

Abstract
Background
Infantile‐onset Pompe disease is a rare and progressive autosomal‐recessive disorder caused by a deficiency of the lysosomal enzyme acid alpha‐glucosidase (GAA). Current treatment involves enzyme replacement therapy (with recombinant human alglucosidase alfa) and symptomatic therapies (e.g. to control secretions). Children who are cross‐reactive immunological material (CRIM)‐negative require immunomodulation prior to commencing enzyme replacement therapy.
Enzyme replacement therapy was developed as the most promising therapeutic approach for Pompe disease; however, the evidence is lacking, especially regarding the optimal dose and dose frequency.
Objectives
To assess the effectiveness, safety and appropriate dose regimen of enzyme replacement therapy for treating infantile‐onset Pompe disease.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register, which is compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the Cochrane Central Register of Controlled Trials (CENTRAL), Embase (Ovid), PubMed and LILACS, and CBM, CNKI, VIP, and WANFANG for literature published in Chinese. In addition, we searched three online registers: WHO International Clinical Trials Registry Platform ClinicalTrials.gov, and www.genzymeclinicalresearch.com. We also searched the reference lists of relevant articles and reviews.
Date of last search of the Group's Inborn Errors of Metabolism Trials Register: 24 November 2016.
Selection criteria
Randomized and quasi‐randomized controlled trials of enzyme replacement therapy in children with infantile‐onset Pompe disease.
Data collection and analysis
Two authors independently selected relevant trials, assessed the risk of bias and extracted data. We contacted investigators to obtain important missing information.
Main results
We found no trials comparing the effectiveness and safety of enzyme replacement therapy to another intervention, no intervention or placebo.
We found one trial (18 participants) that fulfilled the selection criteria, comparing different doses of alglucosidase alfa. The trial provided low‐quality evidence (this was a small trial, there were no numerical results available by dose group, random sequence generation and allocation concealment were unclear, and there was a lack of blinding). The duration of alglucosidase alfa treatment ranged from 52 weeks (the length of the original study) to up to three years (including the extended phase of the trial), with a median duration of treatment being 2.3 years.
The trial only reported that clinical responses including cardiac function and motor development, as well as the proportion of children that were free of invasive ventilation, were similar in the 20 mg/kg every two weeks and the 40 mg/kg every two weeks groups (low‐quality evidence). Long‐term alglucosidase alfa treatment markedly extended survival as well as ventilation‐free survival and improved cardiomyopathy (low‐quality evidence). In relation to the number of children experiencing one or more infusion‐related events, there was no significant difference between dose groups, risk ratio 0.83 (95% confidence interval 0.40 to 1.76) (low‐quality of evidence). However, of note, at 52 weeks, five children in the 20 mg/kg every two weeks dose group experienced a total of 41 mild or moderate (none severe) infusion‐related events and the six children in the 40 mg/kg every two weeks dose group experienced a total of 123 infusion‐related events. By the end of the extended phase of the trial, five children in the 20 mg/kg every two weeks dose group experienced a total of 47 infusion‐related events and the six children in the 40 mg/kg every two weeks dose group experienced a total of 177 infusion‐related events. The trial was supported by the Genzyme Corporation.
Authors' conclusions
The search found no trials comparing the effectiveness and safety of enzyme replacement therapy to another intervention, no intervention or placebo. One small randomized controlled trial provided no robust evidence for which dosing schedule of alglucosidase alfa was more effective to treat infantile‐onset Pompe disease. It is not deemed ethical to proceed with new placebo‐controlled trials, therefore a randomized controlled trial with a large sample size comparing different dosing schedules of enzyme replacement therapy is needed. The main clinical outcomes (i.e. cardiac function, invasive ventilation, survival, motor development, adverse events (e.g. the development of antibodies)) should be standardized when evaluated and reported.
Plain language summary
Enzyme replacement therapies for infantile‐onset Pompe disease
Background
Pompe disease was the first identified lysosomal storage disorder (inherited metabolic diseases that are characterized by an abnormal build‐up of various toxic materials in the body's cells). It causes damage to muscle by the accumulation of glycogen (which is used as an energy source by the body) within the lysosome due to deficiency of an enzyme called acid alpha‐glucosidase (GAA). There is a broad spectrum of Pompe disease types with infantile‐onset (starting during childhood) being the most severe. The usual presenting features of the infantile form are worsening weakness of the heart, muscles used in breathing and skeletal muscle. The tongue and the liver may be enlarged. This review aims to assess the effectiveness and safety of enzyme replacement therapy in children with infantile‐onset Pompe disease.
Search date
The evidence is current to 24 November 2016.
Study characteristics
We searched medical databases for clinical trials of enzyme replacement therapy in children with Pompe disease. We found no randomised trials (where people taking part in the trial have equal chances of being in the treatment or the control group) comparing the effectiveness and safety of enzyme replacement therapy to another intervention, no intervention or placebo (a 'dummy' drug). We found one trial (18 children) that compared two doses of recombinant human alglucosidase alfa (an enzyme replacement therapy): 20 mg/kg every two weeks (low dose) and 40 mg/kg every two weeks (high dose). The duration of treatment ranged from 52 weeks (the length of the original study) to up to three years (including the extension phase of the study), with a median (the middle number) duration of treatment being 2.3 years.
Key results
There were very limited numerical results available by dose group, the trial showed no evidence in favour of a high dose as opposed to a low dose. It described that the clinical responses including cardiac function, motor development (development of a child's muscles and their ability to move around and manipulate their environment), as well as the proportion of children that were free of invasive ventilation,were similar in the two groups (low‐quality evidence). Long‐term alglucosidase alfa treatment markedly extended survival as well as ventilation‐free survival (time without requiring a machine to help with breathing) and improved cardiomyopathy (heart disease) (low‐quality evidence). In relation to the number of children experiencing one or more infusion‐related events, there was no significant difference between the different dose groups (low‐quality evidence), although, of note, at 52 weeks, five children in the low‐dose group experienced a total of 41 mild or moderate (none were severe) infusion‐related events and the six children in the high dose‐group experienced a total of 123 infusion‐related events. By the end of the extension phase, five children in the low‐dose group experienced a total of 47 infusion‐related events and the six children in the high‐dose group experienced a total of 177 infusion‐related events.
New trials should be undertaken with adequate number of participants to detect effectiveness and safety at different doses of alglucosidase alfa. The main clinical outcomes (i.e. cardiac function, time to ventilation, survival, motor development and side effects) should be standardized when evaluated and reported.
Quality of the evidence
The evidence that this small trial provided to the review was of low quality. The trial was supported by the Genzyme Corporation.
