

Abstract
Many individuals live to older ages without clinical impairment. It is unknown whether brain pathologies in these individuals are associated with subtle clinical deficits. We analyzed the brains of 161 clinically normal (CDR=0) older individuals enrolled in the Mayo Clinic Patient Registry or Study of Aging. We assessed for the presence, and burden of beta-amyloid, tau, alpha-synuclein, TDP-43, and vascular pathology. We investigated whether pathologies were associated with antemortem cognitive and motor function, depression, MRI volumetric measures, or the APOE ε4 allele. Eighty-six percent had at least one pathology, and 63% had mixed pathologies. Tau and vascular pathology were associated with poorer memory scores. Tau was also associated with poorer general cognition scores and smaller amygdala, hippocampi and entorhinal cortex volumes. Beta-amyloid neuritic plaque burden was associated with greater depression scores. The presence of a greater number of pathologies was associated with APOE e4 carrier status and with poorer memory performance. Dementia-related pathologies are associated with poorer performance in clinical measures and brain atrophy in the unimpaired elderly.
Keywords: clinically normal aging, neurodegenerative pathology, vascular pathology, cognition, depression, brain volume
1. INTRODUCTION
A proportion of the older adult population performs within a defined normal range on clinical measures of cognition, motor function and depression. Such individuals are referred to, or considered as, normally aged individuals. Many such individuals have died while remaining normal, and have undergone neuropathological examination of their brains. Interestingly, pathological brain examinations of these individuals have demonstrated findings ranging from no/minimal pathology to multiple-mixed pathologies (Bennett et al., 2006; Knopman et al., 2003; Matthews et al., 2009; Rahimi and Kovacs, 2014; SantaCruz et al., 2011; Suemoto et al., 2017). Many of the pathologies identified in these normally aged individuals can also be found in individuals with progressive neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), among others.
There are four main proteins that are associated with neurodegenerative disease. These include beta-amyloid Aβ), tau, alpha-synuclein and the TAR DNA binding protein of 43 kDa (TDP-43). Aβ and tau are considered the two cardinal proteins that characterize Alzheimer’s disease, although alpha-synuclein and TDP-43 are commonly observed (Dickson, 2001; Josephs et al., 2014b); alpha-synuclein associated pathologies characterize the Lewy body spectrum diseases, including PD (Dickson, 2001). Tau and TDP-43 can be associated with frontotemporal lobar degeneration (FTLD) (Mackenzie et al., 2010) and chronic traumatic encephalopathy (CTE) (McKee et al., 2015), and TDP-43 is also associated with ALS (Neumann et al., 2006). Furthermore, at the time of death, it is common to find evidence of vascular pathology in the brains of older individuals (Fernando and Ince, 2004). However, the association between the presence, and burden, of these five pathologies (Aβ, tau, alpha-synuclein, TDP-43, and vascular pathology) and performance on clinical measures of cognitive function, motor function, or depression, in individuals deemed clinically normal (CN) has not been extensively examined. It is also unclear whether a greater number of pathologies are associated with lower performance on clinical measures or with the APOE ε4 allele. Many of the neurodegenerative proteins and vascular pathology are associated with brain atrophy within populations of clinically impaired individuals, but whether they influence brain structure in normal aged individuals is another unknown. Given these gaps in knowledge, we designed a clinico-imaging-pathological study to assess the influence of the neurodegenerative brain proteins and vascular disease in a cohort of CN older subjects who had a postmortem brain examination.
2. METHODS
2.1. Subject selection
We identified 161 subjects in our pathological database that had been recruited and prospectively followed in the Mayo Clinic Alzheimer’s Disease Patient Registry (ADPR) or the Mayo Clinic Study of Aging (MCSA) who underwent a brain autopsy between 1999 and 2015, were CN, and had a Clinical Dementia Rating Scale (Hughes et al., 1982) sum of boxes (CDR-SB) score of 0 at the last assessment before the patient died. For each participant, determination of CN status was based on consensus agreement between the study coordinator, examining physician, and neuropsychologist who evaluated the participant, taking into account education, prior occupation, visual or hearing deficits, and reviewing all other participant clinical information. The ADPR was a community-based study of cognitive aging. The MCSA is a prospective population-based study of cognitive aging in Olmsted County, Minnesota (Roberts et al., 2008), and it has been shown that in the MCSA autopsied participants were very similar to participants in the MCSA who have died without autopsy on a broad array of demographic and clinical features (Graff-Radford et al., 2018). All subjects had also undergone APOE genotyping. Demographic features of all 161 subjects are shown in Table 1. This study was approved by the Mayo Clinic IRB. Prior to death, all subjects or their proxies had provided written consent for brain autopsy examination.
