

Abstract
The tiny green algae belonging to the Chloropicophyceae play a key role in marine phytoplankton communities; this newly erected class of prasinophytes comprises two genera (Chloropicon and Chloroparvula) containing each several species. We sequenced the plastomes and mitogenomes of eight Chloropicon and five Chloroparvula species to better delineate the phylogenetic affinities of these taxa and to infer the suite of changes that their organelle genomes sustained during evolution. The relationships resolved in organelle-based phylogenomic trees were essentially congruent with previously reported rRNA trees, and similar evolutionary trends but distinct dynamics were identified for the plastome and mitogenome. Although the plastome sustained considerable changes in gene content and order at the time the two genera split, subsequently it remained stable and maintained a very small size. The mitogenome, however, was remodeled more gradually and showed more fluctuation in size, mainly as a result of expansions/contractions of intergenic regions. Remarkably, the plastome and mitogenome lost a common set of three tRNA genes, with the trnI(cau) and trnL(uaa) losses being accompanied with important variations in codon usage. Unexpectedly, despite the disappearance of trnI(cau) from the plastome in the Chloroparvula lineage, AUA codons (the codons recognized by this gene product) were detected in certain plastid genes. By comparing the sequences of plastid protein-coding genes from chloropicophycean and phylogenetically diverse chlorophyte algae with those of the corresponding predicted proteins, we discovered that the AUA codon was reassigned from isoleucine to methionine in Chloroparvula. This noncanonical genetic code has not previously been uncovered in plastids.
Keywords: prasinophyte green algae, Picocystis salinarum, chloroplast genome, mitochondrial genome, phylogenomic analyses, AUA codon reassignment
Introduction
Prasinophytes constitute a paraphyletic assemblage of unicellular, morphologically diversified, and predominantly marine green algae at the base of the Chlorophyta (Lemieux et al. 2014; Sym 2015; Leliaert et al. 2016). Apart from the tiny algae belonging to the Mamiellophyceae, which are well known for their important contributions to phytoplankton communities and to primary productivity (Rii et al. 2016), at least four other prasinophyte lineages (Prasinococcales, Pycnococcaceae, Chloropicophyceae, and Picocystophyceae) comprise species of small size (≤5 μm in diameter). A highly reduced, coccoid growth form is thought to confer a distinct advantage to planktonic green algae because the resulting higher surface area-to-volume ratio enhances the efficiency of nutrient uptake and also because a reduced size helps to escape predators and promotes buoyancy (Potter et al. 1997; Grimsley et al. 2015; Sym 2015). Analyses of 18S rRNA metabarcoding data sets have demonstrated that the distribution pattern and habitat of prasinophytes in marine waters vary markedly depending upon the lineage and species examined (Rii et al. 2016; Lopes dos Santos, Gourvil, et al. 2017; Tragin and Vaulot 2018). The members of the three genera found in the order Mamiellales (Mamiellophyceae)—Ostreococcus (the genus containing the smallest known free-living eukaryote, O. tauri), Micromonas and Bathycoccus—are typically found in coastal waters, although some species occur in open oceanic waters (Sym 2015).
More recently, a study based on the analysis of metabarcoding data sets that were produced in the framework of the Tara Oceans expedition and Ocean Sampling Day consortium pointed the key role of the pico/nano-prasinophytes belonging to the lineage sister to the core Chlorophyta (a clade comprising the Chlorodendrophyceae, Pedinophyceae, Trebouxiophyceae, Ulvophyceae, and Chlorophyceae [Lemieux et al. 2014; Leliaert et al. 2016]) and formerly designated as clade VII (Guillou et al. 2004) in marine phytoplankton communities, especially in moderately oligotrophic waters (Lopes dos Santos, Gourvil, et al. 2017; Lopes dos Santos, Pollina, et al. 2017). These algae represent the dominant green algal group in open oceanic waters of tropical regions. This finding provided a fresh impetus to investigate the taxonomy and fundamental aspects of the biology of these algae, such as morphology, ultrastructure, and composition of pigments (Lopes dos Santos et al. 2016; Lopes dos Santos, Gourvil, et al. 2017; Lopes dos Santos, Pollina, et al. 2017). Analysis of the nuclear 18S and plastid 16S rRNA genes from a large sampling of strains and environmental sequences uncovered important diversity within clade VII (Lopes dos Santos, Gourvil, et al. 2017; Lopes dos Santos, Pollina, et al. 2017), leading Lopes dos Santos, Pollina, et al. (2017) to confer the status of class (Chloropicophyceae) to the lineage. Two major clades that received strong support were each erected to the genus level, Chloropicon (clade A) and Chloroparvula (clade B), with each genus displaying multiple subclades (A1–A7 and B1–B3) corresponding to different species. However, the precise relationships among the species could not be resolved unambiguously.
Because the chloroplast and mitochondrial genomes, designated hereafter as plastomes and mitogenomes, can be easily sequenced due to their relatively small size and multiple copies/cell, they are often used to infer deep-level phylogenetic relationships among algae and plants and to clarify phylogenetic issues arising from the analysis of a single or few genes (Gitzendanner et al. 2018; Turmel and Lemieux 2018; Yu et al. 2018). Moreover, comparative analysis of plastomes and mitogenomes provides the opportunity to gain insights into ancestral architectures and evolution of these genomes. Nine plastomes are currently available for prasinophyte picoalgae (Robbens et al. 2007; Turmel et al. 2009; Worden et al. 2009; Moreau et al. 2012; Lemieux et al. 2014). They are smaller in size (<99 kb), have a reduced gene repertoire (ranging from 100 genes in Chloropicon primus to 114 in Picocystis salinarum), and are more gene dense compared to the plastomes of other prasinophytes (e.g. Nephroselmis olivacea [Turmel et al. 1999b] and Pyramimonas parkeae [Turmel et al. 2009; Satjarak and Graham 2017]) and the vast majority of core chlorophytes (Turmel and Lemieux 2018). As revealed by gene mapping on evolutionary trees, the plastome sustained losses of many genes several times independently during the evolution of prasinophyte picoalgae (Turmel and Lemieux 2018). Because the nuclear genomes of mamiellalean picoalgae also feature a reduced size and a high gene density (Grimsley et al. 2015), it appears that miniaturization of both the nuclear genome and plastome occurred along cell reduction during evolution. The Chloropicon primus plastome is currently the sole organelle genome reported for the Chloropicophyceae and at 64.3 kb, it is the smallest plastome documented among photosynthetic green algae (Lemieux et al. 2014). Unlike the plastomes of the picoalgae Ostreococcus tauri, Micromonascommode, and Picocystis salinarum, it has not retained the large inverted repeat (IR) encoding the rRNA genes, a feature commonly found in green algae and land plants (forming together the Viridiplantae or Chloroplastida). Despite the widespread occurrence of the IR, however, multiple independent events of losses were inferred in the Chlorophyta (at least four losses in prasinophytes, seven in the Trebouxiophyceae, three in the Ulvophyceae, and two in the Chlorophyceae) (Turmel and Lemieux 2018). As observed for other picoalgae, the plastome gene order of Chloropicon primus is substantially scrambled relative to most other prasinophyte plastomes (Lemieux et al. 2014). Within the Mamiellales order, however, the Ostreococcus and Micromonas plastomes are highly similar in size and gene content and are essentially colinear.
To date, complete or almost complete mitogenome sequences are available for only five prasinophyte picoalgae, which represent the Mamiellales, Prasinococcales, and Pycnococcaceae (Robbens et al. 2007; Worden et al. 2009; Turmel et al. 2010; Moreau et al. 2012; Pombert et al. 2013). They show important variation in size (24.3 kb in Pycnococcus provasolii to 54.5 kb in Prasinoderma coloniale) and gene content (36 in Pycnococcus to 63 in Ostreococcus and Micromonas), but no general trend toward a reduced gene complement when they are compared to the mitogenomes of Nephroselmis (45.2 kb, 66 genes) (Turmel et al. 1999a), Pyramimonas parkeae (NIES254: 53.4 kb, 58 genes; SCCAP K-0007: 43.3 kb, 59 genes) (Hrda et al. 2017; Satjarak et al. 2017), and Cymbomonas tetramitiformis (73.5 kb, 56 genes) (Satjarak et al. 2017). As documented for Pyramimonas, the mitogenomes of mamiellalean taxa and Prasinoderma carry a large IR with variable gene content. At the level of gene order, mitogenome rearrangements are observed not only across lineages but also within the Mamiellales (Satjarak et al. 2017).
