

Abstract
Self-healing supramolecular hydrogels have emerged as a novel class of biomaterials that combines hydrogels with supramolecular chemistry to develop highly functional biomaterials with advantages including native tissue mimicry, biocompatibility, and injectability. These properties are endowed by the reversibly cross-linked polymer network of the hydrogel. These hydrogels have great potential for realizing yet to be clinically translated tissue engineering therapies. This review presents methods of self-healing supramolecular hydrogel formation and their uses in tissue engineering as well as future perspectives.
Keywords: supramolecular, hydrogels, self-healing, tissue engineering, biomaterials
Graphical Abstract
1. Introduction
In recent decades, increasing demand for biomaterials capable of aiding in regeneration or replacement of damaged tissue motivated the development of new tissue engineering constructs.[1] Due to their unique biocompatibility and biomimetic properties, hydrogels have been widely used in many biomedical applications such as contact lenses, wound dressings, and drug delivery vehicles.[2–4] The properties of hydrogels follow from their structure: namely their highly swollen, hydrophilic 3D crosslinked polymer network that may be either chemically or physically crosslinked to form a material that mimics advantageous properties of the highly hydrated extracellular matrix (ECM) and facilitates nutrient and oxygen transport due to its porous structure.[3,5]
While hydrogels offer many useful properties, the permanent covalent crosslinks in traditional hydrogels are often not ideal for tissue engineering applications, considering the dynamic nature of tissue regeneration and integration with the host. Tissue engineering has yet to achieve anticipated clinical impact, largely due to a failure to integrate with existing vasculature and host tissue.[6] The irreversible covalent crosslinks result in a material that is unable to heal after breaking, making such hydrogels ineffective in sustaining their function or maintaining their desired mechanical properties.[7,8] Covalent crosslinks also constrain the movement of polymer chains in the hydrogel bulk, decreasing their flexibility and creating an elastic material, which contrasts with the viscoelastic and deformable ECM of native tissue.[9] While hydrogels formed by dynamic covalent crosslinks have been investigated for biomedical applications, the reversibility of the linkages can rely on harsh conditions such as elevated temperatures[10,11], low pH[12,13], UV light[14–17] or a combination of these.[18] Such hydrogels are therefore limited to applications that utilize their harsher crosslinking conditions such as cancer therapy, where pH sensitive gelation can occur only in the acidic tumor microenvironment.[19] The tissue-specific microenvironment is an important design component of effective tissue engineering scaffolds as cells are constantly interacting with, remodeling, and moving through the ECM to perform normal functions.[20–22] Recent work has looked into dynamic covalent chemistries including Schiff’s Base, imine crosslinks, and disulfide exchange that would allow for cellular remodeling and tunable cleavage of the hydrazone or disulfide linkers.[8,9,15,23–25] These gels undergo dynamic chemical degradation while maintaining the bulk properties of the hydrogel and have been used primarily as self-healing 3D cell culture substrates. These materials often exhibit slower reversibility than their supramolecular counterparts and can require the addition of a catalyst,[24] but they have still been used for 3D printing as their shear-thinning and self-healing behavior makes them desirable candidates for extrusion based printing. [26,27] Compared to permanently crosslinked hydrogels, the incorporation of cells into dynamic hydrogels is relatively new[9], and work remains to be done towards the development of adaptable covalent hydrogels for tissue engineering.
To achieve properties that remain largely unrealized by covalently crosslinked hydrogels such as self-healing and reversible linkages, supramolecular hydrogels have been developed that combine the unique advantages of hydrogels and supramolecular chemistry.[28] The properties of these hydrogels follow from the dynamic nature of their constituents: supramolecular bonding motifs that rely on hydrogen bonding, electrostatic interactions, π-π bonding, host-guest interactions, hydrophobic interactions, or metal coordination act as dynamic crosslinks between hydrophilic polymers to form hydrogels.[29–35] The variety of supramolecular building blocks offers many tools for development of a diverse range of biomaterials that offer promise as scaffolds for tissue engineering.[36,37]
This review will focus on important work that has been done towards development of self-healing supramolecular hydrogels for tissue engineering applications, with emphasis on supramolecular bonding motifs used for hydrogel formation and recent advances towards their use in regenerative medicine. We will conclude by looking forward at important challenges facing the development of ideal hydrogels as both scaffolds and drug or biologic delivery vehicles to regenerate, maintain, or improve lost or damaged tissue.
2. Supramolecular Bonding Motifs
2.1. Motivation
To advance the field of tissue engineering, new biomaterials capable of effective, controlled delivery of drugs and biologics, and recapitulation of native tissue properties must be developed. Cells exist in vivo within a dynamic, 3D matrix consisting of polysaccharides, ECM biopolymers (proteoglycans and proteins), and soluble factors secreted by nearby cells.[38] This 3D environment is critical to normal cell function, as it enables important cell behaviors including cell adhesion, proliferation, migration, and differentiation.[38–42] Hydrogels have a long history as tools for tissue regeneration and 3D cell culture as they may be engineered to mimic the desired aspects of the native local ECM depending on their intended usage. Early attempts at recapitulation of the ECM included hydrolytically cleavable and metalloproteinase (MMP)-cleavable permanently crosslinked but dynamic hydrogels. For example, a dynamic MMP-cleavable hydrogel was developed to facilitate cell invasion and render the polymer networks degradable by cells to mimic this feature in native tissue, however the enzymatic cleavage of the crosslinks is permanent, leading to degradation of the bulk material.[43,44] It was shown that both hydrolytically and enzymatically degradable hydrogels were favorable over their static covalent counterparts in in vitro cartilage regeneration.[45] Later efforts towards development of dynamic systems included a permanently crosslinked, pH-switchable hydrogel to enable repeated self-healing via hydrogen bonding[46], however, the gelation occurs at pH≤3 and is therefore limited in its application to tissue engineering. Supramolecular hydrogels have emerged as a promising tool for tissue regeneration as they can be biocompatible and recapitulate the viscoelastic nature of the ECM better than their elastic, covalently crosslinked counterparts due to the presence of dynamic linkages. The resulting viscoelastic and dynamic behavior of these linkages are responsible for other advantages such as self-healing and injectability. In addition to offering advantageous scaffolding for tissue engineering, supramolecular hydrogels can be engineered to effectively encapsulate, sequester, protect, and deliver cells, biologics, and drugs for regenerative medicine.
Of particular interest are supramolecular hydrogels that can self-heal after damage either spontaneously or in the presence of a physiological stimulus.[46] This characteristic extends the lifetime of materials and makes them ideal candidates for applications involving repeated mechanical stress[47] or injection.[48,49] The dynamic nature of reversible crosslinks responsible for self-healing behavior also results in shear-thinning (decrease in viscosity as shear stress increases) behavior, leading to injectable hydrogels which have been heavily investigated recently as they represent a critical step towards less invasive delivery of therapies.[48,50] The properties of self-healing supramolecular hydrogels may be selected for and tuned by careful choice of a hydrophilic polymer and the supramolecular bonding motif utilized as a physical crosslinker (Table 1). Ideally, hydrogels for tissue engineering should enable cell infiltration as well as encapsulate and deliver cells and biologics, and be able to autonomously, rapidly, and repeatedly heal in situ at physiological conditions.[51] The most important supramolecular bonding motifs used in hydrogel formation for tissue engineering applications, with an emphasis on recent advances, are described below.
Table 1.
