

Abstract
Transforming growth factor-β (TGF-β) is major inducer of epithelial to mesenchymal transition (EMT), which associates with cancer cell metastasis and resistance to chemotherapy and targeted drugs, through both transcriptional and non-transcriptional mechanisms. We previously reported that in cancer cells, heightened mitogenic signaling allows TGF-β-activated Smad3 to interact with poly(RC) binding protein 1 (PCBP1) and together they regulate many alternative splicing events that favors expression of protein isoforms essential for EMT, cytoskeletal rearrangement, and adherens junction signaling. Here, we show that the exclusion of TGF-β-activated kinase 1 (TAK1) variable exon 12 requires another RNA-binding protein, Fox-1 homolog 2 (Rbfox2), which binds intronic sequences in front of exon 12 independently of the Smad3-PCBP1 complex. Functionally, exon 12-excluded TAK1∆E12 and full length TAK1FL are distinct. The short isoform TAK1∆E12 is constitutively active and supports TGF-β-induced EMT and nuclear factor kappa B (NF-κB) signaling, whereas the full-length isoform TAK1FL promotes TGF-β-induced apoptosis. These observations offer a harmonious explanation for how a single TAK1 kinase can mediate the opposing responses of cell survival and apoptosis in response to TGF-β. They also reveal a propensity of the alternatively spliced TAK1 isoform TAK1∆E12 to cause drug resistance due to its activity in supporting EMT and NF-κB survival signaling.
Introduction
Advanced cancers are well-known to secrete transforming growth factor-β (TGF-β), which, despite its potent growth inhibitory function to normal epithelial cells, promotes epithelial-mesenchymal transition (EMT) and metastasis due to contextual changes that have occurred in the tumor cells (1, 2). Induction of EMT by TGF-β also renders resistance to conventional chemotherapeutics as well as targeted drugs (3, 4), making TGF-β signaling an actively pursued investigational target for intervention in combination with immunotherapy (5). However, the mechanism underlying the conversion of TGF-β into a tumor-promoter still remains incompletely understood.
The general paradigm of TGF-β signaling entails a complex of membrane-bound type I and type II receptors, which upon ligand engagement activate both the canonical Smad-dependent pathway as well as a number of non-canonical non-Smad pathways including mitogen-activated protein kinases (MAPKs) (6, 7). The TGF-β pathway specific Smad2 and Smad3 are activated at the C-terminal phosphorylation site SSXS and induced to accumulate in the nucleus in association with Smad4 to regulate target gene expression. Smad3 is also phosphorylated at several sites in a linker region that bridges its highly conserved MH1 and MH2 domains; our recent data showed that phosphorylation at one of the linker sites, T179, allows TGF-β-activated Smad3 to interact with a RNA binding protein, poly(RC) binding protein 1 (PCBP1, also known as hnRNP E1), in the nucleus (8). The resultant Smad3-PCBP1 complex then binds the variable exon region of CD44 pre-mRNA and suppresses the assembly of the splicing machinery, thereby causing the exclusion of CD44 variable exons to express CD44 standard isoform. The TGF-β-induced alternative splicing has a genome-wide global impact that favors expression of protein isoforms essential for EMT, cytoskeletal rearrangement, and adherens junction signaling (8).
TGF-β-activated kinase 1 (TAK1), also known as MAPK kinase kinase 7 (MAP3K7), is one of the best characterized non-Smad signal transducers critical for TGF-β functions in EMT and apoptosis through activating the c-Jun N-terminal kinase (JNK) and p38 MAPK cascade (9–11). TAK1 also plays an essential role in mediating TGF-β activation of I-kappa B kinase (IKK) and the master transcription factor nuclear factor kappa B (NF-κB) that is required for mounting the EMT response and cell survival (12–15). In analogy to the mechanism defined in interleukin-1/Toll-like receptor pathways, TGF-β-induced activation of TAK1 requires TRAF6, a RING domain ubiquitin ligase that itself is modified by a K63-linked polyubiquitin chain, which acts as a scaffold to recruit TAK1 to the TGF-β receptor complex and triggers TAK1 activation (9, 11, 15). Activity of TAK1 is also regulated by its binding proteins, including TAK1-binding protein 1 (TAB1) that binds constitutively the kinase domain (16, 17), and TAB2 or TAB3 that binds the C-terminal domain and functions as an adaptor linking TRAF6 to TAK1 (18, 19). However, it is unclear how TGF-β utilizes the same TAK1 kinase to elicit the opposing responses of cell survival and apoptosis in different cellular contexts or under the influence of different environmental cues.
Human and mouse TAK1 genes contain 17 exons, including two variable exons 12 and 16, thus giving rise to 4 isoforms, of which variant A with 579 amino acid residues (TAK1∆E12) and variant B with 606 amino acid residues (TAK1FL) are two major forms (20). The short isoform or the exclusion of exon 12 is known to be muscle-specific, but what is astonishing is the fact that the peptide sequence of exon 12 and this alternative splicing event are highly conserved from early deuterostomes to mammals (21). Exclusion of TAK1 exon 12 is also known to occur during EMT or to be induced by TGF-β (8, 21–23), but the functional significance of this ancient splicing event has yet to be described.
Here, we report that the exclusion of TAK1 exon 12 by TGF-β requires Smad3-PCBP1 complex and another independent RNA binding complex containing Fox-1 homolog 2 (Rbfox2). We further show that the exon 12-excluded TAK1∆E12 is required for TGF-β-induced EMT and NF-κB activation, whereas the full length TAK1FL is for promoting apoptosis. Our data also reveal a possibility of reversing the pro-tumorigenic role of TGF-β in cancer cells by blocking the exclusion of TAK1 exon 12.
Results
TGF-β induces the exclusion of TAK1 Exon 12
To investigate the mechanism and functional significance of TGF-β-induced alternative splicing of TAK1, we designed RT-PCR reactions with primers flanking the exon 12 to distinguish the two major mRNA variants (Fig. 1a). Using this approach, we found that the long form TAK1 mRNA containing exon 12 (TAK1FL) is predominantly expressed in cells of the epithelial origin such as normal human HMLE, MCF10A and mouse NMuMG mammary gland epithelial cells, whereas the short form lacking exon 12 (TAK1∆E12) is predominantly expressed in mesenchymal and fibroblast cells, such as human mesenchymal stem cell (hMSC) and mouse embryonic fibroblasts (MEF), as well as highly metastatic human cancer cells, MDA-MB-231, HeLaO3, SCC3 and NCI-H1650 (Fig. 1b). Some cancer cells such as human breast cancer MDA-MB-468, cervical cancer HeLa, as well as mouse breast cancer 4T1 and Met-1 cells express both isoforms. TGF-β-induced exclusion of TAK1 exon 12 was previously reported in NMuMG and HeLa cells (8, 21). We extended this observation to MCF10A and Met-1 cells and found that the shift of exon 12 splicing pattern is a gradual process that reaches its completion in 48–72 hours (Fig. 1c). This kinetic timeline was further supported by measuring exon usage, which is defined as the ratio of a variable exon level to that of the constant exon 2. We found that the usage of Exon 12 decreased from ~ 80% to ~30% in the total pool of TAK1 mRNA upon TGF-β treatment (Fig. 1d). As controls, the usage of constant exon 4 and another alternative exon 16 did not change in response to TGF-β (Fig. 1d). Other ligands including tumor necrosis factor α (TNFα) and interleukin 6 (IL6) had no such effect on exon 12 exclusion (Fig. 1e), indicating that this phenomenon is specific to TGF-β.
Fig. 1. TGF-β induces TAK1 isoform switch in epithelial and breast cancer cells.
