

Abstract
Gulf War Illness (GWI) is a chronic debilitating disease of unknown etiology that affects the brain and has afflicted many veterans of the 1990–91 Gulf War (GW). Here we tested the hypothesis that brain damage may be caused by circulating harmful substances to which GW veterans were exposed but which could not be eliminated due to lack of specific immunity. We assessed the effects of serum from GWI patients on function and morphology of brain cultures in vitro, including cultures of embryonic mouse brain and neuroblastoma N2A line. Blood serum from GWI and healthy GW veterans was added, alone and in combination, to the culture and its effects on the function and morphology of the culture assessed. Neural network function was assessed using electrophysiological recordings from multielectrode arrays in mouse brain cultures, whereas morphological assessments (neural growth and cell apoptosis) were done in neuroblastoma cultures. In contrast to healthy serum, the addition of GWI serum disrupted neural network communication and caused reduced cell growth and increased apoptosis. All of these detrimental effects were prevented or ameliorated by the concomitant addition of serum from healthy GW veterans. These findings indicate that GWI serum contains neuropathogenic factors that can be neutralized by healthy serum. We hypothesize that these factors are persistent antigens circulating in GWI blood that can be neutralized, possibly by specific antibodies present in the healthy serum, as proposed earlier1.
Keywords: Gulf War Illness, Brain cultures, Blood serum, Multielectrode arrays, Neural growth, Apoptosis
Introduction
Gulf War Illness (GWI) is a disease of unknown etiology that has afflicted many veterans of the 1990–91 Gulf War (GW), with a manifestation of chronic physical and neurocognitive complaints2-4. We reported recently that veterans with GWI lack some or all of 6 protective alleles from the Human Leukocyte Antigen (HLA) class 2 genes (DRB1*01:01, DRB1*08:11, DRB1*13:02, DQB1*02:02, DPB1*01:01, DRB1*06:01)5. This protective effect was evident from the analysis of the odds ratios (OR) which were less than 1 for all those 6 alleles (see Table 4 in ref.5) and their collective discriminatory power which was highly statistically significant (P = 1.29 × 10−7, chi-square test, see Table 3 in ref.5). In addition, the protective role of those alleles was further corroborated by the finding of a highly significant negative effect of the number of allele copies carried on the severity of GWI symptoms: the fewer alleles carried, the more severe the symptomatology5. Finally, we found that the presence of DRB1*13:02, one of the 6 protective alleles above, prevented subcortical brain atrophy in GWI1. Since the function of HLA class 2 alleles is specific immunity, namely to match to external antigens and then lead to the production of antibodies neutralizing the offending antigen, we hypothesized that the lack of HLA class 2 protection observed in GWI could have allowed offending antigens to persist. We further hypothesized that such persisting antigens could have come from vaccines and/or toxins to which GW veterans were exposed6, ultimately leading to low level inflammation and chronic disease. We called this the “persistent antigen” hypothesis for GWI1. This hypothesis predicts that such persistent, pathogenic antigens are present in the blood of GWI veterans. It also predicts that healthy GW veterans carrying protective alleles would have specific antibodies in their blood, which could neutralize the hypothesized persistent antigens in GWI serum. As a first step in this direction, here we sought to determine whether neuropathogenic factors are indeed present in GWI serum by assessing the effect of GWI serum on function and morphology of neural cultures in vitro. In addition, we tested whether GWI-induced neurotoxicity could be neutralized by serum from healthy control GW-era veterans.
Materials and methods
Human serum.
Serum from 2 healthy (control) GW-era veterans (participants C1, C2) and 8 veterans with GWI (participants G1, G2, G3, G4, G5, G6, G7, G8) was used; GWI participants met both Fukuda et al. (1998)2 and Steele (2000)4 criteria. Participants provided informed consent and were financially compensated for their time. Study protocols were approved by the appropriate Institutional Review Boards. The experimental design is shown in Table 1A. All participants were in good health, except for the patients with GWI; medications were unremarkable.
Table 1A.
Experimental design of brain and neuroblastoma N2A cultures.
| Source | Serum(ml) | Media (ml) | Total (ml) | |
|---|---|---|---|---|
| Control | GWI | |||
| Control | 0.1 | 0.0 | 0.9 | 1.0 |
| GWI | 0.0 | 0.1 | 0.9 | 1.0 |
| GWI+control | 0.1 | 0.1 | 0.8 | 1.0 |
Participants provided a blood sample that was used for genetic testing. HLA and apolipoprotein E (apoE) genotyping was performed for each participant (see Appendix). Participants were selected randomly from 2 groups: (a) control GW veterans, free of symptoms and carrying 2 of the 6 protective HLA alleles5, and (b) GWI patients with neurocognitive/mood symptoms and lacking any of those alleles. With respect to apoE, both controls and 2 patients carried one apoE4 allele.
Serum cytokines.
The serum of participants C2, G2, G3 and G4 was examined for the presence of cytokines IFN-γ and TNF-α, using ELISA assays with IFN-γ and TNF-α ELISA kits (Aviva Systems Biology, San Diego, CA). For IFN-γ, two replicates of seven serial dilutions from 2000 pg/ml, and for TNF-α seven serial dilutions from 1000 pg/ml were incubated in ELISA microplates, using positive controls provided in the kit and four different samples of serum from participants at two replicates. For detection, biotinylated IFN-γ and TNF-α detector antibodies were used, followed by Horseradish Peroxidase conjugate, then color developing substrate, and finally, and stop solution. Optical Density absorbance was read at 450 nm with a Molecular Devices Spectramax M5 microplate reader (Sunnyvale, CA).
Brain antibodies.