Summary of findings
Summary of findings for the main comparison. Enzyme replacement therapy for infantile‐onset Pompe disease.
| Enzyme replacement therapy for infantile‐onset Pompe disease | ||||||
|
Patient or population: infantile‐onset Pompe disease Settings: hospital in‐patient Intervention: enzyme replacement therapy at 20 mg/kg/2 weeks Control: enzyme replacement therapy 40 mg/kg/2 weeks | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Enzyme replacement therapy (40 mg/kg/2 weeks) | Enzyme replacement therapy (20 mg/kg/2 weeks) | |||||
|
Cardiac function Follow‐up: 52 weeks |
No detailed information comparing the 2 dose groups. Results from initial phase of the trial described that cardiac function was similar in the 2 dose groups and long‐term alglucosidase alfa treatment (both dosing groups) improved cardiomyopathy.1 | 18 (1 RCT) | ⊕⊕⊝⊝ Low2,3 | ‐ | ||
|
Time to ventilation Follow‐up: 52 weeks |
Results from the initial phase of the trial described that the proportion of children that were free of invasive ventilation was similar in the 2 dose groups.1 | 18 (1 RCT) | ⊕⊕⊝⊝ Low2,3 | ‐ | ||
|
Survival Follow‐up: 52 weeks |
"Fifteen of the 18 treated patients reached the age of 18 months by the end of the study; three patients were right‐censored from this analysis because they had not reached the age of 18 months by the end of the study, although they were alive at that time (at ages 15.9 months, 17.9 months, and 14.4 months)." (Kishnani 2007). 1 participant died after receiving alglucosidase alfa for 61 weeks, but before the extension phase of the study began.1 | 18 (1 RCT) | ⊕⊕⊝⊝ Low2,3 | ‐ | ||
|
Motor development Follow‐up: 52 weeks |
Results from the initial trial described that motor development was similar in the 2 dose groups. It was reported in the paper that, "13 of the 18 treated patients acquired substantial motor and functional skills as assessed by the AIMS and Pompe PEDI tests."1 | 18 (1 RCT) | ⊕⊕⊝⊝ Low2,3 | ‐ | ||
|
Quality of life Follow‐up: NA |
Not reported. | 18 (1 RCT) | NA | ‐ | ||
|
Infusion‐related events Follow‐up: 52 weeks |
Study population | RR 0.83 (0.40 to 1.76) | 18 (1 RCT) | ⊕⊕⊝⊝ Low2,4 | ‐ | |
| 667 per 1000 | 553 per 1000 (267 to 1000) | |||||
|
Other adverse events Follow‐up: 52 weeks |
It was stated that at 52 weeks that 16/18 participants (from both dose groups) developed IgG antibodies to alglucosidase alfa, 1 of which developed inhibitory antibodies.1 | 18 (1 RCT) | ⊕⊕⊝⊝ Low2,3 | ‐ | ||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). AIMS: Alberta Infant Motor Scale; CI: confidence interval; IgG: immunoglobulin G; NA: not applicable; Pompe PEDI: Pompe Pediatric Evaluation of Disability Inventory; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1 No data available by dose groups. 2 Study limitations: high risk of bias from lack of blinding and unclear risk from the method of sequence generation, downgrading the quality of evidence. 3 No numerical results available by dose group, downgrading the quality of evidence due to selective reporting. 4 95% CI 0.40 to 1.76, downgrading the quality of evidence due to imprecision.
Background
Description of the condition
Pompe disease was the first recognized lysosomal storage disorder and is the only glycogen storage disease associated with a defect in lysosomal metabolism. It is an autosomal‐recessive disorder caused by a deficiency of the lysosomal enzyme acid alpha‐glucosidase (GAA) leading to glycogen accumulation in multiple tissue types, including skeletal, cardiac, smooth muscle and nervous tissue (Hirschhorn 2001). Symptoms of Pompe disease can manifest at any age. Based on whether clinical symptoms develop prior to, or beyond, one year of age, it is typically classified into infantile‐onset and late‐onset forms (Kishnani 2006a). Infantile‐onset Pompe disease is further subdivided into classical or non‐classical forms based on the presence or absence of cardiomyopathy as a primary feature (Slonim 2000). Children with a small amount of natural GAA enzyme are termed cross‐reactive immunological material (CRIM)‐positive. Children who are unable to produce native enzyme are termed CRIM‐negative. CRIM‐positive and CRIM‐negative status can be determined by western blot analysis (Klinge 2005).
Pompe disease has an estimated frequency of 1 in 40,000 in the African‐American population, 1 in 50,000 in the Chinese population, 1 in 40,000 in the Dutch population and 1 in 146,000 in the Australian population. The infantile‐onset form has an apparent higher incidence among African‐American and Chinese populations (Hirschhorn 2001). Infantile‐onset symptoms are characterized by progressive cardiac, respiratory and skeletal muscle weakness and early death from cardiorespiratory failure. One natural history study reported that the median age of symptom onset in infants with infantile‐onset disease is 1.6 months, with ventilator dependency at 4.7 months and death at 6.0 to 7.7 months (Kishnani 2006a; Van den Hout 2003). Children with classic infantile‐onset Pompe disease rarely survive beyond one year of age.
The clinical diagnosis is traditionally confirmed by the virtual absence (infantile‐onset) or markedly reduced (late‐onset) GAA activity in tissues such as cultured fibroblasts from skin biopsy, muscle biopsy, purified lymphocytes, mononuclear cells and lymphoid cell lines, when combined with clinical and laboratory data. In general, GAA enzyme activities of less than 1% of normal controls are seen in the infantile form (Kishnani 2006a). Both the American College of Medical Genetics (ACMG) and the Japanese guidelines for the management of Pompe disease recommend a GAA assay performed on skin fibroblasts (preferred tissue) or muscle biopsy as the diagnostic 'gold standard' (Eto 2010; Kishnani 2006a). New techniques mean these invasive procedures are not required as enzyme activity can be assessed on leukocytes using fluorometric assay or on dried blood spot using tandem mass spectroscopy (Zhang 2006). Tandem mass spectroscopy has been used for newborn screening for Pompe disease in some countries. Confirmation should be achieved by mutation analysis of the GAA gene (OMIM: 606800), the only gene in which mutations are known to cause Pompe disease (Kroos 2008).
Description of the intervention
There was no disease‐specific treatment for Pompe disease until enzyme replacement therapy (ERT) was approved for human use in 2006 (Chien 2013). The aim of the therapy is to replace the missing enzyme and thus prevent the manifestations of the disease. Results from several different animal studies have shown that children treated early in the disease, prior to the development of extensive tissue damage, are likely to have gains in overall survival and quality of life (Amalfitano 2001; Kishnani 2006b; Kishnani 2009; Klinge 2005). Intravenous ERT with different forms of recombinant human alglucosidase alfa have been administered at doses of 10 mg/kg/week to 40 mg/kg/week. However, it is still controversial whether the dose of ERT affects outcomes. One report looking at treatment with alglucosidase alfa 20 mg/kg every other week found children with a CRIM‐negative status had a good outcome on ERT (Al Khallaf 2013). Another study concluded that treatment with alglucosidase alfa at a higher dose of 40 mg/kg every week was generally well tolerated and led to improved ventilator‐free survival and motor outcomes in children with classic infantile‐onset Pompe disease when compared with treatment at 20 mg/kg every other week (Van Gelder 2013). One further study observed no advantage of different doses of alglucosidase alfa in children with infantile‐onset Pompe disease (Kishnani 2007).
Another factor that may affect outcome is CRIM status, this is because of its influence on the production of specific immunoglobulin G (IgG) antibodies towards alglucosidase alfa, which diminishes its effectiveness (Banugaria 2011). One long‐term retrospective study revealed that CRIM‐negative status was associated with poorer clinical outcome and reduced overall survival in infants with Pompe disease treated with alglucosidase alfa (Nicolino 2009). This is due to CRIM‐negative children being more likely to produce higher levels of anti‐alglucosidase alfa antibodies. Children who are CRIM‐positive also develop antibodies but these tend to be self‐resolving (Kishnani 2007). Other scientists hold the opinion that, irrespective of CRIM status, anti‐alglucosidase alfa antibody titre is the important factor in determining effectiveness of treatment (Van Carin 2015). Immune modulation prior to the start of ERT has been used to try to negate the antibody response in CRIM‐negative children.
There are still limitations to ERT and not all patients are suitable for this very expensive treatment. Further modifications, such as immune modulation, desensitization and an improved uptake formulation are expected to enhance the efficacy of the therapy (Baruteau 2014; Messinger 2012; Sun 2007).