The goals of the present study were to better delineate the phylogenetic affinities among the different species previously described in the Chloropicophyceae and to gain insights into the evolutionary histories of the plastome and mitogenome within this class. We generated the organelle genome sequences of 13 chloropicophycean taxa and compared the newly sequenced plastomes and mitogenomes to one another and to their counterparts in Picocystis salinarum (the unique representative of the Picocystophyceae, a prasinophyte lineage presumed to have emerged just before the Chloropicophyceae). Our phylogenomic analyses produced a robust phylogeny on which the structural changes inferred from genome comparisons were mapped. For each organelle genome, important differences were observed between the Chloropicon and Chloroparvula genera as well as between each genus and Picocystis or any other previously examined prasinophyte. Most unexpected was our discovery of a noncanonical genetic code in the Chloroparvula plastome.
Materials and Methods
Strains and Culture Conditions
Strains of Chloropicon primus (CCMP 1205) and Picocystis salinarum (CCMP 1897) were obtained from the National Center for Marine Algae and Microbiota (NCMA, https://ncma.bigelow.org; last accessed April 8, 2019) and were maintained in K medium at 18 °C. The 12 other chloropicophycean strains originated from the Roscoff Culture Collection (RCC, http://roscoff-culture-collection.org; last accessed April 8, 2019) and were maintained in L1 medium at 22 °C. All algal cultures were grown synchronously under 12-h light/dark cycles.
Genome Sequencing and Annotation
The Chloropicon primus and Picocystis mitogenomes were assembled in the course of sequencing an A + T-rich DNA fraction that also contained the plastome; the methodologies for sequencing these algal mitogenomes were therefore the same as previously described for their plastomes (Lemieux et al. 2014). For the other chloropicophycean strains, the procedure used was as follows. Total cellular DNA was extracted using the EZNA HP Plant DNA Mini kit of Omega Bio-tek (Norcross, GA, USA). Paired-end libraries (inserts of 500 bp) were prepared using the Illumina TruSeq DNA kit and DNA was sequenced on the MiSeq platform (300 cycles). Both library construction and DNA sequencing were performed at the Plateforme d’Analyses Génomiques of Université Laval (Québec, QC, Canada) (http://www.ibis.ulaval.ca/services/an alyse-genomique/; last accessed April 8, 2019). After removing low-quality bases and adapters using AfterQC (Chen et al. 2017), reads were assembled using Spades 3.11.1 (Bankevich et al. 2012). Contigs of plastid and mitochondrial origins were identified by BLASTN and BLASTX searches against a local database of green plant organelle genomes and then were scaffolded in Sequencher 5.4.6 (Gene Codes Corporation, Ann Arbor, MI, USA).
Plastome and mitogenome annotations were performed using a custom-built suite of bioinformatics tools as described previously (Turmel et al. 2017). Genes encoding tRNAs were identified independently using tRNAscan-SE (Lowe and Eddy 1997). Circular maps of organelle genomes were drawn with OGDraw (Lohse et al. 2013). Genome-scale sequence alignments were carried out using LAST 9.5.6 (Frith et al. 2010).
Phylogenomic and Substitution Rate Analyses
Phylogenomic trees were inferred from plastome and mitogenome data sets that each comprised 14 taxa (chloropicophyceans + Picocystis). The plastome data set (PCG12RNA) was prepared from the first and second codon positions of 71 protein-coding genes and the sequences coding for 3 rRNAs and 28 tRNAs, while the mitogenome data set (also designated as PCG12RNA) was derived from the first and second codon positions of 36 protein-coding genes and the sequences coding for 2 rRNAs and 26 tRNAs. Following alignment of the deduced amino acid sequences from the individual protein-coding genes using Muscle 3.7 (Edgar 2004) and conversion of the resulting alignments to codon alignments, poorly aligned and divergent regions in each gene alignment were excluded using Gblocks v0.91b (Castresana 2000) with the -t = c, -b3 = 5, -b4 = 5, and -b5 = half options, and individual alignments were concatenated using Phyutility v2.2.6 (Smith and Dunn 2008), yielding the PCG123 data set. The third codon positions in this data set were excluded using Mesquite v3.51 (Maddison and Maddison 2018) to produce the PCG12 data set, which was then merged with the concatenated alignment of the RNA-coding genes to produce the PCG12RNA data set. To generate this concatenated alignment, individual RNA-coding genes were aligned using Muscle 3.7 (Edgar 2004), ambiguously aligned regions in each gene alignment were removed using TrimAl v1.4 (Capella-Gutierrez et al. 2009) with the options block = 6, gt = 0.9, st = 0.4, and sw = 3, and alignments were concatenated using Phyutility v2.2.6 (Smith and Dunn 2008).
The plastome and mitogenome PCG12RNA data sets were analyzed using maximum likelihood (ML) and Bayesian methods of phylogenetic reconstruction. ML analyses were performed using RAxML v8.2.12 (Stamatakis 2014) and the GTR + Γ4 model of sequence evolution, with each of the data sets being partitioned into gene groups and the model applied to each partition. The partitions included two RNA gene groups (rRNA and tRNA genes) in addition to the protein-coding gene partitions. Confidence of branch points was estimated by bootstrap analysis with 100 replicates. Bayesian analyses were performed using PhyloBayes 4.1 (Lartillot et al. 2009) under the heterogeneous CAT-GTR model. Two independent chains were run for 5,000 cycles and consensus topologies were calculated from the saved trees using the BPCOMP program of PhyloBayes after a burn-in of 1,000 cycles. Under these conditions, the largest discrepancy value observed across all bipartitions in the consensus topologies (maxdiff) was lower than 0.10, indicating that convergence between the chains was achieved.
Synonymous substitutions were estimated from the plastome and mitogenome PCG123 data sets using CODEML in PAML v4.9i (Yang 2007) and the F3X4 codon model (CodonFreq = 2, model = 2 in control file). The topology recovered in the RAxML analyses of the PCG12RNA data set described above was employed as user tree in these computations.
A separate plastome-based phylogenomic tree was inferred using an amino acid data set comprising 167 green plant taxa. This data set, which was derived from 79 protein-coding genes, was assembled and analyzed under ML as described previously (Turmel et al. 2017), except that IQ-Tree 1.6.7 (Nguyen et al. 2015) and ultrafast bootstrap approximation (Hoang et al. 2018) were used for the tree reconstruction and bootstrap analysis, respectively. Taxon sampling included the 129 green algae that were recently examined in the phylogenomic analysis reported by Turmel and Lemieux (2018), 10 land plants as well as 28 additional chlorophytes: 13 prasinophytes (including the 12 chloropicophycean taxa newly sampled in the present study), 1 member of the Trebouxiophyceae and 14 chlorophyceans. The GenBank accessions of the 38 additional plastomes used in our study are as follows: Acorus calamus, NC_007407; Anthoceros formosae, NC_004543; Arabidopsis thaliana, NC_000932; Carteria crucifera, KT624870-KT624932; Chlamydomonas asymmetrica, KT624933-KT625007; Chlamydomonas moewusii, EF587443-EF587503; Chlorogonium capillatum, KT625085-KT625091; Chloromonas radiata, KT625008-KT625084; Chloromonas typhlos, KT624630-KT624716; Golenkinia longispicula, KT625092-KT625150; Haematococcus lacustris, KT625205-KT625298; Hydrodictyon reticulatum, NC_034655; Huperzia lucidula, NC_006861; Lobochlamys culleus, KT625151-KT625204; Lobochlamys segnis, KT624806-KT624869; Marchantia polymorpha, NC_001319; Microglena monadina, KT624717-KT624805; Nicotiana tabacum, NC_001879; Oryza sativa, NC_001320; Pediastrum duplex, NC_034654; Physcomitrella patens, NC_005087; Pinus thunbergii, NC_001631; Pyramimonas parkeae, KX013546; Stephanosphaera pluvialis, KT625299-KT625409; Syntrichia ruralis, NC_012052; Trebouxia aggregata, EU123962-EU124002. Note that although 13 core chlorophyte plastomes were partially sequenced, full sequences are available for most, if not all, of their encoded genes.
