

Abstract
Background
Glucagon‐like peptide analogues are a new class of drugs used in the treatment of type 2 diabetes that mimic the endogenous hormone glucagon‐like peptide 1 (GLP‐1). GLP‐1 is an incretin, a gastrointestinal hormone that is released into the circulation in response to ingested nutrients. GLP‐1 regulates glucose levels by stimulating glucose‐dependent insulin secretion and biosynthesis, and by suppressing glucagon secretion, delayed gastric emptying and promoting satiety.
Objectives
To assess the effects of glucagon‐like peptide analogues in patients with type 2 diabetes mellitus.
Search methods
Studies were obtained from electronic searches of The Cochrane Library (last search issue 1, 2011), MEDLINE (last search March 2011), EMBASE (last search March 2011), Web of Science (last search March 2011) and databases of ongoing trials.
Selection criteria
Studies were included if they were randomised controlled trials of a minimum duration of eight weeks comparing a GLP‐1 analogue with placebo, insulin, an oral anti‐diabetic agent, or another GLP‐1 analogue in people with type 2 diabetes.
Data collection and analysis
Data extraction and quality assessment of studies were done by one reviewer and checked by a second. Data were analysed by type of GLP‐1 agonist and comparison treatment. Where appropriate, data were summarised in a meta‐analysis (mean differences and risk ratios summarised using a random‐effects model).
Main results
Seventeen randomised controlled trials including relevant analyses for 6899 participants were included in the analysis. Studies were mostly of short duration, usually 26 weeks.
In comparison with placebo, all GLP‐1 agonists reduced glycosylated haemoglobin A1c (HbA1c) levels by about 1%. Exenatide 2 mg once weekly and liraglutide 1.8 mg reduced it by 0.20% and 0.24% respectively more than insulin glargine. Exenatide 2 mg once weekly reduced HbA1c more than exenatide 10 μg twice daily, sitagliptin and pioglitazone. Liraglutide 1.8 mg reduced HbA1c by 0.33% more than exenatide 10 μg twice daily. Liraglutide led to similar improvements in HbA1c compared to sulphonylureas but reduced it more than sitagliptin and rosiglitazone.
Both exenatide and liraglutide led to greater weight loss than most active comparators, including in participants not experiencing nausea. Hypoglycaemia occurred more frequently in participants taking concomitant sulphonylurea. GLP‐1 agonists caused gastrointestinal adverse effects, mainly nausea. These adverse events were strongest at the beginning and then subsided. Beta‐cell function was improved with GLP‐1 agonists but the effect did not persist after cessation of treatment.
None of the studies was long enough to assess long‐term positive or negative effects.
Authors' conclusions
GLP‐1 agonists are effective in improving glycaemic control.
Keywords: Humans; Diabetes Mellitus, Type 2; Diabetes Mellitus, Type 2/blood; Diabetes Mellitus, Type 2/drug therapy; Glucagon‐Like Peptide 1; Glucagon‐Like Peptide 1/analogs & derivatives; Glycated Hemoglobin A; Glycated Hemoglobin A/metabolism; Hypoglycemic Agents; Hypoglycemic Agents/therapeutic use; Randomized Controlled Trials as Topic
Glucagon‐like peptide analogues for type 2 diabetes
Glucagon‐like peptide analogues or agonists are a new kind of drug in the treatment of type 2 diabetes that are given by injection under the skin. They regulate glucose levels by stimulating glucose‐dependent insulin secretion and biosynthesis, and by suppressing glucagon secretion, delaying gastric emptying and promoting satiety. Various glucagon‐like peptide‐1 agonists are in use or in the licensing process, including exenatide, liraglutide, albiglutide, taspoglutide, lixisenatide and LY2189265.
Seventeen randomised controlled trials of mostly moderate to high quality randomised approximately 6899 people with type 2 diabetes mellitus. Studies were mostly of short duration, usually 26 weeks. The longest duration study was 30 weeks. Of the seventeen studies, one compared albiglutide with placebo, two compared exenatide 10 µg twice daily against exenatide 2 mg once weekly, one compared exenatide 2 mg once weekly against insulin glargine, one compared exenatide 2 mg once weekly against pioglitazone and sitagliptin, five compared liraglutide with placebo, two compared liraglutide with sulphonylurea, one each compared exenatide twice daily with liraglutide, liraglutide with sitagliptin, liraglutide with rosiglitazone and liraglutide with insulin glargine, two compared taspoglutide with placebo and one each compared lixisenatide with placebo and LY2189265 with placebo. In people already treated with oral anti‐diabetes drugs, addition of glucagon‐like peptide analogues improved blood sugar control in comparison to placebo, rosiglitazone, pioglitazone or sitagliptin, but not always in comparison to insulin (for exenatide) or glimepiride (a sulphonylurea). Glucagon‐like peptide analogous caused more weight loss than any of the comparison treatments. However, more nausea and other gastrointestinal effects such as diarrhoea or vomiting were seen, though these tended to wear off and were not seen in all participants. There was slightly more hypoglycaemia with glucagon‐like analogous than with placebo, but generally less than with other anti‐diabetic treatments. The incidence of hypoglycaemia occurred more frequently in participants taking concomitant sulphonylurea. The studies were not long enough to assess long‐term side effects. None of the studies investigated mortality or morbidity.
Background
Description of the condition
Type 2 diabetes is characterised by hyperglycaemia, associated with insulin resistance and hyperinsulinaemia, but later by progressively impaired insulin secretion in response to glucose load (ingestion of nutrients, i.e. a meal).
A consequence of this is chronic hyperglycaemia (i.e. elevated levels of plasma glucose) with disturbances of carbohydrate, fat and protein metabolism. Long‐term complications of diabetes mellitus include retinopathy, nephropathy and neuropathy. The risk of cardiovascular disease is increased. For a detailed overview of diabetes mellitus, please see under 'Additional information' in the information on the Metabolic and Endocrine Disorders Group in The Cochrane Library (see 'Cochrane Review Groups (CRGs)'). For an explanation of methodological terms, see the main glossary in The Cochrane Library.
Maintenance of tight glucose control is important in preventing complications of diabetes. Traditional treatments for type 2 diabetes aim to control blood glucose and reduce the development of diabetes‐associated secondary complications (Turner 1996). However, there is usually a progressive deterioration in blood glucose control in type 2 diabetes necessitating changes in treatment. People with type 2 diabetes are initially advised on lifestyle changes (weight loss, more exercise and diet) and offered ongoing patient education. If the lifestyle changes fail to control blood glucose, metformin (especially in overweight people) or sulphonylureas (if metformin is contraindicated or not tolerated, or if the person is not overweight) are considered (NICE CG87 2009). When monotherapy with these drugs no longer provides adequate glycaemic control, combination therapy is an option (metformin plus sulphonylurea), but it may only be a matter of time before treatment must be intensified (for example by using insulin therapy or pioglitazone) to control glucose levels adequately. The UKPDS (United Kingdom Prospective Diabetes Study) study has shown that the deterioration in glycaemic control may be attributed to the loss of pancreatic insulin‐secreting beta‐islet cell function (Turner 1996). In addition some of the oral hypoglycaemic agents lead to weight gain and hypoglycaemia, which in turn affects person's compliance and glycaemic control. A glycosylated haemoglobin A1c (HbA1c) level of more than 7% has been taken to indicate inadequate glycaemic control (Nathan 2009) though targets should be individualised.
Description of the intervention
Glucagon‐like peptide analogues or agonists are a new group of drugs that mimic the action of an endogenous hormone called glucagon‐like peptide 1 (GLP‐1). GLP‐1 is an incretin, a gastrointestinal hormone that is released into the circulation in response to ingested nutrients. GLP‐1 regulates glucose levels by stimulating glucose‐dependent insulin secretion and biosynthesis, and by suppressing glucagon secretion, delaying gastric emptying and promoting satiety (Baggio 2004; Nauck 1993). GLP‐1 lowers glucagon secretion in type 2 diabetes in a glucose‐dependent manner thus preventing interference in the normal glucagon counter‐regulatory response to hypoglycaemia (Nauck 2002).
Circulating GLP‐1 undergoes destruction by an enzyme, dipeptidyl‐peptidase IV (DPP‐IV), resulting in a half‐life of 1 to 2 minutes. The natural form is therefore not suitable as a treatment.
Adverse effects of the intervention
Weight gain is a major side effect of some traditional type 2 diabetes therapies such as the sulphonylureas and the glitazones. However, the GLP‐1 analogues have been shown to produce weight loss in people with type 2 diabetes (Amori 2007; Barnett 2009; Monami 2009; NICE CG87 2009; Norris 2009).
As regards adverse effects, nausea is common but wears off with time. No serious adverse effects have yet been proven, but there has been concern about exenatide and liraglutide causing pancreatitis. The manufacturers argue that there is no evidence to explain the pathogenesis of pancreatitis with exenatide and also reports that pancreatitis is common in type 2 diabetes, and therefore is not related to the drug. Studies on rats and mice with doses exceeding the recommended human dose showed histological changes of chronic pancreatitis, but the animals appeared healthy with no behavioural changes suggestive of pain. In addition, the animals were taking food normally and growth was also normal (Butler 2010). The FDA reports that after marketing of exenatide, there have been some cases of acute pancreatitis but that the incidence was low (FDA 2009). The main concern is about the prolonged use of the drug, as there is evidence of chronic low‐grade pancreatitis in rodents and chronic pancreatitis is one of the important causes of pancreatic adenocarcinoma. In the liraglutide development program, it was found that there were more cases of pancreatitis with liraglutide compared with other oral comparators (EMEA 2009), but the absolute risk was low. There are no long term data available to substantiate this. Recently, FDA has issued a warning to remind all the doctors that liraglutide may cause pancreatitis and thyroid carcinoma (Journal Watch 2011).
In addition, there have been reports of thyroid carcinoma in rodents. A two year carcinogenicity study was performed on rats and mice with liraglutide and it was observed that there was proliferation of C‐cells of the thyroid. The changes were dose‐dependent and ranged from mild or moderate hyperplasia to malignancy. Liraglutide induced carcinogenic changes by a non‐genotoxic, specific GLP‐1 receptor mechanism to which rodents are specifically sensitive, whereas monkeys and humans are less sensitive (EMEA 2009). Although humans are not sensitive, the chances of carcinogenic changes with liraglutide cannot be discounted due to lack of evidence. Similarly, a two year carcinogenicity study on rats and mice with exenatide reported incidence of benign thyroid C‐cell adenomas among rats whereas no such cases were found in mice (FDA 2009). The exposure to the drug ranged from 5 to 130 times the recommended maximum human exposure dose.
How the intervention might work
There are currently at least six GLP‐1 analogues. Exenatide (Byetta, Lilly/Amylin) and liraglutide (NN2211, Novo Nordisk) have reached the market. Albiglutide (GlaxoSmithKline), taspoglutide (Ipsen and Roche), lixisenatide (Sanofi‐Aventis) and LY2189265 (Lilly) have been the subject of trials.
Some current glucose lowering treatments cause hypoglycaemia owing to the glucose‐independent effect of the drugs. In contrast, the action of the GLP‐1 analogues is glucose‐dependent, i.e. the higher the plasma glucose level, the greater the effect of GLP‐1 on insulin secretion with the greatest effect in hyperglycaemic conditions, and little or no effect when the blood glucose concentration is less than 3.61 mmol/L (65 mg/dL). This should reduce the occurrence of hypoglycaemia.
Much interest has been raised by the possibility that the GLP‐1 analogues might reduce the loss of beta‐cell mass. Studies in rodents have shown that GLP‐1 increases pancreatic islet beta‐cell mass by enhancing beta‐cell proliferation (Xu 1999), increasing the differentiation of new beta‐cells from progenitor cells in the pancreatic duct epithelium (Abraham 2002) and reducing beta‐cell apoptosis (Farilla 2003; Li 2003). If this applied in humans, use of GLP‐1 analogues may hold the potential to maintain or enhance beta‐cell mass in type 2 diabetes, and prevent progression of the disease.
Current evidence for effectiveness of glucagon‐like peptide analogues in type 2 diabetes
Recent evidence has been summarised in reviews by Shyangdan and colleagues (Shyangdan 2010), Monami and colleagues (Monami 2009), Barnett (Barnett 2009), Amori (Amori 2007), Norris and colleagues (Norris 2009) and in HTA reports for NICE (Shyangdan 2011; Waugh 2010). This Cochrane review is partly based on, and partly an update of, the review by Shyangdan and colleagues (Shyangdan 2010). That review concluded that GLP‐1 agonists are effective in improving glycaemic control when used as third line agents. In contrast to insulin, glitazones and sulphonylureas, GLP‐1 agonists cause weight reduction and the occurrence of hypoglycaemia is less. The risk of hypoglycaemia increased when GLP‐1 agonists were combined with a sulphonylurea but not when given with metformin. GLP‐1 agonists caused gastrointestinal adverse events mainly nausea but this decreased over time.
There have been several other good quality reviews, but these have tended to include all trials. However not all trials are relevant to clinical practice. Some were designed to identify the optimum dosage. Others investigated GLP‐1 analogues against placebo in people on no other glucose lowering drug, whereas in practice, older cheaper drugs with long safety records, such as metformin, should be used first. In the UK, the NICE guideline recommends that the GLP‐1 analogues should be used in triple therapy (NICE CG87 2009; NICE TA203 2010). In the USA, it appears that they are more frequently used in dual therapy.
A long‐acting‐release (LAR) formulation of exenatide has been developed that undergoes slow degradation over a period of weeks and can therefore be administered as a single injection per week. Liraglutide is given only once daily. Newer GLP‐1 analogues include albiglutide, taspoglutide, lixisenatide and LY2189265.
Why it is important to do this review
Conventional treatments used to control hyperglycaemia in type 2 diabetes are unsatisfactory due to weight gain, risk of hypoglycaemia or a decrease in efficacy with disease progression (Pratley 2008). Their glucose‐dependent mechanism of action suggests that the GLP‐1 analogues should not cause hypoglycaemia. In addition it appears that these agents cause weight loss rather than weight gain. Since most people with type 2 diabetes are overweight or obese, this is potentially very important. At present, when people with type 2 diabetes have poor control on a combination of oral agents, the next step is to start a third oral hypoglycaemic agents such as a gliptin or pioglitazone, or a GLP‐1 agonist or insulin (NICE CG87 2009)). The guideline states that GLP‐1 agonists should be continued if it leads to reduction of 1% in HbA1c level and 3% in weight by six months. GLP‐1 agonists cause gastrointestinal adverse events, mainly nausea, leading to discontinuation of the drug in some people. However, there is some evidence that the newer GLP‐1 analogues used once weekly or once every two weeks reduce this adverse event.
Objectives
To assess the effects of glucagon‐like peptide analogues in patients with type 2 diabetes mellitus.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled clinical trials.
Only articles published in full were included, except that meeting abstracts were considered if they contained data on secondary outcomes from a study already published in full, or if there was a published protocol (so that information on the design and quality are available).
Types of participants
Adults (over 18 years of age) with type 2 diabetes.
To be consistent with changes in classification and diagnostic criteria of type 2 diabetes mellitus through the years, the diagnosis should have been established using the standard criteria valid at the time of the beginning of the trial (ADA 1997; ADA 1999; WHO 1998). Ideally, diagnostic criteria should have been described. If necessary, authors' definition of diabetes mellitus were used.
Types of interventions
Trials with a minimum duration of eight weeks of any glucagon‐like peptide 1 (GLP‐1) analogue (exenatide 10 μg twice daily compared against exenatide 2 mg once weekly, exenatide 2 mg once weekly, liraglutide, albiglutide, taspoglutide, lixisenatide and newer GLP‐1 analogues) in combination with metformin or sulphonylurea or both were considered. Studies were also considered if they included additional oral antihyperglycaemic agents, such as thiazolidinediones (TZD). Trials comparing exenatide 10 μg twice daily against placebo or other oral hypoglycaemic agents were not considered. Exenatide 10 μg twice daily is not considered in this review, apart from in comparison with the long‐acting form, having been reviewed elsewhere (Shyangdan 2010) and because it is expected to be replaced by the long‐acting, once weekly form.
Since GLP‐1 agonists are not licensed for use as first line therapy in treatment‐naive patients, the inclusion criteria are based on the comparisons which are considered to be relevant to clinical practice as suggested by the NICE guideline (NICE CG66 2008) and by the ADA/EASD joint statement (Nathan 2009).
Therefore, the following comparisons were excluded:
GLP‐1 used as a monotherapy, whether compared with placebo or another drug.
Use of GLP‐1 in patients naive to treatment, i.e. patients need to have been diagnosed with type 2 diabetes for at least a year and to have been on at least one oral hypoglycaemic drug for six months; where trials did not give sufficient detail, we accepted them if the mean duration of diabetes exceeded two years.
Trials of a GLP‐1 agonists on patients having failed only on a sulphonylurea or a glitazone without having been tried on metformin; in practice, some trials included people who have failed on either metformin or a sulphonylurea, and did not necessarily give results separately. We accepted any such trials if 70% of patients or more had been on metformin.
Trials or arms using non‐standard doses. So most of the data from dose‐ranging studies were not relevant.
Dosages
The standard exenatide regimen is to start with 5 µg twice daily and to increase after a month or so to 10 µg twice daily.
The dose of liraglutide is less clear, with some trials suggesting starting with 0.6 mg, and then increasing in stages to 1.2 mg or 1.8 mg. There are some trials in Japanes patients where liraglutide has been used in the dose of 0.9 mg (Kaku 2010). Otherwise, trials or arms with less than 1.2 mg daily (final dose) were excluded.
For newer GLP‐1 agonists, we only included dosages that are likely to be used in routine care ‐ i.e. those with maximal effects while minimising adverse events.
The following comparisons were included:
1. GLP‐1 agonist as a third line agent. There are two questions of interest to clinicians in this situation. The first is whether the GLP‐1 analogues are effective in improving glycaemic control, without causing adverse effects. The second is whether GLP‐1 analogues are as good as, or better than other options. Since dual therapy is usually metformin and a sulphonylurea, the other options are insulin, a glitazone or a gliptin.
So comparisons are:
1a. Dual therapy + GLP‐1 versus dual therapy + placebo
1b. Dual therapy + GLP‐1 versus same dual therapy + another antihyperglycaemic agent
2. GLP‐1 agonist as a second line agent
The questions are similar to those for third line use:
2a. Monotherapy + GLP‐1 versus same monotherapy + placebo
2b. Monotherapy + GLP‐1 versus same monotherapy + any antihyperglycaemic agent
3. GLP‐1 agonist versus other GLP‐1 agonist
The general principles of inclusion apply here. So, trials were only included if they compared different GLP‐1 analogues as third line or second line agents.
Types of outcome measures
Primary outcomes
glycaemic control as measured by glycated haemoglobin (HbA1c);
hypoglycaemia: graded as mild (symptoms easily controlled by individual), moderate (normal activities interrupted but assistance not required), severe (individual requiring assistance, and associated with blood glucose level less than 50 mg/dL (4 mmol/L) or with prompt recovery after oral carbohydrate or glucagons or intravenous glucose), serious (life threatening or required subject to be admitted to hospital);
weight gain or loss/change in body mass index.
Secondary outcomes
health‐related quality of life (using a validated instrument);
adverse effects (for example congestive heart failure, oedema, pancreatitis, other gastrointestinal effects);
mortality (all‐cause mortality; diabetes‐related mortality (death from myocardial infarction, stroke, peripheral vascular disease, renal disease, hyper‐ or hypoglycaemia or sudden death);
morbidity (both specific to diabetes such as retinopathy or nephropathy, and cardiovascular morbidity, for example angina pectoris, myocardial infarction, heart failure, stroke, peripheral vascular disease);
blood pressure;
fasting blood glucose and post‐prandial glucose;
plasma lipids (triglycerides, total cholesterol, HDL and LDL‐cholesterol);
beta‐cell function.
Covariates, effect modifiers and confounders
age;
ethnicity;
body mass index;
HbA1c at baseline;
diabetes duration.
Search methods for identification of studies
See: Cochrane Metabolic and Endocrine Disorders Group methods.
Electronic searches
We used the following sources for the identification of trials:
The Cochrane Library (issue 1, 2011);
MEDLINE (1996 to March 2011);
EMBASE (1998 to March 2011);
Web of Science (1980 to March 2011).
We also searched databases of ongoing trials:
Current Controlled Trials (www.controlled‐trials.com) and ClinicalTrials.gov
See Appendix 1 for details on all search strategies.
Searching other resources
American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) web sites were searched for recent meeting abstracts
The web sites of the FDA (Food and Drug Administration) and EMEA were searched for information on efficacy and safety
Reference lists
We also looked for additional studies by searching the reference lists of included trials and (systematic) reviews, meta‐analyses and health technology assessment reports identified.
Studies published in any language were to be included.
Data collection and analysis
Selection of studies
To determine the studies to be assessed further, three authors (PR, DS, PS) independently scanned the abstract, titles or both sections of every record retrieved. All potentially relevant articles were investigated as full text. Few differences in opinion existed which were resolved by a third party (NW). There was no article needing the author's clarification for selection. An adapted PRISMA (preferred reporting in systematic review and meta‐analysis (Moher 2009)) flow‐chart of study selection is attached (Figure 1).
Figure 1.

Study flow diagram.
Data extraction and management
Two of the three authors (CC, DS, PS) independently extracted data using a standard data extraction form that was tested, piloted and modified for the current review. Data extraction was checked by a second author (CC, PR, DS). Relevant data on study population, intervention, study design and outcomes were pulled out from included studies. See Characteristics of included studies and Table 21 for details. Few discrepancies were discussed and resolved between two authors. There was no such disagreements needing a third reviewer.
Table 1.
Overview of study populations
|
Characteristic ► GLP analogue ▼ |
[n] screened | [n] randomised | [n] safety | [n] ITT | [n] finishing study | [%] of randomised participants finishing study |
| EXENATIDE | ||||||
| Exenatide versus TZD and DPP‐4 inhibitors | ||||||
| Bergenstal 2010 | I1: ‐ C1: ‐ T: 958 |
I1: 170 C1: 172 C2: 172 T: 514 |
I1: 160 C1: 165 C2: 166 T: 491 |
I1: 160 C1: 165 C2: 166 T: 491 |
I1: 127 C1: 131 C2: 144 T: 402 |
I1: 74.70 C1: 76.16 C2: 83.72 T: 78.21 |
| Exenatide versus insulin glargine | ||||||
| Diamant 2010 | I1: ‐ C1: ‐ T: 659 |
I1: 233 C1: 223 T: 456 |
I1: 233 C1: 223 T: 456 |
I1: 233 C1: 223 T: 456 |
I1: 209 C1: 209 T: 418 |
I1: 89.69 C1: 93.72 T: 91.66 |
| LIRAGLUTIDE | ||||||
| Liraglutide versus placebo and TZD | ||||||
| LEAD 1 (Marre 2009) | I1: ‐ I2: ‐ C1: ‐ C2: ‐ T: 1712 |
I1: 228 I2: 234 C1: 114 C2: 232 T*: 808 |
I1: 228 I2: 234 C1: 114 C2: 231 T*: 807 |
I1: 228 I2: 234 C1: 114 C2: 231 T*: 807 |
I1: 196 I2: 213 C1: 83 C2: 194 T*: 686 |
I1: 85.96 I2: 91.02 C1: 72.80 C2: 83.62 T*: 84.90 |
| Liraglutide versus placebo and SU | ||||||
| LEAD 2 (Nauck 2009) | I1: ‐ I2: ‐ C1: ‐ C2: ‐ T: 1662 |
I1: 241 I2: 242 C1: 122 C2: 244 T*: 849 |
I1: 240 I2: 242 C1:121 C2: 242 T*: 845 |
I1: 240 I2: 242 C1: 121 C2: 242 T*: 845 |
I1: 197 I2: 191 C1: 74 C2: 210 T*: 672 |
I1: 81.74 I2: 78.92 C1: 60.65 C2: 80.06 T*: 79.15 |
| Liraglutide versus placebo and insulin | ||||||
| LEAD 5 (Russell‐Jones 2008) | I1: ‐ C1: ‐ C2: ‐ T: 973 |
I1: 232 C1: 115 C2: 234 T: 581 |
I1: 230 C1: 114 C2: 232 T: 576 |
I1: 230 C1: 114 C2: 232 T: 576 |
I1: 207 C1: 96 C2: 219 T: 522 |
I1: 89.22 C1: 83.48 C2: 93.59 T: 89.84 |
| Liraglutide versus placebo | ||||||
| LEAD 4 (Zinman 2009) | I1: ‐ I2: ‐ C1: ‐ T: 821 |
I1: 178 I2: 178 C1: 177 T: 533 |
I1: 178 I2: 178 C1: 177 T: 533 |
I1: 178 I2: 178 C1: 177 T: 533 |
I1: 153 I2: 133 C1: 121 T: 407 |
I1: 85.95 I2: 74.72 C1: 68.36 T: 76.36 |
| Kaku 2010 | I1: ‐ I2: ‐ C1: ‐ T: 308 |
I1: 88 I2: 88 C1: 88 T: 264 |
I1: 88 I2: 88 C1: 88 T: 264 |
I1: 88 I2: 88 C1: 88 T: 264 |
I1: 83 I2: 84 C1: 74 T: 241 |
I1: 94.32 I2: 95.45 C1: 84.09 T: 91.29 |
| Liraglutide versus SU | ||||||
| Yang 2010 | I1: ‐ I2: ‐ C1: ‐ T: ‐ |
I1: 233 I2: 234 C1: 231 T*: 698 |
I1: 233 I2: 233 C1: 231 T*: 697 |
I1: ‐ I2: ‐ C1: ‐ T*: ‐ |
I1: 187 I2: 175 C1: 215 T*: 577 |
I1: 80.25 I2: 74.79 C1: 93.07 T*: 82.66 |
| Liraglutide versus DPP‐4 inhibitors | ||||||
| Pratley 2010 | I1: ‐ I2: ‐ C1: ‐ T: 1302 |
I1: 225 I2: 221 C1: 219 T: 665 |
I1: 221 I2: 218 C1: 219 T: 658 |
I1: 221 I2: 218 C1: 219 T: 658 |
I1: 169 I2: 191 C1: 194 T: 554 |
I1: 75.11 I2: 86.42 C1: 88.58 T: 83.30 |
| LIXISENATIDE | ||||||
| Lixisenatide versus placebo | ||||||
| Ratner 2010 | I1: ‐ I2: ‐ I3: ‐ I4: ‐ I5: ‐ I6: ‐ I7: ‐ I8: ‐ C1: ‐ T: 1466 |
I1: 55 I2: 52 I3: 55 I4: 54 I5: 53 I6: 56 I7: 54 I8: 54 C1: 109 T: 542 |
I1: 55 I2: 52 I3: 55 I4: 54 I5: 53 I6: 56 I7: 54 I8: 54 C1: 109 T: 542 |
I1: 55 I2: 50 I3: 53 I4: 51 I5: 51 I6: 54 I7: 52 I8: 53 C1: 107 T: 526 |
I1: 53 I2: 47 I3: 46 I4: 45 I5: 51 I6: 51 I7: 46 I8: 47 C1: 103 T: 489 |
I1: 96.36 I2: 90.38 I3: 83.64 I4: 83.33 I5: 96.23 I6: 91.10 I7: 85.18 I8: 87.04 C1: 94.50 T: 90.22 |
| LY2189265 | ||||||
| LY2189265 versus placebo | ||||||
| Umpierrez 2011 | I1: ‐ I2: ‐ I3: ‐ C1: ‐ T: ‐ |
I1: 66 I2: 65 I3: 65 C1: 66 T: 262 |
I1: 66 I2: 65 I3: 65 C1: 66 T: 262 |
I1: 66 I2: 65 I3: 65 C1: 66 T: 262 |
I1: 58 I2: 58 I3: 56 C1: 60 T: 232 |
I1: 87.88 I2: 89.23 I3: 86.15 C1: 90.90 T: 88.55 |
| TASPOGLUTIDE | ||||||
| Taspoglutide versus placebo | ||||||
| Nauck 2009 | I1: ‐ I2: ‐ I3: ‐ I4: ‐ I5: ‐ C1: ‐ T: 572 |
I1: ‐ I2: ‐ I3: ‐ I4: ‐ I5: ‐ C1: ‐ T: 306 |
I1: 50 I2: 49 I3: 50 I4: 50 I5: 49 C1: 49 T:297 |
I1: 50 I2: 49 I3: 50 I4: 50 I5: 49 C1: 49 T:297 |
I1: 49 I2: 45 I3: 44 I4: 46 I5: 46 C1: 47 T: 277 |
I1: ‐ I2: ‐ I3: ‐ I4: ‐ I5: ‐ C1: ‐ T: 90.52 |
| Ratner 2010 | I1: ‐ I2: ‐ I3: ‐ C1: ‐ T: ‐ |
I1: 33 I2: 34 I3: 33 C1: 33 T: 133 |
I1: 32 I2: 33 I3: 32 C1: 32 T: 129 |
I1: ‐ I2: ‐ I3: ‐ C1: ‐ T: 125 |
I1: 32 I2: 31 I3: 27 C1: 27 T: 117 |
I1: 96.97 I2: 91.18 I3: 81.82 C1: 81.82 T: 87.97 |
| ALBIGLUTIDE | ||||||
| Albiglutide versus placebo | ||||||
| Rosenstock 2009 | I1: ‐ I2: ‐ C1: ‐ T: 774 |
I1: 31 I2: 33 C1: 52 T*: 116 |
I1: 31 I2: 32 C1: 51 T*: 114 |
I1: 29 I2: 32 C1: 50 T*: 111 |
I1: 22 I2: 24 C1: 40 T*: 86 |
I3: 70.97 I5: 72.73 C1: 76.92 T*: 74.14 |
| GLP‐1 versus GLP‐1 | ||||||
| Blevins 2011 | I1: ‐ C1: ‐ T: ‐ |
I1: ‐ C1: ‐ T: 303 |
I1: 148 C1: 147 T: 295 |
I1: 148 C1: 147 T: 295 |
I1: 128 C1: 130 T: 258 |
I1: 86.49 C1: 88.43 T: 87.46 |
| Drucker 2008 | I1: ‐ C1: ‐ T: ‐ |
I1: ‐ C1: ‐ T: 254 |
I1: 129 C1: 123 T: 252 |
I1: 129 C1: 123 T: 252 |
I1: 109 C1: 95 T: 204 |
I1: 84.5 C1: 77.23 T: 80.95 |
| LEAD 6 (Buse 2009) | I1: ‐ C1: ‐ T:663 |
I1: 233 C1: 231 T: 464 |
I1: 235 C1: 232 T: 467 |
I1: 233 C1: 231 T: 464 |
I1: 202 C1: 187 T: 389 |
I1: 86.69 C1: 80.95 T: 83.84 |
| Total |
I#: 4051 C#: 2679 T##: 6899 |
I: 3878 C: 2653 T: 6531 |
"‐" denotes not reported
C: control; GLP: glucagon‐like peptide; I: intervention; ITT: intention‐to‐treat; T: Total
“*” indicate totals of the patients whose data were included in this review
‘#’ indicate that the total is missing for some data for both I and C group as they were not reported
‘##’ indicate that this is the actual total number of patients randomised. Please note ‘T’ for all trials were added to get this number
Assessment of risk of bias in included studies
One of the three authors (CC, DS, PS) assessed risk of bias of each trial, the assessment was checked by another author (CC, PR, DS). Any disagreements were resolved by consensus between the authors. There was no requirement of a third party to resolve the problems.
Measures of treatment effect
Dichotomous data were expressed as relative risks with 95% confidence intervals (CI) and continuous data were expressed as mean differences with 95% CIs. Outcomes published in different scales were expressed as standardised mean differences (SMD).
Unit of analysis issues
We planned to take into account the level at which randomisation occurred, such as cross‐over trials, cluster‐randomised trials and multiple observations for the same outcome.
Dealing with missing data
Numbers of patients screened, randomised and analysed as intention‐to‐treat or per‐protocol were recorded, as were descriptions of withdrawals or losses to follow‐up and reasons for withdrawals. Each study was assessed for risk of bias for the issues of incomplete outcome data or missing data by investigating drop‐outs, losses to follow‐up and withdrawn study participants and issues of last‐observation‐carried‐forward (LOCF) and was compared to specification of primary outcome parameters and power calculation.
Assessment of heterogeneity
We identified heterogeneity by visual inspection of the forest plots and by using a standard Chi2 test with a significance level of α = 0.1, in view of the low power of this test. We specifically examined heterogeneity employing the I2 statistic which quantifies inconsistency across studies to assess the impact of heterogeneity on the meta‐analysis (Higgins 2002; Higgins 2003), where an I2 statistic of 75% and more indicates a considerable level of inconsistency (Higgins 2011).
When heterogeneity was found, we planned to determine potential reasons for it by examining individual study and subgroup characteristics.
Assessment of reporting biases
Studies were checked for outcome reporting bias.
Data synthesis
Data were summarised statistically if they were available, sufficiently similar and of sufficient quality. Data were summarised using a random‐effects model. Analyses were done separately for the different drugs and the different comparisons as outlined above.
Subgroup analysis and investigation of heterogeneity
Results for hypoglycaemia were analysed in separate subgroups for studies including and not including sulphonylurea therapy.
Sensitivity analysis
Due to the limited number of studies in each comparison, sensitivity analyses were not carried out. Relevant sensitivity analyses would have especially included analysis by risk of bias.
Results
Description of studies
Results of the search
Seventeen randomised controlled trials fulfilled the inclusion criteria and were included in the review. Four hundred and eighty six records were screened for eligibility. A total of 449 papers were excluded on the basis of title and abstract. Thirty seven full‐text articles were assessed for eligibility, out of which 20 articles, details are shown in Characteristics of excluded studies, were excluded. Of the 17 studies included, four examined exenatide, eight liraglutide (one trial examined exenatide against liraglutide), two taspoglutide and one each examined lixisenatide, albiglutide and LY2189265. Of the exenatide trials, two trials compared exenatide twice daily against once weekly exenatide, one compared once weekly exenatide against insulin glargine and one compared once weekly exenatide against sitagliptin and pioglitazone. The most important studies with liraglutide were against active comparators. The active comparators were exenatide, rosiglitazone, glargine, sitagliptin and glimepiride. Albiglutide and taspoglutide were compared to placebo. Some of the trials also included other comparison groups, as outlined below. For an overview of comparisons please see Table 22.
Table 2.
Overview of comparisons
|
Characteristic ► GLP analogue ▼ |
Intervention | Control | Duration | Quality (of 7) |
| EXENATIDE | ||||
| Exenatide versus TZD | ||||
| Bergenstal 2010 | E QW+M+Placebo | TZD+M+Placebo | 26 weeks | 7 |
| Exenatide versus DPP‐4 inhibitors | ||||
| Bergenstal 2010 | E QW+M+Placebo | DPP‐4+M+Placebo | 26 weeks | 7 |
| Exenatide versus insulin glargine | ||||
| Diamant 2010 | E QW+M/(M+SU) | GLAR+M/(M+SU) | 26 weeks | 6 |
| LIRAGLUTIDE | ||||
| Liraglutide versus placebo | ||||
| LEAD 1 (Marre 2009) | L+SU | SU | 26 weeks | 5 |
| LEAD 2 (Nauck 2009) | L+M | M | 26 weeks | 7 |
| LEAD 5 (Russell‐Jones 2008) | L+M+SU | M+SU | 26 weeks | 6 |
| LEAD 4 (Zinman 2009) | L+M+TZD | M+TZD | 26 weeks | 7 |
| Liraglutide versus insulin | ||||
| LEAD 5 (Russell‐Jones 2008) | L+M+SU | GLAR+M+SU | 26 weeks | 6 |
| Liraglutide versus SU | ||||
| LEAD 2 (Nauck 2009) | L+M | M+SU | 26 weeks | 7 |
| Yang 2010 | L+M+Placebo | SU+M+Placebo | 16 weeks | 5 |
| Liraglutide versus TZD | ||||
| LEAD 1 (Marre 2009) | L+SU | TZD+SU | 26 weeks | 5 |
| Liraglutide versus DPP‐4 inhibitors | ||||
| Pratley 2010 | L+M | DPP‐4+M | 26 weeks | 6 |
| LIXISENATIDE | ||||
| Lixisenatide versus placebo | ||||
| Ratner 2010 | LIXI QD or BID | Placebo | 13 weeks | 7 |
| LY2189265 | ||||
| LY2189265 versus placebo | ||||
| Umpierrez 2011 | LY QW | Placebo | 16 weeks | 6 |
| TASPOGLUTIDE | ||||
| Nauck 2009 | T+M | M | 8 weeks | 7 |
| Ratner 2010 | T+M | Placebo+M | 8 weeks | 5 |
| ALBIGLUTIDE | ||||
| Rosenstock 2009 | A+M | M | 16 weeks | 5 |
| GLP1 versus GLP1 | ||||
| Blevins 2011 | E QW+/‐M+/‐SU+/‐TZD | E BID+/‐M+/‐SU+/‐TZD | 24 weeks | 5 |
| Drucker 2008 | E BID+M | E QW+M | 30 weeks | 4 |
| LEAD 6 (Buse 2009) | L+M/SU | E+M/SU | 26 weeks | 5 |
A: albiglutide; BID: twice daily; DPP‐4: dipeptidyl peptidase‐4 inhibitor; E: exenatide; GLAR: glargine; GLP: glucagon‐like peptide; L: liraglutide; LIXI: lixisenatide; LY: LY2189265; M: metformin; QD: once daily; QW: once weekly; SU: sulphonylurea; T: taspoglutide; TZD: thiazolidinedione.
Included studies
Characteristics of included studies are shown under Characteristics of included studies. Studies were prefixed with the first or first few letters of the drug so that trials appear in the right order in the 'Characteristics of included studies' table.
Albiglutide
Design: The trial by A ‐ Rosenstock 2009 assessing the effects of albiglutide was a multi‐centre and multi‐national double‐blind placebo controlled trial. The primary aim of the study was to study the safety, efficacy and tolerability of incremental doses of albiglutide compared to exenatide or placebo, all in combination with background antihyperglycaemic therapy. The group receiving exenatide was open label and was excluded in the present review as all participants also received metformin, whereas only a proportion of the patients in the other groups did. Study duration was 16 weeks.
Participants: The study included 361 participants with type 2 diabetes with a mean diabetes duration of 4.9 years. Participants had a mean age of between 51 and 56 years. Baseline glycosylated haemoglobin A1c (HbA1c) was between 7.9% and 8.0%, and baseline body mass index (BMI) between 31.2 kg/m2and 33 kg/m2. About a quarter to a third of participants were drug‐naive, while the remainder were receiving metformin monotherapy. Participants were excluded if they had used any other oral antidiabetic agent before the beginning of the study.
Interventions: The trial compared 10 intervention groups. Eight different doses of the drug (4 mg or 15 mg or 30 mg weekly, 15 mg or 30 mg or 50 mg biweekly, and 50 mg or 100 mg monthly) were compared against placebo or exenatide. Only the groups using 30 mg once a week and 30 mg once every two weeks were included in the review for comparison because the trial was partly a dose ranging study and some doses are not relevant to clinical practice. Excluded doses were less effective, caused more adverse effects, or caused more fasting plasma glucose (FPG) fluctuation. Metformin was continued at pre‐study doses.
Outcomes: The primary outcome of the study was HbA1c and FPG changes at the end of the study while the secondary outcomes included fasting fructosamine, C‐peptide, glucagon, insulin, and lipid levels, beta‐cell function and assessment of adverse events. Occurrence and duration of nausea and vomiting, immunogenicity, level of anti‐albiglutide antibodies and pharmacokinetics of albiglutide were assessed. Most of these outcomes were also assessed during the 11 week washout period.
Exenatide
Out of the seventeen trials included, five (one trial (L ‐ LEAD 6 Buse 2009) compared exenatide and liraglutide and therefore it will be considered under liraglutide) examined the safety and efficacy of exenatide.
Design: Three trials were open label (E ‐ Blevins 2011; E ‐ Diamant 2010; E ‐ Drucker 2008) while one was double‐blind (E ‐ Bergenstal 2010). Trial duration ranged from 24 to 30 weeks. All the trials were conducted in multiple settings. Two of them were multinational (E ‐ Bergenstal 2010; E ‐ Diamant 2010). A study by E ‐ Drucker 2008 was carried out in USA and Canada whereas the study by E ‐ Blevins 2011 was conducted in the USA only.
Participants: The trials included a total of 1525 randomised patients. Trial participants had a mean age of between 52 and 58 years. Between 40% and 52% of participants in trials were female. Ethnicity was reported in all the studies and the proportion of Caucasian participants ranged between 30% and 85%. All studies included participants with type 2 diabetes with a mean diabetes duration of between 5 years and 8 years, with most taking oral anti diabetic agents (OADs). Baseline HbA1c was between 8.3% to 8.6% and baseline BMI was between 32 kg/m2and 35 kg/m2. Three trials (E ‐ Blevins 2011; E ‐ Diamant 2010; E ‐ Drucker 2008) gave detailed information on previous treatments. Participants were taking metformin, sulphonylureas or thiazolidinediones either on its own or in combination. The studies by E ‐ Blevins 2011 and E ‐ Drucker 2008 also included between 14% and 21% of participants on diet and exercise only.
Interventions: Two trials (E ‐ Blevins 2011; E ‐ Drucker 2008) compared long acting exenatide i.e. 2 mg once weekly against twice daily exenatide i.e. 10 µg. Twice daily exenatide regimen would start with 5 µg twice a day, increasing to 10 µg twice a day after a few weeks. The study by E ‐ Bergenstal 2010 compared long acting exenatide against sitagliptin 100 mg once daily and pioglitazone 45 mg once daily while the study by E ‐ Diamant 2010 compared long acting exenatide against insulin glargine.
Outcomes: In all the trials, the primary outcome measure was change in HbA1c value from baseline to end of study. Secondary outcome measures included changes in FPG, postprandial glucose (PPG), body weight, hypoglycaemia, blood pressure, lipid profile, beta‐cell function, adverse events, and immunogenicity of exenatide. Quality of life was reported by two trials (E ‐ Blevins 2011; E ‐ Diamant 2010) and none of the trials reported diabetes‐related morbidity (most of them did not last long enough for a meaningful assessment of this outcome).
Liraglutide
Eight trials assessed the safety and efficacy of liraglutide.
Design: Two trials (L ‐ LEAD 6 Buse 2009; L ‐ Pratley 2010) were open label and five trials were double blind (L ‐ Kaku 2010; L ‐ LEAD 1 Marre 2009; L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009; L ‐ Yang 2010). One trial (L ‐ LEAD 5 Russell‐J 2009) included two double blind groups and one open label group. Trial duration was 26 weeks for all trials except one, L ‐ Yang 2010, which was 16 weeks long. All trials were multi‐centre and multi‐national trials except L ‐ Kaku 2010 which was carried out in multiple settings in Japan only. The study by L ‐ Yang 2010 included participants from three Asian countries namely China, South Korea and India.
Participants: The trials included a total of 5086 randomised participants (excluding liraglutide 0.6 mg dose from all other trials except L ‐ Kaku 2010). Liraglutide doses of 0.6 mg and 0.9 mg are standard in Japan and hence L ‐ Kaku 2010 was included in this review. Trial participants had a mean age of between 52.7 years and 61.3 years. Between 33% and 55% of participants in trials were female. Ethnicity was not reported in three of the eight trials (L ‐ LEAD 1 Marre 2009; L ‐ LEAD 5 Russell‐J 2009; L ‐ Yang 2010) and the proportion of Caucasian participants ranged between 81% and 93%. In the study by L ‐ Kaku 2010, all the participants were Japanese. All studies included participants with type 2 diabetes with a mean diabetes duration of between 6.0 years and 11.6 years, with all taking oral antidiabetic medication. Only one trial (L ‐ LEAD 1 Marre 2009) did not clearly report on pre‐study medication, but it can be assumed that the participants were tried on metformin because most of them were from Europe and Asia where metformin is used as a first line medication. Baseline HbA1c values were between 8.1% and 8.6% and baseline BMI was between 29.4 kg/m2 and 33.9 kg/m2. Background antihyperglycaemic medication included sulphonylureas in four trials (L ‐ Kaku 2010; L ‐ LEAD 1 Marre 2009; L ‐ LEAD 5 Russell‐J 2009; L ‐ LEAD 6 Buse 2009), and thiazolidinediones in one trial (L ‐ LEAD 4 Zinman 2009).
Interventions: Two trials (L ‐ LEAD 1 Marre 2009, L ‐ LEAD 2 Nauck 2009) compared five intervention groups, five trials (L ‐ Kaku 2010; L ‐ LEAD 4 Zinman 2009; L ‐ LEAD 5 Russell‐J 2009; L ‐ Pratley 2010; L ‐ Yang 2010) compared three different intervention groups, and L ‐ LEAD 6 Buse 2009 compared two intervention groups. Liraglutide was dosed at 0.6, 0.9, 1.2 or 1.8 mg/day, however, in this review we only consider 1.2 and 1.8 mg/day except L ‐ Kaku 2010. The study by L ‐ Kaku 2010 compared 0.6 or 0.9 mg/day of liraglutide against placebo, with all participants receiving concomitant glimepiride therapy. L ‐ LEAD 1 Marre 2009 compared 1.2 or 1.8 mg/day of liraglutide against placebo or rosiglitazone (4 mg/day), with all groups receiving concomitant glimepiride therapy. L ‐ LEAD 2 Nauck 2009 compared 1.2 or 1.8 mg/day of liraglutide against placebo or glimepiride 4 mg/day, with all groups receiving concomitant therapy with metformin. L ‐ LEAD 4 Zinman 2009 compared 1.2 or 1.8 mg/day of liraglutide against placebo, with all groups receiving concomitant therapy with metformin and rosiglitazone. L ‐ LEAD 5 Russell‐J 2009 compared 1.8 mg/day of liraglutide versus insulin glargine or placebo, with all groups receiving concomitant therapy with metformin and glimepiride. L ‐ LEAD 6 Buse 2009 compared 1.8 mg/day of liraglutide against 10 µg BID exenatide, with all groups remaining on their existing sulphonylurea and/or metformin therapy. L ‐ Pratley 2010 compared 1.2 or 1.8 mg/day of liraglutide against sitagliptin 100 mg/day, with all groups continuing their existing metformin therapy. L ‐ Yang 2010 compared 1.2 or 1.8 mg/day against glimepiride 4 mg/day, with all participants receiving metformin 2000 mg/day. Most trials included a run‐in period used for drug titration.
Outcomes: In all the trials the primary outcome measure was change in HbA1c value from baseline to end of study. Secondary outcome measures included changes in FPG, postprandial glucose (PPG), hypoglycaemia, body weight, adverse events, blood pressure, lipid profile, beta‐cell function and liraglutide immunogenicity. Only one study (L ‐ LEAD 6 Buse 2009) reported health‐related quality of life and none reported about diabetes‐related mortality.
Lixisenatide
One trial assessed the safety and efficacy of lixisenatide.
Design: The trial by Lixi ‐ Ratner 2010 assessing the effects of lixisenatide was a double‐blind, multi‐national, parallel‐group, placebo controlled trial. Trial duration was 13 weeks.
Participants: The trial included a total of 542 randomised participants with type 2 diabetes with a mean duration of diabetes between 6.0 years and 7.2 years. Trial participants had a mean age of between 55.4 years and 56.8 years. Between 40.4% and 63% of participants in the trial were female. The total proportion of Caucasian participants were between 64.8% and 86.8%. All the participants were taking metformin. Baseline HbA1c values were between 7.46% and 7.61% and baseline BMI was between 30.7 kg/m2 and 32.8 kg/m2.
Interventions: The trial compared nine intervention groups. Lixisenatide was dosed at 5 μg, 10 μg, 20 μg, 30 μg once or twice daily and compared against placebo twice daily, with all participants receiving stable dose of metformin. All groups also received diet and lifestyle counselling according to the American Diabetes Association guidelines. It also included an initial 2‐week screening phase followed by a 2‐week, single blind, placebo run in period.
Outcomes: The primary outcome measure was change in HbA1c from baseline to end of study. Secondary outcome measures included the proportion of participants achieving HbA1c level of less than 7% or less than 6.5%, changes in body weight, FPG, 2‐hour post‐prandial glucose, hypoglycaemia, blood pressure, heart rate, electrocardiogram (ECG) and anti‐lixisenatide antibodies. Quality of life and diabetes related morbidity were not reported.
LY2189265
One trial assessed the safety and efficacy of LY2189265.
Design: The trial by LY2189265 ‐Umpierrez 2011 assessing the effects of LY2189265 was a double‐blind, placebo‐controlled randomised trial. It was carried out in multiple settings in US and Puerto Rico. Trial duration was 16 weeks.
Participants: The trial included a total of 262 participants with type 2 diabetes, of which 46% to 56% were female. Mean duration of diabetes was between 7.5 years and 9.0 years. Trial participants had a mean age of between 54 years and 59 years. Between 55% and 61% of participants were Caucasians. Majority of the participants (72.7% to 73.8%) were taking metformin and sulphonylurea in combination. Mean HbA1c was between 8.05% and 8.43% and baseline BMI was between 33.7 kg/m2 and 34.2 kg/m2.
Interventions: The trial compared four intervention groups. LY2189265 (LY) was dosed at 0.5, 1.0 and 2.0 mg. First group (also referred as LY 0.5/1.0 mg) received once weekly subcutaneous injection of LY 0.5 mg in the first four weeks followed by 1.0 mg once weekly injection in the next 12 weeks. Second group (also referred as LY 1.0/1.0 mg) received once weekly subcutaneous injection of LY 1.0 mg for 16 weeks. Third group (also referred as LY 1.0/2.0 mg) received once weekly injection of LY 1.0 mg in the first four weeks followed by once weekly injection of LY 2.0 mg in the next 12 weeks. Placebo was given as weekly injection. All the participants continued their baseline oral antihyperglycaemic drugs.
Outcomes: The primary outcome measure was change in HbA1c from baseline to end of study. Secondary outcome measures included change in FPG, blood glucose response following a solid mixed‐meal test, change in body weight, beta‐cell function, treatment emergent adverse events, and hypoglycaemia.
Taspoglutide
Two trials (T ‐ Nauck 2009; T‐ Ratner 2010) assessed the safety and efficacy of taspoglutide.
Design: Both trials (T ‐ Nauck 2009; T‐ Ratner 2010) assessing the effects of taspoglutide were double blind multi‐centre and multi‐national placebo controlled.
Participants: The trials included a total of 439 participants with type 2 diabetes with a mean diabetes duration of between five years and eight years. Participants had a mean age of between 53 years and 60 years and between 36% and 64% were female. Ethnicity was not reported. Baseline HbA1c was between 7.8% and 8.0%, baseline BMI was between 31.5 kg/m2 and 33.3 kg/m2. All participants had been on metformin monotherapy.
Interventions: T ‐ Nauck 2009 compared six intervention groups. Five different doses of taspoglutide (5 mg or 10 mg or 20 mg once weekly or 10 mg or 20 mg once every two weeks) were compared against placebo. In this review, we only consider 10 mg and 20 mg once weekly and 20 mg once every two weeks as the other doses are unlikely to be relevant to clinical practice. The prestudy drugs were continued at the same dose throughout the study period. The trial by T‐ Ratner 2010 compared four intervention groups. Three different doses of taspoglutide i.e. 20, 30 and 40 mg once weekly were used in the study. Taspoglutide 20 mg was injected once every week in the first 4 weeks and then continued in the same dose for the next four weeks (20/20 mg group) or titrated to 30 mg (20/30 mg group) or to 40 mg (20/40 mg group). All the participants continued metformin in their prestudy dose. The prestudy diet and exercise plan was followed throughout the study. Some participants also received medications for cardiovascular risk factors.
Outcomes: The primary outcome measure in T ‐ Nauck 2009 was change in HbA1c from baseline to end of study while T‐ Ratner 2010 explored gastrointestinal tolerability. It was assessed by comparing the number of participants who withdrew from study because of gastrointestinal adverse events Secondary outcome measures in T ‐ Nauck 2009 included changes in FPG, postprandial glucose, body weight, hypoglycaemia, adverse events, lipid profile, and beta‐cell function. T‐ Ratner 2010 explored changes in FPG, HbA1c, body weight and pharmacokinetic parameters. Quality of life and diabetes‐related morbidity were not reported.
Excluded studies
Studies were excluded because they were not primary trials, because they did not compare clinically relevant interventions, or because patients did not fulfil the inclusion criteria (mainly because of previous medication).
Risk of bias in included studies
Details of risk of bias assessment of the trials are shown in the Characteristics of included studies section, in Figure 2 and Figure 3.
Figure 2.
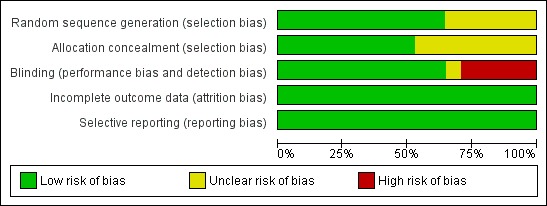
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Figure 3.
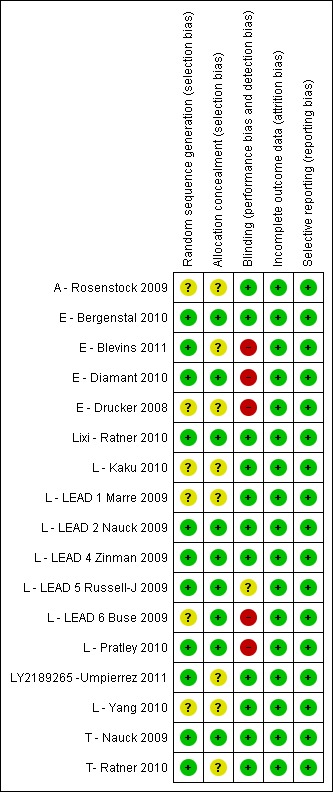
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Out of the seventeen trials, randomisation was adequate in nine, while for the remaining eight (A ‐ Rosenstock 2009; E ‐ Drucker 2008; L ‐ Kaku 2010; L ‐ LEAD 1 Marre 2009; L ‐ LEAD 6 Buse 2009; L ‐ Yang 2010; LY2189265 ‐Umpierrez 2011; T‐ Ratner 2010) the randomisation procedure was not reported or unclear. Ten trials had adequate allocation concealment, while the rest of the trials (A ‐ Rosenstock 2009; E ‐ Drucker 2008; L ‐ Kaku 2010; L ‐ LEAD 1 Marre 2009; L ‐ Yang 2010; LY2189265 ‐Umpierrez 2011; T‐ Ratner 2010) did not report on allocation concealment.
Blinding
Eleven trials were double blind (A ‐ Rosenstock 2009; E ‐ Bergenstal 2010; L ‐ Kaku 2010; L ‐ LEAD 1 Marre 2009; L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009; L ‐ Yang 2010; Lixi ‐ Ratner 2010; LY2189265 ‐Umpierrez 2011; T ‐ Nauck 2009; T‐ Ratner 2010) while five trials (E ‐ Blevins 2011; E ‐ Diamant 2010; E ‐ Drucker 2008; L ‐ LEAD 6 Buse 2009; L ‐ Pratley 2010) were open label. L ‐ LEAD 5 Russell‐J 2009 was a three‐armed placebo controlled blinded study, with liraglutide and liraglutide placebo, and the glargine arm open label.