The possible presence of brain antibodies in the serum of participant G2, whose serum was extensively tested for functional and morphological effects on neural cultures, was investigated using the Encephalopathy, Autoimmune Evaluation, Serum (ENCES) test offered by the Mayo Medical Laboratories (https://www.mayomedicallaboratories.com/test-notifications/attachm.php.id=33779). Tests were carried out to detect the presence in serum of the following antibodies: (1) NMDA-R Ab CBA S (antibody to N-methyl-D-aspartate receptor), (2) Neuronal (V-G) K+ Channel (voltage-gated potassium channel complex), (3) GAD65 (glutamic acid decarboxylase65), (4) GABA-B-R (gamma-aminobutyric acid B receptors), (5) AMPA-R (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor), (6) ANNA-1,2,3 (type 1, 2, 3 antineuronal antibody), (7) AGNA-1 (anti-glial/neuronal nuclear antibody), (8) PCA-1,2-Tr (Purkinje cell cytoplasmic antibody 1, 2 type Tr), (9) amphiphysin (protein associated with the cytoplasmic surface of synaptic vesicles), (10) ACh Receptor (muscle) (acetylcholine receptor), (11) ACh R (ganglionic neuronal acetylcholine receptor), and (12) CRMP-5-IgG (collapsin response mediator protein 5 IgG; collapsins are a family of cytocolic phosphoproteins expressed exclusively in the nervous system). We did not test for other possible brain autoantibodies7.
Neural network function.
We assessed neural network function by recording neural activity (Local Field Potentials, LFP) from embryonic brain cell cultures using multielectrode arrays (MEA) and evaluating the quality of neural communication.
Brain cell cultures.
Cortical cells were isolated by dissociation from cortical tissue of 18-day embryonic day normal mouse brains (strain C57BL/6J; BrainBits, Springfield, IL). The cortical tissue was dispersed by trituration in media (Neurobasal/B27 + 0.5mM Glutamax; Invitrogen, Carlsbad, CA), undispersed tissue was allowed to settle to the tube bottom, and the supernatant was spun in a refrigerated centrifuge at 200 g for 1 min. The pellet was resuspended in 1 ml of Neurobasal media. Cells were stained with 0.4% trypan blue solution (Sigma T8154; Sigma-Aldrich, St. Louis, MO) and counted using a hemocytometer. Each fetal cortex usually yielded 2.3–3 X 106 cells.
MEA.
A MEA (Multi Channel Systems, model MEA120–2-System, Reutlingen, Germany) consists of an array of 60 electrodes embedded on a flat surface surrounded by a circular wall that creates a well around the electrodes. The electrodes are titanium nitride disks, 30 μm in diameter, arranged in an 8 × 8 square array with four missing corners. They are spaced at 200 μm intervals and are attached to gold leads that connect to the 60-channel head-stage amplifier. Approximately 250000 cells in 100 μl of media were pipetted directly onto and around the electrode area of each MEA. Before plating, the MEAs were sterilized with UV light for 1 h, and were then incubated with poly-d-lysine (50 μg/ml) for 2 h. Then, the poly-d-lysine solution was removed, the wells were rinsed with sterile water, and the cells transferred to the MEAs. After plating, the MEAs were covered by a Teflon cover and placed in an incubator for 1 h at 37o C in a humidified incubator containing 5% CO2 to allow the fresh cells, pre-incubated with Neurobasal media, to adhere to the MEAs. After this time, the media were removed and replaced with 1 ml of media + serum, as shown in Table 1A. Fresh media were made weekly, and one-half of the media (0.5 ml) in each MEA was replaced every 3–4 days (Table 1A).
Neural recordings (in duplicates) began on the third Day In Vitro (DIV) and repeated at 6, 10, 13, 17 (or 18) and 20 DIV. In a recording session, MEAs were removed from the incubator and placed on preheated (37° C) stands (the head stage amplifiers) within an enclosed Faraday box to restrict light and eliminate external electrical noise. Following a 2-min stabilization period, electrical activity was recorded simultaneously from all electrodes for 1 min at a sampling frequency of 10 kHz, digitized by a 12-bit A/D converter, and stored on a hard drive for offline processing. After each recording session, MEAs were returned to the incubator and kept there until the next recording session.
Data preprocessing and analysis.
Electrical signals recorded were further amplified (×1200) in the frequency range of 1–3000 Hz by a second-stage amplifier. LFP activity was derived from the data by applying a second-order band-pass Butterworth filter at 0.7–170 Hz to reject low and high frequencies outside the LFP range. The filtered time series were then downsampled to 1 kHz for further analysis. Thus the data from each electrode comprised 60000 time samples. LFP time series were prewhitened using a (25,1,1) ARIMA model8, as described previously9, and the innovations (residuals) retained. All possible pairwise zero-lag crosscorrelations r (Synchronous Neural Interactions, SNI) were computed. Given N = 59 MEA leads recorded from (one lead served as reference), the number of all possible pairwise (k = 2) crosscorrelations computed per MEA was:
| (1) |
Data with artifacts were eliminated from the analysis; 95% of the possible crosscorrelations were valid and retained for further analysis (Table 3). They were Fisher z-transformed10 and their absolute value taken as a measure of the strength of neural interaction:
| (2) |
Finally, for each participant/treatment combination (Table 1, columns in italics) the coefficient of variation of |z| was computed, as a measure of neural network variability11,12:
| (3) |
Eleven such values were computed (Table 1B). Statistical comparisons between serum treatments (Control, GWI, Control+GWI) were assessed using an analysis of variance (ANOVA).
Table 1B.