Gene Order Analyses
Analyses of genome rearrangements were performed using the ProgressiveMauve algorithm of Mauve 2.3.1 (Darling et al. 2010). The number of reversals separating the plastomes and mitogenomes were estimated using GRIMM v2.01 (Tesler 2002) and gene order matrices containing the genes shared by all these genomes (plastomes, 95 genes; mitogenomes, 53 genes). The gene order matrices were also used to infer ML trees using the phylogeny reconstruction option of MLGO (Hu et al. 2014) and confidence of branch points was estimated by 1,000 bootstrap replications. The user tree option of MGR v2.03 (Bourque and Pevzner 2002) was used to infer the number of gene reversals on each branch of the topology recovered from phylogenomic analyses.
Codon Usage and tRNA Analyses
Analysis of codon usage in individual organelle genome was carried out using CUSP in EMBOSS 6.6.0 (Rice et al. 2000) and concatenated sequences of protein-coding genes. CUSP was also used to identify the subset of Chloroparvula plastid genes featuring AUA codons. The positions of these AUA codons as well as the corresponding codons and associated amino acid(s) in green algal orthologs were identified from gene alignments using the “translate as cDNA” option of Jalview (Waterhouse et al. 2009), which dynamically links codon alignments with predicted proteins in a split-frame view. The same program was employed to identify the codons corresponding to universally conserved isoleucine residues in plastome-encoded proteins of phylogenetically diverse green algae.
To determine in which part of the tRNA secondary structure fall the nucleotide differences observed between the Chloroparvula and Chloropicon elongator tRNAMet(CAU) sequences, secondary structures of these tRNAs were determined using tRNAscan-SE (Lowe and Eddy 1997) and a consensus secondary structure was drawn manually.
Results
Of the 13 strains sampled from Chloropicophyceae (table 1), eight belong to the Chloropicon genus; these represent all seven clades identified in this genus and include two distinct isolates of Chloropicon roscoffensis (A4), one from the English Channel (RCC1871) and the other from the Pacific Ocean (RCC2335). The remaining strains represent four separate clades of the Chloroparvula genus (B1–B3 and B) and include two B2-clade members from the Indian (RCC999) and Pacific (RCC696) Oceans. This sampling strategy allowed us to track organelle genome changes on both macro- and micro-evolutionary scales. In addition, the mitogenome sequence of Picocystis salinarum (Picocystophyceae) was generated in order to compare the newly sequenced chloropicophycean mitogenomes with a close relative from a distinct prasinophyte class. Although all chloropicophycean taxa, except Chloropicon primus, were not available as axenic cultures, their entire plastomes and mitogenomes could be assembled with no ambiguity as circular-mapping molecules (supplementary figs. S1 and S2, Supplementary Material online). Considering that an accurate phylogenetic framework is essential to track the suite of genomic changes that took place during chloropicophycean evolution, we will present our phylogenomic analyses before elaborating further on the structural features of the newly sequenced organelle genomes.
Table 1.
Characteristics of Plastomes and Mitogenomes from Chloropicophycean Taxa and Picocystis salinarum
| Taxon | Plastome |
Mitogenome |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Strain ID | Clade | Size (bp) | A + T (%) | Coding (%) | Accession | Size (bp) | A + T (%) | Coding (%) | Accession |
| Chloropicophyceae | ||||||||||
| Chloropicon genus | ||||||||||
| Chloropicon mariensis | RCC998 | A1 | 64,323 | 62.5 | 90.3 | MK085989 | 35,545 | 63.2 | 90.4 | MK086005 |
| Chloropicon primus | CCMP1205 | A2 | 64,335 | 63.3 | 90.1 | KJ746601 | 37,892 | 62.7 | 84.6 | MK085998 |
| Chloropicon sieburthii | RCC287 | A3 | 64,565 | 63.0 | 89.8 | MK085986 | 34,591 | 62.2 | 92.5 | MK086003 |
| Chloropicon roscoffensis | RCC1871 | A4 | 64,534 | 62.9 | 89.9 | MK085991 | 38,736 | 61.9 | 83.1 | MK086004 |
| Chloropicon roscoffensis | RCC2335 | A4 | 64,519 | 62.9 | 89.9 | MK085992 | 38,935 | 61.8 | 82.5 | MK086006 |
| Chloropicon laureae | RCC856 | A5 | 64,740 | 62.8 | 89.6 | MK085987 | 37,769 | 62.8 | 84.4 | MK086001 |
| Chloropicon sp. | RCC4434 | A6 | 67,005 | 64.1 | 86.8 | MK085997 | 41,336 | 61.8 | 74.6 | MK086009 |
| Chloropicon maureeniae | RCC3374 | A7 | 64,094 | 63.3 | 90.6 | MK085994 | 36,041 | 61.4 | 85.5 | MK086008 |
| Chloroparvula genus | ||||||||||
| Chloroparvula pacifica | RCC4656 | B1 | 69,944 | 63.2 | 89.8 | MK085995 | 49,744 | 64.2 | 64.9 | MK086011 |
| Chloroparvula sp. | RCC999 | B2 | 70,659 | 61.3 | 89.3 | MK085990 | 35,287 | 62.9 | 89.1 | MK086002 |
| Chloroparvula sp. | RCC696 | B2 | 70,730 | 61.4 | 89.2 | MK085988 | 35,646 | 62.8 | 88.2 | MK085999 |
| Chloroparvula sp. | RCC4572 | B3 | 70,736 | 61.4 | 89.2 | MK085996 | 35,718 | 62.8 | 88.0 | MK086010 |
| Chloroparvula japonica | RCC2339 | B | 71,262 | 60.3 | 88.8 | MK085993 | 40,432 | 60.1 | 78.3 | MK086007 |
| Picocystophyceae | ||||||||||
| Picocystis salinarum | CCMP1897 | C | 81,133 | 62.8 | 90.4 | NC024828 | 41,858 | 32.3 | 91.9 | MK086000 |
Plastome- and Mitogenome-Based Phylogenomic Analyses
Plastome- and mitogenome-based phylogenomic trees were inferred from 102 and 64 concatenated genes, respectively, using ML and Bayesian methods and Picocystis salinarum as outgroup (fig. 1). Both trees display exactly the same topology and their branches are well resolved, leaving little ambiguity regarding the relative positions of the sampled chloropicophycean taxa. Overall, the relationships are consistent with the trees previously inferred from nuclear 18S and plastid 16S rRNA genes (Lopes dos Santos, Pollina, et al. 2017); however, there are differences in the relative positions of the Chloroparvula sp. B2 and B3 strains and also of Chloropicon mariensis (A5) and Chloropicon laureae (A1). In addition, the position of Chloroparvula japonica (B) relative to other clades within this genus, which received poor support in the tree based on the concatenated rRNA genes, is supported by maximal bootstrap and Bayesian posterior values in the phylogenomic trees.
Fig. 1.

—Organelle phylogenomic trees showing the relationships among chloropicophycean taxa. (A) Plastome-based phylogeny inferred from a data set of 102 genes (first and second positions of 71 protein-coding genes, 3 rRNA genes, and 28 tRNA genes). (B) Mitogenome-based phylogeny inferred from a data set of 64 genes (first and second positions of 36 protein-coding genes, 2 rRNA genes, and 26 tRNA genes). The trees shown here are the best-scoring ML trees that were inferred under the GTR + Γ4 model. Support values are reported on the nodes: from top to bottom are shown the bootstrap support (BS) values for the RAxML GTR + Γ4 analyses and the posterior probability (PP) values for the PhyloBayes CATGTR + Γ4 analyses. Black dots indicate that the corresponding branches received BS and PP values of 100%.
The internal nodes in the Chloropicon lineage are separated by greater distances in the mitogenome tree (fig. 1B) than in the plastome tree (fig. 1A) and similarly, the internal branch in the Chloroparvula lineage that connects the common ancestor of the B2–B3 strains is longer in the mitogenome tree. These branch length differences may reflect differences in mutation rate between the two organelle genomes. As a proxy for comparing the neutral mutation rates of the plastome and mitogenome, we estimated the number of synonymous substitutions per site (dS) among the protein-coding genes of these organelle genomes, using the maximum likelihood approach implemented in PAML (fig. 2). The ratios of mitogenome to plastome dS values observed for the various branches of the phylogeny range from 0.55 to 5.38, with average values of 2.47 and 1.44 for the Chloropicon and Chloroparvula clades, respectively.
Fig. 2.