How the intervention might work
Pompe disease is caused by mutations in the gene that encodes for GAA. Deficiency of this enzyme causes glycogen to accumulate in various tissues. The excessive glycogen in cardiac muscle results in thickening of the ventricular walls and interventricular septum, leading to the hypertrophic cardiomyopathy observed in infantile‐onset disease. Glycogen accumulation is thought to trigger an autophagic process and increase numbers of lysosomes and autophagosomes, resulting in weakness by a mechanical interruption of the contractile apparatus (Fukuda 2006; Reuser 2006). The mechanism of ERT is to replace the missing enzyme with a recombinant form that degrades the alpha‐1,4 and alpha‐1,6 linkages in glycogen and reduces glycogen storage.
Why it is important to do this review
In 2012, the World Health Organization (WHO) appealed for all countries to come together to combat rare diseases (WHO 2012). ERT was developed as the most promising therapeutic approach for Pompe disease; however, the evidence is lacking, especially regarding the optimal dose and dose frequency. This Cochrane Review aimed to determine the effectiveness and safety of ERT for the infantile‐onset form of Pompe disease.
Objectives
To assess the effectiveness, safety and appropriate dose regimen of enzyme replacement therapy for treating infantile‐onset Pompe disease.
Methods
Criteria for considering studies for this review
Types of studies
Randomized and quasi‐randomized controlled trials.
Types of participants
Inclusion criteria include children with: a confirmed diagnosis of classical or non‐classical Pompe disease.
Exclusion criteria include those with:
congenital diseases that are unrelated to Pompe disease and likely to decrease survival rate;
any prior alglucosidase alfa treatment from any source.
Types of interventions
Intervention: ERT alone or with another medical care (e.g. immune tolerance induction, symptomatic medications).
Trials of ERT alone were analyzed separately from those where the participants also received a concomitant medication.
Control: different doses of ERT, another intervention, no intervention or placebo.
Types of outcome measures
Primary outcomes
Cardiac function (as measured by the left ventricular mass z score (LVM‐Z), fractional shortening and ejection fraction).
Time to ventilation.
Survival.
Secondary outcomes
Motor development (as measured by recognized scoring tools such as Alberta Infant Motor Scale, Gross Motor Function Measure 66, etc.).
Quality of life of the parents (as measured by the individual trials).
Infusion‐related events (IREs) (defined as adverse events occurring during or immediately after the alglucosidase alfa infusion, e.g. flushing, nausea, vomiting).
Other adverse events (defined as clinical events occurring after initiation of alglucosidase alfa treatment but excluding IREs, including the development of anti‐alglucosidase alfa antibodies).
Search methods for identification of studies
We attempted to identify all relevant studies regardless of language or publication status (published, unpublished, in press, ongoing).
Electronic searches
The Cochrane Cystic Fibrosis and Genetic Disorders Group's Information Specialist conducted a systematic search of the Group's Inborn Errors of Metabolism Trials Register for relevant trials using the following term: Pompe.
The Inborn Errors of Metabolism Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated with each new issue of the Cochrane Library), weekly searches of MEDLINE and the prospective handsearching of one journal ‐ Journal of Inherited Metabolic Disease. Unpublished work is identified by searching through the abstract books of the Society for the Study of Inborn Errors of Metabolism conference and the SHS Inborn Error Review Series. For full details of all searching activities for the register, see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of last search of the Cystic Fibrosis and Genetic Disorders Group's Inborn Errors of Metabolism Trials Register: 24 November 2016.
The authors searched the following databases and trial registers (with no language or date restrictions) for relevant trials:
CENTRAL (updated with each new issue) in the Cochrane Library (November 2016, Issue 10);
Embase Ovid (1974 to 24 November 2016);
PubMed (www.ncbi.nlm.nih.gov/pubmed; 1946 to 24 November 2016);
LILACS (Latin American and Caribbean Health Science Information database; from 1982 to 24 November 2016);
US National Institutes of Health Ongoing Trials Register CllinicalTrials.gov (www.clinicaltrials.gov; searched 24 November 2016);
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch; searched 24 November 2016);
Genzyme Clinical Research Database (www.genzymeclinicalresearch.com; searched 24 November 2016).
Full search strategies are available in Appendix 1.
The authors also searched the following databases (with no restrictions) for literature published in Chinese:
CBM (Chinese Biomedical Literature Database; 1978 to 24 November 2016);
CNKI (Chinese National Knowledge Infrastructure; 1994 to 24 November 2016);
VIP (VIP Chinese Science & Technology Periodicals; 1989 to 24 November 2016);
WANFANG Digital Periodicals (c.g.wanfangdata.com.cn/periodical.aspx; 1998 to 24 November 2016).
Searching other resources
Grey literature
We contacted specialists in the field and authors of the included trials for any possible unpublished data. We contacted the pharmaceutical company, Genzyme, for information on unpublished or ongoing trials.
Reference lists
We checked the reference lists of related articles and reviews by the above methods.
Data collection and analysis
Selection of studies
Two review authors independently screened the title, abstract and keywords of all trials identified by the search strategy and obtained the full articles for all potentially relevant trials. Two review authors independently assessed the full text and extracted data using a data extraction form. We resolved any disagreements by consensus, consulting with the corresponding author, or asking for advice from the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Data extraction and management
Two review authors independently used a pre‐designed data collection form to extract data from each trial. These extracted data were: characteristics of the trial (author, country, publication year, design, methods of randomization, funding sources, conflicts of interest); participants; interventions and outcomes (types of outcome measures, follow‐up, adverse events). We resolved any discrepancies by discussion.
We planned to measure outcomes at the following time points: one month to up to six months; six months to up to one year; one year to 18 months and at six‐monthly intervals thereafter.
Assessment of risk of bias in included studies
Two review authors independently assessed the risk of bias using a standard form. We contacted the trial authors by telephone or email if important information was unclear. We used the domain‐based evaluation recommended by the Cochrane Handbook for Systematic Reviews of Interventions to address six specific domains: sequence generation, allocation sequence concealment, blinding, incomplete outcome data, selective outcome reporting and other potential sources of bias (Higgins 2011). We created plots of 'Risk of bias' assessment using Review Manager 5 software (RevMan 2014). We resolved any discrepancies by discussion.
Measures of treatment effect
For dichotomous data, we extracted the number of participants experiencing the event and the total number of participants evaluated for that outcome. We used the risk ratio (RR) with 95% confidence intervals (95% CIs) as the effect measure.
For future updates, if we include continuous data, we will calculate the mean difference (MD) or standardized mean difference (SMD) or use any data that could be used to derive the SMD. We will use the MD when outcome measurements in all trials were made on the same scale. We will use the SMD when the same outcome was measured using different instruments. We will calculate the corresponding 95% CI.
For future updates, for time‐to‐event data, which take into account of the number and timing of events, we plan to summarize and analyze data using hazard ratios (HR) with their corresponding 95% CIs (Higgins 2011).
For other outcomes where there were insufficient data to be quantitatively analyzed, we presented the information narratively.
Unit of analysis issues
Given the nature of the intervention, cross‐over trials are not an appropriate trial design. Given the rarity of the disease and that any eligible trials will have a limited sample size, we also do not anticipate including cluster randomized trials or trials with multiple treatment arms.
Dealing with missing data
For future updates of this review, if information and published data of included trials presented in the papers or abstracts are not sufficient to be entered into the review, we will contact authors for further details (e.g. the method of sequence generation, allocation concealment, blinding and withdrawals or missing outcome data).
For the current version of the review, we contacted the trial authors for missing information, but have yet to receive a reply. We carried out an analysis according to the intention‐to‐treat principle.
