

Abstract
Energetically efficient electrocatalysts with high product selectivity are desirable targets for sustainable chemical fuel generation using renewable electricity. Recycling CO2 by reduction to more energy dense products would support a carbon-neutral cycle that mitigates the intermittency of renewable energy sources. Conversion of CO2 to more saturated products typically requires proton equivalents. Complications with product selectivity stem from competitive reactions between H+ or CO2 at shared intermediates. We describe generalized catalytic cycles for H2, CO, and HCO2– formation that are commonly proposed in inorganic molecular catalysts. Thermodynamic considerations and trends for the reactions of H+ or CO2 at key intermediates are outlined. A quantitative understanding of intermediate catalytic steps is key to designing systems that display high selectivity while promoting energetically efficient catalysis by minimizing the overall energy landscape. For CO2 reduction to CO, we describe how an enzymatic active site motif facilitates efficient and selective catalysis and highlight relevant examples from synthetic systems.
Short abstract
Designing energetically efficient and selective electrocatalysts for CO2 reduction to chemical fuels requires an intimate understanding of how CO2, CO, and H+ interact with reduced metal centers.
The electrocatalytic reduction of CO2 is a direct route to sustainable fuel production from renewable electricity.1−3 Although protons are required to convert CO2 to chemical fuels, direct proton reduction to H2 siphons electrons away from CO2 reduction, decreasing the Faradaic yield of carbon-containing products.4,5
Nonselective reduction is commonly the result of the competitive reactions with either H+ or CO2 at key intermediates that ultimately lead to divergent pathways and products (Scheme 1). Some of the earliest work investigating the mechanism of molecular electrocatalysts for CO2 reduction suggested differential reactivity at common intermediates.6 Additional studies quantified the relative reactivity of H+ and CO2 at these proposed electrocatalytic intermediates.7−16 With the resurgence of interest in CO2 reduction over the past decade, new mechanistic studies and catalysts have generated fresh insights into the varying factors that contribute to product selectivity.3,17−29
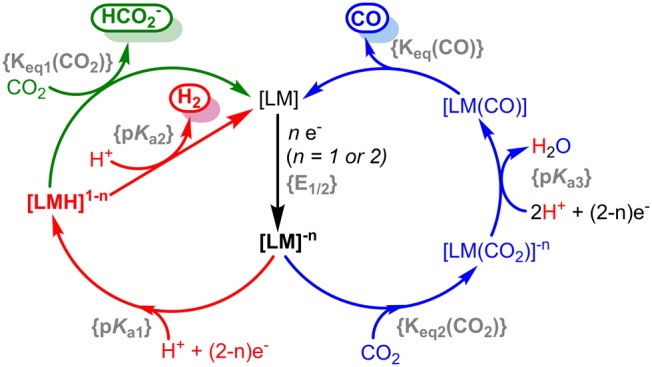
Scheme 1.
Our analysis is focused on the thermodynamic considerations for key steps in the most commonly proposed catalytic cycles for the hydrogen evolution reaction (HER) and carbon dioxide reduction reaction (CO2RR) to formate (HCO2–) and carbon monoxide (CO) by inorganic molecular electrocatalysts. Our evaluation includes general trends in catalyst properties and their broad impact on reactivity. We examine free energy considerations for the reaction of H+ and CO2 with proposed catalyst intermediates and the potential barriers for product release. These considerations provide guidelines for achieving selectivity at divergent reaction paths and are also essential for improving catalytic activity. Although our discussion is not focused on kinetic considerations, we note that intermediate steps in catalysis with high or low free energies intrinsically contribute to kinetic barriers in addition to the overall energetic efficiency (expressed in the overpotential). Thus, a quantitative understanding of the free energy contributions of each step is necessary to flatten the energy landscape and optimize activity.
We note that our analysis utilizes reported catalysts as examples but is not intended to be a complete description of the field. Instead, we refer the reader to more comprehensive reviews of molecular electrocatalysts for CO2 reduction that have recently been published.30−32
Overall Reaction Scheme
A generalized scheme for the catalytic reduction of H+ and CO2 to H2, HCO2–, and CO is shown in Scheme 1. Although other catalytic routes are possible, Scheme 1 represents the most frequently cited mechanisms. Upon electron transfer at a certain redox potential {E1/2}, the reduced intermediate can either protonate to form a metal hydride or directly activate CO2. In the protonation-first pathway (red), a metal hydride is formed which can react either with a second proton to form H2, or with CO2 to produce formate (green). Conversely, CO is typically the product in a CO2-activation-first pathway (blue). Each of these possibilities will be described separately.
Metal Hydride Formation
The protonation-first pathway requires the ability to form a stable metal hydride upon protonation. The free energy of protonation is the difference in pKa between the proton acceptor ({pKa1}, or that of the targeted
| 1 |
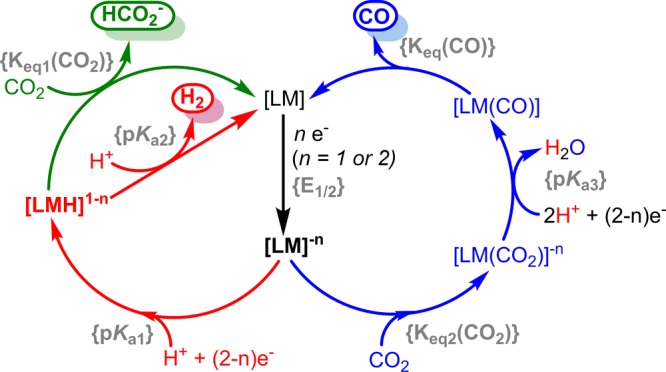
metal hydride intermediate) and proton donor ({pKa(ext)}, or external acid source) as expressed in eq 1. We intuitively expect more electron-rich metal centers to have more negative reduction potentials and be more Brønsted basic (higher metal hydride pKa values).
The measured pKa values of metal hydrides with reported reduction potentials {E1/2} in acetonitrile are plotted in Figure 1.33−53 The series represents a broad span of metal hydrides in different ligand environments (see Tables S1–S3 in the Supporting Information). Following the expected trend, more reducing metal centers are also stronger Brønsted bases. Since pKa is a metric of heterolytic M–H bond free energy, Figure 1 also depicts the linear free energy relationship between redox potential and the bond dissociation free energy of the M–H bond.
Figure 1.
pKa values of metal hydrides plotted versus the reduction potential required to access their conjugate bases. Blue triangles, orange circles, and green diamonds represent d4, d6, and d8 metal hydrides, respectively. Compiled from refs (33−53).
R. H. Morris recently reported a valuable empirical model for calculating metal hydride pKa values based on ligand acidity constants.54,55 A review also compiled experimentally measured and calculated pKa values for a broad range of metal hydrides (as well as dihydrogen complexes).56 Additionally, he notes that since M–H bond dissociation free energies are typically ∼60 kcal/mol,33−37,57,58 the pKa values of metal hydrides are expected to correlate with the redox potential of the conjugate base as seen in Figure 1.54
H2/HCO2– Formation
Upon metal hydride formation, it can react with either another proton or CO2. M. R. Dubois and D. L. Dubois first described how the free energy for the reactions of a metal hydride with H+ or CO2 at a metal hydride is determined by the hydricity (ΔGH–, eq 2) of the metal hydride.59,60 The hydricity is dependent on the two-electron reduction potential and pKa of the transition metal hydride along with the reduction potential for H+/H– in the respective solvent.48,61,62 As a result, hydricity values correlate with the average two-electron reduction potential of the metal (Figure S1).