—Relative synonymous substitution rates in the organelle genomes of the Chloropicophyceae. Plastome and mitogenome trees based on synonymous substitutions were inferred from codon-aligned sequences of concatenated protein-coding genes using CODEML. In both trees, the number of estimated synonymous substitutions per site (dS value) is shown above each branch. In the mitogenome tree, the ratio of the mitogenome to plastome dS values is indicated in blue below each branch.
Given that taxon sampling may influence the branching order of lineages, we tested whether the Chloropicophyceae remain sister to all core chlorophytes when plastome-based trees are constructed using completely sequenced green algal plastomes available in public databases, including those newly sampled in this study. A ML tree was inferred from an amino acid data set derived from 79 genes of 167 green plant taxa representing various lineages of the Chlorophyta and Streptophyta (fig. 3). This tree identified the Chloropicophyceae as sister to all core chlorophytes, with the Picocystophyceae + Pycnococcaceae clade emerging as the preceding lineage. Judging from the depth of the positions occupied by the common ancestor of all Chloropicon and by that of all Chloroparvula species, each of these genera exhibits important genetic diversity (maximal branch lengths of 0.111 and 0.123 substitution/site for the Chloropicon and Chloroparvula clades, respectively); these divergence levels are about 2-fold greater than the level observed for the combined Micromonas and Ostreococcus genera (maximal branch length of 0.065 substitution/site), which together with Bathycoccus constitute the Mamiellales order of the Mamiellophyceae.
Fig. 3.

—Plastome-based phylogenomic tree showing the position of the Chloropicophyceae relative to other classes and major lineages of the Chlorophyta. This ML tree was inferred from a data set of 79 proteins from 167 green plants using IQ-Tree under the GTR+R4 model. BS values are reported on the nodes. The clades that received maximal support were collapsed and represented as triangles with sizes proportional to the number of taxa (indicated in parentheses). Black dots indicate that the corresponding branches received 100% BS.
Plastome Organization
Unlike the 81.1-kb plastome of Picocystis, which contains 114 genes, all chloropicophycean plastomes lack an IR encoding the rRNA genes and also differ by their substantially smaller size and gene repertoire as well as by the absence of introns (table 1, fig. 4A and supplementary fig. S1, Supplementary Material online). At 64,094 bp, the Chloropicon maureeniae (A7) plastome exhibits the smallest size among chloropicophycean plastomes and is also the smallest known among photosynthetic green algae. Plastomes of Chloroparvula species are about 7 kb larger but contain fewer genes (102 versus 97) in comparison to Chloropicon species. Within each of these genera, however, there is very little variation in plastome size, gene content and density of coding sequences (table 1 and fig. 4A). Given that a few genes in the Chloroparvula plastomes, such as the highly variable ycf1 encoding a putative subunit of the plastid protein import apparatus, the TOC/TIC machinery (Kikuchi et al. 2013), exhibit multiple insertions and are expanded relative to the corresponding Chloropicon genes (supplementary fig. S3, Supplementary Material online), all examined chloropicophycean plastomes share similar gene densities.
Fig. 4.

—Changes in gene content and codon frequency among chloropicophycean plastomes and mitogenomes. (A, B) Distribution of variable genes and total number of genes. The distribution of these genes is also shown for other prasinophytes and two early-branching streptophyte algae. The presence of a gene is denoted by a blue box. All chloropicophycean plastomes share the following 95 genes: accD, atpA, B, E, F, H, I, chlB, L, N, clpP, ftsH, infA, minD, petA, B, D, G, L, psaA, B, C, J, psbA, B, C, D, E, F, H, I, J, K, L, N, T, Z, rbcL, rpl2, 5, 14, 16, 19, 20, 23, 32, 36, rpoA, B, C1, C2, rps2, 3, 4, 7, 8, 9, 11, 12, 14, 18, 19, rrf, rrl, rrs, tufA, ycf1, 3, 4, 12, 20, trnA(ugc), C(gca), D(guc), E(uuc), F(gaa), G(gcc), G(ucc), H(gug), I(gau), K(uuu), L(uag), Me(cau), Mf(cau), N(guu), P(ugg), Q(uug), R(acg), R(ucu), S(gcu), S(uga), T(ugu), V(uac), W(cca), Y(gua). All chloropicophycean mitogenomes share the following 53 genes: atp1, 4, 6, 8, 9, cob, cox1, 2, 3, mttB, nad1, 2, 3, 4, 4L, 5, 6, 7, 9, rpl5, 14, 16, rps2, 3, 4, 12, 13, 14, 19, rnl, rns, trnA(ugc), C(gca), D(guc), E(uuc), F(gaa), G(gcc), H(gug), I(gau), K(uuu), L(uag), Me(cau), Mf(cau), N(guu), P(ugg), Q(uug), R(acg), R(ucu), S(gcu), S(uga), V(uac), W(cca), Y(gua). Duplicated gene copies, including those observed for the Chloroparvula pacifica plastid trnP(ugg) and trnW(cca) genes, were counted once. (C, D) Changes in frequency of the AUA codon associated with the presence/absence of trnI(cau). (E, F) Changes in frequencies of the UUA and UUG codons associated with the presence/absence of trnL(uaa) and trnL(caa), respectively.
Ten genes account for the differences in coding capacities of chloropicophycean plastomes and of these, seven are genus specific (fig. 4A). Three protein-coding genes, including tilS, as well as three tRNA genes [trnI(cau), trnL(uaa), and trnS(gaa)] are specific to Chloropicon, whereas only the trnL(caa) gene is specific to Chloroparvula. The fact that both trnI(cau) and tilS are either present or missing is not a coincidence, as tilS encodes the tRNA(Ile)-lysidine synthase that is required to convert to lysidine the cytidine found at the wobble position of the AUA codon-specific tRNAIle(CAU); this conversion changes the amino acid specificity of the tRNA from methionine to isoleucine (Soma et al. 2003). With the notable exception of trnL(uaa), all genes missing in some taxa of the Chloropicophyceae also experienced losses in independent prasinophyte lineages (fig. 4A).
Given that trnI(cau) occurs in all Chloropicon taxa but is absent in all Chloroparvula taxa, one would predict the AUA codon to be less frequent in Chloroparvula than in Chloropicon plastomes or to be even missing entirely. This prediction, which is based on the assumption that no nuclear-encoded tRNAIle(CAU) is targeted to the plastid, was verified by measuring the number of occurrences of the AUA codon in the protein-coding genes of each chloropicophycean plastome. It was found that all five Chloroparvula species, with the exception of Chloroparvula pacifica, exhibit much fewer AUA codons compared to the Chloropicon species (fig. 4C and supplementary table S1, Supplementary Material online). In which genes are located the AUA codons in Chloroparvula? When their positions correspond to universally or almost universally conserved amino acid positions in proteins of phylogenetically diverse green algae, do they generally specify isoleucine or a different amino acid residue? To address these questions, the protein-coding genes of Chloroparvula japonica (RCC2339) and Chloroparvula pacifica (RCC4656) carrying AUA codons were aligned with their orthologs in other green algae and the correspondence between these AUA codons and specific amino acid residues was visualized with the aid of Jalview (Waterhouse et al. 2009). Most of the Chloroparvula genes containing AUA codons encode proteins that are not directly implicated in photosynthesis (e.g. chlB and chlN encode proteins required for light independent chlorophyll synthesis) (fig. 5), and to our surprise, the AUA codons occupying universally conserved positions at the protein level were found to invariably correspond to AUG codons in Chloropicon species as well as in the other green algae examined (fig. 6). As expected, the AUA codons present at universally conserved amino acid positions in the latter algae are decoded as isoleucine residues (supplementary fig S4, Supplementary Material online). These results suggest that in Chloroparvula plastomes, the AUA codon is decoded as methionine and that two codons (AUU and AUC) instead of three, are read as isoleucine. Considering that RNA editing, a process mainly responsible mainly for U→C and C→U changes in organelle transcripts of land plants, has not been detected in green algae (Cahoon et al. 2017), conversion of the AUA codon to AUG at the RNA level is very unlikely to be responsible for decoding of AUA as methionine in Chloroparvula. To explore the molecular basis for the reassignment of AUA to methionine, the Chloroparvula elongator tRNAMet(CAU) gene sequences were aligned with the corresponding sequences of Chloropicon and other green algae and specific mutations were found in the Chloroparvula sequences (fig. 7).
Fig. 5.
