

Abstract
High levels of oxidative stress is detected in neurons affected by many neurodegenerative diseases, including Huntington’s disease (HD). Many of these diseases also show neuronal cell death and axonal transport defects. While nuclear inclusions/accumulations likely cause cell death, we previously showed that cytoplasmic axonal accumulations can also contribute to neuronal death. However, the cellular mechanisms responsible for activating cell death is unclear. One possibility is that perturbations in normal axonal transport alter the function of the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT)-pathway, a signal transduction pathway that promotes survival/growth in response to extracellular signals. To test this proposal in vivo, we expressed active PI3K in the context of pathogenic huntingtin (HTT-138Q) in Drosophila larval nerves, which show axonal transport defects and neuronal cell death. We found that excess expression of active P13K significantly suppressed HTT-138Q-mediated neuronal cell death, but had no effect on HTT-138Q-mediated axonal transport defects. Expression of active PI3K also rescued Paraquat-mediated cell death. Further, increased levels of pSer9 (inactive) glycogen synthase kinase (GSK) 3β was seen in HTT-138Q-mediated larval brains, and in dynein loss of function mutants, indicating the modulation of the pro-survival pathway. Intriguingly, proteins in the PI3K/AKT-pathway showed functional interactions with motor proteins. Taken together our observations suggest that proper axonal transport is likely essential for the normal function of the pro-survival PI3K/AKT-signaling pathway and for neuronal survival in vivo. These results have important implications for targeting therapeutics to early insults during neurodegeneration and death.
Keywords: Axonal transport, cell death, PI3K, motor proteins, Huntingtin, Huntington’s disease
Key messages:
Constitutively active PI3K rescues neuronal cell death induced by excess HTT138Q
Constitutively active PI3K has no effect on axonal blockages induced by excess HTT138Q
Constitutively active PI3K suppresses neuronal cell death induced by Paraquat
Dynein loss of function or excess HTT138Q show increased levels of pGSK3β-ser9.
Components of the PI3K pathway show functional interactions with motor proteins.
Introduction:
Several studies have shown that mutations in both kinesin or dynein, the two motor proteins involved in axonal transport are directly linked to many neurodegenerative diseases. For example, mutations in KIF1B, a kinesin-3 involved in the axonal transport of synaptic vesicles cause peripheral neuropathy in a form of Charcot-Marie-Tooth disease. Mutations in kinesin-1, KIF5A causes a form of Hereditary Spastic Paraplegia, a condition that arises due to axonal degeneration of motor and sensory neurons that is maximal at the distal ends of the longest axons of the CNS. Missense mutations in dynein and dynactin cause ALS-like progressive motor neuron degeneration, with motor neuron cell loss [1]. Defects in axonal transport have also been observed in other neurodegenerative diseases such as Alzheimer’s Disease (AD), Parkinson’s Disease (PD), and Huntington’s Disease (HD), often before protein aggregation, neuronal cell death and behavioral phenotypes [1]. Therefore, long distance transport within narrow caliber axons is likely critical for neuronal homeostasis and survival.
Oxidative stress has also been implicated in many neurodegenerative diseases contributing to neuronal synaptic dysfunction and neuronal loss. Upregulation of reactive oxygen species (ROS) released by microglia and other inflammatory cells can cause axonal degeneration. Work has shown that hydrogen peroxide (H2O2) inhibited the axonal motility of mitochondria and Golgi derived vesicles suggesting that exposure to ROS can disrupt axonal transport contributing to axonal degeneration [2]. Further, ultraviolet stress has been shown to perturb the transport of amyloid precursor protein (APP)-containing vesicles in mammalian neurons [3]. EtOH exposure disrupted the axonal motility of dense core-vesicles in Drosophila larval axons [4]. Together, these studies indicate that the axonal transport pathway is extremely vulnerable as the neuron responses to exogenous stressors. However, the mechanisms of how oxidative stress contributes to neuronal dysfunction and loss are unknown.
We previously showed that pathogenic huntingtin (HTT) containing expanded polyQ repeats disrupted axonal transport and caused axonal accumulations together with neuronal cell death [5]. Neuronal cell death was also induced by targeting pathogenic HTT to the nucleus with no effect on axonal transport, suggesting that defects in transport can lead to cell death. However, the mechanisms of how cell death is initiated or the signaling pathways that are activated are unknown. One pathway that is of particular interest is the PI3K/AKT/GSK3β pro-survival signaling pathway, because it is both sufficient and necessary for trophic factor dependent neuronal survival and synaptic plasticity, and mediates survival signals in a wide range of neuronal cells [6]. Binding of NGF or BDNF to their cognate tyrosine kinase receptor activates the PI3K/AKT pathway and elicits the recruitment of phosphoinositide 3-kinase (PI3K) [7–9]. PI3K in turn with phosphoinositide phosphates PIP2 and PIP3 activate the serine/threonine kinase, AKT. Work has identified GSK3β as a critical downstream effector for the PI3R/AKT survival cascade in primary neurons [10,11]. In response to survival factors, AKT can mediate neuronal survival by repressing the activity of GSK3β by phosphorylation of GSK3β at serine 9 [12]. Indeed, evidence for a substantial pro-survival role for GSK3β, when inhibited by phosphorylation at ser9 has been shown in several cells, including pluripotent stem cells [13–16]. However, when activated by phosphorylation at tyrosine 216, GSK3β can promote apoptosis by inhibiting pro-survival transcription factors (CREB and heat shock factor-1) [17], by facilitating pro-apoptotic transcription factors such as p53 [18] causing neuronal cell death.
Therefore, GSK3β can also induce apoptosis in a wide variety of conditions including DNA damage, hypoxia and endoplasmic reticulum stress [19]. However, while the mechanisms of the PI3K/AKT pro-survival signaling pathway under stress and neurodegeneration appear to be complex, whether disruption of axonal transport inhibits pro-survival signaling is unknown.
Here we test the proposal that perturbations in axonal transport alter the function of the PI3K/AKT pro-survival signaling pathway in vivo in Drosophila. We found that neuronal expression of constitutively active PI3K significantly suppressed neuronal cell death induced by expanded polyQ repeats, while no effect was seen on axonal blockages induced by expanded polyQ. Expression of constitutively active PI3K also suppressed Paraquat-mediated neuronal cell death. Intriguingly, larvae expressing expanded polyQ repeats or larvae carrying a loss of function mutant for dynein showed increased phosphorylation of GSK3β at serine 9. Components of the pro-survival signaling pathway and motor proteins showed functional interactions in the context of axonal transport. Taken together our observations suggest that normal axonal transport is likely essential for the maintenance of the pro-survival PI3K/AKT-signaling pathway and for neuronal survival. This work has important implications for the development of therapeutics targeted to early insults during neuronal dysfunction.
Results:
Neuronal expression of human Huntingtin with expanded polyQ repeats cause axonal transport defects and cell death.
Previous work showed that HTT moves bi-directionally in vivo in larval segmental nerves [20], and in vitro in primary neuronal cultures [21–22]. Larvae expressing human HTT with 15Q repeats (HTT-15Q-normal) show smooth staining in larval segmental nerves with an antibody to a synaptic vesicle protein cysteine string protein (CSP), similar to what is observed in WT larvae (Fig 1A). These larvae also show smooth HTT (mRFF) staining within their segmental nerves (Fig 1A). In contrast, larvae expressing expanded polyQ repeats in the context of HTT (HTT-138Q) show axonal blockages that contain CSP and HTT (mRFP) (Fig1A). Quantification analysis indicates that the extent of axonal blockages is comparable to what was previously seen for loss of function mutations of motor proteins [5, 25, 65–66] (Fig1A, Table 1). These blockages are likely due to disruption of transport as previous work showed decreased APP or HTT vesicle velocities in larval axons expressing expanded polyQ alone or in the context of HTT [5, 20–22]. Further, EM analysis of larval nerves from larvae expressing expanded polyQ repeats showed many types of identifiable axonal cargo (mitochondria, dense core vesicles) within axonal blockages [5].
Figure 1. Expression of human HTT with 138Q repeats cause axonal transport defects and neuronal cell death.
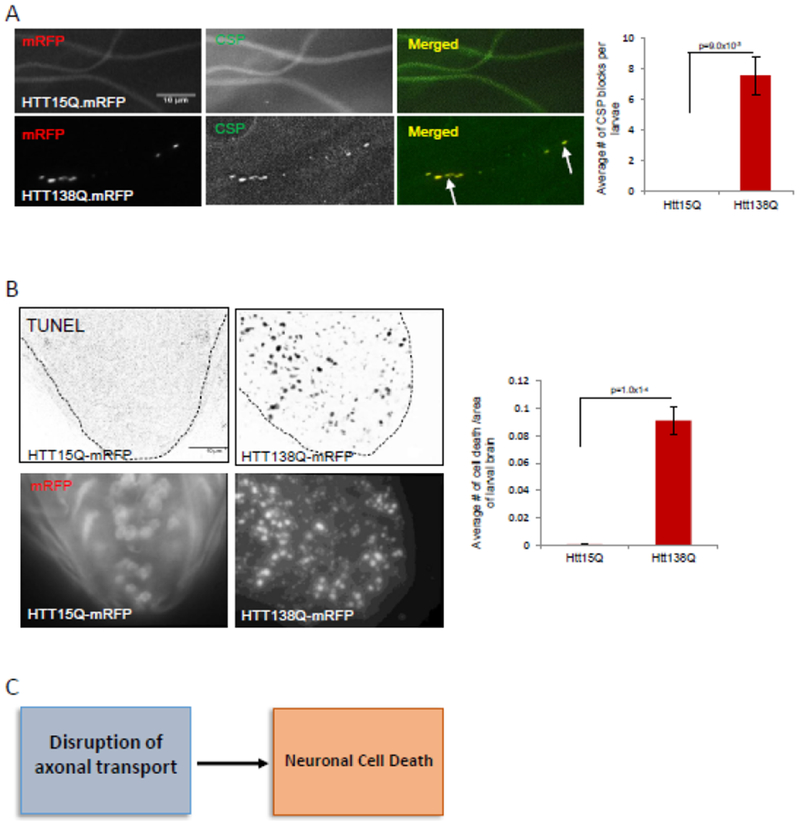
A: Larval segmental nerves from larvae expressing HTT15QmRFP show smooth staining similar to wild type (WT) larvae with the synaptic vesicle marker cysteine string protein (CSP). In contrast, larval segmental nerves from larvae expressing HTT138QmRFP show axonal blockages that contain CSP (arrows). Note that HTT is also present in these blocks. Quantification of axonal blockages indicate significant amounts of axonal blockages in larvae expressing HTT138Q compared to larvae expressing HTT15Q or WT (p=9.0×10−3). N=8 larvae for each genotype. Bar=10μm. B: Larval brains expressing HTT138Q show TUNEL positive cells, in contrast to brains expressing HTT15Q or WT. Quantification analysis indicates a significant amount of neuronal cell death in larval brains expressing HTT138Q compared to larval brains expressing HTT15Q or WT (p=1.0×10−4). Note that larval brains expressing HTT15Q has smooth mRFP staining in the cell bodies while larval brains expressing HTT138Q has mRFP aggregates. N=8 larvae for each genotype. Bar=10μm. C: Flow chart summarizing our observations propose that defects in axonal transport can contribute to neuronal cell death.