Table 1:
Subject demographics, neuropsychological scores and imaging volumes
| All (n=161) | Subjects with imaging (n=102) | |
|---|---|---|
| Demographics | ||
| Female | 86 (53%) | 55 (54%) |
| Age at clinical exam, y | 87 [81, 90] (62, 100) | 88 [81, 91] (73, 100) |
| Age at MRI, y | 87 [81, 90] (73, 99) | |
| Age at death, y | 89 [84, 93] (62, 103) | 91 [84, 94] (74, 103) |
| Post-mortem interval, hrs | 15 [9, 20] (2, 59) | 16 [10, 23] (2, 59) |
| Clinical exam to death, y | 1.5 [0.68, 3.37] (0.04, 9.4) | 1.6 [0.69, 3.31] (0.04, 9.4) |
| MRI to death, y | 3.0 [1.3, 5.5] (0.1, 11.7) | |
| Education, y | 14 [12, 16] (6, 20) | 14 [12, 16] (8, 20) |
| APOE ε4 carrier | 37 (23%) | 21 (21%) |
| Clinical scores | ||
| Mini-Mental State Exam score / 30 | 28 [27, 29] (23, 30) | 28 [27, 29] (23, 30) |
| WMS Logical Memory Delayed Recall / 50 | 17 [12, 24] (1, 35) | 18 [13, 24] (1, 35) |
| Boston Naming / 60 | 55 [51, 57] (24, 60) | 55 [51, 57] (25, 60) |
| Controlled Oral Word Association Test | 36 [28, 45] (9, 74) | 36 [28, 45] (13, 74) |
| WAIS-Block Design / 51 | 20 [15, 25] (0, 51) | 20 [14, 25] (2, 51) |
| AVLT Delayed Recall / 15 | 7 [4, 9] (0, 15) | 6 [4, 9] (0, 15) |
| Trails A / 180 | 47 [36, 62] (22, 125) | 49 [38, 62] (22, 125) |
| Trails B / 300 | 124 [92, 163] (56, 300) | 130 [96, 170] (60, 300) |
| Total UPDRS / 44 | 1 [0, 3] (0, 17) | 1 [0, 3] (0, 17) |
| BDI-II grand total / 63 | 6 [2, 9] (0, 19) | 6 [2, 9] (0, 15) |
| Gray matter volume (cm3) | ||
| TIV (1) | 1.50 [1.40, 1.60] (1.08, 1.87) | |
| Amygdala | 1.98 [1.79, 2.17] (1.51, 2.75) | |
| Hippocampus | 7.06 [6.51, 7.52] (5.18, 8.62) | |
| Entorhinal cortex | 2.86 [2.46, 3.31] (1.61, 4.47) | |
| Lateral frontal | 55.1 [50.6, 60.1] (18.5, 80.8) | |
| Lateral parietal | 34.2 [30.8, 37.8] (12.6, 51.9) | |
| Lateral temporal | 65.0 [59.9, 70.6] (36.5, 88.7) | |
| Medial frontal | 27.4 [25.3, 29.9] (10.8, 38.4) | |
| Medial parietal | 18.5 [16.7, 21.1] (9.0, 28.7) | |
Data shown are Median [quartiles] (range) or n (%)
2.2. Pathological analysis
All 161 subjects had undergone pathological examination according to the recommendations of the Consortium to Establish a Registry for Alzheimer’s disease (CERAD) (Mirra et al., 1991). The left hemi-brain was fixed and paraffin block sections were stained with hematoxylin and eosin and modified Bielschowsky silver stain. Immunohistochemistry was performed using antibodies for Aβ (6F/3D; 1:10; Novocastra Vector Labs, Burlingame, CA), phospho-tau (AT8; 1:1000; Endogen, Woburn, MA), alpha-synuclein (LB509; 1:200; Zymed, San Francisco, CA), and TDP-43 (polyclonal antibody (MC2085) that recognizes a peptide sequence in the 25-kDa C-terminal fragment). For TDP-43 we screened the amygdala and hippocampus to assess for the presence of neuronal cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear inclusions and perivascular inclusions. For each of the 161 cases we determined whether the pathological lesions were negative (absent or scant) or positive (present and more than scant), and we also assigned a stage for each pathological lesion. Aβ was considered positive if there was any evidence of Aβ senile plaques (neuritic, cored or diffuse) in the frontal, temporal, parietal or occipital cortex. Alpha-synuclein was considered positive if Lewy bodies were identified in any brainstem or cortical region. TDP-43 was considered positive if there were any TDP-43 immunoreactive inclusions in amygdala or hippocampus. Tau was considered positive if there were neurofibrillary tangles (NFTs) present beyond the transentorhinal cortex (≥stage II) (Braak and Braak, 1991). We chose to treat tau in this manner as there were only 4 cases without any NFTs. For vascular lesions we reviewed the pathological records at the time of autopsy examination for the presence of microinfarcts and the presence of lacunar (<1cm)/larger infarcts (≥1cm). If any type of infarct was present the case was considered positive for vascular pathology. We did not include cerebral amyloid angiopathy in our vascular pathology variable as this is a different type of vascular disease that is also highly correlated with the presence of Aβ, and would have confounded our analyses. A subject was considered as having none, one, or multiple pathologies if they had none, one, or more than one of the pathologies as defined above. In addition to a designation of present/absent or high/low we also assessed all five pathological lesions based on the distribution/stage as an indirect measure of burden (Table 2). Therefore, each case was assigned a Braak NFT stage (Braak and Braak, 1991) and a CERAD neuritic Aβ stage (Mirra et al., 1991) using modified Bielschowsky silver stain, and a TDP-43 stage (Josephs et al., 2016) using TDP-43 (polyclonal antibody MC2085 that recognizes a peptide sequence in the 25-kDa C-terminal fragment). For Lewy body disease we used the following modified Braak and Braak staging scheme for Lewy bodies (Braak et al., 2003): stage 0= no Lewy bodies, stage 1= Lewy bodies restricted to brainstem; stage 2 = Lewy bodies extending to limbic cortex; and stage 3 =Lewy bodies extending to neocortex. Four stage 0 cases had amygdala only Lewy bodies. For vascular stage we used the following staging scheme: stage 0 = no vascular infarcts; stage 1= microinfarcts or lacunar/large infarcts; stage 2 = both microinfarcts and lacunar/larger infarcts present.
Table 2:
Pathology distribution
| All (n=161) | Subjects with imaging (n=102) | |
|---|---|---|
| β-Amyloid Stage | ||
| 0 | 69 (43%) | 44 (43%) |
| 1 | 50 (31%) | 30 (29%) |
| 2 | 29 (18%) | 19 (19%) |
| 3 | 13 (8%) | 9 (9%) |
| Tau Stage | ||
| 0 | 4 (3%) | 1 (1%) |
| 1 | 20 (12%) | 11 (11%) |
| 2 | 53 (33%) | 36 (35%) |
| 3 | 41 (26%) | 27 (27%) |
| 4 | 25 (16%) | 14 (14%) |
| 5 | 16 (10%) | 11 (11%) |
| 6 | 2 (1%) | 2 (2%) |
| α-Synuclein Stage | ||
| 0 | 139 (87%) | 88 (87%) |
| 1 | 9 (6%) | 7 (7%) |
| 2 | 8 (5%) | 4 (4%) |
| 3 | 4 (3%) | 2 (2%) |
| TDP-43 Stage | ||
| 0 | 126 (81%) | 76 (75%) |
| 1 | 14 (9%) | 13 (13%) |
| 2 | 6 (4%) | 4 (4%) |
| 3 | 2 (1%) | 2 (2%) |
| 4 | 0 (0%) | 0 (0%) |
| 5 | 7 (5%) | 7 (7%) |
| 6 | 0 (0%) | 0 (0%) |
| Vascular Stage | ||
| 0 | 83 (52%) | 49 (49%) |
| 1 | 20 (13%) | 16 (16%) |
| 2 | 42 (26%) | 26 (26%) |
| 3 | 15 (9%) | 10 (10%) |
2.3. MRI analyses
Of the 161 subjects, 102 had completed a volumetric head MRI scan during life. Demographic features of all 102 subjects with MRI scans during life are shown in Table 1. We performed atlas-based parcellation using the automated anatomical labelling (AAL) atlas to measure volumes of the amygdala, hippocampus, entorhinal cortex, lateral frontal lobe (superior frontal, middle frontal, inferior frontal operculum, inferior frontal triangularis), lateral parietal lobe (superior parietal, inferior parietal, supramarginal gyrus, angular gyrus), lateral temporal lobe (superior temporal, middle temporal, inferior temporal), medial frontal lobe (supplemental motor area, frontal superior medial, anterior cingulum), and medial parietal (precuneus, posterior cingulum) lobe at the last MRI scan before death.