The categories of supramolecular interactions and the specific bonding motifs covered in this review.
| Supramolecular Category | Specific Bonding Motif | Section | References |
|---|---|---|---|
| Hydrogen Bonding | Ureidopyrimidinone | 2.2.1 | 31, 48, 56–67 |
| Benzene-1,3,5-tricarboxamide | 2.2.2 | 59, 68–76 | |
| Others | 2.2.3 | 77, 78 | |
| Macrocyclic Host-Guest and Inclusion Complexes | Cyclodextrin Derivatives | 2.3.1 | 80–96, 98,99 |
| Stimuli-Responsive Cyclodextrins | 2.3.2 | 100–105, 106–110 | |
| Hydrophobic Interactions | Peptide Amphiphiles | 2.4.1 | 116–133 |
| Amphiphilic Block Copolymers | 2.4.2 | 134–139 | |
| Electrostatic Interactions | 2.5 | 140–150 | |
| Metal-Ligand Complexation | 2.6 | 20, 151–160 |
2.2. Hydrogen Bonding Motifs
Hydrogen bonding is an important tool for self-assembly in biological systems as it is responsible for nucleobase pairing in DNA and RNA as well as the 3D assembly of proteins.[52] While the strength of a single hydrogen ‘bond’ is relatively weak compared to covalent and some other non-covalent interactions, multivalent molecules greatly increase the degree of association possible between supramolecular moieties.[53] In fact, the cooperative effect of hydrogen bonding is responsible for some of the toughest covalent materials known, including Kevlar and silk.[54,55] Hydrogen bonding has also been utilized to great effect to generate self-healing permanently crosslinked hydrogels, however as was mentioned, the crosslinking occurs at a low pH and therefore is not useful in tissue engineering.[46]
2.2.1. Ureidopyrimidinone
Multiple hydrogen bonding motifs such as the widely reported quadruple hydrogen bonding motif ureidopyrimidinone (UPy), first described in a pioneering work in the 1990s, have been widely utilized in supramolecular hydrogels as a driving force for hydrogel formation.[31,56,57] Multiple hydrogen bonding units act as both hydrogen bond donors and acceptors and intrinsically increase the concentration of hydrogen bonding motifs in solution, leading to increased association between supramolecular building blocks. To achieve hydrogel formation in an aqueous environment, inter- and intramolecular effects must overcome water competition by either increasing the functionality per molecule, the molecule concentration, or using a combination of driving forces. Hydrogel formation is realized when a high enough association constant between supramolecular units is reached. Above a critical concentration, hydrogen bonding, hydrophobic spacers which protect the hydrogen bonding groups from the solvent, and π-π stacking combine to combat competitive water and drive hydrogel formation. [58–61]
Early work on supramolecular polymer networks formed through UPy interactions found that while they exhibited viscoelasticity and shear-thinning behavior, they eroded rapidly in aqueous environments due to oligomer dissociation, making them unfavorable materials for stable hydrogels.[62–64] More recently, work has been done on preparation of biocompatible UPy-based supramolecular hydrogels that are capable of autonomous and spontaneous self-assembly at physiologically relevant conditions for tissue engineering purposes.[48]
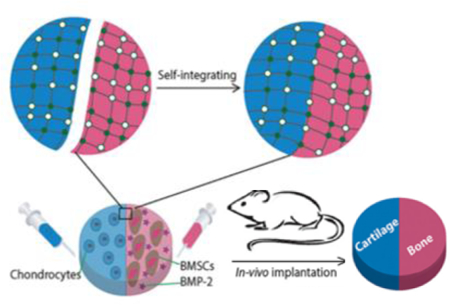
Minimally invasive injectable hydrogels capable of carrying growth factors and cells offer an attractive route to regenerate injured tissues. Injectable, self-healing supramolecular hydrogels utilizing UPy interactions have been used as biologic carriers[61,65] and cell-delivery systems[48], and have been studied in vivo as minimally invasive protein delivery systems for soft tissues including kidney[64], and cardiac[65] regeneration after injury. For example, a pH-switchable, self-healing UPy-modified PEG hydrogel was delivered to the myocardium via local catheter injection to fast-release growth factors to reduce infarct size[65] and subsequently for sustained-release of angiogenic factors to promote cardiac regeneration in a chronic infarct pig model.[66] These hydrogels are also useful for hard tissue engineering such as bone and cartilage[48] regeneration. In contrast to the majority of hydrogels that utilize telechelic UPy units to induce crosslinking a dextran-based hydrogel with multiple pendant UPy units per chain was developed as a cell carrier and drug delivery system that does not require the addition of hydrophobic spacers for gelation. This system has been used to carry chondrocytes, bone marrow stem cells, and bone morphogenetic protein 2 (BMP-2) in an integrated construct to induce seamless osteochondral tissue complex regeneration in an in vivo mouse model (Figure 1).[48]
Figure 1.

Design of a Dextran-UPy hydrogel for multi-tissue regeneration. a) Schematic illustration of Dex-UPy hydrogel formation and the mechanisms of the shear-thinning and self-recovery properties. Top: multiple hydrogen bonds of UPy and their dynamic interactions. Bottom: hydrogel network formation through UPy hydrogen bonds, the shear- thinning under shear stress and self-recovering of the hydrogel. b) Self-integration of the hydrogel pieces to form various structures. Some hydrogel disks were dyed pink with rhodamine and the others were left with the original light-yellow color to visualize the interfaces. Scale bar = 5 mm.
Adapted with permission. [41] 2015, Wiley.
Its self-healing and shear-thinning nature allows possible cell migration and dynamic tissue modeling/remodeling following injection, and it represents an important step towards clinically relevant self-healing hydrogels for multi-tissue organ regeneration. (Figure 2). Another approach to generating shear-thinning supramolecular hydrogels is incorporating UPy modified with bulky, hydrophobic adamantyl groups, instead of alkyl spacers, to create “hydrophobic pockets” that shield the hydrogen bonds from solvent interactions[67] and act to stabilize the structure, resulting in a self-healing and viscoelastic hydrogel that retains its rheological characteristics post injection. This material is likely non-toxic but its suitability for tissue engineering may be limited due to rapid erosion because of progressive solubilization in highly aqueous environments and requires further testing.
Figure 2.

In vivo testing of Dex-UPy hydrogel for bone and cartilage tissue regeneration. a) Schematic illustration of self-integration and the application in cartilage–bone tissue complex regeneration. Hydrogels encapsulating chondrocytes (blue) and BMSCs/BMP-2 (red) were integrated into a construct and then implanted subcutaneously in a nude mouse to form the cartilage–bone tissue complex. (b–e) Subcutaneous implantation of the cell-gel constructs. b) A section of BMSCs/BMP-2 only group, stained with Alizarin red (positive staining represents mineralized bone tissue). c) A section of chondrocytes only group, stained with Alcian blue (positive staining represents cartilage tissue). d) A section of the self-integrated group, stained with both Alizarin red and Alcian blue. e) A magnified image of the interface region of image ‘c’.
Adapted with permission. [41] 2015, Wiley.
2.2.2. Benzene-1,3,5-tricarboxamide
More recently, benzene-1,3,5-tricarboxamide (BTA)[68,69] has been recognized as a useful multiple hydrogen bonding unit for formation of supramolecular hydrogels. BTA is a disk-shaped molecule functionalized with hydrophobic alkyl spacers and oligo(ethylene glycols) to increase solubility in polar solvents, such as water.[68] In water, BTA uses a combination of its three-fold hydrogen bonding, protective hydrophobic pocket, and π-π interactions between the benzene rings to self-assemble into helical fibers (Figure 3).[70,71] At high enough concentrations, BTA based hydrogels form as a result of crosslinking of telechelic BTAs, and much like UPy based hydrogels, are very sensitive to the ratio between hydrophobic and hydrophilic spacers.[59,69,70,72] Recently, this method of self-assembly has received increased attention, and has been combined with principles of DNA-origami to enable precise control over the spatial distribution and recruitment of proteins in adaptive scaffolds. Most recently, self-assembly of BTA monomers were used to form one-dimensional supramolecular polymers that allow facile incorporation of various and DNA-functionalized BTA monomers that enabled efficient and dynamic control of protein activity along the polymer.[72–75] Functional BTAs have been incorporated into hydrogels for siRNA capture that represent a versatile platform for further development of various functionalized BTA polymers.[76] While promising and well characterized, these materials have yet to be tested in vitro for cytotoxicity and have not been developed towards use in regenerative medicine.
Figure 3.
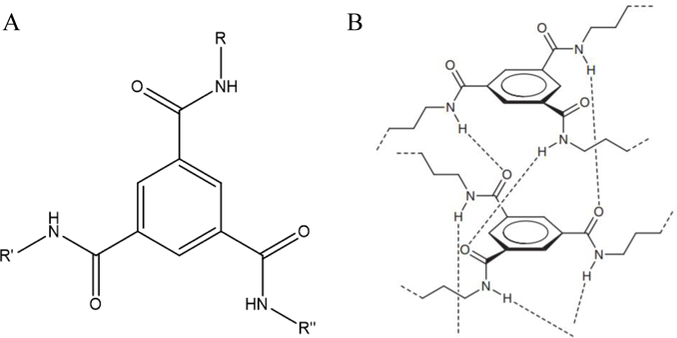
a) Structure of BTA1. R,R’R’’ are alkyl chains/other substituents. b) Three-fold hydrogen intermolecular bonding and helical assembly of BTA.
3B: Adapted with permission from P.J.M. Stals, J.F. Haveman, A.R.A. Palmans, A.P.H.J. Schenning, J. Chem. Educ. 2009, 86, 230. Copyright 2009, American Chemical Society.
