

Abstract
GCN5L1 regulates mitochondrial protein acetylation, cellular bioenergetics, reactive oxygen species (ROS) generation, and organelle positioning in a number of diverse cell types. However, the functional role of GCN5L1 in the heart is currently unknown. As many of the factors regulated by GCN5L1 play a major role in ischemia-reperfusion (I/R) injury, we sought to determine if GCN5L1 is an important nexus in the response to cardiac ischemic stress. Deletion of GCN5L1 in cardiomyocytes resulted in impaired myocardial post-ischemic function and increased infarct development in isolated work-performing hearts. GCN5L1 knockout hearts displayed hallmarks of ROS damage, and scavenging of ROS restored cardiac function and reduced infarct volume in vivo. GCN5L1 knockdown in cardiac-derived AC16 cells was associated with reduced activation of the pro-survival MAP kinase ERK1/2, which was also reversed by ROS scavenging, leading to restored cell viability. We therefore conclude that GCN5L1 activity provides an important protection against I/R induced, ROS-mediated damage in the ischemic heart.
Keywords: GCN5L1, Ischemia Reperfusion, ERK1/2, Reactive Oxygen Species, Ex Vivo Working Heart
INTRODUCTION
The burden of myocardial ischemia-reperfusion (I/R) injury continues to be a leading health problem in the US.1 While progress in reperfusion strategies has improved the immediate mortality of myocardial infarction (MI) patients, the resultant injury to the heart may lead to long-term remodeling and failure. The degree of severity of I/R injury is associated with impaired mitochondrial function and reactive oxygen species generation (ROS) after reperfusion.2–5 Thus, strategies to preserve mitochondrial bioenergetic output and reduce ROS production hold the potential to positively impact cardiac function after I/R injury, and mitigate the progression to infarction-induced heart failure.
Mitochondrial enriched GCN5L1 has recently emerged as a key regulator of mitochondrial protein acetylation, biogenesis and metabolism.6–10 GCN5L1 is expressed in cardiac myocytes, and our laboratory has recently shown an increase in GCN5L1 in the hearts of mice exposed to a high fat diet.10 In the liver, GCN5L1 expression has been associated with the modulation of metabolism and ROS generation.11 However, the role of GCN5L1 in the response of the heart to I/R injury remains unknown.
To address this question, we used a novel conditional knockout mouse model and in vitro tools to examine the role of GCN5L1 under ischemic stress in the heart. Data presented in this manuscript show that the loss or downregulation of GCN5L1 exacerbates I/R injury. We show that hypoxia and reoxygenation results in reduced GCN5L1 expression. We demonstrate using in vitro and ex vivo models that GCN5L1 knockout or knockdown results in decreased cell survival and reduced cardiac function after I/R. In addition, we demonstrate that these changes are accompanied by decreased activation of cardioprotective MAP kinases under ischemic conditions, and that mitigating this defect with ROS scavenging increased both pro-survival signaling and cardiac cell viability. These results indicate that GCN5L1 is a critical modulator of post-ischemic cardiac health and function.
METHODS
Human samples
Failing human heart tissue was obtained from patients (with informed consent) at the University of Pittsburgh Medical Center, and with Institutional Review Board approval. All patients (mean age 58+/− 5.8 yrs [S.D.]; 4M/4F) had end stage chronic ischemic cardiomyopathy, and none were on mechanical support devices. Trans mural samples of the lateral wall of the left ventricle (LV) were obtained from failing human hearts at the time of cardiac transplantation (n=8). Non-failing human heart tissue was obtained at the Cleveland Clinic Foundation with Institutional Review Board approval. Trans mural samples of the lateral wall of the LV were obtained from unmatched donor hearts (n=8) that did not meet criteria for transplantation (donor age 51+/−13.1 yrs [S.D.]; 4M/4F; cause of death, 5 CVA/3 trauma). All cardiac tissue was placed into cold cardioplegic solution (4-8 °C) and rapidly transported to the laboratory. Tissues were then snap frozen in liquid nitrogen and stored at −80 °C until biochemical analyses.
Mouse generation and housing
All mice were bred and housed in accordance with the University of Pittsburgh’s IACUC protocols. Cardiac-specific inducible knockouts were generated by EUCOMM at our request on a C57BL/6J background by crossing αMHC-Cre (Jax B6.FVB(129)-Tg(Myh6-cre/Esr1*)1JMK/J) mice (i.e. αMHC-MerCreMer) to mice with LoxP sites introduced around exon 3 of Bloc1s1 (GCN5L1FL) (Figure 2 A). Cardiomyocyte-specific knockout was induced via tamoxifen injection (single 40 mg/kg IP injection), and confirmed with qPCR and immunoblot (Figure 2 B,C). Conditional knockout mice showed no overt phenotype within the experimental timeframe. Heart weight/body weight ratio and Heart weight/tibia length showed no difference (Supplemental Figures 2,3).
Figure 2. Development of cardiac-specific GCN5L1 knockout (cKO) mice.
