

Abstract
Cardiac ischemia and reperfusion (IR) injury induces excessive emission of deleterious reactive O2 and N2 species (ROS/RNS), including the non-radical oxidant peroxynitrite (ONOO−) that can cause mitochondria dysfunction and cell death. In this study, we explored whether IR injury in isolated hearts induces tyrosine nitration of adenine nucleotide translocase (ANT) and alters its interaction with the voltage-dependent anion channel 1 (VDAC1). We found that IR injury induced tyrosine nitration of ANT and that exposure of isolated cardiac mitochondria to ONOO− induced ANT tyrosine, Y81, nitration. The exposure of isolated cardiac mitochondria to ONOO− also led ANT to form high molecular weight proteins and dissociation of ANT from VDAC1. We also found that IR injury in isolated hearts, hypoxic injury in H9c2 cells, and ONOO− treatment of H9c2 cells and isolated mitochondria, each decreased mitochondrial bound-hexokinase II (HK II), which suggests that ONOO− caused HK II to dissociate from mitochondria. Moreover, we found that mitochondria exposed to ONOO− induced VDAC1 oligomerization which may decrease its binding with HK II. We have reported that ONOO− produced during cardiac IR injury induced tyrosine nitration of VDAC1, which resulted in conformational changes of the protein and increased channel conductance associated with compromised cardiac function on reperfusion. Thus, our results imply that ONOO− produced during IR injury and hypoxic stress impeded HK II association with VDAC1. ONOO− exposure nitrated mitochondrial proteins also led to cytochrome c (cyt c) release from mitochondria. In addition, in isolated mitochondria exposed to ONOO− or obtained after IR, there was significant compromise in mitochondrial respiration and delayed repolarization of membrane potential during oxidative (ADP) phosphorylation. Taken together, ONOO− produced during cardiac IR injury can nitrate tyrosine residues of two key mitochondrial membrane proteins involved in bioenergetics and energy transfer to contribute to mitochondrial and cellular dysfunction.
Keywords: Mitochondria, Adenine Nucleotide Translocase, Voltage-Dependent Anion Channel 1, Hexokinase II, Peroxynitrite, Tyrosine Nitration, Cardiac Ischemia Reperfusion Injury
Introduction
Cardiac ischemia and reperfusion (IR) injury impairs metabolic and/or ion transport via mitochondrial membrane channels and exchangers, specifically, the voltage dependent anion channel (VDAC) and the adenine nucleotide translocase (ANT), located on the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), respectively. Damage to VDAC and ANT due to excessive emission of deleterious reactive O2 and N2 species (ROS/RNS) could interfere with their functional interactions and cause mitochondrial dysfunction and the release of cytochrome c (cyt c), which culminate in cell dysfunction and eventually cell death.
ANT, located in the IMM, mediates the exchange of ATP/ADP between the mitochondrial matrix and the intermembrane space (IMS) (Brand et al., 2005; Palmieri and Pierri, 2010). Of the 4 known isoforms, ANT1 is the dominant isoform in the heart (Palmieri and Pierri, 2010). Under physiological conditions, VDAC and ANT are functionally coupled, which allows for the efficient transfer of metabolites across mitochondrial membranes (Allouche et al., 2012; Camara et al., 2017; Vyssokikh and Brdiczka, 2003). Using surface plasmon resonance (Allouche et al., 2012) ANT and VDAC1 were found to have direct structural interactions that were dependent on the ionic conditions and ionic strength of the experimental buffer. The binding of the ANT inhibitor atractyloside (ATR) or bongkrekic acid (BKA) reduces the interaction of ANT and VDAC1 in vitro (Allouche et al., 2012). It is not known if oxidant stress conditions, such as excess peroxynitrite (ONOO−) production from the reaction of superoxide (O2•−) and nitric oxide (NO•) during I/R injury, induces modification of ANT by tyrosine nitration, and alters its interaction with VDAC1.
VDAC, the most abundant protein in the OMM, plays a crucial role in both mitochondrial metabolism and cell death (Colombini, 2012; Shoshan-Barmatz and Ben-Hail, 2012). In the open state, VDAC favors the transport of anions, such as metabolites, ATP, ADP, and Pi, but it also permits the free diffusion of cations, including Ca2+, K+, and Na+ (Mazure, 2016; O’Rourke, 2007), whereas in the closed state, VDAC favors cationic permeability, notably Ca2+ ions, which in excess, impairs ADP/ATP transport (O’Rourke, 2007). VDAC exists as three isoforms in mammals, VDACs 1, 2 and 3; the protein spans the OMM with 19 β-strands (Messina et al., 2012; Raghavan et al., 2012). Among the three isoforms, VDAC1 is the most abundant in heart mitochondria and it plays a role in modulating cardiac IR injury (Das et al., 2012; McCommis and Baines, 2012; Shoshan-Barmatz and Ben-Hail, 2012; Shoshan-Barmatz et al., 2008). The mechanisms of how VDAC1 regulates cell death in cardiac IR injury remain incompletely understood. There are reports that reducing cytosolic ATP entry into mitochondria via VDAC during ischemia and its subsequent consumption by the F1F0-ATPase might better preserve cellular ATP, thereby reducing glycolysis, ischemic acidosis, and intracellular Ca2+ overload, to culminate in protection against IR injury (Das et al., 2012; McCommis and Baines, 2012; Murphy and Steenbergen, 2008). In addition, the regulation of VDAC1 function by protein-protein interactions with, for example ANT and hexokinase II (HK II), and the effects of deleterious post-translational modifications (dPTMs) on ANT and VDAC may also play a major role in altering cell death pathways.
Studies in cardiac and skeletal myocytes show that VDAC reversibly binds with several cytosolic proteins, including HK II (Das et al., 2012; Perevoshchikova et al., 2010; Zorov et al., 2009). The predominant isoform of hexokinase in the myocardium is HK II, which binds to mitochondria where it acts as an important regulator of mitochondria- mediated cell demise (Azoulay-Zohar et al., 2004; Sun et al., 2008). In this case, the association of HK II with mitochondria inhibits the mitochondrial translocation of Bax, a proapoptotic protein, and the release of cyt c (Majewski et al., 2004; Pastorino et al., 2002), thereby impeding cell apoptosis (Das et al., 2012; Zorov et al., 2009). It has been reported that ischemia (Pasdois et al., 2012) or glucose deprivation in adult hearts or isolated cardiac myocytes (Calmettes et al., 2013) induces HK II dissociation from mitochondria, possibly from VDAC, which destabilizes the mitochondrial contact sites between VDAC and ANT, causing OMM permeabilization and inducing mitochondrial cyt c loss to promote apoptosis (Pasdois et al., 2012). The mechanism that regulates association/dissociation of HK II with/from VDAC is not clear. Suffice it to say, activation of glycogen synthase kinase 3β (GSK-3β) phosphorylates VDAC and this may disrupt the binding of HK II to potentiate cell death (Pastorino et al., 2005). We have reported (Yang et al., 2012) that ONOO− produced during cardiac IR injury induced tyrosine nitration of VDAC1 on at least one specific tyrosine residue, but it is unknown if this also alters the association of HK II with VDAC1.
It is not established if ONOO− also induces tyrosine nitration of ANT and affects VDAC1 and ANT interaction, as well as the interaction of VDAC1 with HK II, all of which are events that may lead to altered mitochondrial and cellular function. Thus, in this study we examined tyrosine nitration of both ANT and VDAC1, and investigated the impact of ONOO− on these two proteins and their functional coupling as well as the effect of VDAC1 interaction with HK II. We hypothesized that ONOO− induces ANT tyrosine nitration and alters its interaction with VDAC1, and impairs the association of HK II with mitochondria, specifically VDAC1. We found that exposure of mitochondria to ONOO− hinders the ability of ANT to interact with VDAC1, and VDAC1 to interact with HK II. Moreover, mitochondrial function is altered by ONOO−. Consequently, mitochondrial exposure to ONOO− triggered disassembly of the HK II-VDAC1-ANT complex, which likely contributes to mitochondrial dysfunction as an underlying factor in cardiac IR injury.
Methods and Materials
Guinea pig heart preparation for ischemia and reperfusion
Guinea pigs were used in this study and the Medical College of Wisconsin Institutional Animal Care and Use Committee approved the project. Hearts were prepared for ex vivo studies as described previously (Stowe et al., 2013; Stowe et al., 2017; Yang et al., 2017). Briefly, guinea pigs were injected intraperitoneally with a combination of heparin (1000 units) to prevent clotting, and ketamine (50 mg/kg) for anesthesia, before sacrifice by decapitation. Hearts were removed and perfused retrograde at constant pressure (55 mmHg) via the aortic root with an oxygenated Krebs-Ringer’s (KR) solution of the following composition (in mM): 138 Na+, 4.5 K+, 1.2 Mg2+, 2.5 Ca2+, 134 Cl−, 15 HCO3−, 1.2 H2PO4−, 11.5 glucose, 2 pyruvate, 16 mannitol, 0.1 probenecid, 0.05 EDTA, and 5 U/L insulin and gassed with 3% CO2, 97% O2 (pH 7.4) at 37°C. Systolic and diastolic left ventricular pressure (LVP), coronary flow (CF), and heart rate (HR) were measured online, and the developed LVP and rate pressure product (RPP) were derived as the difference of systolic LVP and diastolic LVP and the product of the developed LVP and HR, respectively. The aortic inflow line was clamped for 35 min to induce global ischemia followed by unclamping inflow to initiate reperfusion. After 20 min of reperfusion hearts were removed and mitochondria were immediately isolated for further studies.
We evaluated the maximal cardioprotective effects of perfusing the anti-oxidant cocktail (MCGR) on cardiac function after ischemia with extended reperfusion time, 120 min, in isolated guinea pig hearts (n =6 per drug group). Three groups of guinea pigs were perfused either with the MCGR for 5 min before ischemia and for 5 min after ischemia (IR+MCGR), vehicle (IR) or time control (TC), i.e. perfusion without IR. The MCGR cocktail contained 10 µM MnTBAP, a mitochondrial SOD2 mimetic, catalase (30–120 units), 500 µM glutathione, and 100 µM resveratrol.
