

Abstract
Aims: Epidemiologic evidence indicates that diabetes may increase risk of breast cancer (BC) and mortality in patients with cancer. The pathophysiological relationships between diabetes and cancer are not fully understood, and personalized treatments for diabetes-associated BC are urgently needed.
Results: We observed that high glucose (HG), via activation of nuclear phosphatase PP2Cδ, suppresses p53 function, and consequently promotes BC cell proliferation, migration, and invasion. PP2Cδ expression is higher in tumor tissues from BC patients with hyperglycemia than those with normoglycemia. The mechanisms underlying HG stimulation of PP2Cδ involve classical/novel protein kinase-C (PKC) activation and GSK3β phosphorylation. Reactive oxygen species (ROS)/NF-κB pathway also mediates HG induction of PP2Cδ. Furthermore, we identified a 1,5-diheteroarylpenta-1,4-dien-3-one (Compound 23, or C23) as a novel potent PP2Cδ inhibitor with a striking cytotoxicity on MCF-7 cells through cell-based screening assay for growth inhibition and activity of a group of curcumin mimics. Beside directly inhibiting PP2Cδ activity, C23 blocks HG induction of PP2Cδ expression via heat shock protein 27 (HSP27) induction and subsequent ablation of ROS/NF-κB activation. C23 can thus significantly block HG-triggered inhibition of p53 activity, leading to the inhibition of cancer cell proliferation, migration, and invasion. In addition, hyperglycemia promotes BC development in diabetic nude mice, and C23 inhibits the xenografted BC tumor growth.
Conclusions and Innovation: Our findings elucidate mechanisms that may have contributed to diabetes-associated BC progression, and provide the first evidence to support the possible alternative therapeutic approach to BC patients with diabetes.
Keywords: diabetes, high glucose, breast cancer, PP2Cδ, p53, GSK3β, NF-κB
Innovation.
Diabetes increases mortality in patients with breast cancer (BC). We demonstrate for the first time that PP2Cδ mediates high glucose (HG)-stimulated BC progression. The mechanisms underlying HG induction of PP2Cδ involve protein kinase-C (PKC)/GSK3β and reactive oxygen species (ROS)/NF-κB pathways. Moreover, we identified a novel PP2Cδ inhibitor 1,5-diheteroarylpenta-1,4-dien-3-one or Compound 23 (C23), which not only inhibits PP2Cδ activity but also functions as an antioxidant that induces heat shock protein 27 (HSP27) and ablates ROS/NF-κB activation, causing suppression of HG-induced PP2Cδ expression. C23 significantly inhibits the growth of xenografted breast tumor in diabetic mice. We propose that C23 may serve as a unique therapeutic agent for the personalized treatment of BC patients with diabetes.
Introduction
Breast cancer (BC) has a high incidence worldwide. Epidemiologic evidence suggests that women with diabetes have increased risk of BC (7). Moreover, 5-year mortality rates are notably higher in patients with BC and diabetes versus their nondiabetic counterparts (26). Pre-existing diabetes is also related to more advanced stage at presentation (38). In addition, there exist some important distinctions between the BC patients with and without diabetes in the regimen selection and outcomes of cancer therapy (38). Diabetes and cancer share many risk factors, but potential biologic links between the two diseases are not fully understood, and personalized treatments for diabetes-associated BC are therefore urgently needed.
Previous studies (20) suggest that elevated fasting blood glucose instead of insulin is associated with increased risk of breast or colorectal cancer. Accordingly, several studies have substantiated the link between high-normal blood glucose levels and augmented BC risk. Nine of 12 separate studies that explored blood glucose levels in relationship to BC incidence (6, 14, 15, 18, 23–25, 31, 36, 41, 51, 55) indicated an association of higher fasting glucose and poor glycemic control with elevated cancer risks (6, 14, 24, 41). In addition, various evidence demonstrates a robust relationship between blood glucose levels and tumor growth in vivo. For example, calorie restriction-induced decrease in blood glucose in tumor-bearing animals might be responsible for the strikingly extended survival as compared with normal-fed controls (49). Intriguingly, Koroljow employed a hypoglycemic coma induced by insulin to successfully suppress the metastatic tumors in two patients (22). Hyperglycemia can, instead, be used as an indicator of poor survival in patients with a number of cancers (11). One possible mechanism is that hyperglycemia in diabetic patients may provide a favorable environment for the growth and survival of BC cells (3), since the neoplastic cells could use glucose for proliferation via the pentose phosphate pathway (10). In addition, hyperglycemia was reported to confer resistance to chemotherapy in malignant BC cells (56). However, the accurate mechanism(s) remains unknown.
The serine–threonine protein phosphatase PP2Cδ (also known as WIP1 or PPM1D) is a nuclear-type 2C protein phosphatase (PP2C) that is overexpressed and amplified in many types of cancers, including BC and ovarian clear cell adenocarcinoma (29). Upon DNA damage, its transcription is induced in a p53-dependent manner. It dephosphorylates and inactivates several proteins critical for cellular stress responses, including p38 MAPK (50), Chk1 (28), Chk2 (35), and p53 (28). PP2Cδ has been demonstrated to have clear oncogenic properties and to play an important role in tumorigenesis, tumor development and progression (8). Aberrant activation of PP2Cδ is believed to inactivate p53 and RB pathways, leading to stimulation of cell cycle and tumorigenesis (42). Therefore, PP2Cδ is an attractive drug target for the treatment of cancers, and inhibition of its expression or activity could constitute an important new strategy for therapeutic intervention to halt the progression of various cancers.
Here, we demonstrate that PP2Cδ activation plays a role in enhancing the effects of high glucose (HG) on aggressive phenotypes of BC cells. The mechanisms underlying HG stimulation of PP2Cδ involve classical/novel protein kinase-C (PKC)/GSK3β and reactive oxygen species (ROS)/NF-κB pathways. In addition, we identified a novel PP2Cδ inhibitor, 1, 5-diheteroarylpenta-1,4-dien-3-one, or Compound 23 (C23), which not only directly inhibits PP2Cδ activity but also suppresses HG-induced PP2Cδ expression. Using streptozotocin (STZ)-induced diabetic nude mice bearing MCF-7 cells as an animal model, we found that hyperglycemia promoted the development of BC in vivo, and that C23 significantly inhibited the growth of xenografted breast tumor. Our novel results demonstrate for the first time that PP2Cδ plays an important role in the HG-stimulated progression of BC, and C23 may serve as an effective therapeutic agent for the treatment of BC patients with diabetes.
Results
HG induces PP2Cδ, impairs p53 activity, and promotes MCF-7 cell proliferation, invasion, and migration
HG is a risk factor for BC and can promote cancer cell proliferation (34). The tumor suppressor gene p53 is involved in hereditary as well as sporadic BC development and therapeutic responses (48). To determine if HG affects p53, we treated transformed (MCF-7) and normal (MCF-12A) breast epithelial cells with 22.5 mM glucose for up to 48 h and examined p53 acetylation levels. As indicated in Figure 1A and C and Supplementary Figure S1, HG treatment reduced p53 acetylation levels, which are important for their function. To further elucidate the mechanisms underlying HG inhibition of p53, we tested an important nuclear serine–threonine protein phosphatase PP2Cδ (33), which is frequently activated through amplification in primary breast tumors (42). Intriguingly, the decreased p53 acetylation coincided with augmented PP2Cδ protein levels. A similar phenomenon was also observed in ZR-75 cells (p53 wild type) but not in MDA-MB-231 and T-47D cells (Supplementary Figs. S2, S16). A dose–response study in MCF-7 (Fig. 1B, D and Supplementary Fig. S1) shows that decreased p53 acetylation levels were clearly detected with as little as 11 mM glucose, with the lowest levels for 22.5 mM glucose treatment. Thus, 22.5 mM glucose will be adopted in the future studies. To determine whether PP2Cδ expression is induced at mRNA levels in response to HG, we performed qRT-PCR (Fig. 1E). HG significantly promotes PP2Cδ mRNA expression with a maximal 2.5-fold induction as compared with untreated MCF-7 cells.
FIG. 1.

The effects of high glucose on PP2Cδ expression, p53 activity and cell proliferation, migration, and invasion. MCF-7 (A) and normal MCF-12A (C) cells were treated with high glucose (22.5 mM) for 0–48 h with a change of fresh medium every 24 h. In a dose–response study, MCF-7 (B) and MCF-12A (D) cells were treated with different concentrations of glucose as indicated for 24 h. The cells were then lysed. Nuclear fractions were prepared and underwent SDS-PAGE followed by Western blotting with various primary antibodies as shown, then were visualized by the enzyme-linked chemiluminescence system. The bar graphs above are densitometry analyses of the bands. The total amount of TBP is shown for equal loading. Data presented are mean ± SD from three independent experiments, with nontreated controls set as 1. Uncropped blots are shown in Supplementary Figure S1. *p < 0.05 versus control. (E) Induction of PP2Cδ mRNA expression in MCF-7 cells treated with HG analyzed by qRT-PCR. All mRNA were normalized to PUM1 and presented as fold (mean ± SD) over control based on three experiments. *p < 0.05 versus control. (F) MCF-7 cells were transiently transfected with 0.5 μg of pG13-LUC reporter plasmid. About 6 h after transfection, cells were treated HG alone or pretreated with UV for 0.5 h followed by HG treatment. Luciferase activity was determined from the transfected cell extracts. Values (mean ± SD) are expressed as fold over untreated control. *p < 0.05, #p < 0.05 versus their corresponding control groups. (G) MTT cell proliferation assays illustrating the effects of HG on fold cell proliferation compared with a NG condition in MCF-7 cells. *p < 0.05 versus control (n = 3). (H) Wound-healing assay of MCF-7 cells treated with HG. MCF-7 cells were seeded in migration chambers and treated with 22.5 mM HG. Images of the migration area were photographed 0, 24, and 48 h after removal of the migration chamber under a phase contrast microscope. The percentage of the wound closed was quantified from three independent replicates and is expressed as mean ± SD. (I) In vitro invasion assay performed on MCF-7 cells that were treated with HG for 24 and 48 h. Each column (right) represents the mean (±SD) results of three independent experiments. *p < 0.05, versus their corresponding control groups. HG, high glucose; NG, normal glucose; PP2C, type 2C protein phosphatase; PUM1, pumilio RNA-binding family member 1; TBP, TATA-binding protein. Color images are available online.
