

Abstract
Disruption of the BBB is critical to initiation and perpetuation of disease in Multiple Sclerosis (MS). We report an interaction between oligodendroglia and vasculature in MS that distinguishes human white matter injury from normal rodent demyelinating injury. We find perivascular clustering of oligodendrocyte precursor cells (OPCs) in certain active MS lesions, representing an inability to properly detach from vessels following perivascular migration. Perivascular OPCs can themselves disrupt the BBB, interfering with astrocyte end-feet and endothelial tight junction integrity, resulting in altered vascular permeability and an associated CNS inflammation. Aberrant Wnt tone in OPCs mediates their dysfunctional vascular detachment, and also leads to OPC secretion of Wif1, that interferes with Wnt ligand function on endothelial tight junction integrity. Evidence for this defective oligodendroglial-vascular interaction in MS suggests that aberrant OPC perivascular migration not only impairs their lesion recruitment but can also act as a disease perpetuator via disruption of the BBB.
Introduction
MS is an autoimmune disease of the CNS associated with demyelination and axonal loss that eventually leads to neurodegeneration, and exhibits many of the features of an inflammatory autoimmune disorder including breakdown of the BBB. BBB disruption is one of the earliest cerebrovascular abnormalities in MS, allowing unrestricted access of immune cells and blood borne components into the CNS that play a central role in demyelination and axonal damage (1–5). Regeneration of damaged myelin can occur initially, involving a recruitment of migrating OPCs into areas of demyelination from surrounding normal appearing white matter followed by their differentiation into mature oligodendrocytes (6, 7), but remyelination is not durable and can fail at either of these stages (8–10). Recurrence of BBB breakdown may occur at the same or different locations within intervals of weeks or months, with lesions developing irregularly with additional phases of BBB leakage, immunologically mediated demyelination and axonal transection. Contributors to BBB dysregulation during the disease are not fully understood. We report here a pathological finding in MS, suggesting a novel and dysfunctional interaction between OPCs and the vasculature, and questioned its cause and its implication.
Results
Aberrant perivascular clustering of OPCs in multiple sclerosis.
MS is characterized by immune infiltration of cells across the BBB into brain parenchyma, and perivascular accumulation of inflammatory cells. Analysis in certain active MS (Fig. 1a, Suppl. Table 1) lesions however identifies perivascular clusters of cells around CD31+ vasculature (Fig. 1b-j, Fig. 1l), which separate from markers for inflammatory cells (Fig. 1d). These perivascular clusters express the oligodendroglial marker OLIG2 (Fig. 1e-f, Fig. 1h-i) and the marker NKX2.2 (Fig. 1l). They also express the Wnt pathway marker RNF43 (Fig. 1g, Suppl. Fig. 1a-c), previously shown to be a target of Wnt in OPCs that is activated under high Wnt tone (11, 12), and have the typical bipolar morphology of OPCs. Perivascular OPC clusters were not seen in normal appearing white (Fig. 1n) or grey (Fig. 1m) matter from the MS patients analyzed, and whilst isolated OPCs are often seen in close association with blood vessels in the core of chronic MS lesions (Fig. 1j, k), the finding of perivascular clustering is significantly more frequent in active inflammatory areas (Fig. 1o). Cluster frequency, appearance, and size were variable, presumably reflecting significant differences in MS lesion age and history (Fig. 1o, Suppl. Table 1).
Fig. 1: Perivascular clustering of OPCs in multiple sclerosis.
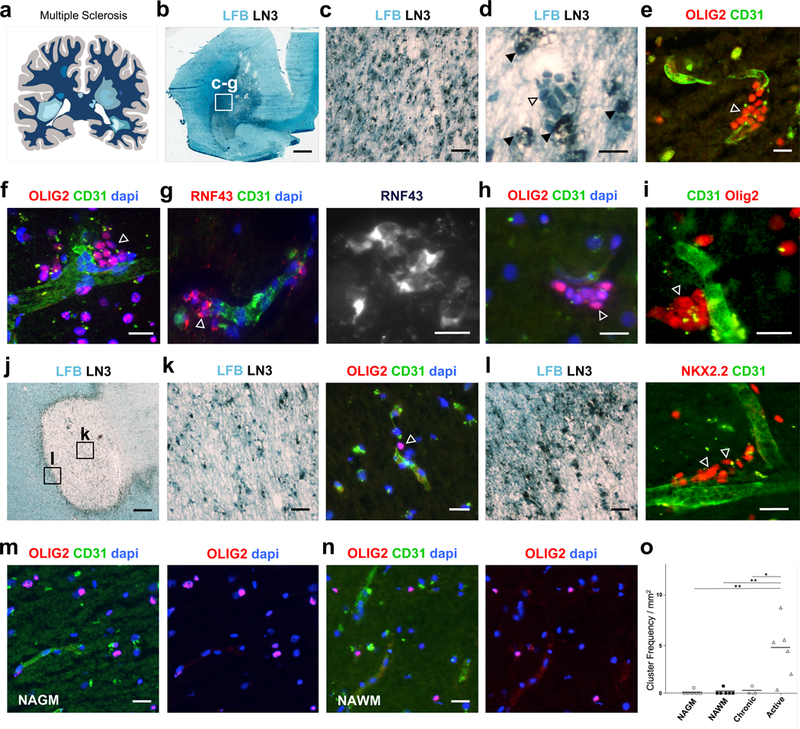
(a) Schematic of human Multiple sclerosis (MS) showing lesions throughout the brain. MS cases assessed are outlined in Suppl. Table 1. (b) Low magnification of MS Case 1 active lesion, with luxol fast blue (LFB) to assess demyelination and LN3 immunohistochemistry to assess inflammatory cell activity. Boxed area shows area of magnified view in (c), and from where images (c-g) were taken. (d) Magnified region from Case 1 showing a cluster of cells (unfilled arrow) which do not co-localize with inflammatory marker LN3 (filled arrows). (e, f) Clustered perivascular OLIG2+ cells (arrows) and (g) clustered RNF43+ cells (arrow) in Case 1 active lesion. (h-i) Clustered OLIG2+ cells (arrows) on blood vessels in active lesions of MS Cases 2 (h) and 3 (i). (j) MS Case 4 chronic active lesion, with boxed areas showing regions where images from (k) and (l) were taken. (k) Chronic core of lesion showing hypocellularity and limited LN3+ activity. Example of isolated OLIG2+ cell (arrow) in close association with blood vessel. (l) Active edge of chronic active lesion with significant LN3+ inflammatory activity and perivascular clustering of NKX2.2+ cells (arrows). (m-n) Clustering of OLIG2+ cells is not seen in normal appearing grey matter (NAGM) or normal appearing white matter (NAWM) from these patients. (o) Frequency of OLIG2+ clusters seen in NAGM, NAWM, and areas of chronic inactive and active lesions from the patients described in Suppl. Table 1. Data were analyzed by one-way ANOVA. The measure of center represents the mean. Active vs. Chronic p= 0.04, Active vs. NAWM p=0.003, Active vs. NAGM p=0.003. Scale bars, 2.2mm (b), 90μm (c, k left panel, l left panel), 20μm (d-i, m,n, k right panel, l right panel), 750μm (j). * P < 0.05, ** P < 0.01.
OPC perivascular clustering represents a defective single cell migration
The underlying cause of this pathological finding in MS is unclear. OPCs are known to associate intimately with vasculature during a developmental phase, where they use blood vessels as a critical physical scaffold for single cell migration (13). They maintain the ability in adulthood to be activated and migrate significant distances in response to injuries, and we find that they retain this capacity for single cell perivascular migration in remyelination (Fig. 2a-d, Suppl. Figs. 2–4). There is a markedly increased association of individual PDGFRα+ (Fig. 2c; 1d vs. 0d p=9.66 E-7) and Olig2+ expressing OPCs with blood vessels at lesion borders at early times during murine remyelination of spinal cord and corpus callosum (Fig. 2a-c, Suppl. Fig. 3a-b). Live imaging of these OPCs in lesioned adult NG2creERT:TdTomato mice, following intracardiac infusion of fluorescein-lectin for vessel labeling, identifies migration at lesion edges limited to an early lesion time window (Fig. 2d, Suppl. Fig. 3d-h, Suppl. Fig. 4). Motile OPCs responding to demyelination at 1.5dpl lesion edges demonstrate single cell perivascular migration, with directed crawling motility along (Fig. 2d, Suppl. Fig. 3h; Suppl. Video 1) and jumping towards/between (Suppl. Fig. 3d, h; Suppl. Video 2) vasculature, whilst tending to move in the direction of the injury (Suppl. Fig. 3g).
Fig. 2: OPC perivascular clusters following demyelination represent a defective single cell migration.
