

Abstract
Three decahydroisoquinoline alkaloids, lepadins I–K, were isolated from a specimen of Didemnum sp. collected in the Bahamas. The structures of the new compounds were assigned by an integrated analaysis of MS, IR1H,13C NMR and 2D NMR spectra. Like previously reported lepadins, the structures of the new compounds contain a decahydroquinoline heterocyclic core in lepadin I, and a new variation, an octahydroquinoline in lepadin J, but differ from earlier reported compounds by acylation of the 3-hydroxyl group by a rare 3’-methylthioacrylate. The absolute configuration of lepadin I was solved by interpretation of NOE measurements, and exciton coupled circular dichroism (ECCD) of the corresponding N-p-bromobenzoyl derivative. The latter constitutes a general method for determination of absolute configuration of the entire lepadin family. The configuration of the remote side-chain secondary carbinol was solved by the modified Mosher’s esters method. Lepadin I inhibited butyrylcholineesterase (BuChE, IC50 3.1 μM), but only weakly inhibited acetylcholineesterase (AChE) (10% at 100 μM).
Graphical Abstract
Introduction
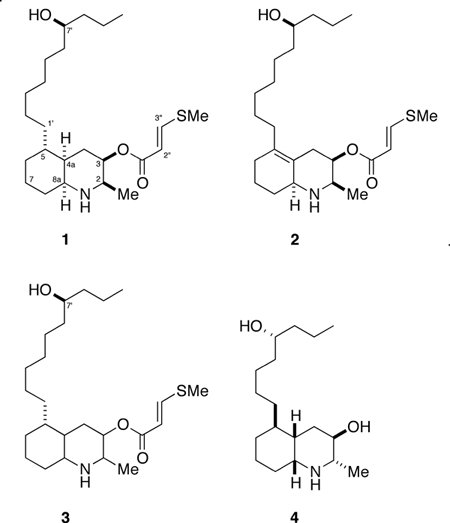
Ascidians express numerous secondary metabolites, and specialize in alkaloids, peptides and other nitrogenous natural products.1 The lepadins are 5-alkyl-3-hydroxydehydroquinoline derivatives found exclusively in ascidians or their predators. Lepadins A-H have been described from various sources including the ascidian species Clavelina lepadiformis from temperate waters, its predator, the flatworm, Prostheceraeus villatus, and other ascidian species within the genera Didemnum and Aplidium.2 Lepadins B and D are free secondary alcohols, but the other members are acylated at the 3-OH group as glycolates or short-chain alkenoate carboxylate esters. Bioactivities of some lepadins have been reported. Lepadins A and B were shown to have moderate cytotoxic effect on cancer cells2b and lepadins D (4), E and F possess antiplasmodial and antitrypanosomal activity.2c Additionally, (−)-lepadin B was shown to be a potent blocker of α7αβ2 and α7 neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes,3 but the broader potential of the alkaloid for nicotinic based therapies has not been evaluated. We describe here, the isolation and structure elucidation of lepadins I (1), J (2) and K (3) and absolute stereoassignment of 1 through development of a general method based on circular dichroism (CD). The new compounds are variants of the decahydroquinoline (azadecalin) core, common to other members of the series, but differ by the presence of a rare 3-methylthioacrylate ester at C-3.
Results and Discussion
The colonial ascidian Didemnum sp., collected at Stirrup Cay in the Bahamas in 2008, was left standing in MeOH (~5 years, –20 ˚C). The MeOH extract was partitioned against solvents of increasing polarity (hexane, CH2Cl2 and n-BuOH). The non-polar CH2Cl2 fraction obtained by solvent partitioning was further separated on a silica column using a stepwise gradient of CH2Cl2 and MeOH. Samples of pure lepadin I (1) and a mixture of lepadin J (2) and lepadin K (3) were obtained after purification by semi-preparative HPLC (aminopropyl column, 10:90 i-PrOH-hexanes, 1% Et2NH) conversion to their corresponding TFA salts and final elution from C18 SPE cartridges (MeOH: H2O).
Lepadin I (1) is an optically active alkaloid (free base: [α]D +4.1±0.6 (c 0.24, MeOH), TFA salt: [α]D +15.0±0.9 (c 0.25, MeOH)) which gave a positive Dragendorff’s test for basic nitrogen and a positive ninhydrin test (pale yellow), suggesting the N atom was associated with a secondary amine. The formula of lepadin I (1), C24H43NO3S, and lepadin J (2), C24H41NO3S, obtained from HRMS measurements, reveals four and five double bond equivalents, respectively and the presence of a sulfur atom. Evidence for the latter was obtained from MS and NMR data (see below). The ESIMS spectrum displayed an unusually intense M+2 ion that was attributed to an isotopomer containing one sulfur atom (34S), and confirmed by matching the experimental and calculated isotopomer patterns of the quasimolecular ion.
Analysis of the1H and13C NMR data for 1 (Table 1) showed the presence of an ester (δC 164.6, s), an acyclic secondary alcohol (δH 3.57, m), an axial esterified cyclic secondary carbinol (δH 5.04, q, J = 2.8 Hz; δC 67.8, CH), a secondary aliphatic methyl group (δH 1.31, d; δC 14.8, CH3) and a linear long-chain alkyl group (terminal Me; δH 0.91, t, J = 6.8 Hz; δC 14.3, CH3). The key feature that distinguished the1H NMR spectrum (CDCl3) of 1 from those of known lepadins was the presence of a highly polarized E-double bond (δH 8.07, d, J = 14.9 Hz; δH 5.74, d, J = 14.9 Hz) associated with the β-methylthioacrylate ester side chain (see below). Comparison of the key NMR chemical data of 1 and 2 with those of other alkaloids from ascidians (e.g. lepadin D, 42c) narrowed the possibilities to new 2,3,5-trisubstituted cis-decahydroquinolines (cis-azadecalins). The substituted side-chain located at C-5 is two carbons longer (C10) than in other lepadins. TOCSY and HMBC experiments revealed a hydroxymethine signal due to a secondary alcohol at C-3’ (CH-OH, δH 3.57, m; δC 71.8, CH) three carbons removed from the chain terminus.
Table 1.