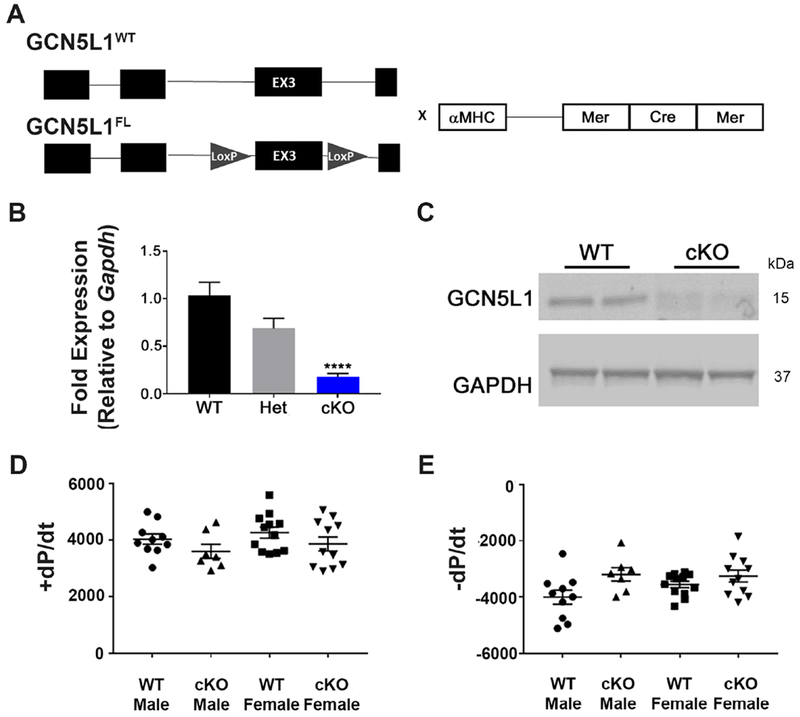
(A) Mice with LoxP sites surrounding exon 3 of GCN5L1 were crossed to αMHC-MerCreMer mice to produce tamoxifen-inducible, cardiomyocyte-specific knockouts of GCN5L1 (GCN5L1 cKO). (B, C) MerCreMer expression in GCN5L1FL/FL cardiomyocytes leads to significant reductions in GCN5L1 transcript and protein expression. (D, E) No overt contractile phenotype is observed in the isolated hearts of cardiac-specific inducible knockouts. N= 5-12. **** = P < 0.0001 vs. control.
Isolated Working heart and Ischemia-Reperfusion
Hearts from anesthetized mice were rapidly excised and cannulated via the aorta in warm oxygenated Krebs-Henseleit buffer (118 mM NaCl, 25 mM NaHCO3, 0.5 mM Na-EDTA [Disodium salt dihydrate], 5 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 11 mM glucose). Retrograde perfusion (i.e. Langendorff) perfusion was initiated to blanch the heart, maintained at a constant aortic pressure of 50 mmHg with a peristaltic pump through a Starling resistor. A small incision was next made in the pulmonary artery to allow perfusate to drain, and the heart was paced at a rate slightly higher than endogenous. The left atrium was then cannulated via the pulmonary vein, and anterograde perfusion was initiated with a constant atrial pressure of 11 mmHg against an aortic workload of 50 mmHg. Left ventricle pressure was measured via Mikro-tip pressure catheter (Millar) carefully inserted into the LV through the aorta. Hearts were paced at a rate slightly above intrinsic rate (~360-500 bpm). The work-performing heart was permitted to equilibrate for 30 minutes to establish baseline functional parameters. Cardiac effluent was collected at intervals to measure LDH release. Global no-flow ischemia was initiated by stopping flow to the left atrium for 20 minutes, followed by reperfusion for 60 minutes. Throughout the experiment, aortic and atrial pressure, left ventricular pressure, cardiac output, and aortic flow were directly measured, and calculated ±dP/dT, coronary flow, and heart rate were monitored. Hearts were reperfused for 60 minutes, as this duration has been shown to be sufficient for effective measurement of functional impairment and tissue damage.12,13 Subsequently, hearts were either removed from the cannula and snap frozen for protein analysis, or perfused with a 1% triphenyltetrazolium chloride (TTC) solution in a phosphate buffer to delineate viable tissue from infarct. For protein analysis, hearts were removed from the cannula after 30 minutes of equilibration (sham), 5 minutes of reperfusion, or 45 minutes of reperfusion. To test the effects of ROS scavenging on recovery, 0.5mM N-acetyl cystine (NAC) was added to reperfusion buffer.
Infarct analysis
Hearts perfused with TTC were incubated at 37 °C for 15 minutes, then soft frozen on dry ice for an hour. Frozen hearts were cut by hand into 5-7 sections, which were then weighed and incubated in formalin overnight. Sections were imaged using AmScopeX MU300 at 2X. White tissue (infarct) was traced using ImageJ and area was calculated as a percentage of the whole section. Infarct volume was calculated by multiplying the infarct area by the weight of each section.
Cell culture and transfection
AC16 cells, a proliferating human cardiac myocyte line derived from fusion of primary adult ventricular myocytes with SV40 fibroblasts,14 were purchased from Millipore. Cells were cultured in DMEM supplemented with 10% FBS and Antibiotic Antimycotic (ThermoFisher). All cultures and experiments were conducted in 25 mM glucose unless otherwise stated. Cells were infected with lentiviral shRNA for GCN5L1 or control scrambled sequence (Sigma-Aldrich) at an MOI of 10. Selection with puromycin eliminated non-transduced cells. Stable knockdown was confirmed and quantitated using RT-qPCR and immunoblotting.
Hypoxia–Reoxygenation of cultured cells
AC16 cells with stable knockdown of GCN5L1 were plated and incubated at 37 °C overnight. Media was replaced with DMEM (normoxia) or Esumi Buffer: 137 mM NaCl, 12 mM KCl, 0.5 mM MgCl2, 0.9 mM CaCl2, 20 mM HEPES; 20 mM 2-deoxy-d-glucose (2-DG), pH 6.2 (hypoxia). Cells were incubated under hypoxic conditions (1% O2, 5% CO2, 94% N2) for indicated time frames, followed by replacement of the media with normoxic buffer and further incubation for specified time frames under normal atmospheric oxygen. For n-acetyl cysteine (NAC) studies, cells were incubated with 0.5 mM NAC on reoxygenation for 30 minutes.
Survival and growth assays
Live cells were quantitated in 96-well plates using CCK-8 kit (Sigma) according to the manufacturer’s instructions. For H/R, absorbances were normalized to simultaneously cultured normoxia controls to determine percent survival.
