

Abstract
Background
Several anticonvulsant drugs are used in the management of neuropathic pain. Oxcarbazepine is an anticonvulsant drug closely related to carbamazepine. Oxcarbazepine has been reported to be efficacious in the treatment of neuropathic pain, but evidence from randomised controlled trials (RCTs) is conflicting. Oxcarbazepine is reportedly better tolerated than carbamazepine. This is the first update of a review published in 2013.
Objectives
To assess the benefits and harms of oxcarbazepine for different types of neuropathic pain.
Search methods
On 21 November 2016, we searched the Cochrane Neuromuscular Specialised Register, CENTRAL, MEDLINE and Embase. We searched the Chinese Biomedical Retrieval System (January 1978 to November 2016). We searched the US National Institutes of Health (NIH) databases and the World Health Organization (WHO) International Clinical Trials Registry Platform for ongoing trials in January 2017, and we wrote to the companies who make oxcarbazepine and to pain experts requesting additional information.
Selection criteria
All RCTs and randomised cross‐over studies of oxcarbazepine for the treatment of people of any age or sex with any neuropathic pain were eligible. We planned to include trials of oxcarbazepine compared with placebo or any other intervention with a treatment duration of at least six weeks, regardless of administration route and dose.
Data collection and analysis
We used standard methodological procedures expected by Cochrane.
Main results
Five multicentre, randomised, placebo‐controlled, double‐blind trials with a total of 862 participants were eligible for inclusion in this updated review. Three trials involved participants with painful diabetic peripheral neuropathy (DPN) (n = 634), one included people with neuropathic pain due to radiculopathy (n = 145), and one, which was newly identified at this update, involved participants with peripheral neuropathic pain of mixed origin (polyneuropathy, peripheral nerve injury or postherpetic neuralgia) (n = 83). Some studies did not report all outcomes of interest. For painful DPN, compared to the baseline, the proportion of participants who reported at least a 50% or 30% reduction of pain scores after 16 weeks of treatment in the oxcarbazepine group versus the placebo group were: at least 50% reduction: 34.8% with oxcarbazepine versus 18.2% with placebo (risk ratio (RR) 1.91, 95% confidence interval (CI) 1.08 to 3.39, number of people needed to treat for an additional beneficial outcome (NNTB) 6, 95% CI 3 to 41); and at least 30% reduction: 44.9% with oxcarbazepine versus 28.6% with placebo (RR 1.57, 95% CI 1.01 to 2.44; NNTB 6, 95% CI 3 to 114; n = 146). Both results were based on data from a single trial, since two trials that found little or no benefit did not provide data that could be included in a meta‐analysis. Although these trials were well designed, incomplete outcome data and possible unblinding of participants due to obvious adverse effects placed the results at a high risk of bias. There was also serious imprecision and a high risk of publication bias. The radiculopathy trial reported no benefit for the outcome 'at least 50% pain relief' from oxcarbazepine. In mixed neuropathies, 19.3% of people receiving oxcarbazepine versus 4.8% receiving placebo had at least 50% pain relief. These small trials had low event rates and provided, at best, low‐quality evidence for any outcome. The proportion of people with 'improved' or 'very much improved' pain was 45.9% with oxcarbazepine versus 30.1% with placebo in DPN (RR 1.46, 95% CI 1.13 to 1.88; n = 493; 2 trials; very‐low‐quality evidence) and 23.9% with oxcarbazepine versus 14.9% with placebo in radiculopathy (RR 1.61, 95% CI 0.81 to 3.20; n = 145).
We found no trials in other types of neuropathic pain such as trigeminal neuralgia.
Trial reports stated that most adverse effects were mild to moderate in severity. Based on moderate‐quality evidence from the three DPN trials, serious adverse effects occurred in 8.3% with oxcarbazepine and 2.5% with placebo (RR 3.65, 95% CI 1.45 to 9.20; n = 634; moderate‐quality evidence). The number needed to treat for an additional harmful (serious adverse effect) outcome (NNTH) was 17 (95% CI 11 to 42). The RR for serious adverse effects in the radiculopathy trial was 3.13 (95% CI 0.65 to 14.98, n = 145). The fifth trial did not provide data.
More people withdrew because of adverse effects with oxcarbazepine than with placebo (DPN: 25.6% with oxcarbazepine versus 6.8% with placebo; RR 3.83, 95% CI 2.29 to 6.40; radiculopathy: 42.3% with oxcarbazepine versus 14.9% with placebo; RR 2.84, 95% CI 1.55 to 5.23; mixed neuropathic pain: 13.5% with oxcarbazepine versus 1.2% with placebo; RR 11.51, 95% CI 1.54 to 86.15).
Authors' conclusions
This review found little evidence to support the effectiveness of oxcarbazepine in painful diabetic neuropathy, neuropathic pain from radiculopathy and a mixture of neuropathies. Some very‐low‐quality evidence suggests efficacy but small trials, low event rates, heterogeneity in some measures and a high risk of publication bias means that we have very low confidence in the measures of effect. Adverse effects, serious adverse effects and adverse effects leading to discontinuation are probably more common with oxcarbazepine than placebo; however, the numbers of participants and event rates are low. More well‐designed, multicentre RCTs investigating oxcarbazepine for various types of neuropathic pain are needed, and selective publication of studies or data should be avoided.
Plain language summary
Oxcarbazepine for neuropathic pain
Review question
What are the benefits and harms of oxcarbazepine in the treatment of pain caused by nerve damage?
Background
Neuropathic pain is pain that arises from damage to the part of the nervous system that carries sensory information (e.g. pain) to the brain. It is difficult to treat because it tends to be severe, long‐lasting and does not respond well to simple painkillers. Some studies have suggested that a medicine called oxcarbazepine, when given on its own, can relieve pain from nerve damage.
Study characteristics
We searched medical databases for clinical trials looking at the potential benefits and harms of oxcarbazepine in different types of neuropathic pain and found five trials. They involved 634 participants with painful diabetic neuropathy (nerve damage), 145 people with neuropathic pain due to radiculopathy (pain that arises at the point where nerves leave the spinal column), and 83 people with peripheral neuropathic pain of various causes (e.g. peripheral nerve injury (injury to nerves that connect the brain and spinal cord to the rest of the body), polyneuropathy (damage or disease affecting several peripheral nerves) and postherpetic neuralgia (pain that occurs after shingles)). The trials all compared oxcarbazepine with placebo (a pretend treatment). Four trials were funded by the manufacturer of oxcarbazepine.
Key results and quality of the evidence
This review found little evidence to support the effectiveness of oxcarbazepine in painful diabetic neuropathy, neuropathic pain from radiculopathy and mixed neuropathies of various causes. Oxcarbazepine may have some effect, but we cannot be confident that the results would be the same with further studies. Side effects, including those that were serious or made people stop taking the medicine were probably more common with oxcarbazepine than placebo. We know of trials that have not reported results, for example in a form of facial pain called trigeminal neuralgia, and some of the trials we found did not report data in a form that we could analyse. We need more well‐designed studies of oxcarbazepine for various types of neuropathic pain, with large numbers of participants spread over different centres (e.g. difference hospitals and clinics), and all relevant data need to be published or presented.
This is the first update of a review published in 2013. Evidence is up to date to November 2016.
Summary of findings
Summary of findings for the main comparison. Oxcarbazepine versus placebo for painful diabetic neuropathy.
| Oxcarbazepine versus placebo for painful diabetic neuropathy | ||||||
| Patient or population: people with painful diabetic neuropathy Settings: hospitals and clinics Intervention: oxcarbazepine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Oxcarbazepine | |||||
| Reduction in participant‐reported pain scores by at least 50% from the baseline VAS Follow‐up: mean 16 weeks | 182 per 1000 | 348 per 1000 (197 to 617) | RR 1.91 (1.08 to 3.39) | 146 (1 RCT) | ⊕⊝⊝⊝ Very low1 | NNTB 6 (95% CI 3 to 41) |
| Reduction in participant‐reported pain scores by at least 30% from the baseline VAS Follow‐up: mean 16 weeks | 286 per 1000 | 449 per 1000 (289 to 698) | RR 1.57 (1.01 to 2.44) | 146 (1 RCT) | ⊕⊝⊝⊝ Very low1 | NNTB 6 (95% CI 3 to 114) |
| Participants' global impression of their change in pain Participant's global assessment of therapeutic effect, 'much' or 'very much' Follow‐up: mean 16 weeks | 301 per 1000 | 439 per 1000 (340 to 566) | RR 1.46 (1.13 to 1.88) | 493 (2 RCTs) | ⊕⊝⊝⊝ Very low2 | NNTB 6 (95% CI 4 to 14) |
| Serious adverse effects Follow‐up: mean 16 weeks | 25 per 1000 | 91 per 1000 (36 to 230) | RR 3.65 (1.45 to 9.20) | 634 (3 RCTs) | ⊕⊕⊕⊝ Moderate3 | NNTH 17 (95% CI 11 to 42) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval;NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; RR: risk ratio; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Downgraded three times: once for inconsistency with other studies not included in the meta‐analysis, once for serious imprecision (small sample size and wide CI) and once for serious study limitations (number of withdrawals not balanced across groups, especially withdrawals due to adverse effects, which led to a high risk of attrition bias. A high risk of publication bias was present. 2Downgraded three times: once for serious study limitations (number of withdrawals not balanced across groups, especially withdrawals due to adverse effects, which led to a high risk of attrition bias), once for inconsistency (a high degree of unexplained heterogeneity between the two included RCTs) and once for publication bias. 3Downgraded once for serious imprecision. Event rate was low.
Background
Description of the condition
Neuropathic pain is defined as "pain initiated or caused by a primary lesion or dysfunction of the nervous system" (Merskey 1994), or more recently as "pain arising as a direct consequence of a lesion or disease affecting the somatosensory system" (Treede 2008). Neuropathic pain is difficult to treat because of its severity, chronicity and resistance to simple analgesics. Neuropathic pain may have its genesis in the brain, spinal cord or peripheral nerves and arises from conditions including radiculopathy (cervical or lumbar), malignancy, infection (e.g. postherpetic neuralgia and HIV‐related neuropathy), injury to the spinal cord, diabetic peripheral neuropathy (DPN), trigeminal neuralgia and complex regional pain syndrome type II (Jensen 2001). The epidemiology of neuropathic pain has not been well described, at least in part because the underlying conditions are so diverse. Current pooled estimates suggest that as much as 3% of the population may be affected by neuropathic pain (Davis 2004; Foley 2003; Heliovaara 1987; Schmader 2002; Verma 2005; Werhagen 2004).
The underlying mechanisms of neuropathic pain are complicated and unclear (Finnerup 2007). We know that the peripheral and central nervous systems are capable of both structural and functional plastic changes in response to injury and experience. Peripheral changes include the sensitisation of nociceptors, which results in decreased activation thresholds and increased pain in response to a given stimulus, and abnormal neuronal sprouting that leads to expansion of nociceptive receptive fields and ectopic firing of dorsal root ganglia cells. Changes in expression of abnormal sodium and calcium channels are considered instrumental in the generation of spontaneous discharges from injured neurons (Callin 2008).
Central changes following peripheral nerve injury include loss of inhibitory effects in the central nervous system and heightened sensitivity of neurons in the spinal cord, which occurs despite reduced peripheral input. Moreover, N‐methyl‐D‐aspartic acid (NMDA) receptors are upregulated and activated, and play an important role in central sensitisation in neuropathic pain. Other important findings include facilitatory descending pathways and the potential relevance of genetic differences between people (Callin 2008; Carpenter 2002; Suzuki 2004).
Neuropathic pain has a profound effect on quality of life and expenditure on health care (Schmader 2002). It remains a major therapeutic challenge despite considerable research efforts over the past few decades. The shared pathophysiology of neuropathic pain and epilepsy supports the rationale for using certain antiepileptic drugs (AEDs) in the treatment of neuropathic pain (Dickenson 2002; Jensen 2002). Several Cochrane Reviews have assessed the effects of individual AEDs in neuropathic pain, including lamotrigine (Wiffen 2013), carbamazepine (Wiffen 2014), gabapentin (Moore 2014), pregabalin (Moore 2009), valproic acid (Gill 2011), phenytoin (Birse 2012), and clonazepam (Corrigan 2012). These replaced an original review of AEDs for neuropathic pain (Wiffen 2010), first published in 2005 and now withdrawn.
Description of the intervention
Oxcarbazepine (10,11‐dihydro‐10‐oxo‐5H,dibenz[b,f]azepine‐5‐carboxamide) is the keto analogue of carbamazepine, a sodium channel modulator used primarily in the treatment of epilepsy and trigeminal neuralgia. It has previously been reported to be better tolerated and safer than carbamazepine, with a lower risk of allergic reactions and drug‐drug interactions (Beydoun 2002a; Dam 1989). Three double‐blind, placebo‐controlled trials have evaluated oxcarbazepine in painful diabetic neuropathy. Two trials showed significant pain reduction in participants treated with oxcarbazepine compared with placebo (Beydoun 2006; Dogra 2005), while the third trial showed no difference in efficacy between oxcarbazepine‐treated and placebo‐treated participants (Grosskopf 2006).
Data on the potential efficacy of oxcarbazepine for treating the pain associated with trigeminal neuralgia were derived from an active‐control double‐blind trial (Lindstrom 1987), and meta‐analyses of three double‐blind comparative trials (Beydoun 2002a; Beydoun 2002b). These showed that oxcarbazepine produced substantial pain relief in most people with trigeminal neuralgia and that there was no significant difference in pain relief between oxcarbazepine and carbamazepine. Additionally, there was a pooled analysis of seven open‐label clinical trials, sharing the same protocol, of oxcarbazepine in different neuropathic pain conditions (Magenta 2005). The results of this analysis suggested that oxcarbazepine administered as monotherapy can relieve pain associated with neuropathies.
Why it is important to do this review
To determine the efficacy of oxcarbazepine for neuropathic pain, we gathered evidence from randomised controlled trials (RCTs) and first published a Cochrane systematic review on this topic in 2013. It concluded that oxcarbazepine was effective in reducing pain in people with DPN, but this conclusion was made based on moderate‐quality evidence from only one RCT (Zhou 2013). We updated this review to consider new, potentially eligible studies.
Objectives
To assess the benefits and harms of oxcarbazepine for different types of neuropathic pain.
Methods
Criteria for considering studies for this review
Types of studies
We included RCTs or randomised cross‐over studies of oxcarbazepine for the treatment of neuropathic pain. Trials were eligible whether published or unpublished. There was no language restriction. We interviewed the study authors by telephone or wrote for clarification if the studies were described as RCTs but there was poor reporting of the methodology. We excluded quasi‐randomised trials and trials described as randomised but which we found not to be adequately randomised or blinded on further examination.
Types of participants
Participants of any age or either sex with any neuropathic pain were eligible.
Types of interventions
Oxcarbazepine versus placebo or any other intervention, regardless of administration route or dose. We allowed cointerventions if they were offered equally to both arms of the trial. The treatment duration needed to be at least six weeks.
Types of outcome measures
We planned to measure outcomes as close to eight weeks (minimum six weeks) as possible. When outcomes were reported after different durations of treatment we extracted the data measured as close to the specified time (eight weeks) as possible, but not less than the minimum.
Primary outcomes
Reduction in participant‐reported pain scores by at least 50% from the baseline, measured on commonly used pain scales such as a visual analogue scale (VAS), verbal rating scale (VRS), numerical rating scale (NRS) (Williamson 2005), and the Faces Pain Scale (FPS) (Bieri 1990).
Secondary outcomes
Reduction in participant‐reported pain scores by at least 30% from the baseline, measured on commonly used pain scales.