MEA experiments (carried out in duplicate). Numbers in parentheses denote total data (crosscorrelations) available (see text for details).
| Experiment | Control | GWI | Combination |
|---|---|---|---|
| 1 | C1(15793) | G1 (16705) | C1+G1 (15643) |
| 2 | C1 (16362) | G2 (15165) | C1+G2 (16198) |
| 3 | C2 (16820) | G3 (16246) | C2+G3 (16820) |
| 4 | G4 (16647) | C2+G4 (16252) |
Bootstrap.
The analysis above yielded one CV value per treatment combination. A bootstrap analysis13 was used to generate confidence intervals for CV and evaluate differences among CVs of the 3 treatments (Control, GWI, Control+GWI) from larger sets. For that purpose, we generated 1000 bootstrap samples with replacement for each of the 11 treatment combinations shown in Table 1B with sizes equal to the N shown in that table. This analysis yielded 1000 CV values for each treatment combination that were then used to perform a nonparametric test comparing the medians of the 3 distributions (Control, GWI, Control+GWI) (Median test, IBM-SPSS).
Assessments in neuroblastoma cultures.
The treatments and cellular measurements in neuroblastoma cultures are shown in Table 1C.
Table 1C.
Cell biology experiments in neuroblastoma cultures.
| Experiment | Control | GWI | Combination | Spreading | Apoptosis |
|---|---|---|---|---|---|
| 1 | C1 | G1 | C1+G1 | X | X |
| 2 | C1 | G2 | C1+G2 | X | X |
| 3 | C1 | G5 | C1+G5 | X | X |
| 4 | C1 | G6 | C1+G6 | X | X |
| 5 | C1 | G7 | C1+G7 | X | |
| 6 | C1 | G8 | C1+G8 | X |
Assessment of cell morphology.
The effects of serum on cell morphology were assessed in neuroblastoma cells seeded in poly-D-lysine coated, 24-well plates at a concentration of 100,000/well in Neurobasal medium containing N2 supplement and L-glutamine (ThermoFisher Scientific, Waltham, MA), in the absence (medium control) or presence of human serum for up to 5 DIV. Human serum was added in 3 combinations (Table 1A). At day 2 DIV photographs were taken from 5–8 fields of each differently treated well using a Motic AE2000-Trinocular inverted microscope (Ted Pella, Redding, Ca) and a Zeiss Axiocam 105 color digital camera (Carl Zeiss Microscopy, LLC, Thornwood, NY). Cell spreading was calculated using ImageJ software (Image Processing and Analysis in JAVA: https://imagej.nih.gov/ij/) by measuring the number of cells with processes relative to the total cell number.
Assessment of apoptosis.
Apoptosis of neuroblastoma cells was evaluated at 2 days post-exposure to human serum, using 4-chamber glass slides (ThermoFisher Scientific, Waltham, MA) coated with poly-D-lysine. Neuro 2A cells were seeded at a concentration of 50000–100000 cells per chamber, in 1 ml of Neurobasal/N2/ L glutamine medium and 10% of healthy, GWI serum, or a combination of both, at 10% concentration each, were added for 2 days. Apoptosis was examined as described previously using the In Situ Cell Death Detection Kit, TMR red (Terminal deoxynucleotidyl transferase (TdT) enzyme and fluorochrome labeling solution, ThermoFisher scientific, Waltham, MA)14. Briefly, cells were fixed in 4% Paraformaldehyde (PFA)/Phosphate Buffer Saline (PBS), then permeabilized with 0.1% Triton X-100 for 2 min on ice, and incubated with 150 μl of TUNEL reaction mixture for 30 min at 37°C in the dark. The cells were then washed with PBS, and Diamond AntiFade medium with 4’,6-diamidino-2-phenylindole (DAPI) stain (ThermoFisher Scientific, Waltham, MA) was used for visualization of nuclei, using the EVOS FL Cell Imaging System (ThermoFisher Scientific, Waltham, MA). Eight to 10 images were obtained from different fields from three experiments for each experimental condition. Apoptosis was calculated with Image J software by measuring the number of TUNEL-labeled cells (red nuclei) relative to the total cell number (DAPI-stained nuclei).
Data analysis.
Standard statistical methods15 were used to analyze the data, as detailed below.
Results
Serum.
No detectable cytokines IFN-γ or TNF-α were found in any sera tested. In addition, no brain antibodies were found in G2 (the only one tested).
Neural network function: MEA.
The effects of GWI serum and Control+GWI combination on MEA network CV are shown in Figure 1, top panel. An ANOVA showed a statistically significant effect of serum treatment (P = 0.014, F-test). Pairwise comparisons in the same analysis showed that Control CV was significantly lower than GWI CV (P = 0.011), whereas it did not differ significantly from the Control+GWI CV (P = 0.939); finally, GWI CV was significantly higher than Control+GWI CV (P = 0.012). The bootstrap analysis (Table 5) revealed a highly significant difference among the median CV of the bootstrapped CV (P < 0.001, Median Test for independent samples, IB-SPSS, version 23). Confidence intervals (99%, Table 2, Figure 1, bottom panel) for the treatment medians of the bootstrap samples were constructed using the bias-corrected and accelerated bootstrap procedure of the IBM-SPSS package (bootstrap sample = 1000,random number generator initialized using the Mersenne Twister). Pairwise comparisons of these medians showed that GWI was significantly higher than Control and Control+GWI (P < 0.001 for both comparisons), whereas Control did not differ significantly from Control+GWI (P = 0.517, Medians Test for independent samples). The effects on CV described above were consistently observed in individual experiments. Altogether, these results demonstrate that GWI serum increased network variability and that this negative effect was prevented by the concomitant addition of Control serum to the culture.
Figure 1.