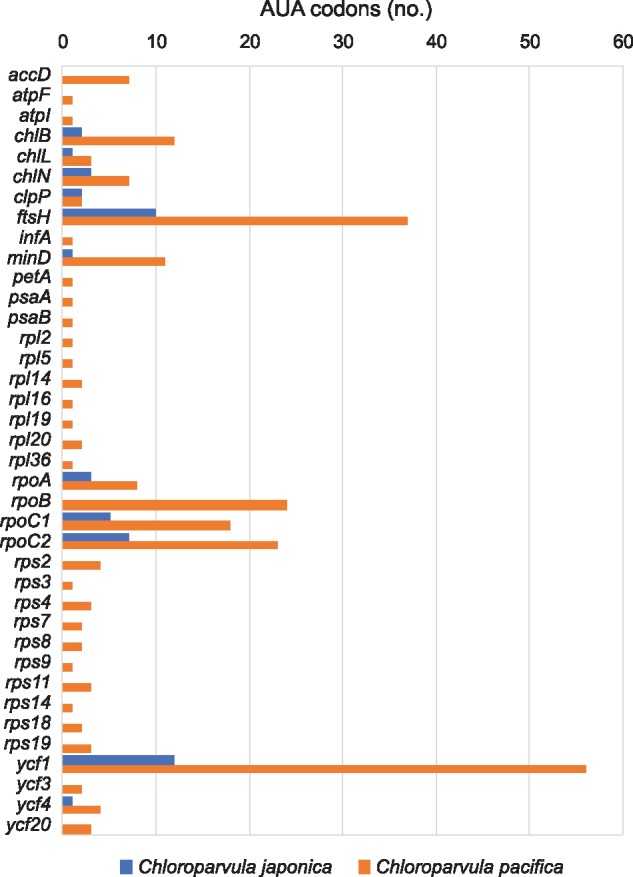
—Number of AUA codons in plastid genes of Chloroparvula japonica and Chloroparvula pacifica.
Fig. 6.

—Partial gene alignments including AUA codons from Chloroparvula plastomes. These codons fall within universally or almost universally conserved sites at the protein level and correspond to AUG codons in orthologous plastid genes of Chloropicon and other green algae. The 21 sites containing AUA codons are distributed among eight protein-coding genes; the number below the gene name indicates the position of the AUA codon in the Chloroparvula pacifica gene.
Fig. 7.

—Evolution of the plastid elongator tRNAMet(CAU) in the Chloropicophyceae. (A) Consensus secondary structure model of the Chloropicon elongator tRNAMet(CAU) showing the variant nucleotides observed in Chloroparvula pacifica. The circled nucleotides in the anticodon stem are conserved in chlorophyte tRNAsMet(CAU). (B) Alignment of trnMe(cau) gene sequences from all examined chloropicophycean taxa. The anticodon sequence is underlined.
With regards to the signature trnL(uaa) and trnL(caa) genes, one also expects to find highly different frequencies for the UUA and UUG codons in Chloropicon and Chloroparvula plastomes, given that the tRNAleu(UAA) recognizes both the UUA and UUG codons (the third position of the latter codon interacting through wobble pairing) and that the tRNAleu(CAA) recognizes solely the UUG codon. As predicted, the UUA codon was found to be prevalent in Chloropicon taxa although the UUG codon was also detected, and contrasting with this observation, the UUG codon was identified almost exclusively in Chloroparvula species (fig. 4E and supplementary table S1, Supplementary Material online). In the case of trnS(gga), its retention in Chloropicon and absence in Chloroparvula have no major impact on codon frequency because the tRNA encoded by this gene has a redundant role compared to that encoded by the universally present trnS(uga), which can recognize all four UCN codons through superwobbling (Alkatib et al. 2012). Similarly, the three tRNA species encoded by genes present in only one or two taxa [tRNAthr(GGU), tRNAarg(CCG), and tRNAarg(CCU) as shown in fig. 4A] are redundant with the products of trnT(ugu), trnR(acg), and trnR(ucu), which recognize four, four, and two codons, respectively, through superwobbling or wobbling (Alkatib et al. 2012). It was found that both trnT(ggu) and trnR(ccg) have several orthologs in other chlorophyte plastomes; however, BLAST searches of the NCBI database using trnR(ccu) identified strong hits only to trnR(ccg) genes from two isolates of the prasinophyte Pyramimonas parkeae, suggesting that the latter tRNA gene arose from duplication of trnR(ucu) and subsequent mutation of the anticodon.
The level of synteny among chloropicophycean plastomes was analyzed using Mauve (fig. 8A). The Picocystis plastome was not included in this analysis because it is extremely rearranged compared to these genomes. The Chloropicon plastomes were found to differ considerably from the Chloroparvula plastomes at the gene organizational level; however, an identical gene order was observed for all strains belonging to the same genus, with the exception of Chloropicon sp. RCC4434 (A6) and Chloroparvula pacifica. The number of gene reversals separating the rearranged plastomes was estimated using GRIMM (supplementary fig. S5A, Supplementary Material online) and MLGO (fig. 8C); in both analyses, the input data was a matrix of signed gene order comprising the 95 genes shared by all taxa, with the output being a distance matrix of the estimated numbers of reversals for all pairwise comparisons in the case of GRIMM and a tree generated with this matrix in the case of MLGO. GRIMM identified more than 50 reversals between the Chloropicon and Chloroparvula plastomes, but only two reversals between the Chloropicon sp. RCC4434 (A6) and all other Chloropicon plastomes and 19 reversals between the Chloroparvula pacifica and all other Chloroparvula plastomes. The results obtained with MLGO were consistent with these results.
Fig. 8.

—Extent of gene rearrangements in chloropicophycean plastomes and mitogenomes. (A, B) Analysis of gene order using Mauve 2.3.1. Locally collinear blocks of genome sequences are represented by boxes of identical color and similarly colored blocks are connected by lines. Blocks above the center line of the aligned regions are in the same orientation as in the reference (RCC1871) genome sequence, while those below this line are in the reverse orientation. (C, D) Phylogenetic relationships inferred from gene order data sets using the tree reconstruction option of MLGO. The plastome and mitogenome data sets included 95 and 53 genes, respectively. Consensus trees of 1000 bootstrap replicates are shown; black dots indicate that the corresponding branches received 100% BS.
Although nearly all plastomes from the same genus were colinear in nucleotide sequence, only those from the most closely related taxa (the Chloropicon roscoffensis taxa and the B2/B3 Chloroparvula sp. strains) could be aligned over their entire length using LAST. The Chloroparvula sp. RCC696 and RCC4572 revealed the lowest density of nucleotide substitutions, followed closely by the two Chloropicon roscoffensis strains (supplementary table S2, Supplementary Material online).
Mitogenome Organization
The 41.9-kb Picocystis mitogenome displays important differences relative to its counterparts in the Chloropicophyceae; notably, it features a very scrambled gene order relative to the latter genomes, an unusually low A + T content (table 1), and a 6.3-kb IR containing the rns, rnl, trnMe(cau), and cob genes (supplementary fig. S6, Supplementary Material online). Although the number of encoded genes is practically the same in all chloropicophycean mitogenomes, noticeable variations are seen at the genome size and gene density levels within each of the two genera (table 1). The Chloropicon pacifica mitogenome is the largest (49.7 kb) and least compact (64.9% coding sequence), followed by the Chloropicon sp. RCC4434 mitogenome, which has 57 genes instead of 61 or 62 as in other chloropicophyceans. Whereas no intron is present in the Picocystis mitogenome, a group I intron encoding a LAGLIDADG endonuclease occurs at site 1931 of the large subunit rRNA gene (rnl) in Chloropicon sieburthii, Chloropicon roscoffensis RCC2335, Chloropicon laureae, and Chloroparvula pacifica; moreover, in the latter strain, another group I intron also encoding a LAGLIDADG endonuclease is found at site 793 of the small subunit rRNA gene (rns) (supplementary fig. S2, Supplementary Material online).
