC free article] [PubMed] [Google Scholar]
Selkoe 2016
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Molecular Medicine 2016;8(6):595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Serrano‐Pozo 2013
- Serrano‐Pozo A, Qian J, Monsell SE, Frosch MP, Betensky RA, Hyman BT. Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. Journal of Neuropathology and Experimental Neurology 2013;72(12):1182‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Siderowf 2014
- Siderowf A, Pontecorvo MJ, Shill HA, Mintun MA, Arora A, Joshi AD, et al. PET imaging of amyloid with Florbetapir F 18 and PET imaging of dopamine degeneration with 18F‐AV‐133 (florbenazine) in patients with Alzheimer's disease and Lewy body disorders. BMC neurology [electronic resource] 2014;14:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smailagic 2015
- Smailagic N, Vacante M, Hyde C, Martin S, Ukoumunne O, Sachpekidis C. 18F‐FDG PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database of Systematic Reviews 2015, Issue 1. [DOI: 10.1002/14651858.CD010632.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sperling 2011
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia: the Journal of the Alzheimer's Association 2011;7(3):280‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Visser 2006
- Visser PJ, Kester A, Jolles J, Verhey F. Ten‐year risk of dementia in subjects with mild cognitive impairment. Neurology 2006;67(7):1201‐7. [DOI] [PubMed] [Google Scholar]
White 2009
- White L. Brain lesions at autopsy in older Japanese‐American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu‐Asia aging study. Journal of Alzheimer's Disease: JAD 2009;18(3):713‐25. [DOI] [PubMed] [Google Scholar]
Whiting 2011
- Whiting PF, Rutjes AWS, Westwood ME, Mallet S, Deeks JJ, Reitsma JB. QUADAS‐2: a revised tool for the quality assessment of diagnostic accuracy studies. Annals of Internal Medicine 2011;155(8):529‐36. [DOI] [PubMed] [Google Scholar]
WHO 2012
- World Health Organization, Alzheimer's Disease International. Dementia: a public health priority. 2012. http://www.who.int/mental_health/publications/dementia_report_2012/en/. World Health Organization, (accessed 23th September 2015).
Winblad 2004
- Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, et al. Mild cognitive impairment‐‐beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. Journal of Internal Medicine 2004;256(3):240‐6. [DOI] [PubMed] [Google Scholar]
Wolz 2016
- Wolz R, Schwarz AJ, Gray KR, Yu P, Hill DL, Alzheimer's Disease Neuroimaging Initiative. Enrichment of clinical trials in MCI due to AD using markers of amyloid and neurodegeneration. Neurology 2016;87(12):1235‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wong 2010
- Wong DF, Rosenberg PB, Zhou Y, Kumar A, Raymont V, Ravert HT, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F‐AV‐45 (Florbetapir F18). Journal of Nuclear Medicine 2010;51(6):913‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2014
- Zhang S, Smailagic N, Hyde C, Noel‐Storr AH, Takwoingi Y, McShane R, et al. 11C‐PIB‐PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD010386.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]