Immunoblotting
Tissue or cells were lysed in 1% CHAPS buffer. Protein was quantitated using a BioDrop μLITE analyzer (BioDrop), and 20 μg of protein was loaded and run on an SDS page gel, then transferred to nitrocellulose membranes. Membranes were blocked using Odyssey blocking buffer and incubated in primary antibodies overnight (GAPDH, 1:1000, ERK1/2 [p42/44], 1:1000, p-ERK1/2 [p-p42/44][Thr202/Tyr204] 1:1000, p38 1:1000, p-p38 [Thr180/Tyr182] 1:1000, JNK 1:1000, pJNK [Thr183/Tyr185] 1:500: Cell Signaling; GCN5L1 1:1000, generated as previously described), followed by incubation at room temperature with fluorescent secondary antibodies for 1 hour (800 nm anti-rabbit, LiCor). Bands were visualized using an Odyssey Imager and quantitated using Image Studio Lite v 5.2 (LiCor).
Carbonylation assay
Cell lysates prepared as described above were tested for protein carbonylation using a Protein Carbonyl Content Assay Kit (Sigma) according to the manufacturer’s instructions.
RT-qPCR
RNA was isolated from tissue or cells using RNEasy kit (Qiagen). RNA was quantitated, and 500 ng-1 μg were used to generate cDNA using Maxima Reverse Transcriptase (ThermoFisher). Quantitative PCR was performed using SYBR-Green (ThermoFisher) and primers for Gcn5l1 and Gapdh for normalization.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 7. Student’s t-tests were used for simple comparisons between groups. One-way Analyses of Variance (ANOVA) were used to compare more than two groups, followed by post-hoc Student’s t-tests. For studies examining multiple timepoints (working heart time course endpoint, LDH assays, Seahorse data) a 2-way ANOVA was used with post-hoc Sidak’s multiple comparisons tests. A P value < 0.05 was taken as significant. All data are represented as the mean ± SEM.
RESULTS
Ischemia reduces GCN5L1 expression in cardiac tissues and cells
GCN5L1 has recently been established as a key regulator of cardiac metabolism.10 Given the importance of metabolic regulation on the progression of ischemic heart disease, we examined whether GCN5L1 expression is altered in ischemic human hearts. We found a significant reduction in GCN5L1 expression in hearts from patients suffering from chronic ischemic injury compared to non-failing hearts (Figure 1 A), suggesting that the etiology of ischemic injury may include the loss of GCN5L1. Since these samples are from end-stage hearts subject to chronic ischemia, they may display compensatory or long-term expression changes unrelated to GCN5l1 expression. We therefore investigated whether acute ischemia could induce the same effects, using an in vitro model of human proliferating cardiomyocyte cells. We chose AC16 cells, a human-origin proliferating cell line. These cells are the product of fusion between adult human ventricular myocytes and SV40 fibroblasts, with gene expression patterns14 and a metabolic profile15 similar to primary human ventricular myocytes. A significant reduction in GCN5L1 protein after four hours of hypoxia followed by reoxygenation was observed (Figure 1 B). These results prompted us to next examine whether this decrease in expression might impact the progression of I/R injury.
Figure 1. Ischemia downregulates GCN5L1 expression.
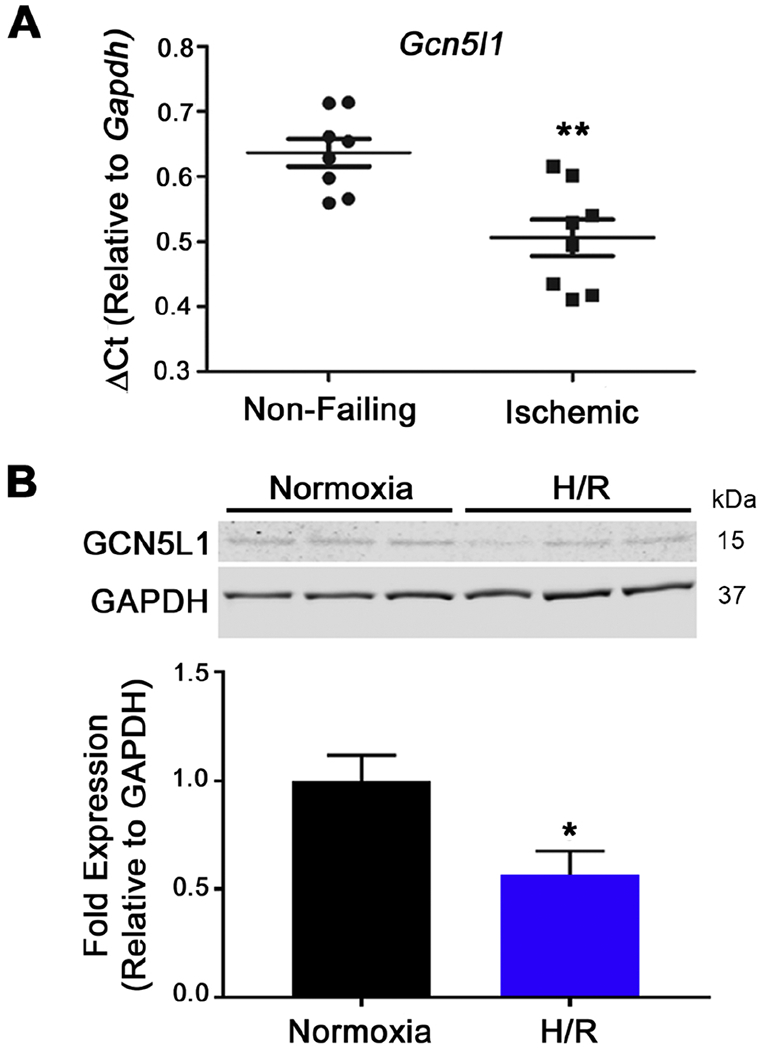
(A) GCN5L1 expression in human heart tissue from non-failing and chronic ischemic failing hearts normalized to Gapdh. N = 8 per group. (B) Immunoblot and quantitation of GCN5L1 protein from a human cardiomyocyte-derived cell line subjected to acute hypoxia and reoxygenation. N=6. * = P < 0.05, ** = P < 0.01, vs. control.