2.4. Clinical and neuropsychological assessments
We analyzed the following clinical and neuropsychological variables for this study: Mini–Mental State Examination (MMSE) (Folstein et al., 1975) to assess general cognitive function, the Wechsler Memory Scale-Revised Logical Memory II delayed recall (Wechsler, 1987) and Auditory Verbal Learning Test (AVLT) (Rey, 1964) to assess verbal episodic memory, the Boston Naming Test (Kaplan et al., 1983) to assess confrontation naming, the Controlled Oral word Association Test (Sumerall et al., 1997) to assess language, the Wechsler Adult Intelligence Scale-R Block Design (Wechsler, 1987) to assess visuo-spatial abilities, Trail Making Test Parts A and B (Reitan, 1958) to assess cognitive speed and executive function, a modified version of the Unified Parkinson’s Disease Rating Scale (UPDRS) (2003) to assess for the presence and severity of Parkinsonism, and the Beck Depression Inventory-II (BDI-II) (Beck et al., 1961) to assess for depression.
2.5. Statistical Analyses
Demographic variables were compared between those positive and negative for Aβ, tau, alpha-synuclein, TDP-43, and vascular disease. Group-wise comparisons of demographics were performed using Fisher’s exact or Wilcoxon rank-sum tests, where appropriate.
Associations between pathological lesion and clinical outcomes were assessed using linear regression models which incorporated all five pathologies as predictors, adjusting for age at clinical exam and years between exam and death. For MRI brain volumes, linear regression models were adjusted for age at MRI scan, duration from MRI scan to death and log-transformed total intracranial volume (log-TIV). Two sets of linear regression models were performed, the first treating the pathological measures as binary variables (i.e. presence/absence) and the second as ordinal variables (i.e. pathological stage). We also compared clinical scores and MRI volumes between subjects with only microinfarcts and subjects with only lacunar/large infarcts using linear regression (subjects with both were excluded).
We assessed the differences in demographics, clinical test scores and brain volumes between those without any pathology and those with at least one type of pathology. Associations with clinical test scores were assessed using linear regression adjusted for age at clinical exam and years from clinical exam to death. Associations with brain volumes were assessed using linear regression adjusted for age at MRI, years from MRI to death, and log-TIV. Logistic regression adjusting for age at death was performed to evaluate the odds of at least one type of pathology for APOE ε4 carriers versus non-carriers. Trend tests were also performed to evaluate the association between number of pathologies present or high (0, 1, 2, 3, 4, 5) and clinical test scores or brain volumes. Associations with clinical test scores were adjusted for age at clinical exam and years from clinical exam to death. Associations with brain volumes were adjusted for age at MRI, years from MRI to death, and log-TIV.
All statistical analyses were performed using R Statistical Software (Foundation for Statistical Computing, Vienna, Austria) version 3.4.4. Results were considered statistically significant at p < 0.05.
3. RESULTS
In our cohort, approximately half (53%) of the subjects were female and 37 (23%) were APOE ε4 carriers (Table 1). The most common pathology was Aβ which was positive in 105 subjects (65%), followed by tau which was positive in 84 subjects (52%). The other pathologies were present in less than 50% of the subjects including alpha-synuclein (n=22, 14%), TDP-43 (n=35, 22%), and vascular pathology (n=77, 48%). Age at death was greater in positive versus negative subjects for tau (median [IQR] 91[87,94] vs. 88 [82,91], p<0.001), alpha-synuclein (93 [88,95] vs. 89 [83,92], p<0.01) and TDP-43 (91 [89,95] vs. 88 [83,92], p<0.001). Aβ(+) subjects were more likely to be APOE ε4 carriers than Aβ(−) subjects (30% vs 9%, p<0.01), as were tau(+) subjects (32% vs 13%, p<0.01). The staging of all five pathologies is shown in Table 2. The most common Aβ stage was 0 with the majority of subjects (n=119; 74%) having no-scant neuritic plaques. For Braak NFT stage, 143 subjects (89%) were ≤ stage IV; only 2 subjects (1%) were stage VI. For alpha-synuclein and TDP-43, the majority of subjects (>80%) were stage 0. For vascular disease, 59 subjects (37%) had only microinfarcts while 23 (15%) had only lacunar/larger infarcts; 8 subjects (5%) had both microinfarcts and lacunar/larger infarcts.
One-hundred thirty eight (86%) of the 161 subjects had at least one pathology and 102 (63%) had multiple pathologies; some as many as four different pathologies (Figure 1). The frequencies of the different combinations of pathologies are show in Table 3. When assessing the co-occurrence of two pathologies, the most common pathology combination was Aβ and tau (40%), followed by Aβ and vascular (28%) and then tau and vascular (20%). Alpha-synuclein and TDP-43 rarely occurred together (3%). When assessing the co-occurrence of three pathologies, the most common combination was Aβ, tau and vascular (16%).