2.2.3. Other Hydrogen Bonding Motifs
Poly(vinyl alcohol) may also form pH-sensitive self-healing hydrogels based on hydrogen bonding and, to a lesser degree, hydrophobic interactions.[77] These materials do not retain their mechanical properties upon re-integration, but hold promise for drug delivery systems for cancer therapy, and require further investigation.[78] While non-UPy hydrogen bonding moieties remain largely un-utilized in tissue engineering applications, they offer alternative routes to formation of self-healing hydrogels, and the novel chemistries described here hold promise for advancing the field in new directions.
2.3. Macrocyclic Host-Guest Interactions and Inclusion Complexes
One of the most studied non-covalent methods for formation of supramolecular hydrogels is host-guest interactions based on macrocyclic compounds such as cyclodextrins (CDs), cucurbit[n]urils, crown ethers, catenanes, and cyclophanes. This review will focus on self-healing supramolecular hydrogels based on CDs and their derivatives. A comprehensive review of supramolecular host-guest systems for biological applications can be found in the literature.[79,80]
2.3.1. Cyclodextrin Derivatives
CDs are a family of oligosaccharides first discovered in the late 19th century and have a long history of use in biological applications due to their superior biocompatibility, unique inclusion capability and ease of functionalization.[81–83] They have a truncated 3D cone shape with a hollow, tapered inner cavity and a fixed length (0.79nm).[84] The width of CDs may be increased with the number of glucose repeat units, connected by α−1,4 glycosidic linkages (Figure 4).[80] Since their discovery, α-, β-, and γ-CDs, composed of 6, 7, and 8 glucose units respectively, have emerged as the most commonly used members of the CD family.[80]
Figure 4.
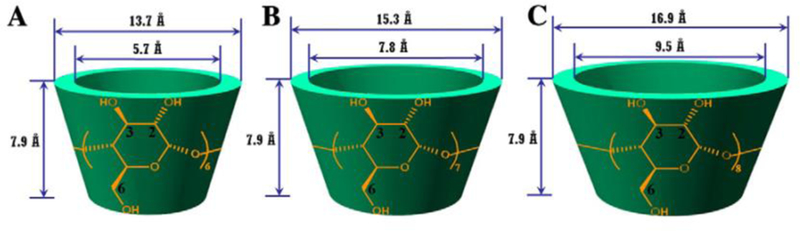
Molecular structures and dimensions of various CDs: A, α-CD; B, β-CD; and C, γ-CD.
Adapted with permission under the terms of 4440310602787 License. [64] 2013, Elsevier B.V.
Members of the CD family have been successfully used as building blocks for self-healing hydrogels capable of stimuli-responsive self-assembly based on host-guest interactions between CDs and polymers.[84] Most often, poly(ethylene oxides) or block copolymers are used to further strengthen the hydrogels with hydrophobic segments that experience intermolecular hydrophobic interactions.[85,86] These supramolecular hydrogels have also been incorporated with covalent bonding to form materials systems with unique chemistry not seen in other supramolecular systems. [87] High molecular weight PEG was first reported to be able to form inclusion complexes with α-CD to assemble into supramolecular hydrogels.[88] Since then, the method of threading CDs onto linear hydrophilic polymers to form a hydrogel has been used to create supramolecular hydrogels (Figure 5a). [89–91] The dominant driving force in most host-guest systems are hydrophobic interactions that combat competitive water to drive self-assembly. Aside from inclusion complex formation, supramolecular hydrogels have been formed through co-assembling hydrophobically-modified polymers with polymers containing pendant CDs (Figure 5b). [85,92] Another promising strategy is the preparation of supramolecular ‘monomers’ with host-guest functionality and subsequent polymerization using various techniques. Host- and guest-containing CD monomers were polymerized via radical polymerization to form poly(acrylamide) chains which then formed inclusion complexes with CDs from nearby polymer chains to assemble a self-healing hydrogel network. [93] Although the crosslinking conditions are mild, and it exhibits an unusual ability to retain its mechanical integrity upon complete rejoining, acrylamide monomers are highly cytotoxic [94], so the utility of the hydrogel may be limited. More recently, a modified β-CD host molecule was developed that forms an inclusion complex to make three-arm host-guest supramolecules.[95] This ‘monomer’ unit was subsequently polymerized using UV light, creating a non-cytotoxic self-healing hydrogel due to reversible self-integration of the free supramolecular host-guest units.[95] These hydrogels have been tested in soft tissue engineering but have yet to be translated to in vivo studies. In another promising application towards soft tissue engineering, a hyaluronic acid-adamantane and β-CD hydrogel construct was tested with secondary autonomous covalent crosslinking for regeneration of myocardial tissue post infarct.[96] After injection, this material acted as a support scaffold after injury and significantly decreased the infarct size while increasing cardiac function in an in vivo mouse model.
Figure 5.
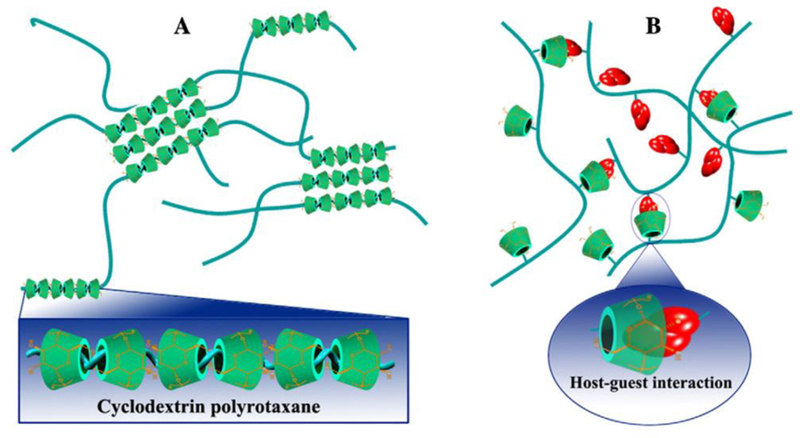
Two different approaches for utilizing host-guest interactions to form supramolecular hydrogels. a) Fabrication of hydrogels based on inclusion complexes between CDs and various polymers that can thread into CD cavities. b) Supramolecular hydrogels formed by physical cross-linking through host–guest interactions between CD-containing polymers and hydrophobically modified polymers.
Adapted with permission under the terms of 4440310602787 License[64] 2013, Elsevier B.V.
CD-containing supramolecular hydrogels are promising alternatives to covalently crosslinked hydrogels as injectable materials for drug and gene delivery. Many hydrogels that require covalent crosslinking for gelation are limited in application as they are not injectable, and it is time-consuming and inefficient to incorporate drugs by sorption, limiting the loading. [97] Additionally, it can require delicate bioactive agents such as drugs, proteins, RNAs, and DNAs be exposed to organic solvents which may alter their chemical integrity. [97] Therefore, the development of a system capable of simultaneous gelation and drug loading in aqueous environment without covalent crosslinking is highly desired. One such system uses supramolecular hydrogel formed by CD-polymer inclusion complexes[98] has been developed as an injectable drug delivery device, but work remains to be done towards its in vivo usage.
Given the biocompatibility, shear-thinning, and self-healing nature of CD-based hydrogels, it is logical to extend their use towards 3D bioprinting. 3D bioprinting combines cells with a shear-thinning and self-recovering hydrogel to mimic native tissue characteristics and has received increased attention in recent years. A host-guest hydrogel using hyaluronate modified with adamantane and β-CD was developed that self-assembles into a shear-thinning hydrogel.[99] To increase its long-term stability and profusion, this hydrogel may be designed to undergo a secondary covalent UV crosslinking while maintaining its injectability. This system is an exciting step towards 3D bioprinting for tissue regeneration, as it was shown to have high (>90%) cell viability and the incorporation of cells did not disrupt or alter the printing process besides necessitating sterility. In the coming years, the use of self-healing supramolecular hydrogels will continue to develop and further bioprinting for tissue engineering.