Exposure of H9c2 cells to hypoxia and ONOO−
H9c2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS) under 95% air, 5% CO2 at 37°C, and subcultured when at 50–60% confluence. To induce hypoxia, H9c2 cells, which were grown on coverslips in 35 mm dishes, were transferred to glucose-free DMEM with 10 mM 2-deoxyglucose, placed in an airtight chamber in a 37°C incubator, and flushed with a hypoxic gas mixture of 95% N2, 5% CO2, as we reported previously (Yang et al., 2017). The cells were exposed to hypoxia for 4 h. For the control, H9c2 cells were incubated in normal DMEM medium containing 10% FBS with 95% air, 5% CO2 at 37°C. After hypoxia, cells were fixed with 4% formaldehyde and examined for HK II interaction with mitochondria by immunofluorescence.
To examine the impact of ONOO− on tyrosine nitration, H9c2 cells were cultured on coverslips in 35 or 60 mm dishes and replenished with fresh culture medium. Various concentrations of ONOO − stock were prepared freshly in 0.1 N NaOH (Murray et al., 2003). ONOO− concentration was determined by absorbance at 302 nm with an extinction coefficient of 1670 M−1 cm−1 in 0.1 N NaOH prior to use (Murray et al., 2003). For exposure to ONOO−, cell medium were changed to 0.25 mM potassium phosphate buffer at pH 7.2, 5 mM MgCl2 and then ONOO− was added to dishes of cells. To achieve the desired [ONOO−], 1 µl of ONOO− stock per ml of medium was added to cell dishes separately. After that cells were incubated for 10 min in the dark and then change the medium to DMEM with 10% fetal bovine serum (FBS) and incubated for 4 h in 95% air, 5% CO2 at 37°C. For the control experiments, degraded ONOO− was added to the cell dishes. The degradation of ONOO− was prepared by leaving a solution of ONOO− overnight at room temperature, which contained similar amounts of nitrite, nitrate and NaOH as in the ONOO− solution, but without the intact oxidant ONOO− (Pesse et al., 2005). After 4 h of treatment, cells in the 60 mm dishes were lysed to examine protein tyrosine nitration by western blot; cells grown on coverslips in 35 mm dishes were fixed with 4% formaldehyde and examined for protein tyrosine nitration by immunofluorescence. To stain mitochondria, cells were incubated with 200 nM MitoTracker™ Red CMXRos (Invitrogen, Life Technologies, Carlsbad, CA) in serum-free medium for 30 min before fixation.
Immunofluorescence staining was used to evaluate cell tyrosine nitration and HK II association/dissociation with mitochondria. Briefly, after hypoxia or treatment with ONOO −, H9c2 cells were fixed with 4% formaldehyde in PBS for 15 min, followed by permeabilization with 100% methanol at −20oC for 10 min, and then with 0.1% Triton X-100 for 15 min. The cells were blocked with blocking solution containing 10% goat serum with 2% BSA for 1 h at room temperature. Primary antibodies, 3-NT (mouse, Millipore, MA), HK II (rabbit, Proteintech, Rosemont, IL) and COX IV (mouse, Cell signaling technology, Danvers, MA) were diluted in blocking solution and incubated with the cells for 2 h at 37°C. Secondary antibodies, anti-mouse conjugated Alexa Fluor 488 (Thermo Fisher Scientific,Waltham, MA), and anti-rabbit conjugated Alexa Fluor 568 (Thermo Fisher Scientific, Waltham, MA) were diluted in blocking reagent and incubated with the cells for 1 h at room temperature. After three more washes with PBS, the cells were stained for nuclei with DAPI (Thermo Fisher Scientific, Waltham, MA, USA). The coverslips were mounted on glass slides with SHUR/Mount™ mounting medium and sealed with nail polish. Images were collected from a Nikon confocal microscope equipped with 63×1.4 NA oil-immersion lens. The fluorescence intensity of the 3-NT signal was quantized by image J. The confocal microscopy images with the same pixels were first adjusted to have the same threshold and then the mean gray values for each cell were measured. For each group, 15 cells were measured and the mean gray value averages were calculated. The co-localization of HK II with COX IV, a mitochondria protein, was determined by line scan analysis with ImageJ to indicate the degree of overlap between the two signals. A line (Green color) across the cell was drawn in the merged figure and the intensity profiles of all pixels of the two signals were plotted with a RGB profile plot program. Confocal microscopy images were analyzed by image J Coloc 2 to quantify the co-localization of HK II and mitochondrial marker COX IV. The auto-thresholded Mander’s split co-localization coefficient (zero: no co-localization; one: perfect co-localization) was used for co-localization parameters. There is one coefficient per channel (HK II or COX IV) that shows the proportion of signal for that channel as co-localizing with another channel. For each group, 10 cells were analyzed and average co-localization coefficients of HK II to COX IV and COX IV to HK II were calculated and plotted in a histogram panel.
Mitochondria isolation
Mitochondria were isolated as described previously (Agarwal et al., 2014b; Aldakkak et al., 2013; Blomeyer et al., 2016; Yang et al., 2012; Yang et al., 2014) with minor modifications. All procedures were carried out at 4°C. Hearts were minced in a chilled isolation buffer containing (in mM): 200 mannitol, 50 sucrose, 5 KH2PO4, 5 MOPS, 1 EGTA, 0.1% fatty acid free BSA, and protease inhibitors at pH 7.15, and then homogenized. The homogenized slurries were centrifuged for 10 min at 8,000 g. After decanting the supernatant, the pellet was re-suspended in isolation buffer and spun at 900 g for 10 min; the supernatant was collected and re-centrifuged at 8000 g. The final mitochondrial pellet following this centrifugation was re-suspended in isolation buffer and purified as described by Graham (Graham, 2001). That is, mitochondria were layered on 30% Percoll in isolation buffer, and then centrifuged for 30 min at 95,000 g. After centrifugation, mitochondria were collected and washed twice with isolation buffer. The purified mitochondria were used for western blot, immunoprecipitation, and enrichment of ANT, or were re-suspended in potassium phosphate buffer for exposure to exogenous ONOO−.
Enrichment of ANT from purified mitochondria
To determine whether IR injury induces ANT tyrosine nitration, ANT was first enriched from isolated mitochondria according to a previous report (Vyssokikh et al., 2001) with minor modifications. In brief, purified heart mitochondria (5 mg) in 1.5 ml of isolation buffer were incubated for 30 min with an equal volume of extraction buffer consisting of (in mM): 40 KH2PO4, 40 KCl, 2 EDTA, and 6% Triton X-100, at pH 6.0. The suspension was centrifuged for 30 min at 24,000 g and the supernatant was loaded on a column filled with 0.25 g of dry hydroxyapatite (Bio-Rad). After elution with extraction buffer, the flow-through fraction was collected and diluted 1:1 with equilibration buffer containing 20 mM Mes/0.2 mM EDTA/0.5% Triton X-100, at pH 6.0. This sample was applied to a 1 ml HiTrap SP cation-exchange column (Pharmacia), washed with equilibration buffer, and then eluted with 0.6 M NaCl in equilibration buffer. 1 ml of fractions were collected, precipitated with trichloroacetic acid, and then analyzed by western blot.
Exposure of mitochondria to exogenous ONOO−
To verify the direct impact of ONOO− on tyrosine nitration of mitochondrial proteins, we exposed isolated mitochondria to various concentrations of ONOO− in vitro according to previous reports (Reynolds et al., 2005; Rowan et al., 2002). Mitochondria (2 mg) isolated from guinea pig hearts were re-suspended in 200 µl solution containing 0.25 mM potassium phosphate buffer at pH 7.2, 5 mM MgCl2. Mitochondria were exposed to ONOO− by directly mixing ONOO− into the mitochondrial suspension (2 mg/200 µl) in 5 boluses of 1 µl each to achieve the various desired concentrations as indicated, and then the mitochondria were incubated for 30 min on ice in the dark; degraded ONOO− or 0.1 N NaOH was used as the control. After incubation, the mitochondrial pellet was washed twice with isolation buffer and then re-suspended in isolation buffer. The mitochondrial suspension was used to measure mitochondrial respiration and membrane potential (ΔΨm), and to assess mitochondrial protein tyrosine nitration, cyt c content, ANT-VDAC1 interaction, and HK II-VDAC1 interaction, by immunoprecipitation or western blot. Since ONOO− was washed out before mitochondria were re-suspended in fresh isolation buffer of pH 7.15, Na-pyruvate was not affected by an alkaline pH effect of ONOO–- in 0.1 N NaOH during assessment of changes in respiration and ΔΨm.
To evaluate the roles of VDAC1, ANT and anti-oxidation on cyt c release following exposure of mitochondria to ONOO−, isolated mitochondria were pre-incubated with DIDS (0.5 mM), an inhibitor of VDAC, bongkrekic acid (50 µM), an inhibitor of ANT, or the cocktail antioxidant MCGR, for 30 min before treatment with ONOO−.
Tyrosine nitrated ANT identification by MS/MS mass spectrometry assay
Mitochondria treated with ONOO− were lysed and the proteins were resolved by SDS-PAGE. Protein bands were stained with Coomassie blue, excised and cut into small pieces and then subjected to ingel digestion by trypsin as described previously (Yang et al., 2012). Extracted tryptic fragments were purified for mass spectrometry analysis with a Thermo Orbitrap Velos instrument. The digests were dissolved in 2 µl of 2% acetonitrile 0.1% formic acid; there were two LC-MS runs of 1.5 µl sample injection each. They were run for 120 min on a water/acetonitrile 0.1% formic acid gradient on a Waters NanoAcquity system with a 10 cm x 75 µm column packed with 3 µm Magic C18AQ stationary phase. The MS/MS data were processed with the Sorcerer Sequest software for searching against a UniProt guinea pig database to which common, ubiquitous laboratory contaminants were added. The results were exported into Scaffold files. The search criteria were set to: minimum peptide count of 2; minimum peptide probability of 0.95; peptide False Discovery Rate <5%; minimum protein probability of 0.95 and protein False Discovery Rate <1%. Those proteins that matched the experimental data and met the search criteria were considered to have a statistically significant probability of being present in the sample. The database searches were conducted with tyrosine nitration as a variable modification.
Cross-linking, immunoprecipitation and western blot
To assess for VDAC1 oligomerization induced by mitochondrial exposure to ONOO−, cross-linking experiments were conducted using the cross linker, EGS (ethylene glycolbis [succinimidylsuccinate]) before lysing mitochondria for western blot as reported previously (Geula et al., 2012). EGS is a water-insoluble, homo-bifunctional N-hydroxysuccinimide ester (NHS ester) that reacts with α-amine groups present on the N-termini of proteins to stabilize protein-protein interactions. In this case, mitochondria treated with ONOO− were re-suspended in PBS at pH 8.3 and the cross-linking reagent EGS was added to the mitochondrial suspension to a final concentration of 0.5 mM and then incubated for 15 min at room temperature. The EGS reaction was quenched by incubation with 10 mM Tris-Cl at pH 7.5 for 15 min at room temperature. After cross-linking, mitochondria were lysed and subjected to SDS-PAGE and immunoblotted with anti-VDAC1 antibody.