Participants' global impression of their change in pain.
Overall quality of life measures, as changes or levels at eight weeks using the Short Form‐36 (SF‐36) Health Survey (Ware 1992), including the physical health summary, mental health summary and pain subscale.
Adverse effects (i.e. classified as any adverse effect; adverse effects leading to withdrawal from treatment; and serious adverse effects that were life‐threatening, required hospitalisation or were fatal).
Search methods for identification of studies
Electronic searches
We searched the Cochrane Neuromuscular Specialised Register (21 November 2016), Cochrane Central Register of Controlled Trials (CENTRAL; 21 November 2016, in the Cochrane Register of Studies Online)), MEDLINE (OvidSP; January 1966 to November 2016), Embase (OvidSP; January 1980 to November 2016) and the Chinese Biomedical Retrieval System (January 1978 to November 2016). We also searched the US National Institutes of Health (NIH) database ClinicalTrials.gov (15 January 2017) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/search/en/) for registered trials (15 January 2017).
The detailed search strategies are in the appendices: Cochrane Neuromuscular Specialised Register (Appendix 1), CENTRAL (Appendix 2), MEDLINE (Appendix 3), Embase (Appendix 4), and the Chinese Biomedical Retrieval System (Appendix 5). In the US NIH database and WHO ICTRP Search Portal, we used the keywords 'oxcarbazepine' OR 'trileptal' OR 'trexapin' to search possible studies for further assessment, but we did not limit by type of participant or trial design.
Searching other resources
We wrote to the companies (e.g. Novartis Pharmaceuticals) who made oxcarbazepine and to pain experts asking for information about other completed or ongoing trials. We examined the Novartis clinical trials results database with the assistance of the Cochrane Neuromuscular Managing Editor. We also searched reference lists of review articles and included studies.
Data collection and analysis
Two review authors (NC and MZ) independently selected the studies for inclusion, assessed their risk of bias and extracted relevant data. For the first version of the review, the Cochrane Neuromuscular Managing Editor also assisted in the identification of trial reports by performing an independent review of English language search results. One review author (NC) entered the data into Review Manager 5 (RevMan 2014), and a second review author (MZ) checked the data entry. The review authors resolved disagreements through discussion.
Selection of studies
Two review authors (NC and MZ) independently scrutinised titles and abstracts identified by the searches to determine which studies might fulfil the selection criteria. We obtained full reports of potentially eligible studies (when available) to determine whether they met inclusion criteria for the review. We excluded studies involving participants with chronic headache and migraine. A third review author (LH) helped to arbitrate if the other two review authors could not reach agreement.
We collated multiple reports of the same study, so that each study, rather than each report, was the unit of interest in the review.
Data extraction and management
Two review authors (NC and MZ) independently extracted data from included trials using a specially designed data extraction form, which they piloted on two studies. They extracted the following data.
Participants: number, age range, gender, type of neuropathic disorder, setting.
Intervention: dosing regimen, duration, route of administration.
Control: placebo, other intervention.
Outcomes: analgesic outcome measures and results, withdrawals and adverse effects (minor and serious).
Design: methods of randomisation, study design (parallel group, cross‐over), treatment duration and duration of study follow‐up, and whether specifically designed to measure pain.
We also included 'Risk of bias' criteria on the data extraction form recording sequence generation, allocation concealment, blinding, incomplete outcome data, selective outcome reporting, and other concerns about bias. One review author (NC) entered data into Review Manager 5 (RevMan 2014) and a second review author (MZ) checked the data entry.
Assessment of risk of bias in included studies
Two review authors (MZ and NC) independently assessed 'Risk of bias' according to the methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). They resolved any disagreement by discussion.
The review authors considered six specific domains (namely sequence generation, allocation concealment, blinding, incomplete outcome data, selective outcome reporting and other bias). They assessed each included study in relation to the criteria as low, high or unclear risk of bias.
Measures of treatment effect
For dichotomous outcomes, we calculated risk ratios (RR) with 95% confidence intervals (CIs) (Deeks 2011).
For continuous data, we planned to calculate the weighted mean difference (WMD) or standardised mean difference (SMD). The included trials reported only dichotomous outcomes.
Unit of analysis issues
We planned to analyse cross‐over trials either using only the data from the first treatment period, or using data from both treatment periods provided that the washout period was adequate. If there had been multiple cross‐over trials, we would have analysed the results with the generic inverse variance method in Review Manager 5 (RevMan 2014). For the only included cross‐over trial in the updated review (Demant 2014), we reported data from both treatment periods as there was an adequate washout period.
Dealing with missing data
We dealt with missing data, such as missing standard deviations, by attempting to contact the trial authors via email or telephone. If participant withdrawal led to missing data, we conducted an intention‐to‐treat (ITT) analysis whenever possible. For dichotomous outcomes, we regarded participants with missing outcome data as treatment failures and included these in the analysis. For continuous outcomes, we planned to carry forward the last recorded value for participants with missing outcome data (Higgins 2011c).
Assessment of heterogeneity
We performed formal statistical testing of heterogeneity between the trials using Review Manager 5 (RevMan 2014). We assessed heterogeneity amongst trials using the Chi² test. We used a P value of 0.10 to determine statistical significance. A P value less than 0.10 might indicate a problem with the heterogeneity of combined data. We also used I² values to quantify inconsistency across studies, which reflected the percentage of the variability in effect estimates that was due to heterogeneity rather than sampling error. An I² value between 30% and 50% might represent moderate heterogeneity, while a value above 50% suggests substantial heterogeneity (Deeks 2011).
Data synthesis
We presented the pooled outcomes data from all similar studies using RRs and 95% CIs.
We reported results as number needed to treat for an additional beneficial outcome (NNTB) for the specified percentage change in pain scores, and number needed to treat for an additional harmful outcome (NNTH) for mild and serious adverse drug reactions.
We planned to perform analyses using a fixed‐effect model and use a random‐effects model to test the sensitivity of the results to the model. Where appropriate, we combined data from included studies using Review Manager 5 (RevMan 2014).
'Summary of findings' table
We included a 'Summary of findings' table for comparisons that included more than one trial to present the main findings of the review, including information about quality of evidence, magnitude of effects and sum of available data on the main outcomes (Schünemann 2011a). We included the following outcomes.
Reduction in participant‐reported pain scores byat least 50% from baseline.
Reduction in participant‐reported pain scores by at least 30% from baseline.
Participants 'much' or 'very much' improved after 16 weeks of treatment.
Serious adverse effects.
We used the five GRADE considerations (study limitations, inconsistency of effect, imprecision, indirectness and publication bias) to assess the quality of a body of evidence (studies that contributed data for the prespecified outcomes) (Ryan 2016). We downgraded the evidence from 'high quality' by one level where one of these factors was present to a serious degree and two levels if very serious. Only RCTs (including randomised cross‐over trials) were eligible for the review and reasons for upgrading (large effect, dose response, and plausible confounding factors) were therefore not applicable. We used methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b; Schünemann 2011b). We used GRADEpro software (GRADEpro 2008; Ryan 2016). We justified all decisions to downgrade or upgrade the quality of the evidence using footnotes and made comments to aid the reader's understanding of the review where necessary.
Subgroup analysis and investigation of heterogeneity
We performed a post hoc subgroup analysis of different doses of oxcarbazepine for painful diabetic neuropathy. If data had been available, we would have undertaken subgroup analysis according to the types of neuropathic pain (peripheral neuropathic pain and central neuropathic pain).
Sensitivity analysis
We planned to conduct sensitivity analyses to examine the effects of excluding studies with a moderate or high risk of bias, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). Since all the included trials had a similar risk of bias, we did not perform a sensitivity analysis.
This review has a published protocol (Zhou 2009). We described deviations from the protocol in Differences between protocol and review.
Results
Description of studies
Results of the search
The electronic searches for this update retrieved 25 references from the Cochrane Neuromuscular Specialised Register, 30 from CENTRAL, 91 from MEDLINE, 123 from Embase, three from Database of Abstracts of Reviews of Effects (DARE), 48 from the Chinese Biomedical Retrieval System, 63 from ClinicalTrials.gov and 70 from the WHO ICTRP. From the searches in other resources, including replies from pharmaceutical companies, company databases and the reference lists of found articles, we retrieved 10 additional references, one of which was an ongoing trial (NCT02219373). One study in peripheral neuropathic pain (NCT01302275) categorised as ongoing in the previous version of this review has since been completed and we included it in the review (Demant 2014). From the total of 404 references (excluding duplicates), we identified 48 records reporting the efficacy or safety, or both, of oxcarbazepine for the treatment of different neuropathic pain conditions. Most records did not meet the entry criteria of the present review because the titles or abstracts indicated that they were open‐label clinical trials comparing the efficacy and safety outcomes at the end of studies with the baseline conditions. Of the 29 remaining references selected for further consideration, we excluded 14 references for 13 studies (see Characteristics of excluded studies table). We included five trials (described in 14 references) that met the eligibility criteria (Beydoun 2006; Demant 2014; Dogra 2005; Grosskopf 2006; Novartis 2004) (see Characteristics of included studies table).
In this update, we found one ongoing trial (NCT02219373). This is a double‐blind RCT to assess the efficacy and safety of gabapentin and oxcarbazepine for the treatment of chronic neuropathic pain in children. The online report provided explicit inclusion and exclusion criteria and the estimated number of participants to be enrolled was 60. Participants in the two experimental groups receive either gabapentin or oxcarbazepine liquids orally and the dose is gradually increased over 13 days according to the participants' weight, while placebo liquid is escalated in similar mode. The primary outcome measure is the frequency of successful treatment two to eight weeks after assignment of intervention. Secondary outcomes are pain scores at rest and evoked manoeuvres, functional disability scores and frequencies of adverse effects. The trial started in October 2014, and was estimated to be completed in October 2016. The completion date has passed and the study status has not been verified in more than two years up to 15 January 2017, neither have we found a published report. See Characteristics of ongoing studies table. Figure 1 shows a flow chart illustrating study selection.
1.
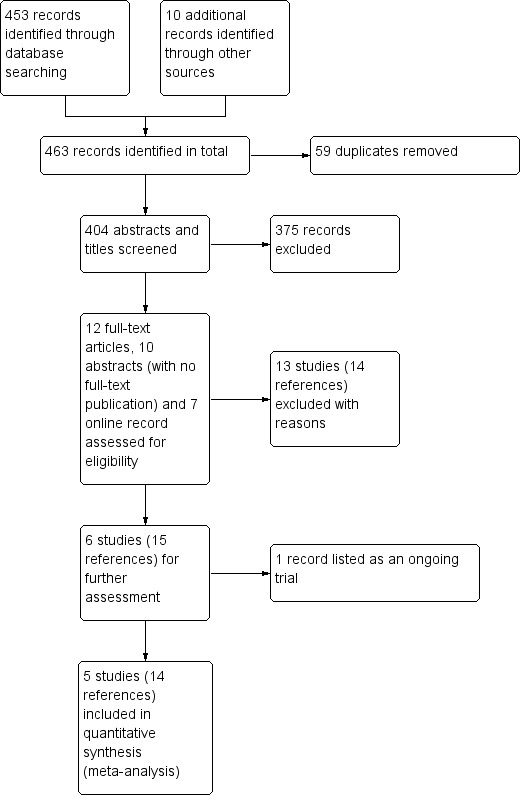
Study flow diagram.
We obtained all data relating to the included studies from published articles or results reported online. We attempted to contact with authors of the included trials, but received no responses.
Included studies
Three included studies involved 634 participants with painful diabetic neuropathy (Beydoun 2006; Dogra 2005; Grosskopf 2006), and one study involved 145 participants with neuropathic pain due to radiculopathy (Novartis 2004). One trial, included for this first time in this updated version, enrolled 83 participants with peripheral neuropathic pain due to polyneuropathy (n = 38), peripheral nerve injury (n = 38) or postherpetic neuralgia (n = 7) (Demant 2014). We identified no eligible trials of oxcarbazepine for other types of neuropathic pain.
Design
Four included trials were from a series of studies supported by the manufacturer of oxcarbazepine (Novartis), so they had similar design, inclusion and exclusion criteria, efficacy and safety assessments (Beydoun 2006; Dogra 2005; Grosskopf 2006; Novartis 2004). They were all placebo‐controlled, double‐blind, parallel‐group trials, conducted in multiple clinics and research centres. The reported study periods differed: a total of 72 weeks for Dogra 2005, and 18 weeks for Beydoun 2006, Grosskopf 2006, and Novartis 2004. Dogra 2005 consisted of a two‐week pre‐randomisation screening phase, an 18‐week double‐blind treatment phase and a 52‐week open‐label extension phase. The double‐blind treatment phase was further divided into a titration period of four weeks and a maintenance period of 12 weeks in each study, and an additional follow‐up period of two weeks for safety monitoring. Beydoun 2006, Grosskopf 2006, and Novartis 2004 consisted of the screening phase (two weeks) and the double‐blind treatment phase (16 weeks).
Demant 2014 was a placebo‐controlled, double‐blind trial, but it used a cross‐over design. The study period comprised a one‐week drug‐free baseline period and a six‐week treatment period before and after cross‐over. Before each baseline week, there was a washout period of at least one week.
Participants
All trials explicitly stated the inclusion and exclusion criteria.
The three studies investigating treatment for painful diabetic neuropathy recruited men and non‐pregnant, non‐lactating women aged 18 years or older with an established clinical diagnosis of type 1 or 2 diabetes mellitus and with a history of neuropathic pain for six months to five years prior to study entry (Beydoun 2006; Dogra 2005; Grosskopf 2006). Stable diabetic control and a certain pain severity and variability were also required.
The radiculopathy trial enrolled participants aged 18 years or older, of both genders, with a diagnosis of radiculopathy and evidence of motor deficit or reflex loss on examination, a history of neuropathic pain for six months or longer, and a VAS score of 50 or higher on a 100‐point scale (Novartis 2004).
Demant 2014 included men and women aged 18 years or older with definite or probable neuropathic pain for more than three months due to polyneuropathy, peripheral nerve injury or postherpetic neuralgia. The median pain intensity had to be at least 4 on an 11‐point NRS (with 0 being "no pain" and 10 "worst possible pain") during the first drug‐free baseline week. Among the randomised participants, 35 had the 'irritable nociceptor' phenotype, while 62 had the 'non‐irritable' nociceptor phenotype based on quantitative sensory testing (QST).
The baseline characteristics of participants were reported to be well matched between groups in each trial in terms of demography, duration of the primary disorders, and duration and severity of neuropathic pain. In total, the studies in this review included 473 males and 389 females, of whom 469 received oral oxcarbazepine, 310 received placebo and 83 received both oxcarbazepine and placebo in order, using a cross‐over design. For the parallel‐group trials, the mean duration of neuropathic pain (2.4 to 7.7 years) and mean VAS score (100‐point scale) of participants before treatment (70.7 to 76.9) in each group were also compared between groups and there were no significant differences.