Incomplete outcome data
All the trials except three (L ‐ Kaku 2010; L ‐ Pratley 2010; L ‐ Yang 2010) used intention‐to‐treat analysis. All the trials reported on rates and reasons for withdrawal. Rates of withdrawal ranged between 1% and 42% (often with more withdrawals in the GLP‐1 agonist groups).
Selective reporting
All the pre‐specified (both primary and secondary) outcomes were reported in all the trials. Ethnicity was not reported in five trials (L ‐ LEAD 1 Marre 2009; L ‐ LEAD 5 Russell‐J 2009; L ‐ Yang 2010; T ‐ Nauck 2009; T‐ Ratner 2010) but other baseline characteristics were reported in the remaining trials.
Other potential sources of bias
A description about the power calculation was unclear in the trial by L ‐ Kaku 2010. However, all the remaining trials had carried out a power calculation. Baseline groups were comparable in all trials.
Effects of interventions
An overview of the results for all comparisons is shown in Appendix 2.
Albiglutide
Albiglutide versus placebo
Results for albiglutide are shown in Data and analyses, 1.1 to 1.12.
HbA1c
A ‐ Rosenstock 2009 found a significant difference (P < 0.05) for HbA1c values between albiglutide and placebo at the end of the study. HbA1c levels decreased by 0.87% (SD 0.65) and 0.79% (SD 0.98) in the participants receiving albiglutide 30 mg weekly and 30 mg every two weeks respectively, whereas the level of HbA1c only decreased by 0.17% (SD 1.01) in the placebo group. The end values of HbA1c were 7.1%, 7.2% and 7.7% for albiglutide 30 mg weekly, albiglutide 30 mg every two weeks and placebo respectively. Similarly, there was a significant difference in the proportion of participants reaching target HbA1c of less than 7% between the albiglutide and placebo groups (52% for albiglutide 30 mg weekly, 50% for 30 mg every two weeks and 20% for placebo). Greater reductions in HbA1c levels were seen in participants with baseline HbA1c values of 8.5% or more but details were not given.
Hypoglycaemia
A definition of hypoglycaemia was not given in the study. No significant difference was found for the incidence of hypoglycaemia among groups. None of the participants in the albiglutide 30 mg weekly group reported hypoglycaemia. Only one patient (3.1%) in the albiglutide 30 mg every two weeks and two patients (3.9%) in the placebo group reported hypoglycaemia during the study period.
Weight change
Weight decreased in both albiglutide and placebo groups. At the end of the study, there was a reduction of 1.4 kg (SD 2.4) in the 30 mg weekly group, of 1.6 kg (SD 2.5) in the 30 mg every two weeks group and of 0.7 kg (SD 2.9) in the placebo group. There were no significant differences in weight reduction between the study groups.
Adverse events
The incidence of adverse events was similar across all groups and ranged between 66.7% and 84.4%. The majority of the adverse events were nausea, vomiting and diarrhoea and the incidence of the first two was more in the participants receiving albiglutide 30 mg once every week while the incidence of diarrhoea was comparatively more in the 30 mg biweekly group. Similarly, the number of participants with a positive immunogenicity test was also higher in albiglutide 30 mg once every week group compared with other groups (6.4% in albiglutide 30 mg weekly versus 3.1% in albiglutide 30 mg every two weeks and 2% with placebo). None of the participants on albiglutide therapy suffered from pancreatitis or any cardiac disorders, however the incidence of skin reactions to the drug was more in the albiglutide group compared to placebo.
Blood pressure
Both albiglutide and placebo groups showed a decrease in systolic and diastolic blood pressure. However, the reduction of blood pressure in the albiglutide groups when compared to placebo group was not significant.
Fasting plasma glucose
Significant reductions (P < 0.05) in fasting plasma glucose were observed in the albiglutide groups compared to placebo. Reduction of 1.44% (SD 2.03) and 1.58% (SD 2.06) were observed with albiglutide 30 mg weekly and 30 mg every two weeks while the reduction was 0.10% (SD 2.90) in the placebo group.
Lipid profiles
No significant changes were seen in the lipid profiles of the participants treated with either albiglutide or placebo.
Beta‐cell function
A significant improvement in beta‐cell function (HOMA‐B ratio) was seen in the participants treated with 30 mg albiglutide weekly (1.4) compared to placebo (1.0) whereas the difference to placebo was not significant with 30 mg albiglutide every two weeks (1.2).
Exenatide
Exenatide versus thiazolidinedione (pioglitazone)
Results for exenatide versus thiazolidinedione are shown in Data and analyses, 2.1 to 2.10. Only one study (E ‐ Bergenstal 2010) compared once weekly exenatide (2 mg) against pioglitazone 45 mg daily.
HbA1c
E ‐ Bergenstal 2010 found a slightly greater reduction in HbA1c with once weekly exenatide than with pioglitazone 45 mg once daily (‐1.5% versus ‐1.2%, P = 0.02).
The proportion of participants achieving target HbA1c level of less 7% was not different between the two treatment groups (60% versus 52%, P = 0.15).
Hypoglycaemia
In the trial, minor hypoglycaemia was defined as any episode where a participant experienced symptoms consistent with hypoglycaemia and a blood glucose level of less than 3 mmol/L. Major hyperglycaemia was defined as any episode resulting in loss of consciousness, seizure or coma that resolved after administration with glucagon or glucose, or any episode with blood glucose level of less than 3.0 mmol/L and a severe impairment that required third‐party assistance to resolve the episode. Incidences of minor hypoglycaemia were similar between the groups, two participants in the exenatide group and one in the pioglitazone group. There were no cases of major hypoglycaemia.
Weight change
Participants taking exenatide once weekly lost weight while those taking pioglitazone gained weight (‐2.3 kg versus + 2.8 kg, P < 0.00001).
Quality of life
In E ‐ Bergenstal 2010, it was found that all the five parameters of weight‐related quality of life and IWQOL total score significantly improved with exenatide (IWQOL total score 5.15, 95% CI 3.11 to 7.19) and not with pioglitazone (1.20, 95% CI ‐0.87 to 3.28). The treatment difference between exenatide and pioglitazone was significant (3.94, 95% CI 1.28 to 6.61, P = 0.0038). The improvement in IWQOL total score with exenatide was consistent with differences in body weight changes.
Adverse events
Withdrawals due to adverse events were increased with once weekly exenatide than pioglitazone (6.9% versus 3.6%). The most commonly reported adverse events with exenatide were nausea and diarrhoea. Withdrawals from the trial were mostly because of these events. Incidences of other adverse events such as headache, urinary tract infection and injection‐site pruritus were similar between the groups. Pioglitazone caused more serious adverse events than exenatide (6% versus 3%). Two other serious events (one in exenatide and other in pioglitazone) led to withdrawals. About half of the participants had low levels of anti‐exenatide antibodies (48%) but in 40% they were not detectable, and there was no relation to glycaemic control and safety.
Blood pressure
Reduction in systolic and diastolic blood pressure was not significantly different between the two groups.
Fasting plasma glucose
E ‐ Bergenstal 2010 found that the reduction in fasting plasma glucose level between once weekly exenatide and pioglitazone was not different (‐1.8 mmol/L versus ‐1.5 mmol/L, P = 0.33).
Post‐prandial glucose
Both exenatide 2 mg once weekly and pioglitazone 45 mg daily led to reduction in post‐prandial glucose levels but the difference was not significant between the two groups.
Lipid profiles
It was found that only pioglitazone led to a significant reduction in triglycerides level. Exenatide 2 mg once weekly led to reduction in total cholesterol and LDL levels. In contrast, pioglitazone led to an increment in these levels. All these changes were not significant. It was also found that all the drugs led to improvement in HDL levels.
Exenatide versus DPP‐4 inhibitors (sitagliptin)
Results for exenatide versus DPP‐4 inhibitors are shown in Data and analyses, 3.1 to 3.10. Only one study (E ‐ Bergenstal 2010) compared once weekly exenatide (2 mg) against sitagliptin 100 mg daily.
HbA1c
E ‐ Bergenstal 2010 found a significantly greater reduction in HbA1c with once weekly exenatide than with sitagliptin 100 mg daily (‐1.5% versus ‐0.9%, P < 0.00001). Similarly, the proportion of participants achieving an HbA1c level of less than 7% was significantly higher with once weekly exenatide than with sitagliptin 100 mg daily (60% versus 35%, P < 0.0001).
Hypoglycaemia
Please see above for definition of hypoglycaemia. The incidence of minor hypoglycaemia was slightly more in the sitagliptin group than the exenatide group (n = 5 versus n = 2). There were no cases of major hypoglycaemia.
Weight change
In E ‐ Bergenstal 2010, once weekly exenatide led to a significantly greater weight loss than sitagliptin 100 mg daily (‐2.3 versus ‐0.8 kg, P = 0.0009).
Quality of life
In E ‐ Bergenstal 2010, all five parameters of weight‐related quality of life and IWQOL total score significantly improved with exenatide (IWQOL total score 5.15, 95% CI 3.11 to 7.19) and sitagliptin (4.56, 95% CI 2.56 to 6.57). The improvement in IWQOL total score with exenatide was consistent with differences in body weight changes.
In E ‐ Bergenstal 2010, overall treatment satisfaction was comparatively higher with exenatide than with sitagliptin (3.96 versus 2.35). The treatment difference between the two was 1.61 (95% CI 0.07 to 3.16, P = 0.0406).
Adverse events
More withdrawals due to adverse events were seen with once weekly exenatide than sitagliptin (6.9% versus 3%). The most commonly reported adverse events with exenatide and sitagliptin were nausea and diarrhoea, while vomiting was more common with exenatide only. Withdrawals from the trial were mostly because of these events. Incidences of other adverse events such as headache, urinary tract infection and injection‐site pruritus were similar between the groups. Incidences of serious adverse events were similar in the exenatide and sitagliptin groups (3% versus 3%). All these events resolved except one in the sitagliptin group, that was fatal. About half (48%) of participants had low levels of anti‐exenatide antibodies (48%) while in 40% they were not detectable, with no relation to glycaemic control and safety.
Blood pressure
At the end of the study, exenatide 2 mg once weekly was found to cause significantly greater reduction in systolic blood pressure than with sitagliptin (treatment difference of ‐4 mm Hg, 95% CI ‐6 to ‐1, P = 0.0055). Reductions in diastolic blood pressure were not different between the groups.
Fasting plasma glucose
E ‐ Bergenstal 2010 found a significant difference in favour of once weekly exenatide compared with sitagliptin 100 mg daily (‐0.90 mmol/L, 95% CI ‐1.50 to ‐0.30, P = 0.0038).
Post‐prandial glucose
Exenatide 2 mg once weekly caused significantly greater reductions in post‐prandial glucose levels at all measurements of the six‐point self‐monitored blood glucose profile than with sitagliptin 100 mg daily (P < 0.05).
Lipid profiles
Exenatide 2 mg once weekly led to reductions in total cholesterol and LDL levels. In contrast, sitagliptin led to an increment. All these changes were not significant. All the drugs led to improvement in HDL levels.
Exenatide versus insulin (glargine)
Results for exenatide versus insulin glargine are shown in Data and analyses, 4.1 to 4.10. The trial by E ‐ Diamant 2010 compared once weekly exenatide against insulin glargine.
HbA1c
Once weekly exenatide led to a slightly greater reduction in HbA1c than with insulin glargine (‐1.5% versus ‐1.3%). The treatment difference between the two group was ‐0.20% (95% CI ‐0.35 to ‐0.05, P = 0.03).
Similarly, the proportion of participants achieving a target HbA1c levels of less than 7% was slightly higher in the once weekly exenatide group than in the insulin glargine group (60% versus 48%, P = 0.03).
Hypoglycaemia
In the study, minor hypoglycaemia was defined as participants experiencing signs or symptoms of hypoglycaemia, with concurrent blood glucose level of less than 3.0 mmol/L that was either self‐treated or resolved independently. Any episode causing loss of consciousness or seizure that resolved after treatment with glucose or any episode with documented blood glucose level of less than 3.0 mmol/L requiring third party assistance was termed as major hypoglycaemia. The number of participants that experienced minor hypoglycaemia was greater in the group taking insulin glargine than those taking exenatide (26% versus 8%). Similarly the number of participants experiencing symptoms of hypoglycaemia but not confirmed by blood glucose measurement was also higher in the group taking insulin glargine (31% versus 13%). Hypoglycaemia occurred most frequently in those taking concomitant sulphonylurea. Major hypoglycaemia occurred in three patients (2 in insulin glargine group and 1 in exenatide group). All three cases were treated with oral carbohydrate administration and did not lead to study discontinuation.
Weight change
Participants taking once weekly exenatide lost significant amounts of weight while those taking insulin glargine gained weight (‐2.6 kg versus +1.4 kg). The treatment difference was ‐4.0 kg (95% CI ‐4.55 to ‐3.45, P < 0.00001).
Quality of Life
It was reported in the trial that a significant improvement for one of the IWQOL‐Lite domains (self esteem) and one EQ‐5D dimensions resulted with once weekly exenatide compared with insulin glargine (no data given). All other domains were similar between the two groups.
Adverse events
The most frequently reported adverse events with exenatide were nausea, diarrhoea, nasopharyngitis, injection‐site reaction and headache while nasopharyngitis and headache were most common with insulin glargine. Gastrointestinal adverse events were mild to moderate in intensity.
Withdrawals due to adverse events were greater in the exenatide group than in the insulin glargine group (4.7% vs. 0.9%). The incidence of serious adverse events was not different between the groups (5% in exenatide group vs. 4% in insulin glargine group). No deaths occurred during the study period. There was one case of oedematous pancreatitis in the exenatide group. It resolved a day after onset and the participant fully recovered. It was found that 68% of participants tested positive for anti‐exenatide antibodies however, these had no effect on treatment response and safety.
Fasting plasma glucose
The reduction in fasting plasma glucose was slightly greater with insulin glargine than with exenatide (‐2.8 mmol/L versus ‐2.1 mmol/L). The treatment difference between the two groups was 0.70 (95% CI 0.14 to 1.26, P = 0.01).
Post‐prandial glucose and glucose profiles
Both treatments reduced post‐prandial glucose at all eight time points. Participants taking once weekly exenatide had significantly lower glucose concentrations after dinner than insulin glargine (P = 0.004) while those on insulin glargine had lower glucose concentrations at 0300 h (P = 0.022) and before breakfast (P < 0.0001). Once weekly exenatide led to greater reduction in post‐prandial glucose excursions compared to insulin glargine after morning (P = 0.001) and evening (P = 0.033) meals.
Liraglutide
Liraglutide versus placebo
Results for liraglutide 0.6 mg versus placebo are shown in Data and analyses, 5.1 to 5.9. Results for liraglutide 0.9 mg verus placebo are shown in Data and analyses, 6.1 to 6.9. Results for liraglutide 1.2 mg versus placebo are shown in Data and analyses, 7.1 to 7.13. Results for liraglutide 1.8 mg versus placebo are shown in Data and analyses, 8.1 to 8.13. Data and analyses, 9.1 to 9.4 show comparisons of 1.2 mg with 1.8 mg liraglutide.
HbA1c
One trial reported change in HbA1c level for 0.6 or 0.9 mg liraglutide versus placebo (L ‐ Kaku 2010). The reduction in HbA1c level at end of the study was significantly greater with 0.9 mg liraglutide than with 0.6 mg liraglutide (‐1.56% versus ‐1.46%) or placebo (‐1.56% versus ‐0.4%). Similarly, the proportion of participants achieving a target HbA1c level of less than 7% was significantly greater with 0.9 mg liraglutide than with 0.6 mg liraglutide (71.3% versus 46.5%, P < 0.05) or placebo (71.3% versus 14.8%, P < 0.0001). Three trials reported HbA1c for 1.2 mg liraglutide versus placebo (L ‐ LEAD 1 Marre 2009; L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009). HbA1c was significantly reduced with 1.2 mg liraglutide compared to placebo. The overall mean difference was ‐1.15 (95% CI ‐1.33 to ‐0.96, P < 0.00001), with no significant heterogeneity of the results. Reductions in HbA1c ranged from ‐1.0% to ‐1.5% in the 1.2 mg liraglutide groups, while HbA1c changes ranged from +0.23% to ‐0.5% in the placebo groups.
The reduction of HbA1c with 1.8 mg liraglutide was similar to that of 1.2 mg liraglutide. Overall, the four studies examining this comparison (L ‐ LEAD 1 Marre 2009; L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009; L ‐ LEAD 5 Russell‐J 2009) found a difference of ‐1.15 (95% CI ‐1.31 to ‐0.99, P < 0.00001) between 1.8 mg liraglutide and placebo. There was no substantial heterogeneity. As with 1.2 mg liraglutide, reductions in HbA1c with 1.8 mg liraglutide ranged from ‐1.0% to ‐1.5%. There was no significant difference between 1.2 mg and 1.8 mg liraglutide in reducing HbA1c (see Analysis 9.1).
Analysis 9.1.
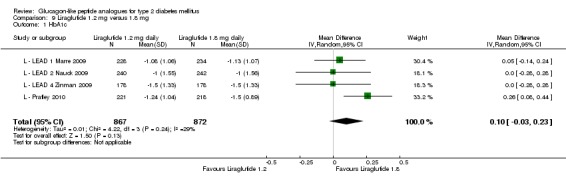
Comparison 9 Liraglutide 1.2 mg versus 1.8 mg, Outcome 1 HbA1c.
The proportion of participants achieving an HbA1c level of 7% or less was also higher with 1.2 mg liraglutide compared with placebo, with 35% to 57.5% reaching the target in the liraglutide groups, and 8% to 28% in the placebo groups. The overall risk ratio for liraglutide 1.2 mg versus placebo was 2.91 (95% CI 1.74 to 4.87, P < 0.0001). There was significant heterogeneity (which disappeared when excluding L ‐ LEAD 4 Zinman 2009).
After treatment with 1.8 mg liraglutide, 42% to 54% reached an HbA1c value of 7% or less. The overall risk ratio for liraglutide 1.8 mg compared with placebo was 3.25 (95% CI 1.97 to 5.36, P < 0.00001), with significant heterogeneity (which disappeared when excluding L ‐ LEAD 4 Zinman 2009). There was no significant difference between 1.2 and 1.8 mg liraglutide (see Analysis 9.2).
Analysis 9.2.
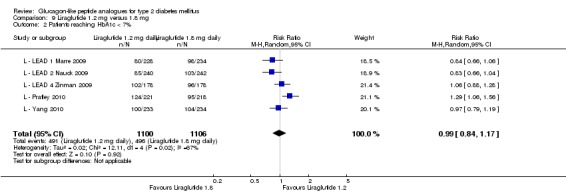
Comparison 9 Liraglutide 1.2 mg versus 1.8 mg, Outcome 2 Patients reaching HbA1c < 7%.
Both L ‐ LEAD 5 Russell‐J 2009 and L ‐ LEAD 2 Nauck 2009 reported the lowest HbA1c values with liraglutide at week 12, with a slight increase towards the end of the studies. However, in the L ‐ LEAD 4 Zinman 2009 study, HbA1c remained steady until the end of the study after the lowest level was observed at week 12. In L ‐ LEAD 1 Marre 2009, the largest decrease in HbA1c with liraglutide was seen in participants previously on monotherapy compared with those on previous combination therapy.
Hypoglycaemia
In the study by L ‐ Kaku 2010, minor hypoglycaemia was defined as an episode that could be self‐treated while those requiring third‐party assistance was considered as major. It was reported that the rate of minor hypoglycaemic episodes (events/patient/year) was higher in the liraglutide groups (2.17 in the 0.6 mg group, 1.96 in the 0.9 mg group) than in the placebo group (1.01). All three trials of 1.2 mg liraglutide reported hypoglycaemia. L ‐ LEAD 1 Marre 2009 and L ‐ LEAD 4 Zinman 2009 defined minor hypoglycaemia as an episode that could be self‐treated while those needing third party assistance or medical interventions were categorised as major. In L ‐ LEAD 1 Marre 2009, the proportion of participants with minor hypoglycaemia was significantly higher (P = 0.048) with liraglutide 1.2 mg compared with placebo whereas in L ‐ LEAD 4 Zinman 2009 and L ‐ LEAD 2 Nauck 2009 no significant difference was seen. Overall, there was no significant difference in minor hypoglycaemia between 1.2 mg liraglutide and placebo (risk ratio 1.54, 95% CI 0.54 to 4.42, P = 0.42), with no significant heterogeneity. Rates of hypoglycaemia were between 0.8% and 9.2% in the liraglutide groups and between 2.6% and 5.1% in the placebo groups.
There were no reports of major hypoglycaemic episodes in participants on either liraglutide 1.2 mg or placebo in any of the studies.
Of the four studies reporting hypoglycaemia with 1.8 mg liraglutide, L ‐ LEAD 1 Marre 2009 found that the incidence of minor hypoglycaemia was higher with liraglutide 1.8 mg compared with placebo (P = 0.0065). Similarly, L ‐ LEAD 4 Zinman 2009 found that the rate of minor hypoglycaemia was significantly higher with liraglutide 1.8 mg compared with placebo (P = 0.0004). In L ‐ LEAD 5 Russell‐J 2009, hypoglycaemia was categorised as major (third party assistance), minor (FPG less than 3.1 mmol/L) and symptoms only. The rate of hypoglycaemia reported was 0.06, 1.2 and 1.0 events/patient‐year (major, minor and symptoms only) in the 1.8 mg liraglutide group and 0, 1.0 and 0.5 events/patient‐year in the placebo group. The proportion of participants with hypoglycaemia was higher with 1.8 mg liraglutide compared to placebo (27.4% versus 16.7%). There was no significant difference in minor hypoglycaemia between 1.8 mg liraglutide and placebo in L ‐ LEAD 2 Nauck 2009. In the other trials, the rate of hypoglycaemia was about between 2.5% and 8% with 1.8 mg liraglutide. Overall, there was significantly more hypoglycaemia with 1.8 mg liraglutide, risk ratio 1.66 (95% CI 1.15 to 2.40, P = 0.007), with no significant heterogeneity.
In two of the trials, no cases of major hypoglycaemia were seen (L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009). In L ‐ LEAD 1 Marre 2009, one major hypoglycaemic episode was reported in a participant on liraglutide 1.8 mg and glimepiride; this was considered to be related to glimepiride and not the study drug and accordingly the dose of glimepiride was reduced. In L ‐ LEAD 5 Russell‐J 2009, five patients had major hypoglycaemic events in the 1.8 mg liraglutide group with only one requiring some medical assistance.
Weight change
In L ‐ Kaku 2010, there was no change in mean body weight with both 0.6 and 0.9 mg dose of liraglutide while a reduction of 1.12 kg in weight was seen with placebo. Two of the three trials showed significantly more weight loss with 1.2 mg liraglutide than with placebo (L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009), while one showed no significant difference (L ‐ LEAD 1 Marre 2009). No significant weight changes in either the 1.2 mg liraglutide group of the placebo group were seen in the study using only sulphonylurea as concomitant antihyperglycaemic therapy (+0.3 kg SD 3.02 with 1.2 mg liraglutide, ‐0.1 kg SD 2.88 with placebo)(L ‐ LEAD 1 Marre 2009). In the other two studies (L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009) the weight loss in the 1.2 mg liraglutide groups was between 1.1 and 1.6 kg greater than in the placebo groups (P < 0.00001 for the combined effect in the two studies). Weight loss in the 1.2 mg liraglutide groups was between 1.0 and 2.6 kg, weight change in the placebo groups ranged between ‐1.5 and +0.6 kg.
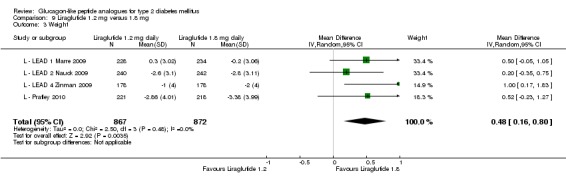
In three of the four studies, the weight reduction with 1.8 mg liraglutide was significantly greater than with placebo (L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009; L ‐ LEAD 5 Russell‐J 2009). There was no difference between the groups in L ‐ LEAD 1 Marre 2009. Overall, the mean difference for 1.8 mg liraglutide versus placebo was ‐1.33 (95% CI ‐2.38 to ‐0.27, P = 0.01) with significant heterogeneity (P < 0.0001) (probably due to different co‐interventions having different effects on weight). Weight loss in the 1.8 mg liraglutide groups was between ‐0.2 and ‐2.8 kg. Overall, weight loss with 1.8 mg liraglutide was 0.48 kg (95% CI 0.08 to 0.88) greater than with 1.2 mg liraglutide (P = 0.02), see Analysis 9.3.
Analysis 9.3.
Comparison 9 Liraglutide 1.2 mg versus 1.8 mg, Outcome 3 Weight.
Adverse events
In L ‐ Kaku 2010, the total numbers of adverse events were similar across all groups (76.1% in 0.6 mg group, 78.4% in 0.9 mg group and 75% in placebo). It was reported that the most common adverse events in the trial were nasopharyngitis, diarrhoea and constipation. The proportions of participants complaining of gastrointestinal adverse events in the first four weeks were higher in the liraglutide groups than the placebo group. The numbers of participants withdrawing from the study were similar across the groups (n = 3 in 0.6 mg group, n = 2 in 0.9 mg group and n = 2 in placebo group). Treatment‐related serious adverse events were seen in eight participants (3 in 0.6 mg group, 2 in 0.9 mg group and 3 in placebo group) but no deaths occurred in the trial.
In all four trials (L ‐ LEAD 1 Marre 2009; L ‐ LEAD 2 Nauck 2009; L ‐ LEAD 4 Zinman 2009; L ‐ LEAD 5 Russell‐J 2009), the most frequently reported adverse events with liraglutide were gastrointestinal events. Nausea occurred in between 10.5% and 29% of participants in the 1.2 mg liraglutide groups and in between 14% and 40% in the 1.8 mg liraglutide groups; vomiting in between 4.5% and 7% with 1.2 mg and between 5% and 17% with 1.8 mg liraglutide; and diarrhoea in around 8% with 1.2 mg and between 10% and 15% with 1.8 mg liraglutide. The corresponding rates in the placebo groups were between around 2% to 4% nausea, 1% to 3.5% vomiting, and 4% to 5.3% diarrhoea. Withdrawals due to adverse events were between 5% and 10% with 1.2 mg liraglutide, between 4% and 15% with 1.8 mg liraglutide and between 1% and 5% in the placebo groups.
Most of the withdrawals were due to gastrointestinal events and occurred during the first four to eight weeks of the studies. Serious adverse events occurred in around 4% of participants with 1.2 mg liraglutide, between 4% and 6% of participants on 1.8 mg liraglutide, and between 3% and 7% of participants on placebo. Only one trial reported a case of pancreatitis, in L ‐ LEAD 2 Nauck 2009, one of the participants on 1.2 mg liraglutide withdrew from the study because of acute pancreatitis. There were two deaths reported in the study considered unrelated to the study drug. There were no deaths in L ‐ LEAD 1 Marre 2009 or L ‐ LEAD 4 Zinman 2009.
None of the studies found any significant differences between treatment groups for physical examination findings, laboratory analyses, ECG, ophthalmology and other adverse events. Between 10% and 13% of participants were positive for anti‐liraglutide antibodies (no difference reported between 1.2 and 1.8 mg), however, this had no effect on the HbA1c response or on adverse events.
Blood pressure
The Japanese study (L ‐ Kaku 2010) reported that neither systolic and diastolic blood pressure changed in any group (no data given). Only one trial (L ‐ LEAD 4 Zinman 2009) reported significantly more reduction in systolic blood pressure with 1.2 or 1.8 mg liraglutide compared to placebo weighted mean difference to placebo between ‐4.5 and ‐5.6 mm Hg), whereas there was no significant difference between either 1.2 or 1.8 mg liraglutide and placebo in systolic blood pressure in L ‐ LEAD 2 Nauck 2009, and a significant difference between 1.8 mg liraglutide and placebo in L ‐ LEAD 5 Russell‐J 2009. Overall, there was no significant difference in systolic blood pressure for 1.2 mg liraglutide versus placebo (mean difference ‐3.26 mm Hg (95% CI ‐7.71 to 1.20, P = 0.15, significant heterogeneity, possibly due to different co‐interventions)), but the results for 1.8 mg liraglutide versus placebo were marginally significant (‐2.42 mm Hg, 95% CI ‐4.90 to 0.05, P = 0.05, no significant heterogeneity). In L ‐ LEAD 1 Marre 2009, there was a reduction in both systolic and diastolic blood pressure with 1.2 and 1.8 mg liraglutide and with placebo but the difference between the groups was not significant (not enough details given to include the data in the statistical summary). There was no reduction in diastolic blood pressure in any of the groups.
There was no significant difference in systolic blood pressure between 1.2 and 1.8 mg liraglutide, see Analysis 9.4.
Analysis 9.4.
Comparison 9 Liraglutide 1.2 mg versus 1.8 mg, Outcome 4 Systolic blood pressure.
Fasting plasma glucose
In L ‐ Kaku 2010, both 0.6 mg (‐2.3 mmol/L) and 0.9 mg (‐2.28 mmol/L) liraglutide significantly reduced fasting plasma glucose levels compared with placebo (‐0.64 mmol/L). Fasting plasma glucose was significantly reduced with liraglutide 1.2 mg or 1.8 mg compared to placebo. The overall mean difference was ‐2.13 (95% CI ‐2.59 to ‐1.68, P < 0.0001, no significant heterogeneity) for 1.2 mg liraglutide versus placebo, and ‐2.21 (95% CI ‐2.49 to ‐1.93, P < 0.00001, no significant heterogeneity) for 1.8 mg liraglutide versus placebo. Changes in fasting plasma glucose ranged between ‐1.6 and ‐2.2 mmol/L with 1.2 mg liraglutide, between ‐1.55 and ‐2.4 mmol/L with 1.8 mg liraglutide and between ‐0.4 and +1.01 mmol/L with placebo.
All studies reported that fasting plasma glucose values decreased within two weeks of commencing liraglutide and remained relatively stable thereafter.
Post‐prandial glucose
L ‐ Kaku 2010 found that both doses of liraglutide (0.6 mg and 0.9 mg) led to significant improvement in the self‐monitored 7‐point plasma glucose profiles compared to placebo (P < 0.0001). In L ‐ LEAD 1 Marre 2009, L ‐ LEAD 2 Nauck 2009, L ‐ LEAD 4 Zinman 2009 post‐prandial glucose values were significantly reduced with 1.2 mg liraglutide versus placebo (reductions by 2.3 to 2.6 mmol/L with liraglutide compared to reductions of 0.4 to 0.8 mmol/L with placebo). Similarly, reductions in post‐prandial glucose values with 1.8 mg liraglutide were significantly larger than with placebo (reductions by 1.8 to 2.7 mmol/L with liraglutide compared to reductions of 0.30 to 0.8 mmol/L with placebo). In L ‐ LEAD 4 Zinman 2009, the post‐prandial increment (post‐meal minus pre‐meal value) was significantly reduced over breakfast in both liraglutide (1.2 and 1.8 mg) groups, but not at the other meals. L ‐ LEAD 5 Russell‐J 2009 reported that significantly more participants on 1.8 mg liraglutide achieved the ADA target for post‐prandial glucose (≤ 10 mmol/L) participants on placebo.
Lipid profiles
In L ‐ Kaku 2010, it was reported that no significant changes occurred in any of the parameters of the lipid profile with any treatment. No data were reported.
Details of effects on lipid profiles were also reported by L ‐ LEAD 4 Zinman 2009. For both liraglutide groups, there was a decrease in free fatty acids (‐0.03 mmol/L SE 0.02 to ‐0.05 mmol/L SE 0.02), whereas levels were increased with placebo (+0.02 mmol/L SE 0.02, P < 0.05). There was significantly more reduction in triglycerides and LDL cholesterol with 1.2 mg liraglutide than with placebo (triglycerides: ‐0.38 mmol/L SE 0.10 versus ‐0.13 mmol/L SE 0.11 with placebo, P < 0.05; LDL cholesterol: ‐0.28 mmol/L SE 0.07 versus ‐0.10 mmol/L SE 0.07 with placebo, P < 0.05). No significant difference to placebo was seen in the 1.8 mg liraglutide group (reduction in triglycerides: ‐0.32 mmol/L SE 0.10; LDL cholesterol: ‐0.23 mmol/L SE 0.07).
Beta‐cell function
L ‐ LEAD 1 Marre 2009, L ‐ LEAD 2 Nauck 2009, L ‐ LEAD 4 Zinman 2009 all reported HOMA‐B and proinsulin‐to‐insulin ratio for 1.2 and 1.8 mg liraglutide versus placebo. All studies showed a significant improvement in HOMA‐B with 1.2 mg liraglutide compared to placebo (improvement between +23% and +28% compared to between ‐4% and +6% with placebo). In L ‐ LEAD 1 Marre 2009, HOMA‐B improvement with 1.8 mg liraglutide was only marginal compared to placebo (P = 0.051), but the difference was significant in the other two studies. The improvement in HOMA‐B all three studies was between +27% and +35%.
All three studies showed a significant improvement in proinsulin‐to‐insulin ratio both with 1.2 and 1.8 mg liraglutide compared to placebo. Changes were between ‐0.03 and ‐0.12 with 1.2 mg liraglutide, between ‐0.09 and ‐0.12 with 1.8 mg liraglutide, and between +0.02 and +0.1 with placebo. There was a significant improvement in proinsulin‐to‐C‐peptide ratio with 1.2 and 1.8 mg liraglutide in L ‐ LEAD 4 Zinman 2009, and with 1.8 mg liraglutide in L ‐ LEAD 5 Russell‐J 2009.
Liraglutide versus insulin (glargine)
Results for liraglutide 1.8 mg versus insulin glargine are shown in Data and analyses, 10.1 to 10.9. This comparison was only carried out by L ‐ LEAD 5 Russell‐J 2009.
HbA1c
HbA1c was significantly more reduced with 1.8 mg liraglutide than with insulin glargine (mean difference ‐0.24%, 95% CI ‐0.39 to ‐0.08, P = 0.0015 according to the original analysis). HbA1c was reduced by 1.33% with liraglutide and by 1.09% with insulin glargine.
Similarly, the proportion of participants achieving the target HbA1c level of less than 7% was higher with liraglutide compared with insulin glargine (53.1% versus 45.8%, P = 0.0139).
Hypoglycaemia
The proportion of participants with minor hypoglycaemia was similar with liraglutide and insulin glargine. Minor hypoglycaemia was seen in 27% of patients in the liraglutide group and 29% in the glargine group. Five patients had major hypoglycaemic events in the liraglutide group with only one requiring medical assistance. There were no reports of major hypoglycaemic episodes in the glargine group.
Weight change
There was significant weight reduction in the liraglutide group (‐1.8 kg SE 0.33) while weight increased the insulin glargine group (+1.6 kg SE 0.33). The mean treatment difference was ‐3.43 kg (95% CI ‐4.00 to ‐2.86, P < 0.0001, according to the original analysis).
Adverse events
The most frequently reported adverse events were nausea, vomiting and diarrhoea, which were more frequent with liraglutide than with insulin glargine (see above). The incidence of gastrointestinal events in the glargine group was similar to that in the placebo group (or even somewhat lower). No other adverse events were seen with glargine (for liraglutide results see above).
Blood pressure
There was a significant reduction of systolic blood pressure with liraglutide (‐4.0 mm Hg) whereas an increase was seen with insulin glargine (+0.54 mm Hg) (treatment difference ‐2.53 mm Hg (95% CI ‐6.82 to ‐2.20, P = 0.0001, according to the original analysis). There was no significant effect on diastolic blood pressure in any of the comparison groups.
Fasting plasma glucose
There was no significant difference in fasting plasma glucose between liraglutide and insulin glargine. Fasting plasma glucose was reduced by 1.55 mmol/L in the liraglutide group and by 1.79 mmol/L in the glargine group.
Post‐prandial glucose
There was no significant difference in post‐prandial glucose between liraglutide and insulin glargine. Post‐prandial glucose was reduced by 1.8 mmol/L in the liraglutide group and by 1.6 mmol/L in the glargine group.
Beta‐cell function
The proinsulin‐to‐insulin ratio was significantly improved with liraglutide compared to insulin glargine (treatment difference ‐0.00366, 95% CI ‐0.0057 to ‐0.00136, P = 0.0019).
Liraglutide versus thiazolidinedione (rosiglitazone)
Results for liraglutide 1.2 mg versus rosiglitazone are shown in Data and analyses, 11.1 to 11.11. Results for liraglutide 1.8 mg versus rosiglitazone are shown in Data and analyses, 12.1 to 12.11. Only one RCT compared liraglutide and rosiglitazone, L ‐ LEAD 1 Marre 2009 (1.2 and 1.8 mg liraglutide). The dose of rosiglitazone was 4 mg.
HbA1c
In L ‐ LEAD 1 Marre 2009, both liraglutide and rosiglitazone reduced HbA1c levels, but the reduction was significantly greater with liraglutide (P < 0.0001). HbA1c reduction was by 1.08% to 1.13% with liraglutide and by 0.44% with rosiglitazone.
HbA1c equal to or less than 7%
The proportion of participants achieving ADA HbA1c target levels was significantly greater with liraglutide compared with rosiglitazone (P ≤ 0.0003). Between 35% and 42% reached an HbA1c level of less than 7% with liraglutide, while only 22% on rosiglitazone did (L ‐ LEAD 1 Marre 2009).
Hypoglycaemia
The incidence of minor hypoglycaemia was significantly higher with liraglutide (8.1% to 9.2%, 0.51 events/patient‐year) than with rosiglitazone (4.3%, 0.12 events/patient‐year) (P = 0.048).There was one report of major hypoglycaemia in a participant on 1.8 mg liraglutide and glimepiride. This was considered to be related to glimepiride and its dose was reduced.
Weight change
There was a significant difference in weight change in favour of both liraglutide groups compared to rosiglitazone (P < 0.0001). Participants on liraglutide had weight changes of between ‐0.2 to +0.3 kg, while weight increased by 2.1 kg in the rosiglitazone group (L ‐ LEAD 1 Marre 2009).
Adverse events
The most frequently reported adverse event was gastrointestinal disorders that included nausea, vomiting and diarrhoea, which was more frequent with liraglutide than with rosiglitazone (see above). No other adverse events were seen with rosiglitazone (for liraglutide results see above) (L ‐ LEAD 1 Marre 2009).
Fasting plasma glucose
The participants on liraglutide had significantly greater reductions in fasting plasma glucose levels compared to those on rosiglitazone (‐1.6 mmol/L for liraglutide versus ‐0.88 mmol/L for rosiglitazone, P ≤ 0.006). Similarly, at the end of study the proportions of participants achieving ADA target fasting plasma glucose values of between 5.0 mmol/L and 7.2 mmol/L were greater with both doses of liraglutide compared with rosiglitazone (37% to 38% with liraglutide versus 26% with rosiglitazone, P ≤ 0.01).
Post‐prandial glucose
The reduction in mean post‐prandial glucose values was significantly greater with both doses of liraglutide compared to rosiglitazone (‐2.5 to ‐2.7 mmol/L with liraglutide versus ‐1.8 mmol/L with rosiglitazone, P < 0.05).
Beta‐cell function
The improvement in mean HOMA‐B was significantly greater with both doses of liraglutide compared with rosiglitazone (P < 0.05). Similarly, the reduction in the proinsulin‐to‐insulin ratio was also greater with liraglutide compared to rosiglitazone (P ≤ 0.02).
Liraglutide versus DPP‐4 inhibitors (sitagliptin)
Results for liraglutide 1.2 mg versus sitagliptin are shown in Data and analyses, 13.1 to 13.11. Results for liraglutide 1.8 mg versus sitagliptin are shown in Data and analyses, 14.1 to 14.11. The comparison between liraglutide and sitagliptin was carried out by L ‐ Pratley 2010. The dose of sitagliptin was 100 mg daily.
HbA1c
In L ‐ Pratley 2010, the reduction in HbA1c with 1.8 mg liraglutide was higher than with 1.2 mg liraglutide (‐1.5% versus ‐1.24%) or sitagliptin 100 mg (‐1.5% versus ‐0.9%). The treatment difference between liraglutide 1.8 mg versus sitagliptin was ‐0.6% (95% CI ‐0.78 to ‐0.42, P < 0.00001) while the difference between liraglutide 1.2 mg versus sitagliptin was ‐0.34% (95% CI ‐0.53 to ‐0.15, P = 0.0006).
HbA1c equal to or less than 7%
L ‐ Pratley 2010 found that the proportion of participants achieving this target was significantly higher with liraglutide than with sitagliptin. Fifty six percent of participants taking liraglutide 1.2 mg, 44% taking liraglutide 1.8 mg and 22% on sitagliptin achieved this target HbA1c level.
Hypoglycaemia
In L ‐ Pratley 2010, the proportion of participants experiencing minor hypoglycaemia was similar in all groups(i.e. 5%). It reported that one participant on 1.2 mg liraglutide had a major hypoglycaemic episode, but none on the 1.8 mg dose or on sitagliptin.
Weight change
The weight change with liraglutide was significantly greater compared to sitagliptin (P < 0.00001). The mean weight loss with liraglutide was between ‐2.86 and ‐3.38 kg, while only a reduction of 0.96 kg occurred with sitagliptin (L ‐ Pratley 2010).
Adverse events
In L ‐ Pratley 2010, the most frequently reported treatment‐emergent adverse events were gastrointestinal related symptoms, that were more frequent with liraglutide than with sitagliptin. Out of all gastrointestinal symptoms, nausea was the most common however, it was transient in nature.The incidence of infections was not different between liraglutide and sitagliptin. The occurrence of serious adverse events were similar between the groups and were thought not to be related to the study drug. Two deaths occurred (one with pancreatic carcinoma taking 1.8 mg liraglutide and one with fatal cardiac arrest taking sitagliptin), both regarded not to be related to the study drug. There were no cases of pancreatitis. No report on anti‐liraglutide antibodies.
Blood pressure
The effect on systolic blood pressure was not different amongst the groups. The treatment difference between liraglutide 1.2 mg and sitagliptin was 0.39 mm Hg (95% CI ‐2.08 to 2.86, P = 0.76) while the difference between liraglutide 1.8 mg and sitagliptin was 0.22 mm Hg (95% CI ‐2.25 to 2.69, P = 0.86). The effect on diastolic blood pressure was significantly greater with sitagliptin compared with liraglutide 1.8 mg (‐1.78 versus +0.07 mm Hg) but not compared with liraglutide 1.2 mg (‐1.78 versus ‐0.71 mm Hg).
Fasting plasma glucose
Reduction in fasting plasma glucose was significantly greater with liraglutide than with sitagliptin. The mean treatment difference between liraglutide 1.2 mg and sitagliptin was ‐1.04 mmol/L (95% CI ‐1.46 to ‐0.62, P < 0.00001) and the difference between liraglutide 1.8 mg versus sitagliptin was ‐1.31 mmol/L (95% CI ‐1.73 to ‐0.89, P < 0.00001).
Postprandial glucose
It was reported that post‐prandial glucose recorded during the study was highly variable, suggesting that most glucose values were not post‐prandial. The authors also added that meal patterns i.e. content and time of day were different across different countries.
Beta cell function
In L ‐ Pratley 2010, both doses of liraglutide led to significant improvements in HOMA of β‐cell function compared with sitagliptin. Similarly liraglutide also led to significant improvements in C‐peptide concentration, and proinsulin‐to‐insulin ratio compared to sitagliptin. The improvement in HOMA index for insulin concentration was not significantly different between the group. Liraglutide 1.8 mg and sitagliptin led to improvement in fasting insulin level but the difference between the two groups was not significant.
Liraglutide versus sulphonylurea (glimepiride)
Results for liraglutide 1.2 and 1.8 mg versus sulphonylurea are shown in Data and analyses, 16.1 to 16.13 and 17.1 to 17.13 respectively. This comparison was carried out by L ‐ LEAD 2 Nauck 2009 (versus 1.2 and 1.8 mg liraglutide) and L ‐ Yang 2010 (versus 1.2 and 1.8 mg liraglutide). The dose of glimepiride was 4 mg daily.
HbA1c
There was no significant difference between 1.2 or 1.8 mg liraglutide and glimepiride. Reductions in HbA1c level with liraglutide ranged between ‐0.97% and ‐1.45% and between ‐0.98% and ‐1.39% with glimepiride.
There was also no significant difference between liraglutide and glimepiride in the proportion of participants reaching an HbA1c of 7% or less (range 35% to 45% with liraglutide and 36% to 44% with glimepiride).
Hypoglycaemia
The incidence of hypoglycaemia was significantly lower with liraglutide compared with glimepiride (0% to 2% with liraglutide versus 17% to 19% with glimepiride. The mean treatment difference between liraglutide 1.2 mg and glimepiride was 0.06 (95% CI 0.00 to 1.72, P = 0.10 with significant heterogeneity, I2 statistic = 82%). This may have been because none of the participants in the study by L ‐ Yang 2010 taking liraglutide 1.2 mg had minor hypoglycaemia. The mean treatment difference between liraglutide 1.8 mg and glimepiride was 0.13, 95% CI 0.07 to 0.25, P < 0.00001. No cases of major hypoglycaemia were reported with either treatment.
Weight change
Participants of the liraglutide groups lost between 2.3 and 2.8 kg, while participants in the glimepiride group gained between 0.08 and 1 kg. It was reported that nausea was not responsible for weight loss in these participants.
Adverse events
The most frequently reported adverse event was gastrointestinal disorders that included nausea, vomiting and diarrhoea, which was more frequent with liraglutide than with rosiglitazone (40% to 44% with liraglutide and 17% with glimepiride reported in L ‐ LEAD 2 Nauck 2009 whereas no data given by L ‐ Yang 2010). It was also reported that the gastrointestinal adverse events with liraglutide was transient and occurred more frequently in the first four weeks and the incidence decreased overtime. The occurrence of serious adverse events were similar between the groups and they were considered not related to the treatment drugs. Withdrawals due to adverse events ranged between 9.4% and 12.9% with liraglutide and 1.3% to 3% with glimepiride.
In L ‐ Yang 2010, anti‐liraglutide antibodies did not have any effect on safety or HbA1c response.
Blood pressure
There was a significant difference in favour of both 1.2 and 1.8 mg liraglutide compared to glimepiride in terms of change in systolic blood pressure. In L ‐ LEAD 2 Nauck 2009, systolic blood pressure was reduced by between 2.3 and 2.8 mm Hg in the liraglutide groups and increased by 0.4 mm Hg in the glimepiride group (P = 0.01). Similarly, in L ‐ Yang 2010, the systolic blood pressure decreased by more than 3 mm Hg in the liraglutide groups while only decreased by 0.91 mm Hg in the glimepiride group. In L ‐ LEAD 2 Nauck 2009, diastolic blood pressure did not change from baseline in any of the intervention groups while in L ‐ Yang 2010 it slightly decreased in all treatment groups
Fasting plasma glucose
There was no significant difference in decrease in fasting plasma glucose from baseline between liraglutide and glimepiride groups (‐1.6 to ‐2.12 mmol/L with liraglutide and ‐1.3 to ‐2.18 mmol/L with glimepiride).
Post‐prandial glucose
There was a reduction in post‐prandial glucose level with all treatment groups (‐2.3 to ‐3.51 mmol/L with liraglutide, ‐2.5 to ‐2.6 mmol/L with glimepiride) but the difference was significant only between liraglutide 1.8 mg and glimepiride (‐3.51 versus ‐2.6 mmol/L, P < 0.0001).
Beta‐cell function
L ‐ LEAD 2 Nauck 2009 reported that there was a significant improvement in mean HOMA‐B value with all treatment groups and the difference was not significant between the treatment groups (+ 23% to + 28% with liraglutide, + 25% with glimepiride). Similar reductions in the proinsulin‐to‐insulin ratio were observed with liraglutide or glimepiride.
L ‐ Yang 2010 also reported improvements in mean HOMA‐B value with all treatment groups but the difference between the groups was not significant. Similarly all treatment groups led to reduction in the proinsulin‐to‐insulin ratio but the difference between the groups was not significant.
Lixisenatide
Lixisenatide versus placebo
Results for lixisenatide versus placebo are shown in Data and analyses, 15.1 to 15.8. The comparison was carried out by only one study (Lixi ‐ Ratner 2010).
HbA1c
A dose‐dependent reduction in HbA1c level was observed with both once daily and twice daily regimen. Reductions in HbA1c level ranged between 0.47% and 0.87% with lixisenatide compared to reduction of 0.18% with placebo.
HbA1c equal to or less than 7%
The proportion of participants achieving the target HbA1c level of 7% or less were significantly (P < 0.05) higher with both once daily (47% to 69%) and twice daily (51% to 77%) lixisenatide compared with placebo (32%).
Hypoglycaemia
There was no dose‐dependent relationship with symptomatic hypoglycaemic episodes. The occurrence of hypoglycaemic episode ranged between 1 and 3 events per group. There were no cases of severe hypoglycaemia.
Weight change
There was a dose dependent reduction in weight with both once daily and twice daily regimen of lixisenatide. Reduction in weight with the once daily lixisenatide ranged between ‐1.94 and ‐3.47 kg while the reduction with twice daily lixisenatide ranged between ‐2.10 and ‐3.89 kg. Participants in the placebo group lost 1.94 kg at end of the study.
Adverse events
Withdrawals due to treatment‐related adverse events ranged from 1.8% to 11.1% in the once daily lixisenatide group and from 0% to 14.8% in the twice daily group. Only 1.8% of participants taking placebo withdrew from the study. The incidence of adverse events was dose‐dependent. Most frequently reported adverse events were gastrointestinal, mainly nausea. It was mild to moderate in intensity and occurred in most cases during the first five weeks of the study. None of the participants had pancreatitis. Eight participants in the lixisenatide group and three in the placebo group experience serious adverse events. One participant taking lixisenatide 30 µg once daily experienced few seconds of loss of consciousness thus withdrew from the study. Another patient taking lixisenatide 10 µg once daily discontinued from the study after experiencing 30 minutes of pruritis all over the body 10 min after the injection during 3rd week of treatment and a second episode of swollen lips/tongue and difficulty in breathing within 10 min of injection. The participant improved after taking oral antihistamine. There were two more cases of urticaria with lixisenatide and three with placebo. The changes in laboratory tests and ECG were not clinically significant (data not given). At the end of study, the proportions of participants with anti‐lixisenatide antibody ranged from 43.1% in the 10 µg once daily group to 71.2% in the 20 µg twice daily group.
Blood pressure
Changes in systolic blood pressure ranged from ‐2 to ‐9 mm Hg in the lixisenatide group while it fell by ‐3 mm Hg in the placebo group. The reduction in diastolic blood pressure with lixisenatide group ranged from ‐2 to ‐4 mm Hg while it reduced by ‐2 mm Hg in the placebo group. In most cases, changes in blood pressure were seen as early as week one.
Fasting plasma glucose
A dose dependent reduction in fasting plasma glucose was observed with both once daily and twice daily regimen of lixisenatide. The reduction with the once daily regimen ranged from ‐0.62 to ‐1.02 mmol/L and ranged from ‐0.19 to ‐1.42 mmol/L with the twice daily regimen. Placebo led to reduction of 0.21 mmol/L.
Post‐prandial glucose
Similarly a dose‐dependent reductions in daily averaged seven‐point self monitored blood glucose and 2 hour post‐prandial plasma glucose concentration occurred with both once daily and twice daily lixisenatide.
LY2189265
LY2189265 versus placebo
Results for LY2189265 versus placebo are shown in Data and analyses, 19.1 to 19.9. The comparison between LY2189265 versus placebo was reported by LY2189265 ‐Umpierrez 2011.
HbA1c
The reduction in HbA1c level with LY2189265 was significantly higher compared to the reduction with placebo at all time points. HbA1c decreased by 1.38% in the 0.5/1.0 group, by 1.32% in the 1.0/1.0 group and by 1.59% in the 1.0/2.0 group (no data given for placebo). There was no significant difference between the LY groups.
HbA1c equal to or less than 7%
The proportions of participants achieving the target HbA1c level across all treatment groups ranged from 49% to 54%.
Hypoglycaemia
The incidence of hypoglycaemia was significantly higher in all the LY groups compared to the placebo group. The numbers of participants experiencing hypoglycaemic episodes with all LY groups were significantly (P < 0.05) higher at 8 weeks compared to placebo but the incidence decreased over time. The difference at end of the study between all LY groups and placebo was not significant (P ≥ 0.17). There were no cases of severe hypoglycaemia.
Weight change
There was a significant weight reduction in all the LY groups compared to placebo group. Reduction in weight with LY ranged from ‐1.44 to ‐2.55 kg with the highest reduction occurring in the LY 1.0/2.0 group. With placebo, weight decreased only by 0.12 kg. Weight reduction was independent of nausea.
Adverse events
The most commonly reported treatment‐related adverse events were gastrointestinal and these events occurred more frequently in the highest LY dose. The occurrence of nausea, diarrhoea and abdominal distension was higher than other adverse events. The proportions of participants experiencing adverse events possibly related to the study drug were 30.8% to 41.5% for the LY groups and 22.7% for placebo. Participants discontinuing the study drug because of adverse events was comparatively greater in the LY groups compared to placebo (6.1% to 6.2% versus 1.5%).
Seven participants experienced serious adverse events (1 in placebo group, 3 in LY 0.5/1.0 group, 2 in LY 1.0/1.0 and 1 in LY 1.0/2.0) and the investigators considered three (hallucination, cryptogenic organising pneumonia and pancreatitis) to be related to the study drug. Two participants had pancreatitis, both in the LY 0.5/1.0 group. First case of pancreatitis was reported at week 16 and the second after week 11. There were no deaths during the study.
Blood pressure
There was reduction in both systolic (‐3.5 mm Hg) and diastolic (‐2.3 mm Hg) blood pressure with placebo. All LY groups had dose‐dependent reductions in systolic blood pressure, ranging from ‐0.6 to ‐3.0 mm Hg, There was no reduction in diastolic blood pressure in any LY group.
Fasting plasma glucose
It was observed that the reduction in fasting plasma glucose level was significantly greater in the LY groups than in the placebo group. Reduction with LY ranged from ‐2.05 to ‐2.65 mmol/L while it fell by 0.49 mmol/L in the placebo group.
Beta‐cell function
There was a significant improvement in HOMA2‐%B in all the LY groups compared to placebo group. The improvement ranged from 32.9% to 45.6% in the LY groups while it increased by 1% in the placebo group. There was no significant change in any LY group for HOMA2‐%S or HOMA2‐%IR.
Taspoglutide
Taspoglutide versus placebo
Results for taspoglutide are shown in Data and analyses, 18.1 to 18.16. The comparison of taspoglutide against placebo was carried out by two trials T ‐ Nauck 2009 and T‐ Ratner 2010.
HbA1c
T ‐ Nauck 2009 found a significant reduction in the level of HbA1c in participants on taspoglutide compared to placebo (P < 0.0001). The reduction was similar in all taspoglutide groups (10 mg weekly ‐1.2%; 20 mg weekly ‐1.2% and 20 mg every two weeks ‐1.0%). The reduction in HbA1c was comparatively larger in participants with higher baseline HbA1c levels (equal to or greater than 8.0%). Similarly T‐ Ratner 2010 also found a significant reduction in HbA1c level taking taspoglutide compared to placebo. The titration of the taspoglutide dose to higher dose did not lead to increased reduction in HbA1c levels (‐1.2% in 20/20 once weekly group; ‐0.9% in 20/30 once weekly group and ‐1.2% in 20/40 once weekly group).