The free energy for protonation of a metal hydride to evolve H2 (ΔG(H2)) is shown in eq 3; it is dependent on its hydricity (ΔGH–), the pKa of the external proton donor, and the heterolytic cleavage energy of H2 (CH2, a solvent-dependent constant). The free energy to reduce CO2 to formate (eq 4) is dependent on the hydricity of the metal hydride “donor” (ΔGH–) and the hydricity of formate (ΔGH–(HCO2–)), the “acceptor”. The free energy of hydride transfer (ΔG(HCO2–)) relates directly to {Keq1(CO2)} in Scheme 1. Several recent perspectives have discussed these relationships in depth.61−63
The pKa2 in Scheme 1 delineates the proton activity in which ΔG(H2) is close to zero, or ergoneutral. Using external acids with a lower pKa than pKa2 will result in H2 evolution
| 2 |
| 3 |
| 4 |
whereas the metal hydride will be stable to protonation with acids of a higher pKa.64−66 Since minimization of free energy leads to efficient catalysis, eq 3 was applied to optimize a class of catalysts for H2 evolution.67 A characteristic of a catalyst with a flattened energetic landscape is reversible reactivity (i.e., hydrogen evolution and oxidation), which was also illustrated in this class.68−70
An interesting aspect of eqs 3 and 4 is that the free energy of protonation of a metal hydride is dependent on the pKa of the proton donor, while the reaction with CO2 is not. As a result, there are conditions in which the reaction of a metal hydride with CO2 is exergonic while protonation to form H2 is endergonic. In these cases, if the pKa of the proton donor is sufficiently low enough to form the metal hydride, selective CO2 reduction can be accessed via thermodynamic considerations alone. These conditions exist because CO2 reduction to formate is a 1H+, 2e– process above the pKa of formic acid, while H+ reduction to H2 is a 2H+, 2e– process across all pKa values. As a result, the thermodynamic potential for each reaction has a differential dependence on proton activity. We recently published a more detailed description on this topic.64
We also note the thermodynamic values in eqs 2 and 4 are solvent-dependent, but do not quantitatively change to the same magnitude in different solvents.41,61,71−76 For example, while the hydricity for metal hydrides and formate decreases from organic solvents to water (or become better donors), formate’s hydricity decreases to a lesser extent. As a result, some metal hydrides that are insufficiently hydridic to reduce CO2 in organic solvents will do so in water.71,72,77
In accordance to the Sabatier principle, the interactions between the catalyst and substrate/product are also important. A significant interaction between the catalyst resting state and formate would make a favorable (negative) contribution to the free energy in eq 4, permitting CO2 reduction with weaker hydride donors. However, the interaction will also inhibit product release and catalyst turnover. Most putative hydride intermediates in successful CO2 reduction catalysts are composed of electron-rich mid or late transition metals18,26,64,78−81 which only weakly bind formate, so product release is not rate-limiting.
CO Production
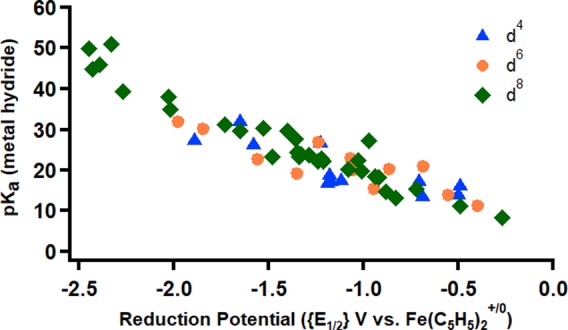
The CO2-activation-first pathway (blue in Scheme 1) requires CO2 activation to outcompete protonation at the reduced metal center. While very little quantitative data exists on CO2 binding constants {Keq2(CO2)} at reduced metal centers, a small but instructive data set exists for Co(I) tetraaminemacrocycles.82 In the absence of ligand steric effects, log{Keq2(CO2)} correlates with the Co(II/I) redox potential (Figure 2).82−84 The relationship is also intuitive, where more electron-rich (reducing) metal centers activate CO2 more strongly. In fact, no single transition metal site is known to react with CO2 at potentials positive of −1.2 V vs Fe(C5H5)2+/0 in organic solvents.30−32
Figure 2.
Relationship between {E1/2} of Co(I) macrocyclic complexes and thermodynamic (log{Keq2}) and kinetic (log k) reactivity with CO2. Data from ref (82).
The negative potentials required to activate CO2 have several undesirable side effects for overall catalyst selectivity, efficiency, and rate. As illustrated in Figure 1, more reducing metal sites are also more Brønsted basic (with the caveat that protonation to form a metal hydride requires two-electron oxidation of the complex, which is not always accessible). Thus, more reducing metal centers favor both the CO2-activation-first and the protonation-first pathways.
Another complicating factor is that in organic solvents the product, CO, is often a better ligand than CO2. Thus, increasing the electron density of the metal for CO2 activation often results in a more stable M–CO complex later in the catalytic cycle, inhibiting turnover. CO release has been shown to be rate-limiting in several known catalysts.84−88 (We note that this is not always the case; an earlier study found CO2 and CO equilibrium binding constants to cobalt macrocycles were competitive in water.)14
As a result, catalyst design for optimal CO2 reduction to CO requires an intimate understanding of how CO2, CO, and H+ interact with reduced metal centers. The importance of these parameters was delineated by Schneider, Fujita, and co-workers in 2012 based on their experimental work with cobalt macrocycles.82,84,87 Their analysis inspired our study on a series of isostructural cobalt pincer complexes, the results of which are summarized in Table 1.89 Cobalt complexes with more electron-donating ligands result in more negative reduction potentials and greater reactivity toward CO2 (see Table 1). We were unable to obtain accurate rate constants, but the overall trend is similar to that observed in the cobalt macrocycles (Figure 2). The Co–(CO) bond strength, measured by the vibrational stretch (ν) of the CO bond by infrared spectroscopy, also increased with decreasing reduction potential.
Table 1. Interaction of CO2, CO, and H+ for an Isostructural Series of Cobalt Pincer Complexes (Data from Ref (89)).
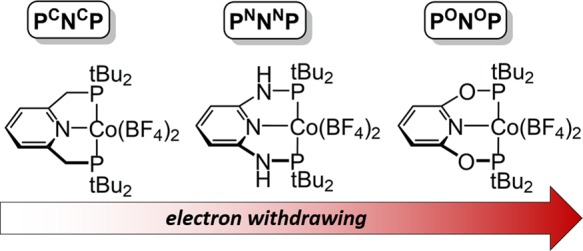
| L | PCNCP | PNNNP | PONOP |
|---|---|---|---|
| E1/2, LCo(II/I)a | –1.03 V | –0.88 V | –0.61 V |
| k[CO2] (s–1), [LCo]b | 102–3 | 102–3 | no reaction |
| [LCo(CO)]+, ν (cm–1) | 1911 | 1923 | 1936 |
| pKa,c [LCo] | 28 | 32 |
vs Fe(C5H5)2+/0 in CH3CN.
Reactivity with CO2 occurs upon reduction of the Co(I) complex, which is electrochemically irreversible. E1/2 for the reversible Co(II/I) couple is provided to illustrate the electronic trend.
Calculated for corresponding protonated complex.
The free energy relationships for metals and their association with CO2, CO, and H+ are comparable to scaling relationships more commonly used for analyzing heterogeneous catalysts. In this case, we find the general trends that relate redox potential with reactivity for the three key substrates follow opposing directions for catalyst optimization. Similar trends were also described for heterogeneous electrocatalysts.90
A generalized energy landscape for a single-site activation of CO2 to CO is depicted in black in Figure 3. A strongly reducing (and thus higher energy) metal site (intermediate A) is utilized to activate the electrophilic carbon in CO2. CO2 can bind to metals in a few different orientations. The η1 coordination is unstable in synthetic transition metal complexes91,92 (although if there is another vacant coordination site, it can bind η2).93−96 The highly nucleophilic oxygen atoms on unstable metal carboxylates (intermediate B) can promote ligand decomposition97 or disproportionation with another equivalent of CO2 to give CO and CO32–.20,98−103 A characteristic of an unstable metal carboxylate is extreme Brønsted basicity (high {pKa3}), which has been observed in some catalytic systems that scavenge protons from adventitious water or electrolyte.104−107 If protonation and reduction of the metal carboxylate (intermediate B) is successful in cleaving a C–O bond, more electron-rich metals will result in a greater energetic barrier for CO release (intermediate C).
Figure 3.
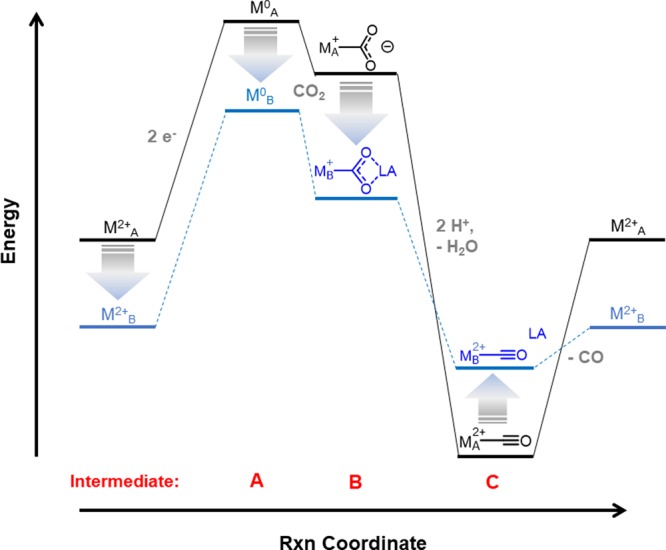
Free energy landscape for a single-site catalyst for CO2 reduction to CO (black) and a catalyst that utilizes a cooperative interaction (blue) to stabilize the metal carboxylate intermediate B.