1H and13C NMR data for lepadin I (1) (TFA salt, CDCl3, 23˚ C)
| 1Ha, δ, mult (J, Hz) | 13Cb δ (mult) | COSYa | NOESYa | HMBCa | |
|---|---|---|---|---|---|
| 1 | |||||
| 2 | 3.49, m | 55.8 (CH) | 9, 3 | 3, 4ax | 9 |
| 3 | 5.04, q (2.8) | 67.8 (CH) | 2, 4ax, 4eq | 2,4ax, 4eq, 9 | 1”, 4, 5, 9 |
| 4ax | 2.48, d (15.6) | 28.9 (CH2) | 3, 4eq, 5 | 3, 4eq, 4a, 1’ | 2, 3, 8a, 4a, 5 |
| 4eq | 1.63, mc | 3, 4ax | 3, 4ax, 4a, | 8a, 4a, 5 | |
| 4a | 1.54, md | 37.5 (CH) | 4eq, 5 | 4ax, 4eq, 8a, 5, 6ax | 3, 8a, 5, |
| 5 | 1.95, m | 34.5 (CH) | 4a, 6ax, | 4a, 1’, 2’, 6ax | 4a, 6, 1’, |
| 6ax | 0.81, qd (12.6, 3.3) | 31.2 (CH2) | 5, 6eq, 7eq, 7ax | 4a, 5, 6eq, 7ax, 7eq, 8ax | 4a, 5, 7,8 |
| 6eq | 1.87, brd (12.6) | 6ax, 7ax, 7eq | 5, 6ax, 1’, 2’ | 4a, 8, | |
| 7eq | 1.54, md | 19.9 (CH2) | 6eq, 7ax, | 6ax, 8eq, 7ax | 8a, 5 |
| 7ax | 1.74, qt (13.5, 3.3) | 6eq, 6ax, 7eq, 8eq, 8ax | 6ax, 8eq, 7eq, | 6, 8 | |
| 8αx | 1.64, mc | 29.3 (CH2) | 8a, 8eq, 7ax | 6ax, 8a, 8eq, | 8a, 4a, 6, 7 |
| 8eq | 2.01, d (14.5) | 8a, 8ax, 7eq | 7ax, 7eq, 8ax | 8a, 4a, 6, 7 | |
| 8a | 3.46, m | 57.4 (CH) | 4a, 8ax, 8eq | 4ax, 4a, 8ax, 8eq | 7 |
| 9 | 1.31, mh | 14.8 (CH3) | 2 | 3, 2” | 2, 3 |
| 1” | 164.6 (Cq) | ||||
| 2” | 5.74, d (14.9) | 111.7 (CH) | 4” | 4” | 2, 1”, 3” |
| 3” | 8.07, d (14.9) | 150.0 (CH) | 3” | 3” | 1”, 2”, 5” |
| 4” | |||||
| 5” | 2.33, s, 3H | 14.2 (CH3) | 3” | ||
| 1’ | 0.92, mf 1.36, me |
33.1 (CH2) | 2’ | 4eq, 5, 6eq | 5, 3’, 2’ |
| 2’ | 1.07, m 1.31, me |
26.1 (CH2) | 1’, 3’ | 5, 6eq | 1’, 3’ |
| 3’ | 1.20, mg | 30.3 (CH2) | 2’, 4’ | 1’, 4’, 5’ | |
| 4’ | 1.24, mg | 30.0 (CH2) | 3’ | 6’, 5’ 3’, | |
| 5’ | 1.26, mg 1.36, me |
25.8 (CH2) | 7’, | ||
| 6’ | 1.39, me | 37.7 (CH2) | 7’ | ||
| 7’ | 3.57, m | 71.8 (CH) | 6’, 8’ | 5’, 9’ | |
| 8’ | 1.39, me | 39.9 (CH2) | 7’, 9’ | ||
| 9’ | 1.35, me 1.44, me |
19.3 (CH2) | 8’, 10’ | 7’, 8’ | |
| 10’ | 0.91, t (6.8)f | 14.3 (CH3) | 9’ | 8’, 9’ | |
| NH | 8.23, bs |
600 MHz, in the presence of Et2NH•TFA buffer (~20 mM).
, 150 MHz, multiplicities assigned by APT and edited HSQC.
overlapped signals. i signal overlapped.
The sulfur–containing functional group in 1–3 was identified as a 3-methylthioacrylate by a process of deduction, including observation of a large red-shifted UV-vis band (λ 270 nm), evaluation of1JCH heteronuclear coupling constants, and matching the1H NMR chemical shifts to those of known compounds. A three-proton singlet (δ 2.34, 3H, s) suggested a methyl group attached to a heteroatom – either S or N – based on the relatively large sp3 one-bond heteronuclear coupling (1JCH = 140 Hz) observed in the1H NMR spectrum of 1 and 2 (‘13C-satellites’) and in coupled1H-13C HSQC spectra. The characteristically high-field13C NMR chemical shift of C-5” in 1 (δ 14.2, q) firmly established the identity of this methyl signal as SMe. The AB vinyl spin-system of 1 and 2 is associated with highly polarized1H NMR chemical shifts (Δδ 2.33 ppm); far larger than the differences expected for vinyl1H signals in a simple α,β-unsaturated alkenoate ester. Furthermore, the polarization is in the opposite direction to the latter compounds, consistent with a β-methylthioacrylate ester.4
The bicyclic ring conformation of 1 was established from NOESY experiment (Figure 1, tm = 300 ms, 600 MHz). Of the two possible chair-chair conformers for the cis-azadecalin core of 1, the one depicted – with a C-8a–N bond axial to the cyclohexane chair and an equatorial side chain subtended from C-5 – is supported by cross peaks between signals for the paired 1,3-syn diaxial protons – H-2ax–H-4ax, H-4aax–H-6ax – and is also the favored conformation of cis-azadecalin ring systems in other lepadins.
Figure 1.

NOESY cross peaks (600 MHz, CDCl3) measured for (a) lepadin I (1), tm = 300 ms, and (b), N-p-bromobenzoyl-lepadin I (6), tm = 400 ms.