Table 1:
Summary of axonal blockages and cell death observations in larvae expressing normal and pathogenic forms of HTT compared to larvae expressing constitutively active and dominant negative P13K. N=(6-10 for each genotype.
| Genotype | Axonal blockages | Cell Death |
|---|---|---|
| Wild type | No | No |
| HTT15Q | No | No |
| HTT138Q | Yes | Yes |
| PI3K.CAAX | No | No |
| PI3K.DN | - | Yes |
| HTT138Q;PI3K.CAAX | Yes | Decreased |
Larvae expressing HTT-138Q show neuronal cell death in contrast to larvae expressing HTT-15Q, as assayed using the TUNEL assay (Fig 1B, Table 1). Quantification analysis show significant amounts of neuronal cell death in HTT-138Q compared to HTT-15Q or WT. These observations are similar to our previous work where both axonal blockages and neuronal cell death were observed in larvae expressing expanded polyQ repeats alone or in the context of HTT or MJD [5]. To identify how axonal transport defects contribute to neuronal cell death, we previously examined larvae expressing expanded polyQ repeats with a nuclear localization sequence (NLS) or a nuclear export sequence (NES). Restricting the expression of pathogenic polyQ to the nucleus using NLS caused neuronal cell death and polyQ protein accumulations within cell bodies, but no axonal blockages were seen within larval segmental nerves [5]. In contrast, restricting the expression of pathogenic polyQ to the cytoplasm using NES showed axonal blockages within larval segmental nerves and neuronal cell death with polyQ accumulations within the cell bodies of larval brains [5]. Taken together these observations postulate that defects in axonal transport can contribute to neuronal cell death (Fig 1C).
Neuronal expression of constitutively active PI3K rescues huntingtin-induced neuronal cell.
To isolate how defects in axonal transport contributes to cell death, we examined the pro-survival phosphotidylinositol 3 kinase (PI3K)/AKT/GSK3β pathway which is a critical signaling cascade that is essential to facilitate cellular survival, proliferation and differentiation. The pro-survival signaling pathway has been implicated in aging and lifespan regulation, and in the proliferation of adult neuronal progenitor cells, as well as synaptic plasticity [36–37, 56]. To evaluate the role of PI3K we examined Drosophila PI3K 92E (Dp110, a class I PI3K). Drosophila has three genes coding for PI3Ks. While the Drosophila class I PI3K gene Dp110 influences growth and cell survival, Drosophila class II and class III PI3K genes have no effect on growth. Work has shown that activation of AKT during Drosophila growth is regulated by PI3K Dp110 [23], similar to what is known in mammals. Overexpression of a constitutively active Dp110 (Dp110-CAAX or P13K.CAAX) in the wing or in the eye imaginal discs enhanced cellular growth, resulting in enlarged cells and organs [23]. However, mutations in Dp110 or expression of dominant negative Dp110 (PI3K.DN) caused larval lethality [23]. Further, Dp110 interacts with key components of the insulin signaling pathway including Chico, PTEN and AKT to control insulin-signaling-dependent cell and organ growth in Drosophila [24].
Since constitutively active P13K (P13K.CAAX) enhanced cellular growth, we tested the hypothesis that expression of P13K.CAAX within neurons in the context of HTT-138Q will promote cell survival, by generating larvae expressing both P13K.CAAX and HTT-138Q in all neurons using the pan-neuronal APPL-GAL4 driver. In control experiments larval brains expressing P13K.CAAX alone did not show cell death and were comparable to WT (Fig 2A, Table 1). These larvae also survived to adults. In contrast, larval brains expressing dominant negative PI3K (PI3K.DN) showed significant amounts of cell death (Fig 2A, Table 1) and failed to survive to adults. Larval brains co-expressing HTT-138Q with PI3K.CAAX also showed cell death. However, quantification analysis revealed a significant decrease in the extent of cell death compared to larvae expressing HTT-138Q alone (Fig 2A, Table 1). Intriguingly, the extent of HTT accumulations within these larval brains were also significantly reduced with the expression of PI3K.CAAX compared to larvae expressing HTT-138Q alone (Fig 2B). However, co-expression of HTT-138Q with P13K.DN caused sever lethality with no larvae observed. Taken together, these observations suggest that while HTT accumulations within cell bodies likely contribute to cell death, excess expression of constitutively active P13K can rescue neuronal cell death (Fig 2C).
Figure 2: Expression of constitutively active PI3K rescues HTT-induced neuronal cell death and HTT aggregates within larval brains.
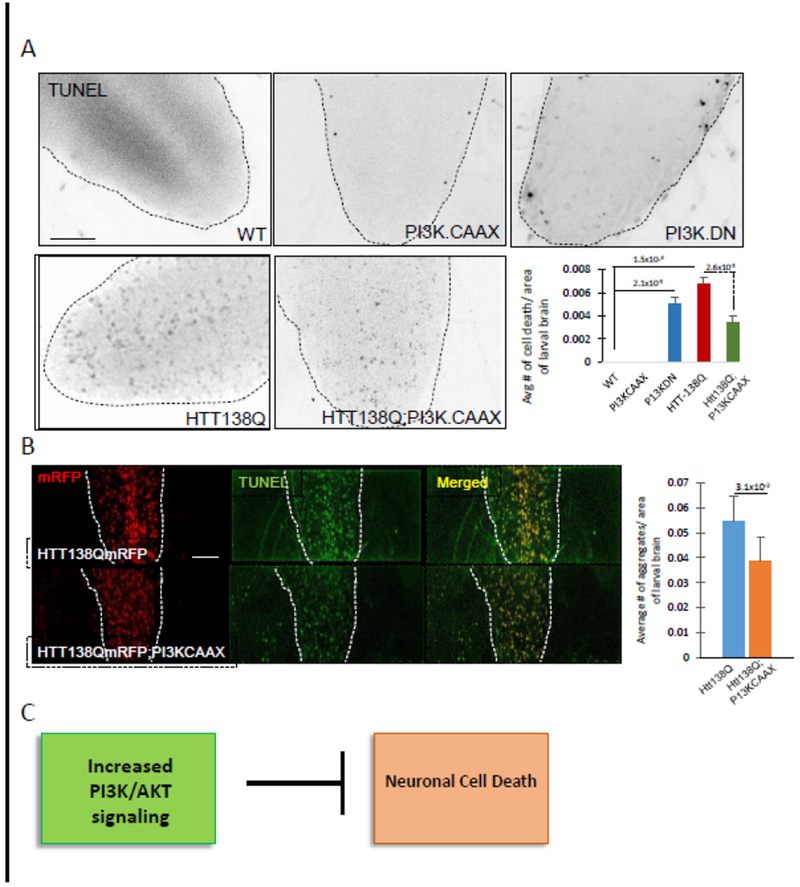
A: Larval brains expressing HTT138QmRFP alone show significant amounts of cell death (p=1.5×10−4). Larval brains expressing both HTT138QmRFP and constitutively active PI3K (HTT138Q;PI3K.CAAX) show decreased amounts of neuronal cell death as compared to HTT138QmRFP alone. Quantification analysis of 8 larval brains for each genotype show that the level of suppression of neuronal cell death is significant in HTT138QmRFP;P13K.CAAX larval brains compared to larval brains expressing HTT138Q alone (p=2.6×10−3). Note that larval brains expressing PI3K.CAAX alone are comparable to WT with little to no TUNEL positive cells, while larval brains expressing a dominant negative form of PI3K (PI3K.DN) shows significant amounts of TUNEL positive cells (p=2.1×10−3). At least 6 larval brains were quantified for each genotype from 3 independent TUNEL assays. B. Expression of HTT138Q causes HTT aggregations within cell bodies in larval brains. Quantification analysis shows that expression of P13K.CAAX in the context of HTT138QmRFP significantly decreases the amount of HTT aggregates in the larval brains (p=3.1×10−2). N=6 larvae for each genotype. Bar=10μm. C: Flow chart summarizing our observations propose that neuronal expression of constitutively active PI3K can block huntingtin-induced neuronal cell.
To test whether constitutively active P13K modifies axonal transport defects we examined the extent of axonal blockages within larval nerves co-expressing HTT-138Q with PI3K.CAAX using the synaptic vesicle marker CSP. Larvae expressing P13K.CAAX alone had smooth CSP stained larval nerves which were comparable to WT. However, larvae expressing HTT-138Q with PI3K.CAAX showed axonal blockages that contained both CSP and HTT, and were similar to larvae expressing HTT-138 alone (Fig 3, Table 1). Quantification analysis failed to show significant changes in the extent of axonal blockages that contained CSP or HTT in larvae expressing HTT-138Q with P13K.CAAX compared to larvae expressing HTT-138Q alone. To examine how constitutively active P13K influences the axonal motility of HTT-138Q in vivo, we evaluated the motility dynamics of HTT-138Q (Fig 4). While large stalled axonal blockages were observed in larval axons expressing HTT-138Q, expression of PI3K.CAAX in the context of HTT-13 8Q did not significantly change the anterograde or retrograde velocities of HTT-138Q-containing vesicles (Fig 4). Therefore, constitutively active P13K has no effect on axonal transport or the motility dynamics of HTT-138Q-containing vesicles in vivo. These observations also suggest that the partial rescue of HTT-138Q-mediated neuronal cell death by excess PI3K.CAAX that we observe is likely to be independent of axonal transport.
Figure 3: Expression of constitutively active PI3K does not affect axonal transport defects induced by HTT138QmRFP.
