

Abstract
DNA lesions interfere with cellular processes such as transcription and replication and need to be adequately resolved to warrant genome integrity. Beyond their primary role in molecule transport, nuclear pore complexes (NPCs) function in other processes such as transcription, nuclear organization and DNA double strand break (DSB) repair. Here we found that the removal of UV-induced DNA lesions by nucleotide excision repair (NER) is compromised in the absence of the Nup84 nuclear pore component. Importantly, nup84Δ cells show an exacerbated sensitivity to UV in early S phase and delayed replication fork progression, suggesting that unrepaired spontaneous DNA lesions persist during S phase. In addition, nup84Δ cells are defective in the repair of replication-born DSBs by sister chromatid recombination (SCR) and rely on post-replicative repair functions for normal proliferation, indicating dysfunctions in the cellular pathways that enable replication on damaged DNA templates. Altogether, our data reveal a central role of the NPC in the DNA damage response to facilitate replication progression through damaged DNA templates by promoting efficient NER and SCR and preventing chromosomal rearrangements.
INTRODUCTION
Living cells experience DNA damage that may lead to mutations and genomic instability if left unrepaired. DNA damage may occur by exogenous agents such as ionizing radiation, UV light and chemicals or by metabolic compounds. Cells possess a number of pathways to warrant genomic integrity, many of which are highly conserved throughout evolution. These pathways and their sensors, which are grouped under the general term of DNA damage response (DDR) coordinate appropriate repair of DNA damage by specialised mechanisms and synchronise the resolution of replication problems with cell cycle progression (1,2).
Nucleotide excision repair (NER) is a versatile DNA repair pathway that acts on helix-distorting lesions like bulky adducts or UV-induced pyrimidine dimers, which are harmful DNA lesions as they block the progression of DNA-dependent processes such as transcription or replication. NER can be divided in two sub-pathways: global genome repair (GG-NER), which act on DNA lesions all over the genome; and transcription-coupled repair (TC-NER), which act specifically on DNA lesions on the transcribed strand (TS) of active genes (3,4). GG- and TC-NER differ in the lesion recognition step, while the core repair reaction is common to both sub-pathways. In TC-NER, stalling of the elongating RNA polymerase (RNAP) at the DNA damage promptly triggers the repair reaction, while a specialized DNA damage recognition complex—the Rad7–Rad16 complex in budding yeast – improves the detection of the helix-distorting DNA lesions in GG-NER.
The presence of an unrepaired DNA lesion may compromise replication progression and cause replication fork (RF) stalling. Such RF stalling, whether at a DNA lesion or at other structural obstacles such as tightly bound proteins, non-canonical DNA structures or R-loops, require specific mechanisms to resume replication and avoid chromosome breakage (5,6). The post-replication repair pathway (PRR; also known as DNA damage tolerance pathway) allows bypass of lesions on the DNA template either by translesion DNA synthesis (TLS) or by a process that relies on transient switching to a non-damaged template. TLS is mainly an error-prone process carried out by specialized low-fidelity polymerases that can replicate across DNA lesions, while template switching is an error-free process. In budding yeast, monoubiquitylation of the PCNA sliding clamp at the conserved Lys164 residue by the Rad18–Rad6 heterodimer triggers TLS, while polyubiquitylation at that same residue by the Rad5 ubiquitin ligase and the Ubc13–Mms2 ubiquitin conjugation complex induces template switching (7,8).
RF blockage can also lead to double-strand breaks (DSBs), the most harmful of the DNA lesions. Two different pathways carry out DSB repair in eukaryotic cells: non-homologous end joining (NHEJ) and homologous recombination (HR). Whereas NHEJ consists of the direct re-ligation of the broken fragments, with minor nucleotide insertions and/or deletions, HR uses a homologous sequence—ideally the sister chromatid—as template to restore the broken DNA in an error-free manner (9,10). To preserve genome stability, HR is strongly repressed in cells that are not in S/G2 phase of the cell cycle and therefore did not yet synthesise a sister chromatid. Notably, resection of the DSB 5′-ends commits its repair to HR. A major player at this initial stage is the MRX complex, which consists of the Mre11, Rad50 and Xrs2 proteins in Saccharomyces cerevisiae (MRE11–RAD50–NBS1 or MRN in vertebrates). The MRX complex binds to the DSB, protects the DNA ends and contributes to 5′-end resection together with other factors. The replication protein A binds to the resulting single-stranded 3′-end and is subsequently substituted by Rad51 to form a nucleoprotein filament competent for homology search and strand invasion. In yeast, Rad51 filament assembly is mediated by Rad52 (11) and several factors not directly involved in DSB repair, including SMC proteins, the Rrm3 helicase, and histone acetylation complexes, all of which contribute to conducting repair to sister chromatid recombination (SCR) (12–14).
Investigating the factors and mechanisms involved in TC-NER, we observed that nup84Δ mutants become hypersensitive to UV and to the UV-mimetic drug 4-nitroquinoline 1-oxide (4-NQO) in the absence of GG-NER in the yeast S. cerevisiae. Nup84 is a core component of the 8-subunit Nup84 sub-complex of the yeast nuclear pore complex (NPC), a highly conserved macromolecular structure composed of about 30 different nucleoporins (15,16). The core function of the NPC is the nucleo-cytoplasmic transport of molecules. However, in the last years NPCs have been implicated in other nuclear functions, including nucleus organization, gene expression and DNA repair (17,18). Thus, a unique irreparable DSB has been shown to relocate to the nuclear periphery in a Nup84-dependent manner (19), and null mutants of the Nup84 sub-complex are sensitive to different genotoxic agents (20,21), undergo genomic instability (22), and show strong negative genetic interactions with mutant alleles of DNA replication and HR factors (23,24). Here, we show that nup84Δ cells are defective in NER and fail to repair UV-induced cyclobutane pyrimidine dimers (CPDs) independently of transcription. nup84Δ cells are also sensitive to other genotoxic compounds and show exacerbated sensitivity to UV in early S phase as well as delayed replication in the absence of exogenous DNA damage. These findings, together with the observation that cells lacking Nup84 are impaired in SCR of replication-born DSBs, provide evidence for a global role of the Nup84 sub-complex in the DDR that is required for NER and SCR. Finally, in the absence of PRR, nup84Δ cells channel DNA damage towards recombinational repair, leading to a synergistic enhancement of gross chromosomal rearrangements (GCRs), consistent with a function of the Nup84 sub-complex in avoiding harmful recombination of DNA breaks with non-sister chromatid templates. Altogether these results support the idea that the NPC may act as a docking site for the coordination of essential DNA metabolic processes, including replication and repair.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains and plasmids used in this study are listed in Supplementary Tables S1 and S2. Strains yHG153-7B and yHG153-8C were obtained by genetic cross between EJY344 (25) and a BY4742-isogenic nup84Δ::KanMX trp1Δ strain. Strains SYRB1-4C and SYRN84B1-10C were obtained by genetic cross between SY2201 (E. Schwob) and WRN84B1α-3D. Strains yHG164-9D and yHG164-9A were obtained by genetic cross between MMII-31 (26) and WRN84B1α-3D. Strains yHG163-7A, yHG163-5D, yHG163-4C and yHG16-1B were generated by genetic cross between RDKY3615 (27) and yHG130-3D. Strains yHG183-4D and yHG183-3A were obtained by genetic cross between WSR-7D and a BY4742-isogenic ulp1ΔN338::HIS3 strain. Strains yHG182-4A and yHG182-4B were obtained by genetic cross between YNN299 (28) and a BY4742-isogenic nup84Δ::KanMX trp1Δ strain. Strain yHG157-3 was generated by insertion of the 3100 bp NcoI fragment isolated from plasmid pCR-ΔN338-ulp1-HIS3 (22) into BY4741. Strain yHG128-3D was obtained by direct deletion into BY4742.