2.3.2. Stimuli-Responsive Cyclodextrin Derivatives
In addition to autonomous assembly via inclusion complex formation, CDs can be utilized in various self-healing stimuli-responsive applications including thermo-responsive, redox and photo-switching, leading to interesting properties such as shape-memory[100] hydrogels and precise spatiotemporal control over size and stiffness of the hydrogel.[101] Stimuli-responsive CDs are useful in the construction of tissue engineering complexes (scaffold, cells, and/or signaling molecules) with various applications including engineering artificial muscle[102], cardiac tissue regeneration[96], anticancer drug delivery[103,104], and delivery of biologics.[105] These functionalities have been used towards development of ‘smart’ hydrogels that respond to their environment, enabling better drug delivery and tissue mimicry. Ferrocene (FC) and its derivatives are widely used as redox-responsive molecules in supramolecular chemistry applications, adding stimuli-responsiveness while preserving the bulk properties.[106] For example, a self-healing supramolecular hydrogel where poly(acrylic acid) (PAA) modified with β-CD and a PAA-FC guest polymer undergoes redox-responsive self-healing based on formation of inclusion complexes has been reported.[102] This work is promising for future applications in stimuli-responsive drug delivery to tissues with a low local pH such as tumors, although cell toxicity and drug encapsulation have not been verified. Separately, a non-cytotoxic hydrogel system that is dually electrochemical and redox responsive was synthesized via formation of host–guest interactions between β-CD and FC and exhibits rapid self-healing capabilities.[107] This work is promising, but may be improved by incorporating controlled drug or biologic delivery systems, as is often required for effective tissue regeneration. Inclusion complexes formed by trans-azobenzene and β-CD as a photo-switchable crosslinker have been used to create a dextran based hydrogel system for light controlled release of DNA and proteins.[105] Azobenzene is a photo-sensitive moiety that enables reversible sol-gel transitions, and although it remains largely unutilized in tissue engineering applications, it was shown in this work to be part of a system that was not harmful to biologics.[108,109] While UV irradiation may be harmful to tissues, the tight spatiotemporal control achieved makes it a promising stimulus for crosslinking in tissue engineering applications.[110] Given their high modularity, CDs are excellent candidates for development of highly stimuli-responsive hydrogels for tissue engineering.
2.4. Hydrophobic Interactions
Hydrophobic interactions are one of the most important driving forces in the supramolecular assembly of tissue engineering complexes.[111] They can form physical crosslinks between polymer chains, which are highly important in self-assembly of biological materials such as fibrillar proteins, or cause the association and subsequent self-assembly of supramolecular polymer fibers.[112] Hydrophobic interactions are driven by the entropic gain that follows from burying the hydrophobic faces of molecules away from the aqueous environment of the swollen hydrogel bulk and the subsequent release of surface-bound water, a large net entropic gain with a small enthalpy penalty.[113] This type of physical crosslinking can be combined with hydrogen bonding as in UPy and BTA where it acts as a protective pocket for the hydrogen bonds, and host-guest interactions. It is often combined with chemical crosslinking to enhance the mechanical properties of the resulting gels, and unlike other supramolecular motifs, can be utilized to generate tough, self-healing hydrogels.[114,115]
2.4.1. Peptides and Peptide Amphiphiles
Due to their inherent bioactivity and biocompatibility, peptides are excellent choices for use in biodegradable supramolecular hydrogel building blocks.[28] Peptide amphiphiles with hydrophilic peptide segments covalently bonded to long hydrophobic polymer tails have been widely used to construct self-assembling, bioactive supramolecular materials.[116,117] Peptide amphiphiles (PA), consisting of a peptide(s) covalently bonded to a carefully designed synthetic polymer, have been developed into self-assembling hydrogels.[118] Supramolecular fibers formed from self-assembly of PAs have been shown to allow bioactive cues such as the fibronectin-derived RGD peptide to freely move along the backbone of the fiber to attain optimal and dynamic RGD spacing, a benefit over covalent polymers, where the spacing and concentration of RGD must be strictly controlled. [119] Due to the biodegradability and non-toxic nature of PA hydrogels, they have been extensively studied for 3D cell culture. For example, di-block co-polypeptide amphiphiles that self-assemble into rapidly self-healing, degradable, and functional hydrogels have been synthesized that have potential in biomedical applications.[120] Once injected, these supramolecular hydrogels aid in establishing a preferential environment for therapeutic cells, and can direct them towards the desired lineage. For example, the self-assembly of amyloid fibers was utilized to develop a tunable, self-healing, thermo-responsive hydrogel that promoted cell attachment, spreading, and differentiation towards a neuronal lineage.[121] This gel is non-toxic in various cell lines, an important step towards utilization in vivo. Additionally, both in vitro and in vivo differentiation of mesenchymal stem cells into chondrocytes without the use of exogenous growth factors was reported in hyaluronate and heparin mimicking PA hydrogels. [122]
Work has been done to improve the encapsulation of hydrophobic drugs in PA-based materials for drug delivery purposes. To this end, aliphatic chains have been added to peptide nanofibers to enhance the solubility of hydrophobic drugs even in the aqueous environment, allowing their use as efficient drug delivery vehicles, however these self-assembling nanofibers were not able to form hydrogels.[123] Later work developed a smart peptide-based logic-gate hydrogel that can sense various biological disease markers and can be programmed for controlled drug release, answering both the need for hydrophobic drug delivery and stimuli-responsive injectable hydrogel scaffolds.[124] Although these works represent important steps towards clinical application of PA hydrogels, the many advances made with PA nanofibers have yet to be incorporated into hydrogels for tissue engineering.
The bioactive properties of self-healing peptide-based hydrogels have also been used to good effect in the regeneration of hard tissues including bone,[125] cartilage,[126] and dental tissue[127], which represents a deviation from traditional approaches to engineering of hard tissues. Self-assembling PAs have been used for dual delivery of dental stem cells and growth factors to regenerate dentin.[118,127] Upon inclusion of a MMP-cleavable linker within the peptide sequence, enhanced viability, spreading, and migration of human mesenchymal stem cells was observed.[128] This material lacks the ability to completely self-heal, as the MMP-cleavable linkers are permanently cut during cell invasion,[118,127] however, PAs with repeated self-healing abilities have also been explored for regeneration of nerves[129] and angiogenesis.[130]
In addition to bone and dental engineering, recent advances have shown the potential for self-healing peptide-based supramolecular hydrogels to improve outcome in cardiovascular disease models by both improving angiogenesis and regenerating or attenuating damage to cardiac tissue following injury. These hydrogels represent a promising low-dose delivery system for growth factors given their many available binding sites and biocompatibility. [29] These materials have also been used for their advantageous mechanical properties and as delivery devices for cells to contribute to modest regeneration of functional cardiovascular tissue. [131,132] While they exhibit many advantageous properties, peptide-based systems also have drawbacks including their high costs, potential difficulty with scale-up, and less-controllable enzymatic degradation.[133]
2.4.2. Amphiphilic Block Copolymers
Although PAs are biocompatible and bioactive, due to the expense of large scale peptide production, it became necessary to develop cost-effective, scalable systems. Amphiphilic block copolymers that self-assemble based on hydrophobic interactions[134] have been utilized for drug delivery[135,136], cell-encapsulation[137], cartilage regeneration[122], and 3D bioprinting.[138] Recently, an injectable star PEG-b-poly (propylene sulfide) scaffold for wound repair was reported.[139] These hydrogel matrices self-assemble in aqueous media and effectively dissolve hydrophobic molecules, enabling local delivery of therapeutic doses of small molecules. Ultimately, these scaffolds improved cellular infiltration, reduced inflammation, and promoted wound closure in vivo, representing one of the few systems whose suitability for clinical translation has been tested.