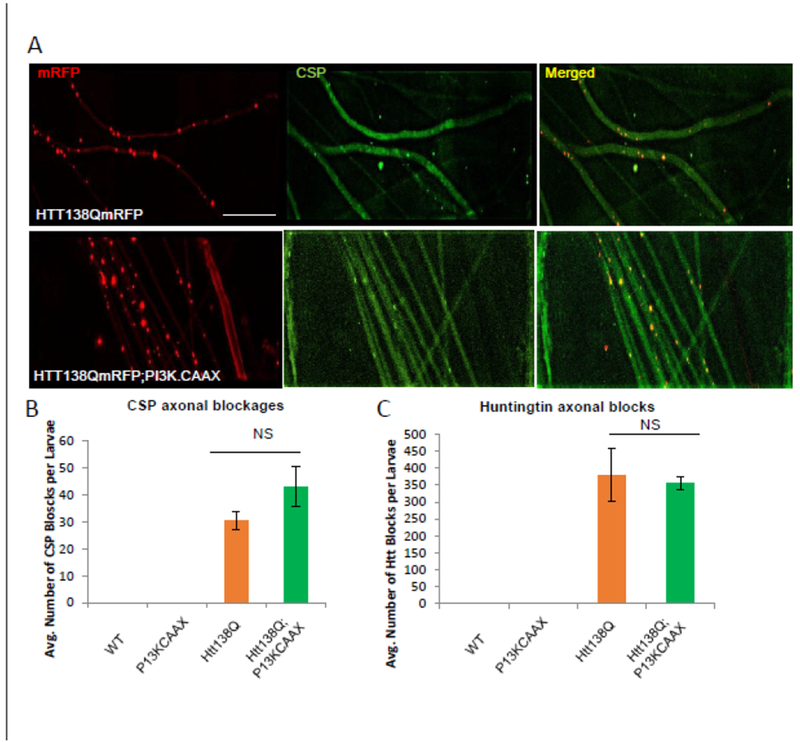
A. Larval nerves expressing HTT138QmRFP alone show axonal blockages containing both huntingtin and the synaptic vesicle marker cysteine string protein (CSP). Note that CSP and HTT co-localize within axonal blocks (yellow dots, merged image.) Larval nerves expressing constitutively active PI3K in the context of HTT138QmRFP also show CSP and HTT containing axonal blockages. B. Quantification analysis reveals that the number of CSP (P=0.06) or huntingtin (p=0.08) positive axonal blockages in larvae expressing HTT138Q;PI3K.CAAX is not significantly different from larvae expressing HTT138Q alone. N= 5 larvae. NS= no significance. Bar=10μm.
Figure 4: Expression of constitutively active PI3K does not effect the in vivo motility dynamics of HTT138QmRFP-containing vesicles.
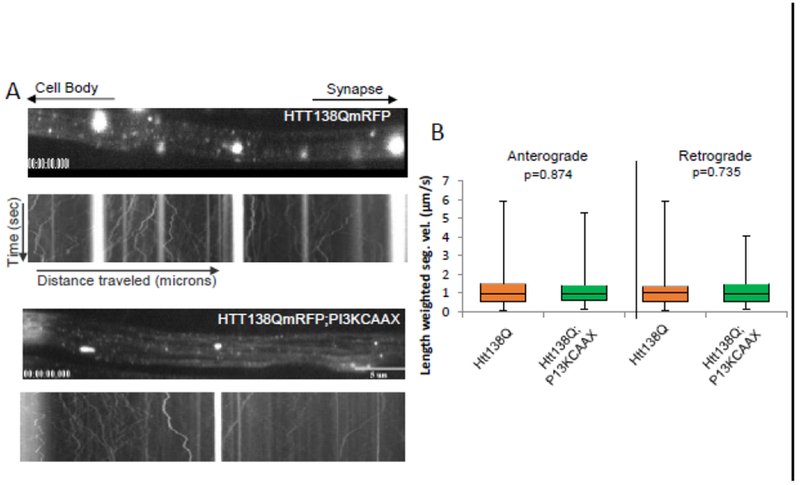
Cell bodies are towards the left and the synapse is towards the right as depicted by the arrows at the top. A representative kymograph from a larval segmental nerve is shown. × axis=distance (μicrons) and Y axis=time (sec). Scale bar =5μm. A. Expression of HTT38QmRFP alone show HTT-containing axonal accumulations, which are stalled, as evident by the bright straight tracks in the kymograph. Expression of PI3K.CAAX with HTT138QmRFP also show axonal blockages that are stalled in the kymograph. B. Box plots of duration-weighted segmental velocities of HTT138QmRFP alone or in the context of PI3K.CAAX does not show significant changes to the anterograde (p=0.874) or retrograde (p=0.735) vesicle velocities. Box plots outline the distribution of duration-weighted segmental velocities each vesicle, for each genotype tested. Over 1000 vesicles are analyzed for each genotype. The horizontal bar represents the median. The upper and lower box edges represent 75% percentile (i.e. upper quartile) and 25% percentile (i.e. lower quartile), respectively. Note that motility analysis was calculated from net anterograde and retrograde moving vesicles and reversing vesicles. For each genotype, a total of 10 larvae were imaged and a total of 40 movies were analyzed as previously done [5,25] using our custom particle tracking program.
Neuronal expression of constitutively active PI3K rescues oxidative stress-mediated cell death
Neurons are post-mitotic and while their ability to regenerate is limited, neurons are highly prone to oxidative stress and its consequences [61]. Indeed, oxidative stress has been associated with aging and neurodegenerative diseases. Oxidative stress can activate phosphorylation of the E3 ubiquitin ligase ZNRF1 which promotes Wallerian degeneration [62]. To test the hypothesis that the PI3K/AKT signaling pathway responds to oxidative stress-mediated neuronal cell death, we fed Paraquat (PQ) to flies from the time larvae hatched from embryos. Paraquat (PQ), an herbicide and a neurotoxicant, identified to be one of the prime risk factors in Parkinson’s disease (PD), is widely used in Drosophila to induce oxidative stress in vivo [26–27]. Paraquat acts as a redox cycler to initially produce superoxide radicals and later other reactive oxygen species (ROS); hydrogen peroxide (H2O2) and hydroxyl radical (OH) [28]. Work has shown mitochondrial dysfunction [27] together with high levels of p-JNK and increased caspase3-like (DEVDase) activity in brains of PQ-exposed WT flies [29]. Extensive loss of dopaminergic neurons, locomotion defects, decreased survival and Parkinsonian behaviors, including dyskinesia, rigidity in limbs and tremor are seen with PQ exposure [26, 29]. While PQ-induced neuronal cell death has been linked to JNK phosphorylation and caspase-3 activation [30], increased levels of p-GSK3β and hyperphosphorylated Tau was also observed after PQ-exposure [31].
We first evaluated the extent of neuronal cell death in WT flies raised in PQ-containing food. Flies were exposed to 0, 10, 20, 30, 50 and 100mM PQ from the time they hatched from the embryo. Equal amounts of embryos were seeded onto food laced with 0, 10, 20, 30, 50 and 100 mM of Paraquat (mixed in fly food) in a fly condo. For each concentration three replicates were done. We found that WT embryos seeded on 30, 50 and 100 mM Paraquat concentrations were lethal, with larvae dying before they reached the 3rd instar stage and none hatched to adults. Therefore, we used 20mM PQ for all our experiments. Work has shown that 20mM PQ had high SOD activity with low mortality rates [63]. SOD is commonly used as a marker to evaluate oxidative stress since SOD catalyzes the breakdown of the superoxide anion, which is up-regulated under conditions of oxidative stress.
Brains from WT PQ-fed (20mM) flies show neuronal cell death in contrast to WT buffer-fed (0mM) fly brains (Fig 5, Table 2). Quantitative analysis of TUNEL positive cells from 10 brains showed significant amounts of cell death in WT PQ-fed fly brains compared to WT buffer-fed fly brains. However, these amounts were not statistically significant and is perhaps due to the variability in the extent of PQ-ingestion between individual animals. Interestingly, PI3K.CAAX expressing flies fed on 20mM PQ-containing food showed significant decreases in the amount of TUNEL positive cells compared to WT flies fed on PQ (Fig 5, Table 2). Taken together these observations demonstrate that expression of PI3K.CAAX is sufficient to suppress oxidative stress induced neuronal cell death.
Figure 5. Neuronal expression of constitutively active PI3K rescues oxidative stress-mediated cell death.
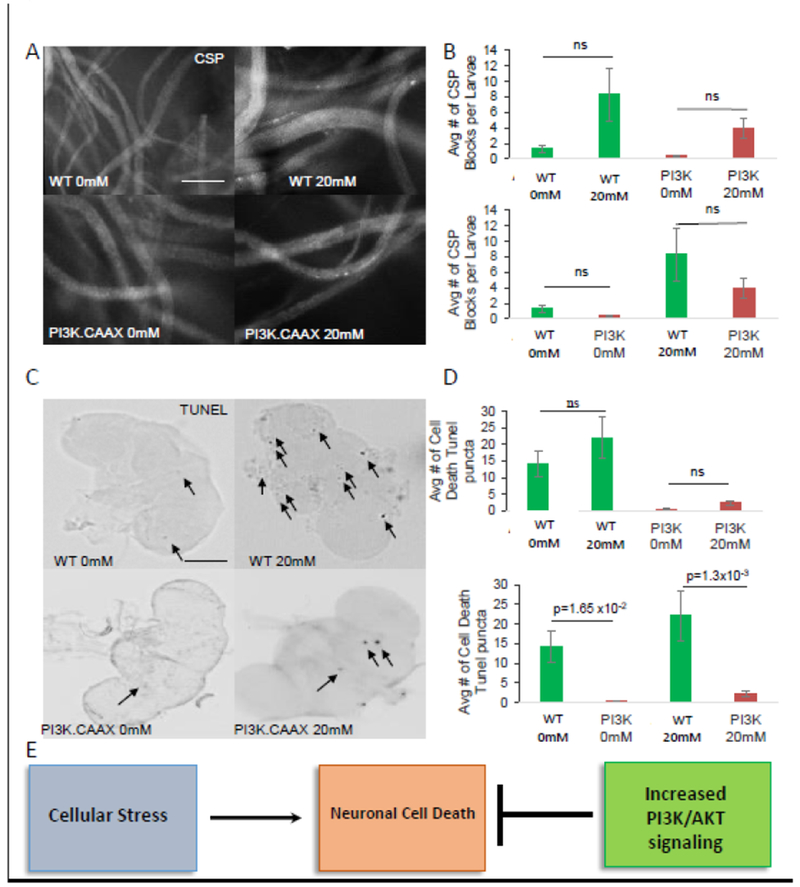
A. Wild type (WT) embryos or embryos expressing constitutively active PI3K (P13K.CAAX) were fed 0mM (buffer only) and 20mM Paraquat (PQ) containing food and raised under these conditions until the time they hatched to adults. Fed larvae were analyzed using the synaptic vesicle protein CSP. Note that all conditions show smoothly stained larval segmental nerves similar to WT. Note that a few CSP positive axonal blockages are observed, in both WT and PI3K.CAAX expressing larvae feeding on 0mM or 20mM PQ. Bar=50μm B. Quantification analysis of 0mM and 20mM PQ fed WT larvae or PI3K.CAAX expressing larvae do not show significant amounts of axonal blocks (p=0.240, p=0.488, p=0.590, p=0.7835 respectively) N=10 larvae, ns=none significant. C. Adult fly heads from WT or PI3K.CAAX expressing flies raised on 0mM and 20mM PQ from the time they hatched from embryos were assayed for cell death using TUNEL. Note that many TUNEL positive cells (arrows) are observed in WT adult brains raised on 20mM PQ compared to 0mM PQ. In contrast, less TUNEL positive cells are observed in adult brains expressing P13K.CAAX raised on 20mM PQ. D. Quantitative analysis of TUNEL positive cells show that WT fly brains raised on 20mM PQ show an increase in TUNEL positive cell death compared to WT adult brains raised on 0 mM PQ. However, this increase was not significant (p=0.253). In contrast, a significant decrease was seen in TUNEL positive cells in adult brains from WT and P13K.CAAX raised on 20mM PQ compared to 0mM PQ (p=1.3×10−3). Note that expression of P13K.CAAX also significantly decreases endogenous TUNEL positive cells (compare adult brains from P13K.CAAX under OmM PQ, p=1.65×l0−2). N=10 brains. Bar=10μm. C: Flow chart summarizing our observations propose that oxidative stress causes neuronal cell death which can be blocked by neuronal expression of constitutively active PI3K.