Given these factors, it is clear that activation of CO2 at more positive potentials confers several benefits. In addition to catalysis at a milder potential, it inhibits the protonation-first pathway by lowering the Brønsted basicity of the metal while favoring product release.
Perhaps it is not surprising that a strategy for activating CO2 at mild potentials can be found in nature, where efficient redox catalysis for energy transduction is a matter of survival. The electrocatalytic activity of Ni-CODH I, a carbon monoxide dehydrogenase of the anaerobic Carboxydothermus hydrogenoformans (Ch), displays reversible electrocatalysis of CO2 to CO at high rates at the thermodynamic potential (no overpotential), or −520 mV vs SHE at pH 7.108 The X-ray crystallographic structure of the enzyme under reducing conditions in the presence of CO2 suggests cooperative binding by Ni and Fe, shown in Figure 4.109 Electrophilic activation of CO2 occurs at the redox active and Lewis basic reduced Ni0, while the adjacent Fe2+ participates in nonredox substrate activation. Thus, the active site capitalizes on a secondary interaction to cooperatively bind CO2 instead of a single metal site, contributing to its high rate and low overpotential.32,109−111
Figure 4.
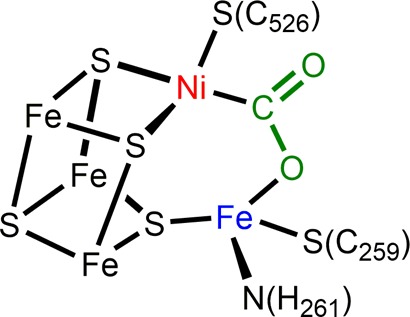
Active site of reduced Ch Ni-CODH II in the presence of CO2 characterized by X-ray crystallography (adapted from ref (109)).
An energetic analysis of a cooperative CO2 activation mechanism is shown in blue in Figure 3. Cooperative activation stabilizes the carboxylate intermediate, making a favorable free energy contribution to CO2 activation. This results in reactivity at more positive potentials, which also destabilizes the subsequent metal carbonyl product, facilitating product release. Not represented in Figure 3 is the protonation-first pathway, but we would expect it to be less favorable as the Brønsted basicity of the reduced metal decreases, contributing to enhanced selectivity.
There is evidence that several synthetic catalysts cooperatively activate CO2, which benefits their rate and/or selectivity. Bimetallic activation of CO2 is proposed in a synthetic dipalladium system.30 Single-site palladium complexes with a triphosphine ligand display a linear free energy relationship (LFER) between Keq(CO2) and redox potential.67 Addition of a hydrogen-bonding interaction or a second metal disrupted the LFER, leading to faster catalytic CO2 reduction at a milder potential. Optimization of the cooperative interaction increased the rate of CO2 to CO catalysis by 3 orders of magnitude compared to the monomer while lowering the overpotential almost 200 mV.59,112−115 Additionally, the Faradaic efficiency improved from 10:90 CO:H2 to 85:15 CO:H2 upon introduction of the second metal center.113,115
However, the two symmetric homobimetallic sites have similar reduction potentials, which results in unproductive metal–metal bond formation, deactivating the catalyst. By using two different metals, the [NiFe] center of Ch Ni-CODH, promotes selective redox chemistry at the Ni site. Several other synthetic transition metal CO2 reduction catalysts have shown substantial evidence for bimetallic CO2 activation.86,97,116−124 A dicobalt carboxylate complex was also structurally characterized from a cobalt macrocyclic catalyst.125 We note several heterobimetallic systems utilize strong oxophilic Lewis acids to activate CO2;116,126 in some cases, the latter can bind the oxygen too tightly for catalyst turnover.
Other successful catalysts attribute improved activity to other types of cooperative CO2 activation. Early mechanistic and computational studies for the catalysts [Ni(cyclam)]+ and cobalt macrocycles indicate the importance of the protons on the macrocycle amines for facilitating CO2 binding.9,127 Savéant and co-workers have also shown that the incorporation of phenol moieties in the secondary coordination sphere of a previously investigated iron porphyrin complex results in a 50-fold rate increase at an overpotential 360 mV lower than the corresponding anisole substituted system.128 Most recently, Dey and co-workers reported a low overpotential electrocatalyst which incorporates a proposed S–H functionality appropriately positioned to stabilize a metal carboxylate.22 A key feature of these hydrogen-bonding interactions is that they are positioned appropriately to facilitate CO2 binding (interaction with an O atom on the carboxylate) without enhancing direct proton delivery to the metal, which would favor the protonation-first pathway. Another system with proximal secondary amines was found to enhance protonation by generating a local hydrogen-bonding environment, even if they do not assist in CO2 binding.129 Although we are not discussing the second protonation event required to liberate water in detail, another possible route to H2 is direct protonation of the metal carboxylic acid. Careful positioning of secondary sphere hydrogen-bonding functionalities is important for circumventing this possibility.
It has been suggested that cationic functionalities also lower the energetic requirement to access a metal carboxylate intermediate. Savéant and coworkers utilized cationic ammonium substituents to promote CO2 reduction through electrostatic stabilization of a bound carboxylate species.130 Iron porphyrin complexes featuring o-NMe3+ substituents function at 230 mV lower potential than the corresponding p-NMe3+ substituted complex, while operating at 100 times the rate. In contrast, catalyst activity was suppressed when the cationic amines were replaced with anionic sulfonate moieties, highlighting the effects of electrostatic interactions on CO2 catalysis.130 Electrostatic interactions have more recently been utilized for a similar beneficial effect in rhenium bipyridine systems.131
In addition to increasing the reaction rate and decreasing the required overpotential for catalysis, CO2 activation involving cooperative interactions can also affect product selectivity. Large enhancements in CO selectivity have been observed in systems featuring hydrogen-bonding,129,132,133 bimetallic,113 and electrostatic interactions.134
Although synthetic catalysts have successfully utilized cooperative CO2 activation to enhance their activity or selectivity, they have yet to achieve the lofty catalytic metrics exhibited by Ni-CODH I. We expect there are more secrets to be discovered for how the active sites of the carbon monoxide dehydrogenases (including the less studied MoCu class) balance key kinetic and thermodynamic factors for efficient, fast, and selective catalysis.
Conclusion
Selectivity for CO2 reduction in the presence of protons is a complex challenge due to multiple possible reaction pathways. To simplify, we have discussed the most commonly cited mechanisms, detailing the thermodynamic parameters involved for each step and how they correlate with redox potential. We emphasize our generalized approach will not apply to all catalyst systems. Instead, we believe our analysis provides a useful framework for thoughtful and creative catalyst design and optimization. In the case of CO2 reduction to CO, it is apparent that several key parameters are inversely related for single-site metals. However, these relationships can be broken using a secondary interaction, mirroring the approach used by a natural enzyme. Although it is not specifically discussed in our analysis, we also believe uncovering strategies for kinetic inhibition for undesirable reactions presents another fruitful area for targeted catalyst design.
Acknowledgments
J.M.B. and J.Y.Y. are supported by NSF Award 1554744. J.Y.Y. is also grateful for support as a Sloan Foundation Fellow and a CIFAR Azrieli Global Scholar.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00095.
The authors declare no competing financial interest.