Gene- and strand-specific repair assays
CPD repair at the RPB2 and GAL10 genes was analysed as described (29). Briefly, cells were grown at 30°C in synthetic complete (SC) medium, irradiated in SD medium w/o amino acids with 150 J/m2 UV-C light (BS03 UV irradiation chamber; Dr. Gröbel UV-Elektronik GmbH), the medium supplemented with amino acids and the cells incubated at 30°C in the dark for recovery. Isolated DNA samples were digested with the indicated restriction enzymes (Roche) and aliquots mock-treated or treated with T4-endonuclease V (T4endoV, Epicentre). Strand-specific probes were obtained by primer extension. Sequences of the primers are listed in Supplementary Table S3. Membranes were analysed and quantified with a PhosphorImager (Fujifilm FLA5100). The remaining intact restriction fragment after treatment with T4endoV corresponds to the fraction of undamaged DNA. The CPD content was calculated using the Poisson expression, -ln(RFa/RFb), where RFa and RFb represent the intact restriction fragment signal intensities of the T4endoV- and mock-treated DNA, respectively. Repair curves were calculated as the fraction of CPDs removed versus time. The initial damage was set to 0% repair.
2D gel electrophoresis
Wild-type and nup84Δ cells (both bar1Δ) growing in YPAD were arrested in G1 with α-factor and released into rich medium containing the indicated HU concentrations. Culture samples were taken at the indicated time points. DNA extraction was performed with the cetyltrimethylammonium bromide method, and neutral–neutral 2D gel electrophoresis was performed as previously described (30).
BrdU incorporation (BrdU-IP)
Analysis of ARS activation by BrdU incorporation was performed as previously described (31). Isogenic wild-type and nup84Δ strains deleted for the BAR1 gene and carrying several copies of the Herpes simplex thymidine kinase (TK) under the control of the constitutive GPD promoter were grown in YPAD, synchronized with α-factor, washed in fresh medium and released into S phase by addition of 1 μg/ml pronase. BrdU (200 μg/ml) was also added to the culture at this point. Immunoprecipitation was performed using anti-BrdU antibody (MBL) as described (31,32). Input and immunoprecipitated DNA were analysed by real time qPCR. Relative BrdU incorporation at a given time point was calculated relative to the input and to the signal without BrdU (time 0) in the same sample. Primer sequences are listed in Supplementary Table S3.
Physical analysis of sister chromatid recombination (SCR)
SCR assays were carried out essentially as described (33). Briefly, cells carrying pRS316TINV were grown to mid-log phase in SC-Ura 3% glycerol-2% lactate, plus 5 μg/ml doxycycline to repress transcription from the Tet promoter; then, 2% galactose was added to induce HO overexpression. Samples were collected at different time points and DNA was purified, digested with SpeI-XhoI and analysed by Southern blot using Hybond N membranes (GE Healthcare). A 32P-labeled 0.22-kb LEU2 probe was obtained by PCR from pRS315. Primers sequences are listed in Supplementary Table S3. Bands were quantified in a Fujifilm FLA-5100.
Recombination and GCR assays
For the recombination assays, cells were cultured in SC medium plates and grown for 3–4 days. Leu+ recombinants resulting from recombination in TINV, L, LYΔNS and L-lacZ systems were selected on SC-Leu plates, while Leu+ recombinants from GL-lacZ system were selected on SGal-Leu plates, to allow the expression of LEU2 from GAL1-10 promoter. For the genetic study of repair of HO-induced DSBs with TINV system, colonies were grown overnight in SC-Ura 3% glycerol-2% lactate medium, plus 5 μg/ml doxycycline to repress transcription from the Tet promoter, and 2% galactose was added to induce HO overexpression for 5 h, as previously described (33,34). For the genetic analyses of SCR with the chromosomal system based on his3-Δ3′ and his3-Δ5′ fragments, cells were transformed with pRS315 or pRS315-ULP1pr-GFP-NSP1-ULP1C and transformants grown in SC-Leu plates for 3–4 days. His+ recombinants were selected on SC-Leu-His plates. Recombination frequencies were obtained by fluctuation tests as the median value of six independent colonies. The final frequency given for each strain and condition is the mean and standard deviation of at least three median values. GCR assays were performed and GCR rates calculated as described (27).
PCNA modification assay
Analysis of PCNA ubiquitylation and sumoylation was performed as described (35). Isogenic wild-type and nup84Δ cells carrying plasmid YEp195-CUP-HisSmt3 or YEp195-CUP-HisUb were grown to mid-log phase and irradiated with 150 J/m2 UV light or left unirradiated. Cells were harvested 30 minutes after UV irradiation and denaturing Ni-NTA pull down was performed to purify SUMO and ubiquitin conjugates. Western blot analysis with anti-Pol30 antibody (kindly provided by H. Ulrich) was performed to visualise the different PCNA species.
Microarray gene expression analyses
Microarray determination of total RNA was performed using the Affymetrix platform as previously described (36). Statistical data analyses were performed using the Limma package (affylmGUI interface) of the R Bioconductor project (http://www.bioconductor.org). Microarray analysis was conducted in triplicate for each strain. Genes were considered significantly up- or down-regulated when their expression values were changed >1.5-fold with a FDR <0.01. Enrichment of Gene Ontology biological process terms was calculated using the AmiGO tool (http://amigo.geneontology.org/amigo), considering enrichment with a p-value<0.05 as statistically significant.
Miscellanea
Analysis of sensitivity to genotoxic agents, Southern, Western, Rad52-YFP foci detection, fluorescence-activated cell sorting analyses (FACS) using a FACScalibur Becton Dickinson machine, and yeast cultures were performed using standard procedures. GraphPad Prism was used to perform the statistical analyses by unpaired student t-test and two-way ANOVA as indicated.
RESULTS
Nup84 is required for DNA repair
Several features of the cellular DDR are linked to the Nup84 sub-complex. Previous studies have shown that this complex plays a role in the response to irreparable DSBs, eroded telomeres, trinucleotide repeats, and in NHEJ (19,22,37,38). With these precedents, and given that nup84Δ mutants show transcription elongation defects (39), we generated nup84Δ rad7Δ double mutants to assess the functionality of TC-NER and explore the role of the Nup84 sub-complex in different repair pathways and replication. For this, we first assayed sensitivity to 4-NQO, UV, methyl methanesulfonate (MMS), hydroxyurea (HU), menadione (Mnd), phleomycin (Phleo), camptothecin (CPT) and caffeine in nup84Δ, rad7Δ, nup84Δ rad7Δ, and wild-type cells (Figure 1A). In agreement with previous data (20,21), the nup84Δ single mutant was itself sensitive to 4-NQO and other genotoxic agents, such as MMS, HU, Mnd, Phleo, CPT, and to caffeine-induced checkpoint override. Interestingly, nup84Δ rad7Δ cells were more sensitive than the single mutants to most of these DNA damaging agents, in particular to UV, 4-NQO, MMS and HU, suggesting that cells lacking Nup84 accumulate DNA damage and also require GG-NER for survival in these conditions. To check whether these phenotypes were common to other nucleoporin mutants or specific of the Nup84 sub-complex, we assayed 4-NQO and UV sensitivities of Nup84 sub-complex subunit Nup133, the functionally related nuclear basket protein Nup60, and the unrelated nucleoporins Nup53 and Nup100 (Supplementary Figure S1A). Consistently, the sensitivity of nup133Δ cells to 4-NQO and UV was exacerbated in the absence of Rad7 as described for nup84Δ cells, while nup60Δ cells were moderately sensitive to 4-NQO and showed increased UV and 4-NQO sensitivities in combination with rad7Δ, although both phenotypes were weaker than those of nup84Δ cells. In contrast, cells lacking Nup53 and Nup100 did not show increased 4-NQO or UV sensitivities at the used concentration, neither alone nor in combination with rad7Δ. These results are consistent with the Nup84 sub-complex having a specific role in coping with DNA damage that is not a general feature of nucleoporins mutants.
Figure 1.