Three genes distinguish the Chloropicon and Chloroparvula genera: trnL(uaa) is specific to Chloropicon, whereas the ribosomal protein-coding gene rpl10 and trnL(caa) are specific to Chloroparvula (fig. 4B). Therefore, as reported above for the plastome, trnL(uaa) and trnL(caa) are mutually exclusive genes in the mitogenome, and the distribution patterns of the UUA and UUG codons in Chloropicon and Chloroparvula taxa are in complete agreement with those observed for the plastome-encoded genes (fig. 4F and supplementary table S1, Supplementary Material online). The rpl6 gene, which is present in all Chloroparvula strains, was identified in the Chloropicon genus but only in the representative of the earliest-diverging lineages (Chloropicon maureeniae and Chloropicon sp. RCC4434); similarly, trnI(cau), a gene present in all Chloropicon taxa, was retained solely in the strain representing the earliest branch of the Chloroparvula genus (Chloroparvula pacifica). In agreement with the distribution of the latter gene, the AUA codon recognized by tRNAile(CAU) is abundant in Chloropicon strains and in Chloroparvula pacifica but was detected at fewer than four sites in other Chloroparvula strains (fig. 4D and supplementary table S1, Supplementary Material online). The trnR(ucg) gene was identified solely in Chloropicon sp. RCC4434; this gene is rarely found in chlorophyte mitogenomes (present only in Nephroselmis olivacea [Turmel et al. 1999a], Gloeotilopsis planctonica [Turmel et al. 2016], and Pseudendoclonium akinetum [Pombert et al. 2004]), and BLAST searches of the NCBI database using the Chloropicon sp. RCC4434 sequence identified trnR(acg) from several chlorophytes as best hits, suggesting that it was possibly derived by duplication of trnR(acg) followed by mutation of the anticodon.
While the mitogenomes of all examined Chloroparvula strains are colinear, differences in gene order are observed for a few strains sampled from the Chloropicon lineage (fig. 8B and D, and supplementary fig. S5B, Supplementary Material online). In particular, the representatives of the two earliest-diverging Chloropicon lineages (Chloropicon sp. RCC4434 and Chloropicon maureeniae), show the most rearrangements compared to the other Chloropicon taxa. Surprisingly, it was found that only three reversals separate the Chloropicon maureeniae and Chloroparvula mitogenomes. With 30–32 reversals, the mitogenome of Chloropicon sp. RCC4434 differs as extensively from the other mitogenomes of the Chloropicon lineage as from the Chloroparvula mitogenomes.
As reported above for the plastomes, the mitogenome sequences of closely related taxa could be aligned entirely or almost entirely using LAST and again, Chloroparvula sp. RCC696 and RCC4572 displayed the lowest density of nucleotide substitutions, followed by the Chloropicon roscoffensis strains (supplementary table S2, Supplementary Material online).
Discussion
Organelle Phylogenomic Analyses Provide a Robust Phylogeny of the Chloropicophyceae
Given their ecological importance in open oceanic waters (Lopes dos Santos, Gourvil, et al. 2017), the tiny green algae from the newly erected prasinophyte class Chloropicophyceae are bound to be the targets of future investigations aimed at better understanding their genetic divergence, biology, habitats, and role in ecosystems. In this context, knowledge about the precise relationships among the various species or clades (A1–A7 and B1–B3) in each of the two genera of this class is essential, as it provides the evolutionary framework required to establish the timing of changes that occurred during species diversification. The plastome- and mitogenome-based phylogenomic trees we report in the present study resolved with robustness the relationships among the Chloropicon and Chloroparvula species (fig. 1). These trees exhibit exactly the same topology but differ from the weakly supported tree of the Chloropicophyceae inferred from concatenated nuclear 18S and plastid 16S rRNA genes (Lopes dos Santos, Pollina, et al. 2017) with regards to the affinities between Chloropicon laureae (A5) and Chloropicon mariensis (A1) as well as the relationships among the three closely related Chloroparvula sp. taxa originally assigned to the B2 and B3 lineages. It should be considered that the Chloropicon species from the A1 and A5 lineages share a common ancestor, a conclusion also congruent with the robust relationships inferred for Chloropicon species from the predicted amino acid sequences of transcripts corresponding to 127 nuclear genes (Lopes dos Santos, Pollina, et al. 2017); however, it remains unclear why the results reported here differ from the 18S + 16S rRNA tree in showing a closer affinity between the B2 RCC696 and B3 RCC4572 strains than between the B2 RCC696 and RCC699 strains (fig. 1). Importantly, the plastome-based phylogenomic analysis including representatives of other Chloroplastida lineages revealed that the Chloropicon and Chloroparvula genera are separated by a very long evolutionary distance and that each chloropicophycean genus shows a substantially greater level of sequence divergence compared to the mamiellalean genera Ostreococcus and Micromonas (fig. 3). The same global phylogenomic tree confirmed that the Chloropicophyceae are sister to all core chlorophytes; however, the prasinophyte lineage that diverged just before the Chloropicophyceae could not be identified, as the relative positions of the Picocystophyceae and Pycnococcaceae were not clearly resolved.
Organelle Genomes Sustained Extensive Changes during the Evolution of the Chloropicophyceae
Consistent with the high level of sequence divergence separating the Chloropicon and Chloroparvula genera, considerable architectural alterations were identified in the plastomes and mitogenomes assembled in the present study, and their mapping on the inferred phylogenomic trees provided significant insights into the evolutionary histories of organelle genomes in the Chloropicophyceae (fig. 9). So far, only a few studies tracked in a comprehensive manner structural genomic changes in the chloroplasts and mitochondria of green algae from the same lineage (Smith and Keeling 2015; Turmel et al. 2016). Although the evolutionary pattern observed for one or the other organelle genome is typically variable across green algal lineages, shared patterns have been observed for the plastome and mitogenome of certain lineages of core chlorophytes, in particular with regards to the co-occurrence of important variations in A + T content and in the proportion of noncoding sequences (Smith and Keeling 2015). In contrast, it is well documented that the plastome and mitogenome of land plants, in particular angiosperms, have their own characteristic pattern, with the mitogenome evolving much more slowly at the sequence level than the plastome and also rearranging more frequently (Mower et al. 2012; Mower and Vickrey 2018).
Fig. 9.

—Predicted evolutionary scenarios for the plastome and mitogenome of the Chloropicophyceae. Gene losses and gene rearrangements are denoted by blue and red vertical lines, respectively. Mutations of tRNA anticodons, changes in codon frequencies, and reassignment of the AUA codon are indicated by yellow lines.
The plastomes and mitogenomes of the Chloropicophyceae experienced similar types of changes, but the tempos of occurrence of these changes differ in the two organelles (fig. 9). In the plastid, numerous gene losses and extensive gene reshuffling accompanied the split of the Chloropicon and Chloroparvula lineages; however, the plastome changed at a much slower pace during species diversification in each genus. In contrast, the mitogenome sustained more gradual changes during the evolution of the Chloropicophyceae and less extensive rearrangements during the emergence of the Chloropicon and Chloroparvula lineages. Differential losses of three mitochondrial genes [rpl10, trnL(uaa), and trnL(caa)] coincided with the divergence of the Chloropicon and Chloroparvula lineages, and further losses of rpl6 and trnI(cau) took place following the branching off of the Chloropicon maureeniae and Chloroparvula pacifica lineages. Remarkably, both the plastome and mitogenome lost three tRNA genes with the same decoding functions [trnI(cau), trnL(uaa), and trnL(caa)], leading to important changes in codon usage (see next section).
Prior to our study, the plastome of Chloropicon primus was the only organelle genome of the Chloropicophyceae that was available for comparison with its homologs in other prasinophyte lineages (Lemieux et al. 2014). Like this plastome, the newly sequenced plastomes of Chloroparvula are extremely divergent from all those previously sampled from prasinophytes, revealing no phylogenetic link with a specific prasinophyte lineage as well as no clue as to what the plastome architecture looked like during the early evolution of the Chloropicophyceae. Considering that the Picocystis salinarum plastome (Lemieux et al. 2014) is substantially more gene rich than the Chloroparvula and Chloropicon plastomes and has retained a rRNA operon-containing IR, it is likely that ancestral traits have massively disappeared soon after the emergence of the Chloropicophyceae. But the situation appears different for the mitogenomes of the Chloropicophyceae, as their gene repertoires are similar to those found in Picocystis (table 1, supplementary fig. S6, Supplementary Material Online), the Mamiellophyceae (Robbens et al. 2007), and Nephroselmidophyceae (Turmel et al. 1999a). As observed for the plastome, however, they are lacking an IR containing the rRNA genes, a feature that is probably an ancestral trait given its presence in Picocystis, the Mamiellophyceae, and Prasinoderma coloniale (Robbens et al. 2007; Pombert et al. 2013).