Immunoprecipitation (IP) was performed as we described before (Yang et al., 2012; Yang et al., 2014). For ANT IP, mitochondria were pelleted and then lysed in PBS containing 1% lauryl maltoside and protease inhibitors on ice for 30 min. For HK II IP, mitochondria were first cross-linked with DSP (dithiobis(succinimidyl propionate)), a DTT cleavable cross linker, for 30 min at room temperature and then lysed with RIPA buffer containing 50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS and protease inhibitors on ice for 30 min. After lysis, the sample was pre-cleared with 30 μl protein G Sepharose-4B beads (Invitrogen) for 1 h at 4°C with constant end-over-end shaking and then centrifuged at 3,000 g for 5 min. The supernatant was collected, and the protein concentration was adjusted to 2 mg/ml with RIPA buffer for HK II IP and with PBS with 1% lauryl maltoside for ANT IP. Anti-HK II or anti-ANT antibody was added to the protein samples individually and incubated with constant end-over-end shaking overnight at 4°C. The next day, after adding 30 μl protein G Sepharose-4B beads, the mixture was incubated for another 2 h under the same conditions as above. The beads were collected and thoroughly washed (five times) with RIPA buffer. The final pellet was re-suspended in 50 μl sample buffer containing 50 mM Tris-Cl, 10% glycerol, 500 mM β-mercaptoethanol, 2% SDS, 0.01% w/v bromophenol blue and protease inhibitors at pH 7.4 and boiled at 95°C for 5 min. The immunoprecipitated proteins were separated using SDS-PAGE and subjected to western blot.
For western blots, mitochondrial protein lysates or immunoprecipitates were resolved by SDS-PAGE. To assess if ANT forms high molecular weight protein complex by interacting with other mitochondrial proteins, non-reducing SDS-PAGE was performed. After gel electrophoresis, proteins were transferred onto PVDF membranes. The membranes were incubated with specific primary antibodies: anti-3-NT (mouse monoclonal, 1:1000, Millipore); anti-ANT (Goat polyclonal, 1:200, Santa Cruz biotechnology); anti-VDAC1 (rabbit polyclonal, 1:1000, Cell Signaling Technology); anti-cyt c (mouse monoclonal, 1:1000, Invitrogen); and anti-HK II (rabbit monoclonal, 1:1000, Cell Signaling Technology). Washed membranes were incubated with the appropriate secondary antibody conjugated to HRP and then immersed in an enhanced chemiluminescence detection solution (GE Healthcare) and exposed to X-Ray film for autoradiography.
Mitochondrial O2 consumption and membrane potential in isolated mitochondria
Mitochondrial bioenergetics were measured following exposure to exogenous ONOO−, or after 35 min ischemia and 20 min reperfusion (I35R20), to determine the functional consequence of tyrosine nitration. O2 consumption rate was measured polarographically using a respirometer (System S200A, Strathkelvin Instruments) as described previously (Agarwal et al., 2014a; Blomeyer et al., 2013; Stowe et al., 2017; Yang et al., 2017). The mitochondria pellet was re-suspended in isolation buffer at 12.5 mg protein/ml; 0.275 mg of mitochondria was added into 0.5 ml respiration buffer containing (in mM):130 KCl, 5 K2HPO4, 20 MOPS, 2.5 EGTA, 1 Na4P2O7, 0.1% BSA, at pH 7.15, to measure O2 consumption. State 2 respiration was initiated with complex I substrate Na-pyruvate (10 mM); state 3 respiration was determined after adding 250 µM ADP, and state 4 respiration was assessed after most of the added ADP was phosphorylated to ATP via oxidative phosphorylation. The respiratory control index (RCI; state 3/state 4 respiration) was determined to evaluate viability and functional coupling of mitochondria.
Mitochondrial membrane potential (ΔΨm) was measured in the mitochondrial suspension using a similar respiration buffer during states 2, 3 and 4 respiration by fluorescence spectrophotometry using the fluorescent dye tetramethyl-rhodamine methyl ester (1 μM TMRM; Molecular Probes, Eugene, OR) at excitations of λ546 nm and λ573 nm and emission λ590 nm, as described before (Agarwal et al., 2014a; Aldakkak et al., 2013). TMRM is a ratiometric, membrane potential sensitive, cationic fluorophore that equilibrates across the IMM based on its electrochemical potential. To assess ΔΨm, the mitochondria pellet was re-suspended in isolation buffer at 12.5 mg protein/ml and then 0.5 mg of mitochondria was added to 1 ml of the respiration buffer. Mitochondria were energized with the complex I substrate Na-pyruvate (10 mM) (Aldakkak et al., 2013; Heinen et al., 2007) and the duration of ΔΨm depolarization resulting from addition of 250 μM ADP (state 3 respiration) was determined. The respiratory uncoupler carbonyl cyanide m-chlorophenyl- hydrazone (4 μM CCCP) was given at the end of each experimental run to fully depolarize ΔΨm.
Statistical analysis
All results are expressed as means ± SEM and were analyzed by one-way ANOVA followed by a post-hoc analysis (Student–Newman–Keuls’ test) to determined statistically significant differences in means among groups. A value of p < 0.05 was considered significant (two-tailed).
Results
Exogenous exposure of mitochondria to ONOO− and IR injury induce tyrosine nitration of ANT
A previous report showed that ONOO− is generated during IR injury and that it causes permanent nitration of tyrosine residues (Szabo et al., 2007). We investigated if ONOO− induces tyrosine nitration of mitochondrial proteins in H9c2 cells. H9c2 cells were exposed to ONOO− for 4 h and then stained with anti-3-NT antibody and Mitotracker red CMXRos, a mitochondrial marker, to verify mitochondrial location. We found that exposure of cells to ONOO− increased protein tyrosine nitration of H9c2 cells (79% ± 2) compared to the control cells (57% ± 1) (Fig. 1A). Co-localization of 3-NT and Mito-tracker red CMXRos indicated that ONOO− caused tyrosine nitration of mitochondrial proteins in H9c2 cells. Western blot analyses also confirmed the protein tyrosine nitration in H9c2 cells exposed to ONOO−, this was not detected when exposed to degraded ONOO− (Fig. 1B). We also conducted control experiments in which 0.1N NaOH was used as a control instead of degraded ONOO −. The results were not significantly different between the 0.1N NaOH and degraded ONOO− groups (data not shown). Thus, in the remaining experiments, 0.1N NaOH was used as the non-treatment control.
Figure 1:

Tyrosine nitration of mitochondrial ANT induced by ONOO−. A: Immunostaining of H9c2 cells treated with 200 µM ONOO− using anti-3-NT and mitochondrial marker MitoTracker™ Red CMXRos. The histogram panel below represents the mean fluorescence intensity from 15 cells in each group. *: p < 0.05 vs. control. B: Western blot analyses of H9c2 cells treated with 0 to 500 µM ONOO− using anti-3-NT and β-tubulin (loading control) antibodies. The histogram panel below represents the mean relative 3-NT band intensity (normalized to β-tubulin) from three independent experiments. *: p 0.05 vs. control. C: Western blot analyses of protein tyrosine nitration in isolated mitochondria treated with 0 to 500 µM ONOO− using anti-3-NT and COX IV (loading control) antibodies. Histogram panel represents the mean relative 3-NT band intensity (normalized to COX IV) from three time experiments. *: p < 0.05 vs. control. D: Coverage of ANT residues detected by mass spectrometric assay. Highlighted yellow are detected peptides; highlighted green are detected nitrated tyrosine Y81. E: Spectrum of peptide displaying the nitration of Y81. F: Western blot analyses of tyrosine nitration of enriched ANT and mitochondrial lysate isolated from mitochondria treated with 200 µM ONOO− in the absence or presence of MCGR using anti-3-NT antibody (3-NT panel); ANT band confirmed the loading of similar amount of enriched ANT and mitochondrial lysate (ANT panel). The histogram panel below represents the mean relative 3-NT band intensity (normalized to ANT) from three independent experiments. *: p < 0.05 vs. presence of MCGR for mitochondrial lysate; #: p < 0.05 vs. presence of MCGR for enriched ANT.
We also determined whether exposure of isolated mitochondria to exogenous ONOO− can induce tyrosine nitration of ANT and VDAC1. Mitochondria were treated with various concentrations of ONOO− and then lysed and the proteins subjected to western blot and mass spectrometric analysis. We found that ONOO− treatment induced tyrosine nitration of proteins in isolated mitochondria (Fig. 1C), including a 32 kDa protein that we identified previously as VDAC1 (Yang et al., 2012). In addition, mass spectrometric analysis indicated that for ANT (Fig. 1D), 125 of 298 amino acids (42% coverage) were identified, of which 7 of the 12 known tyrosine residues were detected; one of these, Y81 (highlighted in green) was nitrated (Figs. 1D,E).
We showed previously that resveratrol reduced tyrosine nitration of VDAC1 protein induced by ischemia and reperfusion in the ex vivo isolated heart model and reduced protein nitration after ONOO− exposure to recombinant VDAC1 in vitro (Yang et al., 2012). Moreover, we also have reported that when compared to MnTBAP treated hearts, MCG treated hearts had better normalization of the mitochondrial redox state (NADH and FAD), decreased O2•− levels, lower m[Ca2+] during and after ischemia, as well as higher contractile and relaxant indices on reperfusion and a reduction in infarct size (Camara, AJP 2007). Based on these prior studies, we combined MCG and resveratrol (MCGR cocktail) to attempt to maximize the cardioprotection against heart IR injury and to determine if the MCGR cocktail attenuated the effects of ONOO− on ANT and VDAC1. ANT was enriched after mitochondrial exposure to ONOO− in the presence or absence of MCGR, and tyrosine nitration of ANT was examined by western blot. Fig. 1F shows that compared to ONOO− alone treatment, mitochondria treated with MCGR before adding ONOO− exhibited weaker anti 3-NT band intensities in some proteins with band sizes of approximately 37 and 30 kDa. Fig. 1F also shows that the size of the 30 kDa protein was similar in size to that of enriched ANT; this suggests that MCGR attenuated nitration of ANT induced by exposure of mitochondria to ONOO−.