HbA1c equal to or less than 7%
T ‐ Nauck 2009 found a significant difference in the proportion of participants achieving target HbA1c level of less than 7% between intervention groups, with 79%, 81%, and 63% of participants achieving this target with 10 mg weekly, 20 mg weekly and 20 mg every two weeks taspoglutide, respectively, and 17% with placebo (P < 0.0001 versus placebo). Similarly, the proportion of participants achieving the target HbA1c level was higher with taspoglutide than with placebo (72% in the 20/20 mg once weekly group, 53% in the 20/30 mg once weekly group, 70% in the 20/40 mg once weekly group and 19% in the placebo group).
Hypoglycaemia
In T ‐ Nauck 2009, no definition of hypoglycaemia was given. Also no data were reported for the separate comparison groups however it was reported that 6 patients had 7 hypoglycaemic events, 2 of which were asymptomatic. In addition, it was also reported that there were no cases of severe hypoglycaemia in the taspoglutide group.
No definition of hypoglycaemia was given in T‐ Ratner 2010. Incidence of hypoglycaemia was comparatively similarly between all the treatment groups. There were no cases of severe hypoglycaemia during the study.
Weight change
The weight reduction was comparatively greater in the taspoglutide groups compared with the placebo group. The reduction was significant in the 10 mg once weekly taspoglutide group (‐2.1 kg, P = 0.02 versus placebo) and the 20 mg every two weekly taspoglutide group (‐1.9 kg, P = 0.01 versus placebo) compared to placebo group (‐0.8 kg). However there was no significant difference between the 20 mg once weekly taspoglutide group compared against placebo group. The mean difference was ‐1.07 kg (95% CI ‐2.93 to 0.79, P = 0.26) with significant heterogeneity (I2 statistic = 88%). The titration of 20 mg weekly dose to 30 mg once weekly led to significant weight reduction compared to placebo (‐3 versus ‐2 kg, P = 0.03) but not when the dose was titrated to 40 mg weekly (‐2.7 versus ‐2 kg, P = 0.17) (T‐ Ratner 2010).
Adverse events
In T ‐ Nauck 2009, the most commonly reported adverse events were gastrointestinal (nausea, vomiting and diarrhoea) which occurred more frequently in the taspoglutide groups than with placebo. In the taspoglutide groups, nausea occurred in 24% of patients on 10 mg weekly, in 52% on 20 mg weekly, and in 41% on 20 mg every two weeks (versus in 6% on placebo). Vomiting occurred in 4% of patients on 10 mg taspoglutide weekly, in 22% on 20 mg weekly, and in 24% on 20 mg every two weeks (versus in 4% on placebo). Diarrhoea occurred in 10% of patients on 10 mg taspoglutide weekly, in 10% on 20 mg weekly, and in 18% on 20 mg every two weeks (versus in 8% on placebo). Nausea was commoner at the beginning of treatment and decreased over the course of the study. Similarly, in T‐ Ratner 2010, the most frequently reported adverse events were gastrointestinal events, mainly nausea, diarrhoea and dyspepsia. The occurrence of these events was higher in the participants taking taspoglutide than those on placebo. The overall incidence did not change with titration to higher dose. It was also reported that nausea was mild to moderate in intensity and occurred more frequently in the first few weeks. The occurrence of nausea and vomiting reduced over time, with the greatest reduction seen in participants who remained on the 20 mg taspoglutide throughout the study. Injection site reactions were higher with taspoglutide than with placebo.
In T ‐ Nauck 2009, six patients experienced serious adverse events and that led to discontinuation in two of them in the placebo group however, the investigators considered this not to be related to the study drug. There were reports of mild to moderate injection site reactions which did not lead to any discontinuation. There were no significant differences in headache and no clinically relevant abnormalities in ECG, vital signs, and laboratory parameters.
In T‐ Ratner 2010, two serious adverse events were reported however both of them were considered not to be related to the study drug.
Fasting plasma glucose
In T ‐ Nauck 2009, fasting plasma glucose was significantly reduced in all considered taspoglutide groups compared to placebo (P = 0.02 to P < 0.0001). The reduction in the 10 mg weekly and the 20 mg once weekly groups was ‐2.5 mmol/L, ‐1.4 mmol/L in the 20 mg every two weeks group, and ‐0.78 mmol/L in the placebo group. Fasting plasma glucose fluctuated more in once every two week regimens than in the weekly regimens.
Fasting plasma glucose significantly reduced in all the taspoglutide groups compared to placebo. The reduction in the 20/20 mg, 20/30 mg and 20/40 taspoglutide group was ‐2.3 mmol/L, ‐1.6 mmol/L and ‐2.2 mmol/L respectively while the reduction in the placebo group was ‐2.2 mmol/L.
Post‐prandial glucose
The mean percent decrease from baseline in plasma glucose 120 min after a mixed meal was larger in the once weekly taspoglutide groups (‐22% with 10 mg weekly, ‐18% with 20 mg weekly) compared to 20 mg once every two weeks (‐5.5%) and placebo (‐10.5%). Similarly, the percentage change from baseline in glucose AUC (area under the curve over 240 min) was comparatively larger in the once weekly taspoglutide groups (‐27.5% with 10 mg weekly and ‐22.2% with 20 mg weekly) compared to 20 mg once every two weeks (‐9.2%) and placebo (‐7.2%).
The median percentage change in plasma insulin levels at 120 min was +44.4% with 20 mg once weekly taspoglutide group, +28.5% with 10 mg once weekly, ‐13% with 20 mg every two weeks and ‐15.3% with placebo. This outcome was not reported in T‐ Ratner 2010.
Lipid profiles
There was a decline in triglyceride levels with taspoglutide that appeared to be dose‐related and there was also some decrease in total cholesterol levels. None of the other lipid parameters showed any consistent changes. Lipid profile was not reported in T‐ Ratner 2010.
Beta‐cell function
There were statistically significant decreases in the fasting proinsulin‐to‐insulin molar ratio in the once weekly taspoglutide groups (‐0.12 with 10 mg once weekly, ‐0.17 with 20 mg once weekly, P < 0.01) compared to placebo. Changes with 20 mg taspoglutide every two weeks (‐0.055) and placebo (+0.002) were non‐significant. This outcome was not reported by T‐ Ratner 2010.
GLP‐1 agonist versus GLP‐1 agonist
Results for different GLP‐1 agonists or different GLP‐1 formulations compared with each other are shown in Data and analyses, 20.1 to 20.16. L ‐ LEAD 6 Buse 2009 compared 1.8 mg once daily liraglutide with 10 µg exenatide twice daily, E ‐ Blevins 2011 and E ‐ Drucker 2008 compared 10 µg exenatide BID with 2 mg exenatide once weekly.
HbA1c
In the direct comparison of exenatide with liraglutide (L ‐ LEAD 6 Buse 2009), HbA1c was significantly more reduced with liraglutide (‐1.22% versus ‐0.79%, mean difference 0.33 (95% CI 0.11 to 0.55, P < 0.0001). Similarly, the proportion of participants that reached the target HbA1c level of 7% or less was significantly higher with liraglutide (54%) than with exenatide (43%).
When comparing 10 µg exenatide twice daily with 2 mg exenatide once weekly(E ‐ Blevins 2011, E ‐ Drucker 2008), HbA1c was significantly more reduced with the weekly regimen. The mean treatment difference was 0.55% (95% CI 0.26 to 0.84, P = 0.0002) with slight heterogeneity of 55%. Similarly, the proportion of participants achieving a target HbA1c level of 7% or less was higher with once weekly exenatide. The mean difference was 0.65 (95% CI 0.42 to 1.01, P = 0.06) with significant heterogeneity (I2 statistic = 84%).
Hypoglycaemia
The proportion of participants who had minor hypoglycaemia was significantly higher with the exenatide group than with liraglutide (34% versus 26%, P = 0.0131). Participants on metformin as background therapy had fewer episodes of minor hypoglycaemia in both the liraglutide (6%) and the exenatide (11%) group compared to the participants taking sulphonylurea with or without metformin (33% for liraglutide versus 42% for exenatide). Two participants in the exenatide group receiving sulphonylurea as concomitant medication reported major hypoglycaemic episodes but no major episodes were reported in the liraglutide group.
There was no significant difference in hypoglycaemia between participants on exenatide twice daily or exenatide once a week. It was reported by E ‐ Drucker 2008 that participants on sulphonylurea background therapy had more episodes of minor hypoglycaemia compared to the participants without sulphonylurea background therapy (with SU: 14.5% with exenatide once weekly, 15.4% with exenatide twice a day; without SU: 0% with exenatide once weekly, 1.1% with exenatide twice a day). Similarly, E ‐ Blevins 2011 found that the participants taking concomitant sulphonylureas therapy experienced hypoglycaemia (four in the twice daily group while five in the once weekly group). No major hypoglycaemic episodes were reported in both studies.
Weight change
There was no significant difference in weight loss between liraglutide and exenatide. Weight loss with exenatide was ‐2.87 kg (SE 0.33) and weight loss with liraglutide was ‐3.24 kg (SE 0.33). The proportion of participants who lost weight was similar between the groups (78% with liraglutide versus 76% with exenatide).
Similarly, there was no significant difference in weight loss between 10 µg exenatide BID and 2 mg exenatide QW. Weight loss ranged from ‐1.4 to ‐3.7 kg in the once weekly group and ‐2.3 to ‐3.6 kg in the twice daily group. In E ‐ Drucker 2008, more than 75% of the participants lost weight in both groups (76% with exenatide once weekly versus 79% with exenatide twice daily). Weight decreased in participants who reported no episodes of nausea throughout the study (70%) .In E ‐ Blevins 2011, 77% of participants in the exenatide once weekly group and 63% in the twice daily group lost weight.
Treatment satisfaction / Quality of life
Overall satisfaction was significantly higher with liraglutide than with exenatide (Diabetes Treatment satisfaction Questionnaire, 15.18 SE 0.58 with liraglutide, 13.30 SE 0.58 with exenatide, P = 0.0004).
There was no significant difference in treatment satisfaction between exenatide once a week compared with exenatide twice daily (Diabetes Treatment Satisfaction Questionnaire); there was also no significant difference in weight‐related quality of life (IWQOL‐Lite).
Adverse events
The overall rate of adverse events was slightly higher with exenatide than with liraglutide (79% versus 75%), but there were more serious and severe adverse events with liraglutide (serious: 5.1% versus 2.6%; severe: 7.2% versus 4.7%), only one of these (severe hypoglycaemia with exenatide) was considered treatment‐related. There were slightly more withdrawals due to adverse events with exenatide than with liraglutide (13% versus 10%). The most commonly reported adverse event was nausea followed by diarrhoea and vomiting. The rate of gastrointestinal events was similar between exenatide and liraglutide groups. However, nausea was reported to be less persistent with liraglutide. There were no reports of acute pancreatitis in either group. One episode of mild chronic pancreatitis was reported in the liraglutide group but the investigators confirmed it not to be related to the study drug.
When comparing exenatide 10 µg twice daily with exenatide 2 mg once weekly, there was slightly more nausea (34.5% to 35% versus 14% to 26.4%) and vomiting (8.9% to 18.6% versus 4.7% to 10.8%) with the twice daily regimen. Nausea was predominantly mild in intensity and there were no cases of severe nausea with exenatide once a week. Participants with nausea lost somewhat more weight, but weight also decreased in participants with no nausea. Injection site pruritus occurred in 5.4% to 17.6% of the once weekly group and 1.4% to 2.4% of the twice daily group, however injection site bruising was somewhat more frequent with twice daily exenatide compared with once weekly exenatide (10.3% versus 4.7%). The proportion of participants with serious adverse events was low in both groups (2% to 5.4% for once weekly and 3.4% to 4% for twice daily) and none were considered to be related to the study drug. Withdrawal because of adverse events was similar between the groups (5% to 6.1% for once weekly versus 5% to 5.4% for twice daily exenatide).
Blood pressure
Systolic and diastolic blood pressure decreased with both exenatide and liraglutide but there was no significant difference between the treatment groups. There was a reduction of 2 mm Hg SD 17.93 in systolic blood pressure with exenatide and of 2.51 mm Hg SD 17.55 with liraglutide.
Similarly, a reduction in systolic and diastolic blood pressure with both exenatide 10 µg BID and exenatide 2 mg QW was not significantly different between the groups. The reduction in systolic blood pressure ranged from 2.9 to 4.7 mm Hg with exenatide once weekly and 1.2 to 3.4 mm Hg with exenatide twice daily.
Fasting plasma glucose
Fasting plasma glucose was significantly more reduced with liraglutide than with exenatide (‐1.61 mmol/L versus ‐0.60 mmol/L), with a mean difference between the groups of 1.01 mmol/L (95% CI 0.46 to 1.56, P < 0.0001).
Fasting plasma glucose was significantly was also significantly lower with 2 mg exenatide once weekly than with 10 µg exenatide BID (‐1.9 to ‐2.3 mmol/L versus ‐0.7 to ‐1.4 mmol/L), with a mean difference between the groups of 1.18 mmol/L (95% CI 1.02 to 1.33, P < 0.00001).
Post‐prandial glucose
Exenatide reduced the post‐prandial plasma glucose increment (as obtained from self‐monitored 7‐point plasma glucose measurements) more than did liraglutide after breakfast and dinner (treatment difference breakfast: 1.33 mmol/L, 95% CI 0.80 to 1.86, P < 0.0001; dinner: 1.01 mmol/L, 95% CI 0.44 to 1.57, P = 0.0005); treatment difference after lunch was not significant.
Both exenatide once a week and exenatide twice daily led to significant improvements in 7‐point self‐monitored blood glucose profiles. A meal tolerance test was carried out in 51 participants and 2 h post‐prandial values were significantly more reduced with exenatide twice a day than with exenatide once weekly (‐6.9 mmol/L SE 0.5 versus ‐5.3 mmol/L, P = 0.0124).
Lipid profiles
There were significantly greater reductions in triglycerides and free fatty acids with liraglutide than with exenatide (triglycerides: ‐0.41 mmol/L SE 0.1 versus ‐0.23 mmol/L SE 0.1, P = 0.0485; free fatty acids: ‐0.17 mmol/L SE 0.02 versus ‐0.10 mmol/L SE 0.02, P = 0.0014), and increases in VLDL cholesterol were significantly smaller with liraglutide (+0.20 mmol/L SE 0.04 versus +0.27 mmol/L SE 0.04, P = 0.0277). There were no significant differences in any other lipid parameters.
When comparing exenatide twice a day and exenatide once weekly, there were significantly greater reductions from baseline in total and LDL‐cholesterol with the once a week regimen (The mean difference between the two groups for total cholesterol was 0.31 mmol/L (95% CI 0.10 to 0.51, P = 0.003) with heterogeneity of 61% and the mean difference for LDL cholesterol was 0.20 mmol/L (95% CI 0.09 to 0.30) with no heterogeneity. There were no significant differences in any other lipid parameters.
Beta‐cell function
HOMA‐B improved significantly more with liraglutide than with exenatide (+32.1% versus +2.7% (both different from exenatide studies), P < 0.0001). There were no significant changes in proinsulin‐insulin ratio with either treatment and no difference between the groups in this parameter.
Beta‐cell function was not reported for exenatide twice daily versus once weekly.
Discussion
Summary of main results
For overview of results please see Appendix 2. Seventeen randomised controlled trials of mostly low risk of bias including relevant analyses for 6899 participants were included in the analysis. Of these, one compared albiglutide with placebo, two compared exenatide twice daily against once weekly exenatide, one compared once weekly exenatide against insulin glargine, one compared exenatide once weekly against sitagliptin and pioglitazone, five compared liraglutide with placebo, two compared liraglutide with glimepiride (sulphonylurea), and one each compared exenatide with liraglutide, liraglutide with sitagliptin, liraglutide with rosiglitazone and liraglutide with insulin glargine, two compared taspoglutide with placebo and one each compared lixisenatide with placebo and LY2189265 with placebo. In comparison with placebo, all glucagon‐like peptide‐1 (GLP‐1) agonists significantly reduced glycosylated haemoglobin A1c (HbA1c) (treatment difference between 0.47% and 1.56%) and increased the proportion of participants reaching an HbA1c of 7% or less. Results for mild hypoglycaemia were variable, with more patients on exenatide and concomitant sulphonylurea and more participants on 1.8 mg liraglutide experiencing mild hypoglycaemia than patients on placebo. There were no differences in severe hypoglycaemia. Patients using GLP‐1 agonists lost significantly more weight than patients in the placebo groups. GLP‐1 agonists caused gastrointestinal adverse effects, mainly nausea, as well as vomiting and diarrhoea; however, studies generally reported that these effects were strongest at the beginning and then subsided, and that weight loss also occurred in patients not experiencing nausea. Fasting blood glucose was reduced significantly more with GLP‐1 agonists. Where reported, GLP‐1 agonists reduced post‐prandial glucose and glucose variability. No significant difference in blood pressure was seen in comparison with placebo and some improvement in lipid parameters was seen, but this outcome was reported infrequently and results were inconsistent. Beta‐cell function was improved with GLP‐1 agonists (HOMA‐B, proinsulin‐to‐insulin ratio).
Only one study assessed albiglutide but overall, the effects of this agent on HbA1c and weight appeared to be slightly less than that of the other agents. Lixisenatide was found to be superior against placebo in terms of HbA1c, weight, weight, blood pressure and FPG. One study that assessed efficacy of LY2189265 and found that it was superior to placebo with respect to HbA1c, weight, systolic blood pressure (SBP), fasting plasma glucose (FPG) and beta‐cell function. Taspoglutide was found to be superior than placebo in terms of HbA1c, weight (with some doses), FPG, post‐prandial glucose (PPG), beta‐cell function and some improvement in triglyceride levels.
Both once weekly exenatide and liraglutide were superior to insulin glargine on most outcomes. There was slightly greater reduction in HbA1c level with exenatide than with glargine (‐1.5% versus ‐1.3%) while there was a small but statistically significantly larger reduction in HbA1c with liraglutide (0.24%). The incidence of hypoglycaemia was less with once weekly exenatide than with insulin glargine (18% less). No significant difference was seen in hypoglycaemia between liraglutide and exenatide. Long acting exenatide was superior to insulin glargine in terms of the proportion of participants achieving HbA1c level < 7%, weight and health‐related quality of life. It was also superior to sitagliptin and pioglitazone in terms of HbA1c, weight, FPG, health‐related quality of life, PPG and improvement in some parameters of lipid profile. Exenatide 2 mg weekly was superior to exenatide 10 µg twice daily with respect to HbA1c, proportion of participants reaching the target HbA1c level of 7% or less, fasting plasma glucose, frequency of gastrointestinal adverse events and improvement of some lipid parameters; however, post‐prandial glucose was significantly more reduced with the twice daily regimen.
Liraglutide was superior to glimepiride in terms of hypoglycaemia, weight reduction and systolic blood pressure. Liraglutide was superior to rosiglitazone 4 mg with respect to HbA1c, weight reduction, fasting and post‐prandial plasma glucose values and beta‐cell function; there was slightly more minor hypoglycaemia with liraglutide (4%). Liraglutide was superior to sitagliptin in terms of HbA1c, weight, FPG and beta‐cell function. In a head‐to‐head comparison of exenatide and liraglutide, liraglutide was superior to exenatide with respect to HbA1c, hypoglycaemia, patient satisfaction, less persistent nausea, systolic blood pressure, improvement of some lipid parameters, and improvement in beta‐cell function. None of the studies was long enough to assess long‐term positive or negative effects.
Overall completeness and applicability of evidence
Most of the studies evaluated exenatide or liraglutide. So far, few studies on other GLP‐1 agonists have been published, although a number are in progress (see Characteristics of ongoing studies). There were only three head‐to‐head comparisons of different GLP‐1 agonists (one study compared liraglutide versus exenatide twice daily and other two compared exenatide twice daily vs exenatide once weekly), so no firm conclusions could be made on the relative effectiveness of the different agents. Once weekly exenatide was better than twice daily exenatide with respect to most outcomes especially HbA1c, FPG and frequency of gastrointestinal adverse events. The improvement in HbA1c with once weekly exenatide was 0.4%. In a head to head comparison of twice daily exenatide and liraglutide, liraglutide was superior to exenatide. This trial was sponsored by the manufacturer of liraglutide however, the findings appear plausible given the pharmacodynamics of the two drugs, with liraglutide having a more prolonged action with less 'peak and trough effect'. These findings suggest that the once weekly exenatide may be better than liraglutide. Hence, in future GLP‐1 analogues may be given once weekly or once every two weeks.
In our meta‐analysis, we found that there was no significant difference between the 1.2 mg and the 1.8 mg liraglutide in terms of HbA1c and systolic blood pressure. However, weight reduction with the 1.8 mg liraglutide was slightly greater than the 1.2 mg liraglutide.
Only one exenatide trial and one liraglutide trial carried out comparisons with insulin glargine, the most commonly used basal insulin in the UK. In addition, the liraglutide trial used the 1.8 mg dose. Therefore there is a lack of trials comparing GLP‐1 agonists against insulin glargine. There are also no trials comparing GLP‐1 agonists against NPH insulin, the more cost‐effective insulin in type 2 diabetes (Waugh 2010).
Quality of the evidence
Studies were mainly of good to high quality. However, four studies had small comparison groups (fewer than 50 participants) (A ‐ Rosenstock 2009; Lixi ‐ Ratner 2010; T ‐ Nauck 2009; T‐ Ratner 2010); six studies had a duration of between eight and 16 weeks (A ‐ Rosenstock 2009; L ‐ Yang 2010; Lixi ‐ Ratner 2010; LY2189265 ‐Umpierrez 2011; T ‐ Nauck 2009; T‐ Ratner 2010), and the remaining trials were between 24 to 30 weeks long ‐ so there is insufficient evidence regarding long‐term outcomes. The studies were not long enough to entirely remove concerns about pancreatitis and renal failure with exenatide (FDA 2008; FDA 2009 (kidney function); FDA 2009 (safety update) or pancreatitis and thyroid carcinoma with liraglutide (EMEA 2009). The FDA has recently issued a reminder to physicians that there is a potential risk of thyroid carcinoma and pancreatitis with liraglutide (Journal Watch 2011). It is however difficult to prove if exenatide and liraglutide are responsible for pancreatitis because the incidence of it is increased in type 2 diabetes (Butler 2010; Girman 2010). One study followed up a cohort of patients taking exenatide and other hyperglycaemic drugs and concluded that there was no association of exenatide use and risk of acute pancreatitis (Dore 2011).
All the studies included in this review had industry connections.
In some studies there were some uncertainties or inequalities regarding previous or concomitant anti‐diabetic treatment. Patients in L ‐ LEAD 1 Marre 2009 were assumed to have been on previous metformin therapy, but this was not reported by the study. In A ‐ Rosenstock 2009, all patients in a concomitant exenatide group were on metformin, whereas only a proportion of patients in the other groups were, so this group could not be included in the analysis.
A range of studies had substantial losses to follow‐up (10% and more but less than 20%), with more withdrawals often occurring in the GLP‐1 groups (mainly due to adverse events).
Potential biases in the review process
Due to the limited number of studies in each categories no sensitivity or subgroup analyses were carried out and while some outcomes showed some heterogeneity in the overall results, some sources of heterogeneity could either not be identified or could only be speculated on.
Agreements and disagreements with other studies or reviews
Other published reviews included slightly different studies:
The review by Shyangdan 2010 included a search up to July 2010 and included 28 studies. It however included all the exenatide studies (including twice daily dosage). This Cochrane review is an update of that review and excludes all the studies comparing the twice daily exenatide against placebo or other oral hypoglycaemic agents, but not the ones against other GLP‐1 agonists.
The review by Norris 2009 included a search up to 2008, and included eight trials, including two excluded by the current review as less than 70% of patients had been on previous metformin therapy. The authors concluded that exenatide 10 µg twice daily reduced HbA1c to a similar extent as insulin or oral anti‐diabetic agents and that there was a beneficial effect on weight loss. Hypogycaemia with exenatide occurred mainly in participants also taking a sulphonylurea.
The review by Amori 2007 included data up to 2007 only, including seven studies on exenatide and two on liraglutide. The review included comparisons that are not clinically relevant (such as monotherapy versus placebo and 0.6 mg liraglutide). The authors found a moderate effect of GLP‐1 analogues on HbA1c (‐1%), a beneficial effect on weight and post‐prandial glucose, but increased gastrointestinal adverse events. In placebo‐controlled trials, increased hypoglycaemia with exenatide was seen mainly in trials using concomitant sulphonylurea.
Twenty‐one RCTs were included in the review by Monami 2009, including six unpublished studies. The search included studies up to 2008. Again, the authors found that GLP‐1 agonists reduced HbA1c by around 1%, and also reduced post‐prandial glucose and weight. There was no evidence of increased cardiovascular risk, but gastrointestinal adverse effects were common. Increased hypoglycaemia with exenatide versus placebo was only seen in trials using concomitant sulphonylurea.
Authors' conclusions
The evidence to date shows that the glucagon‐like peptide 1 (GLP‐1) analogues can provide a useful improvement in glucose control when added to dual treatment with oral drugs, and that at least in the short term, they can be an alternative to starting insulin. How long this effect would last, is not known. If we assume that the disease will steadily progress, as shown in UKPDS 16, then some of the benefit will be lost since the beta‐cells will no longer be there to release insulin. Other benefits such as delayed gastric emptying may continue, which may help control post‐prandial hyperglycaemia.
The glucose‐dependent nature of the insulin release means that hypoglycaemia should be less of a problem, but the differences in the trials were not marked. Hypoglycaemia was seen mainly when GLP‐1 analogues were used in combination with sulphonylureas.
Weight loss is a useful feature in the trials, though perhaps seen less in routine care (Loh and Clement 2007) .
The drawbacks are the need for injections, once a day with liraglutide and twice daily with exenatide, the high rate of side‐effects, especially nausea, and the cost. However, newer GLP‐1 agonists can be given once weekly or once every two weeks.
Injecting of a foreign peptide could lead to antibody formation, but studies measuring antibody formation noted that although antibodies were detected, these did not appear to reduce efficacy or have any safety effects.
More high quality trials are needed that:
compare one GLP‐1 agonist against other GLP‐1 agonists, with the emphasis on long‐acting agents;
measure health‐related quality of life and treatment satisfaction;
measure long‐term outcomes (longer than one year follow‐up) in terms of diabetes‐related morbidity and mortality and adverse events, and to indicate duration of efficacy in a progressive disease;
examine diverse populations, including adolescents and older adults;
use active controls.
Feedback
Comments and Criticisms, 8 June 2013
Summary
Shyangdan et al. (2011) concludes in the abstract of their review that GLP‐1 analogues are effective in improving glycemic control1. Although this statement is technically correct, we believe this one‐line conclusion is a misleading representation of the known clinical benefits and safety of these new agents. The authors also suggest the utilization of GLP‐1 analogues provides a “useful improvement in glucose control” despite acknowledging there is no morbidity and mortality data available. They recognize studies of longer duration are needed to find an effect in these outcomes; however, this message is obfuscated in the abstract as well as the author’s implications for practice.
This is important to address as discordance between the abstract and full text has been frequently recognized in the literature and has been suggested to mislead readers in the interpretation of the results2‐4.
Additionally, there remains a lack of convincing, hard outcome data for GLP‐1 analogues. In fact, a recent meta‐analysis has been unable to find a statistically significant difference in cardiovascular (CV) disease reduction5.
Focusing predominantly on improvements in surrogate markers such as HbA1c could lead to unintentional, increased CV harm or in this instance, pancreatitis and pancreatic cancer. Rosiglitazone, for example, was effective for decreasing HbA1c but there are concerns regarding increased risk of myocardial infarction6. Thus, we feel the lack of outcome data should be emphasized and should be at the fore‐front of the reader’s take home messages.
In this same analysis, there were also two issues which caught our attention. The first is found in the risk of bias assessment (figure 3) which suggested all the included trials were at low risk of attrition bias. Conversely, the authors specified that a range of studies had substantial losses to follow‐up with more withdrawals occurring in the GLP‐1 groups. We recognize these trials reported reasons for dropouts and utilized LOCF; however, the assessment for this risk of bias goes beyond how the study authors checked for and handled missing data. The disproportionate loss between the different arms could imbalance the groups and if the dropouts occur early in the study period, LOCF could lead to an underestimation of the potential harms of GLP‐1 analogues. These dropouts seemed likely to occur early as the review authors themselves recognize nausea, vomiting and diarrhea were “strongest at the beginning and then subsided”.
Second, the author included one trial of 0.6 to 0.9 mg of liraglutide in Japanese patients and excluded other trials with doses less than 1.2 mg per day on the basis of these doses being standard in Japan. We acknowledge the exclusion of the few studies based on this criteria did not impact the results in any significant fashion; however, the rationale and justification of such an exclusion is lacking. In order to appropriately gauge the safety and efficacy of liraglutide across multiple doses, all collected information for a particular dosage should be included in the analysis.
References:
1. Shyangdan DS, Royle P, Clar C, Sharma P, Waugh N, Snaith A. Glucagon‐like peptide analogues for type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2011, Issue 10. Art. No.: CD006423/ DOI: 10.1002/14651858.CD006423.pub2.
2. Altwairgi AK, Booth CM, Hopamn WM, Baetz TD. Discordance between conclusions stated in the abstract and conclusions in the article: analysis of published randomized controlled trials of systemic therapy in lung cancer. J Clin Oncol 2012;30(28):3552‐7.
3. Bernal‐Delgado E, Fisher ES. Abstracts in high profile journals often fail to report harm. BMC Med Res Methodol 2008; 8:14. doi: 10.1186/1471‐2288‐8‐14.
4. Boutron I, Dutton S, Ravaud P, Altman DG. Reporting and interpretation of randomized controlled trials with statistically nonsignifcant results for primary outcomes. JAMA 2010;303(20):2058‐64.
5. Sun F, Yu K, Wu S, Zhang Y, Yang Z et al. Cardiovascular safety and glycemic control of glucagon‐like peptide‐1 receptor agonists for type 2 diabetes mellitus: A pairwise and network meta‐analysis. Diabetes Res Clin Pract 2012;98(3):386‐95.
6. Nissen SE, Wolski K. Rosiglitazone revisited: an updated meta‐analysis of risk for myocardial infarction and cardiovascular mortality. Arch Intern Med 2010;170(14):1191‐201.
Reply
Disagree. As the commentators admit, we have noted the lack of long‐term outcomes.
Disagree, this is an unjustified assertion.
Agreed, as we said. Note that it is not surprising that a meta‐analysis showed no difference. This is to be expected given the duration of use. Note the time it took for UKPDS to show a significant reduction in CVD.
Disagree. The evidence on pancreatitis has emerged since we produced this review, and it will be mentioned when it is updated. This criticism is unjustified. There is no evidence for CV harm. Indeed, the evidence for pancreatitis (mainly Singh et al 2013) is not entirely convincing. The suggestion that the incretin drugs increase the risk of pancreatic cancer is unproven. If there is a risk, it will take years to prove it because it is probably very small.
Disagree. This paragraph is confused. Early drop‐outs, will not be exposed to any long‐term harm – they stop taking the drugs. Though we agree that LOCF is unsatisfactory and have said so on various occasions in other HTAs.
Disagree. As explained, that dose was included only for Japanese patients in whom it appears to be standard. It is not a standard dose in other ethnic groups so information should not be included. As stated, the review aimed to be relevant to clinical practice. It therefore focused on clinically relevant dosage. To do as the commentators suggest, would mean including lots of data from early dose‐ranging studies. These are clinically irrelevant.
The letter submitted reads more like an attempt to pick fault with the published review, rather than constructive criticism. Some of the language used is intemperate and inappropriate ‐ “obfuscation”, “to mislead readers”.
Note from the Coordinating Editor
Numbering introduced by the Feedback Editor for better readability.
The Cochrane Metabolic and Endocrine Disorders Review Group now has a policy that a set of patient‐important outcomes whether investigated or not have to be specified in major sections of the Cochrane review like the abstract and plain language summary. However, this policy was not in place when Cochrane review authors published this review.
Contributors
I certify that I have no affiliations with or involvement in any organisation or entity with a direct financial interest in the subject matter of my criticisms.
Nichoe Huan, Bsc(Pharm) (lawrence.huan@vch.ca )
Krystin Boyce, Bsc, Bsc(Pharm)
Lora Wang, Bsc(Pharm)
Aaron M Tejani, Bsc(Pharm), PharmD
Reply by the review authors
Acknowledgements
None.
Appendices
Appendix 1. Search strategies
| Search terms |
| Unless otherwise stated, search terms are free text terms; MeSH = Medical subject heading (MEDLINE medical index term); exP = exploded MeSH; the dollar sign ($) stands for any character(s); the question mark (?) substitutes one or no characters; tw = text word; pt = publication type; sh = MeSH; adj = adjacent (i.e. number of words within range of search term). MEDLINE 1. exp Glucagon‐Like Peptides/ 2. (glucagon like peptide* or GLP‐1).tw. 3. (exenatide or liraglutide or albiglutide or taspoglutide or lixisenatide).tw. 4. randomized controlled trial.pt. 5. random*.tw. 6. 1 or 3 or 2 7. 4 or 5 8. 6 and 7 Embase 1. exp Glucagon‐Like Peptide 1/ 2. (Glucagon‐Like Peptide 1 or GLP‐1).tw. 3. (exenatide or liraglutide or albiglutide or taspoglutide or lixisenatide).tw. 4. randomized controlled trial/ 5. (randomised or randomized).tw. 6. 1 or 3 or 2 7. 4 or 5 8. 6 and 7 The Cochrane Library (exenatide or liraglutide or albiglutide or taspoglutide or lixisenatide or glucagon like peptide or GLP‐1):ti Web of Science databases ‐ Science Citation Index Expanded; Social Sciences Citation Index (SSCI); Conference Proceedings Citation Index‐ Science Title=(glucagon like peptide* or GLP‐1 or exenatide or liraglutide or albiglutide or taspoglutide) Refined by: Document Type=( MEETING ABSTRACT ) |
Appendix 2. Overview of results
|
Study ID ► Characteristic ▼ |
Albiglutide versus placebo | Exenatide QW versus pioglitazone | Exenatide QW versus insulin glargine | Exenatide QW versus sitagliptin | Exenatide BID versus exenatide QW | Liraglutide versus placebo | Exenatide versus Liraglutide | Liraglutide versus insulin | Liraglutide versus glimepiride | Liraglutide versus TZD | Liraglutide versus DPP‐4 inhibitors | Lixisenatide versus placebo | LY2189265 versus placebo | Taspoglutide versus placebo |
| n studies | 1 | 1 | 1 | 1 | 1 | 0.6 mg: 1; 0.9 mg: 1; 1.2 mg: 3; 1.8 mg: 4 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 2 |
| HbA1c | versus placebo ‐0.62% to ‐0.7% | ‐0.3% versus pioglitazone | ‐0.2% versus glargine | ‐0.6% versus sitagliptin | favours exenatide QW 0.55% (95% CI 0.26 to 0.84, P = 0.0002) | ‐1.06 to ‐1.16% more with 0.6 and 0.9 mg dose versus placebo; ‐1. to ‐1.23% with 1.2 and 1.8 mg dose versus placebo | favours liraglutide 0.33% (95% CI 0.11 to 0.55, P < 0.0001) | favours liraglutide ‐0.24% (95% CI ‐0.39 to ‐0.08, P = 0.0015) | no significant difference | favours liraglutide ‐0.64% to ‐0.69% | favours liraglutide ‐0.34% to ‐0.60% | versus placebo ‐0.29% to ‐0.69% | ‐1.32% to ‐1.59%; no data for placebo | significant reduction with taspoglutide; titration of taspoglutide dose did not lead to increased reduction. |
| HbA1c ≤ 7% | 30% to 32% more than placebo | 5% more than pioglitazone | 28% more than glargine | 25% more than sitagliptin | 16% to 28% more with exenatide QW | 31.7% to 56.5% more with 0.6 and 0.9 mg dose versus placebo; 26% to 34% more than placebo with 1.2 and 1.8 mg | 11% more with liraglutide | 7% more with liraglutide | no significant difference | 13% to 20% more with liraglutide | 22% to 34% more than sitagliptin | 15% to 45% more than placebo | 49% to 54% across all groups | 34% to 64% more with taspoglutide; titration of taspoglutide dose did not lead to increment |
| Hypoglycaemia | no difference versus placebo | no difference versus pioglitazone | minor hypoglycaemia and symptoms only hypoglycaemia 18% less than glargine. 2 major hypoglycaemia in glargine and 1 in exenatide QW | hypoglycaemia more with sitagliptin. No cases of severe hypoglycaemia | no significant difference; more hypoglycaemia in participants receiving concomitant sulphonylurea treatment | rate of hypoglycaemia more with 0.6 and 0.9 mg dose; no significant difference with 1.2 mg liraglutide, 0 to 11% more patients with hypoglycaemia in 1.8 mg liraglutide than placebo group (RR 1.66) | 8% more patients with hypoglycaemia in exenatide than liraglutide group (RR 1.32) | no significant difference | 15% to 17% more patients with hypoglycaemia in glimepiride group than liraglutide | 4% more patients with hypoglycaemia in liraglutide group than rosiglitazone (RR 2.01) | no difference | dose‐dependent relation (1 to 3 events of hypoglycaemia per group) | significantly higher in all LY groups | unclear in one study; other study says that the incidence was similar between the groups. |
| Weight | versus placebo ‐0.7 kg to ‐0.9 kg | ‐5.1 kg versus pioglitazone | ‐4 kg versus glargine | ‐1.5 kg versus sitagliptin | no significant difference | no difference between 0.6/0.9 mg verus placebo; versus placebo ‐0.75 to ‐1.3 kg with 1.2 and 1.8 mg dose | no significant difference | favours liraglutide ‐3.43 kg (95% CI ‐4.00 to ‐2.86, P < 0.0001) | versus glimepiride ‐2.68 to ‐3.8 kg | favours liraglutide ‐1.8 to ‐2.3 kg | favours liraglutide ‐1.9 to ‐2.42 kg | versus placebo 0 to ‐1.95 kg | versus placebo ‐1.32 to ‐2.43 kg | significant reduction in weight when the 20 mg once weekly dose was titrated to 30 mg weekly but not when titrated to 40 mg once weekly |
| QoL | ‐ | significant improvement with exenatide; not with pioglitazone | significant improvement with exenatide for one of IWQOL‐Lite and one EQ‐5D dimensions; no difference for others | significant improvement with exenatide QW | no significant difference in treatment satisfaction | ‐ | greater treatment satisfaction with liraglutide | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Adverse events | ||||||||||||||
| Nausea | 25% to 26% | 19% more with exenatide24% | 12% more with exenatide | 14% more with exenatide | 8.1% to 21% more with the twice daily dose | 10.5% to 40% | nausea less persistent with liraglutide | see before | see before | see before | see before | dose‐dependent; 2.7% to 30.6% more than placebo | more common with LY; 6% to 9.3% more than placebo | 24% to 52% |
| Vomiting | 9% to 13% | 811% more with exenatide | 5% more with exenatide | 8% more with exenatide | 4.2% to 7.8% more with the twice daily dose | 4.5% to 17% | no obvious difference | see before | see before | see before | see before | 2.7% to 17.6% more than placebo | more common in higher dose of LY | 9% to 21% |
| Diarrhoea | 16% to 22% | 118% more with exenatide | 3% more with exenatide | 9% more with exenatide | 0.4% to 5.2% more with exenatide QW | 8% to 15% | no obvious difference | see before | see before | see before | see before | more common with lixisenatide | more common in higher dose of LY | 10% to 27% |
| Systolic blood pressure | no difference | no difference | ‐ | ‐4 mm Hg versus sitagliptin | no significant difference | no significant difference | no significant difference | favours liraglutide ‐2.53 mm Hg (95% CI ‐6.82 to ‐2.20, P = 0.0001) | versus glimepiride ‐2.7 to ‐3.2 mm Hg | ‐ | no difference | versus placebo +1 to ‐6 mm Hg | dose dependent reduction (‐0.6 to ‐3 mm Hg) | ‐ |
| FPG | versus placebo ‐1.3 to ‐1.5 mmol/L | versus pioglitazone ‐0.3 mmol/L | favour glargine; 0.70 mmol/L (95% CI 0.14 to 1.26, P = 0.01) | favour exenatide QW: ‐0.90 mmol/L (95% CI ‐1.50 to ‐0.30, P = 0.0038) | favours exenatide QW compared to exenatide BID 1.18 mmol/L (95% CI 1.02 to 1.33, P < 0.00001) | with 0.6 and 0.9 mg versus placebo ‐1.64 to ‐1.66 mmol/L; with 1.2 and 1.8 mg versus placebo ‐2.0 to ‐2.2 mmol/L | favours liraglutide 1.01 mmol/L (95% CI 0.46 to 1.56, P < 0.0001) | no significant difference | no significant difference | favours liraglutide ‐0.7 mmol/L | favours liraglutide ‐1.04 to ‐1.31 mmol/L | dose‐dependent reduction; versus placebo +0.02 to ‐1.21 mmol/L | versus placebo ‐1.56 to ‐2.16 mmol/L | significant reduction with taspoglutide |
| PPG | ‐ | no difference | exenatide QW led to lower glucose concentration after dinner and also post‐prandial glucose excursion after morning and evening meals was lower with exenatide QW. | significant reduction at all time points with exenatide QW | meal test (n = 51), 2 h PPG significantly more reduced with exenatide twice daily than once weekly; | versus placebo ‐1.8 to ‐2.3 mmol/L | post‐prandial plasma glucose increment reduced more by exenatide than liraglutide after breakfast and dinner | no significant difference | no significant difference | favours liraglutide ‐0.7 to ‐0.9 mmol/L | recorded values during the study were highly variable therefore not post‐prandial in most cases. Not reported | dose‐dependent reduction | ‐ | more improvement with taspoglutide versus placebo |
| Lipid profiles | no difference versus placebo | pioglitazone decreased triglycerides significantly; no difference for other parameters. | ‐ | exenatide QW reduced total cholesterol and LDL; all led to improvement in HDL levels | significantly greater reductions in total cholesterol and LDL with exenatide QW than exenatide BID | one study, triglycerides and LDL reduced with 1.2 but not 1.8 mg liraglutide | triglycerides and free fatty acids more reduced with liraglutide than exenatide | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | dose‐related decline in triglycerides, P not reported |
| Beta‐cell function | improved, HOMA‐B ratio 1.2 to 1.4 | ‐ | ‐ | ‐ | ‐ | improved with liraglutide | more improvement in HOMA‐B with liraglutide | more improvement in proinsulin‐to‐insulin ratio with liraglutide | no significant difference | more improvement with liraglutide | significant improvement in HOMA‐B, C‐peptide concentration and proinsulin‐to‐insulin ratio with liraglutide | ‐ | significant improvement in HOMA2‐%B; 31.9 to 44.6% more than placebo | improved in weekly taspoglutide regimens |
|
Footnotes "‐" denotes not reported BID: twice daily; CI: confidence interval; DPP‐4: dipeptidyl peptidase‐4 inhibitor; FPG: fasting plasma glucose; HbA1c: glycosylated haemoglobin A1c; HDL: high density lipoprotein; HOMA‐B: homeostatic model assessment‐beta‐cell function; IWQOL: impact of weight on quality of life questionnaire; LDL: low density lipoprotein; LY: LY2189265; n: number; PPG: post‐prandial glucose; QoL: quality of life; QW: once weekly; RR: relative risk;TZD: thiazolidinedione. | ||||||||||||||
Data and analyses
Comparison 1.
Albiglutide versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | Other data | No numeric data | ||
| 2 HbA1c ‐ with plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 ALBI 30 mg weekly | 1 | 79 | Mean Difference (IV, Random, 95% CI) | ‐0.7 [‐1.07, ‐0.33] |
| 2.2 ALBI 30 mg every 2 weeks | 1 | 82 | Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐1.06, ‐0.18] |
| 3 HbA1c < 7% | Other data | No numeric data | ||
| 4 Hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change ‐ with plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 ALBI 30 mg weekly | 1 | 79 | Mean Difference (IV, Random, 95% CI) | ‐0.7 [‐1.89, 0.49] |
| 5.2 ALBI 30 mg every 2 weeks | 1 | 82 | Mean Difference (IV, Random, 95% CI) | ‐0.9 [‐2.08, 0.28] |
| 6 Weight change (kg) | Other data | No numeric data | ||
| 7 Adverse events | Other data | No numeric data | ||
| 8 Blood pressure (mm Hg) | Other data | No numeric data | ||
| 9 Fasting plasma glucose (mmol/L) | Other data | No numeric data | ||
| 10 Lipid profiles | Other data | No numeric data | ||
| 11 Beta‐cell function | Other data | No numeric data | ||
| 12 Subgroups | Other data | No numeric data |
Analysis 1.1.
Comparison 1 Albiglutide versus placebo, Outcome 1 HbA1c (%).
HbA1c (%)
| Study | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|
| A ‐ Rosenstock 2009 | ‐0.87% (SD 0.65) 7.1% P < 0.05 versus placebo |
‐0.79% (SD 0.98) 7.2% P < 0.05 versus placebo |
‐0.17% (SD 1.01) 7.7% |
Analysis 1.2.
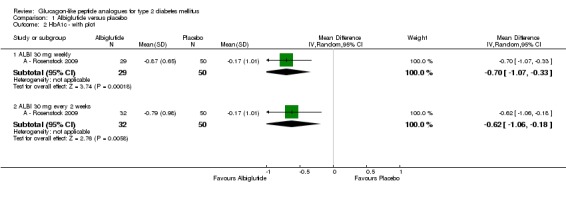
Comparison 1 Albiglutide versus placebo, Outcome 2 HbA1c ‐ with plot.
Analysis 1.3.
Comparison 1 Albiglutide versus placebo, Outcome 3 HbA1c < 7%.
HbA1c < 7%
| Study | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|
| A ‐ Rosenstock 2009 | 52% (P value not given) |
50% (P value not given) |
20% |
Analysis 1.4.
Comparison 1 Albiglutide versus placebo, Outcome 4 Hypoglycaemia.
Hypoglycaemia
| Study | Definition | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|---|
| A ‐ Rosenstock 2009 | not given | n = 0, P = NS | n=1 (3.1%), P = NS | n = 2 (3.9%) |
Analysis 1.5.
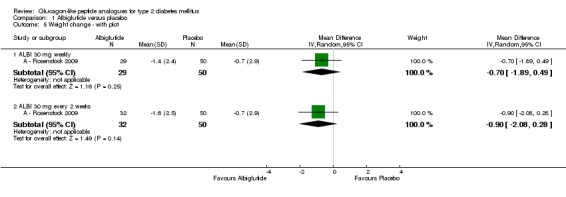
Comparison 1 Albiglutide versus placebo, Outcome 5 Weight change ‐ with plot.
Analysis 1.6.
Comparison 1 Albiglutide versus placebo, Outcome 6 Weight change (kg).
Weight change (kg)
| Study | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|
| A ‐ Rosenstock 2009 | ‐1.4 kg (SD 2.4), P = NS | ‐1.6 kg (SD 2.5), P = NS | ‐0.7 kg (SD 2.9) |
Analysis 1.7.
Comparison 1 Albiglutide versus placebo, Outcome 7 Adverse events.
Adverse events
| Study | Description | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|---|
| A ‐ Rosenstock 2009 | any adverse event | 83.9% | 84.4% | 66.7% |
| A ‐ Rosenstock 2009 | withdrawals | 32.2% | 42.8% | 23.5% |
| A ‐ Rosenstock 2009 | adverse events similar across groups for: abdominal pain, headache, dizziness, hyperglycaemia, nasopharyngitis, influenza, upper respiratory tract infection, back pain no systemic allergic reactions to albiglutide |
|||
| A ‐ Rosenstock 2009 | nausea | n = 8 (25.8%) | n = 8 (25%) | n = 6 (11.8%) |
| A ‐ Rosenstock 2009 | vomiting | n = 4 (12.9%) | n = 3 (9.4%) | n = 1 (2%) |
| A ‐ Rosenstock 2009 | diarrhoea | n = 5 (16.1%) | n = 7 (21.9%) | n = 2 (3.9%) |
| A ‐ Rosenstock 2009 | cardiac disorders | none | none | n = 1 |
| A ‐ Rosenstock 2009 | pancreatitis | none | none | none |
| A ‐ Rosenstock 2009 | skin reactions | 15 events in 7 patients | 11 events in 6 patients | 3 events in 3 patients |
| A ‐ Rosenstock 2009 | positive immunogenicity test | 2 of 31 (6.4%) | 1 of 32 (3.1%) | 1 of 51 (2%) |
Analysis 1.8.
Comparison 1 Albiglutide versus placebo, Outcome 8 Blood pressure (mm Hg).
Blood pressure (mm Hg)
| Study | Description | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|---|
| A ‐ Rosenstock 2009 | systolic BP (mm Hg) | ‐5.8 (SD 11.2), P = NS | ‐7.4 (SD 14.2), P = NS | ‐0.7 (SD 13.9) |
| A ‐ Rosenstock 2009 | diastolic BP (mm Hg) | ‐1.9 (SD 8.1), P = NS | ‐4.4 (SD 8.9), P = NS | ‐1.0 (SD 8.2) |
Analysis 1.9.
Comparison 1 Albiglutide versus placebo, Outcome 9 Fasting plasma glucose (mmol/L).
Fasting plasma glucose (mmol/L)
| Study | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo | FPG fluctuations |
|---|---|---|---|---|
| A ‐ Rosenstock 2009 | ‐1.44% (SD 2.03), P < 0.05 versus placebo | ‐1.58% (SD 2.06), P < 0.05 versus placebo | ‐0.10% (SD 2.90) | according to figure 1C in the paper, the once weekly dosing schedules seemed to cause less fluctuation in FPG than the less frequent dosing schedules; however, data are only shown for 50 mg every 2 weeks and not for 30 mg every 2 weeks COMMENT: paper says "%" but should probably be "mmol/L"?? |
Analysis 1.10.
Comparison 1 Albiglutide versus placebo, Outcome 10 Lipid profiles.
Lipid profiles
| Study | Description | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|---|
| A ‐ Rosenstock 2009 | triglycerides (mmol/L) | +0.1 (SD 0.9), P = NS | ‐0.3 (SD 1.0), P = NS | ‐0.4 (SD 1.7) |
| A ‐ Rosenstock 2009 | total cholesterol (mmol/L) | +0.01 (SD 0.6), P = NS | ‐0.18 (SD 0.45), P = NS | +0.1 (SD 0.77) |
| A ‐ Rosenstock 2009 | HDL (mmol/L) | ‐0.05 (SD 0.17), P = NS | ‐0.03 (SD 0.14), P = NS | ‐0.002 (SD 0.13) |
| A ‐ Rosenstock 2009 | LDL (mmol/L) | +0.003 (SD 0.6), P = NS | ‐0.06 (SD 0.39), P = NS | +0.19 (SD 0.6) |
| A ‐ Rosenstock 2009 | free fatty acids (mmol/L) | +0.05 (SD 0.17), P = NS | ‐0.01 (SD 0.25), P = NS | +0.08 (SD 0.2) |
Analysis 1.11.
Comparison 1 Albiglutide versus placebo, Outcome 11 Beta‐cell function.
Beta‐cell function
| Study | Description | ALBI 30 mg weekly | ALBI 30 mg every 2 weeks | Placebo |
|---|---|---|---|---|
| A ‐ Rosenstock 2009 | HOMA‐B endpoint | 95.4 | 74.7 | 50.4 |
| A ‐ Rosenstock 2009 | HOMA‐B ratio at endpoint | 1.4, P < 0.05 versus placebo | 1.2, P = NS | 1.0 |
Analysis 1.12.
Comparison 1 Albiglutide versus placebo, Outcome 12 Subgroups.
Subgroups
| Study | Outcome | Results / comments |
|---|---|---|
| A ‐ Rosenstock 2009 | HbA1c | Numerically greater reductions were seen in participants with baseline HbA1c ≥ 8.5% (no details given) |
Comparison 2.
Exenatide 2 mg once weekly versus thiazolidinedione
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia (minor) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Weight change (kg) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5 Quality of life | Other data | No numeric data | ||
| 6 Adverse events | Other data | No numeric data | ||
| 7 Blood pressure (mm Hg) | Other data | No numeric data | ||
| 8 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9 Post‐prandial glucose / glucose profiles | Other data | No numeric data | ||
| 10 Lipid profiles | Other data | No numeric data |
Analysis 2.1.
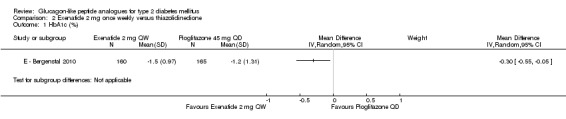
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 1 HbA1c (%).
Analysis 2.2.
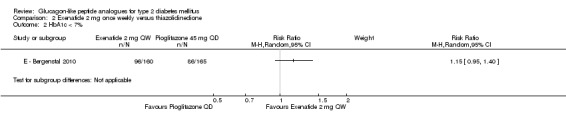
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 2 HbA1c < 7%.
Analysis 2.3.
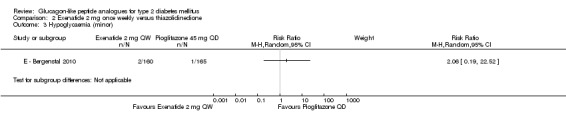
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 3 Hypoglycaemia (minor).
Analysis 2.4.
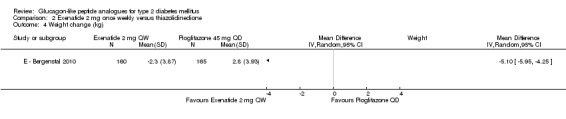
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 4 Weight change (kg).
Analysis 2.5.
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 5 Quality of life.
Quality of life
| Study | Description | Exenatide 2 mg QW | Pioglitazone 45 mg QD | Difference between groups |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | IWQOL total score | 5.15, 95% CI 3.11 to 7.19 | 1.20, 95% CI ‐0.87 to 3.28 | EX vs PIO: 3.94, 95% CI 1.28 to 6.61, P = 0.0038 |
| E ‐ Bergenstal 2010 | Overall treatment satisfaction | 3.96, 95% CI 2.78 to 5.15 | NR | NR |
Analysis 2.6.
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 6 Adverse events.
Adverse events
| Study | Description | Exenatide 2 mg QW | Pioglitazone 45 mg QD |
|---|---|---|---|
| E ‐ Bergenstal 2010 | Withdrawals due to adverse events | 6.9% | 3.6% |
| E ‐ Bergenstal 2010 | Nausea | 24% | 5% |
| E ‐ Bergenstal 2010 | Diarrhoea | 18% | 7% |
| E ‐ Bergenstal 2010 | Vomiting | 11% | 3% |
| E ‐ Bergenstal 2010 | Urinary tract infection | 6% | 4% |
| E ‐ Bergenstal 2010 | Headache | 9% | 4% |
| E ‐ Bergenstal 2010 | Injection‐site pruritus | 5% | 1% |
| E ‐ Bergenstal 2010 | Serious adverse events | 3% | 6% |
| E ‐ Bergenstal 2010 | Anti‐Exenatide antibodies | either low (< 1/625; n = 74, 48%) or not detectable (n = 61, 40%) titres | ‐ |
| E ‐ Bergenstal 2010 | Severe hypoglycaemia | 0 | 0 |
Analysis 2.7.