Interventions
After the screening phase, eligible participants were randomised to placebo or to oxcarbazepine treatment with a given target dose. There was only one active group in Dogra 2005 (oxcarbazepine 1800 mg/day), Grosskopf 2006 (oxcarbazepine 1200 mg/day), Novartis 2004 (oxcarbazepine 1800 mg/day) and Demant 2014 (up to oxcarbazepine 2400 mg/day). Beydoun 2006 gave one of three doses of oxcarbazepine (300 mg, 600 mg or 900 mg) twice a day to participants in the different experimental groups. For the present analyses, 83 participants with painful diabetic neuropathy were analysed in the oxcarbazepine 600 mg/day group, 158 in the 1200 mg/day group and 157 in the 1800 mg/day group. A total of 71 participants with pain due to radiculopathy were analysed in the oxcarbazepine 1800 mg/day group, and 83 with painful polyneuropathy, nerve injury pain or postherpetic neuralgia were treated with up to 2400 mg/day dose (up to 1800 mg/day for participants aged 70 years or older). At the beginning of the treatment phase, participants took a small initial dose of study medication (300 mg/day) and then the oxcarbazepine was titrated over three to four weeks according to tolerability or the given maximum dose. During the subsequent maintenance period, treatment remained at the dose reached at the end of the titration period. Participants in the control group received matching placebo tablets. The mean (± standard deviation) oxcarbazepine dose during the maintenance period was 1455 ± 389 mg/day for Dogra 2005, 1091 ± 222 mg/day for Grosskopf 2006, and 1684 ± 181 or 1846 ± 152 mg/day for Demant 2014 (only subgroup data were available). Paracetamol was allowed in all trials as rescue medication, some other analgesics or drugs such as benzodiazepines, non‐steroidal anti‐inflammatory drugs, opioids or stable doses of selective serotonin reuptake inhibitors were permitted during the study phase in one trial (Novartis 2004), and tramadol in another (Demant 2014). The other three trials allowed none of these supplementary medications (Dogra 2005; Beydoun 2006; Grosskopf 2006).
Outcomes
Variables were evaluated after treatment for six weeks in Demant 2014, 16 weeks in Beydoun 2006 and Grosskopf 2006, and 18 weeks in Dogra 2005. Withdrawals were recorded in detail. There were high percentages of dropouts, and common reasons for discontinuation included adverse effects, protocol violations and unsatisfactory responses to treatment. Most efficacy analyses were performed with an ITT analysis, which comprised all randomised participants regardless of completeness of treatment or follow‐up, based on the last observation carried forward (LOCF) principle.
The outcome measures were similar among the four trials from the same series of studies. They all used a VAS for participants to report pain severity. For the three trials investigating painful diabetic neuropathy (Beydoun 2006; Dogra 2005; Grosskopf 2006) and the one trial of peripheral neuropathic pain (Demant 2014), the primary outcome was change in mean pain scores between baseline and each end of double‐blind treatment. The other trial compared the mean VAS score between groups during the last week of double‐blind treatment (Novartis 2004). Other efficacy variables used in the five trials included: participant's global assessment of therapeutic effect (GATE), Participant‐rated Global Impression of Change (PGIC), onset of therapeutic effect, rate of responders, pain relief, effect on evoked pain, durability of treatment effect, and sleep disturbances. Quality of life was assessed by SF‐36 and the Profile of Mood States (POMS) on up to five occasions, or Neuropathic Pain Symptom Inventory (NPSI) throughout the double‐blind phase. In addition, all included trials reported adverse effects during the treatment phase and clearly described serious adverse effects and adverse effects leading to discontinuation.
Excluded studies
We excluded 13 studies on inspection of the full text (Besi 2015; Beydoun 2002; Beydoun 2007; Brainer‐Lima 2003; Gong 2010; Hu 2010; Li 2011; Liebel 2001; Lindström 1987; Min 2016; Rémillard 1994; Venancio‐Ramirez 2004; Zhou 2010). Reasons for exclusion were: trials only reported the results in abstract form but they did not refer to any useable data of the prespecified outcome measures of our protocol; we contacted the sponsor company but they could not find the full text or any useful data (Beydoun 2002; Brainer‐Lima 2003; Liebel 2001); open‐label studies evaluating the changes between the pre‐ and post‐treatment conditions, without control groups (Beydoun 2007; Rémillard 1994); not blinded or of an adequately randomised design (Besi 2015; Gong 2010; Hu 2010; Li 2011; Zhou 2010); or short follow‐up duration, less than the minimum we specified in our protocol (Lindström 1987; Min 2016; Venancio‐Ramirez 2004). See Characteristics of excluded studies table.
Risk of bias in included studies
The five included trials all had a moderately large sample of participants and standardised protocols. All included studies were multicentre, placebo‐controlled, double‐blind trials.
Allocation
Based on the published methods, we considered allocation sequence generation and concealment to be well performed to minimise bias in four trials (Dogra 2005; Beydoun 2006; Grosskopf 2006; Demant 2014). For the only trial involving participants with radiculopathy, we were unable to obtain detailed information about the methods of allocation (Novartis 2004) (Characteristics of included studies table).
Blinding
In the double‐blind phase of the four trials of similar design, matched placebos were used and administered in the same way with the same initiated doses and increments (Beydoun 2006; Dogra 2005; Grosskopf 2006; Novartis 2004). The participants and personnel involved in the study performance and assessment were likely to have been blinded. In Demant 2014, participants were asked to guess which treatment they had received after each treatment period, and the correct guess rate was very high (67/83 after the first treatment period and 70/83 after the second treatment period). This might be the result of many participants experiencing adverse effects with oxcarbazepine, which could lead to unblinding. We agree with the trial authors' evaluation that the influence could go in both directions; some participants might think the treatment was ineffective because of the obvious adverse effects, while others might overrate the effect knowing they were on active treatment. It is difficult to evaluate the extent of possible bias in each direction (Demant 2014). Such a risk of bias may also apply to the other four included studies, but no relevant data were reported, so we considered these trials at unclear risk of performance bias for blinding of participants (Characteristics of included studies table).
Incomplete outcome data
All five trials clearly reported withdrawals after randomisation. The main reasons for discontinuation, in both the oxcarbazepine and placebo groups, included adverse effects, lack of efficacy and protocol violations, but the numbers of participants with incomplete outcome data and the reasons for withdrawal were imbalanced across groups (Beydoun 2006; Demant 2014; Dogra 2005; Grosskopf 2006; Novartis 2004). To deal with missing outcome data, the trial authors performed ITT analyses using the LOCF approach, whereby all withdrawn participants had the mean weekly VAS score from their last week of treatment carried forward for assessment. However, participants might have withdrawn before they found any obvious effect, so imputing outcomes in this way for missing participants (especially for early withdrawals) could have led to an underestimation of the true intervention effect.
There were no detailed descriptions of the results for completers and dropouts in the reports, so we could not conduct an ITT analysis by regarding participants with missing outcome data as failures with no response. Since the dropout rates were high or very high in all groups (19.1% to 56.3%), and they were significantly higher in the oxcarbazepine groups than the control groups (Beydoun 2006; Demant 2014; Dogra 2005; Grosskopf 2006; Novartis 2004), a potential risk of bias against oxcarbazepine due to incomplete outcome data should be considered (Higgins 2011b). This led us to downgrade the quality of the evidence.
Selective reporting
All outcomes specified in the protocols were reported in the published reports, so the four studies published in full text were probably free from selective reporting bias (Beydoun 2006; Demant 2014; Dogra 2005; Grosskopf 2006). For the one trial that was only published in abstract form, all expected outcomes in the protocol were reported online (Novartis 2004). To avoid publication bias, we included the unpublished data from this trial.
Other potential sources of bias
We identified no other potential source of bias.
Overall risk of bias
We rated all five trials as at a high overall risk of bias mainly because of a large and imbalanced proportion of missing outcome data across groups, especially those due to adverse effects of the interventions (Figure 2).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1
We planned to evaluate the effects of treatment after eight weeks, but we could not extract the relevant data because 1. in the cross‐over trial, oxcarbazepine or placebo was used for only six weeks in each treatment period (Demant 2014); 2. the other four included trials reported outcomes 16 weeks after starting the treatment; and 3. the rates of change in the measured outcomes per week were not constant (Beydoun 2006; Dogra 2005; Grosskopf 2006; Novartis 2004). Therefore, we extracted and analysed data measured at the end of the follow‐up period in each study, and subgrouped outcomes according to different follow‐up durations where possible. We analysed the effects of four daily doses (600 mg, 1200 mg, 1800 mg and 2400 mg) of oral oxcarbazepine versus placebo for painful diabetic neuropathy; neuropathic pain due to radiculopathy; or due to polyneuropathy, peripheral nerve injury or postherpetic neuralgia. There were no eligible trials of oxcarbazepine at other doses, other administration routes, compared to any other intervention or for other types of neuropathic pain.
Four included trials measured pain severity on a 100‐unit VAS, where 'no pain' was at the left end and 'worst pain imaginable' at the right end (Beydoun 2006; Dogra 2005; Grosskopf 2006; Novartis 2004). The other trial used an 11‐point NRS to rate participants' pain intensity, with 0 being 'no pain' and 10 'worst possible pain' (Demant 2014). Authors did not always report the proportion of participants whose pain scores were reduced by at least 50% or at least 30% from the baseline, which were the primary and first secondary outcome measures in our review. One of the primary efficacy variables in the trial was the mean change in pain score from baseline to the end of double‐blind treatment, but there was extensive variability in the results. Since different types of neuropathy have different mechanisms and characteristics that could affect treatment efficacy, we presented results for each condition separately where possible, with a separate heading for neuropathic pain of mixed aetiologies because the results for each different condition were not available from Demant 2014.
Oxcarbazepine versus placebo for painful diabetic neuropathy
Three trials investigated the effects of oxcarbazepine in painful diabetic neuropathy (Beydoun 2006; Dogra 2005; Grosskopf 2006).
Primary outcome
Reduction in participant‐reported pain scores by at least 50% from the baseline
Dogra 2005 showed that the proportion of participants experiencing at least 50% pain relief was 24/69 (34.8%) in the oxcarbazepine 1800 mg/day group (34.8%) versus 14/77 (18.2%) in the placebo group (RR 1.91, 95% CI 1.08 to 3.39; P = 0.03; n = 146; Analysis 1.1; Figure 3). The NNTB was 6 (95% CI 3 to 41).
1.1. Analysis.
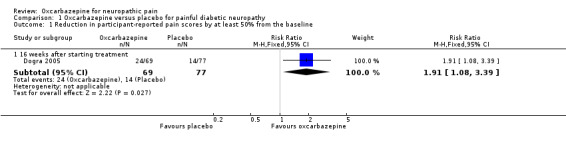
Comparison 1 Oxcarbazepine versus placebo for painful diabetic neuropathy, Outcome 1 Reduction in participant‐reported pain scores by at least 50% from the baseline.
3.
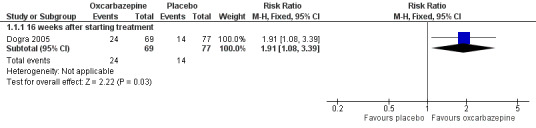
Forest plot of comparison: 1 Oxcarbazepine versus placebo for painful diabetic neuropathy, outcome: 1.1 Reduction in participant‐reported pain scores by at least 50% from the baseline.
There were no data from Beydoun 2006 or Grosskopf 2006 for this outcome, so we were unable to resolve the discrepancies between the included trials. The included trial had significantly higher withdrawal rates from the oxcarbazepine group than the control group, so pooled results may also be at risk of bias from incomplete outcome data.
We assessed the quality of the evidence as very low (imprecision, publication bias, inconsistency and study limitations were all present to a serious degree) (Table 1).
Secondary outcomes
Reduction in participant‐reported pain scores by at least 30% from the baseline
Dogra 2005 provided data for comparisons between baseline and the 16th treatment week. A higher percentage of participants in the oxcarbazepine group experienced at least 30% pain relief compared with placebo (31/69 (44.9%) participants with oxcarbazepine versus 22/77 (28.6%) with placebo; RR 1.57, 95% CI 1.01 to 2.44; P = 0.04; n = 146; Analysis 1.2). The NNTB was 6 (95% CI 3 to 114).
1.2. Analysis.
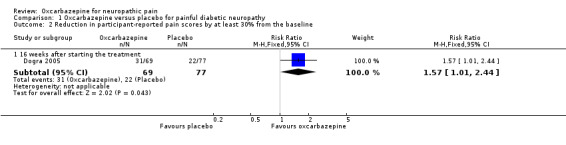
Comparison 1 Oxcarbazepine versus placebo for painful diabetic neuropathy, Outcome 2 Reduction in participant‐reported pain scores by at least 30% from the baseline.
Beydoun 2006 and Grosskopf 2006 did not provide data for reduction in participant‐reported pain scores by at least 30% from the baseline.
We assessed the quality of this evidence as very low (imprecision, publication bias, inconsistency and study limitations were all present to a serious degree) (Table 1).
Participants' global impression of their change in pain
Two studies reported data for participants' global impression of their change in pain (Beydoun 2006; Dogra 2005); the other study reported only that there was no significant difference (Grosskopf 2006).
The two studies providing data used GATE to assess participants' global impression of change in neuropathic pain, applying a 7‐point Likert scale with scores ranging from ‐3 (very much improved) to 3 (very much deteriorated). We compared the proportion of participants in the oxcarbazepine and placebo groups who reported all degrees of improvement (‐3 to ‐1 points for GATE) and who were 'much' or 'very much' improved (‐3 to ‐2 points for GATE) after 16 weeks of treatment. The meta‐analysis was at potential risk of bias from imbalanced incomplete outcome data and inconsistent placebo responses (Table 1).
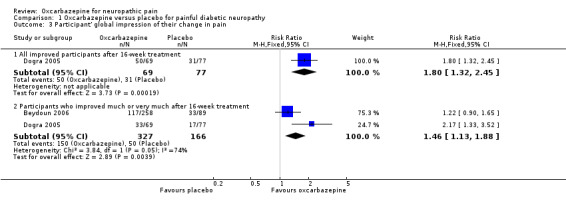
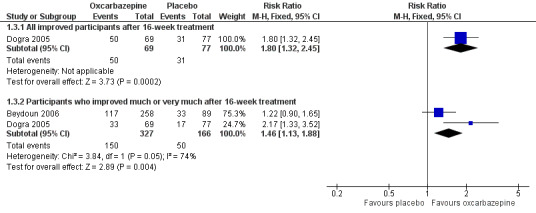
In Dogra 2005, more participants who received oxcarbazepine felt globally improved compared with participants who received placebo (50/60 (72.5%) with participants versus 31/77 (40.3%) with placebo; RR 1.80, 95% CI 1.32 to 2.45; P = 0.0002; n = 146).
Pooling relevant data from Beydoun 2006 and Dogra 2005, pain was 'much' or 'very much' improved in 150/327 (45.9%) participants receiving oxcarbazepine versus 50/166 (30.1%) participants receiving placebo (RR 1.46, 95% CI 1.13 to 1.88; P = 0.004; n = 493; Analysis 1.3; Figure 4). The corresponding NNTB score change across all dosages of oxcarbazepine was 6 (95% CI 4 to 14). However, there was a high degree of heterogeneity between the two included studies (I² = 74%, Chi² = 3.84, degrees of freedom (df) = 1, P = 0.05).
1.3. Analysis.
Comparison 1 Oxcarbazepine versus placebo for painful diabetic neuropathy, Outcome 3 Participant' global impression of their change in pain.
4.
Forest plot of comparison: 1 Oxcarbazepine versus placebo for painful diabetic neuropathy, outcome: 1.3 Participant' global impression of their change in pain.
Overall, we judged these data at very low quality because of serious study limitations (attrition bias), inconsistency and a serious risk of publication bias.
Overall quality of life measures
All studies performed the SF‐36 Health Survey on different occasions during the double‐blind phase including at the eighth week, which was when we specified to measure this outcome in our protocol (Zhou 2009). However, the scores were not recorded for each follow‐up period and some information could only be obtained at week 16 or at the final visit. In Dogra 2005, the aggregate mental health score was significantly different between oxcarbazepine‐treated and placebo‐treated participants (in favour of oxcarbazepine, P = 0.03). In Beydoun 2006 and Grosskopf 2006, the trial authors stated only that there were no significant differences between the oxcarbazepine groups and placebo groups for the quality of life questionnaire.