To further substantiate that HG inhibits p53 function, we performed PG13-luc p53 reporter gene assays, demonstrating that HG suppressed p53 transcriptional activity under basal and UV-stimulated conditions (Fig. 1F). To examine the effects of HG on cancer cell growth, MCF-7 cells were treated with 22.5 mM glucose for 24 and 48 h before the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The results revealed that prolonged HG exposure (for 48 h) promoted MCF-7 cell proliferation (Fig. 1G). We then investigated the effects of HG on MCF-7 cell migration and invasion by performing wound-healing and transwell assays, which indicated that treatment with HG for 24 and 48 h significantly enhanced cancer cell migration and invasion (Fig. 1H, I).
GSK3β and NF-κB are involved in HG induction of PP2Cδ
Next, we sought to determine the mechanism on how HG induces PP2Cδ expression. Glycogen synthase kinase 3 (GSK3) is important in a number of central intracellular signaling pathways such as glucose regulation, and involves in diverse cellular processes including cellular proliferation, migration, inflammation, and survival (46). NF-κB is a transcription factor, playing crucial roles in inflammation, cell proliferation and survival (4). Hence, we investigated the role that GSK3β and NF-κB serve in HG-induced BC cell proliferation. HG treatment led to increased phosphorylation of GSK3β at Ser 9, which resulted in GSK3β inactivation and NF-κB activation in a time- and dose-dependent manner (Fig. 2A, B and Supplementary Fig. S3). Furthermore, inhibition of GSK3β with a specific inhibitor BIO and 1-Azakenpaullone significantly increased PP2Cδ expression (Fig. 2C and Supplementary Figs. S3, S4, and S17), suggesting that GSK3β is a negative regulator of PP2Cδ. Given that HG induced PP2Cδ accompanied by inactivation of GSK3β, we hypothesize that GSK3β, at least partially, mediates HG induction of PP2Cδ. In addition, inhibition of NF-κB with the chemical inhibitor of IKKβ, IMD-0354, and parthenolide partially blocked HG-induced increase in PP2Cδ protein levels (Fig. 2C and Supplementary Figs. S3, S4, and S17), implying that NF-κB also partially mediates PP2Cδ induction by HG. This result is consistent with previous studies that NF-κB may promote tumorigenesis by positively regulating PP2Cδ (27). Taken together, we demonstrate that GSK3β and NF-κB are involved in the regulation of PP2Cδ induced by HG.
FIG. 2.

HG increases PP2Cδ protein levels partially via classical/novel PKC activation, suppression of GSK3β, and activation of NF-κB in cultured MCF-7 cells. MCF-7 cells were treated with (A) HG (22.5 mM) for the times indicated and (B), various concentrations of HG for 24 h. The cells were then lysed, and 100 μg of the cell lysates underwent SDS-PAGE followed by Western blotting with various primary antibodies as shown. NF-κB activation was analyzed by measuring inhibitory κB-α factor (IκB-α) degradation and subsequent translocation of NF-κB (p65) to the nucleus using Western blot assays. (C) Western blot of nuclear lysates from MCF-7 cells treated with BIO (2.5 μM) or IMD-0354 (2 μM) in the presence or absence of HG for 24 h. Antibodies against PP2Cδ and TBP for endogenous control were used. In separate sets of experiments shown in (D), 2 μg of protein extracts underwent GSK3β activity assays. The data represent mean ± SD from three separate experiments. *p < 0.05 versus control; #p < 0.05 versus HG. (E) MCF-7 cells were treated with 10 μM H89, 10 μM Ro31-8220, and 10 μM LY294002 in the presence of HG for 24 h. Whole-cell extracts were prepared and analyzed by Western blotting with antibodies against GSK3β and phosphor-GSK3β. (F) MCF-7 cells were transfected with Control siRNA, PKA Cα siRNA, or Akt siRNA for 24 h followed by incubation with HG for 24 h. (G) MCF-7 cells stably expressing Control shRNA or PKC shRNA were incubated with HG for 24 h. (H) Cells were treated with a variety of PKC inhibitors with differing specificities, namely 2 μM GF 109203X, 2 μM Go6976, 20 μM Rottlerin, and 50 μM ATM, in the presence of HG for 24 h. GSK3β and phosphor-GSK3β protein expression were analyzed by immunoblotting. Uncropped blots are shown in Supplementary Figure S3. *p < 0.05 versus control; #p < 0.05 versus HG. ATM, aurothiomalate; DMSO, dimethyl sulfoxide; GSK3β, glycogen synthase kinase 3; PKC, protein kinase-C.
PKC activation mediates HG-induced GSK3β phosphorylation and inactivation
We next explored the underlying mechanisms on how HG affects GSK3β. GSK-3β can be phosphorylated on serine 9 catalyzed by a number of kinases such as PKA, PKC, and Akt (19). To identify the specific kinases that can catalyze the GSK-3 phosphorylation stimulated by HG, we investigated the effect of different kinase inhibitors (H89, Ro31–8220, and LY294002) on phospho-GSK-3 levels in MCF-7 cells. The data showed that inhibition of Akt by the inhibitor LY294002 or of PKA by H89 did not reduce HG-induced phosphorylation of GSK-3β. It is thus concluded that Akt and PKA are not the primary GSK-3 kinases responsible for HG-induced GSK-3 phosphorylation. On the contrary, Ro31–8220, a global PKC inhibitor, abolished HG-stimulated GSK-3 phosphorylation (Fig. 2E and Supplementary Fig. S4), suggesting PKC as a critical GSK-3β kinase. Moreover, siRNA knockdown of Akt or PKA did not affect HG-induced phosphorylation of GSK-3β (Fig. 2F and Supplementary Fig. S4), whereas shRNA knockdown of PKC eliminated HG-stimulated GSK-3 phosphorylation (Fig. 2G and Supplementary Fig. S4), further confirming that PKC is the critical kinase essential to GSK-3 phosphorylation elicited by HG.
The above-mentioned data spurred us to further identify the specific PKC isoforms involved in phospho-GSK-3β in response to enhanced glucose metabolism. Specifically, MCF-7 cells were treated with a variety of PKC inhibitors with differing specificities, namely GF 109203X, selective for α and β1 isoforms; Go6976, specific for PKCα; rottlerin, specific for PKCδ; and aurothiomalate (ATM), a small molecule inhibitor of atypical PKC (aPKC). With the exception of ATM, which inhibits mainly aPKC (9), each elicited at least partial repression of GSK-3β phosphorylation, implicating that conventional and novel PKC isoforms (cPKC/nPKC) instead of aPKC may play primary roles in phosphorylation of GSK-3 in response to HG (Fig. 2H and Supplementary Fig. S4).
We then evaluate whether enhanced PKC activity in breast epithelial cells is required for PP2Cδ induction, antiapoptotic glucose signaling, and increased survival by examining the effect of PKC inhibition on PP2Cδ and cell death upon UV damage. As shown in Supplementary Figure S5A, B and S18, PKC inhibition by inhibitor Ro31 partially suppressed HG-triggered PP2Cδ induction under both basal and UV exposure conditions. In addition, HG significantly ablated UV-induced apoptosis, and inhibition of either NF-κB or PKC partially abolished the protective effect of HG on UV-induced apoptosis, as measured by flow cytometer and cleaved caspase 3 assays (Supplementary Figs. S5C, D and S18).
HG-induced ROS generation is involved in NF-κB activation and PP2Cδ induction
Our previous data indicated that in MCF-7 cells, HG treatment elevated NF-κB activity in a time- and dose-dependent manner. Furthermore, inhibition of NF-κB with its chemical inhibitors partially blocked HG-induced increase in PP2Cδ protein levels, suggesting that NF-κB also partially mediates PP2Cδ induction by HG. These results are consistent with those reported in the previous studies, in which NF-κB was suggested to promote tumorigenesis by upregulating PP2Cδ (27). Next, we extended our analysis to determine whether ROS mediates HG-induced NF-κB activation. MCF-7 cells were treated with HG for 24 h in the presence or absence of structurally different cell-permeable antioxidants and ROS scavengers, dimethylthiourea (DMTU), catalase, and manganese(III) tetrakis (4-benzoic acid) porphyrin (MnTBAP). Intracellular ROS was assessed by the assay of carbonyl groups in proteins (Fig. 3A) and the fluorescence intensity of dichlorofluorescein (DCF) emission (Supplementary Fig. S6). Consistent with a previous study where ROS was shown to be a crucial mechanism of PP2Cδ activation (12), we observed greater ROS production, NF-κB and PP2Cδ activation in MCF-7 cells treated with HG than those in the control group (Fig. 3A, C and Supplementary Fig. S9). The ROS scavengers significantly blocked HG-induced ROS formation and partially offset HG induction of NF-κB and PP2Cδ, suggesting that ROS and NF-κB activation at least partly mediate HG-induced PP2Cδ activation. Next, to substantiate the results obtained from DCF, we further examined intracellular H2O2 levels by using the fluorescent sensor HyPer-3 with a high degree of sensitivity and specificity. The source of cellular ROS production induced by HG was further examined by inhibition of NADPH oxidase (NOX) and superoxide dismutase (SOD). We found that MCF-7 cells produced a significant amount of H2O2 when treated with HG. HG-induced H2O2 production was repressed with diphenyleneiodonium chloride (DPI), a selective inhibitor of NOXes and diethyldithiocarbamic acid (DDC), an SOD inhibitor (Fig. 3B), suggesting that the H2O2 source is NOX enzymes and superoxide (O2•−). We reason that HG, via activation of NOX, induces intracellular generation of O2•−, which is catalytically converted to H2O2 by SOD. H2O2 is more detrimental than O2•− owing to its longer biological life span and ability to diffuse across lipid bilayers (37). In other words, O2•− signal can be preserved by dismutation into H2O2, which can hence accumulate and act as an intracellular messenger molecule in the response to HG.