Supplementary Material
References
- Lewis N. S.; Nocera D. G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (43), 15729–15735. 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B. Powering the planet with solar fuel. Nat. Chem. 2009, 1, 7. 10.1038/nchem.141. [DOI] [PubMed] [Google Scholar]
- Kang P.; Chen Z.; Brookhart M.; Meyer T. J. Electrocatalytic reduction of carbon dioxide: let the molecules do the work. Top. Catal. 2015, 58 (1), 30–45. 10.1007/s11244-014-0344-y. [DOI] [Google Scholar]
- Seh Z. W.; Kibsgaard J.; Dickens C. F.; Chorkendorff I.; Nørskov J. K.; Jaramillo T. F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355 (6321), eaad4998 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Hu Y.; Ma L.; Zhu G.; Wang Y.; Xue X.; Chen R.; Yang S.; Jin Z. Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Advanced Science 2018, 5 (1), 1700275. 10.1002/advs.201700275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher B. J.; Eisenberg R. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 1980, 102 (24), 7361–7363. 10.1021/ja00544a035. [DOI] [Google Scholar]
- Tait A. M.; Hoffman M. Z.; Hayon E. The reactivity of cobalt(I) complexes containing unsaturated macrocyclic ligands in aqueous solution. J. Am. Chem. Soc. 1976, 98 (1), 86–93. 10.1021/ja00417a015. [DOI] [Google Scholar]
- Gangi D. A.; Durand R. R. Binding of carbon dioxide to cobalt and nickel tetra-aza macrocycles. J. Chem. Soc., Chem. Commun. 1986, 9, 697–699. 10.1039/c39860000697. [DOI] [Google Scholar]
- Beley M.; Collin J. P.; Ruppert R.; Sauvage J. P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108 (24), 7461–7467. 10.1021/ja00284a003. [DOI] [PubMed] [Google Scholar]
- Grant J. L.; Goswami K.; Spreer L. O.; Otvos J. W.; Calvin M. Photochemical reduction of carbon dioxide to carbon monoxide in water using a nickel(II) tetra-azamacrocycle complex as catalyst. J. Chem. Soc., Dalton Trans. 1987, (9), 2105–2109. 10.1039/dt9870002105. [DOI] [Google Scholar]
- Ishida H.; Tanaka H.; Tanaka K.; Tanaka T. Selective formation of HCOO– in the electrochemical CO2 reduction catalysed by [Ru(bpy)2(CO)2]2+(bpy = 2,2′-bipyridine). J. Chem. Soc., Chem. Commun. 1987, 2, 131–132. 10.1039/C39870000131. [DOI] [Google Scholar]
- Collin J. P.; Jouaiti A.; Sauvage J. P. Electrocatalytic properties of (tetraazacyclotetradecane)nickel(2+) and Ni2(biscyclam)4+ with respect to carbon dioxide and water reduction. Inorg. Chem. 1988, 27 (11), 1986–1990. 10.1021/ic00284a030. [DOI] [Google Scholar]
- Creutz C.; Schwarz H. A.; Wishart J. F.; Fujita E.; Sutin N. A dissociative pathway for equilibration of a hydrido CoL(H)2+ complex with carbon dioxide and carbon monoxide. Ligand binding constants in the macrocyclic [14]-dienecobalt(I) system. J. Am. Chem. Soc. 1989, 111 (3), 1153–1154. 10.1021/ja00185a069. [DOI] [Google Scholar]
- Creutz C.; Schwarz H. A.; Wishart J. F.; Fujita E.; Sutin N. Thermodynamics and kinetics of carbon dioxide binding to two stereoisomers of a cobalt(I) macrocycle in aqueous solution. J. Am. Chem. Soc. 1991, 113 (9), 3361–3371. 10.1021/ja00009a022. [DOI] [Google Scholar]
- Fujita E.; Haff J.; Sanzenbacher R.; Elias H. High electrocatalytic activity of RRSS-[NiIIHTIM](ClO4)2 and [NiIIDMC](ClO4)2 for carbon dioxide reduction (HTIM = 2,3,9,10-Tetramethyl-1,4,8,11-tetraazacyclotetradecane, DMC = C-meso-5,12-Dimethyl-1,4,8,11-tetraazacyclotetradecane). Inorg. Chem. 1994, 33 (21), 4627–4628. 10.1021/ic00099a011. [DOI] [Google Scholar]
- Kelly C. A.; Blinn E. L.; Camaioni N.; D’Angelantonio M.; Mulazzani Q. G. Mechanism of CO2 and H+ reduction by Ni(cyclam)+ in aqueous solution. A pulse and continuous radiolysis study. Inorg. Chem. 1999, 38 (7), 1579–1584. 10.1021/ic980902p. [DOI] [Google Scholar]
- Smieja J. M.; Benson E. E.; Kumar B.; Grice K. A.; Seu C. S.; Miller A. J. M.; Mayer J. M.; Kubiak C. P. Kinetic and structural studies, origins of selectivity, and interfacial charge transfer in the artificial photosynthesis of CO. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (39), 15646–15650. 10.1073/pnas.1119863109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri A.; Berben L. A. Tailoring electrocatalysts for selective CO2 or H+ reduction: iron carbonyl clusters as a case study. Inorg. Chem. 2016, 55 (2), 378–385. 10.1021/acs.inorgchem.5b02293. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan S.; Chidsey C. E. D. Initiation of the electrochemical reduction of CO2 by a singly reduced ruthenium(II) bipyridine complex. Inorg. Chem. 2017, 56 (14), 8326–8333. 10.1021/acs.inorgchem.7b01004. [DOI] [PubMed] [Google Scholar]
- Nichols A. W.; Chatterjee S.; Sabat M.; Machan C. W. Electrocatalytic reduction of CO2 to formate by an iron schiff base complex. Inorg. Chem. 2018, 57 (4), 2111–2121. 10.1021/acs.inorgchem.7b02955. [DOI] [PubMed] [Google Scholar]
- Fogeron T.; Todorova T. K.; Porcher J.-P.; Gomez-Mingot M.; Chamoreau L.-M.; Mellot-Draznieks C.; Li Y.; Fontecave M. A bioinspired nickel(bis-dithiolene) complex as a homogeneous catalyst for carbon dioxide electroreduction. ACS Catal. 2018, 8 (3), 2030–2038. 10.1021/acscatal.7b03383. [DOI] [Google Scholar]
- Dey S.; Ahmed M. E.; Dey A. Activation of Co(I) state in a cobalt-dithiolato catalyst for selective and efficient CO2 reduction to CO. Inorg. Chem. 2018, 57 (10), 5939–5947. 10.1021/acs.inorgchem.8b00450. [DOI] [PubMed] [Google Scholar]
- Cometto C.; Chen L.; Anxolabéhère-Mallart E.; Fave C.; Lau T.-C.; Robert M.. Molecular electrochemical catalysis of the CO2-to-CO conversion with a Co complex: A cyclic voltammetry mechanistic investigation. Organometallics 2018, in press. 10.1021/acs.organomet.8b00555 [DOI] [Google Scholar]
- Yoo C.; Kim Y.-E.; Lee Y. Selective transformation of CO2 to CO at a single nickel center. Acc. Chem. Res. 2018, 51 (5), 1144–1152. 10.1021/acs.accounts.7b00634. [DOI] [PubMed] [Google Scholar]
- Cometto C.; Chen L.; Lo P.-K.; Guo Z.; Lau K.-C.; Anxolabéhère-Mallart E.; Fave C.; Lau T.-C.; Robert M. Highly selective molecular catalysts for the CO2-to-CO electrochemical conversion at very low overpotential. Contrasting Fe vs Co quaterpyridine complexes upon mechanistic studies. ACS Catal. 2018, 8 (4), 3411–3417. 10.1021/acscatal.7b04412. [DOI] [Google Scholar]
- Kanega R.; Onishi N.; Wang L.; Himeda Y. Electroreduction of carbon dioxide to formate by homogeneous Ir catalysts in water. ACS Catal. 2018, 8 (12), 11296–11301. 10.1021/acscatal.8b02525. [DOI] [Google Scholar]
- Nie W.; McCrory C. C. L. Electrocatalytic CO2 reduction by a cobalt bis(pyridylmonoimine) complex: effect of acid concentration on catalyst activity and stability. Chem. Commun. 2018, 54 (13), 1579–1582. 10.1039/C7CC08546J. [DOI] [PubMed] [Google Scholar]