The1H NMR and13C NMR spectrum of lepadin J (2) (Table 2) showed similar features to those of 1, except for the absence of two sp3 CH signals (DEPT) which were replaced by two quaternary C signals, (13C NMR) corresponding to a tetrasubstituted C=C double bond.5 This double bond was located at C-4a–C-5 based on HMBC evidence; correlations were observed from H-3, H-4, H-8 to C-4a and H-4 to C-5. Additional evidence for double bond placement at C-4a-C-5 was provided by the1H NMR spectrum of 2; the H-4 and H-1’ signals were deshielded compared to 1, consistent with allylic protons.
Table 2.
1H and13C NMRa data for lepadin J (2) (TFA salt, CDCl3, 23˚ C)
| Position | 1H (mult, J)a | 13C (mult)b | COSY | HMBC |
|---|---|---|---|---|
| 1 | 8.03, brs | 2, 8a | ||
| 2 | 3.53, m | 54.0 (CH) | 1, 9 | |
| 3 | 5.07, m | 68.4 (CH) | 2, 4 | 4a |
| 4 | 3.12, m; 2.12, m |
31.1 (CH2) | 4a, 5 | |
| 4a | 117.3 (C) | |||
| 5 | 142.4 (C) | |||
| 6 | 2.01, m; 1.85, m | 32.8 (CH2) | 7 | |
| 7 | 1.30, m | 22.4 (CH2) | 6 | |
| 8 | 2.05, m; 1.77, m | 26.4 (CH2) | 8a | 4a |
| 8a | 3.73, m | 55.3 (CH) | 8 | |
| 9 | 1.29, m | 14.6 (CH3) | 1, 2 | |
| 1” | 164.3 (C) | |||
| 2” | 5.74, d (14.8) | 111.3 (CH) | 3” | 1” |
| 3” | 8.08, dd (14.8, 1.5) | 149.8 (CH) | 2” | 1”, 2”, 5” |
| 4” | ||||
| 5” | 2.33, d (1.5) | 13.6 (CH3) | ||
| 1’ | 2.02, m; 1.85, m | 27.1 (CH2) | ||
| 2’ | 1.31, m; 1.20, m | 27.9 (CH2) | ||
| 3’ | 1.24, mc | 29.3 (CH2)c | ||
| 4’ | 1.24, mc | 29.3 (CH2)c | ||
| 5’ | 1.38, m; 1.27, mc | 25.2 (CH2)c | ||
| 6’ | 1.40, m | 37.1 (CH2) | 7’ | |
| 7’ | 3.57, m | 71.2 (CH) | 6’, 8’ | |
| 8’ | 1.41, m | 39.3 (CH2) | 7’ | |
| 9’ | 1.43, m; 1.33, m | 18.5 (CH2) | 10’ | |
| 10’ | 0.91, t (6.7) | 13.6 (CH3) | 9’ | 8’, 9’ |
| NH | 8.23, bs |
, 600 MHz.
, 150 MHz, multiplicities assigned by edited HSQC.
, interchangeable assignments.
The1H NMR and13C NMR spectrum of lepadin K (3) (Table 3) supported a stereoisomer of 1. The major differences in the1H NMR spectra of 3 and 1 were the signals due to H-4 and those of the cyclohexane ring. Absence of a COSY correlation between H-2 and H-3 (3J~ 0 Hz) supported the same C-2 and C-3 configurations and a cis azadecalin configuration as 1. Attempted purification of 3 led to material decomposition, therefore the relative stereochemistry of the remaining stereocenters remain ambiguous.
Table 3.
1H and13C NMRa data for lepadin K (3) (TFA salt, CDCl3, 23˚ C)
| Position | 1H (mult, J)a | 13C (mult)b | COSY | HMBC |
|---|---|---|---|---|
| 1 | 8.22, brs | 2, 8a | ||
| 2 | 3.35, m | 53.1 (CH) | 1 | |
| 3 | 5.04, m | 67.2 (CH) | 2, 4 | 4a |
| 4 | 2.24, m; 1.89, m | 25.8 (CH2) | 4a | 2, 3, 8a |
| 4a | 1.58, m | 41.1 (CH) | 4, 5, 8a | |
| 5 | 2.24 | 39.9 (CH) | 4a, 6 | |
| 6 | 2.05, m; 1.10, m | 28.9 (CH2) | 5 | |
| 7 | 1.77, m; 1.47, m | 19.3 (CH2) | 8 | |
| 8 | 2.13, m; 1.92, m | 29.3 (CH2) | 7 | 4a |
| 8a | 3.68, m | 59.3 (CH) | 4a | |
| 9 | 1.28, m | 14.6 (CH3) | 2 | |
| 1” | 164.3 (C) | |||
| 2” | 5.72, d (14.9) | 111.3 (CH) | 3” | 1” |
| 3” | 8.00, dd (14.9) | 149.5 (CH) | 2” | 1”, 2”, 5” |
| 4” | ||||
| 5” | 2.32, s | 13.6 (CH3) | ||
| 1’ | 1.47, m; 0.96, m | 33.0 (CH2) | ||
| 2’ | 1.31, m; 1.20, m | 27.9 (CH2) | ||
| 3’ | 1.24, mc | 29.3 (CH2)c | ||
| 4’ | 1.24, mc | 29.3 (CH2)c | ||
| 5’ | 1.38, m; 1.27, mc | 25.2 (CH2)c | ||
| 6’ | 1.40, m | 37.1 (CH2) | 7’ | |
| 7’ | 3.57, m | 71.2 (CH) | 6’, 8’ | |
| 8’ | 1.41, m | 39.3 (CH2) | 7’ | |
| 9’ | 1.43, m; 1.33, m | 18.5 (CH2) | 10’ | |
| 10’ | 0.91, t (6.7) | 13.6 (CH3) | 9’ | 8’, 9’ |
. 600 MHz.
, 150 MHz, multiplicities assigned by edited HSQC.
, interchangeable assignments.