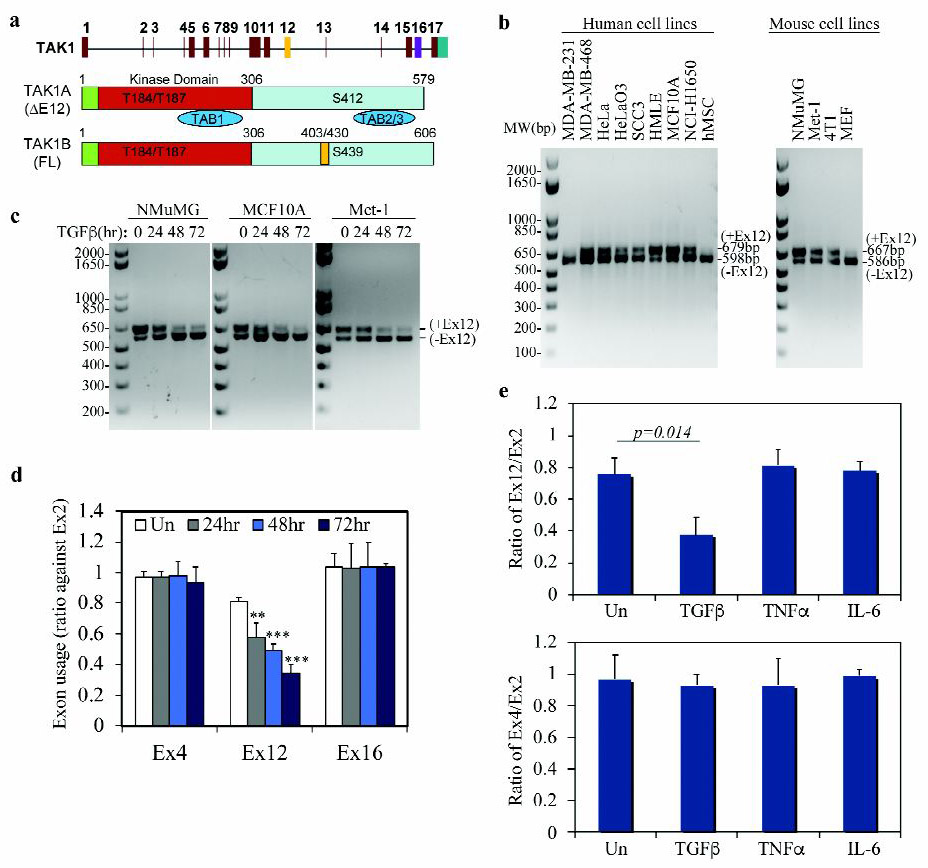
(a) Schematic diagram of structure of the TAK1 gene (top) and alternatively spliced mRNA transcripts of TAK1∆E12 with exon 12 exclusion and TAK1FL containing all exons.
(b) Detection of TAK1 isoforms with or without Exon 12 in various human and mouse cells using a RT-PCR reaction with one pair of primers.
(c) RT-PCR gel pictures showing that isoform switching of TAK1 in mammary epithelial NMuMG and MCF10A cells, and in breast cancer Met-1 cells treated with TGF-β.
(d) TGF-β specifically induces TAK1 exon 12 exclusion. qRT-PCR analysis of exon expression ratio of TAK1 variable exon 12 or 16 to standard exon 2 in NMuMG cells treated with TGF-β. Expression ratio of standard exon 4 to standard exon 2 was used as a control. Data are shown as mean ± SD (n=3), statistically significant difference between treated and untreated (Un) samples is indicated, **p<0.01, *** p<0.001.
(e) Exclusion of TAK1 exon 12 is specifically induced by TGF-β but not by TNFα or IL-6. qRT-PCR analysis of the exon ratio of TAK1 variable exon 12 to standard exon 2 (top), or exon 16 to standard exon 2 (bottom) in NMuMG cells treated with TGF-β, TNFα or IL6 for 72 h. Data are shown as mean ± SD (n=3), statistically significant difference between treated and untreated (Un) samples is indicated.
Exclusion of TAK1 Exon 12 requires two separate RNA-binding complexes containing Smad3-PCBP1 and Rbfox2, respectively
Previously, we showed that Smad3 and PCBP1 are both required for TGF-β-induced TAK1 exon 12 exclusion in HeLa cells (8). Using two independent siRNAs specific for Smad3 and PCBP1, we confirmed here that Smad3 and PCBP1 are also required for exon 12 exclusion in NMuMG cells (Fig. 2a, 2b). In the intron leading to exon 12, there are four copies of Rbfox2 binding sequence (U)GCAUG between the 3’ splice site and the branch point (24). Rbfox2 is an RNA binding protein that determines the selection of variable exons depending on the relative location of the binding sites; it promotes exon exclusion if the binding site is upstream of an alternative exon or inclusion if the binding site is downstream (24, 25). Using siRNAs specific for Rbfox2, we found that Rbfox2 is also required for the TGF-β-induced exclusion of exon 12 (Fig. 2a, 2b). Interestingly, Rbfox2 expression is induced by TGF-β (Fig. 2c) and the temporal pattern of induction is in accordance with the time course of TGF-β-induced change in exon 12 usage (Fig. 2d). Moreover, TGF-β-induced Rbfox2 expression is dependent on Smad3 not PCBP1, since it was blocked only by the Smad3-specific siRNA (Fig. 2e). As we demonstrated previously, besides its canonical function in transcriptional regulation, Smad3 also binds directly to pre-mRNA and regulates alternative splicing (8). Using RNA-immunoprecipitation (RIP), we confirmed that Smad3 as well as PCBP1 indeed binds TAK1 pre-mRNA and the binding was augmented by TGF-β (Fig. 2f, 2g). Likewise, the binding of Rbfox2 to TAK1 pre-mRNA was also enhanced by TGF-β (Fig. 2h). Also akin to the binding of CD44 pre-mRNA, the association of PCBP1 and Smad3 with TAK1 pre-mRNA is of the cooperative nature as knocking-down PCBP1 reduced the binding by Smad3 while knocking-down Smad3 reduced the binding by PCBP1 reciprocally (Fig. 2f, 2g). Nevertheless, the interaction between Rbfox2 and TAK1 pre-mRNA did not show inter-dependence with that of Smad3/PCBP1 as knocking-down Smad3 or PCBP1 had no effect on the Rbfox2 binding to TAK1 pre-mRNA (Fig. 2h) and vice versa (Fig. 2f, 2g). In contrast, the binding of U2 snRNP auxiliary factor 2 (U2AF2), an essential 3’ splice site selecting factor, to TAK1 pre-mRNA was blocked by TGF-β, most likely due to a concerted action of Smad3, PCBP1, and Rbfox2 that excluded its presence (Fig. 2i). Taken together, these results indicate that the exclusion of TAK1 exon 12 requires two separate Smad3-PCBP1 and Rbfox2 complexes, and Smad3 plays a dual role in this regulation by first mediating the TGF-β induction of Rbfox2 and then acting with the other two proteins to deny the binding of U2AF to TAK1 pre-mRNA.
Fig. 2. TGF-β-mediated TAK1 exon 12 exclusion requires Smad3, PCBP1 and Rbfox2.
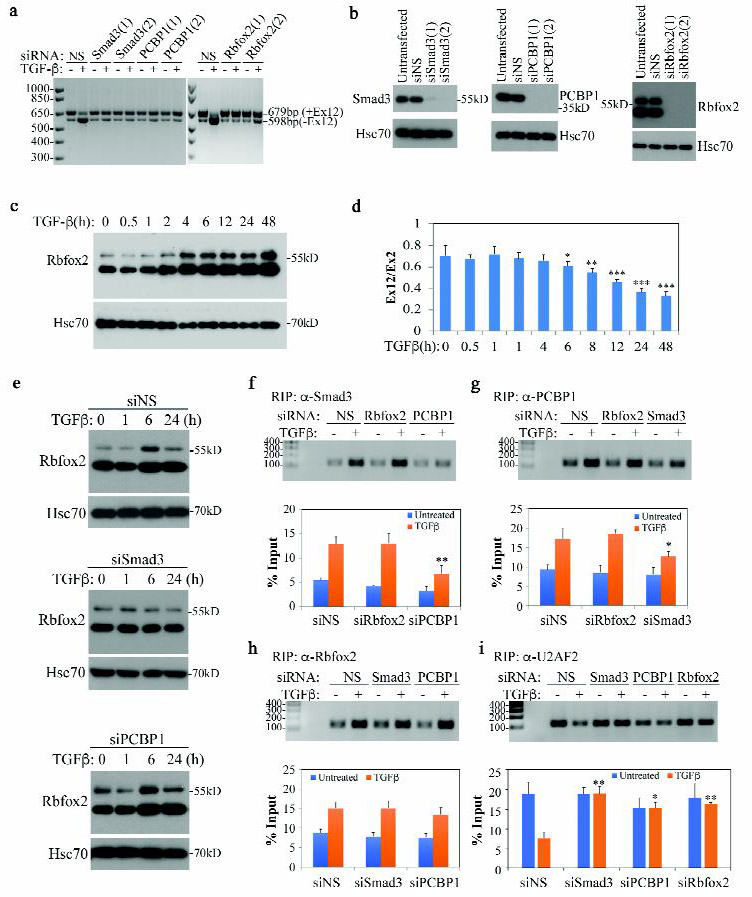
(a) RT-PCR detection of TAK1 isoforms in NMuMG cells transfected with siRNA and treated with TGF-β for 72 h. NS indicates Non-specific siRNA.