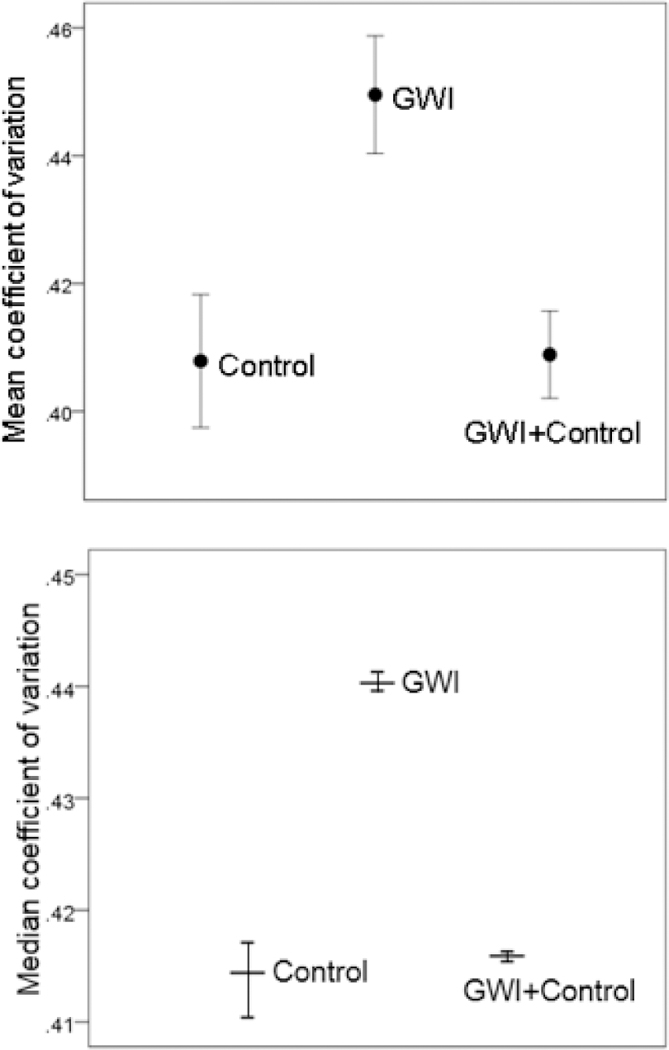
A, mean ± SEM coefficient of variation in MEAs for the 3 treatments (Table 1B). N = 3 for control, N = 4 for GWI, and N = 4 for GWI+Control. B, median coefficient of variation from the bootstrap analysis ± 99% confidence intervals. (Table 2; see text for details.)
Table 2.
Results of the bootstrap analysis. CV, coefficient of variation; CI, confidence interval.
| Treatment combination |
CV observed |
Mean CV Bootstrap |
Median CV bootstrap |
Lower 99% median CI |
Upper 99% median CI |
|
|---|---|---|---|---|---|---|
| 1 | C1a | 0.4102 | 0.4071 | 0.4071 | .4069 | 0.4074 |
| 2 | G1 | 0.4729 | 0.4687 | 0.4688 | 0.4684 | 0.4691 |
| 3 | C1a+G1 | 0.4237 | 0.4205 | 0.4204 | 0.4201 | 0.4207 |
| 4 | C1b | 0.3888 | 0.3889 | 0.3890 | 0.3887 | 0.3893 |
| 5 | G2 | 0.4342 | 0.4342 | 0.4342 | 0.4340 | 0.4344 |
| 6 | C1b+G2 | 0.3939 | 0.3939 | 0.3939 | 0.3937 | 0.3941 |
| 7 | C2 | 0.4246 | 0.4200 | 0.4200 | 0.4198 | 0.4202 |
| 8 | G3 | 0.4556 | 0.4571 | 0.4555 | 0.4543 | 0.4564 |
| 9 | C2+G3 | 0.4165 | 0.4177 | 0.4177 | 0.4174 | 0.4179 |
| 10 | G4 | 0.4355 | 0.4358 | 0.4354 | 0.4348 | 0.4360 |
| 11 | C2+G4 | 0.4013 | 0.3996 | 0.3981 | 0.3968 | 0.3997 |
Cell morphology (Figure 2, top panel).
Figure 2.
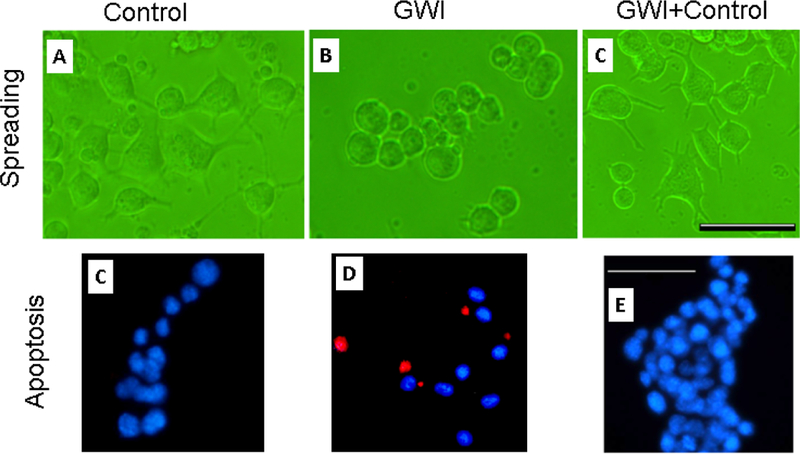
Representative fields of Neuro 2A cells cultured for 2 days in the presence of control (healthy serum, participant C1), GWI serum (participant G2), and healthy and GWI serum combination (C1+G2). Horizontal bars are 200 μm.