Assessment of heterogeneity
We planned to use the Chi² test and visual assessment of the forest plots to observe whether statistical heterogeneity existed between the identified trials. A large Chi² statistic (or a low P value) provides evidence of heterogeneity of intervention effects. Otherwise, if CIs for the results of individual studies (generally depicted graphically using horizontal lines) have poor overlap, this generally indicates the presence of statistical heterogeneity. We plan to quantify heterogeneity using the I² statistic, which illustrates the percentage of the variability in effect estimates resulting from heterogeneity rather than sampling error. We considered there to be no important heterogeneity if the I² statistic was below 40%, a moderate degree if the I² statistic was between 30% and 60%, a substantial degree if the I² statistic was between 50% and 90% and a considerable degree if the I² statistic was between 75% and 100% (Higgins 2011).
Assessment of reporting biases
For future updates, if we include 10 or more trials, we will use funnel plots to assess biases including publication bias and other reporting biases. If there are biases, the funnel plots may be asymmetrical. However, it is notable that there are a number of explanations for the asymmetry of a funnel plot, including true heterogeneity, poor methodological quality (high risk of bias) of smaller studies, publication bias and language bias (Sterne 2011).
Data synthesis
We conducted data analyses using Review Manager 5 software (RevMan 2014). We used a fixed‐effect model to present the data from the included trial. For future updates, if ore trials are included and in the absence of substantial statistical heterogeneity (I² statistic of 50% or less), we plan to use a fixed‐effect model to combine trials. However, if we identify substantial heterogeneity (I² statistic greater than 50%), we will use a random‐effects model. If we are unable to explain this heterogeneity by use of subgroup analyses, or by clinical or methodological features of the trials, we will present a narrative summary.
Subgroup analysis and investigation of heterogeneity
For future updates of this review, if more trials are included considering that the following factors may affect clinical outcomes, we plan to undertake the following subgroup analyses.
Children with CRIM‐negative versus children with CRIM‐positive status.
Children given immunomodulation versus children not given immunomodulation.
Dosage and frequency of dosing (e.g. 20 mg/kg every other week versus 40 mg/kg every other week).
Age at commencement of treatment.
Sensitivity analysis
For future updates, if more trials are included, we will perform sensitivity analyses to assess the robustness of the meta‐analysis. We will implement sensitivity analysis by excluding trials with a high risk of bias in relation to the method of randomization, allocation concealment and the rates of withdrawal for each outcome.
'Summary of findings' table
We created a 'Summary of findings' table for all seven outcomes listed above (Types of outcome measures). We used the GRADE approach to rate the quality of evidence and grade strength of recommendations (Guyatt 2011). In the GRADE approach, randomized controlled trials start as high‐quality evidence. Five factors may lead to downgrading the quality of evidence, which are risk of bias, inconsistency, indirectness, imprecision and publication bias. Recommendations are characterized as strong or weak according to the quality of the supporting evidence. Methods and recommendations are described in Chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We used GRADEpro software (GRADEpro 2011). We justified all decisions to downgrade or upgrade the quality of evidence using footnotes and we made comments to aid the reader's understanding of the review where necessary.
Results
Description of studies
Results of the search
We identified 1589 references from the electronic searches and 50 from other sources. After initial assessment, 15 references were considered as potentially eligible after screening. Following the full‐text review of the 15 articles, we excluded nine studies (10 references) and one is awaiting assessment. One trial (four references) is included in this review (Figure 1).
1.
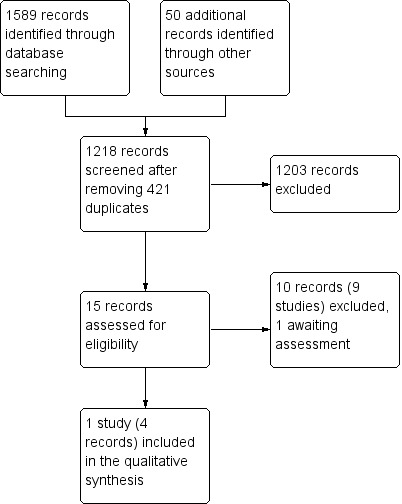
Study flow diagram.
Included studies
The included trial randomized 18 participants with infantile‐onset Pompe disease to different doses of alglucosidase alfa (see below). The trial also compared a historical control group which was not relevant to this review (Kishnani 2007).
Recombinant human acid alpha‐glucosidase versus placebo or no intervention
We found no randomized controlled trials comparing alglucosidase alfa versus placebo or no intervention.
Different doses of recombinant human acid alpha‐glucosidase
The Kishnani study was a multicentre, multinational (six centres in the USA, five in Europe, one in Taiwan and one in Israel), open‐label, dose‐ranging study (Kishnani 2007). Participants were diagnosed at six months of age and younger and exhibited severe GAA deficiency and cardiomyopathy. The study involved 18 participants in the initial phase of the study and 16 surviving participants in the extension phase. Participants recruited to the prospective arm were randomised into two groups of nine to receive different dosing schedules of alglucosidase alfa (Myozyme®); intravenous infusions of 20 mg/kg every other week or 40 mg/kg every other week for a minimum of 52 weeks. Analyses were from 52 weeks after the last participants were randomized to treatment (randomized phase) to three years (extension phase of study), with a median duration of treatment of 2.3 years. We only extracted prospective data (and not data from the historical control group) with respect to changes from baseline after 52 weeks of treatment. Given the participants were enrolled over a period of one year, safety data ranged from 52 weeks for the last baby randomized to treatment to 106 weeks for the first baby treated with alglucosidase alfa. The trial was supported by the Genzyme Corporation.
Excluded studies
We excluded nine studies from the review (Case 2015; Chakrapani 2010; Chen 2011; Cook 2006; Kishnani 2006b; Laforêt 2008; Nicolino 2009; Levine 2008; Prater 2013). The reasons for exclusion were that the studies were not randomized or were retrospective.
Studies awaiting assessment
A further trial, published as a conference abstract only, is awaiting assessment; we tried to contact the authors, but to date have not received a response. If there are further publications relating to this trial, we will assess it for eligibility (Van Gelder 2013).
Risk of bias in included studies
See the 'Risk of bias' figure (Figure 2).
2.
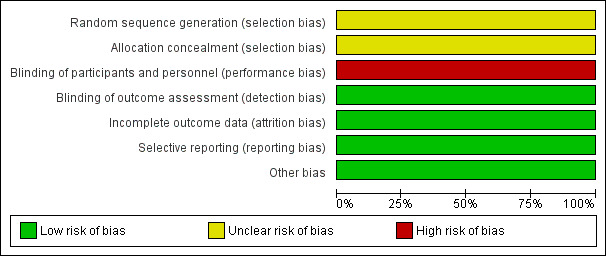
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Sequence generation
The trial did not describe generation of the random sequence. Therefore, we classified it at unclear risk of bias (Kishnani 2007).
Allocation concealment
The report did not describe concealment of allocation and we assessed the trial as at an unclear risk of bias (Kishnani 2007).
Blinding
Clinicians and participants were not blinded. Therefore, we judged this trial to be at a high risk of performance bias (Kishnani 2007). However, for blinding of outcome assessors, the paediatric cardiologist and pathologist were blinded to dose and participant, and therefore, the risk of detection bias was low.
Incomplete outcome data
Withdrawals were less than 20%. We assessed the trial as at a low risk of attrition bias (Kishnani 2007).
Selective reporting
The trial reported all the significant and non‐significant results, we therefore judged it at low risk of reporting bias (Kishnani 2007).
Other potential sources of bias
We identified no other potential sources of bias.
Effects of interventions
See: Table 1
Recombinant human alglucosidase alfa ‐ 20 mg/kg every two weeks versus 40 mg/kg every two weeks
The included study compared alglucosidase alfa at 20 mg/kg every two weeks to alglucosidase alfa at 40 mg/kg every two weeks (Kishnani 2007). The study described results between the alglucosidase alfa treatment group and comparative information with the historical control group. Only the IREs could be quantitatively analyzed.