(b) Western blot showing Smad3, PCBP1 and Rbfox2 knockdown in NMuMG cells.
(c) Western blot showing Rbfox2 expression is induced by TGF-β in NMuMG cells.
(d) qRT-PCR analysis of TAK1 exon 12 ratio to TAK1 standard exon 2 in NMuMG cells treated with TGF-β at various time points. Data are shown as mean ± SD (n=3), statistically significant difference between treated and untreated samples is indicated, *p<0.05, **p<0.01, *** p<0.001.
(e) Western blot showing that Smad3 but not PCBP1 is required for TGF-β-induced Rbfox2 expression in NMuMG cells.
(f-i) Recruiting Smad3, PCBP1, Rbfox2 and U2AF2 to TAK1 pre-RNA by TGF-β. Top panels: standard PCR and gel analysis of RIP samples with a TAK1 pre-mRNA primer set, TGF-β treatment was for 2 h; bottom panel: qRT-PCR analysis of samples used in top panels. Data are shown as mean ± SD (n=3), statistically significant difference comparing siRNA transfected to control siNS transfected TGF-β-treated sample is indicated, *p<0.05, **p<0.01.
The exon 12 excluded TAK1 isoform is constitutively active
TAK1 is a key kinase of the non-canonical, Smad-independent TGF-β signaling pathway that activates JNK, p38 MAPK, and NF-κB to elicit apoptotic and EMT responses (9, 11, 15). Activation of TAK1 is marked by several phosphorylation events at residues T184, T187, and S439 (or S412 in the exon 12 skipped form) (26–28). In mouse NMuMG cells, TGF-β treatment induced rapid phosphorylation at these sites, which coincided with the activation of downstream JNK and p38 MAPK (Fig. 3a). To address the regulation and function of the two splicing variants, we generated a stable line of NMuMG cells over-expressing shTAK1 and found that removing TAK1 blocked the activation of JNK and p38 MAPK (Fig. 3a), reaffirming its essential role in the non-canonical TGF-β signaling. We next stably expressed TAK1FL and TAK1∆E12 in shTAK1 cells. Remarkably, while phosphorylation at T184/T187 and S439 of the TAK1FL was fully inducible by TGF-β, those sites of the short form, TAK1∆E12, were constitutively phosphorylated (Fig. 3a). Consequentially, re-expressing TAK1FL restored the TGF-β inducibility of JNK and p38 MAPK, whereas re-expressing TAK1∆E12 resulted in their constitutive activation (Fig. 3a). Likewise, using this reconstituted NMuMG cell system, we demonstrated that TAK1 is also required for the TGF-β-induced phosphorylation and degradation of IκB, but only TAK1∆E12 exhibited the ability to restore the regulation of IκB (Fig. 3b) and induction of NF-κB-mediated transcription by TGF-β as monitored by the NF-κB-luc reporter assay (Fig. 3c) or qRT-PCR analyses of several NF-κB target genes, including the IκBα, Csf2, anti-apoptotic Bcl-xl and EMT-inducing Snail1 genes (Fig. 3d).
Fig. 3. TAK1∆E12 is constitutively active.
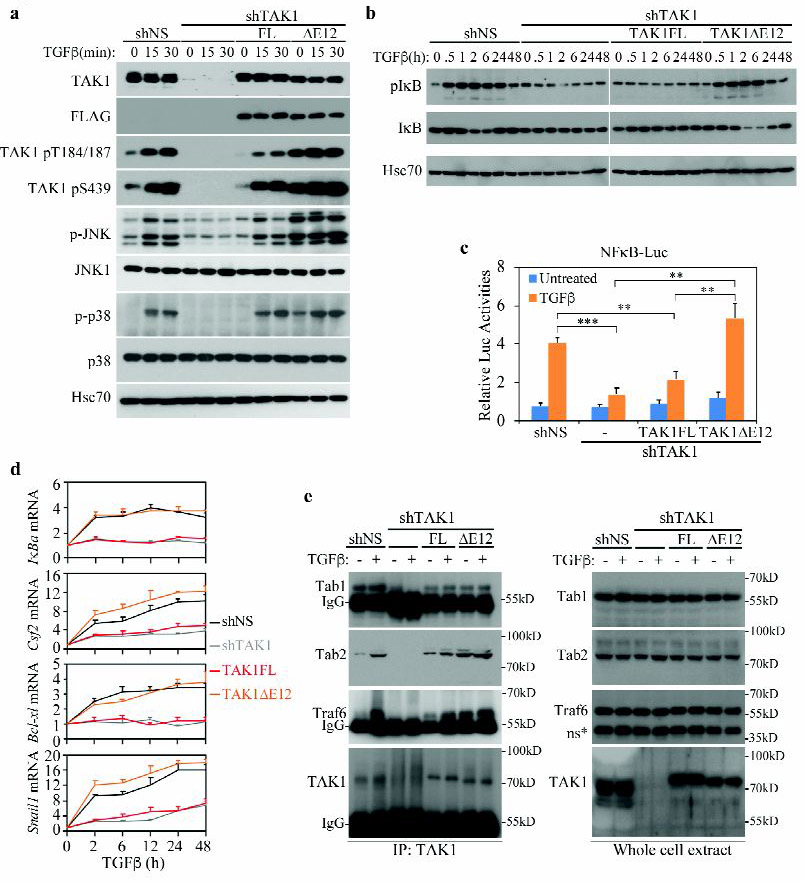
(a) Effects of TAK1FL and TAK1∆E12 isoforms on the TGF-β-induced activation of TAK1, JNK and p38 in NMuMG cells. NMuMG cells were stably transfected with shNS (Non-specific) or shTAK1, and TAK1 expression was rescued in shTAK1 cells with stably transfection of a Flag-tagged TAK1FL or TAK1∆E12 construct. The levels of phospho- or total-TAK1, JNK and p38 in cell lysates were analyzed by Western blots, Hsc70 blot was used as a loading control.
(b) Effects of TAK1FL and TAK1∆E12 isoforms on the TGF-β-induced activation of IKK and in NMuMG cells. IKK activation was analyzed by Western blot for phospho-IκB and degradation of total IκB levels.
(c) TAK1∆E12 but not TAK1FL fully rescued TGF-β-induced NF-κB activity. NMuMG cells that stably expressing various TAK1 constructs were transfected with NFκB-Luc reporter, and luciferase activities were measured after treated with ± TGF-β for overnight. Data are represented as mean ± S.D. (n=3), statistically significant difference is indicated, **p<0.01, ***p<0.001.
(d) qRT-PCR analyses of IκBα, Csf2, Bcl-xl, and Snail1 mRNAs in NMuMG cells. TAK1FL or TAK1∆E12 was stably expressed in shTAK1 cells. Relative levels of expression were normalized to untreated cells. Data are shown as mean ± S.D. (n=3).
(e) IP-Western blot analyses of the ability of TAK1 to bind Tab1, Tab2 or Traf6 in NMuMG cells after TGF-β treatment for 30 min. NMuMG cells expressing different vectors as indicated were subjected to anti-TAK1 IP, then blotted for associated Tab1, Tab2 and Traf6. Expression of Tab1, Tab2, Traf6 and TAK1 in whole cell lysates were shown at right. *ns indicates non-specific band.