Table 2:
Summary of axonal blockages and cell death observations from wild type and constitutively active larvae and adult flies fed on Paraquat-laced food. N=6-10 for each genotype.
| PQ Treatment | Axonal blockages | Cell Death |
|---|---|---|
| Wild type 0mM | No | No |
| Wild type 20mM | No | Yes |
| PI3K.CAAX 0mM | No | No |
| PI3K.CAAX 20mM | No | Decreased |
To evaluate how PQ-mediated oxidative stress affects axonal transport, we examined PQ-feed larvae using the synaptic vesicle antibody CSP. Larval segmental nerves from larvae fed 20mM PQ were smoothly stained and were similar to buffer-fed larvae (Fig5, Table 2). These observations are consistent with a recent study that showed that 20mM PQ treatment of larvae expressing ANF-GFP had no effect on the motility of dense-core vesicles [64]. Therefore, PQ-mediated oxidative stress does not affect axonal transport.
pGSKβ-ser9 levels are enhanced in larvae expressing HTT138Q and in larvae carrying dynein mutants
While our observations indicate that expression of constitutively active P13K is sufficient to rescue HTT-138Q-mediated or oxidative stress induced cell death, it is unclear whether cell death under these situations directly result due to deficiencies in the P13K/AKT pathway. Since GSK3β is a major target of PI3K/AKT in neurons and inhibition of GSK3β activity can protect neurons from cell death [48, 10], we used ser9 phosphorylation of GSK3β (pGSK3β-ser9) to probe for the modulation of the PI3K/AKT signaling pathway as previously done [10–16,32–33, 54]. In the PI3K/AKT pro-survival cascade, in response to survival factors, AKT represses the activity of GSK3β by phosphorylation of GSK3β at serine 9 to mediate neuronal survival. Therefore, we predict that larvae expressing constitutively active PI3K (P13K.CAAX) should increase the level of pGSK3β-ser9 indicating the activation of the pro-survival pathway. Indeed, western blot analysis of larval brains from larvae expressing PI3K.CAAX show increased amounts of pGSK3β-ser9 compared to WT larvae (Fig 6A). Quantification analysis of the intensity of pGSK3β-ser9 as a ratio of total GSK3β from 3 independent blots show a significant increase in pGSK3β-ser9 in larval brains expressing P13K.CAAX (Fig 6B). These increases were not due to increases in the level of total GSK3β (Fig 6C). Since PI3K.CAAX expressing larvae did not show cell death and were viable as adults (Fig 2A), perhaps neuronal death directly results from the disruption of the PI3K/AKT pro-survival pathway.
Figure 6. The protein level of p-GSK3β-ser9 is increased in both motor protein mutant larvae and larvae expressing HTT-138Q.
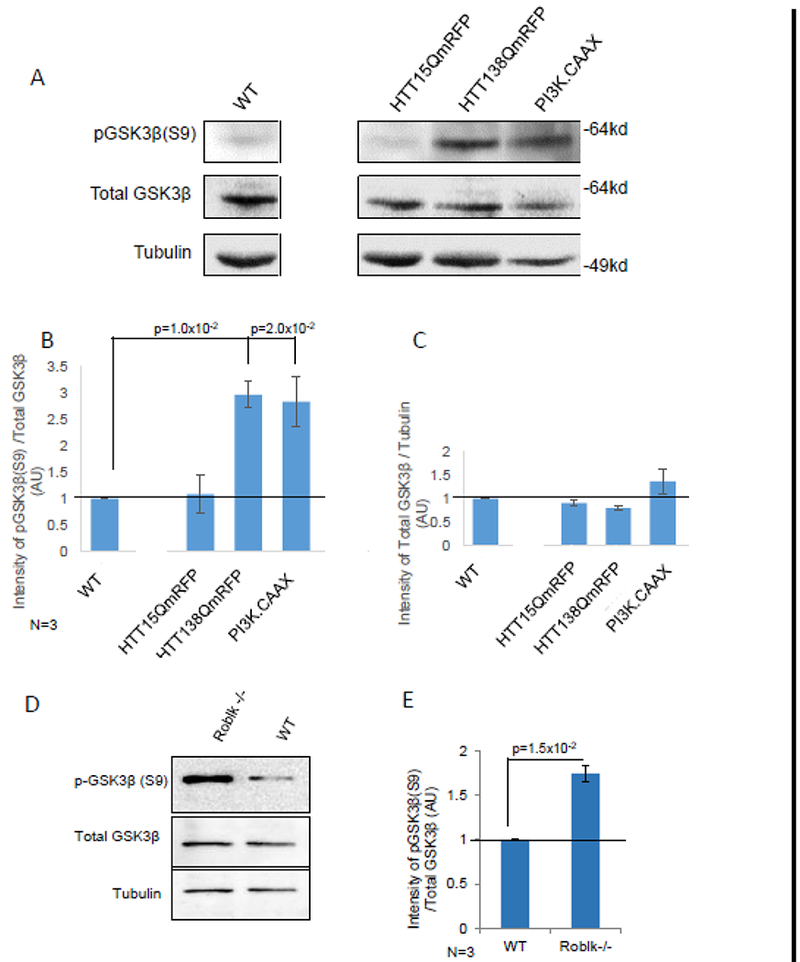
A. representative western blot of larval brains from WT and larvae expressing HTT15Q, HTT138Q and PI3K.CAAX alone were probed with antibodies against p-GSK3β-ser9, a marker for the activation of the PI3K/AKT pathway and total GSK3β. Tubulin was used as a loading control. B-C. Quantitative analysis of 3 independent blots show significant increases in the level of pGSK3β-ser9 in HTT138Q (p=1.0×l0−2) and PI3K.CAAX (p=2.0×l0−2). The intensity of pGSK3β-ser9 was normalized to total GSK3b. The intensity of total GSK3β was also normalized to tubulin to identify changes in the total levels of GSK3β. Note that no significant changes were observed. D. A representative western blot of larval brains from homozygous loss of function dynein mutants (Roblk−/−). These larvae are sickly and die at 2nd-3rd instar larval stage. E. Quantitative analysis of 3 independent blots show significant increases in the level of pGSK3β-ser9 in Roblk−/− (p=1.5×l0−2). The intensity of pGSK3β-ser9 was normalized to total GSK3β. The level of total GSK3β and Tubulin are unchansed. AU=Arbitrary units.
To test the hypothesis that HTT-138Q-mediated neuronal cell death was due to deficiencies in the P13K/AKT pathway we examined the level of pGSK3β-ser9 using western blot analysis. Strikingly, we found that larvae expressing HTT-138Q showed increased levels of pGSK3β-ser9 compared to WT larvae (Fig 6A). The extent of pGSK3β-ser9 seen in HTT-138Q larvae was similar to what we observed for larvae expressing P13K.CAAX alone. Quantification analysis of 3 independent western blots indicate a significant increase in the intensity of pGSK3β-ser9 as a ratio of total GSK3β in larval brains expressing HTT138Q compared to WT (Fig 6B). Note that the increased level of pGSK3β-ser9 was not due to increases in total GSK3β levels (Fig 6C). Therefore, while the pro-survival pathway is likely activated in larval brains expressing HTT-138Q, the presence of neuronal cell death in these brains indicate that this pathway is still defective.
Since larvae expressing HTT-138Q showed axonal blockages, we next tested the hypothesis that defects in axonal transport could modulate the function of the PI3K/AKT pathway. We previously showed that homozygous Roblk−/− larvae contain axonal blockages within their larval segmental nerves, neuronal cell death within their larval brains, and are lethal at late larval stages [25]. Intriguingly, Roblk−/− larvae show significant increases in the level of pGSK3β-ser9 compared to the levels seen in WT (Fig 6D-E). Therefore, taken together other observations suggest that defects in axonal transport can modulate the P13K/AKT-mediated pro-survival pathway.