Loss of GCN5L1 predisposes ex vivo hearts to ischemia-reperfusion injury
To test the effect of GCN5L1 loss on recovery from I/R injury, mice with inducible cardiomyocyte-specific knockout of GCN5L1 (GCN5L1 cKO) were generated (Figure 2 A). Loss of GCN5L1 mRNA and protein expression was confirmed in cardiac lysates after tamoxifen injection (Figure 2 B,C), while no change in GCN5L1 protein expression was observed in other tissues examines (Supplemental Figure 1). There were no changes in overt phenotype or cardiac size ratio observed after knockout induction within the time frame studied (Figure 2 D,E and Supplemental Figure 2). Echocardiographical analysis revealed no difference in cardiac function or morphology in vivo (Supplemental Figure 3).
To test whether GCN5L1 loss results in changes in cardiac function, hearts from GCN5L1 cKO mice were evaluated using an ex vivo working heart procedure. Hearts were then subjected to global no-flow ischemia followed by reperfusion (Figure 3 A). While wildtype hearts showed a significant degree of recovery after reperfusion, hearts lacking GCN5L1 expression failed to recover in both male and female groups (Figure 3 B,C). A significant difference in ±dP/dt was observed as early as 15 minutes after reperfusion in both male and female cohorts (Figure 3 B,C). The overall recovery of function in GCN5L1 knockouts was significantly lower than wildtype counterparts, with knockouts exhibiting decreased cardiac work (Figure 3 D), decreased contractility (+dP/dt, Figure 3 E) and decreased relaxation (−dP/dt, Figure 3 F) as a percentage of baseline.
Figure 3. Loss of GCN5L1 expression reduces functional recovery after ischemia-reperfusion (I/R) injury.

(A) Experimental I/R protocol. (B, C) Work performed by isolated hearts after I/R is significantly reduced in cKO hearts from males and females. (D, E) After 60 minutes reperfusion, workload and contractility (+dP/dt) are significantly reduced in both cKO males and females. (F) Relaxation rate (−dP/dt) is significantly decreased in cKO hearts after reperfusion in females and trends towards a decrease in males. N = 4-6 mice per group. * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001 vs. control.
As a decrease in function after I/R injury may be the result of reversible stunning or irreversible infarct development, we next investigated whether tissue death was altered by GCN5L1 knockout. After I/R injury, infarct staining revealed larger infarct volumes in GCN5L1 knockouts for both males and females (Figure 4 A,B). Infarct sizes reported in both human and mouse males are larger than females, and we observed this pattern as expected.16–18 Comparison of the changes in infarct volume between wildtype and knockout mice was found to be similar in both males and females (Figure 4 C), indicating that the effects of GCN5L1 loss are not dependent on the sex of the mouse. Furthermore, significant release of lactate dehydrogenase into the perfusate was observed in male GCN5L1 knockout mice but not wildtype cohorts (Figure 4 D,E), while levels were too low to detect in either female cohort. These data suggest the loss of cell membrane integrity, and support the hypothesis that loss of GCN5L1 predisposes these hearts to irreversible tissue death post I/R.
Figure 4. GCN5L1 loss promotes infarct development and LDH release from the myocardium.
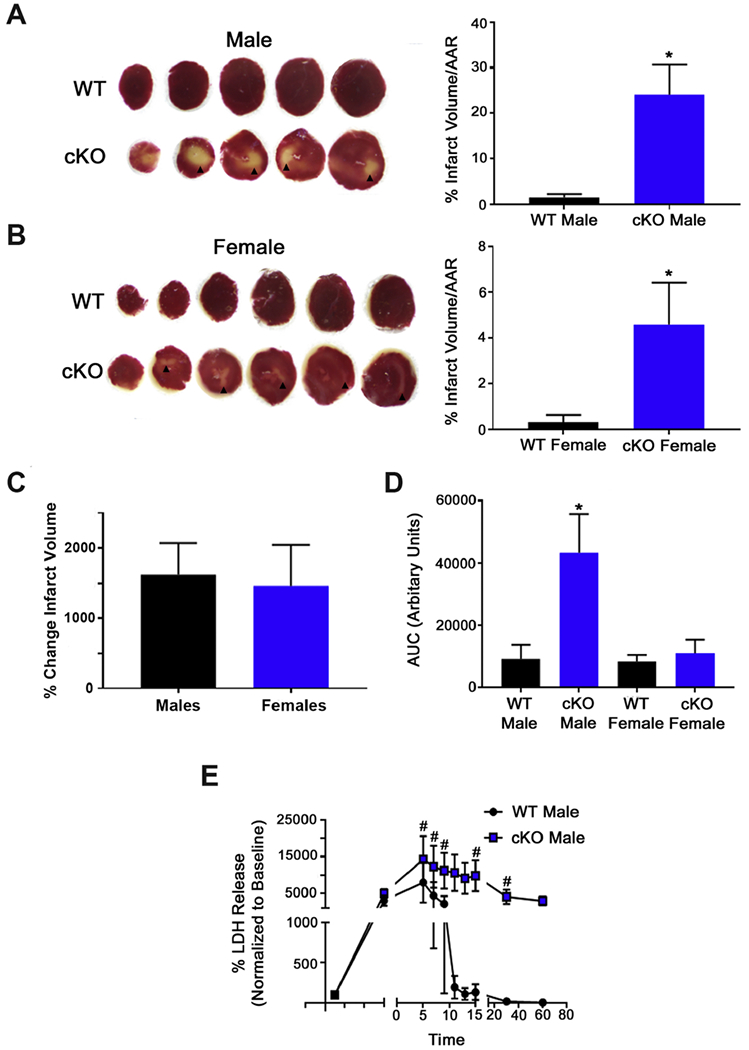
(A, B) Representative TTC-stained hearts from male and female mice. Viable tissue is stained red. Arrowheads indicate infarcted myocardium. (C) Comparison of the change in infarct size caused by GCN5L1 loss in males and females. (D) Total cumulative LDH release (area under the curve) in male and female mice. (E) LDH release from male mice over the course of reperfusion. N = 4-6 mice per group. * = P < 0.05 vs. WT; # = P < 0.05 vs. baseline