In addition to showing that exogenous ONOO− nitrates ANT on tyrosine, we also showed that ANT was tyrosine nitrated following IR injury. Mitochondrial proteins from time control (TC) and IR-injured hearts were subjected to immunoprecipitation using anti-ANT antibody followed by western blot with anti-3-NT antibody. Using this approach, we at first did not observe anti-3-NT signals (data not shown), possibly because ANT tyrosine nitration was below the detectable range of the immunoprecipitation and western blot analysis. So, alternatively, after isolating mitochondria from IR-injured or TC hearts, we first enriched ANT with hydroxyapatite and HiTrap SP cation-exchange column and subjected the enriched ANT to gel electrophoresis. The gels were stained with Coomassie blue (Fig. 2A) or subjected to western blot (Fig. 2B) using anti-3-NT antibody. As shown in Figs. 2B and C, compared to TC, after 35 min ischemia and 20 min reperfusion (I35R20), there was an increase in the ANT anti-3-NT band intensity, indicating tyrosine nitration of ANT. Moreover, in addition to the 30 kDa band, bands approximating 60 kDa also appeared in the anti-3-NT signal. This 60 kDa band may be due to other oxidative modifications of the protein caused by exposure to ONOO −. The formation of a protein complex with ANT likely would not be detectable under the denaturing condition of the SDS-PAGE.
Figure 2:
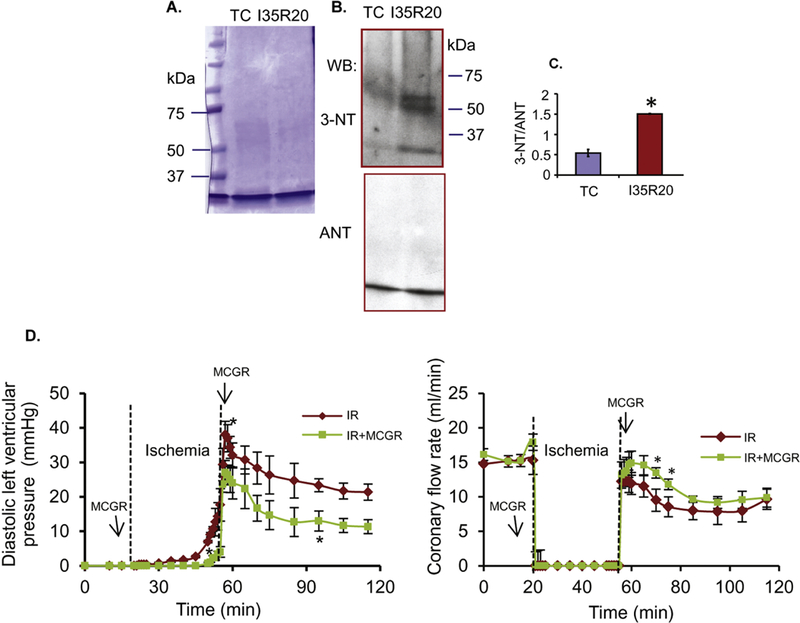
Tyrosine nitration of mitochondrial ANT induced by IR injury. A: Coomassie blue stain of enriched ANT from mitochondria isolated from hearts subjected to time control (TC) or 35 min ischemia and 20 min reperfusion (I35R20). B: Western blot of enriched ANT with 3-NT antibody; ANT was used as loading control. C: Data summary of 3-NT band intensity to ANT band ratio for TC and I35R20; *p < 0.05 vs. TC. D: Time course of changes in diastolic LVP (left panel) and coronary flow rate (right panel) during 35 min ischemia and 20 min reperfusion alone (IR) or in the presence of MCGR (IR+MCGR). *: p < 0.05 vs. IR.
As shown in Fig. 2D, compared to IR only (vehicle), perfusion of MCGR before and after ischemia resulted in a significantly lower diastolic LVP during late ischemia and most of reperfusion (Fig. 2D left panel); coronary flow was higher in the MCGR treated group than in the IR alone (vehicle) during the early part of reperfusion (Fig. 2D right panel). The time control (TC) maintained the levels of these variables for the duration of the perfusion (data not shown).
ANT dissociates from VDAC1 after ex vivo cardiac I/R injury and in vitro exposure of mitochondria to ONOO−
We postulated that ONOO− generated during IR injury (Yang et al., 2012) or exposure of mitochondria to exogenous ONOO− might affect the interaction between ANT and VDAC1 via tyrosine nitration. Thus, mitochondria isolated from IR or TC hearts were lysed, subjected to immunoprecipitation with anti-ANT antibody, and then immunoblotted with anti-VDAC1 and ANT antibodies. Indeed, we found that IR reduced the association of ANT with VDAC1 when compared to TC (Figs. 3A,B lanes TC and I35R20).
Figure 3:
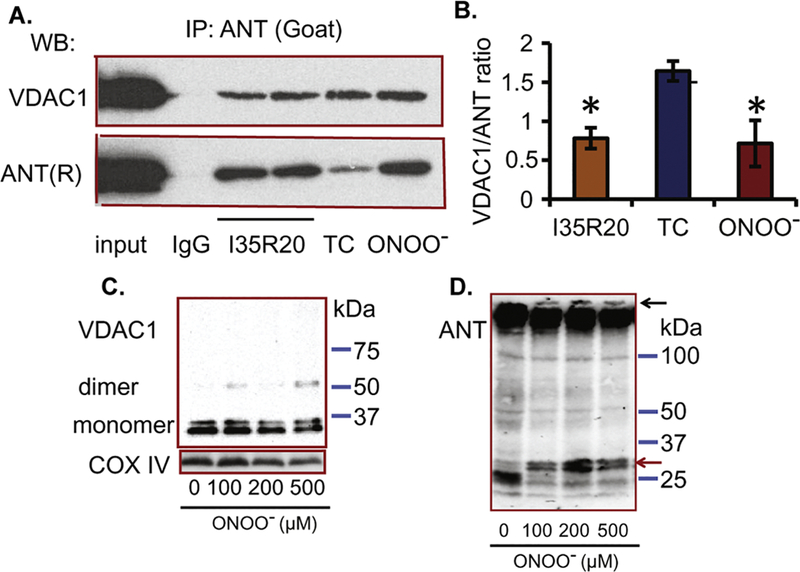
Dissociation of VDAC1 and ANT after I35R20 or ONOO− treatment. A: Immunoprecipitation of mitochondrial proteins with anti-ANT antibody (goat) followed by western blot with VDAC1 (upper panel) and ANT (rabbit (R), low panel) antibodies; ONOO−, mitochondria treated with 200 μM ONOO−; TC, time control; I35R20, 35 min ischemia and 20 min reperfusion; IgG, negative control. B: Summary of VDAC1/ANT ratio from 3 experiments. *p < 0.05 vs. TC. C: Evaluation of VDAC1 oligomerization based on EGS cross-linking and western blot using anti-VDAC1 and COX IV (loading control) antibodies in isolated mitochondria treated with 0 to 500 µM ONOO−. D: Evaluation of ANT disulfide- linked protein complexes in isolated mitochondria treated with 0 to 500 µM ONOO− using SDS-PAGE under non-reducing conditions followed by immunoblotting with anti-ANT antibody.
Next, we examined if in vitro application of ONOO− to mitochondria induced ANT dissociation from VDAC1. To do so, isolated mitochondria were treated with ONOO− and then subjected to immunoprecipitation with anti-ANT antibody followed by immunoblotting with anti-VDAC1 and ANT antibodies. We found that ONOO− treatment decreased the binding of VDAC1 to ANT (Fig. 3A,B, ONOO− lane); this suggests that mitochondria exposure to ONOO− induced the dissociation of VDAC1 from ANT, possibly because ONOO− nitrated the two proteins.
We further investigated whether VDAC1 formed oligomers and whether ANT formed protein complexes following exposure to ONOO−. Mitochondria were exposed to ONOO− (in vitro model) in the presence of the cross-linker EGS to stabilize the oligomeric state before western blot. Fig. 3C shows that exposure of mitochondria to ONOO− at concentrations of 100, 200 or 500 µM produced protein bands at molecular weights about two times that of VDAC1, thus suggesting oligomerization of VDAC1, which is consistent with the results obtained from recombinant VDAC1 exposed to ONOO− (Yang et al., 2012). In contrast, by adding the cross-linker EGS to mitochondria as was used for VDAC1, we found that the ANT western blot displayed smears and multiple bands in both control and ONOO− exposed mitochondria (data not shown), with no clear evidence of oligomerization. Imaizumi et al. (Imaizumi and Aniya, 2011) have previously reported that after exposure of rat liver mitoplasts to ONOO−, ANT formed a high molecular weight protein complex more than 150 kDa with mitochondrial membrane bound glutathione transferase (mtMGST1) and cyclophilin D (CypD). Using a similar approach, i.e. non-reducing gel electrophoresis followed by western blot, we found that compared to the control, treatment of isolated mitochondria with ONOO− displayed distinct bands with greater than 150 kDa proteins, suggesting that ANT formed a high molecular weight protein complex (Fig. 3D, dark arrow). The increase in high molecular weight ANT in the non-reducing condition (Fig. 3D, dark arrow) suggests that ONOO− induced a disulfide- linked complex of ANT with other proteins, which may include mtMGST1 and CypD as reported by Imaizumi et al. (Imaizumi and Aniya, 2011). Moreover, ONOO− treatment decreased the intensity of an approximately 30 kDa ANT band and increased the intensity of an approximately 35 kDa ANT band (Fig. 3D, red arrow). Whether this increased intensity of the 35 kDa band after ONOO− treatment is related to an ANT conformational change or to ANT forming a complex with other proteins needs to be determined.
HK II dissociates from mitochondria after ex vivo cardiac IR injury and after in vitro exposure of mitochondria to ONOO−
Our results above (Fig. 3) showed that mitochondria exposure to ONOO− facilitates dissociation of ANT and VDAC1, which may also facilitate dissociation of HK II from VDAC1. Thus, we next investigated whether exposure to ONOO− endogenous or exogenous impacted VDAC1 association with HK II, a key anti-apoptotic protein. We first examined whether IR injury induced dissociation of HK II from mitochondria and whether it could be reversed by MCGR. Guinea pig hearts were treated with or without MCGR before being subjected to IR injury. Immediately after the 20 min reperfusion, heart mitochondrial and cytosolic fractions were isolated, lysed, and subjected to western blot with HK II antibody. We found that compared to TC, IR injury resulted in a decrease in HK II associated with mitochondria and an increase in the cytosolic fraction, whereas MCGR partially maintained the HK II association with mitochondria and reduced the cytosolic fraction (Fig. 4A). As shown in Fig. 2D, hearts that showed reduced mitochondrial-HK II association displayed reduced cardiac functional recovery compared to TC (time control) hearts with high mitochondrial-HK II association. MCGR-treated hearts trended to have better cardiac function. These results suggest that dissociation of HK II from mitochondria during IR may be related to IR-induced superoxide (O2•-) and ONOO−production.