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 7 Blood pressure (mm Hg).
Blood pressure (mm Hg)
| Study | Description | Exenatide 2 mg QW | Pioglitazone 45 mg QD | Difference between groups |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | Systolic blood pressure | NR | NR | EX vs. PIO: P = NS |
| E ‐ Bergenstal 2010 | Diastolic blood pressure | NR | NR | EX vs. PIO: P = NS |
Analysis 2.8.
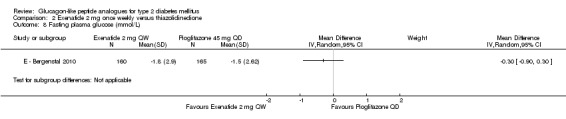
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 8 Fasting plasma glucose (mmol/L).
Analysis 2.9.
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 9 Post‐prandial glucose / glucose profiles.
Post‐prandial glucose / glucose profiles
| Study | Description | Exenatide 2 mg QW | Pioglitazone 45 mg QD | Description |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | Self‐monitored blood glucose | NR | NR | EX vs PIO: P = NS |
Analysis 2.10.
Comparison 2 Exenatide 2 mg once weekly versus thiazolidinedione, Outcome 10 Lipid profiles.
Lipid profiles
| Study | Description | Exenatide 2 mg QW | Sitagliptin 100 mg QD | Difference between groups |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | Fasting triglycerides (mmol/l) | NR | NR | EX vs. SITA: P = NS; PIO vs. SITA: P = 0.0062 |
| E ‐ Bergenstal 2010 | Total cholesterol (mmol/l) | NR | NR | EX vs. SITA: P = NS; PIO vs. SITA: P = NS |
| E ‐ Bergenstal 2010 | HDL (mmol/l) | NR | NR | EX vs. SITA: P = NS; PIO vs. SITA: P < 0.0001 |
| E ‐ Bergenstal 2010 | LDL (mmol/l) | NR | NR | EX vs. SITA: P = NS; PIO vs. SITA: P = NS |
Comparison 3.
Exenatide 2 mg once weekly versus DPP‐4 inhibitors
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia (minor) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Weight change (kg) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5 Quality of life | Other data | No numeric data | ||
| 6 Adverse events | Other data | No numeric data | ||
| 7 Blood pressure (mm Hg) | Other data | No numeric data | ||
| 8 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9 Post‐prandial glucose / glucose profiles | Other data | No numeric data | ||
| 10 Lipid profiles | Other data | No numeric data |
Analysis 3.1.
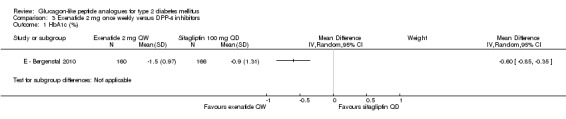
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 1 HbA1c (%).
Analysis 3.2.
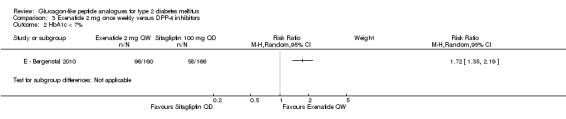
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 2 HbA1c < 7%.
Analysis 3.3.
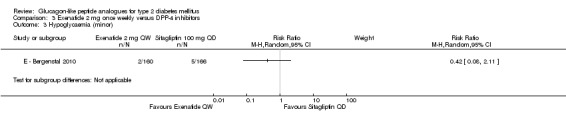
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 3 Hypoglycaemia (minor).
Analysis 3.4.
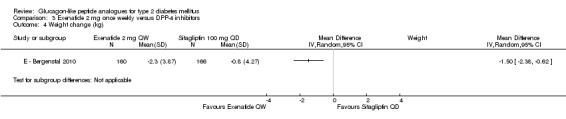
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 4 Weight change (kg).
Analysis 3.5.
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 5 Quality of life.
Quality of life
| Study | Description | Exenatide 2 mg QW | Sitagliptin 100 mg QD | Difference between groups |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | IWQOL total score | 5.15, 95% CI 3.11 to 7.19 | 4.56, 95% CI 2.56 to 6.57 | EX vs PIO: 3.94, 95% CI 1.28 to 6.61, P = 0.0038 |
| E ‐ Bergenstal 2010 | Overall treatment satisfaction | 3.96, 95% CI 2.78 to 5.15 | 2.35, 95% CI 1.19 to 3.51 | EX vs. SITA: 1.61, 95% CI 0.07 to 3.16, P = 0.0406 |
Analysis 3.6.
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 6 Adverse events.
Adverse events
| Study | Description | Exenatide 2 mg QW | Sitagliptin 100 mg QD |
|---|---|---|---|
| E ‐ Bergenstal 2010 | Withdrawals due to adverse events | 6.9% | 3% |
| E ‐ Bergenstal 2010 | Nausea | 24% | 10% |
| E ‐ Bergenstal 2010 | Diarrhoea | 18% | 10% |
| E ‐ Bergenstal 2010 | Vomiting | 11% | 2% |
| E ‐ Bergenstal 2010 | Urinary tract infection | 6% | 5% |
| E ‐ Bergenstal 2010 | Headache | 9% | 9% |
| E ‐ Bergenstal 2010 | Injection‐site pruritus | 5% | 5% |
| E ‐ Bergenstal 2010 | Serious adverse events | 3% | 3% |
| E ‐ Bergenstal 2010 | Anti‐Exenatide antibodies | either low (< 1/625; n = 74, 48%) or not detectable (n = 61, 40%) titres | ‐ |
| E ‐ Bergenstal 2010 | Severe hypoglycaemia | 0 | 0 |
Analysis 3.7.
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 7 Blood pressure (mm Hg).
Blood pressure (mm Hg)
| Study | Description | Exenatide 2 mg QW | Sitagliptin 100 mg QD | Difference between groups |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | Systolic blood pressure | NR | NR | EX vs. SITA: ‐4 mm Hg (95% CI ‐6 to ‐1); P = 0.0055 |
| E ‐ Bergenstal 2010 | Diastolic blood pressure | NR | NR | EX vs. SITA: P = NS |
Analysis 3.8.
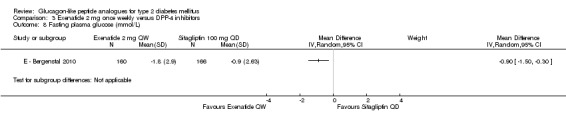
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 8 Fasting plasma glucose (mmol/L).
Analysis 3.9.
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 9 Post‐prandial glucose / glucose profiles.
Post‐prandial glucose / glucose profiles
| Study | Description | Exenatide 2 mg QW | Sitagliptin 100 mg QD | Description |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | Self‐monitored blood glucose | NR | NR | EX vs SITA: P < 0.05 |
Analysis 3.10.
Comparison 3 Exenatide 2 mg once weekly versus DPP‐4 inhibitors, Outcome 10 Lipid profiles.
Lipid profiles
| Study | Description | Exenatide 2 mg QW | Sitagliptin 100 mg QD | Difference between groups |
|---|---|---|---|---|
| E ‐ Bergenstal 2010 | Fasting triglycerides (mmol/l) | NR | NR | EX vs. SITA: P = 0.9718; PIO vs. SITA: p=0.0062 |
| E ‐ Bergenstal 2010 | Total cholesterol (mmol/l) | NR | NR | EX vs. SITA: P = 0.3424; PIO vs. SITA: P = 0.3424 |
| E ‐ Bergenstal 2010 | HDL (mmol/l) | NR | NR | EX vs. SITA: P = 0.9546; PIO vs. SITA: P < 0.0001 |
| E ‐ Bergenstal 2010 | LDL (mmol/l) | NR | NR | EX vs. SITA: P = 0.6113; PIO vs. SITA: P = 0.9965 |
Comparison 4.
Exenatide 2 mg once weekly versus insulin glargine
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia (symptoms only) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Hypoglycaemia (minor) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5 Severe hypoglycaemia | Other data | No numeric data | ||
| 6 Weight change (kg) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7 Adverse events | Other data | No numeric data | ||
| 8 Quality of life | Other data | No numeric data | ||
| 9 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10 Postprandial glucose / glucose profiles | Other data | No numeric data |
Analysis 4.1.
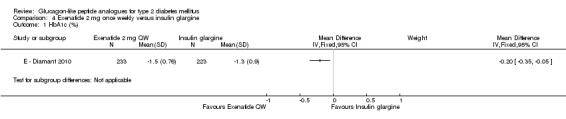
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 1 HbA1c (%).
Analysis 4.2.
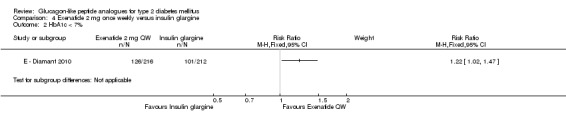
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 2 HbA1c < 7%.
Analysis 4.3.
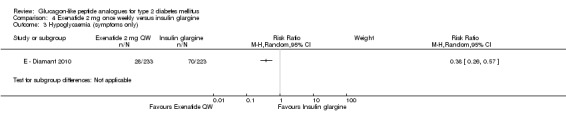
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 3 Hypoglycaemia (symptoms only).
Analysis 4.4.
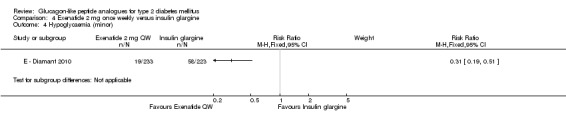
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 4 Hypoglycaemia (minor).
Analysis 4.5.
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 5 Severe hypoglycaemia.
Severe hypoglycaemia
| Study | Description | Exenatide 2 mg QW | Insulin glargine |
|---|---|---|---|
| E ‐ Diamant 2010 | Taking metformin only | 1 (0.4%) | 1 (0.4%) |
| E ‐ Diamant 2010 | Taking both metformin and sulphonylureas | None | 1 (0.4%) |
Analysis 4.6.
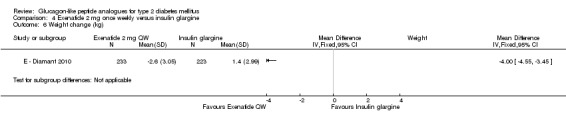
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 6 Weight change (kg).
Analysis 4.7.
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 7 Adverse events.
Adverse events
| Study | Description | Exenatide 2 mg QW | Insulin glargine |
|---|---|---|---|
| E ‐ Diamant 2010 | withdrawal due to adverse events | 11 (4.7%) | 2 (0.9%) |
| E ‐ Diamant 2010 | nausea | 30 (13%) | 3 (1%) |
| E ‐ Diamant 2010 | vomiting | 10 (4%) | 3 (1%) |
| E ‐ Diamant 2010 | diarrhoea | 20 (9%) | 8 (4%) |
| E ‐ Diamant 2010 | Nasopharyngitis | 30 (13%) | 39 (17%) |
| E ‐ Diamant 2010 | Headache | 23 (10%) | 16 (7%) |
| E ‐ Diamant 2010 | Injection‐site reaction | 30 (13%) | 4 (2%) |
| E ‐ Diamant 2010 | Patients with one or more serious adverse events Pancreatitis |
11 (5%) 1 (0.4%) |
10 (4%) None |
| E ‐ Diamant 2010 | Deaths | None | None |
| E ‐ Diamant 2010 | Anti‐exenatide antibodies | 127/233 (54.5%) tested positive for anti‐exenatide antibodies. No effect on HbA1c | ‐ |
Analysis 4.8.
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 8 Quality of life.
Quality of life
| Study | Description | Exenatide 2 mg QW | Insulin glargine |
|---|---|---|---|
| E ‐ Diamant 2010 | IWQOL‐Lite (self‐esteem) | Significant improvement compared with insulin glargine; no data given |
Analysis 4.9.
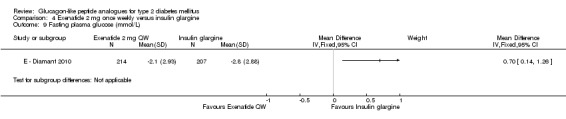
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 9 Fasting plasma glucose (mmol/L).
Analysis 4.10.
Comparison 4 Exenatide 2 mg once weekly versus insulin glargine, Outcome 10 Postprandial glucose / glucose profiles.
Postprandial glucose / glucose profiles
| Study | Description | Exenatide 2 mg QW | Insulin glargine | Comment |
|---|---|---|---|---|
| E ‐ Diamant 2010 | 8‐point SMBG | NR | NR | Both treatments reduced PPG at all eight timepoints (all P < 0.0001) 0300 hour and before breafast: participants receiving insulin glargine had lower glucose concentrations than exenatide at 0300 hour (P = 0.022) and before breakfast (P < 0.0001) Dinner, morning and evening meals: participants receiving exenatide had lower glucose concentrations after dinner than insulin glargine (P = 0.004). Morning and evening meals: participants receiving exenatide had lower postprandial glucose excursions than insulin glargine after morning (P = 0.001) and evening meals (P = 0.033). |
Comparison 5.
Liraglutide 0.6 mg daily versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia rate (events/patient‐year) | Other data | No numeric data | ||
| 4 Severe hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change (kg) | Other data | No numeric data | ||
| 6 Adverse events | Other data | No numeric data | ||
| 7 Blood pressure (mm Hg) | Other data | No numeric data | ||
| 8 Fasting plasma glucose (mmol/L) | Other data | No numeric data | ||
| 9 Post‐prandial glucose / glucose profiles | Other data | No numeric data |
Analysis 5.1.
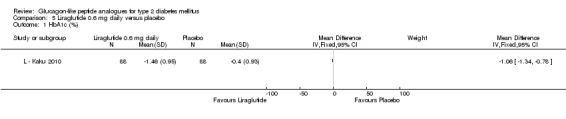
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 1 HbA1c (%).
Analysis 5.2.
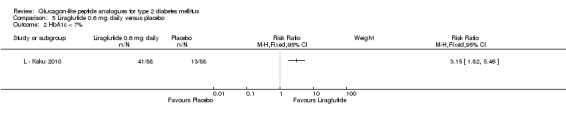
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 2 HbA1c < 7%.
Analysis 5.3.
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 3 Hypoglycaemia rate (events/patient‐year).
Hypoglycaemia rate (events/patient‐year)
| Study | Liraglutide 0.6 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | 2.17 | 1.01 |
Analysis 5.4.
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 4 Severe hypoglycaemia.
Severe hypoglycaemia
| Study | Liraglutide 0.6 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | None | None |
Analysis 5.5.
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 5 Weight change (kg).
Weight change (kg)
| Study | Liraglutide 0.6 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | +0.06. P < 0.0001 versus placebo | ‐1.12 |
Analysis 5.6.
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 6 Adverse events.
Adverse events
| Study | Description | Liraglutide 0.6 mg daily | Placebo |
|---|---|---|---|
| L ‐ Kaku 2010 | Withdrawals due to adverse events | 3 (3%) | 2 (2%) |
| L ‐ Kaku 2010 | Overall adverse events | 67 (76.1%) | 66 (75%) |
| L ‐ Kaku 2010 | Gastrointestinal adverse events | More subjects in the two liraglutide groups reported gastrointestinal adverse events during the first 4 weeks of the trial than subjects on placebo. No major differences in gastrointestinal adverse events across groups. | |
| L ‐ Kaku 2010 | Serious adverse events | 3 (3%) | 2 (2%) |
| L ‐ Kaku 2010 | Pancreatitis | None | None |
| L ‐ Kaku 2010 | Deaths | None | None |
Analysis 5.7.
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 7 Blood pressure (mm Hg).
Blood pressure (mm Hg)
| Study | |
|---|---|
| L ‐ Kaku 2010 | SBP did not change in both groups; P = NS between groups DBP did not change in both groups; P = NS between groups |
Analysis 5.8.
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 8 Fasting plasma glucose (mmol/L).
Fasting plasma glucose (mmol/L)
| Study | Liraglutide 0.6 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | ‐2.3, P < 0.0001 versus placebo | ‐0.64 |
Analysis 5.9.
Comparison 5 Liraglutide 0.6 mg daily versus placebo, Outcome 9 Post‐prandial glucose / glucose profiles.
Post‐prandial glucose / glucose profiles
| Study | Description | Liraglutide 0.6 mg daily | Placebo |
|---|---|---|---|
| L ‐ Kaku 2010 | 7‐point SMBG profile | ‐2.66, P < 0.0001 versus placebo | ‐0.35 |
Comparison 6.
Liraglutide 0.9 mg daily versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia rate (events/patient‐year) | Other data | No numeric data | ||
| 4 Severe hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change (kg) | Other data | No numeric data | ||
| 6 Adverse events | Other data | No numeric data | ||
| 7 Blood pressure (mm Hg) | Other data | No numeric data | ||
| 8 Fasting plasma glucose (mmol/L) | Other data | No numeric data | ||
| 9 Postprandial glucose / glucose profiles | Other data | No numeric data |
Analysis 6.1.
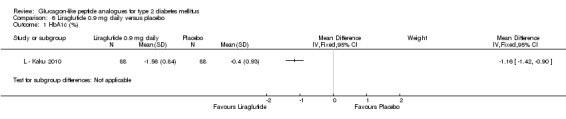
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 1 HbA1c (%).
Analysis 6.2.
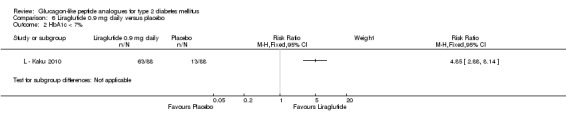
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 2 HbA1c < 7%.
Analysis 6.3.
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 3 Hypoglycaemia rate (events/patient‐year).
Hypoglycaemia rate (events/patient‐year)
| Study | Liraglutide 0.9 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | 1.96 | 1.01 |
Analysis 6.4.
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 4 Severe hypoglycaemia.
Severe hypoglycaemia
| Study | Liraglutide 0.9 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | None | None |
Analysis 6.5.
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 5 Weight change (kg).
Weight change (kg)
| Study | Liraglutide 0.9 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | ‐0.37, P = 0.0071 versus placebo | ‐1.12 |
Analysis 6.6.
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 6 Adverse events.
Adverse events
| Study | Description | Liraglutide 0.9 mg daily | Placebo |
|---|---|---|---|
| L ‐ Kaku 2010 | Withdrawals due to adverse events | 2 (2%) | 2 (2%) |
| L ‐ Kaku 2010 | Overall adverse events | 69 (78.4%) | 66 (75%) |
| L ‐ Kaku 2010 | Gastrointestinal adverse events | More subjects in the two liraglutide groups reported gastrointestinal adverse events during the first 4 weeks of the trial than subjects on placebo. No major differences in gastrointestinal adverse events across groups. | |
| L ‐ Kaku 2010 | Serious adverse events | 2 (2%) | 2 (2%) |
| L ‐ Kaku 2010 | Pancreatitis | None | None |
| L ‐ Kaku 2010 | Deaths | None | None |
Analysis 6.7.
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 7 Blood pressure (mm Hg).
Blood pressure (mm Hg)
| Study | |
|---|---|
| L ‐ Kaku 2010 | SBP did not change in both groups; P = NS between groups DBP did not change in both groups; P = NS between groups |
Analysis 6.8.
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 8 Fasting plasma glucose (mmol/L).
Fasting plasma glucose (mmol/L)
| Study | Liraglutide 0.9 mg daily | Placebo |
|---|---|---|
| L ‐ Kaku 2010 | ‐2.28, P < 0.0001 versus placebo | ‐0.64 |
Analysis 6.9.
Comparison 6 Liraglutide 0.9 mg daily versus placebo, Outcome 9 Postprandial glucose / glucose profiles.
Postprandial glucose / glucose profiles
| Study | Description | Liraglutide 0.9 mg daily | Placebo |
|---|---|---|---|
| L ‐ Kaku 2010 | 7‐point SMBG profile | ‐2.89, P < 0.0001 versus placebo | ‐0.35 |
Comparison 7.
Liraglutide 1.2 mg versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c | 3 | 1058 | Mean Difference (IV, Random, 95% CI) | ‐1.15 [‐1.33, ‐0.96] |
| 2 HbA1c < 7% | 3 | 1058 | Risk Ratio (M‐H, Random, 95% CI) | 2.91 [1.74, 4.87] |
| 3 Hypoglycaemia | 3 | 1058 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.54, 4.42] |
| 4 Weight change | 3 | 1058 | Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.95, 0.45] |
| 5 Adverse events | Other data | No numeric data | ||
| 6 Systolic blood pressure | 2 | 716 | Mean Difference (IV, Random, 95% CI) | ‐3.26 [‐7.71, 1.20] |
| 7 Fasting plasma glucose (mmol/L) | 3 | 1058 | Mean Difference (IV, Random, 95% CI) | ‐2.13 [‐2.59, ‐1.68] |
| 8 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 9 Triglycerides (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10 Total cholesterol (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11 HDL‐cholesterol (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12 LDL‐cholesterol (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 13 Beta‐cell function | Other data | No numeric data |
Analysis 7.1.
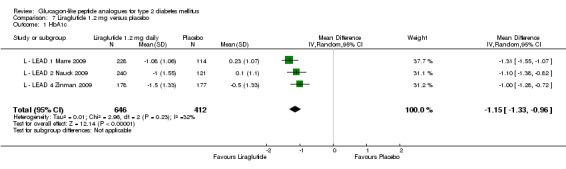
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 1 HbA1c.
Analysis 7.2.
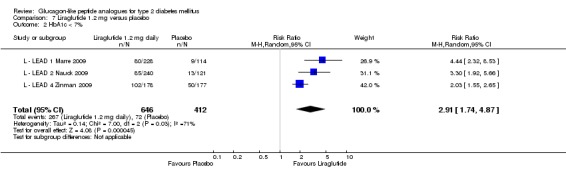
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 2 HbA1c < 7%.
Analysis 7.3.
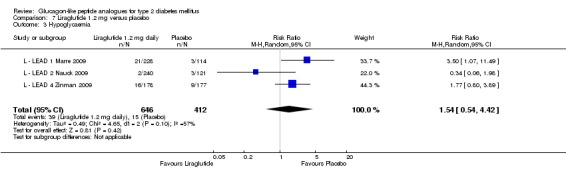
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 3 Hypoglycaemia.
Analysis 7.4.
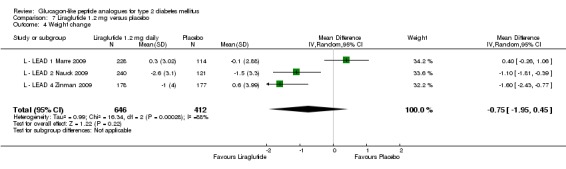
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 4 Weight change.
Analysis 7.5.
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 5 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.2 mg daily | Placebo |
|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | withdrawal due to adverse events | 5% | 5% |
| L ‐ LEAD 1 Marre 2009 | no significant differences across groups for: blood pressure; no significant changes is: ophthalmoscopy, biochemistry, urinalysis, haematology, ECG no deaths |
||
| L ‐ LEAD 1 Marre 2009 | nausea | 10.5% (highest) | 1.8% (lowest) |
| L ‐ LEAD 1 Marre 2009 | vomiting | 4.4% | |
| L ‐ LEAD 1 Marre 2009 | diarrhoea | 7.9% | |
| L ‐ LEAD 1 Marre 2009 | serious adverse events (mostly judged to be unlikely to be related to study medication) | 4% | 3% |
| L ‐ LEAD 1 Marre 2009 | liraglutide auto‐antibodies | 9 to13%, no effect on HbA1c | |
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to adverse events | 10% | 2% |
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to nausea/vomiting/diarrhoea | 5% | 0 |
| L ‐ LEAD 2 Nauck 2009 | no significant differences across groups for: physical examination findings, laboratory analyses, ECG, ophthalmoscopy | ||
| L ‐ LEAD 2 Nauck 2009 | any GI event | 40% | 17% |
| L ‐ LEAD 2 Nauck 2009 | nausea | 16% | 3 to 4% |
| L ‐ LEAD 2 Nauck 2009 | vomiting | 5 to 7% | 1% |
| L ‐ LEAD 2 Nauck 2009 | diarrhoea | 8% | 4% |
| L ‐ LEAD 2 Nauck 2009 | serious adverse events: 2 deaths unrelated to liraglutide treatment; 1 participant in 1.2 mg liraglutide group withdrawn due to acute pancreatitis | ||
| L ‐ LEAD 2 Nauck 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 2 Nauck 2009 | auto‐immune response | NR | NR |
| L ‐ LEAD 4 Zinman 2009 | withdrawal due to adverse events | 6% | 3% |
| L ‐ LEAD 4 Zinman 2009 | withdrawal due to nausea/vomiting/diarrhoea | 3% | 0 |
| L ‐ LEAD 4 Zinman 2009 | no significant differences across groups for: physical examination findings, laboratory analyses, ECG, ophthalmoscopy, cardiovascular adverse events | ||
| L ‐ LEAD 4 Zinman 2009 | any GI event | 45% | 19% |
| L ‐ LEAD 4 Zinman 2009 | nausea | 29% | NR |
| L ‐ LEAD 4 Zinman 2009 | vomiting | 7% | NR |
| L ‐ LEAD 4 Zinman 2009 | diarrhoea | NR | NR |
| L ‐ LEAD 4 Zinman 2009 | serious adverse events | 8 events in 8 participants | 13 events in 12 participants |
| L ‐ LEAD 4 Zinman 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 4 Zinman 2009 | auto‐immune response | 4.1% (no effect on HbA1c) |
Analysis 7.6.
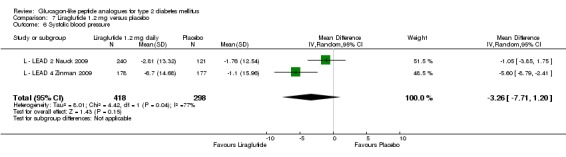
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 6 Systolic blood pressure.
Analysis 7.7.
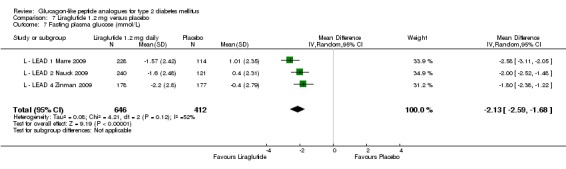
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 7 Fasting plasma glucose (mmol/L).
Analysis 7.8.
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 8 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.2 mg daily | Placebo | Comments |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | average of values obtained 90 min after breakfast, lunch and evening meal | ‐2.5 mmol/L | ‐0.4 mmol/L | Liraglutide 1.2 mg versus placebo: P < 0.0001 |
| L ‐ LEAD 2 Nauck 2009 | from self‐monitored 7‐point plasma glucose measurements | ‐2.3 mmol/L | ‐0.6 mmol/L | Liraglutide 1.2 mg versus placebo: P < 0.001 |
| L ‐ LEAD 4 Zinman 2009 | from self‐monitored 7‐point plasma glucose measurements | ‐2.6 mmol/L | ‐0.8 mmol/L | Liraglutide 1.2 mg versus placebo: P < 0.001; the postprandial increment (postmeal value minus premeal) was significantly reduced over breakfast with liraglutide treatment (‐0.9, ‐0.8, ‐0.3 mmol/L respectively; P < 0.05 for both liraglutide groups versus placebo) but not for lunch and dinner. |
Analysis 7.9.
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 9 Triglycerides (mmol/L).
Analysis 7.10.
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 10 Total cholesterol (mmol/L).
Analysis 7.11.
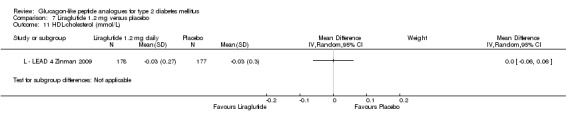
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 11 HDL‐cholesterol (mmol/L).
Analysis 7.12.
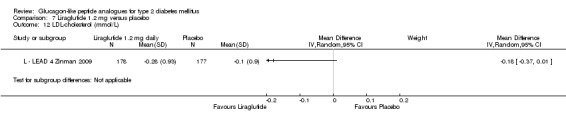
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 12 LDL‐cholesterol (mmol/L).
Analysis 7.13.
Comparison 7 Liraglutide 1.2 mg versus placebo, Outcome 13 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.2 mg daily | Placebo |
|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | HOMA‐B (%) | 99% SE 184.3 (+28%), P = 0.01 versus placebo | 52% SE 107.3 (‐4%) |
| L ‐ LEAD 1 Marre 2009 | proinsulin‐to‐insulin ratio | 0.33 SE 0.2 (‐0.12), p ≤ 0.02 versus placebo | 0.46 SE 0.29 (+0.02) |
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 2 Nauck 2009 | HOMA‐B (%) | +23% | ‐2% |
| L ‐ LEAD 2 Nauck 2009 | proinsulin‐to‐insulin ratio | ‐0.1, P < 0.0001 versus placebo | +0.1 |
| L ‐ LEAD 2 Nauck 2009 | proinsulin‐to‐C‐peptide ratio | ||
| L ‐ LEAD 4 Zinman 2009 | HOMA‐B (%) | +27% SD 59, P < 0.05 vs placebo | 6% SD 60 |
| L ‐ LEAD 4 Zinman 2009 | proinsulin‐to‐insulin ratio | ‐0.029 SD 0.35, P < 0.05 vs placebo | 0.036 SD 39 |
| L ‐ LEAD 4 Zinman 2009 | proinsulin‐to‐C‐peptide ratio | ‐0.007 SD 0.01, P < 0.05 vs placebo | ‐0.002 SD 0.01 |
Comparison 8.
Liraglutide 1.8 mg versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c | 4 | 1410 | Mean Difference (IV, Random, 95% CI) | ‐1.15 [‐1.31, ‐0.99] |
| 2 HbA1c < 7% | 4 | 1410 | Risk Ratio (M‐H, Random, 95% CI) | 3.25 [1.97, 5.36] |
| 3 Hypoglycaemia | 4 | 1410 | Risk Ratio (M‐H, Random, 95% CI) | 1.66 [1.15, 2.40] |
| 4 Weight change | 4 | 1410 | Mean Difference (IV, Random, 95% CI) | ‐1.33 [‐2.38, ‐0.27] |
| 5 Adverse events | Other data | No numeric data | ||
| 6 Systolic blood pressure | 3 | 1062 | Mean Difference (IV, Random, 95% CI) | ‐2.42 [‐4.90, 0.05] |
| 7 Fasting plasma glucose (mmol/L) | 4 | 1410 | Mean Difference (IV, Random, 95% CI) | ‐2.21 [‐2.49, ‐1.93] |
| 8 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 9 Triglycerides (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10 Total cholesterol (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11 HDL‐cholesterol (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12 LDL‐cholesterol (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 13 Beta‐cell function | Other data | No numeric data |
Analysis 8.1.
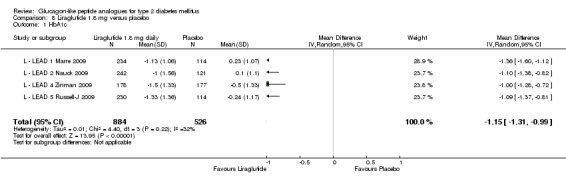
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 1 HbA1c.
Analysis 8.2.

Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 2 HbA1c < 7%.
Analysis 8.3.
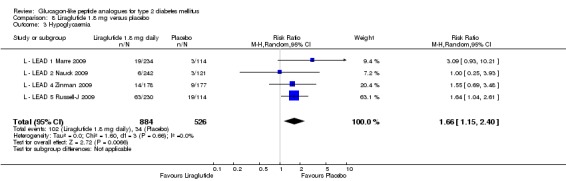
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 3 Hypoglycaemia.
Analysis 8.4.
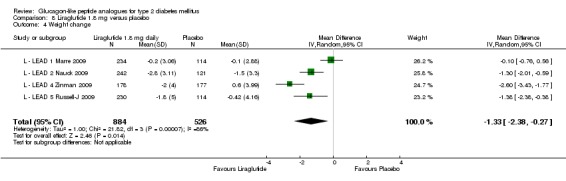
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 4 Weight change.
Analysis 8.5.
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 5 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.8 mg daily | Placebo |
|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | withdrawal due to adverse events | 4% | 5% |
| L ‐ LEAD 1 Marre 2009 | no significant differences across groups for: blood pressure; no significant changes is: ophthalmoscopy, biochemistry, urinalysis, haematology, ECG no deaths |
||
| L ‐ LEAD 1 Marre 2009 | nausea | 1.8% (lowest) | |
| L ‐ LEAD 1 Marre 2009 | vomiting | ||
| L ‐ LEAD 1 Marre 2009 | diarrhoea | ||
| L ‐ LEAD 1 Marre 2009 | serious adverse events (mostly judged to be unlikely to be related to study medication) | 5% | 3% |
| L ‐ LEAD 1 Marre 2009 | liraglutide auto‐antibodies | 9 to13%, no effect on HbA1c | |
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to adverse events | 12% | 2% |
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to nausea/vomiting/diarrhoea | 8% | 0 |
| L ‐ LEAD 2 Nauck 2009 | no significant differences across groups for: physical examination findings, laboratory analyses, ECG, ophthalmoscopy | ||
| L ‐ LEAD 2 Nauck 2009 | any GI event | 44% | 17% |
| L ‐ LEAD 2 Nauck 2009 | nausea | 19% | NR |
| L ‐ LEAD 2 Nauck 2009 | vomiting | 5 to 7% | 1% |
| L ‐ LEAD 2 Nauck 2009 | diarrhoea | 15% | 4% |
| L ‐ LEAD 2 Nauck 2009 | serious adverse events: 2 deaths unrelated to liraglutide treatment | ||
| L ‐ LEAD 2 Nauck 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 2 Nauck 2009 | anti‐liraglutide antibodies | NR | NR |
| L ‐ LEAD 4 Zinman 2009 | withdrawal due to adverse events | 15% | 3% |
| L ‐ LEAD 4 Zinman 2009 | withdrawal due to nausea/vomiting/diarrhoea | 11% | 0 |
| L ‐ LEAD 4 Zinman 2009 | no significant differences across groups for: physical examination findings, laboratory analyses, ECG, ophthalmoscopy, cardiovascular adverse events | ||
| L ‐ LEAD 4 Zinman 2009 | any GI event | 56% | 19% |
| L ‐ LEAD 4 Zinman 2009 | nausea | 40% | NR |
| L ‐ LEAD 4 Zinman 2009 | vomiting | 17% | NR |
| L ‐ LEAD 4 Zinman 2009 | diarrhoea | NR | NR |
| L ‐ LEAD 4 Zinman 2009 | serious adverse events | 10 events in 7 participants | 13 events in 12 participants |
| L ‐ LEAD 4 Zinman 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 4 Zinman 2009 | anti‐liraglutide antibodies | 6.7% (no effect on HbA1c) | |
| L ‐ LEAD 5 Russell‐J 2009 | withdrawal due to adverse events | 5% | 0.9% |
| L ‐ LEAD 5 Russell‐J 2009 | no significant differences across groups for: nasopharyngitis, headache; no pancreatitis | ||
| L ‐ LEAD 5 Russell‐J 2009 | nausea | 13.9% | 3.5% |
| L ‐ LEAD 5 Russell‐J 2009 | diarrhoea | 10.% | 5.3% |
| L ‐ LEAD 5 Russell‐J 2009 | dyspepsia | 6.5% | 0.9% |
| L ‐ LEAD 5 Russell‐J 2009 | vomiting | 6.5% | 3.5% |
| L ‐ LEAD 5 Russell‐J 2009 | serious adverse events | 4% | 7% |
| L ‐ LEAD 5 Russell‐J 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 5 Russell‐J 2009 | anti‐liraglutide antibodies | 9.8% (no effect on HbA1c) | |
| L ‐ LEAD 5 Russell‐J 2009 |
Analysis 8.6.
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 6 Systolic blood pressure.
Analysis 8.7.
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 7 Fasting plasma glucose (mmol/L).
Analysis 8.8.
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 8 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.8 mg daily | Placebo | Comments |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | average of values obtained 90 min after breakfast, lunch and evening meal | ‐2.7 mmol/L | ‐0.4 mmol/L | Liraglutide 1.8 mg versus placebo: P < 0.0001 |
| L ‐ LEAD 2 Nauck 2009 | from self‐monitored 7‐point plasma glucose measurements | ‐2.6 mmol/L | ‐0.6 mmol/L | Liraglutide 1.8 mg versus placebo: P < 0.001 |
| L ‐ LEAD 4 Zinman 2009 | from self‐monitored 7‐point plasma glucose measurements | ‐2.7 mmol/L | ‐0.8 mmol/L | Liraglutide 1.8 mg versus placebo: P < 0.001; the postprandial increment (postmeal value minus premeal) was significantly reduced over breakfast with liraglutide treatment (‐0.9, ‐0.8, ‐0.3 mmol/L respectively; P < 0.05 for both liraglutide groups versus placebo) but not for lunch and dinner. |
| L ‐ LEAD 5 Russell‐J 2009 | from self‐monitored 7‐point plasma glucose measurements | ‐1.81 mmol/L | ‐0.03 mmol/L | Liraglutide 1.8 mg versus placebo: P < 0.0001; there was a statistically significantly higher likelihood of achieving ADA targets for PPG (≤ 10 mmol/l) (P < 0.0001) with liraglutide versus placebo. |
Analysis 8.9.
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 9 Triglycerides (mmol/L).
Analysis 8.10.
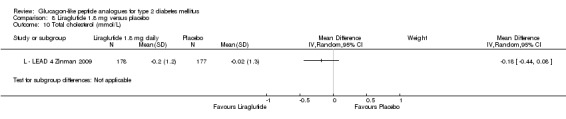
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 10 Total cholesterol (mmol/L).
Analysis 8.11.
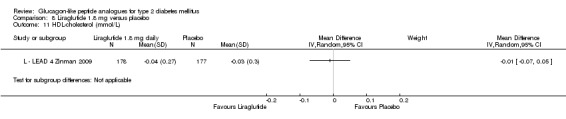
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 11 HDL‐cholesterol (mmol/L).
Analysis 8.12.
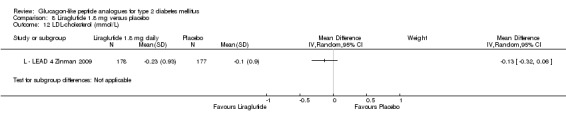
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 12 LDL‐cholesterol (mmol/L).
Analysis 8.13.
Comparison 8 Liraglutide 1.8 mg versus placebo, Outcome 13 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.8 mg daily | Placebo |
|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | HOMA‐B (%) | 91% SE 108.2 (+35%), P = 0.051 versus placebo | 52% SE 107.3 (‐4%) |
| L ‐ LEAD 1 Marre 2009 | proinsulin‐to‐insulin ratio | 0.36 SE 0.2 (‐0.12), p ≤ 0.02 versus placebo | 0.46 SE 0.29 (+0.02) |
| L ‐ LEAD 1 Marre 2009 | |||
| L ‐ LEAD 2 Nauck 2009 | HOMA‐B (%) | +28% | ‐2% |
| L ‐ LEAD 2 Nauck 2009 | proinsulin‐to‐insulin ratio | ‐0.1, P < 0.0001 versus placebo | +0.1 |
| L ‐ LEAD 2 Nauck 2009 | proinsulin‐to‐C‐peptide ratio | ||
| L ‐ LEAD 4 Zinman 2009 | HOMA‐B (%) | +27% SD 56, P < 0.05 vs placebo | 6% SD 60 |
| L ‐ LEAD 4 Zinman 2009 | proinsulin‐to‐insulin ratio | ‐0.085 SD 3.47, P < 0.05 vs placebo | 0.036 SD 39 |
| L ‐ LEAD 4 Zinman 2009 | proinsulin‐to‐C‐peptide ratio | ‐0.008 SD 0.01, P < 0.05 vs placebo | ‐0.002 SD 0.01 |
| L ‐ LEAD 5 Russell‐J 2009 | proinsulin‐to‐C‐peptide ratio | ‐0.00671 (95% CI: ‐0.00964, ‐0.00377, P < 0.0001) versus placebo | |
| L ‐ LEAD 5 Russell‐J 2009 | |||
| L ‐ LEAD 5 Russell‐J 2009 |
Comparison 9.
Liraglutide 1.2 mg versus 1.8 mg
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c | 4 | 1739 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.03, 0.23] |
| 2 Patients reaching HbA1c < 7% | 5 | 2206 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.84, 1.17] |
| 3 Weight | 4 | 1739 | Mean Difference (IV, Random, 95% CI) | 0.48 [0.16, 0.80] |
| 4 Systolic blood pressure | 4 | 1739 | Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐1.48, 1.04] |
Comparison 10.
Liraglutide versus insulin glargine
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Weight change | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5 Adverse events | Other data | No numeric data | ||
| 6 Systolic blood pressure | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 9 Beta‐cell function | Other data | No numeric data |
Analysis 10.1.
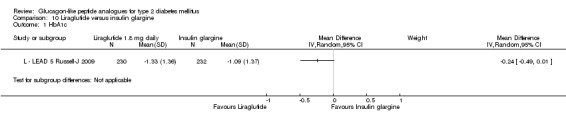
Comparison 10 Liraglutide versus insulin glargine, Outcome 1 HbA1c.
Analysis 10.2.
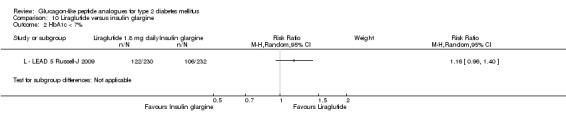
Comparison 10 Liraglutide versus insulin glargine, Outcome 2 HbA1c < 7%.
Analysis 10.3.
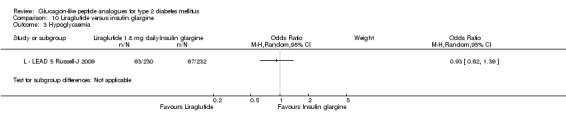
Comparison 10 Liraglutide versus insulin glargine, Outcome 3 Hypoglycaemia.
Analysis 10.4.
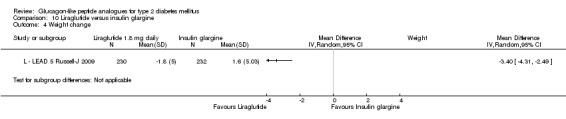
Comparison 10 Liraglutide versus insulin glargine, Outcome 4 Weight change.
Analysis 10.5.
Comparison 10 Liraglutide versus insulin glargine, Outcome 5 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.8 mg daily | Insulin glargine |
|---|---|---|---|
| L ‐ LEAD 5 Russell‐J 2009 | withdrawal due to adverse events | 5% | 2% |
| L ‐ LEAD 5 Russell‐J 2009 | no significant differences across groups for: nasopharyngitis, headache; no pancreatitis | ||
| L ‐ LEAD 5 Russell‐J 2009 | nausea | 13.9% | 1.3% |
| L ‐ LEAD 5 Russell‐J 2009 | diarrhoea | 10.% | 1.3% |
| L ‐ LEAD 5 Russell‐J 2009 | dyspepsia | 6.5% | 1.7% |
| L ‐ LEAD 5 Russell‐J 2009 | vomiting | 6.5% | 0.4% |
| L ‐ LEAD 5 Russell‐J 2009 | serious adverse events | 4% | 7% |
| L ‐ LEAD 5 Russell‐J 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 5 Russell‐J 2009 | anti‐liraglutide antibodies | 9.8% (no effect on HbA1c) |
Analysis 10.6.
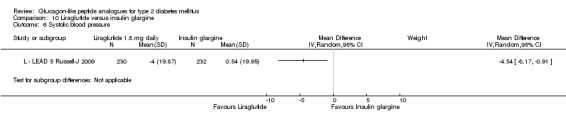
Comparison 10 Liraglutide versus insulin glargine, Outcome 6 Systolic blood pressure.
Analysis 10.7.
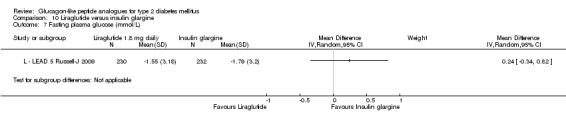
Comparison 10 Liraglutide versus insulin glargine, Outcome 7 Fasting plasma glucose (mmol/L).
Analysis 10.8.
Comparison 10 Liraglutide versus insulin glargine, Outcome 8 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.8 mg daily | Insulin glargine | Comments |
|---|---|---|---|---|
| L ‐ LEAD 5 Russell‐J 2009 | from self‐monitored 7‐point plasma glucose measurements | ‐1.81 mmol/L | ‐1.61 mmol/L | no significant difference |
Analysis 10.9.
Comparison 10 Liraglutide versus insulin glargine, Outcome 9 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.8 mg |
|---|---|---|
| L ‐ LEAD 5 Russell‐J 2009 | proinsulin‐to‐C‐peptide ratio | ‐0.00366 (95% CI ‐0.0057 to ‐0.00136, P = 0.0019) versus insulin glargine; ‐0.00671 (95% CI ‐0.00964 to ‐0.00377, P < 0.0001) versus placebo |
Comparison 11.
Liraglutide 1.2 mg daily versus thiazolidinedione
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia (mild/moderate/overall) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Severe hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6 Adverse events | Other data | No numeric data | ||
| 7 Systolic blood pressure (mm Hg) | Other data | No numeric data | ||
| 8 Diastolic blood pressure (mm Hg) | Other data | No numeric data | ||
| 9 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 11 Beta‐cell function | Other data | No numeric data |
Analysis 11.1.
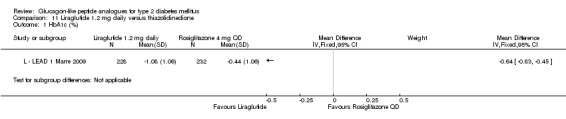
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 1 HbA1c (%).
Analysis 11.2.
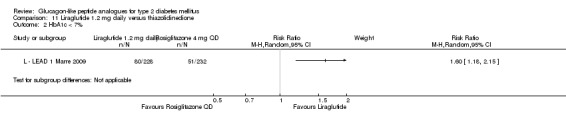
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 2 HbA1c < 7%.
Analysis 11.3.
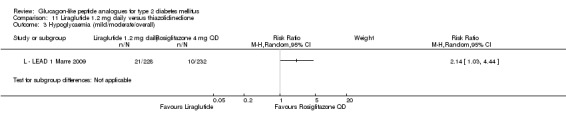
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 3 Hypoglycaemia (mild/moderate/overall).
Analysis 11.4.
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 4 Severe hypoglycaemia.
Severe hypoglycaemia
| Study | Liraglutide 1.2 mg daily | Rosiglitazone 4 mg QD |
|---|---|---|
| L ‐ LEAD 1 Marre 2009 | NR | NR |
Analysis 11.5.
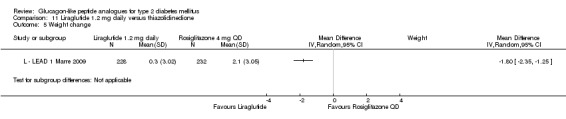
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 5 Weight change.
Analysis 11.6.
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 6 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.2 mg daily | Rosiglitazone 4 mg QD |
|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | withdrawal due to adverse events | 11 (5%) | 7 (3%) |
| L ‐ LEAD 1 Marre 2009 | overall adverse events | NR | NR |
| L ‐ LEAD 1 Marre 2009 | nausea | 24 (10.5%) | NR |
| L ‐ LEAD 1 Marre 2009 | vomiting | 10 (4.4%) | NR |
| L ‐ LEAD 1 Marre 2009 | diarrhoea | 18 (7.9%) | NR |
| L ‐ LEAD 1 Marre 2009 | serious adverse events (mostly judged to be unlikely to be related to study medication) | 9 (4%) | 7 (3%) |
| L ‐ LEAD 1 Marre 2009 | liraglutide auto‐antibodies | 9 to 13%, no effect on HbA1c | |
| L ‐ LEAD 1 Marre 2009 | deaths | None | None |
| L ‐ LEAD 1 Marre 2009 | pancreatitis | None | None |
| L ‐ LEAD 1 Marre 2009 |
Analysis 11.7.
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 7 Systolic blood pressure (mm Hg).
Systolic blood pressure (mm Hg)
| Study | Description | Liraglutide 1.2 mg daily | Rosiglitazone 4 mg QD | p values |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | Change from baseline | ‐2.6 to ‐2.8 | 0.9 to 2.3 | P = NS between groups |
Analysis 11.8.
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 8 Diastolic blood pressure (mm Hg).
Diastolic blood pressure (mm Hg)
| Study | Description | Liraglutide 1.2 mg daily | Rosiglitazone 4 mg QD | p values |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | Change from baseline | ‐0.7 to ‐1.4 | ‐0.7 to ‐1.4 | P = NS between groups |
Analysis 11.9.
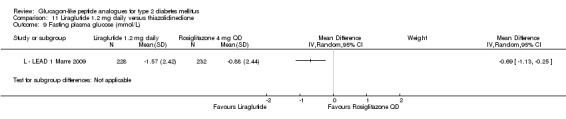
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 9 Fasting plasma glucose (mmol/L).
Analysis 11.10.
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 10 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.2 mg daily | Rosiglitazone 4 mg QD | Comments |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | average of values obtained 90 min after breakfast, lunch and evening meal | ‐2.5 mmol/L | ‐1.8 mmol/L | P = 0.043 1.2 mg versus rosiglitazone |
Analysis 11.11.
Comparison 11 Liraglutide 1.2 mg daily versus thiazolidinedione, Outcome 11 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.2 mg | TZD |
|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | HOMA‐B (%) | 99% SE 184.3 (+28%) | 59% SE 63.3 (+13%) |
| L ‐ LEAD 1 Marre 2009 | proinsulin‐to‐insulin ratio | 0.33 SE 0.2 (‐0.12) | 0.40 SE 0.2 (‐0.05) |
Comparison 12.
Liraglutide 1.8 mg daily versus thiazolidinedione
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 Severe hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change (kg) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6 Adverse events | Other data | No numeric data | ||
| 7 Systolic blood pressure (mm Hg) | Other data | No numeric data | ||
| 8 Diastolic blood pressure (mm Hg) | Other data | No numeric data | ||
| 9 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 11 Beta‐cell function | Other data | No numeric data |
Analysis 12.1.
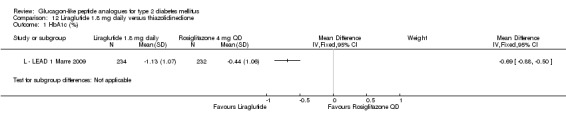
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 1 HbA1c (%).
Analysis 12.2.
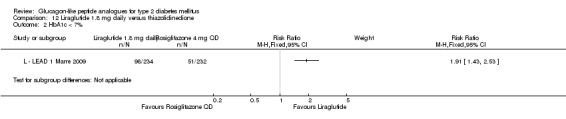
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 2 HbA1c < 7%.
Analysis 12.3.
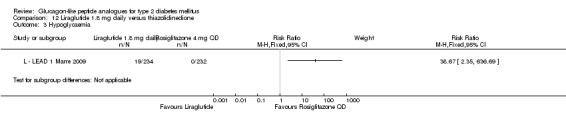
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 3 Hypoglycaemia.
Analysis 12.4.
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 4 Severe hypoglycaemia.
Severe hypoglycaemia
| Study | Liraglutide 1.8 mg daily | Rosiglitazone 4 mg QD |
|---|---|---|
| L ‐ LEAD 1 Marre 2009 | 1 | NR |
Analysis 12.5.
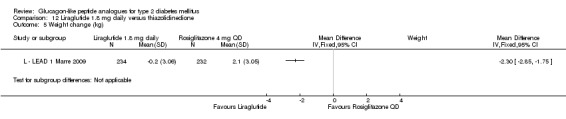
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 5 Weight change (kg).
Analysis 12.6.
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 6 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.8 mg daily | Rosiglitazone 4 mg QD |
|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | withdrawal due to adverse events | 9 (4%) | 7 (3%) |
| L ‐ LEAD 1 Marre 2009 | overall adverse events | NR | NR |
| L ‐ LEAD 1 Marre 2009 | nausea | NR | NR |
| L ‐ LEAD 1 Marre 2009 | vomiting | NR | NR |
| L ‐ LEAD 1 Marre 2009 | NR | NR | |
| L ‐ LEAD 1 Marre 2009 | serious adverse events (mostly judged to be unlikely to be related to study medication) | 12 (5%) | 7 (3%) |
| L ‐ LEAD 1 Marre 2009 | liraglutide auto‐antibodies | 9 to 13%, no effect on HbA1c | |
| L ‐ LEAD 1 Marre 2009 | deaths | None | None |
| L ‐ LEAD 1 Marre 2009 | pancreatitis | None | None |
Analysis 12.7.
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 7 Systolic blood pressure (mm Hg).
Systolic blood pressure (mm Hg)
| Study | Description | Liraglutide 1.8 mg daily | Rosiglitazone 4 mg QD | p values |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | Change from baseline | ‐2.6 to ‐2.8 | 0.9 to 2.3 | P = NS between groups |
Analysis 12.8.
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 8 Diastolic blood pressure (mm Hg).
Diastolic blood pressure (mm Hg)
| Study | Description | Liraglutide 1.8 mg daily | Rosiglitazone 4 mg QD | p values |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | Change from baseline | ‐0.7 to 1.4 | ‐0.7 to ‐1.4 | P = NS between groups |
Analysis 12.9.
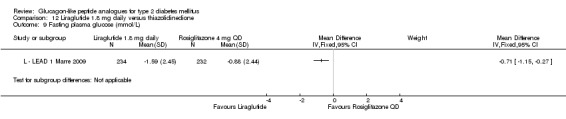
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 9 Fasting plasma glucose (mmol/L).
Analysis 12.10.
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 10 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.8 mg daily | Rosiglitazone 4 mg QD | Comments |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | average of values obtained 90 min after breakfast, lunch and evening meal | ‐2.7 mmol/L | ‐1.8 mmol/L | P = 0.0022 1.8 mg versus rosiglitazone |
Analysis 12.11.
Comparison 12 Liraglutide 1.8 mg daily versus thiazolidinedione, Outcome 11 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.8 mg daily | Rosiglitazone 4 mg QD | Difference between groups |
|---|---|---|---|---|
| L ‐ LEAD 1 Marre 2009 | HOMA‐B (%) | 91% SE 108.2 (+35%) | 59% SE 63.3 (+13%) | 30, 95% CI 2.00 to 58.6, p ≤ 0.05 |
| L ‐ LEAD 1 Marre 2009 | proinsulin‐to‐insulin ratio | 0.36 SE 0.2 (‐0.12) | 0.40 SE 0.2 (‐0.05) | ‐0.05, 95% CI ‐0.10 to ‐0.01, p ≤ 0.05 |
Comparison 13.
Liraglutide 1.2 mg daily versus DPP‐4 inhibitors
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia (mild/moderate/overall) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Severe hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change (kg) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6 Adverse events | Other data | No numeric data | ||
| 7 Systolic blood pressure (mm Hg) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8 Diastolic blood pressure (mm Hg) | Other data | No numeric data | ||
| 9 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 11 Beta‐cell function | Other data | No numeric data |
Analysis 13.1.
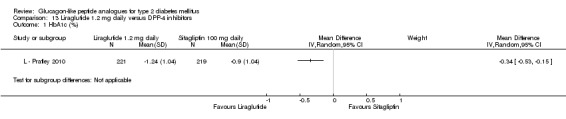
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 1 HbA1c (%).
Analysis 13.2.
