

Abstract
Mechanisms mediating clearance of hepatitis delta virus (HDV) are poorly understood. This study analyzed in detail profound down-regulation of HDV infection in the woodchuck model. Super-infection with HDV of woodchucks chronically infected with HBV-related woodchuck hepatitis virus produced two patterns. In the first, HDV viremia had a sharp peak followed by a considerable decline, and initial rise of HDV virions’ infectivity followed by abrupt infectivity loss. In the second, HDV titer rose and later displayed plateau-like profile with high HDV levels; and HDV infectivity became persistently high when HDV titer reached the plateau. The infectivity loss was not due to defects in the virions’ envelope, binding to anti-envelope antibodies, or mutations in HDV genome, but it correlated with profound reduction of the replication capacity of virion-associated HDV genomes. Subsequent finding that in virions with reduced infectivity most HDV RNAs were not full-length genomes suggests possible HDV clearance via RNA fragmentation.
Keywords: hepatitis delta virus, HDV-host interactions, regulation of HDV infection outcome
INTRODUCTION
Human hepatitis delta virus (HDV) is a sub-viral agent of hepatitis B virus (HBV). HDV takes only the envelope proteins from HBV in order to assemble its own virions and infect hepatocytes via the HBV receptor, sodium taurocholate cotransporting polypeptide (NTCP) (1–3). HDV does not use HBV replication machinery, and acquires all other needed factors from the host. The genome of HDV is an approximately 1,700 nucleotides long single stranded circular negative sense RNA. Viral genome replicates through its exact complement antigenome via RNA-directed RNA synthesis, which is catalyzed by cellular DNA-dependent RNA polymerase II. Both genome and antigenome have ribozymes (4–5). HDV encodes only one protein, the delta antigen. There are two forms of delta antigen - small ( AgS) and large ( AgL). The AgL has extended C-terminus as compared to AgS. This extension is the result of RNA editing of the antigenome performed by the host adenosine deaminase acting on RNA, or ADAR-1. Deamination converts the adenosine at position 1012 into inosine, which results in the change of the amber termination codon to tryptophan codon thus extending the C-terminus of delta antigen. The AgS is essential for viral RNA replication. Large delta antigen does not support replication, but it binds the small cytosolic loop, which is located in the S domain on all HBV envelope proteins, and thus facilitates assembly of HDV virions (2, 4–7). HBV encodes three envelope proteins, the small (S), middle (M) and large (L). The M has additional N-terminal PreS2 domain as compared to the S, while the L has an additional N-terminal PreS1 domain as compared to the M. The PreS1 domain interacts with the HBV receptor. The expression of the L and S is sufficient to from infectious HBV and HDV particles (2, 8–13). The sequence of the small cytosolic loop or HDV-binding site (HDV-BS) is very conserved among the orthohepadnaviruses, which infect humans, higher primates and some rodents (2, 14–16). This includes woodchuck hepatitis virus (WHV), which is an orthohepadnavirus that is closely related to HBV. WHV is often used as the HBV surrogate. HBV and WHV have similar genomes, virions, mechanisms of replication, infection profiles, and induce hepatocellular carcinoma (HCC). WHV-infected woodchucks are an invaluable model to study HBV infection and related pathogenesis, and to test anti-HBV and anti-HCC strategies. The woodchuck model is also used to study mechanisms of the immune response to HBV and HDV, and to develop immunotherapies against HBV. The studies that used recently resolved woodchuck blood and liver transcriptomes further showed considerable similarities in hepadnavirus infection courses and in associated pathogenesis in humans and woodchucks thus increasing significance and biological relevance of the woodchuck experimental model (16–26). In addition, WHV also serves as a helper virus for HDV instead of HBV in order to conduct the studies that employ co- or super-infection strategies in the woodchuck model, and analyze the mechanisms of transient and chronic HDV infection in vivo (16, 27).
HDV remains a significant human pathogen that causes transient and chronic liver infections. There are about twenty million chronic HBV/HDV carriers worldwide. Most HBV/HDV co-infections are clinically resolved. More than eighty percent of HDV super-infections, when chronic HBV carriers are super-infected with HDV, progress to chronic HDV infection (1, 28–32). HDV can accelerate liver disease and cause faster/more frequent cirrhosis. It increases HCC risk approximately 3-fold as compared to chronic carriers of HBV only. The only approved anti-HDV therapy, interferon (IFN) treatment, is unsatisfactory. Anti-HBV drugs (nucleos(t)ide analogs) do not block HDV infection. No drugs directly targeting HDV exist in clinical practice (5, 28–36).
The mechanisms of (i) HDV-host interactions and (ii) immune response to HDV are not well understood. It was found that anti-Ag antibodies may persist for years, but they do not regulate HDV infection. An HDV-specific cytotoxic T cell response, and oligospecific T helper cell immune response were found in HDV-infected individuals that were negative for HDV (by PCR). As anticipated, these T cell responses were absent in patients with persistent HDV viremia (18, 36–38). The mechanisms determining the outcomes of HDV infection, i.e., whether initial infection will become transient or chronic, including critical determinants of establishment and maintenance of chronic HDV infection, remain poorly understood.
The present study analyzed in detail the mechanism of profound down-regulation of HDV infection observed when two chronic WHV carrier woodchucks were super-infected with identical HDV inoculum. The first observed pattern of the super-infection displayed an early peak of serum HDV titer that was followed by substantial decrease in concentration of serum virus. The second pattern reflected the rise of HDV titer with subsequent establishment of plateau-like serum HDV profile. In case of the first pattern, infectivity of HDV virions initially rose and became considerably high at the time when the titer peaked at week +4 after HDV inoculation, but then it experienced a rapid and abrupt drop to undetectable levels within two weeks. In the second case, HDV infectivity also increased initially, reached considerably high levels by week +8, and then remained at high levels until the end of the study at week +14. We conducted extensive investigation into the reasons of such dramatically different profiles of HDV infectivity. The loss of the infectivity of HDV virions occurred not because (i) the virions’ envelopes were defective, (ii) most of serum HDV particles were bound to the antibodies against WHV envelope proteins, or (iii) virion-associated HDV RNA genomes were mutated. We found that the loss of infectivity correlated with the suppression of the replication capacity of virion-associated HDV genomes. Subsequent experiments suggested that in the virions with reduced infectivity/replication capacity of HDV genomes, the majority of HDV RNA molecules were not intact full-length viral genomes, but rather fragmented RNAs. We speculated that this apparent host-mediated fragmentation of HDV RNA genomes could represent at least one mechanism used by the host in order to facilitate resolution of HDV infection. This is the only study that analyzed the patterns of HDV infectivity over a period of several months after the super-infection, and found specific defects in the virus life cycle indicating that host response to HDV infection may employ the sensors of foreign (viral) RNA, and action of cellular RNase(s). These findings were especially interesting, because the mechanisms determining the outcomes of HDV infection are poorly understood. Furthermore, profound reduction of the replication capacity of virion-associated HDV RNA genomes observed early in infection could be an early predictor of subsequent HDV clearance.
In addition, this study for the first time (i) demonstrated the existence of circulating HDV-antibody complexes, which is the phenomenon similar to the one that was previously observed for HBV and WHV (25, 39–46), and (ii) found that in the same serum samples the fraction of antibody-bound HDV virions was always lower than that of antibody-bound WHV virions and did not depend on the time after HDV super-infection, serum HDV titer or the HDV/WHV ratio in serum samples. The latter results suggest that HDV and WHV virions could be recognized differently by the same antibodies against the envelope proteins regardless of the fact that both viruses are coated with the envelope proteins of WHV. Such lack in recognition/binding of HDV by anti-envelope antibodies may be related to the high chronicity rates observed as a result of HDV super-infection (32).
MATERIALS AND METHODS
Super-infection of chronic WHV carrier woodchucks with HDV.
The woodchucks were housed at Northeastern Wildlife Inc. (Harrison, ID), which is a NIH/OLAW-assured animal facility. All animal work was conducted according to the protocols approved by Northeastern Wildlife’s Institutional Animal Care and Use Committee. Chronic WHV carrier woodchucks were generated by inoculation of neonates with strain WHV7 at three days post-birth (47). Two chronic WHV carriers woodchucks, female F6438 and male M6593, were inoculated intravenously with woodchuck serum derived WHV-enveloped HDV (wHDV) inoculum (~8.3 ×109 HDV genome equivalents (GE)/woodchuck) as described previously (27). The woodchucks were monitored for 14 weeks after the super-infection. The liver biopsies were collected at weeks −1 (prior to the super-infection), and +6, and +12 post-inoculation with wHDV. The serum samples were collected weekly starting with week −1. Sera and liver tissues were also harvested at necropsy at week +14.
Measurements of WHV relaxed circular DNA (rcDNA) in sera.
The isolation of serum WHV rcDNA was conducted as described previously with modifications (24). Each aliquot of serum (50 l) was mixed with 450 l of lysis buffer (0.01 M Tris-HCl, pH 8.0, 0.01 M EDTA, 0.1 M NaCl, 0.2% SDS) containing 1 mg/ml of Pronase (Roche), and incubated at 37 C for two hours. After the incubation, DNA was extracted first with 500 l of phenol and then with a mixture of chloroform/isoamyl alcohol (24:1). The precipitation of DNA was done by adding 2.5 l of 10 µg/µl dextran solution in DNase-/RNase-free water, 0.1 volume of 3.0 M sodium acetate, pH 5.5 and 2 volumes of 100% ethanol. The numbers of WHV rcDNA copies were measured by real-time PCR (qPCR). The calibration curve for the assay was obtained using 10-fold dilution series of the construct pUC-CMV-WHV linearized with NheI. The curve spanned the range from 20 to 2.0×107 GE of WHV (48–49). The qPCR used primers 23 (2504-AGAAGACGCACTCCCTCTCCT-2524) and 24 (2579-TGGCAGATGGAGATTGAGAGC-2559), and TaqMan probe 28 (2531-/6-FAM/AGAAGATCTCAATCACCGCGTCGCAG/3BHQ_1/-2556). The 5-end of the probe was labeled with 6-carboxyfluorescein (6-FAM), while its 3 -end contained a Black Hole Quencher 1 (BHQ_1). The positions of the primers and probes used in this study are based on sequence of strain WHV7 (26). The qPCR was conducted using the TaqMan Gene Expression Mastermix on a 7500 RT-PCR System (Applied Biosystems) (48–49).
Measurements of intrahepatic replicative intermediate DNA (RI-DNA) and covalently closed circular DNA (cccDNA) of WHV.
The pieces of liver tissues were homogenized in glass grinder using 0.6 to 1.5 ml of TE buffer (10:10) (10 mM Tris-HCl pH 7.6, 10 mM EDTA) (48–49). Next, 210 l of the resulting tissue homogenate were combined with 390 l of TE buffer (10:10) and 600 l of a buffer that contained 10 mM Tris-HCl, pH 7.6, 10 mM EDTA, 0.2% SDS and 4 mg/ml Pronase (Roche). The mixture was incubated for two hours at 37 C. Total DNA was then extracted twice with an equal volume of phenol/chloroform; and after that DNA was precipitated by adding 2 volumes of 100% ethanol and 0.1 volumes of 3.0 M sodium acetate, pH 5.5. The DNA sedimentation was achieved by centrifugation for 30 min at 13,000 rpm at 4 C. The precipitated DNA pellets were first washed with 100% ethanol, and then re-dissolved in 25–50 µl of DNase-/RNase-free water (48–49). The measurements of WHV RI-DNA copy numbers were performed by qPCR as it was described for rcDNA.