Figure 4:
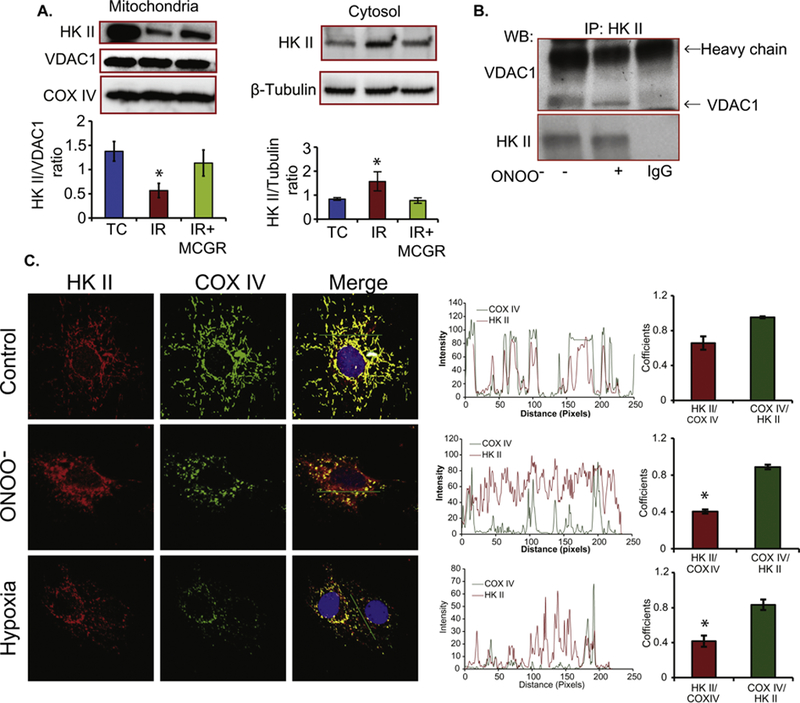
Dissociation of HK II from mitochondria. A: Western blot of mitochondrial and cytosolic proteins from TC, I35R20 (IR) and I35R20+MCGR (IR+MCGR) with anti-HK II antibody. The histogram panel below shows summary of HK II/VDAC1 ratio (for mitochondrial fraction) and HK II/β-tubulin ratio (for cytosolic fraction) from 3 experiments. *: p < 0.05 vs. TC. B: Immunoprecipitation of mitochondrial proteins with HK II antibody followed by western blot with VDAC1 and HK II antibod-ies. Mitochondria were treated with or without ONOO−. C: Immunostaining of H9c2 cells treated with 200 µM ONOO− or exposed to 4 h hypoxia with HK II and COX IV antibodies. COX IV was used as mitochondrial marker. The intensity profile panels, alongside the immunostaining shows the degree of overlapping of HK II with COX IV. The histogram represents the mean Mander’s split co-localization coefficients for HK II to COX IV and COX IV to HK II in control, ONOO− exposed, and hypoxia treated cells, in that order. The mean Mander’s split co-localization coefficients was calculated from n=10 cells. *: p < 0.05 HK II/COX IV in ONOO− or hypoxia group vs. control group.
To further confirm the role of ONOO− exposure to dissociate HK II from VDAC1, we incubated freshly isolated mitochondria with 200 µM ONOO−. After incubation, mitochondria were lysed and subjected to immunoprecipitation with anti-HK II. The anti-HK II immunoprecipitates were subjected to western blot with antibodies against VDAC1 and HK II. We found that compared to control/vehicle (NaOH), ONOO− treatment significantly reduced the interaction of HK II with VDAC1 (Fig. 4B); this supported our thesis that tyrosine nitration decreases the association of HK II with VDAC1.
We then tested if exposure to ONOO− promoted dissociation of HK II from mitochondria in H9c2 cells. We treated H9c2 cells either with ONOO−, or exposed the cells to hypoxia, and then determined the distribution of HK II in mitochondria and cytosol by immunofluorescence staining of the cells with HK II and COX IV, a mitochondrial inner membrane protein. We found that both ONOO − and hypoxia induced mitochondrial fragmentation (Fig. 4C COX IV panels) and increased HK II content in cytosol compared to the vehicle (Fig. 4C HK II and merger panels). Mander’s split co-localization coefficients of HK II/COX IV (Fig. 4C histogram panel) for control, ONOO−, and hypoxia treated H9c2 cells were 65% ±7, 41% ±7, 40% ±2 respectively, which suggests that ONOO− and hypoxia reduced co-localization of HK II to mitochondria.
Exposure to ONOO− induced mitochondrial cytochrome c release
Cyt c release from mitochondria following damage to mitochondrial membranes is a key factor in initiating apoptotic cell death. We measured cyt c release from isolated mitochondria to determine if exposure to ONOO− stimulated mitochondrial cytochrome c release and whether this was mediated via modification of ANT and VDAC1 and that oxidative stress contribute to the release. Freshly isolated mitochondria from guinea pig hearts were pretreated with or without the antioxidant cocktail MCGR, the VDAC inhibitor DIDS, or the ANT inhibitor BKA, followed by exposure to exogenous ONOO−. We observed that mitochondria exposed to ONOO− had decreased cyt c content. MCGR, DIDS, and BKA had no significant effect on mitochondrial cyt c content (Fig. 5A). Moreover, and in agreement with the above (Fig. 5A), ONOO−-induced cyt c release from mitochondria was significantly attenuated by DIDS, BKA and MCGR (Fig. 5B). These results suggest that VDAC1 and ANT are involved in the permeabilization of mitochondrial membranes during oxidative/nitrosative (ROS/RNS) stress, as shown by release of cyt c, a harbinger of cell death.
Figure 5:
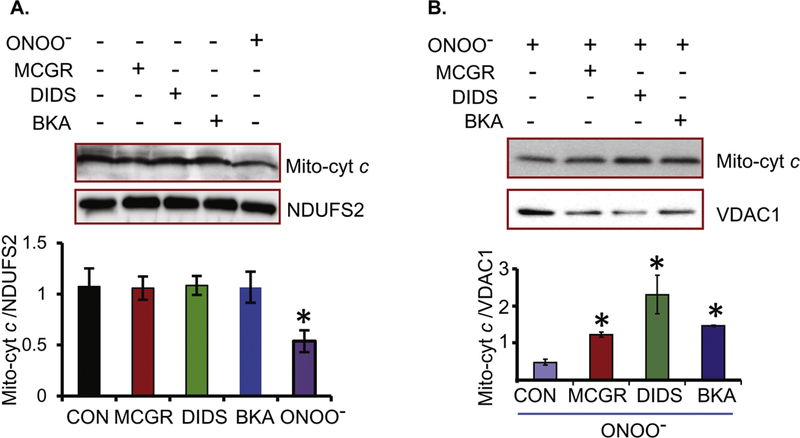
Cytochrome c release from isolated mitochondria. A: Western blot of mitochondrial proteins treated without (CON) or with ONOO−, MCGR, DIDS or BKA using anti-cyt c antibody; *: p < 0.05 vs. CON. NDUFS2, complex I subunit 2, was used as loading control. B: Western blot of mitochondrial protein treated with ONOO− (CON), or incubated with MCGR, DIDS or BKA before treatment with ONOO−; *: p < 0.05 vs. ONOO− alone. VDAC1 was used as loading control.
Exposure of mitochondria to ONOO− decreases respiration rate and delays recovery of membrane potential during state 3 respiration
We observed that exposure of mitochondria to ONOO− induced oligomerization of VDAC1, formation of high molecular weight protein complex of ANT, dissociation of ANT and VDAC1, and dissociation of HK II from VDAC1; each of these changes might be associated with impaired mitochondrial function. We measured mitochondrial respiration rates and calculated RCIs (rate of state 3 respiration/rate of state 4 respiration) as described in Materials and Methods. Freshly isolated mitochondria from guinea pig hearts were treated with various concentrations of ONOO− or 0.1 N NaOH (vehicle/control) for 30 min. In addition, mitochondria were isolated from hearts after I35R20 to further assess changes in bioenergetics. We found (Fig. 6A,B) that compared to the control (NaOH), exposure of mitochondria to 100 and 200 µM ONOO− decreased RCIs to values similar to RCIs of hearts subjected to I35R20, i.e. 10.2 ± 2.5 for control versus 1.6 ± 0.4 for 200 µM ONOO−, and 3.5 ± 0.3 for I35R20 (TC RCI: 11.1 ± 0.9 ). RCIs were not significantly different from the controls for the 50 and 75 µM ONOO− treatments (data not shown).