TAK1 is a crucial tertiary MAP3K whose multifaceted functions require formation of kinase complexes with TRAF6 as well as TABs (16, 17, 19, 29). Activation of TAK1 by TGF-β depends on the assembly of these complexes, which are promoted by the K63-linked poly-ubiquitin modification of TRAF6 (9, 11). Residues encoded by TAK1 exon 12 are in juxtaposition to the essential S439 phosphorylation site, making alternative splicing of this exon a possible regulatory mechanism for controlling interaction with TAB2, which binds in the C-terminal region of TAK1 (Fig. 1a). Indeed, our data showed that whereas the binding of TAB1 to the kinase domain of TAK1 was not influenced by the presence or absence of exon 12, removing it rendered TAK1∆E12 to interact with TAB2 and TRAF6 constitutively rather than inducible by TGF-β in TAK1-depleted NMuMG cells (Fig. 3e). The constitutive interaction of TAK1∆E12 with TRAF6 and TAB2 correlated well with the constitutive activation of JNK and p38 MAPK herein (Fig. 3a), thus implying a direct causal relationship. Finally, although TAK1∆E12 is capable of constitutively activating JNK and p38MAPK, it nevertheless requires TGF-β input to restore NF-κB activation in TAK1-depleted NMuMG cells (Fig. 3b).
TAK1FL and TAK1∆E12 differentially regulate apoptosis and EMT
Having uncovered the difference in TGF-β-induced phosphorylation, binding of TRAF6, and abilities to activate downstream pathways of JNK, p38 MAPK and NF-κB between these two splicing variants of TAK1, we next asked if they regulate the apoptosis and EMT responses differently. Once again in the TAK1-depeleted stable NMuMG cells, Annexin V staining showed that removing TAK1 by shRNA drastically reduced the size of Annexin V positive cell population upon TGF-β treatment, whereas re-expressing of TAK1FL restored it (Fig. 4a, 4b). In contrast, re-expressing of the TAK1∆E12 did not show such an effect, indicating that only the TAK1FL supports TGF-β-stimulated apoptosis. In keeping with this notion, Western blot analysis showed that only TAK1FL, but not TAK1∆E12, allowed the TGF-β-induced caspase-3 cleavage (Fig. 4c). It is known that TGF-β-induced EMT requires inputs from JNK and p38 MAPK pathway as well as NF-κB activation (12, 30–32). Since the TAK1∆E12 isoform is highly conducive to the activation of these two pathways by TGF-β, it is likely that it also supports the EMT response. Indeed, TAK1 depletion by shRNA in NMuMG cells prevented the appearance of the mesenchymal-specific F-actin stress fiber formation and the disruption of the regular, cobble stone-like E-cadherin staining pattern at the cell-cell junction upon TGF-β treatment (Fig. 4d). Re-expressing TAK1FL did not alter this outcome but re-expressing TAK1∆E12 without TGF-β treatment partially blurred the clear E-cadherin bordering pattern; with TGF-β, it generated the full-fledged EMT response (Fig. 4d). Western blot analyses of epithelial marker E-cadherin and mesenchymal marker N-cadherin showed reciprocal changes in the protein levels, thus confirming the above immunofluorescence imaging observation (Fig. 4e). Taken together, our data indicate that while the long form TAK1FL supports induction of apoptosis by TGF-β, the short form TAK1∆E12 mediates EMT.
Fig. 4. TAK1FL and TAK1∆E12 differentially regulate TGF-β-induced apoptosis and EMT.
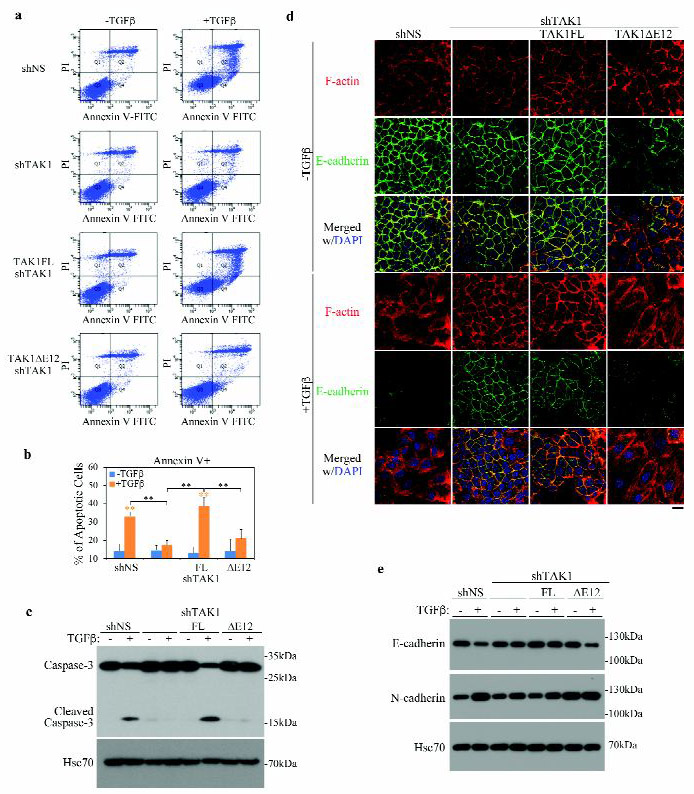
(a) Effect of TAK1FL and TAK1∆E12 isoforms on the TGF-β-induced apoptosis in NMuMG cells. Apoptosis were detected by FACS analysis of Annexin V and PI staining of serum-starved NMuMG cells after treated with ± TGF-β for 24 h.
(b) Quantitation of apoptotic (Annexin V+) cells in A. Data are represented as mean ± S.D. (n=3), statistically significant difference is indicated, **p<0.01.
(c) Western blot analyses of the cleaved caspase-3 in NMuMG cells after treatment with ± TGF-β for overnight.
(d) Effect of TAK1FL and TAK1∆E12 isoforms on the TGF-β-induced EMT in NMuMG cells. Immunofluorescence staining of F-actin (phalloidin) and E-cadherin in NMuMG cells after TGF-β treatment for 2 days. Bar = 20 μm.
(e) Western blot analyses of expression of epithelial marker E-cadherin and mesenchymal marker N-cadherin in NMuMG cells carrying various TAK1 vectors after TGF-β treatment for 2 days.
Within different cellular context, activation of the NF-κB signaling is known to be a deciding factor as to whether TGF-β induces EMT or apoptosis (12–14). Thus, the differential activities of TAK1FL and TAK1∆E12 likely hinge on the unique ability of TAK1∆E12 to activate the NF-κB pathway. Indeed, treating NMuMG cells with chemical inhibitors of either JNK, p38, or NF-κB blocked the TGF-β-induced EMT (Fig. 5a). Moreover, these inhibitors also showed efficacy in blocking EMT in the reconstituted TAK1∆E12 cells (Fig. 5a), reaffirming the essential requirement of NF-κB signaling in EMT. On the other hand, re-expressing TAK1FL in TAK1-depleted NMuMG cells restored the capacity of TGF-β to induce apoptosis, with or without the NF-κB inhibitor (Fig. 5b), implying that NF-κB was not activated by the full length TAK1. In contrast, the TAK1∆E12 isoform did not support the TGF-β-induced apoptosis, but blocking NF-κB signaling promoted apoptosis in an unimpeded manner (Fig. 5b), implying TAK1∆E12 activated the protective NF-κB signaling against the TGF-β induced cell death in the reconstituted cells.
Fig. 5. NF-κB mediates TGF-β-induced EMT and anti-apoptotic responses.

(a) Effect of chemical inhibitors of JNK, p38 and NF-κB on the TGF-β-induced EMT in NMuMG cells. Immunofluorescence staining of F-actin (phalloidin) and E-cadherin in NMuMG cells after TGF-β treatment for 2 days. Bar = 20 μm.
(b) Cell death was measured with the Cell Death Elisa kit in NMuMG cells after treatment with or without TGF-β for 24 h. When indicated, NF-κB inhibitor was added to cells 2 h before TGF-β addition. Data are represented as mean ± S.D. (n=3), statistically significant differences is indicated, *p<0.05, ***p<0.001; ns, not significant.