Nup84 is required for the repair of DNA damage. (A) Sensitivity of wild-type (WT, BY4741), rad7Δ (yHG72-1B), nup84Δ (YDL116W) and nup84Δ rad7Δ (yN84R7-24A) cells to 4-nitroquinoline 1-oxide (4-NQO), UV light, methyl methanesulfonate (MMS), hydroxyurea (HU), menadione (Mnd), phleomycin (Phleo), camptothecin (CPT) and caffeine. 10-fold serial dilutions of exponentially growing cells are shown. (B) Representative results of Southern analysis showing the repair of a 4.4-kb (NsiI/PvuI) RPB2 fragment in WT (BY4741) and nup84Δ (YDL116W) cells on the transcribed strand (TS) and the non-transcribed strand (NTS) (left). Non-irradiated DNA and DNA not treated with T4endoV were used as controls. Graphical representation of the quantified results is shown (right). Average values and standard deviations of independent experiments are plotted (N ≥ 3). P values are shown (two-way ANOVA). The initial damage generated was on average 0.51 CPD/kb in the TS and 0.56 CPD/kb in the NTS. (C) Representative results of Southern analysis showing the repair of a 1.6-kb (EcoRI/SalI) GAL10 fragment in WT (BY4741) and nup84Δ (YDL116W) cells (left). Analysis was performed on the TS in conditions in which GAL10 was not transcribed (glucose). The initial damage was 0.54 CPD/kb on average. Other details as in B. (D) Recombination analysis in WT (WRB1-1A) and nup84Δ (WRN84B1α-3D) cells carrying the L and LYΔNS plasmid systems (left) or the L-lacZ (Transcription ON) and GL-lacZ (Transcription OFF) systems grown in glucose-containing medium (right). Average and standard deviation of fluctuation tests consisting in the median value of six independent colonies each one are shown (N ≥ 3). *P < 0.05; **P < 0.01 (unpaired Student's t-test). A scheme of the system is shown on top of each panel.
Increased UV-sensitivity in the absence of GG-NER is commonly associated with defects in TC-NER. We therefore tested the ability of nup84Δ cells to repair UV-induced DNA damage in the TS and the non-transcribed strand (NTS) DNA strands of a 4.4-kb restriction fragment containing the constitutively expressed RPB2 gene. Notably, the CPD repair efficiency of nup84Δ cells were much lower than in the wild type for both DNA strands, even four hours after UV irradiation (51% versus 86% in the TS, 34% versus 67% in the NTS, respectively; Figure 1B), indicating that Nup84 is required for both TC- and GG-NER. Consistently, repair in the glucose-repressed GAL10 gene was also strongly impaired in nup84Δ cells (8% vs 31% in the wild type; Figure 1C). Altogether, these results indicate that Nup84 is necessary for efficient repair of UV-induced DNA damage independently of transcription.
Consistent with an accumulation of unrepaired DNA damage, mutants of the Nup84 sub-complex lead to increased recombination frequencies (22). To assess whether transcription stimulates recombination in nup84Δ cells, we used two plasmid-based recombination systems carrying direct repeats of a 0.6 kb internal fragment from truncated LEU2 genes without intervening sequence (L) or separated by a 3.7 kb-long intervening sequence (LYΔNS) (Figure 1D). The presence of a long intervening sequence between the repeats enhanced the recombination frequency in the same proportion in nup84Δ and in wild-type cells (3.6- and 3.3-fold increase in LYΔNS vs. L, respectively). Next, provided that Nup84 has been previously involved in transcription elongation (39), we wondered whether the hyper-recombination was transcription-dependent. We used recombination systems in which the lacZ gene is located between the leu2 direct repeats and transcription is regulated by the GAL1-10 promoter (GL-lacZ) or by the endogenous LEU2 promoter (L-lacZ), thus allowing to compare the recombination frequencies obtained without and with transcription, respectively, by growing the cells in glucose-containing media. As shown in Figure 1D, a similar increase in transcription-induced recombination was obtained for nup84Δ and wild-type cells (2.6- and 2.9-fold increase, respectively). Therefore, these results indicate that genome instability in nup84Δ cells arise independently of transcription.
Finally, since the Nup84 sub-complex has been linked to sumoylation homeostasis by controlling the proper NPC localisation of the SUMO-specific protease Ulp1 (22) and many repair factors, including NER, are sumoylated in response to DNA damage (25,40), we asked whether the role of Nup84 in DNA repair may rely on sumoylation defects. Previously, different constructs have been used either to enrich Ulp1 at the NPC by over-expressing a GFP-tagged version of Ulp1 (GFP-Ulp1; (22)) or to artificially tether Ulp1 to the NPC by expressing a GFP-tagged chimeric protein containing the catalytic domain of Ulp1 fused to the nucleoporin Nsp1 from the ULP1 promoter (GFP-Nsp1-Ulp1C; (41)). To confirm the functionality of these formerly characterized constructs, we assayed whether their expression complemented drug sensitivity and sumoylation defects of the ulp1ΔN338 mutant, which encodes a defective Ulp1 protein unable to anchor to the NPC (Supplementary Figure S2). The results indicate that although GFP-Ulp1 over-expression led to a better complementation than GFP-Nsp1-Ulp1C expression, it also alters the sumoylation pattern in wild-type cells. To test whether nup84Δ sensitivity to genotoxic agents in GG-NER-defective background could be suppressed by artificial localisation of the Ulp1 SUMO protease to the NPC, we analysed 4-NQO, UV, MMS and HU sensitivity in wild-type, nup84Δ, rad7Δ and nup84Δ rad7Δ cells expressing GFP-Nsp1-Ulp1C (Figure 2A) or over-expressing GFP-Ulp1 (Supplementary Figure S3A). Tethering of the Ulp1 catalytic domain to the NPC led to a weak suppression of both nup84Δ and nup84Δ rad7Δ sensitivity to 4-NQO, UV and MMS but did not reduce HU sensitivity. In the case of GFP-Ulp1 over-expression, partial suppression of the sensitivity to 4-NQO, UV, MMS and HU were observed. Consistently, we found that siz1Δ siz2Δ cells, which are defective in SUMO ligation and moderately deficient in NER (25), and the ulp1ΔN338 mutant were sensitive to 4-NQO and UV to a lesser extent than nup84Δ cells, whereas sensitivity of nup84Δ siz1Δ siz2Δ triple mutants was very similar to nup84Δ sensitivity (Supplementary Figure S3B). Altogether, our results suggest that Nup84 integrity is required for DNA repair beyond SUMO homeostasis.
Figure 2.

nup84Δ sensitivity to UV is exacerbated during S phase independently of SUMO homeostasis. (A) Sensitivity of WT (BY4741), rad7Δ (yHG72-1B), nup84Δ (YDL116W) and nup84Δ rad7Δ (yN84R7-24A) cells with and without expression of a GFP-Nsp1-Ulp1C fusion protein to 4-NQO, UV light, MMS and HU. 10-fold serial dilutions of exponentially growing cells are shown. (B) UV survival curves of asynchronously growing (dash lines) or early S phase-enriched (S; plain lanes) WT (BY4741), nup84Δ (YDL116W), rad1Δ (YPL022W) and rad52Δ (YML032C) cells. Average and standard deviation are plotted (N ≥ 3). Statistical analyses were performed between both growth conditions for each genotype. **P < 0.01; ***P < 0.001 (Two-way ANOVA). (C) UV survival curves of asynchronously growing (dash lines) or early S phase-enriched (S; plain lanes) WT (BY4741) and nup84Δ (YDL116W) cells transformed with an empty vector (–) or a plasmid expressing the GFP-Nsp1-Ulp1C fusion protein (N ≥ 3). Statistical analyses were performed between GFP-Nsp1-Ulp1C expression and empty vector for each genotype and growth condition. *P < 0.05. Other details as in B.
Absence of Nup84 sensitizes cells to DNA damage during S phase and causes replication impairment
Strikingly, nup84Δ cells show strong CPD-removal defects but only mild UV sensitivity (see Figure 1). This apparent discrepancy prompted us to quantify UV survival both in asynchronously growing cells and in cells irradiated shortly upon release from α-factor-induced G1 arrest. NER-deficient rad1Δ, HR-deficient rad52Δ and wild-type cells were used as controls. Interestingly, both nup84Δ and rad52Δ cells were much more sensitive to UV irradiation in early S-enriched than in asynchronously growing cells, in contrast to wild-type and rad1Δ cells, which were equally UV sensitive in either condition (Figure 2B). Artificial NPC tethering of Ulp1 using the GFP-Nsp1-Ulp1C fusion protein led to a partial suppression of UV sensitivity in asynchronously growing nup84Δ cells (Figure 2C), in agreement with the partial suppression of UV sensitivity observed in plates (see Figure 2A). Importantly, it did not affect UV survival in early S-enriched nup84Δ cells or in wild-type cells (Figure 2C). Over-expression of GFP-Ulp1 led to similar results in nup84Δ cells, as UV sensitivity was partially suppressed in asynchronous but not in early S-enriched cells (Supplementary Figure S3C). However, expression of the GFP-Ulp1 construct in wild-type cells led to a slight increase in UV sensitivity, indicating that Ulp1 over-expression interferes with the DDR, as suggested by the alteration of the pattern of sumoylated proteins in MMS-treated cells (see Supplementary Figure S2B). Together, these results indicate that cells lacking Nup84 are sensitized to DNA damage mostly during S phase, and that this increased sensitivity in replicating cells is independent of Ulp1 localization at the NPC.