- Göttle A. J.; Koper M. T. M. Determinant role of electrogenerated reactive nucleophilic species on selectivity during reduction of CO2 catalyzed by metalloporphyrins. J. Am. Chem. Soc. 2018, 140 (14), 4826–4834. 10.1021/jacs.7b11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage N. P.; Dulaney H. A.; Huckaba A. J.; Jurss J. W.; Delcamp J. H. Electrocatalytic reduction of CO2 to CO with Re-pyridyl-NHCs: Proton source influence on rates and product selectivities. Inorg. Chem. 2016, 55 (12), 6085–6094. 10.1021/acs.inorgchem.6b00626. [DOI] [PubMed] [Google Scholar]
- Francke R.; Schille B.; Roemelt M. Homogeneously catalyzed electroreduction of carbon dioxide—Methods, mechanisms, and catalysts. Chem. Rev. 2018, 118 (9), 4631–4701. 10.1021/acs.chemrev.7b00459. [DOI] [PubMed] [Google Scholar]
- Takeda H.; Cometto C.; Ishitani O.; Robert M. Electrons, photons, protons and earth-abundant metal complexes for molecular catalysis of CO2 reduction. ACS Catal. 2017, 7 (1), 70–88. 10.1021/acscatal.6b02181. [DOI] [Google Scholar]
- Appel A. M.; Bercaw J. E.; Bocarsly A. B.; Dobbek H.; DuBois D. L.; Dupuis M.; Ferry J. G.; Fujita E.; Hille R.; Kenis P. J. A.; Kerfeld C. A.; Morris R. H.; Peden C. H. F.; Portis A. R.; Ragsdale S. W.; Rauchfuss T. B.; Reek J. N. H.; Seefeldt L. C.; Thauer R. K.; Waldrop G. L. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113 (8), 6621–6658. 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berning D. E.; Noll B. C.; DuBois D. L. Relative hydride, proton, and hydrogen atom transfer abilities of [HM(diphosphine)2]PF6 complexes (M = Pt, Ni). J. Am. Chem. Soc. 1999, 121 (49), 11432–11447. 10.1021/ja991888y. [DOI] [Google Scholar]
- Raebiger J. W.; DuBois D. L. Thermodynamic studies of HRh(depx)2 and [(H)2Rh(depx)2](CF3SO3): Relationships between five-coordinate monohydrides and six-coordinate dihydrides. Organometallics 2005, 24 (1), 110–118. 10.1021/om049437w. [DOI] [Google Scholar]
- Roberts J. A. S.; Appel A. M.; DuBois D. L.; Bullock R. M. Comprehensive thermochemistry of W–H bonding in the metal hydrides CpW(CO)2(IMes)H, [CpW(CO)2(IMes)H]•+, and [CpW(CO)2(IMes)(H)2]+. Influence of an N-heterocyclic carbene ligand on metal hydride bond energies. J. Am. Chem. Soc. 2011, 133 (37), 14604–14613. 10.1021/ja202830w. [DOI] [PubMed] [Google Scholar]
- Chen S.; Rousseau R.; Raugei S.; Dupuis M.; DuBois D. L.; Bullock R. M. Comprehensive thermodynamics of nickel hydride Bis(diphosphine) complexes: A predictive model through computations. Organometallics 2011, 30 (22), 6108–6118. 10.1021/om200645x. [DOI] [Google Scholar]
- van der Eide E. F.; Helm M. L.; Walter E. D.; Bullock R. M. Structural and spectroscopic characterization of 17- and 18-electron piano-stool complexes of chromium. Thermochemical analyses of weak Cr–H bonds. Inorg. Chem. 2013, 52 (3), 1591–1603. 10.1021/ic302460y. [DOI] [PubMed] [Google Scholar]
- Choi J.; Pulling M. E.; Smith D. M.; Norton J. R. Unusually weak metal–hydrogen bonds in HV(CO)4(P–P) and their effectiveness as H• donors. J. Am. Chem. Soc. 2008, 130 (13), 4250–4252. 10.1021/ja710455c. [DOI] [PubMed] [Google Scholar]
- Tilset M.; Parker V. D. Solution homolytic bond dissociation energies of organotransition-metal hydrides. J. Am. Chem. Soc. 1989, 111 (17), 6711–6717. 10.1021/ja00199a034. [DOI] [Google Scholar]
- Estes D. P.; Vannucci A. K.; Hall A. R.; Lichtenberger D. L.; Norton J. R. Thermodynamics of the metal–hydrogen bonds in (η5-C5H5)M(CO)2H (M = Fe, Ru, Os). Organometallics 2011, 30 (12), 3444–3447. 10.1021/om2001519. [DOI] [Google Scholar]
- Matsubara Y.; Fujita E.; Doherty M. D.; Muckerman J. T.; Creutz C. Thermodynamic and kinetic hydricity of ruthenium(II) hydride complexes. J. Am. Chem. Soc. 2012, 134 (38), 15743–15757. 10.1021/ja302937q. [DOI] [PubMed] [Google Scholar]
- Ciancanelli R.; Noll B. C.; DuBois D. L.; DuBois M. R. Comprehensive thermodynamic characterization of the metal–hydrogen bond in a series of cobalt-hydride complexes. J. Am. Chem. Soc. 2002, 124 (12), 2984–2992. 10.1021/ja0122804. [DOI] [PubMed] [Google Scholar]
- Fang M.; Wiedner E. S.; Dougherty W. G.; Kassel W. S.; Liu T.; DuBois D. L.; Bullock R. M. Cobalt complexes containing pendant amines in the second coordination sphere as electrocatalysts for H2 production. Organometallics 2014, 33 (20), 5820–5833. 10.1021/om5004607. [DOI] [Google Scholar]
- Hu Y.; Norton J. R. Kinetics and Thermodynamics of H–/H•/H+ transfer from a rhodium(III) hydride. J. Am. Chem. Soc. 2014, 136 (16), 5938–5948. 10.1021/ja412309j. [DOI] [PubMed] [Google Scholar]
- Barrett S. M.; Pitman C. L.; Walden A. G.; Miller A. J. M. Photoswitchable hydride transfer from iridium to 1-methylnicotinamide rationalized by thermochemical cycles. J. Am. Chem. Soc. 2014, 136 (42), 14718–14721. 10.1021/ja508762g. [DOI] [PubMed] [Google Scholar]
- Price A. J.; Ciancanelli R.; Noll B. C.; Curtis C. J.; DuBois D. L.; DuBois M. R. HRh(dppb)2, a powerful hydride donor. Organometallics 2002, 21 (22), 4833–4839. 10.1021/om020421k. [DOI] [Google Scholar]
- Lilio A. M.; Reineke M. H.; Moore C. E.; Rheingold A. L.; Takase M. K.; Kubiak C. P. Incorporation of pendant bases into Rh(diphosphine)2 complexes: Synthesis, thermodynamic studies, and catalytic CO2 hydrogenation activity of [Rh(P2N2)2]+ complexes. J. Am. Chem. Soc. 2015, 137 (25), 8251–8260. 10.1021/jacs.5b04291. [DOI] [PubMed] [Google Scholar]
- Curtis C. J.; Miedaner A.; Ellis W. W.; DuBois D. L. Measurement of the hydride donor abilities of [HM(diphosphine)2]+ complexes (M = Ni, Pt) by heterolytic activation of hydrogen. J. Am. Chem. Soc. 2002, 124 (9), 1918–1925. 10.1021/ja0116829. [DOI] [PubMed] [Google Scholar]
- Wiese S.; Kilgore U. J.; DuBois D. L.; Bullock R. M. [Ni(PMe2NPh2)2](BF4)2 as an electrocatalyst for H2 production. ACS Catal. 2012, 2 (5), 720–727. 10.1021/cs300019h. [DOI] [Google Scholar]
- Berning D. E.; Miedaner A.; Curtis C. J.; Noll B. C.; Rakowski DuBois M. C.; DuBois D. L. Free-energy relationships between the proton and hydride donor abilities of [HNi(diphosphine)2]+ complexes and the half-wave potentials of their conjugate bases. Organometallics 2001, 20 (9), 1832–1839. 10.1021/om0100582. [DOI] [Google Scholar]
- Curtis C. J.; Miedaner A.; Raebiger J. W.; DuBois D. L. Periodic trends in metal hydride donor thermodynamics: Measurement and comparison of the hydride donor abilities of the series HM(PNP)2+ (M = Ni, Pd, Pt; PNP = Et2PCH2N(Me)CH2PEt2). Organometallics 2004, 23 (3), 511–516. 10.1021/om0342816. [DOI] [Google Scholar]
- Raebiger J. W.; Miedaner A.; Curtis C. J.; Miller S. M.; Anderson O. P.; DuBois D. L. Using ligand bite angles to control the hydricity of palladium diphosphine complexes. J. Am. Chem. Soc. 2004, 126 (17), 5502–5514. 10.1021/ja0395240. [DOI] [PubMed] [Google Scholar]