Figure 6:
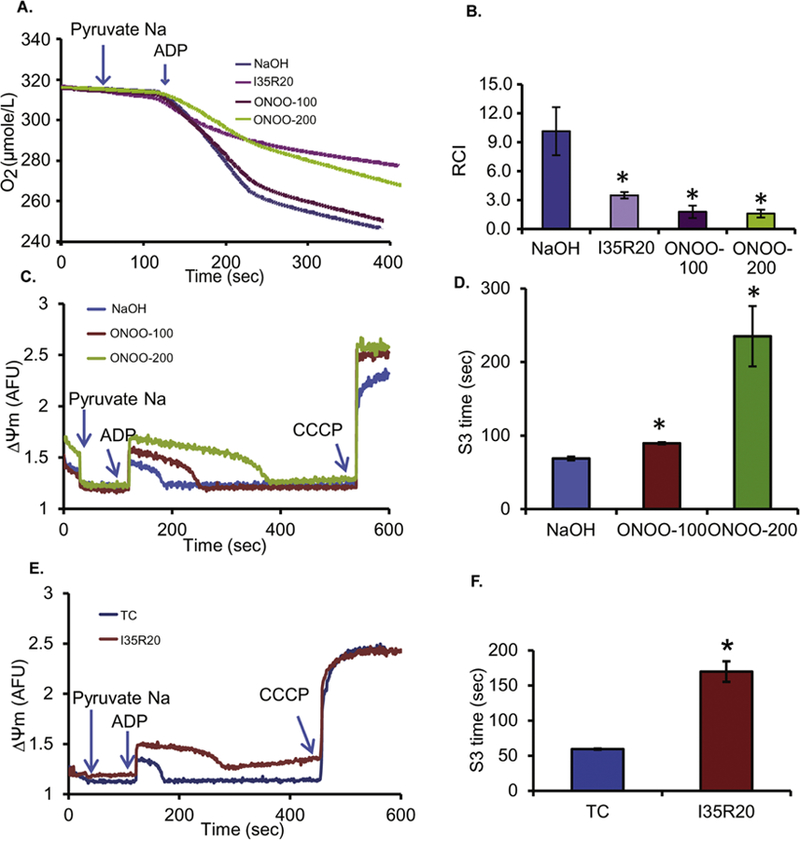
Lower respiration control index and delayed ΔΨm repolarization (state 3 respiration) after ONOO− treatment or 35 min of ischemia and 20 min of reperfusion (I35R20). A: Representative traces of respiration in mitochondria subjected to treatment with NaOH (control), 100 or 200 µM ONOO−, or I35R20. B: Summary of the RCIs for control, I35R20 and ONOO−-treated mitochondria; *:p < 0.05 vs. control. C: Time course of ΔΨm with NaOH treatment (control) or treatment with 100 or 200 µM ONOO−. D: Summary of effects of ONOO− compared to control on duration of recovery from ADP- induced state 3 ΔΨm (S3 time, sec) depolarization; *: p < 0.05 vs. control. E: Time course of ΔΨm from mitochondria isolated from ex vivo hearts subjected to either time control (TC) or I35R20. F: Summary of effects of TC or I35R20 on duration of recovery from ADP-induced state 3 ΔΨm (S3 time, sec) depolarization; *: p < 0.05 vs. control.
Another indicator of bioenergetics we utilized is recovery of ΔΨm after depolarization with ADP (state 3 respiration). The effects of 0.1 N NaOH (control) and 100 and 200 µM ONOO− on ΔΨm in Na-pyruvate energized mitochondria are shown in Fig. 6C and summarized in Fig. 6D. Our results show that ONOO− did not affect ΔΨm during resting state 2 respiration. In both NaOH (control) and ONOO − treated mitochondria, we observed that adding ADP initiated a sudden, partial, and reversible ΔΨ m depolarization. When compared to the control (NaOH), exposure to 100 and 200 µM ONOO− significantly increased the duration of state 3 ΔΨm depolarization, i.e. from 68 ± 2 sec (control) to 89 ± 1 sec and 235 ± 41 sec with NaOH, 100 and 200 µM ONOO−, respectively. This is consistent with the prolonged state 3 respiration, i.e. slower phosphorylation of the added ADP after mitochondria treatment with exogenous ONOO−. Similar to the ONOO− treated isolated mitochondria, mitochondria isolated after I35R20, when compared to their comparable time controls, showed markedly delayed repolarization of state 3, i.e. from 59 ± 2 sec (time control) to 170 ± 15 sec with I35R20; this is consistent with the protracted state 3 respiration (Figs. 6E,F).
Discussion
ONOO− levels increase during cardiac IR injury (Novalija et al., 2002; Yang et al., 2012; Zweier et al., 2001) and a reduction in injury is associated with a decrease in its release (Novalija et al., 2002; Yang et al., 2012; Zweier et al., 2001). ONOO− is a strong oxidant of mitochondrial proteins and lipids (Burwell and Brookes, 2008). Thus, mitochondria are both a site for ONOO− production and a target of ONOO−-induced dPTMs (Burwell and Brookes, 2008). Irreversible dPTMs of specific mitochondrial proteins, notably nitration of tyrosine residues by ONOO−, likely enhance mitochondrial dysfunction during IR injury. Our prior study (Yang et al., 2012) showed tyrosine nitration of VDAC1 induced after IR injury. In the present study, we found that ex vivo cardiac IR injury also led to an increase in tyrosine nitration of ANT (Fig. 2). During oxidative/nitrosative stress after IR injury, an increase in ONOO− production (Yang et al., 2012; Zweier et al., 2001) could, in part, be responsible for the dissociation of VDAC1 and ANT, and dissociation of HK II from VDAC1 (Fig. 3,4). In vitro treatment of mitochondria with ONOO− led to ANT tyrosine nitration (Fig. 1), dissociation of both ANT (Fig. 3A) and HK II (Fig. 4A, C) from VDAC1, induced cyt c release from mitochondria (Fig. 5), reduced RCI, and delayed ΔΨm repolarization after transient depolarization with ADP (state 3 respiration) (Fig. 6).The ONOO−-induced cyt c release from mitochondria was attenuated by MCGR, BKA and DIDS (Fig. 5B), indicating that oxidative stress was involved, and ONOO− might have altered ANT and VDAC1.
As in isolated mitochondria from guinea pig hearts, H9c2 cells treated with ONOO− or hypoxia also displayed dissociation of HK II from mitochondria (Fig. 4C). In the ex vivo isolated hearts, when compared to time controls (TC), 35 min ischemia and 20 min reperfusion significantly increased diastolic LVP and decreased coronary flow rate (Fig. 2D), which coincided with indicators of mitochondrial dysfunction, i.e., reduction of RCI (Fig. 6A,B), delayed ΔΨm repolarization (Fig. 6E,F), VDAC1 dissociation from ANT (Fig. 3), and HK II dissociation from VDAC1 (Fig. 4). Altogether, these data indicate that mitochondrial dysfunctions induced by ONOO− produced during IR injury, likely resulted in alteration of metabolite fluxes across the IMM and OMM, which led to compromise of cardiac function on reperfusion (Fig. 2D). Although we have focused on the important roles of VDAC1 and ANT nitration on their associations with each other and with other proteins; however, it must be noted that cardiac IR injury can lead to nitration of many other mitochondrial and non-mitochondrial proteins.
ANT tyrosine nitration and interaction with VDAC1
In the present study, we have demonstrated that cardiac IR injury and exposure of mitochondria to exogenous ONOO− led to tyrosine nitration of ANT (Figs. 1,2), which is consistent with decreased mitochondrial bioenergetics (Fig. 6). Moreover, our data also demonstrate that BKA, which locks ANT in the m-conformation, resulted in reduced cyt c release from mitochondria treated with ONOO− (Fig. 5); this suggests that ANT may be involved in permeabilization of mitochondria during exposure to ONOO−. Consistent with our current observation that cardiac IR injury can lead to nitration of ANT, ANT tyrosine nitration has also been reported (Martin et al., 2009) in spinal cord nerve cells of SOD1 G93A mutant mice, which exhibited mitochondrial dysfunction and enhanced free radical production (Yim et al., 1996). Cortical ablation in mice has been shown to cause neuronal apoptosis, increased ROS and ONOO− production, and nitration of ANT and CyPD (Martin et al., 2011).
The mechanism of ONOO− induced dissocation of ANT and VDAC1 is unknown. It is well recognized that tyrosine nitration renders the neutral tyrosine residue a charged residue; this might alter the tertiary structure of a protein (Batthyany et al., 2017; Szabo et al., 2007). This suggests that IR-induced oxidative/nitrosative stress or exogenous ONOO− treatment nitrates ANT and VDAC1 and may alter their structure and possibly impair their interaction. We found that in ONOO− treated mitochondria (Fig. 3C), and in recombinant VDAC1 exposed to ONOO− (Yang et al., 2012), the VDAC1 dimer band intensity increased, while the VDAC1 monomer band intensity decreased (Fig. 3C); this suggests ONOO− induced VDAC1 oligomerization. Moreover, by using non-reducing SDS-PAGE followed by western blot with ANT antibody as reported by Imaizumi et al. (Imaizumi and Aniya, 2011), we showed that compared to the control (NaOH), ONOO− induced an increase in a 35 kDa ANT band with a simultaneous decrease in the 30 kDa ANT band (Fig. 3D). However, the reason for the shift in the mass of ANT after ONOO− is not understood. In addition, our results also show that under the non-reducing condition, ANT forms high molecular weight protein complex (Fig. 3D). Interestingly, Imaizumi et al. reported that in ONOO− treated mitoplasts, ANT together with CypD and mtMGST1 formed a disulfide-linked high molecular weight protein complex (Imaizumi and Aniya, 2011). Although we did not determine if ANT forms a high molecular weight protein complex with CypD, our data are consistent with their results (Imaizumi and Aniya, 2011). Overall, our findings imply that ONOO− causes VDAC1 oligomerization and ANT to form high molecular weight complexes with other proteins that together may impair the VDAC1-ANT interaction. From our proteomics results, even though we did not have full coverage of the proteins, we confirmed the nitration of ANT and VDAC1. Whether nitration alone of either ANT or VDAC1 is sufficient to induce a change in ANT-VDAC1 interaction is yet to be determined. Other oxidative modifications induced by ONOO− may also play a role.
The bovine ADP/ATP carrier (ANT) structure was elucidated in the presence of carboxyatractylo-side (CATR) by X-ray crystallography. It showed that ANT consists of 6 transmembrane (TM) helices labeled H1 to H6, which are connected by loops M1, M2, and M3 on the matrix side and loops C1 and C2 on the mitochondrial intermembrane space (IMS) side (Eva Pebay-Peyroula1, 2003). The TM helices 1 to 4 form the cavity which binds to ADP and CATR. ANT Y81, which was nitrated during exposure of mitochondria to ONOO−, is in the H2 helix. The H2 helix was reported to be important in the process of substrate binding and translocation (Kedrov et al., 2010; Kihira et al., 2006). Further experiments are needed to determine if Y81 nitration alters H2 conformation and or the binding of the ADP/ATP to ANT. One report (Ferrer-Sueta et al., 2018) suggests that nitration of tyrosine residues may induce local conformational changes in specific regions of a given protein. The binding of VDAC1 and ANT in vitro depends on the ANT1 conformation (Allouche et al., 2012). Therefore, an alteration in the tertiary protein structure by tyrosine nitration may alter the interaction of ANT with VDAC1.
HK II and VDAC1 interactions
In addition to its cytoprotective effects in cell survival, HK II plays an important role in glucose metabolism by catalyzing the initial step in glycolysis (Camara et al., 2010). The association of HK II with VDAC influences mitochondrial metabolism and plays a role in mitochondria-induced apoptosis (Pastorino and Hoek, 2008). Others (Gurel et al., 2009; Pasdois et al., 2011, 2013), using isolated Langendorff-perfused rat hearts, have reported that 30 min global ischemia significantly reduced HK II activity and induced dissociation of HK II from mitochondria. Consistent with these results, our current observation also showed that 35 min global ischemia and 20 min reperfusion caused a significant dissociation of HK II from mitochondria (Fig. 4A), which correlated with impaired cardiac function (i.e. increased diastolic LVP and decreased coronary flow rate) when compared to time controls (TC) (Fig. 2D).