Figure 1:

Plot showing the different types of pathologies for each subject grouped by number of pathologies
Table 3:
Proportion of individuals having each combination of either two or three pathologies in the entire cohort of 161 CN subjects
| Pathology 1 | Pathology 2 | Pathology 3 | |||
|---|---|---|---|---|---|
| Tau | α-Synuclein | TDP-43 | Vascular | ||
| Combinations of any two pathologies | |||||
| β-Amyloid | 40% | 12% | 14% | 28% | NA |
| Tau | 11% | 11% | 20% | NA | |
| α-Synuclein | 3% | 5% | NA | ||
| TDP-43 | 8% | NA | |||
| Combinations of any three pathologies | |||||
| β-Amyloid | 10% | NA | α-Synuclein | ||
| β-Amyloid | 8% | 2% | NA | TDP-43 | |
| β-Amyloid | 16% | 4% | 6% | NA | Vascular |
| Tau | 2% | NA | TDP-43 | ||
| Tau | 3% | 4% | NA | Vascular | |
| α-Synuclein | 1% | NA | Vascular | ||
In the linear regression models investigating each type of pathology adjusting for the other pathologies, we found that tau, both presence and burden, was independently associated with poorer MMSE, WMS delayed recall, AVLT delayed recall scores (Figure 2, Figure 4), and smaller amygdala and entorhinal cortex volumes (Figure 3, Figure 5). Tau burden (i.e. NFT Braak stage) was also associated with smaller hippocampal volumes (Figure 5). We observed an association between higher Aβ CERAD stage and higher BDI-II scores (Figure 4). We also found that the presence and burden of vascular pathology was associated with poorer AVLT scores (Figure 2, Figure 4). None of the clinical and MRI outcome measures differed between subjects with microinfarcts compared to subjects with lacunar/large infarcts.
Figure 2:
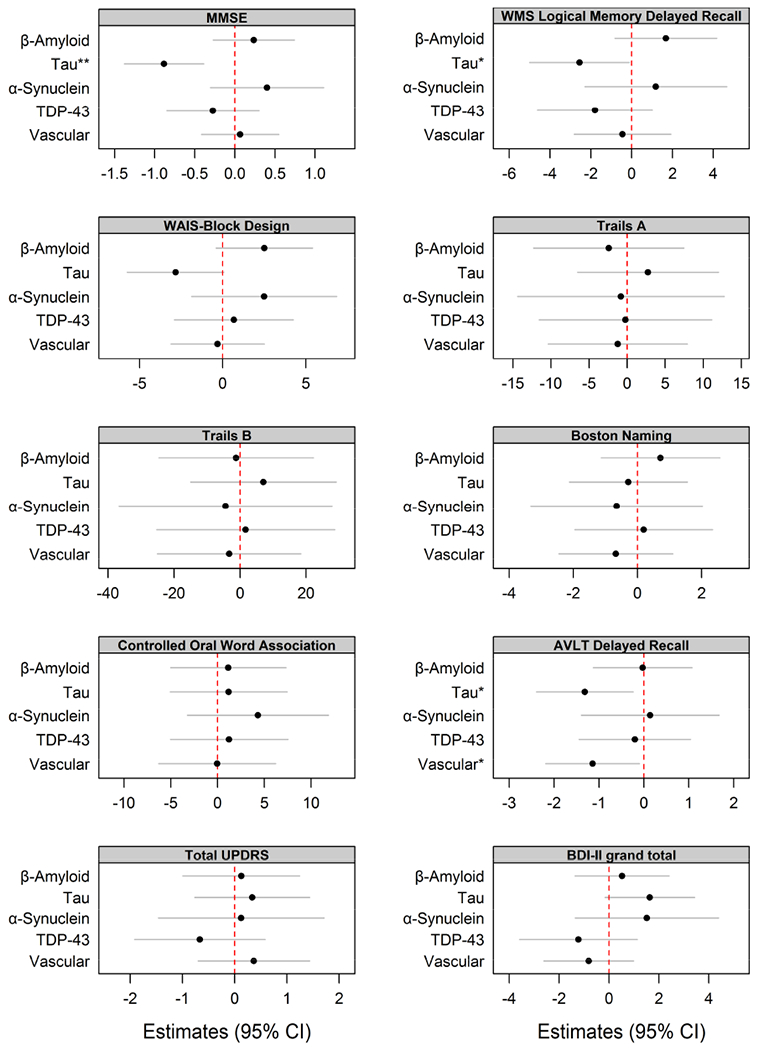
Plots of estimates from age-adjusted linear regression models with neuropsychological tests as an outcome with pathological measures assessed as binary variables. Models were adjusted for age at neurological exams. (*p<0.05, **p<0.01, ***p<0.001.)
Figure 4:

Plots of estimates from age-adjusted linear regression models with neuropsychological tests as an outcome with pathological measures assessed as ordinal variables. Models were adjusted for age at neurological exams. (*p<0.05, **p<0.01, ***p<0.001.)
Figure 3:

Plots of estimates from age-adjusted linear regression models with gray matter volumes (cm3) as an outcome (N=102) with pathological measures assessed as binary variables. Models were adjusted for age at MRI scan, years from MRI scan to death and log-transformed TIV (*p<0.05, **p<0.01, ***p<0.001).
Figure 5:

Plots of estimates from age-adjusted linear regression models with gray matter volumes (cm3) as an outcome (N=102) with pathological measures assessed as ordinal variables. Models were adjusted for age at MRI scan, years from MRI scan to death and log-transformed TIV (*p<0.05, **p<0.01, ***p<0.001).
The linear regression models also showed some associations that could be considered counterintuitive. These counterintuitive results were almost exclusively observed when pathologies were treated as burden, rather than presence/absence. That is, we observed greater burden of pathology to be associated with better performance on clinical measures or larger MRI volumes. For alpha-synuclein, we observed a higher lesion burden to be associated with better performance on the WAIS block design (Figure 4) and an association between the presence of, and higher lesion burden of pathology and larger entorhinal cortex volumes (Figure 3, Figure 5). For neuritic Aβ, we observed an association between higher CERAD Aβ stage and better performance on the AVLT delayed recall (Figure 4) and larger hippocampal volumes (Figure 5). For vascular pathology, we observed an association between a higher burden of vascular pathology and larger amygdala and entorhinal cortex volumes (Figure 5). For TDP-43, we observed a higher lesion burden to be associated with larger medial frontal lobe volumes (Figure 5).
In investigating associations with having at least one pathology as compared to none, we found subjects with at least one pathology were older compared to subjects with no pathology (Table 4). After adjusting for age there was an association between the presence of at least one pathology and APOE ε4 (Table 4). The odds of having at least one pathology being an APOE ε4 carrier was 5.3 [(1.3, 39.4); p = 0.05] times higher than the odds of having at least one pathology without being an APOE ε4 carrier. There was no association between the presence of having at least one pathology and any clinical measure or brain volume (Table 4). In our trend test assessing the association between a greater number of pathologies and clinical and imaging measures, we found that having a greater number of pathologies was associated with poorer AVLT delayed recall score (estimate (95% CI) −0.62 (−1.1, −0.04) (Table 5).