Increased ROS production underpins increased I/R injury in GCN5L1 knockout hearts
As reactive oxygen species damage is a central feature of reperfusion injury, we assessed post I/R oxidative stress by measuring protein carbonylation in wildtype and knockout hearts. Since there were no sex differences in the response to I/R in GCN5L1 cKO mice (Figures 3 and 4), we exclusively used male mice to reduce animal numbers. Carbonyl content was significantly elevated in lysates of both sham and I/R GCN5L1 cKO hearts, suggesting that protein oxidation and therefore oxidative stress is elevated in knockout mice (Figure 5 A). We next sought to replicate our ex vivo findings in a cell model so that we could interrogate the signaling pathways involved. We chose AC16 cells, a human-origin proliferating cell line with gene expression patterns14 and metabolic profile15 similarto primary human ventricular myocytes. Cells were subjected to hypoxia/reoxygenation (H/R) and glucose deprivation to mimic ischemia. To directly test the impact of reduced GCN5L1 on the outcome of H/R on cell survival, AC16 cells were transduced with lentiviral particles containing shRNA to silence GCN5L1 expression, or control shRNA. Transduction and knockdown of GCN5L1 was confirmed by immunoblotting (Figure 5 B).
Figure 5. Oxidative damage is increased in GCN5L1 cKO hearts.

(A) Protein carbonylation is elevated in both sham and I/R-subjected GCN5L1 cKO hearts. ** = P < 0.01 vs. wildtype. N = 5 mice per group. (B) Immunoblot confirmation of GCN5L1 knockdown (KD) in AC16 human ventricular myocytes using stably-expressed shRNA. N = 6. (C) Mitochondrial ROS production in control and GCN5L1 KD cells in normoxic and H/R conditions. (D-E) Seahorse XF analyses reveal differences in uncoupled oxygen consumption rates (OCR) and spare respiratory capacity. N = 36-41 replicates. (F) Incubation with mitoTEMPOL restores cell survival of KD cells in response to H/R injury. N = 6 per condition. ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001 vs. normoxia, # = P < 0.05 vs. control.
Similar to our ex vivo results, protein carbonylation was increased in GCN5L1 knockdown cells, however this did not reach significance (Supplemental Figure 4). Furthermore, MitoSOX staining revealed significantly elevated ROS production in the mitochondria of GCN5L1 knockdown cells in response to hypoxia and reoxygenation (Figure 5C). Our laboratory has previously identified several proteins modified by GCN5L1 that impact fatty acid oxidation and glycolysis,10,19 which may impair mitochondrial function when altered and result in elevated ROS production. Seahorse analysis of knockdown cells did indeed reveal substantially altered respiration with a greatly reduced spare capacity (Figures 5 D and E). Finally, treatment of the cells with mitochondrial ROS scavenger mitoTEMPOL rescued survival in knockdown cells (Figure 5 F), suggesting that mitochondrial-derived ROS is a major source of cellular damage in this system.
To verify that ROS drives the observed phenotype in GCN5L1 knockout mice, hearts were subjected to I/R injury, and n-acetyl cysteine (NAC) was administered to wildtype and knockout hearts on reperfusion. NAC treatment restored post-ischemic function in GCN5L1 cKO hearts, returning contractile function to levels indistinguishable from wildtype mice (Figure 6 A-E). In addition, although a trend towards increased infarct size remains in the knockout in the presence of NAC, this difference is no longer significant (Figure 6 F), and the mean is considerably reduced from infarct levels seen in the absence of NAC (treated = 8.001 +/− 4.5, vs. untreated = 24.02 +/−6.619). Relative to untreated hearts, NAC improved post-ischemic function in GCN5L1 cKO hearts by ~250-400% (Figure 6 G-I, Supplemental Figure 5).
Figure 6. Reducing ROS damage in GCN5L1 cKO hearts restores post-ischemic functional recovery.