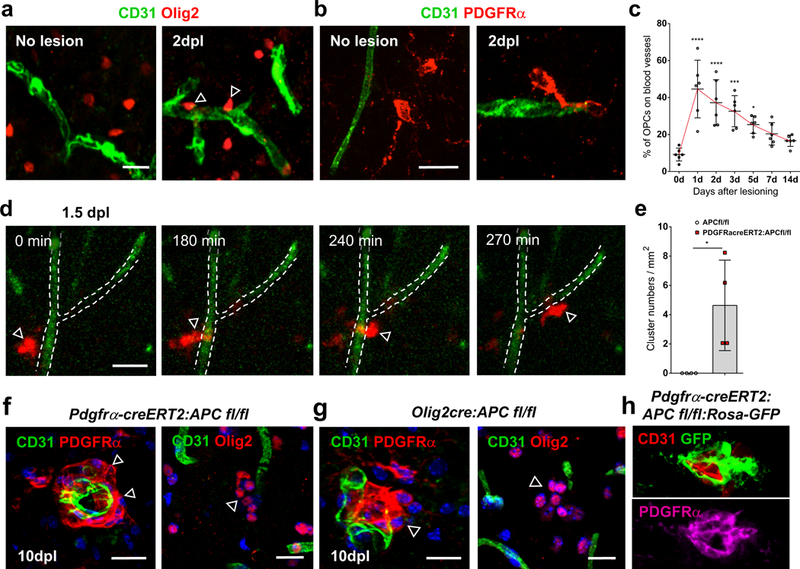
OPCs use single cell perivascular migration for their recruitment to areas of demyelination. (a-b) OPCs show increased association with vasculature at lesion edges following focal demyelination in mice. Double staining for Olig2+ (a) or PDGFRα+ (b) cells and CD31+ vasculature before lysolecithin lesioning (“No lesion”), and at lesion edges at 2 days post lesioning (dpl) in mouse corpus callosum and in spinal cord (Suppl. Fig. 3a). (c) Percentage of PDGFRα+ OPC cell bodies directly in contact with blood vessels at lesion edges at different days post lesioning in mouse spinal cord dorsal funiculus (n=6 animals at each time, all statistical analyses compared to 0d, 1d vs. 0d p=9.66 E-7, 2d vs. 0d p=6.94 E-5, 3d vs. 0d p=0.0003, 5d vs. 0d p= 0.0161, 7d vs. 0d ns p=0.1477, 14d vs. 0d ns p=0.5022). Data were analyzed by unpaired two-sided Student’s t test. (d) Time lapse imaging in slice cultures of adult spinal cord dorsal funiculus from 1.5dpl lysolecithin lesioned NG2creERT:TdTomato mice, following intracardiac infusion of fluorescein-lectin for vessel labeling. A TdTomato-expressing cell (red, arrow) migrates along a vessel (green, outlined by dotted line) at the edge of a 1.5dpl lesion (Suppl. Video 1). (e) Frequency of PDGFRα+ OPC clusters at 10dpl lesion edges in PDGFRα-creERT2:APC fl/fl mice compared to APCfl/fl controls (n=4 animals, p=0.0240). Data were analyzed by unpaired two-sided Student’s t test. (f-h) Perivascular clustering is mediated by high Wnt tone in OPCs during murine remyelination, as evidenced by PDGFRα+ and Olig2+ cell clusters in PDGFRα-creERT2:APC fl/fl (f) and Olig2cre:APC fl/fl (g) mice (which are not seen in control mice, e), and by GFP+/ PDGFRα+ double positive cell clusters in PDGFRα-creERT2:APC fl/fl:Rosa-GFP (h), at 10dpl lesion edges following focal lysolecithin injection into spinal cord dorsal funiculus white matter. Scale bars, 20μm in all panels. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Values are mean ± s.d.
OPCs use single cell perivascular migration for their recruitment into areas of demyelination, and their perivascular clustering is therefore not a feature that is seen in wildtype murine white matter injury. OPC association with blood vessels during murine developmental migration is mediated by a Wnt-driven upregulation of Cxcr4 in OPCs (13), and the finding of individual perivascular OLIG2+ cells expressing CXCR4 in human white matter injury (Suppl. Fig. 3i) suggests a conserved mechanism of attraction to the endothelium. To address whether the perivascular clustering of OPCs in MS represents a dysfunctional migration, and an involvement for overactive Wnt signaling in this aberrant vascular interaction, we performed focal demyelination in adult inducible PDGFRα-creERT2:APCfl/fl and Olig2-cre:APCfl/fl mice, which have excessive Wnt activation in OPCs due to the conditional loss of the obligate pathway repressor adenomatous polyposis coli (APC). We find that OPCs accumulate as perivascular clusters at 10dpl lesion edges during remyelination in both PDGFRα-creERT2:APCfl/fl (Fig. 2f) and Olig2-cre:APCfl/fl (Fig. 2g) mice. These clusters are not seen in control littermate remyelination (Fig. 2a-b, e), and have excessive Wnt tone evidenced by significantly increased Axin2 mRNA expression (Suppl. Fig. 5a-d) and GFP expression in PDGFRα-creERT2:APCfl/fl:Rosa-GFP (Fig. 2h, Suppl. Fig. 5e-f). We find reduced OPC numbers at early 3dpl time points in lesion centers in both PDGFRα-creERT2:APC fl/fl (Suppl. Fig. 6a, c)(p=0.0003) and Olig2cre:APC fl/fl mice (Suppl. Fig. 6a, b)(p=0.0013), without effects on OPC proliferation (Suppl. Fig. 6e-h), suggesting that an increased OPC association with vasculature (Suppl. Fig. 6d), in the form of perivascular clusters (Fig. 2e), leads to a reduced ability for proper recruitment to the lesion core. These results indicate that OPC perivascular clustering following demyelination represents an aberrant OPC migration on vessels and is mediated by defective Wnt signaling.
OPC perivascular clusters disrupt the BBB.
We questioned how this dysfunctional OPC perivascular migration and vascular detachment failure affects the integrity of the underlying vessel. Injury models themselves cause significant vascular damage. We therefore required a non-injury model to identify the specific effects of an OPC perivascular cluster in the absence of any injury-induced vascular disruption. We used Olig2-cre: APC fl/fl mice, which have defective OPC vascular detachment postnatally, with perivascular accumulations appearing in a non-injury setting that closely resemble the clusters seen in both human white matter injury (Suppl. Fig. 7) and following demyelination in PDGFRα-creERT2:APCfl/fl and Olig2-cre:APCfl/fl. In this non-injury setting, we find that aberrant oligodendroglial-vascular interactions interfere with several aspects of blood brain barrier integrity. OPC perivascular clusters interfere with astrocyte-vascular interactions by altering the positioning of astrocyte endfeet on vasculature. Use of Aldh1l1-GFP mice, (which label astrocytes and their end-feet) (Fig. 3a), shows that, when crossed into Olig2-cre: APC fl/fl mice, there are significant astrocyte end feet placement deficits (p=1.56 E-7) in areas where OPCs are clustered around blood vessels (Fig. 3b, c, g). 3D reconstruction of astrocytes and OPCs on vessels in Olig2-cre:APC fl/fl: TdTomato: Aldh1l1-GFP mice suggests a competition for space on the vessel surface and a physical displacement of astrocyte processes from vasculature at sites of OPC clustering (Fig. 3d-f). This is also seen as significant gaps in staining for Aquaporin-4 (Aqp4) (Fig. 3h-i, Suppl. Fig. 8)(p=1.47 E-6), a water channel highly enriched in astrocyte end-feet at the BBB. 3D reconstruction of perivascular clusters from 10dpl lesion edges in PDGFRa–creERT2:APC fl/fl mice following focal demyelination (Suppl. Fig. 9) suggests a similar mechanism of endfoot displacement whereby the processes of perivascular clustered OPCs can actually exist between endothelial cells and overlying astrocyte endfeet.
Fig. 3: OPC perivascular clusters physically displace astrocyte end feet from vessels.