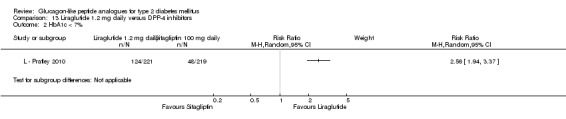
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 2 HbA1c < 7%.
Analysis 13.3.
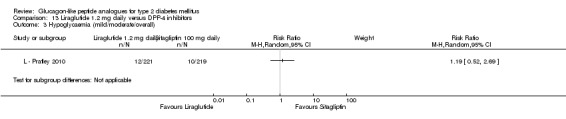
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 3 Hypoglycaemia (mild/moderate/overall).
Analysis 13.4.
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 4 Severe hypoglycaemia.
Severe hypoglycaemia
| Study | Liraglutide 1.2 mg daily | Sitagliptin 100 mg daily |
|---|---|---|
| L ‐ Pratley 2010 | 1 (0.45%) | None |
Analysis 13.5.
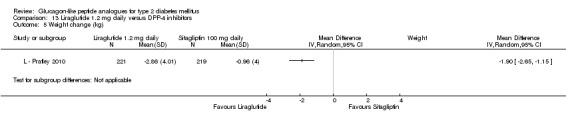
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 5 Weight change (kg).
Analysis 13.6.
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 6 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.2 mg daily | Sitagliptin 100 mg daily |
|---|---|---|---|
| L ‐ Pratley 2010 | withdrawal due to adverse events | 14 (6.3%) | 4 (1.8%) |
| L ‐ Pratley 2010 | overall adverse events | 146 (66%) | 127 (58%) |
| L ‐ Pratley 2010 | nausea | 46 (21%) | 10 (5%) |
| L ‐ Pratley 2010 | vomiting | 17 (8%) | 9 (4%) |
| L ‐ Pratley 2010 | diarrhoea | 16 (7%) | 10 (5%) |
| L ‐ Pratley 2010 | other gastrointestinal adverse events | 17 (8%) | 6 (3%) |
| L ‐ Pratley 2010 | serious adverse events (mostly judged to be unlikely to be related to study medication) | 6 (3%) | 4 (2%) |
| L ‐ Pratley 2010 | liraglutide auto‐antibodies | NR | NR |
| L ‐ Pratley 2010 | deaths | 0 | 1 (< 1%) unrelated to drug |
| L ‐ Pratley 2010 | pancreatitis | None | None |
Analysis 13.7.
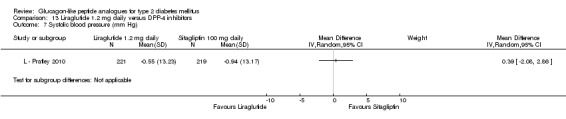
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 7 Systolic blood pressure (mm Hg).
Analysis 13.8.
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 8 Diastolic blood pressure (mm Hg).
Diastolic blood pressure (mm Hg)
| Study | Description | Liraglutide 1.2 mg daily | Sitagliptin 100 mg daily | p values |
|---|---|---|---|---|
| L ‐ Pratley 2010 | Change from baseline | ‐0.71 | ‐1.78 | P = NS between groups |
Analysis 13.9.
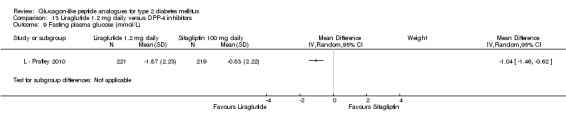
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 9 Fasting plasma glucose (mmol/L).
Analysis 13.10.
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 10 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.2 mg daily | Sitagliptin 100 mg daily | Comments |
|---|---|---|---|---|
| L ‐ Pratley 2010 | PPG | NR | NR | Reported in the paper that ' PPG was highly variable suggesting that glucose values were not PPG in many cases |
Analysis 13.11.
Comparison 13 Liraglutide 1.2 mg daily versus DPP‐4 inhibitors, Outcome 11 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.2 mg daily | Sitagliptin 100 mg daily | Difference between groups |
|---|---|---|---|---|
| L ‐ Pratley 2010 | HOMA‐B (%), mean change from baseline | 27.23 % (95% CI 19.73 to 34.73) | 4.18 % (95% CI ‐3.27 to 11.62) | 23.05% (95% CI 12.95 to 33.15), P < 0.0001 |
| L ‐ Pratley 2010 | HOMA‐IR (%), mean change from baseline | ‐1.06 % (95% CI ‐1.70 to ‐0.42) | ‐0.94 % (95% CI, ‐1.58 to ‐0.30) | ‐0.12% (95% CI ‐0.99 to 0.75), P = 0.7834 |
| L ‐ Pratley 2010 | Fasting insulin (pmol/L) | 5.12 (95% CI ‐4.34 to 14.59) | ‐6.77 (95% CI ‐16.18 to 2.64) | 11.89 (95% CI ‐0.84 to 24.63), P = 0.0672 |
| L ‐ Pratley 2010 | Fasting C‐peptide (nmol/L) | 0.09 (95% CI 0.03 to 0.15) | ‐0.04 (95% CI ‐0.10 to 0.02) | 0.13 (95% CI 0.05 to 0.21), P = 0.0011 |
| L ‐ Pratley 2010 | Fasting proinsulin‐to‐insulin ratio | ‐0.08 (95% CI ‐0.11 to ‐0.05) | ‐0.03 (95% CI ‐0.06 to ‐0.00) | ‐0.05 (95% CI ‐0.09 to ‐0.01), P = 0.0121 |
Comparison 14.
Liraglutide 1.8 mg daily versus DPP‐4 inhibitors
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3 Hypoglycaemia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Severe hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change (kg) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6 Adverse events | Other data | No numeric data | ||
| 7 Systolic blood pressure (mm Hg) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8 Diastolic blood pressure (mm Hg) | Other data | No numeric data | ||
| 9 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 11 Beta‐cell function | Other data | No numeric data |
Analysis 14.1.
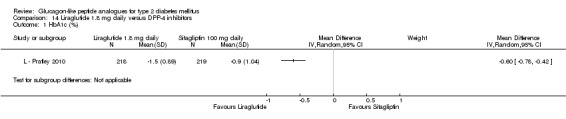
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 1 HbA1c (%).
Analysis 14.2.
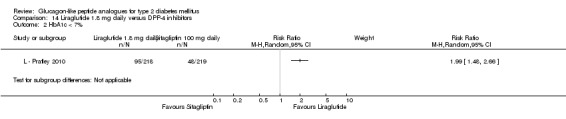
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 2 HbA1c < 7%.
Analysis 14.3.
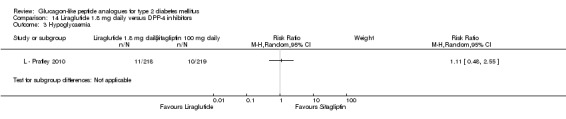
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 3 Hypoglycaemia.
Analysis 14.4.
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 4 Severe hypoglycaemia.
Severe hypoglycaemia
| Study | Liraglutide 1.8 mg daily | Sitagliptin 100 mg daily |
|---|---|---|
| L ‐ Pratley 2010 | NR, presumably none | NR, presumably none |
Analysis 14.5.
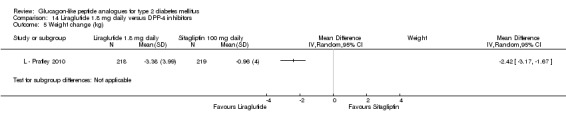
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 5 Weight change (kg).
Analysis 14.6.
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 6 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.8 mg daily | Sitagliptin 100 mg daily |
|---|---|---|---|
| L ‐ Pratley 2010 | withdrawal due to adverse events | 15 (6.9%) | 4 (1.8%) |
| L ‐ Pratley 2010 | overall adverse events | 159 (73%) | 127 (58%) |
| L ‐ Pratley 2010 | nausea | 59 (27%) | 10 (5%) |
| L ‐ Pratley 2010 | vomiting | 21 (10%) | 9 (4%) |
| L ‐ Pratley 2010 | diarrhoea | 25 (11%) | 10 (5%) |
| L ‐ Pratley 2010 | other gastrointestinal adverse events | 11 (5%) | 6 (3%) |
| L ‐ Pratley 2010 | serious adverse events (mostly judged to be unlikely to be related to study medication) | 6 (3%) | 4 (2%) |
| L ‐ Pratley 2010 | liraglutide auto‐antibodies | NR | NR |
| L ‐ Pratley 2010 | deaths | 1 (< 1%) unrelated to drug | 1 (< 1%) unrelated to drug |
| L ‐ Pratley 2010 | pancreatitis | None | None |
Analysis 14.7.
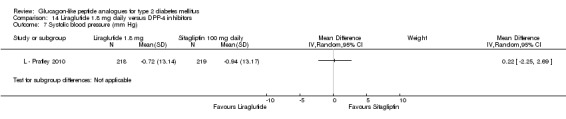
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 7 Systolic blood pressure (mm Hg).
Analysis 14.8.
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 8 Diastolic blood pressure (mm Hg).
Diastolic blood pressure (mm Hg)
| Study | Description | Liraglutide 1.8 mg daily | Sitagliptin 100 mg daily | p values |
|---|---|---|---|---|
| L ‐ Pratley 2010 | Change from baseline | 0.07 | ‐1.78 | P = 0.0210 versus SITA |
Analysis 14.9.
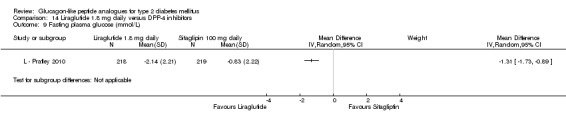
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 9 Fasting plasma glucose (mmol/L).
Analysis 14.10.
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 10 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.8 mg daily | Sitagliptin 100 mg daily | Comments |
|---|---|---|---|---|
| L ‐ Pratley 2010 | PPG | NR | NR | Reported in the paper that ' PPG was highly variable suggesting that glucose values were not PPG in many cases |
Analysis 14.11.
Comparison 14 Liraglutide 1.8 mg daily versus DPP‐4 inhibitors, Outcome 11 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.8 mg daily | Sitagliptin 100 mg daily | Difference between groups |
|---|---|---|---|---|
| L ‐ Pratley 2010 | Fasting insulin (pmol/L) | 1.29 (95% CI ‐8.04 to 10.62) | ‐6.77 (95% CI ‐16.18 to 2.64) | 8.06 (95% CI ‐4.51 to 20.63), P = 0.2083 |
| L ‐ Pratley 2010 | 0.09 (95% CI 0.03 to 0.15) | ‐0.04 (95% CI ‐0.10 to 0.02) | 0.14 (95% CI 0.06 to 0.21), P = 0.0008 | |
| L ‐ Pratley 2010 | Fasting insulin (pmol/L) | 1.29 (95% CI ‐8.04 to 10.62) | ‐6.77 (95% CI ‐16.18 to 2.64) | 8.06 (95% CI ‐4.51 to 20.63), P = 0.2083 |
| L ‐ Pratley 2010 | 0.09 (95% CI 0.03 to 0.15) | ‐0.04 (95% CI ‐0.10 to 0.02) | 0.14 (95% CI 0.06 to 0.21), P = 0.0008 | |
| L ‐ Pratley 2010 | ‐0.10 (95% CI ‐0.12 to ‐0.07) | ‐0.03 (95% CI ‐0.06 to ‐0.00) | ‐0.07 (95% CI ‐0.10 to ‐0.03), P = 0.0004 |
Comparison 15.
Lixisenatide versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | Other data | No numeric data | ||
| 2 HbA1c < 7% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 LIXI 5 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 LIXI 10 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 LIXI 20 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 LIXI 30 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 LIXI 5 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 LIXI 10 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.7 LIXI 20 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.8 LIXI 30 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Symptomatic hypoglycaemia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 LIXI 5 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 LIXI 10 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 LIXI 20 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 LIXI 30 µg QD | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 LIXI 5 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 LIXI 10 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 LIXI 20 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 LIXI 30 µg BID | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Weight change (kg) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4.1 LIXI 5 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 LIXi 10 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 LIXI 20 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 LIXI 30 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 LIXI 5 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 LIXI 10 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 LIXI 20 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.8 LIXI 30 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events | Other data | No numeric data | ||
| 6 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6.1 LIXI 5 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 LIXI 10 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 LIXI 20 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.4 LIXI 30 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.5 LIXI 5 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.6 LIXI 10 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.7 LIXI 20 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.8 LIXI 30 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Post‐prandial glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7.1 LIXI 5μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 LIXI 10μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 LIXI 20μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 LIXI 30μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 LIXI 5μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.6 LIXI 10μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.7 LIXI 20μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.8 LIXI 30μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Average self‐monitored 7‐point blood glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8.1 LIXI 5 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 LIXI 10 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 LIXI 20 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 LIXI 30 μg QD | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 LIXI 5 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.6 LIXI 10 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.7 LIXI 20 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.8 LIXI 30 μg BID | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Analysis 15.1.
Comparison 15 Lixisenatide versus placebo, Outcome 1 HbA1c (%).
HbA1c (%)
| Study | LIXI 5 μg QD | LIXI 10 μg QD | LIXI 20 μg QD | LIXI 30 μg QD | LIXI 5 μg BID | LIXI 10 μg BID | LIXI 20 μg BID | LIXI 30 μg BID | Placebo | p values |
|---|---|---|---|---|---|---|---|---|---|---|
| Lixi ‐ Ratner 2010 | ‐0.47 | ‐0.5 | ‐0.69 | ‐0.76 | ‐0.65 | ‐0.78 | ‐0.75 | ‐0.87 | ‐0.18 | change from baseline to end: LIXI: P < 0.05 for all doses |
Analysis 15.2.

Comparison 15 Lixisenatide versus placebo, Outcome 2 HbA1c < 7%.
Analysis 15.3.
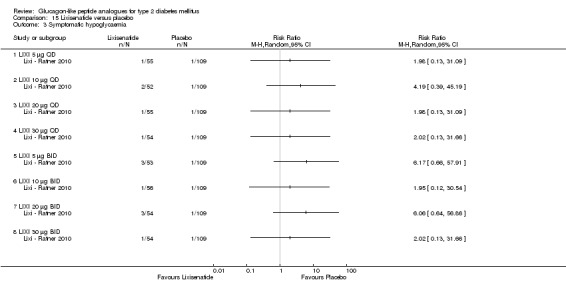
Comparison 15 Lixisenatide versus placebo, Outcome 3 Symptomatic hypoglycaemia.
Analysis 15.4.
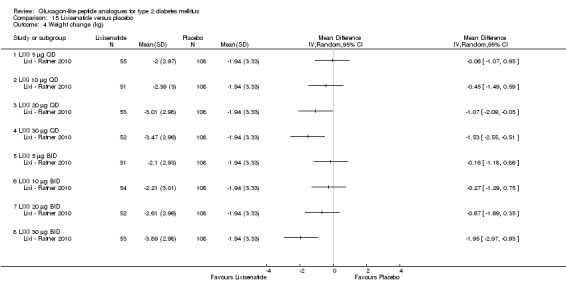
Comparison 15 Lixisenatide versus placebo, Outcome 4 Weight change (kg).
Analysis 15.5.
Comparison 15 Lixisenatide versus placebo, Outcome 5 Adverse events.
Adverse events
| Study | Description | LIXI 5 μg QD | LIXI 10 μg QD | LIXI 20 μg QD | LIXI 30 μg QD | LIXI 5 μg BID | LIXI 10 μg BID | LIXI 20 μg BID | LIXI 30 μg BID | Placebo |
|---|---|---|---|---|---|---|---|---|---|---|
| Lixi ‐ Ratner 2010 | Withdrawals due to adverse events | 1 (1.8%) | 2 (3.8%) | 3 (5.5%) | 6 (11.1%) | 0 | 2 (3.6%) | 8 (14.8%) | 5 (9.3%) | 2 (1.8%) |
| Lixi ‐ Ratner 2010 | Any treatment‐emergent adverse events | 31 (56.4%) | 26 (50.0%) | 37 (67.3%) | 42 (77.8%) | 30 (56.6%) | 32 (57.1%) | 38 (70.4%) | 40 (74.1%) | 65 (59.6%) |
| Lixi ‐ Ratner 2010 | Nausea | 4 (7.3%) | 6 (11.5%) | 14 (25.5%) | 19 (35.2%) | 4 (7.5%) | 8 (14.3%) | 12 (22.2%) | 18 (33.3%) | 5 (4.6%) |
| Lixi ‐ Ratner 2010 | Vomiting | 2 (3.6%) | 3 (5.8%) | 3 (5.5%) | 10 (18.5%) | 3 (5.7%) | 4 (7.1%) | 5 (9.3%) | 2 (3.7%) | 1 (0.9%) |
| Lixi ‐ Ratner 2010 | Diarrhoea | 3 (5.5%) | 4 (7.7%) | 5 (9.1%) | 4 (7.4%) | 3 (5.7%) | 4 (7.1%) | 6 (11.1%) | 14 (25.9%) | 8 (7.3%) |
| Lixi ‐ Ratner 2010 | Headache | 7 (12.7%) | 3 (5.8%) | 7 (12.7%) | 7 (13.0%) | 7 (13.2%) | 5 (8.9%) | 6 (11.1%) | 4 (7.4%) | 11 (10.1%) |
| Lixi ‐ Ratner 2010 | Dizziness | 1 (1.8%) | 4 (7.7%) | 4 (7.3%) | 6 (11.1%) | 3 (5.7%) | 5 (8.9%) | 2 (3.7%) | 5 (9.3%) | 7 (6.4%) |
| Lixi ‐ Ratner 2010 | Any serious treatment‐emergent adverse events | 0 | 1 (1.9%) | 1 (1.8%) | 3 (5.6%) | 0 | 1 (1.8%) | 2 (3.7%) | 0 | 3 (2.8%) |
| Lixi ‐ Ratner 2010 | Pancreatitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Lixi ‐ Ratner 2010 | Severe hypoglycaemia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Analysis 15.6.

Comparison 15 Lixisenatide versus placebo, Outcome 6 Fasting plasma glucose (mmol/L).
Analysis 15.7.
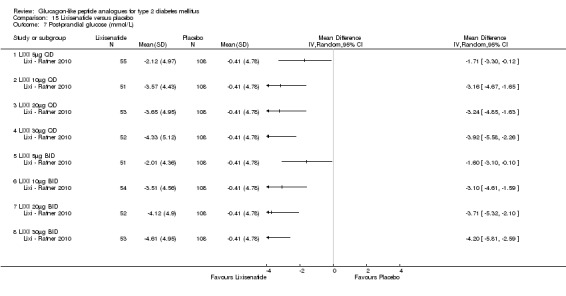
Comparison 15 Lixisenatide versus placebo, Outcome 7 Post‐prandial glucose (mmol/L).
Analysis 15.8.
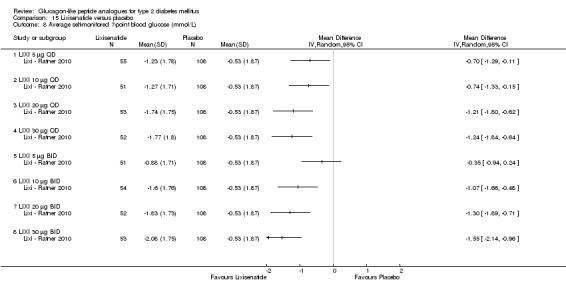
Comparison 15 Lixisenatide versus placebo, Outcome 8 Average self‐monitored 7‐point blood glucose (mmol/L).
Comparison 16.
Liraglutide 1.2 mg versus SU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2 HbA1c (%) | Other data | No numeric data | ||
| 3 HbA1c < 7% | 2 | 946 | Risk Ratio (IV, Random, 95% CI) | 0.98 [0.84, 1.14] |
| 4 Hypoglycaemia | 2 | 946 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 1.72] |
| 5 Weight change | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6 Weight change (kg) | Other data | No numeric data | ||
| 7 Adverse events | Other data | No numeric data | ||
| 8 Systolic blood pressure | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9 Blood pressure (mm Hg) | Other data | No numeric data | ||
| 10 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11 Fasting plasma glucose (mmol/L) | Other data | No numeric data | ||
| 12 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 13 Beta‐cell function | Other data | No numeric data |
Analysis 16.1.
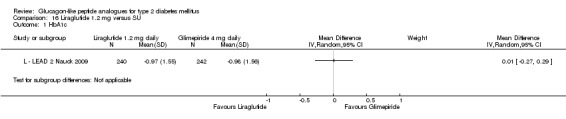
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 1 HbA1c.
Analysis 16.2.
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 2 HbA1c (%).
HbA1c (%)
| Study | Description | Liraglutide 1.2 mg daily | Glimepiride 4 mg daily | Difference between groups |
|---|---|---|---|---|
| L ‐ Yang 2010 | Change in HbA1c | ‐1.36 | ‐1.39 | 0.03, 95% CI ‐0.14 to 0.20 |
Analysis 16.3.
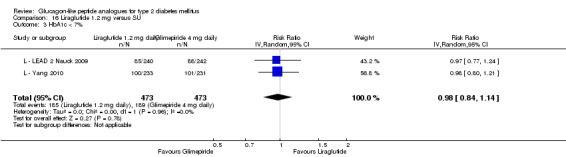
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 3 HbA1c < 7%.
Analysis 16.4.
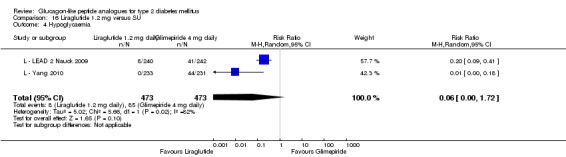
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 4 Hypoglycaemia.
Analysis 16.5.
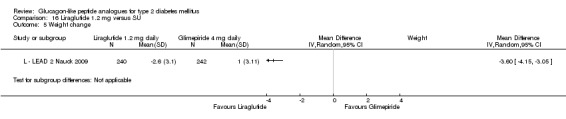
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 5 Weight change.
Analysis 16.6.
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 6 Weight change (kg).
Weight change (kg)
| Study | Description | Liraglutide 1.2 mg daily | Glimepiride 4 mg daily | p value |
|---|---|---|---|---|
| L ‐ Yang 2010 | Change from baseline | ‐2.35 SD 2.4 | 0.08 | LIR 1.2 vs. GLIM: P < 0.0001 |
Analysis 16.7.
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 7 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.2 mg daily | Glimepiride 4 mg daily |
|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to adverse events | 10% | 3% |
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to nausea/vomiting/diarrhoea | 5% | 0 |
| L ‐ LEAD 2 Nauck 2009 | no significant differences across groups for: physical examination findings, laboratory analyses, ECG, ophthalmoscopy | ||
| L ‐ LEAD 2 Nauck 2009 | any GI event | 40% | 17% |
| L ‐ LEAD 2 Nauck 2009 | nausea | 16% | 3 to 4% |
| L ‐ LEAD 2 Nauck 2009 | vomiting | 5 to7% | 1% |
| L ‐ LEAD 2 Nauck 2009 | diarrhoea | 8% | 4% |
| L ‐ LEAD 2 Nauck 2009 | serious adverse events: 2 deaths unrelated to liraglutide treatment; 1 participant each in 1.2 mg liraglutide group and in glimepiride group withdrawn due to acute pancreatitis | ||
| L ‐ LEAD 2 Nauck 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 2 Nauck 2009 | anti‐liraglutide antibodies | NR | NR |
| L ‐ Yang 2010 | withdrawal due to adverse events | 9.4% | 1.3% |
| L ‐ Yang 2010 | serious adverse events | 1.7 to 3.4% | 1.7 to 3.4% |
| L ‐ Yang 2010 | pancreatitis | none | none |
| L ‐ Yang 2010 | deaths | none | none |
| L ‐ Yang 2010 | anti‐liraglutide antibodies | n = 8 from all liraglutide groups | ‐ |
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 |
Analysis 16.8.
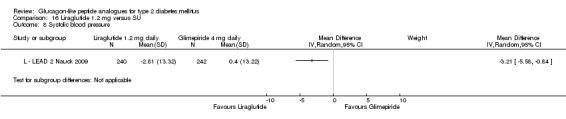
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 8 Systolic blood pressure.
Analysis 16.9.
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 9 Blood pressure (mm Hg).
Blood pressure (mm Hg)
| Study | Description | Liraglutide 1.2 mg daily | Glimepiride 4 mg daily | p values |
|---|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | Systolic blood pressure, change from baseline | ‐2.81 SD 13.32 | 0.4 SD 13.22 | NR |
| L ‐ LEAD 2 Nauck 2009 | Diastolic blood pressure, change from baseline | No change | No change | NR |
| L ‐ Yang 2010 | Systolic blood pressure, change from baseline | Reduction of more than 3 mm Hg | Reduction of 0.91 mm Hg | Reduction in the liraglutide 1.2 group was significantly higher than that in the glimepiride; p value = NR |
| L ‐ Yang 2010 | Diastolic blood pressure, change from baseline | A slight decrease in mean diastolic BP was observed | A slight decrease in mean diastolic BP was observed | NR |
Analysis 16.10.
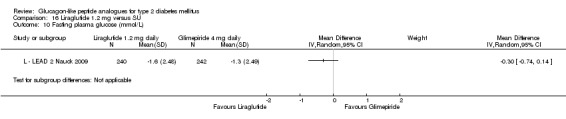
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 10 Fasting plasma glucose (mmol/L).
Analysis 16.11.
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 11 Fasting plasma glucose (mmol/L).
Fasting plasma glucose (mmol/L)
| Study | Description | Liraglutide 1.2 mg daily | Glimepiride 4 mg daily | p values |
|---|---|---|---|---|
| L ‐ Yang 2010 | Change from baseline | ‐2.05 | ‐2.18 | NR |
Analysis 16.12.
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 12 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.2 mg daily | Glimepiride 4 mg daily | Comments |
|---|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | Self‐monitored 7‐point plasma glucose measurements | ‐2.3 mmol/L | ‐2.5 mmol/L | no significant difference between groups |
| L ‐ Yang 2010 | Self‐monitored 7‐point plasma glucose measurements | ‐3.03 mmol/L | ‐2.6 mmol/L | LIR 1.2 vs. GLIM: P = NS |
Analysis 16.13.
Comparison 16 Liraglutide 1.2 mg versus SU, Outcome 13 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.2 mg daily | Glimepiride 4 mg daily | Difference between groups |
|---|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | HOMA‐B (%) | +23% | +25% | No significant difference between liraglutide and glimepiride groups |
| L ‐ LEAD 2 Nauck 2009 | proinsulin‐to‐insulin ratio | ‐0.1, P < 0.0001 versus placebo | similar to liraglutide groups but no value stated | No significant difference between liraglutide and glimepiride groups |
| L ‐ Yang 2010 | HOMA‐B | Increases between 14 and 21% points observed in all treatment groups. | see previous column | No significant difference between liraglutide and glimepiride groups |
| L ‐ Yang 2010 | Proinsulin‐to‐insulin ratio | A slight decrease between 0.06 and 0.11 observed in all treatment groups. | see previous column | No significant difference between liraglutide and glimepiride groups |
Comparison 17.
Liraglutide 1.8 mg daily versus SU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2 HbA1c (%) | Other data | No numeric data | ||
| 3 HbA1c < 7% | 2 | 949 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.94, 1.26] |
| 4 Hypoglycaemia | 2 | 949 | Risk Ratio (M‐H, Random, 95% CI) | 0.13 [0.07, 0.25] |
| 5 Weight change (kg) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6 Weight change (kg) | Other data | No numeric data | ||
| 7 Adverse events | Other data | No numeric data | ||
| 8 Systolic blood pressure (mm Hg) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9 Blood pressure (mm Hg) | Other data | No numeric data | ||
| 10 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 11 Fasting plasma glucose (mmol/L) | Other data | No numeric data | ||
| 12 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 13 Beta‐cell function | Other data | No numeric data |
Analysis 17.1.
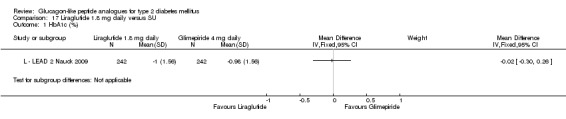
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 1 HbA1c (%).
Analysis 17.2.
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 2 HbA1c (%).
HbA1c (%)
| Study | Description | Liraglutide 1.8 mg daily | Glimepiride 4 mg daily | Difference between groups |
|---|---|---|---|---|
| L ‐ Yang 2010 | Change in HbA1c | ‐1.45 | ‐1.39 | ‐0.06, 95% CI ‐0.23 to 0.11 |
Analysis 17.3.
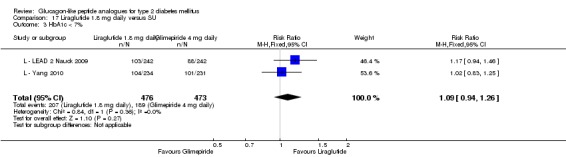
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 3 HbA1c < 7%.
Analysis 17.4.
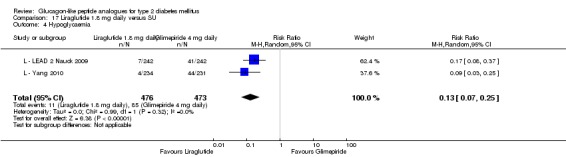
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 4 Hypoglycaemia.
Analysis 17.5.
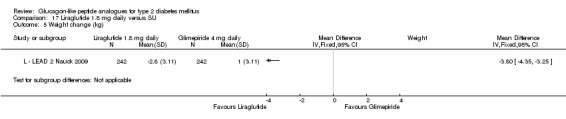
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 5 Weight change (kg).
Analysis 17.6.
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 6 Weight change (kg).
Weight change (kg)
| Study | Description | Liraglutide 1.8 mg daily | Glimepiride 4 mg daily | p value |
|---|---|---|---|---|
| L ‐ Yang 2010 | Change from baseline | ‐2.44 SD 2.6 | 0.08 | LIR 1.8 vs. GLIM: P < 0.0001 |
Analysis 17.7.
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 7 Adverse events.
Adverse events
| Study | Description | Liraglutide 1.8 mg daily | Glimepiride 4 mg daily |
|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to adverse events | 12% | 3% |
| L ‐ LEAD 2 Nauck 2009 | withdrawal due to nausea/vomiting/diarrhoea | 8% | 0 |
| L ‐ LEAD 2 Nauck 2009 | no significant differences across groups for: physical examination findings, laboratory analyses, ECG, ophthalmoscopy | ||
| L ‐ LEAD 2 Nauck 2009 | any GI event | 44% | 17% |
| L ‐ LEAD 2 Nauck 2009 | nausea | 19% | 3 to 4% |
| L ‐ LEAD 2 Nauck 2009 | vomiting | 5 to 7% | 1% |
| L ‐ LEAD 2 Nauck 2009 | diarrhoea | 15% | 4% |
| L ‐ LEAD 2 Nauck 2009 | serious adverse events: 2 deaths unrelated to liraglutide treatment; 1 participant each in 1.2 mg liraglutide group and in glimepiride group withdrawn due to acute pancreatitis | ||
| L ‐ LEAD 2 Nauck 2009 | injection site reactions | NR | NR |
| L ‐ LEAD 2 Nauck 2009 | anti‐liraglutide antibodies | NR | NR |
| L ‐ Yang 2010 | withdrawal due to adverse events | 12.9% | 1.3% |
| L ‐ Yang 2010 | serious adverse events | 1.7 to 3.4% | 1.7 to 3.4% |
| L ‐ Yang 2010 | pancreatitis | none | none |
| L ‐ Yang 2010 | deaths | none | none |
| L ‐ Yang 2010 | anti‐liraglutide antibodies | n = 8 from all liraglutide groups | ‐ |
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 | |||
| L ‐ Yang 2010 |
Analysis 17.8.
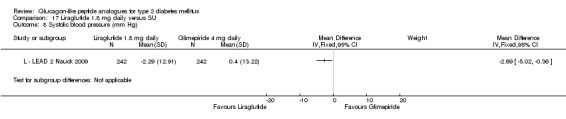
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 8 Systolic blood pressure (mm Hg).
Analysis 17.9.
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 9 Blood pressure (mm Hg).
Blood pressure (mm Hg)
| Study | Description | Liraglutide 1.8 mg daily | Glimepiride 4 mg daily | p values |
|---|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | Systolic blood pressure, change from baseline | ‐2.29 SD 12.91 | 0.4 SD 13.22 | NR |
| L ‐ LEAD 2 Nauck 2009 | Diastolic blood pressure, change from baseline | No change | No change | NR |
| L ‐ Yang 2010 | Systolic blood pressure, change from baseline | Reduction of more than 3 mm Hg | Reduction of 0.91 mm Hg | Reduction in the liraglutide 1.2 group was significantly higher than that in the glimepiride; p value = NR |
| L ‐ Yang 2010 | Diastolic blood pressure, change from baseline | A slight decrease in mean diastolic BP was observed | A slight decrease in mean diastolic BP was observed | NR |
Analysis 17.10.
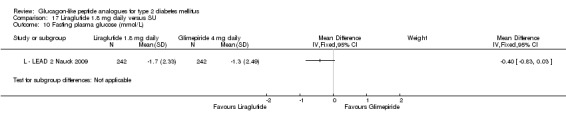
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 10 Fasting plasma glucose (mmol/L).
Analysis 17.11.
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 11 Fasting plasma glucose (mmol/L).
Fasting plasma glucose (mmol/L)
| Study | Description | Liraglutide 1.8 mg daily | Glimepiride 4 mg daily | p values |
|---|---|---|---|---|
| L ‐ Yang 2010 | Change from baseline | ‐2.12 | ‐2.18 | NR |
Analysis 17.12.
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 12 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Liraglutide 1.8 mg daily | Glimepiride 4 mg daily | Comments |
|---|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | Self‐monitored 7‐point plasma glucose measurements | ‐2.6 mmol/L | ‐2.5 mmol/L | no significant difference between groups |
| L ‐ Yang 2010 | Self‐monitored 7‐point plasma glucose measurements | ‐3.51 mmol/L | ‐2.6 mmol/L | LIR 1.8 vs. GLIM: P < 0.0001 |
Analysis 17.13.
Comparison 17 Liraglutide 1.8 mg daily versus SU, Outcome 13 Beta‐cell function.
Beta‐cell function
| Study | Description | Liraglutide 1.8 mg daily | Glimepiride 4 mg daily | Difference between groups |
|---|---|---|---|---|
| L ‐ LEAD 2 Nauck 2009 | HOMA‐B (%) | +28% | +25% | No significant difference between liraglutide and glimepiride groups |
| L ‐ LEAD 2 Nauck 2009 | Proinsulin‐to‐insulin ratio | ‐0.1, P < 0.0001 versus placebo | similar to liraglutide groups but no value stated | No significant difference between liraglutide and glimepiride groups |
| L ‐ Yang 2010 | HOMA‐B | Increases between 14% and 21% points observed in all treatment groups. | see previous column | No significant difference between liraglutide and glimepiride groups |
| L ‐ Yang 2010 | Proinsulin‐to‐insulin ratio | A slight decrease between 0.06 and 0.11 observed in all treatment groups. | see previous column | No significant difference between liraglutide and glimepiride groups |
Comparison 18.
Taspoglutide versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c (%) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Taspoglutide 10 mg once weekly verus placebo | 1 | 98 | Mean Difference (IV, Random, 95% CI) | ‐1.0 [‐1.19, ‐0.81] |
| 1.2 Taspoglutide 20 mg once weekly versus placebo | 2 | 163 | Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.16, ‐0.58] |
| 1.3 Taspoglutide 20 mg once every 2 weeks versus placebo | 1 | 98 | Mean Difference (IV, Random, 95% CI) | ‐0.8 [‐0.99, ‐0.61] |
| 2 HbA1c (%) | Other data | No numeric data | ||
| 3 HbA1c < 7% | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Taspoglutide 10 mg once weekly versus placebo | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 4.88 [2.55, 9.33] |
| 3.2 Taspoglutide 20 mg once weekly versus placebo | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 4.41 [2.70, 7.22] |
| 3.3 Taspoglutide 20 mg once every two weeks versus placebo | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 3.88 [1.99, 7.56] |
| 4 HbA1c < 7% | Other data | No numeric data | ||
| 5 Hypoglycaemia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Taspoglutide 20/20 mg once weekly versus placebo | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 15.30] |
| 5.2 Taspoglutide 20/30 mg once weekly versus placebo | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 2.91 [0.32, 26.53] |
| 5.3 Taspoglutide 20/40 mg once weekly versus placebo | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.97] |
| 6 Hypoglycaemia | Other data | No numeric data | ||
| 7 Weight change (kg) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Taspoglutide 10 mg once weekly versus placebo | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐1.3 [‐2.13, ‐0.47] |
| 7.2 Taspoglutide 20 mg once weekly versus placebo | 2 | 153 | Mean Difference (IV, Random, 95% CI) | ‐1.07 [‐2.93, 0.79] |
| 7.3 Taspoglutide 20 mg once every two weeks versus placebo | 1 | 80 | Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐1.93, ‐0.27] |
| 8 Weight change | Other data | No numeric data | ||
| 9 Adverse events | Other data | No numeric data | ||
| 10 Adverse events | Other data | No numeric data | ||
| 11 Fasting plasma glucose (mmol/L) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.1 Taspoglutide 20/20 mg once weekly versus placebo | 1 | 64 | Mean Difference (IV, Random, 95% CI) | ‐1.70 [‐2.53, ‐0.87] |
| 11.2 Taspoglutide 20/30 mg once weekly versus placebo | 1 | 65 | Mean Difference (IV, Random, 95% CI) | ‐1.0 [‐1.83, ‐0.17] |
| 11.3 Taspoglutide 20/40 mg once weekly versus placebo | 1 | 64 | Mean Difference (IV, Random, 95% CI) | ‐1.6 [‐2.43, ‐0.77] |
| 12 Fasting plasma glucose | Other data | No numeric data | ||
| 13 Postprandial glucose and insulin | Other data | No numeric data | ||
| 14 Lipid profiles | Other data | No numeric data | ||
| 15 Beta‐cell function | Other data | No numeric data | ||
| 16 Subgroup | Other data | No numeric data | ||
| 16.1 Participants with HbA1c ≥8% | Other data | No numeric data |
Analysis 18.1.
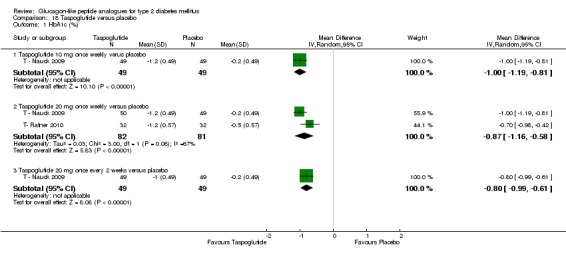
Comparison 18 Taspoglutide versus placebo, Outcome 1 HbA1c (%).
Analysis 18.2.
Comparison 18 Taspoglutide versus placebo, Outcome 2 HbA1c (%).
HbA1c (%)
| Study | 20/30 mg (n = 33) | 20/40 mg (n = 32) | Placebo (n = 32) |
|---|---|---|---|
| T‐ Ratner 2010 | ‐0.90 SD 0.57, P < 0.0001 versus placebo | ‐1.20 SD 0.57, P < 0.0001 versus placebo | ‐0.50 SD 0.57 |
Analysis 18.3.
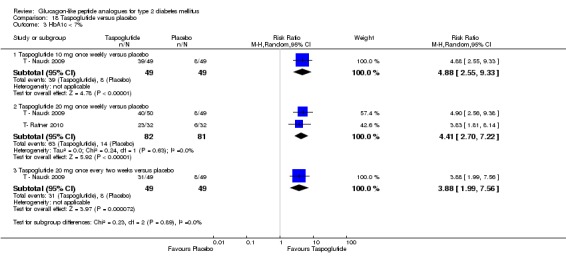
Comparison 18 Taspoglutide versus placebo, Outcome 3 HbA1c < 7%.
Analysis 18.4.
Comparison 18 Taspoglutide versus placebo, Outcome 4 HbA1c < 7%.
HbA1c < 7%
| Study | 20/30 mg once weekly | 20/40 mg once weekly | Placebo |
|---|---|---|---|
| T‐ Ratner 2010 | 53% | 70% | 19% |
Analysis 18.5.
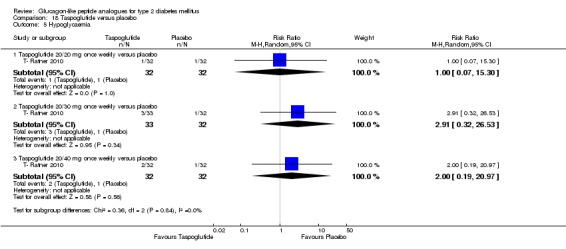
Comparison 18 Taspoglutide versus placebo, Outcome 5 Hypoglycaemia.
Analysis 18.6.
Comparison 18 Taspoglutide versus placebo, Outcome 6 Hypoglycaemia.
Hypoglycaemia
| Study | |
|---|---|
| T ‐ Nauck 2009 | hypoglycaemia not defined; no data reported for the separate comparison groups; overall, there were 7 hypoglycaemic events in 6 patients, 2 of which were asymptomatic; there were no cases of severe hypoglycaemia in the taspoglutide group |
Analysis 18.7.
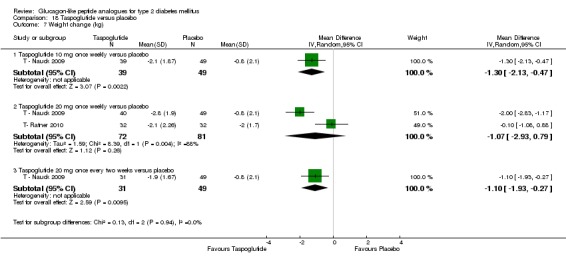
Comparison 18 Taspoglutide versus placebo, Outcome 7 Weight change (kg).
Analysis 18.8.
Comparison 18 Taspoglutide versus placebo, Outcome 8 Weight change.
Weight change
| Study | 20/30 mg once weekly | 20/40 mg once weekly | Placebo |
|---|---|---|---|
| T‐ Ratner 2010 | ‐3 SD 1.72, P = 0.03 versus placebo | ‐2.7 SD 2.26, P = 0.17 versus placebo | ‐2 SD 1.70 |
Analysis 18.9.
Comparison 18 Taspoglutide versus placebo, Outcome 9 Adverse events.
Adverse events
| Study | Description | 20/20 mg once weekly | 20/30 mg once weekly | 20/40 mg once weekly | Placebo |
|---|---|---|---|---|---|
| T‐ Ratner 2010 | Withdrawal due to adverse events | n = 3 | n = 6 | n = 6 | n = 1 |
| T‐ Ratner 2010 | Nausea | 38% | 52% | 34% | 13% |
| T‐ Ratner 2010 | Headache | 16% | 6% | 9% | 13% |
| T‐ Ratner 2010 | Diarrhoea | 13% | 21% | 9% | 9% |
| T‐ Ratner 2010 | Vomiting | 13% | 27% | 13% | 0 |
| T‐ Ratner 2010 | Dyspepsia | 19% | 15% | 16% | 0 |
| T‐ Ratner 2010 | Abdominal distension | 9% | 12% | 3% | 0 |
| T‐ Ratner 2010 | Injection site reactions | 69% | 52% | 59% | 13% |
| T‐ Ratner 2010 | serious adverse events | 0 | 0 | n = 1, unrelated to study drug | n = 1, unrelated to study drug |
| T‐ Ratner 2010 | severe hypoglycaemia | 0 | 0 | 0 | 0 |
Analysis 18.10.
Comparison 18 Taspoglutide versus placebo, Outcome 10 Adverse events.
Adverse events
| Study | Description | 10 mg weekly | 20 mg weekly | 20 mg every 2 weeks | Placebo |
|---|---|---|---|---|---|
| T ‐ Nauck 2009 | withdrawal due to adverse event | n = 2 | n = 3 | n = 1 | n = 0 |
| T ‐ Nauck 2009 | adverse events similar across groups for: headache no clinically relevant abnormalities in ECG, vital signs, laboratory parameters |
||||
| T ‐ Nauck 2009 | nausea | n = 12 (24%) | n = 26 (52%) | n = 20 (41%) | n = 3 (6%) |
| T ‐ Nauck 2009 | vomiting | n = 2 (4%) | n = 11 (22%) | n = 12 (24%) | n = 2 (4%) |
| T ‐ Nauck 2009 | diarrhoea | n = 5 (10%) | n = 5 (10%) | n = 9 (18%) | n = 4 (8%) |
| T ‐ Nauck 2009 | serious adverse events: 6 patients, 2 of which in placebo group considered to be unrelated to study treatment |
||||
| T ‐ Nauck 2009 | mild and moderate injection site reactions which did not result in treatment discontinuation |
Analysis 18.11.
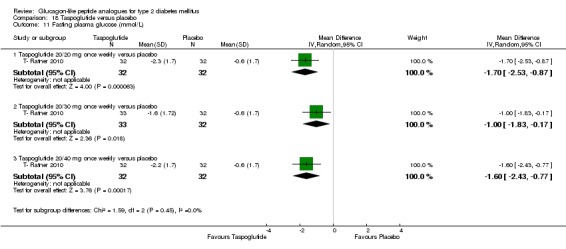
Comparison 18 Taspoglutide versus placebo, Outcome 11 Fasting plasma glucose (mmol/L).
Analysis 18.12.
Comparison 18 Taspoglutide versus placebo, Outcome 12 Fasting plasma glucose.
Fasting plasma glucose
| Study | 10 mg once weekly | 20 mg once weekly | 20 mg every 2 weeks | Placebo | FPG fluctuations |
|---|---|---|---|---|---|
| T ‐ Nauck 2009 | ‐2.5 mmol/L, P < 0.0001 | ‐2.5 mmol/L, P < 0.0001 | ‐1.4 mmol/L, P = 0.02 | ‐0.78 mmol/L | the fluctuation in FPG was less for the weekly regimens than for the dosing once every 2 weeks: 0.6 mmol/L amplitude between week 2 and week 8 for both 10 mg and 20 mg per week versus 1.3 mmol/L amplitude between week 2 and week 8 for 20 mg every 2 weeks |
Analysis 18.13.
Comparison 18 Taspoglutide versus placebo, Outcome 13 Postprandial glucose and insulin.
Postprandial glucose and insulin
| Study | Description | 10 mg once weekly | 20 mg once weekly | 20 mg every 2 weeks | Placebo |
|---|---|---|---|---|---|
| T ‐ Nauck 2009 | plasma glucose 120 min after a mixed meal (% change from baseline) | ‐22.0% | ‐18.0% | ‐5.5% | ‐10.5% |
| T ‐ Nauck 2009 | glucose AUC (% change from baseline) | ‐27.5% (SE 3.2) | ‐22.2% (SE 3.3) | ‐9.2% (SE 5.0) | ‐7.2% (SE3 .5) |
| T ‐ Nauck 2009 | mean % change in plasma insulin at 120 min | +28.5% | +44.9% | ‐13% | ‐15.3% |
Analysis 18.14.
Comparison 18 Taspoglutide versus placebo, Outcome 14 Lipid profiles.
Lipid profiles
| Study | Description | 10 mg once weekly | 20 mg once weekly | 20 mg every 2 weeks | Placebo |
|---|---|---|---|---|---|
| T ‐ Nauck 2009 | Triglycerides (mmol/L) | ‐0.29 mmol/L | ‐0.54 mmol/L | ‐0.30 mmol/L | +0.10 mmol/L |
| T ‐ Nauck 2009 | Total cholesterol (mmol/L) | ‐0.31 mmol/L | ‐0.23 mmol/L | ‐0.31 mmol/L | +0.18 mmol/L |
| T ‐ Nauck 2009 | HDL (mmol/L) | ‐0.05 mmol/L | ‐0.03 mmol/L | +0 mmol/L | +0 mmol/L |
| T ‐ Nauck 2009 | LDL (mmol/L) | ‐0.21 mmol/L | +0.03 mmol/L | ‐0.16 mmol/L | +0.13 mmol/L |
Analysis 18.15.
Comparison 18 Taspoglutide versus placebo, Outcome 15 Beta‐cell function.
Beta‐cell function
| Study | Description | 10 mg once weekly | 20 mg once weekly | 20 mg every 2 weeks | Placebo |
|---|---|---|---|---|---|
| T ‐ Nauck 2009 | proinsulin‐to‐insulin ratio | ‐0.12, P = 0.0076 | ‐0.166, P = 0.0003 | ‐0.055, P = NS | 0.002 |
Analysis 18.16.
Comparison 18 Taspoglutide versus placebo, Outcome 16 Subgroup.
Subgroup
| Study | 10 mg once weekly | 20 mg once weekly | 20 mg every 2 weeks | Placebo |
|---|---|---|---|---|
| Participants with HbA1c ≥8% | ||||
| T ‐ Nauck 2009 | ‐1.5%, P < 0.0001 versus placebo | ‐1.4%, P < 0.0001 versus placebo | ‐1.3%, P < 0.0001 versus placebo | ‐0.3% |
Comparison 19.
LY2189265 versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c | Other data | No numeric data | ||
| 2 HbA1c < 7% | Other data | No numeric data | ||
| 3 Hypoglycaemia | Other data | No numeric data | ||
| 3.1 LY 0.5/1.0 QW | Other data | No numeric data | ||
| 4 Weight change (kg) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 LY 0.5/1.0 QW | 1 | 130 | Mean Difference (IV, Random, 95% CI) | ‐1.32 [‐2.40, ‐0.24] |
| 4.2 LY 1.0/1.0 QW | 1 | 128 | Mean Difference (IV, Random, 95% CI) | ‐1.22 [‐2.30, ‐0.14] |
| 4.3 LY 1.0/2.0 QW | 1 | 128 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐3.52, ‐1.34] |
| 5 Adverse events | Other data | No numeric data | ||
| 6 Systolic blood pressure (mm Hg) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6.1 LY 0.5/1.0 QW | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 LY 1.0/1.0 QW | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 LY 1.0/2.0 QW | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Diastolic blood pressure (mm Hg) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7.1 LY 0.5/1.0 QW | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 LY 1.0/1.0 QW | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 LY 1.0/2.0 QW | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Fasting plasma glucose (mmol/L) | Other data | No numeric data | ||
| 9 Beta‐cell function | Other data | No numeric data |
Analysis 19.1.
Comparison 19 LY2189265 versus placebo, Outcome 1 HbA1c.
HbA1c
| Study | Description | LY 0.5/1.0 QW | LY 1.0/1.0 QW | LY 1.0/2.0 QW | Placebo | p values |
|---|---|---|---|---|---|---|
| LY2189265 ‐Umpierrez 2011 | Change in HbA1c | ‐1.38 SE 0.12 | ‐1.32 SE 0.12 | ‐1.59 SE 0.12 | Not reported | All LY groups vs. placebo: P < 0.001; Between LY groups: P > 0.05 |
Analysis 19.2.
Comparison 19 LY2189265 versus placebo, Outcome 2 HbA1c < 7%.
HbA1c < 7%
| Study | Description | Result |
|---|---|---|
| LY2189265 ‐Umpierrez 2011 | HbA1c < 7% | 49 to 54% of participants achieved a target HbA1c level of < 7% at 16 weeks |
Analysis 19.3.
Comparison 19 LY2189265 versus placebo, Outcome 3 Hypoglycaemia.
Hypoglycaemia
| Study | Description | LY 0.5/1.0 | LY 1.0/1.0 | LY 1.0/2.0 | Placebo |
|---|---|---|---|---|---|
| LY 0.5/1.0 QW | |||||
| LY2189265 ‐Umpierrez 2011 | Oveall hypoglycaemic episodes | n = 183 | n = 237 | n = 164 | n = 84 |
| LY2189265 ‐Umpierrez 2011 | number of hypoglycaemic episodes at 4 weeks | n = 68 | n = 85 | n = 56 | n = 16 |
| LY2189265 ‐Umpierrez 2011 | number of hypoglycaemic episodes at 8 weeks | n = 59 | n = 55 | n = 47 | n = 18 |
| LY2189265 ‐Umpierrez 2011 | number of hypoglycaemic episodes at 16 weeks | n = 19 | n = 35 | n = 23 | n = 16 |
| LY2189265 ‐Umpierrez 2011 | severe hypoglycaemia | 0 | 0 | 0 | 0 |
Analysis 19.4.
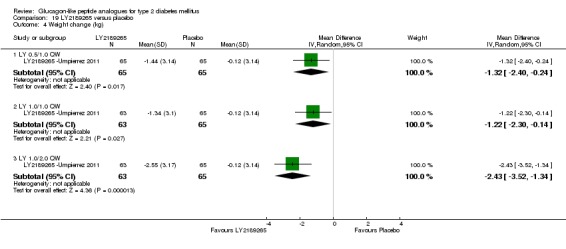
Comparison 19 LY2189265 versus placebo, Outcome 4 Weight change (kg).
Analysis 19.5.
Comparison 19 LY2189265 versus placebo, Outcome 5 Adverse events.
Adverse events
| Study | Description | LY 0.5/1.0 QW | LY 1.0/1.0 QW | LY 1.0/2.0 QW | Placebo | comments |
|---|---|---|---|---|---|---|
| LY2189265 ‐Umpierrez 2011 | withdrawal due to adverse events | 3 (4.5%) | 4 (6.2%) | 4 (6.2%) | 1 (1.5%) | |
| LY2189265 ‐Umpierrez 2011 | nausea | 9 (13.6%) | 11 (16.9%) | 9 (13.8%) | 5 (7.6%) | Nausea occurred more commonly in participants receiving LY. |
| LY2189265 ‐Umpierrez 2011 | diarrhoea | 5 (7.6) | 4 (6.2%) | 9 (13.8%) | 5 (7.6%) | Higher LY dosage was generally associated with a higher prevalence of adverse events. |
| LY2189265 ‐Umpierrez 2011 | abdominal distention | 3 (4.5%) | 5 (7.7%) | 9 (13.8%) | 4 (6.1%) | Higher LY dosage was generally associated with a higher prevalence of adverse events. |
| LY2189265 ‐Umpierrez 2011 | vomiting | 3 (4.5%) | 1 (1.5%) | 7 (10.8%) | 2 (3.0%) | Higher LY dosage was generally associated with a higher prevalence of adverse events. |
| LY2189265 ‐Umpierrez 2011 | Pancreatitis | 2 | Both cases related to study drug. One participant approximately 5 months after last dose of LY 0.5/1.0, later this was regarded as serious. Second participant after the 11th weekly dose of LY 0.5/1.0 had approximately 1.5‐ and 2.5‐fold increase in amylase and lipase levels respectively with no obvious symptom or abnormality in the pancreas during abodominal CT scan. | |||
| LY2189265 ‐Umpierrez 2011 | possibly LY related adverse events | 22 (33.3%) | 20 (30.8%) | 27 (41.5%) | 15 (22.7%) | |
| LY2189265 ‐Umpierrez 2011 | serious adverse events | 3 (4.5%) | 2 (3.1%) | 1 (1.5%) | 1 (1.5%) | three serious adverse events related to study drug and included hallucination, cryptogenic organizing pneumonia and pancreatitis |
| LY2189265 ‐Umpierrez 2011 | deaths | 0 | 0 | 0 | 0 |
Analysis 19.6.
Comparison 19 LY2189265 versus placebo, Outcome 6 Systolic blood pressure (mm Hg).
Analysis 19.7.
Comparison 19 LY2189265 versus placebo, Outcome 7 Diastolic blood pressure (mm Hg).