Table 4:
Comparisons between subjects without pathology and subjects who had at least one type of pathology
| No pathology (N = 23) | At least one pathology (N = 138) | |
|---|---|---|
| Female, N (%) | 13 (57%) | 73 (53%) |
| Age at clinical exam, yrs | 84 (77, 86) | 87 (83, 90)** |
| Age at MRI, yrs | 82 (77, 87) | 87 (82, 90) |
| Age at death, yrs | 84 (78, 89) | 90 (85, 94)** |
| MRI to death, yrs | 4.0 (1.3, 5.9) | 3.0 (1.4, 5.6) |
| Post-mortem interval, hrs | 14 (8, 20) | 15 (10, 20) |
| Clinical exam to death, yrs | 1.0 (0.5, 1.6) | 1.6 (0.7, 3.5)* |
| Education, yrs | 14 (12, 16) | 14 (12, 16) |
| APOE ε4 carrier, N (%) | 2 (9%) | 35 (25%)* |
| 23 | 3 (13%) | 19 (14%) |
| 24 | 1 (4%) | 2 (1%) |
| 33 | 18 (78%) | 84 (61%) |
| 34 | 1 (4%) | 29 (21%) |
| 44 | 0 (0%) | 4 (3%) |
| Clinical scores | No pathology (N = 23) | At least one pathology (N = 138) |
| Mini-Mental State Exam score / 30 | 29 (27, 29) | 28 (27, 29) |
| WMS Logical Memory Delayed Recall / 50 | 21 (16, 25) | 17 (12, 23) |
| Boston Naming / 60 | 56 (54, 58) | 54 (51, 57) |
| Controlled Oral Word Association Test | 34 (25, 43) | 36 (29, 45) |
| WAIS-Block Design / 51 | 20 (16, 27) | 20 (14, 25) |
| AVLT Delayed Recall / 15 | 8 (4, 11) | 6 (4, 8) |
| Trails A / 180 | 47 (36, 56) | 46 (36, 64) |
| Trails B / 300 | 114 (83, 140) | 126 (95, 170) |
| Total UPDRS / 44 | 1 (0, 1) | 1 (0, 2) |
| BDI-II / 63 | 4 (2, 6) | 6 (2, 9) |
| Gray matter volumes (cm3) | No pathology (N = 12) | At least one pathology (N = 90) |
| TIV (1) | 1.57 (1.46, 1.60) | 1.49 (1.39, 1.60) |
| Amygdala | 2.07 (1.93, 2.11) | 1.96 (1.78, 2.17) |
| Hippocampus | 7.35 (6.70, 7.48) | 6.97 (6.42, 7.52) |
| Entorhinal cortex | 3.03 (2.77, 3.37) | 2.82 (2.44, 3.30) |
| Lateral frontal | 55 (51, 71) | 55 (50, 60) |
| Lateral parietal | 37 (32, 43) | 34 (31, 38) |
| Lateral temporal | 68 (63, 77) | 64 (60, 70) |
| Medial frontal | 29 (26, 33) | 27 (25, 30) |
| Medial parietal | 20 (17, 22) | 18 (17, 21) |
Unless indicated otherwise, data shown are median (IQR).
= p<0.05
= p<0.01
= p<0.001.
APOE= apolipoprotein ; AVLT = auditory verbal learning test; BDI = Becks depression inventory; TIV = total intracranial volume; UPDRS = Unified Parkinson’s disease rating scale (modified version used) ; WAIS = Wechsler Adult Intelligent scale
Table 5:
Trend test of number of pathologies
| Trend test | ||
|---|---|---|
| Est. (95% CI) | p-values | |
| Clinical scores | ||
| MMSE / 30 | −0.12 (−0.34, −0.01) | 0.29 |
| WMS Logical Memory Delayed Recall / 50 | −0.30 (−1.4, −0.10) | 0.53 |
| Boston Naming / 60 | −0.10 (−0.89, −0.15) | 0.81 |
| Controlled Oral Word Association | 0.65 (−2.4, 0.72) | 0.67 |
| WAIS-Block Design / 51 | 0.22 (−1.0, −0.23) | 0.73 |
| AVLT Delayed Recall / 15 | −0.62 (−1.1, −0.04) | 0.01 |
| Trails A / 180 | −0.30 (−4.2, 2.1) | 0.88 |
| Trails B / 300 | 0.38 (−8.9, 5.2) | 0.94 |
| Total UPDRS / 44 | 0.24 (−0.29, 0.17) | 0.37 |
| BDI-II / 63 | 0.60 (−0.18, 0.20) | 0.13 |
| Gray matter volumes (cm3) | ||
| Amygdala | −0.007 (−0.04, 0.03) | 0.68 |
| Hippocampus | 0.03 (−0.06, 0.10) | 0.50 |
| Entorhinal cortex | 0.04 (−0.03, 0.10) | 0.26 |
| Lateral frontal | −0.60 (−2.0, 0.90) | 0.43 |
| Lateral parietal | −0.40 (−1.0, 0.40) | 0.32 |
| Lateral temporal | −0.40 (−1.0, 0.70) | 0.47 |
| Medial frontal | −0.20 (−0.90, 0.50) | 0.55 |
| Medial parietal | −0.20 (−0.70, 0.20) | 0.29 |
APOE= apolipoprotein ; AVLT = auditory verbal learning test; BDI = Becks depression inventory; UPDRS = Unified Parkinson’s disease rating scale (modified version used) ; WAIS = Wechsler Adult Intelligent scale
4. DISCUSSION
We studied a unique cohort of 161 prospectively followed, cognitively and neurologically normal subjects with a CDR-SB score of 0 at the time of death. We found that the majority had at least one of the dementia related pathologies and we found evidence that some of these pathologies influence clinical and anatomic outcomes in these people. We also found associations that could be considered counterintuitive whereby a higher burden/distribution of protein was associated with better clinical performance and/or larger brain volumes.