Adverse effects
Adverse effects often started during the titration period and resolved before studies finished. Frequently reported adverse effects included dizziness, headache, nausea, somnolence, fatigue, vomiting, back pain, diarrhoea, tremor, skin rash and blurred vision. Most adverse effects were mild to moderate in intensity and resulted in dosage adjustment.
Adverse effects leading to withdrawal
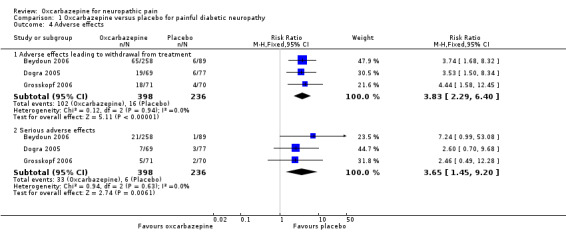
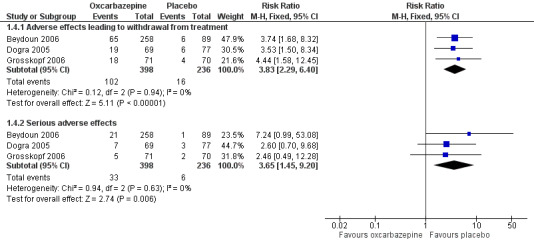
Pooled data from three studies showed that more participants receiving oxcarbazepine experienced adverse effects leading to withdrawal than participants receiving placebo (102/398 (25.6%) with oxcarbazepine group versus 16/236 (6.8%) with placebo group discontinued treatment prematurely owing to adverse effects (RR 3.83, 95% CI 2.29 to 6.40; P < 0.00001; n = 634; Analysis 1.4) (Beydoun 2006; Dogra 2005; Grosskopf 2006).
1.4. Analysis.
Comparison 1 Oxcarbazepine versus placebo for painful diabetic neuropathy, Outcome 4 Adverse effects.
Serious adverse effects
Serious adverse effects occurred in a small number of participants; some effects were considered related to treatment, such as erythema multiforme, Steven Johnson syndrome, severe fatigue, weakness, light‐headedness and headache leading to hospitalisations. Although the trials each reported no significant difference between oxcarbazepine and placebo for serious adverse events, our meta‐analysis of the results showed a higher frequency of serious adverse events in the oxcarbazepine group (RR 3.65, 95% CI 1.45 to 9.20; P = 0.006; n = 634; Analysis 1.4; Figure 5; Table 1). The NNTH, defined as number with a serious adverse drug reaction was 17 (95% CI 11 to 42). The I² statistic was 0% for both of the above two pooled results, indicating no important heterogeneity across included trials.
5.
Forest plot of comparison: 1 Oxcarbazepine versus placebo for painful diabetic neuropathy, outcome: 1.4 Adverse effects.
We judged this evidence as moderate quality. The low event rate reduced the precision of the effect estimate.
Post‐hoc subgroup analysis
Different daily doses of oxcarbazepine versus placebo for painful diabetic neuropathy (600 mg/day, 1200 mg/day, 1800 mg/day)
Participants' global impression of their change in pain
We performed a subgroup analysis with participants with painful diabetic neuropathy receiving different doses of oxcarbazepine (600 mg/day, 1200 mg/day or 1800 mg/day). There was suggestion of a dose‐effect relationship in one dose‐ranging study (Beydoun 2006). Although Demant 2014 used a higher daily dose (up to oxcarbazepine 1800 mg/day to 2400 mg/day), we did not extract data from this trial for dose‐effect analysis because of the many differences in pain aetiology, study design and treatment duration.
Pooled data from two studies showed that the percentage of participants who improved 'much' or 'very much' in pain (‐3 to ‐2 points for GATE) was greater in the 1800 mg/day oxcarbazepine group (76/157 (48.4%) with oxcarbazepine 1800 mg/day versus 50/166 (30.1%) with placebo; RR 1.64, 95% CI 1.01 to 2.67; P = 0.05; n = 323) (Beydoun 2006; Dogra 2005). In Beydoun 2006, the results showed no difference between oxcarbazepine 600 mg/day or 1200 mg/day and placebo (600 mg/day: 30/83 (36.1%) with oxcarbazepine 600 mg/day versus 33/89 (37.1%) with placebo; RR 0.97, 95% CI 0.66 to 1.45; P = 0.90; n = 176; 1200 mg/day: 44/87 (50.6%) with oxcarbazepine 1200 mg/day versus 33/89 (37.1%) with placebo; RR 1.36, 95% CI 0.97 to 1.92; P = 0.07; n = 176; Analysis 2.1; Figure 6). NNTB score change could not be calculated for the oxcarbazepine 600 mg/day group because the event rate was lower than that of the placebo group. The NNTB score change was 7 (95% CI 4 to 97) in the oxcarbazepine 1200 mg/day group and 6 (95% CI 4 to 13) in the oxcarbazepine 1800 mg/day group.
2.1. Analysis.
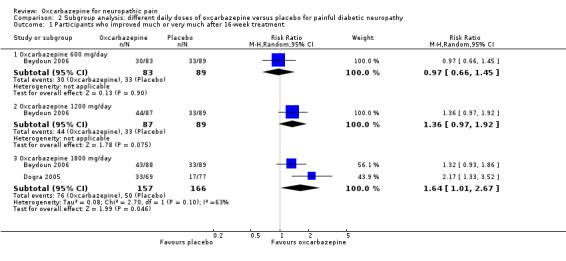
Comparison 2 Subgroup analysis: different daily doses of oxcarbazepine versus placebo for painful diabetic neuropathy, Outcome 1 Participants who improved much or very much after 16‐week treatment.
6.
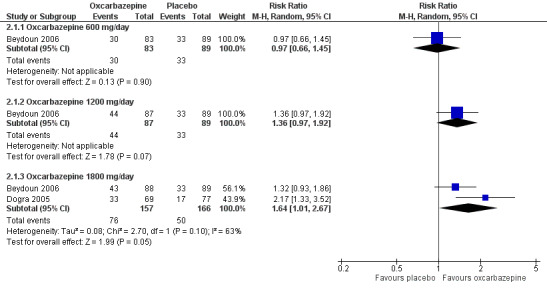
Forest plot of comparison: 4 Subgroup analysis: different daily doses of oxcarbazepine versus placebo for painful diabetic neuropathy, outcome: 4.1 Participants who improved much or very much after 16‐week treatment.
Adverse effects leading to withdrawal from treatment
Adverse effects leading to discontinuation occurred more frequently with the higher daily doses of oxcarbazepine (1200 mg/day: 38/158 with oxcarbazepine 1200 mg/day versus 10/159 with placebo; RR 3.83, 95% CI 1.97 to 7.41; P < 0.0001; n = 317; Beydoun 2006; Grosskopf 2006; 1800 mg/day: 55/157 with oxcarbazepine 1800 mg/day versus 12/166 with placebo; RR 4.83, 95% CI 2.68 to 8.70; P < 0.00001; n = 323; Beydoun 2006; Dogra 2005). In Beydoun 2006, the result for adverse effects leading to withdrawal between oxcarbazepine 600 mg/day and placebo was imprecise and did not rule out equivalence (RR 1.61, 95% CI 0.60 to 4.32; P = 0.35; n = 172) (Analysis 2.2). Although the rates of adverse effects increased with dose, the CIs were wide and overlapping.
2.2. Analysis.
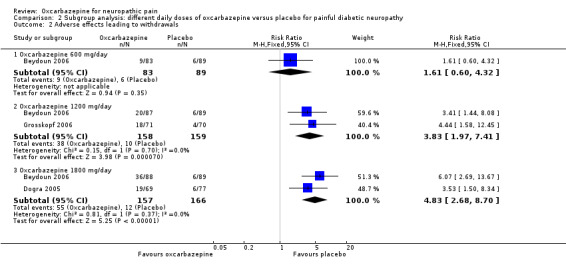
Comparison 2 Subgroup analysis: different daily doses of oxcarbazepine versus placebo for painful diabetic neuropathy, Outcome 2 Adverse effects leading to withdrawals.
Serious adverse effects
Comparisons of serious adverse effects in the oxcarbazepine and placebo groups showed a similar trend towards a dose‐dependent relationship with oxcarbazepine 1800 mg/day having the highest percentage of serious adverse effects (oxcarbazepine 1800 mg/day: 17/157 (10.8%); n = 323; Beydoun 2006; Dogra 2005; oxcarbazepine 1200 mg/day: 14/158 (8.9%); n = 317; Beydoun 2006; Grosskopf 2006; oxcarbazepine 600 mg/day: 2/83 (2.4%); n = 172; Beydoun 2006). Compared to the placebo group, more people receiving the two higher doses of oxcarbazepine had adverse effects (Analysis 2.3). The NNTH score was 78 (95% CI 19 to ∞) for oxcarbazepine 600 mg/day, 14 (95% CI 8 to 49) for oxcarbazepine 1200 mg/day and 12 (95% CI 7 to 33) for oxcarbazepine 1800 mg/day.
2.3. Analysis.
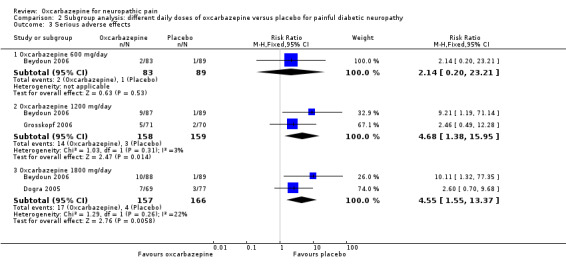
Comparison 2 Subgroup analysis: different daily doses of oxcarbazepine versus placebo for painful diabetic neuropathy, Outcome 3 Serious adverse effects.
Oxcarbazepine versus placebo for neuropathic pain due to radiculopathy
We found one trial including 145 participants with radiculopathy comparing oxcarbazepine versus placebo (Novartis 2004).
Primary outcome
Reduction in participant‐reported pain scores by at least 50% from the baseline
Seven of 70 (10.0%) participants receiving oxcarbazepine had at least 50% pain relief versus 14/74 (18.9%) participants receiving placebo (RR 0.53, 95% CI 0.23 to 1.23; P = 0.14; n = 144; Analysis 3.1).
3.1. Analysis.
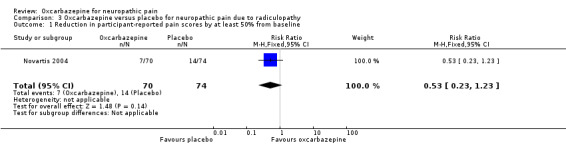
Comparison 3 Oxcarbazepine versus placebo for neuropathic pain due to radiculopathy, Outcome 1 Reduction in participant‐reported pain scores by at least 50% from baseline.
Low event rates and the small size of the study made these results very imprecise. The trial was at high risk of attrition bias.
Secondary outcomes
Reduction in participant‐reported pain scores by at least 30% from the baseline
The trial did not report reduction in participant‐reported pain scores by at least 30% from the baseline.
Participants' global impression of their change in pain
The number of participants improved (all degrees) was 35/71 (49.3%) in the oxcarbazepine group versus 27/74 (36.5%) in the placebo group (RR 1.35, 95% CI 0.92 to 1.98; P = 0.12; n = 145) and the number of participants who 'much' or 'very much' improved was 17/71 (23.9%) in the oxcarbazepine group versus 11/74 (14.9%) in the placebo group (RR 1.61, 95% CI 0.81 to 3.20; P = 0.17; n = 145; Analysis 3.2).
3.2. Analysis.
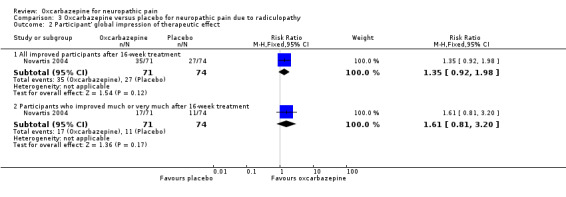
Comparison 3 Oxcarbazepine versus placebo for neuropathic pain due to radiculopathy, Outcome 2 Participant' global impression of therapeutic effect.
These results were imprecise and, although they favoured oxcarbazepine, did not rule out a lack of effect. The trial was at high risk of attrition bias.
Overall quality of life measures
The trial did not report overall quality of life measures.
Adverse effects
Any adverse effect
In Novartis 2004, the proportion of participants who experienced any type of adverse effect was 68/71 (95.8%) in the oxcarbazepine group and 58/74 (78.4%) in the placebo group, with therefore a slightly higher rate of adverse effects with oxcarbazepine (RR 1.22, 95% CI 1.07 to 1.39; P = 0.002; n = 145).
Adverse effects leading to withdrawal from treatment
The proportion of participants who withdrew due to adverse effects was also higher in the oxcarbazepine group than in the placebo group (30/71 (42.3%) participants with oxcarbazepine versus 11/74 (14.9%) participants with placebo; RR 2.84, 95% CI 1.55 to 5.23; P = 0.0008; n = 145).
Serious adverse effects
There were six serious adverse effects (8.5%) in the oxcarbazepine group and two serious adverse effects (2.7%), including one death, in the control group (RR 3.13, 95% CI 0.65 to 14.98; P = 0.15; n = 145) (Analysis 3.3). The size of the trial and the low event rate made this result very imprecise. The trial was at high risk of attrition bias.
3.3. Analysis.
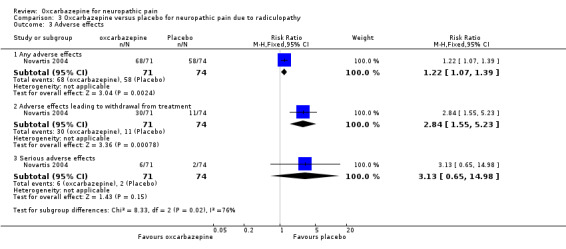
Comparison 3 Oxcarbazepine versus placebo for neuropathic pain due to radiculopathy, Outcome 3 Adverse effects.
Oxcarbazepine versus placebo for neuropathic pain of various origins
We found one trial comparing oxcarbazepine versus placebo for neuropathic pain of various origins. Demant 2014 was a cross‐over study of 83 participants with peripheral neuropathic pain due to polyneuropathy, surgical or traumatic nerve injury, or postherpetic neuralgia. We attempted to contact the authors to obtain separate data for each treated condition but without success. Therefore, we presented combined results of participants with different types of neuropathic pain.
The investigators performed "repeated‐measurements analysis of variance (ANOVA) with treatment and phenotype‐treatment interaction as factors, baseline value as covariate, patient as random effect, and period as fixed effect for continuous outcomes". They used Fisher's exact test or McNemar's change test for dichotomous outcomes.
Primary outcome
Reduction in participant‐reported pain scores by at least 50% from the baseline
Demant 2014 reported that 16/83 (19.3%) participants had a 50% or more reduction in total pain score on oxcarbazepine and 4/83 (4.8%) participants receiving placebo. The NNTB was 7 (95% CI 4 to 22). The trial was at high risk of attrition bias and unblinding.
Secondary outcome measures
Reduction in participant‐reported pain scores by at least 30% from the baseline
The trial did not report reduction in participant‐reported pain scores by at least 30% from the baseline.
Participants' global impression of their change in pain
In Demant 2014, investigators used PGIC to record participants' global impression of change, applying a 7‐point scale but scoring in the opposite direction to GATE (3 meant much improved, while ‐3 meant much worse). The trial authors reported no statistically significant difference between oxcarbazepine and placebo based on PGIC (score difference 0.23, P = 0.277). We could not obtain any relevant data from the trial for meta‐analysis.
Overall quality of life measures
Demant 2014 reported that health‐related quality of life was unaltered by oxcarbazepine, but details were not available.