The relative configurations of 1 and 2 were derived from a combination of J coupling analysis and 2D NOESY experiments (Figure 1). Scalar coupling (J = 4.8 Hz) and an NOE cross peak between1H NMR signals for H-4a and H-8a indicated a cis-decalin structure for 1. (2R*,3R*,4aR*,5S*,8aS*) (C-2,3 cis; C4a5-cis; C-4a,8a cis). NOESY correlations between H-2 and H-8a showed these hydrogens adopted a diaxial disposition. The small coupling constant (3J = 2.8 Hz) between H-2 and H-3 revealed that they adopted a syn configuration and H-3 occupied the equatorial position. By considering the information above, the protons H-2, H-3, H-4a and H-8a were determined to be all syn-configured.6
Assignment of the geometry of the C-2”–C-3” double bond of 1 is equivocal because the magnitude of the vicinal coupling constant lies close to the edge of the ranges for both E and Z double bonds. To securely assign the geometry by1H NMR comparison with a simple model compound, a mixture of geometrical isomers of β-methylthioacrylic acid (5, E:Z 40:1) was prepared by addition of NaSMe to freshly distilled propiolic acid (Figure 2).
Figure 2.

Preparation of geometrical isomers of 3-methylthioacrylic acid (5).
The chemical shifts and coupling constants of 1 showed an excellent match with those of the major isomer E-5 (δ 7.87, d, J = 14.7 Hz, H-3; 5.64, d, J = 14.7 Hz; 2.35, s,1JCH = 161.5 Hz), while the minor isomer Z-5 exhibited a smaller vicinal coupling constant (δ 7.46 (d, 1H, J = 10.2 Hz, H-3); 5.85 (d, 1H, J = 10.2 Hz, H-2). Therefore the C-2”–C-3” double bond of 1 has an E-configuration.
The reported lepadins exhibit heterogeneous relative and absolute configurations. The ring junction of the decahydroquinoline core in 1 has the same configuration as (+)-lepadin G2d, but is antipodal to (–)-lepadins A2a and B,2b,7,8 and diastereomeric to those of other lepadins. No conclusive relational assignments of configuration can be made by comparisons of [α]D, rendering the assignment of absolute configuration a non-trivial problem. Not all lepadins – compounds 1 and 2 included – are crystalline; consequently, X-ray is of limited help, and no generally useful chiroptical property resides in these alkaloid structures for the independent assignment of their absolute stereostructures. The only definitive configurational assignments in the lepadin series have been made by total synthesis. For example, asymmetric syntheses of lepadins B,7 D, E and H8 and also of F (3)9,10 and G9 allowed assignment of absolute configurations by comparisons of optical rotations of synthetic and natural products.
We reasoned that the absolute configuration of 1 could be assigned by exciton coupled circular dichroism (ECCD), exploiting the pendant methythioacrylate at C-3 – a red-shifted, relatively strong chromophore (λmax 270 nm) – and introduction of a second chromophore, specifically an N-p-bromobenzoyl group. N-Acylation of 1 (Scheme 1, p-Br-C6H4COCl, Et3N, DMAP, CH2Cl2) gave the corresponding p-bromobenzamide 6. Most of the ring1H NMR signals of tertiary amide 6 (600 MHz, CDCl3) were doubled (4:3 mixture of rotamers). Further examination of the1H NMR and NOESY data (Figure 1b) of 6 indicated that the azadecalin ring system had flipped to the alternate chair-chair conformation, most likely to relieve allylic strain about the tertiary amide. The1H NMR spectrum of 6 shows a broader multiplet structure for H-3 (δ 4.91, m, w1/2 = 20.8 Hz), compared to the narrower H-3 multiplet of 1 (δ 5.04, q, J = 2.8 Hz), as a consequence of the larger H-3ax-H-4ax coupling in addition to ax-eq couplings. The NOESY correlations of 6 (Figure 1b) were fully consistent with flipped azadecalin chair-chair conformation, including a prominent H-4ax-H3-9 cross peak. Molecular modeling of 6 (Figure 3) clearly supported the alternate chair-chair conformation with the C-8a–N bond disposed equatorially to the cyclohexane ring.11 This conclusion was upheld by DFT calculations (B3LYP/6–31+G(d)) of the minimized energy and geometry of 6 (Figure 1). Four low-lying conformations were discovered for 6, each with essentially the same conformation of the heterocyclic ring, and differing only in the rotational conformers about the amide group and the relatively free rotating 3-methylthioacrylate group.
Scheme 1.

Conversion of lepadin I (1) to the corresponding benzamide, 6, and Mosher’s esters 7a,b (S and R configurations at C*, respectively), and anisotropic1H NMR shifts (Δδ = δS – δR in parts per-billion, ppb. See Ref. 12). Due to1H signal overlap, signal assignments labeled * (confirmed by TOCSY, see SI) are interchangeable.
Figure 3.

(a) Energy minimized molecular model of 6 (Gaussian, B3LYP/6–31+G(d), See Supporting Information). (b) Helicity of the N–(C=O)-Ar and C-3–O bond vectors subtends a negative angle (approximately θ = –66.5˚) in 6 corresponding to the 2R,3R,4aR,5S,8aS configuration of natural 1.
Unlike 1, which exhibited only a weak simple Cotton effect, CE, λ 270 nm Δε +0.56), the CD spectrum of 6 (Figure 4) showed a strongly negative bisignate CE (λ 239 nm (Δε +4.7), 252 (0), 272 (–9.4)) characteristic of exciton coupling between the benzamide and the methythioacrylate chromophores. The electronic transition dipole moments of methylthioacrylate and p-bromobenzamide chromophores lie approximately parallel to the N-(C=O) and C-3-O bond vectors, respectively, and subtend an angle of ±66.5˚, the sign being dependent upon absolute helicity (Figure 3). The observed negative split Cotton effect is consistent with a helicity of –66.5˚; consequently, the absolute configuration of the heterocyclic core of natural 1 is (2R,3R,4aR,5S,8aS) as depicted.
Figure 4.

CD spectra of lepadin I (1), and its p-bromobenzamide 6 (MeOH, 23 ˚C).