FIG. 3.

HG increases PP2Cδ protein levels partially via ROS induction and activation of NF-κB in cultured MCF-7 cells. (A) MCF-7 cells were treated with HG in the presence or absence of 10 mM DMTU, 100 μM MnTBAP, or 500 U/mL PEG-catalase for 24 h. The intracellular ROS was detected by the assay of carbonyl groups in proteins. (B) Fluorescence image (488-nm excitation) of MCF-7 cells expressing HyPer-3. HyPer-3-expressing MCF-7 cells were exposed to 100 μM H2O2 (positive control) for 1 min, or HG (22 mM) in the presence of DPI (5 μM), or DDC (1 mM) for 24 h. Intracellular H2O2 were detected under imaging system of HyPer-3. The scale bar denotes 20 μm. Right panel indicates F488/F404 ratio. *p < 0.05 versus control; #p < 0.05 versus HG. (C) p-Ser536-p65, p65, PP2Cδ, and actin were detected by Western blotting with specific antibodies. (D) MCF-7 cells were treated with HG or 22.5 mM of 3-O-methyl-D-glucose, and HG in the presence of 22.5 mM 2-DG or 10 μM cytochalasin B. p-Ser536-p65, p65, PP2Cδ, and actin were detected by Western blotting with specific antibodies. The data represent mean ± SD from three separate experiments. Uncropped blots are shown in Supplementary Figure S6. *p < 0.05 versus control; #p < 0.05 versus HG. DDC, diethyldithiocarbamic acid; DMTU, dimethylthiourea; DPI, diphenyleneiodonium chloride; MnTBAP, manganese(III) tetrakis (4-benzoic acid) porphyrin; ROS, reactive oxygen species. Color images are available online.
At this point, we tried to determine that glucose metabolism, but not glucose autooxidation, is necessary for HG-induced ROS generation and NF-κB activation. We added 2-DG, an inhibitor of glucose metabolism, 3-O-methyl-D-glucose, an autooxidizable glucose analog that enters into the cell but is not metabolized, or cytochalasin B, a glucose transporter inhibitor, to the media to observe if they can induce intracellular ROS and NF-κB activation. Our results showed that HG instead of 3-O-methyl-D-glucose induced NF-κB and PP2Cδ activation, and that 2-DG and cytochalasin B partially abolished HG induction of NF-κB and PP2Cδ (Fig. 3D and Supplementary Fig. S9). Overall, these studies lead to the identification of the roles of ROS in HG-induced NF-κB and PP2Cδ activation, and provide important clues on the influences of ROS on central cellular processes such as proliferation and apoptosis, which are implicated in the development of cancer.
PP2Cδ is responsible for HG-induced suppression of p53 acetylation
The tumor suppressor p53 is a crucial transcription factor in cellular stress response pathways. Acetylation of p53 is a vital revocable enzymatic process that occurs upon DNA damage and genotoxic stress, and is crucial for p53 stability, sequence-specific DNA binding, and transcriptional activity. Thus, p53 acetylation is indispensable for its activation. We therefore investigated if PP2Cδ mediates HG-induced suppression of p53 acetylation. As indicated in Figure 4A and Supplementary Figure S10, consistent with previous studies (45), UV damage dramatically increased p53 acetylation at K373. HG treatment promoted both basal and UV-induced PP2Cδ protein expression and activity, leading to impairment of p53 acetylation and transcription of its target genes p21 and Noxa (Fig. 4A–C and Supplementary Fig. S10). These effects of HG were blocked by CCT007093, a specific PP2Cδ inhibitor (43), suggesting that HG insult restrained p53 function via PP2Cδ induction.
FIG. 4.

PP2Cδ inhibition abolishes HG-induced suppression of p53 acetylation. (A) MCF-7 cells were either untreated (Mock) or subjected to DNA damage (UV) in the presence or absence of HG or CCT007093 (15 μM), and cell lysates underwent immunoblotting for each of the proteins as indicated. Uncropped blots are shown in Supplementary Figure S7. (B) PP2Cδ activity was measured by a malachite green-based assay with a synthetic phosphopeptide used as substrates as described in Materials and Methods section. Data presented are mean ± SD from three independent experiments and represented as percentage of control value. (C) The p21 and Noxa mRNA for each treatment were analyzed by RT-qPCR. All mRNA are normalized to PUM1 and presented as fold (mean ± SD) over untreated cells based on three experiments. *p < 0.05 versus control; †p < 0.05 versus HG; #p < 0.05 versus UV; $p < 0.05 versus UV+HG.
Development of C23, a curcumin-based small molecule PP2Cδ inhibitor
When overexpressed, PP2Cδ damages the fundamental tumor surveillance networks that are commonly found in various cancers. Accordingly, employing a pharmacological inhibitor of PP2Cδ has been proposed to be an innovative chemotherapeutic method to recuperate the intrinsic tumor surveillance in various cancers. Rayter et al. (43) identified a specific chemical PP2Cδ inhibitor, CCT007093. However, CCT007093 has significant off-target effects and might have limitations as a drug development candidate (43). Thus, we sought to develop a more effective and selective PP2Cδ inhibitor to achieve the anticipated chemotherapeutic potential and to circumvent off-target effects.
A group of curcumin mimics (Compounds 1–33) (Supplementary Fig. S11) have been demonstrated by Chen and coworkers as very promising anticancer agents due to their impressive cytotoxicity in a panel of cancer cell lines (47). However, the underlying mechanism of action is unexplored yet. The structural similarity between this group of compounds and CCT007093 spurred us to investigate the inhibitory ability of compounds 1–33 on PP2Cδ. Structurally, CCT007093 and compounds 1–33 have a similar central five-carbon monoketone linker and two identical aromatic terminal rings. Compounds 1–33 are structurally different from CCT007093 by possessing a unique nitrogen-containing moiety in the terminal heteroaromatic rings. Compounds 7 (C7) and 23 are able to elicit more powerful inhibition of PP2Cδ in cells (as assessed by phospho-P38 measurement) than other compounds (Fig. 5A and Supplementary Figs. S7 and S12) and CCT007093 (Fig. 6A and Supplementary Fig. S13). More interestingly, the much stronger inhibitory ability of C7 and C23 on PP2Cδ in cells is fully mirrored by their impressive cytotoxicity. The growth inhibition assay screening identified that C7 and C23 have a striking cytotoxicity on MCF-7 cells (IC50: 3.36 and 0.98 μM) but not MCF-12A cells versus CCT007093 (IC50: 511878348.2 μM) (Fig. 5B, D). Considering that MTT is a redox-sensitive probe, the MTT assay may exaggerate results obtained using redox perturbing compounds. Therefore, we further employed the trypan blue exclusion assay to substantiate the results obtained from the MTT assay. After incubation with varying concentrations of CCT, C23, and C7 for 5 days, the cells were treated with trypan blue, and the viable cells were counted (Fig. 5E). The results are consistent with those from the MTT assay (Fig. 5D), confirming the remarkable cytotoxicity of C23 against MCF-7 cells.
FIG. 5.

C23 elicits more effective inhibition of PP2Cδ than other compounds and shows impressive cytotoxicity against MCF-7 cells. (A) Proteins were extracted from MCF-7 cells that were treated with 2.5 μM different compounds (1–33) for 12 h. They were analyzed by Western blotting with antibodies against PP2Cδ or phosphor-p38 and p38. Vinculin was used as a loading control. (B) IC50 values exhibited by the test compounds 1–33 for cytotoxicity against MCF-7 cells by the MTT assay method. (C) Predicted binding modes of inhibitors to PP2Cδ. Protein surface is drawn in white. Oxygen atoms are shown in red and nitrogen atoms in blue. Carbon atoms of C23, C7, CCT007093, and protein are shown in cyan, slate, green, and white, respectively. Side chains of crucial residues in the binding site are shown as sticks and labeled. Hydrogen bonds between inhibitors and PP2Cδ are depicted in yellow dotted lines. Figures were generated by PyMol. (D) IC50 curves for MCF7 cells treated with CCT007093, C23 and C7. Percentage cell growth inhibition is plotted against the treatment concentrations. (E) Determination of the cytotoxicity by the trypan blue exclusion assay. The cells were incubated with different concentrations of compounds for 5 days, and the viable cells were counted after trypan blue staining. Data are expressed as mean ± SD from triplicates. *p < 0.05 versus control. (F) MCF-7 cells were either untreated or subjected to HG challenge in the presence or absence of CCT007093 (15 μM) or C23 (2.5 μM), and cell lysates underwent immunoblotting for the proteins as indicated. Uncropped blots are shown in Supplementary Figure S9. (G) PP2Cδ activity was measured from cells treated as (E). *p < 0.05 versus control; #p < 0.05 versus CCT007093; $p < 0.05 versus HG+CCT007093. (H) The phosphatase activity of PP2Cδ, PP2Cβ, and PP2A was measured from cells treated with C23. *p < 0.05 versus control; #p < 0.05 versus PP2Cδ. C7, Compound 7; C23, Compound 23 or 1,5-diheteroarylpenta-1,4-dien-3-one; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. Color images are available online.