Adverse effects
Among the mixed group of 83 participants in this trial, the trialists reported that adverse effects were the reason for all premature trial withdrawals (13/96 (13.5%) with oxcarbazepine versus 1/85 (1.2%) with placebo) (RR 11.51, 95% CI 1.54 to 86.15). They also found that most participants experienced adverse effects during oxcarbazepine treatment (frequency 94%, 95% CI 89% to 99%), but any adverse effect or at least one adverse effect was experienced by 36% of participants during placebo treatment (95% CI 26% to 47%). The trial authors listed several most frequently occurring adverse effects, and most were mild. No details were available to compare serious adverse effects between treatment and placebo groups (Demant 2014). The trial was at high risk of attrition bias and unblinding.
Discussion
Summary of main results
We included five RCTs, involving 862 participants; all of which compared oral oxcarbazepine with placebo for the treatment of neuropathic pain. Three trials investigated people with painful diabetic neuropathy, one involved participants with neuropathic pain due to radiculopathy and one studied neuropathic pain of mixed aetiologies. Four trials had similar designs, methods, treatment and follow‐up periods, which allowed for the pooling of some, but not all, relevant efficacy and safety data (Beydoun 2006; Dogra 2005; Grosskopf 2006; Novartis 2004). The results indicated a possible slight effect for oxcarbazepine in relieving painful DPN (measured as at least 50% and at least 30% pain relief from baseline on a VAS) but this evidence was of very low quality. Oxcarbazepine also increased numbers of people 'much' or 'very much' improved according to the participant's global impression of change, but with high heterogeneity (Table 1). We downgraded the quality of the evidence for each outcome measure to very low according to GRADE considerations, because of the small number of included studies and participants, inconsistency among studies, imprecision, attribution bias and because we strongly suspected publication bias (Table 1). As a result, we are uncertain whether oral oxcarbazepine is effective for the treatment of painful diabetic neuropathy.
Results from the subgroup analyses of different daily doses in DPN suggested a trend towards a dose‐effect relationship for global impression of improvement in pain and adverse effects. The highest daily dose of oxcarbazepine (1800 mg/day) had the greatest effect on pain relief but also led to the most adverse effects, especially serious ones.
In people with neuropathic pain of mixed aetiologies (polyneuropathy, surgical or traumatic nerve injury, or postherpetic neuralgia), oxcarbazepine also increased the number of people with at least 50% pain relief from baseline compared to placebo. For participants with radiculopathy, data from one trial showed that oxcarbazepine neither relieved pain nor improved participants' global impression of change compared to placebo. However, for each condition, data were available from only a single trial with a small sample size, which also led the quality of evidence to be low.
The benefits of oxcarbazepine were seen in two studies that used a higher dose (1800 mg/day to 2400 mg/day) of oxcarbazepine (Demant 2014; Dogra 2005). The other two studies included diabetic neuropathy trials found no significant differences between each dose of oxcarbazepine and placebo (Beydoun 2006; Grosskopf 2006), although there was a statistical trend favouring the two higher doses (1200 mg/day and 1800 mg/day) (Beydoun 2006). Neither of these studies were included a in meta‐analysis, which is thus heavily biased towards the reports that found benefit from oxcarbazepine. The investigators themselves put forward a number of factors to explain the differences in outcome among the trials. Besides the higher dose that was used in Dogra 2005, its lower placebo response might at least partially contribute to the statistically significant findings. In the placebo group of Dogra 2005, the mean change of VAS scores from the baseline to treatment end was ‐14.7 units, and about 22% of participants experienced an obvious or significant improvement; the corresponding values were ‐19.1 units and 37.3% for Beydoun 2006 and ‐22.0 units for Grosskopf 2006 (percentage of improved participants was not reported). This phenomenon can be partly explained by the substantial variation in placebo effect among people with painful diabetic neuropathy (Backonja 2003; Beydoun 2006; Eisenberg 2007). Furthermore, the effect of withdrawals due to adverse effects should also be considered. In Dogra 2005, 6/77 (7.8%) participants withdrew from the placebo group because of adverse effects, compared to 19/69 (27.5%) participants who withdrew from the oxcarbazepine 1800 mg/day group. The percentage of withdrawals was lower in Demant 2014, at 13/96 (13.5%), even with a higher daily dose of up to 1800 mg/day to 2400 mg/day. Withdrawals were comparable in the placebo group of Beydoun 2006 (6/89 (6.7%) participants) but withdrawals in the oxcarbazepine 1800 mg group were much higher (36/88 (40.9%) participants). Although analyses were based on ITT populations, the impact of different withdrawal rates on the treatment‐effect estimation cannot be overlooked for reasons mentioned in the Risk of bias in included studies section.
Most adverse effects related to the study drugs were reported as mild to moderate in severity; however, the proportions of both any effects and of effects leading to withdrawal were higher in the oxcarbazepine group than the placebo group. The frequency of such adverse effects appeared to be dose related, based on our analyses.
Overall completeness and applicability of evidence
The RCTs in the present review included participants with common causes of neuropathic pain, including painful diabetic neuropathy, radiculopathy and peripheral neuropathic pain due to mixed aetiologies (polyneuropathy, peripheral nerve injury and postherpetic neuralgia). Relevant data for any outcome measure were only available from at most three trials, and we found no trials in people with other causes of neuropathic pain. The included trials all compared oral oxcarbazepine with placebo for the treatment of neuropathic pain. Although sharing a similar design, the included trials reported some outcomes in different ways; this prevented pooling of data from the three diabetic neuropathy trials (Beydoun 2006; Dogra 2005; Grosskopf 2006). In fact, their results (such as reduction in pain intensity) were inconsistent. For some outcome measures, we could only obtain data from the single trial in which results favoured oxcarbazepine (Dogra 2005). The two trials that reported no benefit did not provide data for the primary outcome or for some secondary outcomes (e.g. participant‐reported pain reduction scores of at least 30% from the baseline; all degrees of improvement in participants' global impression of their change in pain) when assessing the effects of oxcarbazepine in people with diabetic neuropathy.
Most of our results came from one or two trials. We found no results of some trials published only as abstracts. For example, we were unable to obtain data from studies assessing the effectiveness of oxcarbazepine in participants with trigeminal neuralgia, since they were only published in abstract form or in reviews (Beydoun 2002a; Beydoun 2002b; Carrazana 2003; Nasreddine 2007). It is strongly suspected that some publication bias may be present. Overall the results of the review should be applied with caution.
It has been suggested that some human leukocyte antigen (HLA) alleles (e.g. HLA‐B*1502 and HLA‐A*3101) are associated with carbamazepine‐induced skin‐related adverse drug reactions that are sometimes serious and potentially life‐threatening in certain populations (Illing 2013). This potential association was not investigated in included trials in this review. The data available were insufficient to assess rare adverse effects.
As specified in the newly included trial, pain phenotype based on putative mechanism of pain might also predict response to treatment. Demant 2014 compared the effect of oxcarbazepine in participants with and without the irritable nociceptor phenotype, as defined by hypersensitivity and preserved small nerve fibre function. The investigators found that oxcarbazepine was more efficacious for relief of peripheral neuropathic pain among participants with the irritable nociceptor phenotype (Demant 2014). Therefore, the investigators hypothesised that the inconsistent results of studies investigating analgesic effects of anticonvulsants with sodium channel blocking effects in people with neuropathic pain might be partly attributable to such pain phenotypes. These phenotypes were not distinguished in the other trials included in the present review, so we could not perform further analyses to test the hypothesis.
Quality of the evidence
Although the placebo response in neuropathic pain can vary considerably among trials, and is difficult to regulate and control, RCTs still provide the most powerful evidence on which to base conclusions about the effect of treatment (GRADE). The five included trials were well designed and conducted in multiple centres. However, risks of bias remain, mainly from incompleteness of outcome data and possible unblinding of participants due to obvious adverse effects. Two trials of oxcarbazepine in DPN remain unpublished and this introduces a potential risk of publication bias, particularly as the trials that did not provide data reported little or no benefit from oxcarbazepine. The considerable inconsistencies in placebo responses and outcomes of participants' global impression in the diabetic neuropathy trials could undermine the reliability of the combined results. Serious imprecision also exists, because the sample size and event numbers were small and the CI included both little or no effect and substantial effects. Mean pain severity at randomisation also differed from trial to trial but was not analysed further in this review because of limited data availability. These differences might impact the results to some extent. For these reasons, we rated the evidence quality for our main outcome measures as very low according to GRADE criteria (Table 1; GRADE).
Potential biases in the review process
To reduce the risk of attrition bias, we performed meta‐analyses on an ITT population using the LOCF approach; imputing outcomes for missing participants in this way could have led to an underestimation of the true effect. We could not obtain enough data to conduct an ITT analysis by regarding participants with missing outcome data as failures with no response.
We have searched systematically, including published and unpublished studies, without language restrictions. However, all the included studies were published in English, and most results came from small studies, and we found no results of some trials published only as abstracts. Therefore, publication bias should still be possible.
Agreements and disagreements with other studies or reviews
The efficacy of oxcarbazepine has also been investigated for types of neuropathic pain other than those assessed in the present review, for example trigeminal neuralgia. Several RCTs have reported comparable effectiveness of oxcarbazepine and carbamazepine in relieving pain and improving quality of life in participants with trigeminal neuralgia (Beydoun 2002a; Beydoun 2002b; Carrazana 2003; Nasreddine 2007). These studies have only been published as a meta‐analysis in abstract form (Beydoun 2002a; Beydoun 2002b) or in reviews (Carrazana 2003; Nasreddine 2007), in which detailed descriptions and data were unavailable. Thus, they could not be included in the current review. One postherpetic neuralgia study showed a significant decrease in VAS score and improvements in participants' quality of life but had an open‐label design (Criscuolo 2005). As a result, we cannot draw any conclusion about oxcarbazepine for the treatment of these types of neuropathic pain.
In agreement with the conclusions of our systematic review, the latest European Federation of Neurological Societies (EFNS) guidelines do not recommend oxcarbazepine for people with diabetic neuropathic pain because of insufficient and conflicting evidence (Attal 2006; Attal 2010). However, these guidelines recommend oxcarbazepine as one of the first‐line drugs for classical trigeminal neuralgia. In one retrospective study of a large cohort of participants, investigators concluded that the efficacy of oxcarbazepine was comparable to that of carbamazepine in treating trigeminal neuralgia, and that oxcarbazepine had a greater tolerability (Di Stefano 2014). In the absence of eligible RCTs, we were unable to draw any conclusions in this review about the use of oxcarbazepine for this type of neuropathic pain.
Authors' conclusions
Implications for practice.
This review found little evidence to support the effectiveness of oxcarbazepine in painful diabetic neuropathy, neuropathic pain from radiculopathy and a mixture of neuropathies. Some very‐low‐quality evidence suggests efficacy but small trials, low event rates, heterogeneity in some measures and a high risk of publication bias means that we have very low confidence in the measures of effect. Adverse effects, serious adverse effects and adverse effects leading to discontinuation are probably more common with oxcarbazepine than placebo, but the numbers of participants or event rates are low.
Implications for research.
More well‐designed, large, multicentre, randomised placebo‐ or active‐controlled trials investigating oxcarbazepine for neuropathic pain are needed. The response in neuropathic pain of other causes, and in participants with different pain phenotypes, should be investigated. A dose‐ranging design should be considered to determine whether a dose‐response relationship is present. The speed and period of titration, the observation and management of intolerance, and the most appropriate target dose might be studied further. Selective publication of studies or data should be avoided.
What's new
| Date | Event | Description |
|---|---|---|
| 27 February 2017 | New citation required and conclusions have changed | We included one new trial, revised results accordingly, reappraised and downgraded some of the GRADE assessments, and revised the conclusions. |
| 21 November 2016 | New search has been performed | We updated the literature searches. We found 8 new possibly appropriate references and incorporated them in the review. |
Acknowledgements
We are grateful for the assistance of Cochrane Neuromuscular.
This project was supported by the National Institute for Health Research (NIHR) via Cochrane Infrastructure funding to Cochrane Neuromuscular. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service or the Department of Health. Cochrane Neuromuscular is also supported by the Medical Research Council (MRC) Centre for Neuromuscular Diseases.
The Information Specialist of Cochrane Neuromuscular, Angela Gunn, provided literature searches.