The above-described assignment can be generalized method for assignments of other stereochemically heterogeneous lepadines, all of which share a C-3 secondary hydroxyl or acyloxy group (the latter can be converted to the former by saponification). Simple derivatization of the core 3-hydroxyperhydroquinoline structure as a bis-N,O-diarenoyl derivative (e.g. by dibenzoylation) will give rise to ECCD that can be interpreted in a manner similar to that of 6, assuming the N-benzamide ‘anchors’ the azadecalin in the preferred conformer (viz. Figure 5). Interpretation of the sign of the helicity for N-Bz and O-Bz electronic transition dipole moments in the idealized model will predict the sign of the split-Cotton effect as illustrated for the four possible C-2–C-3 diastereomers (Figure 6).
Figure 5.

Model for prediction of absolute configurations of N,O-diarenoyl lepadin derivatives from ECCD. (a) 2R,3S and 2R,3R – both negative split Cotton effects, and (b) 2S,3S and 2S,3R – both positive split Cotton effects.
Figure 6.

Structures of cytosaminomycin A (8), entadamides A (9) and B (10), planchonelline (11) and umbraculumin C (12).
The remaining asymmetric center, at the secondary carbinol C-7’, was solved independently by application of the advanced Mosher’s ester method.12 Acylation of 5 (Scheme 1, R- or S-MTPA-Cl, Et3N, DMAP, CH2Cl2) gave, after HPLC purification, the corresponding Mosher’s esters, (S)-7a and (R)-7b, respectively. The anisotropic1H NMR shifts (Δδ) about the C-7’ MTPA group in 7a,b (Scheme 1) conformed to the Mosher’s ester model12 and therefore, the configuration is 7S in 1. Therefore, the complete configuration of 1 is 2R,3R,4aR,5S,8aS,7’S. Given that lepadins I–K were obtained from the same ascidian sample, it is likely 1-3 share the 7S’ configuration.
The free acid 5 was first reported as a secondary metabolite from fermentations of Streptomyces lincolnensis var. lincolnensis,13 and derivatives have been described since from other Streptomyces (e.g. the modified cytosine nucleoside, cytosaminomycin A (814), Figure 6). Derivatives of 5 are less common in plants. The first reported natural product 3-methylthioacrylate derivatives are petasol esters B and C, isolated from extracts of butterbur, Petasites officinalis.15 Subsequently, 3-methylthioacrylate esters S-petasin16b and related sesquiterpenes16a were isolated from other Petasites spp. Entadamides A (9) and B (10), from the seeds of the tropical legume, Entada phaseolloides, are 3-methylthioacrylamides of ethanolamine.17
The pyrrolizidine alkaloid, planchonelline (11), was purified from Planchonella thyrsoidea collected in Papua New Guinea.18 Compounds 1-3 constitute the second finding of methythioacrylate esters from a marine invertebrate; Cimino and coworkers earlier reported the 1,2-diacyl-sn-glycerol, umbraculumin C (12), from the skin glands of the Mediterranean opisthobranch, Umbraculum mediterraneum.19
The origin of 5 for biosynthesis 1–3, 8-12 – which conceivably may proceed through either oxidative deamination and S-methylation of L-cysteine (Scheme 2), or more likely, oxidative degradation of L-methionine – has been studied only in Streptomcyes.
Scheme 2.

Possible origin of 3-methylthioacrylic acid (5) in the biosynthesis of 1–3 and 8-12. The ordering of reactions is arbitrary.
Visser and Meyer showed, in-time dependent methionine-supplemented fermentation of S. lincolnensis, the appearance of 5 in the culture tracked with the concentration of methionine and production of lincomycin; therefore, 5 may be a shunt metabolite of secondary metabolite production. Surette and Vining demonstrated that cultures of S. lincolnensis, supplemented with either S-[14CH3]-methionine or S-[14CH3]-3-methylthiopropionic acid (but not S-[14CH3]-3-methylthiopropionamide) delivered [14C]-5.20
The acrylate ester present in 1–3 appears to be a strong electrophile; a consequence of a polarized double bond, made more so by the presence of the 3-methythio group, and evidenced by the large differences in the1H and13C NMR chemical shifts for the α and β CH signals. Consequently, 1–3 may be competent Michael acceptors and possible alkylating agents. For example, natural products 9 and 10 are potent inhibitors of 5-lipoxygenase of RBL-1 cells (IC50 ~0.1 μg.mL–1), a property suggestive of the electrophilic nature of derivatives of 5, and touted as the core of “a new type of anti-inflammatory drug”.21,22 The activity of 1 was briefly examined in an anti-cholinesterase reporter assay using a modification of Ellman’s photometric method23 with physostigmine as a positive control IC50 = 1.48 μM). Compound 1 showed no significant activity (IC50 >100 μM) toward acetylcholineesterase (AChE), but modest inhibition of butyrylcholineesterase activity (BuChE, IC50 = 3.1 μM) which is indicative of non-specific activity.
In conclusion, three new decahydroquinolines, lepadins I (1), J (2) and K (3), embodying a rare 3-methylthioacrylate, were isolated from a Bahamian Didemnum sp. A general method was developed for assignment of absolute configuration of 3-hydroxylated perhydroquinolines – the core heterocycle of the lepadin family – based on exciton coupled CD. Compound 1 showed modest inhibitory activity against butyrylcholineesterase, and essentially no activity against acetylcholinesterase.
Experimental Section
General Experimental Procedures.
General experimental procedures are described elsewhere.24 Sub-milligram reaction % yields were determined by the1H NMR method of quantitation by solvent13C satellite (QSCS)25 and external calibration against 3α-bromocholestane recorded under the same conditions.13C NMR signal multiplicities (CH3, CH2, CH, Cq) were determined from edited HSQC or APT experiments. Optical rotations were measured in a Jasco P-2000 digital polarimeter using a 1.00 cm quartz cell (fill volume = 125 μL) at the emission D line of Na˚. HRMS measurements were made on an Agilent 6350 TOFMS instrument using ESI in positive ion mode.
Animal Material.
The red-orange encrusting ascidian Didemnum sp. (08–13–072) was collected, in June of 2008, by hand using scuba at Stirrup Cay at a depth of approximately –10 m. A type sample is archived at UC San Diego. The material was stored in MeOH at –20 ˚C (five years) and the MeOH-extract concentrated and solvent-partitioned in the following manner.
Extraction and Isolation of Lepadins I–K (1–3).