Neuro 2A cells in healthy serum spread, extending elongated processes 2 days post-serum exposure (Figure 2, top left panel), with >70% of cells forming processes. The presence of GWI serum resulted in cell aggregation and rounding (Figure 2, top middle panel), and only <20% cells developed processes. However, the simultaneous presence of healthy and GWI serum (Figure 2, top right panel) resulted in >60% cells spread cells with processes. The results of quantitative statistical analysis are shown in Figure 3, top panel. An ANOVA showed a highly statistically significant effect of treatment on percent cellular spreading (P = 1.7×10–32, F-test). With pairwise comparisons, GWI spreading was significantly lower than the Control (P = 1.18×10–20) and the GWI+Control (P = 6.34×10–19), whereas Control and GWI+Control did not differ significantly (P = 0.242).
Figure 3.
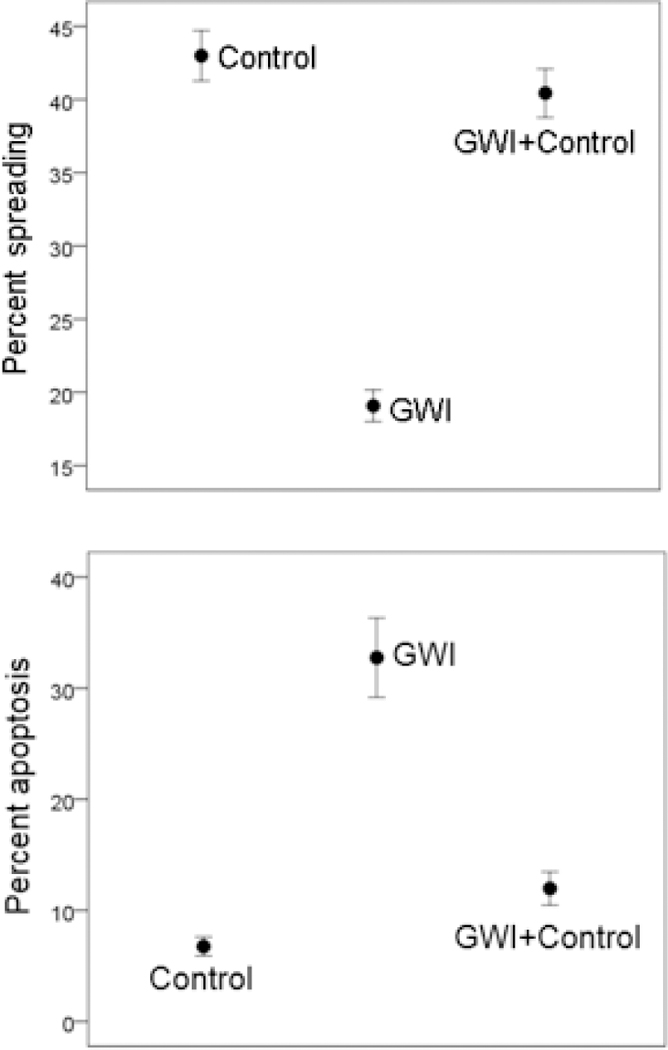
A, mean ± SEM percent cell spreading in the 3 treatments. N = 30 measurements for Control, 45 for GWI, and 35 for GWI+Control. B, mean ± SEM percent cell apoptosis (TUNEL assay) in the 3 treatments. N = 17 measurements for Control, 28 for GWI, and 26 for GWI+Control.
Cell apoptosis (Figure 2, bottom panel).
Neuro 2A cells cultured for 2 days in the presence of healthy serum (Figure 2, bottom left panel) were <5% apoptotic with TUNEL, whereas >25% cells in GWI serum were apoptotic (Figure 2, bottom middle panel. The simultaneous presence of healthy and GWI serum effectively protected the cells from apoptosis (Figure 2, right bottom panel), since <7% percent of cells were apoptotic. An ANOVA showed a highly statistically significant effect of treatment on percent apoptosis (P = 6.45×10–11, F-test). In pairwise comparisons, GWI apoptosis was significantly higher than the Control (P = 4.42×10–10) and the GWI+Control (P = 8.74×10–9), whereas Control and GWI+Control did not differ significantly (P = 0.152).
Discussion
General.
Neurological symptoms indicating functional and structural brain abnormalities often dominate chronic illness in GWI, and include difficulty in memory and concentration, trouble finding words, blurred vision, tremors, numbness, headaches and mood alteration among others4. Structural16 and functional17 brain abnormalities in GWI have been described, but no causes have been identified. Current hypotheses include exposures to toxins (see 18 for an extensive review) and presence of persistent antigens1 that could not be eliminated due to lack of HLA protection5. Although these hypotheses are not mutually exclusive, our persistent antigen hypothesis rests on more direct evidence, namely (a) the clear dependence of GWI symptom severity on the counts of HLA protective alleles5, (b) the differential distribution of functional GWI brain abnormalities, depending on the protective HLA allele counts19, and (c) the protective effect of one of those alleles (DRB1*13:02) in preventing subcortical brain atrophy in GWI1,17. In the present study, we tested directly for possible harmful effects of hypothesized persistent antigens in the blood of GWI patients and for a possible beneficial role of the blood of healthy GW-era veterans. Given that persistent antigens might be remnants of vaccines that could not be eliminated in GWI but which were successfully eliminated in healthy GW veterans by the production of specific antibodies, we reasoned that such antibodies might neutralize GWI antigens and prevent harmful effects. Indeed, this is what we found in all tests we performed.
Network variability.