During cccDNA isolation, 210 l of the above mentioned liver tissue homogenate were mixed with 930 l of TE buffer (10:10) and 60 l of 10% SDS. The resulting mixture was vortexed, and then 300 l of 2.5 M KCl were added to the mixture. The obtained mixture was vortexed again and then incubated for at least 30 min at room temperature. Next, the mixture was centrifuged for half an hour at 13,000 rpm at 4 C. The generated supernatant was extracted first with phenol. The second extraction was done with phenol/chloroform mixture. The DNA then was isolated from the obtained aqueous phase by (i) adding two volumes of 100% ethanol and 2 l of dextran solution (10 µg/µl) in DNase-/RNase-free water, (ii) incubating the resulting mixture overnight at room temperature, with (iii) subsequent centrifugation to precipitate DNA. The obtained DNA pellet was washed twice with 70% ethanol, and then once with 100% ethanol. The DNA pellet was re-suspended in 50 l of DNase-/RNase-free water. Next, 40 l of resulting DNA solution were incubated overnight at 37 C in a 500 l reaction mixture that contained 1x NEB (New England Biolabs) buffer 4, 1 mM ATP, 50 U of Plasmid-Safe ATP-Dependent DNase (Epicentre), and 40 g/ml of RNase A. The cccDNA was next isolated by extraction with phenol and then - with phenol/chloroform mixture followed by ethanol precipitation (48–49). The qPCR for quantification of cccDNA levels employed primers 290 (1701-GGTCCGTGTTGCTTGGTCT-1719) and 291 (1977-GGACATGGAACACAGGCAAAAA CA-1954), and TaqMan probe 292 (1846-/6-FAM/AATGGGAGGAGGGCAGCATTGATCCT/3BHQ_1/-1871). A calibration curve covering the range of concentrations from 20 to 2.0×107 GE of WHV was generated using 10-fold dilution series of pUC-CMV-WHV linearized with NheI. The concentration of DNA in total DNA preparation was determined using the DNA-binding dye Hoechst 33258, and then it was used to normalize the copy numbers of RI-DNA and cccDNA per µg of total DNA (48–49).
Analysis of pre-genomic RNA (pgRNA) in liver tissue samples.
Total RNA was isolated from liver tissue samples with TRI reagent (Molecular Research Center) according to the instructions of the manufacturer and then treated using Turbo DNase (Life Technologies). The primary goal of DNase treatment was to eliminate WHV DNA replication intermediates. After the DNase treatment, RNA was re-extracted with TRI reagent. The resulting RNA was reverse transcribed using WHV-specific primer 850 (2602-TGTACCCATTGAAGATCAGCAGTT-2579) and High Capacity cDNA Reverse Transcription Kit (Life Technologies). The pgRNA copy numbers were quantified via qPCR that employed the primers 23 and 24, and TaqMan probe 28. The WHV7 RNA standard for the quantification was in vitro transcribed from the plasmid pJSWHV7–2C6 linearized with XhoI, and gel-purified. The calibration curve for qPCR was generated using a 10-fold dilution series of the WHV7 RNA standard within a range of 20 to 2.0×106 GE of WHV RNA (26). The pgRNA levels were expressed per µg of total RNA.
Quantification of HDV genomes.
The genomic (G) RNA of HDV was quantified in total RNA isolated with TRI reagent from the sera, liver tissues or infected primary woodchuck hepatocytes as described previously (27, 50). The HDV G RNA was reverse transcribed using the reverse primer 22 (393-CCTAGCATCTCCTCCTATCGCTAT-360). The same reverse primer, forward primer 21 (312-GGACCCCTTCAGCGAACA-329), and TaqMan probe 27 (393-/6-FAM/CCTAGCATCTCC TCCTATCGCTAT/3BHQ_1/-360) were employed for qPCR. The HDV numbering in this study was according to Kuo et. al (51). The calibration curve was generated using the in vitro transcribed and gel-purified HDV G RNA standard, and it covered the range of G RNA amounts between 20 and 2×106 GE of HDV (27, 50, 52).
Fraction of serum WHV and HDV virions bound to anti-WHsAg antibodies.
The measurements of the virions bound to anti-WHsAg antibodies were done as described previously (48–49). Aliquots of serum were incubated with 100 μl of Pansorbin (Calbiochem) in Williams’ E Medium (in a total volume of 1 ml) at 4°C for 3 hours using a rocking platform. During the incubation, the WHV and HDV virions that were bound to anti-WHsAg antibodies became attached to Pansorbin, and were precipitated by subsequent brief centrifugation for one min at 13,000 rpm. The precipitated immune complexes were washed with ice-cold PBS containing 0.5% Nonidet P-40 (Fisher) four times, and then subjected to isolation of total DNA (for antibody-bound WHV) or RNA (for antibody-bound HDV). The procedures for isolation of either DNA or RNA from sera and quantification of WHV rcDNA or HDV G RNA are described in the preceding sections.
Preparation of primary woodchuck hepatocytes (PWH) and in vitro infection of PWH with serum-derived wHDV.
Primary woodchuck hepatocytes (PWH) were prepared using a modification of previously published protocols (53–54). PWH were isolated from the whole liver of WHV-negative adult woodchucks. Briefly, a three-step collagenase perfusion method was used. All perfusion solutions were maintained at 37°C using a heated water bath and the perfusion was carried out using a peristaltic pump. The liver was perfused ex situ via the hepatic vein at a constant flow rate 30–40 ml/min for 10–15 min with Calcium and Magnesium free Hank’s Balanced Salt Solution (HBSS) containing 0.1 mM EGTA followed by a washout step using Calcium and Magnesium free HBSS without EGTA. The final perfusion step used EMEM (Mediatech) containing 25 mM HEPES buffer (Mediatech) and 0.025 mg/ml of Liberase™ (Roche). This step continued until liver revealed the signs of digestion (approximately 10–20 min.). The liver was then placed in a small sterile beaker with cold EMEM containing 25 mM HEPES and chopped using sterile scissors to release the liver cells. The cell suspension was filtered through nylon gauze (100 μm) and collected in 50 ml conical tubes. The cells were centrifuged for 5 min at 50 x g and 4˚C and then resuspended in a fresh cold EMEM with 25 mM HEPES. This step was repeated 3 times in order to isolate the hepatocyte fraction. Hepatocyte viability was evaluated by the trypan blue exclusion method and the number of hepatocytes isolated was determined using a hemacytometer. The prepared PWH were resuspended in Williams E Medium supplemented with the primary hepatocyte maintenance supplement CM4000 (Thermo Fisher Scientific), seeded at about 2×105 cells/well onto 48-well Corning™ BioCoat™ Collagen I plates (Corning Life Sciences), and infected with serum wHDV next day essentially as described previously (50, 52). The infections were performed at multiplicity of infection (MOI) of 2.0 HDV GE/cell in the equal volume of serum in the presence of 5% of polyethylene glycol (PEG) 8000 (Sigma). The PWH were incubated overnight with the infection mixture containing wHDV. Next morning the media was replaced with regular hepatocyte media (Williams E Medium-CM4000), and infected PWH were maintained in culture for 9 days. At day 9 post-infection, PWH were harvested and total RNA was isolated with TRI reagent and HDV G RNA was measured by qPCR (27, 50, 52).
Treatment of HDV virions using benzonase nuclease.
To ascertain whether virion-associated serum HDV G RNA was protected by the virions’ envelopes, the aliquots of serum containing wHDV (10 µl) were incubated with 100 U of benzonase nuclease (EMD Millipore) in 50 µl volume reaction mixture containing 50 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 100 µg/ml BSA for 22 hours at 37°C according to the manufacturer’s instructions. The reaction was stopped by adding EDTA at a final concentration of 2 mM to inactivate the benzonase. Aliquots of serum, for which benzonase treatment was omitted, were handled similarly. The preparation of “naked” wHDV RNA isolated from serum was used as the control for efficiency of the treatment with benzonase. After the incubation, the total RNA was extracted with TRI reagent and HDV G RNA was quantified by qPCR (27, 50, 52).
Analysis of replication capacity of HDV RNA.
Total RNA from sera or liver tissue samples was isolated with TRI reagent. Using Lipofectamine 2000 (Invitrogen), the isolated RNA was transfected into Ag-293 cells that express the small delta antigen of HDV ( AgS) in the presence of tetracycline (55). The 48-well plates were used for seeding of Ag-293 cells. We used 0.1 to 1.0 HDV GE/cell during the transfection. All wells were transfected with the same total amount of RNA. In case of serum RNA, total RNA from Huh7 cells was used to bring the total RNA amounts to 0.5 µg/well. In case of liver RNA, we used RiboMinus™ Eukaryote Kit (Life technologies) according to the instructions of the manufacturer in order to remove the cellular ribosomal RNA. If needed, the total RNA from the uninfected woodchuck liver was used to bring the total RNA amounts to 2 µg/well during the transfection. The cells were incubated with transfection media overnight, and then the media was replaced with regular cell media containing 1 µg/ml of tetracycline. The cells were kept in the presence of tetracycline for 48 hours, after which the tetracycline was omitted. For the next seven days the transfected cells were cultured in the absence of tetracycline. Next, the total RNA was isolated using TRI reagent, and HDV G RNA was measured by qPCR (27, 50, 52).
Sequence analysis of serum-associated HDV RNAs.
The sequencing of serum HDV G RNA was performed via RT-PCR using three overlapping PCR amplicons. The amplicon 1 spanned the positions 1325 to 1640, while the amplicon 2 – 1094 to 1404, and the amplicon 3 – 770 to 1122. The cDNA synthesis was done using the reverse primer 1064 (27-GTCTCCTCGCTCGGAACTTG-8) and cDNA Reverse Transcription Kit (Applied Biosystems). The primers 1078 (1303-CTCTCCTTGTCGGTGAATCCTC-1324) and 1081 (1663-GAGTCCAGCAGTCTCCTCTTTAC-1641) were used for the amplicon 1. The primers 1083 (1075-GGGACTCCCTGCAGATTGG-1093) and 1086 (1428-TCTCGGAAAGAAGGATAAGGATGG-1405) were used for the amplicon 2, while the primers 1051 (749-CTCGGTAATGGCGAATGGGAC-769) and 1071 (1142-AACCCCCTCGAAGGTGGATC-1123) were used for the amplicon 3. The Klentaq DNA polymerase (AB Bioscience) was used for all PCR reactions. All PCR reactions were conducted using 2.5 µl out of 20 µl of the cDNA reaction mixture as a template. The final PCR reaction products were gel-purified, cloned into the pCR®II-TOPO vector (Invitrogen), and resulting plasmids were subjected for sequencing.
Analysis of the effects of mutations on the replication capacity of HDV genomes.
Using the QuickChange Lightning site-directed mutagenesis kit II (Agilent Technologies), the mutations of interest found in serum HDV G RNA by sequencing were introduced into the construct pDL542 that expresses a greater than unit-length HDV RNA genome, which does not produce Ag but can replicate if functional AgS is provided in-trans (56). Using Lipofectamine 2000, a mutation-bearing construct was transfected into AgS-293 cells (55), and then replication capacity of mutated HDV G RNA was analyzed by providing AgS in-trans in the presence of tetracycline. The cells were harvested at day 9 post-transfection, and accumulation of HDV G RNA was measured using qPCR.
Analysis of the mutations in small delta antigen on its ability to support HDV RNA replication.
The mutations of interest found in the AgS open reading frame were introduced into the plasmid pDL444 that expresses wild type AgS (57) using the site-directed mutagenesis approach described in the previous section. Next, a mutation-bearing version of pDL444 was transfected along with unmodified pDL542 (in 1:1 ratio) into Huh7 cells. In this case, the replication of HDV RNA was expected to be observed if mutated AgS could support it. Nine days later, the cells were harvested using TRI reagent, and HDV G RNA was quantified by qPCR.
Northern analysis of serum HDV RNAs.
The Northern analysis was performed as described previously (58–59). Briefly, total RNA isolated from serum samples was glyoxalated, and analyzed using 1.7% agarose gel. The RNAs were then transferred from the gel to the charged nylon Zeta-probe membrane (Bio-Rad). The HDV G RNA was detected using 32P-labeled ribo-probe generated by in vitro transcription using the plasmid pTW107 linearized with HindIII as a template. The hybridization with the HDV G RNA-specific probe was conducted overnight at 75 C in ULTRAhyb™ Ultrasensitive Hybridization Buffer (Thermo Fisher Scientific), containing 0.1 mg/ml of sheared salmon sperm DNA. Radioactively-labeled bands were visualized and analyzed using a Typhoon FLA 9000 biomolecular imager (GE Healthcare). The quantifications were performed using ImageJ software.