Since the accumulation of unrepaired DNA damage might compromise the progression of RFs, providing a rationale for the high sensitisation of nup84Δ cells to damage in S phase, we addressed DNA replication in Nup84-deficient cells. First, we analysed S-phase progression by FACS and found that nup84Δ cells entered S phase more slowly than the wild type after release from G1 arrest (Figure 3A and Supplementary Figure S4A). We then analysed the accumulation of replication intermediates by 2D-gel electrophoresis, which provides a much more conclusive analysis of replication at the molecular level, in cells released from G1 arrest in medium with low HU concentrations to slowdown replication and allow a better detection of intermediates. Replication intermediates, mainly consisting in bubble-shaped and simple Y arcs, were analysed on a DNA fragment comprising the early replication origin ARS508 and the proximal SPF1 gene (Figure 3B). These intermediates were visible 15 min after G1 release in wild-type cells, reaching a peak at 30 min and being barely detectable after 1 h at 20 mM HU (Figure 3C). However, replication intermediates were not detected until 30 min after G1 release in nup84Δ cells and remained present after 60 min. Similar results were obtained at a higher concentration of HU (40 mM) (Supplementary Figure S4B–D). Our data thus suggest that replication onset is delayed in the absence of Nup84. To confirm these results with a different approach, we monitored BrdU incorporation by ChIP (BrdU-IP) at different time points after release from α-factor-induced G1 arrest in the vicinity of the early-firing origins ARS508 and ARS1211 at which replication proceeds in head-on orientation with the transcribed SPF1 gene and TRX1-PDC1 locus, respectively. Consistent with previous results (31,42,43), BrdU incorporation levels in wild-type cells were higher at the proximity of the replication origin than further away, peaking 20 minutes after G1 release (Figure 3D). In nup84Δ cells, BrdU incorporation was significantly reduced in the vicinity of either replication origins, peaking at a later time point and at lower levels as compared to the wild type (Figure 3D), and suggesting that RFs progress less efficiently. Altogether, these results indicate that replication is compromised in the absence of Nup84.
Figure 3.
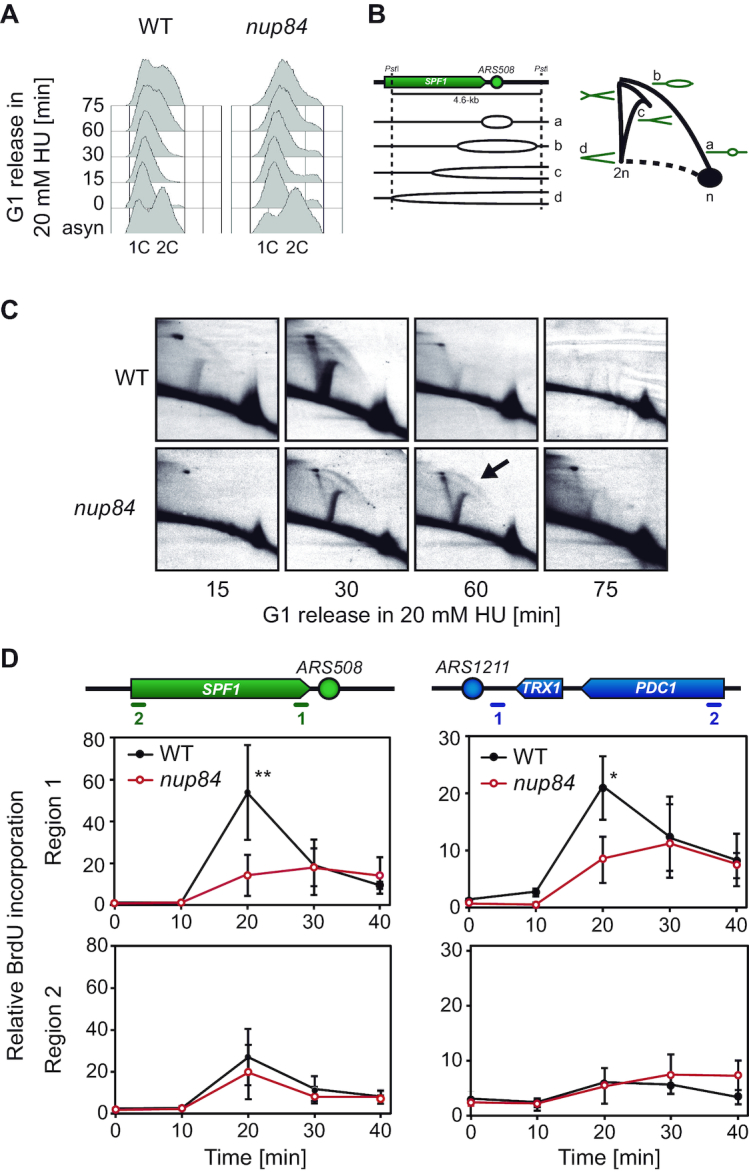
Replication impairment in nup84Δ cells. (A) Flow cytometry analyses of wild type (WT, WRB1-1A) and nup84Δ (WRN84B1-10C) cells released from α-factor-induced G1 arrest into HU-containing medium (20 mM). (B) Diagram depicting the appearance of different replication intermediates at the analysed ARS508-containing fragment and their migration in 2D-gel electrophoresis analyses. (C) Analysis of replication intermediates at the early origin ARS508 by 2D-gel electrophoresis in WT (WRB1-1A) and nup84Δ (WRN84B1-10C) cells released from α-factor-induced G1 arrest into HU-containing medium (20 mM). Replication intermediates were analysed at different time points after G1 release. (D) BrdU incorporation in WT (SYRB1-4C) and nup84Δ (SYRN84B1-10C) strains. Cells were synchronized in G1 with α-factor and released into fresh medium. BrdU immunoprecipitation was performed at different time points after G1 release, and the resulting DNA analysed by real time qPCR at the early replication origins ARS508 and ARS1211. Quantification of BrdU incorporation relative to input DNA and to time 0 (no BrdU) is plotted for each region. Average and standard deviation are shown (N = 3). *P < 0.05; **P < 0.01 (unpaired Student's t-test). Schemes of the analysed region are shown on top of the graphs.
Defective DSB repair by sister chromatid recombination in nup84 cells
When RF progression is blocked in yeast, the activation of the S-phase checkpoint mediated by phosphorylation of the Rad53 kinase protects the fork and promotes replication restart. To assess the functional relevance of the S-phase checkpoint in cell proliferation of nup84Δ cells, we generated double mutants carrying the non-phosphorylatable rad53K227A allele and nup84Δ. We found that the combination of nup84Δ mutation with rad53K227A caused a synthetic growth defect that was exacerbated upon DNA damage induced by 4-NQO, UV, MMS and HU treatments (Figure 4A), suggesting that cells lacking Nup84 rely on a functional S-phase checkpoint to eliminate or bypass DNA damage that compromise replication. Analysis of Rad53 phosphorylation by Western blotting confirmed that the DNA damage checkpoint is activated in nup84Δ cells in the absence of exogenous damage (Figure 4B). This behaviour is shared with the nup133Δ and nup60Δ mutants affected in a Nup84 sub-complex component and a functionally related nucleoporin, respectively, but not with the unrelated nucleoporin mutants nup53Δ and nup100Δ (Supplementary Figure S1B). Altogether, these results are consistent with the idea that nup84Δ cells accumulate DNA damage that interferes with replication.
Figure 4.