GWI serum induced a significant increase in neural network variability, which was prevented by the concomitant addition of serum from healthy GW-era (control) veterans (Figure 1A,B). In a previous study12, we reported that the addition of apolipoprotein E4 (apoE4), a known harmful agent, to MEA cultures resulted in increased network variability. Thus, the addition of GWI serum also produced a detrimental effect on the network. Remarkably, this was prevented by healthy serum. We interpret these results in the context of our “persistent antigen” hypothesis for GWI1, which postulates the existence of antigens in GWI that could not be eliminated due to lack of relevant HLA class 2 alleles5. The fact that healthy serum prevented the detrimental GWI effect suggests that this serum might contain substances that counteract the negative GWI effect. We hypothesize that these substances are antibodies against the persistent GWI antigens: such antibodies would neutralize the antigens and thus prevent their harmful effect. Furthermore, we hypothesize that GWI antigens are probably fragments of agents contained in vaccines administered to GW veterans against which GWI veterans could not make antibodies due to their specific HLA class 2 makeup. These hypotheses remain to be tested.
Cell morphology and apoptosis.
GWI serum effects were detrimental on all aspects of cell morphology and survival: It stunned neural cell growth manifested as reduced cell spreading without process formation. In the combined presence of healthy and GWI serum, cell processes formed in the same way as in heathy serum, and minimal cell clusters were observed (Figure 2, top). Altered cell morphology changes were accompanied by significant apoptosis in the presence of GWI serum. (Figure 2, bottom), an effect prevented by healthy serum. These observations were supported by the results of quantitative analyses (Figure 3A,B).
Overall then, these results document the detrimental effect of GWI serum on neural structure and function in culture. They also document the impressive prevention of those effects by the concomitant addition to the culture of serum from healthy GW-era veterans. These findings support at least in part, our persistent antigen hypothesis, as a pivotal factor other than the toxic exposure hypothesis. In fact, such persistent pathogens could account for current symptoms and signs of GWI persisting 27 years later, including low-grade inflammation20 and immune dysfunction21. Over the years, they could have induced other abnormalities, such as epigenetic modifications22,23.
The possible nature of harmful GWI agents.
(a) Our first thought was that these agents might include brain autoantibodies. We explored this possibility by testing for the presence of a wide variety of 12 such antibodies, offered by the ENCES test of the Mayo Medical Laboratories, in one GWI patient whose serum had shown detrimental effects on neural network communication and cell morphology: no such antibody was detected. Hence, we did not test further sera with this test. (We did not test for other brain antibodies7.) (b) Another possibility was that the harmful effects could be due to circulating cytokines interferon gamma (INF-γ) and tumor necrosis alpha (TNF-α) for the following reasons. IFN-γ is a master regulator in inflammation and immunity being considered a prime pro-inflammatory cytokine24-26 (Zhang, 2007, Pollard et al. 2013, Mitagami et al. 2015). TNF-α is a prominent cytokine in chronic inflammatory multi-system diseases27,28. To explore this possibility, we tested the sera we had available at the time (one control and 3 GWI sera) for those cytokines using ELISA, with negative results. Hence, we did not test further sera with this test. (c) Although chemical exposures could have caused brain damage at the time of exposure, to account for observed neuropsychological deficits in GWI18, it is unlikely that such exposures of could have resulted in residual harmful substances, still circulating in the blood; we did not look for such substances. In addition, it is difficult to explain with this hypothesis the prevention of the harmful GWI effect by healthy serum. (d) Finally, we entertained the hypothesis that the harmful GWI effects might be caused by fragments of pathogens contained in the vaccines administered to GW veterans. Those vaccines contained the following inactivated (or attenuated) pathogens6,29: adenovirus (types 4 and 7), anthrax, botulinum toxoid, cholera, hepatitis B, influenza, measles, meningococcus (A,C,Y,W135), mumps, plague, polio, rabies, rubella, smallpox, tetanus-diphtheria, typhoid, varicella, and yellow fever. Our hypothesis above1 is based on the following considerations. (i) Vaccines can have long-lasting adverse effects in susceptible individuals, resulting from molecular mimicry or persistent antigen30. (ii) Patients with GWI are deficient in 6 HLA class 2 alleles (see above) 5. HLA class 2 alleles are necessary for specific immunity, i.e. the production of antibodies to neutralize specific pathogen. In the absence of specific alleles, matching specific antigens, such antigens may persist, potentially causing low-grade inflammation; interestingly, elevation of inflammatory markers has been described in GWI20. (iii) The presence of DRB1*13:02, one of the 6 GWI protective alleles above, was found to prevent subcortical brain atrophy in GWI1. (iv) Brain dysfunction in GWI is HLA-associated31,32. There are two sequelae of our “persistent antigen” hypothesis1: first, non-neutralized vaccine fragments in GWI serum are, at least in part, to blame for the harmful effects on neural cultures and, second, serum from healthy GW veterans who received the same vaccines but did not develop GWI because they had the requisite HLA protection (as they possessed at least one of the 6 alleles above), might contain antibodies to the hypothesized persistent antigens that could neutralize them, if added to GWI serum. In fact, this is what we observed in this study. This effect is reminiscent of older classical treatment of infectious diseases by administering to the patient serum from a patient who successfully fought the disease, as seen most recently in the Ebola virus outbreak33.
These findings open the possibility of successful, immunotherapy-based intervention in GWI, as follows: if the cause is pathogenic antigens, then the administration of specific antibodies (to those antigens) might be effective in neutralizing them and bring the GWI process to a halt. Such an approach would be in keeping with a recently advocated resurgence of refined antiserum treatment using monoclonal antibodies34. Given that GWI affects several systems and that, typically, symptom severities in these systems are significantly correlated, such specific immunotherapy could be beneficial to overall GWI, and not only GWI’s brain manifestations.