2.5. Electrostatic (Ionic) Interactions
Ionically cross-linked hydrogels are biocompatible and self-healing based on the mild conditions and reversibility of their gelation, which is due to the electrostatic interaction between oppositely charged ions within the constituent materials.[140] Alginate, a naturally occurring, anionic, and hydrophilic polymer forms a crosslink in the presence of cations, most typically Ca2+, and is the most well-studied ionically cross-linked polymer for tissue engineering.[141–144] Initial hurdles to its use, including control of gelation rate, and lack of uniform structure have been addressed.[140,145] A comprehensive review of ionically cross-linked alginate hydrogels and their applications to biomedical engineering can be found in the literature.[141]
Various groups have introduced ionically cross-linked hydrogels with increased mechanical strength and toughness, addressing a known drawback of these types of hydrogels.[146,147] Recently, hyaluronate-based hydrogels have been explored as scaffolds for injectable cell delivery, an important area of development in tissue engineering.[148] Other materials such as PEG and clay have also been used to prepare ionically crosslinked hydrogels with unique properties and potential applications in tissue engineering, however, it should be noted that most ionically crosslinked hydrogels are not inherently self-healing and often require many modifications to achieve this property.[149,150]
2.6. Metal Coordination
Metal-ligand coordination plays a critical role in adhesion, self-assembly, and toughness in many biological materials.[151] Metal–ligand complexation has been widely utilized in the formation of supramolecular self-healing hydrogels owing to their high stability, rapid self-assembly, relatively high bonding energies, and the ease of incorporating biocompatible polymers.[152–154] A supramolecular metal-ligand complex is formed between a transition metal ion and the electron-donating organic ligand to enable a rapidly reversible and spontaneous linkage, the stability of which varies based on metal-ion concentration and pH.[152,155] With careful selection of the metal-ligand pair, supramolecular hydrogels with many advantageous near-covalent mechanical properties may be developed.[20] Researchers have sought to mimic the viscoelasticity and the partial recovery of the Young’s Modulus that leads to the acellular self-healing abilities displayed by mussel byssal threads.[153] The histidine-metal coordination acted as a sacrificial bond to dissipate energy and reformed upon removal of the applied load. Apart from histidine, the pH-induced Fe3+-catechol crosslinking pair has been used to generate self-healing supramolecular hydrogels that achieve near-covalent elastic moduli.[156] This work establishes a method for decoration of hydrophilic polymers with metal-coordinating ligands but does not offer insight into their suitability for biomedical applications. Subsequent work in the area resulted in histidine-based metal coordination hydrogels made with PEG which establishes a robust self-healing biomaterial with mechanical properties comparable to native soft tissues.[153] These monovalent systems were not designed for tissue engineering, and as such their biocompatibility has not been fully evaluated and their stability in vivo is likely low, making them less useful for longer-term applications.[157] Recently, an injectable hydrogel that uses reversible poly-histidine-Ni coordination bonds has been used to make a multivalent biomaterial stable enough for use in sustained drug delivery systems but its in vivo suitability is yet to be proven.[158]
Other ligands for metal coordination hydrogels have been developed including terpyridine, a pyridine derivative commonly used to form directed and stable but reversible complexes with a variety of metal ions in biocompatible hydrogels, however, these remain largely unproven in tissue engineering systems.[159] Although not intended for tissue engineering, a conductive, self-healing hybrid hydrogel has been reported which has potential for prostheses given its conductivity, enhanced mechanical properties, and elasticity.[160] Metal-ligand coordination-based hydrogels offer unique materials with potential to achieve high elastic moduli while maintaining their self-healing properties and represent an interesting class of biomaterials for tissue engineering.
3. Conclusions and Outlook
Recent innovations in supramolecular chemistry and biology, including a more complete investigation of ECM properties on cell fate and function, scaffold effects, and growth factors, have been incorporated into supramolecular hydrogel design to develop novel biomaterials with self-healing capabilities that are very useful in tissue engineering. These materials have been explored for 3D culture of various cell types as well as regeneration of dental and bone tissue in addition to soft tissue engineering including dental pulp, cartilage, and cardiovascular tissues.
Looking forward, major advances in self-healing supramolecular hydrogels will come from the continued incorporation of novel chemistries into biocompatible hydrogels that can advantageously recapitulate native tissue architecture and offer controlled delivery of cells and signaling cues. Specifically, supramolecular ‘monomer’ units will continue to be developed that direct spatiotemporal control of drugs and biologics. Structural, mechanical, and dynamic behavior is a hallmark of native tissue, and one of the most difficult aspects to mimic given the large range of cell types and tissue systems. One could envision that future ‘smart’ materials would be able to not only react to stimuli but respond and adapt to mechanical and biological changes in the scaffold, controlling release of drugs and biologics in advantageous spatiotemporal patterns over a controlled timeframe. In addition to materials chemistry, cell adhesion is critical to cell infiltration and therefore to tissue integration, a goal for many tissue engineering constructs.
The mechanical properties of these soft hydrogels may need to be improved for applications that require stiffer scaffolds, a slower rate of erosion and clearance, repeated self-healing or a combination thereof. This may be achieved by the advent of biomaterials that repeatedly self-heal under physiological conditions, allowing sustained release of drugs and biologics as well as the retention of bulk properties.
Many currently available supramolecular hydrogels, although they have been extensively characterized, have yet to be tested in vivo, and in some cases, have not yet been evaluated in vitro, meaning their suitability for clinical application is to be determined. The development of future hydrogel biomaterials for tissue engineering purposes should include studies on biocompatibility and host integration, including potential immune response, to enable regeneration or complete replacement of lost or damaged tissues, eventually leading to improved patient outcomes and quality of life.
Acknowledgements:
The authors gratefully acknowledge financial support from the US National Institutes of Health (NIDCR R01 DE022327 and NHLBI R01 HL136231: P.X.M.).
Author Photograph(s)