Primary outcomes
1. Cardiac function
The study reported no detailed data comparing the two dose groups. Results from the initial trial described that cardiac function was similar in the two dose groups and long‐term alglucosidase alfa treatment (both dosing groups) improved cardiomyopathy (low‐quality evidence).
2. Time to ventilation
The study reported no data about time to ventilation comparing the two dose groups. Results from the initial phase of the trial described that the proportion of children that were free of invasive ventilation was similar in the two dose groups (low‐quality evidence).
3. Survival
The study reported no data on survival comparing the two dose groups. The original paper stated that "Fifteen of the 18 treated patients reached the age of 18 months by the end of the study; three patients were right‐censored from this analysis because they had not reached the age of 18 months by the end of the study, although they were alive at that time (at ages 15.9 months, 17.9 months, and 14.4 months)" (Kishnani 2007). One participant died after receiving alglucosidase alfa for 61 weeks, but before the extension study began. In the extension study, four participants died before the end of the study. Results revealed that long‐term alglucosidase alfa treatment (both dosing groups) markedly extended overall survival and ventilation‐free survival (low‐quality evidence). It is important to note, as the trial authors stated "This is in striking contrast to what was observed in the untreated historical control group, in which only one of 61 patients survived to the age of 18 months (1.9%; 95% CI: 0% to 5.5%)".
Secondary outcomes
1. Motor development
The study reported no data about motor development between the two dose groups. Results from the initial trial described that motor development was similar in the two dose groups. It reported that "13 of the 18 treated patients acquired substantial motor and functional skills as assessed by the AIMS [Alberta Infant Motor Scale] and Pompe PEDI [Pompe Pediatric Evaluation of Disability Inventory] tests" (low‐quality evidence).
2. Quality of life of the parents
The study did not report data for this outcome.
3. Infusion‐related events
In the initial phase of the study, five participants in the 20 mg/kg every two weeks dose group experienced a total of 41 IREs and the six participants in the 40 mg/kg every two weeks dose group experienced a total of 123 IREs. All the IREs were mild or moderate in intensity; none was severe. The IREs could be controlled by slowing or interrupting infusions. In the extension study, five participants in the 20 mg/kg every two weeks dose group experienced a total of 47 IREs and the six participants in the 40 mg/kg every two weeks dose group experienced a total of 177 IREs. In relation to the number of participants experiencing one or more event. the meta‐analysis showed no significant difference between groups, RR 0.83 (95% CI 0.40 to 1.76) (low‐quality evidence) (Analysis 1.1).
1.1. Analysis.
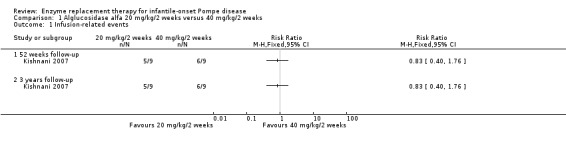
Comparison 1 Alglucosidase alfa 20 mg/kg/2 weeks versus 40 mg/kg/2 weeks, Outcome 1 Infusion‐related events.
4. Other adverse events
There was no information or data for the different dose groups for other adverse events reported; however, the trial report stated that at 52 weeks, 16/18 participants (from both dose groups) developed IgG antibodies to alglucosidase alfa, one of whom developed inhibitory antibodies (low‐quality evidence).
Discussion
Summary of main results
We did not find any randomised controlled trials comparing enzyme replacement therapy to another intervention, no intervention or placebo . We included one trial comparing different doses of alglucosidase alfa. Treatment doses were 20 mg/kg every two weeks and 40 mg/kg every two weeks. The duration of alglucosidase alfa treatment ranged from 52 weeks (randomized phase) to three years (extension study), with a median duration of treatment of 2.3 years. Results suggested the higher alglucosidase alfa dose may be beneficial but this was not demonstrated statistically. The trial reported that clinical responses including cardiac function and motor development, as well as the proportion of children that were free of invasive ventilation, were similar in the 20 mg/kg every two weeks and the 40 mg/kg every two weeks groups (low‐quality evidence). Results revealed that long‐term alglucosidase alfa treatment (both dosing groups) markedly improved cardiomyopathy and extended overall survival and ventilation‐free survival (low‐quality evidence). In relation to survival, it is important to note, as the trial authors stated "This is in striking contrast to what was observed in the untreated historical control group, in which only one of 61 patients survived to the age of 18 months (1.9%; 95% CI: 0% to 5.5%)". While there was no significant difference between dose groups in the number of children experiencing IREs, the number of individual IREs experienced in the higher dose group was greater than experienced in the lower dose group at both 52 weeks and at the end of the extension study. ERT appeared to be relatively well‐tolerated; IREs were mild or moderate and could be controlled by slowing or interrupting infusions.
Due to the low numbers of participants, it is impossible to draw conclusions in regard to the effectiveness of treatment between CRIM‐positive and CRIM‐negative children (only 3 out of 18 children in the included study were CRIM negative).
Overall completeness and applicability of evidence
Alglucosidase alfa was approved in 2006 with the recommended dose of 20 mg/kg every two weeks. Since then, studies of the effectiveness and safety of alglucosidase alfa have been undertaken. Some studies showed the prognosis of infantile‐onset Pompe disease has changed and life expectancy has increased. Other studies reported progression of weakness and decline in function after ERT, prompting controversy about the long‐term effect of alglucosidase alfa and the optimum dose.
The objective of this review was to evaluate the effectiveness and safety of ERT for treating infantile‐onset Pompe disease. We found no randomized controlled trials comparing ERT with placebo, which is common for clinical studies of rare diseases. The ERT doses varied and many important outcomes were presented only narratively by the trial authors.
Quality of the evidence
The evidence that this trial provided to the review was of low quality (no numerical results were available by dose group, random sequence generation and allocation concealment were unclear, and there was a lack of blinding). In addition, the sample size was small, making the 95% CIs wide. See Table 1.
Potential biases in the review process
The review was limited by the small sample size and the inclusion of narrative outcome reporting, but this is common in the investigation of rare diseases.
Agreements and disagreements with other studies or reviews
This was the first systematic review of ERT for infantile‐onset Pompe disease. Previous studies have simply summarized evidence based on sporadic studies or case reports. In this review, there were no detailed data available between the two dose groups in relation to cardiac function, time to ventilation, survival and motor development. However, when compared to a historical control group of 61 children, results from the initial study demonstrated that ERT reduced the risk of death by 99%, reduced the risk of death or invasive ventilation by 92% and reduced the risk of death or any type of ventilation by 88% in 52 weeks. Alglucosidase alfa treatment reduced the risk of death by 95%, reduced the risk of death or invasive ventilation by 91% and reduced the risk of death or any type of ventilation by 87% in three years (results from this comparison with a historical control group were not included in the results section of this review).
In a further open‐label, multicenter study, bi‐weekly infusions with alglucosidase alfa from 20 mg/kg to 40 mg/kg for up to 168 weeks also prolonged survival and invasive ventilation‐free survival (reduced the risk of death by 79% and the risk of invasive ventilation by 58%, P < 0.05, respectively), as compared to the untreated reference cohort (Nicolino 2009). With the promising results in survival from non‐randomised trials, the accumulation of glycogen in the central nervous system becomes a concern. In one study including five children with infantile‐onset Pompe disease (the largest sample size of emerging case reports), results revealed that improvement in brain myelination could be seen in the five cases who survived by effective ERT treatment. However, whether ERT has a direct therapeutic effect on the brain is still unclear(Chien 2006).