Grants
Funded by the University of Minnesota (the American Legion Brain Sciences Chair) and the U.S. Department of Veterans Affairs. The sponsors had no role in the current study design, analysis or interpretation, or in the writing of this paper. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Abbreviations
- ACh Receptor (muscle)
antibody to acetylcholine receptor
- ACh R
antibody to ganglionic neuronal acetylcholine receptor
- AGNA-1
anti-glial/neuronal nuclear antibody
- AMPA-R
antibody to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- Amphiphysin
antibody to protein associated with the cytoplasmic surface of synaptic vesicles
- ANNA-1,2,3
type 1, 2, 3 antineuronal antibody
- ANOVA
Analysis of variance
- CRMP-5-IgG
antibody to collapsin response mediator protein 5 IgG
- CV
Coefficient of variation
- DAPI
4’,6-diamidino-2-phenylindole stain
- DIV
Days in vitro
- ENCES
Encephalopathy Autoimmune Evaluation Serum
- GABA-B-R
antibody to gamma-aminobutyric acid B receptors
- GAD65
antibody to glutamic acid decarboxylase 65
- GW
Gulf War
- GWI
Gulf War Illness
- HLA
Human Leukocyte Antigen
- IFN-γ
Interferon-γ
- LFP
Local Field Potentials
- MEA
multielectrode arrays
- NMDA-R Ab CBA S
antibody to N-methyl-D-aspartate receptor
- Neuronal (V-G) K+ Channel
antibody to voltage-gated potassium channel complex
- PFA
Paraformaldehyde
- PBS
Phosphate Buffer Saline
- PCA-1,2-Tr
Purkinje cell cytoplasmic antibody 1, 2 type Tr
- TNF-α
Tumor Necrosis Factor-α
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- UV
ultraviolet
Appendix
HLAgenotyping.
DNA isolation was carried out from 3 ml of whole blood drawn in EDTA tubes, using a commercially available kit (ArchivePure cat. 2300730) from 5Prime (distributed by Fisher Scientific or VWR) with an expected yield of 50–150μg of DNA. The purified DNA samples were sent to Histogenetics (http://www.histogenetics.com/) for high-resolution HLA Sequence-based Typing (SBT; details are given in https://bioinformatics.bethematchclinical.org/HLA-Resources/HLA-Typing/High-Resolution-Typing-Procedures/ and https://bioinformaticsbethematchclinical.org/WorkArea/DownloadAsset. aspx?id=6482). Their sequencing DNA templates are produced by locus- and group-specific amplifications that include exon 2 and 3 for class I (A, B, C) and exon 2 for class II (DRB1, DRB3/4/5, DQB1, and DPB1) and reported as Antigen Recognition Site (ARS) alleles as per ASHI recommendation35.
ApoE genotyping.
DNA samples were genotyped using PCR amplification followed by restriction enzyme digestion36. Each amplification reaction contained PCR buffer with 15 mmol/L MgCL2 ng amounts of genomic DNA, 20 pmol apoE forward (5N TAA GCT TGG CAC GGC TGT CCA AGG A 3N) and reverse (5T ATA AAT ATA AAA TAT AAA TAA CAG AAT TCG CCC CGG CCT GGT ACA C 3N) primers, 1.25 mmol/L of each deoxynucleotide triphosphate, 10% dimethylsulfoxide, and 0.25 μL Amplitaq DNA polymerase. Reaction conditions in a thermocycler included an initial denaturing period of 3 min at 95 C, 1 min at 60 C, and 2 min at 72 C; followed by 32 cycles of 1 min at 95 C, 1 min at 60 C, and 2 min at 72 C; and a final extension of 1 min at 95 C, 1 min at 60 C, and 3 min at 72C. PCR products were digested with HhaI and separated on a 4% Agarose gel which was stained with Ethidium Bromide. Known apoE isoform standards were included in the analysis.
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared.
References
- 1.James LM, Christova P, Engdahl BE, et al. Human leukocyte antigen (HLA) and GulfWar Illness (GWI): HLA-DRB1*13:02 spares subcortical atrophy in Gulf War veterans. EbioMedicine. 2017; 26:126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fukuda K, Nisenbaum R, Stewart G, et al. Chronic multisymptom illness affecting Air Force veterans of the Gulf War. JAMA. 1998; 280: 981–8. [DOI] [PubMed] [Google Scholar]
- 3.Kang HK, Li B, Mahan CM, et al. Health of US veterans of 1991 Gulf War: a follow-up survey in 10 years. J Occup Environ Med. 2009; 51: 401–10. [DOI] [PubMed] [Google Scholar]
- 4.Steele L Prevalence and patterns of Gulf War illness in Kansas veterans: association of symptoms with characteristics of person, place, and time of military service. Am J Epidemiol. 2000; 152: 992–1002. [DOI] [PubMed] [Google Scholar]
- 5.Georgopoulos AP, James LM, Mahan MY, et al. Reduced Human Leukocyte Antigen (HLA) protection in Gulf War Illness (GWI). EBioMedicine. 2016; 3:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Institute of Medicine National Research Council. Gulf War and Health: Volume 1 Depleted Uranium, Pyridostigmine Bromide, Sarin, and Vaccines. Washington, DC: National Academies Press, 2000. [PubMed] [Google Scholar]
- 7.Abou-Donia MB, Conboy LA, Kokkotou E, et al. Screening for novel central nervous system biomarkers in veterans with Gulf War Illness. Neutoxicol Teratol. 2017; 61: 31–46. [DOI] [PubMed] [Google Scholar]
- 8.Box GEP, Jenkins GM. Time Series Analysis: Forecasting and Control. San Francisco, Holden-Day, United States, 1976. [Google Scholar]