Tak1∆E12 supports TGF-β-induced of EMT and invasion in Met-1 breast cancer cells.
As shown in Fig. 1a, several lines of cancer cells express equal amount of the two TAK1 isoforms; these include the Met-1 mouse mammary tumor cells derived from MMTV-PyMT mouse breast cancer model (33), while highly metastatic cancer cells, such as MDA-MB-231 cells, express predominantly the alternatively spliced TAK1∆E12 isoform. To determine if this splicing variant is essential for the TGF-β-induced EMT response in cancer cells in which other genetic lesions should have created a conducive cellular context, we generated two independent lines of TAK1 knockout (KO) Met-1 cells using the CRISPR/Cas9 strategy with sgRNA targeting either TAK1 exon 2 or exon 5 (Fig. 6a). Once again, TGF-β-induced of EMT was disrupted in both TAK1 KO cells, and only the short TAK1∆E12 isoform, not the full length TAK1FL, restored the ability of TAK1KO cells to undergo TGF-β-induced EMT (Fig. 6b). Similarly, Western analyses of epithelial maker E-cadherin, mesenchymal markers N-cadherin, Vimentin, and Snail corroborated this conclusion (Fig. 6c). Furthermore, removal of TAK1 drastically reduced the ability of Met-1 cells to migrate and invade through the matrigel membrane of the Boyden transwell invasion system, which was rescued by re-expressing the TAK1∆E12 but not the TAK1FL (Fig. 6d). These data clearly established the role of the alternative splicing in conferring the ability to induce EMT and invasion by TGF-β through expression of the short TAK1∆E12 isoform.
Fig. 6. TAK1∆E12 induces EMT and invasion responses by TGF-β in breast cancer Met-1 cells.

(a) Generation of Met-1 cells with TAK1 deletion by Crispr/Cas9 and reconstitution of Met-1 cells that stably expressing TAK1FL and TAK1∆E12. Two clones that with sgRNA targeting either exon 2 (Tak1 KO-2) or Exon 5 (TAK1 KO-1) were selected, then TAK1FL or TAK1∆E12 was stably expressed in TAK1 KO-1 Met-1 cells. Scr indicates the cells that were transfected with scramble sgRNA.
(b) TAK1∆E12 promotes EMT responses in Met-1 cells. Cells were treated with TGF-β for 3 days, then immunostained for F-actin (phalloidin) and E-cadherin. Bar = 20 μm.
(c) Western blot analyses of expression of the epithelial marker E-cadherin and mesenchymal marker N-cadherin, Vimentin and Snail in Met-1 cells carrying various TAK1 vectors after TGF-β treatment for 3 days.
(d) TAK1∆E12 is required for TGF-β-mediated migration and invasion. Quantitation of migration and invasion were performed by counting cells that across membrane. Data are represented as mean ± S.D. (n=3), statistically significant difference comparing to TGF-β-treated Scr cells or TAK1∆E12-expressing TAK1 KO-1 cells is indicated using black or red asterisks respectively. **p<0.01, ***p<0.001.
TAK1∆E12 confers Met-1 cells the resistance to Cyclophosphamide and Afatinib
In cancer clinics, the ability of tumor cells to undergo EMT is a major cause of resistance to conventional chemotherapy as well as targeted drugs (3, 34). Compared to normal NMuMG cells, the mouse breast cancer Met-1 cells exhibited complete resistance to TGF-β-induced apoptosis and hyper-responsiveness to NF-κB cell survival signaling induced by TGF-β (Fig. 7a, 7b). Met-1 cells express roughly equal amount of TAK1FL and the alternatively spliced TAK1∆E12 (Fig. 1b, 1c and Fig. 6a). Removing total TAK1 by CRISPR/Cas9 in Met-1 cells and replacing it with TAK1∆E12 showed that it is the short isoform that is responsible for mediating TGF-β-induced NF-κB survival signaling (Fig. 7c), just as in NMuMG cells (Fig. 3d). These observations indicate that to Met-1 cells, TGF-β has already masqueraded into a tumor-promoter, to the extent that it was able to counter-act the cytotoxic effect of cyclophosphamide (CTX), a commonly used chemotherapy drug, and Afatinib, a new generation EGFR inhibitor (Fig. 7d). Notably, TAK1KO cells abolished the protective effect of TGF-β against the drug treatment (Fig. 7e). Re-expressing TAK1FL in TAK1KO Met-1 cells not only restored the ability of TGF-β to induce apoptosis, it also reversed the protective role of TGF-β to augment CTX and Afatinib (Fig. 7f). In contrast to TAK1FL, re-expressing TAK1∆E12 restored the protective effect of TGF-β against drug treatment (Fig. 7g). This TGF-β induced drug resistance coincides with the ability of TGF-β to induce NF-κB signaling (Fig. 7c) and EMT (Fig. 6b, 6c), which are both mediated by TAK1∆E12. To demonstrate the role of NF-κB signaling in TGF-β-induced drug resistance, we treated Met-1 cells with NF-κB inhibitor and found that it induced cell death and diminished the TGF-β-induced resistance to CTX and Afatinib in both control scramble and TAK1∆E12 reconstituted Met-1 cells (Fig. 7h, 7i).
Fig. 7. TAK1FL but not TAK1∆E12 increases sensitivity of Met-1 cells towards Cyclophosphamide and Afatinib.
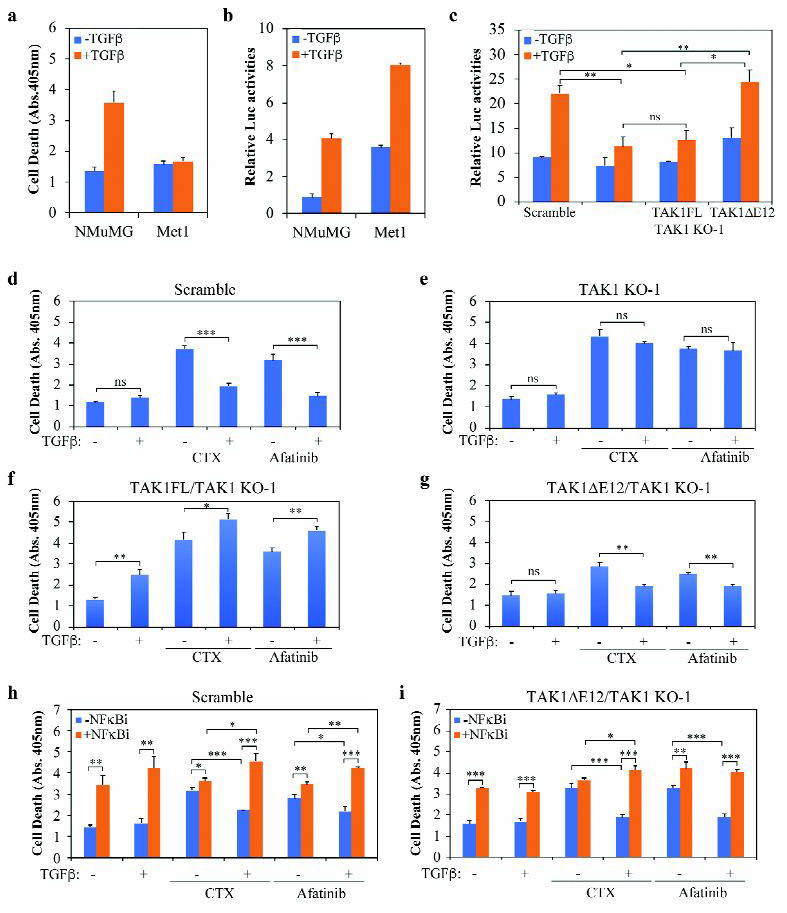
(a) Cell death was measured with the Cell Death Elisa kit after treating cells with or without TGF-β for 24 h.
(b) NFκB-Luc reporter assay in cells treated with or without TGF-β for 24 h.
(c) NFκB-Luc reporter assay in Met-1 cells expressing different TAK1 constructs. The TGF-β treatment was for 24 h.
(d-g) Cell death was measured with the Cell Death Elisa kit in Met-1 cells expressing different Tak1 constructs. Cells were treated with or without TGF-β, Cyclophosphamide (CTX) (5 mM) and/or Afatinib (0.5 μM) for 24 h.