An aliquot of the total MeOH extract of the ascidian (30 mL) was adjusted to 9:1 MeOH/H2O and partitioned with hexanes (30 mL). The aqueous-MeOH layer was adjusted to 40% H2O and further extracted with CHCl2 (2 × 35 mL). The CHCl2-MeOH fraction (99 mg) was concentrated and partially purified on silica column (1.5 × 9 cm) using stepwise gradient of CHCl2/MeOH (100:0–0:100 v/v). The vanillin positive bands were combined, resulting in five fractions. Fraction five (34.0 mg) was subjected to semi-preparative HPLC (Microsorb NH2 column, 4.6 mm × 250 mm with an isocratic solvent hexane:i-PrOH:Et2NH, 90:10:1, v/v/v). Lepadin I (1) was obtained as the TFA salt together with Et2NH•TFA (25.4 mg) (r.t. = 7.3 min) and a mixture (1.2 mg) of lepadin J (2), K (3) and Et2NH•TFA. Further purification of the latter mixture by HPLC (Microsorb NH2 column, 4.6 mm × 250 mm; gradient: 0–2 min: 9% i-PrOH-hexane (1% i-Pr2NH), gradient to 15 min: 10.5% i-PrOH/hexane (1% i-Pr2NH); 1 mL/min) gave additional lepadin J (2) (0.2 mg, r.t. = 10.3 min), however, lepadin K (3) could not be recovered.
Lepadin I (1).
Colorless oil. Free base: [α]D23 +4.1±0.6 (c 0.24, MeOH), TFA salt: [α]D23 +15.0±0.9 (c 0.25, MeOH). UV (MeOH) λmax 275 (ε 3500). CD (MeOH) λ 269 (Δε +0.56). FTIR (ATR, ZnSe). 3300–2900 (bs), 1672 (s), cm–1.1H NMR ,13C NMR (see Table 1). HRESIMS m/z 426.3037 [M+H]+ (calcd for C24H44NO3S, 426.3036).
Lepadin J (2).
Colorless oil.1H NMR,13C NMR (see Table 2). FTIR (ATR, ZnSe). 3800–2900 (bs), 1672 (s), cm–1. (see Table 2). HRESIMS m/z 424.2886 [M+H]+ (calcd for C24H42NO3S, 424.2880).
Lepadin K (3).
1H NMR,13C NMR (see Table 3). ESIMS m/z 426.3 [M+H]+ (calcd for C24H44NO3S, 426.3).
3-Methythioacrylic acid (5)
Sodium thiomethoxide (123.2 mg, 1.74 mmol) was added to stirred, freshly distilled propiolic acid (123.0 mg, 1.74 mmol). After 15 minutes, methanol was added and stirring continued for 15 h during which time a flocculent white precipitate formed. Aqueous citric acid (10% w/v, 0.2 mL) was added and the mixture extracted with a mixture of diethyl ether and hexane (1:1, 2 × 1 mL). The organic layer was dried (MgSO4) and concentrated to give a pale yellow solid which was shown by1H NMR to be exclusively 3-methylthioacrylic acid (80.0 mg, 39%, 5, E:Z = 40:1).1H NMR (CDCl3) major isomer E-5. δ 7.87 (d, J = 14.7 Hz,1JCH = 168.5 Hz, H-3); 5.64 (d, J = 14.7 Hz,1JCH = 165.8 Hz, H-2); 2.35 (s, 3H,1JCH = 136 Hz, SMe), consistent with literature values.1813C NMR (CDCl3) δ 170.2 (s, C-1), 150.3 (CH), 112.2 (CH), 14.51 (CH3).
When the addition reaction was repeated with CH3SH (NaSMe and 0.86 equ AcOH in MeOH), the major product was Z-5 (E:Z = 1:10), albeit isolated in low yield (5%).
minor isomer Z-5. δ 7.46 (d, 1H, J = 10.2 Hz, H-3); 5.85 (d, 1H, J = 10.2 Hz, H-2) consistent with literature values.17a
Lepadin I p-bromobenzamide (6)
A solution of p-bromobenzoyl chloride in CH2Cl2 (1M, 25 μL, 25 μmol) was added to a stirred solution of 1 (1 mg, 2.3 μmol) in CH2Cl2 (ca. 0.2 mL) containing a crystal of DMAP and triethylamine (10 μL, 69 μmol). The mixture was stirred for 6 h at room temperature then concentrated under a stream of N2. The residue was applied to a ‘pencil column’ packed with silica (8.5 × 50 mm) and eluted with 1:19 MeOH-CH2Cl2 followed by 1:4 MeOH-CH2Cl2. The fraction containing 6 eluted after 9 column volumes and was further purified by HPLC (silica, 250 × 10 mm, 40:60 EtOAc-hexanes, 4.0 mL.min–1, UV detection λ = 250 nm) to give pure benzamide 6 (396 μg, 28%). [α]D23 –3.2 (c 1.58, MeOH); UV (MeOH) λmax 272 nm (log10ε 4.50). CD (MeOH, 23 ˚C) λ 239 nm (Δε +4.7), 252 (0), 272 (–9.4).1H NMR (600 MHz, CDCl3, 4:3 mixture of amide rotamers) δ major [minor] 7.68 (d, 1H, J = 14.9 Hz, H-3”, [7.77]), 7.53 (d, J = 8.7 Hz, 2H, p-Br-Bz), 7.22 (d, J = 8.7 Hz, 2H, p-Br-Bz), 5.54 (d, J = 14.9 Hz, H-2”, [5.64]), 4.91 (m, 1H, H-3, [5.02]), 4.72 (m, 1H, H-8a, [3.71]), 4.15 (pent, 1H, J = 6.8 Hz, H-2, [5.02]), 3.61 (m, 1H, H-7’), 2.30 (s, 3H, SMe, [2.35]), 2.12 (m, 1H, H-4ax, [2.12]), 1.87 (m, 1H, H-8, [1.86]), 1.83 (m, 1H, H-4a, [1.54]), 1.68 (m, 1H, H-8, [1.39]), 1.64 (m, 1H, H-4eq, [1.69]), 1.13 (d, 3H, J = 6.8 Hz, C-2 Me, [1.33]), 0.93 (t, 3H, J = 7.3 Hz, H-10’). HRESIMS m/z 630.2222, 632.2207 [M+Na+] (calcd for C31H4679BrNO4SNa, 630.2223).