- Miedaner A.; Raebiger J. W.; Curtis C. J.; Miller S. M.; DuBois D. L. Thermodynamic studies of [HPt(EtXantphos)2]+ and [(H)2Pt(EtXantphos)2]2+. Organometallics 2004, 23 (11), 2670–2679. 10.1021/om034238i. [DOI] [Google Scholar]
- Morris R. H. Estimating the acidity of transition metal hydride and dihydrogen complexes by adding ligand acidity constants. J. Am. Chem. Soc. 2014, 136 (5), 1948–1959. 10.1021/ja410718r. [DOI] [PubMed] [Google Scholar]
- Sung M. M. H.; Jdanova S.; Morris R. H. Ligand acidity constants as calculated by density functional theory for PF3 and N-Heterocyclic carbene ligands in hydride complexes of Iron(II). J. Organomet. Chem. 2019, 880, 15–21. 10.1016/j.jorganchem.2018.10.024. [DOI] [Google Scholar]
- Morris R. H. Brønsted–Lowry acid strength of metal hydride and dihydrogen complexes. Chem. Rev. 2016, 116 (15), 8588–8654. 10.1021/acs.chemrev.5b00695. [DOI] [PubMed] [Google Scholar]
- Wang D.; Angelici R. J. Metal–hydrogen bond dissociation enthalpies in series of complexes of eight different transition metals. J. Am. Chem. Soc. 1996, 118 (5), 935–942. 10.1021/ja9441930. [DOI] [Google Scholar]
- Fu X.; Wayland B. B. Thermodynamics of rhodium hydride reactions with CO, aldehydes, and olefins in water: Organo-rhodium porphyrin bond dissociation free energies. J. Am. Chem. Soc. 2005, 127 (47), 16460–16467. 10.1021/ja054548n. [DOI] [PubMed] [Google Scholar]
- Rakowski Dubois M.; Dubois D. L. Development of molecular electrocatalysts for CO2 reduction and H2 production/oxidation. Acc. Chem. Res. 2009, 42 (12), 1974–1982. 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]
- DuBois D. L.; Berning D. E. Hydricity of transition-metal hydrides and its role in CO2 reduction. Appl. Organomet. Chem. 2000, 14 (12), 860–862. 10.1002/1099-0739(200012)14:12<860::AID-AOC87>3.0.CO;2-A. [DOI] [Google Scholar]
- Wiedner E. S.; Chambers M. B.; Pitman C. L.; Bullock R. M.; Miller A. J. M.; Appel A. M. Thermodynamic hydricity of transition metal hydrides. Chem. Rev. 2016, 116 (15), 8655–8692. 10.1021/acs.chemrev.6b00168. [DOI] [PubMed] [Google Scholar]
- Waldie K. M.; Ostericher A. L.; Reineke M. H.; Sasayama A. F.; Kubiak C. P. Hydricity of transition-metal hydrides: Thermodynamic considerations for CO2 reduction. ACS Catal. 2018, 8 (2), 1313–1324. 10.1021/acscatal.7b03396. [DOI] [Google Scholar]
- Bullock R. M.; Appel A. M.; Helm M. L. Production of hydrogen by electrocatalysis: making the H–H bond by combining protons and hydrides. Chem. Commun. 2014, 50 (24), 3125–3143. 10.1039/C3CC46135A. [DOI] [PubMed] [Google Scholar]
- Ceballos B. M.; Yang J. Y. Directing the reactivity of metal hydrides for selective CO2 reduction. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (50), 12686–12691. 10.1073/pnas.1811396115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay C.; Ceballos B. M.; Yang J. Y., pH-dependent reactivity of a water-soluble nickel complex: Hydrogen evolution vs selective electrochemical hydride generation. Organometallics 2018, in press. 10.1021/acs.organomet.8b00558 [DOI] [Google Scholar]
- Tsay C.; Yang J. Y. Electrocatalytic hydrogen evolution under acidic aqueous conditions and mechanistic studies of a highly stable molecular catalyst. J. Am. Chem. Soc. 2016, 138 (43), 14174–14177. 10.1021/jacs.6b05851. [DOI] [PubMed] [Google Scholar]
- DuBois D. L. Development of molecular electrocatalysts for energy storage. Inorg. Chem. 2014, 53 (8), 3935–3960. 10.1021/ic4026969. [DOI] [PubMed] [Google Scholar]
- Smith S. E.; Yang J. Y.; DuBois D. L.; Bullock R. M. Reversible electrocatalytic production and oxidation of hydrogen at low overpotentials by a functional hydrogenase mimic. Angew. Chem., Int. Ed. 2012, 51 (13), 3152–3155. 10.1002/anie.201108461. [DOI] [PubMed] [Google Scholar]
- Priyadarshani N.; Dutta A.; Ginovska B.; Buchko G. W.; O’Hagan M.; Raugei S.; Shaw W. J. Achieving reversible H2/H+ interconversion at room temperature with enzyme-inspired molecular complexes: A mechanistic study. ACS Catal. 2016, 6 (9), 6037–6049. 10.1021/acscatal.6b01433. [DOI] [Google Scholar]
- Dutta A.; Appel A. M.; Shaw W. J. Designing electrochemically reversible H2 oxidation and production catalysts. Nature Reviews Chemistry 2018, 2 (9), 244–252. 10.1038/s41570-018-0032-8. [DOI] [Google Scholar]
- Taheri A.; Thompson E. J.; Fettinger J. C.; Berben L. A. An iron electrocatalyst for selective reduction of CO2 to formate in water: Including thermochemical insights. ACS Catal. 2015, 5 (12), 7140–7151. 10.1021/acscatal.5b01708. [DOI] [Google Scholar]
- Ceballos B. M.; Tsay C.; Yang J. Y. CO2 reduction or HCO2- oxidation? Solvent-dependent thermochemistry of a nickel hydride complex. Chem. Commun. 2017, 53 (53), 7405–7408. 10.1039/C7CC02511D. [DOI] [PubMed] [Google Scholar]
- Creutz C.; Chou M. H. Rapid transfer of hydride ion from a ruthenium complex to C1 species in water. J. Am. Chem. Soc. 2007, 129 (33), 10108–10109. 10.1021/ja074158w. [DOI] [PubMed] [Google Scholar]
- Muckerman J. T.; Achord P.; Creutz C.; Polyansky D. E.; Fujita E. Calculation of thermodynamic hydricities and the design of hydride donors for CO2 reduction. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (39), 15657–15662. 10.1073/pnas.1201026109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brereton K. R.; Pitman C. L.; Cundari T. R.; Miller A. J. M. Solvent-dependent thermochemistry of an iridium/ruthenium H2 evolution catalyst. Inorg. Chem. 2016, 55 (22), 12042–12051. 10.1021/acs.inorgchem.6b02223. [DOI] [PubMed] [Google Scholar]
- Tsay C.; Livesay B. N.; Ruelas S.; Yang J. Y. Solvation effects on transition metal hydricity. J. Am. Chem. Soc. 2015, 137 (44), 14114–14121. 10.1021/jacs.5b07777. [DOI] [PubMed] [Google Scholar]
- Burgess S. A.; Appel A. M.; Linehan J. C.; Wiedner E. S. Changing the mechanism for CO2 hydrogenation using solvent-dependent thermodynamics. Angew. Chem., Int. Ed. 2017, 56 (47), 15002–15005. 10.1002/anie.201709319. [DOI] [PubMed] [Google Scholar]
- Loewen N. D.; Neelakantan T. V.; Berben L. A. Renewable formate from C–H bond formation with CO2: Using iron carbonyl clusters as electrocatalysts. Acc. Chem. Res. 2017, 50 (9), 2362–2370. 10.1021/acs.accounts.7b00302. [DOI] [PubMed] [Google Scholar]
- Kang P.; Zhang S.; Meyer T. J.; Brookhart M. Rapid selective electrocatalytic reduction of carbon dioxide to formate by an iridium pincer catalyst immobilized on carbon nanotube electrodes. Angew. Chem., Int. Ed. 2014, 53 (33), 8709–8713. 10.1002/anie.201310722. [DOI] [PubMed] [Google Scholar]
- Kang P.; Cheng C.; Chen Z.; Schauer C. K.; Meyer T. J.; Brookhart M. Selective electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complexes. J. Am. Chem. Soc. 2012, 134 (12), 5500–5503. 10.1021/ja300543s. [DOI] [PubMed] [Google Scholar]