FIG. 6.

C23 inhibits HG-induced cancer cell progression via PP2Cd inhibition mediated by HSP27 induction. (A) MCF-7 cells were either untreated or subjected to HG challenge in the presence or absence of CCT007093 or C23, and cell lysates underwent immunoblotting for the proteins as indicated. (B) The intracellular oxidative stress was detected by the assay of carbonyl groups in proteins. *p < 0.05 versus control; #p < 0.05 versus HG. (C) MCF-7 cells were transiently transfected with 0.5 μg of pG13-LUC reporter plasmid. About 6 h after transfection, cells were treated as (A). Luciferase activity was determined from the transfected cell extracts. Values (mean ± SD) are expressed as fold over untreated control. *p < 0.05 versus control; #p < 0.05 versus CCT007093; $p < 0.05 versus HG+CCT007093. (D) MCF-7 cells were transfected with Control siRNA or HSP27 siRNA for 24 h followed by incubation with HG or HG/C23 combination for 24 h. Cell lysates underwent immunoblotting for the proteins as indicated. Uncropped blots are shown in Supplementary Figure S10. *p < 0.05 versus control; #p < 0.05 versus HG/Control siRNA; $p < 0.05 versus control/HSP27 siRNA; ♣p < 0.05 versus HG/Comp23/Control siRNA. The intracellular oxidative stress (E) and p53 transcription activity (F) were detected by the assay of carbonyl groups in proteins and luciferase reporter gene (PG13-Luc) assay from cells treated as (D). *p < 0.05 versus control; #p < 0.05 versus HG/Control siRNA; $p < 0.05 versus HG/Comp23/Control siRNA. (G) MCF-7 cells transfected with Control siRNA or HSP27 siRNA were incubated with HG or HG/C23 combination for 48 h, and subjected to wound-healing assay. MTT cell proliferation assay (H) and in vitro invasion assay (I) were performed on MCF-7 cells treated as (G). *p < 0.05 versus control; #p < 0.05 versus HG/Control siRNA; $p < 0.05 versus HG/Comp23/Control siRNA. HSP27, heat shock protein 27. Color images are available online.
To provide insights into how C23, C7, and CCT007093 interact with the phosphatase PP2Cδ, we performed docking simulation for binding modes of these inhibitors to the PP2Cδ phosphatase domain. Our results show that they are buried into the catalytic site composed of Asp105, Arg110, Arg243, Arg258, Arg259, Gln265, Phe268, Asp314, Arg364, Asp366, and Asn367 (Fig. 5C). The carbonyl oxygen atom and the nitrogen atom on the heteroaromatic ring of C23 form two hydrogen bonds with side chains of Arg258 and Asp105, respectively. This leads to a stable binding model of C23 which is consistent with its strongest inhibition on PP2Cδ. Similarly, the nitrogen atom on the heteroaromatic ring of C7 forms a stronger hydrogen bond with the side chain of Arg259 than the carbonyl oxygen of CCT007093 with the hydrogen atom of the main chain of Asn367. Therefore, C7 can inhibit PP2Cδ more effectively than CCT007093, probably due to stronger hydrogen bonding and van der Waals force. Accordingly, C23 can exert more potent inhibition of PP2Cδ in cells, as assessed by phospho-P38 and PP2Cδ phosphatase activity measurement, than CCT007093 under basal and HG conditions (Fig. 5F, G and Supplementary Fig. S12). Next, to further elucidate C23's specificity in PP2Cδ inhibition and possible off‐target effect, we used PP2Cβ and PP2A as controls to compare effects of C23 on these phosphatases. PP2Cβ (PPM1B) and PP2Cδ are members of the protein phosphatase 2C (PP2C) family of isoenzymes, which are magnesium or manganese dependent and structurally different from PP2A protein phosphatases. Herein, we demonstrate that C23 has more potent inhibitory effect on phosphatase activity of PP2Cδ than PP2Cβ, with no effect on PP2A (Fig. 5H), suggesting its good specificity against PP2Cδ.
Compound 23, via heat shock protein 27 activation, suppresses HG-induced PP2Cδ expression, cancer cell proliferation, migration, and invasion
In addition to inhibiting the activity of PP2Cδ, C23 inhibits HG-induced PP2Cδ expression (Fig. 6A). Our data have ascertained this mechanism by showing that C23 dramatically induced protein expression of heat shock protein 27 (HSP27), which functions as an antioxidant during oxidative stress, repressing the levels of ROS by enhancing levels of intracellular glutathione and reducing the levels of iron (2). Moreover, HG-induced NF-κB activation was inhibited by C23 (Fig. 6A and Supplementary Fig. S13), accompanied by significant decrease in cellular oxidative stress (Fig. 6B). Accordingly, C23 was able to improve p53 function in cells, as assessed by p53 acetylation (Fig. 6A) and luciferase reporter gene (PG13-Luc) assay (Fig. 6C), than CCT007093 under basal and HG conditions. To further substantiate the antioxidant effect of C23, we assayed intracellular GSH levels. HG lowered the GSH levels in MCF-7 cells. C23 slightly increased GSH levels in MCF-7 cells, but the difference was not statistically significant. However, C23 restored HG-induced decrease in GSH levels (Supplementary Fig. S8).
To further substantiate the role of HSP27 in C23 suppression of HG-induced PP2Cδ expression, its expression was knocked down by using HSP27 siRNA (Fig. 6D). C23 significantly abolished HG-induced oxidative stress and PP2Cδ expression, which was abrogated by HSP27 knockdown, implicating that HSP27 plays an important role in C23 suppression of HG-induced oxidative stress and PP2Cδ expression (Fig. 6D, E and Supplementary Fig. S13). Accordingly, the rescue effects of C23 on HG-induced inhibition of p53 activity were apparently abolished by HSP27 siRNA treatment (Fig. 6D, F). Therefore, HSP27 plays an important role in C23 inhibition of HG-induced BC cell migration, proliferation, and invasion (Fig. 6G–I).
Pharmacological targeting of PP2Cδ with C23 blocks the growth of MCF-7 xenografts in diabetic nude mice
We then explored whether the PP2Cδ pathway observed in cell culture might modulate BC progression in diabetic BC xenograft nude mice models. To simulate hyperglycemic condition-induced epigenetic alterations in oncogenic pathways in cancer, a nude mouse model of STZ-induced diabetes was used in this study (16). The therapeutic efficacy of C23 was examined in these mice bearing established MCF-7 xenografts (Supplementary Fig. S14). STZ-induced hyperglycemia dramatically stimulated tumor growth in vivo. This observation is consistent with several lines of evidence showing a robust correlation between blood glucose levels and tumor growth in vivo (11, 18, 22, 49). C23 therapy significantly decreased tumor burden, as shown by end-point tumor volumes (Fig. 7A, B) and weights (Fig. 7C). In addition, immunohistochemical (IHC) staining from harvested tumors showed noteworthy induction of PP2Cδ and decrease in p53 acetylation in STZ-treated mice. C23 administration significantly abolished all these effects, accompanied by induction of HSP27 (Fig. 7D). These data suggest that PP2Cδ and p53 are responsible for the BC progression in diabetic mice. Thus, pharmacological targeting of PP2Cδ pathway may be efficacious against diabetes-associated BC in vivo.
FIG. 7.

C23 inhibits HG-induced breast cancer progression in vivo. (A) Representative images of MCF-7 xenografts isolated from mice treated with vehicle, STZ (150 mg/kg), C23 (5 mg/kg, i.p.), or STZ + C23. n = 6 mice/group; n = 2 independent experiments. (B) Dynamic tumor growth from mice treated as (A). (C) Tumor weights from A. Values represent mean ± SD; *p < 0.05 versus control; #p < 0.05 versus STZ. (D) Representative images and quantification of IHC staining of tumor samples from A. Staining was analyzed with a system based on the percentage of positively stained cells and the staining intensity. Integrated optical density of all the positive staining in each image was determined, and its ratio to total area of each photograph was calculated as density. Values represent mean ± SD, n = 6 mice/group. *p < 0.01 versus control; #p < 0.01 versus STZ. (E) Scatter dot plots of fasting blood glucose levels in breast cancer patients with and without LN metastasis. Mean values ± standard deviation are also denoted. The average fasting blood glucose levels were significantly higher in breast cancer patients with LN metastasis compared with those without LN metastasis (7.24 ± 0.48 mM, n = 42 and 5.71 ± 0.27 mM, n = 38, respectively, p < 0.001). (F) The expression of PP2Cδ and Acetyl-p53 in representative cases of primary breast tumors and adjacent normal tissue specimens from 56 normoglycemic and 24 hyperglycemic (fasting blood glucose levels ≥7.0 mM) patients. Original magnification, × 400. *p < 0.01 versus corresponding controls. (G) The identified signaling pathway in this study indicated that C23 represses hyperglycemia-induced tumor growth via inhibiting PP2Cδ activity and expression. IHC, immunohistochemical; LN, lymph node; STZ, streptozotocin. Color images are available online.