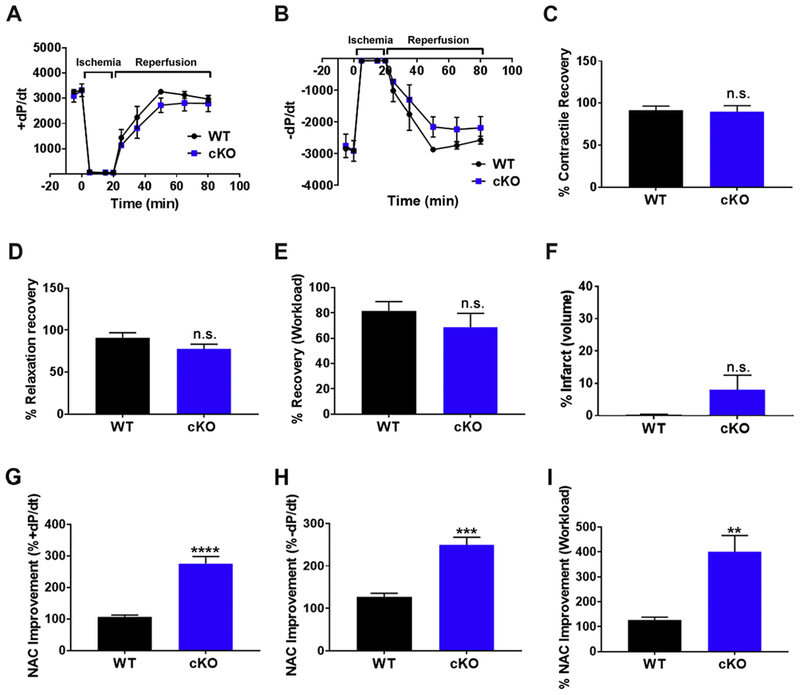
(A, B) +dP/dt and −dP/dt recovery from I/R injury is restored in GCN5L1 cKO mice in the presence of ROS-scavenger N-acetyl cysteine (NAC). (C-E) Recovery after reperfusion in the presence of NACasa percentage of +dP/dt, −dP/dt, and workload. (F) Infarct volume is reduced three-fold in cKO mice in the presence of NAC following I/R injury, and is no longer significantly different from wildtype mice. (G-I) Changes in post-ischemic function relative to untreated hearts from Figure 3. N = 5 mice per group. ** = P < 0.01, *** = P < 0.001, **** = P <0.0001 vs. WT.
ROS scavenging restores pro-survival ERK1/2 signaling and viability in GCN5L1-depleted cells after H/R
Mitogen-activated protein kinases (MAPKs) have been associated with survival signaling in the heart in the context of I/R injury, and Wang et al. have reported altered ERK1/2 signaling through ROS-induced activation in hepatic cells when GCN5L1 expression is reduced.11 We determined that activated pERK1/2 is reduced in GCN5L1 cKO hearts after IR injury (Figure 7 A). Switching to our cell model, we next sought to determine if the decrease in ERK activation observed in GCN5L1 knockout hearts was observed in vitro. We observed a significant decrease in ERK1/2 phosphorylation at basal levels in knockdown cells (Figure 7 B-E). Total levels of ERK1/2, as well as MAPKs p38, and JNK protein were not found to be different between control cells and knockdown cells under normoxic conditions (Supplemental Figure 6).
Figure 7. ROS scavenging restores pro-survival ERK1/2 signaling and cell viability in cardiac-derived GCN5L1 knockdown (KD) cells.
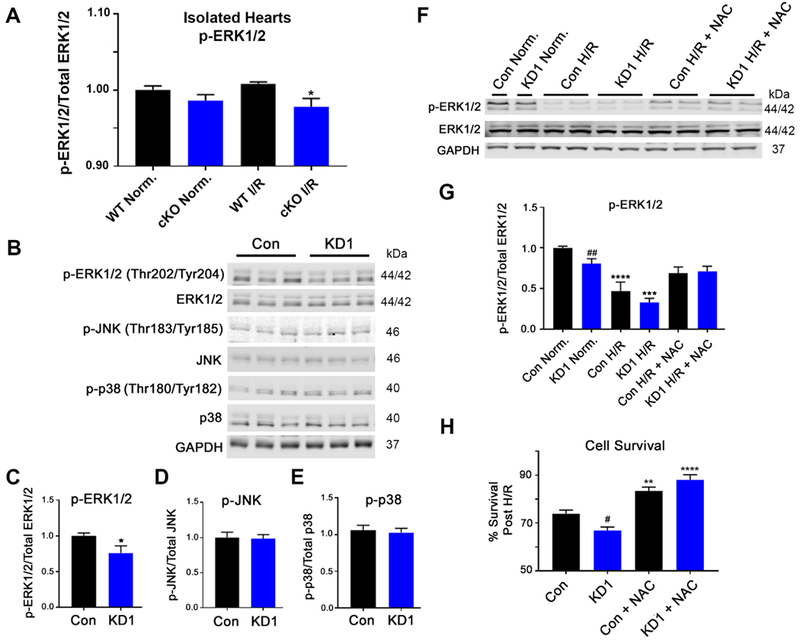
(A) ERK1/2 signaling is reduced in GCN5L1 cKO hearts following I/R injury. (B-E) Basal ERK1/2 signaling is reduced in GCN5L1 KD cells, while there is no change in the other MAP kinase proteins JNK or p38. N = 8-12 replicates per group. * = P < 0.05 vs. control. (F, G) ERK1/2 phosphorylation is significantly blunted in GCN5L1 KD cells at baseline, and in control and KD cells in response to H/R. Treatment with NAC restored ERK1/2 signaling in KD cells to baseline levels. (H) As with the isolated cKO hearts, treatment with NAC restored cell via bility in GCN5L1 KD cells subjected to H/R. N = 8-12 replicates per group. ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001 vs. normoxia, # = P < 0.05, ## = P < 0.01 vs. control.
To determine whether the defects in pro-survival signaling was ROS-dependent, phosphorylation of ERK1/2 was measured after H/R in combination with the ROS scavenger NAC. As shown previously, ERK1/2 activation was blunted at baseline in GCN5L1 KD cells, and in both groups following H/R. However, NAC treatment re-established ERK1/2 signaling in both control and knockdown cells subject to H/R treatment, bringing GCN5L1 KD cells back to activity levels seen under normoxic conditions (Figure 7 F,G). Finally, we determined whether preventing ROS accumulation rescues the GCN5L1 knockdown phenotype after H/R. NAC incubation increased survival in both groups subject to H/R, eliminating the difference between knockdown and control cells seen in untreated cells (Figure 7 H). This suggests that amelioration of ROS damage in GCN5L1-deficient cells aids cell survival in response to ischemia, and this effect may occur through the promotion of pro-survival ERK signaling.