Components of the P13K/AKT pathway are functionally linked to molecular motors
One proposal for a link between axonal transport defects and the modulation of the P13K/AKT pro-survival pathway is that components of the PI3K/AKT pro-survival pathway use the axonal transport pathway for proper localization within neurons. In this context we predict that excess of PI3K or AKT with genetic reduction of motor proteins should disrupt transport causing axonal blockages due to the titration of available motors away from other axonal cargoes. To test this prediction, we generated larvae expressing P13K.CAAX or AKT in the context of heterozygous kinesin-1 (KLC−/+) or dynein (Roblk−/+). In contrast to larvae homozygous for KLC (−/−) or Roblk (−/−) that showed CSP containing axonal blockages and larval lethality [5,25,65–66], larvae heterozygous for KLC (−/+) or Roblk (−/+) show smooth CSP staining within larval segmental nerves and are comparable to WT (Fig 7A). Larvae expressing P13K.CAAX or AKT alone also show smooth CSP staining within larval segmental nerves similar to WT (Fig 7A), indicating that expression of these components alone does not cause axonal transport defects. However, larvae expressing P13K.CAAX or AKT with reduction of either KLC−/+ or Roblk−/+ show CSP containing axonal blockages (Fig 7A). Quantification analysis reveal significant amounts of axonal blockages in larvae expressing PI3K.CAAX or AKT with KLC−/+ or Roblk−/+ compared to larvae expressing P13K.CAAX or AKT.EXEL alone (Fig7A). Therefore, while P13K and AKT likely do not directly interact with motor proteins, these likely use the axonal transport pathway for their localization within neurons and are perhaps packaged in an endosomal cargo complex.
Figure 7. Components of the P13K/AKT pathway genetically interact with molecular motors.
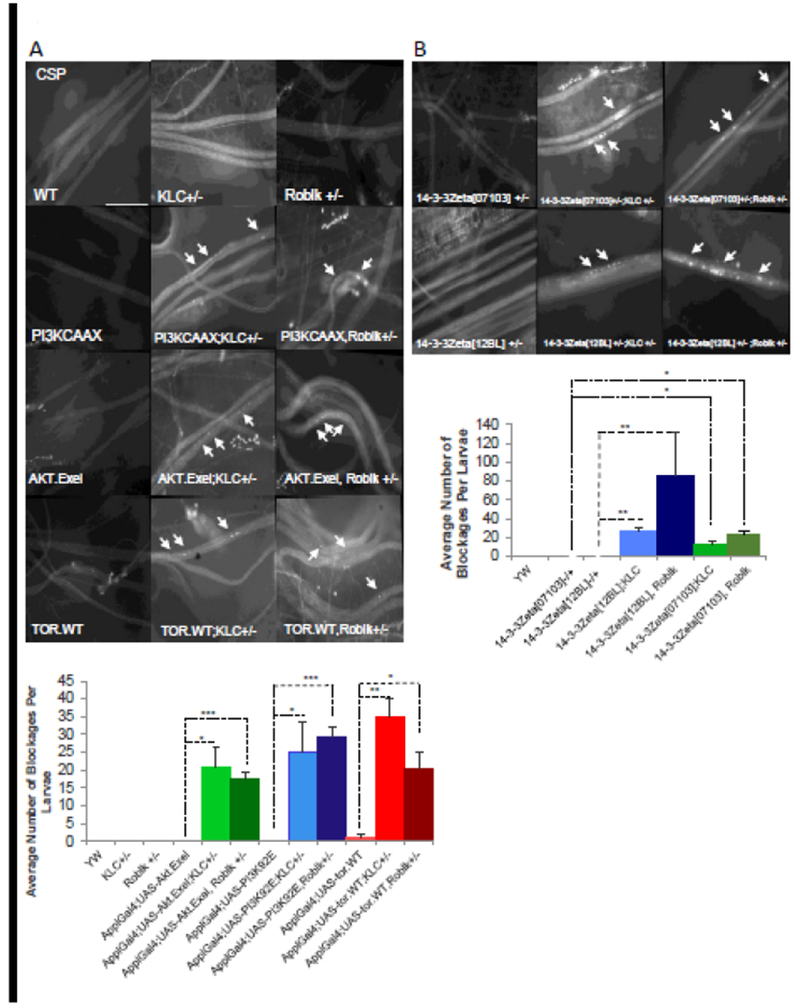
A. WT larval nerves are smoothly stained with CSP, the synaptic vesicle marker. Larval nerves from heterozygous kinesin (KLC−/+) or dynein (Roblk−/+) larvae are also smoothly stained with CSP. Larval segmental nerves from larvae expressing PI3K, AKT, or Tor alone also show smooth CSP staining. In contrast, larval nerves expressing PI3K, AKT or Tor in the context of KLC−/+ or Roblk−/+ show CSP containing axonal blockages (arrows). Quantitative analysis reveals that the extent of axonal blockages is significantly increased in larvae expressing PI3K, AKT or Tor in the context of KLC−/+ or Roblk−/+ (*p<0.05 **p<0.005 ***p<0.0005). N= 6 larvae. Scale Bar = 50 μm. B. Larval nerves from larvae heterozygous for loss of function of 14-3-3zeta (14-3-3zeta[07103]−/+, 14-3-3zeta12BL−/+] show smooth CSP staining. Larvae that are 14-3-3zeta−/+;KLC−/+ or 14-3-3zeta−/+;Roblk−/+ show CSP positive axonal blockages. Quantitative analysis reveals that the number of axonal blockages is significant in these larvae compared to larvae that are WT, 14-3-3zeta−/+, KLC−/+ or Roblk−/+ (*p<0.05, **p<0.005). N= 5 larvae. Scale Bar = 50 μm.
During cell growth TOR and 14.3.3 proteins are important downstream targets of PI3K and AKT. Activation of TOR can regulate both nutrients and growth factor signaling [7] and 14-3-3 can inhibit apoptosis [67]. In response to survival signals, 14-3-3 positively regulates AKT downstream signaling by binding and sequestering pro-apoptotic proteins away from their interaction partners and sites of action [67,68]. 14-3-3 can also interact with PI3K [69–73]. To further test the proposal that the P13K/AKT pro-survival pathway uses the axonal transport pathway, we first examined larvae expressing TOR.WT in the context of reduction of either KLC−/+ or Roblk−/+. While larvae expressing TOR.WT show smooth CSP staining within larval segmental nerves, larvae expressing TOR.WT with reduction of either KLC−/+ or Roblk−/+ show CSP containing axonal blockages. Quantification analysis reveal significant amounts of axonal blockages in larvae expressing TOR.WT with KLC−/+ or Roblk−/+ compared to larvae expressing TOR.WT alone (Fig7). We next tested larvae carrying heterozygous mutations for 14.3.3zeta (using two 14.3.3zeta mutant lines 14.3.3zeta12BL or 14.3.3zeta07013). While larvae carrying homozygous mutations for 14.3.3zeta12BL or 14.3.3zeta07013 were lethal, larvae heterozygous for 14.3.3zeta12BL−/+ or 14.3.3zeta07013−/+ show smooth CSP staining within larval segmental nerves (Fig 7B). In contrast, larvae heterozygous for both 14.3.3zeta and kinesin (14.3.3zeta12BL;KLC−/+ or 14.3.3zeta07013;KLC−/+) show CSP containing axonal blockages (Fig 7B). Larvae heterozygous for both 14.3.3zeta and dynein (14.3.3z12BL;Roblk−/+ or 14.3.3zeta07013;Roblk−/+) also show CSP containing axonal blockages (Fig 7B). Quantification analysis reveal significant amounts of axonal blockages (Fig 7B). Taken together our observations indicate that the P13K/AKT pro-survival pathway is functionally linked to axonal transport. Perhaps PI3K, AKT, and downstream effectors of the P13K/AKT pro-survival pathway use the axonal transport pathway for proper localization within neurons for normal function.
Discussion:
We have identified that perturbations in normal axonal transport alter the function of the PI3K/AKT pro-survival signaling pathway in vivo in Drosophila. Our observations lead us to two main conclusions: 1) neuronal expression of constitutively active PI3K is sufficient to suppress neuronal cell death induced by pathogenic HTT or oxidative stress, but has no effect on axonal transport defects mediated by pathogenic HTT, 2) disruption of axonal transport modulates the function of P13K/AKT pro-survival pathway likely via perturbation of normal transport of components in the P13K/AKT pathway. Therefore, these findings provide new insight into the pathological mechanisms of neuronal disease propagation, with important implications for targeting therapeutics against neuronal cell death and degradation.
The pro-survival PI3K/AKT pathway is a multistep signaling cascade that is highly conserved and tightly regulated. PI3K facilitates crucial cellular survival, proliferation and differentiation. It has been implicated in aging and lifespan regulation, in the proliferation of adult neuronal progenitor cells, as well as in maintaining synaptic plasticity [36–37, 56]. PI3Ks operate downstream of receptor tyrosine kinases (RTKs) and G protein coupled receptors (GPCRs). They are responsible for propagating an array of signals due to numerous growth factors and cytokines to mediate intracellular communications by generating phospholipids, which in turn activate AKT and other effectors including GSK3β, TOR and 14.3.3. Active AKT (phosphorylation at T308 and S473) facilitates the phosphorylation of GSK3β at serine 9, repressing the activity of GSK3β to promote cell survival and growth. While the role of the PI3K/AKT pro-survival pathway in the context of neurodegeneration has not been well established, our observations demonstrate that this pathway is modulated during conditions of neurodegeneration and during the disruption of axonal transport. Expression of pathogenic HTT increased the level of pGSK3β-ser9, which is widely used to assess the modulation of the PI3K/AKT signaling pathway [32–33, 54]. Increased levels of pGSK3β-ser9 was also seen in larvae expressing PI3K-CAAX alone (Fig 6). Similarly, homozygous dynein mutations also showed increased levels of pGSK3β-ser9. Our findings are strikingly similar to several previous work which showed that the PI3K/AKT pathway is activate during neuronal dysfunction. Aβ oligomer treated neurons exhibited elevated levels of activated AKT, mTOR and PI3K [38], and AKT or mTOR inhibitors blocked Aβ oligomer-induced neuronal cell cycle events [38]. Increases in both AKT and GSK3β phosphorylation were also seen during iron-induced neurotoxicity which was proposed to be via the regulation of transcriptional activity [39]. The PI3K/AKT pathway was also shown to be active in MPP+ induced apoptosis in human neuroblastoma SH-EP1 cells, and was found to play a key role in IGF-mediated cell survival [40]. Therefore, taken together our findings propose that under conditions of neurodegeneration and axonal transport defects the PI3K/AKT pro-survival pathway is likely activated perhaps to promote cell survival and growth.