- 9.Christopoulos VN, Boeff DV, Evans CD, et al. A network analysis of developing brain cultures. J Neural Eng. 2012; 9: 046008. [DOI] [PubMed] [Google Scholar]
- 10.Fisher RA. Statistical Methods for Research Workers, 13th edition. Edinburgh, Oliver and Boyd, 1958 [Google Scholar]
- 11.Leuthold AC, Mahan MY, Stanwyck JJ, et al. The number of cysteine residues per mole in apolipoprotein E affects systematically synchronous neural interactions in women’s healthy brains. Exp Brain Res. 2013; 226: 525–536. [DOI] [PubMed] [Google Scholar]
- 12.Christopoulos V, Georgopoulos A, Georgopoulos AP. The effect of apolipoprotein E4 on synchronous neural interactions in brain cultures. Exp Brain Res. 2015; 233: 1977–82. [DOI] [PubMed] [Google Scholar]
- 13.Efron B, Tibshirani R. An Introduction to the Bootstrap. New York, Chapman & Hall, 1993. [Google Scholar]
- 14.Kaminari A, Giannakas N, Tzinia A, et al. Overexpression of matrix metalloproteinase-9 (MMP-9) rescues insulin-mediated impairment in the 5XFAD model of Alzheimer’s disease. Scientific Reports. 2017; 7(1): 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snedecor GW, Cochran WG. Statistical methods. Ames, IA: Iowa State University Press, 1989. [Google Scholar]
- 16.Christova P, James LM, Engdahl BE, et al. Subcortical brain atrophy in Gulf War Illness. Exp Brain Res. 2017; 235: 2777–86. [DOI] [PubMed] [Google Scholar]
- 17.Engdahl BE, James LM, Miller RD, et al. A Magnetoencephalographic (MEG) study of Gulf War Illness (GWI). EBioMedicine. 2016; 12:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White RF, Steele L, O’Callaghan JP, et al. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: Effects of toxicant exposures during deployment. Cortex. 2016; 74: 449–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.James LM, Engdahl BE, Leuthold AC, et al. Brain correlates of Human Leukocyte Antigen (HLA) protection in Gulf War Illness (GWI). EBioMedicine. 2016; 13:72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson GJ, Slater BC, Leis LA, et al. Blood biomarkers of chronic inflammation in Gulf War Illness. PLoS One. 2016;11(6):e0157855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broderick G, Ben-Hamo R, Vashishtha S, et al. Altered immune pathway activity under exercise challenge in Gulf War Illness: an exploratory analysis. Brain Behav Immun. 2013; 28: 159–69. [DOI] [PubMed] [Google Scholar]
- 22.Marsit CJ. Influence of environmental exposure on human epigenetic regulation. J Exp Biol. 2015; 218: 71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piercea LM, Kurataa WE, Matsumotoa KW, et al. Long-term epigenetic alterations in a rat model of Gulf War Illness. NeuroToxicology. 2016; 55: 20–32. [DOI] [PubMed] [Google Scholar]
- 24.Yin Zhang J. and yang interplay of IFN-γ in inflammation and autoimmune disease. J Clin Investigation. 2007; 117(4): 871–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitagami Y, Yasunaga J, Kinosada H, et al. Interferon-γ promotes inflammation and development of T-cell lymphoma in HTLV-1 bZIP factor transgenic mice. PLoS Pathog. 2015; 11(8): e1005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollard KM, Cauvi DM, Toomey CB, et al. Interferon-γ and systemic autoimmunity. Discov Med. 2013; 16(87): 123–131. [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008; 214: 149–160. [DOI] [PubMed] [Google Scholar]
- 28.Popa C, Mihai G, Netea MG, et al. The role of TNF-α in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res. 2007; 48: 751–762. [DOI] [PubMed] [Google Scholar]
- 29.DOD (U.S. Department of Defense). Information Paper: Vaccine Use During the Gulf War. 2000; Washington, DC:Office of the Special Assistant for Gulf War Illnesses; http://www.gulflink.osd.mil/va/ [Google Scholar]
- 30.Institute of Medicine. Adverse effects of vaccines: Evidence and causality. 2012; Washington, DC: National Academies Press. [PubMed]
- 31.James LM, Engdahl BE, Leuthold AC, et al. Brain correlates of Human Leukocyte Antigen (HLA) protection in Gulf War Illness (GWI). EBioMedicine. 2016; 13:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georgopoulos AP, James LM, Carpenter AF, et al. Gulf War illness (GWI) as a neuroimmune disease. Exp Brain Res. 2017; 235(10): 3217–3225. [DOI] [PubMed] [Google Scholar]
- 33.Kreil TR. Treatment of Ebola virus infection with antibodies from reconvalescent donors. Emerg Infect Dis. 2015; 21(3): 521–523. doi: 10.3201/eid2103.141838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marston HD, Paules CI, Fauci AS. Monoclonal antibodies for emerging infectious diseases — borrowing from history. New England J Med. 2018; 378: 1469–1472. [DOI] [PubMed] [Google Scholar]
- 35.Cano P, Klitz W, Mack SJ, et al. Common and well-documented HLA alleles: report of the Ad-Hoc committee of the American society for histocompatiblity and immunogenetics. Hum Immunol. 2007; 68: 392–417. [DOI] [PubMed] [Google Scholar]
- 36.Reymer WA, Groenemeyer BE, Van de Burg R. Apolipoprotein E genotyping on agarose gels. Clin Chem. 1995; 41: 1046–1047. [PubMed] [Google Scholar]