RESULTS
Serum and intrahepatic markers of HDV and WHV infections.
Two chronic WHV carrier woodchucks super-infected with identical HDV inoculum displayed two very different patterns of HDV infection (Fig. 1). For M6593, serum HDV titer increased to levels above 108 HDV GE/ml by week +4 after HDV inoculation, and then significantly declined below 105 HDV GE/ml by week +14, which could suggest subsequent resolution of HDV infection. At the same time, the titer of serum WHV did not change significantly during the entire monitoring period and was close to or above 1010 of WHV GE/ml (Fig. 1). For F6438, HDV titer increased gradually to 5.95×109 HDV GE/ml by week +10, and remained plateau-like and relatively high above the level of 9.0×108 GE/ml until the end of the study (Fig. 1). This profile of serum HDV could be consistent with subsequent establishment of chronic HDV infection. In this woodchuck, the levels of serum WHV fluctuated somewhat more than in case of M6593, but still remained relatively high at all times between 2.86×109 and 2.07×1010 GE/ml. The present study was focused on analysis of the mechanisms mediating the observed different profiles of HDV infection in the super-infection settings in vivo, and especially on understanding of mechanism(s) facilitating profound down-regulation of HDV infection observed for M6593 (Fig. 1).
Fig 1. Serum titers of WHV and HDV in chronic WHV carrier woodchucks super-infected with HDV.
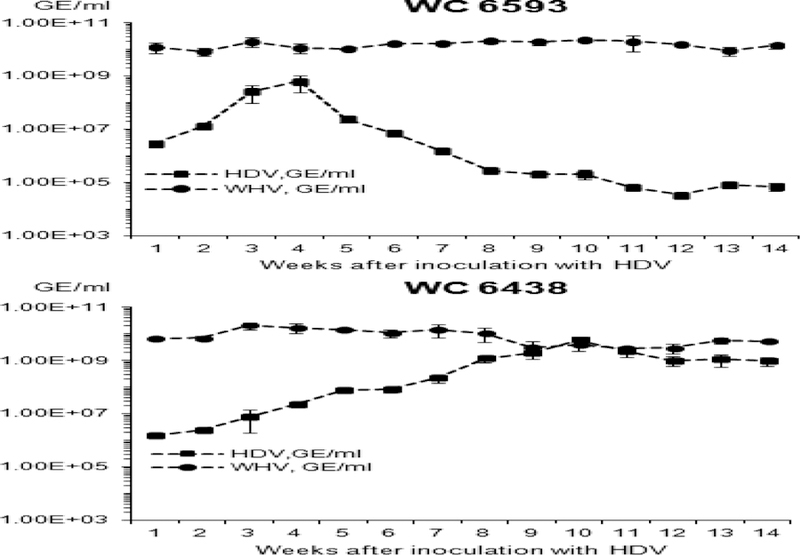
Two woodchucks chronically infected with WHV were super-infected with wHDV and monitored for 14 weeks. The Y axis reflects serum concentration of WHV (black circles) or HDV (black rectangles) in logarithmic scale. The X axis represents the time (week) after super-infection with HDV. The top panel shows the results for woodchuck M6593, while the bottom panel - for woodchuck F6438.
We next analyzed the intrahepatic markers of WHV and HDV replication. The results are summarized in Fig. 2. In agreement with the observed patterns of serum HDV titers, F6438 demonstrated much higher levels of accumulation of HDV genomic (G) RNA than those measured for M6593. By week +6, they reached about 106 HDV GE/µg of total RNA, and then were within the range of (2.5 to 3.0)x107 HDV GE/µg of total RNA (Fig. 2A). In case of M6593, the levels of HDV G RNA dropped from 2.38×104 GE/µg to 6.19×102 GE/µg of total RNA from week +6 to week +12. For each woodchuck, we also analyzed two different areas of the liver collected at necropsy (week +14), and for M6593, in one area HDV levels were about 2.2×102 GE/µg of total RNA, while in a different area the amounts of HDV genomes were about 100-fold higher (Fig. 2A), which shows that HDV infection of the liver is not uniform, and sometimes the accumulation of HDV RNA may differ significantly between different liver areas.
Fig. 2. Intrahepatic replication markers of HDV and WHV in woodchucks M6593 and F6438.
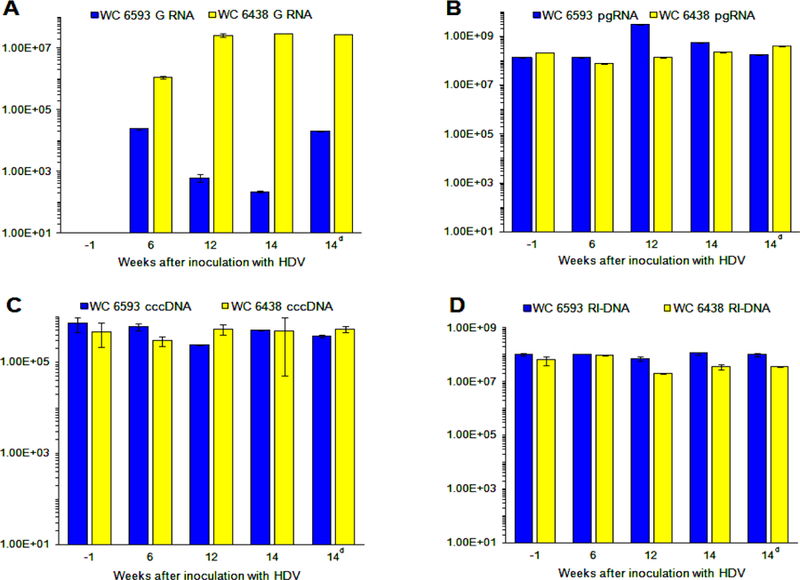
The intrahepatic levels of HDV G RNA (panel A), and pgRNA (panel B), cccDNA (panel C) and RI-DNA (panel D) of WHV were measured using specific qPCR assays. The details are described in Materials and Methods. The X axis on every panel represents time in weeks relatively to the time of HDV super-infection. (Thus, for example, week -1 means 1 week prior to the super-infection, while week 12 means 12 weeks after the super-infection). The Y axis represents the corresponding replication marker values in logarithmic scale. The HDV RNA levels are expressed in HDV GE/µg of total RNA, the pgRNA levels are shown in WHV GE/µg of total RNA, while the levels of RI-DNA and cccDNA are shown in WHV GE/µg of total DNA. For the latter two cases, the concentration of DNA in a total DNA preparation was used for calculations. The data for WC 6593 are shown as blue bars, while the data for WC 6438 are shown as yellow bars. During analysis of tissues harvested at necropsy (week +14), two different areas of harvested livers were analyzed and compared (the data for the second area is marked as 14d on time scale (d - duplicate)).
For M6593, the pgRNA amounts ranged between 1.33×108 and 5.69×108 WHV GE/µg of total RNA, while only at week +12, a somewhat higher pgRNA levels of 3.12×109 GE/µg of total RNA were measured (Fig. 2B). The levels of cccDNA and RI-DNA of WHV did not display significant changes, and were (2.40 to 7.20)x105 WHV GE/µg of total DNA and (0.73 to 1.16)x108 WHV GE/µg of total DNA, respectively, for the entire monitoring period (Figs. 2C and 2D). Similarly, in case of F6438, the levels of the three WHV replication markers did not significantly change during the entire study. The pgRNA levels were in the range from 7.53×107 to 3.87×108 WHV GE/µg of total RNA (Fig. 2B), the amounts of cccDNA were in the range of (2.97 to 5.40)x105 WHV GE/µg of total DNA (Fig. 2C), and the numbers of RI-DNA were in the range of (2.01 to 9.61)x107 WHV GE/µg of total DNA (Fig. 2D). In summary, these results suggested that concomitant HDV infection did not have a significant influence on the overall levels of intrahepatic WHV genome replication (Fig. 2).
Specific infectivity of serum HDV virions.
Our further investigation was driven by the overall hypothesis that the host may down-regulate HDV super-infection by altering the HDV life cycle: i.e., by facilitating the emergence of defective virus. With this in mind, we investigated a number of virus properties starting with the analysis of infectivity of serum HDV. Using the livers of WHV-negative adult woodchucks, we prepared the cultures of primary woodchuck hepatocytes (PWH), and infected plated PWH in vitro with aliquots of serum HDV collected from M6593 and F6438 at different times after HDV super-infection. Nine days later, total RNA was isolated from infected PWH, and amounts of accumulated HDV G RNA were quantified by qPCR. Based on the qPCR results, we calculated the specific infectivity (SI) of HDV. The SI is a normalized parameter that does not depend on HDV titer. It shows how many new HDV genomes/per hepatocyte were produced as a result of HDV infection per each HDV genome that was used per hepatocyte in the inoculum (50). Thus, the SI characterizes the potential of HDV virions in the inoculum to induce productive infection in PWH. The results are summarized in Fig. 3. For M6593, for which the down-regulation of HDV infection was observed, the infectivity initially rose, and reached relatively high levels by week +4. Then, within one week, the infectivity dramatically dropped more than 50-fold down to barely detectable levels, and already at week +6 HDV virions appeared non-infectious. The very low HDV titers at later times precluded the measurements of the infectivity in the samples collected after week +6 (Fig. 3, top panel). The observed loss of HDV infectivity correlated with decline of HDV titers, and may suggest that host-mediated suppression of HDV spread and super-infection could be important for resolution of HDV infection.
Fig. 3. Infectivity of serum HDV virions harvested from woodchucks M6593 and F6438.
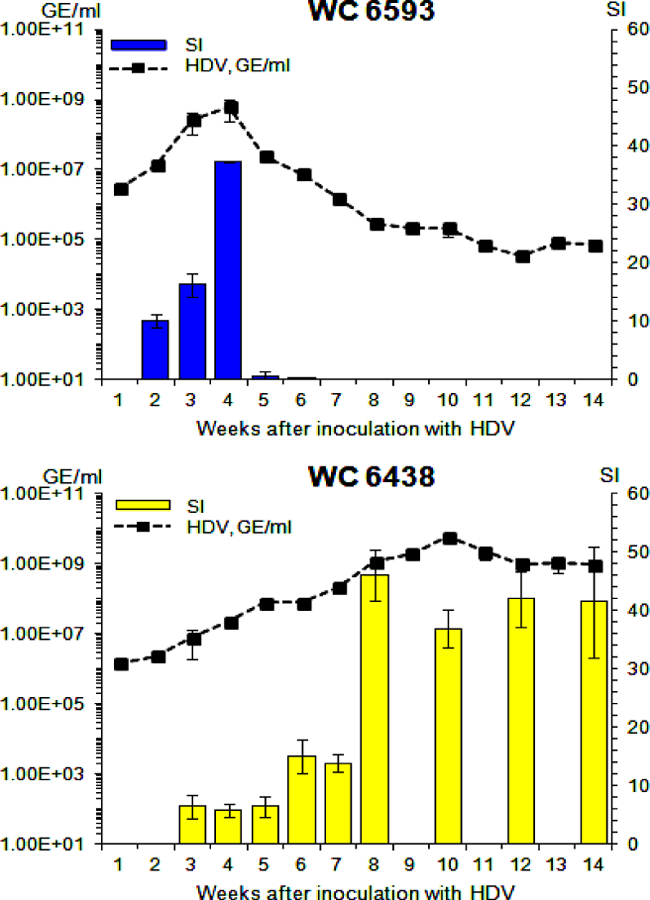
The infectivity of serum HDV virions collected at different time points was analyzed using in vitro infection of primary woodchucks hepatocytes. The details are described in Materials and Methods and in Results. The left Y axis reflects serum concentration of HDV (black rectangles) in logarithmic scale. The right Y axis represents the values of specific infectivity (SI), which is a normalized parameter that shows how many new HDV genomes/hepatocyte were produced as a result of infection per each HDV genome used per hepatocyte in the inoculum. The SI values are shown as blue (M6593) or yellow (F6438) bars. The X axis represents the time (week) after super-infection with HDV. The top panel shows the results for woodchuck M6593, while the bottom panel -for woodchuck F6438.