It has been consistently shown that even if older adults die without a clinical diagnosis of dementia or other impairment, it is likely that they will have some form of dementia-related pathology. In fact, in our cohort, 86% of CN elderly had at least one dementia-related pathology. As observed in our cohort, the most common dementia-related pathologies observed in CN older adults were AD-related pathologies (Aβ and tau)(Rahimi and Kovacs, 2014). The next most common pathology in our cohort was vascular pathology, then TDP-43 and, lastly, alpha-synuclein. Similar to AD-related pathologies, vascular pathology is common in CN older adults, but prevalence estimates are broad, with estimates ranging from 33-80% in community-based samples (Brayne et al., 2010; Fernando and Ince, 2004; Matthews et al., 2009). TDP-43 prevalence estimates are also wide-ranging. A meta-analysis found that TDP-43 was present in 37% of CN older adults in North America (Nascimento et al., 2018). However, among community-dwelling CN older adults, TDP-43 was present in 1% of subjects (Robinson et al., 2018), while another study of the oldest old (90 years and older), found TDP-43 to be present in 18% of subjects without dementia (Keage et al., 2014). Hence, the prevalence of TDP-43 seems to be highly dependent on the characteristics of the cohort, and is likely also dependent on the regions assessed pathologically for its presence (amygdala vs. hippocampus) (Josephs et al., 2014a). Alpha-synuclein was, indeed, relatively rare in CN older adults in other studies, observed in 5-12% of cases (Brayne et al., 2010; Knopman et al., 2003; Petrovitch et al., 2005).
Mixed pathologies were very common in our cohort with more than 60% of the subjects having two or more pathologies, a third having at least three pathologies, and almost 10% having four pathologies. It is apparent that some combinations of pathologies are more common than others, with combinations involving Aβ, tau and vascular pathology being the most common. These findings are quite striking given that the subjects in our cohort were cognitively normal with a CDR-SB score of 0 at the time of last evaluation prior to death. Mixed pathologies have been reported in elderly cohorts consisting of individuals with a range of clinical diagnoses, including dementia (Rahimi and Kovacs, 2014). However, mixed pathologies have not been commonly reported in CN subjects (Bennett et al., 2006; Knopman et al., 2003; SantaCruz et al., 2011; Suemoto et al., 2017) , especially when defined as CDR-SB =0. These findings suggest that cognitive reserve may be playing a role in sparing these individuals from developing clinical symptoms of the disease prior to death (Stern, 2009). Alternatively, it is also likely that it is the burden of the pathologies that is key, as opposed to the number of pathologies, when the burdens are low. While tau and Aβ were the most common proteins in our cohort, only around 10% had the highest levels of tau burden/distribution (Braak NFT stage V-VI) or Aβ(+) neuritic plaque burden/distribution (CERAD stage 3), as others have found (Bennett et al., 2006; Knopman et al., 2003). Similar to another study (SantaCruz et al., 2011), we found the majority of those with the highest levels of tau were Braak NFT stage V, with only 2 subjects (1%) being Braak stage VI.
In our cohort of CN subjects, we found strong associations between tau and clinical measures of episodic memory function, as well as general cognition. These associations were observed when we assessed the presence/absence of tau or tau NFT stage. Hence, the presence of tau and a higher NFT stage were both associated with poorer episodic memory and general cognition. The association between tau and poorer memory is likely related to the association of tau and smaller volumes of medial temporal lobe structures, given that these structures are strongly associated with episodic memory. This is consistent with the fact that these structures are involved in early Braak NFT stages (Braak and Braak, 1991). Another notable finding in our study was the association between the presence, and greater burden, of vascular pathology and lower episodic memory performance. We did not observe any association between vascular pathology and volume of medial temporal lobe structures, suggesting that the influence of vascular pathology on episodic memory is likely independent of medial temporal atrophy. It is possible that the influence from vascular pathology was related to lesions in subcortical grey matter structures, such as the thalamus that has been reported to be associated with episodic memory loss (Aggleton et al., 2016). In the regression analysis when we treated the variables as ordinal, we observed an association between a higher Aβ burden of neuritic plaques and higher scores on the BDI-II scale. This was not observed in our binary analysis where we investigated the presence/absence of any Aβ, i.e. neuritic, diffuse and cored plaques, perhaps suggesting the relationship is specific to neuritic plaque burden. Work in mice has linked Aβ to depression, with evidence that increased Aβ production is initiated by a stress response linked to depression (Green et al., 2006).
Although several previous autopsy studies have examined the association of dementia pathologies with clinical measures of cognition in cohorts that include CN individuals, few have investigated this association in a cohort of purely CN individuals (Bennett et al., 2006; Bennett et al., 2012; Boyle et al., 2018; Price et al., 2009; SantaCruz et al., 2011; Suemoto et al., 2017). In our cohort, all subjects were clinically diagnosed as being CN and also had a CDR-SB score of 0 at the evaluation closest to death. Hence, our cohort is relatively unique even among studies with CN individuals. One study that assessed the association of Aβ(+) neuritic and diffuse plaques with cognition in CN subjects identified an association between the presence of neuritic plaques and lower performance in general cognition and memory, after accounting for NFTs (Malek-Ahmadi et al., 2016). Another study that assessed the association of alpha-synuclein pathologies with cognition did not find any association with cognition (Markesbery et al., 2009). Two other studies found an association between higher burden of Alzheimer’s pathologies (i.e. Aβ and/or tau) and lower performance on memory measures (Bennett et al., 2006; Schmitt et al., 2000). One study did find an association between Aβ and global cognition with only a trend for tangles (Bennett et al., 2012). In our study, however, we identified an association between tau, but not Aβ, and global cognition similar to another study (Price et al., 2009). The difference in findings could be due to different inclusion criteria or to different methods of analysis. Most autopsy clinico-pathologic and imaging pathologic association studies typically combine subjects with and without normal cognition, exclude subjects with dementia but including subjects with varying degrees of mild cognitive impairment or report on cohorts in which all the subjects were CN at the time of enrollment but of which some later became impaired. For example, a recent longitudinal study that enrolled CN subjects and followed them to death found associations between clinical trajectories and pathological variables (Nguyen et al., 2018). Yet, while 100% of the cohort was normal at enrollment, 71% were cognitively impaired at death making this cohort very different from ours where 100% of subjects had a CDR sum of boxes of 0 at death.
Our results demonstrate that tau and vascular pathology were the only pathologies that influenced cognition in CN subjects. While we did not conduct an analysis in which the relative effect of each pathology was calculated, other groups have attempted this type of analysis with one study reporting that tau had the biggest effect on cognition, followed by vascular pathology (Suemoto et al., 2017). However, the cohort in that study included both cognitively impaired and unimpaired subjects and vascular pathology was defined slightly differently from in our study. Another study found that the presence of AD pathology (tau and Aβ) was the biggest contributor to cognitive decline, followed by vascular pathology, but again in a cohort that included both cognitively impaired and unimpaired subjects (Boyle et al., 2018).