(h-i) Cell death was measured in Met-1 cells as above, NF-κB inhibitor was added to cells 2 h before TGF-β, CTX) or Afatinib treatment as indicated.
All bar graphs are displayed as mean ± S.D. (n=3), statistically significant difference is indicated, *p<0.05, **p< 0.01, ***p<0.001; ns, not significant.
TAK1∆E12-mediated drug resistance applies to human MDA-MB-231 cells
To demonstrate the relevance of our finding in human cells, we examined the TGF-β- mediated drug resistance in MDA-MB-231 cells, a line of commonly used human breast cancer cells that are negative for estrogen and progesterone receptors as well as HER-2/Neu amplification. Patients with such triple negative breast cancers (TNBC) are routinely treated with chemotherapy, but their prognostic outcomes are usually very poor relative to other subtypes of breast cancers (35). Like their mouse counterpart, Met-1 breast cancer cells, and in reference to normal human mammary gland epithelial MCF-10A cells, MDA-MB-231 cells also exhibited complete resistance to TGF-β-induced apoptosis and high basal NF-κB pathway activity that was further inducible by TGF-β (Fig. 8a). Exon usage analyses showed that Exon 12 was utilized less than 12% in the total pool of TAK1 mRNA in MDA-MB-231 cells comparing to nearly 70% in MCF10A cells (Fig. 8b) while Western blot analysis detected only TAK1∆E12 (Fig. 8c), indicating that it is the predominant TAK1 isoform. TGF-β treatment further reduced Exon 12 usage in both cell lines (Fig. 8b). To create a pair of cell lines with isogenic background, we deleted the TAK1 gene in MDA-MB-231 cells using the CRISPR/Cas9 approach with a sgRNA targeting human TAK1 exon 2 and re-expressed either TAK1FL or TAK1∆E12 in these endogenous TAK1-deleted cells (Fig. 8c). Once again, just as what we described in the murine cells, only the TAK1∆E12 isoform supported TGF-β-induced NF-κB activation (Fig. 8d). Likewise, TGF-β displayed a protective role against the cytotoxicity of CTX in scramble control MDA-MB-231 cells in TAK1-dependent manner (Fig. 8e). while TAK1FL restored TGF-β-induced apoptosis, even augmented the apoptotic response in the presence of CTX, the short TAK1∆E12 isoform restored TGF-β-induced resistance to CTX (Fig. 8e). Thus, the differential functions of TAK1 alternative splicing products in mediating TGF-β-induced cell death and drug resistance also applies to human breast cancers.
Fig. 8. TAK1∆E12 mediates NF-κB activation and chemo-resistance in MDA-MB-231 cells.
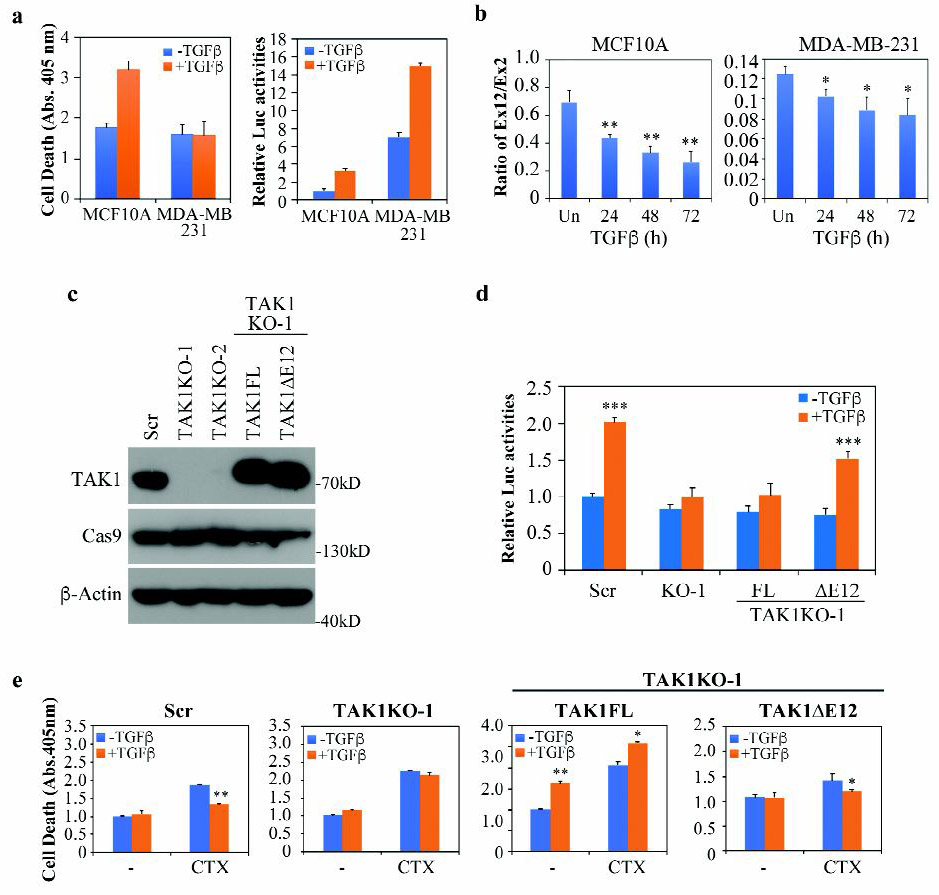
(a) Cell death and NFκB-Luc reporter assay were measured after treating cells with or without TGF-β for 24 h.
(b) qRT-PCR analysis of TAK1 exon 12 usage in cells treated with TGF-β at various time points. Data is shown as mean ± SD (n=3), statistically significant difference between treated and untreated samples is indicated, *p<0.05, **p<0.01.
(c) Generation of MDA-MB-231 cells with TAK1 deletion by Crispr/Cas9. Two independent clones that with sgRNA targeting exon 2 were selected, then TAK1FL or TAK1∆E12 was stably expressed in TAK1 KO-1 cells. Scr indicates the cells that were transfected with scramble sgRNA.
(d) NFκB-Luc reporter assay in MDA-MB-231 cells expressing different TAK1 constructs. The TGF-β treatment was for 24 h.
(e) Cell death was measured with the Cell Death Elisa kit in MDA-MB-231 cells expressing different TAK1 constructs. Cells were treated with or without TGF-β and/or Cyclophosphamide (CTX) (10 mM) for 24 h.
All bar graphs are displayed as mean ± S.D. (n=3), statistically significant differences is indicated, *p<0.05, **p< 0.01, ***p<0.001.
Discussion
The acquired ability to induce EMT and protect tumor cells from committing programmed cell death lies at the center of TGF-β conversion from a tumor suppressor to a tumor promoter in advanced cancers (1, 36). This dichotomous functional switch is achieved through changes at levels of receptor signaling, transcriptional network, translational control, as well as non-coding RNA mechanism that collectively govern the TGF-β signaling outputs. We recently reported that in cancer cells, phosphorylation at T179 of Smad3 with input from the heightened mitogenic pathways allows Smad3 to interact with RNA-binding protein PCBP1 (8). In the presence of TGF-β, the Smad3-PCBP1 complex is recruited onto alternatively spliced mRNA transcripts in the nucleus, resulting in the exclusion of variable exons (8). We now extended that observation to TAK1 and showed that both Smad3-PCBP1 and the RNA-binding complex containing Rbfox2 are required for directing alternative splicing of TAK1 pre-mRNA. Our data further revealed distinct functions of the full length and exon 12-excluded TAK1 isoforms in eliciting various signaling responses to TGF-β, and suggested blocking TGF-β-induced alternative splicing of TAK1 as a viable strategy to combat resistance to cancer therapeutic drugs.