HK II and VDAC1 interaction is regulated by either changes in HK II or VADC1 protein structure and function (Pastorino and Hoek, 2008). For HK II, the presence of the N-terminal domain of the HK protein is necessary for HK II binding to mitochondria. During increases in metabolism, the concentration of the metabolite of HK II, glucose-6-phosphate, increases; this induces a conformational change in HK II, which displaces it from the mitochondrion (Pastorino and Hoek, 2008). For VDAC1, glutamate 72 is essential for HK II binding (Zaid et al., 2005); other charged residues within the putative first cytoplasmic loop of VDAC1 are also required for HKII binding (Pastorino and Hoek, 2008); VDAC1 oligomerization induced by IR facilitates the separation of HK II from mitochondria (Zhou et al., 2017). However, it is not known if HK II dissociation from mitochondria is related to VDAC1 tyrosine nitration induced by ONOO−. In our study, we furnish evidence that in vitro ONOO− exposure causes HK II to dissociate from mitochondria based on our results from H9c2 cells and isolated mitochondria (Fig. 4). In addition, our results show that exposure to ONOO− enhanced the oligomerization of VDAC1 (Fig. 3C). Our prior study (Yang et al., 2012) showed there was tyrosine nitration of Y65 or Y67; these residues are located in the first cytoplasmic loop of VDAC1 and close to the glutamate 72 residue and other charged residues. Thus, tyrosine nitration of VDAC1 may contribute to a change in VDAC1/HK II interaction.
ONOO− exposure and mitochondrial membrane permeabilization
A previous study (Pastorino et al., 2002) that dissociation of HK II from VDAC induced by ischemia was associated with cyt c release. Several mechanisms have been proposed to describe how cyt c is released from mitochondria. It was reported (Vyssokikh and Brdiczka, 2003) that cyt c release from mitochondria requires a two-step process: 1) Bax induces a small fraction of cyt c release from the inter-membrane space; 2) a massive release of cyt c results from mPTP opening, followed by mitochondrial swelling and membrane disruption (Halestrap et al., 2002). It has been shown that HK II interaction with VDAC prevents the binding of the pro-apoptotic protein Bax to VDAC. Therefore, the dissociation of HK II from OMM increases Bax binding to VDAC, promotes the release of cyt c, and the concomitant activation of the cytosolic apoptotic cascade (Pastorino et al., 2002). Native Bax is usually present in the cytosol and when activated, it translocates to mitochondria (Lalier et al., 2007). In our study, ONOO− treatment stimulated cyt c release from isolated mitochondria, which may be independent of Bax translocation to mitochondria. This suggests that other processes aside from Bax, such as ONOO− mediated mitochondrial membrane permeablization might be involved in cyt c release from mitochondria.
Based on our experiments, ONOO−-mediated mitochondrial cyt c release may be attributed to permeabilization of the mitochondrial membranes. Specifically, release of cyt c from mitochondria induced by ONOO− treatment was attenuated by pretreatment with DIDS, an inhibitor of VDAC, and by BKA, an inhibitor of ANT (Fig. 5). Furthermore, the ex vivo and in vitro studies showed that ONOO− induced tyrosine nitration of ANT and VDAC1 (Figs. 1, 2), which led to VDAC1 oligomerization (Fig. 3C) and formation of a yet undetermined high molecular weight complex that includes ANT (Fig. 3D), possibly linked by disulfide bonds, and to dissociation of VDAC1 from ANT (Fig. 3A). Involvement of permeabilization of mitochondrial membranes is further confirmed from our observation that ONOO− induced cyt c release from isolated mitochondria was attenuated by MCGR, a cocktail of antioxidants. This suggests that ROS and/or ONOO− induced permeabilization of the mitochondrial membrane; this could be followed by mitochondrial swelling and functional disruption. Consistent with our findings, it was reported (Madesh and Hajnoczky, 2001) in permeabilized HepG2 cells that O2•− elicited a rapid and massive release of cyt c that was abrogated by inhibiting VDAC with DIDS. Moreover, in VDAC reconstituted liposomes, cyt c released after exposure to O2•− was reduced by DIDS (Madesh and Hajnoczky, 2001); this suggests that the release of cyt c mediated by O2•− was attributable in part to VDAC-dependent selective permeabilization of the OMM, which as we noted above, is consistent with our results.
Effect of ONOO− on mitochondrial bioenergetics
We found that exposure of isolated healthy cardiac mitochondria to low [ONOO−] (50 and 75 µM) did not significantly change RCI when compared to controls (NaOH), whereas exposure to high [ONOO−] (100 and 200 µM) significantly decreased RCI to levels similar to those obtained in mitochondria isolated from hearts after IR injury (Figs. 6A,B). Moreover, the 100 and 200 µM ONOO− treatments delayed the time to ΔΨm repolarization after adding ADP (state 3 respiration) compared to the controls (NaOH) (Figs. 6C,D). A similar delay in ΔΨm repolarization was observed in mitochondria isolated from hearts after I35R20 (Figs. 6E,F). These data suggest that ONOO−, from either endogenous or exogenous sources, directly impairs mitochondrial respiration, and that this may be a contributing factor in cardiac dysfunction. It has been reported (Brown and Borutaite, 2007), that ONOO− weakens function of mitochondrial proteins, including complexes I and III, in part, by dPTM such as tyrosine nitration and S-nitrosation that could also impact mitochondrial function. In our present study using isolated mitochondria, respiration and ΔΨm were measured after mitochondria were exposed to ONOO− for 30 min, which induced tyrosine nitration of mitochondrial proteins (Figs. 1C), notably of ANT (Figs. 1D,E). The results implied that ONOO− (100 and 200 µM) treatment caused, at least in part, a decrease in RCI and a delay of ΔΨm repolarization time that were likely due to the tyrosine nitration of mitochondrial proteins, including ANT and VDAC1, again, in agreement with a previous report (Brown and Borutaite, 2007).
Our results demonstrate that after in vitro exposure to ONOO−, the slower rate of oxidative phosphorylation was associated with tyrosine nitration of VDAC1 and ANT and the concomitant dissociation of VDAC1 and ANT. As noted earlier, this may lead to altered flux of substrates and metabolites across the OMM and IMM and impact mitochondrial function. Under physiological conditions, the transfer of energy metabolites across mitochondria depends on the integrity of the transporters (VDAC) and exchangers (ANT) and their interaction with creatine kinase (Camara et al., 2017). The present study (Figs. 3,4) and other studies (Vyssokikh and Brdiczka, 2003) demonstrate that dPTMs, especially tyrosine nitration, can induce a change in VDAC structure, which could alter its interaction with the IMM protein, ANT, and the cytosolic protein, HK II. Thus, these modifications could hinder the normal transport or exchange of metabolites, including the transport of cytosolic ADP/ATP and Pi, and the exchange of matrix ATP for cytosolic ADP, which leads to impaired oxidative phosphorylation. This notion is supported by the protracted state 3 respiration (Figs. 6A,B) and delayed repolarization of ΔΨm (Figs. 6C-F) and impaired cardiac function after I35R20 (Table 1).
Summary and conclusion
In summary, we detected tyrosine nitration of ANT and VDAC1 following exposure of isolated mitochondria or H9c2 cells to ONOO−. Similar results were also obtained following IR injury in isolated hearts. Furthermore, exposure to ONOO− resulted in the dissociation of HK II from VDAC1, and a reduction in the interaction of VDAC1 with ANT. Isolated mitochondria exposed to ONOO − exhibited compromised bioenergetics. Based on these findings, ONOO− leads to mitochondrial dysfunction and also induces cyt c release. These events may be associated with the impact of ONOO− on VDAC1 and ANT, although further studies will be required to confirm this notion. Additionally, nitration of VDAC1 and ANT may be contributors to mitochondrial dysfunction, though other potential modifications induced by ONOO− exposure should be considered. MCGR was cardioprotective against IR injury in an isolated heart setting and also preserved HK II binding to mitochondria. This suggests that MCGR-preserved HK II association to mitochondria contributes to this cardioprotection. Therefore, preventing ONOO− generation and preserving the integrity of these mitochondrial proteins and their interactions with each other and with cytosolic proteins, e.g. HK II, could represent novel strategies in mitigating cardiac IR injury.
Highlights:
Peroxynitrite (ONOO−) induces tyrosine nitration of ANT and VDAC1, dissociation of ANT with VDAC1, and of HK II with VDAC1; this enhances cytochrome c release from mitochondria, impairs mitochondrial bioenergetics, and compromises cardiac function after IR.
Acknowledgements
This work was supported in part by the National Institutes of Health (R01 HL-131673–01A1) and the Veterans Administration (BX-002539–01) United States.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
disclosures
The authors have nothing to disclose concerning any conflict of interest.