Appendices
Appendix 1. NMD Register (CRS) search strategy
#1 oxcarbazepine OR trileptal* OR trexapin [REFERENCE] [STANDARD] #2 "neuropathic pain" OR "neuropathy pain" [REFERENCE] [STANDARD] #3 "cervical radiculopathy" or "lumber radiculopathy" [REFERENCE] [STANDARD] #4 MeSH DESCRIPTOR Radiculopathy [REFERENCE] [STANDARD] #5 diabet* [REFERENCE] [STANDARD] #6 MeSH DESCRIPTOR Diabetic Neuropathies Explode All [REFERENCE] [STANDARD] #7 "peripheral neuropath*" or "HIV neuropath*" [REFERENCE] [STANDARD] #8 postherpetic NEAR3 neuralgia [REFERENCE] [STANDARD] #9 MeSH DESCRIPTOR Spinal Cord Injuries Explode All [REFERENCE] [STANDARD] #10 "spinal cord injur*" or "trigeminal neuralgia" OR "trigeminus neuralgia" [REFERENCE] [STANDARD] #11 "complex regional pain syndrome" [REFERENCE] [STANDARD] #12 MeSH DESCRIPTOR Complex Regional Pain Syndromes Explode All [REFERENCE] [STANDARD] #13 #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 [REFERENCE] [STANDARD] #14 #1 and #13 [REFERENCE] [STANDARD] #15 (#1 and #13) AND (INREGISTER) [REFERENCE] [STANDARD]
Appendix 2. Cochrane Register of Controlled Trials (CENTRAL) search strategy
#1 (oxcarbazepine OR trileptal* OR trexapin):TI,AB,KY #2 ("neuropathic pain" OR "neuropathy pain"):TI,AB,KY #3 ("cervical radiculopathy" or "lumber radiculopathy"):TI,AB,KY #4 MESH DESCRIPTOR radiculopathy #5 diabet*:TI,AB,KY #6 MESH DESCRIPTOR Diabetic Neuropathies EXPLODE ALL TREES #7 ("peripheral neuropath*" or "HIV neuropath*"):TI,AB,KY #8 (postherpetic NEAR3 neuralgia):TI,AB,KY #9 MESH DESCRIPTOR Spinal Cord Injuries EXPLODE ALL TREES #10 "spinal cord injur*" or "trigeminal neuralgia" OR "trigeminus neuralgia" #11 "complex regional pain syndrome" #12 MESH DESCRIPTOR Complex Regional Pain Syndromes EXPLODE ALL TREES #13 #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 #14 #1 AND #13 #15 neuromusc:cc #16 #14 NOT #15
Appendix 3. MEDLINE (OvidSP) search strategy
Database: Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE(R) Daily and Ovid MEDLINE(R) <1946 to Present> Ovid MEDLINE(R) 1946 to November Week 2 2016 Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (469735) 2 controlled clinical trial.pt. (95071) 3 randomized.ab. (403958) 4 placebo.ab. (191867) 5 drug therapy.fs. (2035584) 6 randomly.ab. (285175) 7 trial.ab. (426009) 8 groups.ab. (1757094) 9 or/1‐8 (4181337) 10 exp animals/ not humans.sh. (4669151) 11 9 not 10 (3603015) 12 (oxcarbazepine or trileptal$ or trexapin).mp. (1784) 13 Radiculopathy/ (4487) 14 Diabetic Neuropathies/ (13937) 15 Neuralgia, Postherpetic/ (891) 16 Spinal Cord Injuries/ (34456) 17 Trigeminal Neuralgia/ (6262) 18 exp Complex Regional Pain Syndromes/ (5293) 19 (neuropath$ pain or cervical radiculopathy or lumber radiculopathy or diabetic neuropath$).mp. (32327) 20 (peripheral neuropath$ or postherpetic neuralgia or HIV neuropath$ or spinal cord injur$ or trigeminal neuralgia or complex regional pain syndrome).mp. (73928) 21 or/13‐20 (108008) 22 11 and 12 and 21 (101) 23 remove duplicates from 22 (94)
Appendix 4. Embase (OvidSP) search strategy
Database: Embase <1980 to 2016 Week 47> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure.sh. (53825) 2 double‐blind procedure.sh. (136941) 3 single‐blind procedure.sh. (27251) 4 randomized controlled trial.sh. (461742) 5 (random$ or crossover$ or cross over$ or placebo$ or (doubl$ adj blind$) or allocat$).tw,ot. (1342982) 6 trial.ti. (213901) 7 or/1‐6 (1499488) 8 (animal/ or nonhuman/ or animal experiment/) and human/ (1720673) 9 animal/ or nonanimal/ or animal experiment/ (3736832) 10 9 not 8 (3030962) 11 7 not 10 (1386306) 12 limit 11 to embase (493694) 13 (oxcarbazepine or trileptal$ or trexapin).mp. (9161) 14 radiculopathy/ (9102) 15 radicular pain/ (3453) 16 diabetic neuropathy/ (20865) 17 postherpetic neuralgia/ (4974) 18 spinal cord injury/ (48084) 19 trigeminus neuralgia/ (9462) 20 exp complex regional pain syndrome/ (8371) 21 (neuropath$ pain or cervical radiculopathy or lumbar radiculopathy or diabetic neuropath$ or peripheral neuropath$).mp. (94563) 22 (postherpetic neuralgia or HIV neuropath$ or spinal cord injur$ or trigeminal neuralgia or complex regional pain syndrome).mp. (72140) 23 or/14‐22 (175466) 24 12 and 13 and 23 (42) 25 remove duplicates from 24 (42) 26 11 and 13 and 23 (129) 27 remove duplicates from 26 (127)
Appendix 5. Chinese Biomedical Retrieval System Database search strategy
(n.b. all of the search terms were translated to Chinese terms when we conducted the searches) 1. neuropathic pain 2. radiculopathy 3. cervical radiculopathy 4. lumbar radiculopathy 5. diabetic neuropathy 6. peripheral neuropathy 7. postherpetic neuralgia or PHN 8. HIV neuropathy 9. Spinal Cord Injuries 10.trigeminal neuralgia 11.complex regional pain syndrome or CRPS 12.1‐11/or 13.oxcarbazepine 14.trileptal 15.qulai or renao or wanyi (trade names of oxcarbazepine in Chinese) 16.13‐15/or 17.random 18.control 19.clinical trial 20.blind procedure 21.placebo 22.17‐21/or 23.12 and 16 and 22
Data and analyses
Comparison 1. Oxcarbazepine versus placebo for painful diabetic neuropathy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Reduction in participant‐reported pain scores by at least 50% from the baseline | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 16 weeks after starting treatment | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.08, 3.39] |
| 2 Reduction in participant‐reported pain scores by at least 30% from the baseline | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 16 weeks after starting the treatment | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [1.01, 2.44] |
| 3 Participant' global impression of their change in pain | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 All improved participants after 16‐week treatment | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [1.32, 2.45] |
| 3.2 Participants who improved much or very much after 16‐week treatment | 2 | 493 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [1.13, 1.88] |
| 4 Adverse effects | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Adverse effects leading to withdrawal from treatment | 3 | 634 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.83 [2.29, 6.40] |
| 4.2 Serious adverse effects | 3 | 634 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.65 [1.45, 9.20] |
Comparison 2. Subgroup analysis: different daily doses of oxcarbazepine versus placebo for painful diabetic neuropathy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participants who improved much or very much after 16‐week treatment | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Oxcarbazepine 600 mg/day | 1 | 172 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.66, 1.45] |
| 1.2 Oxcarbazepine 1200 mg/day | 1 | 176 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.97, 1.92] |
| 1.3 Oxcarbazepine 1800 mg/day | 2 | 323 | Risk Ratio (M‐H, Random, 95% CI) | 1.64 [1.01, 2.67] |
| 2 Adverse effects leading to withdrawals | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Oxcarbazepine 600 mg/day | 1 | 172 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.60, 4.32] |
| 2.2 Oxcarbazepine 1200 mg/day | 2 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.83 [1.97, 7.41] |
| 2.3 Oxcarbazepine 1800 mg/day | 2 | 323 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.83 [2.68, 8.70] |
| 3 Serious adverse effects | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Oxcarbazepine 600 mg/day | 1 | 172 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.14 [0.20, 23.21] |
| 3.2 Oxcarbazepine 1200 mg/day | 2 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.68 [1.38, 15.95] |
| 3.3 Oxcarbazepine 1800 mg/day | 2 | 323 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.55 [1.55, 13.37] |
Comparison 3. Oxcarbazepine versus placebo for neuropathic pain due to radiculopathy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Reduction in participant‐reported pain scores by at least 50% from baseline | 1 | 144 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.23, 1.23] |
| 2 Participant' global impression of therapeutic effect | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 All improved participants after 16‐week treatment | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.92, 1.98] |
| 2.2 Participants who improved much or very much after 16‐week treatment | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.81, 3.20] |
| 3 Adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Any adverse effects | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [1.07, 1.39] |
| 3.2 Adverse effects leading to withdrawal from treatment | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.84 [1.55, 5.23] |
| 3.3 Serious adverse effects | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.13 [0.65, 14.98] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Beydoun 2006.
| Methods | Multicentre, double‐blind, placebo‐controlled, randomised, parallel‐group, dose‐ranging study. 2‐week pre‐randomisation screening phase and 16‐week double‐blind phase (including a 4‐week titration period and 12‐week maintenance period). |
|
| Participants | 347 participants with painful diabetic neuropathy. Randomised in a 1:1:1:1 ratio to oxcarbazepine 600 mg/day (300 mg bid; n = 83), 1200 mg/day (600 mg bid; n = 87), 1800 mg/day (900 mg bid; n = 88) or placebo (N = 89). Gender (% male): oxcarbazepine 600 mg 60%; oxcarbazepine 900 mg 52%; oxcarbazepine 1200 mg 53%; placebo 56%. Mean age, years (SD): oxcarbazepine 600 mg: 61.1 (10.7); oxcarbazepine 900 mg: 60.3 (10.3); oxcarbazepine 1200 mg: 59.3 (9.3); placebo: 62.1 (10.3). Inclusion criteria: men and non‐pregnant women; aged ≥ 18 years; diagnosis of diabetes mellitus (type 1 or 2) and pain attributed to diabetic neuropathy for 6 months to 5 years; pain rating score ≥ 50 units on a 100‐unit VAS at screening visit; stable glycaemic control (HbA1c ≤ 11% at baseline); and baseline serum sodium ≥ 35 mmol/L or higher; VAS must have averaged at least 40 units during pre‐randomisation phase, with < 25% variability in the 7 days prior to randomisation. Exclusion criteria: other types of pain; clinically significant medical or psychiatric illnesses; history of hyponatraemia or non‐compliance; drug or alcohol abuse in preceding year; amputations other than toes; treatment with lithium or monoamine oxidase inhibitors; previous treatment with oxcarbazepine; history of sensitivity to carbamazepine or its metabolites. |
|
| Interventions |
Experimental groups: oxcarbazepine 300 mg/day, oxcarbazepine 1200 mg/day, oxcarbazepine 1800 mg/day. Participants received the Initiated with oxcarbazepine 300 mg (1 tablet) or matching placebo daily for 3 days, then 1 tablet bid from 4th day. Thereafter, dose was increased by 1 tablet every 5 days until the target dose of 600 mg/day, 1200 mg/day or 1800 mg/day was reached. Control group: placebo. Dose reductions due to tolerability problems were allowed at any time during the study. |
|
| Outcomes |
Primary outcome: change in mean VAS score from baseline to end of study (16 weeks). Secondary outcomes: participants' Global Assessment of the Therapeutic Effect, onset of therapeutic effect, durability of treatment effect, sleep disturbances and quality of life. Safety assessed during screening and double‐blind treatment phases, consisting of monitoring vital signs, neurological and physical examinations, laboratory parameters, spontaneous reports of adverse effects and serious adverse effects. |
|
| Funding | Funded by Novartis Pharmaceuticals Inc. | |
| Conflicts of interest | None known. | |
| Notes | Trial conducted between November 2001 and September 2003 at 37 centres across US. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Study design was similar to Dogra 2005 and funded by same pharmaceutical company, so likely that randomisation was adequate. |
| Allocation concealment (selection bias) | Low risk | Study design was similar to Dogra 2005 and funded by same pharmaceutical company, so likely that allocation concealment was performed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated that there was a double‐blind phase, and matching placebos were used, so participants and personnel involved in the study were likely to be blinded. Obvious adverse effects experienced during oxcarbazepine treatment might have unblinded some participants, but relevant data were not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Stated that there was a double‐blind phase, and personnel involved in the study were likely to be blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Missing data of both groups clearly described and an ITT analysis performed. Number of withdrawals not balanced across doses of oxcarbazepine and placebo groups (oxcarbazepine 600 mg/day: 16/83; oxcarbazepine 1200 mg/day: 34/87; oxcarbazepine 1800 mg/day: 48/88; placebo: 17/89 participants), especially those due to adverse effects (oxcarbazepine 600 mg/day: 9/83, oxcarbazepine 1200 mg/day: 20/87, oxcarbazepine 1800 mg/day: 36/88, and placebo: 6/89 participants). |
| Selective reporting (reporting bias) | Low risk | Outcomes listed in methods section were all reported. |
| Other bias | Low risk | No other potential bias found. |
Demant 2014.
| Methods | Randomised, double‐blind, placebo‐controlled trial. Randomisation took place within two phenotype groups: 'irritable nociceptor' or 'non‐irritable nociceptor'. 1‐week baseline period and a 6‐week treatment period before and after cross‐over. Before each baseline week, there was a medication tapering‐off or washout period. |
|
| Participants | 97 participants; 83 included in ITT population; 39 completed study per protocol. Inclusion criteria: aged ≥ 18 years; both genders; definite or probable neuropathic pain; diagnosis of polyneuropathy, postherpetic neuralgia or peripheral nerve injury; pain duration > 3 months; pain rating at baseline ≥ 4 points on 11‐point numeric rating scale; informed consent obtained. Exclusion criteria: other non‐neuropathic pain condition; allergy to oxcarbazepine; renal or hepatic impairment; epilepsy; depression and other serious psychiatric disorders; serious medical condition; previous treatment for neuropathic pain that could not be stopped; pregnancy; people expected to be unable to comply with study protocol; treatment with anticonvulsants, antidepressants or opioids. |
|
| Interventions |
Experimental group: oxcarbazepine 300 mg capsules gradually increased during 21 days from 300 mg/day up to 2400 mg/day, and maintained at 2400 mg/day for remaining weeks. For participants who experienced unacceptable adverse effects, dose could be lowered to 1800‐2100 mg/day or stay at 1800 mg/day. In participants aged ≥ 70 years, dose increased only to 1800 mg/day, and there was a lower dose of 1200‐1500 mg/day in cases of serious adverse effects. Control group: identical placebo capsules increased gradually from 1 capsule daily up to 8 capsules daily. Drug or placebo capsules bid. Up to paracetamol 3 g and tramadol 50 mg daily were allowed as rescue medication. |
|
| Outcomes |
Primary outcome: total pain rated on numeric rating scale. Secondary outcomes: response defined as ≥ 50% reduction in pain score; Neuropathic Pain Symptom Inventory; Patient Global Impression of Change; rating of evoked pain; sleep disturbance; quality of life; use of rescue medication. |
|
| Funding | Sponsored by Odense University Hospital. | |
| Conflicts of interest | None known. | |
| Notes | Recruitment from March 2011 to April 2011. Trial conducted in 2 closely collaborating centres in Denmark: the Department of Neurology at Odense University Hospital and the Danish Pain Research Center at Aarhus University Hospital. ClinicalTrials.gov identifier: NCT01302275. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants' treatment sequence within each phenotype group was assigned using a computer‐generated randomisation list with a block size of 4. |
| Allocation concealment (selection bias) | Low risk | Quote: "the randomization plan was generated by a person in the hospital pharmacy at Odense University Hospital who was not otherwise involved in the conduct of the trial." In addition: "sealed opaque envelopes with the treatment sequence for each patient," were present at the study sites in case of emergency. Allocation concealment was likely to have been performed. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Used matched placebo capsules; report stated that all participants, investigators and other personnel were blinded to individual treatment assignments. Trial authors considered that adverse effects experienced during oxcarbazepine treatment might have unblinded many participants, as reflected in a high percentage of participants correctly guessing which treatment they had received in each period. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All personnel involved in the study, including those who assessed outcomes, were blinded to individual treatment assignments. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawal rate due to adverse effects was high. 39 participants completed the study per protocol. The results relied on an ITT population and last observation carried forward data, which might lead to overestimation of efficacy. |
| Selective reporting (reporting bias) | Low risk | Outcomes listed in methods were all reported. |
| Other bias | Low risk | No other potential bias found. |
Dogra 2005.
| Methods | Multicentre, placebo‐controlled, double‐blind, parallel‐group study with 3 phases. 2‐week pre‐randomisation screening phase, 18‐week double‐blind treatment phase (including 4‐week titration period, 12‐week maintenance period and 2‐week follow‐up period), and 52‐week open‐label extension phase. Primary efficacy analysis performed on ITT population. |
|
| Participants | 289 participants screened, 146 participants randomised to either oxcarbazepine (69 participants) or placebo (77 participants). Gender (% male): oxcarbazepine 54%; placebo 62%. Mean age, years (SD): oxcarbazepine 59.7 (10.4); placebo 60.5 (8.1). Inclusion criteria: male or female outpatients; aged ≥ 18 years; established clinical diagnosis of diabetes mellitus (type 1 or 2); stable diabetic control as evidenced by HbA1c ≤ 11% at baseline, mean HbA1c over the 6 months prior to study entry within 1 unit (%) of baseline; history of neuropathic pain between 6 months and 5 years in duration; pain rating ≥ 50 units on VAS at first screening visit; mean pain score 50 units over 4 of last 7 days prior to randomisation; ≤ 25% variation in severity of pain in 7 days prior to randomisation, as assessed from electronic diary information recorded daily during screening phase. Exclusion criteria: presence of other pain that could confound assessment of neuropathic pain of diabetic origin; currently or had previously taken oxcarbazepine; presence of skin lesions that could affect ability to assess their neuropathic pain, or if they had undergone amputations (other than toes); history of renal insufficiency (creatinine clearance < 30 mL/minute); hyponatraemia (serum sodium < 135 mmol/L); chronic infectious disease; known hypersensitivity to oxcarbazepine or carbamazepine. 40 participants withdrawn during double‐blind phase due to adverse effects, protocol violation, unsatisfactory effect, participant withdrew consent, lost to follow‐up or administrative reasons. However, all were included in ITT analyses. |
|
| Interventions |
Experimental group: oxcarbazepine 300 mg/day orally, increased 3 days later to 300 mg bid, and then titrated in 300 mg increments every 5 days as tolerated to maximum dose 900 mg bid (1800 mg/day) over 4‐week titration period. Then followed 12‐week maintenance period. Mean oxcarbazepine dose during the maintenance period 1445 mg/day. Control group: matched placebos administered in same regimen. |
|
| Outcomes |
Primary outcome: mean daily VAS score for pain severity during the last week of double‐blind treatment compared with baseline. Secondary outcomes: participant's global assessment of therapeutic effect, onset of therapeutic effect, durability of treatment effect, sleep disturbances and quality of life. Safety assessed during screening and double‐blind treatment phases, including monitoring vital signs, neurological and physical status, laboratory parameters, spontaneous reports of adverse effects and serious adverse effects. |
|
| Funding | Funded by Novartis Pharmaceuticals Inc. | |
| Conflicts of interest | None known. | |
| Notes | Conducted in 22 diabetes and neurology pain clinics and research centres in the USA and Canada. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised in a 1:1 ratio to each group; "the randomisation scheme was created by Novartis Drug Supply Management using a validated system that automates the random assignment of treatment groups to randomisation numbers." |
| Allocation concealment (selection bias) | Low risk | Stated, "the randomization scheme was created by Novartis Drug Supply Management using a validated system that automates the random assignment of treatment groups to randomization numbers. The randomization numbers were assigned to centres in blocks," so allocation concealment was likely to have been performed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | In double‐blind phase, matched placebos were used and administered in same way with same initiated doses and increments. Obvious adverse effects experienced during oxcarbazepine treatment might have unblinded some participants, but relevant data were not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Stated they used double‐blind design, and personnel involved in study was likely to be blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Numbers of withdrawals not balanced across groups (25/69 participants with oxcarbazepine vs 15/77 participants with placebo), especially those due to adverse effects (19/69 participants with oxcarbazepine vs 6/77 participants with placebo). |
| Selective reporting (reporting bias) | Low risk | Outcomes listed in the methods section were all reported. |
| Other bias | Low risk | No other potential bias found. |
Grosskopf 2006.