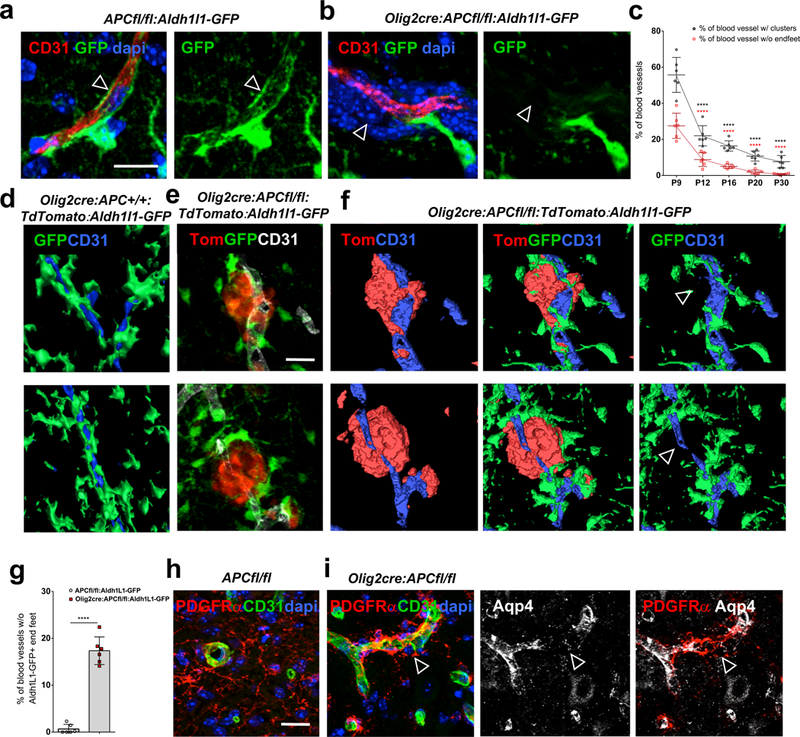
(a) GFP labels astrocytes and their end feet (arrow, in close association with CD31+ vasculature) in P9 corpus callosum of APCfl/fl:Aldh1l1-GFP control mice, but when crossed into Olig2cre: APC fl/fl mice (b), end feet are displaced (arrow in b) from vessels in areas with OPC perivascular clusters (stained with dapi). (c) Percentage of blood vessels with OPC clusters (P12 vs. P9 p=2.51 E-8, P16 vs. P9 p=1.93 E-8, P20 vs. P9 p=9.92 E-8, P30 vs. P9 p=1.04 E-7) and percentage without astrocyte endfeet (P12 vs. P9 p=5.85 E-8, P16 vs. P9 p=2.44 E-8, P20 vs. P9 p=1.96 E-8, P30 vs. P9 p=1.81 E-8) at different postnatal times in CC of Olig2cre:APCfl/fl:Aldh1l1-GFP (n=6 animals at each time, all statistical analyses compared to P9). Data were analyzed by unpaired two-sided Student’s t test. d) 3D reconstruction of astrocytes (in green) covering vessels (in blue) in CC of P9 Olig2cre:APC+/+:TdTomato:Aldh1l1-GFP control mice. (e) P9 CC from Olig2cre:APCfl/fl:TdTomato:Aldh1l1-GFP showing perivascular OPC clusters (red), vessels (white) and astrocytes (green). (f) 3D reconstruction of images in (e) showing OPCs (red), astrocytes (green) and vessels (blue). Astrocyte processes are completely displaced from areas with OPC perivascular clusters (arrows). (g) Quantitative analysis of percentage loss of Aldh1L1-GFP+ astrocytic endfeet coverage of vessels in P9 CC of Olig2cre:APCfl/fl:Aldh1l1-GFP versus APCfl/fl:Aldh1l1-GFP controls (n=6 animals, p=1.56 E-7). Data were analyzed by unpaired two-sided Student’s t test. (h-i) PDGFRα+ OPC perivascular clusters are seen in P9 spinal cord of Olig2-cre:APC fl/fl mice (i), but not in APCfl/fl controls (h), and lead to gaps in astrocyte end foot marker Aquaporin 4 (Aqp4)(arrows in i, quantified in Suppl. Fig. 8e). Scale bars, 20μm in all panels. **** P < 0.0001. Values are mean ± s.d.
In addition to astrocyte disruption, OPC perivascular clusters also cause endothelial cell (EC) dysfunction. Staining for Plasmalemma vesicle-associated protein (PLVAP), a structural component of endothelial fenestrae, but gaps in staining for claudin5 (Cldn5) (Fig. 4a-c), a structural component of EC tight junctions, identifies areas of non-intact BBB around OPC clusters (Fig. 4a-c). In contrast to astrocyte coverage, pericyte coverage of vessels seems unaffected by OPC clustering (Suppl. Fig. 10), but does not rule out functional deficits in these cells. The effects on astrocyte end foot placement and EC tight junction integrity suggested possible effects on the barrier function of the BBB. We assessed leakage of the soluble 340kDa serum glycoprotein fibrinogen from vessels, as well as leakage of a 10-kDa Dextran-Tetramethylrhodamine tracer 6 hours post tail vein injection. Both of these are detectable only within the lumen of vessels in control mice (Fig. 4d, g), but there are significant increases in the amount seen in CNS parenchyma around OPC clusters (arrows in Fig. 4e, h) in Olig2-cre:APC fl/fl mice (Fig. 4f p=0.0026, Fig. 4i p=6.46 E-6), suggesting extravasation of blood components and further supporting the notion that BBB integrity is affected at these areas.
Fig. 4: OPC perivascular clusters disrupt the BBB.
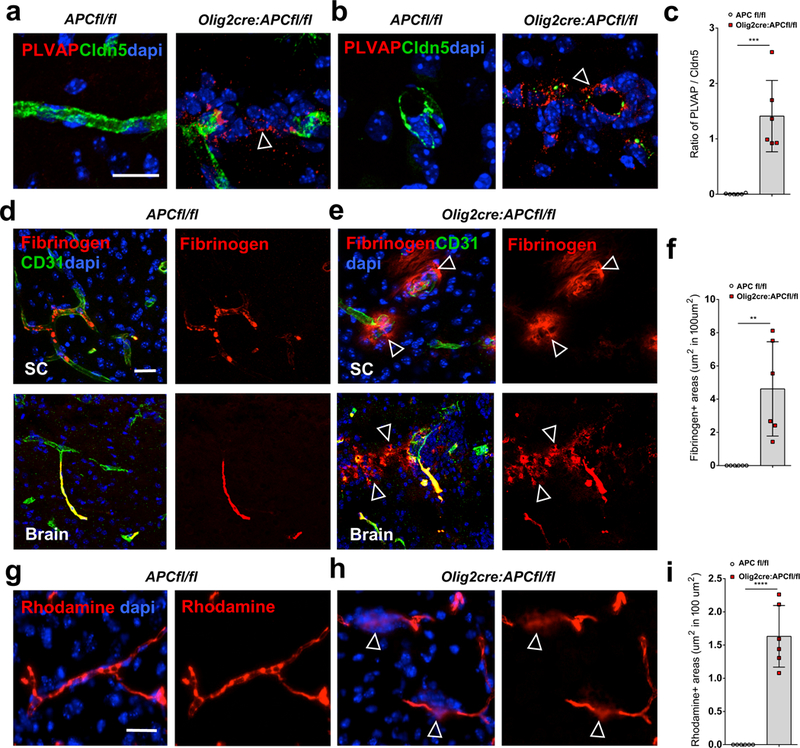
(a-b) Staining for plasmalemma vesicle-associated protein (PLVAP) and claudin5 (Cldn5) in P9 spinal cord of Olig2-cre:APC fl/fl and APCfl/fl control mice around vessels in longitudinal (a) and transverse (b) views. A PLVAP+/claudin5- state around OPC clusters in Olig2-cre: APC fl/fl mice (arrows) identifies areas of immature BBB. (c) Ratio of PLVAP:Cldn5 expression on endothelium in areas with OPC perivascular clusters in Olig2-cre:APC fl/fl spinal cord compared to comparable regions without clusters in APCfl/fl controls (n=6 animals, p=3.29 E-4). Data were analyzed by unpaired two-sided Student’s t test. (d-e) Staining shows fibrinogen only within the lumen of CD31+ blood vessels in P9 APCfl/fl control spinal cord (SC) and brain (d), but leaking into parenchyma (arrows) around OPC clusters (stained with dapi) in Olig2-cre:APC fl/fl (e). (f) Quantification of area of fibrinogen staining outside blood vessels in CNS parenchyma per mm2 in SC of Olig2-cre:APC fl/fl and APCfl/fl controls (n=6 animals, p=0.0026). Data were analyzed by unpaired two-sided Student’s t test. (g-h) Dextran- tetramethylrhodamine (rhodamine) injection into P9 Olig2-cre:APC fl/fl tail vein (h) shows extravasation into brain parenchyma (arrows in h) around OPC perivascular clusters (stained with dapi), which is not seen in APCfl/fl control mice (g). (i) Quantification of area of rhodamine staining outside blood vessels in CNS parenchyma per mm2 in SC of Olig2-cre:APC fl/fl and APCfl/fl controls (n=6 animals, p=6.46 E-6). Data were analyzed by unpaired two-sided Student’s t test. Scale bars, 20μm in all panels. ** P < 0.01, *** P < 0.001, **** P < 0.0001. Values are mean ± s.d.
OPC perivascular clusters trigger a CNS inflammation.