However, there is also evidence that the PI3K/AKT pathway is inhibited under conditions of neurodegeneration. In DA neuronal cell systems, enhanced expression of miR-126 impaired IGF-1 signaling and increased vulnerability to the neurotoxin 6-OHDA by downregulating factors in IGF-1/PI3K signaling, implicating the inhibition of this pathway in DA neuron loss and in PD pathogenesis [42]. In Alzheimer’s disease brains, the level and activity of pAKT was decreased indicating that excess levels of Aβ interferes with AKT activation [43]. Therefore, while the importance of this pathway during neuronal death and disease is evident, the mechanisms of how the PI3K/AKT pathway is affected during neuronal toxicity and degeneration are likely to be complex.
One possible explanation for the discrepancies in the role of the PI3K/AKT pathway during neuronal death and degeneration is that, initially this pathway could be activated as a result of early insults such as axonal transport defects, but is later deactivated due to overpowering pro-apoptotic mechanisms. Perhaps defects in axonal transport initiates a signaling cascade which at first enhances the production of pro-survival factors at the cell body. However, as problems escalate within the neuronal cell, i.e., increase in the aggregation of toxic proteins within axonal projections and in cell bodies perhaps due to continued perturbations in axonal transport, pro-apoptosis mechanisms are likely activated which counteract PI3K/AKT-mediated pro-survival. Consistent with this proposal, our observations demonstrate that homozygous dynein mutant larvae show increased levels of pGSK3β-ser9, similar to larvae expressing PI3K.CAAX. Expression of pathogenic HTT also showed increased levels of pGSK3β-ser9. However, in contrast to larvae expressing PI3K.CAAX, both homozygous dynein mutant larvae or larvae expressing pathogenic HTT were lethal at larval stages and did not eclose to adults, indicating that the level of activation of the pro-survival pathway is likely not sufficient for complete rescue of the organism. This prediction is consistent with our finding that, although excess expression of PI3K.CAAX significantly suppressed neuronal cell death mediated by pathogenic HTT or oxidative stress, the extent of excess P13K.CAAX that was supplied was unable to completely rescue cell death. Our findings are also consistent with previous work which showed that constitutive activation of PI3K is sufficient to stimulate some but not all of the effects of insulin that were shown to be dependent on PI3K activation [51]. Further, constitutively activated PI3K also accelerated proliferation in tumor initiation [52]. Mice expressing constitutively active PI3K showed increased levels of PKB/AKT phosphorylation and enhanced Leukocyte proliferation and survival [53]. PI3K/AKT signaling was shown to regulate axonal regeneration in mammals [55]. Further, down regulation of the P13K/AKT pathway by loss-of-function of PI3K extended lifespan and significantly delayed polyQ aggregation and toxicity [41], via a mechanism that is thought to promote autophagy for the clearance of abnormal protein aggregates [41]. Therefore, while it is clear that the activation of the P13K/AKT pathway stimulates survival, however, during extreme conditions of degeneration the activities of P13K/AKT-mediated pro-survival may not be able to fully function in order to rescue death.
It is possible that the balance between the active/inactive states of GSK3β mediated by P13K/AKT signaling plays a critical role in the maintenance of pro-survival/pro-apoptosis during neuronal disease. Indeed, the active/inactive states of GSK3β mediated by phosphorylation of Tyr-216 or Ser 9 has previously been shown to play important roles in the balance between promoting or inhibiting apoptosis [17,12]. PI3K is also central to this balance as it can both repress cell death and induce cell death by inhibiting GSK3β activity [12, 47]. Overexpression of GSK3β in fibroblasts and neuronal PC12 cells resulted in apoptosis [48], and promoted apoptosis in neuronal SH-SY5Y cells [49]. Inhibition of GSK3β, or expression of the inactive GSK3β -K85R mutant, reduced the number of sympathetic neurons undergoing cell death mediated by loss of PI3K signaling [9]. Cell death induced by PI3K inhibition was rescued by the expression of dominant negative GSK3β, indicating that suppression of GSK3β Try-216 activity is directly linked to the pro-survival effects of P13K [48]. Further, small molecule GSK3β inhibitors protected cerebellar granule neurons from death by inhibiting PI3K signaling [50]. While further study is needed to better understand such GSK3β mechanisms mediated by PI3/AKT signaling in the context of pathogenic HTT in vivo, it is clear that a balance in the active/inactive states of GSK3β is likely critical for P13K/AKT-mediated functions in the maintenance of pro-survival/pro-apoptosis.
Our observations also suggest that the PI3K/AKT-mediated pro-survival activities likely acts downstream of axonal transport defects (Fig 8). Axonal transport defects induced by dynein loss of function or excess of pathogenic HTT activate the P13K/AKT pathway (Fig 6). While excess constitutively active PI3K suppressed neuronal cell death seen in larvae expressing pathogenic HTT, no effect was seen on the pathogenic HTT-mediated axonal blockages (Fig 2). Excess constitutively active PI3K also suppressed oxidative stress-mediated neuronal cell death (Fig 5). Further, although components of the PI3K/AKT pathway showed functional interactions with molecular motor proteins (Fig 7), it is unclear whether PI3K and the components of the PI3K/AKT pathway directly bind molecular motors for their own motility and/or for their proper localization within axons. It is possible that a moving signaling PI3K/AKT endosomal complex exists within axons, similar to what has been previously observed with NGF/trkA or JIP3/INK-mediated injury signaling complexes [44–46]. Although future work is required to investigate the presence of such a complex in vivo, our observations propose that the axonal transport pathway and the function of the PI3K/AKT pro-survival pathway are tightly coupled during neurodegeneration. Therefore, a better understanding of the mechanisms of how pro-survival activities can be maintained under conditions of neuronal disease propagation will have important implications for targeting therapeutics against neuronal cell death and degradation.
Figure 8: Proposed model for the activation of the PI3K/AKT-mediated pro-survival pathway which likely acts downstream of axonal transport defects.
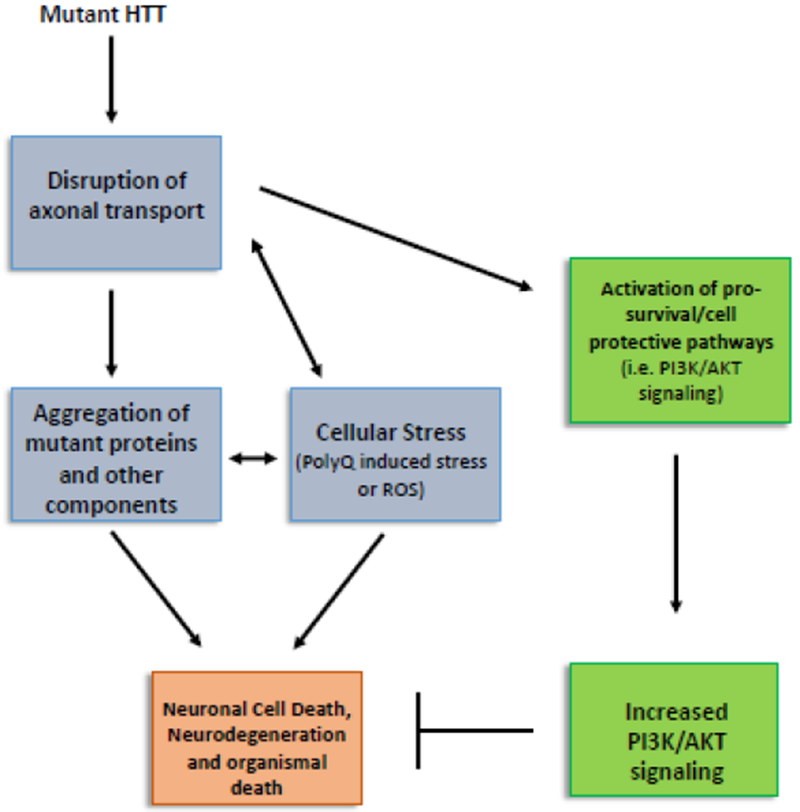
Our observations suggest that the PI3K/AKT-mediated pro-survival activities likely occur downstream of axonal transport defects. While HTT138Q mediated defects in axonal transport or Paraquat exposure (oxidative stress) causes neuronal cell death, excess constitutively active P13K can only suppress HTT138Q or Paraquat mediated neuronal cell death (Fig 2–5). Further, defects in axonal transport mediated by loss of dynein motors or by excess expression of HTT138Q activates the PI3K/AKT pathway as assayed by p-GSK3β-serS9 (Fig 6).
Methods
Drosophila Genetics
Fly stocks and crosses were maintained at room temperature unless otherwise stated. Males containing the neuronal driver APPL-GAL4;B3/Pin were crossed with either virgin female UAS-HTT-15Q, UAS-HTT-138Q, (gift from Dr Littleton), UAS-PI3K-CAAX, UAS-PI3K-DN, UAS-AKT-Exel, or UAS-TOR-WT (Bloomington Flybase). To reduce functional kinesin or dynein motors, male UAS-PI3K lines were crossed with virgin female APPL-GAL4;klc8ex94/B3 or APPL-GAL4;roblk/B3. Loss of function mutants 14-3-3zeta(07103)/cyO or 14-3-3zeta(12BL)/cyO were crossed with virgin female APPL-GAL4;klc8ex94/B3 or APPL-GAL4;roblk/B3. Crosses were maintained at 29°C for protein overexpression.
Paraquat feeding
Embryos were harvested from the APPL-GAL4 fly stock (WT) and from the cross APPL-GAL4;UAS-PI3K-CAAX. Equal amounts of embryos were seeded into food laced with 0, 10, 20, 30, 50 and 100 mM of Paraquat mixed in fly food in a fly condo. For each concentration three replicates were done for each genotype. The fly condo was maintained in 29°C for protein expression. At least 10 larvae from each concentration from each genotype were analyzed for axonal transport defects. At least 10 adult flies from each concentration from each genotype were analyzed for cell death. Note that in all cases larvae and flies for each genotype were continuously fed in Paraquat. 30, 50 and 100 mM Paraquat concentrations were lethal, with larvae dying before they reached the 3rd instar stage and no adults were seen.
In Vivo Vesicle imaging
Larvae are dissected and imaged as detailed previously [57, 58]. A Nikon Eclipse TE-2000E inverted fluorescence microscope and Metamorph Imaging software were used not in vivo imaging of vesicles within larval axons. Time-lapse movies were collected using a CoolSnap HQ camera (Roper Scientific, Surrey, BC, Canada). 5-10 larvae were dissected per genotype. 150-frame movies were recorded consecutively for 30 min from each larva at 100×/1.40 NA (90 μm field-of-view) oil objective with a 2×2 binning factor yielding a 0.126 micron/pixel spatial resolution and a 200 msec exposure for each frame. Movies were cropped and oriented for analysis in Metamorph (MDS Analytical Technologies, Sunnyvale, CA) and analyzed using a MATLAB 2010b-based (Mathworks) custom single particle tracking program [58,60]. Segmental velocities were defined as the mean velocity of a trajectory segment uninterrupted by a pause, reversal, or movie termination event. Duration-weighted segmental velocity evaluates the average velocity behavior that vesicles exhibit per time spent moving.