Reference
- Agarwal B, Dash RK, Stowe DF, Bosnjak ZJ, Camara AK, 2014a. Isoflurane modulates cardiac mitochondrial bioenergetics by selectively attenuating respiratory complexes. Biochimica et biophysica acta 1837, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal B, Stowe DF, Dash RK, Bosnjak ZJ, Camara AK, 2014b. Mitochondrial targets for volatile anesthetics against cardiac ischemia-reperfusion injury. Front Physiol 5, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldakkak M, Stowe DF, Dash RK, Camara AK, 2013. Mitochondrial handling of excess Ca2+ is substrate-dependent with implications for reactive oxygen species generation. Free Radic Biol Med 56, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allouche M, Pertuiset C, Robert JL, Martel C, Veneziano R, Henry C, dein OS, Saint N, Brenner C, Chopineau J, 2012. ANT-VDAC1 interaction is direct and depends on ANT isoform conformation in vitro. Biochemical and biophysical research communications 429, 12–17. [DOI] [PubMed] [Google Scholar]
- Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V, 2004. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. The Biochemical journal 377, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batthyany C, Bartesaghi S, Mastrogiovanni M, Lima A, Demicheli V, Radi R, 2017. Tyrosine-Nitrated Proteins: Proteomic and Bioanalytical Aspects. Antioxidants & redox signaling 26, 313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomeyer CA, Bazil JN, Stowe DF, Dash RK, Camara AK, 2016. Mg2+ differentially regulates two modes of mitochondrial Ca2+ uptake in isolated cardiac mitochondria: implications for mitochondrial Ca2+ sequestration. Journal of bioenergetics and biomembranes 48, 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomeyer CA, Bazil JN, Stowe DF, Pradhan RK, Dash RK, Camara AK, 2013. Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na+-induced Ca2+ release. Journal of bioenergetics and biomembranes 45, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ, 2005. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. The Biochemical journal 392, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Borutaite V, 2007. Nitric oxide and mitochondrial respiration in the heart. Cardiovascular research 75, 283–290. [DOI] [PubMed] [Google Scholar]
- Burwell LS, Brookes PS, 2008. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxidants & redox signaling 10, 579–599. [DOI] [PubMed] [Google Scholar]
- Calmettes G, John SA, Weiss JN, Ribalet B, 2013. Hexokinase-mitochondrial interactions regulate glucose metabolism differentially in adult and neonatal cardiac myocytes. The Journal of General Physiology 142, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara AK, Lesnefsky EJ, Stowe DF, 2010. Potential therapeutic benefits of strategies directed to mitochondria. Antioxidants & redox signaling 13, 279–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara AKS, Zhou Y, Wen PC, Tajkhorshid E, Kwok WM, 2017. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front Physiol 8, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombini M, 2012. VDAC structure, selectivity, and dynamics. Biochimica et biophysica acta 1818, 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Steenbergen C, Murphy E, 2012. Does the voltage dependent anion channel modulate cardiac ischemia-reperfusion injury? Biochimica et biophysica acta 1818, 1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Sueta G, Campolo N, Trujillo M, Bartesaghi S, Carballal S, Romero N, Alvarez B, Radi R, 2018. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem Rev 118, 1338–1408. [DOI] [PubMed] [Google Scholar]
- Geula S, Ben-Hail D, Shoshan-Barmatz V, 2012. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. The Biochemical Journal 444, 475–485. [DOI] [PubMed] [Google Scholar]
- Graham JM, 2001. Purification of a crude mitochondrial fraction by density-gradient centrifugation. Current Protocols in Cell Biology Chapter 3, Unit 3 4. [DOI] [PubMed]
- Gurel E, Smeele KM, Eerbeek O, Koeman A, Demirci C, Hollmann MW, Zuurbier CJ, 2009. Ischemic preconditioning affects hexokinase activity and HKII in different subcellular compartments throughout cardiac ischemia-reperfusion. J Appl Physiol (1985) 106, 1909–1916. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, McStay GP, Clarke SJ, 2002. The permeability transition pore complex: another view. Biochimie 84, 153–166. [DOI] [PubMed] [Google Scholar]
- Heinen A, Camara AK, Aldakkak M, Rhodes SS, Riess ML, Stowe DF, 2007. Mitochondrial Ca 2+-induced K+ influx increases respiration and enhances ROS production while maintaining membrane potential. Am J Physiol Cell Physiol 292, C148–156. [DOI] [PubMed] [Google Scholar]
- Imaizumi N, Aniya Y, 2011. The role of a membrane-bound glutathione transferase in the peroxynitrite-induced mitochondrial permeability transition pore: formation of a disulfide-linked protein complex. Archives of Biochemistry and Biophysics 516, 160–172. [DOI] [PubMed] [Google Scholar]
- Kedrov A, Hellawell AM, Klosin A, Broadhurst RB, Kunji ER, Muller DJ, 2010. Probing the interactions of carboxy-atractyloside and atractyloside with the yeast mitochondrial ADP/ATP carrier. Structure 18, 39–46. [DOI] [PubMed] [Google Scholar]
- Kihira Y, Hashimoto M, Shinohara Y, Majima E, Terada H, 2006. Roles of adjoining Asp and Cys residues of first matrix-facing loop in transport activity of yeast and bovine mitochondrial ADP/ATP carriers. J Biochem 139, 575–582. [DOI] [PubMed] [Google Scholar]
- Lalier L, Cartron PF, Juin P, Nedelkina S, Manon S, Bechinger B, Vallette FM, 2007. Bax activation and mitochondrial insertion during apoptosis. Apoptosis : an international journal on programmed cell death 12, 887–896. [DOI] [PubMed] [Google Scholar]
- Madesh M, Hajnoczky G, 2001. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. The Journal of Cell Biology 155, 1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N, 2004. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Molecular Cell 16, 819–830. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Adams NA, Pan Y, Price A, Wong M, 2011. The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q, 2009. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Experimental Neurology 218, 333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure NM, 2016. News about VDAC1 in Hypoxia. Frontiers in Oncology 6, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCommis KS, Baines CP, 2012. The role of VDAC in cell death: friend or foe? Biochimica et Biophysica Acta 1818, 1444–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina A, Reina S, Guarino F, De Pinto V, 2012. VDAC isoforms in mammals. Biochimica et Biophysica Acta 1818, 1466–1476. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C, 2008. Mechanisms underlying acute protection from cardiac ischemia -reperfusion injury. Physiological Reviews 88, 581–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA, 2003. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. The Journal of Biological Chemistry 278, 37223–37230. [DOI] [PubMed] [Google Scholar]
- Novalija E, Varadarajan SG, Camara AK, An J, Chen Q, Riess ML, Hogg N, Stowe DF, 2002. Anesthetic preconditioning: triggering role of reactive oxygen and nitrogen species in isolated hearts. American journal of physiology. Heart and Circulatory Physiology 283, H44–52. [DOI] [PubMed] [Google Scholar]
- O’Rourke B, 2007. Mitochondrial ion channels. Annual Review of Physiology 69, 19–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri F, Pierri CL, 2010. Mitochondrial metabolite transport. Essays in Biochemistry 47, 37–52. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Parker JE, Griffiths EJ, Halestrap AP, 2011. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. The Biochemical Journal 436, 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdois P, Parker JE, Griffiths EJ, Halestrap AP, 2013. Hexokinase II and reperfusion injury: TAT-HK2 peptide impairs vascular function in Langendorff-perfused rat hearts. Circulation Research 112, e3–7. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Parker JE, Halestrap AP, 2012. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J Am Heart Assoc 2, e005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB, 2008. Regulation of hexokinase binding to VDAC. Journal of Bioenergetics and Biomembranes 40, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB, Shulga N, 2005. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Research 65, 10545–10554. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Shulga N, Hoek JB, 2002. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. The Journal of Biological Chemistry 277, 7610–7618. [DOI] [PubMed] [Google Scholar]
- Perevoshchikova IV, Zorov SD, Kotova EA, Zorov DB, Antonenko YN, 2010. Hexokinase inhibits flux of fluorescently labeled ATP through mitochondrial outer membrane porin. FEBS Letters 584, 2397–2402. [DOI] [PubMed] [Google Scholar]
- Pesse B, Levrand S, Feihl F, Waeber B, Gavillet B, Pacher P, Liaudet L, 2005. Peroxynitrite activates ERK via Raf-1 and MEK, independently from EGF receptor and p21Ras in H9c2 cardiomyocytes. Journal of Molecular and Cellular Cardiology 38, 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan A, Sheiko T, Graham BH, Craigen WJ, 2012. Voltage-dependant anion channels: novel insights into isoform function through genetic models. Biochimica et Biophysica Acta 1818, 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Berry RW, Binder LI, 2005. Site-specific nitration and oxidative dityrosine bridging of the tau protein by peroxynitrite: implications for Alzheimer’s disease. Biochemistry 44, 1690–1700. [DOI] [PubMed] [Google Scholar]
- Rowan WH 3rd, Sun P, Liu L, 2002. Nitration of annexin II tetramer. Biochemistry 41, 1409–1420. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Ben-Hail D, 2012. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12, 24–34. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Keinan N, Zaid H, 2008. Uncovering the role of VDAC in the regulation of cell life and death. Journal of Bioenergetics and Biomembranes 40, 183–191. [DOI] [PubMed] [Google Scholar]
- Stowe DF, Gadicherla AK, Zhou Y, Aldakkak M, Cheng Q, Kwok WM, Jiang MT, Heisner JS, Yang M, Camara AK, 2013. Protection against cardiac injury by small Ca 2+-sensitive K+ channels identified in guinea pig cardiac inner mitochondrial membrane. Biochimica et Biophysica Acta 1828, 427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowe DF, Yang M, Heisner JS, Camara AKS, 2017. Endogenous and Agonist-induced Opening of Mitochondrial Big Versus Small Ca2+-sensitive K+ Channels on Cardiac Cell and Mitochondrial Protection. J Cardiovasc Pharmacol 70, 314–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H, 2008. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol 28, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Ischiropoulos H, Radi R, 2007. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6, 662–680. [DOI] [PubMed] [Google Scholar]
- Vyssokikh MY, Brdiczka D, 2003. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochimica Polonica 50, 389–404. [PubMed] [Google Scholar]
- Vyssokikh MY, Katz A, Rueck A, Wuensch C, Dorner A, Zorov DB, Brdiczka D, 2001. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. The Biochemical Journal 358, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Camara AK, Wakim BT, Zhou Y, Gadicherla AK, Kwok WM, Stowe DF, 2012. Tyrosine nitration of voltage-dependent anion channels in cardiac ischemia-reperfusion: reduction by peroxynitrite scavenging. Biochimica et Biophysica Acta 1817, 2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Camara AKS, Aldakkak M, Kwok WM, Stowe DF, 2017. Identity and function of a cardiac mitochondrial small conductance Ca2+-activated K+ channel splice variant. Biochimica et Biophysica Acta 1858, 442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Stowe DF, Udoh KB, Heisner JS, Camara AK, 2014. Reversible blockade of complex I or inhibition of PKC-beta reduces activation and mitochondria translocation of p66Shc to preserve cardiac function after ischemia. PloS one 9, e113534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER, 1996. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci U S A 93, 5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V, 2005. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ 12, 751–760. [DOI] [PubMed] [Google Scholar]
- Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y, 2017. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ, 2009. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovascular research 83, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier JL, Fertmann J, Wei G, 2001. Nitric oxide and peroxynitrite in postischemic myocardium. Antioxidants & Redox Signaling 3, 11–22. [DOI] [PubMed] [Google Scholar]
- Camara AK, Aldakkak M, Heisner JS, Rhodes SS, Riess ML, An J, Heinen A, Stowe DF ROS scavenging before 27 degrees C ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state Am J Physiol Cell Physiol, 292 (2007), pp. C2021–2031. [DOI] [PubMed] [Google Scholar]