OPC effects on the barrier function of the BBB trigger a CNS inflammatory reaction. We found increased numbers of cells expressing Iba1 (which stains microglia and macrophages) which have a less processed and more globose morphology (Suppl. Fig. 11a-b). Pronounced CD11c and F4/80 staining in cells around perivascular OPCs (Fig. 5a-b, d; Suppl. Fig. 11b), that is not present in control littermate mouse brains (Fig. 5a-b, d; Suppl. Fig. 11a), also identifies activated microglia/macrophages. These cells expressed high levels of iNOS rather than Arg-1 (Suppl. Fig. 11b), suggesting an M1-polarized population. Use of Cx3cr1-GFP: CCR2-RFP mice crossed into the Olig2cre:APC fl/fl suggests that this represents predominantly a microglial activation (Fig. 5c, Suppl. Fig. 11c-d) rather than recruitment of activated macrophages from the circulation. However, small numbers of RFP+ macrophages (in the CCR2-RFP mice, Suppl. Fig. 11d-e) and CD3 expressing T cells, including CD4 and CD8 subsets, were observed within the perivascular clusters (Fig. 5e-g), suggesting extravasation of inflammatory cells into surrounding brain parenchyma in addition to protein leakage. Effects on the barrier function of the BBB lead to injury to both surrounding axons and perivascular OPCs. SMI-32+ axonal spheroids, indicating swellings of damaged axons, are significantly increased (p=0.0286) and identify axonal degeneration adjacent to perivascular clusters (Fig. 5h-j). OPCs themselves also undergo significant cell death within clusters (p=0.0009)(Fig. 5l-n), leading to a resolution over time of the aggregations (Fig. 5k; Suppl. Fig. 12a-c), with reinvestment of astrocyte endfeet (Fig. 3c, Suppl. Fig. 12b) on blood vessels and reduction in microglial activation (Suppl. Fig. 12d).
Fig. 5: OPC perivascular clusters trigger a CNS inflammation.
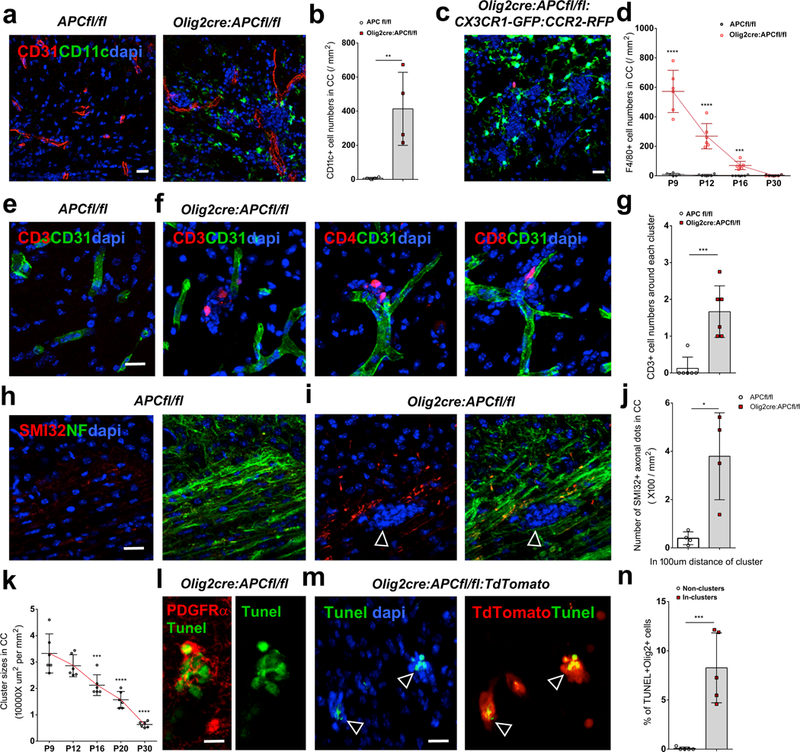
(a-b) Marked upregulation of CD11c expression in P9 Olig2-cre:APC fl/fl brain compared to controls around OPC perivascular clusters (stained with dapi), quantified in (b)(n=4 animals; p=0.009). Data were analyzed by unpaired two-sided Student’s t test. (c) As Cd11c can detect some other immune cells in addition to activated microglia and macrophages, we made use of CX3CR1-GFP: CCR2-RFP mice (which label microglia green, and macrophages red) crossed into Olig2cre:APC fl/fl, identifying these cells as predominantly activated microglia around OPC clusters. (d) Frequency of F4/80 expressing cells at different postnatal times in CC in Olig2-cre:APC fl/fl versus controls (n=6 animals at each time, all statistical analyses compare Olig2cre:APC fl/fl to controls at each time, P9 p=2.28 E-6, P12 p=1.94 E-5, P16 p=1.39 E-4, P30 p=0.5490). Data were analyzed by unpaired two-sided Student’s t test. (e-f) Extravasation of small numbers of CD3+, CD4+ and CD8+ T cells within OPC perivascular clusters in P9 Olig2 cre: APC fl/fl mouse brain (f) which are not seen in APCfl/fl controls (e). (g) Quantification of CD3+ cell numbers per perivascular cluster in Olig2-cre:APC fl/fl versus controls (n=6 animals, p=0.0006). Data were analyzed by unpaired two-sided Student’s t test. (h-i) SMI-32+ (non phosphorylated neurofilament) axonal spheroids co-localize with the neurofilament marker NF200 (NF), indicating swellings of damaged axons adjacent to an OPC perivascular cluster (arrows in i, stained with dapi) in Olig2cre:APC fl/fl P9 brain, which are not seen in controls (h). (j) Quantification of SMI32+/NF200+ axonal dots within 100μm distance of clusters in Olig2cre:APCfl/fl P9 CC versus similar areas in APCfl/fl (n=4 animals, p=0.0286). Data were analyzed by unpaired two-sided Student’s t test. (k) Quantification of OPC perivascular cluster size in Olig2 cre: APC fl/fl: TdTomato corpus callosum at times between P9 and P30 (n=6 animals, all statistical analyses compared to P9, P12 vs. P9 p=0.3846, P16 vs. P9 p=0.0007, P20 vs. P9 p=3.37 E-6, P30 vs. P9 p=2.55 E-8). Data were analyzed by unpaired two-sided Student’s t test. (l-m) Tunel staining in P9 mouse brain co-localizes with (l) PDGFRα (in Olig2cre:APC fl/fl) and with (m) TdTomato (in Olig2cre:APC fl/fl: TdTomato, arrows) indicating OPC death within perivascular clusters. (n) Percentage of TdTomato+ cells that are TUNEL+ in perivascular clusters versus non clusters in Olig2cre:APCfl/fl:TdTomato (n=6 animals, p=0.0009). Data were analyzed by unpaired two-sided Student’s t test.Scale bars, 10μm (l), 20μm (all other panels). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Values are mean ± s.d.
To further assess the contribution of OPC perivascular clusters as a cause of BBB disruption, we performed an in vivo pharmacological blockade of the chemokine mediating their vascular association. Postnatal treatment of Olig2cre:APCfl/fl mice intravenously with the Cxcr4-Sdf1 inhibitor AMD3100 leads to significant reversal of OPC cluster size on vasculature (p=1.04 E-5)(Suppl. Fig. 13a-c). This led to concomitant reductions in the number of vessels lacking Aqp4 (p=0.0001)(Suppl. Fig. 13g-i) and end feet coverage (p=0.0008)(Suppl. Fig. 13d-f), and significant normalization of fibrinogen extravasation (p=0.0023)(Suppl. Fig. 13j-l) and CD11c+ numbers (p=0.0044)(Suppl. Fig. 13m-o). These results identify a requirement for the physical presence of OPC clusters on blood vessels for effects on vascular integrity, and show that failure of OPC detachment from vasculature can itself disrupt the BBB and trigger CNS inflammation, in an otherwise non-inflammatory setting.
Wif1 production by Wnt activated perivascular OPCs disrupts EC tight junctions.