Previous studies suggest that glucose is a principal driving force for the growth of cancer cells (3). Next, we sought to substantiate the promoting role of hyperglycemia in cancer progression by comparing the blood glucose levels of BC cases with and without lymph node (LN) metastasis. Of the 80 BC cases from Tongji Hospital, Tongji University School of Medicine, there were 42 cases that had spread to LNs. The average fasting blood glucose levels were significantly higher in BC patients with LN metastasis than in those without LN metastasis (7.24 ± 0.48 mM and 5.71 ± 0.27 mM, respectively, p < 0.001) (Fig. 7E). These clinical data showed that blood glucose levels were positively correlated with progression of BCs, and thus hyperglycemia could be involved in the mechanisms of BC development. In an attempt to explore the clinical relevance of PP2Cδ pathway in BC progression, we measured the expression levels of PP2Cδ and p53 acetylation in paired samples of primary BCs and adjacent normal tissue specimens from 56 normoglycemic (i.e., fasting blood glucose levels <5.6 mM) and 24 hyperglycemic (≥7.0 mM) patients via tissue microarray IHC staining. The levels of PP2Cδ are dramatically higher, and those of p53 acetylation are lower in the primary cancers and adjacent normal tissues from hyperglycemic versus normoglycemic patients (Fig. 7F). These data are in accordance with our results obtained from BC cell lines and xenograft models, and demonstrate that the induction of PP2Cδ and inhibition of p53 function are important for cancer development, particularly in diabetic patients who generally have elevated blood glucose levels. In short, our findings highlight the clinical relevance of PP2Cδ overactivation under the hyperglycemic conditions that contribute to compromised p53 function in BC progression.
Discussion
It is well known that hyperglycemia/HG can advance the progression of a number of cancers, including BC. Despite its important clinical significance, the precise underlying molecular mechanisms remain poorly understood (15). In this study, we demonstrate that HG, via activation of PP2Cδ and compromise of p53 function, promotes aggressive BC cell phenotypes associated with cancer progression (migration, invasion, and cancer cell survival). The mechanisms underlying HG stimulation of PP2Cδ involve classical/novel PKC activation, and its downstream target protein GSK3β phosphorylation and inactivation. In addition, HG-induced ROS generation and subsequent NF-κB activation play a partial role in HG induction of PP2Cδ (Fig. 7G). Given that PP2Cδ has been demonstrated to be induced by a variety of stresses, some of which are p53 independent (50), this study leads a step forward in terms of new knowledge and concepts of its activation. Findings in this study provide novel insights into the mechanism of PP2Cδ induction in response to HG, and may pave an avenue for a new understanding regarding the internal links between diabetes and cancer.
Due to the clear oncogenic properties and its important role in tumorigenesis, tumor development and progression (8), PP2Cδ has emerged as an attractive drug target for the treatment of cancers, and inhibition of its expression or activity could constitute an important new strategy for therapeutic intervention to halt the progression of various cancers. Utilizing the AlphaScreen format, Rayter et al. (43) identified a specific chemical PP2Cδ inhibitor, CCT007093. This system is also the first reported use for detecting protein phosphatase activity in a high-throughput screen. Here, we sought to develop a more effective and selective PP2Cδ inhibitor to achieve the anticipated chemotherapeutic potential and to circumvent off-target effects. The structure similarity between a group of curcumin mimics (Compounds 1–33) and CCT007093 spurred us to investigate the inhibitory ability of compounds 1–33 on PP2Cδ. They have a similar central five-carbon monoketone linker and two identical aromatic terminal rings (Supplementary Fig. S4). This group of compounds was synthesized according to the procedure previously described by us (47). The growth inhibition assay screening identified that C7 and C23 have a striking cytotoxicity on MCF-7 cells (IC50: 2.10 and 0.98 μM) instead of MCF-12A cells versus CCT007093 (IC50: 511878348.2 μM) (Fig. 4E). More interestingly, the impressive cytotoxicity of C7 and C23 is fully mirrored by their much stronger inhibitory ability on PP2Cδ in cells (as assessed by phospho-P38 measurement) than CCT007093 (Fig. 4F and Supplementary Fig. S5). In addition, in vitro phosphatase assay substantiates that C23 directly inhibits PP2Cδ (Fig. 4G). Docking simulation for binding modes of these inhibitors to PP2Cδ phosphatase domain suggests that the carbonyl oxygen and the nitrogen on the heteroaromatic ring of C23 form two hydrogen bonds with side chains of Arg258 and Asp105, respectively. This leads to a stable binding model of C23 which is consistent with its stronger inhibition on PP2Cδ. Therefore, the mechanism(s) underlying PP2Cδ inhibition by C23 might involve its potent binding to the PP2Cδ catalytic site composed of Asp105, Arg110, Arg243, Arg258, Arg259, Gln265, Phe268, Asp314, Arg364, Asp366, and Asn367, leading to PP2Cδ inhibition by stopping a substrate from entering the enzyme's active site and/or interfering with the catalytic reaction of the enzyme.
In addition to inhibiting the activity of PP2Cδ, C23 inhibits HG-induced PP2Cδ expression. This action was found to be mediated by significant induction of HSP27, and subsequent ablation of HG-induced ROS production and NF-κB activation. This result is consistent with previous studies that have demonstrated NF-κB may promote tumorigenesis by positively regulating PP2Cδ (27), and that ROS-dependent phenomena, including NF-κB activation, are decreased by the expression of HSP27, which is a novel regulator of intracellular redox state. Upon heat shock, HSP27 was originally characterized as a protein chaperone facilitating the accurate refolding of damaged proteins (44). Further investigation of HSP27 demonstrated that it responds to cellular stress conditions instead of heat shock; for example, oxidative stress and chemical stress. In response to oxidative stress, HSP27 functions as an antioxidant, repressing ROS production via enhancing the levels of intracellular glutathione and decreasing the levels of intracellular iron (2). Kanagasabai et al. suggested that ectopic expression of Hsp27 notably depleted both mutant p53 and NF-κB (p65), reversed the drug resistance in Adriamycin (doxorubicin)-resistant MCF-7 cells (MCF-7/adr), and promoted cell death via increasing G2/M population and apoptosis. They proposed that clinical change in Hsp27 or NF-κB level should be a potential approach to avoid drug resistance in BC (21). Our current findings suggest that HSP27 plays a partial role in C23 inhibition of HG-induced BC cell migration, proliferation, and invasion (Fig. 6). Importantly in vivo, pharmacological targeting of PP2Cδ with C23 blocks the growth of MCF-7 xenografts in diabetic nude mice. Together, our findings demonstrate that a plausible mechanism of action for C23 in inhibition of HG-induced BC progression is to suppress PP2Cδ by directly inhibiting the activity of PP2Cδ and impeding HG-induced PP2Cδ expression via HSP27 induction.
In conclusion, we demonstrate that activation of PP2Cδ plays an important role in the HG-stimulated progression of BCs. The mechanisms underlying HG stimulation of PP2Cδ involve classical/novel PKCs/GSK3β and ROS/NF-κB pathways. Moreover, we identified a novel PP2Cδ inhibitor, C23, which not only directly inhibits PP2Cδ activity but also suppresses HG-induced PP2Cδ expression via HSP27 activation. Our data suggest that C23 may serve as a unique therapeutic agent for the treatment of BC patients with diabetes.
Materials and Methods
Cell culture, treatment, and standard assays
Human cell lines, MCF-12A, MCF-10A, MCF-7, MDA-MB-231, T-47D, ZR-75 cells were obtained from American Type Culture Collection (Rockville, MD). These cells were authenticated by Laragen, Inc. (Culver City, CA), by short tandem repeat profiling and monitoring cell morphology and biological behavior, and tested to exclude mycoplasma contamination before experiments. The normal glucose-cultured MCF-7 and MDA-MB-231 cells were used for experiments after 2 months of incessant culture in normal glucose (5.5 mM) medium. After 5 h of serum starvation, the cells were treated with normal glucose (5.5 mM glucose +17 mM mannitol) or HG (22.5 mM glucose) for 0–48 h. For the dose course assays, some cells were exposed to HG (media that contained 11 or 22.5 mM glucose) for 24 h. Some of the cells that were exposed to HG (22.5 mM) were also incubated with a specific inhibitor of GSK3β, BIO (2.5 μM) or chemical inhibitor of IKKβ, IMD-0354 (2 μM) for 24 h. Standard cell culture, nuclear fractionation, and in vitro invasion assay were carried out as described previously (52–54, 57).
Western blot and immunoprecipitation
Whole-cell extract or nuclear fractions were analyzed by standard methods, and antibodies to PP2Cδ, p53, GSK3β, phospho-GSK3β (S9), NFκB-p65, phospho-p65 (S536), IκBα, PKC, p38, phospho-p38, HSP27, and TATA-binding protein, vinculin or actin as loading controls. Densitometry was performed using Scion Image software (Scion Corp., Frederick, MD). For p53 acetylation determination, the cell lysate was immunoprecipitated with antiacetylated p53 antibodies (anti-p53-acetyl K373 or acetyl K382), and then immunoblotted with anti-p53 antibody (DO-1).