The situation was quite different for F6438. In this case, the infectivity initially increased, and reached relatively high levels by week +8, which were somewhat higher than the SI level found for M6593 at week +4. Interestingly, the infectivity remained relatively high with minor fluctuations thereafter (Fig. 3, bottom panel). The high SI levels coincided with serum HDV titer reaching the plateau-like high levels. These data suggest that maintenance of sufficiently high infectivity supporting continuous virus spread and super-infection might be critical for establishment and maintenance of chronic HDV infection.
Overall, the results indicated that serum HDV virions harvested from M6593 at week +5 and thereafter were defective in terms of infectivity. The infectivity of the virions could be regulated (i) by binding to antibodies against the envelope proteins, (ii) by the properties of the envelope proteins, and (iii) by the properties of the HDV ribonucleoprotein (RNP) residing within the virions, which consists of HDV RNA genome bound to a certain number of Ag molecules (1, 4). Our further efforts were focused on investigating the mechanism(s) facilitating profoundly different profiles of HDV infectivity as summarized in Fig. 3.
Fraction of antibody-bound WHV and HDV virions.
We used immuno-precipitation with pansorbin to quantify the percentage of serum virions bound to anti-envelope antibodies (i.e., anti-WHsAg antibodies) as previously described (48–49). The results are summarized in Table 1. First, we analyzed the samples collected from M6593 and F6438. We found that in all cases the fraction of antibody-bound HDV virions was always lower than that of antibody-bound WHV virions. This tendency did not depend on the time after HDV super-infection, the titer of HDV, and WHV/HDV ratio in sera (Table 1, Fig. 1). To investigate whether the observed phenomenon is common for chronic WHV carrier woodchucks super-infected with HDV, we analyzed additional serum samples from our collection harvested from three other woodchucks. The outcomes of the analysis confirmed our initial observations. Overall, in the same serum samples the fraction of antibody-bound HDV was 2.7 to 66.8-fold lower than the fraction of antibody-bound WHV virions (Table 1). There was only one exception noted. For woodchuck 6438 at week +2, the fraction of antibody-bound HDV virons was 8.17% that was less than 2-fold lower than that of antibody-bound WHV (Table 1). Thus, in all other cases, the percentage of antibody-bound HDV virions did not exceed 3.4%, while the same serum samples could contain up to 53.87% of WHV in complexes with antibodies (Table 1). These observations were surprising, considering that both viruses were coated with WHV envelope proteins. Possible explanations of the data shown in Table 1 are presented in the Discussion. The generated data strongly suggest that the small fraction of HDV virions bound to anti-WHsAg antibodies (Table 1) indicates that the antibodies did not down-regulate the overall infectivity of the pools of serum HDV virions in the settings of super-infection. Furthermore, the results indicate that binding of HDV virions to anti-WHsAg antibodies likely have very little or no effect on the outcome of HDV super-infection.
Table 1.
Fractions of serum WHV and HDV virions bound to anti-WHsAg antibodies.
| Woodchuck | Time after wHDV super-infection (week) | Fraction of virions bound to anti-WHsAg antibodies,% a | ||
|---|---|---|---|---|
| WHV | HDV | Fold difference WHV/HDV b | ||
| 6593 | +2 | 53.87 ± 2.29 | 1.98 ± 0.72 | 27.21 |
| +4 | 32.18 ± 2.34 | 3.40± 1.10 | 9.45 | |
| +6 | 26.40 ± 1.22 | 3.28 ± 1.59 | 8.04 | |
| +8 | 28.97 ± 0.68 | und c | ND d | |
| +11 | 27.02 ±2.17 | und c | ND d | |
| 6438 | +2 | 14.01 ± 3.04 | 8.17 ± 4.44 | 1.71 |
| +4 | 19.04 ± 1.24 | 0.67 ± 0.24 | 28.42 | |
| +8 | 11.37 ± 3.13 | 1.99 ± 0.35 | 5.72 | |
| +10 | 4.17 ± 1.10 | 1.53 ± 0.39 | 2.73 | |
| +12 | 4.03 ± 1.18 | 1.48 ± 0.18 | 2.73 | |
| 7807 | +3 | 51.27 ± 3.85 | 1.85 ± 0.30 | 27.65 |
| +6 | 32.92 ± 3.52 | 0.51 ± 0.09 | 64.94 | |
| 7724 | +2 | 10.11 ± 4.65 | 0.15 ± 0.09 | 66.83 |
| +4 | 7.54 ± 3.11 | 0.16 ± 0.08 | 45.96 | |
| +5 | 15.08 ± 3.46 | 0.36 ± 0.17 | 42.07 | |
| +6 | 12.63 ± 2.17 | 1.12 ± 0.2 | 11.30 | |
| 4166 | N/A e | 11.10 ± 0.62 | 2.63 ± 0.49 | 4.22 |
The fraction of antibody-bound virions represents a percentage of the virions in the context of the immune complexes with anti-WHsAg antibodies out of total number of serum virions of WHV or HDV.
The fold difference represents the ratio of the fraction of antibody-bound WHV/fraction of antibody-bound HDV in the same serum sample.
“Und” stands for “undetectable”, which means that in this case antibody-bound virions could not be detected by our assay.
“ND” stands for “not determined”, because in this case the antibody-bound HDV could not be detected, and as a consequence the fold-difference although is very significant, cannot be quantified.
N/A stands for “not available”, i.e., the information at what time point the serum from woodchuck 4166 was collected was not available to us.
Integrity of the envelopes of HDV virions.
Another possibility was that HDV virions in M6593 could have defective envelopes that are impaired in terms of protecting HDV RNP from the outside environment within sera. This hypothesis was addressed using the treatment of serum virions with benzonase nuclease. The results of the treatment are presented in Fig. 4. The virions collected at weeks +3, +6 and +14 from M6593 and F6438 demonstrated significant resistance to benzonase nuclease, and thus well protected virion-associated HDV genomes (Fig. 4). The data therefore show that the loss of HDV infectivity (Fig. 3) could not be explained by defects in the outer envelope of circulating HDV virions.
Fig. 4. Analysis of the integrity of the envelopes of HDV virions harvested from woodchucks M6593 and F6438.
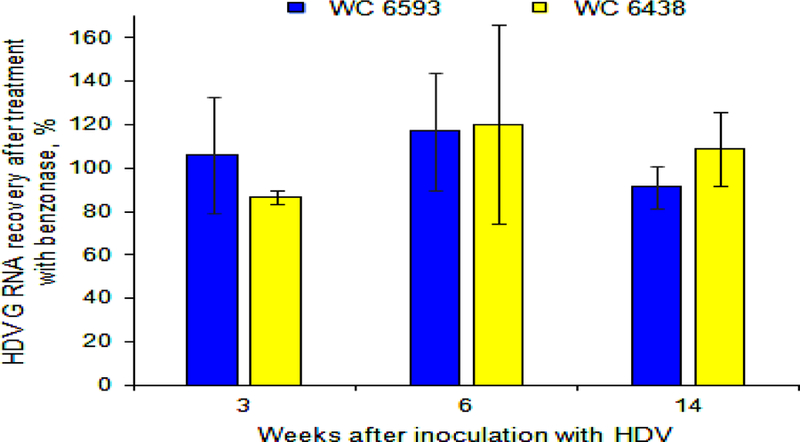
The integrity of the envelopes of HDV virions was analyzed using the treatment with benzonase endonuclease. The details are described in Materials and Methods and in Results. The X axis represents time in weeks after the HDV super-infection. The Y axis represents the percentage of HDV G RNA recovery, which was calculated relatively to HDV titer value in the corresponding sample that was not treated with benzonase, and thus was used as 100% value. The data for WC 6593 are shown as blue bars, while the data for WC 6438 are shown as yellow bars. As a control, we used HDV G RNA that was extracted from serum sample collected from F6438 at week +14. In this case, after the treatment with benzonase, only 0.04% of G RNA was recovered (not shown).
Replication capacity of virion-associated and intrahepatic HDV RNA genomes.
We next examined the replication capacity of the viron-associated HDV RNA genomes. The RNA from virions was isolated using TRI reagent, and then transfected into Ag-293 cells. The HDV RNA replication was started by providing wild type (wt) AgS in the presence of tetracycline for 48 hours. Then, tetracycline was omitted, and HDV genomes had to rely on their own replication capacity and production of necessary functional AgS as a consequence of HDV genome replication for the next 7 days. Nine days post-transfection, the content of HDV G RNA in Ag-293 cells was determined by qPCR. The replication capacity was defined as a normalized parameter that shows how many new HDV genomes/per cell were produced as a result of the transfection per every HDV genome that was used per cell during the transfection procedure. As a control, we used in vitro transcribed and gel-purified unit length (i.e., full genome size) HDV RNA (58). The replication capacity measured for the control RNA was considered as 100% value. The results are shown in Fig. 5. For M6593, at weeks +2, +3, and +4, the replication capacity of the genomes was relatively high (Fig. 5, top panel), which was consistent with the levels of observed infectivity (Fig. 3). Beginning at week +5, the replication capacity was significantly reduced to 13.4%, and then further decreased to about 2.4% by week +14 (Fig. 5, top panel). The loss of replication capacity correlated with the previously observed loss of HDV infectivity at the same time points (Fig. 3). The obtained results suggest that the loss of infectivity, at least in large part, was likely mediated by the reduced ability of HDV genomes to replicate (i.e., support the RNA-directed RNA synthesis).
Fig. 5. Replication capacity of virion-associated HDV RNA genomes harvested from woodchucks M6593 and F6438.
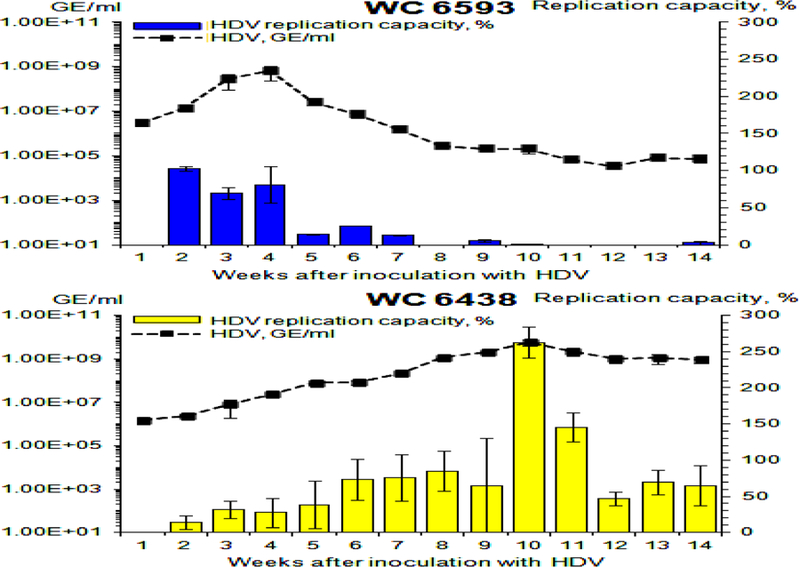
The replication capacity of HDV genomes was analyzed using transfection of HDV RNA isolated from serum virions into Ag-293 cells. The details are described in Materials and Methods. The left Y axis reflects serum concentration of HDV (black rectangles) in logarithmic scale. The right Y axis represents the values of replication capacity, which is a normalized parameter that shows how many new HDV genomes/cell were produced as a result of transfection per each HDV genome used per cell in the transfection mixture. In vitro transcribed and gel-purified unit length HDV RNA (58) was used a positive control. The level of the replication capacity measured for this control RNA was considered as 100% value. The values of the replication capacity (%) are shown as blue (M6593) or yellow (F6438) bars. The X axis represents the time (week) after super-infection with HDV. The top panel shows the results for woodchuck M6593, while the bottom panel - for woodchuck F6438.
A very different pattern of replication capacity of HDV genomes was found for F6438. The replication capacity rose and exceeded 70% by week +6, and remained sufficiently high thereafter (Fig. 5, bottom panel) to support the relatively high infectivity levels (Fig. 3). The highest levels of replication capacity were observed for weeks +10 and +11. They exceeded 250% and 140%, respectively, at these times (Fig. 5, bottom panel). Clearly, in this case, the levels of the HDV genome replication capacity correlated again with the levels of the infectivity of HDV virions. These results suggest that the host may suppress HDV infection by profoundly reducing the replication capacity of at least virion-associated HDV genomes.