OPC perivascular clusters cause disruption of cells at the vascular surface, and their physical presence on vessels is required for BBB effects. We questioned how these perivascular OPCs might further contribute to effects on the barrier function of the BBB via local effects resulting from physical proximity to the endothelium. Excessive Wnt signaling in OPCs mediates their perivascular accumulation. To uncover factors upregulated in clustered perivascular OPCs that might have effects on neighboring endothelium, we performed RNAseq profiling of spinal cord white matter from P4 and P9 Olig2cre:APC fl/fl mice (Suppl. Fig. 14a). Differential expression analysis compared with APCfl/fl controls identified 270 genes significantly altered (FDR < 0.05) at P4 (43 up-regulated and 227 down-regulated genes in Olig2cre:APC fl/fl)(Suppl. Fig. 14b), and 4,505 genes in P9 (2,092 up and 2,413 down in Olig2cre:APC fl/fl). We identified Wif1 (Wnt inhibitory factor 1) as the most upregulated secreted factor in this tissue at both time points (FDR < 2.13E-63 at P4, FDR < 3.79E-89 at P9)(Suppl. Fig. 14c). We found Wif1 mRNA expression markedly increased in Wnt-activated perivascular OPCs in spinal cord (Fig. 6a, Suppl. Fig. 15g, p=2.67 E-5) and corpus callosum (Fig. 6b, d) of Olig2cre:APC fl/fl mice, in cultured OPCs from these same mice (Fig. 6c, p=5.43 E-5), and in wildtype OPCs treated with Wnt3a (Fig. 6c, p=0.0093). Wif1 protein was also significantly increased on western blot in OPCs isolated from Olig2cre:APC fl/fl compared to controls (Suppl. Fig. 15a-b)(p=0.0286). WIF1 is also expressed by OPCs in MS lesions. We find WIF1 mRNA expression in OLIG2+ clusters in MS (Fig. 6e-f), and evidence of WIF1 protein expressed by cells around the vasculature in active MS lesions (Fig. 6g, Suppl. Fig. 1d). Wif1 is directly upregulated by Wnt signaling in a number of cell types, acting as a negative feedback control mechanism of the pathway (12). It is a secreted protein that functions as a paracrine inhibitor of the pathway by binding, and inhibiting the activity of, extracellular Wnt ligands (14). As Wnt ligands are important for the induction and maintenance of barrier properties in endothelial cells, establishing a paracrine loop that maintains the BBB (15–18), we reasoned that high local production of Wif1 by Wnt-activated perivascular OPCs might affect endothelial cell tight junction integrity. We found that treatment of primary CNS endothelial cells with Wif1 significantly (p=0.0235) reduced their expression of Claudin5 tight junctions (Fig. 6h-i, Suppl. Fig. 15c-d), and counteracted the effects of Wnt ligands (p=3.03 E-6) in inducing barrier markers in these cells (Fig. 6h, i, m, Suppl. Fig. 15c-d). This was accompanied by a reduction in Wnt tone in endothelial cells (evidenced by significantly reduced Axin2 expression, p=0.0338), and increased expression of the endothelial fenestrae marker PLVAP (p=1.46 E-11)(Fig. 6m). Similar effects on tight junction formation, barrier marker expression, and Wnt tone in endothelial cells were observed when treated with conditioned medium from Wnt hyperactive (Olig2cre:APC fl/fl) OPCs (Fig. 6j, k, n; Suppl. Fig. 15e-f). Selective depletion of secreted Wif1 protein from this conditioned medium, using anti-Wif1 antibody mediated pull down (Fig. 6l), prevented these effects on endothelium (Fig. 6j, k, n; Suppl. Fig. 15e-f), identifying it as the mediator of these effects on tight junction marker expression and Wnt pathway tone in ECs. Wif1 also functionally regulates the barrier properties of endothelium (Fig. 6o), as Wif1 treatment of ECs in an in vitro BBB permeability assay significantly increases the permeability to a 10-kDa dextran-tetramethylrhodamine dye and significantly counteracts the action of Wnt ligands to reduce barrier permeability. Evidence of WIF1 expression in OLIG2+ clusters in human white matter injury (Fig. 6e-f) suggests that high local production of this Wnt inhibitor by perivascular OPCs may also contribute to dysfunction of EC barrier properties in MS.
Fig. 6: Wif1 production by Wnt activated perivascular OPCs disrupts EC tight junctions.
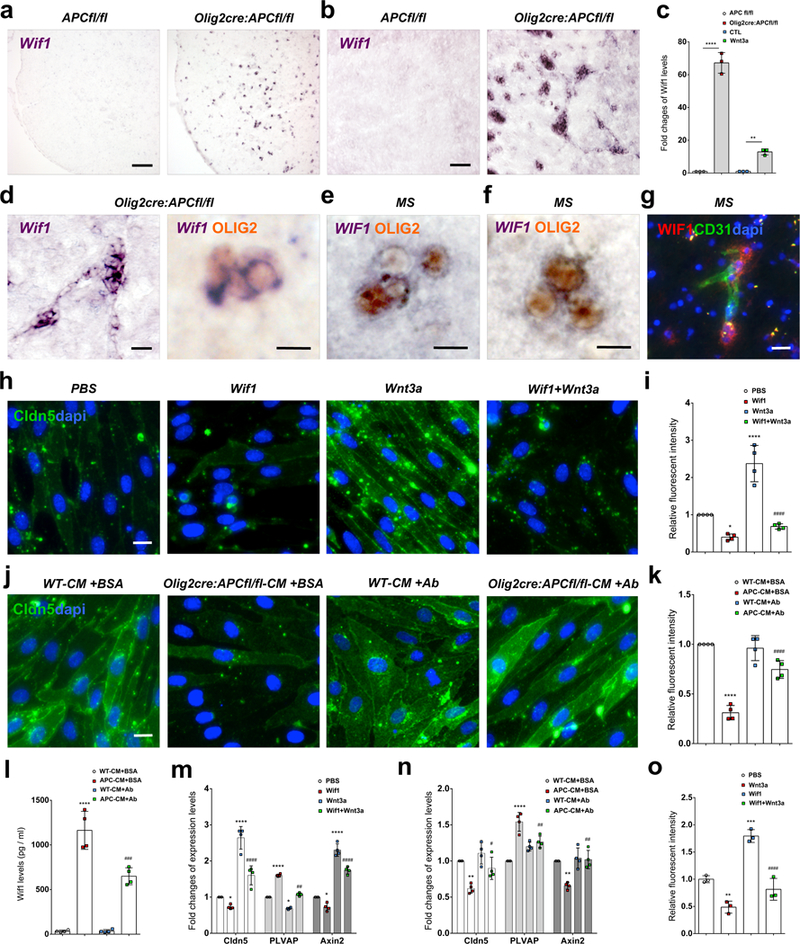
(a-c) Wif1 mRNA is highly upregulated by Wnt activation in OPCs in Olig2cre:APCfl/fl mice in vivo in P9 mouse (a) spinal cord and (b) corpus callosum and (c) in vitro (fold change by qPCR) in isolated OPCs from Olig2cre:APCfl/fl compared to APCfl/fl (n=3 independent experiments, ****p=5.43E-5), and in isolated WT OPCs treated with Wnt3a compared to untreated CTL (n=3 independent experiments, **p=0.0093). Data were analyzed by unpaired two-sided Student’s t test (c). (d) Significantly upregulated Wif1 mRNA in vivo co-localizes with Olig2 protein in clustered cells in Olig2cre:APCfl/fl corpus callosum. (e-f) WIF1 is expressed by clustered OLIG2+ cells in MS active lesion white matter (f). (g) WIF1 protein expression by cells around CD31+ vasculature (arrows) in MS lesion white matter. (h) Tight junctions stained for Claudin5 (Cldn5) protein in isolated CNS endothelial cells (ECs) treated with control PBS, Wif1, Wnt3a, or Wif1+Wnt3a for 2 days, and (i) Claudin5 fluorescent intensity sums (as fold change compared to PBS control) in EC cultures under these treatment conditions (n=4 independent experiments; PBS vs. Wif1 *p=0.0235; PBS vs. Wnt3a ****p=2.59 E-5; Wnt3a vs. Wif1+Wnt3a ####p=3.03 E-6). Data were analyzed by one-way ANOVA. (j) Claudin5+ tight junctions in isolated ECs treated for 2 days with conditioned medium from APCfl/fl or Olig2cre:APC fl/fl OPCs (WT-CM+BSA and Olig2cre:APC fl/fl-CM+BSA respectively) or conditioned medium from the same OPCs depleted of Wif1 protein by overnight anti-Wif1 antibody (+Ab) treatment and agarose bead pull down, and (k) Claudin5 fluorescent intensity sums (as fold change compared to WT-CM+BSA control) in EC cultures under these treatment conditions (n=4 independent experiments; WT-CM+BSA vs. APC-CM+BSA ****p=3.13 E-7; APC-CM+BSA vs. APC-CM+Ab ####p=5.49 E-5). Data were analyzed by one-way ANOVA. (l) ELISA OD values for Wif1 protein concentrations in conditioned medium from these groups (n=4 independent experiments; WT-CM+BSA vs. APC-CM+BSA ****p=6.79 E-8; WT-CM+AB vs. APC-CM+Ab ###p=0.0002). Data were analyzed by one-way ANOVA. (m-n) Fold change of mRNA in cultured ECs (by qPCR relative to control) for tight junction marker Claudin5, endothelial fenestrae marker PLVAP, and Wnt pathway activation marker Axin2 in the Wif1/Wnt3a treatment groups from Fig.4h (m), and the OPC conditioned medium treatment groups from Fig. 4j (n). (m) n=4 independent experiments; Claudin5: PBS vs. Wif1 *p=0.0381; PBS vs. Wnt3a ****p=3.86 E-7; Wnt3a vs. Wif1+Wnt3a ####p=6.16 E-5; PLVAP: PBS vs. Wif1 ****p=1.46 E-11; PBS vs. Wnt3a *p=0.0137; Wnt3a vs. Wif1+Wnt3a ##p=0.0017; Axin2: PBS vs. Wif1 *p=0.0338; PBS vs. Wnt3a ****p=8.97 E-9; Wnt3a vs. Wif1+Wnt3a ####p=8.07 E-5. Data were analyzed by one-way ANOVA. (n) n=4 independent experiments; Claudin5: WT-CM+BSA vs. APC-CM+BSA **p=0.0019; APC-CM+BSA vs. APC-CM+Ab #p=0.018; PLVAP: WT-CM+BSA vs. APC-CM+BSA ****p=3.93 E-6; APC-CM+BSA vs. APC-CM+Ab ##p=0.00217; Axin2: WT-CM+BSA vs. APC-CM+BSA **p=0.00164; APC-CM+BSA vs. APC-CM+Ab ##p=0.00103; In these three biomarkers, WT-CM+BSA vs. WT-CM+Ab has no significant difference. Data were analyzed by one-way ANOVA. (o) In vitro BBB endothelial permeability assay with control PBS, Wif1, Wnt3a, or Wif1+Wnt3a (n=3 independent experiments; PBS vs. Wnt3a **p=0.0063; PBS vs. Wif1 ***p=0.0004; Wif1 vs. Wif1+Wnt3a ####p=8.22 E-5). Data were analyzed by one-way ANOVA. Scale bars, 90μm (a, b), 20μm (d left panel, g), 10μm (d right panel, e, f, h, j). Values are mean ± s.d.