RT-PCR and RT-qPCR
Total RNA was extracted using RNA-Bee reagent (Amsbio LLC, Cambridge, MA) according to manufacturer's protocol. RT-PCR was performed using SuperScript One-Step RT-PCR kit (Invitrogen). Real-time quantitative PCR was performed using iQ SYBR Green Supermix and iScript cDNA synthesis kit on Bio-Rad iQ5 Real Time System (Bio-Rad) according to the manufacturer's protocol. Primer sequences for each gene are available on request. The pumilio RNA-binding family member 1 (PUM1), which is an internal and stably expressed gene, served as a reference gene (30), and each sample was normalized on the basis of its PUM1 content.
GSK3β activity assays
MCF-7 cells were treated with HG and BIO for 24 h. GSK-3β activity was detected from immunoprecipitated cell lysates as described previously (40).
Intracellular GSH assay
The reduced intracellular glutathione was assayed using a Glutathione Assay Kit from Cayman Chemical (Ann Arbor, MI) according to the manufacturer's instructions. The measurement is based on quantification of GSH using glutathione reductase with an optimized enzymatic recycling method.
ROS assay
To determine the intracellular ROS formation, the fluorescent probe 2′,7′-dihydrodichlorofluorescein diacetate (DCFDA; Molecular Probes) was employed in MCF-7 cells according to the manufacturer's instruction and as described previously (54). OxyICC™ Oxidized Protein Detection Kit (fluorescent ICC) was used to detect carbonyl modification of cellular proteins caused by oxidative stress using fluorescent immunocytochemistry. Intracellular H2O2 were measured by digital imaging using pC1-HyPer-3, a gift from Vsevolod Belousov (Addgene plasmid # 42131), referring to described methods (5). In brief, MCF-7 cells were transfected by 1 μg of DNA per one 35-mm dish with FuGene6 transfection reagent. After 16 h, cells were exposed to HG (22 mM) in the presence of the NAPDH oxidase inhibitor DPI (5 μM) or the SOD inhibitor DDC (1 mM) for 24 h. Cells exposed to 100 μM H2O2 for 1 min were used as a positive control. The fluorescence of transfected cells was visualized by using a fluorescence microscope (Leica DM IRB) with an L 20 × /0.40 COAA PH 1 objective (Leica, Germany). Images were acquired and analyzed with PROGRES GRYPHAX Microscope Camera Software (JENOPTIK AG, Germany).
P53 reporter luciferase assays
A p53-responsive reporter plasmid containing 13 copies of the p53-binding consensus sequence, pG13-LUC, was a gift from Bert Vogelstein (Addgene plasmid # 16442). Cells were transfected with pG13-LUC plasmid, and luciferase assays were performed as previously described (17).
siRNA and shRNA gene silencing
For PKA and Akt knockdown, MCF-7 cells were transfected with human-specific PKA Cα siRNA, Akt siRNA, or scrambled siRNA (Cell Signaling Technology, Inc.) for 48 h using BioT transfection reagent (Bioland Scientific LLC) according to the manufacturer's instructions. Human PKC shRNAs (sc-29449-SH) and control shRNA lentiviral particles were obtained from Santa Cruz Biotechnology. For optimal shRNA plasmid transfection efficiency, Santa Cruz Biotechnology's shRNA Plasmid Transfection Reagent and shRNA Plasmid Transfection Medium were used according to manufacturer's Transfection Protocol.
Intracellular phosphatase activity
The intracellular PP2Cδ and PP2Cβ phosphatase activities were assayed using DuoSet IC (IntraCellular) Phosphatase Activity Assay Development Systems from R&D Systems (Minneapolis, MN), according to the manufacturer's instructions. In brief, 96-well cell culture plates were coated with the PP2Cδ or PP2Cβ antibody (Novus Biologicals). Following experimental treatment and lysis, lysate samples containing phosphatase were added. Cellular PP2Cδ present was bound to the well of a microplate by the immobilized antibody and assayed for its ability to release phosphate from a synthetic phosphopeptide substrate (DLDVPIPGRFDRRVS(PO3)VAAE Ser/Thr Phosphatase Substrate I). Cellular PP2A activity was determined using a PP2A immunoprecipitation phosphatase assay kit (Upstate) according to the manufacturer's protocol.
Apoptosis assay
MCF-7 cells were treated with IMD-0354 (2 μM), Ro31 (10 μM), HG (22.5 mM), or a combination of HG with IMD or Ro31. Two hours after the start of the treatments, cells were exposed or not to 20 mJ/cm2 ultraviolet (UV). Subsequently, cells were incubated with the mentioned conditions for 40 h, and then harvested. Apoptosis was determined by measuring the membrane changes (phosphatidylserine based) with an APC Annexin V Apoptosis Detection Kit with PI (BioLegend, Inc., San Diego, CA) as described previously (39). Fluorescence was detected using the BD FACSCanto™ flow cytometer (Becton Dickenson, Mountain View, CA) and FlowJo software version 10.4 (TreeStar, Ashland, OR). The experiments were repeated three times.
Growth inhibition assay
The growth inhibition assays were performed by culturing cells in 96-well plates. The following day, the cells were treated with ½ serial dilutions of the drugs or the solvent (dimethyl sulfoxide [DMSO]) alone for the control. The amount of solvent was the same in all the conditions, and each condition was in duplicate or triplicate. For each drug, the highest concentration is mentioned in the related figure and the Results section. After 5 days incubation, the effect of the drugs on cell proliferation was determined by either MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma-Aldrich, St. Louis, MO) or bioluminescence assay (Cell Titer Glo, Promega, Madison, WI). After incubation of the cell with MTT (1 mg/mL), the developed color was dissolved in DMSO and read by a plate reader. The bioluminescence assays were performed according to the manufacturer's recommendation. The growth inhibition rates were determined by comparing the readout for each drug dilution versus the control, and from the growth inhibition curves the IC50s were calculated. Each assay was performed at least twice. For the trypan blue exclusion assay, MCF-7 cells were seeded in 12-well plates and allowed to grow for 16 h. The cells were then treated with vehicle (DMSO) or various concentrations of CCT, C23, and C7 for 5 days. The cells were stained with trypan blue (0.4%, w/v), and the number of viable cells was counted under a microscope.
Migration assays
Cell migration assays were performed with migration culture dish inserts from Ibidi (Martinsried, Germany). Cells were cultured in the chambers of the dish insert and treated. Twenty-four hours after maintenance of cells, the insert was removed, and culture medium was refreshed to start the treatment and migration process. Images of the migration area were photographed 0, 24, and 48 h after removal of the migration chamber under a Leica DMIBR microscope and a Gryphax Microscope camera (JENOPTIK, Berlin, Germany). The percentage of the wound closed was quantified from three independent replicates, and is expressed as mean ± SD.
Homology modeling of human PPM1D structure
The X-ray crystal structure of the serine/threonine phosphatase domain of human PPM1B (Protein Data Bank entry 2P8E) (1) was used as a template to construct the phosphatase domain (residues 99–378) of human PPM1D structure model. The sequence of the human PPM1D was retrieved from UniProt. According to the secondary structure information of the template, the sequence alignments were adjusted manually to be more reasonable. The 3D model of human PPM1D was generated using MODELLER (version 9.12) (32).
Docking
Grid-based ligand docking from energetics (GLIDE) software (13) (Schrödinger suite 2009) was used for predicting binding models of CCT007093, C7 and C23 based on human PPM1D structure model.
The modeled human PPM1D structure was prepared using the “Protein Preparation Wizard” implemented in Schrödinger suite. Hydrogen atoms and charges were added during a brief relaxation performed with the “Interactive Optimizer” option, and a restrained partial minimization was terminated when the root-mean-square deviation reached a maximum value of 0.3 Å to relieve steric clashes. Amino acid residues located within 20 Å from the centroid of manganese binding residues (Asp105, Asp314, and Asp366) were defined as the binding pocket for docking simulation. Three-dimensional structures of CCT007093, C7, and C23 were prepared with “LigPrep” tool of Schrödinger suite.
In the docking process, the inhibitors were docked into the binding site using GLIDE 5.5 with extraprecision (XP) to generate the poses. The final binding model was selected based on the Glide scoring function (G-Score) as well as visual inspection.
Synthesis of drug-loaded poly(lactic-co-glycolic acid) nanoparticles
To improve aqueous solubility and bioavailability of C23, we developed poly(lactic-co-glycolic acid) nanoparticles (PLGA-NPs) of C23. Drug-loaded PLGA-NPs (Akina Polyscitech, Inc.) were synthesized by standard single emulsion techniques. In brief, a 10% w/w drug was added to 3% w/v PLGA 50:50 in dichloromethane (DCM, Millipore Sigma) and sonicated using a microtip sonicator at 20 watts for 2 min to form an oil phase. This mixture was then added dropwise to 15 mL of 5% w/v polyvinyl alcohol (PVA, Millipore Sigma) and emulsified using an ultrasonicator for 10 min at 30 watts. The suspension was stirred overnight to evaporate the solvent, followed by ultracentrifugation and lyophilization to collect the drug-loaded NPs.