Following the divergence of the Chloropicon and Chloroparvula lineages, the plastome maintained a nearly identical size and gene density in each genus, whereas the mitogenome fluctuated at these levels (table 1). It is not clear what are the causes of these differences: we cannot exclude the possibility that there is an elevated pressure to keep the plastome more reduced and gene-dense than the mitogenome, but there might also exist differences related to the type and rate of errors associated with the mechanisms ensuring DNA integrity in the two organelles. As proposed for land plants (Christensen 2014), DNA repair may be more prone to errors in the mitogenome than the plastome in the Chloropicophyceae, thus possibly accounting for the lower gene density found in the mitogenome. Considering that gene rearrangements are the products of illegitimate recombination events involving different intergenic regions, organelle genomes with longer intergenic regions, which is the case here for the mitogenome, are expected to have more opportunity to recombine. Consistent with this idea, the mitogenomes of representatives from several Chloropicon clades exhibited substantial gene reversals (fig. 9). The mitogenome of Chloropicon sp. RCC4434, which is the longest in the Chloropicon lineage, was the most rearranged relative to its counterparts; however, the Chloroparvula mitogenomes, including the largest identified in the Chloropicophyceae (Chloroparvula pacifica mitogenome) were all colinear. The seemingly higher rate of mitogenome recombination in the Chloropicon genus may be due to differences in the efficiency of the nuclear-encoded enzymes catalyzing recombination or to the presence of more favorable sequences serving as DNA substrates for these enzymes.
Our estimates of synonymous substitutions in the organelles of the Chloropicophyceae indicate that the mitogenome tends to exhibit a slightly higher neutral mutation rate than the plastome. The mitogenome/plastome dS ratio showed a 10-fold variation among the branches of the tree topology (fig. 2), with average values of 2.47 and 1.44 for the Chloropicon and Chloroparvula clades, respectively. Higher synonymous substitution rates were also estimated for the mitogenome versus the plastome in most algal species previously examined, contrasting with the radically opposite trend observed in most land plant taxa (Smith 2015). Only four branches in the tree of the Chloropicophyceae displayed a lower substitution rate for the mitogenome than the plastome (fig. 2). In the genus Ostreococcus, the range of variation of the mitogenome/plastome dS ratio was comparable to that reported here for the Chloropicophyceae: a population genetics study with 13 strains of Ostreococcus tauri revealed that the plastome has a 1.33-fold higher level of synonymous nucleotide diversity relative to the mitogenome (Blanc-Mathieu et al. 2013), whereas an independent study based on the divergence between two different Ostreococcus species showed that the dS level of the mitogenome exceeded more than 5-fold that of the plastome (Smith 2015).
Adaptation of Codon Usage in Chloroparvula Plastomes and Mitogenomes, with Reassignment of the AUA Codon to Methionine in the Plastome
Losses of genes from organelle genomes may have diverse consequences depending on the function of the encoded products. Before being completely extinguished, organelle genes essential for cell survival are generally transferred to the nucleus where they are endowed with the presequence necessary for organelle targeting of their encoded products or alternatively, they are replaced by existing nuclear genes of similar function whose products are retargeted to the organelle. Such events of gene transfer to the nucleus are predicted for the photosynthetic gene psbM and the ribosomal-protein genes that were found to be missing in some strains of the Chloropicophyceae (fig. 4A and B), but firm evidence for their occurrence will await characterization of nuclear genomes from these strains. While tRNA genes with dispensable or redundant function can disappear from the cell without any further impact, loss of genes encoding tRNAs needed for reading specific codons leads to adaptation of codon usage (fig. 4C–F). To our knowledge, our study provides the first evidence for codon adaptation resulting from losses of trnI(cau) and trnL(uaa) in algae. These two genes are found in a large proportion of the green algal organelle genomes currently available: trnI(cau) is present in 121/129 plastomes and 29/36 mitogenomes examined, while trnL(uaa) is present in 129/129 plastomes and 33/36 mitogenomes. They suffered convergent loss from both the plastome and mitogenome in the Chloroparvula lineage and these events were intimately associated with the disappearance of the original AUA isoleucine and UUA leucine codons recognized by their encoded tRNAs. These codons vanished through mutations mainly to AUU isoleucine and UUG leucine codons, respectively (supplementary table S1, Supplementary Material Online).
The status of the unassigned AUA codons was temporary following the loss of trnI(cau) and tilS in plastomes, as new AUA codons were created by mutations of AUG codons in the Chloroparvula lineage and translated to methionine residues following the emergence of a modified tRNAMet(CAU). The frequent occurrence of these codons at universally or almost universally conserved sites exhibiting AUG codons in plastomes of phylogenetically diverse green algae together with the complete lack of AUA codons at highly conserved codon sites decoded as isoleucine provide compelling evidence not only for the evolution of a variant genetic code in which both AUA and AUG are read as methionine (fig. 6 and supplementary fig. S4, Supplementary Material Online) but also for the mechanism proposed above, which is also known as the codon-capture hypothesis (Osawa et al. 1989; Watanabe and Yokobori 2011). This variant code is used in the mitochondria of most metazoans and some yeasts (Sacharomyces and Torulopsis) and the codon capture hypothesis was also favored to explain the reassignment of AUA to methionine in these mitochondria (Osawa et al. 1989). To our knowledge, this is the first time that this noncanonical genetic code is reported in plastids. In green algal plastomes, a variant genetic code has been previously documented only for the chlorophyte Jenufa minuta (Sphaeropleales, Chlorophyceae), where UGA codons, which are normally stop codons, specify tryptophan (Turmel and Lemieux 2018).
Efficient decoding of the reintroduced AUA codons as methionine required alterations of an existing tRNAMet(CAU) in metazoan mitochondria (Watanabe and Yokobori 2011). Indeed, it has been shown that a modified nucleotide in the anticodon loop at the wobble position 34 (5-formylcytidine) or at position 37 (N6-threonylcarbamyladenosine) enables the tRNAMet(CAU) to pair with both AUA and AUG codons. Although no information is currently available about the presence of modified nucleotides at these positions in plastid tRNAsMet(CAU), alignments of the Chloroparvula tRNA gene sequences with the corresponding sequences of Chloropicon and other green algae unveiled specific substitutions in the former sequences that map to three of the five base pairings forming the stem of the anticodon arm (fig. 7). The changes in the C29: G41 and U31: G29 pairs are especially noteworthy considering that these base pairs are very conserved among chlorophyte tRNAsMet(CAU). Perhaps, these mutations may influence the anticodon–codon interaction and allow the wobble position of the anticodon to recognize both purine nucleotides at the third codon position. Obviously, to decipher the mechanism that mediated the reassignment of the AUA codon in the Chloropicophyceae, it will be important to investigate whether the Chloroparvula and Chloropicon tRNAsMet(CAU) differ with respect to modified nucleotides in the anticodon loop.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada (http://www.nserc-crsng.gc.ca/index_eng.asp; last accessed April 8, 2019) (grant RGPIN-2017-04506 to C.L.). We thank Daniel Vaulot and Ehsan Kayal for helpful discussions.
Footnotes
Data deposition: This project has been deposited at GenBank under the accessions MK085986-MK086011.