Replication-born DSBs are particularly harmful in the absence of Nup84. (A) Sensitivity of wild type (WT, WRB1-1A), nup84Δ (WRN84B1-10C), rad53K227A (CY7031) and nup84Δ rad53K227A (yHG159-15C) to 4-NQO, UV, MMS and HU. 10-fold serial dilutions of exponentially growing cells are shown. (B) Western blot analysis of Rad53 in wild-type (WT, BY4741) and nup84Δ (YDL116W) cell cultures. HU-treated cells (50 mM HU for 2 h) are shown as control of Rad53 phosphorylation. (C) Sensitivity of wild type (WT, WRB1-1A), nup84Δ (WRN84B1α-3D), rad3-102 (yHG164-9D) and nup84Δ rad3-102 (yHG164-9A) to 4-NQO and UV. 10-fold serial dilutions of exponentially growing cells are shown.
To investigate the relationship between the Nup84 sub-complex and HR, which is a major repair pathway in replicating cells, we combined deletions of the HR gene RAD52 with nup84Δ, nup133Δ and nup60Δ mutations. Consistent with previous reports (22–24), synthetic negative interactions were observed between nucleoporin and HR genes and the recovered nup84Δ rad52Δ and nup133Δ rad52Δ double mutants showed reduced growth (Supplementary Figure S5A). Notably, deletion of RAD52 led to increased sensitivity to UV, 4-NQO, MMS or HU treatment in nup84Δ, nup133Δ and nup60Δ cells. FACS analyses of untreated cultures suggest that nup84Δ rad52Δ, nup133Δ rad52Δ and nup60Δ rad52Δ mutants tend to accumulate in S/G2 (Supplementary Figure S5B). Consequently, we assessed the impact of replication-born DNA breaks on nup84Δ cells. For this, we first generated double mutants with the rad3-102 allele, which blocks the NER reaction at a post-incision stage and leads to a replication-born DSB at the site of the original DNA lesion (26). The nup84Δ rad3-102 double mutants were more sensitive to 4-NQO and UV than the respective single mutants (Figure 4C), consistent with the idea that nup84Δ cells are sensitive to replication-born DSBs. Since SCR is the major DSB repair pathway during replication (33,44), we assayed the efficiency of repair of replication-born DSBs by SCR in nup84Δ mutants. For this we used the plasmid-based leu2 inverted-repeat recombination system TINV containing a 24-bp mini HO site (Figure 5A). Consistent with the recombination analyses with direct-repeat systems (Figure 1 and (22)), nup84Δ cells show high levels of spontaneous recombination in the absence of HO induction (3.9-fold above wild type), as shown in Figure 5B. Over-expression of the HO endonuclease, which generates preferentially single-stranded DNA breaks at this site that may be converted into DSBs during replication (45), leads to a strong increase in recombination in wild-type cells (300-fold) that likely reflects the repair of replication-born DSBs repaired by unequal SCR during S phase. In nup84Δ cells, HO-induced recombination was severely impaired (6.9-fold decrease over wild type and only 10-fold increase over spontaneous frequency) after 5 h of HO expression, suggesting that Nup84 might be required for the repair of HO-induced replication-born DSBs by SCR. Consistently, detailed molecular analysis of the kinetics of repair of replication-born DSBs revealed a strong reduction in the efficiency of SCR in nup84Δ mutants, with similar efficiency of HO cleavage (Figure 5C and Supplementary Figure S6A). DSBs peaked 3 h after HO induction (about 40% of cut molecules), while SCR values were close to 4% in nup84Δ and reached 12% in wild-type cells 9 h after HO induction. These results indicate that Nup84 is necessary for the repair of replication-born DSBs by SCR. To assess whether nup84Δ SCR defects occurred as a consequence of Ulp1 mislocalisation, we analysed genetically the frequency of SCR using a specific chromosomal repeat recombination system (28) in wild-type and nup84Δ cells with and without expression of GFP-Nsp1-Ulp1C (Supplementary Figure S6B and C). Our results confirmed that nup84Δ is deficient in repair by SCR and indicated that artificial tethering of Ulp1 to the NPC leads to a partial suppression of the observed SCR defects. We then analysed SCR at the molecular level in the ulp1ΔN338 mutant, which lacks the NPC anchoring domain (Supplementary Figure S6D). In contrast to nup84Δ cells, ulp1ΔN338 cells do not show defects in SCR as compared to isogenic wild-type cells. Altogether, these data suggest that the localization of Ulp1 at the NPC per se is not required for SCR, while forcing its NPC anchorage in the absence of a functional Nup84 sub-complex can partially compensate for the strong SCR defect. The absence of Nup84 thus amplifies the threat of replication progression through damaged DNA, including UV-induced damage, and promotes a high sensitization of these cells to UV lesions during S-phase.
Figure 5.

DSB repair by sister chromatid recombination is defective in nup84Δ cells. (A) Scheme of pRS316TINV plasmid carrying two inverted leu2-repeats. Fragments generated by HO cleavage and XhoI/SpeI digestion, as detected by the LEU2 probe represented as a black line, are indicated with their corresponding sizes. (B) Genetic analysis of recombination in wild type (WT, WSR-7D) and nup84Δ (WSRN84-3D) cells carrying the TINV system. Cells were grown in glucose (–HO) or in galactose (+HO) for 5 hours to measure spontaneous and HO-induced recombination, respectively. Average and standard deviation of fluctuation tests consisting in the median value of six independent colonies each one are shown (N ≥ 3). **P < 0.01; ***P < 0.001 (unpaired Student's t-test). (C) Representative Southern blot showing the physical analysis of HO-induced DSB formation and its repair kinetics in WT (WSR-7D) and nup84Δ (WSRN84-3D) cells (left). Quantification of DSBs (1.4-kb plus 2.4-kb bands) and sister chromatid recombination (SCR; 4.7-kb band) related to total plasmid DNA (right). Average and standard deviation are shown (N = 3). P values are shown (two-way ANOVA).
Absence of Nup84 bypasses post-replicative repair in favour of pathways resulting in gross chromosomal rearrangements
Replication and PRR are regulated through ubiquitylation and sumoylation of the PCNA sliding clamp at specific residues (7,8). To gain insights into PCNA modifications in the absence of Nup84, we used affinity chromatography under denaturing conditions to isolate ubiquitin and SUMO conjugates from total cell lysate from untreated or UV-irradiated wild-type and nup84Δ cultures. While the pattern of ubiquitylation and sumoylation in wild-type cells was similar to published data obtained in the absence of exogenous DNA damage and upon MMS treatment (35), nup84Δ cells showed a strongly altered pattern of both modifications (Figure 6A). Notably, DNA damage-dependent PCNA modifications such as mono- and poly-ubiquitylation were detected in untreated nup84Δ cells, consistent with an accumulation of endogenous damage. In addition, PCNA species that are detected at low level in UV-irradiated wild-type cells were detected as major species in nup84Δ cells independently of UV-damage.
Figure 6.

Nup84 is required for appropriate post replication repair. (A) Pattern of PCNA modifications in wild-type (WT, BY4741) and nup84Δ (YDL116W) cells in untreated or UV-irradiated (150 J/m2) cultures. Ubiquitylated (U) and sumoylated (S) forms of endogenous PCNA were affinity purified from cells expressing His-tagged ubiquitin and Smt3 and detected by anti-PCNA Western blot. Migration of the different species as described in (35) and unspecific signals are indicated. U1, U2 and U3: mono- di- and tri-ubiquitylated PCNA species, respectively; S127 and S164: sumoylation on K127 and K164 residue, respectively; PCNA: unmodified PCNA; *: unspecific signal. (B) Sensitivity of wild type (WT, BY4741), nup84Δ (YDL116W), rad5Δ (YLR032W), nup84Δ rad5Δ (yHG132-13C), rad18Δ (yHG128-3D) and nup84Δ rad18Δ (yHG130-3D) to 4-NQO, UV and HU. 10-fold serial dilutions of exponentially growing cells are shown. (C) Recombination analysis of WT, nup84Δ, rad5Δ, nup84Δ rad5Δ, rad18Δ and nup84Δ rad18Δ cells described in A using the plasmid borne LYΔNS direct-repeat system (N ≥ 4). A scheme of the recombination system is shown. *P < 0.05; **P < 0.01; ***P < 0.001 (unpaired Student's t-test) (D) Percentage of S/G2 cells with Rad52-YFP foci in WT, nup84Δ, rad5Δ, nup84Δ rad5Δ, rad18Δ and nup84Δ rad18Δ cells described in A (N ≥ 3). Statistical analyses as in B. (E) Growth in galactose and glucose-containing media of rad52Δ cells expressing wild type RAD52 under the control of the GAL1 promoter in combination with the nup84Δ, rad5Δ and rad18Δ mutations obtained by genetic crosses between strains BYR52α (transformed with pDML5) and yHG130-3D or yHG132-13C. (F) Gross chromosomal rearrangement (GCR) rates obtained by scoring Canr 5-FOAr cells in WT (yHG163-7A), nup84Δ (yHG163-5D), rad18Δ (yHG163-4C) and nup84Δ rad18Δ (yHG163-3A) cells (N ≥ 11). 95% confidence intervals for the median and fold change relative to WT are shown. A scheme of the system is depicted on top.