Quite surprisingly, we found many counterintuitive results in our linear regression analyses that used pathological stage, whereby higher stage in all pathologies except for tau were associated with better cognition or larger MRI volumes. While these findings need to be replicated in a different cohort, it is intriguing to postulate that this may support the theory of cognitive or brain reserve (Stern, 2009). Specifically, our findings may support the notion that having cognitive reserve or starting off with larger brain volumes helps to protect the individual from dropping below the threshold in which signs and symptoms develop in the presence of high neuropathological stages. Alternatively, the findings could be driven by the fact that we restricted our cohort to CN subjects and excluded subjects with cognitive impairment. For example, considering the counterintuitive relationship between Aβ and hippocampal volume and given that greater Braak NFT stage was related to smaller hippocampal volumes; if a subject has high NFT stage and high Aβ stage then it may be that the only way they could remain CN is if they have preserved hippocampal volume, since the presence of hippocampal atrophy in addition to high tau and Aβ stage would likely render them cognitively impaired. We refer to this as the “Triangular effect”. Following the same thinking, the results could also be telling us that a relationship between tau and hippocampal volume alone is not sufficient to render a subject cognitively impaired in the absence of a relationship between other pathologies and medial temporal volumes. Supportive of this hypothesis was the fact that the trend test showed that performance on the AVLT was related to the number of pathologies present in the brain. In addition to the tau relationships, one of the other pathologies may need to exert an influence in order for a subject to become cognitively impaired, i.e. the effects of multiple pathologies are necessary. However, it is important to remember that the neuropathological stages themselves are not perfect surrogates of pathological burden. The fact that we did not see these counterintuitive associations when we analyzed the presence/absence of pathologies may tell us that the binary analysis lacks sensitivity.
We found that APOE ε4 carriers were more likely to have at least one type of pathology, compared to non-carriers. This may be driven mainly by the well-known association between APOE ε4 and Aβ, particularly in younger individuals. With-that-said, we cannot exclude the possibility that APOE ε4 is also playing a role in the deposition of the other four pathologies. Indeed, we and others recently reported an association between the APOE ε4 allele and Lewy body disease (Dickson et al., 2018) and between APOE4 and TDP-43(Wennberg et al., 2018; Yang et al., 2018), independent of Aβ pathology.
There are limitations of the study that should be mentioned. Although the CDR sum boxes was determined to be 0 at the time of last examination before death it is possible that it could have changed between that time and death which was approximately 1.5 years. Another limitation is the fact that we did not consider the location of the vascular infarcts which may have provided more granularity in the results.
Although past imaging studies have shown that CN aged individuals have dementia pathology, including some of the pathologies examined in this study and vascular pathology, it is important to consider that because these studies are conducted in vivo, it is not known whether or not the individuals will develop dementia before death. Our study indicates that even if an individual dies CN, dementia-related protein deposition and vascular pathologies are associated with poorer memory performance and depression. Moreover, we found that the APOE ε4 allele appears to be a risk factor for having at least one of the five types of pathology measured here. Furthermore, although imaging technology is quite advanced, there is still debate as to whether we are able to precisely measure the correct forms of Aβ and tau in vivo and how much off-target binding contributes noise (Klunk, 2018). Additionally, we are not currently able to measure alpha-synuclein and TDP-43 using imaging. Therefore, histological studies at autopsy are very important and may provide us with a more accurate measure of these proteins.
Highlights.
Clinically normal elderly may have dementia-related pathology and subtle deficits
Tau and vascular pathology are associated with poorer memory scores
Tau is also associated with smaller medial temporal lobe volumes
Tau may be necessary but not sufficient to cause dementia
APOE ε4 appears to be a risk factor for more than one pathology
Acknowledgements
This study was funded by the following grants from the US National Institutes of Health (National Institute on Aging): R01 AG037491, P50 AG16574 and U01 AG006786. We thank the families of the patients who donated their brains to science and thus allowed completion of this study.
Disclosure Statement
KAJ, DWD, RCP, LP, MEM, MMM, KK, JLW, JEP, AMW, NT, and SDW receive research support from the National Institutes of Health (NIH). LP has a US patent #9,448,232 entitled: “Methods and materials for detecting C9ORF72 hexanucleotide repeat expansion positive frontotemporal lobar degeneration or C9ORF72 hexanucleotide repeat expansion positive amyotrophic lateral sclerosis”. DSK serves on a Data Safety Monitoring Board for DIAN; is an investigator in clinical trials sponsored by Biogen, Lilly Pharmaceuticals and the University of Southern California; and receives research support from the NIH. RCP serves as a consultant for Roche, Merck, Genentech, Biogen,& Eli Lilly& Co, and receives research support from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggleton JP, Pralus A, Nelson AJ, Hornberger M, 2016. Thalamic pathology and memory loss in early Alzheimer’s disease: moving the focus from the medial temporal lobe to Papez circuit. Brain 139(Pt 7), 1877–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J, 1961. An inventory for measuring depression. Arch Gen Psychiatry 4, 561–571. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS, 2006. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66(12), 1837–1844. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA, 2012. Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 72(4), 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA, 2018. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 83(1), 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4), 239–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E, 2003. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2), 197–211. [DOI] [PubMed] [Google Scholar]
- Brayne C, Ince PG, Keage HA, McKeith IG, Matthews FE, Polvikoski T, Sulkava R, 2010. Education, the brain and dementia: neuroprotection or compensation? Brain 133(Pt 8), 2210–2216. [DOI] [PubMed] [Google Scholar]
- Dickson DW, 2001. Alpha-synuclein and the Lewy body disorders. Curr Opin Neurol 14(4), 423–432. [DOI] [PubMed] [Google Scholar]