In addition, we analyzed the replication capacity of intrahepatic HDV genomes using the same approach as for serum HDV G RNAs (Fig. 6). The obstacle here was a very low content of intrahepatic HDV G RNA in liver tissues harvested from M6593 (Fig. 2), which interfered with isolating enough HDV genomes for transfection. We tried to overcome this problem by eliminating the ribosomal RNA from the final RNA preparations using RiboMinus™ Eukaryote Kit (Life technologies). This was not an issue for F6438, which had sufficient amounts of intrahepatic HDV G RNA (Fig. 2). As mentioned above the positive control here was in vitro transcribed and gel purified HDV G RNA (58). The results are presented in Fig. 6. The HDV genomes isolated from the liver of F6438 had sufficiently high replication capacity at all time points tested. In case of M6593, the genomes isolated from the liver biopsy collected at week +6 showed somewhat reduced replication capacity (Fig. 6). This may indicate that defects in HDV genomes, at least in part, may occur in hepatocytes, which, however, does not eliminate the possibility that the genomes were also altered within the virions. However, since, we were able to measure the replication capacity of HDV genomes only for one time point for M6593 (i.e., for week +6), the making of generalizing conclusions at this time appears premature. Thus, analysis of a considerable number of intrahepatic RNA samples from woodchucks resolving HDV infection, is warranted for the follow-up studies.
Fig. 6. Replication capacity of intrahepatic HDV RNA genomes harvested from woodchucks M6593 and F6438.
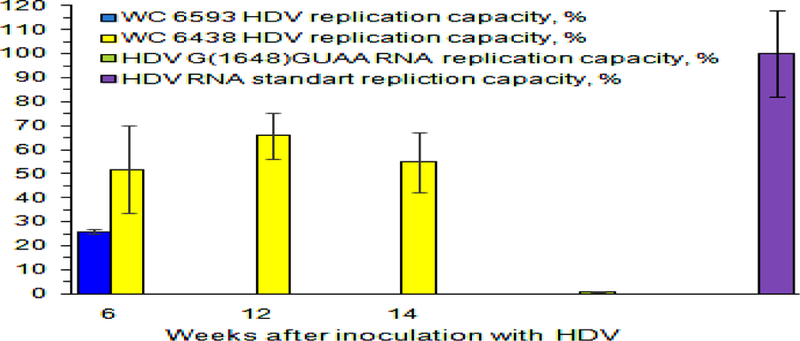
Y axis represents replication capacity of HDV genomes, %. The values of the replication capacity (%) are shown as colored bars. Replication capacity is a normalized parameter, which shows how many HDV genomes/cell were produced as a result of transfection per every HDV genome that was used per cell during the transfection procedure. The value of the replication capacity obtained for in vitro made gel-purified unit length HDV G RNA was used as 100% value. Intrahepatic RNA was isolated from the liver tissue samples obtained from F6438 at weeks +6, +12, and +14 and analyzed. Due to very low intrahepatic HDV G RNA content, the amounts of HDV G RNA that are sufficient for transfection could only be isolated for the week +6 sample of M6593. The time after HDV inoculation (week) is shown at the bottom. As a negative control, the in vitro made gel-purified HDV RNA bearing the mutation G(1648)GUAA, which replication capacity was expected to be profoundly reduced (59), was used during the transfection.
Analysis of mutations in serum HDV genomes.
Because the loss of replication capacity was found for HDV genomes harvested from M6593 (Fig. 5), we next addressed whether any mutations in HDV G RNA were responsible for the reduced replication capacity. An extensive sequencing of the genomes isolated from serum samples of M6593, F6438, and from the serum HDV inoculum was conducted. Three mutations were found that were unique for M6593, observed at different time points, and were not found in F6438 and in the inoculum (Table 2) with one exception. The mutation U(1221)C was also detected at week +2 in 80% of HDV genomes in F6438. Other changes that occurred at low frequency were not considered for further examination. In addition, there were no areas, in which mutations were clustered. All three abovementioned nucleotide changes occurred in the region of the open reading frame (ORF) for AgS. The first change U(1221)C did not lead to an amino acid change, and was observed at all time points tested from week +2 to week +14. The fraction of the genomes with this change varied from 15% to 91.7% (Table 2). The second mutation U(1262)C caused the amino acid substitution Lys(113)Glu, while the third change U(1469)A caused the change Ile(44) Leu. The change U(1262)C was observed at weeks +6, +11 and +14, but it was not detected at earlier times and at week +8. The fraction of HDV genomes with this change ranged between 10 and 35%. The mutation U(1469)A was also observed at weeks +6, +11 and +14, and the fraction of HDV genomes bearing this mutation varied from 8 to 55.5% (Table 2). These mutations could potentially alter the ability of HDV G RNA to support RNA-directed RNA synthesis, and/or produce mutated AgS that is impaired in terms of supporting the replication of the viral genome. We therefore examined both these possibilities.
Table 2.
Unique mutations observed in serum HDV harvested from M6593 a.
| Mutation b | Frequency of the occurrence of the mutation, % c | ||||||
|---|---|---|---|---|---|---|---|
| Time after HDV super-infection, week | |||||||
| +2 | +4 | +5 | +6 | +8 | +11 | +14 | |
| U(1221)C | 91.7 | 20 | 30 | 15 | 75 | 50 | 45.5 |
| Glu(126)Glu d | |||||||
| U(1262)C | 0 | 0 | 0 | 35 | 0 | 10 | 18.2 |
| Lys(113)Glu | |||||||
| U(1469)A | 0 | 0 | 0 | 55.5 | 0 | 8.3 | 50 |
| Ile(44)Leu | |||||||
These mutations were not found in the HDV genomes recovered from F6438 or the inoculum with one exception. The mutation U(1221)C was also detected in serum HDV genomes harvested at week +2 from F6438 at the level of 80% (see text).
All mutations are shown as the nucleotide changes on genomic RNA of HDV.
This parameter shows the percentage of the genomes bearing a particular mutation in the entire pool of recovered HDV G RNA sequences.
This nucleotide change did not result in an amino acid change.
First, we analyzed the ability of mutated HDV RNA genomes to support RNA-directed RNA synthesis. As the base construct, we used plasmid pDL542 that drives the expression of a greater than unit length HDV G RNA, which does not facilitate the production of delta antigen, but can replicate if functional AgS is provided in trans (56). Therefore, the mutations in context of this RNA can be tested independently of their potential to produce mutated AgS. For each abovementioned mutation, a mutation-bearing version of pDL542 was transfected into AgS-293 cells, and the replication of HDV genome was initiated by providing wt AgS from a fragment of integrated HDV DNA in the presence of tetracycline. The cells were harvested at day 9 post-transfection, and the levels of accumulated HDV G RNA were measured by qPCR. As a control, unmodified pDL542 was used. The results are summarized in Fig. 7. The data demonstrate that none of the three mutations significantly impaired the ability of HDV G RNA to support RNA-directed RNA replication.
Fig. 7. Replication capacity of HDV genomes bearing the mutations recovered from M6593.
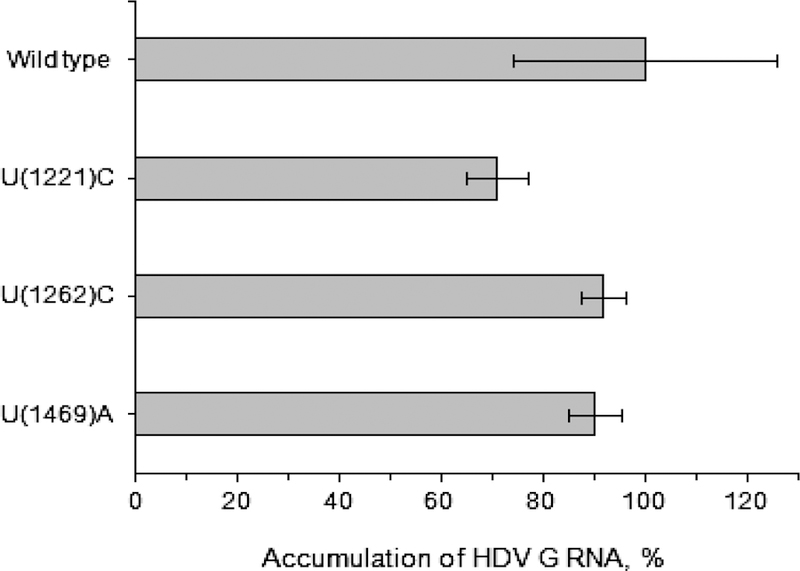
The details are described in Materials and Methods and in the text. The mutations were placed into the HDV genome in the context of pDL542, and then the construct containing the mutation was transfected into AgS-293 cells. In these settings, the HDV genome (with or without the mutation) doesn’t make AgS, but it is replication-competent, and will support RNA-directed RNA replication, if functional AgS is provided in-trans. In this case, wt AgS was provided from the integrated fragment of HDV DNA in the presence of tetracycline. The described settings allowed us to analyze the influence of an individual mutation on the ability of HDV genome to support RNA-directed RNA synthesis separately from the effect that AgS bearing the mutation might have on HDV G RNA replication.
We then examined the properties of AgS variants bearing the mutations. Each mutation was separately introduced into vector pDL444 that drives the expression of wt AgS. The first mutation U(1221)C that does not cause an amino acid change was introduced into pDL444 as an additional control. The ability of mutated AgS to support HDV genome replication was assayed as described previously (56–57). Each mutated version of pDL444 was co-transfected along with unmodified pDL542 at a 1:1 ratio into Huh7 cells. In these settings, only a potential effect of each mutation on the properties of AgS (separately from potential influence on the ability of RNA genome to support RNA-directed RNA synthesis) was evaluated. The cells were harvested at day 9 post-transfection, and HDV G RNA was quantified by qPCR. The results are summarized in Table 3. Only mutant Lys(113)Glu was found to be poorly supporting HDV G replication, while the other two mutants supported the replication similarly to that of wt AgS. The AgS bearing the mutation Lys(113)Glu supported HDV G replication about 10-fold poorer than wt AgS (Table 3). However, it needs to be considered that the highest frequency of the appearance of the corresponding nucleotide change was 35% at week +6 (Table 2). Therefore, since we already demonstrated that the genome with the mutation is replication-competent just as wt genome (Fig. 7), then in order to affect HDV G replication, the mutated AgS has to “poison” multimers of delta antigen similarly to the large delta antigen, which if present at 5% level in a pool along with 95% of wt AgS can profoundly inhibit HDV G replication (60–61). Therefore, we examined different ratios of the plasmids coding for mutant AgS (i.e., mutated pDL444) and wt AgS (i.e., unmodified pDL444) in terms of their ability to support HDV G replication. In this case, pDL542 was co-transfected into Huh7 cells at a ratio of 1:1 with a combination of the two plasmids, unmodified pDL444 plus mutated pDL444 version. The rest of the experiment was conducted as described above, when pDL542 was co-transfected with a single mutated version of pDL444. The results are presented in Table 3. At the mutant:wt ratio of 80:20 (representing the mass ratio between the two corresponding plasmids), the replication of HDV G RNA was reduced by ~50% as compared to that facilitated by wt AgS. When the mutant-expressing plasmid was present at level of 40%, the level of the G replication was about 76%. For the other combinations, when the levels of mutated plasmid were between 5% and 20%, the levels of HDV G replication were between 68% and 84%. The data therefore suggest that the mutated AgS does not efficiently alter the ability of the multimers of delta antigen to support HDV genome replication. Taken together, we concluded that the loss of the replication capacity of HDV genomes observed for M6593 could not be explained by mutations in HDV G RNA.