Mosher’s Esters 7a,b – Assignment of the Absolute Configuration at C-7’
(R)- and (S)-α-Methoxyl-α-(trifluoromethyl)phenylacetyl chloride (3 μL, 16 μmol) was added to separate solutions of 6 (100 μg, 0.16 μmol) and DMAP (0.1 mg, 0.8 μmol) in pyridine-d5 (30 μL). The reactions, monitored by1H NMR, were complete in 1 h. The mixtures were dried under reduced pressure, re-dissolved in EtOAc, and the solutions sequentially washed with 0.5 M HCl, saturated NaHCO3 and brine, then dried over anhydrous Na2SO4. After removal of solvent, the residues were separately purified by HPLC (silica, 250 × 4.6 mm, 20:80 EtOAc-hexanes, 1.5 mL.min–1, UV detection λ = 250 nm) to give pure (S)-7a (35 μg) or (R)-7b (38 μg), respectively.
7a: (S)-Mosher ester:1H NMR (600 MHz, CDCl3, 4:3 mixture of amide rotamers), partial side-chain assignments: δ 0.85 (t, J = 6.8 Hz, H-10’); 1.26, m and 1.19, m (H-9’); 1.58, m and 1.53, m, H-8’); 5.11 (m, H-7’); 1.66, m and 1.60, m (H-6’); 1.28, m, 1.20, m; 1.31, m; 1.27, m, 1.29 (H-5’ to H-3’). The remainder of the1H NMR signals of 7a, exclusive of the MTPA group, were identical to those of 6. HRESIMS m/z 846.2622 [M+Na+] (calcd for C41H5379BrF3NO6SNa, 846.2621).
7b: (R)-Mosher ester:1H NMR (600 MHz, CDCl3, 4:3 mixture of amide rotamers) δ 0.92 (t, J = 6.8 Hz, H-10’); 1.35, m and 1.19, m (H-9’); 1.67, m and 1.58, m (H-8’); 5.11, m (H-7’); 1.57 (m, H-6’); 1.21, m; 1.15, m; 1.17, m; 1.23, m, 1.14, m (H-5’ to H-3’). The remainder of the1H NMR signals of 7b, exclusive of the MTPA group, were identical to those of 6. HRESIMS m/z 846.2625 [M+Na+] (calcd for C41H5379BrF3NO6SNa, 846.2621).
Supplementary Material
Acknowledgement.
We thank Javier Turon and Suzanna Lopez-Gentil for the gift of the ascidian sample, Y. Su measurement of HRMS, A. Renshaw and M. Jamison for assistance with 500 MHz NMR measurements. Acquisitions of the Agilent TOF mass spectrometer and the 500 MHz NMR spectrometer were made possible with funds from the NIH Shared Instrument Grant program (S10RR025636) and the NSF Chemical Research Instrument Fund (CHE0741968), respectively. We are grateful for funding that supports this work from the NIH (AI100776–05 and AT009783–01 to TFM).
Footnotes
Supporting Information Available.1H NMR for 1-3,13C NMR and APT spectra for 1, 1D and 2D NMR data for 6-7a,b and B3LP/6–31+G(d) calculations for 6 are available free of charge via Internet at http://pubs.acs.org.
References:
- (1).(a) Molinski TF Marine Pyridoacridine Alkaloids: Structure, Synthesis and Biological Activity. Chem. Rev 1993, 93, 1825–1838. [Google Scholar]; (b) Ireland CM; Roll DM; Molinski TF; McKee TC; Zabriskie TM; Swersey JC, Uniqueness of the Marine Chemical Environment: Categories of Marine Natural Products from Invertebrates In Biomedical Importance of Marine Organisms, Fautin DG, Ed. California Academy of Sciences: San Francisco, 1988; Vol. 13, pp 41–57.. [Google Scholar]
- (2).(a) Lepadin A , Clavelina lepadiformis: Steffan, B. Lepadin A, A Decahydroquinoline Alkaloid from the Tunicate Clavelina lepadiformis. Tetrahedron 1991, 47, 8729–8732. [Google Scholar]; (b) Lepadins B,and C Clavelina lepadiformis and Prostheceraeus villatus: Kubanek, J.; Williams, D. E.; de Silva, E. D.; Allen, T.; Andersen, R. J. Cytotoxic Alkaloids from the Flatworm Prostheceraeus villatus and its Tunicate prey Clavelina lepadiformis. Tetrahedron Lett 1995, 36, 6189–6192. [Google Scholar]; (c) Lepadins D-F, Didemnum sp.: Wright, A. D.; Goclik, E.; König, G. M.; Kaminsky, R. Lepadins D-F. Antiplasmodial and Antitrypanosomal Decahydroquinoline Derivatives from the Tropical Marine Tunicate Didemnum sp . J. Med. Chem 2002, 45, 3067–3072. [DOI] [PubMed] [Google Scholar]; (d) Lepadins F,G and H, Aplidium tabascum: Davis, R. A.; Carroll, A. R.; Quinn, R. J. Lepadins F-H, New cis-Decahydroquinoline Alkaloids from the Australian Ascidian Aplidium tabascum. J. Nat. Prod 2002, 65, 454–457. [DOI] [PubMed] [Google Scholar]
- (3).Tsuneki H; Toyooka N; Sasaoka T; Nemoto H; Dani JA; Kimura I Marine Alkaloids (–)-Pictamine and (–)-Lepadin B Block Neuronal Nicotinic Acetylcholine Receptors. Biol. Pharm. Bull 2005, 28, 611–614. [DOI] [PubMed] [Google Scholar]
- (4).Caserio MC; Pratt RE; Holland RJ The Nature of Sulfur Bonding in α,β-Unsaturated Sulfides and Sulfonium Salts. J. Am. Chem. Soc 1966, 88, 5747–5753. [Google Scholar]
- (5).Lepadin J (2) co-occurred with an inseparable, isomeric minor component which we surmise is the corresponding Δ4a,8a double bond isomer.