Since ubiquitylation of PCNA in response to DNA damage is dependent on the PRR factors Rad18 and Rad5, we generated double mutants to explore the functional relevance of PRR in the absence of Nup84. Strikingly, both the nup84Δ rad5Δ and the nup84Δ rad18Δ double mutants were less sensitive to 4-NQO, UV and HU than the rad5Δ and rad18Δ single mutants, respectively (Figure 6B). Interestingly, these results suggest that PRR is partially bypassed in nup84Δ cells, possibly due to the channelling of DNA lesions to alternative repair pathways, such as recombination with non-sister templates. This idea is consistent with the hyper-recombination phenotype of nup84Δ mutants, which is further increased in nup84Δ rad18Δ double mutants (Figure 6C). Notably, the percentage of S/G2 cells with Rad52-YFP foci, an indicator of recombinational DSB repair sites (46), also increased significantly in nup84Δ rad18Δ double mutants (Figure 6D). This suggests that, in the absence of PRR, DNA damage accumulated in nup84Δ cells may be channelled to Rad52-dependent HR repair during replication. This would imply that HR would become relevant for cell viability under these conditions. Consistently, nup84Δ rad18Δ rad52Δ triple mutants expressing RAD52 from the GAL1 promoter were not viable in glucose-containing plates in which Rad52 expression is repressed (Figure 6E). It is likely that most DNA lesions accumulated in nup84Δ cells result in DNA breaks in S phase, consistent with the possibility that most bulky DNA lesions such as those produced by UV would cause RF breakage that would rely on SCR for their repair. Given the SCR defects of nup84Δ cells (see Figure 5), DNA lesions would be channelled toward recombination with DNA sequences others than the sister chromatid. In haploid yeast cells such recombination events would have to occur with ectopic regions, with the potential to generate large deletions or GCRs. To test this hypothesis, we used a previously characterized genomic system to score GCRs as the loss of a fragment of chromosome V containing the contra-selectable CAN1 and URA3 genes (27) in nup84Δ, rad18Δ and nup84Δ rad18Δ cells (Figure 6F). The absence of Nup84 increased GCRs (19-fold over wild type) as previously described (19) and, in agreement with our hypothesis, a synergistic increase was observed in the nup84Δ rad18Δ double mutant (235-fold over wild type). Thus, the Nup84 sub-complex may prevent recombination events with sequences different to the sister chromatid, therewith preventing GCRs and genome instability.
The transcription profile of nup84Δ cells is consistent with DNA damage accumulation
Since our results indicate that different DNA repair mechanisms, including NER and SCR, are affected in nup84Δ mutants, we wondered whether these phenotypes could result from a deficient expression of specific DNA repair genes caused by nup84Δ. Therefore, we performed microarray analysis of gene expression from total RNA in wild-type and nup84Δ cells. We selected the genes whose expression in the mutant was 1.5-fold above or below the wild-type levels (false discovery rate < 0.01), and analysed the gene ontology term enrichment of the different biological processes. No significant enrichment of DNA metabolism genes was found among the down-regulated genes (Supplementary Table S4), implying that the different DNA repair defects of nup84Δ are not a secondary effect caused by transcription impairment. Nevertheless, several genes involved in PRR (RAD18), HR (RAD51, RMI1 and SHU2), S-phase checkpoint (DUN1 and MRC1), and DNA damage response (DDR48 and UBC5) were up-regulated in nup84Δ cells (Supplementary Table S4 and Supplementary Dataset S1). Therefore, this transcription profile is consistent with the activation of a cellular response to an over-accumulation of DNA damage that compromise replication.
DISCUSSION
The NPC has been previously linked to nuclear functions distinct from transport, including the regulation of gene expression and DNA repair (17,18). In this study, we have found that yeast cells lacking the Nup84 outer ring nucleoporin show NER and SCR defects. In addition, nup84Δ mutants show exacerbated UV sensitivity in early S phase, replication defects, increased spontaneous recombination frequencies and genetic interactions with HR mutants. These results indicate that spontaneous DNA lesions accumulate in nup84Δ cells, and that these cells are particularly vulnerable to DNA damage while replicating. Consistently, replication-associated PRR is partially bypassed in the absence of Nup84, leading to an increase in GCR events. Altogether, our results show that cell cycle progression and replication dynamics are compromised in the absence of Nup84, leading to concomitant accumulation of spontaneous DNA damage and alteration of the DDR.
We found that nup84 mutants are defective in the repair of UV-induced DNA lesions and particularly sensitive to UV during S-phase. Genetic assays showed that mutants of the outer ring Nup84 sub-complex and the FG nucleoporin Nup60 accumulate DSBs and are defective in DSB repair by NHEJ due to de-localization of the SUMO-specific protease Ulp1 from the NPC (22). Artificial anchoring of the Ulp1 protease to the NPC partially suppressed the sensitivity of asynchronously growing nup84 cells to 4-NQO, UV and MMS (see Figure 2 and Supplementary Figure S3). Thus, in nup84 cells, Ulp1 mislocalisation might impair the correct activity of NER and other repair factors, which are sumoylated in response to UV or MMS treatment (25,40), and could therewith contribute to the observed repair defects. Interestingly however, localisation of Ulp1 at the NPC did not have any impact on the survival of nup84 cells irradiated with UV light in early S-phase (see Figure 2 and Supplementary Figure S3), indicating that the participation of NPC-anchored Ulp1 in the repair of photolesions seems to be restricted to non-replicating cells. Although NER is a repair pathway that acts throughout the cell cycle, it is conceivable that NER activity needs to be coordinated with G1 or S-phase specific repair pathways. Consistent with this idea, S-phase repair, including NER, requires a proficient replication stress response as deficiencies in the intra-S checkpoint Mec1/ATR kinase or the HR factor Rad52 abrogate repair of CPDs and bulky lesions uniquely during S phase (47,48). Thus, the strong deficiency in CPD repair observed in Nup84-deficient cells may rely on the combination of inaccurate SUMO-homeostasis and alterations in S phase-specific response to DNA damage.
Our replication analyses revealed that replication onset and progression are impaired in nup84 cells even in the absence of genotoxic agents (Figure 3 and Supplementary Figure S4). As a consequence of repair defects, spontaneous DNA lesions generated by internal metabolites and ROS may accumulate in the absence of Nup84. Such unrepaired endogenous damage could in turn impair replication progression; eventually leading to further DSBs in the absence of an appropriate cellular response to RF stalling. A large body of evidence pinpoints toward an accumulation of DSBs in nup84 cells, including elevated number of cells with Rad52 foci (22), synthetic growth defects with HR mutants (22,24) and increased spontaneous recombination frequencies in different reporter systems (Figure 1; (19,22)). Consistent with the idea that DSBs occur as a consequence of RF stalling at unrepaired endogenous DNA lesions, rad52 nup84 mutants accumulate in S/G2 (Supplementary Figure S4) and the S-phase checkpoint kinase Rad53 is phosphorylated in nup84 mutants (Figure 4). It is also conceivable that impaired replication progression itself generates DSBs in nup84 cells. In either case, our results indicate that the cellular response to RF stalling at DNA damage is defective in the absence of the Nup84 sub-complex.