Table 3.
| Mutation | mutant:wt ratiod | Accumulation of HDV G RNA at day 9 post-transfection, % c |
|---|---|---|
| Wild type e | N/A | 100 ± 3.79 |
| U(1221)C | 100:0 | 78.81 ± 10.04 |
| Glu(126)Glu f | ||
| U(1262)C | ||
| Lys(113)Glu | 5:95 | 69.43 ±11.23 |
| 10:90 | 68.04 ± 43.30 | |
| 20:80 | 83.94 ± 10.16 | |
| 40:60 | 76.53 ±37.75 | |
| 80:20 | 52.61 ± 5.22 | |
| 100:0 | 12.46 ± 2.19 | |
| U(1469)A | 100:0 | 109.65 ± 1.19 |
| Ile(44)Leu | ||
The tested mutations were found in the HDV genomes recovered from M6593 (see Table 2).
The replication capacity was measured using co-transfection of mutation-bearing plasmid pDL444 and unmutated vector pDL542 into Huh7 cells. In these settings, the replication of HDV G RNA that is transcribed from pDL542 and cannot make delta antigen (56) depends on the AgS that is provided from the mutated version of pDL444.
The percentage is calculated relatively to the levels of accumulated HDV G RNA, which replication was supported by wild type AgS supplied from unmodified vector pDL444.
The combination of wild type (wt) and mutated form of AgS in different ratios was only examined for the change U(1262)C that causes the amino acid substitution Lys(113)Glu. In this case, co-transfection was performed using a ratio of 1:1 of pDL542:(pDL444 mutant + pDL444 wt). The shown in the table ratios of mutant:wt represent the mass ratios between pDL444 mutant and pDL444 wt plasmids.
The wild type AgS was supplied from the plasmid pDL444.
This silent mutation (in terms of amino acid change) was used as an additional control.
Integrity of virion-associated HDV RNA genomes.
Our next hypothesis was that the integrity of the virion-associated HDV G RNA could be compromised, which could be responsible for the observed loss of the replication capacity. Therefore, we examined serum HDV RNA genomes using Northern blot analysis. We isolated total RNA from serum samples collected at different times after HDV super-infection, and assayed the isolated RNA using the loading of the same amounts of HDV G RNA (i.e., 106 HDV GE (as measured by qPCR)) per lane in the Northern gel. The serum HDV concentration between weeks +3 and +14 for F6438 was sufficiently high to achieve the loading level of 106 HDV GE/lane, while very low HDV titer observed for M6593 at later times during the infection precluded the analysis of the time points after week +6 (Fig. 1). The results of the Northern analysis are presented in Fig. 8. During the quantification of the levels of full-length (i.e. full genome size unit length) HDV G RNA (fl-G-RNA), for each animal the value of 100% was assigned to the highest level of full-length G RNA observed during the time course (Fig. 8). For M6593, the amounts of fl-G-RNA varied at different time points. Initially, the fraction of fl-G-RNA increased from 48% (week +2) to 100% (week +3), and then it was about 84% at week +4. Later on it significantly dropped to ~7% by week +5, and somewhat increased to ~34% at week +6 (Fig. 8). The levels of fl-G-RNA between weeks +2 and +4 were consistent with the considerable levels of the replication capacity and infectivity measured at the same time points (Figs. 5 and 3, respectively). The subsequent reduction of the accumulation of fl-G-RNA in virions was consistent with the trends of reduced levels of the replication capacity (Fig. 5), and infectivity (Fig. 3). The levels of fl-G-RNA more closely correlated with the levels of the replication capacity, which were reduced at later times as compared to weeks +2 to +4, but at week +6 they were somewhat higher than at week +5 (Fig. 5). The profoundly reduced infectivity at weeks +5 and +6 (Fig. 3) may suggest that the numbers of virion-associated fl-G-RNA were not sufficient to support the infectivity of the virions or that there were additional reasons (apart from reduced levels of fl-G-RNA) mediating the reduction of the infectivity. The levels of fl-G-RNA varied as well at different times for F6438. There was a correlation between the increase in fl-G-RNA numbers from ~18% (week +3) to 100% (week +10), and then the reduction to ~41% (week +12) and to 63% at week +14 (Fig. 8) with the trends in the corresponding levels of the infectivity (Fig. 3) and replication capacity (Fig. 5). However, a more closer correlation was again observed between the levels of fl-G-RNA (Fig. 8) and the levels of replication capacity (Fig. 5). The differences observed for the infectivity (Fig. 3) and levels of fl-G-RNA (Fig. 8), like, for example, moderate levels of infectivity were found when the level of fl-G-RNA was just ~11% (week +5), and a more than 3-fold difference in infectivity was found between weeks +7 and +8 (Fig. 3), while the fl-G-RNA levels measured at weeks +7 and +8 were very close (Fig. 8), may represent differences between the assays, and/or suggest that there was more than one factor (besides the number of fl-G-RNA) that determined the level of the infectivity. In addition, it should be emphasized that if, for example, fl-G-RNA lost just a small fragment of about 100–150 nucleotides as a result of RNA fragmentation, such cleaved RNA is expected still to migrate practically as intact fl-G-RNA under conditions of our Northern blot assay. Therefore, in some situations the amounts of fl-G-RNA as calculated based on the Northern blot image could have been overestimated. However, the overall tendency was that the decreased levels of fl-G-RNA in virions correlated with decreased levels of the replication capacity and infectivity at least most of the time. The Northern analysis results also at least in large part explain the initial rise in the infectivity values at early weeks after the HDV super-infection for both animals (Fig. 3), and at least for F6438 explain the rise of replication capacity at the same times (Fig. 5). However, as mentioned above, the differences between different assays should be taken into consideration during the comparisons. In summary, the generated data support the interpretation that for both woodchucks, there was apparently host-mediated fragmentation of the virion-associated HDV RNA genomes (Fig. 8) that influenced the levels of the replication capacity (Fig. 5) and infectivity (Fig. 3). Consistent with this interpretation were the findings that at a number of time points the numbers of HDV G RNAs measured by qPCR were clearly higher than the levels of fl-G-RNA measured by Northern blot for both woodchucks. This is also consistent with the nature of the two assays. Thus, qPCR actually analyzes a relatively short intact region of HDV G RNA (27, 50, 52) that could be present on intact HDV genomes and on RNA fragments bearing the intact qPCR amplicon, while the Northern blot allows to analyze for fl-G-RNA species.
Fig. 8. Northern analysis of serum HDV RNA genomes harvested from woodchucks M6593 and F6438.
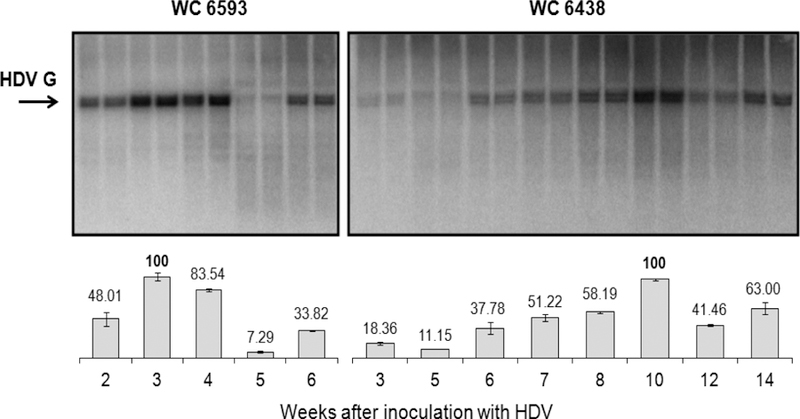
The HDV G RNA isolated from serum virions was quantified by qPCR and 106 HDV GE were loaded onto each lane of the Northern gel. The details are described in Materials and Methods. The top panel shows the gel images for samples from woodchuck 6593 (left) and woodchuck 6438 (right). The position of the full genome size unit length (i.e., full-length) HDV G RNA is shown by the arrow. The bottom panel shows the calculations of the fractions of the full-length HDV G RNA in each sample. The value of 100% was assigned to the highest number of full-length HDV G RNA observed for each animal. At the bottom of the Figure, the time after the HDV super-infection (week) is indicated. Each time point is represented in the Northern gels by two adjacent lanes. The abovementioned calculations represent the average number obtained for such two lanes (i.e., duplicate samples).
DISCUSSION
The mechanism(s) that determine the outcomes of HDV super-infection are far from being understood. This study examined in detail the down-regulation of HDV infection, which was observed when two chronic WHV carrier woodchucks super-infected with identical HDV inoculum displayed two very different patterns of HDV infection. One woodchuck had initial brief increase of serum HDV titer, which then significantly declined to very low levels and remained low for the rest of the study (Fig. 1). This down-regulation of HDV infection was associated with low levels of accumulated intrahepatic HDV G RNA (Fig. 2). A gradual increase of HDV titer that reached high levels and displayed plateau-like pattern at later times in the infection was observed for the second animal. This pattern was associated with considerably higher levels of accumulation of intrahepatic HDV genomes (Fig. 2). Subsequent experiments showed that when HDV infection was down-regulated, the infectivity of circulating HDV virions was profoundly reduced within a short period of time (Fig. 3). Further experiments demonstrated that the loss of infectivity was not due to the binding to anti-WHsAg antibodies (Table 1), or defects of the virions’ envelopes that could expose HDV RNPs (Fig. 4). The loss of infectivity, however, correlated with the loss of replication capacity of virion-associated viral genomes (Fig. 5), which was not mediated by mutations in HDV genomes (Fig. 7, Tables 2&3), but correlated considerably well with the reduced levels of full-length HDV RNA genomes residing within the virions (Fig. 8).
Furthermore, during preparation of this manuscript, we completed a longer time course of the HDV super-infection using another pair of chronic WHV carrier woodchucks. These chronic WHV carriers were super-infected with 500 µl of serum HDV that was harvested from animal F6438 at week +10 (i.e., each woodchuck was inoculated with ~2.98×109 GE of HDV (Fig. 1)), and then were monitored for 24 weeks after the super-infection. One of the woodchucks, F9101, demonstrated another case of down-regulation of HDV infection, while animal M9001 showed a profile of HDV infection with persistent relatively high HDV viremia (Fig. 9). Importantly, the new case of down-regulation of HDV infection was again associated with early and profound reduction of the replication capacity of virion-associated HDV genomes in F9101, while in M9001, the replication capacity rose and then stayed at relatively high levels (Fig. 9). Clearly, the observed overall tendencies were very similar to those found for the pair M6593/F6438, which was analyzed in detail in this study. The additional data (Fig. 9) strongly suggest that (i) down-regulation of HDV infection via reduction of the replication capacity of HDV genomes may represent at least one of the common host-mediated mechanisms related to resolution/control of HDV infection, (ii) this mechanism likely does not depend on the gender (i.e., the infection was down-regulated in M6593 and F9101), and (iii) this mechanism may not depend on the inoculum, because two different woodchuck pairs were infected with related, but not identical HDV inocula.
Fig. 9. Replication capacity of virion-associated HDV RNA genomes harvested from woodchucks F9101 and M9001.
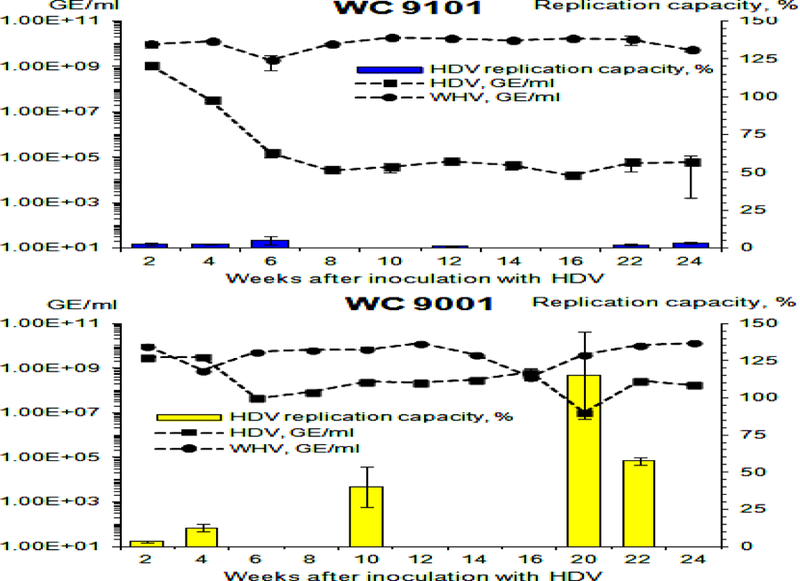
The replication capacity of HDV genomes was analyzed using transfection of HDV RNA isolated from serum virions into Ag-293 cells. The experiments were conducted basically as it was done for Fig. 5 with one modification. After tetracycline was omitted, the cells were cultured further for additional nine (and not seven) days. The left Y axis reflects serum concentration of HDV (black rectangles) and WHV (black circles) in logarithmic scale. The right Y axis represents the values of replication capacity, which is a normalized parameter that shows how many new HDV genomes/cell were produced as a result of transfection per each HDV genome used per cell in the transfection mixture. As a positive control, in vitro transcribed and gel-purified unit length HDV G RNA (58) was used. The level of the replication capacity measured for this control HDV G RNA was used as 100% value. The replication capacity values (%) are shown as blue (F9101) or yellow (M9001) bars. The X axis represents the time (week) after super-infection with HDV. The top panel shows the results for woodchuck F9101, while the bottom panel - for woodchuck M9001.