- (6).For lepadins J and K, no useful through space NOEs were observed. For K, strong NOE correlations were observed between H3 and H4, but no NOE was observed between H-3 and H-4a.
- (7).Toyooka N; Okumura M; Takahata H; Nemoto H Construction of 4a,8a-cis-Octahydroquinolin-7-one Core Using an Intramolecular Aldol Type of Cyclization: An Application to Enantioselective Total Synthesis of Lepadin B. Tetrahedron 1999, 55, 10673–10684. [Google Scholar]
- (8).Pu X; Ma D Total Synthesis of Lepadins B, D, E, and H; Determination of the Configuration of the Latter Three Alkaloids. Angew. Chem. Int. Ed 2004, 43, 4322–4225. [DOI] [PubMed] [Google Scholar]
- (9).Niethe A; Fischer D; Blechert S Total Synthesis of ent-Lepadin F and G by a Tandem Ene-Yne-Ene Ring Closing Metathesis. J. Org. Chem 2008, 73, 3088–3093. [DOI] [PubMed] [Google Scholar]
- (10).Li G; Hsung RP; Slafer BW; Sagamanova IK Total Synthesis of (+)-Lepadin F. Org. Lett 2008, 10, 4991–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).The cyclohexane chair, with an equatorial C-8a–N, bond alleviates non-bonded torsional strain of the pyramidalized amide (C=O)-N bond, a phenomenon that Ma and coworkers had also observed in the corresponding trifluoroacetamide of lepadin F derivatives (seeRef. 8).
- (12).Ohtani I; Kusumi T; Kashman Y; Kakisawa H High-Field FT NMR Application of Mosher’s Method. The Absolute Configurations of Marine Terpenoids. J. Am. Chem. Soc 1991, 113, 4092–4096. [Google Scholar]
- (13).Visser J; Meyer HF trans-3-(Methythio)-acrylic Acid, a New Metabolic Product from Streptomyces lincolnensis. J. Antibiotics 1969, 22, 510. [DOI] [PubMed] [Google Scholar]
- (14).Shiomi K; Haneda K; Tomoda H; Iwai Y; Omura S Cytosaminomycins, New Anticoccidial Agents Produced by Streptomcyes sp. KO8119. II. Structure Elucidation of Cytosaminomycins A, B, C and D. J. Antibiotics 1994, 47, 783–786. [DOI] [PubMed] [Google Scholar]
- (15).Stoll A; Morf R; Rheiner A; Renz J Über Inhaltsstoffe aus Petasites officianalis Moench I. Petasin und die Petasolester B und C. Experientia 1956, 12, 360–362. [DOI] [PubMed] [Google Scholar]
- (16).(a) Naya K; Hayashi I; Takagi S; Nakamura M; Kobayashi M Bull. Chem. Soc. Jap 1972, 45, 3673–3685. [Google Scholar]; (b) Sugama K; Hayashi K; Nakagawa T; Mitsuhashi H; Yoshida N Phytochemistry 1983, 22, 1619–1622 [Google Scholar]
- (17).(a) Ikegami F; Shibasaki I; Ohmiya S; Ruangruangsi N; Murakoshi I Entadamide A, a New Sulfur-Containing Amide from Entada phaseoloides Seeds. Chem. Pharm. Bull 1985, 33, 5154–5154. [Google Scholar]; (b) Ikegami F; Ohmiya S; Ruangrungsi N; Sakai S-I; Murakoshi I Entadamide B, a Second New Sulfur-Containing Amide from Entada phaseoloides. Phytochemistry 1987, 26, 1525–1526. [Google Scholar]
- (18).Hart N; Lamberton J Pyrrolizidine Alkaloids from Planchonella Species (Family Sapotaceae). Aust. J. Chem 1966, 19, 1259–1264. [Google Scholar]
- (19).(a) Cimino G; Crispino A; Spinella A; Sodano G Two Ichthyotoxic Diacylglycerols from the Opisthobranch Mollusc Umbraculum mediterraneum. Tetrahedron Lett 1988, 29, 3613–3616. The absolute configuration of 11 was solved, independently, through chemical interconversion [Google Scholar]; (b) Gavagnin M; Spinella A; Cimino G; Sodano G Stereochemistry of Ichthyotoxic Diacylglycerols from Opisthobranch Molluscs. Tetrahedron Lett. 1990, 31, 6093–6094 [Google Scholar]; (c) De Medeiros EF; Herbert JM; Taylor RJK The Synthesis and Absolute Configuration of the Novel Ichthyotoxic Diacyiglycerols, Umbraculumin A and Umbraculumin C. J.C.S. Perkin Trans 1 1991, 2725–2730. [Google Scholar]
- (20).Surette R; Vining LC Formation of 3-Methythioacrylic Acid from Methionine by Streptomyces lincolnensis. Isolation of a Peroxidase. J. Antibiotics 1976, 29, 646–652. [DOI] [PubMed] [Google Scholar]
- (21).Ikegami F; Sekine T; Aburada M; Fujii Y; Komatsu Y; Murakoshi I Synthesis of Entadamide A and Entadamide B Isolated from Entada phaseoloides and Their Inhibitory Effects on 5-Lipoxygenase. Chem. Pharm. Bull 1989, 37, 1932–1933. [DOI] [PubMed] [Google Scholar]
- (22).Vinyl sulfonium salts are strong Michael acceptors and have been proposed as useful moieties for design of enzyme inhibitors. Zhao G; Zhou ZS Vinyl sulfonium as novel proteolytic enzyme inhibitor. Bioorg. Med. Chem. Lett 2001, 2331–2335. [DOI] [PubMed] [Google Scholar]
- (23).Ellman GL; Courtney KD; Andres V Jr. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol 1961, 7, 88–95. [DOI] [PubMed] [Google Scholar]
- (24).Dalisay DS; Rogers EW; Edison A; Molinski TF Structure Elucidation at the Nanomole Scale. 1. Trisoxazole Macrolides and Thiazole-Containing Cyclic Peptides from the Nudibranch Hexabranchus sanguineus. J. Nat. Prod 2009, 72, 732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dalisay DS; Molinski TF NMR Quantitation of Natural Products at the Nanomole Scale. J. Nat. Prod 2009, 72, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.