TAK1 situates at the cellular hub to which various cytokines and developmental signaling pathways converge (37). Formation of TAK1-TABs-TRAF complexes constitutes necessary steps towards its activation. The 27 amino acid residues encoded by exon 12 are in juxtaposition to the critical activating S439 residue and near the docking sites for TAB2/3 binding in the C-terminus. Currently, only information regarding the crystal structure of a chimeric protein containing the TAK1 kinase domain and the TAK1-binding domain of TAB1 is available (38), so it is not yet clear how this peptide segment alters TAK1 function. One possibility is that it impedes the interaction between TAK1 and TAB2 since without it, TAK1∆E12 constitutively interacts with TAB2 and TRAF6 (Fig. 3d). Moreover, the Exon 12 sequence could also function as an inhibitory motif that blocks NF-κB activation by either a direct interference or recruiting other negative regulators. Regardless, the fact that alternative splicing of TAK1 exon 12 was conserved in ancient early deuterostomes underscores the importance of this regulation.
TGF-β-induced EMT is known to require inputs from MAPKs, including JNK and p38 MAPK (30–32). Although our data showed that TAK1FL is capable to mediate TGF-β activation of JNK and p38 MAPK, it cannot induce EMT. Even TAK1∆E12, which is constitutively active in activating JNK and p38 MAPK, still depends on TGF-β to restore the full-fledged EMT response in TAK1-depleted cells. One functional distinction between these two alternatively spliced isoforms is the ability of TAK1∆E12 to activate NF-κB signaling, which is known to be essential for TGF-β induction of EMT (12). Our data showed a parallel dependence on TGF-β for TAK1∆E12 to activate NF-κB signaling (Fig. 3c, 7c) and induce EMT (Fig. 4d, 4e, 6b, 6c). Clearly, the unique role of TAK1∆E12 in inducing EMT lies in its ability to activate NF-κB signaling. The NFκB pathway is known to promote cell survival and protect cell against cytotoxicity of cancer therapies (38, 39). Recently, many studies indicated that resistance to conventional chemotherapy and targeted drugs correlates closely with the ability of tumor cells to undergo EMT (3, 34, 40). The dual role of the NF-κB pathway in promoting cell survival and EMT might be one mechanism underlying the correlation between EMT and drug resistance.
The revelation that the alternatively spliced TAK1∆E12 isoform elicits both NF-κB signaling and EMT responses, whereas the TAK1FL only induces apoptosis, suggests that by shedding TAK1 exon 12 TGF-β is converted into a tumor promoter. In this regard, it may be possible to increase the sensitivity of tumor cells to cancer chemotherapies by blocking the exclusion of exon 12. Previous attempts that targeted the TGF-β pathway for cancer treatment primarily focused on inhibiting TGF-β receptors or Smad activities (5). However, these strategies yielded mixed results, likely because they in effect blocked both tumor promoting as well as suppressing roles of TGF-β. In light of our current findings, one potential approach would be targeting Rbfox2, which is required for the TGF-β-induced TAK1 exon 12 exclusion in addition to Smad3 and PCBP1. Unlike Epithelial splicing regulatory proteins (ESRPs) whose expression are restricted to epithelial cells and down-regulation of which abrogates epithelial tissue-specific splicing events, Rbfox2 is more abundant in mesenchymal tissues and a major contributor to mesenchymal splicing preferences (22, 23). Thus, inhibiting Rbfox2 could specifically abrogate alternative splicing events that promote EMT. Indeed, depleting Rbfox2 in mesenchymal cell lineages has been shown to elicit phenotypic changes towards an epithelial morphology (23). Alternatively, one could use splice-switching oligonucleotides (SSOs) to specially target desired splicing silencers or enhancers around the TAK1 exon 12. These SSOs fall under the category of antisense oligonucleotides that interfere with splicing events by targeting the splicing in a sequence-specific manner (41). As a proof of concept, such SSOs were successfully used to induce TAK1 isoform switching from TAK1FL to TAK1∆E12 in mouse livers (42). It would be interesting to design specific SSOs that could induce TAK1 isoform switching from TAK1∆E12 to TAK1FL, and test if they are effective in overcoming drug resistance through blocking NF-κB signaling and EMT while leaving the tumor suppressing activity of TGF-β intact.
Materials and Methods
Cell lines and culture conditions
NMuMG (ATCC, Manassas, VA: CRL-1636), MDA-MB-231 (ATCC: HTB-26), and MDA-MB-468 (ATCC: HTB132) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). MCF10A (ATCC: CRL-10317) cells were cultured in DMEM/F12 Ham’s Mixture plus 5% horse serum, EGF 20 ng/ml, insulin 10 μg/ml, hydrocortisone 0.5 mg/ml, and cholera toxin 100 ng/ml. Lung cancer NCI-H1650 cells were cultured in RPMI-1640 medium plus 10% FBS. Mouse breast cancer Met-1 and 4T1 cells, as well as human oral cancer SCC3 cells and immortalized mesenchymal stem cells (hMSC) were provided by Drs. K Hunter, L Yang, R. Weigert, T. Misteli (National Cancer Institute), respectively, and cultured in DMEM plus 10% FBS. The culture conditions for HeLa, OSCC3, MEF, HMEC were described previously (8, 43). All culture media contain penicillin-streptomycin (100 unit/ml) and the cells were grown at 37 °C and 5% CO2. Mycoplasma was tested before cells were used. For short time treatment (≤ 24 h), cells were stimulated with 4 ng/ml TGF-β1 (Peprotech, Rocky Hill, NJ) after starvation in media supplemented with 0.2% FBS for 4 h. For long time treatment (> 24 h), cells were not pre-starved of serum.
Plasmid Construction, transfection and inhibitors
For knocking down TAK1 in NMuMG cells, pSuper retro-neo-GFP vector (OligoEngine, Seattle, WA) was used to construct shTAK1 plasmid that targets 3’-UTR of mouse TAK1 gene. Flag-tagged mouse TAK1FL or TAK1∆E12 plasmids were constructed by insert TAK1 cDNA from MGC clone 5989 (Image: 3499247) or HA-TAK1 (gift from Dr. K. Matsumoto) (10) to pLNCX vector, respectively. For CRISPR/Cas9 mediated editing of the TAK1 gene, plasmid lentiCRISPRv2 (Addgene 52961) was used to deliver individual sgRNAs (single guide RNA). Sequences of primers used in the construction of shRNA, cDNA and CRISPR/Cas9 vectors are listed in Supplementary Table S1. Stocks of lentiviruses were made after co-transfecting lentiCRISPRv2, pVSVG and psPAX2 plasmids into HEK293T cells. Lipofectamine 2000 and 3000 (Life Technologies, Grand island, NY) was used to transfect plasmid DNA according to the manufacturer’s instructions. G418 or puromycin (Life Technologies) was used to select stable clones. JNK inhibitor, SP600125, and p38 inhibitor, SB202190 were obtained from EMD Millipore (Burlington, MA), NF-kB inhibitor, BMS345541, was obtained from Abcam (Cambridge, MA)
Quantitative RT-PCR
RNA was isolated with RNeasy mini kit (Qiagen, Germantown, MD) per manufacturer’s instructions. Precipitated RNA was eluted in nuclease-free water and quantified using a Nanodrop. High Capacity Reverse Transcription Kit (Life Technologies) was used to generate cDNA from RNA (500 ng-2000 ng). Quantitative PCR was performed with Power SYBR Green PCR Master Mix (Life Technologies) using specific oligonucleotide sequences (Table S1). All samples were run in three experimental replicates. Fold-changes were calculated using the 2^-ddCt method. The calculated threshold values were determined by the maximum curvature and Ct was calculated as CtControl – Ctsample. All qRT-PCR values were normalized with Rpl19.
RNA immunoprecipitation (RIP)
RIP was done using the RIP-Assay Kit (MBL International, Woburn, MA). Briefly, cells grown on 150 cm dishes were washed with ice cold PBS and about 1×107 cells were lysed in lysis buffer. Precleared lysate was incubated with RIP certified antibody immobilized on magnetic Dynabeads Protein G (Life Technologies) and incubated for 4 hours at 4 °C. RNA was then isolated, incubated with DNase I to eliminate contaminating genomic DNA and subjected for RT-PCR and qRT-PCR analyses.
Western blot, immunoprecipitation and immunofluorescence
Western blot, immunoprecipitation and immunofluorescence were performed as described (8). Antibodies used were described in supplemental Table S2.
Luciferase reporter assay
Reporter assays were performed in 6 well plates after transfecting NFκB-Luc (0.5 μg) and pRL-TK (0.2 μg) reporter plasmids. Luciferase activities were determined using Dual Luciferase Reporter Assay System (Promega, Madison, WI).