A. Peter X Ma is the Richard H Kingery Endowed Collegiate Professor of Biologic and Materials Sciences at the University of Michigan. His research is in the areas of biomaterials, biomedical polymers, controlled release, tissue engineering, and regenerative medicine.
Dr. Ma received a Whitaker Foundation Biomedical Engineering Young Investigator Award and DuPont Young Professor Award. He was named one of the Top 100 materials scientists in the world by Thomson Reuters. He is an elected Fellow of: the American Institute for Medical and Biological Engineering, Biomaterials Science and Engineering, Materials Research Society and American Association for the Advancement of Science.

B. Laura Saunders obtained a BS in Chemical Engineering from the University at Buffalo in 2016 and is currently pursuing her PhD in Macromolecular Science and Engineering at the University of Michigan. Her research project focuses on development of targeted polymeric vectors for nucleotide delivery for cardiac regeneration and hydrogel-based injectable drug delivery systems.
Contributor Information
Laura Saunders, Prof., Macromolecular Science and Engineering Center, University of Michigan, Ann Arbor, MI, USA
Peter X. Ma, Prof., Macromolecular Science and Engineering Center, University of Michigan, Ann Arbor, MI, USA, Biologic and Materials Science, University of Michigan, Ann Arbor, MI, USA, Biomedical Engineering, University of Michigan, Ann Arbor, MI, USA, Materials Science and Engineering, University of Michigan, Ann Arbor, MI, USA, mapx@umich.edu
References
- [1].Vacanti JP, Langer R, Lancet 1999, 354 (suppl I): 32–34. [DOI] [PubMed] [Google Scholar]
- [2].Peppas NA, Hilt JZ, Khademhosseini A, Langer R, Adv. Mater 2006, 18, 1345–1360. [Google Scholar]
- [3].Caló E, Khutoryanskiy VV, Eur. Polym. J 2015, 65, 252–267. [Google Scholar]
- [4].Wichterle O, Lím D, Nature 1960, 185, 117–118. [Google Scholar]
- [5].Lee KY, Mooney DJ, Chem. Rev, 2001, 101 (7), 1869–1880. [DOI] [PubMed] [Google Scholar]
- [6].Telemeco TA, Ayres C, Bowlin GL, Wnek GE, Boland ED, Cohen N, Baumgarten CM, Mathews J, Simpson DG, Acta Biomater. I 2005, doi: 10.1016/j.actbio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- [7].Wojtecki RJ, Meador MA, Rowan SJ, Nat. Mater 2011, 10, 14–27. [DOI] [PubMed] [Google Scholar]
- [8].Wang H, Heilshorn SC, Adv. Mater 2015, 27, 3717–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rosales AM, Anseth KS, Nat. Rev. Mater 2016, 1, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Adzima BJ, Kloxin CJ, Bowman CN, Adv. Mater 2010, 22, 2784–2787. [DOI] [PubMed] [Google Scholar]
- [11].Chen X, Dam MA, Ono K, Mal A, Shen H, Nutt SR, Sheran K, Wudl F Science 2002. 295, 1698–1702. [DOI] [PubMed] [Google Scholar]
- [12].Wu J, Su ZG, Ma GH, Int. J. Pharm 2006, 315, 1–11. [DOI] [PubMed] [Google Scholar]
- [13].Zhang R, Tang M, Bowyer A, Eisenthal R, Hubble J, Biomaterials 2005, 26, 4677–4683. [DOI] [PubMed] [Google Scholar]
- [14].Kloxin AM, Tibbitt MW, Anseth KS, Nat. Protoc 2010, 5, 1867–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fairbanks BD, Singh SP, Bowman CN, Anseth KS, Macromolecules 2011, 44 2444–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Angeles L, Boulder C, Science 2009, 324, 59–64.19342581 [Google Scholar]
- [17].Scott TF, Schneider AD, Cook WD, Bowman CN, Science 2005, 308, 1615–1617. [DOI] [PubMed] [Google Scholar]
- [18].Rowan SJ, Cantrill SJ, Cousins GRL, Sanders JKM, Stoddart JF, Angew. Chem. Int. Ed 2002, 41, 898–952. [DOI] [PubMed] [Google Scholar]
- [19].Qu J, Zhao X, Ma PX, Guo B, Acta Biomater 2017), 58, 168–180. [DOI] [PubMed] [Google Scholar]
- [20].Appel EA, del Barrio J, Loh XJ, Scherman OA, Chem. Soc. Rev 2012, 41, 6195. [DOI] [PubMed] [Google Scholar]
- [21].Lutolf MP, Nat. Publ. Gr 2009, 8, 451–453. [DOI] [PubMed] [Google Scholar]
- [22].Jin Y, Yu C, Denman RJ, Zhang W, Chem. Soc. Rev 2013, 42, 6634–6654. [DOI] [PubMed] [Google Scholar]
- [23].Boehnke N, Cam C, Bat E, Segura T, Maynard HD, Biomacromolecules 2015, 16, 2101–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McKinnon DD, Domaille DW, Cha JN, Anseth KS Adv. Mat 2014, 26 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hozumi T, Kageyama T, Ohta S, Fukuda J, Ito T, Biomacromolecules 2018, 19, 288–297. [DOI] [PubMed] [Google Scholar]
- [26].Shi L, Carstensen H, Holzl K, Lunzer M, Li H, Hilborn J, Ovsianikov A, Ossipov DA, Chem. Mater 2017, 29, 5816–5823. [Google Scholar]
- [27].Wang LL, Highley CB, Yeh YC, Galarraga JH, Uman S, Burdick JA, J. Biomed. Mater. Res. Part A 2018, 106, 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dong R, Pang Y, Su Y, Zhu X, Biomater. Sci 2015, 3, 937–954. [DOI] [PubMed] [Google Scholar]
- [29].Webber MJ, Appel EA, Meijer EW, Langer R, Nat. Mater 2015, 15, 13–26. [DOI] [PubMed] [Google Scholar]
- [30].Seiffert S, Sprakel J, Chem. Soc. Rev 2012, 41, 909–930. [DOI] [PubMed] [Google Scholar]
- [31].Sijbesma RP, Beijer FH, Brunsveld L, Folmer BJB, Hirschberg JHKK, Science 1997, 278, 1601–1604. [DOI] [PubMed] [Google Scholar]
- [32].Lehn J‐M, Angew. Chemie Int. Ed. English 1988, 27, 89–112. [Google Scholar]
- [33].Aida T, Meijer EW, Stupp SI, Science 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dong R, Zhou Y, Huang X, Zhu X, Lu Y, Shen J, Adv. Mater 2015, 27, 498–526. [DOI] [PubMed] [Google Scholar]
- [35].Hou S, Ma PX, Chem. Mater 2015, 27, 7627–7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Langer R & Tirrell DA Nature 2004, 428, 487–492. [DOI] [PubMed] [Google Scholar]
- [37].Caliari SR, Burdick JA, Nat. Methods 2016, 13, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chan G, Mooney DJ, Trends Biotechnol 2008, doi: 10.1016/j.tibtech.2008.03.011. [DOI] [PubMed] [Google Scholar]
- [39].Hoffman BD, Grashoff C, Schwartz MA, Nature 2011. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677–689. [DOI] [PubMed] [Google Scholar]
- [41].Ma PX, Adv. Drug Deliv. Rev 2008, 60, 184–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ma PX, Mater. Today 2004, 7, 30–40. [Google Scholar]
- [43].Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA, Proc. Natl. Acad. Sci 2003, 100, 5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zisch AH, Lutolf MP, Hubbell JA, Cardiovasc. Pathol 2003, 12, 295–310. [DOI] [PubMed] [Google Scholar]
- [45].Chung C, Beecham M, Mauck RL, Burdick JA, Biomaterials 2009, 30, 4287–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Phadke A, Zhang C, Arman B, Hsu C-C, Mashelkar RA, Lele AK, Tauber MJ, Arya G, Varghese S, Proc. Natl. Acad. Sci 2012, 109, 4383–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dong R, Zhao X, Guo B, Ma PX, ACS Appl. Mater. Interfaces 2016, 8, 17138–17150. [DOI] [PubMed] [Google Scholar]
- [48].Hou S, Wang X, Park S, Jin X, Ma PX, Adv. Healthc. Mater 2015, 4, 1491–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].White SR, Blaiszik BJ, Kramer SLB, Olugebefola SC, Moore JS, Sottos NR, Am. Sci 2011, 99, 392–399. [Google Scholar]
- [50].Burdick JA, Anseth KS, Biomaterials 2002, 23, 4315–4323. [DOI] [PubMed] [Google Scholar]
- [51].Wang Y, Adokoh CK, Narain R, Expert Opin. Drug Deliv 2018, 15, 77–91. [DOI] [PubMed] [Google Scholar]
- [52].Roh YH, Ruiz RCH, Peng S, Lee JB, Luo D, Chem. Soc. Rev 2011, 40, 5730–5744. [DOI] [PubMed] [Google Scholar]
- [53].Hirschberg JHKK, Brunsveid L, Ramzi A, Vekemans JAJM, Sijbesma RP, Meijer EW, Nature 2000, 407, 167–170. [DOI] [PubMed] [Google Scholar]