Discussion
Recruitment of OPCs into areas of demyelination is a critical initial step for successful myelin regeneration, and dysfunction of this process contributes to remyelination failure in MS (10). We show here that OPCs utilize single cell perivascular migration on microvessels for recruitment into murine demyelinated injury. This perivascular migration following injury appears to be a recapitulation of their developmental migration, where OPCs require a vascular scaffold for their dispersal through the CNS (13). They show a significantly increased association with vasculature at lesion edges at early time points during remyelination, along with morphological changes more akin to their developmental counterparts. Live imaging studies in murine remyelination suggest a limited time window of OPC migration into lesions around 1.5dpl, with the majority of OPC numbers in lesions made up from proliferation once they have migrated into the intralesional environment.
Completion of OPC perivascular migration requires their detachment from vessels to allow for oligodendrocyte differentiation and interaction with axonal targets. We find in several MS cases, in lesion areas with active inflammation, that OPCs can be found clustered on vasculature, representing a defect in single cell perivascular migration and inability to detach from blood vessels. We do not see this OPC perivascular clustering in wildtype murine remyelination, and it therefore seems to represent a feature that distinguishes human from murine white matter injury. This OPC perivascular clustering is mediated by aberrant Wnt tone in remyelination, as evidenced in PDGFRα-creERT2:APCfl/fl and Olig2-cre:APCfl/fl. Importantly, it affects both proper OPC dispersal into remyelinating lesions, and exposes them to further damage from blood-borne inflammatory insults.
Failure of OPC detachment from vasculature following migration represents a pathological oligodendroglial-vascular interaction and detrimentally affects the underlying vessel. We find in both development and remyelination that OPC perivascular clusters disrupt other cells at the microvessel surface, causing physical displacement of astrocyte end feet from vessels, with the processes of clustered OPCs seeming to exist between endothelium and overlying astrocyte endfeet. Whilst pericyte coverage seems unaltered, it will be important to assess how these cells might be functionally affected, considering critical roles in formation of the BBB (19) and emerging roles in CNS remyelination (20). In order to determine the specific effects of these OPC clusters on the integrity of underlying vasculature, we had to make use of a non injury model, as injury models by their very nature induce significant vascular disruption. We find, in an otherwise non inflammatory setting, that OPC perivascular clusters themselves cause endothelial disruption, and BBB integrity defects that trigger a subsequent CNS inflammation. Vascular integrity defects lead to damage to surrounding axons and perivascular OPCs, identifying OPC clusters themselves as contributors to pathology.
Pharmacological reversal of OPC perivascular clustering with AMD3100 (which inhibits the OPC-vascular Cxcr4/Sdf1 interaction without altering OPC Wnt tone) in vivo in Olig2-cre:APCfl/fl shows that these clusters are required for detrimental BBB effects, and moreover that their physical presence on vessels is necessary for these effects, presumably through disruption of cells at the vascular surface. In addition, we identify Wif1, previously reported to disrupt the BBB in Wnt-driven medulloblastoma (21), as a factor secreted by OPCs in Wnt-driven perivascular clusters that may contribute to endothelial dysfunction in MS. Wif1 secreted by Wnt activated OPCs acts to counteract the effects of Wnt ligands in promoting endothelial tight junction markers, acts to increase permeability of endothelium, and also to reduce Wnt tone in endothelial cells. Considering that Wnt tone in CNS vessels has been implicated in the restoration of BBB integrity in MS, and also in limiting immune cell infiltration into the CNS (22), the local production of Wif1 by dysregulated perivascular OPCs could further contribute to pathology in MS. There may be a variety of contributors to BBB dysregulation in MS that are not fully understood. It will be important to establish the contribution of this aberrant oligodendroglial-vascular interaction to the failed OPC recruitment seen in many MS lesions, and to perpetuation of disease through ongoing disruption of the BBB.
Methods
Mice
Animal husbandry and procedures were performed according to UCSF guidelines under IACUC approved protocols. Mouse ages were identified in main text and figure legends. Both mouse genders were used in this study. Mice were randomly assigned to the different experimental groups.
NG2-CreERT:TdTomato
This mouse has been described previously (23). We used 8-week old mice to label OPCs in slice cultures. Before slice culture, the mice were given tamoxifen (T-5648, Sigma) gavage with 1mg of tamoxifen twice a day for 5 consecutive days (10mg in total).
Olig2-cre
A multi-functional mouse line was constructed previously (24) by inserting tva, an avianspecific retroviral receptor, and an IRES-cre recombinase cassette into the endogenous Olig2 locus by homologous recombination. This Olig2-cre allows for cre mediated activity in oligodendrocyte lineage cells.
PDGFRα-creERT2
These mice have been described previously (25). A tamoxifen inducible cre recombinase gene was inserted into exon 2. These mice were used to cross with APC floxed mice. In order to knock out APC, the mouse was given tamoxifen gavage as described above.
APC floxed
These mice have been described previously (26). A conditional (floxed) allele of APC (adenomatous polyposis coli) was provided by Dr. R. Fodde (Leiden University). Through intercrosses with Olig2-cre or PDGFRα-CreERT2 recombinase we can achieve conditional knockout of APC in OPCs. We used a breeding strategy of crossing either Olig2cre:APCfl/fl or PDGFRα-CreERT2:APCfl/fl mice with APCfl/fl mice, thus generating Olig2cre:APCfl/fl or PDGFRα-CreERT2:APCfl/fl offspring and APCfl/fl control littermates. Control littermates are written as ‘APCfl/fl’ in figures.
Aldh1l1-GFP
These mice have been described previously (27). The diffuse expression pattern of the Aldh1L1-GFP throughout the CNS is consistent with pan-astrocyte expression, which allows us to visualize the astrocyte processes and end feet in our study.
Cx3cr1-GFP:CCR2-RFP
These mice have been described previously (28). The Cx3cr1-GFP specifically labels microglia in the CNS, and CCR2-RFP labels macrophages. By using this reporter mouse, we are able to distinguish infiltrating macrophages from resident microglia within tissue sections.
Immunohistochemistry
The method has been described previously (29). Primary antibodies were to the following proteins: PDGFRα (1:200, rat, 558774, BD Biosciences), PDGFRα (1:8000, rabbit, gift from W. Stallcup, Sanford Burnham Prebys), Olig2 (1:2000, rabbit, gift from C.D. Stiles, Harvard), CD31 (1:200, rat, 553370, BD Biosciences), CD31 (1:200, rabbit, ab28346, Abcam), RNF43 (1:200, rabbit, ab84125, Abcam), WIF1 (1:200, rabbit, ab71204, Abcam), Caspase-3 (1:200, rabbit, 9661, Cell signaling), Aqp4 (1:100, mouse, ab9512, Abcam), PLVAP (1:100, rat, 553849, BD Pharmingen), Claudin5 (1:200, mouse, 352588, ThermoFisher), Fibrinogen (1:200, mouse, ab58207, Abcam), Iba1 (1:500, rabbit, SAG4318, Wako), CD11c (1:500, rabbit, MBS767644, MyBioSource), Arg1 (1:100, goat, sc-18355, Santa Cruz), iNOS (1:200, rabbit, PA3030A, ThermoFisher), CD3 (1:100, rabbit, ab5690, Abcam), CD4 (1:200, rabbit, ab183685, Abcam), CD8 (1:100, rat, ab22378, Abcam), F4/80 (1:500, rat, ab6640, Abcam), Ki67 (1:200, rabbit, ab16667, Abcam), SMI32 (1:1000, mouse, NE1023, Millipore), NF200 (1:1000, rabbit, N4142, Sigma), Wif1 (1:1000, rabbit, ab155101, Abcam).