Analysis 19.8.
Comparison 19 LY2189265 versus placebo, Outcome 8 Fasting plasma glucose (mmol/L).
Fasting plasma glucose (mmol/L)
| Study | Description | LY 0.5/1.0 QW | LY 1.0/1.0 QW | LY 1.0/2.0 QW | Placebo | p values |
|---|---|---|---|---|---|---|
| LY2189265 ‐Umpierrez 2011 | Change in FPG | ‐2.10 | ‐2.05 | ‐2.65 | ‐0.49 | All LY groups vs. placebo: P < 0.001; Between LY groups: P > 0.05 |
Analysis 19.9.
Comparison 19 LY2189265 versus placebo, Outcome 9 Beta‐cell function.
Beta‐cell function
| Study | Description | LY 0.5/1.0 QW | LY 1.0/1.0 QW | LY 1.0/2.0 QW | Placebo | p values |
|---|---|---|---|---|---|---|
| LY2189265 ‐Umpierrez 2011 | β‐cell function (HOMA2‐%B) | 39.2 SE 45.6% | 44.3 SE 93.9% | 45.6 SE 55.2% | 1.0 SE 41.1% | All LY groups vs. placebo: P < 0.01 |
| LY2189265 ‐Umpierrez 2011 | Insulin sensitivity (HOMA2‐%S) | Not reported | Not reported | Not reported | Not reported | No statistically significant change in all LY groups |
| LY2189265 ‐Umpierrez 2011 | Insulin resistance (HOMA2‐%IR) | Not reported | Not reported | Not reported | Not reported | No statistically significant change in all LY groups |
Comparison 20.
GLP‐1 agonist versus GLP‐1 agonist
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HbA1c | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | 0.33 [0.11, 0.55] |
| 1.2 versus Exenatide 2 mg once weekly | 2 | 547 | Mean Difference (IV, Random, 95% CI) | 0.55 [0.26, 0.84] |
| 2 HbA1c < 7% | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus Liraglutide | 1 | 464 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.66, 0.96] |
| 2.2 versus Exenatide 2 mg once weekly | 2 | 511 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.42, 1.01] |
| 3 Hypoglycaemia | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus Liraglutide | 1 | 467 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.99, 1.75] |
| 3.2 versus Exenatide 2 mg once weekly | 1 | 295 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.45, 2.86] |
| 4 Hypoglycaemia | Other data | No numeric data | ||
| 5 Weight change | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | 0.37 [‐0.55, 1.29] |
| 5.2 versus Exenatide 2 mg once weekly | 1 | 295 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐1.29, 1.49] |
| 6 Weight change | Other data | No numeric data | ||
| 7 Treatment satisfaction | Other data | No numeric data | ||
| 7.1 versus Liraglutide | Other data | No numeric data | ||
| 7.2 versus Exenatide 2 mg once weekly | Other data | No numeric data | ||
| 8 Adverse events | Other data | No numeric data | ||
| 8.1 versus Liraglutide | Other data | No numeric data | ||
| 8.2 versus Exenatide 2 mg once weekly | Other data | No numeric data | ||
| 9 Systolic blood pressure | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | 0.51 [‐2.72, 3.74] |
| 9.2 versus Exenatide 2 mg once weekly | 2 | 547 | Mean Difference (IV, Random, 95% CI) | 1.49 [‐0.71, 3.69] |
| 10 Fasting plasma glucose (mmol/L) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | 1.01 [0.46, 1.56] |
| 10.2 versus Exenatide 2 mg once weekly | 2 | 547 | Mean Difference (IV, Random, 95% CI) | 1.18 [1.02, 1.33] |
| 11 Post‐prandial glucose (mmol/L) | Other data | No numeric data | ||
| 11.1 versus Liraglutide | Other data | No numeric data | ||
| 11.2 versus Exenatide 2 mg once weekly | Other data | No numeric data | ||
| 12 Triglycerides (mmol/L) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | 0.18 [‐0.10, 0.46] |
| 12.2 versus Exenatide 2 mg once weekly | 2 | 547 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.04, 0.06] |
| 13 Total cholesterol (mmol/L) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 13.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | 0.11 [‐0.09, 0.31] |
| 13.2 versus Exenatide 2 mg once weekly | 2 | 547 | Mean Difference (IV, Random, 95% CI) | 0.31 [0.10, 0.51] |
| 14 HDL‐cholesterol (mmol/L) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 14.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.07, 0.05] |
| 14.2 versus Exenatide 2 mg once weekly | 2 | 547 | Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.03, 0.04] |
| 15 LDL‐cholesterol (mmol/L) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.1 versus Liraglutide | 1 | 464 | Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.13, 0.21] |
| 15.2 versus Exenatide 2 mg once weekly | 2 | 547 | Mean Difference (IV, Random, 95% CI) | 0.20 [0.09, 0.30] |
| 16 Beta‐cell function | Other data | No numeric data |
Analysis 20.1.
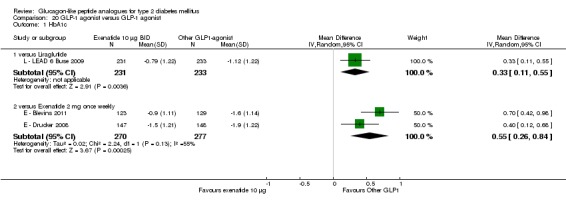
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 1 HbA1c.
Analysis 20.2.
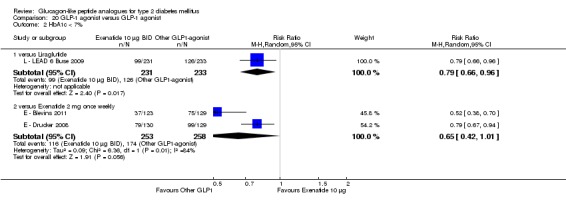
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 2 HbA1c < 7%.
Analysis 20.3.
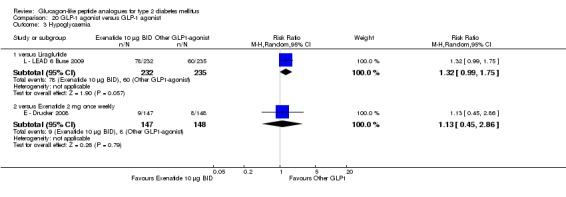
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 3 Hypoglycaemia.
Analysis 20.4.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 4 Hypoglycaemia.
Hypoglycaemia
| Study | Description | Exenatide 10 µg BID | Exenatide 2 mg QW |
|---|---|---|---|
| E ‐ Blevins 2011 | Minor hypoglycaemia occured only among participants using a concomitant SU (n = 74) | n = 4 | n = 5 |
| E ‐ Blevins 2011 | Major hypoglycaemia | None | None |
Analysis 20.5.
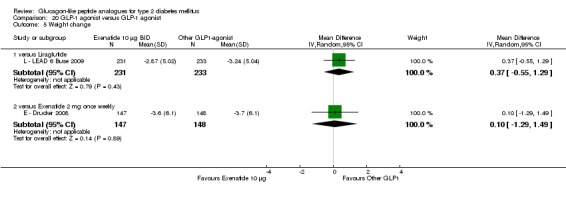
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 5 Weight change.
Analysis 20.6.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 6 Weight change.
Weight change
| Study | Description | Exenatide 10 µg BID | Exenatide 2 mg QW | Difference between groups |
|---|---|---|---|---|
| E ‐ Blevins 2011 | Change in weight (kg) | ‐1.4 | ‐2.3 | ‐0.95 (95% CI ‐1.9 to 0.01) |
| E ‐ Blevins 2011 | Proportion of participants experiencing weight loss by end of study, n (%) | 77 (63) | 99 (77) | NR |
Analysis 20.7.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 7 Treatment satisfaction.
Treatment satisfaction
| Study | Description | Exenatide 10 µg BID | Other GLP1 agonist |
|---|---|---|---|
| versus Liraglutide | |||
| L ‐ LEAD 6 Buse 2009 | Diabetes Treatment Satisfaction Questionnaire (6 of 8 items) | 13.3 SD6.94 | 15.18 SD 7.36, P = 0.0004 versus exenatide 10 µg BID |
| versus Exenatide 2 mg once weekly | |||
| E ‐ Drucker 2008 | Diabetes Treatment Satisfaction Questionnaire (8 of 8 items) | 29.97 | 31.17, P = NS versus exenatide 10 µg BID |
Analysis 20.8.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 8 Adverse events.
Adverse events
| Study | Description | Exenatide 10 µg BID | Other GLP1 agonist |
|---|---|---|---|
| versus Liraglutide | |||
| L ‐ LEAD 6 Buse 2009 | withdrawal due to adverse events | 13% | 10% |
| L ‐ LEAD 6 Buse 2009 | overall rate of adverse events | 78.9% | 74.9% |
| L ‐ LEAD 6 Buse 2009 | no significant differences across groups for: infections, headache, back pain, metabolism an nutrition disorders, general disorders and administration‐site conditions; 1 episode of mild pancreatitis in liraglutide group (considered to be chronic and unrelated to treatment | ||
| L ‐ LEAD 6 Buse 2009 | nausea | 28.0% | 25.5% |
| L ‐ LEAD 6 Buse 2009 | diarrhoea | 12.1% | 12.3% |
| L ‐ LEAD 6 Buse 2009 | dyspepsia | 4.7% | 8.9% |
| L ‐ LEAD 6 Buse 2009 | vomiting | 9.9% | 6.0% |
| L ‐ LEAD 6 Buse 2009 | constipation | 2.6% | 5.1% |
| L ‐ LEAD 6 Buse 2009 | serious adverse events (only on event considered to be related to study medication (exenatide, severe hypoglycaemia)) | 2.6% | 5.1% |
| L ‐ LEAD 6 Buse 2009 | severe adverse events | 4.7% | 7.2% |
| versus Exenatide 2 mg once weekly | |||
| E ‐ Blevins 2011 | withdrawal due to adverse events | 5% | 5% |
| E ‐ Blevins 2011 | nausea | 35%; events: n = 51 | 14%; events: n = 21 |
| E ‐ Blevins 2011 | vomiting | 8.9% | 4.7% |
| E ‐ Blevins 2011 | diarrhoea | 4.1% | 9.3% |
| E ‐ Blevins 2011 | headache | 8.1% | 4.7% |
| E ‐ Blevins 2011 | dizziness | 6.5% | 2.3% |
| E ‐ Blevins 2011 | upper respiratory tract infection | 4.1% | 7.0% |
| E ‐ Blevins 2011 | injection site erythema | 2.4% | 5.4% |
| E ‐ Blevins 2011 | serious adverse events | 4%; one fatal myocardial infarction | 2%, one participant with a history of dyslipidemia was hospitalised and withdrew due to a diagnosis of pancreatitis. |
| E ‐ Blevins 2011 | anti‐exenatide antibody levels | 51% participants positive for treatment‐emergent antibodies to exenatide | 73% participants positive for treatment‐emergent antibodies to exenatide |
| E ‐ Drucker 2008 | withdrawal due to adverse events | 5.4% | 6.1% |
| E ‐ Drucker 2008 | no clinically significant abnormalities in vital signs, ECG reports, haematological, chemistry or urinalysis values; no major hypoglycaemia | ||
| E ‐ Drucker 2008 | nausea (modestly greater weight loss in people with nausea, but participants without nausea also lost weight) |
34.5% | 26.4% |
| E ‐ Drucker 2008 | diarrhoea | 13.1% | 13.5% |
| E ‐ Drucker 2008 | vomiting | 18.6% | 10.8% |
| E ‐ Drucker 2008 | constipation | 6.2% | 10.8% |
| E ‐ Drucker 2008 | injection site pruritus | 1.4% | 17.6% |
| E ‐ Drucker 2008 | injection site bruising | 10.3% | 4.7% |
| E ‐ Drucker 2008 | serious adverse events (none considered to be related to study treatment) | 3.4% | 5.4% |
| E ‐ Drucker 2008 | anti‐exenatide antibody levels | significantly more anti‐exenatide antibodies with exenatide once weekly, but mostly not detectable or low titre | |
Analysis 20.9.
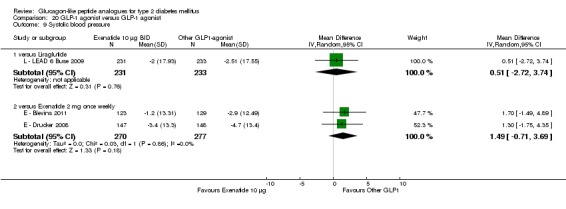
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 9 Systolic blood pressure.
Analysis 20.10.
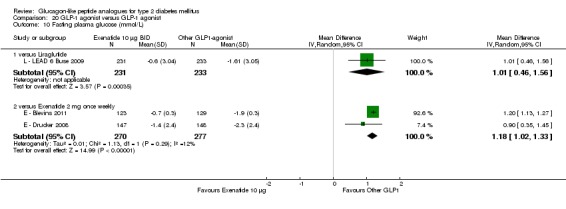
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 10 Fasting plasma glucose (mmol/L).
Analysis 20.11.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 11 Post‐prandial glucose (mmol/L).
Post‐prandial glucose (mmol/L)
| Study | Description | Exenatide 10 µg BID | Other GLP1 agonist | Comments |
|---|---|---|---|---|
| versus Liraglutide | ||||
| L ‐ LEAD 6 Buse 2009 | from self‐monitored 7‐point plasma glucose measurements | after breakfast: 8.9 SE 0.4 mmol/L after dinner: 7.8 SE 0.4 mmol/L |
after breakfast: 9.7 SE 0.5 mmol/L after dinner: 8.2 SE 0.3 mmol/L |
Exenatide reduced post‐prandial plasma glucose increment more than did liraglutide after breakfast and dinner; treatment difference after lunch was not significant. |
| L ‐ LEAD 6 Buse 2009 | ||||
| versus Exenatide 2 mg once weekly | ||||
| E ‐ Drucker 2008 | from self‐monitored 7‐point plasma glucose measurements | both treatments reduced post‐prandial values compared to baseline | ||
| E ‐ Drucker 2008 | 2 h post‐prandial glucose, meal tolerance test (n = 51) | ‐6.9 mmol/L SE 0.5 | ‐5.3 mmol/L SE 0.5 | P = 0.0124 |
Analysis 20.12.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 12 Triglycerides (mmol/L).
Analysis 20.13.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 13 Total cholesterol (mmol/L).
Analysis 20.14.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 14 HDL‐cholesterol (mmol/L).
Analysis 20.15.
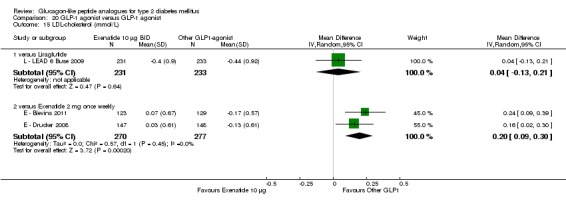
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 15 LDL‐cholesterol (mmol/L).
Analysis 20.16.
Comparison 20 GLP‐1 agonist versus GLP‐1 agonist, Outcome 16 Beta‐cell function.
Beta‐cell function
| Study | Description | Exenatide 10 µg BID | Liraglutide 1.8 mg daily |
|---|---|---|---|
| L ‐ LEAD 6 Buse 2009 | HOMA‐B (%) | +2.74% SD 103 | +32.12% SD 103, P < 0.0001 versus exenatide 10 µg BID |
| L ‐ LEAD 6 Buse 2009 | proinsulin‐to‐insulin ratio | ‐0.02 SD 0.46 | 0.00 SD 0.46, P = NS |
What's new
| Date | Event | Description |
|---|---|---|
| 8 June 2013 | Feedback has been incorporated | Feedback arrived at 8 June 3013 |
History
| Date | Event | Description |
|---|---|---|
| 5 October 2011 | Amended | This review version is identical with the previously published one. The only correction that is being made is in the authors contact details. |
Differences between protocol and review
Authors have changed since publication of the protocol.
Contact person has changed since publication of the protocol.
Studies with a minimum duration of eight weeks were included.
Outcome costs was deleted and blood pressure introduced.
Number of glucagon‐like peptide 1 (GLP‐1) agonists increased from two to six. Exenatide twice daily rendered obsolete by once weekly form so review focusses on latter.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods |
TRIAL DESIGN: Randomised double‐blind, placebo‐controlled parallel group phase II trial, multi‐centre (118 sites) DURATION OF INTERVENTION: 16 weeks DURATION OF FOLLOW‐UP: 11 week washout phase to assess safety and immunogeneity RUN‐IN PERIOD: None SETTING: Not reported (NR) COUNTRY: US (106 sites), Mexico (9 sites), Chile (2 sites), Dominican Republic (1 site) |
|
| Participants |
INCLUSION CRITERIA: Patients with type 2 diabetes diagnosed ≥ 3 months before screening, men and women aged 18 to 75 years, drug‐naive (diet and exercise) or treated with metformin monotherapy and stable for > 3 months before prescreening; BMI ≥ 20 and ≤ 40 kg/m2, HbA1c at screening ≥ 7 and ≤ 10%; only participants treated with metformin monotherapy were eligible for the exenatide arm (consistent with labelling). EXCLUSION CRITERIA: Any oral antidiabetic monotherapy (except metformin) ≤ 3 months prior to screening or insulin < 1 month prior to screening and not used for > 7 days; history of pancreatitis, cardiovascular, cerebrovascular, renal or hepatobiliary diseases; fasting serum triglycerides ≥ 9 mmol/L at screening; or haematological profiles considered to be clinically significant; use of lipid lowering medications must have been maintained at same dose for 3 months prior to treatment, no prescription or over the counter weight loss drugs 3 months prior to enrolment. AGE: mean 54.0 to 55.5 years (SD 9.7 to 10.6) SEX: 45.1% to 74.2% female (74.2% in ALBI 30 mg weekly group, 50% in ALBI 30 mg every 2 weeks group, 45.1% in placebo group) DIABETES DURATION: mean 4.9 years [range 3.9 to 5.5 years (SD 3.0 to 5.4)] ETHNICITY: Caucasian (43.8% to 71%) (87.1% and 12.9% of participants were from U.S. and Latin American clinics respectively) HbA1c (%): ALBI Weekly 30 mg: 8.0 (SD 0.9), Placebo: 7.9 (SD 0.9), ALBI every 2 weeks 30 mg: 8.0 (SD 1.0) BMI (kg/m2): ALBI weekly 30 mg: 33.0 (SD 3.9), ALBI every 2 weeks 30 mg: 31.2 (SD 4.1), Placebo: 31.8 (SD 5.4) PREVIOUS THERAPY: Diet and exercise only: 29.0% to 34.4%, MET: 65.6% to 71.0% NUMBERS: Randomised: 361, received treatment (and included in the safety analysis): 356, efficacy analysis: 345; ITT: placebo: 51, exenatide: 35; ALBI weekly 4 mg: 35, 15 mg: 35, 30 mg: 31; ALBI every 2 weeks 15 mg: 33, 30 mg: 32, 50 mg: 35; ALBI monthly 50 mg: 35, 100 mg: 34 |
|
| Interventions |
COMPARISON: Albiglutide (ALBI, 8 doses/schedules) +/‐ Metformin (MET) VERSUS Exenatide (EX) + MET VERSUS Placebo +/‐ MET NO. OF COMPARISON GROUPS: 10 For the current review, the following groups were excluded: EX (as this included co‐medication with metformin in all patients, whereas only 65.6% to 74.3% of the patients in the other groups received metformin, so this was not really a comparison of exenatide versus albiglutide); ALBI 4 or 15 mg weekly or 15 mg every two weeks (smaller effect on HbA1c), and ALBI 50 mg every two weeks or 50 or 100 mg monthly (no improvement in HbA1c compared to groups with largest effect, significantly more adverse events) DOSE ALBI: 30 mg weekly or 30 mg every 2 weeks injected subcutaneously to the abdomen DOSE PLACEBO: Placebo injections DOSE MET: Not reported, presumably pre study levels OTHER TREATMENT: Not reported |
|
| Outcomes |
PRIMARY OUTCOMES: Change from baseline in HbA1c at week 16 SECONDARY OUTCOMES: Fasting plasma glucose (FPG), fasting fructosamine, C‐peptide, glucagon, insulin, lipid profiles, beta‐cell function (homeostasis model) OTHER OUTCOMES: Adverse event assessments and safety analyses (nausea and vomiting, immunogenicity), 11 week washout post‐intervention (HbA1c, FPG, ALBI concentrations, fasting fructosamine, C‐peptide, glucagon, insulin, lipid profiles, immunogenicity) |
|
| Notes |
AIM: To evaluate the efficacy, safety and tolerability of incremental doses of albiglutide, administered using three dosing schedules in patients with type 2 diabetes inadequately controlled with diet and exercise or metformin monotherapy. SOURCE OF FUNDING: GlaxoSmithKline, Middlesex, UK OTHER: Conflict of interest: One author has received research grants and consulting honoraria for serving on scientific advisory boards from GlaxoSmithKline. Another author has received research grants from and acted as a consultant for GlaxoSmithKline, MB FY, and MS are employees/stockholders of GlaxoSmithKline. SAMPLE SIZE: With 30 participants planned in each arm, a two‐sided 95% CI for each group mean response had a half‐width of 0.36% on HbA1c scale, assuming a standard deviation of 1%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details given |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind (study personnel and patients) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comparisons made on the intent to treat population, using last observation carried forward, adequate description of withdrawals or losses to follow‐up (28% withdrawals or losses to follow‐up) |
| Selective reporting (reporting bias) | Low risk | All pre‐specified (primary and secondary) outcomes were reported |
| Methods |
TRIAL DESIGN:Randomised, double‐blind, double‐dummy, superiority trial DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: NR RUN‐IN PERIOD: NR SETTING: 72 hospitals and clinics COUNTRY: USA, India and Mexico. |
|
| Participants |
INCLUSION CRITERIA: participants aged between 18 years and older; type 2 diabetes but otherwise healthy; treated with a stable metformin regimen for at least 2 months before screening; HbA1c of 7.1% to 11.0%; BMI 25 to 45 kg/m2; FPG < 15.5 mmol/L; not taking or has been stable on other medications of a minimum of 2 months‐hormone replacement therapy, OCPs, antihypertensives, lipid‐lowering agents, thyroid replacement therapy, antidepressant agents and drugs known to affect body weight including prescription medications, sibutramine, topiramate and over‐the‐counter antiobesity agents. EXCLUSION CRITERIA: pregnant or lactating women; clinically significant medical conditions like hepatic disease, renal disease, cardiovascular disease, gastroparesis, malignancy, macular edema or chronic infections; drugs or alcohol abuse; fasting triglycerides ≥ 600 mg/dL; previously exposed to exenatide LAR (long acting release); blood donated within 60 days of screening or is planning to do during study; major surgery or a blood transfusion within 2 months of screening; currently taking drugs that are excluded or is expected to be treated with one; received any investigational drug within 1 month of screening; known allergies or hypersensitivity to any component of study treatment; previous experience of clinically significant adverse events with TZD or DPP‐4 inhibitors; immediate family or affiliation with the personnel; employed by Amylin, Eli Lilly or Alkermes AGE: 52 to 53 years (SD 10 to 11) SEX: 44% to 52% female [44% in EX 2 mg group; 48% in sitagliptin (SITA) 100 mg group; 52% in pioglitazone (PIO) 45 mg group] DIABETES DURATION: 5 to 6 years (SD 4 to 5) ETHNICITY: 30% to 39% White; 8% to 12% Black; 27% to 31% Hispanic; 23% to 25% Asian; 0% to 2% Native American; 1% to 2% other HbA1c (%): EX 2 mg: 8.6 SD 1.2; SITA 100 mg: 8.5 SD 1.2; PIO 45 mg: 8.5 SD 1.1 BMI (kg/m2): EX 2 mg: 32 SD 5; SITA 100 mg: 32 SD 5; PIO 45 mg: 32 SD 6 PREVIOUS THERAPY: NR NUMBERS: Screened: 958; randomised: 514 (EX 2 mg: 170, SITA 100 mg: 172, PIO 45 mg: 172); evaluable patient: 387 (EX 2 mg: 122, SITA 100 mg: 137, PIO 45 mg: 128); ITT: 491 (EX 2 mg: 160, SITA 100 mg: 166, PIO 45 mg: 165) |
|
| Interventions |
COMPARISON: EX 2 mg once weekly + MET + placebo, versus SITA 100 mg once daily + MET + placebo, versus PIO 45 mg once daily + MET + placebo NO. OF COMPARISON GROUPS: 3 DOSE EX: EX 2 mg once weekly injection plus oral placebo once daily DOSE SITA: SITA 100 mg orally once daily plus placebo once weekly injection DOSE PIO: PIO 45 mg orally once daily plus placebo once weekly injection DOSE MET: Mean oral dose between 1480 to 1583 mg OTHER TREATMENT: NR |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c SECONDARY OUTCOMES: Proportion of participants achieving the HbA1c target of 6.5% or lower, or 7.0% or lower; FPG ≤ 7 mmol/L; six‐point self‐monitored blood glucose profile; body weight; fasting lipid profile; fasting insulin profile; SBP and DBP; cardiovascular risk markers (urinary albumin‐to‐creatinine ratio, serum adiponectin, B‐type natriuretic peptide, high‐sensitivity C‐reactive protein, and plasminogen activator inhibitor‐1); patient‐reported outcomes from the Impact of Weight on Quality of Life Questionnaire‐Lite (IWQOL), Psychological General Well‐being (PGWB) index, the Diabetes Treatment Satisfaction Questionnaire (DTSQ), and EuroQol‐5 dimensions (EQ‐5D). OTHER OUTCOMES: Safety and tolerability; exenatide antibodies; hypoglycaemia |
|
| Notes |
AIM: To compare the efficacy, safety, and tolerability of three recommended therapies for patients not sufficiently controlled on metformin. SOURCE OF FUNDING: Amylin Pharmaceuticals and Eli Lilly OTHER: Conflict of interest: First author's institution has received consultancy fees or research grant support, or both, with receipt of travel and accomodation expenses in some cases, from different pharmaceutical companies. This author also owns stock in Merck. Another author is a member of the scientific advisory board for Amylin Pharmaceuticals, is a consultant for Amylin Pharmaceuticals, Eli Lilly and AstraZeneca, is on the speaker's bureau of Amylin Pharmaceuticals, Eli Lilly, Merck, Novo Nordisk, and Sanofi‐Aventis and has received travel and accomodation expenses from Amylin Pharmaceuticals. Some authors are employees and stockholders of Amylin Pharmaceuticals. One author is an employee and stockholder of Eli Lilly. All research activity and advisory or consultancy services were done under contract with the non‐profit International Diabetes Center at Park Nicollet. SAMPLE SIZE: Estimated that 500 participants would provide at least 90% power to detect a statistically significant difference between exenatide and sitagliptin or pioglitazone, and assumptions of a difference of 0.5% between groups, a common SD of 1.2% and an early withdrawal of 10%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation; done centrally by UBC Clinical Technologies via an interactive voice response system and was independent of the sponsor, investigators, study‐site staff and participants; allocated in a 1:1:1 ratio |
| Allocation concealment (selection bias) | Low risk | Interactive voice response system to conceal allocation |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat analysis; missing data were imputed by last‐observation‐carried forward method; adequate description of withdrawals and losses to follow up |
| Selective reporting (reporting bias) | Low risk | All the predefined and prespecified outcomes were reported |
| Methods |
TRIAL DESIGN: Randomised, open‐label, comparator‐controlled study DURATION OF INTERVENTION: 24 weeks DURATION OF FOLLOW‐UP: NR RUN‐IN PERIOD: NR SETTING: 43 sites COUNTRY: United States |
|
| Participants |
INCLUSION CRITERIA: At least 18 years of age and diagnosed with type 2 diabetes, otherwise healthy, and treated for at least 2 months with diet and exercise alone or with a stable, maximally effective regimen of metformin, sulphonylurea (SU), thiazolidinedione, or a combination of these medications; HbA1c of 7.1% to 11.0%, FPG less than 280 mg/dL (15.5 mmol/L), BMI from 25 to 45 kg/m2. EXCLUSION CRITERIA: Patients using concomitant weight loss agents. AGE: 55 to 56 years (SD 10 to 11) SEX: 40% to 45% female (40% in EX 2 mg once weekly group; 45% in EX 10 µg twice daily group) DIABETES DURATION: 7 SD5 years ETHNICITY: 55% to 63% Caucasian; 5% to 7% Black; 4% Asian; 29% to 33% Hispanic HbA1c (%): EX 10 µg BID: 8.4 SD 1.2; EX 2 mg QW: 8.5 SD 1.1 BMI (kg/m2): EX 10 µg BID: 33.0 SD 5.3; EX 2 mg QW: 33.6 SD 5.5 PREVIOUS THERAPY: Diet and exercise only: 16% to 21%; single oral antidiabetic therapy: 43% to 50%; combination oral antidiabetic therapy: 28% to 40% (alone or in combination of: 71% to 80% MET; 28% to 31% SU; 10% to 17% TZD) NUMBERS: EX 10 µg BID: 123; EX 2 mg QW: 129 |
|
| Interventions |
COMPARISON: EX 2 mg QW +/‐ MET +/‐ SU +/‐ TZD or EX 10 µg BID +/‐ MET +/‐ SU +/‐ TZD NO. OF COMPARISON GROUPS: 2 DOSE EX QW: Subcutaneous injection of exenatide 2 mg once weekly or exenatide 10 µg twice daily. lead‐in for 2 mg EX QW: 2 mg once weekly for 24 weeks lead‐in for 10 µg EX BID: 5 µg EX twice daily for 4 weeks, then 10 µg EX twice daily for the remainder of the 20 weeks OTHER TREATMENT: Patients not allowed to change their oral antidiabetic, lipid‐lowering, and antihypertensive medications during the study, unless instructed otherwise by the investigator. |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c from baseline to week 24. SECONDARY OUTCOMES: Body weight, FPG, proportion of subjects achieving HbA1c targets of less than 7% and 6.5% or less at week 24, proportion of patients achieving FPG target of 126 mg/dL (7.0 mmol/L) or less at week 24, SBP, DBP, fasting lipid concentrations, OTHER OUTCOMES: Safety and tolerability; antibody titres. |
|
| Notes |
AIM: To compare the effects of exenatide once weekly and exenatide twice daily on glycaemic control, body weight, and safety. SOURCE OF FUNDING: Amylin Pharmaceuticals OTHER: Two authors have received research grants from Amylin Pharmaceuticals, Inc. and serve as advisers and speaker's bureau members for Amylin Pharmaceuticals, Inc. First author has also served as an adviser and speaker's bureau member for Eli Lilly & Co. Some authors are employees and stockholders of Amylin Pharmaceuticals, Inc. One author is an employee and stockholder of Eli Lilly & Co. SAMPLE SIZE: A sample size of approximately 250 patients (ratio of 1:1) was estimated to provide 90% power to demonstrate that EX QW was non‐inferior to EX BID by a 0.4% difference in the HbA1c change from baseline to week 24, using a one‐sided, two‐sample t test with a significance level of 0.025 and assuming a greater (0.1%) reduction in HbA1c by EX QW compared with EX BID, a 15% withdrawal rate, and a common SD of 1.1%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised 1:1 to treatment with EX BID or EX QW, with randomisation performed centrally via an interactive voice or web response system. Randomization was stratified according to concomitant SU use at screening and baseline HbA1c stratum (< 9.0% or ≥ 9.0%). |
| Allocation concealment (selection bias) | Unclear risk | Randomization performed centrally via an interactive voice or web response system. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label; Sponsor personnel remained blinded to HbA1c and FPG data throughout treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | intent‐to‐treat (ITT) population consisted of all randomised patients receiving at least one dose of randomised study medication; Missing post baseline efficacy data were imputed using the last observation carried forward (LOCF) approach; adequate description of withdrawals and loss to follow‐up. |
| Selective reporting (reporting bias) | Low risk | All the prespecified and predefined outcomes were reported. |
| Methods |
TRIAL DESIGN: Phase 3, parallel, open‐label, randomised controlled trial DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: NR RUN‐IN PERIOD: NR SETTING: 72 sites COUNTRY: USA (and Puerto Rico), the European Union, Russia, Australia, Korea, Taiwan, and Mexico. |
|
| Participants |
INCLUSION CRITERIA: Participants with type 2 diabetes aged 18 years or older (no upper limit specified) with sub optimum glycaemic control despite maximum tolerated doses of metformin or combined metformin and sulphonylurea treatment for 3 months or longer; HbA1c between 7.1% and 11.0%, inclusive, BMI between 25 kg/m2 and 45 kg/m2, stable bodyweight for 3 months or more; participants have had to be treated with a stable dose of metformin of 1500 mg or more per day for 8 or more weeks before screening. EXCLUSION CRITERIA: more than three episodes of major hypoglycaemia within 6 months of screening; treatment within 4 weeks of screening with systemic glucocorticoids; and treatment for longer than 2 weeks with insulin, thiazolidinediones, α‐glucosidase inhibitors, meglitinides, exenatide twice‐a‐day formulation, dipeptidyl peptidase‐4 inhibitors or pramlintide acetate within 3 months of screening. Prescription and non‐prescription weight‐loss drugs were excluded within 3 months of screening and during the entire 26‐week study. AGE: 58 SD 9 to 10 years SEX: 45% to 48% female (EX 2 mg: 48%, GLAR: 45%) DIABETES DURATION: 7.8 to 8.0 years (SD 6.0) ETHNICITY: < 1% to 1% African American; 82% to 85% White; 6% Asian; 9% to 12% Hispanic HbA1c (%): EX: 8.3 SD 1.1; GLAR: 8.3 SD 1.0 BMI (kg/m2): 32 SD 5.0 for both groups PREVIOUS THERAPY: 70% MET; 30% MET + SU NUMBERS: Screened: 659; randomised: 456 (EX: 233, GLAR: 223) |
|
| Interventions |
COMPARISON: EX 2 mg once weekly + 70% MET/30% MET + SU VERSUS GLAR + MET/MET + SU NO. OF COMPARISON GROUPS: 2 DOSE EX: EX 2 mg injected into abdominal subcutaneous tissue at randomisation and once a week (within 2 days of date of first injection) thereafter. DOSE GLAR: GLAR implemented by INITIATE (Initiate Insulin by Aggressive Titration and Education) dosing algorithm; GLAR treatment started with 10 IU per day, measured FBG concentrations every morning and the dose of insulin adjusted to achieve a target glucose of 4.0 to 5.5 mmol/L. Participants and investigators were asked to adhere to titration targets however there was no central supervision to enforce titration. Insulin was injected at the same time every day, preferably at bedtime. Mean doses of GLAR increased from a baseline of 10 IU per day to 31 IU per day at endpoint (last measurement brought forward). DOSE MET: MET was continued in their stable dose until week 26. Mean doses of MET ˜ 2000 mg throughout study, DOSE SU: If a participant taking metformin and sulphonylureas had confirmed hypoglycaemia, the dose of sulphonylurea was reduced. 46 (21%) of 223 patients had a reduction in SU dose. BOTH GROUPS: Specific instructions for eight‐point self‐monitored blood‐glucose profiles (measured before and 2 hour after morning, midday, and evening meals, at bedtime, and at 0300 hours) were given to both treatment groups. OTHER TREATMENT: NR |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c at week 26 compared with baseline. SECONDARY OUTCOMES: Proportion of participants achieving HbA1c targets (< 7.0% and < 6.5%), fasting serum glucose concentrations, self‐monitored blood glucose concentrations, bodyweight, fasting serum lipid concentrations, urinary albumin‐to‐creatinine ratio, high‐sensitivity C‐reactive protein, homeostasis model assessment of β‐cell function and insulin sensitivity, alanine aminotransferase, and 1,5‐anhydroglucitol (a short term marker for glycaemic control). Administered five health outcomes questionnaires: Impact of weight on quality of life‐lite (IWQOL‐Lite), EuroQol instrument (EQ‐5D), binge eating scale (BES) and diabetes treatment satisfaction questionnaire (status version; DTSQs). OTHER OUTCOMES: Adverse events, clinical laboratory assessments, vital signs, and hypoglycaemia. |
|
| Notes |
AIM: To test the hypothesis that improvement in HbA1c concentration achieved with once‐weekly exenatide is better than that achieved with the existing standard second‐line treatment for patients not responding to oral blood‐glucose lowering agents, insulin glargine titrated to glucose targets. SOURCE OF FUNDING: Amylin and Eli Lilly OTHER: Conflict of interest: First author is a consultant and speaker for Eli Lilly, Novo Nordisk, and Merck, Sharp and Dohme; and a consultant of Sanofi‐Aventis. Through this author, the VU University Medical Centre in Amsterdam has received research grants from Amylin Pharmaceuticals, Eli Lilly, Novo Nordisk, Merck, Sharp and Dohme, Novartis and Takeda. Another author has served on advisory panels for Abbott Laboratories, AstraZeneca, Bristol‐Myers Squibb, Eli Lilly, Novo Nordisk and Sanofi‐Aventis, and has received honoraria as member of the speakers's bureau for Abbott Laboratories, AstraZeneca, Bristol‐Myers Squibb and Eli Lilly. Another author has served on an advisory panel for Eli Lilly, has received travel grants from Novo Nordisk, Eli Lilly, Merck, Sharp and Dohme and Servier and has received research funding support from Sanofi‐Aventis. Some authors are employees of Eli Lilly. One author is an employees of Amylin Pharmaceuticals. SAMPLE SIZE: A sample size of 205 patients per treatment was needed to achieve 92% power to detect a difference of 0.4% in change in HbA1c from baseline. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | One to one allocation and block (size four) randomisation, stratified according to country and oral blood glucose lowering treatment (70% metformin only; 30% metformin plus sulphonylurea). Computer‐generated randomisation sequence administered by the sponsor via an automated voice‐response system. |
| Allocation concealment (selection bias) | Low risk | Computer‐generated randomisation sequence administered by the sponsor via an automated voice‐response system. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label (study participants and clinical investigators were not blinded); investigators analysing data were blinded to treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Modified intention‐to‐treat analysis, details of withdrawals and losses to follow up |
| Selective reporting (reporting bias) | Low risk | All the predefined and prespecified outcomes were reported |
| Methods |
TRIAL DESIGN: Randomised comparator‐controlled, open label trial, non‐inferiority study DURATION OF INTERVENTION: 30 weeks DURATION OF FOLLOW‐UP: No post‐intervention follow‐up SETTING: Not reported COUNTRY: Canada/USA |
|
| Participants |
INCLUSION CRITERIA: People with type 2 diabetes, aged at least 16 years old, HbA1c between 7% and 11%, FPG < 16 mmol/L, BMI between 25 and 45 kg/m2, therapy with diet modification and exercise, or pharmacological treatment with MET, a SU, a TZD, or any combination of two of these agents; weight stable (weight did not vary > 10%) for 6 months prior screening, no abnormal results of clinical significance on blood testing. EXCLUSION CRITERIA: Patients who had used meglitinides, alpha‐glucosidase inhibitors, insulin therapy, weight‐loss drugs, corticosteroids, drugs known to affect gastrointestinal motility, or any investigational drug; any exposure to exenatide or a GLP‐1 analogue; or evidence of clinically significant medical conditions that might preclude safe participation in the study. AGE: 55 SD 10 years SEX: 45% to 49% female DIABETES DURATION: 6 to 7 years (SD 5 to 6) ETHNICITY: 73% to 83% White, 6% to 13% Black, 11% to 14% Hispanic, 0% to 1% Asian HbA1c (%) : EX 2 mg QW: 8.3 SD 1.0; EX 10 µg BID: 8.3 SD 1.0 BMI (kg/m2): EX 2 mg QW: 35 SD 5; EX 10 µg BID: 35 SD 5 PREVIOUS THERAPY: Monotherapy: 43% to 46%, combination therapy: 36% to 39%; all MET: 69% to 77%, all SU: 37%, all TZD: 15% to 17%; diet/exercise only: 14% to 16% NUMBERS: Randomised: 303, Analysed as ITT: 295 (8 withdrew before lead‐in); EX 2 mg QW: 148, EX 10 µg BID: 147 |
|
| Interventions |
COMPARISON: EX twice daily + previous therapy VERSUS EX once weekly + previous therapy NO. OF COMPARISON GROUPS: 2 RUN‐IN: None DOSE EX: subcutaneous injection of exenatide 2 mg once a week or 10 µg twice a day lead‐in for 2 mg QW: 3 days 5 µg EX BID, then 2 mg QW lead‐in for 10 µg EX BID: 5 µg EX BID for 28 days, then 10 µg EX BID for the remainder of the 30 weeks PREVIOUS THERAPY: Diet/exercise or metformin (MET), sulphonylurea (SU), or thiazolidinedione (TZD) as monotherapy or combination of any two; see above for combinations used, doses of MET, SU and TZD were not reported; to avoid hypoglycaemia, SU dose was reduced to minimum labelled dose until week 10, then up‐titrated to reach target FPG of ≤ 6 mmol/L OTHER TREATMENT: Not reported |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c at the end of the study i.e. 30 weeks (non‐inferiority within 0.4%) SECONDARY OUTCOMES: Safety and tolerability, body weight, fasting plasma glucose (FPG), postprandial glucose (PPG), fasting glucagon, fasting lipids, blood pressure, proportion of patients achieving HbA1c concentrations of ≤ 7.0%, ≤ 6.5%, ≤ 6.0%, overall and by baseline HbA1c strata; HbA1c by antibody titre; bodyweight in the presence and absence of nausea OTHER OUTCOMES: Treatment‐emergent adverse events (defined as those occurring on or after receiving the first injection of study medication): patients who lost glucose control (1.5% increase in HbA1c or HbA1c of ≥11.5% at or after week 14; patients with loss of glucose control withdrawn from the study); hypoglycaemic episodes: minor (symptoms of hypoglycaemia and a plasma glucose < 3 mmol/L) and major (loss of consciousness, seizure, or coma; third party assistance to resolve or administration of glucose or glucagon; and a plasma glucose < 3 mmol/L); vital signs, ECG reports, or haematological, chemistry, or urinalysis values |
|
| Notes |
AIM: To compare the efficacy and safety of exenatide once a week to that of exenatide given twice daily, over 30 weeks, in patients with type 2 diabetes. SOURCE OF FUNDING: Amylin Pharmaceuticals and Eli Lilly and Company. OTHER: Conflict of interest: One author has been a consultant for and received lecture honoraria from Amylin, Eli Lilly and Novo Nordisk. SAMPLE SIZE: A sample size of 300 patients was estimated to provide 90% power to test the hypothesis that the treatments were non‐inferior with respect to HbA1c control. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method not stated; randomisation was stratified according to concomitant sulphonylurea use at screening and HbA1c strata (< 9.0% vs ≥ 9.0%) |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label; the investigators, sponsor, patients, and all personnel involved with the study were not blinded to the identity of the study medication; blinding of HbA1c and FPG results |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data were imputed as the last observation carried forward, ITT analysis, adequate description of withdrawals and losses to follow‐up (12% withdrawals, no significant difference between groups) |
| Selective reporting (reporting bias) | Low risk | Included all expected and prespecified outcomes |
| Methods |
TRIAL DESIGN: Double‐blind, multicenter, randomised, parallel‐group, three arm trial DURATION OF INTERVENTION: 24 weeks DURATION OF FOLLOW‐UP: This trial was part of a 52 week, multicenter, double‐blind, randomised, parallel‐group trial in which the initial 24 week double‐blind period was followed by a 28 week open label period RUN‐IN PERIOD: 4 week preceeded randomisation, after which subjects were stratified according to their pretrial SU therapy. SETTING: 49 centres COUNTRY: Japan |
|
| Participants |
INCLUSION CRITERIA: Japanese men and women ≥ 20 years of age with type 2 diabetes mellitus currently treated with an SU [glibenclamide (1.25 to 10 mg), gliclazide (40 to 160 mg) or glimepiride (1 to 6 mg) for ≥ 8 weeks, HbA1c between 7.0% to < 10%, BMI < 35.0 kg/m2. EXCLUSION CRITERIA: Treated with insulin within 12 weeks, were receiving or expecting to receive systemic corticosteroids, or had known hypoglycaemia unawareness or recurrent major hypoglycaemia, impaired renal or hepatic function, significant cardiovascular disease (heart failure, coronary artery disease or uncontrolled hypertension) or non stabilized proliferative retinopathy or maculopathy. AGE: 58.6 SD 9.7 to 61.3 SD 11.0 years SEX: 33% to 40% female (LIR 0.6 mg: 40%; LIR 0.9 mg: 33%; Placebo: 35%) DIABETES DURATION: 9.3 SD 5.8 to 11.6 SD 7.7 years ETHNICITY: All Japanese patients. HbA1c (%): LIR 0.6 mg: 8.6 SD 0.91; LIR 0.9 mg: 8.21 SD 0.78; Placebo: 8.45 SD 0.99 BMI (kg/m2): LIR 0.6 mg: 25.3 SD 3.6; LIR 0.9 mg: 24.4 SD 3.4; Placebo: 24.9 SD 4.0 PREVIOUS THERAPY: Treated with SU [glibenclamide (1.25 to 10 mg), gliclazide (40 to 160 mg) or glimepiride (1 to 6 mg) NUMBERS: Screened: 308; Randomised: 264 (LIR 0.6 mg: 88; LIR 0.9 mg: 88; Placebo: 88); Fully analysis set: 264 (LIR 0.6 mg: 88; LIR 0.9 mg: 88; Placebo: 88); per‐protocol set: 235 (LIR 0.6 mg: 79; LIR 0.9 mg: 83; Placebo: 73) |
|
| Interventions |
COMPARISON: Liraglutide (LIR) 0.6 mg/0.9 mg + SU VERSUS Placebo + SU NO. OF COMPARISON GROUPS: 3 DOSE LIR: LIR 0.6 mg or 0.9 mg once daily: LIR doses were up titrated from 0.3 mg/day (50 µl) to 0.6 mg/day (100 µl) after the first week, with an additional increase to 0.9 mg/day (150 µl) for the 0.9 mg cohort after the second week. LIR was injected once daily in the morning or evening s.c. into the upper arm, thigh or abdomen. DOSE SU: SU continued in prestudy dose. OTHER TREATMENT: NR |
|
| Outcomes |
PRIMARY OUTCOMES: HbA1c level at 24 weeks SECONDARY OUTCOMES: 7‐point self‐measured PPG profiles, body weight, FPG, mean PPG, lipid profile and biomarkers for cardiovascular effects, proportions of subjects reaching HbA1c ≤ 7% or ≤ 6.5% OTHER OUTCOMES: Incidence of hypoglycaemic episodes (self‐treated hypoglycaemic episodes were classified as minor, while those requiring third party assistance were considered as major and the remainder as symptoms‐only), incidence of adverse events, vital signs and clinical laboratory assessments. |
|
| Notes |
AIM: the efficacy and safety of two doses of liraglutide (0.6 and 0.9 mg/day) over 24 weeks compared with placebo, in each case as add‐on to SU monotherapy SOURCE OF FUNDING: Novo Nordisk OTHER: Conflict of interest: NR SAMPLE SIZE: No information. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Subjects were stratified according to pretrial SU therapy. Insufficient information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy endpoints: Full analysis set; Per protocol set; LOCF; adequate description of adverse events, withdrawals and losses to follow‐up |
| Selective reporting (reporting bias) | Low risk | All predefined and prespecified outcomes were reported |
| Methods |
TRIAL DESIGN: Randomised double‐blind, double dummy, active control, five armed parallel trial, multi‐centre (116 sites) DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: No post‐intervention follow‐up RUN‐IN PERIOD: 2 weeks SETTING: NR COUNTRY: 21 countries (mainly in Europe and Asia) |
|
| Participants |
INCLUSION CRITERIA: Patients with type 2 diabetes, OADs for ≥ 3 months, aged 18 to 80 years old, HbA1c 7% to 11% (previous OAD monotherapy) or 7% to 10% (previous OAD combination therapy), BMI ≤ 45 kg/m2 EXCLUSION CRITERIA: Using insulin within 3 months, impaired liver or renal function, uncontrolled hypertension (≥ 180/100 mm Hg), cancer, use of any drug apart from OAD likely to affect glucose concentrations AGE: 54.7 SD 10.0 to 57.7 SD 9.0 years SEX: 47% to 55% female DIABETES DURATION: (median, 25th and 75th percentile) 6.5 (3.7, 10.5) to 6.7 (4.0, 10.7) years ETHNICITY: NR HbA1c (%): LIR 1.2 mg: 8.5 SD 1.1; LIR 1.8 mg: 8.5 SD 0.9; Placebo: 8.4 SD 1.0; ROS (rosiglitazone): 8.4 SD 1.0 BMI (kg/m2): LIR 1.2 mg: 29.8 SD 5.1; LIR 1.8 mg: 30 SD 5.1; Placebo: 30.3 SD 5.4; ROS: 29.4 SD 4.8 PREVIOUS THERAPY: Previously on mono‐therapy: 27% to 32%, on combination therapy: 68% to 73% NUMBERS: 1712 screened; Randomised: 1041 (1 to 37 participants per centre); LIR 0.6 mg: 233; LIR 1.2 mg: 228; LIR 1.8 mg: 234; Placebo: 114; ROS: 232 |
|
| Interventions |
COMPARISON: LIR (3 doses) + Glimepiride (SU) VERSUS Placebo + SU VERSUS ROS (TZD) + SU NO. OF COMPARISON GROUPS: 5 (LIR 0.6 mg not considered in the present review) DOSE LIR: 1.2 mg and 1.8 mg: LIR up‐titrated weekly in 0.6 mg increments until the allocated dose reached; injected subcutaneously once daily DOSE SU: Forced glimepiride titration for 2 weeks and then 2 weeks maintenance period; glimepiride 2 to 4 mg/day DOSE TZD: 4 mg/day rosiglitazone OTHER TREATMENT: NR |
|
| Outcomes |
PRIMARY OUTCOMES: HbA1c (change from baseline to end of treatment) SECONDARY OUTCOMES: Proportion of participants reaching targeted goals of HbA1c (≤ 7%, ≤ 6.5%), FPG (5 to ≤ 7.2 mmol/L), PPG (10 mmol/L); body weight, FPG (fasting plasma glucose), PPG (post prandial glucose), beta‐cell function, blood pressure; superiority of liraglutide to placebo and non‐inferiority to rosiglitazone was tested OTHER OUTCOMES: Hypoglycaemic episodes based on PG levels (< 3.1 mmol/L) (minor: self‐treated; major: requiring third party assistance), liraglutide antibodies including cross‐reacting and neutralizing antibodies, tolerability (gastrointestinal complaints), pulse, adverse events, vital signs, ECG, biochemical and haematological parameters, calcitonin |
|
| Notes |
AIM: To compare efficacy and safety of liraglutide and glimepiride combination therapy with either placebo or rosiglitazone added to glimepiride. SOURCE OF FUNDING: Novo Nordisk OTHER: Conflict of interest: One author had received lecture fees from Novo Nordisk, Servier, MSD. The second author had received honoraria, grants and lecture fees from Novo Nordisk. The remaining authors had no conflict of interest SAMPLE SIZE: A combined power (calculated as the product of the marginal powers for HbA1c and body weight) of at least 85% was required. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information; participants were stratified according to previous treatment (monotherapy or combination therapy) |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comparisons made on the intent to treat population, missing data imputed using last observation carried forward, adequate description of withdrawals and losses to follow‐up (overall 14% withdrawals, 9 to 27% in the individual groups) |
| Selective reporting (reporting bias) | Low risk | All pre‐specified (primary and secondary) outcomes were reported |
| Methods |
TRIAL DESIGN: RCT, double dummy, active control, parallel group trial (part of a phase 3 clinical development program for liraglutide), multicenter (170 sites) DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: No post‐intervention follow‐up RUN‐IN PERIOD: 6 weeks SETTING: Not reported COUNTRY: Multinational (21 countries) |
|
| Participants |
INCLUSION CRITERIA: People with type 2 diabetes, age 18 to 80 years old, HbA1c between 7% and 11% (prestudy OAD monotherapy for ≥ 3 months) or between 7% and 10% (prestudy combination OAD therapy for ≥ 3 months), BMI ≤ 40 kg/m2 EXCLUSION CRITERIA: Patients who had used insulin during the previous 3 months (except short term treatment) AGE: mean 56 to 57 years (SD 9) SEX: 40% to 46% female DIABETES DURATION: mean 7 to 8 years (SD 5) ETHNICITY: 88% to 89% White, 2% to 4% Black, 7% to 9% Asian, 1% to 3% other HbA1c (%): LIR 1.2 mg: 8.3 SD 1, LIR 1.8 mg: 8.4 SD 1, SU: 8.4 SD 1, Placebo: 8.4 SD 1.1 BMI (kg/m2): LIR 1.2 mg: 31.1 SD 4.8, LIR 1.8 mg: 30.9 SD 4.6, SU: 31.2 SD 4.6, Placebo: 31.6 SD 4.4 PREVIOUS THERAPY: 65% combination therapy, 35% monotherapy (88% metformin) NUMBERS: Randomised: 1091 (4 withdrew consent before treatment), Analysed as ITT: 1087; LIR 0.6 mg: 242, LIR 1.2 mg: 240, LIR 1.8 mg: 242, SU: 242, Placebo: 121 |
|
| Interventions |
COMPARISON: Liraglutide (LIR) (3 doses) + Metformin (MET) VERSUS Glimepiride (SU) + MET VERSUS Placebo + MET NO. OF COMPARISON GROUPS: 5 RUN‐IN: Forced titration period of metformin for three weeks (dose increased up to 2000 mg/day: 1000 mg in the morning and 1000 mg in the evening) followed by a 3 week metformin maintenance period before randomisation. DOSE LIR: 0.6, 1.2 or 1.8 mg/day injected subcutaneously once daily (0.6 mg not included in this review). LIR titrated after randomisation for 2 to 3 weeks (increase by 0.6 mg/day per week) DOSE MET: 1500 to 2000 mg/day DOSE SU: 4 mg glimepiride OD with the first meal of the day. Glimepiride (SU) titrated after randomisation for 2 to 3 weeks (1 mg in week 1, 2 mg week 2, 4 mg week 3) OTHER TREATMENT: not reported |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c at the end of the study SECONDARY OUTCOMES: Body weight, fasting plasma glucose (FPG), postprandial glucose (PPG) (7 point plasma glucose profiles: before each meal, 90 min after breakfast), beta cell function (based on fasting insulin, fasting C‐peptide, fasting proinsulin‐to‐insulin ratio and the homeostasis model assessment index of beta cell function (HOMA‐B)) OTHER OUTCOMES: Adverse events, vital signs, electrocardiogram, biochemical and hematology measures, and subject‐reported hypoglycaemic episodes (based on symptoms and plasma glucose <3.1 mmol/L) |
|
| Notes |
AIM: To study the efficacy and safety of Liraglutide as a combination therapy with metformin as compared with placebo and glimepiride in addition to metformin SOURCE OF FUNDING: Novo Nordisk (presumably) OTHER: Conflict of interest: Some authors are members of advisory board and have received honoraria from Novo Nordisk, and work for Novo Nordisk. SAMPLE SIZE: The combined power (calculated as the product of the marginal powers for A1C and weight) was at least 85%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned as 2:2:2:1:2. Telephone or web based randomisation. Patients randomly assigned to the lowest available randomisation number and stratified with respect to their previous use of OAD monotherapy or combination therapy |
| Allocation concealment (selection bias) | Low risk | Telephone or web based randomisation |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data were imputed as the last observation carried forward, ITT analysis, adequate description of withdrawals and losses to follow‐up (19% withdrawals and losses to follow‐up) |
| Selective reporting (reporting bias) | Low risk | Included all expected outcomes, including those prespecified |
| Methods |
TRIAL DESIGN: RCT (1:1:1), placebo control, parallel group, multicenter (96 sites) DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: No post‐intervention follow‐up RUN‐IN PERIOD: 6 to 9 weeks SETTING: Not reported COUNTRY: USA and Canada |
|
| Participants |
INCLUSION CRITERIA: people with type 2 diabetes, 18 to 80 years of age HbA1c between 7% to 11% (prestudy OAD monotherapy for ≥ 3 months or 7% to 10% (prestudy OAD combination therapy for ≥ 3 months, BMI ≤ 45kg/m2; eligibility for randomisation: participants tolerating maximum doses of OAD (metformin and rosiglitazone) and with FPG values of 135 to 230 mg/dL (7.5 to 12.8 mmol/L) after 6 weeks treatment with titrated dose EXCLUSION CRITERIA: Participants using insulin during the previous 3 months (except short term treatment) AGE: mean 55 years (SD 10) SEX: 38% to 49% female DIABETES DURATION: mean 9 years (SD 6) ETHNICITY: 81% to 84% White, 10% to 15% Black, 1% to 3% Asian, 1% American Indian, 2% to 3% other HbA1c (%): LIR 1.2 mg: 8.5 SD 1.2, LIR 1.8 mg: 8.6 SD 1.2, Placebo: 8.4 SD 1.2 BMI (kg/m2): LIR 1.2 mg: 33.2 SD 5.4, LIR 1.8 mg: 33.5 SD 5.1, Placebo: 33.9 SD 5.2 PREVIOUS THERAPY: 17% monotherapy, 83% combination therapy NUMBERS: 533 randomised; LIR 1.2 mg:178; LIR 1.8 mg:178; Placebo:177 |
|
| Interventions |
COMPARISON: Liraglutide (LIR) (2 doses) + Metformin (MET) + Rosiglitazone (TZD) VERSUS Placebo + MET + TZD NO. OF COMPARISON GROUPS: 3 RUN‐IN: Treatment with other OADs except MET and TZD were discontinued prior to randomisation. MET dose started at 500 mg and was titrated up to 2000 mg/day. Rosiglitazone (TZD) dose started at 4 mg and was titrated up to 8 mg/day. DOSE LIR: LIR 1.2 mg/day or 1.8 mg/day injected subcutaneously once daily. LIR started with 0.6 mg/day for a week and increased up to 1.2 mg/day and then to 1.8 mg/day after an additional week for those randomised to highest dose DOSE MET: 2000 mg/day (1000 mg in the morning and 1000 mg in the evening) DOSE TZD: Rosiglitazone 8 mg/day (4 mg in the morning and 4 mg in the evening) OTHER TREATMENT: None |
|
| Outcomes |