Thus, importantly, the generated data suggest that the down-regulation (and possibly the subsequent clearance) of HDV infection (which would represent the least pathogenic outcome of initial HDV super-infection) was mediated at least in large part by host-caused defects in the HDV life cycle (Figs. 3, 5, 8, and 9). Apparently, the primary target of the host response was HDV RNA that was likely at least partially fragmented in the virions (Fig. 8), which led to the reduced replication capacity of virion-associated HDV genomes that in turn was likely the primary cause of the loss of infectivity of serum HDV virions (Figs. 3 and 5). On a contrary, when HDV infection was not suppressed, the replication capacity and infectivity remained sufficiently high (which considerably correlated with the fraction of full-length HDV genomes in the virions) (Figs 3, 5, 8 and 9). Such not suppressed HDV infection most likely would progress to chronic HDV infection (which would be more pathogenic outcome of initial HDV super-infection). Furthermore, both the replication capacity and infectivity control the size of the HDV reservoir in infected livers. Since as mentioned above they both were not considerably altered in the infection pattern that displayed persistently high HDV titer at later times after the super-infection (Figs. 3, 5 and 9), it can be speculated that replication capacity of HDV genomes and virions’ infectivity, which are very important for support of continuous virus spread and super-infection, may represent critical determinants of the establishment and maintenance of chronic (persistent) HDV infection. We may further speculate that disruption of continuous HDV spread and super-infection may interfere with the maintenance of chronic HDV infection, and might represent a strategy for considerable suppression and possibly even resolution of persistent HDV infection.
The data further suggest that while HDV clearance or progression to chronic infection takes several months after the super-infection (16), the host senses HDV early in the super-infection settings (Figs. 3, 5, 8 and 9), and it is likely that the magnitude of the host response over a period of several months “decides” the outcome of HDV infection. Thus, the data in Figs. 5 and 9 show that the replication capacity of HDV genomes was profoundly suppressed but not completely abolished and that low HDV titers remained, which would suggest that the pressure from the host has to continue for several months in order to sufficiently reduce HDV reservoir that would likely lead to HDV clearance. The Northern blot results suggested that the apparent fragmentation of HDV RNA likely takes place independently of the final infection outcome, and its extent varies at different times (Fig. 8). However, what shifts the equilibrium during virus-host interactions in favor of HDV clearance or persistence remains unknown. Furthermore, if the above interpretations are correct, and the observed mechanism of down-regulation of HDV infection may represent at least one of the common pathways of HDV clearance, then at least the loss of the replication capacity of virion-associated HDV genomes could represent a potential early predictor of the outcomes of HDV super-infection.
The potential of linking HDV RNA fragmentation (leading to the loss of the replication capacity) to HDV clearance warrants the analysis of the immune response to HDV infection with a focus on host RNases and immune factors that sense viral RNA and trigger virus RNA-specific regulatory pathways. The question of where HDV RNA gets fragmented - inside the cells and/or inside the virions remains unsolved. Since apparently fragmented RNAs are present in the virions, it is possible that at least a substantial fraction of HDV RNA genomes may be synthesized, processed and circularized, and form complexes with multiple copies of delta antigens. The formed RNPs then may undergo RNase cleavage that may result in only several cuts per HDV G RNA molecule, in which case the RNA still maintains its rod-like structure via multiple Watson-Crick pairings (1, 4) and RNP is not dismembered and could be packaged into the virions. It is likely that more extensive cleavage of the RNA to multiple short fragments could cause disassembly of the HDV RNPs. Alternatively, the host RNase could be packaged along with HDV RNP into virions, and could cleave HDV RNA either during the assembly or later within the released virions.
A separate issue is the low fraction of circulating HDV virions bound to antibodies against the envelope proteins. This was an unexpected finding, because both HDV and the helper hepadnavirus are coated with the same hepadnavirus envelope proteins (1, 2). The data indicate that the envelope proteins covering the virions of WHV and HDV may bear different protein modifications that could account for the differences in the interactions with anti-envelope antibodies. It needs to be taken into account that HBV and HDV could use different compartments for assembly and different routes for egress (62). It was suggested that HBV egress employs multivesicular body (MVB), while genome-free subviral particles (SVPs) that consist of the envelope proteins could use a different egress route (63–64). HBV likely utilizes ESCRTIII/Vps4B during budding and egresses via the exosome pathway. Some ESCRT-II factors were shown to be critical for HBV egress (65–67). Whether HDV and SVPs egress via the same route still remains unclear (62, 68–69). The use of different sites for assembly and different egress routes could explain how the envelope proteins covering HDV virions may have protein modifications that differ from those that are present on the proteins covering the hepadnavirus virions. We observed low fractions of antibody-bound HDV virions in five chronic WHV carrier woodchucks that were super-infected with HDV, which suggests that it could be a common feature of HDV super-infection. Based on the data from other labs (39–46), it became apparent that anti-envelope antibodies could help to control the levels of the hepadnavirus during the chronic infection and maintain hepadnavirus-host equilibrium, but likely do not facilitate the clearance of hepadnavirus. Our results suggest that anti-envelope antibodies do not regulate HDV infection in settings of the super-infection. Moreover, it appears that a critical line of the host defense that is mediated by anti-envelope antibodies is absent during HDV super-infection, which could be related somehow to the high rates of HDV chronicity resulting from the super-infection in humans (32). This being the case would also suggest that the outcome of HDV super-infection is likely to be determined by the actions of T cells rather than those of B cells. Furthermore, it would be of significant interest to determine whether the antibodies that are induced by immunization with a vaccine containing HBV surface antigen (HBsAg) can bind HDV as efficiently as HBV. This would seriously advance the understanding of how the abovementioned antibodies protect against both HBV and HDV infections. If the antibodies bind both viruses equally well, the protection mechanism is most likely mediated by formation of the immune complexes leading to the elimination of the viruses. If only HBV and not HDV is efficiently recognized and bound by the antibodies, then the protection is likely mediated by an indirect mechanism that prevents HBV from establishing its reservoir in infected livers, and therefore leads to the absence of a continuous source of HBsAg that is very likely needed to mediate HDV persistence.
Several limitations of the present study should be acknowledged. First, although (i) the detailed analysis of the down-regulation of HDV super-infection led to a number of interesting findings that suggest that the host mediates the suppression of HDV infection likely via fragmentation of HDV G RNA, which leads to a profound reduction of viral genome replication capacity and virions’ infectivity, and (ii) examination of the second pair of super-infected woodchucks further confirmed that the loss of the replication capacity may represent a common mechanism of the host-mediated suppression of HDV infection, the analysis of a larger number of chronic WHV carriers super-infected with HDV need to be conducted in order to better understand the mechanisms leading either to HDV clearance or to HDV persistence. In addition, the monitoring period after the HDV super-infection should be increased to allow the infection to be cleared or to progress to chronicity. It also needs to be determined whether the mechanisms of HDV clearance/persistence suggested by our results represent major common mechanisms, or there could exist alternative mechanism(s) that, for example, may mediate HDV clearance without altering HDV properties.
Second, the very low levels of intrahepatic HDV RNA genomes observed in M6593 made it difficult to analyze their properties. Although, our data (Fig. 6) indicate that the reduced replication capacity of HDV genomes may also be a property of intrahepatic HDV genomes, we refrained from making a firm conclusion, because we were only able to analyze a tissue sample collected at one time point after the super-infection. For a firm conclusion, the number of liver biopsies collected at very early times after HDV super-infection would need to be increased. The subsequent analysis of these biopsies would help to understand when and where the fragmentation of HDV RNA/suppression of the replication capacity takes place (i.e., in infected hepatocytes and/or in virions).
Third, although our results suggest that the host facilitates the fragmentation of HDV RNA, we have not specifically assayed for HDV RNA fragments. One strategy to address that could be to use the technology of SMRT (single molecule real time) RNA sequencing offered by Pacific Biosciences, which allows unbiased long-read sequencing of full-length RNA molecules (70), which, however, should not use the poly(A) selection, because HDV G RNAs and their fragments do not bear poly(A) tails. Such approach if successful may help to identify the situations, in which the samples are enriched for fragmented HDV RNAs, and to provide important clues regarding the mechanism(s) that generate the HDV RNA fragments. Furthermore, to better understand the mechanism of the loss of full-length HDV G RNA from the virions, it would be useful to analyze HDV G RNA for the presence of m6A RNA modification, which could reduce the stability of the m6A-bearing RNA, as it was found for HBV RNAs (71).
ACKNOWLEDGEMENTS
SOG and SM were supported by NIH grant R01CA166213. SOG was also supported by NIH grant R21AI097647 and by the University of Kansas School of Medicine Bridging Grant. We thank Jeffrey Glenn for helpful discussion. We acknowledge help of Megan Dudek.