| Methods | Multicentre, double‐blind, placebo‐controlled study. 2‐week pre‐randomisation screening phase and a 16‐week double‐blind treatment phase (including 4‐week titration period and 12‐week maintenance period). |
|
| Participants | 141 participants randomised to oxcarbazepine (n = 71) or placebo (n = 70). Gender (% male): oxcarbazepine 40 (56%); placebo 38 (54%); all 78 (55%). Mean age, years (SD): oxcarbazepine 60.8 (10.6); placebo 61.4 (10.6); all 61.1 (10.5). Inclusion criteria: males and non‐pregnant, non‐lactating females aged ≥ 18 years with established clinical diagnosis of diabetes mellitus type 1 or 2; history of neuropathic pain for 6 months to 5 years prior to study entry; stable diabetic control defined as HbA1c ≤ 11% at baseline and mean HbA1c within 1% of baseline over 6 months prior to study entry; pain rating ≥ 50 units on 100‐unit VAS at first screening visit, and mean VAS score ≥ 40 units over 4 of the last 7 days prior to randomisation and with < 25% variability during the last 7 days prior to randomisation. Exclusion criteria: pain that could confound assessment of neuropathic pain of diabetic origin; previous or current oxcarbazepine treatment; skin conditions that could affect assessment of pain; amputations (other than toes); renal insufficiency; serum sodium < 135 mmol/L; chronic infectious disease and known hypersensitivity to oxcarbazepine or carbamazepine. |
|
| Interventions |
Experimental group: oxcarbazepine 300 mg/day orally titrated over 4 weeks to tolerability or maximum dose of 600 mg bid (1200 mg/day), and dose remained unchanged throughout the maintenance period, except for dose reductions in the event of poor tolerability. Mean oxcarbazepine dose during maintenance period 1091 mg/day. Control group: matched placebos administered in same way with same initiated doses and increments. |
|
| Outcomes |
Primary outcome: mean VAS score for pain severity for the final week of double‐blind treatment compared with baseline. Second outcomes: participants' global assessment of therapeutic effect, onset of therapeutic effect, durability of therapeutic effect, sleep disturbances and quality of life. |
|
| Funding | Funded by Novartis Pharmaceuticals Inc. | |
| Conflicts of interest | None known. | |
| Notes | Conducted in 22 centres in the USA, Germany and the UK. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stated that participants were randomly assigned in a 1:1 ratio to each group; detailed method of sequence generation not specified in this article, but it was reported that design of study was similar to Dogra 2005, so randomisation numbers were likely to have been used as well. |
| Allocation concealment (selection bias) | Low risk | Method of allocation concealment not described, but it was reported that design of this study was similar to Dogra 2005, so allocation concealment was likely to have been performed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated to be double‐blind study, and matching placebos used, so participants and personnel involved in study were likely to be blinded. Obvious adverse effects experienced during oxcarbazepine treatment might have unblinded some participants, but relevant data were not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Stated to be double‐blind study, and personnel involved in study likely to have been blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Missing data in both groups were clearly described and ITT analysis performed. But numbers of withdrawals not balanced across groups (29/71 participants with oxcarbazepine vs 17/70 participants with placebo), especially those due to adverse effects (18/71 with participants with oxcarbazepine vs 4/70 participants with placebo). |
| Selective reporting (reporting bias) | Low risk | Outcomes listed in the methods section were all reported. |
| Other bias | Low risk | No other potential bias found. |
Novartis 2004.
| Methods | Multicentre, double‐blind, randomised, placebo‐control study. 2‐week pre‐randomisation screening phase and 16‐week double‐blind treatment phase (including 4‐week titration period and 12‐week maintenance period). |
|
| Participants | 145 participants randomised to oxcarbazepine (n = 71) or placebo (n = 74). Gender (female:male): oxcarbazepine 43:28; placebo 36:38. Mean age, years (SD): oxcarbazepine 53.8 (13.67); placebo 55.1 (12.33). Inclusion criteria: men or women aged ≥ 18 years with diagnosis of radiculopathy and evidence of motor deficit or reflex loss on examination, history of neuropathic pain for ≥ 6 months and VAS score ≥ 50. Exclusion criteria: prior oxcarbazepine experience; history of hyponatraemia or renal insufficiency (creatinine clearance < 30 mL/minute); known sensitivity to carbamazepine or being treated with lithium or monoamine oxidase inhibitors. |
|
| Interventions |
Experimental group: initiated with oxcarbazepine 300 mg/day orally and titrated over 4 weeks to a maximum target dose of 1800 mg/day, and final dose remained unchanged throughout maintenance period. Control group: matching placebo tablets administered in same way. |
|
| Outcomes |
Primary outcome: mean VAS score for pain severity during last week of double‐blind treatment. Secondary outcomes: participants' global assessment of therapeutic effect; durability of treatment effect; onset of therapeutic effect; proportion of days rescue medication administered during the double‐blind phase and mean daily dose of rescue medication administered during the double‐blind phase. |
|
| Funding | None known. | |
| Conflicts of interest | Funded by Novartis Pharmaceuticals Inc. | |
| Notes | Conducted in 40 centres: 3 in Great Britain and 37 in the US. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Relevant information could not be obtained. |
| Allocation concealment (selection bias) | Unclear risk | Relevant information could not be obtained. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Stated to be double‐blind, and used matching placebos. Likely to have been blinded. Obvious adverse effects experienced during oxcarbazepine treatment might have unblinded some participants, but relevant data were not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Stated to be double‐blinded, and personnel involved in study were likely to be blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Missing data in both groups clearly described and ITT analysis performed, but data of 1 participant in oxcarbazepine group was not included in ITT analysis and reason was not clarified. Numbers of withdrawals not balanced across groups (40/71 participants with oxcarbazepine vs 33/74 participants with placebo), especially those due to adverse effects (30/71 participants with oxcarbazepine vs 11/74 with placebo) or lack of efficacy (4/71 participants with oxcarbazepine vs 15/74 participants with placebo). |
| Selective reporting (reporting bias) | Low risk | No data from this trial published, but all outcomes listed in the methods section were reported on the Novartis website (Novartis database). |
| Other bias | Low risk | No other potential bias found. |
bid: twice daily; HbA1c: haemoglobin A1c; ITT: intention to treat; n: number of participants; SD: standard deviation; VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Besi 2015 | Clinical prospective observational exploratory survey of people with idiopathic trigeminal neuralgia and its variants. |
| Beydoun 2002 | Abstract of a double‐blind RCT comparing efficacy and safety of oxcarbazepine vs carbamazepine for the treatment of 46 participants with new‐onset trigeminal neuralgia. Published abstract provided no data for our prespecified outcomes. Attempts to obtain the full text or data from the authors and pharmaceutical company were unsuccessful. |
| Beydoun 2007 | Report of 2 open‐label studies (involving 497 and 97 participants) to evaluate long‐term safety and tolerability of oxcarbazepine for diabetic neuropathy without a control group. |
| Brainer‐Lima 2003 | Abstract. Attempts to obtain the full text or data from the authors and pharmaceutical company were unsuccessful. |
| Gong 2010 | Non‐blind trial comparing efficacy and safety of oxcarbazepine with carbamazepine and topiramate in 99 participants with trigeminal neuralgia. |
| Hu 2010 | Non‐blind trial of 369 participants with trigeminal neuralgia comparing efficacy and safety of oxcarbazepine to carbamazepine. |
| Li 2011 | Non‐blind trial of 78 participants with trigeminal neuralgia comparing efficacy and safety of oxcarbazepine to carbamazepine. |
| Liebel 2001 | Abstract. We were unable to obtain data for our prespecified outcomes from abstract. We were unable to find the full text or retrieve any useful data from the authors and pharmaceutical company. |
| Lindström 1987 | Double‐blind cross‐over trial of 15 participants with trigeminal neuralgia receiving oxcarbazepine or carbamazepine for only 3 weeks. Trial had short follow‐up, less than the minimum (6 weeks) we specified in protocol. |
| Min 2016 | Randomised cross‐over trial of 55 participants with neuropathic pain receiving oxcarbazepine or pregabalin. Trial had short follow‐up (1‐2 weeks), less than the minimum (6 weeks) we specified in protocol. |
| Rémillard 1994 | Open‐label study with 20 participants investigating efficacy of oxcarbazepine for intractable trigeminal neuralgia without a control group. |
| Venancio‐Ramirez 2004 | Randomised trial of gabapentin and oxcarbazepine in 60 participants with postherpetic neuropathy. 4‐week observation period was too short to meet our inclusion criteria. |
| Zhou 2010 | Open‐label trial of 64 participants with trigeminal neuralgia comparing the efficacy of oxcarbazepine to carbamazepine. |
RCT: randomised controlled trial.
Characteristics of ongoing studies [ordered by study ID]
NCT02219373.
| Trial name or title | Gabapentin and Oxcarbazepine for Chronic Neuropathic Pain in Children and Adolescents: a Double‐blind, Randomized Clinical Effectiveness Study. |
| Methods | Randomised, double‐blind, 2‐group parallel design, with cross‐over assignment of interventions. |
| Participants | Estimated enrolment: 60 participants. Inclusion criteria:
Exclusion criteria: unstable psychiatric illness (suicidal ideation, disorganised behaviour); uncontrolled seizure disorder; chronic headaches only; abdominal pain only; prior experience with anticonvulsants for pain; children with syndrome of inappropriate antidiuretic hormone secretion. |
| Interventions | 3 intervention groups:
|
| Outcomes | Primary outcomes: 2‐point reduction in mean daily pain scores, at least 30% reduction relative to baseline and global overall impression of strong benefit. Secondary outcomes: pain scores at rest and evoked manoeuvres, functional disability scores, and frequency of adverse effects (including tolerability, depression and anxiety, and neuropsychological measures). |
| Starting date | October 2014. |
| Contact information | Monique Ribeiro, MD. Tel: +1 617 3557040. Email: Monique.Ribeiro@childrens.harvard.edu. Kimberly Lobo, MPH. Tel: +1 857 2183556. Email: Kimberly.Lobo@childrens.harvard.edu. |
| Notes | USA study, sponsored by Boston Children's Hospital. Informed consent by parents. Study estimated to be completed in October 2016, which has passed and study status has not been verified since August 2014. |
Differences between protocol and review
This review has a published protocol (Zhou 2009).
We searched the Chinese Biomedical Retrieval System in addition to other databases.
We did not search the Trials Register of the Cochrane Pain, Palliative and Supportive Care Group.
In the updated version, we added a lower limit for treatment duration (at least six weeks) to the inclusion criteria, since the previous version of review specified this minimum duration under 'Types of outcome measures,' but not in the 'Types of interventions' section.
In the updated version, we set clear values for the assessment of heterogeneity according to Deeks 2011.
We added a 'Summary of findings' table to the review, which was not mentioned in the protocol, and described the GRADE methodology.
For the 'Risk of bias' assessments, we used 'high,' 'low' or 'unclear' risk of bias and assessed performance bias and detection bias separately according to Higgins 2011b.
We performed a post hoc subgroup analysis for different doses of oxcarbazepine
Changes in authorship have taken place since publication of the first version.
Contributions of authors
NC and MZ: searched for studies and extracted data for the update; drafted the updated version of review.
LH: supervised and revised the text.
CZ: carried out analyses.
All authors were involved in writing the protocol, full review and updated review.
Declarations of interest
MZ: none known.
NC: none known.
LH: none known.
MY: none known.
CZ: none known.
FW: none known.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Beydoun 2006 {published and unpublished data}
- Multicenter, double‐blind, randomized placebo‐control, parallel group study to evaluate the safety and efficacy of three doses of oxcarbazepine in patients with neuropathic pain due to diabetic neuropathy, 2003. www.novctrd.com/CtrdWeb/product.nov?diseaseid=141&productid=330 (accessed 2 October 2015).
- Beydoun A, Shaibani A, Hopwood M, Wan Y. Oxcarbazepine in painful diabetic neuropathy: results of a dose‐ranging study. Acta Neurologica Scandinavica 2006;113(6):395‐404. [PUBMED: 16674606] [DOI] [PubMed] [Google Scholar]
- Beydoun A, Wan Y, Hopwood M, Liebel J. Results of a randomized, placebo‐controlled trial suggest oxcarbazepine has a therapeutic effect in the treatment of painful diabetic neuropathy. European Journal of Neurology 2004;11 Suppl 2:101. [Google Scholar]
Demant 2014 {published and unpublished data}
- Oxcarbazepine for the treatment of chronic peripheral neuropathic pain (IMIOXC). https://clinicaltrials.gov/ct2/show/study/NCT01302275?term=NCT01302275&rank=1 2014 (accessed 2 October 2015).
- Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double‐blind, placebo‐controlled phenotype‐stratified study. Pain 2014;155(11):2263‐73. [PUBMED: 25139589] [DOI] [PubMed] [Google Scholar]
Dogra 2005 {published and unpublished data}
- Multicenter, double‐blind, randomized placebo‐control, parallel‐group study to evaluate the safety and efficacy of oxcarbazepine in patients with neuropathic pain due to diabetic neuropathy, 2005. www.novctrd.com/CtrdWeb/product.nov?diseaseid=141&productid=330 (accessed 2 October 2015).
- Dogra S, Beydoun S, Hopwood M, Mazzola J, Wan Y. Oxcarbazepine in painful diabetic neuropathy: a randomized, placebo‐controlled study. Journal of Pain 2004;5(3 Suppl 1):S6. [DOI] [PubMed] [Google Scholar]
- Dogra S, Beydoun S, Mazzola J, Hopwood M, Wan Y. Oxcarbazepine in painful diabetic neuropathy: a randomized, placebo‐controlled study. European Journal of Pain 2005;9(5):543‐54. [PUBMED: 16139183] [DOI] [PubMed] [Google Scholar]
- Dogra S, Beydoun S, Mazzola J, Hopwood M, Wan Y, Liebel J. Oxcarbazepine provides pain relief in patients with painful diabetic neuropathy: a randomized, placebo‐controlled study. European Journal of Neurology 2004;11 Suppl 2:92. [Google Scholar]
Grosskopf 2006 {published and unpublished data}
- Multicenter, double‐blind, randomized, placebo‐control, parallel study to evaluate the safety and efficacy of oxcarbazepine in patients with neuropathic pain due to diabetic neuropathy, 2003. www.novctrd.com/CtrdWeb/product.nov?diseaseid=141&productid=330 (accessed 2 October 2015).