TUNEL assay
Cell death was analyzed by first dissecting adult brains. Briefly, brains were dissected in dissection buffer (2× stock contains 128 mM NaCl, 4 mM MgCl2, 2 mM KCl, 5 mM HEPES, and 36 mM sucrose, pH 7.2) for 20 min. Brains were subsequently fixed in 4% paraformaldehyde in PBS for 30 min at 25°C. After washing in PBS, cells were permeablized with 5% saponin for 30 min at 25°C. After washing in PBS, brains were mounted in Vectashield mounting medium (Vector Labs) for imaging. TUNEL assay was performed using the In Situ Cell Death Detection Kit (Roche Life Science) per manufacturer’s instructions. Incubation in DNAse I was used as a positive control, while incubations in the labeling solution only was used as a negative control. The number of puncta in each brain was quantified in ImageJ (NIH) using the Threshold tool and Analyze Particles tool. At least 10 adult brains were imaged and quantified from each genotype.
Larval immunohistochemistry
Third instar larvae were dissected, fixed, and immunostained as described [25,59]. Briefly, larvae were dissected in dissection buffer (2X stock contains 128 mM NaCl, 4 mM MgCl2, 2 mM KCl, 5 mM HEPES, and 36 mM sucrose, pH 7.2). Following dissection, larvae were treated with 0, 10, or 50 mM EtOH at 25°C for 20 min. Larvae were fixed in 4% formaldehyde and incubated with primary antibodies against either rabbit monoclonal cysteine string protein (DCSP-3, 1:10, Developmental Studies Hybridoma Bank) overnight. As needed larvae were also incubated with the neuronal membrane marker Texas Red-conjugated horse radish peroxidase (HRP) and secondary antibody (Alexa anti-rabbit 488, 1:100, Invitrogen) for 2 hrs at room temperature, mounted using Vectashield mounting medium (Vector Labs). Images were obtained using a Nikon TE-2000E inverted microscope at 60×. At least 5-10 larvae were imaged from each genotype.
Statistical analysis
For immunofluorescence analysis of axonal blockages or cell death, statistical analysis was performed in Excel (Microsoft Corp.), using the two-sample two-sided Student’s t-test. Differences were considered significant at a significance level of 0.05, which means a 95% statistically significant correlation for 6-10 individual larvae from several independent crosses. For western blots, quantification analysis was performed using Image Lab software. Data obtained from Image Lab was analyzed in Excel (Microsoft Corp) using two-sided Student’s t-test. Additionally, Bonferroni’s test and Tukey’s Honestly Significant Difference test was performed in Minitab 18. Both of the methods are pair-wise multiple comparison procedures specifically designed to compare each treatment with a control [93, 94]. Statistical analysis for in vivo motility of vesicles were done as detailed in [58,60]. Briefly, to select the appropriate statistical test, transport parameter distributions were first checked for normality using the nortest package of R: Lilliefors test and Anderson-Darling test. Statistical significance of normal distributions was calculated by a two-sample two-tailed Student’s t-test while the non-normal segmental velocity distributions were compared using the nonparametric Wilcoxon-Mann-Whitney rank sum test. Percent of cargo population tended to follow normal distributions. Duration-weighted segmental velocity, flux, and run length often followed a mixture of normal distributions or a non-normal distribution and therefore both the two-tailed Student’s t-test or the nonparametric Wilcoxon-Mann-Whitney rank sum test was used accordingly.
Acknowledgements
We thank the members of the Gunawardena lab for their constructive discussions. This work was supported by grants from the National Institute of Health (R03 NS084386 and R03 NS092024), and BrightFocus Foundation (A2018509S) to SG. TH was supported by fellowships from the Honors College Academic Enrichment Fund, a Honors College Research Scholarship and a Center for Undergraduate Research and Creative Activities (CURCA) fellowship from SUNY at Buffalo. CT was supported by fellowships from CURCA. JW was supported by a College for Arts and Sciences Dissertation Scholarship and a Marc Diamond Research Fellowship. SG thanks Priyantha Karunaratne for constant support.
Footnotes
Competing Interests:
The authors declare no conflicts of interest or competing financial interests.
References:
- 1:White JA II, Banerjee R, Gunawardena S. (2016) Axonal transport and neurodegenerative disease: How marine drugs can be used a therapeutics. Marine Drugs 19,14(5) pii: E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2:Fang C, Bourdette D, Banker G. (2012) Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases. Molecular Neurodegeneration 7, 29. doi: 10.1186/1750-1326-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3:Almenar-Queralt A, Falzone TL, Shen Z, Lillo C, Killian RL, Arreola AS, Niederst ED, Ng KS, Kim SN, Briggs SP, Williams DS, Goldstein LS. (2014) UV irradiation accelerates amyloid precursor protein (APP) processing and disrupts APP axonal transport. J Neurosci. 34(9):3320–39. doi: 10.1523/JNEUROSCI.1503-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4:Iacobucci GJ, Gunawardena S. (2017). Ethanol stimulates the in vivo axonal movement of neuropeptide-dense core vesicles in Drosophila motor neurons. J. Neurochemistry In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5:Gunawardena S, Her L, Laymon RA, Brusch RG, Niesman IR, Sintasath L, Bonini NM and Goldstein LSB. (2003) Disruption of axonal transport by loss of huntingtin or expression of poly Q protein in Drosophila. Neuron 40, 25–40. [DOI] [PubMed] [Google Scholar]
- 6:Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. (199) Regulation of neuronal survival by the serine-threonine protein kinase AKT. Science 275, 661–665. [DOI] [PubMed] [Google Scholar]
- 7:Manning BD, Cantley LC. 2007) AKT/PKB signaling: Navigating downstream. Cell 129: 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8:Hemmings BA, Restuccia DF. (2015) The PI3K-PKB/Akt Pathway Cold Spring Harb Perspect Biol, 7:a026609 doi: 10.1101/cshperspect.a026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9:Crowder RJ, Freeman RS. (1998) Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 18(8):2933–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10:Crowder RJ, Freeman RS. (2000) Glycogen synthase kinase-3 beta activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J Biol Chem. 3;275(44):34266–71. [DOI] [PubMed] [Google Scholar]
- 11:Xia Y, Wang CZ, Liu J, Anastasio NC, Johnson KM. (2010) Brain-derived neurotrophic factor prevents phencyclidine-induced apoptosis in developing brain by parallel activation of both the ERK and PI-3K/Akt pathways. Neuropharmacology. 58(2):330–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12:Maurer U, Preiss F, Brauns-Schubert P, Schlicher L, Charvet C. (2014) GSK-3 - at the crossroads of cell death and survival. J Cell Sci. 127(Pt 7):1369–78. [DOI] [PubMed] [Google Scholar]
- 13:Dickey EJ, Long SN, Hunt RW. Hypoxic ischemic encephalopathy--what can we learn from humans? (2011) J Vet Intern Med. 25(6):1231–40. doi: 10.1111/j.1939-1676.2011.00818.x. [DOI] [PubMed] [Google Scholar]
- 14:Sun AY, Wang Q, Simonyi A, Sun GY. (2008) Botanical phenolics and brain health. Neuromolecular Med. 10(4):259–74. doi: 10.1007/s12017-008-8052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15:Kotliarova S, Pastorino S, Kovell LC, Kotliarov Y, Song H, Zhang W, Bailey R, Maric D, Zenklusen JC, Lee J, Fine HA. (2008) Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 68(16):6643–51. doi: 10.1158/0008-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16:Romorini L, Garate X, Neiman G, Luzzani C, Furmento VA, Guberman AS, Sevlever GE, Scassa ME, Miriuka SG. (2006) AKT/GSK3β signaling pathway is critically involved in human pluripotent stem cell survival. Sci Rep. 6:35660. doi: 10.1038/srep35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17:Grimes CA, Jope RS (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 65(4):391–426. [DOI] [PubMed] [Google Scholar]
- 18:Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GV, Jope RS (2002) Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc Natl Acad Sci U S A. 99(12):7951–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19:Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D. (2012) GSK-3β: A Bifunctional Role in Cell Death Pathways Int J Cell Biol 930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20:White JA II, Anderson E, Zimmerman K, Zheng K, Rouhani R, Gunawardena S. (2015) Huntingtin differentially regulates the axonal transport of a sub-set of Rab-containing vesicles in vivo. Human Molecular Genetics. 24(25):7182–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21:Zala D, Colin E, Rangone H, Liot G, Humbert S, Saudou F. (2008) Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. 17(24):3837–46. [DOI] [PubMed] [Google Scholar]
- 22:Her LS, Goldstein LS. (2008) Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci. 10;28(50):13662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23:Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. (1996) The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 15(23): 6584–94. [PMC free article] [PubMed] [Google Scholar]
- 24:Scanga SE, Ruel L, Binari RC, Snow B, Stambolic V, Bouchard D, Peters M, Calvieri B, Mak TW, Woodgett JR, Manoukian AS. (2000) The conserved PI3’K/PTEN/Akt signaling pathway regulates both cell size and survival in Drosophila. Oncogene. 19(35):3971–7. [DOI] [PubMed] [Google Scholar]
- 25:Gunawardena S and Goldstein LSB. (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 32:389–401. [DOI] [PubMed] [Google Scholar]
- 26:Cassar M, Issa AR, Riemensperger T, Petitgas C, Rival T, Coulom H, Iché-Torres M, Han KA, Birman S. (2015) A dopamine receptor contributes to paraquat-induced neurotoxicity in Drosophila. Hum Mol Genet. 24(1):197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27:Hosamani R; Muralidhara. (2013) Acute exposure of Drosophila melanogaster to paraquat causes oxidative stress and mitochondrial dysfunction. Arch Insect Biochem Physiol. 83(1):25–40. [DOI] [PubMed] [Google Scholar]
- 28:Lascano R, Muñoz N, Robert G, Rodriguez M, Melchiorre M, Trippi V, Quero G, (2012) Paraquat: An Oxidative Stress Inducer in “Herbicides - Properties, Synthesis and Control of Weeds”, edited by Mohammed Naguib Abd El-Ghany Hasaneen, ISBN 978-953-307-803-8, January 13, 2012 [Google Scholar]