- [54].Keten S, Xu Z, Ihle B, Buehler MJ, Nat. Mater 2010, 9, 359–367. doi: 10.1038/nmat2704. [DOI] [PubMed] [Google Scholar]
- [55].Dobb MG, Johnson DJ, Saville BP, J Polym Sci Polym Phys Ed 1977, 15, 2201–2211. [Google Scholar]
- [56].Beijer FH, Sijbesma RP, Kooijman H, Spek AL, Meijer EW, J. Am. Chem. Soc 1998, 120, 6761–6769. doi: 10.1021/ja974112a. [DOI] [Google Scholar]
- [57].Beijer FH, Kooijman H, Spek AL, Sijbesma RP, Meijer EW, Angew. Chemie Int. Ed 1998, 37, 75–78. [Google Scholar]
- [58].Goor OJGM, Hendrikse SIS, Dankers PYW, Meijer EW, Chem. Soc. Rev 2017, 46, 6621–6637. [DOI] [PubMed] [Google Scholar]
- [59].Muylaert DEP, van Almen GC, Talacua H, Fledderus JO, Kluin J, Hendrikse SIS, van Dongen JLJ, Sijbesma E, Bosman AW, Mes T, Thakkar SH, Smits AIPM, Bouten CVC, Dankers PYW, Verhaar MC, Biomaterials 2016, 76, 187–195. [DOI] [PubMed] [Google Scholar]
- [60].Wong CTS, Foo P, Seok J, Mulyasasmita W, Parisi-amon A, Heilshorn SC, Proc. Natl. Acad. Sci 2009, 106, 22067–22072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dankers PYW, Hermans TM, Baughman TW, Kamikawa Y, Kieltyka RE, Bastings MMC, Janssen HM, Sommerdijk NAJM, Larsen A, Van Luyn MJA, Bosman AW, Popa ER, Fytas G, Meijer EW, Adv. Mater 2012, 24, 2703–2709. [DOI] [PubMed] [Google Scholar]
- [62].Lange RFM, van Gurp M, Meijer EW, J. Polym. Sci., Part A-1: Polym. Chem 1999, 37, 3657–3670. [Google Scholar]
- [63].Ligthart GBWL, Ohkawa H, Sijbesma RP, Meijer EW, J. Am. Chem. Soc 2005, 127, 810–811. [DOI] [PubMed] [Google Scholar]
- [64].Dankers PYW, Van Luyn MJA, Huizinga-Van Der Vlag A, Petersen AH, Koerts JA, Bosman AW, Popa ER, Eur. Polym. J 2015, 72, 484–493. [Google Scholar]
- [65].Bastings MMC, Koudstaal S, Kieltyka RE, Nakano Y, Pape ACH, Feyen DAM, van Slochteren FJ, Doevendans PA, Sluijter JPG, Meijer EW, Chamuleau SAJ, Dankers PYW, Adv. Healthc. Mater 2014, 3, 70–78. [DOI] [PubMed] [Google Scholar]
- [66].Koudstaal S, Bastings MMC, Feyen DAM, Waring CD, Van Slochteren FJ, Dankers PYW, Torella D, Sluijter JPG, Nadal-Ginard B, Doevendans PA, Ellison GM, Chamuleau SAJ, J. Cardiovasc. Transl. Res 2014, 7, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chirila TV, Lee HH, Oddon M, Nieuwenhuizen MML, Blakey I, Nicholson TM, J. Appl. Polym. Sci 2014, 131, 1–12. [Google Scholar]
- [68].Bakker MH, Dankers PYW, Elsevier Ltd, 2018. doi: 10.1016/B978-0-08-102015-9.00009-5. [DOI] [Google Scholar]
- [69].Leenders CMA, Mes T, Baker MB, Koenigs MME, Besenius P, Palmans ARA, Meijer EW, Mater. Horizons 2014, 1, 116–120. [Google Scholar]
- [70].Leenders CMA, Albertazzi L, Mes T, Koenigs MME, Palmans ARA, Meijer EW, Chem. Commun 2013, 49, 1963–1965. [DOI] [PubMed] [Google Scholar]
- [71].Stals PJM, Haveman JF, Palmans ARA, Schenning APHJ, J. Chem. Educ 2009, 86, 230. [Google Scholar]
- [72].Lou X, Lafleur RPM, Leenders CMA, Schoenmakers SMC, Matsumoto NM, Baker MB, Van Dongen JLJ, Palmans ARA, Meijer EW, Nat. Commun 2017, 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wijnands SPW, Engelen W, Lafleur RPM, Meijer EW, Merkx M, Nat. Commun 2018, doi: 10.1038/s41467-017-02559-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Albertazzi L, Van Der Hofstad RW, Meijer EW, Science 2014, 491, 10–15. [DOI] [PubMed] [Google Scholar]
- [75].Baker MB, Gosens RPJ, Albertazzi L, Matsumoto NM, Palmans ARA, Meijer EW, ChemBioChem 2016, 17, 207–213. [DOI] [PubMed] [Google Scholar]
- [76].Bakker MH, Lee CC, Meijer EW, Dankers PYW, Albertazzi L, ACS Nano 2016, 10, 1845–1852. [DOI] [PubMed] [Google Scholar]
- [77].Wei Z, Yang JH, Zhou J, Xu F, Zrínyi M, Dussault PH, Osada Y, Chen YM, Chem. Soc. Rev 2014, 43, 8114–8131. [DOI] [PubMed] [Google Scholar]
- [78].Chang G, Chen Y, Li Y, Li S, Huang F, Shen Y, Xie A, Carbohydr. Polym 2015, 122, 336–342. [DOI] [PubMed] [Google Scholar]
- [79].Ma X, Zhao Y, Chem. Rev 2015, 115, 7794–7839. [DOI] [PubMed] [Google Scholar]
- [80].Zhang J, Ma PX, Adv. Drug Deliv. Rev 2013, 65, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Davis ME, Brewster ME, Nat. Rev. Drug Discov 2004, 3, 1023–1035. [DOI] [PubMed] [Google Scholar]
- [82].Villiers A, 1891. doi: 10.1016/S0360-0564(08)60302-8. [DOI] [Google Scholar]
- [83].Schardinger F, 1903, doi: 10.1007/BF02067497. [DOI] [Google Scholar]
- [84].Challa R, Ahuja A, Ali J, Khar RK, AAPS PharmSciTech 2005, 6, E329–E357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yuen F, Tam KC, Soft Matter 2010, 6, 4613–4630. [Google Scholar]
- [86].Deng W, Yamaguchi H, Takashima Y, Harada A, Angew. Chem., Int. Ed 2007, 46, 5144–5147. [DOI] [PubMed] [Google Scholar]
- [87].Rodell CB, Dusaj NN, Highley CB, Burdick JA, Adv. Mater 2016. doi: 10.1002/adma.201602268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Li J, Harada A, Kamachi M, Polym. J 1994, 26, 1019–1026. [Google Scholar]
- [89].Wenz G, Keller B, Angew. Chem., Int. Ed. Engl 1992, 31, 197–199. [Google Scholar]
- [90].Miyauchi M, Kawaguchi Y, Harada A, J. Incl. Phenom 2004, 50, 57–62. [Google Scholar]
- [91].Li J, NPG Asia Mater 2010, 2, 112–118. [Google Scholar]
- [92].Li J, Jun X, Adv. Drug Delivery Rev 2013, 25, 2849–2853. [Google Scholar]
- [93].Kakuta T, Takashima Y, Nakahata M, Otsubo M, Yamaguchi H, Harada A, Adv. Mater 2013, 25, 2849–2853. [DOI] [PubMed] [Google Scholar]
- [94].Kandow CE, Georges PC, Janmey PA, Beningo KA, Methods Cell Biol 2007, 83, 29–46. [DOI] [PubMed] [Google Scholar]
- [95].Wang Z, Ren Y, Zhu Y, Hao L, Chen Y, An G, Wu H, Shi X, Mao C, Angew. Chemie Int. Ed 2018. doi: 10.1002/anie.201804400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Rodell CB, J.W.M. Jr, Dorsey SM, Wade RJ, Wang LL, Woo YJ, Burdick JA, Adv. Funct. Mater 2015. doi: 10.1002/adfm.201403550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Song X, Li J, 2018, doi: 10.1007/978-3-662-57511-6. [DOI] [Google Scholar]
- [98].Khodaverdi E, Gharechahi M, Alibolandi M, Tekie FM, Khashyarmanesh B, Hadizadeh F, Int. J. Pharm. Investig 2016, 6, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Highley CB, Rodell CB, Burdick JA, Adv. Mater 2015, 27, 5075–5079. [DOI] [PubMed] [Google Scholar]
- [100].Miyamae K, Nakahata M, Takashima Y, Harada A, Angew. Chemie - Int. Ed 2015, 54, 8984–8987. [DOI] [PubMed] [Google Scholar]
- [101].Zheng Z, Hu J, Wang H, Huang J, Yu Y, Zhang Q, Cheng Y, ACS Appl. Mater. Interfaces 2017, 9, 24511–24517. [DOI] [PubMed] [Google Scholar]
- [102].Nakahata M, Takashima Y, Yamaguchi H, Harada A, Nat. Commun 2011, 2, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Song X, Wen Y, ling Zhu J, Zhao F, Zhang ZX, Li J, Biomacromolecules 2016, 17, 3957–3963. [DOI] [PubMed] [Google Scholar]
- [104].Mealy JE, Rodell CB, Burdick JA, J. Mater. Chem. B 2015, 3, 8010–8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Peng K, Tomatsu I, Kros A, Chem. Commun 2010, 46, 4094–4096. [DOI] [PubMed] [Google Scholar]
- [106].Harada A, Takashima Y, Nakahata M, Acc. Chem. Res 2014, 47, 2128–2140. [DOI] [PubMed] [Google Scholar]
- [107].Peng L, Zhang H, Feng A, Huo M, Wang Z, Hu J, Gao W, Yuan J, Polym. Chem 2015. 6, 3652–3659. [Google Scholar]
- [108].Chen H, Ma X, Wu S, Tian H, Angew. Chemie 126 (2014) 14373–14376. [Google Scholar]
- [109].Dong R, Liu Y, Zhou Y, Yan D, Zhu X, Polym. Chem 2011, 2, 2771–2774. [Google Scholar]
- [110].Yagai S, Kitamura A, Chem. Soc. Rev 2008, 37, 1520–1529. [DOI] [PubMed] [Google Scholar]