Footnotes
Declarations of interests: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Taylor J 2000. Hepatitis delta virus. Virology 344:71–76. [DOI] [PubMed] [Google Scholar]
- 2.Sureau C 2006. The role of the HBV envelope proteins in the HDV replication cycle. p. 113–131. In: Casey JL (ed). Hepatitis delta virus. Current Topics in Microbiology and Immunology 1st ed. vol 307 Springer-Verlag, Berlin, Heidelberg, Germany. [DOI] [PubMed] [Google Scholar]
- 3.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium turocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 3 10.7554/eLife.00049. [DOI] [PubMed] [Google Scholar]
- 4.Taylor JM. 2006. Structure and replication of hepatitis delta virus RNA. Curr. Top. Microbiol. Immunol 307:1–23. [DOI] [PubMed] [Google Scholar]
- 5.Sureau C, Negro F. 2016. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol 64(1 Suppl):S102–S116. [DOI] [PubMed] [Google Scholar]
- 6.Wong SK, Lazinski DW. 2002. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 99:15118–15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casey JL. 2006. RNA editing in hepatitis delta virus. Curr. Top. Microbiol. Immunol 307:67–89. [DOI] [PubMed] [Google Scholar]
- 8.Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev 64:51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeger C, Mason WS. 2015. Molecular biology of hepatitis B virus infetcion. Virology 479–480:672–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanchet M, Sureau C. 2006. Analysis of the cytosolic domains of the hepatitis B virus envelope proteins for their function in viral particles assembly and infectivity. J. Virol 80:11935–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanchet M, Sureau C. 2007. Infectivity determinants of the hepatitis B virus pre-S domain are confined to the N-terminal 75 amino acid residues. J. Virol 81:5841–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sureau C, Guerra B, Lee H. 1994. The middle hepatitis B virus envelope protein is not necessary for infectivity of hepatitis delta virus. J. Virol 68:4063–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W 2015. The hepatitis B virus receptor. Annu. Rev. Cell. Dev. Biol 31:125–147. [DOI] [PubMed] [Google Scholar]
- 14.Schaefer S 2005. Hepatitis B virus: significance of genotypes. J. Viral. Hepat 12:111–124. [DOI] [PubMed] [Google Scholar]
- 15.Schaefer S 2007. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J. Gastroenterol 13:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casey JL. 2006. The woodchuck model of HDV infection. Curr. Top. Microbiol. Immunol 307:211–225. [DOI] [PubMed] [Google Scholar]
- 17.Menne S, Cote PJ. 2007. The woodchuck as an animal model for pathogenesis and therapy of chronic hepatitis B virus infection. World J. Gastroenterol 13:104–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fiedler M, Roggendorf M. 2006. Immunology of HDV infection. Curr. Top. Microbiol. Immunol 307:187–209. [DOI] [PubMed] [Google Scholar]
- 19.Tennant BC, Toshkov IA, Peek SF, Jacob JR, Menne S, Hornbuckle WE, Schinazi RD, Corba BE, Cote PJ, Gerin JL. 2004. Hepatocellular carcinoma in the woodchuck model of hepatitis B virus infection. Gastroenterology 127:S283–S293. [DOI] [PubMed] [Google Scholar]
- 20.Menne S, Tennant BC. 1999. Unraveling hepatitis B virus infection of mice and men (and woodchucks and ducks). Nat. Med 5:1125–1126. [DOI] [PubMed] [Google Scholar]
- 21.Fletcher SP, Chin DJ, Cheng DT, Ravindran P, Bitter H, Gruenbaum L, Cote PJ, Ma H, Klumpp K, Menne S. 2013. Identification of an intrahepatic transcriptional signature associated with self-limiting infection in the woodchuck model of hepatitis B. Hepatology 57:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fletcher SP, Chin DJ, Li Y, Iniguez AL, Taillon B, Swinney DC, Ravindran P, Cheng DT, Bitter H, Lopatin Y, Ma H, Klumpp K, Menne S. 2012. Transcriptomic analysis of the woodchuck model of chronic hepatitis B. Hepatology 56:820–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fletcher SP, Chin DJ, Gruenbaum L, Bitter H, Rasmussen E, Ravindran P, Swinney DC, Birzele F, Schmucki R, Lorenz SH, Kopetzki E, Carter J, Triyatni M, Thampi LM, Yang J, AIDeghither D, Murredu MG, Cote P, Menne S. 2015. Intrahepatic transcriptional signature associated with response to interferon- treatment in the woodchuck model of chronic hepatitis B. PLoS Pathog 11:e1005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou T, Saputelli J, Aldrich CE, Deslauriers M, Condreay LD, Mason WS. 1999. Emergence of drug-resistant populations of woodchuck hepatitis virus in woodchucks treated with the antiviral nucleoside lamivudine. Antimicrob. Agents Chemother 43:1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glebe D, Lorenz H, Gerlich WH, Butler SD, Tochkov IA, Tennant BC, Cote P, Menne S. 2009. Correlation of virus and host response markers with circulating immune complexes during acute and chronic woodchuck hepatitis virus infection. J. Virol 83:1579–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodrigues L, Freitas N, Kallakury BV, Menne S, Gudima SO. 2015. Superinfection with woodchuck hepatitis virus strain WHVNY of the livers chronically infected with strain WHV7. J. Virol 89: 384–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freitas N, Salisse J, Cunha C, Toshkov I, Menne S, Gudima SO. 2012. Hepatitis delta virus infects the cells of hepadnavirus-induced hepatocellular carcinoma in woodchucks. Hepatology 56:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wedemeyer H 2011. Hepatitis D revival. Liver Int 31(Suppl 1): 140–144. [DOI] [PubMed] [Google Scholar]
- 29.Rizzetto M. 2015. Hepatitis D Virus: Introduction and epidemiology. Cold Spring Harb. Perspect. Med 5:a021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor JM. 2102 Virology of hepatitis D virus. Semin. Liver Dis 32:1950200. [DOI] [PubMed] [Google Scholar]
- 31.Lai MM. 1995. Molecular biologic and pathogenetic analysis of hepatitis delta virus. J. Hepatol 22 (1 Suppl):127–131. [PubMed] [Google Scholar]
- 32.Koytak ES, Yurdaydin C, Glenn JS. 2007. Hepatitis d. Curr. Treat. Options. Gastroenterol 10:456–463. [DOI] [PubMed] [Google Scholar]
- 33.Fattovich G, Stroffolini T, Zagni I, Donato F. 2004. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 127(5 Suppl 1): S35–S50. [DOI] [PubMed] [Google Scholar]
- 34.Fattovich G, Giustina G, Christensen E, Pantalena M, Zagni I, Realdi G, Schalm SW. 2000. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut 46:420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yurdaydin C 2012. Treatment of chronic delta hepatitis. Semin. Liver. Dis 32:237–244. [DOI] [PubMed] [Google Scholar]
- 36.Grabowski J, Wedemeyer H. 2010. Hepatitis delta: immunopathogenesis and clinical challenges. Dig. Dis 28:133–138. [DOI] [PubMed] [Google Scholar]
- 37.Roggendorf M 2012. Perspectives for vaccine against hepatitis delta virus. Semin. Liver Dis 32:256–261. [DOI] [PubMed] [Google Scholar]
- 38.Park SH, Rehermann B. 2014. Immune response to HCV and other hepatitis viruses. Immunity 40:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerlich WH. 2007. The enigma of concurrent hepatitis B surface antigen (HBsAg) and antibodies to HBsAg. Clin. Infect. Dis 2007 44:1170–1172. [DOI] [PubMed] [Google Scholar]
- 40.Hayashi J, Noguchi A, Nakashima K, Morofuji M, Kashiwagi S. 1990. Frequency of concurrence of hepatitis B virus surface antigen and antibody in a large number of carriers in Okinawa, Japan. Gastroenterol. Jpn 25:593–597. [DOI] [PubMed] [Google Scholar]
- 41.Kim SA, Lee SI, Choi IH, Shin JS, Uhm JR, Kim SJ, Choi HJ. 1990. Circulating immune complexes and cell-mediated immunity in patients with hepatitis B virus associated liver disease. Yonsei Med. J 31:347–358. [DOI] [PubMed] [Google Scholar]
- 42.Zhang JM, Xu Y, Wang XY, Yin YK, Wu XH, Weng XH, Lu M. 2007. Coexistence of hepatitis B surface antigen (HBsAg) and heterologus subtype-specific antibodies to HBsAg among patients with chronic hepatitis B virus infection. Clin. Infect. Dis 44:1161–1169. [DOI] [PubMed] [Google Scholar]
- 43.Madalinski K, Burczynska B, Heermann KH, Uy A, Gerlich WH. 1991. Analysis of viral proteins in circulating immune complexes from chronic carriers of hepatitis B virus. Clin. Exp. Immunol 84:493–500. [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai JF, Margolis HS, Jeng JE, Ho MS, Chang WY, Hsieh MY, Lin ZY, Tsai JH. 1998. Immunoglobulin- and hepatitis B surface antigen-specific circulating immune complexes in chronic hepatitis B virus infection. Clin. Immunol. Immunopathol 86:246–251. [DOI] [PubMed] [Google Scholar]
- 45.Lada O, Benhamou Y, Poynard T, Thibault V. 2006. Coexistence of hepatitis B virus antigen (HBsAg) and anti-HBs antibodies in chronic hepatitis B virus carriers: influence of “a” determinant variants. J. Virol 80:2968–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shiels MT, Taswell HF, Czaja AJ, Nelson C, Swenke P. 1987. Frequency and significance of concurrent hepatitis B surface antigen and antibody in acute and chronic hepatitis B. Gastroenterology 93: 675–680. [DOI] [PubMed] [Google Scholar]
- 47.Cote PJ, Korba BE, Miller RH, Jacob JR, Baldwin BH, Hornbuckle WE, Purcell RH, Tennant BC, Gerin JL. 2000. Effects of age and viral determinants on chronicity as an outcome of experimental woodchuck hepatitis virus infection. Hepatology 31: 190–200. [DOI] [PubMed] [Google Scholar]
- 48.Freitas N, Lukash T, Dudek M, Litwin S, Menne S, Gudima SO. 2015. Capacity of a natural strain of woodchuck hepatitis virus, WHVNY, to induce acute infection in naive adult woodchucks. Virus Res 205:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freitas N, Lukash T, Rodrigues L, Litwin S, Kallakury BV, Menne S, Gudima SO. 2015. Infection patterns induced in naive adult woodchucks by virions of woodchuck hepatitis virus collected during either the acute or chronic phase of infection. J. Virol 89:8749–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gudima SO, He Y, Chai N, Bruss V, Urban S, Mason W, Taylor J. 2008. Primary human hepatocytes are susceptible to infection by hepatitis delta virus assembled with envelope proteins of woodchuck hepatitis virus. J. Virol 82:7276–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuo M, Goldberg J, Coates L, Mason W, Taylor J. 1988. Molecular cloning of hepatitis delta virus RNA from an infected woodchuck liver: sequence, structure, and applications. J. Virol 62:1855–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gudima SO, He Y, Meier A, Chang J, Chen R, Jarnik M, Nicolas E, Bruss V, Taylor J. 2007. Assembly of hepatitis delta virus: particle characterization including the ability to infect primary human hepatocytes. J. Virol 81:3608–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strom SC, Pisarov LA, Dorko K, Thompson MT, Schuetz JD, Schuetz EG. 1996. Use of human hepatocytes to study P450 gene induction. Methods Enzymol 272:388–401. [DOI] [PubMed] [Google Scholar]
- 54.Berry MN, Friend DS. 1969. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J. Cell Biol 43:506–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang J, Gudima SO, Tarn C, Nie X, Taylor JM. 2005. Development of a novel system to study hepatitis delta virus genome replication. J. Virol 79:8182–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lazinski DW, Taylor JM. 1994. Expression of hepatitis delta virus RNA deletions: cis and trans requirements for self-cleavage, ligation and RNA packaging. J. Virol 68:2879–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lazinski DW, Taylor JM. 1993. Relating structure to function in the hepatitis delta virus antigen. J. Virol 67:2672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gudima SO, Chang J, Taylor JM. 2004. Features affecting the ability of hepatitis delta virus RNAs to initiate RNA-directed RNA synthesis. J. Virol 78:5737–5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gudima S, Dingle K, Wu TT, Moraleda G, Taylor J. 1999. Characterization of the 5’ ends for polyadenylated RNAs synthesized during the replication of hepatitis delta virus. J. Virol 73:6533–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chao M, Hsieh SY, Taylor JM.1990. Role of two forms of hepatitis delta antigen: evidence for a mechanism of self-limiting genome replication. J. Virol 64:5066–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moraleda G, Dingle K, Biswas P, Chang J, Zuccola H, Hogle J, Taylor J. 2000. Interactions between hepatitis delta virus proteins. J. Virol 74:5509–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang CJ, Sung SY, Chen DS, Chen PJ. 1996. N-linked glycosylation of hepatitis B surface antigens is involved but not essential in the assembly of hepatitis delta virus. Virology 220:28–36. [DOI] [PubMed] [Google Scholar]
- 63.Watanabe T, Sorensen EM, Naito A, Schott M, Kim S, Ahlquist P. 2007. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci 104:10205–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patient R, Hourioux C, Roingeard P. 2009. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell. Microbiol 11:1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prange R 2012. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol 201:449–461. [DOI] [PubMed] [Google Scholar]
- 66.Stieler JT, Prange R. 2014. Involvement of ESCRT-II in hepatitis B virus morphogenesis. PLoS One 9:e91279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lambert C, Doring T, Prange R. 2007. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vsp4, and gamma 2-adaptin. J. Virol 81:9050–9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Julithe R, Abou-Jaode G, Sureau C. 2014. Modification of the hepatitis B virus envelope protein glycosylation pattern interferes with secretion of viral particles, infectivity, and susceptibility to neutralizing antibodies. J.Virol 88:9049–9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le Duff Y, Blanchet M, Sureau C. 2009. The pre-S1 and antigenic loop infectivity determinants of the hepatitis B virus envelope proteins are functionally independent. J. Virol 83: 12443–12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Treutlein B, Gokce O, Quake SR, Sudhof TC. 2014. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. USA 111:E1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Imam H, Khan M, Gokhale NS, McIntyre ABR, KIm GW, Jang JY, Kim SJ, Mason CE, Horner SM, Siddiqui A. 2018. N6-methyladnosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc. Natl. Acad. Sci. USA 115:8829–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]