PRIMARY OUTCOMES: HbA1c (change from randomisation to end of study) SECONDARY OUTCOMES: Change in following parameters: body weight, FPG (Fasting plasma glucose), PPG (Postprandial glucose) (from 7‐point plasma glucose profiles), beta‐cell function (based on fasting insulin, fasting C‐peptide, fasting pro‐insulin to insulin ratio, and the homeostasis model assessment (HOMA) for beta cells (HOMA‐B) and insulin resistance (HOMA‐IR)), blood pressure, lipids OTHER OUTCOMES: Safety variables including adverse events, vital signs, ECG, biochemical and haematology measures, and subject reported hypoglycaemic episodes (plasma glucose < 3.1 mmol/L) Superiority of liraglutide tested |
|
| Notes |
AIM:To study the efficacy and safety of Liraglutide as a combination therapy with metformin and rosiglitazone in type 2 diabetes SOURCE OF FUNDING: Novo Nordisk (presumably) OTHER: Conflict of interest Novo Nordisk, Denmark SAMPLE SIZE: The combined power (calculated as the product of the marginal powers for HbA1c and weight) was > 95%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised as 1:1:1. Telephone or web based randomisation; protocol states: "Randomisation will be carried out centrally using a randomisation system, IVRS/IWRS" [Interactive voice (or web) response system] |
| Allocation concealment (selection bias) | Low risk | Telephone or web based randomisation |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data were imputed as the last observation carried forward (LOCF), ITT analysis, adequate description of withdrawals and losses to follow‐up (24% withdrawals and losses to follow‐up) |
| Selective reporting (reporting bias) | Low risk | Included all prespecified outcomes |
| Methods |
TRIAL DESIGN: RCT (2:1:2), parallel group, placebo‐controlled, multicenter (107 sites) DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: 1 week post‐intervention follow‐up (but no data given) RUN‐IN PERIOD: 6 weeks SETTING: Not reported COUNTRY: Multinational (17 countries) |
|
| Participants |
INCLUSION CRITERIA: People with type 2 diabetes, 18 to 80 years, treated with OAD for at least 3 months before screening, HbA1c level between 7.5% to 10% (monotherapy) or 7% to 10% (combination therapy), BMI ≤ 45kg/m2 EXCLUSION CRITERIA: Patients who had used insulin within 3 months prior to the trial; patients with impaired renal or hepatic function, cardiovascular disease, cancer, hypertension, retinopathy, maculopathy; pregnant patients; those with recurrent hypoglycaemia or hypoglycaemia awareness; those who were seropositive for hepatitis B antigen or hepatitis C antibody; or who have used any other drugs except OAD that could affect blood glucose levels AGE: mean 57.5 years (SD10) SEX: 40% to 51% female DIABETES DURATION: mean 9.2 to 9.7 years (SD 6) ETHNICITY: Not reported HbA1c (%): LIR: 8.3 SD 0.9, Placebo: 8.3 SD 0.9, GLAR (insulin glargine): 8.2 SD 0.9 BMI (kg/m2): LIR: 30.4 SD5 .3, Placebo: 31.3 SD 5.0, GLAR: 30.3 SD 5.3 EXISTING THERAPY: 5 to 6% monotherapy, 94% to 95% combination treatment NUMBERS: 581 randomised, ITT population 576; LIR: 230, Placebo: 114, GLAR: 232 |
|
| Interventions |
COMPARISON: Liraglutide (LIR) + Metformin (MET) + Glimepiride (SU) VERSUS Placebo + MET + SU VERSUS GLAR + MET + SU NO. Of COMPARISON GROUPS: 3 RUN‐IN: Forced MET and SU dose escalation over 3 weeks followed by 3 weeks maintenance DOSE LIR: 1.8 mg OD. Dose escalation starting at 0.6 mg and increasing weekly by 0.6 mg over 2 weeks. DOSE MET: 2 gm (1 gm BID) DOSE SU: Glimepiride 4 mg OD, reduction to 2 mg allowed in case of adverse events or hypoglycaemia DOSE GLAR: Insulin glargine (open label). Insulin was titrated according to patient‐driven algorithm (average dose at the end of study was 24 IU/day) OTHER TREATMENT: None |
|
| Outcomes |
PRIMARY OUTCOMES: HbA1c (change from baseline to end) SECONDARY OUTCOMES: Change in weight, FPG, eight point plasma glucose (PG) profiles, beta‐cell function, and blood pressure OTHER OUTCOMES: Safety variables like hypoglycaemic episodes, adverse events |
|
| Notes |
AIM: To compare the efficacy and safety of liraglutide to insulin glargine all as add on to combination therapy of metformin and glimepiride SOURCE OF FUNDING: Novo Nordisk (presumably) OTHER: Conflict of interest: Novo Nordisk, Amylin Pharmaceuticals SAMPLE SIZE: Study was powered to determine a 3% difference in weight with a combined power > 85%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised as 2:1:1 using a telephone or web‐based randomisation system |
| Allocation concealment (selection bias) | Low risk | Central randomisation using a telephone or web‐based randomisation system |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Investigators, participants, and study monitors were blinded to liraglutide, open label glargine |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data were imputed as the last observation carried forward (LOCF), ITT analysis, adequate description of withdrawals and losses to follow‐up (9.4% withdrawals and losses to follow‐up) |
| Selective reporting (reporting bias) | Low risk | Included all prespecified outcomes |
| Methods |
TRIAL DESIGN: RCT, open label, active comparator, parallel group, multi‐centre (132 sites) DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: No post‐intervention follow‐up RUN‐IN PERIOD: None COUNTRY: 15 countries |
|
| Participants |
INCLUSION CRITERIA: People with type 2 diabetes, 18 to 80 years old, HbA1c between 7% and 11%, BMI ≤ 45 kg/m2, stable on treatment with maximally tolerated dose of metformin, sulphonylurea, or both, for 3 months or more EXCLUSION CRITERIA: Previous insulin treatment (except short term treatment), previous treatment with exenatide or liraglutide, impaired renal or liver function, diseases like retinopathy or maculopathy or related to cardiovascular requiring acute treatment, uncontrolled hypertension, cancer AGE: mean 56 to 57 years (SD 10) SEX: 45% to 51% female DIABETES DURATION: mean 7.9 to 8.5 years (SD 6) ETHNICITY: 91% to 93% White, 1% to 5% Asian, 12% to 13% Black, 3% to 4% other HbA1c (%): LIR: 8.2 SD 1.0, EX: 8.1 SD 1.0 BMI (kg/m2): LIR: 32.9 SD 5.5, EX: 32.9 SD 5.7 PREVIOUS THERAPY: 62% to 64% MET plus SU, 27% MET monotherapy, 9% to 10% SU monotherapy NUMBERS: 464 randomised; LIR: 233; EX: 231; (exposed LIR: 235; EX: 232) |
|
| Interventions |
COMPARISON: LIR + existing therapy MET and/or SU VERSUS EX + existing MET and/or SU therapy NO. OF COMPARSION GROUPS: 2 RUN‐IN: None DOSE LIR: Liraglutide 1.8 mg OD; 2 week dose escalation starting at 0.6 mg/day increasing weekly by 0.6 mg up to 1.8 mg DOSE EX: Exenatide 10 μg BID; 4 week dose escalation starting at 5 μg BID increasing to 10 μg BID at 4 weeks DOSE MET: Prestudy dose DOSE SU: Prestudy dose, in case of unacceptable hypoglycaemia SU dose could be reduced to no less than 50% of starting dose OTHER TREATMENT: None |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c % from baseline to end point SECONDARY OUTCOMES: Reduction in FPG levels (mmol/L), % of patients who achieved a target HbA1c level ≤ 7%, % of patients who achieved a target HbA1c level ≤ 6.5%, mean change in body weight %, mean changes in self measured 7‐point plasma glucose profiles, % change in HOMA‐B from baseline, mean changes in glucagon, blood pressure and lipid profiles, overall treatment satisfaction (Diabetes Treatment Satisfaction questionnaire) assessed in a subgroup of patients OTHER OUTCOMES: Safety variables including adverse events, vital signs, electrocardiogram, biochemical and haematological measures, and patient reported hypoglycaemic episodes were assessed. |
|
| Notes |
AIM: To compare the effectiveness of liraglutide to exenatide as an add on therapy to metformin and/or sulphonylurea. SOURCE OF FUNDING: Novo Nordisk OTHER: Conflict of interest: main author is a member of advisory board and have received honoraria from Novo Nordisk and various other pharmaceutical companies, and work as consultant for various other pharmaceutical companies. SAMPLE SIZE: 85% power to detect an HbA1c difference of 0.4% between groups. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned (1:1) to the lowest available number of the numbers allocated to the site |
| Allocation concealment (selection bias) | Low risk | Randomisation was done with telephone based or web based system |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Last observation carried forward data with repeated measures analysis and multiple imputation methods, ITT analysis, adequate description of withdrawals and losses to follow‐up (17% withdrawals and losses to follow‐up) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Methods |
TRIAL DESIGN: Active comparator, parallel‐group, open label trial; non‐inferiority and superiority comparison DURATION OF INTERVENTION: 26 weeks DURATION OF FOLLOW‐UP: After the 26 week study, participants could continue into a 12 month follow‐up trial RUN‐IN PERIOD: SETTING: 158 office‐based sites COUNTRY: Multinational (11 European countries: Croatia, Germany, Ireland, Italy, Netherlands, Romania, Serbia, Slovakia, Slovenia, Spain and UK; the USA and Canada) |
|
| Participants |
INCLUSION CRITERIA: Participants aged between 18 to 80 years; type 2 diabetes mellitus, HbA1c of 7.5% to10.0%; BMI ≤ 45 kg/m2; treated with metformin ≥ 1500 mg daily for 3 months or longer. EXCLUSION CRITERIA: Previous treatment with any antihyperglycaemic drug apart from metformin within 3 months of the trial; recurrent major hypoglycaemia or hypoglycaemia unawareness; present use of any drug except metformin that could affect glucose; contraindications to trial drugs; impaired renal or hepatic function; clinically significant cardiovascular disease; or cancer. AGE: 55.0 SD 9.0 to 55.9 SD9.6 years SEX: 45% to 48% female (LIR 1.2 mg: 48%; LIR 1.8 mg: 48%; SITA: 45%) DIABETES DURATION: 6.0 SD 4.5 to 6.4 SD 5.4 years ETHNICITY: 82% to 91% White (15% to 17% Hispanic or Latino); 5% to 10% Black; 1% to 2% Asian or Pacific Islander; 4% to 5% Other HbA1c (%): LIR 1.2 mg: 8.4 SD 8.0; LIR 1.8 mg: 8.4 SD 0.7; SITA: 8.5 SD 0.7 BMI (kg/m2): LIR 1.2 mg: 32.6 SD 5.2; LIR 1.8 mg: 33.1 SD 5.1; SITA: 32.6 SD 5.4 PREVIOUS THERAPY: MET NUMBERS: Assessed: 1302; randomised: 665 (LIR 1.2 mg: 225; LIR 1.8 mg: 221; SITA: 219); full analysis set: 658 (LIR 1.2 mg: 221; LIR 1.8 mg: 218; SITA: 219); fully analysis/safety analysis set: 658 (LIR 1.2 mg: 221; LIR 1.8 mg: 218; SITA: 219) |
|
| Interventions |
COMPARISON: LIR 1.2 mg or 1.8 mg once daily + MET VERSUS SITA 100 mg daily + MET NO. OF COMPARISON GROUPS: 3 DOSE LIR: LIR started at 0.6 mg/day and escalated by 0.6 mg/week to the allocated dose; s.c. with a pen device. DOSE SITA: SITA was started and maintained at 100 mg/day. DOSE MET: Background treatment with MET remained stable. OTHER TREATMENT: NR |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c from baseline to week 26 SECONDARY OUTCOMES: Superiority and non‐inferiority comparisons; proportions of participants reaching HbA1c targets of less than 7.0% (ADA) or of 6.5% or lower (AACE, IDF, NICE); FPG; PPG; bodyweight; β‐cell function; fasting lipid profile; cardiovascular risk markers (high sensitivity C‐reactive protein, plasminogen activator inhibitor type 1, N‐terminal pro‐B‐type natriuretic peptide, adiponectin, interleukin‐6, tumour necrosis factor α, and von Willebrand factor); BP; HR; physical measures (waist circumference, waist‐to‐hip ratio); treatment satisfaction; and a composite endpoint of proportions of participants with HbA1c < 7% with no hypoglycaemia, and weight change of 0kg or less. OTHER OUTCOMES: Adverse events, self‐reported hypoglycaemia, and selected haematological and biochemical measures including calcitonin. Minor hypoglycaemic episodes (PG < 3.1 mmol/L); Major hypoglycaemic episodes (third‐party assistance irrespective of glucose concentrations). |
|
| Notes |
AIM: To compare the efficacy and safety of treatment with liraglutide or sitagliptin for 26 weeks in individuals with type 2 diabetes who did not achieve adequate glycaemic control with metformin. SOURCE OF FUNDING: Novo Nordisk OTHER: Conflict of interest: Authors have received grants and consultancy fees from different pharmaceutical companies. SAMPLE SIZE: To show that 1.8 mg liraglutide plus metformin was non‐inferior to 100 mg sitagliptin with metformin with a margin of 0.4%, 163 participants per group were needed for 85% power and with a predicted withdrawal of 25%, 217 participants per group were needed. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated by Novo Nordisk; participants randomly assigned in a 1:1:1 ratio, stratified by country, to receive 1.2 mg or 1.8 mg s.c. liraglutide once daily or 100 mg oral sitagliptin once daily. |
| Allocation concealment (selection bias) | Low risk | Computer‐generated; consecutive allocation of the randomisation code to individual participants was concealed by use of a telephone‐based (interactive voice response system) or web‐based (interactive web response system) randomisation system. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label; data were masked from the statistician until database release |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Primary efficacy analyses: full analysis set with missing values imputed by last observation carried forward; Secondary efficacy analyses: full analysis set, apart from treatment satisfaction analyses, in which missing data were no imputed; Superiority: full analysis set; Non‐inferiority: full analysis and per‐protocol sets. Adequate description of withdrawals and loss to follow up. |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Methods |
TRIAL DESIGN: Double‐blind, double dummy, randomised, four‐arm, active control trial DURATION OF INTERVENTION: 16 weeks DURATION OF FOLLOW‐UP: RUN‐IN PERIOD: Eligible participants discontinued their pretrial OADs except metformin and entered a 3‐week run‐in with forced escalation of metformin to 2000 mg/day, followed by another 3‐week metformin maintenance period. SETTING: China (17 sites), South Korea (10 sites) and India (24 sites) COUNTRY: China, South Korea and India |
|
| Participants |
INCLUSION CRITERIA: Participants diagnosed with type 2 diabetes and treated with one or more oral antidiabetic drugs (OADs) for at least 3 months, aged 18 to 80 years (18 to 75 years for Chinese subjects), with HbA1c ≥ 7.0% and ≤ 11.0% for subjects on OAD monotherapy or ≥ 7.0% and ≤ 10.0% for subjects on OAD combination therapy and BMI ≤ 45.0 kg/m2. EXCLUSION CRITERIA: Participants treated with insulin within the last 3 months. AGE: 52.7 SD 9.1 to 53.6 SD9.7 years SEX: 41.6% to 46.2% female (LIR 1.2 mg: 45.1%; LIR 1.8 mg: 46.2%; GLIM (Glimepiride): 41.6%) DIABETES DURATION: 7.2 SD 5.2 to 7.8 SD 6.1 years ETHNICITY: NR HbA1c (%): LIR 1.2 mg: 8.6 SD 1.1; LIR 1.8 mg: 8.6 SD 1.1; GLIM: 8.5 SD 1.1 BMI (kg/m2): LIR 1.2 mg: 25.4 SD 3.7; LIR 1.8 mg: 25.8 SD 3.8; GLIM: 25.3 SD 3.7 PREVIOUS THERAPY: OAD monotherapy: 29.4% to 32.1%; OAD combination: 67.9% to 70.6% NUMBERS: Randomised: 926; Exposed: 928 (LIR 1.2 mg: 233; LIR 1.8 mg: 233; GLIM: 231); per‐protocol set: 562 (LIR 1.2 mg: 179; LIR 1.8 mg: 172; GLIM: 211) |
|
| Interventions |
COMPARISON: LIR 0.6 mg/1.2 mg/1.8 mg + MET + GLIM Placebo VERSUS GLIM + MET + LIR placebo NO. OF COMPARISON GROUPS: 3 (LIR 0.6 mg arm excluded) DOSE LIR 1.2 mg/ LIR 1.8 mg: dose of LIR was increased from 0.6 mg/day to the respective target dose level (in steps of 0.6 mg/day per week) LIR was injected once daily at any time of the day. DOSE GLIM: Subjects were instructed to start dose escalation of GLIM by daily administration of a 1 mg capsule. Further dose escalation took place over a 2‐week period, incrementing to a maximum dose of 4 mg/day. DOSE LIR PLACEBO: dose of LIR placebo was increased from 0.6 mg/day to the respective target dose level (in steps of 0.6 mg/day per week) LIR was injected once daily at any time of the day. DOSE GLIM PLACEBO: start dose escalation of GLIM placebo by daily administration of a 1 mg capsule. Further dose escalation took place over a 2‐week period, incrementing to a maximum dose of 4 mg/day. OTHER TREATMENT: |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c from baseline to the end of the trial SECONDARY OUTCOMES: Changes in body weight, FPG, 7‐point self‐measured plasma glucose profile [before each main meal (breakfast, lunch and dinner), 90 min after start of each main meal, and at bedtime], blood pressure (BP) and β‐cell function measured by the homeostasis model assessment index of β‐cell function and the pro‐insulin to insulin ratio. OTHER OUTCOMES: Safety variables included AEs, physical examination, pulse rate, electrocardiogram, haematology, biochemistry and urine measures, formation of liraglutide antibodies and subject reported hypoglycaemic events (based on symptoms and plasma glucose < 3.1 mmol/L). Minor hypoglycaemic events were self‐treated; major events required third‐party assistance. |
|
| Notes |
AIM: To assess and compare the efficacy and safety of liraglutide with those of glimepiride, both in combination with metformin for the treatment of type 2 diabetes in Asian population from China, South Korea and India. SOURCE OF FUNDING: Novo Nordisk OTHER: Conflict of interest: Two authors are employees of Novo Nordisk. Other authors have no competing interests. SAMPLE SIZE: In order to be able to show that liraglutide was non‐inferior to glimepiride when using a 1 : 1 : 1 : 1 randomisation and a non‐inferiority margin of 0.4% (difference in HbA1c reduction) with a power of at least 85%, the sample size needed was 168 subjects per group. Assuming a drop out rate of 25%, the total number of subjects to be randomised was 896 (224 subjects for each dose of the liraglutide + metformin group and 224 in glimepiride + metformin group). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Inadequate information; participants were stratified with respect to their pretrial OAD therapy (monotherapy or combination) and randomised into 1:1:1:1 ratio |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Full analysis set (FAS) with LOCF; adequate description of adverse events, withdrawals and loss to follow‐up. |
| Selective reporting (reporting bias) | Low risk | All the pre‐defined and pre‐specified outcomes were reported. |
| Methods |
TRIAL DESIGN: Multinational, randomised, double‐blind, parallel‐group, placebo controlled trial DURATION OF INTERVENTION: 13 weeks DURATION OF FOLLOW‐UP: RUN‐IN PERIOD: Initial 2 week screening phase, then, a 2 week, single‐blind, placebo run‐in period. SETTING: 133 centres COUNTRY: Multinational |
|
| Participants |
INCLUSION CRITERIA: Participants with type 2 diabetes at least 1 year's duration aged 30 to 75 years and inadequately controlled (HbA1c ≥ 7.0 and < 9.0%) on stable metformin monotherapy (≥ 1000 mg day) for at least 3 months prior to screening. EXCLUSION CRITERIA: History of gastrointestinal disease with prolonged nausea and vomiting during the previous 6 months; history of chronic pancreatitis or stomach/gastric surgery; severe cardiovascular events during the previous 6 months; or hepatic or renal disease at screening [serum creatinine ≥ 114.4 µmol/L for males and ≥ 106.8 µmol/L for females. AGE: 55.4 SD 9.2 to 56.8 SD 7.8 years SEX: 40.4% to 63% female DIABETES DURATION: 6.0 SD 4.8 to 7.2 SD 4.9 years ETHNICITY: 64.8% to 86.8% Caucasian, 1.8% to 16.7% Black, 9.3% to 21.8% Other HbA1c (%): LIXI 5 μg QD: 7.58 SD 0.7; LIXI 10 μg QD: 7.52 SD 0.6; LIXI 20 μg QD: 7.58 SD 0.7; LIXI 30 μg QD: 7.52 SD 0.7; LIXI 5 μg BID: 7.60 SD 0.6; LIXI 10 μg BID: 7.54 SD 0.6; LIXI 20 μg BID: 7.61 SD 0.7; LIXI 30 μg BID: 7.46 SD 0.5; Placebo: 7.53 SD 0.6 BMI (kg/m2): LIXI 5 μg QD: 30.7 SD 4.6; LIXI 10 μg QD: 31.9 SD 4.0; LIXI 20 μg QD: 32.0 SD 4.3; LIXI 30 μg QD: 31.6 SD 3.6; LIXI 5 μg BID: 31.6 SD 4.2; LIXI 10 μg BID: 32.8 SD 4.4; LIXI 20 μg BID: 32.7 SD 4.4; LIXI 30 μg BID: 32.3 SD 4.5; Placebo: 31.7 SD 4.2 PREVIOUS THERAPY: MET NUMBERS: Screened: 1466; randomised: 542 (LIXI 5 μg QD: 55; LIXI 10 μg QD: 52; LIXI 20 μg QD: 55; LIXI 30 μg QD: 54; LIXI 5 μg BID: 53; LIXI 10 μg BID: 56; LIXI 20 μg BID: 54; LIXI 30 μg BID: 54; Placebo: 55) |
|
| Interventions |
COMPARISON: Lixisenatide (LIXI) 5 μg, 10 μg, 20 μg and 30 μg QD or BID VERSUS Placebo NO. OF COMPARISON GROUPS: 9 DOSE LIXI: Subcutaneous injections of LIXI doses of 5, 10, 20 or 30 μg administered once daily or, twice daily within 1 hour before breakfast DOSE PLACEBO: One of four volume‐matched placebo treatments administered twice daily. DOSE MET: Stable doses of MET OTHER TREATMENT: All patients received diet and lifestyle counselling according to the American Diabetes Association guidelines. |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c from baseline to end of study. SECONDARY OUTCOMES: Percentage of patients achieving an HbA1c ≤ 7.0 or ≤ 6.5%, changes in body weight, FPG, and 2 h post‐prandial plasma glucose after a standardized breakfast. Measurement of anti‐lixisenatide antibody levels; safety and tolerability. OTHER OUTCOMES: Assessed by physical examination, adverse event reporting, blood pressure, heart rate, 12‐lead ECG and standard laboratory measurements. Symptomatic hypoglycaemia was defined as symptoms consistent with hypoglycaemia, with an accompanying blood glucose < 3.3 mmol/L or prompt recovery with carbohydrate. |
|
| Notes |
AIM: To evaluate thoroughly the dose–response effect of lixisenatide using once‐ or twice‐daily regimens (5 to 30 µg once or twice daily) on HbA1c changes over 13 weeks in metformin‐treated patients with Type 2 diabetes. SOURCE OF FUNDING: sanofi‐aventis, the manufacturer of lixisenatide. OTHER: Conflict of interest: First author has received research support from different pharmaceutical companies and has acted as a consultant for some of the companies. Another author has served on advisory boards and received honorarium or consulting fees from different pharmaceutical companies. He has also received research grants from Merck, Pfizer, sanofi‐aventis, Novo Nordisk, Bristol‐Myers Squibb, Eli Lilly, GlaxoSmithKline, Forest, Takeda, Novartis, AstraZeneca, Amylin, Johnson & Johnson, Daiichi Sankyo, Boehringer Ingelheim and MannKind. One author is an employee of sanofi‐aventis. SAMPLE SIZE: Sample sizes of 50 patients in each active treatment group and 100 patients in the placebo group were calculated to provide a statistical power of 81% to detect a 0.6% (6.6 mmol/mol) difference in HbA1c between an active treatment and placebo assuming a standard deviation of 1.2% (13.1 mmol/L). Statistical significance was assumed at the 5% level. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised to one of 12 treatment arms (2:2:2:2:2:2:2:2:1:1:1:1) using interactive voice response system. |
| Allocation concealment (selection bias) | Low risk | Interactive voice response system. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The study drug, added‐on to stable metformin, was double‐blind regarding active treatment or placebo and open‐label regarding the treatment volume. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis; adequate description of withdrawals and losses to follow up. |
| Selective reporting (reporting bias) | Low risk | All the prespecified and predefined outcomes were reported. |
| Methods |
TRIAL DESIGN: Randomised, placebo‐controlled, double‐blind study DURATION OF INTERVENTION: 16 weeks DURATION OF FOLLOW‐UP: SETTING: multiple (36 sites in United States and 3 sites in Puerto Rico) COUNTRY: United States; Puerto Rico |
|
| Participants |
INCLUSION CRITERIA: Participants at least 18 years of age with type 2 diabetes; BMI between 27 and 40 kg/m2; HbA1c > 7.0% but ≤ 10.5%; stable weight for at least 3 months at entry; receiving stable therapy for at least 3 months with an oral antihyperglycaemic medications from each of the two different classes (sulphonylurea, biguanide, thiazolidinedione or DPP‐IV inhibitors). EXCLUSION CRITERIA: previous use of GLP‐1 or a GLP‐1 analogue, or current use of insulin or weight‐loss medication; history of clinically significant gastric emptying abnormality, cardiovascular disorders or uncontrolled diabetes requiring hospitalisation more than once in the previous 6 months. AGE: 54 SD 11 to 59 SD 12 years SEX: 46% to 56% of female DIABETES DURATION: 7.5 SD 5.4 to 9.0 SD 7.6 years ETHNICITY: 55% to 61% Caucasians; 29% to 39% Hispanic; HbA1c (%): LY 0.5/1.0 QW: 8.25 SD 0.9; LY 1.0/1.0 QW: 8.25 SD 1.0; LY 1.0/2.0 QW: 8.43 SD 1.0; Placebo: 8.05 SD 0.8 BMI (kg/m2): LY 0.5/1.0 QW: 33.7 SD 4.1; LY 1.0/1.0 QW: 33.9 SD 4.0; LY 1.0/2.0 QW: 34.2 SD 4.1; Placebo: 33.9 SD 4.3 PREVIOUS THERAPY: 72.7% to 73.8% MET + SU; 12.3% to 13.6% MET + TZD; 7.6% to 9.1% MET + DPP‐IV: 4.5% to 6.2% other; 9.1% to 13.8% discontinued NUMBERS: LY 0.5/1.0 QW: 66; LY 1.0/1.0 QW: 65; LY 1.0/2.0 QW: 65; Placebo: 66 |
|
| Interventions |
COMPARISON: LY 0.5/1.0 QW or LY 1.0/1.0 QW or LY 1.0/2.0 QW VERSUS Placebo NO. OF COMPARISON GROUPS: 4 LEAD‐ IN PERIOD: All participants under went two weeks lead‐in period of placebo injection DOSE LY 0.5/1.0: Once weekly subcutaneous injection of LY 0.5 mg for 4 weeks followed by 1.0 mg for 12 weeks. DOSE LY 1.0/1.0: Once weekly subcutaneous injection of LY 1.0 for 16 weeks DOSE LY 1.0/2.0: Once weekly subcutaneous injection of LY 1.0 for 4 weeks then 2.0 mg for 12 weeks. DOSE Placebo: Once weekly subcutaneous injection of placebo for 16 weeks OTHER TREATMENT: Participants continued their baseline oral antihyperglycaemic medications regimen. |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c SECONDARY OUTCOMES: Changes in FPG, blood glucose responses following a solid mixed‐meal test, body weight, HOMA‐2 algorithm to assess β ‐cell function (%‐B), insulin sensitivity (%‐S) and resistance (%‐IR) OTHER OUTCOMES: Safety and tolerability that includes treatment emergent adverse events, hypoglycaemia, vital signs and laboratory tests. |
|
| Notes |
AIM: To evaluate the efficacy and tolerability of once‐weekly LY2189265 (LY), a GLP‐1 IgG4‐Fc fusion protein, in participants with type 2 diabetes failing oral antihyperglycaemic medications. SOURCE OF FUNDING: Eli Lilly and Company OTHER: First author has received research support from different pharmaceutical companies and has acted as a consultant for some of the companies. Another author has served on advisory boards and received honorarium or consulting fees from different pharmaceutical companies. He has also received research grants from some of these companies. Two authors are employees and shareholders of Eli Lilly and Company. One author was a shareholder, and during the conduct of the study and preparation of the paper, an employee, of Eli Lilly and Company. SAMPLE SIZE: Sixty patients per arm were estimated to provide 90% power at a 2‐sided α of 0.05 to detect a 0.9% change from baseline in HbA1c relative to placebo, assuming a standard deviation of 1.3 and 20% dropout rate. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was carried out using a computer‐generated random sequence and was stratified according to oral antihyperglycaemic medication. |
| Allocation concealment (selection bias) | Unclear risk | Inadequate information |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All efficacy and safety analyses were performed using the intent‐to‐treat population Adequate description of withdrawals and losses to follow up |
| Selective reporting (reporting bias) | Low risk | All the prespecified and predefined outcomes were reported. |
| Methods |
TRIAL DESIGN: RCT, parallel group, placebo controlled, multi‐centre, phase 2b trial DURATION OF INTERVENTION: 8 weeks DURATION OF FOLLOW‐UP: 4 weeks post‐intervention RUN‐IN PERIOD: Up to 3 week screening period COUNTRY: Germany and Switzerland |
|
| Participants |
INCLUSION CRITERIA: Participants with type 2 diabetes, on metformin monotherapy (≥ 1500 mg/day) for at least 3 months before screening, age 18 to 75 years, HbA1c between 7.0% and 9.5%, BMI > 25 and ≤ 45 kg/m2 and stable weight (± 10%) for at least 3 months before screening EXCLUSION CRITERIA: Type 1 diabetes; patient on any OAD other than metformin during the prior three months (except insulin); previous exposure to any GLP‐1 analogues; impaired liver or kidney function, GI disease, uncontrolled hypertension or stroke or myocardial infarction AGE: mean 53 to 57 years (SD 6 to 11) SEX: 39% to 64% female DIABETES DURATION: mean 5 to 6 years (SD 4 to 5) ETHNICITY: Not reported HbA1c (%): Placebo: 8.0 SE 0.1, TAS 10 mg QW: 7.9 SE 0.1, TAS 20 mg QW: 7.8 SE 0.1, TAS 20 mg Q2W: 7.9 SE 0.1 BMI (kg/m2): Placebo: 31.8 SE 4.9, TAS 10 mg QW: 32.6 SE 4.7, TAS 20 mg QW: 32.4 SE 5.2, TAS 20 mg Q2W: 33.2 SE 5.1 PREVIOUS THERAPY: Metformin monotherapy (mean 1888 mg to 2019 mg) NUMBERS: 306 randomised (safety database 297); Placebo: 49, TAS 5 mg QW: 50, TAS 10 mg QW: 49, TAS 20 mg QW: 50, TAS 10 mg Q2W: 50, TAS 20 mg Q2W: 49 |
|
| Interventions |
COMPARISON: Taspoglutide (TAS) (5 dose schedules) + Metformin (MET) VERSUS Placebo + MET NO. OF COMPARISON GROUPS: 6 For the current review, the following groups were excluded: 5 mg weekly and 10 mg every 2 weeks as the effect of those groups were less favourable than those of the other groups. RUN‐IN: Screening only DOSE TAS: Taspoglutide 10 mg or 20 mg once weekly, or 20 mg once every 2 weeks DOSE MET: Pre‐study MET regimen OTHER TREATMENT: Patients continued their prestudy diet and exercise plan throughout the study; patients were on a variety of other medication, such as ACE inhibitors, thiazide diuretics, angiotensin receptor blockers, statins, beta‐blockers |
|
| Outcomes |
PRIMARY OUTCOMES: Change in HbA1c from baseline to end of the study (assessed 1 week after 8 consecutive weeks of treatment) SECONDARY OUTCOMES: % of patients achieving HbA1c ≤ 7% and ≤ 6.5%; changes in following parameters from baseline: FPG, body weight, fructosamine, C‐peptide, fasting insulin, pro‐insulin, pro‐insulin‐to‐insulin molar ratio, fasting glucagon, lipids OTHER OUTCOMES: Safety variables including adverse events, vital signs, physical examination, clinical laboratory tests, electrocardiogram, local tolerance at the injection site, anti‐taspoglutide antibodies |
|
| Notes |
AIM: To assess the efficacy and safety of taspoglutide in patient with type 2 diabetes inadequately controlled with metformin therapy. SOURCE OF FUNDING: Hoffmann‐La Roche, Switzerland. OTHER: Association of some of the authors with Roche SAMPLE SIZE: Sample size provided 90% power to detect a 1% difference in HbA1c. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were randomly assigned by central randomisation system |
| Allocation concealment (selection bias) | Low risk | Patients were randomly assigned by interactive voice response system |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data were imputed as the last observation carried forward, ITT analysis (ITT population comprised of all patients who were randomly assigned, received at least one dose of study medication, and had a baseline and at least post‐baseline HbA1C assessment), adequate description of withdrawals and losses to follow‐up (6.5% withdrawals / losses to follow‐up) |
| Selective reporting (reporting bias) | Low risk | Included all expected outcomes, including those prespecified |
| Methods |
TRIAL DESIGN: Randomized, double‐blind, placebo‐controlled phase II trial DURATION OF INTERVENTION: 8 weeks DURATION OF FOLLOW‐UP: 4 weeks follow up RUN‐IN PERIOD: NR SETTING: 27 sites COUNTRY: Australia, France, Germany, Mexico, Peru and USA |
|
| Participants |
INCLUSION CRITERIA: Men and post‐menopausal or surgically sterilised women aged 18 to 75 years with type 2 diabetes mellitus, treated with a stable daily dose of metformin monotherapy for at least 3 months before screening; dose of metformin not adjusted during the study; HbA1c between 7.0% and 9.5%; FPG > 7 mmol/L and ≤ 13.3 mmol/L; BMI > 25 kg/m2 and ≤ 45.0 kg/m2; weight ≤ ± 10% for at least 3 months before screening. EXCLUSION CRITERIA: Subjects with serious co‐morbidities or abnormalities in laboratory tests; those who had previously been treated with GLP‐1 receptor agonists (including GLP‐1 itself) at any time, or with other glucose‐lowering medications (apart from metformin) or weight‐loss medications within 12 to 6 weeks respectively. AGE: 55 SE 2.0 to 60 SE 2.0 years SEX: 53% to 59% female (TAS 20/20 mg QW: 53%; TAS 20/30 mg QW; 55%; TAS 20/40 mg QW: 59%; Placebo: 59%) DIABETES DURATION: 6 SE 1.0 to 8 SE 1.0 years ETHNICITY: NR HbA1c (%): TAS 20/20 mg QW: 8.0 SE0.1; TAS 20/30 mg QW: 8.0 SE0.1; TAS 20/40 mg QW: 7.8 SE0.1; Placebo: 7.8 SE0.1 BMI (kg/m2): TAS 20/20 mg QW: 33.3 SE0.9; TAS 20/30 mg QW: 31.6 SE1.0; TAS 20/40 mg QW: 31.5 SE0.9; Plaebo: 33.2 SE1.0 PREVIOUS THERAPY: MET NUMBERS: Randomised: 133 (TAS 20 mg QW: 32, TAS 20/30 mg QW: 33; TAS 20/40 QW: 32; Placebo: 32); safety population: 129; ITT population: 125 |
|
| Interventions |
COMPARISON: TAS + MET VERSUS Placebo + MET NO. OF COMPARISON GROUPS: 4 DOSE TAS: 20 mg taspoglutide once weekly s.c. for 4 weeks followed by 4 weeks of 20 mg once weekly (20/20) or titration up to 30 mg once weekly (20/30) or 40 mg once weekly (20/40) taspoglutide; DOSE PLACEBO: placebo s.c. once weekly DOSE MET: pre‐study metformin regimen throughout the study DIET and EXERCISE: pre‐study diet and exercise plan throughout the study OTHER TREATMENT: Some patients received medications for cardiovascular risk factors: statins (22%); ACE‐inhibitors (21%), fibrates (5%). ACE, thiazide diuretics, thyroid hormones and/or lipid‐lowering medications were permitted but only with doses stable for at least 6 weeks prior to screening. |
|
| Outcomes |
PRIMARY OUTCOMES: GI tolerability, assessed by comparing the number of subjects who withdrew from study because of GI adverse events. SECONDARY OUTCOMES: FPG, HbA1c, body weight and pharmacokinetic parameters. OTHER OUTCOMES: NR |
|
| Notes |
AIM: To investigate the safety and tolerability of up titration to high doses of taspoglutide in patients with type 2 diabetes mellitus. SOURCE OF FUNDING: Hoffmann‐La Roche OTHER: Conflict of interest: Two authors have received consulting and advisory board honoraria from F. Hoffmann‐La Roche as well as from other pharmaceutical companies developing incretin‐based therapies whose products may be perceived as competitive to taspoglutide. One author has received consulting and research fees from F. Hoffman‐La Roche and Ispen. Some authors are employees of F. Hoffman‐La Roche. SAMPLE SIZE: The paper says 'it was planned to enrol approximately 120 subjects into the study assigned randomly and equally to each of the treatment groups. This sample size was determined by practical considerations rather than formal calculations'. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central system using a stratified randomisation procedure based on disease severity (HbA1c < 8.0% or ≥ 8.0%) to avoid imbalances between treatment groups. |
| Allocation concealment (selection bias) | Unclear risk | Central system; insufficient information |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Descriptive statistics were used to report the safety results; data on secondary endpoints were analysed for the ITT population using the last observation carried forward; description of withdrawals and loss to follow up given. |
| Selective reporting (reporting bias) | Low risk | All prespecified and predefined outcomes reported. |
BMI: Body mass index, HbA1c: Glycosylated haemoglobin, ALBI: Albiglutide, MET: Metformin, EX: Exenatide, TZD: Thiazolidinedione, SU: Sulphonylurea, DPP‐4: Dipeptidyl peptidase‐4 inhibitor, SITA: Sitagliptin, PIO: Pioglitazone, GLAR: Insulin glargine, GLP: Glucagon like peptide, LIR: Liraglutide, ROS: Rosiglitazone, GLIM: Glimepiride, LIXI: Lixisenatide, LY: LY2189265, TAS: Taspoglutide, OCP: Oral contraceptive pill, LAR: Long acting release, ACE: Angiotensin‐converting enzyme, FPG: Fasting plasma glucose, PPG: Post‐prandial glucose, PG: Plasma glucose, HOMA‐B: Homeostasis model assessment‐beta cell function, ITT: Intention‐to‐treat, LOCF: Last observation carried forward, AE: Adverse event, ECG: Electrocardiography, GI: Gastrointestinal, SE: Standard error, SD: standard deviation, QD: Once daily, BID: Twice daily, QW: Once weekly, NR: Not reported, IWQOL: Impact of weight on quality of life questionnaire‐lite, PGWB: Psychological general well‐being index, DTSQ: Diabetes treatment satisfaction questionnaire, and EQ‐5D: EuroQol‐5 dimensions.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Astrup 2009 | Participants without type 2 diabetes mellitus |
| Barnett 2007 | Patients failing on either metformin therapy or sulphonylurea treatment were included, but less than 70% of patients were on metformin therapy (only 55%). |
| Blonde 2006 | Combined analysis of three different trials, so patients included for analysis not actually randomised. |
| Bode 2010 | Monotherpay compared to glimepiride only. |
| Brixner 2009 | All patient received exenatide. |
| Buse 2004 | Include patients failing on sulphonylurea only. |
| Feingloss 2005 | Liraglutide doses used in the study are clinically not relevant. |
| Gallwitz 2010 | Combined analysis of 3 LEAD trials (LEAD 1, 2 and 4). |
| Garber 2009 (LEAD 3) | Previous treatment with OADs discontinued at randomisation and compared to monotherapy only. |
| Kapitza 2009 | Single‐dose administration of taspoglutide with follow up duration of less than 8 weeks. |
| Kim 2007 | Patients on diet/exercise or metformin were included, but less than 70% were on metformin therapy (60%). |
| Madsbad 2004 | Liraglutide doses used in the study are clinically not relevant. |
| Malloy 2009 | Participants aged less than 18 years of age. |
| Moretto 2008 | Monotherapy compared to placebo only. |
| Nauck 2009 | Combined analysis of subset of participants from two different trials, so patients included for analysis not actually randomised. |
| Okerson 2010 | Combined analysis of six different trials. |
| Riddle 2006 | Combined analysis of two different trials, so patients included for analysis not actually randomised. |
| Seino 2008 | Liraglutide doses used in the study are clinically not relevant. |
| Trescoli‐Serrano 2005 | Published as abstract only. |
| Vilsboll 2007 | Monotherapy and compared to placebo only. |
LEAD: Liraglutide Effect and Action in Diabetes, OAD: Oral antidiabetic drug
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Efficacy and safety of albiglutide in treatment of type 2 diabetes |
| Methods |
TRIAL DESIGN: Phase III, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study DURATION OF INTERVENTION: two years COUNTRY: USA, Germany, HongKong, Mexico, Peru, Phillipines, Russian Federation, South Africa, Spain, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Albiglutide: Alibiglutide + metformin + placebo sitagliptin + placebo glimepiride Sitagliptin: sitagliptin + metformin + placebo albiglutide + placebo glimepiride Glimepiride: glimepiride + metformin + placebo albiglutide + placebo sitagliptin Metformin: metformin + placebo albiglutide + placebo sitagliptin + placebo glimepiride |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c SECONDARY OUTCOMES: change in FPG and body weight |
| Starting date | February 2009, estimated completion date December 2012 (last updated on June 2, 2011) |
| Contact information | GlaxoSmithKline |
| Notes | ClinicalTrials.gov Identifier: NCT00838903; other study ID number: 112753 |
| Trial name or title | A study to determine the safety and efficacy of albiglutide in patients with type 2 diabetes |
| Methods |
TRIAL DESIGN: Phase III, randomised, open‐label, parallel‐group study DURATION OF INTERVENTION: one year COUNTRY: USA, Russian Federation, South Africa, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Albiglutide: albiglutide weekly injection Insulin glargine: insulin glargine |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c SECONDARY OUTCOMES: change in FPG and body weight |
| Starting date | February 2009, estimated completion date December 2012 (last updated on June 9, 2011) |
| Contact information | GlaxoSmithKline |
| Notes | ClinicalTrials.gov identifier: NCT00838916; other study ID number: 112754 |
| Trial name or title | Safety and efficacy study of albiglutide in type 2 diabetes |
| Methods |
TRIAL DESIGN: Phase III, randomised, double‐blind, placebo‐controlled, parallel‐group study DURATION OF INTERVENTION: one year COUNTRY: USA, Mexico |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Albiglutide: albiglutide weekly injection Placebo: matching albiglutide placebo weekly injection Albiglutide uptitration: albiglutide uptitration at week 12 |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c SECONDARY OUTCOMES: change in FPG and body weight |
| Starting date | January 2009, estimated completion date October 2012 (last updated on June 9, 2011) |
| Contact information | GlaxoSmithKline |
| Notes | ClinicalTrials.gov identifier: NCT00849017; other study ID number: 112756 |
| Trial name or title | Safety and efficacy of albiglutide in type 2 diabetes |
| Methods |
TRIAL DESIGN: Phase III, randomised, double‐blind, placebo‐controlled, parallel‐group trial DURATION OF INTERVENTION: one year COUNTRY: USA, India, Peru, South Africa, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Albiglutide: albiglutide weekly injection + pioglitazone (with or without metformin) Placebo: placebo albiglutide weekly injection + pioglitazone (with or without metformin) |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c SECONDARY OUTCOMES: change in FPG and body weight |
| Starting date | January 2009, estimated completion date October 2012 (last updated on June 9, 2011) |
| Contact information | GlaxoSmithKline |
| Notes | ClinicalTrials.gov identifier: NCT00849056; other study ID number: 112755 |
| Trial name or title | Dose ranging study of albiglutide in Japanese subjects |
| Methods |
TRIAL DESIGN: Phase II, double‐blind, randomised, placebo‐controlled, multicenter, 4‐parallel‐group, dose ranging study DURATION OF INTERVENTION: 16 weeks COUNTRY: Japan |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Albiglutide: subcutaneous injection albiglutide 15 mg or 30 mg weekly or 30 mg every two weeks Placebo: subcutaneous injection of placebo to match albiglutide |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c SECONDARY OUTCOMES: change in HbA1c over time, change in FPG and body weight; Proportion of subjects who achieve HbA1c treatment goal; pharmacokinetic levels of albiglutide |
| Starting date | April 2010, estimated completion date March 2011 (last updated on November 18, 2010) |
| Contact information | GlaxoSmithKline |
| Notes | ClinicalTrials.gov Identifier: NCT01098461; other study ID number: 110932 |
| Trial name or title | A study to determine the efficacy and safety of albiglutide as compared with liraglutide. |
| Methods |
TRIAL DESIGN: Phase III, randomised, open‐label, parallel‐group, multicenter study DURATION OF INTERVENTION: 32 weeks COUNTRY: USA, Australia, Israel, Korea, Peru, Phillipines, Spain, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Albiglutide: once weekly injection Liraglutide: liraglutide daily subcutaneous injection, starting at 0.6 mg, then up‐titrating to 1.2 mg then 1.8 mg in accordance with prescribing information. |
| Outcomes |
PRIMARY OUTCOME: Evaluation of change from baseline of HbA1c levels of albiglutide as compared with liraglutide. SECONDARY OUTCOMES: HbA1c change from baseline over time; proportion of subjects at an HbA1c treatment goal of < 7.0% and/or < 6.5%; FPG, body weight, time for hyperglycaemia rescue |
| Starting date | May 2010, estimated completion date September 2011 (last updated on June 9, 2011) |
| Contact information | GlaxoSmithKline |
| Notes | ClinicalTrials.gov identifier: NCT01128894 |
| Trial name or title | Efficacy of exenatide once weekly and once‐daily insulin glargine in patients with type 2 diabetes treated with metformin alone or in combination with sulfonylurea (DURATION ‐ 3) |
| Methods |
TRIAL DESIGN: Phase III, open label, randomised, parallel assignment study (extension study) DURATION OF INTERVENTION: 26 weeks COUNTRY: USA, Australia, Belgium, Czech Republic, Denmark, France, Germany, Greece, Hungary, Korea, Mexico, Netherlands, Puerto Rico, Russian Federation, Spain, Taiwan, |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Exenatide: exenatide 2 mg once weekly subcutaneous injection Insulin glargine: variable dose once daily subcutaneous injection, |
| Outcomes |
PRIMARY OUTCOME: Estimate the difference in change in HbA1c from baseline to treatment endpoint between 2.0 mg exenatide once weekly and insulin glargine QD in patients with type 2 diabetes and inadequate glycaemic control using Met alone or in combination with SU. SECONDARY OUTCOMES:
|
| Starting date | April 2008, estimated completion date January 2012 (last updated on December 3, 2010) |
| Contact information | Eli Lilly and Company |
| Notes | ClinicalTrials.gov identifier: NCT00641056; other study ID number: H8O‐MC‐GWBR (DURATION ‐ 3) |
| Trial name or title | A study to examine the effects of exenatide once‐weekly injection on glucose control and safety in Asian subjects |
| Methods |
TRIAL DESIGN: Phase III, randomised, open‐label trial DURATION OF INTERVENTION: 26 weeks COUNTRY: China, India, Japan, Korea, Taiwan |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Exenatide QW: 2 mg once weekly subcutaneous injection Exenatide BID: 5 μg subcutaneous injection twice a day in the first four weeks then, 10 μg subcutaneous injection twice a day (22 weeks) |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c SECONDARY OUTCOMES: proportion of patients achieving HbA1c ≤ 7% and ≤ 6.5%; fasting serum glucose; body weight; parameters related to glycaemic control, including fasting & postprandial plasma glucose & 6‐point SMBG profiles; serum lipids; incidence and rate of hypoglycaemic events; safety, tolerability, and treatment‐emergent events; beta‐cell function (HOMA‐B) and insulin sensitivity (HOMA‐S) |
| Starting date | July 2009, estimated completion date June 2011 (last updated on December 16, 2010) |
| Contact information | Eli Lilly and Company |
| Notes | ClinicalTrials.gov Identifier: NCT00917267; other study ID number: H8O‐MC‐GWCK |
| Trial name or title | Study to evaluate the efficacy and safety of exenatide once‐weekly injection compared to once‐daily insulin in type 2 diabetes mellitus |
| Methods |
TRIAL DESIGN: Phase III, randomised, open‐label study DURATION OF INTERVENTION: 26 weeks COUNTRY: Japan |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Exenatide: 2 mg once weekly subcutaneous injection Insulin glargine: subcutaneous injection, titrated to achieve fasting serum glucose target, once a day |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c SECONDARY OUTCOMES: proportion of subjects achieving HBA1c ≤ 7% or ≤ 6.5%; fasting serum glucose; body weight; 1,5‐anhydroglucitol; self‐monitored blood glucose profile at 7 time points; serum lipids; hypoglycaemia; vital signs; waist & hip circumference; waist‐hip ratio; safety & tolerability; patient‐reported health outcomes |
| Starting date | July 2009, estimated completion date June 2011 (last updated on February 28, 2011) |
| Contact information | Eli Lilly and Company |
| Notes | ClinicalTrials.gov Identifier: NCT00935532; other study ID number: H8O‐JE‐GWBX |
| Trial name or title | Efficacy of once‐weekly exenatide versus once or twice daily insulin detemir in patients with type 2 diabetes |
| Methods |
TRIAL DESIGN: Phase III, randomised, open‐label trial DURATION OF INTERVENTION: 26 weeks COUNTRY: Ireland, UK, |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Exenatide: 2 mg once weekly subcutaneous injection Insulin detemir: subcutaneous injection, with dosage titrated according to the detemir label and published titration schedule, once or twice a day |
| Outcomes |
PRIMARY OUTCOME: To test the hypothesis that exenatide given once weekly is superior to a titration of insulin detemir given once or twice daily assessed by the proportion of patients who have achieved HbA1c concentration ≤ 7.0% with weight loss (≥ 1.0 kg) at endpoint. SECONDARY OUTCOMES: Proportion of patients who have achieved HbA1c ≤ 7.0% with weight loss (≥ 1.0 kg) at 12 weeks; proportion of patients who have achieved HbA1c ≤ 7.4% with weight loss (≥ 1.0 kg) at endpoint; proportion of patients who have achieved HbA1c ≤ 7.0% and ≤ 7.4%, with minimal weight gain (≤ 1 kg) at endpoint; change in HbA1c and body weight; proportion of patients achieving HbA1c ≤ 7.4%, ≤ 7.0% and ≤ 6.5% at endpoint; change in fasting serum glucose; 7‐point self‐monitored blood glucose (SMBG) profile; changes in CV risk parameters; incidence and rate of hypoglycaemic events; safety and tolerability |
| Starting date | October 2009, estimated completion date September 2011 (last updated on January 13, 2011) |
| Contact information | Eli Lilly and Company |
| Notes | ClinicalTrials.gov Identifier: NCT01003184; other study ID number: H8O‐EW‐GWDL |
| Trial name or title | Safety and efficacy of exenatide once weekly versus liraglutide in subjects with type 2 diabetes |
| Methods |
TRIAL DESIGN: Phase III, randomised, parallel assignment, open‐label trial DURATION OF INTERVENTION: 26 weeks COUNTRY: Argentina, Austria, Australia, Belgium, Canada, Czech Republic, France, Germany, Greece, Hungary, India, Israel, Italy, Korea, Mexico, Poland, Romania, Slovakia, South Africa, Spain, Taiwan |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Exenatide: 2 mg once weekly subcutaneous injection Liraglutide: subcutaneous injection, forced titration to 1.8 mg, once daily |
| Outcomes |
PRIMARY OUTCOME: change in HbA1c from baseline to treatment endpoint SECONDARY OUTCOMES: proportion of subjects achieving HbA1c < 7%; FPG, body weight, lipid profile, safety and tolerability, hypoglycaemia and blood pressure |
| Starting date | January 2010, estimation completion date January 2011 (last updated on January 18, 2011) |
| Contact information | Amylin Pharmaceuticals, Inc. and Eli Lilly and Company |
| Notes | ClinicalTrials.gov Identifier: NCT01029886 ; other study ID number: H8O‐MC‐GWDE |
| Trial name or title | Exenatide study of cardiovascular event lowering trial (EXSCEL): a trial to evaluate cardiovascular outcomes after treatment with exenatide once weekly in patients with type 2 diabetes mellitus |
| Methods |
TRIAL DESIGN:Phase III, randomised, double‐blind, placebo‐controlled trial DURATION OF INTERVENTION: 5.5 years COUNTRY: USA, Argentina, Australia, Austria, Brazil, Bulgaria, Chile, Colombia, Czech Republic, France, Germany, HongKong, Hungary, India, Israel, Italy, Korea, Latvia, Lithuania, Malaysia, Mexico, New Zealand, Peru, Phillipines, Poland, Romania, Russian Federation, Slovakia, Taiwan, Ukraine, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Exenatide: 2 mg once weekly subcutaneous injection Placebo: matching volume of placebo, once weekly subcutaneous injection |
| Outcomes |
PRIMARY OUTCOME: Time to first confirmed cardiovascular event in the primary composite cardiovascular endpoint SECONDARY OUTCOMES: Time to all‐cause mortality; time to first confirmed cardiovascular event for each component of the primary composite endpoint; time to hospitalisation for acute coronary syndrome; time to hospitalisation for heart failure |
| Starting date | June 2010; estimation completion date March 2017 (last updated on June 3, 2011) |
| Contact information | Amylin Pharmaceuticals, Inc. |
| Notes | ClinicalTrials.gov Identifier: NCT01144338; other study ID number: BCB109 |
| Trial name or title | The effect of insulin detemir in combination with liraglutide and metformin compared to liraglutide and metformin in participants with type 2 diabetes. A 26 week, randomised, open‐label, parallel‐group, multicenter, multinational trial with a 26 week extension |
| Methods |