DISCUSSION
We demonstrate here for the first time that GCN5L1 is necessary for the recovery of the heart from I/R injury. We show that loss of GCN5L1 results in decreased post-ischemic performance, and increased infarct development. These results are also observed in vitro, where a myocardial cell line with stable knockdown of GCN5L1 exhibits reduced survival after hypoxia and reoxygenation. These changes are accompanied by impaired activation of the MAPK ERK1/2. Reduced activation of ERK1/2 was found to be ROS-dependent, suggesting that GCN5L1 may protect cardiomyocytes from H/R injury by suppression of ROS-inhibited ERK1/2 activity (Summarized in Figure 8).
Figure 8. Hypothesized mechanism of action by GCN5L1 in cardiac cells following I/R injury.
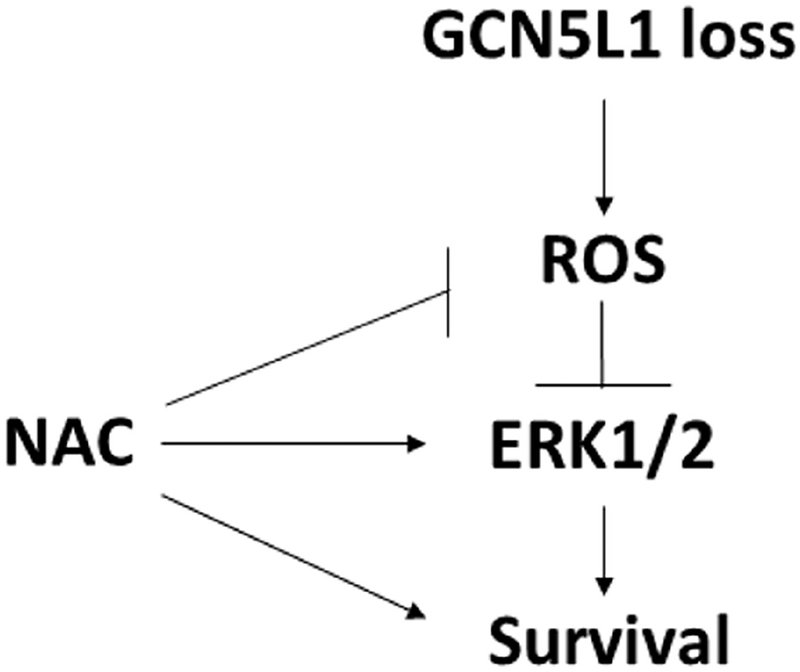
Loss of GCN5L1 disrupts mitochondrial function leading to increased ROS production. Treatment of cells with antioxidants rescues GCN5L1-deficient cells after ischemic injury.
We also demonstrate for the first time that GCN5L1 protein expression is downregulated after chronic ischemia in human cardiac ventricular tissue and acute hypoxia in cultured ventricular myocytes. The loss of GCN5L1 protein in tissue collected from patients exhibiting cardiac ischemia resulting in heart failure implicates GCN5L1 as a potential mediator of long-term injury. In vitro studies were subsequently used to narrow down the time frame under which GCN5L1 expression is downregulated, with significant changes in protein expression after merely four hours of reperfusion. These data suggest that decreased GCN5L1 levels may determine the severity of I/R injury at early stages of reperfusion, and strategies to prevent or compensate for this decrease may result in the prevention of injury. The mechanism by which GCN5L1 is downregulated in the heart remains unknown. We have previously shown that changes in nutritive status alters GCN5L1 expression in the heart,10 and the loss of substrate which occurs during ischemia may potentially trigger a compensatory downregulation of GCN5L1. GCN5L1 protein abundance was greatly increased in mouse embryonic fibroblast cells treated with the lysosomal inhibitor Bafilomycin A1,7 suggesting that protein degradation may be involved in regulating GCN5L1 levels. Further study is needed to determine whether this is the case in the heart.
One potential effect of increased ROS expression is altered activation of ERK1/2. Wang and colleagues have recently reported that loss of GCN5L1 in hepatocytes similarly promotes ROS production, leading to ERK1/2 activation.11 In contrast, we observed a ROS-dependent decrease in ERK1/2 phosphorylation in cardiac cells, as other groups have reported,19–21 suggesting that ROS normally inhibited by GCN5L1 expression do not activate the same pathways in cardiomyocytes and in hepatocytes. These data indicate that a decrease in basal ERK1/2 phosphorylation in the presence of H/R-induced ROS may play a role in increased toxicity observed after H/R in knockdown cardiac derived cells. Furthermore, our findings suggest that the loss of GCN5L1 may impact the survival of cardiomyocytes by reducing normal pro-survival kinase signaling. As with the previous report in hepatocytes,11 the molecular target of GCN5L1 in terms of elevated ROS remains to be determined, and further studies will be required to identify how GCN5L1 loss leads to increased production in cardiac cells.
While GCN5L1 was initially identified and extensively characterized as an acetyltransferase-related protein regulating numerous aspects of mitochondrial metabolism6–10, recent work has shown that a subset of this protein has other functions when localized to different cellular compartments.22 In the heart, previous studies have shown that GCN5L1 is required for the acetylation of proteins related to mitochondrial fatty acid oxidation.8–10 This is also true of the liver, where loss of GCN5L1 acetylation activity leads to increased fatty acid oxidation via reduced inhibition of the fatty acid oxidation (FAO) enzyme HADHA.23 In mouse embryonic stem cells, GCN5L1 is required for the acetylation of Kif1-Binding Protein (KBP), an interactor of the mitochondrial-associated kinesin Kif1Bα.24 Outside of the mitochondria, GCN5L1 has been implicated in the positioning of synaptic vesicles and the rupture of lysosomes in response to NLRP3 inflammasome activation.25,26 More recently, GCN5L1 has been shown to be a crucial mediator of tubulin acetylation via its interaction with the α-Tubulin acetyltransferase, αTAT1.27 These recent studies show that GCN5L1 may have multiple functions in different cell types that extend beyond its well characterized role in mitochondrial protein acetylation, and studies are ongoing to further develop our understanding of GCN5L1 biology in the heart.