Some historical studies have not found an association between CRIM status and motor development (Amalfitano 2001; Van den Hout 2000). In more recent years, it is has become recognized that CRIM status does influence the effectiveness of treatment because of its relationship to the development of anti‐alglucosidase alfa IgG antibodies. This recognition has led to the use of immune modulation therapy prior to ERT in CRIM‐negative children (Berrier 2015; Kazi 2016). This review only included one randomized controlled trial, with too few participants to assess effects of CRIM status. Future studies should focus on developing a better understanding of CRIM status and antibody formation in response to ERT.
Authors' conclusions
Implications for practice.
The search found no trials comparing the effectiveness and safety of enzyme replacement therapy (ERT) to another intervention, no intervention or placebo. One small randomized trial provided no robust evidence for which dosing schedule (20 mg/kg every two weeks or 40 mg/kg every two weeks) of ERT was more effective for infantile‐onset Pompe disease. Results revealed that long‐term alglucosidase alfa treatment (both dosing groups) markedly improved cardiomyopathy and extended overall survival and ventilation‐free survival (low‐quality evidence).
Implications for research.
This review highlights the need for continued research into the use of ERT for infantile‐onset Pompe disease. It is not deemed ethical to proceed with new trials including a placebo group and we therefore suggest a randomized controlled trial comparing different dosing schedules. Main clinical outcomes (i.e. cardiac function, time to ventilation, survival, motor development, adverse events (e.g. the development of antibodies)) should be standardized, measured and fully reported.
What's new
| Date | Event | Description |
|---|---|---|
| 14 December 2017 | Amended | A duplicated sentence has been removed from the 'Plain language summary'. |
Notes
This review was managed by the Cochrane Cystic Fibrosis and Genetic Disorders Group in collaboration with Cochrane Neuromuscular.
Acknowledgements
This review was developed in collaboration with Cochrane Neuromuscular.
The editorial process was primarily managed by the Cochrane Cystic Fibrosis and Genetic Disorders Group and the editorial team from Cochrane Neuromuscular provided comments and peer review at title proposal, protocol and review stages.
The authors are grateful to their affiliated institutions and organizations, and thank the referees and editors for their comments and encouragement. We would like to thank Tracey Remmington and Nikki Jahnke from the Cystic Fibrosis & Genetics Disorders Group for their outstanding help.
The authors are grateful for the contribution to the protocol from Yi Liang (previous co‐author).
This project was supported by the National Institute for Health Research (NIHR) via Cochrane Infrastructure funding to Cochrane Neuromuscular. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service, or the Department of Health. Cochrane Neuromuscular is also supported by the MRC Centre for Neuromuscular Diseases.
Appendices
Appendix 1. Search strategies of English databases
| Databases | Search strategies |
| CENTRAL | #1 glycogen storage disease type II [MeSH descriptor] OR Pompe* disease [All fields] OR acid maltase deficienc* [All fields] OR Generalized Glycogenosis [All fields] OR GAA Deficienc* [All fields] OR glycogen storage disease type II [All fields] OR Alpha‐1,4‐Glucosidase Deficienc* [All fields] #2 Enzyme Replacement Therapy [MeSH descriptor] OR Enzyme Replacement Therap*[ All fields] OR alglucosidase alfa[All fields] OR Myozyme[All fields] OR ERT[All fields] OR GAA[All fields] #3 #1 AND #2 |
| PubMed | #1 glycogen storage disease type II [mesh] OR Pompe* disease [All fields] OR acid maltase deficienc* [All fields] OR Generalized Glycogenosis [All fields] OR GAA Deficienc* [All fields] OR glycogen storage disease type II [All fields] OR Alpha‐1,4‐Glucosidase Deficienc* [All fields] #2 Enzyme Replacement Therapy [mesh] OR Enzyme Replacement Therap*[All fields] OR alglucosidase alfa[All fields] OR Myozyme[All fields] OR ERT[All fields] OR GAA[All fields] #3 #1 AND #2 |
| Embase (Ovid) | #1 glycogen storage disease type 2 [Map Term] OR Pompe* disease [All fields] OR acid maltase deficienc* [All fields] OR Generalized Glycogenosis [All fields] OR GAA Deficienc* [All fields] OR glycogen storage disease type II [All fields] OR Alpha‐1,4‐Glucosidase Deficienc* [All fields] #2 Enzyme Replacement Therapy [Map Term] OR Enzyme Replacement Therap*[ All fields] OR alglucosidase alfa[All fields] OR Myozyme[All fields] OR ERT[All fields] OR GAA[All fields] #3 #1 AND #2 |
| LILACS | #1 Pompe$ disease [All fields] OR acid maltase deficienc$ [All fields] OR Generalized Glycogenosis [All fields] OR GAA Deficienc$ [All fields] OR glycogen storage disease type II [All fields] OR Alpha‐1,4‐Glucosidase Deficienc$ [All fields] #2 Enzyme Replacement Therap$ [ All fields] OR alglucosidase alfa[All fields] OR Myozyme[All fields] OR ERT[All fields] OR GAA[All fields] #3 #1 AND #2 |
| WHO ICTRP | Pompe |
| ClinicalTrials.gov | Pompe |
| Genzyme | Pompe |
Appendix 2. Search strategies of Chinese databases
| Databases | Search strategies |
| CBM | 全部字段:"庞贝病" OR "糖原贮积病II型" OR "α‐葡糖苷酶缺乏症" |
| CNKI | FT="庞贝病"+"糖原贮积病II型"+"α‐葡糖苷酶缺乏症" |
| VIP | U="庞贝病" OR "糖原贮积病II型" OR "α‐葡糖苷酶缺乏症" |
| WANFANG | 全部字段:"庞贝病" OR "糖原贮积病II型" OR "α‐葡糖苷酶缺乏症" |
Data and analyses
Comparison 1. Alglucosidase alfa 20 mg/kg/2 weeks versus 40 mg/kg/2 weeks.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Infusion‐related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 52 weeks follow‐up | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 3 years follow‐up | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Kishnani 2007.
| Methods | Multicentre, multinational, open‐label randomized trial. It also compared a historical control group which was not relevant to this review. Initial phase (52 weeks of treatment) involved 18 participants, while the extension phase involved 16 surviving participants (up to a total of 3 years of treatment). |
|
| Participants | Initial phase: 18 participants (11 boys, 7 girls) with IPD. Mean (SD) age at first infusion: 4.6 (1.67) months. Eligible participants had documented symptoms, skin fibroblast GAA activity 1% of normal mean and hypertrophic cardiomyopathy (left ventricular mass index 65 g/m² by echocardiogram); aged ≤ 26 weeks at enrolment. Exclusion criteria: respiratory insufficiency (O₂ saturation ≤ 90% or CO₂ partial pressure 55 mmHg (venous) or 40 mmHg (arterial) in room air or any ventilator use), major congenital anomaly or clinically significant intercurrent illness unrelated to Pompe disease, or any prior alglucosidase alfa treatment. Extension phase: same randomized group, 16 surviving participants. |
|
| Interventions | Participants randomized in 1:1 ratio.