TRIAL DESIGN: Randomised, open label, active control, parallel group, phase III trial, multicenter and multinational DURATION OF INTERVENTION: 26 weeks with a 26 week extension COUNTRY: Europe and North America |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Insulin detemir: Liraglutide 1.8 mg/day and insulin detemir (dose titrated based on fasting plasma glucose) both as injection subcutaneously and metformin at least 1.5 g every day. Liraglutide: Liraglutide 1.8 mg/day subcutaneous injection and metformin at least 1.5 g every day (randomised treatment arm without intensification with insulin detemir despite HbA1c equal to or greater than 7.0%) Liraglutide: Liraglutide 1.8 mg/day subcutaneous injection and metformin at least 1.5 g every day (non‐randomised trial arm with participants continuing liraglutide and metformin treatment without intensification with insulin detemir. Participants with HbA1c less than 7.0% after 12 weeks of run‐in will continue in the trial in this arm) |
| Outcomes |
PRIMARY OUTCOME:Change in HbA1c from baseline at 26 weeks. SECONDARY OUTCOMES:All these outcomes measured at weeks 26 and 52. Change in fasting plasma glucose concentrations, 7 point plasma glucose profile, fasting insulin, fasting proinsulin, fasting C‐peptide, lipids, body weight, waist and hip circumference, adverse events and hypoglycaemic events. |
| Starting date | March 2010, completed (last updated April 2011) |
| Contact information | Novo Nordisk |
| Notes | Study ID number: NN2211‐1842, EudraCT No: 2007‐005317‐19, ClinicalTrials.gov Identifier: NCT00856986 |
| Trial name or title | Efficacy assessment of insulin glargine versus liraglutide after oral agent failure (EAGLE) |
| Methods |
TRIAL DESIGN: Multicenter, international, randomised (1:1), parallel‐group, open‐label, comparative, phase IV study DURATION OF INTERVENTION: 24 weeks comparative period; 24 weeks extension period COUNTRY:USA, Austria, Brazil, Canada, Czech Republic, Finland, France, Ireland, Mexico, Netherlands, Slovakia, Spain, Sweden |
| Participants |
INCLUSION CRITERIA (comparative period):
INCLUSION CRITERIA (extension period):Patients treated with liraglutide (at the maximal tolerated dosage), having a mean FPG ≥ 250 mg/dL at visit 10 (Week 12) or visit 11 (Week 18), or a HbA1c ≥ 7% at visit 12 (Week 24). Dosage of metformin compliant with the inclusion criteria of visit 1 (i.e. maximum tolerated dosage, with a minimum daily dosage of 1g), and maintained stable during the comparative period. |
| Interventions |
Liralutide: 1.8 mg once a day Insulin glargine: 100 U/mL once a day |
| Outcomes |
PRIMARY OUTCOME: percentage of patients reaching HbA1c < 7 % SECONDARY OUTCOMES: Percentage of patients whose HbA1c has decreased but remains ≥ 7%; percentage of patients whose HbA1c has increased; HbA1c change; PPG; FPG, vital signs; hypoglycaemia; dose of insulin glargine or liraglutide |
| Starting date | July 2010, estimated completion date June 2012 (Last Updated on June 14, 2011) |
| Contact information | Contact‐us@sanofi‐aventis.com |
| Notes | ClinicalTrials.gov Identifier: NCT01117350; other study ID numbers: LANTU_C_03680, 2010‐018437‐21, U1111‐1116‐9684 |
| Trial name or title | Comparison of two treatment regimens (sitagliptin versus liraglutide) on participants who failed to achieve good glucose control on metformin alone (MK‐0431‐403) |
| Methods |
TRIAL DESIGN: Phase III, multicenter, randomised, open‐label clinical trial DURATION OF INTERVENTION: 26 weeks COUNTRY: USA, Canada, France, Hungary, Italy, Lithuania, Puerto Rico, Slovenia, Sweden, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Liraglutide: 0.6 mg by subcutaneous (pen) injection, once daily, on days 1 to 7; in subsequent weeks, the dose may be up‐titrated to 1.8 mg once daily. Sitagliptin: 100 mg tablet, orally, once daily. Glimepiride: starting dose of 1 mg tablet (up‐titrated as needed), once daily, as needed, after week 12 therapy. Metformin: metformin tablets at a dose of ≥ 1500 mg per day |
| Outcomes |
PRIMARY OUTCOME: Glycaemic control SECONDARY OUTCOMES: Percentage of patients reaching haemoglobin A1C goals (< 7.0% and < 6.5%); FPG |
| Starting date | March 2011, estimated completion date May 2012 (last updated on May 3, 2011) |
| Contact information | Toll Free Number 1‐888‐577‐8839 (Merck) |
| Notes | ClinicalTrials.gov identifier: NCT01296412 |
| Trial name or title | Dual action of liraglutide and insulin degludec in type 2 diabetes: a trial comparing the efficacy and safety of insulin degludec/liraglutide, insulin degludec and liraglutide in subjects with type 2 diabetes (DUAL™ I) |
| Methods |
TRIAL DESIGN: Phase III, randomised, parallel three‐arm, open‐label, multi‐centre, multinational treat‐to‐target trial DURATION OF INTERVENTION: 26 weeks COUNTRY: USA, Australia, Canada, Finland, Germany, Hungary, India, Ireland, Italy, Malaysia, Mexico, Puerto Rico, Russian Federation, Singapore, Slovakia, South Africa, Spain, Taiwan, Thailand, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Insulin degludec/liraglutide: insulin degludec/liraglutide treatment will be initiated and titrated (individually adjusted) twice weekly according to the mean SMPG (fasting). Insulin degludec/liraglutide is injected subcutaneously (under the skin) once daily. Insulin degludec: insulin degludec treatment will be initiated with 10 U and titrated (individually adjusted) twice weekly according to the mean SMPG (fasting). Insulin degludec is injected subcutaneously (under the skin) once daily. Liraglutide: liraglutide will be started with 0.6 mg and subsequent 0.6 mg weekly dose escalation to 1.8 mg. Liraglutide dose of 1.8 mg/day will be continued for the remaining part of the trial. Liraglutide is injected subcutaneously (under the skin) once daily. Subjects should continue their pre‐trial treatment with metformin or metformin + pioglitazone throughout the entire trial |
| Outcomes |
PRIMARY OUTCOME: Change from baseline in HbA1c SECONDARY OUTCOMES: body weight, hypoglycaemia, meal test, daily insulin dose |
| Starting date | May 2011, estimated completion date October 2012 (last updated on May 23, 2011) |
| Contact information | Klaus Kjær Laigaard (Novo Nordisk) |
| Notes | ClinicalTrials.gov Identifier: NCT01336023; other study ID numbers: NN9068‐3697, U1111‐1119‐1174, 2010‐021560‐15 |
| Trial name or title | GLP‐1 agonist AVE0010 versus exenatide in patients with type 2 diabetes for glycaemic control and safety evaluation, on top of metformin (GETGOAL‐X) |
| Methods |
TRIAL DESIGN: Phase III, randomised, open‐label, active‐controlled, 2‐arm parallel‐group, multicenter study DURATION OF INTERVENTION: 24 weeks COUNTRY: |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Lixisenatide (AVE0010) at least 24 weeks of treatment, extension period of variable duration Exenatide: at least 24 weeks of treatment extension period of variable duration |
| Outcomes |
PRIMARY OUTCOME: Absolute change from baseline in HbA1c SECONDARY OUTCOMES: body weight, FPG, treatment satisfaction |
| Starting date | June 2008, completed (last updated on November 25, 2010) |
| Contact information | Sanofi‐Aventis |
| Notes | ClinicalTrials.gov identifier: NCT00707031 other study ID numbers: EFC6019, EudraCT 2007‐005883‐28 |
| Trial name or title | GLP‐1 agonist AVE0010 in patients with type 2 diabetes for glycaemic control and safety evaluation, on top of sulphonylurea (GETGOAL‐S) |
| Methods |
TRIAL DESIGN: Phase III, randomised, double‐blind, placebo‐controlled, 2‐arm parallel‐group, multicenter study DURATION OF INTERVENTION: 24 weeks COUNTRY: USA, Bulgaria, Czech Republic, Egypt, Germany, India, Israel, Japan, Korea, Netherlands, Romania, Russian Federation, Taiwan, Thailand, Tunisia, Turkey |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Lixisenatide (AVE0010) at least 24 weeks of treatment, extension period of variable duration Placebo: at least 24 weeks of treatment, extension period of variable duration |
| Outcomes |
PRIMARY OUTCOME: Absolute change from baseline in HbA1c SECONDARY OUTCOMES: body weight, FPG, 2‐hours post‐prandial plasma glucose, glucagon, insulin, pro‐insulin, C‐peptide |
| Starting date | July 2008, completed (last updated on January 25, 2011) |
| Contact information | Sanofi‐Aventis |
| Notes | ClinicalTrials.gov Identifier: NCT00713830, other study ID numbers: EFC6015, EudraCT 2007‐005881‐11 |
| Trial name or title | GLP‐1 agonist AVE0010 in patients with type 2 diabetes for glycaemic control and safety evaluation, on top of pioglitazone (GETGOAL‐P) |
| Methods |
TRIAL DESIGN: Phase III, randomised, double‐blind, placebo‐controlled, 2‐arm parallel‐group, multicenter study DURATION OF INTERVENTION:24 weeks COUNTRY:USA, Austria, Canada, France, Germany, Greece, Guatemala, India, Mexico, Peru, Puerto Rico, Romania, Turkey |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Lixisenatide: AVE0010 Placebo: |
| Outcomes |
PRIMARY OUTCOME: Glycaemic control SECONDARY OUTCOMES: body weight, FPG, fasting insulin levels |
| Starting date | September 2008, estimated completion date June 2011 (last updated on June 22, 2010) |
| Contact information | Sanofi‐Aventis |
| Notes | ClinicalTrials.gov Identifier: NCT00763815 |
| Trial name or title | A randomised, placebo‐controlled, 2‐arm parallel‐group, multicenter study with a 24‐week double‐blind treatment period assessing the efficacy and safety of lixisenatide in patients with type 2 diabetes insufficiently controlled with insulin glargine and metformin |
| Methods |
TRIAL DESIGN: Randomised, placebo‐controlled parallel group phase III trial, multi‐centre DURATION OF INTERVENTION: 24 weeks COUNTRY: USA, Canada, Sweden, Estonia |
| Participants |
INCLUSION CRITERIA: At screening:
At the end of the run in phase and before randomisation:
|
| Interventions |
Lixisenatide: Lixisenatide once daily on top of insulin glargine (both injected in the morning within 1 hour prior to breakfast) and metformin (at least 1.5 g/day) Placebo: Placebo once daily on top of insulin glargine (both injected in the morning within 1 hour prior to breakfast) and metformin (at least 1.5 g/day) |
| Outcomes |
PRIMARY OUTCOME: Glycaemic control SECONDARY OUTCOMES: percentage of patients reaching HbA1c < 7% and ≤ 6.5 %, on plasma glucose (fasting, post‐prandial during a standardised meal challenge test, 7‐point self monitored profiles), body weight, insulin glargine doses; safety and tolerability, treatment satisfaction (diabetes treatment satisfaction questionnaire) |
| Starting date | October 2009, estimated completion August 2011 |
| Contact information | GV‐Contact‐us@sanofi‐aventis.com |
| Notes | Study ID Numbers: EFC10781, EudraCT: 2008‐007335‐40; ClinicalTrials.gov identifier: NCT00975286 |
| Trial name or title | A randomised, double‐blind, double‐dummy, 2‐arm parallel‐group, multicenter 24‐week study comparing the efficacy and safety of ave0010 to sitagliptin as add‐on to metformin in obese type 2 diabetic patients younger than 50 and not adequately controlled with metformin |
| Methods |
TRIAL DESIGN: Randomised double‐blind, placebo‐controlled parallel group phase III trial, multi‐centre DURATION OF INTERVENTION: 24 weeks COUNTRY: USA, Australia, Canada, Chile, Mexico, Russian Federation |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Lixisenatide: Injection of lixisenatide once a day in the morning within 1 hour prior to breakfast (first 2 weeks of double‐blind period: titration 10 to 15 µg, then 15 to 20 µg) and one capsule of sitagliptin placebo intake in the morning with or without food. On top of metformin background therapy Sitagliptin: One capsule of sitagliptin intake in the morning with or without food and lixisenatide matched placebo injection once a day in the morning within 1 hour prior to breakfast. On top of metformin background therapy. |
| Outcomes |
PRIMARY OUTCOME: Percentage of patients with HbA1c values < 7% and a weight loss of at least 5% of baseline body weight SECONDARY OUTCOMES: Absolute change in HbA1c values, percentage of patients with HbA1c values ≤ 6.5%, absolute change in body weight, change in fasting plasma glucose, change in plasma glucose and in beta‐cell function during a test meal, change in insulin resistance assessed by HOMA‐IR, change in beta‐cell function assessed by HOMA‐beta, percentage of patients requiring rescue therapy during the double‐blind treatment period; safety and tolerability |
| Starting date | August 2009, completed (last updated on May 6, 20 2011) |
| Contact information | GV‐Contact‐us@sanofi‐aventis.com |
| Notes | Study ID Numbers: EFC10780, EudraCT: 2008‐007 334‐22 |
| Trial name or title | A randomised controlled clinical trial in type 2 diabetes comparing semaglutide to placebo and liraglutide |
| Methods |
TRIAL DESIGN: Phase II, multi‐centre, multi national, double‐blind, placebo‐controlled, randomised, nine armed parallel group, dose finding trial DURATION OF INTERVENTION: 12 weeks COUNTRY: Austria, Bulgaria, Finland, Former Serbia and Montenegro, France, Germany, Hungary, India, Italy, South Africa, Spain, Switzerland, Turkey, UK |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Semaglutide: 0.1 or 0.2 or 0.4 or 0.8 mg once weekly s.c. injection; 0.8 mg or 1.6 mg with titration, once weekly s.c. injection Placebo: 0.1 or 0.2 or 0.4 mg once weekly s.c. injection; 0.8 mg or 1.6 mg with titration, once weekly s.c. injection Liraglutide: 1.2 or 1.8 mg with titration, once daily, s.c. injection |
| Outcomes |
PRIMARY OUTCOME: Change in HbA1c SECONDARY OUTCOMES: Percentage of subjects with an adverse events; percentage of subjects with hypoglycaemic episode; ECG, vital signs; safety laboratory parameters; percentage of subjects developing anti‐semaglutide antibodies and calcitonin |
| Starting date | June 2008, Final data collection date for primary outcome measure February 2009 (last updated on March 16, 2011) |
| Contact information | Novo Nordisk |
| Notes | ClinicalTrials.gov Identifier: NCT00696657; other study ID numbers: NN9535‐1821, 2007‐003956‐12 |
| Trial name or title | Randomised, active controlled, open label study to compare taspoglutide with exenatide as add‐on treatment to metformin and/or thiazolidinediones in patients with type 2 diabetes mellitus |
| Methods |
TRIAL DESIGN: Randomised, open label, active controlled, parallel assigned, phase III trial, multi centre, multinational, safety and efficacy study DURATION OF INTERVENTION: 3+ years COUNTRY: USA, Europe, America, South Africa, Asia |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Taspoglutide: 10 mg subcutaneous injection once weekly Taspoglutide: 10 mg subcutaneous injection once weekly for 4 weeks followed by 20 mg subcutaneous once weekly Exenatide: Exenatide 5 mg twice daily for 4 weeks followed by 10 mg twice daily [should presumably read "µg"?] |
| Outcomes |
PRIMARY OUTCOME: Change in HbA1c at 24 weeks SECONDARY OUTCOMES: All the outcomes measured at 24 weeks. Fasting body weight, proportion of participants reaching target HbA1c ≤ 7.0%, ≤ 6.5%, relative change in glucose, insulin, C‐peptide and glucagon values during a meal tolerance test in a subset of patients, beta cell function (proinsulin/insulin ratio) |
| Starting date | July 2008, estimated completion April 2012 (last updated on March 15, 2011) |
| Contact information | Hoffmann‐La Roche |
| Notes | Study ID numbers:BC21625, 2008‐001856‐36, ClinicalTrials.gov Identifier: NCT00717457 |
| Trial name or title | A multicenter, randomised, double‐blind, placebo‐controlled study to assess the safety, tolerability and effect of taspoglutide on glycaemic control compared to placebo in patients with type 2 diabetes mellitus inadequately controlled with metformin plus pioglitazone |
| Methods |
TRIAL DESIGN: Randomised, double‐blind, placebo‐controlled, phase III trial, multicenter, multinational, safety and efficacy study DURATION OF INTERVENTION: 1 to 2 years COUNTRY: USA, Europe and America |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Taspoglutide: 10 mg subcutaneous injection once weekly, metformin > 1.5 g/day, pioglitazone ≥ 30 mg/day Taspoglutide: 10 mg subcutaneous injection once weekly for 4 weeks followed by 20 mg subcutaneous once weekly, metformin as prescribed and placebo orally once daily, metformin > 1.5 g/day, pioglitazone ≥ 30 mg/day Placebo: Placebo subcutaneous once weekly, metformin > 1.5 g/day, pioglitazone ≥ 30 mg/day |
| Outcomes |
PRIMARY OUTCOME: Absolute change from baseline in HbA1c at 24 weeks SECONDARY OUTCOMES: Change from baseline in fasting plasma glucose, change from baseline in body weight, responder rates for HbA1c (target ≤ 7.0%, ≤ 6.5%), responder rates for body weight and beta cell function at 24 weeks; safety: adverse events, vital signs, physical examination, clinical laboratory tests, ECG and anti‐taspoglutide antibodies throughout the study |
| Starting date | October 2008, estimated completion September 2010 (last updated on March 15, 2011) |
| Contact information | Hoffmann‐La Roche |
| Notes | Study ID numbers: BC20963, 2008‐001744‐39, ClinicalTrials.gov Identifier: NCT00744367 |
| Trial name or title | A multicenter, randomised, double‐dummy, placebo and active‐controlled study to assess the safety, tolerability and effect of taspoglutide on glycaemic control compared to sitagliptin and placebo in patients with type 2 diabetes mellitus inadequately controlled with metformin |
| Methods |
TRIAL DESIGN: Randomised, double blind, placebo and active controlled, phase III trial, multicenter, multinational, safety and efficacy study DURATION OF INTERVENTION: 2+ years COUNTRY: USA, Europe, Asia, America, Australia |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Taspoglutide: 10 mg subcutaneous injection once weekly, metformin as prescribed and placebo per orally once daily Taspoglutide: 10 mg subcutaneous injection once weekly for 4 weeks followed by 20 mg subcutaneous once weekly, metformin as prescribed and placebo per orally once daily Sitagliptin: Sitagliptin 100 mg per orally once daily, placebo subcutaneous once weekly and metformin as prescribed Placebo: Placebo subcutaneous once daily, metformin as prescribed and placebo per orally once daily |
| Outcomes |
PRIMARY OUTCOME:Mean changes in HbA1c at 24 weeks SECONDARY OUTCOMES:Change from baseline in fasting plasma glucose, change from baseline in body weight, responder rates for HbA1c (target ≤ 7.0%, ≤ 6.5%), responder rates for body weight, change from baseline in lipid profile and beta cell function at 24 weeks; safety: adverse events, vital signs, physical examination, clinical laboratory tests, ECG and anti‐taspoglutide antibodies throughout study |
| Starting date | October 2008, estimated completion May 2012 (last updated on March 15, 2011) |
| Contact information | Hoffmann‐La Roche |
| Notes | Study ID numbers: BC21713, 2008‐001854‐42, ClinicalTrials.gov Identifier: NCT00754988 |
| Trial name or title | A multicenter, randomised, open‐label, active‐controlled study to compare the safety, tolerability and effect on glycaemic control of taspoglutide versus insulin glargine in insulin‐naïve type 2 diabetic patients inadequately controlled with metformin and sulphonylurea combination therapy |
| Methods |
TRIAL DESIGN: Randomised, open‐label, active controlled, parallel assignment, multicenter, multinational, phase III trial DURATION OF INTERVENTION: 2+ years COUNTRY: USA, Australia, Asia, Europe |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Taspoglutide: 10 mg subcutaneous injection once weekly and metformin as prescribed Taspoglutide: 10 mg subcutaneous injection once weekly for 4 weeks followed by 20 mg subcutaneous once weekly and metformin as prescribed Insulin glargine: Starting dose at 10 IU daily and metformin as prescribed |
| Outcomes |
PRIMARY OUTCOME: Absolute change from baseline in HbA1c at 24 weeks SECONDARY OUTCOMES: Change from baseline in fasting plasma glucose, change from baseline in body weight, responder rates for HbA1c (target ≤ 7.0%, ≤ 6.5%), incidence of hypoglycaemia and change from baseline in lipid profile at 24 weeks; relative change in glucose, insulin, C‐peptide and glucagon during a meal tolerance test at 24 weeks; safety: adverse events, vital signs, physical examination, clinical laboratory tests, ECG and anti‐taspoglutide antibodies measured throughout study |
| Starting date | October 2008, estimated completion June 2012 (last updated on March 15, 2011) |
| Contact information | Hoffmann‐La Roche |
| Notes | Study ID numbers: BC20965, 2008‐001855‐23, ClinicalTrials.gov Identifier: NCT00755287 |
| Trial name or title | A randomised, double‐blind, placebo‐controlled study to assess the effect of taspoglutide on glycaemic control, and its safety and tolerability, in obese patients with type 2 diabetes mellitus inadequately controlled with metformin monotherapy. |
| Methods |
TRIAL DESIGN: Randomised, double‐blind, placebo‐controlled, parallel assigned, phase III trial, multicenter, multinational, safety and efficacy study DURATION OF INTERVENTION: 52 weeks (12 months) COUNTRY: USA, Europe |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Taspoglutide: 10 mg subcutaneous injection once weekly for 4 weeks followed by 20 mg subcutaneous once weekly Placebo: Subcutaneous once weekly |
| Outcomes |
PRIMARY OUTCOME: Absolute change from baseline in HbA1c at 24 weeks SECONDARY OUTCOMES: Change from baseline in body weight, % of patients achieving ≥ 5% weight loss at weeks 24; % of patients achieving target HbA1c ≤ 6.5%, ≤ 7.0%, change from baseline in fasting plasma glucose, change from baseline in lipid profile, relative change in glucose, insulin, C‐peptide and glucagon during a meal tolerance test and beta cell function at 24 weeks; safety: adverse events,clinical laboratory tests, vital signs, physical examination, ECG, anti‐taspoglutide antibodies at planned clinical visits for 12 months |
| Starting date | January 2009, completed (last updated on April 18, 2011) |
| Contact information | Hoffmann‐La Roche |
| Notes | Study ID numbers: BC22092, 2008‐005809‐20, ClinicalTrials.gov Identifier: NCT00823992 |
| Trial name or title | A multicenter, randomised, double blind (double dummy), active controlled study to compare the safety, tolerability and effect on glycaemic control of taspoglutide versus pioglitazone in type 2 diabetes patients inadequately controlled on therapy with sulphonylurea or metformin plus sulphonylurea |
| Methods |
TRIAL DESIGN: Randomised, double blind (participant, investigator), three arm, phase III trial, parallel assignment, multi centre, multinational, safety and efficacy study DURATION OF INTERVENTION: 104 weeks (24 months) COUNTRY: USA, Europe, North and South America |
| Participants |
INCLUSION CRITERIA:
|
| Interventions |
Taspoglutide: 10 mg subcutaneous injection once weekly Taspoglutide: 10 mg subcutaneous injection once weekly for 4 weeks followed by 20 mg subcutaneous once weekly Pioglitazone: 30 mg orally once daily for 4 weeks followed by 45 mg once daily |
| Outcomes |
PRIMARY OUTCOME: Absolute change in HbA1c from baseline at 24 weeks SECONDARY OUTCOMES: Proportion of participants achieving target HbA1c ≤ 6.5%, ≤ 7.0% at weeks 24, 52 and 104; absolute/percentage change from baseline in body weight, responder rates for body weight, absolute/percentage change from baseline in waist and hip circumference, absolute/percentage change from baseline in fasting plasma glucose at weeks 24, 52 and 104; adverse events, laboratory parameters, cardiovascular events at each clinic visit up to 106 weeks |
| Starting date | May 2009, completed (last updated on June 15, 2011) |
| Contact information | Hoffmann‐La Roche |
| Notes | Study ID numbers: BC21893, 2009‐009157‐24, ClinicalTrials.gov Identifier: NCT00909597 |
BG: biguanide; BMI: body mass index; , CV: cardiovascular; DPP‐4: dipeptidyl peptidase‐4 inhibitor; ECG: electrocardiography; FPG: fasting plasma glucose; HbA1c: glycosylated haemoglobin A1c; HOMA‐B: homeostatic model assessment‐beta cell function; HOMA‐IS: homeostatic model assessment‐insulin sensitivity; HOMA‐IR: homeostatic model assessment‐insulin resistance; Met: metformin; OAD: oral antidiabetic drug; PG: plasma glucose; PPG: post‐prandial glucose; QD: once daily; SMPG: self‐measured plasma glucose; SU: sulphonylurea; TZD: thiazolidinedione; WHO: World Health Organization.
Contributions of authors
DEEPSON SHYANGDAN: Data extraction, quality assessment of studies, data summary and analysis, writing of main text
PAMELA ROYLE: Searching for studies, study selection, checking of data, data extraction, quality assessment of studies, writing of main text
CHRISTINE CLAR: Data extraction, quality assessment of studies, data summary and analysis, writing of main text
PAWANA SHARMA: Data extraction, quality assessment of studies, writing of protocol
NORMAN WAUGH: Study selection, general supervision of the review, writing of main text
AILSA SNAITH: Author of first version of the review (exenatide and liraglutide only) and of protocol
Sources of support
Internal sources
University of Aberdeen, Department of Public Health, UK.
University of Warwick, Warwick Evidence, UK.
External sources
No sources of support supplied
Declarations of interest
None known.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
- Rosenstock J, Reusch J, Bush M, Yang F, Stewart M. Potential of albiglutide, a long‐acting GLP‐1 receptor agonist, in type 2 diabetes. Diabetes Care 2009;32(10):1880‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergenstal RM, Wysham C, MacConell L, Malloy J, Walsh B, Yan P, et al. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION‐2): a randomised trial. Lancet Aug 2010;376:431‐9. [DOI] [PubMed] [Google Scholar]
- Blevins T, Pullman J, Malloy J, Yan P, Taylor K, Schulteis C, et al. DURATION‐5: Exenatide once weekly resulted in greater improvements in glycemic control compared with exenatide twice daily in patients with type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism 2011;96(5):1301‐10. [DOI] [PubMed] [Google Scholar]
- Diamant M, Van GL, Stranks S, Northrup J, Cao D, Taylor K, et al. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION‐3): an open‐label randomised trial. Lancet 2010;375:2234‐43. [DOI] [PubMed] [Google Scholar]
- Best JH, Boye KS, Rubin RR, Cao D, Kim TH, Peyrot M. Improved treatment satisfaction and weight‐related quality of life with exenatide once weekly or twice daily. Diabetic Medicine 2009;26(7):722‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Best JH, Rubin RR, Zhuang D, Velez F, Kim T, Peyrot M. Higher diabetes treatment satisfaction associated with improved glucose control for both exenatide once weekly and twice daily. Diabetes 2008;57:A520. [Google Scholar]; Drucker DJ, Buse JB, Taylor K, Kendall DM, Trautmann M, Zhuang D, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open‐label, non‐inferiority study.[see comment]. Lancet 2008;372(9645):1240‐50. [DOI] [PubMed] [Google Scholar]
- Ratner RE, Rosenstock J, Boka G, DRI6012 Study Investigators. Dose‐dependent effects of the once‐daily GLP‐1 receptor agonist lixisenatide in patients with Type 2 diabetes inadequately controlled with metformin: a randomized, double‐blind, placebo‐controlled trial. Diabetic Medicine 2010;27(9):1024‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku K, Rasmussen MF, Clauson P, Seino Y. Improved glycaemic control with minimal hypoglycaemia and no weight change with the once‐daily human glucagon‐like peptide‐1 analogue liraglutide as add‐on to sulphonylurea in Japanese patients with type 2 diabetes. Diabetes Obesity and Metabolism 2010;12:341‐7. [DOI] [PubMed] [Google Scholar]
- Marre M, Shaw J, Brändle M, Bebakar WMW, Kamaruddin NA, Strand J, et al. Liraglutide, a once‐daily human GLP‐1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with type 2 diabetes (LEAD‐1 SU). Diabetic Medicine 2009;26:268‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, et al. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)‐2 study. Diabetes Care 2009;32(1):84‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinman B, Gerich J, Buse JB, Lewin A, Schwartz S, Raskin P, et al. Efficacy and safety of the human glucagon‐like peptide‐1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD‐4 Met+TZD). Diabetes Care 2009;32(7):1224‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell‐Jones D, Vaag A, Schmitz O, Sethi BK, Lalic N, Antic S, et al. Liraglutide vs insulin glargine and placebo in combination with metformin and sulfonylurea therapy in type 2 diabetes mellitus (LEAD‐5 met+SU): a randomised controlled trial. Diabetologia 2009;52:2046‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buse J, Sesti G, Schmidt WE, Montanya E, Chang CT, Xu Y, et al. Switching from twice‐daily exenatide to once‐daily liraglutide improves glycemic control in T2D on oral agents. American Diabetes Association. 2009:591‐P. [DOI] [PMC free article] [PubMed] [Google Scholar]; Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26‐week randomised, parallel‐group, multinational, open‐label trial (LEAD‐6).[see comment]. Lancet 2009;374(9683):39‐47. [DOI] [PubMed] [Google Scholar]
- Pratley RE, Nauck M, Bailey T, Montanya E, Cuddihy R, Filetti S, et al. Liraglutide versus sitagliptin for patients with type 2 diabetes who did not have adequate glycaemic control with metformin: a 26‐week, randomised, parallel‐group, open‐label trial. Lancet 2010;375:1447‐56. [DOI] [PubMed] [Google Scholar]
- Umpierrez GE, Blevins T, Rosenstock J, Cheng C, Anderson JH, Bastyr EJ 3rd, EGO Study Group. The effects of LY2189265, a long‐acting glucagon‐like peptide‐1 analog, in a randomized, placebo‐controlled, double‐blind study of overweight/obese patients with type 2 diabetes: the EGO study. Diabetes, Obesity and Metabolism Jan 2011;13:418‐25. [DOI] [PubMed] [Google Scholar]
- Yang W, Chen L, Ji Q, Liu X, Ma J, Tandon N, et al. Liraglutide provides similar glycaemic control as glimepiride (both in combination with metformin) and reduces body weight and systolic blood pressure in Asian population with type 2 diabetes from China, South Korea and India: a 16‐week, randomized, double‐blind, active control trial. Diabetes, Obesity and Metabolism Jan 2011;13(1):81‐88. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Ratner RE, Kapitza C, Berria R, Boldrin M, Balena R. Treatment with the human once‐weekly glucagon‐like peptide‐1 analog taspoglutide in combination with metformin improves glycemic control and lowers body weight in patients with type 2 diabetes inadequately controlled with metformin alone: a double‐blind placebo‐controlled study. Diabetes Care 2009;32(7):1237‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner R, Nauck M, Kapitza C, Asnaghi V, Boldrin M, Balena R. Safety and tolerability of high doses of taspoglutide, a once‐weekly human GLP‐1 analogue, in diabetic patients treated with metformin: a randomized double‐blind placebo‐controlled study. Diabetic Medicine 2010;27:556‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
- Astrup A, Rossner S, Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. NN8022‐1807 Study Group. Effects of liraglutide in the treatment of obesity: a randomised, double‐blind, placebo‐controlled study. Lancet 2009 Nov 7;374:1606‐16. [DOI] [PubMed] [Google Scholar]
- Barnett AH, Burger J, Johns D, Brodows R, Kendall DM, Roberts A, et al. Tolerability and efficacy of exenatide and titrated insulin glargine in adult patients with type 2 diabetes previously uncontrolled with metformin or a sulfonylurea: a multinational, randomized, open‐label, two‐period, crossover noninferiority trial. Clinical Therapeutics 2007;29:2333‐48. [DOI] [PubMed] [Google Scholar]; Secnik K, Hayes C, Matza L, Kim S, Oglesby A, Yurgin N, et al. Improved treatment satisfaction in patients with type‐2 diabetes treated with exenatide or insulin glargine. Value in Health 2005;8:A155‐A156. [Google Scholar]; Trautmann M, Burger J, Johns D, Brodows R, Okerson T, Roberts A, et al. Less hypoglycemia with exenatide versus insulin glargine, despite similar Hba(1c) improvement, in patients with T2dm adjunctively treated with metformin. Diabetes 2007;56:A45. [Google Scholar]; Trautmann M, Burger J, Johns D, Brodows R, Okerson T, Roberts A, et al. Relationship between hypoglycaemia, background oral antidiabetic agent, and glycaemic control in patients treated with exenatide or insulin glargine. Diabetologia 2007;50:S338. [Google Scholar]
- Blonde L, Klein EJ, Han J, Zhang B, Mac SM, Poon TH, et al. Interim analysis of the effects of exenatide treatment on A1C,weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes, Obesity and Metabolism 2006;8:436‐47. [DOI] [PubMed] [Google Scholar]
- Bode BW, Testa MA, Magwire M, Hale PM, Hammer M, Blonde L, et al. LEAD‐3 Study Group. Patient‐reported outcomes following treatment with the human GLP‐1 analogue liraglutide or glimepiride in monotherapy: results from a randomized controlled trial in patients with type 2 diabetes. Diabetes Obesity and Metabolism 2010 Jul;12(7):604‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brixner DI, McAdam‐Marx C, Ye X, Boye KS, Nielsen LL, Wintle M, et al. Six‐month outcomes on A1C and cardiovascular risk factors in patients with type 2 diabetes treated with exenatide in an ambulatory care setting. Diabetes Obesity and Metabolism 2009 Dec;11(12):1122‐30. [DOI] [PubMed] [Google Scholar]
- Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin‐4) on glycemic control over 30 weeks in sulfonylurea‐treated patients with type 2 diabetes. Diabetes Care 2004;27(11):2628‐35. [DOI] [PubMed] [Google Scholar]
- Feinglos MN, Saad MF, Pi‐Sunyer FX, An B, Santiago O, Liraglutide Dose‐Response Study Group. Effects of liraglutide (NN2211), a long‐acting GLP‐1analogue, on glycaemic control and bodyweight in subjects with type 2 diabetes. Diabetic Medicine 2005;22:1016‐23. [DOI] [PubMed] [Google Scholar]
- Gallwitz B, Vaag A, Falahati A, Madsbad S. Adding liraglutide to oral antidiabetic drug therapy: onset of treatment effects over time. International Journal of Clinical Practice 2010 Jan;64(2):267‐76. [DOI] [PubMed] [Google Scholar]
- Garber A, Henry R, Ratner R, Garcia‐Hernandez PA, Rodriguez‐Pattzi H, Olvera‐Alvarez I, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD‐3 Mono): a randomised, 52‐week, phase III, double‐blind, parallel‐treatment trial.[see comment]. Lancet 2009;373(9662):473‐81. [DOI] [PubMed] [Google Scholar]
- Kapitza C, Heise T, Birman P, Jallet K, Ramis J, Balena R. Pharmacokinetic and pharmacodynamic properties of taspoglutide, a once‐weekly, human GLP‐1 analogue, after single‐dose administration in patients with Type 2 diabetes. Diabetic Medicine 2009 Nov;26(11):1156‐64. [DOI] [PubMed] [Google Scholar]
- Kim D, MacConell L, Zhuang D, Kothare PA, Trautmann M, Fineman M, et al. Effects of once‐weekly dosing of a long‐acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care 2007;30:1487‐93. [DOI] [PubMed] [Google Scholar]
- Madsbad S, Schmitz O, Ranstam J, Jakobsen G, Matthews DR, NN2211‐1310 International study group. Improved glycemic control with no weight increase in patients with type 2 diabetes after once‐daily treatment with the long‐acting glucagon‐like peptide 1 analog liraglutide (NN2211). Diabetes Care 2004;27:1335‐42. [DOI] [PubMed] [Google Scholar]
- Malloy J, Capparelli E, Gottschalk M, Guan X, Kothare P, Fineman M. Pharmacology and tolerability of a single dose of exenatide in adolescent patients with type 2 diabetes mellitus being treated with metformin: a randomized, placebo‐controlled, single‐blind, dose‐escalation, crossover study. Clinical Therapeutics 2009 Apr;31(4):806‐15. [DOI] [PubMed] [Google Scholar]
- Moretto TJ, Milton DR, Ridge TD, Macconell LA, Okerson T, Wolka AM, et al. Efficacy and tolerability of exenatide monotherapy over 24 weeks in antidiabetic drug‐naive patients with type 2 diabetes: a randomized, double‐blind, placebo‐controlled, parallel‐group study. Clinical Therapeutics 2008;30:1448‐60. [DOI] [PubMed] [Google Scholar]
- Harder H, Nielsen L, Thi TDT, Astrup A. The effect of liraglutide, a long‐acting glucagon‐like peptide 1 derivative, on glycemic control, body composition, and 24‐h energy expenditure in patients with type 2 diabetes. Diabetes Care 2004;27:1915‐21. [DOI] [PubMed] [Google Scholar]; Nauck M, Marre M. Adding liraglutide to oral antidiabetic drug monotherapy: efficacy and weight benefits. Postgraduate Medicine 2009;121(3):5‐15. [DOI] [PubMed] [Google Scholar]
- Okerson T, Yan P, Stonehouse A, Brodows R. Effects of exenatide on systolic blood pressure in subjects with type 2 diabetes. American Journal of Hypertension 2010 Mar;23(3):334‐39. [DOI] [PubMed] [Google Scholar]
- Riddle MC, Henry RR, Poon TH, Zhang B, Mac MS, John HH, et al. Exenatide elicits sustained glycaemic control and progressive reduction of body weight in patients with type 2 diabetes inadequately controlled by sulphonylureas with or without metformin. Diabetes/Metabolism Research and Reviews 2006;22:483‐91. [DOI] [PubMed] [Google Scholar]
- Seino Y, Rasmussen MF, Zdravkovic M, Kaku K. Dose‐dependent improvement in glycemia with once‐daily liraglutide without hypoglycemia or weight gain: A double‐blind, randomized, controlled trial in Japanese patients with type 2 diabetes. Diabetes Research and Clinical Practice 2008;81(2):161‐8. [DOI] [PubMed] [Google Scholar]
- Trescoli‐Serrano C, Rivas M, Fajardo‐Montanana C, Pino I, Garcia‐Zarco M, Martorrell‐Barraquet A, et al. Clinical site experience comparing insulin glargine with exenatide treatment in type 2 diabetic patients. Diabetologia 2005;48:A3. 16163567 [Google Scholar]
- Courreges JP, Vilsboll T, Zdravkovic M, Le‐Thi T, Krarup T, Schmitz O, et al. Beneficial effects of once‐daily liraglutide, a human glucagon‐like peptide‐1 analogue, on cardiovascular risk biomarkers in patients with type 2 diabetes. Diabetic Medicine 2008;25(9):1129‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]; Horowitz M. Patient‐reported rating of gastrointestinal adverse effects during treatment of type 2 diabetes with the once‐daily human GLP‐1 analogue, liraglutide. Diabetes, Obesity and Metabolism 2008;10:593‐02. [DOI] [PubMed] [Google Scholar]; Valentine WJ, Hammer M, Palmer AJ, Ray JA. Projecting the long‐term clinical benefits of treatment with the GLP‐1 analogue liraglutide in subjects with type 2 diabetes. Diabetes 2007;56:A568. [Google Scholar]; Vilsboll T, Brock B, Perrild H, Levin K, Lervang HH, Kolendorf K, et al. Liraglutide, a once‐daily human GLP‐1 analogue, improves pancreatic B‐cell function and arginine‐stimulated insulin secretion during hyperglycaemia in patients with type 2 diabetes mellitus. Diabetic Medicine 2008;25(2):152‐56. [DOI] [PubMed] [Google Scholar]; Vilsboll T, Zdravkovic M, Le‐Thi T, Krarup T, Schmitz O, Courreges JP, et al. Liraglutide, a long‐acting human glucagon‐like peptide‐1 analog, given as monotherapy significantly improves glycemic control and lowers body weight without risk of hypoglycemia in patients with type 2 diabetes. Diabetes Care 2007;30:1608‐10. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
- NCT00838903. Efficacy and safety of albiglutide in treatment of type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT00838903.
- NCT00838916. A study to determine the safety and efficacy of albiglutide in patients with type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT00838916.
- NCT00849017. Safety and efficacy study of albiglutide in type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT00849017.
- NCT00849056. Safety and efficacy of albiglutide in type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT00849056.
- NCT01098461. Dose ranging study of albiglutide in Japanese subjects. http://clinicaltrials.gov/ct2/show/NCT01098461.
- NCT01128894. A study to determine the efficacy and safety of albiglutide as compared with liraglutide.. http://clinicaltrials.gov/ct2/show/NCT01128894 accessed 17 June 2011.
- NCT00641056. Efficacy of exenatide once weekly and once‐daily insulin glargine in patients with type 2 diabetes treated with metformin alone or in combination with sulfonylurea (DURATION ‐ 3). http://clinicaltrials.gov/ct2/show/NCT00641056.
- NCT00917267. A study to examine the effects of exenatide once‐weekly injection on glucose control and safety in Asian subjects. http://clinicaltrials.gov/ct2/show/NCT00917267.
- NCT00935532. Study to evaluate the efficacy and safety of exenatide once‐weekly injection compared to once‐daily insulin in type 2 diabetes mellitus. http://clinicaltrials.gov/ct2/show/NCT00935532.
- NCT01003184. Efficacy of once‐weekly exenatide versus once or twice daily insulin detemir in patients with type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT01003184.
- NCT01029886. Safety and efficacy of exenatide once weekly versus liraglutide in subjects with type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT01029886 accessed 17 June 2011.
- NCT01144338. Exenatide study of cardiovascular event lowering trial (EXSCEL): a trial to evaluate cardiovascular outcomes after treatment with exenatide once weekly in patients with type 2 diabetes mellitus. http://clinicaltrials.gov/ct2/show/NCT01144338.
- NCT00856986. The effect of insulin detemir in combination with liraglutide and metformin compared to liraglutide and metformin in subjects with type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT00856986 accessed 17 June 2011.
- NCT01117350. Efficacy assessment of insulin glargine versus liraglutide after oral agent failure (EAGLE). http://clinicaltrials.gov/ct2/show/NCT01117350 accessed 17 June 2011.
- NCT01296412. Comparison of two treatment regimens (sitagliptin versus liraglutide) on participants who failed to achieve good glucose control on metformin alone (MK‐0431‐403). http://clinicaltrials.gov/ct2/show/NCT01296412 accessed 17 June 2011.
- NCT01336023. Dual action of liraglutide and insulin degludec in type 2 diabetes: a trial comparing the efficacy and safety of insulin degludec/liraglutide, insulin degludec and liraglutide in subjects with type 2 diabetes (DUAL™ I). http://clinicaltrials.gov/ct2/show/NCT01336023 accessed 17 June 2011.
- NCT00707031. GLP‐1 agonist AVE0010 versus exenatide in patients with type 2 diabetes for glycemic control and safety evaluation, on top of metformin (GETGOAL‐X). http://clinicaltrials.gov/ct2/show/NCT00707031 accessed 17 June 2011.
- NCT00713830. GLP‐1 agonist AVE0010 in patients with type 2 diabetes for glycemic control and safety evaluation, on top of sulfonylurea (GETGOAL‐S). http://clinicaltrials.gov/ct2/show/NCT00713830 accessed 17 June 2011.
- NCT00763815. GLP‐1 agonist AVE0010 in patients with type 2 diabetes for glycemic control and safety evaluation, on top of pioglitazone (GETGOAL‐P). http://clinicaltrials.gov/ct2/show/NCT00763815 accessed 17 June 2011.
- NCT00975286. 24‐week treatment with lixisenatide in type 2 diabetes insufficiently controlled with metformin and insulin glargine. http://clinicaltrials.gov/ct2/show/NCT00975286 accessed 17 June 2011.
- NCT00976937. 24‐week study comparing lixisenatide to sitagliptin as add‐on to metformin in obese type 2 diabetic patients younger than 50. http://clinicaltrials.gov/ct2/show/NCT00976937 accessed 17 June 2011.
- NCT00696657. A randomised controlled clinical trial in type 2 diabetes comparing semaglutide to placebo and liraglutide. http://clinicaltrials.gov/ct2/show/NCT00696657 accessed 17 June 2011.
- NCT00717457. A study of taspoglutide versus exenatide for the treatment of patients with type 2 diabetes mellitus inadequately controlled with metformin, thiazolidinedione or a combination of both. http://clinicaltrials.gov/ct2/show/NCT00717457 accessed 17 June 2011.
- NCT00744367. A study of taspoglutide versus placebo for the treatment of patients with type 2 diabetes mellitus inadequately controlled with metformin plus pioglitazone. http://clinicaltrials.gov/ct2/show/NCT00744367 accessed 17 June 2011.
- NCT00754988. A study of taspoglutide versus sitagliptin for the treatment of patients with type 2 diabetes mellitus inadequately controlled with metformin. http://clinicaltrials.gov/ct2/show/NCT00754988 accessed 17 June 2011.
- NCT00755287. A study of the safety, tolerability and effect on glycemic control of taspoglutide versus insulin glargine in insulin naive type 2 diabetic patients inadequately controlled with metformin plus sulphonylurea. http://clinicaltrials.gov/ct2/show/NCT00755287 accessed 17 June 2011.
- NCT00823992. A study of taspoglutide versus placebo for the treatment of obese patients with type 2 diabetes mellitus inadequately controlled with metformin monotherapy. http://clinicaltrials.gov/ct2/show/NCT00823992 accessed 17 June 2011.
- NCT00909597. A study of taspoglutide versus pioglitazone in patients with type 2 diabetes. http://clinicaltrials.gov/ct2/show/NCT00909597 accessed 17 June 2011.
Additional references
- Abraham EJ, Leech CA, Lin JC, Zulewski H, Habener JF. Insulinotropic hormone glucagon‐like peptide‐1 differentiation of human pancreatic islet‐derived progenitor cells into insulin‐producing cells. Endocrinology 2002;143(8):3152‐61. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. Report on the Expert Committee on the Diagnosis and Classication of Diabetes Mellitus. Diabetes Care 1997;20(Suppl 1):S5‐20. [Google Scholar]
- The Expert Committee on the Diagnosis and Classication of Diabetes Mellitus. Report of the Expert Committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 1999;22(Suppl 1):S5‐19. [DOI] [PubMed] [Google Scholar]
- Amori RE, Lau J, Pittas, AG. Efficacy and safety of incretin therapy in type 2 diabetes. Journal of the American Medical Association 2007;298(2):194‐206. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Huang Q, Brown TJ, Drucker DJ. A recombinant human glucagon‐like peptide (GLP)‐1‐albumin protein (albugon) mimics peptidergic activation of GLP‐1 receptor‐dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes 2004;53(9):2492‐500. [DOI] [PubMed] [Google Scholar]
- Barnett AH. New treatments in type 2 diabetes: a focus on the incretin‐based therapies. Clinical Endocrinology 2009;70:343‐53. [DOI] [PubMed] [Google Scholar]
- Butler PC, Matveyenko AV, Dry S, Bhushan A, Elashoff R. Glucagon‐like peptide‐1 therapy and the exocrine pancreas: innocent bystander or friendly fire?. Diabetologia 2010;53:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore DD, Bloomgren GL, Wenten M, Hoffman C, Clifford CR, Quinn SG, et al. A cohort study of acute pancreatitis in relation to exenatide use. Diabetes, Obesity and Metabolism Jun 2011;13(6):559‐66. [DOI] [PubMed] [Google Scholar]
- EMEA. Assessment report for Victoza (liraglutide). http://www.emea.europa.eu/humandocs/PDFs/EPAR/victoza/H‐1026‐en6.pdf accessed 10 March 2010.
- Farilla L, Bulotta A, Hirshberg B, Li CS, Khoury N, Noushmehr H, et al. Glucagon‐like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets.[see comment]. Endocrinology 2003;144(12):5149‐58. [DOI] [PubMed] [Google Scholar]
- FDA. Information for Healthcare Professionals: Exenatide (marketed as Byetta). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm accessed 10 March 2010.
- FDA. Byetta (exenatide) prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/021773s9s11s18s22s25lbl.pdf accessed 10 March 2010.
- FDA. Information for Healthcare Professionals: Reports of Altered Kidney Function in patients using Exenatide (Marketed as Byetta). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm188656.htm accessed 10 March 2010.
- FDA. Byetta Safety Update for Healthcare Professionals. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm190406.htm accessed 10 March 2010.
- Girman CJ, Kou TD, Cai B, Alexander CM, O'Neill EA, Williams‐Herman DE, et al. Patients with type 2 diabetes mellitus have higher risk for acute pancreatitis compared with those without diabetes. Diabetes Obesity and Metabolism Sep 2010;12(9):766‐71. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21:1539‐58. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analysis. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
- Journal Watch. FDA Reminds Clinicians of Liraglutide's Risks for Pancreatitis and Cancer. http://firstwatch.jwatch.org/cgi/content/full/2011/614/2?q=topic_diabetes accessed 17 June 2011.
- Kaku K, Rasmussen MF, Clauson P, Seino Y. Improved glycaemic control with minimal hypoglycaemiaand no weight change with the once‐daily humanglucagon‐like peptide‐1 analogue liraglutide as add‐onto sulphonylurea in Japanese patients with type 2 diabetes. Diabetes, Obesity and Metabolism 2010;12:341–7. [DOI] [PubMed] [Google Scholar]
- Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon‐like peptide‐1 receptor signaling modulates beta cell apoptosis. The Journal of Biological Chemistry 2003;278(1):471‐8. [DOI] [PubMed] [Google Scholar]
- Loh JA, Clement SC. Efficacy of exenatide therapy over 12 months in a "real world" setting. Diabetes 2007;56:A152. [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA group. Preferred reporting items for systematic reviews and meta‐analysis: the PRISMA statement. British Medical Journal 2009;339:b2535. [PMC free article] [PubMed] [Google Scholar]
- Monami M, Marchionni N, Mannucci E. Glucagon like peptide‐1 receptor agonists in type 2 diabetes: a meta‐analysis of randomized clinical trials. European Journal of Endocrinology 2009;160:909‐17. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, et al. American Diabetes Association, European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009;32(1):193‐03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon‐like peptide 1 (7‐36 amide) in type 2 (non‐insulin‐dependent) diabetic patients. Diabetologia 1993;36(8):741‐4. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, et a. Effects of glucagon‐like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycaemic clamp experiments in healthy volunteers. The Journal of Clinical Endocrinology and Metabolism 2002;87(3):1239‐46. [DOI] [PubMed] [Google Scholar]
- National Collaborating Centre for Chronic Conditions. Type 2 diabetes: national clinical guideline for management in primary and secondary care. London: Royal College of Physicians2008; Vol. http://www.nice.org.uk/nicemedia/pdf/CG66FullGuideline0509.pdf.
- National Institute for Health and Clinical Excellence. Type 2 diabetes: the management of type 2 diabetes. NICE clinical guideline 872009; Vol. http://www.nice.org.uk/cg87.
- National Institute for Health and Clinical Excellence. Liraglutide for the treatment of type 2 diabetes mellitus. NICE technology appraisal guidance 203. http://guidance.nice.org.uk/TA2032010.
- Norris SL, Lee N, Thakurta S, Chan BKS. Exenatide efficacy and safety: a systematic review. Diabetic Medicine 2009;26:837‐46. [DOI] [PubMed] [Google Scholar]
- Pratley RE. Overview of glucagon‐like peptide‐1 analogs and dipeptidyl peptidase‐4 inhibitors for type 2 diabetes. Medscape Journal of Medicine 2008;10(7):171. [PMC free article] [PubMed] [Google Scholar]
- Shyangdan DS, Royle PL, Clar C, Sharma P, Waugh NR. Glucagon‐like peptide analogues for type 2 diabetes mellitus: systematic review and meta‐analysis. BMC Endocrine Disorders 2010;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyangdan D, Cummins E, Royle P, Waugh N. Liraglutide for the treatment of type 2 diabetes. Health Technology Assessment 2011;15(Suppl. 1):75‐84. [DOI] [PubMed] [Google Scholar]
- Turner RC, Holman RR. The UK Prospective Diabetes Study. UK Prospective Diabetes Study Group. Annals of Medicine 1996;28(5):439‐44. [DOI] [PubMed] [Google Scholar]
- UKPDS Study Group. UKPDS 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. UK Prospective Diabetes Study Group. Diabetes 1995;44:1249‐58. [PubMed] [Google Scholar]
- Waugh N, Cummins E, Royle P, Clar C, Marien M, Richter B, et al. Newer agents for blood glucose control in type 2 diabetes: systematic review and economic evaluation. Health Technology Assessment July 2010;14(36):1‐248. [DOI] [PubMed] [Google Scholar]
- Alberti KM, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its compliactions. Part I: diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabetic Medicine 1998;15:539‐53. [DOI] [PubMed] [Google Scholar]
- Xu G, Stoffers DA, Habener JF, Bonner‐Weir S. Exendin‐4 stimulates both beta‐cell replication and neogenesis, resulting in increased beta‐ cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999;48(12):2270‐6. [DOI] [PubMed] [Google Scholar]