In Situ Hybridization
The technique has been described previously (29). Briefly, DIG labeled antisense Axin2, Cxcr4, Wif1, or PDGFRα probes were used. The targeted mRNA-expressing cells were visualized as a dark purple deposition with NBT/BCIP–alkaline phosphatase combination. Cell counting was performed under a 10× objective lens for each sample using an Image Pro Plus software.
Lysolecithin lesioning
The method has been described previously (29). Demyelinated lesions were produced in the dorsal funiculus spinal cord white matter of 8- to 10-week-old Olig2-cre:APCfl/fl, PDGFRα-creERT2:APCfl/fl, and APCfl/fl littermate mice. Anesthesia was induced and maintained with inhalational isoflurane and oxygen supplemented with 0.05 ml of buprenorphine (Vetergesic; 0.05 mg/ml) given subcutaneously. Having exposed the spinal vertebrae at the level of T12/T13, tissue was cleared overlying the intervertebral space, and the dura was pierced with a dental needle just lateral to midline. A Hamilton needle was advanced through the pierced dura at an angle of 45°, and 0.5 μl 1% lysolecithin (L-α- lysophosphatidylcholine) was injected into the dorsal funiculus white matter.
Live imaging of remyelination in slice culture
Demyelination was produced in mouse spinal cords in vivo, as described above. Live imaging was performed on slices taken from spinal cord lesions at various times after lysolecithin induced demyelination in vivo. Images shown in Figure 2 and Supplementary Figures 2 and 3 were from slices of cord taken at 1.5 days post lesioning (dpl). To label the shape of the vasculature we injected Fluorescent-lectin (100ul, Vector Labs DL-1174) via tail vein injection. After 10 minutes allowing dye circulation, spinal cords were dissected immediately in ice-cold artificial cerebrospinal fluid (ACSF). Spinal cords were then embedded in 5% low melting point agarose in ACSF, and vibratome-sliced at 300μm. Slices were transferred onto Millicell-CM slice culture inserts (Millipore) over culture medium (FluoroBrite DMEM with 25% horse serum, 25% HBSS, 1% N2 supplement, 1% penicillin and streptomycin) in glass bottom 6-well plates and incubated at 37 °C, 5% CO2 for at least 1 hour. The plate was then transferred to an inverted Leica TCS SP5X confocal with an on-stage incubator (while streaming 5% CO2, 5% O2), and imaged using a 10x immersion objective (NA=0.3) at specified interval and total imaging time in texts with intermittent repositioning of the focal planes. Lesion boundaries were assessed using dapi staining following the completion of live imaging. The time-lapse images were processed in Imaris (Bitplane).
Human MS
Human multiple sclerosis post-mortem tissue blocks were provided by the UK Multiple Sclerosis Tissue Bank at Imperial College London. All tissues were collected following fully informed consent by the donors via a prospective donor scheme following ethical approval by the London Multicentre Research Ethics committee (MREC 02/2/39). Multiple sclerosis lesions were characterized as described previously (30), using luxol fast blue (LFB) to assess demyelination, SMI-31 immunohistochemistry to assess preservation of axons, and LN3 immunohistochemistry to assess inflammatory cell activity. Lesions with florid parenchymal and perivascular inflammatory cell infiltration, myelin fragmentation, and demyelination with indistinct margins were classified as active plaques. Chronic active plaques were classified as those with extensive demyelination, well-demarcated borders and abundant inflammatory cells at the lesion edge. Chronic inactive plaques were classified as those with extensive demyelination and well-demarcated borders, but few or no inflammatory cells in any part of the demyelinated area. ‘Chonic’ lesions quantified in Fig. 1o and Suppl. Fig. 1a were chronic inactive areas. Areas of normal appearing white matter ‘NAWM’ and normal appearing grey matter ‘NAGM’ from each patient were also assessed. Areas of NAGM and NAWM did not demonstrate increased inflammatory activity, but it is not possible to completely rule out other subtle pathology in these areas. Cases assessed are described in Suppl. Table. 1.
Dextran-Tetramethylrhodamine tracer to assess in vivo barrier function of BBB
Dextran-tetramethylrhodamine tracer (Dextran-Tetramethylrhodamine, 10-kDa, D1817, ThermoFisher) was injected into mice through tail vein injection, and allowed to circulate for 6 hours. Brain and spinal cord were then dissected immediately and fixed by immersion fixation overnight in 4% PFA to immobilize the tracers at the end of the experiment. The Dextran-tetramethylrhodamine tracer leakage was assessed within frozen sections.
In vitro BBB permeability assay
Primary cultured endothelial cells were cultured on the apical surface of a 3μm pore size, collagen type I and fibronectin precoated Transwell filter inserts (CLS3472, Corning). After treatments, 100 mg/ml 10-kDa dextran-tetramethylrhodamine was added to upper chamber. The endothelial cell in vitro barrier was then incubated at 37°C for 1 hour. The relative fluorescence intensity of Rhodamine that had passed across the barrier into the lower chamber was measured with a FilterMax F5 microplate reader. Results shown are the mean fluorescence intensity of three independent experiments. The permeability of blank insert was used as the positive control.
OPC culture and collection of conditioned medium
Mouse OPCs were isolated by immunopanning from P8 APC fl/fl mouse and Olig2cre:APC fl/fl mouse cortices as previously described (31). Briefly, mouse brain cerebral hemispheres were minced and dissociated with papain at 37℃ for 60 min. After trituration, cells were incubated at room temperature sequentially on three immunopanning dishes: Ran-2, GalC, and O4. The purified mouse OPCs were plated onto 10 cm dishes and cultured for another 4 days. The cultured medium was filtered with a 0.22μm filter (Millipore), then collected as conditioned medium for downstream experiments or stored at −80℃. The conditioned medium from APC fl/fl control OPC cultures was labeled as WT-CM, the conditioned medium from Olig2cre:APC fl/fl OPC cultures was labeled as APC-CM.
Wif1 depletion from conditioned medium and Wif1 ELISA
To deplete Wif1 from OPC conditioned medium, APCfl/fl control or Olig2cre:APC fl/fl OPC conditioned medium were incubated with Wif1 antibody (1:100, rabbit, ab155101, Abcam) at 4℃ overnight followed by pull down with agarose beads (sc-2003, Santa Cruz). BSA was used as the non-antibody control. The purified Wif1-depleted conditioned media were then used for endothelial cell treatment. To clarify the concentrations of Wif1 protein in the conditioned media before and after Wif1 depletion, four groups of conditioned media (WT-CM+BSA, Olig2cre:APC fl/fl-CM+BSA, WT-CM+Ab and Olig2cre:APC fl/fl-CM+Ab) were examined using the Wif1 specific ELISA kit (mouse WIF1 PicoKine Elisa Kit, EK1523, Boster) according to the manufacturer’s instructions. The OD values were determined by measuring the absorbance at 450nm using the microplate reader (Bio-RAD, Model 680). Independent experiments were performed in triplicate.
Western blot
Western blot was also used to quantity Wif1 and Claudin5 expression levels. Protein samples were separated on 10 % SDS-PAGE gels, transferred to nitrocellulose membranes and probed with antibody against Wif1 (1:1000, rabbit, ab155101, Abcam), or Claudin5 (1:500, mouse, 352588, ThermoFisher). Mouse heart tissue lysate was used as the positive control. Quantification of band intensity was analyzed using the Image Pro Plus software.
Endothelial cell culture and treatment
For in vitro endothelial cell (EC) treatment, primary cultured rat endothelial cells were isolated from P14 SD rat cortices as previously described (32), and cultured on collagen type I and fibronectin pre-coated coverslips for immunostaining and 6 cm dishes for qPCR. Cultured ECs were treated with recombinant Wif1 (10ug/ml, ab208465, Abcam), recombinant Wnt3a (100ng/ml, 315–20, Peprotech) or Wif1 with Wnt3a for 2 days. PBS was used as control. To test the effects of conditioned media on ECs, cultured ECs were treated with conditioned media with or without Wif1 depletion (25% fresh medium+ 75% conditioned medium) for 2 days.
qPCR
Total ribonucleic acid (RNA) was isolated using RNeasy Plus Mini Kit (74134, Qiagen). Quantitative polymerase chain reaction (qPCR) was performed with the C1000 Touch™ Real-time PCR Detection System (Bio-Rad) and GoTaq® qPCR Master Mix (Promega). The oligonucleotide primers, amplification procedure, and melt curve analysis were performed. For each sample, independent repeats were performed in triplicate.
mRNAseq of Olig2cre:APCfl/fl mice
Whole spinal cord was dissected from three Olig2cre:APCfl/fl mice and three APCfl/fl littermate control mice at both postnatal day 4 (P4) and P9 time points. An RNAseq library was prepared using QuantSeq kit (Lexogen) according to the manufacturer’s instructions and sequenced 50-bp single-end on the HiSeq 4000 (Illumina). We trimmed adapter from sequence reads using trim galore v0.4.4 with cutadapt v1.14, and used FastQC v0.11.6 to assess quality control for fastq files. Sequence reads were mapped to the mouse genome reference (GRCm38) with gencode annotation (M14) and we counted number of reads using STAR aligner v2.5.0a. A differential expression analysis was performed on read counts using DESeq2. Based on PCA plot (Suppl. Fig. 14a) and sample-to-sample distance heatmap, we removed outlier samples which were clustered with different condition. Pair-wise comparisons were performed between Olig2cre:APCfl/fl and APCfl/fl littermate controls, and between P4 and P9. We obtained FDR adjusted p-value and log2 fold change. Genes were selected by significance threshold of FDR < 0.05, log2FC > 1 or log2FC < −1.