- [111].Whitesides GM, Grzybowski B, Science 2002, 295, 2418–2421. [DOI] [PubMed] [Google Scholar]
- [112].Lupas A, Methods Enzymol 1996, 266, 513–525. [DOI] [PubMed] [Google Scholar]
- [113].Smith GF, 2009, doi: 10.1016/S0079-6468(09)04801-2. [DOI] [Google Scholar]
- [114].Mihajlovic M, Staropoli M, Appavou MS, Wyss HM, Pyckhout-Hintzen W, Sijbesma RP, Macromolecules 2017, 50, 3333–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Tuncaboylu DC, Sari M, Oppermann W, Okay O, Macromolecules 2011, 44, 4997–5005. [Google Scholar]
- [116].Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA, Science 1998, 281, 389–392. [DOI] [PubMed] [Google Scholar]
- [117].Fichman G, Gazit E, Acta Biomater 2014, 10, 1671–1682. [DOI] [PubMed] [Google Scholar]
- [118].Galler KM, Cavender A, Yuwono V, Dong H, Shi S, Schmalz G, Hartgerink JD, D’Souza RN, Tissue Eng. Part A 2008, 14, 2051–2058. [DOI] [PubMed] [Google Scholar]
- [119].Huang J, Gräter SV, Corbellini F, Rinck S, Bock E, Kemkemer R, Kessler H, Ding J, Spatz JP, Nano Lett 2009, 9, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Deming TJ, Nature 2002, 417, 424–428. [DOI] [PubMed] [Google Scholar]
- [121].Jacob RS, Ghosh D, Singh PK, Basu SK, Jha NN, Das S, Sukul PK, Patil S, Sathaye S, Kumar A, Chowdhury A, Malik S, Sen S, Maji SK, Biomaterials 2015, 54, 97–105. [DOI] [PubMed] [Google Scholar]
- [122].Ustun Yaylaci S, Sardan Ekiz M, Arslan E, Can N, Kilic E, Ozkan H, Orujalipoor I, Ide S, Tekinay AB, Guler MO, Biomacromolecules 2016, 17, 679–689. [DOI] [PubMed] [Google Scholar]
- [123].Zhang P, Cheetham AG, Lin YA, Cui H, ACS Nano 2013, 7, 5965–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ikeda M, Tanida T, Yoshii T, Kurotani K, Onogi S, Urayama K, Hamachi I, Nat. Chem 2014, 6, 511–518. [DOI] [PubMed] [Google Scholar]
- [125].Lee SS, Hsu EL, Mendoza M, Ghodasra J, Nickoli MS, Ashtekar A, Polavarapu M, Babu J, Riaz RM, Nicolas JD, Nelson D, Hashmi SZ, Kaltz SR, Earhart JS, Merk BR, Mckee JS, Bairstow SF, Shah RN, Hsu WK, Stupp SI, Adv. Healthc. Mater 2015, 4, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Fernández-Colino A, Arias FJ, Alonso M, Carlos Rodríguez-Cabello J, Biomacromolecules 2014, 15, 3781–3793. [DOI] [PubMed] [Google Scholar]
- [127].Galler KM, Hartgerink JD, Cavender AC, Schmalz G, D’Souza RN,, Tissue Eng. Part A 2012, 18, 176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Galler KM, Aulisa L, Regan KR, D’Souza RN, Hartgerink JD, J. Am. Chem. Soc 2010, 132, 3217–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Black KA, Lin BF, Wonder EA, Desai SS, Chung EJ, Ulery BD, Katari RS, Tirrell MV, Tissue Eng. Part A 2015, 21, 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Hosseinkhani H, Hosseinkhani M, Khademhosseini A, Kobayashi H, Tabata Y, Biomaterials 2006, 27, 5836–5844. [DOI] [PubMed] [Google Scholar]
- [131].Hendriks J, Riesle J, van Blitterswijk CA, Tissue Eng J. Regen. Med 2010, 4, 524–531.. [Google Scholar]
- [132].Padin-Iruegas ME, Misao Y, Davis ME, Segers VFM, Esposito G, Tokunou T, Urbanek K, Hosoda T, Rota M, Anversa P, Leri A, Lee RT, Kajstura J, 2010. doi: 10.1161/CIRCULATIONAHA.109.852285.Cardiac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Kisiday J, Jin M, Kurz B, Hung H, Semino C, Zhang S, Grodzinsky AJ, Proc. Natl. Acad. Sci 2002, 99, 9996–10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Deming TJ, Soft Matter 2005, 1, 28–35. [DOI] [PubMed] [Google Scholar]
- [135].Li J, Li X, Ni X, Wang X, Li H, Leong KW, Biomaterials 2006, 27, 4132–4140. [DOI] [PubMed] [Google Scholar]
- [136].Jeong B, Bae YH, Lee DS, Kim SW, Nature 1997, 388, 860–862. [DOI] [PubMed] [Google Scholar]
- [137].Zhang J, Tokatlian T, Zhong J, Ng QKT, Patterson M, Lowry WE, Carmichael ST, Segura T, Adv. Mater 2011, 23, 5098–5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Murphy R, Walsh DP, Hamilton CA, Cryan SA, In Het Panhuis M, Heise A, Biomacromolecules 2018, 19, 2691–2699. [DOI] [PubMed] [Google Scholar]
- [139].Zhu S, Li S, Escuin-Ordinas H, Dimatteo R, Xi W, Ribas A, Segura T, J. Control Release 2018, 282, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kuo CK, Ma PX, Biomaterials 2001, 22, 511–521. [DOI] [PubMed] [Google Scholar]
- [141].Ma P, (Ed.)Elisseeff J(Ed.), Tabata Y, Tranquillo R, Anseth K, Mooney D, Zhang S, Bhatia S, Zhao X, Atala A, Obradovic B, Lee S, Alhadlaq A, Anusaksathien O, Barr J, Baxter B, Chen V, jin QM, Patel P, Tessmar J, Vunjak-Novakovic G, Zielinski B, Lohmann C, Huang YC, Tsang V, Carlsson D, Cui FZ, Kleinman T, Golenser J, Grassl E, Guo C, Holland T, Jiang X, Khan Y, Lavik E, Lee J, Lewis A, Li F, Mukherjee P, Ng S, Prisk V, Radisic M, Rowe S, Salem A, Shah D, Sheardown H, Shimmura S, Spirio L, Wei G, Williams C, Yeo Y, de Groot K, West J, Park K, Li S, Nerem R, Spector M, Griffith M, Patrick C Jr., Giannobile W, Randolph M, Mao J, Gill T, Chang T, Langer R, Leong K, Heller J, Hollinger J, Domb A, Colton C, Yannas I, Whitesides G, Mikos A, Laurencin C, Mooney M, Chu TM, Marra K, Huard J (2006). Scaffolding In Tissue Engineering Boca Raton: CRC Press. [Google Scholar]
- [142].Park H, Kang SW, Kim BS, Mooney DJ, Lee KY, Macromol. Biosci 2009, 9, 895–901. [DOI] [PubMed] [Google Scholar]
- [143].Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ, Biomaterials 2008, 29, 3671–3682. [DOI] [PubMed] [Google Scholar]
- [144].Zhao L, Weir MD, Xu HHK, Biomaterials 2010, 31, 6502–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Kuo CK, Ma PX, J. Biomed. Mater. Res. - Part A 2008, 84, 899–907. [DOI] [PubMed] [Google Scholar]
- [146].Ren Z, Zhang Y, Li Y, Xu B, Liu W, J. Mater. Chem. B 2015, 3, 6347–6354. [DOI] [PubMed] [Google Scholar]
- [147].Sun JY, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ, Suo Z, Nature 2012, 489, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Park H, Woo EK, Lee KY, J. Control. Release 2014, 196, 146–153. [DOI] [PubMed] [Google Scholar]
- [149].Han YL, Yang Y, Liu S, Wu J, Chen Y, Lu TJ, Xu F, Biofabrication 2013. doi: 10.1088/1758-5082/5/3/035004. [DOI] [PubMed] [Google Scholar]
- [150].Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T, Nature 2010, 463, 339–343. [DOI] [PubMed] [Google Scholar]
- [151].Haas KL, Franz KJ, Chem. Rev 2009, 109, 4921–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Atwood JL, Steed JW, Supramolecular Chemistry, Wiley Ltd 2000, 2, 41–44. [Google Scholar]
- [153].Fullenkamp DE, He L, Barrett DG, Burghardt WR, Messersmith PB, Macromolecules 2013, 46, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Varghese S, Lele A, Mashelkar R, J. Polym. Sci. Part A 2006, 44, 666–670. [Google Scholar]
- [155].Hogg R, Wilkins RG, Int. Conf. Co-Ord. Chem. Detroit, 1962.
- [156].Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH, Proc. Natl. Acad. Sci 2011, 108, 2651–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Wang C, Stewart RJ, Kopeček J, Nature 1999, 397, 417–420. [DOI] [PubMed] [Google Scholar]
- [158].Tang Q, Zhao D, Zhou Q, Yang H, Peng K, Zhang X, Macromol. Rapid Commun 2018, 39, 1800109. [DOI] [PubMed] [Google Scholar]
- [159].Hofmeier H, Hoogenboom R, Wouters MEL, Schubert US, J. Am. Chem. Soc 2005, 127, 2913–2921. [DOI] [PubMed] [Google Scholar]
- [160].Shi Y, Wang M, Ma C, Wang Y, Li X, Yu G, Nano Lett 2015, 15, 6276–6281. [DOI] [PubMed] [Google Scholar]