Nanoparticle characterization
The formulated nanoparticles were characterized for their size, size distribution, and surface charges using a ZetaPALS zeta potential analyzer (Brookhaven Instruments, Inc.). The morphology of the NPs was determined by scanning electron microscopy (Hitachi S-4800 II FE SEM). Drug loading efficiency for these nanoparticles was determined by direct method. In brief, 1 mg of NPs was dissolved in 1 mL of DCM to precipitate the polymer. Four milliliters of 100% ethanol (Decon Laboratories, Inc.) was then added, following which the mixture was centrifuged to separate the polymer (in the DCM, lower phase) and drug (in ethanol, upper phase). The amount of drug loaded onto the NPs was measured by its absorbance at 322 nm using a UV–Vis spectrophotometer (Infinite M200 plate reader, Tecan, San Jose, CA) and determined by the following formula:
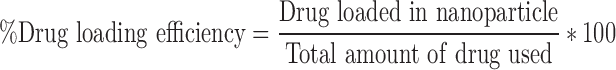 |
To perform the drug release kinetics of the NPs, drug-loaded nanoparticles were resuspended in 1 × phosphate buffered saline (PBS, pH 7.4) at a 1 mg/mL concentration and added to a dialysis bag (100 kDa molecular weight cutoff, Spectrum Laboratories, Rancho Dominguez, CA) for ∼21 days at 37°C. At predetermined time points, 1 mL of the dialysates was collected and replenished with fresh PBS. The amount of drug released was determined by its absorbance at 322 nm using a UV–Vis spectrophotometer. A standard curve of either known drug 1 or C23 concentrations was used to determine the respective released drug amounts.
In vivo model of diabetes-induced BC progression
Female athymic nude mice (Foxn1nu) (4 weeks of age) were purchased from The Jackson Laboratory (Bar Harbor, ME). E2 pellets obtained from Innovative Research of America (Sarasota, FL) were implanted 7days before cancer cell injection to enhance the endogenous E2 production. The cultured MCF-7 cells in the logarithmic phase were collected and diluted to 1 × 107 cells/mL. Then, cancer cell suspension (in Matrigel) was inoculated into the second mammary fat pad on the right side of mice (100 μL/injection). About 22 days after cancer cell injection, the mice were injected with a single dose of 150 mg/kg STZ (Zanosar Teva Pharmaceuticals, Irvine, CA) intraperitoneally. Body weight or blood glucose level was measured once before and daily after STZ administration until a diabetic state (a glucose concentration >16.7 mM) was substantiated by OneTouch Ultra Mini Blood Glucose Monitoring System.
About 12 days after STZ injection, some animals were administered with C23 or its vehicle. To improve aqueous solubility and bioavailability of C23, we developed PLGA-NPs of C23. The drug-loaded PLGA-NP size was 187 ± 61 nm with zeta potential of −18 mV. The drug loading efficiency was 62%, resulting in 85 μg of C23 in each milligram of PLGA-NP. The in vitro time course of drug release kinetics is shown in Supplementary Figure S15A, and the representative scanning electron microscopy (SEM) images of PLGA-NP are shown in Supplementary Figure S15B. The 100 μL C23 formulation was administered to mice (5 mg/kg, i.p.) every other day. Control mice were treated with vehicle by the same route. Electronic calipers were employed to determine the length and width of each tumor twice a week. The equation: volume = length × width2 was used to calculate tumor volumes. At the end of the experiment, the animals were sacrificed, and the tumors harvested. The fresh tumor tissues were instantaneously placed in 4% paraformaldehyde for further IHC analysis. All animal studies were performed in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of Charles Drew University of Medicine and Science.
Human tissue samples and IHC analysis
We retrieved tumor tissue specimens from breast tissue originating from primary tumor resections of patients diagnosed with BC between 2012 and 2015 at Tongji Hospital, Tongji University School of Medicine. A retrospective analysis of patient data was carried out, and age, gender, height, weight, body mass index (BMI), blood glucose, tumor location, tumor type, metastasis, and patient survival were documented. The formalin-fixed, paraffin-embedded primary BCs and adjacent normal tissue specimens from 80 BC cases were selected to construct the tissue arrays, and IHC analyses were carried out with a standard immunostaining protocol as previously described (57). The study complies with the regulations of the local ethics committee.
Statistical analysis
Statistical analysis in this study was performed using SPSS version 18.0 software (SPSS, Inc.). Results were expressed as “mean value ± SD.” The significance of mean values between two groups was determined by Student's t-test. All differences were two sided. The significance of the results from patient specimens was analyzed by the χ2 test or the Pearson correlation coefficient test. A p value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported in part by NIH-NIMHD U54MD007598, NIH/NCI1 U54CA14393, U56 CA101599-01; Department-of-Defense Breast Cancer Research Program Grant BC043180, NIH/NCATS CTSI UL1TR000124 to J.V. Vadgama, and Accelerating Excellence in Translational Science Pilot Grants G0812D05, NIH/NCI SC1CA200517 to Y. Wu. We are grateful to the CSUPERB New Investigator Award and the Research Development Award to Q.-H. Chen. This study was partially supported by NIH Grant 2G12MD007595.
Abbreviations Used
- aPKC
atypical PKC
- ATM
aurothiomalate
- BC
breast cancer
- C7
Compound 7
- C23
Compound 23 or 1,5-diheteroarylpenta-1,4-dien-3-one
- DCF
dichlorofluorescein
- DCM
dichloromethane
- DDC
diethyldithiocarbamic acid
- DMSO
dimethyl sulfoxide
- DMTU
dimethylthiourea
- DPI
diphenyleneiodonium chloride
- GLIDE
grid-based ligand docking from energetics
- GSK3
glycogen synthase kinase 3
- HG
high glucose
- HSP27
heat shock protein 27
- IHC
immunohistochemical
- LN
lymph node
- MnTBAP
manganese(III) tetrakis (4-benzoic acid) porphyrin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NOX
NADPH oxidase
- O2•−
superoxide
- PBS
phosphate buffered saline
- PKC
protein kinase-C
- PLGA-NPs
poly(lactic-co-glycolic acid) nanoparticles
- PP2C
type 2C protein phosphatase
- PUM1
pumilio RNA-binding family member
- ROS
reactive oxygen species
- SEM
scanning electron microscopy
- SOD
superoxide dismutase
- STZ
streptozotocin
- TBP
TATA-binding protein
Author Disclosure Statement
No competing financial interests exist.
References
- 1. Almo SC, Bonanno JB, Sauder JM, Emtage S, Dilorenzo TP, Malashkevich V, Wasserman SR, Swaminathan S, Eswaramoorthy S, Agarwal R, Kumaran D, Madegowda M, Ragumani S, Patskovsky Y, Alvarado J, Ramagopal UA, Faber-Barata J, Chance MR, Sali A, Fiser A, Zhang ZY, Lawrence DS, and Burley SK. Structural genomics of protein phosphatases. J Struct Funct Genomics 8: 121–140, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arrigo AP, Virot S, Chaufour S, Firdaus W, Kretz-Remy C, and Diaz-Latoud C. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid Redox Signal 7: 414–422, 2005 [DOI] [PubMed] [Google Scholar]
- 3. Beckner ME, Stracke ML, Liotta LA, and Schiffmann E. Glycolysis as primary energy source in tumor cell chemotaxis. J Natl Cancer Inst 82: 1836–1840, 1990 [DOI] [PubMed] [Google Scholar]
- 4. Bhatelia K, Singh A, Tomar D, Singh K, Sripada L, Chagtoo M, Prajapati P, Singh R, Godbole MM, and Singh R. Antiviral signaling protein MITA acts as a tumor suppressor in breast cancer by regulating NF-kappaB induced cell death. Biochim Biophys Acta 1842: 144–153, 2014 [DOI] [PubMed] [Google Scholar]
- 5. Bilan DS, Pase L, Joosen L, Gorokhovatsky AY, Ermakova YG, Gadella TW, Grabher C, Schultz C, Lukyanov S, and Belousov VV. HyPer-3: a genetically encoded H(2)O(2) probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chem Biol 8: 535–542, 2013 [DOI] [PubMed] [Google Scholar]
- 6. Bjorge T, Lukanova A, Jonsson H, Tretli S, Ulmer H, Manjer J, Stocks T, Selmer R, Nagel G, Almquist M, Concin H, Hallmans G, Haggstrom C, Stattin P, and Engeland A. Metabolic syndrome and breast cancer in the me-can (metabolic syndrome and cancer) project. Cancer Epidemiol Biomark Prev 19: 1737–1745, 2010 [DOI] [PubMed] [Google Scholar]
- 7. Boyle P, Boniol M, Koechlin A, Robertson C, Valentini F, Coppens K, Fairley LL, Boniol M, Zheng T, Zhang Y, Pasterk M, Smans M, Curado MP, Mullie P, Gandini S, Bota M, Bolli GB, Rosenstock J, and Autier P. Diabetes and breast cancer risk: a meta-analysis. Br J Cancer 107: 1608–1617, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, Kallioniemi A, Fornace AJ, Jr., and Appella E. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet 31: 210–215, 2002 [DOI] [PubMed] [Google Scholar]
- 9. Butler AM, Scotti Buzhardt ML, Erdogan E, Li S, Inman KS, Fields AP, and Murray NR. A small molecule inhibitor of atypical protein kinase C signaling inhibits pancreatic cancer cell transformed growth and invasion. Oncotarget 6: 15297–15310, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dang CV. and Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci 24: 68–72, 1999 [DOI] [PubMed] [Google Scholar]
- 11. Derr RL, Ye X, Islas MU, Desideri S, Saudek CD, and Grossman SA. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J Clin Oncol 27: 1082–1086, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Douarre C, Mergui X, Sidibe A, Gomez D, Alberti P, Mailliet P, Trentesaux C, and Riou JF. DNA damage signaling induced by the G-quadruplex ligand 12459 is modulated by PPM1D/WIP1 phosphatase. Nucleic Acids Res 41: 3588–3599, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, and Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47: 1739–1749, 2004 [DOI] [PubMed] [Google Scholar]