- Grosskopf J, Mazzola J, Wan Y, Hopwood M. A randomized, placebo‐controlled study of oxcarbazepine in painful diabetic neuropathy. Acta Neurologica Scandinavica 2006;114(3):177‐80. [PUBMED: 16911345] [DOI] [PubMed] [Google Scholar]
- Riviere M‐E, Wan Y, Hopwood M, Liebel J. Oxcarbazepine in the treatment of painful diabetic neuropathy: a randomized, placebo‐controlled study. European Journal of Neurology 2004;11 Suppl 2:101. [Google Scholar]
Novartis 2004 {published and unpublished data}
- Multicenter, double‐blind, randomized, placebo‐control, parallel‐group study to evaluate the safety and efficacy of oxcarbazepine in patients with neuropathic pain due to radiculopathy, 2005. www.novctrd.com/CtrdWeb/product.nov?diseaseid=429&productid=330 (accessed 2 October 2015).
- Harden RN, Urniasz KL, Hopwood M, Wan Y. Efficacy and safety of oxcarbazepine in patients with neuropathic pain due to radiculopathy: a double‐blind, randomized, placebo‐controlled study. Neurology 2005;64 Suppl 1:A120‐1. [Google Scholar]
References to studies excluded from this review
Besi 2015 {published data only}
- Besi E, Boniface DR, Cregg R, Zakrzewska JM. Comparison of tolerability and adverse symptoms in oxcarbazepine and carbamazepine in the treatment of trigeminal neuralgia and neuralgiform headaches using the Liverpool Adverse Events Profile (AEP). Journal of Headache and Pain 2015;16:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Beydoun 2002 {published data only}
- Beydoun A, D'Souza J. Oxcarbazepine versus carbamazepine in patients with new onset trigeminal neuralgia result of double blind comparative trial [abstract no.P06.116]. Neurology 2002;58 Suppl 3:A471. [Google Scholar]
Beydoun 2007 {published data only}
- Beydoun S, Alarcón F, Mangat S, Wan Y. Long‐term safety and tolerability of oxcarbazepine in painful diabetic neuropathy. Acta Neurologica Scandinavica 2007;115(4):284‐8. [DOI] [PubMed] [Google Scholar]
Brainer‐Lima 2003 {published data only}
- Brainer‐Lima P, Naylor F. Oxcarbazepine for cancer‐related neuropathic pain and opiate detoxification. 22nd Annual Meeting of the American Pain Society; 2003 March 20‐23; Chicago (IL). 2003.
Gong 2010 {published data only}
- Gong JJ. A randomised controlled trial of 3 drugs for the treatment of idiopathic trigeminal neuralgia [3种药物治疗原发性三叉神经痛的随机对照试验]. Journal of China Traditional Chinese Medicine Information 2010;2(12):74. [Google Scholar]
Hu 2010 {published data only}
- Hu YJ. The comparative efficacy of oxcarbazepine and carbamazepine for trigeminal neuralgia [奥卡西平与卡马西平治疗三次神经痛的疗效对比]. Shandong Medical Journal 2010;50(25):107. [Google Scholar]
Li 2011 {published data only}
- Li G, Zha Q, Chen JZ, Cao QH, Peng WJ. Clinical efficacy of oxcarbazepine for primary trigeminal neuralgia [奥卡西平治疗原发性三叉神经痛临床观察]. Zhejiang Journal of Integrated Traditional Chinese and Western Medicine 2011;21(3):167‐8. [Google Scholar]
Liebel 2001 {published data only}
- Liebel J. Results of a double‐blind trial comparing oxcarbazepine (OXC) vs carbamazepine (CBZ) in newly diagnosed, untreated patients with trigeminal neuralgia. Journal of the Neurological Sciences 2001;187 Suppl 1:S5. [Google Scholar]
Lindström 1987 {published data only}
- Lindström P. The analgesic effect of carbamazepine in trigeminal neuralgia. Pain 1987;28 Suppl 4:S85. [DOI] [PubMed] [Google Scholar]
Min 2016 {published and unpublished data}
- Symptom‐based treatment for neuropathic pain in spinal cord injured patients, randomized clinical trial, 2014. clinicaltrials.gov/ct2/show/study/NCT02180880?term=oxcarbazepine&rank=34 (accessed 2 October 2015).
- Min K, Oh Y, Lee SH, Ryu JS. Symptom‐based treatment of neuropathic pain in spinal cord‐injured patients: a randomized crossover clinical trial. American Journal of Physical Medicine & Rehabilitation 2016;95(5):330‐8. [DOI] [PubMed] [Google Scholar]
Rémillard 1994 {published data only}
- Rémillard G. Oxcarbazepine and intractable trigeminal neuralgia. Epilepsia 1994;35 Suppl 3:S28‐9. [Google Scholar]
Venancio‐Ramirez 2004 {published data only}
- Venancio‐Ramirez L, Hernández‐Santos JR, Tenopala‐Villegas S, Torres‐Huerta JC, Rivera‐León G, Canseco‐Aguilar C. Comparison of oxcarbazepine and gabapentin at standard dose in treatment of pain for postherpetic neuropathy. Revista Mexicana de Anestesiologia 2004;27(3):129‐33. [Google Scholar]
Zhou 2010 {published data only}
- Zhou JF. A clinical research on the pharmacological treatment of trigeminal neuralgia [药物治疗三叉神经痛的临床研究]. Medical Information Operations Sciences Fascicule 2010;1:89. [Google Scholar]
References to ongoing studies
NCT02219373 {unpublished data only}
- NCT02219373. Gabapentin and oxcarbazepine for chronic neuropathic pain in children and adolescents: a clinical effectiveness study. clinicaltrials.gov/ct2/show/NCT02219373 (accessed 15 January 2017).
Additional references
Attal 2006
- Attal N, Cruccu G, Haanpää M, Hansson P, Jensen TS, Nurmikko T, et al. EFNS Task Force. EFNS guidelines on pharmacological treatment of neuropathic pain. European Journal of Neurology 2006;13(11):1153‐69. [DOI] [PubMed] [Google Scholar]
Attal 2010
- Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS, et al. European Federation of Neurological Societies. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2009 revision. European Journal of Neurology 2010;17(9):1113‐e88. [DOI: 10.1111/j.1468-1311.2010.02999.x] [DOI] [PubMed] [Google Scholar]
Backonja 2003
- Backonja M, Glanzman RL. Gabapentin dosing for neuropathic pain: evidence from randomized placebo‐controlled clinical trials. Clinical Therapeutics 2003;25(1):81‐104. [DOI] [PubMed] [Google Scholar]
Beydoun 2002a
- Beydoun A, Schmidt D, Souza J. Oxcarbazepine versus carbamazepine in trigeminal neuralgia: a meta‐analysis of three double‐blind comparative trials. Neurology 2002;58 Suppl 3:A131. [Google Scholar]
Beydoun 2002b
- Beydoun A, Schmidt D, D'Souza J, on behalf of the oxcarbazepine study group. Meta‐analysis of comparative trials of oxcarbazepine versus carbamazepine in trigeminal neuralgia. 21st American Pain Society Annual Meeting; 2002 Mar 14‐17; Baltimore (MD).
Bieri 1990
- Bieri D, Reeve RA, Champion GD, Addicoat L, Ziegler JB. The Faces Pain Scale for the self‐assessment of the severity of pain experienced by children: development, initial validation and preliminary investigation for ratio scale properties. Pain 1990;41(2):139‐50. [DOI] [PubMed] [Google Scholar]
Birse 2012
- Birse F, Derry S, Moore RA. Phenytoin for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD009485.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Callin 2008
- Callin S, Bennett MI. Assessment of neuropathic pain. Continuing Education in Anaesthesia, Critical Care and Pain 2008;8(6):210‐3. [Google Scholar]
Carpenter 2002
- Carpenter KJ, Dickenson AH. Molecular aspects of pain research. Pharmacogenomics Journal 2002;2(2):87‐95. [DOI] [PubMed] [Google Scholar]
Carrazana 2003
- Carrazana E, Mikoshiba I. Rationale and evidence for the use of oxcarbazepine in neuropathic pain. Journal of Pain and Symptom Management 2003;25(5 Suppl):S31‐5. [DOI] [PubMed] [Google Scholar]
Corrigan 2012
- Corrigan R, Derry S, Wiffen PJ, Moore RA. Clonazepam for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD009486.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Criscuolo 2005
- Criscuolo S, Auletta C, Lippi S, Brogi F, Brogi A. Oxcarbazepine monotherapy in postherpetic neuralgia unresponsive to carbamazepine and gabapentin. Acta Neurologica Scandinavica 2005;111(4):229‐32. [DOI] [PubMed] [Google Scholar]
Dam 1989
- Dam M, Ekberg R, Loyning Y, Waltimo O, Jakobsen L. A double‐blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Research 1989;3(1):70‐6. [DOI] [PubMed] [Google Scholar]
Davis 2004
- Davis MP, Walsh D. Epidemiology of cancer pain and factors influencing poor pain control. American Journal of Hospice and Palliative Care 2004;21(2):137‐42. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Di Stefano 2014
- Stefano G, Cesa S, Truini A, Cruccu G. Natural history and outcome of 200 outpatients with classical trigeminal neuralgia treated with carbamazepine or oxcarbazepine in a tertiary centre for neuropathic pain. Journal of Headache and Pain 2014;15:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dickenson 2002
- Dickenson AH, Matthews EA, Suzuki R. Neurobiology of neuropathic pain: mode of action of anticonvulsants. European Journal of Pain 2002;6 Suppl A:51‐60. [DOI] [PubMed] [Google Scholar]
Eisenberg 2007
- Eisenberg E, River Y, Shifrin A, Krivoy N. Antiepileptic drugs in the treatment of neuropathic pain. Drugs 2007;67(9):1265‐89. [DOI] [PubMed] [Google Scholar]
Finnerup 2007
- Finnerup NB, Sindrup SH, Jensen TS. Chronic neuropathic pain: mechanisms, drug targets and measurement. Fundamental Clinical Pharmacology 2007;21(1):129‐36. [DOI] [PubMed] [Google Scholar]
Foley 2003
- Foley KM. Opioids and chronic neuropathic pain. New England Journal of Medicine 2003;348(13):1279‐81. [DOI] [PubMed] [Google Scholar]
Gill 2011
- Gill D, Derry S, Wiffen PJ, Moore RA. Valproic acid and sodium valproate for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD009183.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro 2008 [Computer program]
- GRADE Working Group, McMaster University. GRADEpro. Version 3.2 for Windows. Hamilton (ON): GRADE Working Group, McMaster University, 2008.
Heliovaara 1987
- Heliovaara M, Impivaara O, Sievers K, Melkas T, Knekt P, Korpi J, et al. Lumbar disc syndrome in Finland. Journal of Epidemiology and Community Health 1987;41(3):251‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Altman DG, Sterne JAC. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JPT, Deeks JJ, Altman DG. Chapter 16: Special topics in statistics. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Illing 2013
- Illing PT, Vivian JP, Purcell AW, Rossjohn J, McCluskey J. Human leukocyte antigen‐associated drug hypersensitivity. Current Opinion in Immunology 2013;25(1):81‐9. [DOI] [PubMed] [Google Scholar]
Jensen 2001
- Jensen TS, Gottrup H, Sindrup SH. The clinical picture of neuropathic pain. European Journal of Pharmacology 2001;429(1‐3):1‐11. [DOI] [PubMed] [Google Scholar]
Jensen 2002
- Jensen TS. Anticonvulsants in neuropathic pain: rationale and clinical evidence. European Journal of Pain 2002;6 Suppl A:61‐8. [DOI] [PubMed] [Google Scholar]
Lindstrom 1987
- Lindstrom P. The analgesic effect of carbamazepine in trigeminal neuralgia. Pain 1987;30:S85. [DOI] [PubMed] [Google Scholar]
Magenta 2005
- Magenta P, Arghetti S, Palma F, Jann S, Sterlicchio M, Bianconi C, et al. Oxcarbazepine is effective and safe in the treatment of neuropathic pain: pooled analysis of seven clinical studies. Neurological Sciences 2005;26(4):218‐26. [DOI] [PubMed] [Google Scholar]
Merskey 1994
- Merskey H, Bogduk N. Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms. 2nd Edition. Seattle (WA): IASP Press, 1994. [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2014
- Moore RA, Wiffen PJ, Derry S, Toelle T, Rice AS. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD007938.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nasreddine 2007
- Nasreddine W, Beydoun A. Oxcarbazepine in neuropathic pain. Expert Opinion on Investigational Drugs 2007;16(10):1615‐25. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ryan 2016
- Ryan R, Hill S, Cochrane Consumers and Communication Group. How to GRADE the quality of the evidence, Version 1.0, June 2016. cccrg.cochrane.org/author‐resources (accessed prior to 16 November 2017).
Schmader 2002
- Schmader KE. Epidemiology and impact on quality of life of postherpetic neuralgia and painful diabetic neuropathy. Clinical Journal of Pain 2002;18(6):350‐4. [DOI] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and 'Summary of findings' tables. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Suzuki 2004
- Suzuki R, Rygh LJ, Dickenson AH. Bad news from the brain: descending 5‐HT pathways that control spinal pain processing trends. Trends in Pharmacological Sciences 2004;25(12):613‐7. [DOI] [PubMed] [Google Scholar]
Treede 2008
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008;70(18):1630‐5. [DOI] [PubMed] [Google Scholar]
Verma 2005
- Verma S, Estanislao L, Simpson D. HIV‐associated neuropathic pain: epidemiology, pathophysiology and management. CNS Drugs 2005;19(4):325‐34. [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item Short‐Form Health Survey (SF‐36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473‐83. [PubMed] [Google Scholar]
Werhagen 2004
- Werhagen L, Budh CN, Hultling C, Molander C. Neuropathic pain after traumatic spinal cord injury? Relations to gender, spinal level, completeness, and age at the time of injury. Spinal Cord 2004;42(12):665‐73. [DOI] [PubMed] [Google Scholar]
Wiffen 2010
- Wiffen PJ, McQuay HJ, Edwards J, Moore RA. WITHDRAWN: Anticonvulsant drugs for acute and chronic pain. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD001133.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2013
- Wiffen PJ, Derry S, Moore RA. Lamotrigine for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2013, Issue 12. [DOI: 10.1002/14651858.CD006044.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2014
- Wiffen PJ, Derry S, Moore RA, Kalso EA. Carbamazepine for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD005451.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Williamson 2005
- Williamson A, Hoggart B. Pain: a review of three commonly used pain rating scales. Journal of Clinical Nursing 2005;14(7):798‐804. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Zhou 2009
- Zhou M, He L, Yang M, Chen N, Guo J, Li Q, Yang X, Yang J, Zhu C. Oxcarbazepine for neuropathic pain. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007963] [DOI] [PubMed] [Google Scholar]
Zhou 2013
- Zhou M, Chen N, He L, Yang M, Zhu C, Wu F. Oxcarbazepine for neuropathic pain. Cochrane Database of Systematic Reviews 2013, Issue 3. [DOI: 10.1002/14651858.CD007963.pub2] [DOI] [PubMed] [Google Scholar]