In the extension phase there were 8 surviving participants in each dose group. |
|
| Outcomes | Efficacy outcomes included survival, ventilator use, cardiac function, motor development and cognitive development. Safety outcomes included adverse events and development of anti‐alglucosidase alfa IgG antibodies. In the initial phase of the study, survival and ventilation data were analyzed up to 18 months of age, as compared to survival of the historical control group; all other efficacy data were analyzed with respect to changes from baseline after 52 weeks of treatment. Safety data were analyzed for the duration of treatment. Because participants were enrolled over a period of 1 year, safety data ranged from 52 weeks for the last patient randomized to treatment to 106 weeks for the first participant treated with alglucosidase alfa. In the extension phase, outcomes were reported up to a period of 3 years. |
|
| Notes | Genzyme Corporation supplied alglucosidase alfa, which contains alglucosidase alfa produced in Chinese hamster ovary cells. The majority of the investigators from the trial could not avoid a conflict of interest as they had received research or grant support from the Genzyme Corporation. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Simple randomization scheme used to assign participants to the 2 dose groups. |
| Allocation concealment (selection bias) | Unclear risk | No detail provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Clinicians and participants were not blinded to dose of alglucosidase alfa. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Echocardiograms and glycogen content in muscle biopsy samples were centrally read by a paediatric cardiologist and a pathologist who were blinded to dose, participant and time point. Motor and cognitive evaluations were centrally scored by a non‐blinded central clinician. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant died before end of study in the 20 mg/kg/every other week group. |
| Selective reporting (reporting bias) | Low risk | All outcomes reported. |
| Other bias | Low risk | We identified no other potential sources of bias. |
GAA: acid alpha‐glucosidase; IgG: immunoglobulin G; IPD: infantile‐onset Pompe disease; IV: intravenous; SD: standard deviation.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Case 2015 | Participants received prior alglucosidase alfa treatment for > 6 months. |
| Chakrapani 2010 | Not prospective. Described 20 people with IPD who treated from 2000 to 2009 in the UK. |
| Chen 2011 | Not randomized. Explored left ventricular reverse remodelling after ERT in 9 infants and children with Pompe cardiomyopathy, and 36 healthy control participants. |
| Cook 2006 | Not randomized. Evaluated 24‐hour ambulatory electrocardiograms at baseline and during ERT in 12 participants with IPD from 2003 to 2005. |
| Kishnani 2006b | Not randomized. Open‐label, multinational, multicentre study examining safety and efficacy of alglucosidase alfa in treating 8 participants with IPD. |
| Laforêt 2008 | Participants aged > 8 years (juveniles and adults), not infantile onset. |
| Levine 2008 | Not randomized. Described cardiac response of infants with Pompe disease treated with ERT. |
| Nicolino 2009 | Not randomized. Open‐label, multicentre study examining safety and efficacy of alglucosidase alfa in treating 21 participants with IPD. Survival results were compared with an untreated historic reference cohort. |
| Prater 2013 | Not prospective. Characterized the long‐term impact of ERT with alglucosidase alfa on lysosomal glycogen accumulation and autophagy in some of the oldest survivors (8 people with classic IPD). |
ERT: enzyme replacement therapy IPD: infantile‐onset Pompe disease
Characteristics of studies awaiting assessment [ordered by study ID]
Van Gelder 2013.
| Methods | No information provided. |
| Participants | 10 children with infantile‐onset Pompe disease. |
| Interventions | Myozyme® treatment at 40 mg/kg every week compared to 20 mg/kg every other week for ≥ 1 year. |
| Outcomes | Survival, ventilator‐free survival, left ventricular mass index, motor outcome and infusion‐associated reactions. |
| Notes |
Differences between protocol and review
Objectives
We included a post hoc change to the objectives of the review. It was not clearly outlined in the protocol that as well as effectiveness and safety, we also planned to assess the appropriate dose regimen; this has now been clarified.
Types of outcomes
Amendments made to 'Respiratory function' (changed to 'Time to ventilation') and 'Quality of Life' ('Quality of life of parents') outcomes as more appropriate given very young children are the eligible participants.
Contributions of authors
| Roles and responsibilities | |
| Task | Who undertook the task? |
| Protocol stage: draft the protocol | Min Chen |
| Review stage: select which trials to include (2 people + 1 arbiter) | Min Chen, Shuyan Quan + Ling‐li Zhang |
| Review stage: extract data from trials (2 people) | Min Chen, Shuyan Quan |
| Review stage: enter data into Review Manager 5 | Min Chen |
| Review stage: carry out the analysis | Min Chen, Ling‐li Zhang |
| Review stage: interpret the analysis | Min Chen, Ling‐li Zhang |
| Review stage: draft the final review | Min Chen, Ling‐li Zhang |
| Update stage: update the review | Ling‐li Zhang |
Sources of support
Internal sources
-
Evidence based establishment of evaluation index system for pediatric rational drug use in China (No. 81373381); New fund of West China Second University Hospital(No.kx034), China.
The funder has no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group and Cochrane Neuromuscular.
Declarations of interest
All authors: none known.
Edited (no change to conclusions)
References
References to studies included in this review
Kishnani 2007 {published data only}
- Kishnani P, Byrne B, Nicolino M, Mandel H, Leslie N, Hwu LH, et al. Enzyme replacement therapy (ERT) with recombinant human acid alpha glucosidase (rhGAA) in infantile onset Pompe disease (IOPD). Journal of Inherited Metabolic Disease 2005;28(Suppl 1):195. [Abstract no: 387‐O; CENTRAL: 1000729; CRS: 5500131000000071] [Google Scholar]
- Kishnani PS, Byrne B, Nicolino M, Mandel H, Leslie N, Hwu WL, et al. Enzyme replacement therapy with recombinant human acid alpha glucosidase (rhGAA) in infantile onset Pompe disease (IOPD). Neuromuscular Disorders : NMD 2005;15(9‐10):712. [Abstract no: G.P.10.09; CENTRAL: 636009; CRS: 5500050000000494] [Google Scholar]
- Kishnani PS, Corzo D, Leslie ND, Gruskin D, Ploeg A, Clancy JP, et al. Early treatment with alglucosidase alpha prolongs long‐term survival of infants with Pompe disease. Pediatric Research 2009; Vol. 66, issue 3:329‐35. [DOI] [PMC free article] [PubMed]
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, et al. Recombinant human acid [alpha]‐glucosidase: major clinical benefits in infantile‐onset Pompe disease. Neurology 2007;68(2):99‐109. [CENTRAL: 574244; CRS: 5500131000000335; JID:: 0401060; PUBMED: 17151339] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Case 2015 {published data only}
- Case LE, Bjartmar C, Morgan C, Casey R, Charrow J, Clancy JP, et al. Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease. Neuromuscular Disorders 2015;25(4):321‐32. [DOI] [PubMed] [Google Scholar]
Chakrapani 2010 {published data only}
- Chakrapani A, Vellodi A, Robinson P, Jones S, Wraith JE. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. Journal of Inherited Metabolic Disease 2010;33(6):747‐50. [DOI] [PubMed] [Google Scholar]
Chen 2011 {published data only}
- Chen CA, Chien YH, Hwu WL, Lee NC, Wang JK, Chen LR, et al. Left ventricular geometry, global function, and dyssynchrony in infants and children with Pompe cardiomyopathy undergoing enzyme replacement therapy. Journal of Cardiac Failure 2011;17(11):930‐6. [DOI] [PubMed] [Google Scholar]
Cook 2006 {published data only}
- Cook AL, Kishnani PS, Carboni MP, Kanter RJ, Chen YT, Ansong AK, et al. Ambulatory electrocardiogram analysis in infants treated with recombinant human acid α‐glucosidase enzyme replacement therapy for Pompe disease. Genetics in Medicine 2006;8(5):313‐7. [DOI] [PubMed] [Google Scholar]
Kishnani 2006b {published data only}
- Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, et al. Chinese hamster ovary cell‐derived recombinant human acid alpha‐glucosidase in infantile‐onset Pompe disease. Journal of Pediatrics 2006;149(1):89‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Laforêt 2008 {published data only}
- Laforêt P, Clemens PR, Corzo D, Escolar D, Florence J, Ploeg A, et al. Safety and efficacy results from a randomized, double‐blind, placebo‐controlled study of alglucosidase alfa for the treatment of Pompe disease in juveniles and adults. Neuromuscular Disorders : NMD 2008;18(9‐10):832‐3. [CENTRAL: 1215751; CRS: 5500135000001638] [Google Scholar]
- Ploeg A, Clemens PR, Corzo D, Escolar D, Florence J, Laforet P, et al. Safety and efficacy results from a randomized, double‐blind, placebo‐controlled study of alglucosidase alpha for the treatment of Pompe's disease in juveniles and adults. European Journal of Neurology 2008;15(Suppl 3):412‐3, Abstract no: LBN103. [CENTRAL: 691433; CRS: 5500050000000495] [Google Scholar]
Levine 2008 {published data only}
- Levine JC, Kishnani PS, Chen YT, Herlong JR, Li JS. Cardiac remodeling after enzyme replacement therapy with acid α‐glucosidase for infants with Pompe disease. Pediatric Cardiology 2008;29(6):1033‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nicolino 2009 {published data only}
- Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, et al. Clinical outcomes after long‐term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genetics in Medicine 2009;11(3):210‐9. [DOI] [PubMed] [Google Scholar]
Prater 2013 {published data only}
- Prater SN, Patel TT, Buckley AF, Mandel H, Vlodavski E, Banugaria SG, et al. Skeletal muscle pathology of infantile Pompe disease during long‐term enzyme replacement therapy. Orphanet Journal of Rare Diseases 2013;8(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Van Gelder 2013 {published data only}
- Gelder C, Plug I, Kroos M, Reuser A, Ploeg A. A higher dose of enzyme therapy in patients with classic infantile Pompe disease seems to improve ventilator‐free survival and motor function. BMC Musculoskeletal Disorders 2013;14(Suppl 2):19. [Google Scholar]
Additional references
Al Khallaf 2013
- Al Khallaf HH, Propst J, Geffrard S, Botha E, Ali Pervaiz M. CRIM‐negative Pompe disease patients with satisfactory clinical outcomes on enzyme replacement therapy. JIMD Reports 2013;9:133‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Amalfitano 2001
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, et al. Recombinant human acid alpha‐glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genetics in Medicine 2001;3(2):132‐8. [PubMed] [Google Scholar]
Banugaria 2011
- Banugaria SG, Prater SN, Ng YK, Kobori JA, Finkel RS, Ladda RL, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genetics in Medicine 2011;13(8):729‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Baruteau 2014
- Baruteau J, Broomfield A, Crook V, Finnegan N, Harvey K, Burke D, et al. Successful desensitisation in a patient with CRIM‐positive infantile‐onset Pompe disease. JIMD Reports 2014;12:99‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Berrier 2015
- Berrier KL, Kazi ZB, Prater SN, Bali DS, Goldstein J, Stefanescu MC, et al. CRIM‐negative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genetics in Medicine 2015;17(11):912‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chien 2006
- Chien YH, Lee NC, Peng SF, HWU WL. Brain development in infantile‐onset Pompe disease treated by enzyme replacement therapy. Pediatric Research 2006;60(3):349‐52. [DOI] [PubMed] [Google Scholar]
Chien 2013
- Chien YH, Hwu WL, Lee NC. Pompe disease: early diagnosis and early treatment make a difference. Pediatrics and Neonatology 2013;54(4):219‐27. [DOI] [PubMed] [Google Scholar]
Eto 2010
- Eto Y. The Japanese guideline for diagnosis and management of glycogen storage disease type II (Pompe disease). 3rd European Symposium on Steps Forward in Pompe Disease; 2009 Nov 20‐21; Munich (Germany). 2010:S73‐4.
Fukuda 2006
- Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Plotz PH, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Annals of Neurology 2006;59(4):700‐8. [DOI] [PubMed] [Google Scholar]
GRADEpro 2011 [Computer program]
- GRADE Working Group, McMaster University. GRADEpro. Version 3.6 for Windows. Hamilton (ON): GRADE Working Group, McMaster University, 2011.
Guyatt 2011
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction ‐ GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hirschhorn 2001
- Hirschhorn R, Reuser AJJ. The metabolic and molecular bases of inherited disease. Glycogen storage disease type II: acid alpha‐glucosidase (acid maltase) deficiency. hommbid.mhmedical.com/content.aspx?bookid=474§ionid=45374135&Resultclick=2 (accessed 7 November 2014).
Kazi 2016
- Kazi ZB, Prater SN, Kobori JA, Viskochil D, Bailey C, Gera R, et al. Durable and sustained immune tolerance to ERT in Pompe disease with entrenched immune responses. JCI Insight 2016;1(11):e86821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kishnani 2006a
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case L, et al. Pompe disease diagnosis and management guideline. Genetics in Medicine 2006;8(5):267‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kishnani 2009
- Kishnani PS, Corzo D, Leslie ND, Gruskin D, Ploeg A, Clancy JP, et al. Early treatment with alglucosidase alpha prolongs long‐term survival of infants with Pompe disease. Pediatric Research 2009; Vol. 66, issue 3:329‐35. [DOI] [PMC free article] [PubMed]
Klinge 2005
- Klinge L, Straub V, Neudorf U, Schaper J, Bosbach T, Görlinger K, et al. Safety and efficacy of recombinant acid alpha‐glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscular Disorders 2005;15(1):24‐31. [DOI] [PubMed] [Google Scholar]
Kroos 2008
- Kroos M, Pomponio RJ, Van VL, Palmer RE, Phipps M, Vander HR, et al. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Human Mutation 2008;29(6):E13‐26. [DOI] [PubMed] [Google Scholar]
Messinger 2012
- Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM‐negative infantile Pompe disease. Genetics in Medicine 2012;14(1):135‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reuser 2006
- Reuser AJJ, Drost MR. Lysosomal dysfunction, cellular pathology and clinical symptoms: basic principles. Acta Paediatrica 2006;95(S451):77‐82. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Slonim 2000
- Slonim AE, Bulone L, Ritz S, Goldberg T, Chen A, Martiniuk F. Identification of two subtypes of infantile acid maltase deficiency. Journal of Pediatrics 2000;137(2):283‐5. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne J, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Sun 2007
- Sun B, Bird A, Young SP, Kishnani PS, Chen YT, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. American Journal of Human Genetics 2007;81(5):1042‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Carin 2015
- Gelder CM, Hoogeveen‐Westerveld M, Kroos MA, Plu I, Ploeg AT, Reuser AJ. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. Journal of Inherited Metabolic Disease 2015;38(2):305‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van den Hout 2000
- Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme‐Dijkhuis A, Ploeg AT. Recombinant human α‐glucosidase from rabbit milk in Pompe patients. Lancet 2000;356(9227):397‐8. [DOI] [PubMed] [Google Scholar]
Van den Hout 2003
- Hout HM, Hop W, Diggelen OP, Smeitink JA, Smit GP, Poll‐The BT, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112(2):332‐40. [DOI] [PubMed] [Google Scholar]
WHO 2012
- World Health Organization. Bulletin of the World Health Organization: coming together to combat rare diseases. www.who.int/bulletin/volumes/90/6/12‐020612/en/ (accessed 2 December 2013). [DOI] [PMC free article] [PubMed]
Zhang 2006
- Zhang H, Kallwass H, Young SP, Carr C, Dai J, Kishnani PS, et al. Comparison of maltose and acarbose as inhibitors of maltase‐glucoamylase activity in assaying acid α‐glucosidase activity in dried blood spots for the diagnosis of infantile Pompe disease. Genetics in Medicine 2006;8(5):302‐6. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Chen 2015
- Chen M, Zhang L, Liang Y. Enzyme replacement therapy for infantile‐onset Pompe disease. Cochrane Database of Systematic Reviews 2015, Issue 3. [DOI: 10.1002/14651858.CD011539] [DOI] [PMC free article] [PubMed] [Google Scholar]