- 14. Garmendia ML, Pereira A, Alvarado ME, and Atalah E. Relation between insulin resistance and breast cancer among Chilean women. Ann Epidemiol 17: 403–409, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Gezgen G, Roach EC, Kizilarslanoglu MC, Petekkaya I, and Altundag K. Metabolic syndrome and breast cancer: an overview. J BUON 17: 223–229, 2012 [PubMed] [Google Scholar]
- 16. Graham ML, Janecek JL, Kittredge JA, Hering BJ, and Schuurman HJ. The streptozotocin-induced diabetic nude mouse model: differences between animals from different sources. Comp Med 61: 356–360, 2011 [PMC free article] [PubMed] [Google Scholar]
- 17. Granja AG, Nogal ML, Hurtado C, Salas J, Salas ML, Carrascosa AL, and Revilla Y. Modulation of p53 cellular function and cell death by African swine fever virus. J Virol 78: 7165–7174, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jee SH, Ohrr H, Sull JW, Yun JE, Ji M, and Samet JM. Fasting serum glucose level and cancer risk in Korean men and women. JAMA 293: 194–202, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Jope RS. and Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Kabat GC, Kim MY, Strickler HD, Shikany JM, Lane D, Luo J, Ning Y, Gunter MJ, and Rohan TE. A longitudinal study of serum insulin and glucose levels in relation to colorectal cancer risk among postmenopausal women. Br J Cancer 106: 227–232, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kanagasabai R, Krishnamurthy K, Druhan LJ, and Ilangovan G. Forced expression of heat shock protein 27 (Hsp27) reverses P-glycoprotein (ABCB1)-mediated drug efflux and MDR1 gene expression in Adriamycin-resistant human breast cancer cells. J Biol Chem 286: 33289–33300, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koroljow S. Two cases of malignant tumors with metastases apparently treated successfully with hypoglycemic coma. Psychiatr Q 36: 261–270, 1962 [DOI] [PubMed] [Google Scholar]
- 23. Larsson SC, Mantzoros CS, and Wolk A. Diabetes mellitus and risk of breast cancer: a meta-analysis. Int J Cancer 121: 856–862, 2007 [DOI] [PubMed] [Google Scholar]
- 24. Lawlor DA, Smith GD, and Ebrahim S. Hyperinsulinaemia and increased risk of breast cancer: findings from the British Women's Heart and Health Study. Cancer Causes Contr 15: 267–275, 2004 [DOI] [PubMed] [Google Scholar]
- 25. Liao S, Li J, Wei W, Wang L, Zhang Y, Li J, Wang C, and Sun S. Association between diabetes mellitus and breast cancer risk: a meta-analysis of the literature. Asian Pac J Cancer Prev 12: 1061–1065, 2011 [PubMed] [Google Scholar]
- 26. Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, and Hux JE. The impact of diabetes on survival following breast cancer. Breast Cancer Res Treat 109: 389–395, 2008 [DOI] [PubMed] [Google Scholar]
- 27. Lowe JM, Cha H, Yang Q, and Fornace AJ, Jr., Nuclear factor-kappaB (NF-kappaB) is a novel positive transcriptional regulator of the oncogenic Wip1 phosphatase. J Biol Chem 285: 5249–5257, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu X, Nannenga B, and Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 19: 1162–1174, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, and Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev 27: 123–135, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lyng MB, Laenkholm AV, Pallisgaard N, and Ditzel HJ. Identification of genes for normalization of real-time RT-PCR data in breast carcinomas. BMC Cancer 8: 20, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manjer J, Kaaks R, Riboli E, and Berglund G. Risk of breast cancer in relation to anthropometry, blood pressure, blood lipids and glucose metabolism: a prospective study within the Malmo Preventive Project. Eur J Cancer Prev 10: 33–42, 2001 [DOI] [PubMed] [Google Scholar]
- 32. Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, and Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29: 291–325, 2000 [DOI] [PubMed] [Google Scholar]
- 33. Moorhead GB, Trinkle-Mulcahy L, and Ulke-Lemee A. Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol 8: 234–244, 2007 [DOI] [PubMed] [Google Scholar]
- 34. Muti P, Quattrin T, Grant BJ, Krogh V, Micheli A, Schunemann HJ, Ram M, Freudenheim JL, Sieri S, Trevisan M, and Berrino F. Fasting glucose is a risk factor for breast cancer: a prospective study. Cancer Epidemiol Biomark Prev 11: 1361–1368, 2002 [PubMed] [Google Scholar]
- 35. Oliva-Trastoy M, Berthonaud V, Chevalier A, Ducrot C, Marsolier-Kergoat MC, Mann C, and Leteurtre F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 26: 1449–1458, 2007 [DOI] [PubMed] [Google Scholar]
- 36. Osaki Y, Taniguchi S, Tahara A, Okamoto M, and Kishimoto T. Metabolic syndrome and incidence of liver and breast cancers in Japan. Cancer Epidemiol 36: 141–147, 2012 [DOI] [PubMed] [Google Scholar]
- 37. Paravicini TM. and Touyz RM. Redox signaling in hypertension. Cardiovasc Res 71: 247–258, 2006 [DOI] [PubMed] [Google Scholar]
- 38. Peairs KS, Barone BB, Snyder CF, Yeh HC, Stein KB, Derr RL, Brancati FL, and Wolff AC. Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol 29: 40–46, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rahman AA, Makpol S, Jamal R, Harun R, Mokhtar N, and Ngah WZ. Tocotrienol-rich fraction, [6]-gingerol and epigallocatechin gallate inhibit proliferation and induce apoptosis of glioma cancer cells. Molecules 19: 14528–14541, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rao R, Zhang MZ, Zhao M, Cai H, Harris RC, Breyer MD, and Hao CM. Lithium treatment inhibits renal GSK-3 activity and promotes cyclooxygenase 2-dependent polyuria. Am J Physiol Ren Physiol 288: F642–F649, 2005 [DOI] [PubMed] [Google Scholar]
- 41. Rapp K, Schroeder J, Klenk J, Ulmer H, Concin H, Diem G, Oberaigner W, and Weiland SK. Fasting blood glucose and cancer risk in a cohort of more than 140,000 adults in Austria. Diabetologia 49: 945–952, 2006 [DOI] [PubMed] [Google Scholar]
- 42. Rauta J, Alarmo EL, Kauraniemi P, Karhu R, Kuukasjarvi T, and Kallioniemi A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat 95: 257–263, 2006 [DOI] [PubMed] [Google Scholar]
- 43. Rayter S, Elliott R, Travers J, Rowlands MG, Richardson TB, Boxall K, Jones K, Linardopoulos S, Workman P, Aherne W, Lord CJ, and Ashworth A. A chemical inhibitor of PPM1D that selectively kills cells overexpressing PPM1D. Oncogene 27: 1036–1044, 2008 [DOI] [PubMed] [Google Scholar]
- 44. Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo AP, Buchner J, and Gaestel M. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem 274: 18947–18956, 1999 [DOI] [PubMed] [Google Scholar]
- 45. Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, and Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 12: 2831–2841, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salim T, Sand-Dejmek J, and Sjolander A. The inflammatory mediator leukotriene D induces subcellular beta-catenin translocation and migration of colon cancer cells. Exp Cell Res 321: 255–266, 2013 [DOI] [PubMed] [Google Scholar]
- 47. Samaan N, Zhong Q, Fernandez J, Chen G, Hussain AM, Zheng S, Wang G, and Chen QH. Design, synthesis, and evaluation of novel heteroaromatic analogs of curcumin as anti-cancer agents. Eur J Med Chem 75: 123–131, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scata KA. and El-Deiry WS. p53, BRCA1 and breast Cancer chemoresistance. Adv Exp Med Biol 608: 70–86, 2007 [DOI] [PubMed] [Google Scholar]
- 49. Seyfried TN, Sanderson TM, El-Abbadi MM, McGowan R, and Mukherjee P. Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br J Cancer 89: 1375–1382, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, and Imai K. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J 19: 6517–6526, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vona-Davis L, Howard-McNatt M, and Rose DP. Adiposity, type 2 diabetes and the metabolic syndrome in breast cancer. Obes Rev 8: 395–408, 2007 [DOI] [PubMed] [Google Scholar]
- 52. Wu Y, Sarkissyan M, McGhee E, Lee S, and Vadgama JV. Combined inhibition of glycolysis and AMPK induces synergistic breast cancer cell killing. Breast Cancer Res Treat 151: 529–539, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu Y, Yu X, Yi X, Wu K, Dwabe S, Atefi M, Elshimali Y, Kemp KT, 2nd, Bhat K, Haro J, Sarkissyan M, and Vadgama JV. Aberrant Phosphorylation of SMAD4 Thr277-Mediated USP9x-SMAD4 Interaction by Free Fatty Acids Promotes Breast Cancer Metastasis. Cancer Res 77: 1383–1394, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu Y, Zhou H, Wu K, Lee S, Li R, and Liu X. PTEN phosphorylation and nuclear export mediate free fatty acid-induced oxidative stress. Antioxid Redox Signal 20: 1382–1395, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xue F. and Michels KB. Diabetes, metabolic syndrome, and breast cancer: a review of the current evidence. Am J Clin Nutr 86: s823–s835, 2007 [DOI] [PubMed] [Google Scholar]
- 56. Zeng L, Biernacka KM, Holly JM, Jarrett C, Morrison AA, Morgan A, Winters ZE, Foulstone EJ, Shield JP, and Perks CM. Hyperglycaemia confers resistance to chemotherapy on breast cancer cells: the role of fatty acid synthase. Endocr Relat Cancer 17: 539–551, 2010 [DOI] [PubMed] [Google Scholar]
- 57. Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, Sawaya R, Xie K, Yung WK, Medema RH, He X, and Huang S. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 20: 427–442, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.