A unique irreparable HO-induced DSB has been shown to re-localise to the nuclear periphery in a Mec1- and Nup84-dependent manner, and this NPC localisation to rely on physical interaction between the Slx5/Slx8 SUMO-targeted ubiquitin ligases and the Nup84 sub-complex (19). Further work inferred a role for the inner nuclear membrane protein Mps3, the HR factor Rad51 and SUMO-modified histone variant H2A.Z in this process (49,50). Similarly, eroded telomeres undergoing replication repair were shown to localise at nuclear pores (38). In subtelomeric regions mutations of the Nup84 sub-complex were shown to decrease the repair efficiency of induced DSBs (51), while proximity to the nuclear periphery favours the repair of induced DSBs by break induced replication (52). These data suggest that the Nup84 sub-complex plays a role in the regulation of DSB repair in sub-telomeric regions and may act as a docking site for the repair of persistent, particularly harmful, artificially induced DSBs. Interestingly, using both a system in which a specific single-stranded break is induced at an inefficient HO cleavage site (33) and an SCR-specific recombination assay (28), our results show that the absence of Nup84 leads to a severe defect in the repair of replication-born DSBs by SCR (see Figure 5 and Supplementary Figure S6). This SCR deficiency could be due to defects in the choice of the repair template during HR in nup84 cells, in which non-sister recombination would be erroneously favoured as has been proposed for cells deficient in histone H3K56 acetylation (13) but it could also reflects a more efficient repair capability of DNA breaks re-located in proximity to the nuclear pore, as described for the recombinational repair of heterochromatin in Drosophila cells (53). Interestingly, strong SCR defects were observed in a null mutation of the Wss1 metalloprotease (13), which can act both as SUMO ligase and DNA-dependent protease and shows genetic interactions with Slx5 and Slx8 (54–56). In addition, proper SUMO homeostasis has been recently proposed to inhibit alternative pathways for the repair of protein-DNA crosslinks (57). Altogether, these findings open the possibility that the Nup84 sub-complex might be required for the regulation of protein activities acting in DSB repair by SCR, rather than being directly involved in the mechanism of DSB repair. Our findings that artificial NPC tethering of Ulp1 partially suppresses the strong SCR defects of nup84 cells (see Supplementary Figure S6) further supports this view.
Analysing alternative replication-associated pathways, we have uncovered that deletion of NUP84 alleviate the sensitivity of PRR mutants rad5 and rad18 to genotoxic insult (see Figure 6), suggesting that blocked RFs might be channelled to other repair pathways in the absence of Nup84. This effect may conceivably be mediated through a misregulation of post-transcriptional protein modifications, which participate in the regulation of replication progression. PCNA sumoylation occurs both during normal unperturbed DNA replication and in response to DNA damage, and PRR pathways are controlled by ubiquitylation of PCNA (7,8). Indeed, the pattern of PCNA post-translational modifications is altered in nup84 as compared to wild-type cells (Figure 6), consistent with an accumulation of endogenous damage in untreated cells and alterations in the cellular response to UV irradiation.
Interestingly, it has been proposed that damaged RFs might relocate to the NPC, as an early ARS treated with HU and MMS moves to the nuclear periphery (19). In the same line, expanded CAG repeats were shown to localize transiently to the nuclear periphery in replicating cells and the absence of Nup84 to lead to triplet repeat instability (37). Consistent with the idea that PRR is partially bypassed in the absence of Nup84 in favor of alternative repair pathways, nup84 rad18 double mutants show increased recombination frequency, Rad52-foci accumulation and GCR rate than either one of the single mutants (see Figure 6). The low repair efficiency of spontaneous DNA lesions in nup84 cells would explain the negative impact on RF progression. The conversion of such lesions into DNA breaks after RF breakage and a diminished DSB repair efficiency may explain their channelling away of PRR. The finding that NUP84 deletion increases GCR events (see Figure 6 and (19)) is consistent with this view.
The results of this study allow us to propose a model in which the Nup84 sub-complex acts as a platform to coordinate the DDR throughout the cell cycle and to ensure proper replication through damaged DNA (Figure 7). In non-replicating cells, Nup84-dependent activity of the Ulp1 SUMO protease would ensure efficient NER. In replicating cells, the Nup84 sub-complex would coordinate the progression of RFs with DNA repair mechanisms, playing a key role in the maintenance of genome integrity. In the absence of Nup84, cell viability would be reduced in response to genotoxic treatments and GCR increased as a consequence from the sealing of DSBs by a non-HR mechanism. Although further work will be necessary to decipher the specific functions of the Nup84 sub-complex in DNA replication and repair, this study reveals its central role in the DDR and opens perspectives to understand the mechanisms enabling the coordination between different processes and their relevance in the maintenance of genome stability in eukaryotes.
Figure 7.

Model for the role of Nup84 in the DNA damage response. In non-replicating cells, Nup84 is important for efficient repair by nucleotide excision repair (NER). Part of this function is mediated by the correct localization of the Ulp1 SUMO protease at the nuclear pore. In replicating cells, Nup84 is required for the appropriate regulation of homologous recombination and DNA damage tolerance pathway therewith ensuring error-free repair by SCR and replication progression.
DATA AVAILABILITY
Expression data can be accessed at Gene Expression Omnibus under accession number GSE113118.
Supplementary Material
ACKNOWLEDGEMENTS
We thank B. Palancade, H. Ulrich, E. Johnson, R. Rothstein, E. Schwob and F. Winston for gently providing reagents and R. Wellinger for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council [ERC2014 AdG669898 TARLOOP to A.A.]; Spanish Ministry of Economy and Competitiveness [BFU2016-75058-P to A.A.]; Junta de Andalucía [PA12-BIO1238 to A.A.]; European Union Regional Funds (FEDER). Funding for open access charge: European Research Council [ERC2014 AdG669898 TARLOOP]; Spanish Ministry of Economy and Competitiveness [BFU2016-75058-P].
Conflict of interest statement. None declared.