- Roy S.; Sharma B.; Pécaut J.; Simon P.; Fontecave M.; Tran P. D.; Derat E.; Artero V. Molecular cobalt complexes with pendant amines for selective electrocatalytic reduction of carbon dioxide to formic acid. J. Am. Chem. Soc. 2017, 139 (10), 3685–3696. 10.1021/jacs.6b11474. [DOI] [PubMed] [Google Scholar]
- Ogata T.; Yanagida S.; Brunschwig B. S.; Fujita E. Mechanistic and kinetic studies of cobalt macrocycles in a photochemical CO2 reduction system: Evidence of Co-CO2 adducts as intermediates. J. Am. Chem. Soc. 1995, 117 (25), 6708–6716. 10.1021/ja00130a009. [DOI] [Google Scholar]
- Schmidt M. H.; Miskelly G. M.; Lewis N. S. Effects of redox potential, steric configuration, solvent, and alkali metal cations on the binding of carbon dioxide to cobalt(I) and nickel(I) macrocycles. J. Am. Chem. Soc. 1990, 112 (9), 3420–3426. 10.1021/ja00165a027. [DOI] [Google Scholar]
- Fujita E.; Creutz C.; Sutin N.; Szalda D. J. Carbon dioxide activation by cobalt(I) macrocycles: factors affecting carbon dioxide and carbon monoxide binding. J. Am. Chem. Soc. 1991, 113 (1), 343–353. 10.1021/ja00001a048. [DOI] [Google Scholar]
- Froehlich J. D.; Kubiak C. P. The homogeneous reduction of CO2 by [Ni(cyclam)]+: Increased catalytic rates with the addition of a CO scavenger. J. Am. Chem. Soc. 2015, 137 (10), 3565–3573. 10.1021/ja512575v. [DOI] [PubMed] [Google Scholar]
- Isse A. A.; Gennaro A.; Vianello E.; Floriani C. Electrochemical reduction of carbon dioxide catalyzed by [CoI(salophen)Li]. J. Mol. Catal. 1991, 70 (2), 197–208. 10.1016/0304-5102(91)80161-U. [DOI] [Google Scholar]
- Schneider J.; Jia H.; Muckerman J. T.; Fujita E. Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal centre of CO2reductioncatalysts. Chem. Soc. Rev. 2012, 41 (6), 2036–2051. 10.1039/C1CS15278E. [DOI] [PubMed] [Google Scholar]
- Balazs G. B.; Anson F. C. Effects of CO on the electrocatalytic activity of Ni (cyclam)2+ toward the reduction of CO2. J. Electroanal. Chem. 1993, 361 (1–2), 149–157. 10.1016/0022-0728(93)87049-2. [DOI] [Google Scholar]
- Shaffer D. W.; Johnson S. I.; Rheingold A. L.; Ziller J. W.; Goddard W. A.; Nielsen R. J.; Yang J. Y. Reactivity of a series of isostructural cobalt pincer complexes with CO2, CO, and H+. Inorg. Chem. 2014, 53 (24), 13031–13041. 10.1021/ic5021725. [DOI] [PubMed] [Google Scholar]
- Hansen H. A.; Varley J. B.; Peterson A. A.; Nørskov J. K. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 2013, 4 (3), 388–392. 10.1021/jz3021155. [DOI] [PubMed] [Google Scholar]
- Leitner W. The coordination chemistry of carbon dioxide and its relevance for catalysis: a critical survey. Coord. Chem. Rev. 1996, 153 (0), 257–284. 10.1016/0010-8545(95)01226-5. [DOI] [Google Scholar]
- Gibson D. H. The organometallic chemistry of carbon dioxide. Chem. Rev. 1996, 96 (6), 2063–2096. 10.1021/cr940212c. [DOI] [PubMed] [Google Scholar]
- Aresta M.; Nobile C. F.; Albano V. G.; Forni E.; Manassero M. New nickel-carbon dioxide complex: synthesis, properties, and crystallographic characterization of (carbon dioxide)-bis(tricyclohexylphosphine)nickel. J. Chem. Soc., Chem. Commun. 1975, 15, 636–637. 10.1039/C39750000636. [DOI] [Google Scholar]
- Dohring A.; Jolly P. W.; Kruger C.; Romão M. J. The Ni(0)-CO2 system: Structure and reactions of [Ni(PCy3)2(n2-CO2)]. Z. Naturforsch., B: J. Chem. Sci. 1985, 40B, 484–488. 10.1515/znb-1985-0408. [DOI] [Google Scholar]
- Mason M. G.; Ibers J. A. Reactivity of some transition metal systems toward liquid carbon dioxide. J. Am. Chem. Soc. 1982, 104 (19), 5153–5157. 10.1021/ja00383a026. [DOI] [Google Scholar]
- Aresta M.; Nobile C. F. (Carbon dioxide)bis(trialkylphosphine)nickel complexes. J. Chem. Soc., Dalton Trans. 1977, (7), 708–711. 10.1039/dt9770000708. [DOI] [Google Scholar]
- Hammouche M.; Lexa D.; Momenteau M.; Saveant J. M. Chemical catalysis of electrochemical reactions. Homogeneous catalysis of the electrochemical reduction of carbon dioxide by iron(″0″) porphyrins. Role of the addition of magnesium cations. J. Am. Chem. Soc. 1991, 113 (22), 8455–8466. 10.1021/ja00022a038. [DOI] [Google Scholar]
- Maher J. M.; Cooper N. J. Reduction of carbon dioxide to carbon monoxide by transition-metal dianions. J. Am. Chem. Soc. 1980, 102 (25), 7604–7606. 10.1021/ja00545a055. [DOI] [Google Scholar]
- Lee G. R.; Maher J. M.; Cooper N. J. Reductive disproportionation of carbon dioxide by dianionic carbonylmetalates of the transition metals. J. Am. Chem. Soc. 1987, 109 (10), 2956–2962. 10.1021/ja00244a017. [DOI] [Google Scholar]
- Chatt J.; Kubota M.; Leigh G. J.; March F. C.; Mason R.; Yarrow D. J. A possible carbon dioxide complex of molybdenum and its rearrangement product di-[small micro]-carbonato-bis{carbonyltris(dimethylphenylphosphine)molybdenum}: X-ray crystal structure. J. Chem. Soc., Chem. Commun. 1974, 24, 1033–1034. 10.1039/C39740001033. [DOI] [Google Scholar]
- Karsch H. H. Funktionelle trimethylphosphinderivate, III. Ambivalentes verhalten von tetrakis(trimethylphosphin)eisen: Reaktion mit CO2. Chem. Ber. 1977, 110 (6), 2213–2221. 10.1002/cber.19771100619. [DOI] [Google Scholar]
- Evans G. O.; Walter W. F.; Mills D. R.; Streit C. A. Reactions of carbon dioxide with metal carbonyl anions. J. Organomet. Chem. 1978, 144 (2), C34–C38. 10.1016/S0022-328X(00)84173-X. [DOI] [Google Scholar]
- Machan C. W.; Chabolla S. A.; Yin J.; Gilson M. K.; Tezcan F. A.; Kubiak C. P. Supramolecular assembly promotes the electrocatalytic reduction of carbon dioxide by Re(I) bipyridine catalysts at a lower overpotential. J. Am. Chem. Soc. 2014, 136 (41), 14598–14607. 10.1021/ja5085282. [DOI] [PubMed] [Google Scholar]
- Sullivan B. P.; Bolinger C. M.; Conrad D.; Vining W. J.; Meyer T. J. One- and two-electron pathways in the electrocatalytic reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine). J. Chem. Soc., Chem. Commun. 1985, 20, 1414–1416. 10.1039/C39850001414. [DOI] [Google Scholar]
- Bolinger C. M.; Story N.; Sullivan B. P.; Meyer T. J. Electrocatalytic reduction of carbon dioxide by 2,2’-bipyridine complexes of rhodium and iridium. Inorg. Chem. 1988, 27 (25), 4582–4587. 10.1021/ic00298a016. [DOI] [Google Scholar]
- Sampson M. D.; Froehlich J. D.; Smieja J. M.; Benson E. E.; Sharp I. D.; Kubiak C. P. Direct observation of the reduction of carbon dioxide by rhenium bipyridine catalysts. Energy Environ. Sci. 2013, 6 (12), 3748–3755. 10.1039/c3ee42186d. [DOI] [Google Scholar]
- Yang W.; Sinha Roy S.; Pitts W. C.; Nelson R. L.; Fronczek F. R.; Jurss J. W. Electrocatalytic CO2 reduction with Cis and Trans conformers of a rigid dinuclear rhenium complex: Comparing the monometallic and cooperative bimetallic pathways. Inorg. Chem. 2018, 57 (15), 9564–9575. 10.1021/acs.inorgchem.8b01775. [DOI] [PubMed] [Google Scholar]