- Dickson DW Heckman MG, Murray ME, Soto AI, Walton RL, Diehl NN, van Gerpen JA, Uitti RJ, Wszolek ZK, Ertekin-Taner N Knopman DS, Petersen RC, Graff-Radford NR, Boeve BF, Bu G, Ferman TJ, Ross OA, 2018. APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91(12), e1182–e1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando MS, Ince PG, 2004. Vascular pathologies and cognition in a population-based cohort of elderly people. J Neurol Sci 226(1-2), 13–17. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR, 1975. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12(3), 189–198. [DOI] [PubMed] [Google Scholar]
- Graff-Radford J, Raman MR, Rabinstein AA, Przybelski SA, Lesnick TG, Boeve BF, Murray ME, Dickson DW, Reichard RR, Parisi JE, Knopman DS, Petersen RC, Jack CR Jr., Kantarci K, 2018. Association Between Microinfarcts and Blood Pressure Trajectories. JAMA Neurol 75(2), 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM, 2006. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci 26(35), 9047–9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL, 1982. A new clinical scale for the staging of dementia. Br J Psychiatry 140, 566–572. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, Petersen RC, Dickson DW, 2014a. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 127(3), 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, Liesinger AM, Petersen RC, Parisi JE, Dickson DW, 2016. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131(4), 571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF, Ivnik RJ, Smith GE, Jack CR Jr., Parisi JE, Petersen RC, Dickson DW, 2014b. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127(6), 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S, 1983. Boston Naming Test. Lee & Febiger, Philadelphia. [Google Scholar]
- Keage HA, Hunter S, Matthews FE, Ince PG, Hodges J, Hokkanen SR, Highley JR, Dening T, Brayne C, 2014. TDP-43 pathology in the population: prevalence and associations with dementia and age. J Alzheimers Dis 42(2), 641–650. [DOI] [PubMed] [Google Scholar]
- Klunk WE, 2018. Molecular imaging: What is right and what is an illusion? Alzheimers Dement (Amst) 10, 217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, Smith GE, Dickson DW, Johnson KA, Petersen LE, McDonald WC, Braak H, Petersen RC, 2003. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 62(11), 1087–1095. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM, 2010. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119(1), 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek-Ahmadi M, Perez SE, Chen K, Mufson EJ, 2016. Neuritic and Diffuse Plaque Associations with Memory in Non-Cognitively Impaired Elderly. J Alzheimers Dis 53(4), 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery WR, Jicha GA, Liu H, Schmitt FA, 2009. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol 68(7), 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews FE, Brayne C, Lowe J, McKeith I, Wharton SB, Ince P, 2009. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med 6(11), e1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stein TD, Kiernan PT, Alvarez VE, 2015. The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25(3), 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L, 1991. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41(4), 479–486. [DOI] [PubMed] [Google Scholar]
- Nascimento C, Di Lorenzo Alho, Bazan Conceicao Amaral C, Leite REP, Nitrini R, Jacob-Filho W, Pasqualucci CA, Hokkanen SRK, Hunter S, Keage H, Kovacs GG, Grinberg LT, Suemoto CK, 2018. Prevalence of transactive response DNA-binding protein 43 (TDP-43) proteinopathy in cognitively normal older adults: systematic review and meta-analysis. Neuropathol Appl Neurobiol 44(3), 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM, 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796), 130–133. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Mattek N, Woltjer R, Howieson D, Silbert L, Hofer S, Kaye J, Dodge H, Erten-Lyons D, 2018. Pathologies Underlying Longitudinal Cognitive Decline in the Oldest Old. Alzheimer Dis Assoc Disord 32(4), 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovitch H, Ross GW, Steinhorn SC, Abbott RD, Markesbery W, Davis D, Nelson J, Hardman J, Masaki K, Vogt MR, Launer L, White LR, 2005. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol 57(1), 98–103. [DOI] [PubMed] [Google Scholar]
- Price JL, McKeel DW Jr., Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC, 2009. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 30(7), 1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi J, Kovacs GG, 2014. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 6(9), 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitan RM, 1958. Validity of the trail making test as an indicator of organic brain damage. Perceptual and Motor Skills 8, 271–276. [Google Scholar]
- Rey A, 1964. L’examen psychologique dans les cas d’encephalopathie traumatique. Archives de Psychologie 28, 286–340. [Google Scholar]
- Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, Ivnik RJ, Tangalos EG, Petersen RC, Rocca WA, 2008. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 30(1), 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AC, Davidson YS, Horan MA, Pendleton N, Mann DMA, 2018. Pathological Correlates of Cognitive Impairment in The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age. J Alzheimers Dis [DOI] [PubMed] [Google Scholar]
- SantaCruz KS, Sonnen JA, Pezhouh MK, Desrosiers MF, Nelson PT, Tyas SL, 2011. Alzheimer disease pathology in subjects without dementia in 2 studies of aging: the Nun Study and the Adult Changes in Thought Study. J Neuropathol Exp Neurol 70(10), 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR, 2000. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology 55(3), 370–376. [DOI] [PubMed] [Google Scholar]
- Stern Y, 2009. Cognitive reserve. Neuropsychologia 47(10), 2015–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suemoto CK, Ferretti-Rebustini RE, Rodriguez RD, Leite RE, Soterio L, Brucki SM, Spera RR, Cippiciani TM, Farfel JM, Chiavegatto Filho A, Naslavsky MS, Zatz M, Pasqualucci CA, Jacob-Filho W, Nitrini R, Grinberg LT, 2017. Neuropathological diagnoses and clinical correlates in older adults in Brazil: A cross-sectional study. PLoS Med 14(3), e1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumerall SW, Timmons PL, James AL, Ewing MJ, Oehlert ME, 1997. Expanded norms for the Controlled Oral Word Association Test. J Clin Psychol 53(5), 517–521. [DOI] [PubMed] [Google Scholar]
- UPDRS, 2003. The Unified Parkinson’s Disease Rating Scale (UPDRS): status and recommendations. Mov Disord 18(7), 738–750. [DOI] [PubMed] [Google Scholar]
- Wechsler D, 1987. Wechsler Memory Scale - Revised Manual. Psychological Corporation, San Antonio, TX. [Google Scholar]
- Wennberg AM, Tosakulwong N, Lesnick TG, Murray ME, Whitwell JL, Liesinger AM, Petrucelli L, Boeve BF, Parisi JE, Knopman DS, Petersen RC, Dickson DW, Josephs KA, 2018. Association of Apolipoprotein E epsilon4 With Transactive Response DNA-Binding Protein 43. JAMA Neurol 75(11), 1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, Yu L, White CC, Chibnik LB, Chhatwal JP, Sperling RA, Bennett DA, Schneider JA, De Jager PL, 2018. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: a community-based cohort study. Lancet Neurol 17(9), 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]