REFERENCES
- 1. Giglia-Mari G., Zotter A., Vermeulen W.. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011; 3:a000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prakash S., Prakash L.. Nucleotide excision repair in yeast. Mutat. Res. 2000; 451:13–24. [DOI] [PubMed] [Google Scholar]
- 4. Scharer O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013; 5:a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aguilera A., Garcia-Muse T.. Causes of genome instability. Annu. Rev. Genet. 2013; 47:1–32. [DOI] [PubMed] [Google Scholar]
- 6. Zeman M.K., Cimprich K.A.. Causes and consequences of replication stress. Nat. Cell Biol. 2014; 16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoege C., Pfander B., Moldovan G.L., Pyrowolakis G., Jentsch S.. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002; 419:135–141. [DOI] [PubMed] [Google Scholar]
- 8. Stelter P., Ulrich H.D.. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003; 425:188–191. [DOI] [PubMed] [Google Scholar]
- 9. Jasin M., Rothstein R.. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013; 5:a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heyer W.D., Ehmsen K.T., Liu J.. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010; 44:113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krogh B.O., Symington L.S.. Recombination proteins in yeast. Annu. Rev. Genet. 2004; 38:233–271. [DOI] [PubMed] [Google Scholar]
- 12. De Piccoli G., Cortes-Ledesma F., Ira G., Torres-Rosell J., Uhle S., Farmer S., Hwang J.Y., Machin F., Ceschia A., McAleenan A. et al.. Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat. Cell Biol. 2006; 8:1032–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Munoz-Galvan S., Jimeno S., Rothstein R., Aguilera A.. Histone H3K56 acetylation, Rad52, and non-DNA repair factors control double-strand break repair choice with the sister chromatid. PLoS Genet. 2013; 9:e1003237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Munoz-Galvan S., Garcia-Rubio M., Ortega P., Ruiz J.F., Jimeno S., Pardo B., Gomez-Gonzalez B., Aguilera A.. A new role for Rrm3 in repair of replication-born DNA breakage by sister chromatid recombination. PLoS Genet. 2017; 13:e1006781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knockenhauer K.E., Schwartz T.U.. The nuclear pore complex as a flexible and dynamic gate. Cell. 2016; 164:1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alber F., Dokudovskaya S., Veenhoff L.M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B.T. et al.. The molecular architecture of the nuclear pore complex. Nature. 2007; 450:695–701. [DOI] [PubMed] [Google Scholar]
- 17. Ptak C., Wozniak R.W.. Nucleoporins and chromatin metabolism. Curr. Opin. Cell Biol. 2016; 40:153–160. [DOI] [PubMed] [Google Scholar]
- 18. Geli V., Lisby M.. Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. Bioessays. 2015; 37:1287–1292. [DOI] [PubMed] [Google Scholar]
- 19. Nagai S., Dubrana K., Tsai-Pflugfelder M., Davidson M.B., Roberts T.M., Brown G.W., Varela E., Hediger F., Gasser S.M., Krogan N.J.. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science. 2008; 322:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bennett C.B., Lewis L.K., Karthikeyan G., Lobachev K.S., Jin Y.H., Sterling J.F., Snipe J.R., Resnick M.A.. Genes required for ionizing radiation resistance in yeast. Nat. Genet. 2001; 29:426–434. [DOI] [PubMed] [Google Scholar]
- 21. Chang M., Bellaoui M., Boone C., Brown G.W.. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:16934–16939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palancade B., Liu X., Garcia-Rubio M., Aguilera A., Zhao X., Doye V.. Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Mol. Biol. Cell. 2007; 18:2912–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pan X., Ye P., Yuan D.S., Wang X., Bader J.S., Boeke J.D.. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006; 124:1069–1081. [DOI] [PubMed] [Google Scholar]
- 24. Loeillet S., Palancade B., Cartron M., Thierry A., Richard G.F., Dujon B., Doye V., Nicolas A.. Genetic network interactions among replication, repair and nuclear pore deficiencies in yeast. DNA Repair (Amst). 2005; 4:459–468. [DOI] [PubMed] [Google Scholar]
- 25. Silver H.R., Nissley J.A., Reed S.H., Hou Y.M., Johnson E.S.. A role for SUMO in nucleotide excision repair. DNA Repair (Amst.). 2011; 10:1243–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moriel-Carretero M., Aguilera A.. A postincision-deficient TFIIH causes replication fork breakage and uncovers alternative Rad51- or Pol32-mediated restart mechanisms. Mol. Cell. 2010; 37:690–701. [DOI] [PubMed] [Google Scholar]
- 27. Putnam C.D., Kolodner R.D.. Determination of gross chromosomal rearrangement rates. Cold Spring Harb. Protoc. 2010; 2010:pdb.prot5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fasullo M.T., Davis R.W.. Recombinational substrates designed to study recombination between unique and repetitive sequences in vivo. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:6215–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaillard H., Wellinger R.E., Aguilera A.. Methods to study transcription-coupled repair in chromatin. Methods Mol. Biol. 2015; 1288:273–288. [DOI] [PubMed] [Google Scholar]
- 30. Dandjinou A.T., Larrivee M., Wellinger R.E., Wellinger R.J.. Two-dimensional agarose gel analysis of DNA replication intermediates. Methods Mol. Biol. 2006; 313:193–208. [DOI] [PubMed] [Google Scholar]
- 31. Gaillard H., Aguilera A.. Cleavage factor I links transcription termination to DNA damage response and genome integrity maintenance in Saccharomyces cerevisiae. PLoS Genet. 2014; 10:e1004203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hecht A., Grunstein M.. Mapping DNA interaction sites of chromosomal proteins using immunoprecipitation and polymerase chain reaction. Methods Enzymol. 1999; 304:399–414. [DOI] [PubMed] [Google Scholar]
- 33. Gonzalez-Barrera S., Cortes-Ledesma F., Wellinger R.E., Aguilera A.. Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol. Cell. 2003; 11:1661–1671. [DOI] [PubMed] [Google Scholar]
- 34. Gonzalez-Barrera S., Garcia-Rubio M., Aguilera A.. Transcription and double-strand breaks induce similar mitotic recombination events in Saccharomyces cerevisiae. Genetics. 2002; 162:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davies A.A., Ulrich H.D.. Detection of PCNA modifications in Saccharomyces cerevisiae. Methods Mol. Biol. 2012; 920:543–567. [DOI] [PubMed] [Google Scholar]
- 36. Gomez-Gonzalez B., Garcia-Rubio M., Bermejo R., Gaillard H., Shirahige K., Marin A., Foiani M., Aguilera A.. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 2011; 30:3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Su X.A., Dion V., Gasser S.M., Freudenreich C.H.. Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev. 2015; 29:1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khadaroo B., Teixeira M.T., Luciano P., Eckert-Boulet N., Germann S.M., Simon M.N., Gallina I., Abdallah P., Gilson E., Geli V. et al.. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol. 2009; 11:980–987. [DOI] [PubMed] [Google Scholar]
- 39. Tous C., Rondon A.G., Garcia-Rubio M., Gonzalez-Aguilera C., Luna R., Aguilera A.. A novel assay identifies transcript elongation roles for the Nup84 complex and RNA processing factors. EMBO J. 2011; 30:1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Psakhye I., Jentsch S.. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell. 2012; 151:807–820. [DOI] [PubMed] [Google Scholar]
- 41. Texari L., Dieppois G., Vinciguerra P., Contreras M.P., Groner A., Letourneau A., Stutz F.. The nuclear pore regulates GAL1 gene transcription by controlling the localization of the SUMO protease Ulp1. Mol. Cell. 2013; 51:807–818. [DOI] [PubMed] [Google Scholar]
- 42. Garcia-Benitez F., Gaillard H., Aguilera A.. Physical proximity of chromatin to nuclear pores prevents harmful R loop accumulation contributing to maintain genome stability. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:10942–10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Herrera-Moyano E., Mergui X., Garcia-Rubio M.L., Barroso S., Aguilera A.. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014; 28:735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kadyk L.C., Hartwell L.H.. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics. 1992; 132:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cortes-Ledesma F., Aguilera A.. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep. 2006; 7:919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lisby M., Rothstein R., Mortensen U.H.. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:8276–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Belanger F., Angers J.P., Fortier E., Hammond-Martel I., Costantino S., Drobetsky E., Wurtele H.. Mutations in replicative stress response pathways are associated with S Phase-specific defects in nucleotide excision repair. J. Biol. Chem. 2016; 291:522–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Auclair Y., Rouget R., Affar el B., Drobetsky E.A.. ATR kinase is required for global genomic nucleotide excision repair exclusively during S phase in human cells. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:17896–17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kalocsay M., Hiller N.J., Jentsch S.. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell. 2009; 33:335–343. [DOI] [PubMed] [Google Scholar]
- 50. Oza P., Jaspersen S.L., Miele A., Dekker J., Peterson C.L.. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009; 23:912–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Therizols P., Fairhead C., Cabal G.G., Genovesio A., Olivo-Marin J.C., Dujon B., Fabre E.. Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. J. Cell Biol. 2006; 172:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chung D.K., Chan J.N., Strecker J., Zhang W., Ebrahimi-Ardebili S., Lu T., Abraham K.J., Durocher D., Mekhail K.. Perinuclear tethers license telomeric DSBs for a broad kinesin- and NPC-dependent DNA repair process. Nat. Commun. 2015; 6:7742. [DOI] [PubMed] [Google Scholar]
- 53. Ryu T., Spatola B., Delabaere L., Bowlin K., Hopp H., Kunitake R., Karpen G.H., Chiolo I.. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015; 17:1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stingele J., Schwarz M.S., Bloemeke N., Wolf P.G., Jentsch S.. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 2014; 158:327–338. [DOI] [PubMed] [Google Scholar]
- 55. Balakirev M.Y., Mullally J.E., Favier A., Assard N., Sulpice E., Lindsey D.F., Rulina A.V., Gidrol X., Wilkinson K.D.. Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic SUMO conjugates. Elife. 2015; 4:e06763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mullen J.R., Chen C.F., Brill S.J.. Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligase. Mol. Cell Biol. 2010; 30:3737–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sharma P., Mullen J.R., Li M., Zaratiegui M., Bunting S.F., Brill S.J.. A lysine desert protects a novel domain in the Slx5-Slx8 SUMO targeted Ub ligase to maintain sumoylation levels in saccharomyces cerevisiae. Genetics. 2017; 206:1807–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Expression data can be accessed at Gene Expression Omnibus under accession number GSE113118.