- Parkin A.; Seravalli J.; Vincent K. A.; Ragsdale S. W.; Armstrong F. A. Rapid and efficient electrocatalytic CO2/CO interconversions by Carboxydothermus Hydrogenoformans CO dehydrogenase I on an electrode. J. Am. Chem. Soc. 2007, 129 (34), 10328–10329. 10.1021/ja073643o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung J.-H.; Dobbek H. Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 2007, 318 (5855), 1461–1464. 10.1126/science.1148481. [DOI] [PubMed] [Google Scholar]
- Can M.; Armstrong F. A.; Ragsdale S. W. Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and Acetyl-CoA synthase. Chem. Rev. 2014, 114 (8), 4149–4174. 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A. Bioinorganic modeling chemistry of carbon monoxide dehydrogenases: description of model complexes, current status and possible future scopes. Dalton Trans 2014, 43, 12135–12145. 10.1039/C4DT00729H. [DOI] [PubMed] [Google Scholar]
- Raebiger J. W.; Turner J. W.; Noll B. C.; Curtis C. J.; Miedaner A.; Cox B.; DuBois D. L. Electrochemical reduction of CO2 to CO catalyzed by a bimetallic palladium complex. Organometallics 2006, 25 (14), 3345–3351. 10.1021/om060228g. [DOI] [Google Scholar]
- Steffey B. D.; Curtis C. J.; DuBois D. L. Electrochemical reduction of CO2 catalyzed by a dinuclear palladium complex containing a bridging hexaphosphine ligand: Evidence for cooperativity. Organometallics 1995, 14 (10), 4937–4943. 10.1021/om00010a066. [DOI] [Google Scholar]
- Dubois D. L. Development of transition metal phosphine complexes as electrocatalysts for CO2 and CO reduction. Comments Inorg. Chem. 1997, 19 (5), 307–325. 10.1080/02603599708032743. [DOI] [Google Scholar]
- DuBois D. L.; Miedaner A.; Haltiwanger R. C. Electrochemical reduction of carbon dioxide catalyzed by [Pd(triphosphine)(solvent)](BF4)2 complexes: synthetic and mechanistic studies. J. Am. Chem. Soc. 1991, 113 (23), 8753–8764. 10.1021/ja00023a023. [DOI] [Google Scholar]
- Krogman J. P.; Foxman B. M.; Thomas C. M. Activation of CO2 by a heterobimetallic Zr/Co complex. J. Am. Chem. Soc. 2011, 133 (37), 14582–14585. 10.1021/ja2071847. [DOI] [PubMed] [Google Scholar]
- Fachinetti G.; Floriani C.; Zanazzi P. F. Bifunctional activation of carbon dioxide. Synthesis and structure of a reversible carbon dioxide carrier. J. Am. Chem. Soc. 1978, 100 (23), 7405–7407. 10.1021/ja00491a045. [DOI] [Google Scholar]
- Gambarotta S.; Arena F.; Floriani C.; Zanazzi P. F. Carbon dioxide fixation: bifunctional complexes containing acidic and basic sites working as reversible carriers. J. Am. Chem. Soc. 1982, 104 (19), 5082–5092. 10.1021/ja00383a015. [DOI] [Google Scholar]
- Cooper O.; Camp C.; Pécaut J.; Kefalidis C. E.; Maron L.; Gambarelli S.; Mazzanti M. Multimetallic cooperativity in uranium-mediated CO2 activation. J. Am. Chem. Soc. 2014, 136 (18), 6716–6723. 10.1021/ja5017624. [DOI] [PubMed] [Google Scholar]
- Lim C.-H.; Holder A. M.; Hynes J. T.; Musgrave C. B. Roles of the Lewis acid and base in the chemical reduction of CO2 catalyzed by frustrated Lewis pairs. Inorg. Chem. 2013, 52 (17), 10062–10066. 10.1021/ic4013729. [DOI] [PubMed] [Google Scholar]
- Hattori T.; Suzuki Y.; Miyano S. Lewis acid-mediated carboxylation of aryl- and allylsilanes with carbon dioxide. Chem. Lett. 2003, 32 (5), 454–455. 10.1246/cl.2003.454. [DOI] [Google Scholar]
- Menard G.; Stephan D. W. CO2 reduction via aluminum complexes of ammonia boranes. Dalton Trans 2013, 42 (15), 5447–5453. 10.1039/c3dt00098b. [DOI] [PubMed] [Google Scholar]
- Fachinetti G.; Floriani C.; Zanazzi P. F.; Zanzari A. R. Bifunctional model complexes active in carbon dioxide fixation: synthesis and x-ray structure of bimetallic cobalt(I)-alkali cation-Schiff base complexes. Inorg. Chem. 1979, 18 (12), 3469–3475. 10.1021/ic50202a035. [DOI] [Google Scholar]
- Bhugun I.; Lexa D.; Savéant J.-M. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins. Synergistic effect of Lewis acid cations. J. Phys. Chem. 1996, 100 (51), 19981–19985. 10.1021/jp9618486. [DOI] [Google Scholar]
- Fujita E.; Szalda D. J.; Creutz C.; Sutin N. Carbon dioxide activation: thermodynamics of carbon dioxide binding and the involvement of two cobalt centers in the reduction of carbon dioxide by a cobalt(I) macrocycle. J. Am. Chem. Soc. 1988, 110 (14), 4870–4871. 10.1021/ja00222a079. [DOI] [Google Scholar]
- Pinkes J. R.; Steffey B. D.; Vites J. C.; Cutler A. R. Carbon dioxide insertion into the iron-zirconium and ruthenium-zirconium bonds of the heterobimetallic complexes Cp(CO)2MZr(Cl)Cp2: direct production of the.mu.-.eta.1(C):.eta.2(O,O’)-CO2 compounds Cp(CO)2MCO2Zr(Cl)Cp2. Organometallics 1994, 13 (1), 21–23. 10.1021/om00013a009. [DOI] [Google Scholar]
- Fujita E.; Creutz C.; Sutin N.; Brunschwig B. S. Carbon dioxide activation by cobalt macrocycles: evidence of hydrogen bonding between bound CO2 and the macrocycle in solution. Inorg. Chem. 1993, 32 (12), 2657–2662. 10.1021/ic00064a015. [DOI] [Google Scholar]
- Costentin C.; Drouet S.; Robert M.; Savéant J.-M. A local proton source enhances CO2 electroreduction to CO by a molecular Fe catalyst. Science 2012, 338 (6103), 90–94. 10.1126/science.1224581. [DOI] [PubMed] [Google Scholar]
- Chapovetsky A.; Welborn M.; Luna J. M.; Haiges R.; Miller T. F.; Marinescu S. C. Pendant hydrogen-bond donors in cobalt catalysts independently enhance CO2 reduction. ACS Cent. Sci. 2018, 4 (3), 397–404. 10.1021/acscentsci.7b00607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcarate I.; Costentin C.; Robert M.; Savéant J.-M. Through-space charge interaction substituent effects in molecular catalysis leading to the design of the most efficient catalyst of CO2-to-CO electrochemical conversion. J. Am. Chem. Soc. 2016, 138 (51), 16639–16644. 10.1021/jacs.6b07014. [DOI] [PubMed] [Google Scholar]
- Sung S.; Kumar D.; Gil-Sepulcre M.; Nippe M. Electrocatalytic CO2 reduction by imidazolium-functionalized molecular catalysts. J. Am. Chem. Soc. 2017, 139 (40), 13993–13996. 10.1021/jacs.7b07709. [DOI] [PubMed] [Google Scholar]
- Froehlich J. D.; Kubiak C. P. Homogeneous CO2 reduction by Ni(cyclam) at a glassy carbon electrode. Inorg. Chem. 2012, 51 (7), 3932–3934. 10.1021/ic3001619. [DOI] [PubMed] [Google Scholar]
- Chapovetsky A.; Do T. H.; Haiges R.; Takase M. K.; Marinescu S. C. Proton-assisted reduction of CO2 by cobalt aminopyridine macrocycles. J. Am. Chem. Soc. 2016, 138 (18), 5765–5768. 10.1021/jacs.6b01980. [DOI] [PubMed] [Google Scholar]
- DeLuca E. E.; Xu Z.; Lam J.; Wolf M. O., Improved electrocatalytic CO2 reduction with palladium bis(NHC) pincer complexes bearing cationic side chains. Organometallics. 2018 10.1021/acs.organomet.8b00649. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.