

Abstract
Development of selective kinase inhibitors that target the ATP binding site continues to be a challenge largely due to similar binding pockets. Palbociclib is a cyclin-dependent kinase inhibitor that targets the ATP binding site of CDK4 and CDK6 with similar potency. The enzymatic function associated with the kinase can be effectively probed using kinase inhibitors however the kinase independent functions cannot. Herein, we report a palbociclib based PROTAC that selectively degrades CDK6 while sparing the homolog CDK4. We used competition studies to characterize the binding and mechanism of CDK6 degradation.
Graphical Abstract
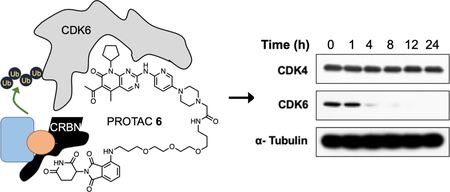
Cyclin dependent kinases are a family of serine-threonine kinases that exert a variety of functions, which include regulation of cell cycle (CDK1, 2, 4, 6 and 11) and gene expression (CDK7, and 9)1–9. CDK4/6 is a therapeutic target for cancer and palbociclib is the first CDK4/6 selective inhibitor that was approved by the FDA in 2015 for cancer therapy5. CDK4/6 is catalytically inactive, and upon binding to cyclin D, CDK4/6 is activated resulting in phosphorylation of RB family of proteins. This leads to the release of RB mediated inhibition of E2F transcription factors. E2F activates many cell cycle genes including cyclin E that binds to and activates CDK2, which in turn hyperphosphorylates RB proteins. This feedback loop ensures the irreversible progression of the cell cycle from G1 to S phase10. Knock out studies have shown that cyclin D1 and CDK4/6 may be dispensable in normal cells; however, they are critical for tumor growth11.
Palbociclib inhibits the kinase activity of CDK4-cyclin D1 and CDK6-cyclin D3 complexes. Palbociclib is an ATP competitive kinase inhibitor that binds to the hinge region of CDK4/6 and inhibits phosphorylation of downstream substrates. In addition to kinase-dependent cell cycle regulation function of CDK6, a recent report suggests CDK6 plays a role in transcriptional regulation through a kinase-independent mechanism12. The available CDK4/6 inhibitors can be used to probe the role of kinase dependent functions of CDK4/6. However, the lack of selectivity and their inability to target the non-kinase domain makes them unsuitable to probe the above-mentioned kinase independent function of CDK6.
To address this, we employed the emerging proteolysis targeting chimera (PROTAC) based strategy to develop CDK6 selective degrader that will target both kinase-dependent and kinase-independent CDK6 function. PROTAC is a heterobifunctional molecule wherein one fragment interacts with the protein of interest and the other binds to a component of an E3-ubiquitin ligase and the two are connected via a linker. PROTAC facilitates the formation of a ternary complex by binding to both the target protein and either a component of E3 ubiquitin ligase or the E2 ligase. The resulting ternary complex facilitates poly-ubiquitination of the target protein, which is subsequently degraded by the proteasome8, 13–20. Recent studies with BET degraders demonstrated improved inhibition of cancer cell growth and the induction of apoptosis when compared to the corresponding BET inhibitors15, 16, 18, 21, 22.
Although the kinase fold of CDK4 and CDK6 are identical, the distribution of surface exposed lysine residues, which is required for ubiquitination by an E3 ligase in CDK4 and CDK6 are different (Supplementary Figure S1). We hypothesized that a PROTAC strategy might yield a selective CDK6 degrader. X-ray crystal structure (pdb: 5L2I) of palbociclib (1) bound to CDK6 showed that nitrogen atoms in the amino-pyrimidine core of palbocilcib interacts with the hinge region residues of CDK6 and the piperazine ring is solvent exposed23. Structure-activity relationship (SAR) studies demonstrated that modifications on the piperazine ring did not result in loss of CDK4/6 binding affinity24. Thus, we speculated that the nitrogen atom of the piperazine ring is ideally positioned to conjugate the linker to generate bifunctional PROTAC molecules (Figure 1).
Figure 1:

Binding of Palbociclib to CDK6 (PDB code 5L2I). The terminal piperazine ring is solvent exposed and was used to design to heterobifunctional PROTACs.
We synthesized a set of five PROTAC molecules by conjugating palbociclib (1) to phthalimide based cereblon E3 ligase ligands (pomalidomide) via flexible linkers with varying lengths and composition (Figure 2).
Figure 2:

Design of palbociclib-based PROTACs.
The synthetic route to access PROTACs (2 – 6) is summarized in Scheme 1. Briefly, a reaction of palbociclib (1) with t-butyl 2-bromoacetate in N-Methyl-2-pyrrolidone (NMP) solvent resulted in intermediate 7. Condensation of 4-fluoroisobenzofuran-1,3-dione (8)16 with commercially available 3-aminopiperidine-2,6-dione hydrochloride afforded previously reported intermediate 925. Displacement of the fluoro group with different alkyl amines yielded intermediate 1025, 1126 and 1915 which were treated with 7 to generate PROTACs 2, 3 and 6 in two steps respectively. Condensation of commercially available 4-nitroisobenzofuran-1,3-dione (12) with 3-aminopiperidine-2,6-dione hydrochloride resulted in the formation of 2-(2,6-dioxopiperidin-3-yl)-4-nitroisoindoline-1,3-dione which was subsequently reduced under H2 over 5% Pd/C to yield 13. Treatment of 6-bromohexanoic acid with thionyl chloride resulted in 6-bromohexanoyl chloride which then refluxed with 13 followed by a Finkelstein reaction to yield 14. Alkylation of palbociclib (1) with 14 resulted in PROTAC 4. A reaction of 2-(2-(2-chloroethoxy)ethoxy)ethan-1-ol (15) with p-toluenesulfonyl chloride yielded 2-(2-(2-chloroethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (16). Alkylation of the hydroxyl group of 17 with 2-(2-(2-chloroethoxy)ethoxy)ethyl 4-methylbenzene sulfonate (16) followed by a Finkelstein reaction yielded 18. Alkylation of 1 with intermediate 18 yielded PROTAC 5.
Scheme 1:

(i) t-butyl 2-bromoacetate, NMP, 70 °C, 3h; (ii) 3-aminopiperidine-2,6-dione hydrochloride, KOAc, Acetic acid, reflux, 24h; (iii) t-butyl (4-aminobutyl)carbamate, DMA, DIEA, 80 °C, 2h; (iv) 7, TFA, DCM, 0 °C, 3h; (v) HATU, HOBT, DIEA, DMF, 2h; (vi) t-butyl (8-aminooctyl)carbamate, DMA, DIEA, 80 °C, 2h; (vii) 3-aminopiperidine-2,6-dione hydrochloride, KOAc, Acetic acid, reflux, 24h; (viii) H2, 5% Pd/C, DMF, 16h; (ix) (a) 6-bromohexanoic acid, thionyl chloride, 0 °C, 2h, (b) 13, NEt3, THF, 80 °C, 16h; (x) NaI, acetone, reflux, 24h; (xi) 1, NEt3, NMP, 70 °C, 12h; (xii) p-toluenesulfonyl chloride, KOH, DCM; (xiii) 2-(2-(2-chloroethoxy)ethoxy)ethan-1-ol, tosyl chloride, K2CO3,DMF, 70 °C, 3h; (xiv) NaI, acetone, reflux, 24h; (xv) t-butyl (3-(2-(2-(3-aminopropoxy)ethoxy) ethoxy)propyl)carbamate, DMA, DIEA, 80 °C, 2h.
To identify a CDK6 selective degrader we subjected MiaPaCa2 cells to 500 nM of PROTACs 2 – 6 and assessed degradation of CDK family of kinases by Western blot analysis (Figure 3). Interestingly, shorter linker length (6–11 atoms) PROTAC 2 – 5 exhibited partial degradation of CDK6 whereas PROTAC 6 with longer linker (17 atoms) selectively degraded CDK6 (Figure 3A) while sparing other members of the CDK (2, 4, 5, 7 and 9) family and Rb (a non-CDK). Degrader 2, 4, 5 induced partial degradation of CDK4 but robust degradation of CDK6. Palbociclib inhibits both CDK4 and CDK6, however, Longer linker length PROTAC 6 selectively degrade CDK6. This could be because of the significant modifications to palbociclib in PROTAC 6 that resulted in the loss of CDK4 binding. To assess if the lack of CDK4 degradation is due to lack of binding of 6 to CDK4 we subjected PROTAC 6 to in vitro cell-free kinase assay (Figure 3B)
Figure 3:

Effects of palbociclib-based degraders in MiaPaCa2 cells. (A) Western blot analyses of a panel of kinases with lysates generated from MiaPaCa2 cells treated with 0.5 μM of degrader analogs for 4h (CDK4, 6 and RB) 24h (CDK2, 5, 7 and 9 for 24h). (B) Cell-free assay showing inhibition of CDK4 and CDK6 with PROTAC 6. (C) Dose-response studies with different degraders in MiaPaCa2 cells when treated for 24h.
PROTAC 6 inhibited both CDK4 and CDK6 in vitro with similar potency. This suggests that a stable ternary complex could not be formed with CDK4 thus preventing its degradation. However, the other possibility that cannot be rule out is the rapid deubiquitination of CDK4 as a contributing factor for the absence of CDK4 degradation. Next, we performed a dose-dependent study with PROTACs 2 - 6 to estimate relative potency. Consistent with the single dose screens, we observed potent and selective degradation of CDK6 only with PROTAC 6. Together, these studies identified PROTAC 6 as a potent and selective degrader of CDK6.
We next evaluated PROTAC 6 in a dose-response study at 4 and 24 hours in HPNE and MiaPaCa2 cell lines (Figure 4). At both time points, we observed nearly quantitative degradation of CDK6 in both cell lines at 100 nM while CDK4 levels remained unchanged. At the 4h time point in both cell lines we observed reduced degradation of CDK6 at 5 and 10μM. This is because at higher concentrations of the PROTAC the corresponding binary complexes CDK6-PROTAC 6 and CRBN-PROTAC 6 are formed, which prevents the formation of the ternary complex that is required for degradation. This observation is consistent with previously reported PROTAC studies and is commonly known as the “hook effect” (Figure 4A and4B). Next, we subjected PROTAC 6 (500 nM) to a time-dependent study and probed for CDK6 in HPNE and MaiPaCa2 cells. We observed a time-dependent decrease in CDK6 levels as early as 4 hours and complete degradation by 8h and remained so until the 24h time point.
Figure 4:

Effect of PROTAC 6 in HPNE and MiaPaCa2 cell lines. Dose-response studies with different concentrations of PROTAC 6 at 4 and 24h in (A) HPNE and (B) MiaPaCa2 cells. Time-course studies with degrader 6 at different intervals in (C) HPNE and (D) MiaPaCa2 cells
To evaluate the mechanism of action of PROTAC 6, we conducted a series of competition experiments. First, we conducted a competition experiment with PROTAC 6 and the CDK4/6 ligand, palbociclib. Briefly, MiaPaCa2 cells were treated with 10 μM of palbociclib, PROTAC 6 (1μM) individually or in combination and incubated for 24 hours. The lysates were then probed for CDK4 and CDK6 using Western blot analysis. We observed a reduction of CDK6 levels in PROTAC 6 treated sample and no such effect was observed in both palbociclib or palbociclib + PROTAC 6 treated lysates. Since palbociclib saturated CDK6 binding sites in the combination treatment, PROTAC 6 failed to degrade CDK6 (Figure 5A). Since PROTAC strategy requires formation of ternary complex, next, we probed competition with CRBN ligand. MiaPaCa2 cells were treated with increasing concentrations of pomalidomide (0.1 – 10μM) alone and in combination with PROTAC 6 (1μM) for 24 hours, and the lysates were subjected to Western blot analyses (Figure 5B). Pomalidomide by itself did not affect CDK6 and at 5 and 10μM pomalidomide was able to block PROTAC 6 mediated degradation of CDK6. These competitions studies demonstrate the need for simultaneous engagement of CDK6 and CRBN by PROTAC 6 for productive degradation.
Figure 5:

Mechanism of action of degrader 6. (A) Western blot analyses showing inhibition of CDK6 degradation upon simultaneous treatment of palbociclib (10 μM) and PROTAC 6 for 24h. (B) Western blot analysis showing inhibition of CDK6 degradation upon simultaneous treatment of increasing concentrations of pomalidomide and PROTAC 6 for 24h. (C) Western blot analysis showing inhibition of CDK6 degradation when pre-incubation with 10 μM of proteasome inhibitor MG132 for 1h followed by treatment of PROTAC 6 for 8h.
It has been suggested that PROTAC based strategy involves ubiquitination of protein of interest followed by its proteasomal degradation. To probe this, we subjected MiaPaCa2 cells to proteasome inhibitor MG132 for 1 hour followed by PROTAC 6 for 8 hours. As anticipated, CDK6 degradation was abrogated in the presence of MG132, which is consistent with previously reported studies with other PROTACs (Figure 5C)25. Collectively, these studies elucidate the mechanism of action i.e., the formation of the ternary complex (CDK6•PROTAC 6•CRBN) and a proteasomal based degradation of CDK6 by PROTAC 6.
Our results in pancreatic cell lines are consistent with a recent report from the Gray and Winter lab that showed a palbociclib-based CDK6 PROTAC with a different linker degraded CDK6 in acute myeloid leukemia cell lines27,28 (while this manuscript was in preparation). The need for CDK6 selective PROTACs reported by the Winter/Gray lab and PROTAC 6 reported here are highlighted by two recent reports. Kollmann et al. characterized a kinase-independent role for CDK6, which cannot be probed using the current CDK4/6 inhibitors12. Yang et al. showed that abemaciclib treatment mediated resistance is due to elevated CDK6 levels29. CDK6 selective PROTACs will serve as effective compounds that can be used to dissect the kinase independent function of CDK6 and as viable hits for optimization as therapeutics to overcome inhibitor resistance.
In conclusion, we report the development of a palbociclib-based PROTAC (6) as a selective CDK6 degrader. Although Palbociclib (1) inhibits CDK4 and CDK6 with equal potency, PROTAC 6 selectively degrades CDK6 while sparing other members of the CDK family. A PROTAC strategy efficiently converted a non-selective CDK4/6 inhibitor to a selective CDK6 degrader. Competition studies confirmed the need for the formation of a ternary complex as a prerequisite for efficient CDK6 degradation.
Supplementary Material
Acknowledgements:
This work was supported in part by NIH grants CA197999, and CA036727. We would like to thank Ed Ezell for NMR, and the Natarajan lab members for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found, in the online version.
References
- 1.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature reviews Drug discovery. 2015;14(2): 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellan C, De Falco G, Lazzi S, et al. CDK9/CYCLIN T1 expression during normal lymphoid differentiation and malignant transformation. The Journal of pathology. 2004;203(4): 946–952. [DOI] [PubMed] [Google Scholar]
- 3.De Falco G, Bellan C, D’Amuri A, et al. Cdk9 regulates neural differentiation and its expression correlates with the differentiation grade of neuroblastoma and PNET tumors. Cancer biology & therapy. 2005;4(3): 277–281. [DOI] [PubMed] [Google Scholar]
- 4.Simone C, Giordano A. Abrogation of signal-dependent activation of the cdk9/cyclin T2a complex in human RD rhabdomyosarcoma cells. Cell death and differentiation. 2007;14(1): 192–195. [DOI] [PubMed] [Google Scholar]
- 5.Sonawane YA, Taylor MA, Napoleon JV, Rana S, Contreras JI, Natarajan A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. Journal of medicinal chemistry. 2016;59(19): 8667–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robb CM, Kour S, Contreras JI, et al. Characterization of CDK(5) inhibitor, 20–223 (aka CP668863) for colorectal cancer therapy. Oncotarget. 2018;9(4): 5216–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rana S, Sonawane YA, Taylor MA, Kizhake S, Zahid M, Natarajan A. Synthesis of aminopyrazole analogs and their evaluation as CDK inhibitors for cancer therapy. Bioorganic & medicinal chemistry letters. 2018;28(23–24): 3736–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robb CM, Contreras JI, Kour S, et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chemical communications. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contreras JI, Robb CM, King HM, et al. Chemical Genetic Screens Identify Kinase Inhibitor Combinations that Target Anti-Apoptotic Proteins for Cancer Therapy. ACS chemical biology. 2018;13(5): 1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nature reviews Cancer. 2002;2(12): 910–917. [DOI] [PubMed] [Google Scholar]
- 11.Malumbres M, Sotillo R, Santamaria D, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118(4): 493–504. [DOI] [PubMed] [Google Scholar]
- 12.Kollmann K, Heller G, Schneckenleithner C, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer cell. 2013;24(2): 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15): 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winter GE, Buckley DL, Paulk J, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241): 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai L, Zhou B, Yang CY, et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer research. 2017;77(9): 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou B, Hu J, Xu F, et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. Journal of medicinal chemistry. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wurz RP, Dellamaggiore K, Dou H, et al. A “Click Chemistry Platform” for the Rapid Synthesis of Bispecific Molecules for Inducing Protein Degradation. Journal of medicinal chemistry. 2017. [DOI] [PubMed] [Google Scholar]
- 18.Raina K, Lu J, Qian Y, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(26): 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lebraud H, Wright DJ, Johnson CN, Heightman TD. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS central science. 2016;2(12): 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang K, Song Y, Xie H, et al. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorganic & medicinal chemistry letters. 2018;28(14): 2493–2497. [DOI] [PubMed] [Google Scholar]
- 21.Saenz DT, Fiskus W, Qian Y, et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zorba A, Nguyen C, Xu Y, et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(31): E7285–E7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu H, Schulze-Gahmen U. Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. Journal of medicinal chemistry. 2006;49(13): 3826–3831. [DOI] [PubMed] [Google Scholar]
- 24.Toogood PL, Harvey PJ, Repine JT, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. Journal of medicinal chemistry. 2005;48(7): 2388–2406. [DOI] [PubMed] [Google Scholar]
- 25.Zhou B, Hu J, Xu F, et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. Journal of medicinal chemistry. 2018;61(2): 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Remillard D, Buckley DL, Paulk J, et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angewandte Chemie. 2017;56(21): 5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brand M, Jiang B, Bauer S, et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell chemical biology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao B, Burgess K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chemical communications. 2019;55(18): 2704–2707. [DOI] [PubMed] [Google Scholar]
- 29.Yang C, Li Z, Bhatt T, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36(16): 2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.