Code availability
R code used for the mRNAseq analysis can be found on the following github page. https://github.com/baranzini-lab/RNAseq_QuantSeq_Fancy. Raw sequence data (fastq) for the mRNAseq data are available on DASH data share (https://doi.org/10.7272/Q63N21KB).
Quantifications
OPC quantifications:
For the OPC-vessel association analysis, PDGFRα/CD31 double staining was used to detect the association of OPCs with blood vessels. The percentage of PDGFRα+ cells with their cell bodies directly in contact with blood vessels was quantified.
To quantify OPC process and blood vessel correlation, we used the image-Pro Plus 5 software to measure the value of the angle between OPC leading process and blood vessel. This measurement was only done for OPCs in which the cell body was on blood vessel and leading process was clearly seen. Oriana software was used to draw the vector map for summarizing the correlation of OPC and blood vessel.
For the OPC leading process direction analysis, we used the image-Pro Plus 5 software to draw a vector between the OPC cell body and the end of the leading process in the direction of the OPC’s leading process. This leading process direction analysis was only done for these OPCs in which the cell body was on a blood vessel and leading process was clearly seen. Oriana software was used to draw the vector maps for summarizing the directionality of OPC migration.
For the qualification of OPC migration in slice cultures, the directionality of OPC migration was measured by Image-Pro Plus 5 and Oriana as described previously. Speed of migration was measured by dividing the total length of OPC movement by the total time. Vector map shows the directions of OPC movement.
For OPC cluster quantification, a ‘cluster’ constituted 4+ cells in cell body contact.
Quantifications of astrocytic endfeet coverage
To quantify Aldh1L1-GFP astrocytic endfeet coverage, images of areas with blood vessels covered by OPC clusters in Olig2cre:APCfl/fl:Aldh1l1-GFP mice and blood vessels with no clusters in similar positions of APCfl/fl:Aldh1l1-GFP control mice were captured for analysis. When the GFP+ process was wrapped on/ along CD31+ vessels, it was considered as an astrocytic endfoot. Quantifications of length of blood vessels and GFP+ endfeet were done using image-Pro Plus 5 software. AQP4+ endfeet coverage quantification was performed in a similar way.
Quantifications of fibrinogen and dye leakage
To identify vascular leakage of fibrinogen or dye, we performed CD31 staining of blood vessels to mark out the edges of vasculature. To quantify the extent of leakage in Olig2cre:APCfl/fl, we measured the fibrinogen or dye immunoreactive areas outside these blood vessels. Similar areas in non-cre APCfl/fl mice were used as control. The fluorescent area quantifications were measured as described below.
Quantification of Claudin5/PLVAP distribution
To quantify blood vessel distribution levels of Claudin5 versus PLVAP, double staining of Claudin5 /PLVAP was performed in non-cre APCfl/fl control mice and Olig2cre:APCfl/fl mice. Regions with OPC perivascular clusters were imaged in Olig2cre:APCfl/fl alongside comparable areas in control APC:fl/fl mice. The lengths of Claudin5 and PLVAP positive endothelium in these imaged areas were measured using image-Pro Plus 5 software.
Quantification of pericyte coverage
To quantify the pericyte coverage, double staining of CD31 / PDGFRβ was performed in non-cre APCfl/fl control mice and Olig2cre:APCfl/fl mice. The length of blood vessels covered with pericytes was measured using image-Pro Plus 5 software.
Fluorescent intensity and area quantifications
To quantify fluorescent positive area, intensity and Western-blot positive band, the positive areas were automatically selected in image-Pro Plus 5 software. The areas of interest (AOI) were separated by setting threshold at least two times the background. Cell counting and fluorescence intensity analyses were conducted on six randomly chosen fields for each sample using an Image Pro Plus image analysis system.
Statistical analysis
Statistical significance between groups was determined with GraphPad Prism software. The unpaired t-test was used to determine the significance between two experimental groups. One-way analysis of variance (ANOVA) was used to determine the significances among three and four groups. Data distribution was assumed to be normal, but this was not formally tested. A probability of P<0.05 was considered statistically significant. All significant statistical results are indicated within the figures with the following conventions: * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Error bars represent ± s.d. No statistical methods were used to pre-determine sample sizes. The sample size per group was determined from previous publications with similar methodology. Control mice and their littermate mutant mice were collected from different litters, and randomly selected for each experiment. For lesioning experiments using adult control mice, those mice were randomly assigned into different time points as indicated. Investigators were blinded to group allocation during data analysis. All experiments were performed at least 3 times, and the findings were replicated in individual mice and cell cultures in each experiment.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31871045) and National Science Foundation of Chongqing (cstc2018jcyjAX0702) to J.N., the US National Institutes of Health (R01–5R01NS088155) to S.E.B. (S.E.B. is the Heidrich Family and Friends Endowed Chair in Neurology at UCSF), and from the US National Institutes of Health (1R01NS097551–01A1) to S.P.J.F. S.P.J.F. is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society.
Footnotes
Data Availability
The data that support the findings of this study are available from the corresponding author upon request. Raw sequence data (fastq) for the mRNAseq data are available on DASH data share (https://doi.org/10.7272/Q63N21KB).
Accession Codes
Raw sequence data (fastq) for the mRNAseq data are available on DASH data share (https://doi.org/10.7272/Q63N21KB).
Competing interests
The authors declare no competing interests.
References
- 1.Compston A, Coles A, Multiple sclerosis. Lancet 372, 1502–1517 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Maggi P et al. , The formation of inflammatory demyelinated lesions in cerebral white matter. Ann Neurol 76, 594–608 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez JI, Cayrol R, Prat A, Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta 1812, 252–264 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Minagar A, Alexander JS, Blood-brain barrier disruption in multiple sclerosis. Mult Scler 9, 540–549 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Obermeier B, Daneman R, Ransohoff RM, Development, maintenance and disruption of the blood-brain barrier. Nat Med 19, 1584–1596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franklin RJM, Ffrench-Constant C, Regenerating CNS myelin - from mechanisms to experimental medicines. Nat Rev Neurosci 18, 753–769 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Gallo V, Deneen B, Glial development: the crossroads of regeneration and repair in the CNS. Neuron 83, 283–308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang A, Tourtellotte WW, Rudick R, Trapp BD, Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. The New England journal of medicine 346, 165–173 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Kuhlmann T et al. , Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131, 1749–1758 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Boyd A, Zhang H, Williams A, Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol 125, 841–859 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen MA et al. , Fibrinogen Activates BMP Signaling in Oligodendrocyte Progenitor Cells and Inhibits Remyelination after Vascular Damage. Neuron 96, 1003–1012 e1007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fancy SP et al. , Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat Neurosci 17, 506–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai HH et al. , Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 351, 379–384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsieh JC et al. , A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature 398, 431–436 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Liebner S et al. , Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol 183, 409–417 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daneman R et al. , Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A 106, 641–646 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y et al. , Canonical WNT signaling components in vascular development and barrier formation. J Clin Invest 124, 3825–3846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang J et al. , Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat Med 23, 450–460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daneman R, Zhou L, Kebede AA, Barres BA, Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De La Fuente AG et al. , Pericytes Stimulate Oligodendrocyte Progenitor Cell Differentiation during CNS Remyelination. Cell Rep 20, 1755–1764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phoenix TN et al. , Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 29, 508–522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lengfeld JE et al. , Endothelial Wnt/beta-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc Natl Acad Sci U S A 114, E1168–E1177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu X et al. , Age-dependent fate and lineage restriction of single NG2 cells. Development 138, 745–753 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuller U et al. , Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14, 123–134 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivers LE et al. , PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 11, 1392–1401 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robanus-Maandag EC et al. , A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis 31, 946–952 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Cahoy JD et al. , A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28, 264–278 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizutani M et al. , The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol 188, 29–36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnett HA et al. , bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science 306, 2111–2115 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Lock C et al. , Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8, 500–508 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Mei F et al. , Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med 20, 954–960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sauer I, Dunay IR, Weisgraber K, Bienert M, Dathe M, An apolipoprotein E-derived peptide mediates uptake of sterically stabilized liposomes into brain capillary endothelial cells. Biochemistry 44, 2021–2029 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.