Supplementary Material
-
-
GCN5L1 is a key regulator of mitochondrial protein acetylation, biogenesis and organelle positioning.
-
-
Loss of GCN5L1 in the heart promotes mitochondrial reactive oxygen species production, leading to cell death.
-
-
Treatment of GCN5L1-depleted hearts or cardiac cells with antioxidants prevents cell death following ischemic injury.
-
-
Loss of protective ERK1/2 signaling is evident in GCN5L1-depleted cardiac cells, and antioxidant treatment restores both MAPK signaling and cell survival.
ACKNOWLEDGEMENTS
This work was supported by NIH T32HL110849 to J.R.M., and by NIH K22HL116728, R56HL132917, R01HL132917 to I.S. We thank Christine Moravec (Cleveland Clinic Foundation) for access to stored tissue samples, Christopher O’Donnell (University of Pittsburgh) for equipment access, and Bonnie Lemster (University of Pittsburgh) for sample curation and processing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
None to report.
References
- 1.Benjamin EJ et al. Heart Disease and Stroke Statistics—2017 Update: A Report From the American Heart Association. Circulation 135, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brookes PS, Yoon Y, Robotham JL, Anders MW & Sheu S-S Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. - Cell Physiol 287, C817–C833 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Rivas GJ & Torre-Amione G Abnormal mitochondrial function during ischemia reperfusion provides targets for pharmacological therapy. Methodist Debakey Cardiovasc. J 5, 2–7 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Shintani-Ishida K, Inui M & Yoshida K-I Ischemia-reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. J. Mol. Cell. Cardiol 53, 233–239 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Alam MR, Baetz D & Ovize M Cyclophilin D and myocardial ischemia–reperfusion injury: A fresh perspective. J. Mol. Cell. Cardiol 78, 80–89 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Scott I, Webster BR, Li JH & Sack MN Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem. J 443, 655–661 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott I et al. GCN5-like Protein 1 (GCN5L1) Controls Mitochondrial Content through Coordinated Regulation of Mitochondrial Biogenesis and Mitophagy. J. Biol. Chem 289, 2864–2872 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alrob OA et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc. Res 103, 485–97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukushima A et al. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am. J. Physiol. Heart Circ. Physiol 311, H347–63 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Thapa D et al. Acetylation of mitochondrial proteins by GCN5L1 promotes enhanced fatty acid oxidation in the heart. Am. J. Physiol. Heart Circ. Physiol 313, H265–H274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L et al. GCN5L1 modulates cross-talk between mitochondria and cell signaling to regulate FoxO1 stability and gluconeogenesis. Nat. Commun 8, 523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrera R, Benhabbouche S, Bopassa JC, Li B & Ovize M One Hour Reperfusion is Enough to Assess Function and Infarct Size With TTC Staining in Langendorff Rat Model. Cardiovasc. Drugs Ther 23, 327–331 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Kim JH et al. Cardiodynamics and infarct size in regional and global ischemic isolated heart model: comparison of 1 hour and 2 hours reperfusion. Korean Circ. J 42, 600–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson MM et al. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol 39, 133–147 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Truong J, Mailloux RJ & Chan HM Impact of methylmercury exposure on mitochondrial energetics in AC16 and H9C2 cardiomyocytes. Toxicol. Vitr 29, 953–961 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Moore LG, McMurtry IF & Reeves JT Effects of sex hormones on cardiovascularand hematologic responses to chronic hypoxia in rats. Proc. Soc. Exp. Biol. Med 158, 658–62 (1978). [DOI] [PubMed] [Google Scholar]
- 17.GAO X, XU Q, KIRIAZIS H, DART A & DU X Mouse model of post-infarct ventricular rupture: time course, strain- and gender-dependency, tensile strength, and histopathology. Cardiovasc. Res 65, 469–477 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Ostadal B, Netuka I, Maly J, Besik J & Ostadalova I Gender Differences in Cardiac Ischemic Injury and Protection—Experimental Aspects. Exp. Biol. Med 234, 1011–1019 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Dhingra S, Sharma AK, Singla DK & Singal PK p38 and ERK1/2 MAPKs mediate the interplay of TNF-α and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am. J. Physiol. Circ. Physiol 293, H3524–H3531 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Thuc LC et al. Mitochondrial KATP channels-derived reactive oxygen species activate pro-survival pathway in pravastatin-induced cardioprotection. Apoptosis 15, 669–678 (2010). [DOI] [PubMed] [Google Scholar]
- 21.XU W et al. Exogenous hydrogen sulfide protects H9c2 cardiac cells against high glucose-induced injury by inhibiting the activities of the p38 MAPK and ERK1/2 pathways. Int. J. Mol. Med 32, 917–925 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Scott I, Wang L, Wu K, Thapa D & Sack MN GCN5L1/BLOS1 Links Acetylation, Organelle Remodeling, and Metabolism. Trends Cell Biol 28, 346–355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thapa D et al. The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial β-oxidation enzyme HADHA. J. Biol. Chem 293, 17676–17684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donato V et al. The TDH-GCN5L1-Fbxo15-KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat Cell Biol 19, 341–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niwa S et al. BORC Regulates the Axonal Transport of Synaptic Vesicle Precursors by Activating ARL-8. Curr. Biol 27, 2569–2578.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaidt MM et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 111, 1110–1124.e18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu K et al. GCN5L1 interacts with αTAT1 and RanBP2 to regulate hepatic α-tubulin acetylation and lysosome trafficking. J. Cell Sci jcs.221036 (2018). doi: 10.1242/jcs.221036 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.