- 29:Shukla AK, Pragya P, Chaouhan HS, Tiwari AK, Patel DK, Abdin MZ, et al. (2014) Heat Shock Protein-70 (Hsp-70) Suppresses Paraquat-Induced Neurodegeneration by Inhibiting JNK and Caspase-3 Activation in Drosophila Model of Parkinson’s Disease. PLoS ONE 9(6): e98886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30:Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK (2004) The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem 279: 32626–32632. [DOI] [PubMed] [Google Scholar]
- 31:Wills J, Credle J, Oaks AW, Duka V, Lee J-H, Jones J, et al. (2012) Paraquat, but Not Maneb, Induces Synucleinopathy and Tauopathy in Striata of Mice through Inhibition of Proteasomal and Autophagic Pathways. PLoS ONE 7(1): e30745 10.1371/journal.pone.0030745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32:Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, Cohen P (1994) The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem. J. 303, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33:Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, Barkley CR, Wu J, Nandula S, Dutta B, Xie Y, Chin YR, Kim DI, Ferris JS, Gruvberger-Saal SK, Laakso M, Wang X, Memeo L, Rojtman A, Matos T, Yu JS, Cordon-Cardo C, Isola J, Terry MB, Toker A, Mills GB, Zhao JJ, Murty VV, Hibshoosh H, Parsons R. (2009) 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 69(15):6299–306. doi: 10.1158/0008-5472.CAN-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34:Wang Y, Hao Y, & Alway SE (2011) Suppression of GSK-3β activation by M-cadherin protects myoblasts against mitochondria-associated apoptosis during myogenic differentiation. Journal of Cell Science, 124(22), 3835–3847. 10.1242/jcs.086686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35:Uranga RM, Katz S, & Salvador GA (2013) Enhanced Phosphatidylinositol 3-kinase (PI3K)/Akt Signaling Has Pleiotropic Targets in Hippocampal Neurons Exposed to Iron-induced Oxidative Stress. The Journal of Biological Chemistry, 288(27), 19773–19784. 10.1074/jbc.M113.457622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36:Rodgers EE, Theibert AB. (2002) Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci. Jun-Aug;20(3-5):187–97. [DOI] [PubMed] [Google Scholar]
- 37:Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J. (2014) The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 26(12):2694–701. doi: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 38:Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. (2009) The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener 16;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39:Uranga RM, Katz S, Salvador GA. (2013) Enhanced phosphatidylinositol 3-kinase (PI3K)/Akt signaling has pleiotropic targets in hippocampal neurons exposed to iron-induced oxidative stress. J Biol Chem. 288(27):19773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40:Wang L, Yang HJ, Xia YY, et al. (2010) Insulin-like growth factor 1 protects human neuroblastoma cells SH-EP1 against MPP+-induced apoptosis by AKT/GSK-3beta/JNK signaling. Apoptosis. 15:1470–9. [DOI] [PubMed] [Google Scholar]
- 41:Morley JF, Brignull HR, Weyers JJ, Morimoto RI. (2002) The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci USA 99: 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42:Kim W, Lee Y, McKenna ND, et al. (2014) miR-126 contributes to Parkinson’s disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol Aging. 35:1712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43:Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW. (2009) The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol Biol Cell. 20(5):1533–44. doi: 10.1091/mbc.E08-07-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44:Cavalli V, Kujala P, Klumperman J, Goldstein LS. (2005) Sunday Driver links axonal transport to damage signaling. J Cell Biol. 168(5):775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45:Drerup CM, Nechiporuk AV. (2013) JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS Genet. 2013;9(2):e1003303. doi: 10.1371/journal.pgen.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46:Klinedinst S, Wang X, Xiong X, Haenfler JM, Collins CA. (2013) Independent pathways downstream of the Wnd/DLK MAPKKK regulate synaptic structure, axonal transport, and injury signaling. J Neurosci. 33(31):12764–78. doi: 10.1523/JNEUROSCI.5160-12.2013. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47:Nakano N, Matsuda S, Ichimura M, Minami A, Ogino M, Murai T, Kitagishi Y. (2017) PI3K/AKT signaling mediated by G protein-coupled receptors is involved in neurodegenerative Parkinson’s disease. Int J Mol Med. 39(2):253–260. doi: 10.3892/ijmm.2016.2833. [DOI] [PubMed] [Google Scholar]
- 48:Pap M, Cooper GM. (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 273(32):19929–32. [DOI] [PubMed] [Google Scholar]
- 49:Bijur GN, Briggs B, Hitchcock CL, Williams MV. (1999) Ascorbic acid-dehydroascorbate induces cell cycle arrest at G2/M DNA damage checkpoint during oxidative stress. Environ Mol Mutagen. 33(2):144–52. [DOI] [PubMed] [Google Scholar]
- 50:Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD Reith AD (2010) Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 77, 94–102. doi: 10.1046/j.1471--4159.2001.t01--1--00251.x [DOI] [PubMed] [Google Scholar]
- 51:Frevert EU, Bjørbaek C, Venable CL, Keller SR, Kahn BB. (1998) Targeting of constitutively active phosphoinositide 3-kinase to GLUT4-containing vesicles in 3T3-L1 adipocytes.J Biol Chem. 273(39):25480–7. [DOI] [PubMed] [Google Scholar]
- 52:Sheen MR, Marotti JD, Allegrezza MJ, Rutkowski M, Conejo-Garcia JR, Fiering S. (2016) Constitutively activated PI3K accelerates tumor initiation and modifies histopathology of breast cancer. Oncogenesis. 5(10):e267. doi: 10.1038/oncsis.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53:Costa C, Barberis L, Ambrogio C, Manazza AD, Patrucco E, Azzolino O, Neilsen PO, Ciraolo E, Altruda F, Prestwich GD, Chiarle R, Wymann M, Ridley A, Hirsch E. (2007) Negative feedback regulation of Rac in leukocytes from mice expressing a constitutively active phosphatidylinositol 3-kinase gamma. Proc Natl Acad Sci U S A. 104(36):14354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54:Cross DA, Alessi DR, Cohen P, Andjelkovich M and Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 55:Saijilafu Hur EM, Liu CM, Jiao Z, Xu WL, Zhou FQ. (2013) PI3K–GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nat. Commun. 4:2690 doi: 10.1038/ncomms3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56:Rodgers EE, Theibert AB (2002) Functions of PI 3-kinase in development of the nervous system. Int. J. Dev. Neurosci. 20:187–197. [DOI] [PubMed] [Google Scholar]
- 57:Kuznicki ML, Gunawardena S. (2010) In vivo visualization of synaptic vesicles within Drosophila larval segmental axons. Journal of Visualized Experiments. 44:2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58:Gunawardena S, Yang G, Goldstein LSB. (2013) Presenilin controls kinesin-1 and dynein activity during axonal transport. Human Molecular Genetics 22(19):3828–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59:Fye S, Dolma K, Kang MJ, Gunawardena S. (2010) Visualization of larval segmental nerves in 3rd instar Drosophila larval preparations. Journal of Visualized Experiments. 43:2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60:Reis GF, Yang G, Szpankowski L, Weaver C, Shah SB, Robinson JT, Hays TS, Danuser G and Goldstein LS (2012) Molecular motor function in axonal transport in vivo probed by genetic and computational analysis in Drosophila. Molecular biology of the cell, 23, 1700–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61:Mattson MP, Magnus T. (2006) Ageing and neuronal vulnerability. Nat Rev Neurosci. 7(4):278–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62:Wakatsuki S, Saitoh F, Araki T. (2011) ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Bio 13(12):1415–23. [DOI] [PubMed] [Google Scholar]
- 63:Rzezniczak TZ, Douglas LA, Watterson JH, Merritt TJ.(2011) Paraquat administration in Drosophila for use in metabolic studies of oxidative stress. Anal Biochem. 419(2):345–7. [DOI] [PubMed] [Google Scholar]
- 64:Liao PC, Tandarich LC, Hollenbeck PJ. (2017) ROS regulation of axonal mitochondrial transport is mediated by Ca2+ and JNK in Drosophila. PLoS One. 12(5):e0178105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65:Martin M, Iyadurai SJ, Gassman A, Gindhart JG Jr, Hays TS, Saxton WM. (1999) Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 10(11):3717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66:McGrail M, Gepner J, Silvanovich A, Ludmann S, Serr M, Hays TS. (1995) Regulation of cytoplasmic dynein function in vivo by the Drosophila Glued complex. J Cell Biol. 131(2):411–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67:van Hemert MJ, Steensma HY, van Heusden GP. (2001) 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays. 23(10):936–46. [DOI] [PubMed] [Google Scholar]
- 68:Porter GW, Khuri FR, Fu H. (2006) Dynamic 14-3-3/client protein interactions integrate survival and apoptotic pathways. Semin Cancer Biol. 16:193–202. [DOI] [PubMed] [Google Scholar]
- 69:Bonnefoy-Bérard N, Liu YC, von Willebrand M, Sung A, Elly C, Mustelin T, Yoshida H, Ishizaka K, Altman A. (1995) Inhibition of phosphatidylinositol 3-kinase activity by association with 14-3-3 proteins in T cells. Proc Natl Acad Sci U S A. 92(22):10142–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70:Craparo A, Freund R, Gustafson TA. (1997) 14-3-3 (epsilon) interacts with the insulin-like growth factor I receptor and insulin receptor substrate I in a phosphoserine-dependent manner. J Biol Chem. 272:11663–11669. [DOI] [PubMed] [Google Scholar]
- 71:Lonic A, Barry EF, Quach C, Kobe B, Saunders N, Guthridge MA. (2008) Fibroblast growth factor receptor 2 phosphorylation on serine 779 couples to 14-3-3 and regulates cell survival and proliferation. Mol Cell Biol. 28:3372–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72:Munday AD, Berndt MC, Mitchell CA. (2000) Phosphoinositide 3-kinase forms a complex with platelet membrane glycoprotein Ib-IX-V complex and 14-3-3zeta. Blood. 96:577–584. [PubMed] [Google Scholar]
- 73:Oksvold MP, Huitfeldt HS, Langdon WY. (2004) Identification of 14-3-3zeta as an EGF receptor interacting protein. FEBS Lett. 569:207–210. [DOI] [PubMed] [Google Scholar]