Apoptosis assay
Cells plated in 6-well plates were incubated with or without 4 ng/ml TGF-β in 0.2% FBS medium for 24 h. Then the cells were harvested and washed in cold PBS. Assay was performed using FITC Annexin V Apoptosis Detection kit (BD Biosciences, San Jose, CA) according to manufacturer’s instructions. Each sample was stained with FITC-Annexin (5.0 μl) and propidium iodide (5.0 μl), and percentage of apoptotic cells were evaluated after analysis using FACSCAN flow cytometer (Becton-Dickinson FACS Canto II). Apoptosis assay was also performed with the Cell Death Detection ELISA assay kit (Roche), which quantitatively determine DNA fragmentation using both anti-histone and peroxidase-conjugated anti-DNA antibodies. Whenever indicated, cyclophosphamide (CTX) (Sigma-Aldrich, St Louis, MO, C0768) and/or Afatinib (Santa Cruz Biotechnology, Dallas, TX, CAS439081–18-2) were added 30 min before TGF-β stimulation. For each treatment, triplicate experiments were performed.
Migration and invasion assay
Invasion across a basement membrane was performed using Corning Biocoat™ Growth Factor Reduced Matrigel Invasion Chamber with 8.0 mm PET membrane in 24-well plate (Corning Life Sciences, Corning, NY) according to manufacturer’s instructions. Briefly, a volume of 2.5 × 104 cells in medium that were treated with indicated factors for 24 h were added to each invasion chamber. After 18–24 h, invaded cells located on the underside of chambers were fixed with formaldehyde for 10 min and stained with trypan blue before counting. Transwell migration assays were performed essentially as the invasion assays above except that a 8-μm Biocoat Cell Culture Control Inserts was used instead of the Matrigel inserts, and the migrating cells were counted after incubation of 1.0 × 104 cells for 8 h.
Statistical analysis
Two-tailed student’s t-test was used for statistical analysis. All bar graphs are displayed as mean ± S.D of 3 biological replicates.
Supplementary Material
Supplemental Table S1: Primer, siRNA, shRNA, sgRNA sequences
Supplemental Table S2: Antibody information
Acknowledgment
We thank Drs. K. Matsumoto for providing HA-TAK1 (∆E12) plasmids; ZG Liu for NFκB-Luc reporter construct; K Hunter, L. Yang and T. Misteli for Met-1, 4T1 and hMSC cells respectively. This research was supported by the intramural research program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Conflict of Interest
The authors have declared that no conflict of interest exists.
Competing Interests statement: the authors declare no competing financial interests.
References
- 1.Seoane J, Gomis RR. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harbor perspectives in biology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr Opin Cell Biol. 2014;31:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang S, Holzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell. 2012;151(5):937–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akhurst RJ. Targeting TGF-beta Signaling for Therapeutic Gain. Cold Spring Harbor perspectives in biology. 2017;9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hata A, Chen YG. TGF-beta Signaling from Receptors to Smads. Cold Spring Harbor perspectives in biology. 2016;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang YE. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harbor perspectives in biology. 2017;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tripathi V, Sixt KM, Gao S, Xu X, Huang J, Weigert R, et al. Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-beta. Mol Cell. 2016;64(3):549–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10(10):1199–207. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270(5244):2008–11. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31(6):918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114(4):569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arsura M, Panta GR, Bilyeu JD, Cavin LG, Sovak MA, Oliver AA, et al. Transient activation of NF-kappaB through a TAK1/IKK kinase pathway by TGF-beta1 inhibits AP-1/SMAD signaling and apoptosis: implications in liver tumor formation. Oncogene. 2003;22(3):412–25. [DOI] [PubMed] [Google Scholar]
- 14.Gingery A, Bradley EW, Pederson L, Ruan M, Horwood NJ, Oursler MJ. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Experimental cell research. 2008;314(15):2725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamidi A, von Bulow V, Hamidi R, Winssinger N, Barluenga S, Heldin CH, et al. Polyubiquitination of transforming growth factor beta (TGFbeta)-associated kinase 1 mediates nuclear factor-kappaB activation in response to different inflammatory stimuli. J Biol Chem. 2012;287(1):123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kishimoto K, Matsumoto K, Ninomiya-Tsuji J. TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J Biol Chem. 2000;275(10):7359–64. [DOI] [PubMed] [Google Scholar]
- 17.Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, et al. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science. 1996;272(5265):1179–82. [DOI] [PubMed] [Google Scholar]
- 18.Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, Matsumoto K. Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J. 2003;22(23):6277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5(4):649–58. [DOI] [PubMed] [Google Scholar]
- 20.Dempsey CE, Sakurai H, Sugita T, Guesdon F. Alternative splicing and gene structure of the transforming growth factor beta-activated kinase 1. Biochimica et biophysica acta. 2000;1517(1):46–52. [DOI] [PubMed] [Google Scholar]
- 21.Venables JP, Vignal E, Baghdiguian S, Fort P, Tazi J. Tissue-specific alternative splicing of Tak1 is conserved in deuterostomes. Mol Biol Evol. 2012;29(1):261–9. [DOI] [PubMed] [Google Scholar]
- 22.Venables JP, Brosseau JP, Gadea G, Klinck R, Prinos P, Beaulieu JF, et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol Cell Biol. 2013;33(2):396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS genetics. 2011;7(8):e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nature structural & molecular biology. 2009;16(2):130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22(18):2550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scholz R, Sidler CL, Thali RF, Winssinger N, Cheung PC, Neumann D. Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process. J Biol Chem. 2010;285(33):25753–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakurai H, Miyoshi H, Mizukami J, Sugita T. Phosphorylation-dependent activation of TAK1 mitogen-activated protein kinase kinase kinase by TAB1. FEBS letters. 2000;474(2–3):141–5. [DOI] [PubMed] [Google Scholar]
- 28.Ouyang C, Nie L, Gu M, Wu A, Han X, Wang X, et al. Transforming growth factor (TGF)-beta-activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase A catalytic subunit alpha (PKACalpha) and X-linked protein kinase (PRKX). J Biol Chem. 2014;289(35):24226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–51. [DOI] [PubMed] [Google Scholar]
- 30.Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. Embo J. 2002;21(14):3749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002;115(Pt 15):3193–206. [DOI] [PubMed] [Google Scholar]
- 32.Alcorn JF, Guala AS, van der Velden J, McElhinney B, Irvin CG, Davis RJ, et al. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-beta1. J Cell Sci. 2008;121(Pt 7):1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borowsky AD, Namba R, Young LJ, Hunter KW, Hodgson JG, Tepper CG, et al. Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis. 2005;22(1):47–59. [DOI] [PubMed] [Google Scholar]
- 34.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massague J TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirata Y, Takahashi M, Morishita T, Noguchi T, Matsuzawa A. Post-Translational Modifications of the TAK1-TAB Complex. Int J Mol Sci. 2017;18(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown K, Vial SC, Dedi N, Long JM, Dunster NJ, Cheetham GM. Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. Journal of molecular biology. 2005;354(5):1013–20. [DOI] [PubMed] [Google Scholar]
- 39.Brown M, Cohen J, Arun P, Chen Z, Van Waes C. NF-kappaB in carcinoma therapy and prevention. Expert Opin Ther Targets. 2008;12(9):1109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Disterer P, Kryczka A, Liu Y, Badi YE, Wong JJ, Owen JS, et al. Development of therapeutic splice-switching oligonucleotides. Hum Gene Ther. 2014;25(7):587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou D, Shao Q, Fan X, Wu P, Lin W, Wei H, et al. Regulation of Tak1 alternative splicing by splice-switching oligonucleotides. Biochemical and biophysical research communications. 2018;497(4):1018–24. [DOI] [PubMed] [Google Scholar]
- 43.Tang LY, Heller M, Meng Z, Yu LR, Tang Y, Zhou M, et al. Transforming Growth Factor-beta (TGF-beta) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J Biol Chem. 2017;292(10):4302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: Primer, siRNA, shRNA, sgRNA sequences
Supplemental Table S2: Antibody information