Literature Cited
- Alkatib S, et al. 2012. The contributions of wobbling and superwobbling to the reading of the genetic code. PLoS Genet. 811:e1003076.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 195:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc-Mathieu R, Sanchez-Ferandin S, Eyre-Walker A, Piganeau G.. 2013. Organellar inheritance in the green lineage: insights from Ostreococcus tauri. Genome Biol Evol. 58:1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque G, Pevzner PA.. 2002. Genome-scale evolution: reconstructing gene orders in the ancestral species. Genome Res. 121:26–36. [PMC free article] [PubMed] [Google Scholar]
- Cahoon AB, Nauss JA, Stanley CD, Qureshi A.. 2017. Deep transcriptome sequencing of two green algae, Chara vulgaris and Chlamydomonas reinhardtii, provides no evidence of organellar RNA editing. Genes 8:80. [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T.. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2515:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 174:540–552. [DOI] [PubMed] [Google Scholar]
- Chen S, et al. 2017. AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinformatics 18(Suppl 3):80.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AC. 2014. Genes and junk in plant mitochondria—repair mechanisms and selection. Genome Biol Evol. 66:1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT.. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 56:e11147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 325:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Hamada M, Horton P.. 2010. Parameters for accurate genome alignment. BMC Bioinformatics 11:80.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitzendanner MA, Soltis PS, Yi T-S, Li D-Z, Soltis DE.. 2018. Plastome phylogenetics: 30 years of inferences into plant evolution. Adv Bot Res. 85:293–313. [Google Scholar]
- Grimsley N, Yau S, Piganeau G, Moreau H.. 2015. Typical features of genomes in the Mamiellophyceae In: Ohtsuka S, Suzaki T, Horiguchi T, Suzuki N, Not F, editors. Marine Protists: Diversity and Dynamics. Tokyo: Springer Japan; p. 107–127. [Google Scholar]
- Guillou L, et al. 2004. Diversity of picoplanktonic prasinophytes assessed by direct nuclear SSU rDNA sequencing of environmental samples and novel isolates retrieved from oceanic and coastal marine ecosystems. Protist 1552:193–214. [DOI] [PubMed] [Google Scholar]
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS.. 2018. UFBoot2: improving the Ultrafast Bootstrap Approximation. Mol Biol Evol. 352:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrda S, Hroudova M, Vlcek C, Hampl V.. 2017. Mitochondrial genome of prasinophyte alga Pyramimonas parkeae. J Eukaryot Microbiol. 643:360–369. [DOI] [PubMed] [Google Scholar]
- Hu F, Lin Y, Tang J.. 2014. MLGO: phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinformatics 15:354.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, et al. 2013. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 3396119:571–574. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S.. 2009. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2517:2286–2288. [DOI] [PubMed] [Google Scholar]
- Leliaert F, et al. 2016. Chloroplast phylogenomic analyses reveal the deepest-branching lineage of the Chlorophyta, Palmophyllophyceae class. nov. Sci Rep. 6:25367.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux C, Otis C, Turmel M.. 2014. Six newly sequenced chloroplast genomes from prasinophyte green algae provide insights into the relationships among prasinophyte lineages and the diversity of streamlined genome architecture in picoplanktonic species. BMC Genomics 15:857.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Drechsel O, Kahlau S, Bock R.. 2013. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41(Web Server issue):W575–W581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes dos Santos A, Gourvil P, Rodriguez F, Garrido JL, Vaulot D.. 2016. Photosynthetic pigments of oceanic Chlorophyta belonging to prasinophytes clade VII. J Phycol. 521:148–155. [DOI] [PubMed] [Google Scholar]
- Lopes dos Santos A, Gourvil P, Tragin M, Noël M-H, Decelle J, Romac S, Vaulot D.. 2017. Diversity and oceanic distribution of prasinophytes clade VII, the dominant group of green algae in oceanic waters. ISME J. 112:512–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes Dos Santos A, et al. 2017. Chloropicophyceae, a new class of picophytoplanktonic prasinophytes. Sci Rep. 71:14019.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR.. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 255:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR.. 2018. Mesquite: a modular system for evolutionary analysis. Version 3.51. http://www.mesquiteproject.org; last accessed April 8, 2019.
- Moreau H, et al. 2012. Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 138:R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower JP, Sloan DB, Alverson AJ.. 2012. Plant mitochondrial genome diversity: the genomics revolution In: Wendel JH, editor. Plant Genome Diversity Volume 1: Plant Genomes, Their Residents, and Their Evolutionary Dynamics. New York: Springer; p. 123–124. [Google Scholar]
- Mower JP, Vickrey TL.. 2018. Structural diversity among plastid genomes of land plants. Adv Bot Res. 85:263–292. [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 321:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa S, Ohama T, Jukes TH, Watanabe K, Yokoyama S.. 1989. Evolution of the mitochondrial genetic code. II. Reassignment of codon AUA from isoleucine to methionine. J Mol Evol. 295:373–380. [DOI] [PubMed] [Google Scholar]
- Pombert JF, Otis C, Lemieux C, Turmel M.. 2004. The complete mitochondrial DNA sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) highlights distinctive evolutionary trends in the chlorophyta and suggests a sister-group relationship between the Ulvophyceae and Chlorophyceae. Mol Biol Evol. 215:922–935. [DOI] [PubMed] [Google Scholar]
- Pombert JF, Otis C, Turmel M, Lemieux C.. 2013. The mitochondrial genome of the prasinophyte Prasinoderma coloniale reveals two trans-spliced group I introns in the large subunit rRNA gene. PLoS One 812:e84325.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter D, Lajeunesse TC, Saunders GW, Anderson RA.. 1997. Convergent evolution masks extensive biodiversity among marine coccoid picoplankton. Biodiv Conserv. 6:99–107. [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 166:276–277. [DOI] [PubMed] [Google Scholar]
- Rii YM, et al. 2016. Diversity and productivity of photosynthetic picoeukaryotes in biogeochemically distinct regions of the South East Pacific Ocean. Limnol Oceanography. 613:806–824. [Google Scholar]
- Robbens S, et al. 2007. The complete chloroplast and mitochondrial DNA sequence of Ostreococcus tauri: organelle genomes of the smallest eukaryote are examples of compaction. Mol Biol Evol. 244:956–968. [DOI] [PubMed] [Google Scholar]
- Satjarak A, Burns JA, Kim E, Graham LE.. 2017. Complete mitochondrial genomes of prasinophyte algae Pyramimonas parkeae and Cymbomonas tetramitiformis. J Phycol. 533:601–615. [DOI] [PubMed] [Google Scholar]
- Satjarak A, Graham LE.. 2017. Comparative DNA sequence analyses of Pyramimonas parkeae (Prasinophyceae) chloroplast genomes. J Phycol. 532:415–424. [DOI] [PubMed] [Google Scholar]
- Smith DR. 2015. Mutation rates in plastid genomes: they are lower than you might think. Genome Biol Evol. 75:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ.. 2015. Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci U S A. 11233:10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SA, Dunn CW.. 2008. Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 245:715–716. [DOI] [PubMed] [Google Scholar]
- Soma A, et al. 2003. An RNA-modifying enzyme that governs both the codon and amino acid specificities of isoleucine tRNA. Mol Cell 123:689–698. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 309:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sym SD. 2015. Basal lineages of green algae – their diversity and phylogeny In:Ohtsuka S, Suzaki T, Horiguchi T, Suzuki N, Not F, editors. Marine Protists: Diversity and Dynamics. Tokyo: Springer Japan; p. 89–105. [Google Scholar]
- Tesler G. 2002. GRIMM: genome rearrangements web server. Bioinformatics 183:492–493. [DOI] [PubMed] [Google Scholar]
- Tragin M, Vaulot D.. 2018. Green microalgae in marine coastal waters: the Ocean Sampling Day (OSD) dataset. Sci Rep. 81:14020.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Gagnon MC, O'Kelly CJ, Otis C, Lemieux C.. 2009. The chloroplast genomes of the green algae Pyramimonas, Monomastix, and Pycnococcus shed new light on the evolutionary history of prasinophytes and the origin of the secondary chloroplasts of euglenids. Mol Biol Evol. 263:631–648. [DOI] [PubMed] [Google Scholar]
- Turmel M, Lemieux C.. 2018. Evolution of the plastid genome in green algae. Adv Bot Res. 85:157–193. [Google Scholar]
- Turmel M, et al. 1999a. The complete mitochondrial DNA sequences of Nephroselmis olivacea and Pedinomonas minor. Two radically different evolutionary patterns within green algae. Plant Cell 11:1717–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 1999b. The complete chloroplast DNA sequence of the green alga Nephroselmis olivacea: insights into the architecture of ancestral chloroplast genomes. Proc Natl Acad Sci U S A. 9618:10248–10253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2010. A deviant genetic code in the reduced mitochondrial genome of the picoplanktonic green alga Pycnococcus provasolii. J Mol Evol. 702:203–214. [DOI] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2016. Mitochondrion-to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns in Gloeotilopsis green algae (Ulotrichales, Ulvophyceae). Genome Biol Evol. 89:2789–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C.. 2017. Divergent copies of the large inverted repeat in the chloroplast genomes of ulvophycean green algae. Sci Rep. 71:994.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Yokobori S.. 2011. tRNA modification and genetic code variations in animal mitochondria. J Nucleic Acids 2011:623095.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ.. 2009. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 259:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden AZ, et al. 2009. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 3245924:268–272. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 248:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yu M, et al. 2018. Evolution of the plastid genomes in diatoms. Adv Bot Res. 85:129–155. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.